putting the anion into the cage – fluoride inclusion in the smallest trisimidazolium macrotricycle
TRANSCRIPT

FULL PAPER
DOI: 10.1002/ejoc.201100902
Putting the Anion into the Cage – Fluoride Inclusion in the SmallestTrisimidazolium Macrotricycle
Valeria Amendola,[a] Massimo Boiocchi,[b] Luigi Fabbrizzi,*[a] and Nadia Fusco[a]
Keywords: Supramolecular chemistry / Anions / Receptors / Cage compounds / Inclusion compounds / Hydrogen bonds /Fluorides / Charge transfer
The trisbenzimidazolium cyclophane receptor 23+ incorpo-rates the F– anion in MeCN solution, which was inferred byspectrophotometric and 1H NMR titration experiments, withan association constant logK � 7. On the basis of geometricconsiderations, it is assumed that F– lies in the middle of thetriangle 3C, whose vertices are the carbon atoms of the threeimidazolium C–H fragments, and profits from three C–H···FH-bonds. No other anion is encapsulated by 23+ for reasonsof size. The parent trisbenzimidazolium tripodal receptor 43+
does not exert size exclusion selectivity and forms 1:1 com-plexes not only with F–, but also with other mono- (Br–) and
Introduction
The interaction of concave receptors with convex sub-strates is the basic process of molecular recognition.[1]
Whatever the nature of the interaction, selectivity mostlydepends upon size and shape complementarity between thereceptor and the substrate. Examples of size dependentselectivity refer to the interaction of alkali metal ions withcrown ethers[2] and cryptands in MeOH.[3] Cage-shapedcryptands provide higher affinity and selectivity in metalcomplexation with respect to crown ethers, due to their highdegree of preorganisation and rigidity. Preorganised, rigidreceptors are highly desired for anion recognition becausethe energy of the interaction (hydrogen bonding and/orelectrostatic) is intrinsically low and a cage-shaped systemcan afford additional stability to the receptor–anion com-plex.[4] It is not accidental that the first studies on anionrecognition involved positively-charged, cage-shaped recep-tors by Park and Simmons[5] (diammonium), Lehn[6] (tetra-ammonium) and Schmidtchen[7] (tetraalkylammonium). Avariety of cages of different cavity size have been reported,which are capable of H-bond donation from N–H frag-ments, either charged[8] (ammonium), neutral[9] (amide) or
[a] Dipartimento di Chimica Generale, Università di Pavia,Via Taramelli 12, 27100 Pavia, ItalyFax: +39-0382-528544E-mail: [email protected]
[b] Centro Grandi Strumenti, Università di Pavia,Via Bassi, 27100 Pavia, ItalySupporting information for this article is available on theWWW under http://dx.doi.org/10.1002/ejoc.201100902.
© 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2011, 6434–64446434
polyatomic anions (NO3–). X-ray diffraction studies on
[4···Br]2+ and [4···NO3]2+ complexes indicated that, due to thesteric restraints of the tripodal receptor, the anion is not posi-tioned in the middle of the 3C triangle but stays below it,profiting from H-bonds. Cage effects are observed in thehigher thermodynamic stability of the [2···F]2+ complex withrespect to [4···F]2+ and in its resistance to excess F–. In fact,on addition of excess fluoride, the tripodal [4···F]2+ complexdecomposes with deprotonation of a C–H fragment and for-mation of the very stable HF2
– complex.
from both N–H (ammonium) and C–H (pyridine) frag-ments.[10] As a general principle, a cage receptor providinga rigid spheroidal cavity can include or exclude sphericalanions (e.g. halides) simply on the basis of their radius (sizeexclusion selectivity).
In this context, an interesting case is presented by tris-imidazolium cyclophanes of the type 13+, in which threehighly polarised C–H fragments point towards the cavity,as documented by X-ray diffraction studies on crystallinesalts of 1a3+[11] and 1b3+.[12] Imidazolium is a subunit fre-quently used in the design of anion receptors because ofits ability as a H-bond donor and to establish electrostaticinteractions with the substrate.[13] A number of investi-gations have been carried out on the interaction of imid-azolium containing receptors with mono- and polyatomicanions in solution, and the equilibrium constants of thecomplexation equilibria have been determined through ti-tration experiments involving different spectroscopic tech-niques (UV/Vis, NMR, emission).[14] However, no system-atic studies have been reported on anion complexation by13+ cages in solution, which may have led to the conclusionthat the cage is too small to incorporate anions, even thesmallest: fluoride. In fact, in the structurally investigated13+ salts, the counterions were well outside the cavity, asexpected on the basis of their size (Br– for 1a3+,[11] Br– and[FeIIICl4]– for 1b3+).[12] However, the possibility of the in-clusion of an anion into a 13+ cage can be assessed throughsimple geometrical considerations.
In order to profit from three H-bond interactions, aspherical anion X– included into 13+ should be positioned
Fluoride Inclusion in the Smallest Trisimidazolium Macrotricycle
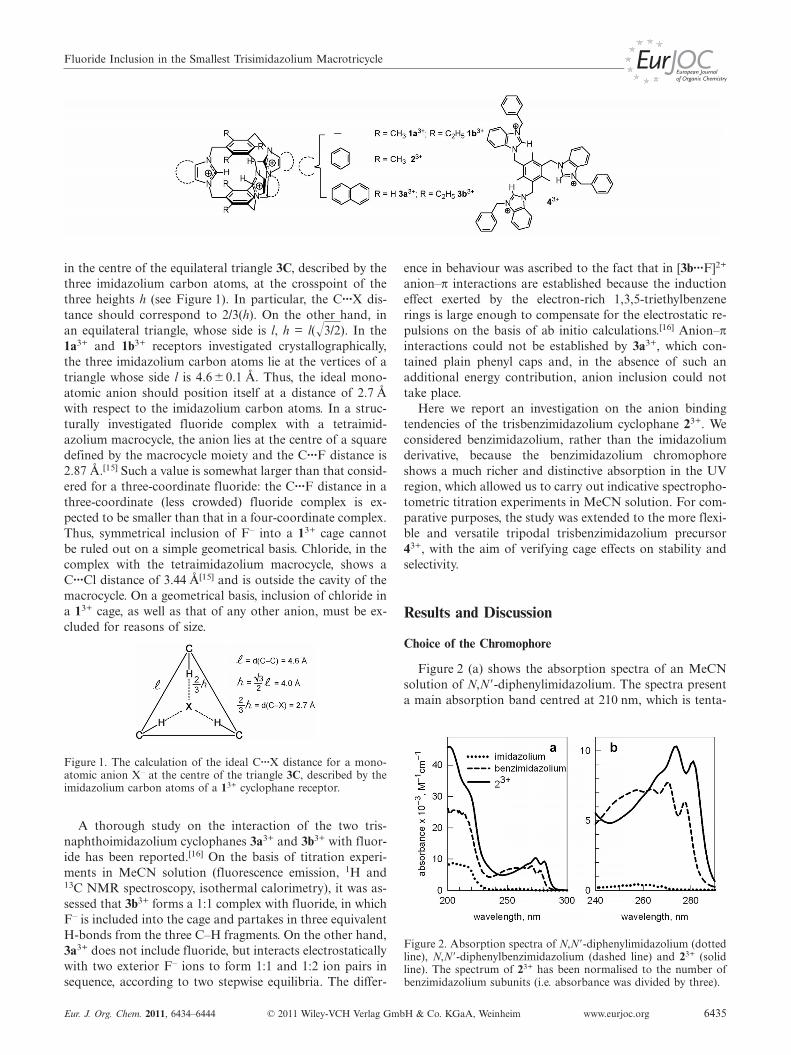
in the centre of the equilateral triangle 3C, described by thethree imidazolium carbon atoms, at the crosspoint of thethree heights h (see Figure 1). In particular, the C···X dis-tance should correspond to 2/3(h). On the other hand, inan equilateral triangle, whose side is l, h = l(�3/2). In the1a3+ and 1b3+ receptors investigated crystallographically,the three imidazolium carbon atoms lie at the vertices of atriangle whose side l is 4.6�0.1 Å. Thus, the ideal mono-atomic anion should position itself at a distance of 2.7 Åwith respect to the imidazolium carbon atoms. In a struc-turally investigated fluoride complex with a tetraimid-azolium macrocycle, the anion lies at the centre of a squaredefined by the macrocycle moiety and the C···F distance is2.87 Å.[15] Such a value is somewhat larger than that consid-ered for a three-coordinate fluoride: the C···F distance in athree-coordinate (less crowded) fluoride complex is ex-pected to be smaller than that in a four-coordinate complex.Thus, symmetrical inclusion of F– into a 13+ cage cannotbe ruled out on a simple geometrical basis. Chloride, in thecomplex with the tetraimidazolium macrocycle, shows aC···Cl distance of 3.44 Å[15] and is outside the cavity of themacrocycle. On a geometrical basis, inclusion of chloride ina 13+ cage, as well as that of any other anion, must be ex-cluded for reasons of size.
Figure 1. The calculation of the ideal C···X distance for a mono-atomic anion X– at the centre of the triangle 3C, described by theimidazolium carbon atoms of a 13+ cyclophane receptor.
A thorough study on the interaction of the two tris-naphthoimidazolium cyclophanes 3a3+ and 3b3+ with fluor-ide has been reported.[16] On the basis of titration experi-ments in MeCN solution (fluorescence emission, 1H and13C NMR spectroscopy, isothermal calorimetry), it was as-sessed that 3b3+ forms a 1:1 complex with fluoride, in whichF– is included into the cage and partakes in three equivalentH-bonds from the three C–H fragments. On the other hand,3a3+ does not include fluoride, but interacts electrostaticallywith two exterior F– ions to form 1:1 and 1:2 ion pairs insequence, according to two stepwise equilibria. The differ-
Eur. J. Org. Chem. 2011, 6434–6444 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 6435
ence in behaviour was ascribed to the fact that in [3b···F]2+
anion–π interactions are established because the inductioneffect exerted by the electron-rich 1,3,5-triethylbenzenerings is large enough to compensate for the electrostatic re-pulsions on the basis of ab initio calculations.[16] Anion–πinteractions could not be established by 3a3+, which con-tained plain phenyl caps and, in the absence of such anadditional energy contribution, anion inclusion could nottake place.
Here we report an investigation on the anion bindingtendencies of the trisbenzimidazolium cyclophane 23+. Weconsidered benzimidazolium, rather than the imidazoliumderivative, because the benzimidazolium chromophoreshows a much richer and distinctive absorption in the UVregion, which allowed us to carry out indicative spectropho-tometric titration experiments in MeCN solution. For com-parative purposes, the study was extended to the more flexi-ble and versatile tripodal trisbenzimidazolium precursor43+, with the aim of verifying cage effects on stability andselectivity.
Results and Discussion
Choice of the Chromophore
Figure 2 (a) shows the absorption spectra of an MeCNsolution of N,N�-diphenylimidazolium. The spectra presenta main absorption band centred at 210 nm, which is tenta-
Figure 2. Absorption spectra of N,N�-diphenylimidazolium (dottedline), N,N�-diphenylbenzimidazolium (dashed line) and 23+ (solidline). The spectrum of 23+ has been normalised to the number ofbenzimidazolium subunits (i.e. absorbance was divided by three).

V. Amendola, M. Boiocchi, L. Fabbrizzi, N. FuscoFULL PAPERtively assigned to the charge transfer transition hν1, illus-trated in Figure 3. A very weak absorption is observed ataround 260 nm.
Figure 3. Expected charge transfer transitions in imidazolium andbenzimidazolium. For each state, whether ground or excited, onlyone mesomeric form of two is shown (with the exception of theexcited state of imidazolium).
The spectrum of an MeCN solution of N,N�-diphenyl-benzimidazolium is shown as a dashed line in Figure 2 (a).It presents an intense band at ca. 210 nm, analogous to thatof the imidazolium derivative, and a strong absorption at270–280 nm, with a defined vibrational structure. The hν2
transition responsible for such an absorption, must involvethe fused phenyl ring, and is tentatively assigned to thecharge transfer process illustrated in Figure 3. Note that inthe excited state originated by hν1, a double positive chargeis delocalised over the imidazolium ring, whereas in the ex-cited state generated by hν2, the double negative charge isequally distributed over the N-heterocycle and the benzenering. The latter situation is more energetically favourable,which explains why hν2 takes place at a distinctly higherwavelength. The spectrum of 23+, solid lines in Figure 2 (aand b), is very similar to that of the reference chromophoreN,N�-dimethylbenzimidazolium, presenting bands originat-ing from the hν1 and hν2 transitions.
Figure 4. (a) Absorption spectra taken from the titration of an MeCN solution of [2](PF6)3 (2�10–4 m) with DBU; (b) lines: concentrationprofiles of the species present at the equilibrium over the course of the titration {% concentrations calculated with respect to the analyticalconcentration of [2](PF6)3}; symbols: titration profiles at selected wavelengths. Vertical axes on the right indicate the absorbances at 274and 293 nm in sequence; LH3
3+ = 23+.
www.eurjoc.org © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2011, 6434–64446436
Acid–Base Behaviour of 23+
Benzimidazolium is a weak Brønsted acid, which, on de-protonation of the C–H fragment, gives the correspondingcarbene (Scheme 1).
Scheme 1. The acidic dissociation equilibrium of a benzimidazol-ium fragment.
In order to ascertain the acidic behaviour of 23+ (LH33+),
a solution of 23+ in MeCN was titrated with the strong base1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, B). Figure 4 (a)shows the spectra obtained over the course of the titrationof a solution of [2](PF6)3 (2� 10–4 m) with a standard solu-tion of DBU.
It is observed that, on the addition of base, the typicalbands of the benzimidazolium subunit in the range 270–280 nm undergo a moderate blueshift and their intensityprogressively decreases. Moreover, a new band develops at300–310 nm. The best fit of the spectrophotometric ti-tration data over the 260–320 nm interval was obtained byassuming the occurrence of the proton transfer equilibriumto which the logK value of 4.25� 0.01 corresponded, seeEquation (1).
LH33+ + B p LH2
2+ + BH+ (1)
In Figure 4 (b), the concentration profiles of the two spe-cies LH3
3+ and LH22+ over the course of the titration are
reported as solid and dashed lines, respectively. Signifi-cantly, absorbance measured at selected wavelengths(274 nm, monitoring the disappearance of LH3
3+, and293 nm, monitoring the formation of the conjugated base
Fluoride Inclusion in the Smallest Trisimidazolium Macrotricycle
LH22+) satisfactorily superimpose on the concentration
profiles (see Figure 4, b).The neutralisation of 23+ with DBU was also investigated
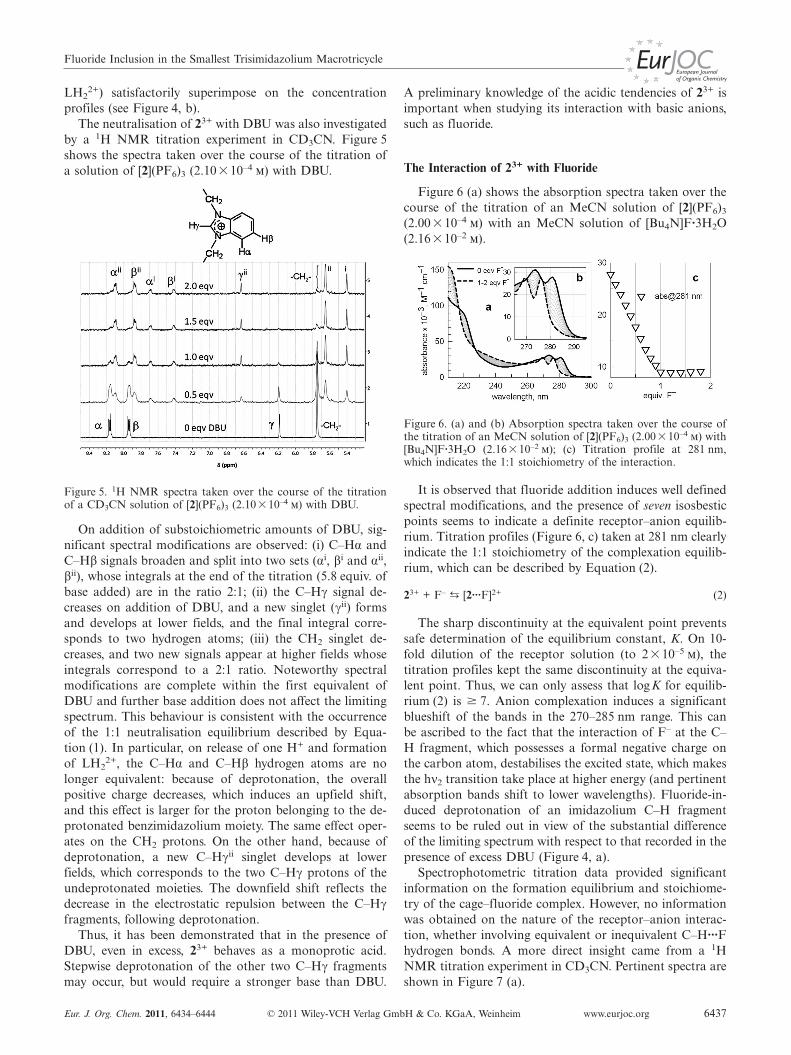
by a 1H NMR titration experiment in CD3CN. Figure 5shows the spectra taken over the course of the titration ofa solution of [2](PF6)3 (2.10�10–4 m) with DBU.
Figure 5. 1H NMR spectra taken over the course of the titrationof a CD3CN solution of [2](PF6)3 (2.10�10–4 m) with DBU.
On addition of substoichiometric amounts of DBU, sig-nificant spectral modifications are observed: (i) C–Hα andC–Hβ signals broaden and split into two sets (αi, βi and αii,βii), whose integrals at the end of the titration (5.8 equiv. ofbase added) are in the ratio 2:1; (ii) the C–Hγ signal de-creases on addition of DBU, and a new singlet (γii) formsand develops at lower fields, and the final integral corre-sponds to two hydrogen atoms; (iii) the CH2 singlet de-creases, and two new signals appear at higher fields whoseintegrals correspond to a 2:1 ratio. Noteworthy spectralmodifications are complete within the first equivalent ofDBU and further base addition does not affect the limitingspectrum. This behaviour is consistent with the occurrenceof the 1:1 neutralisation equilibrium described by Equa-tion (1). In particular, on release of one H+ and formationof LH2
2+, the C–Hα and C–Hβ hydrogen atoms are nolonger equivalent: because of deprotonation, the overallpositive charge decreases, which induces an upfield shift,and this effect is larger for the proton belonging to the de-protonated benzimidazolium moiety. The same effect oper-ates on the CH2 protons. On the other hand, because ofdeprotonation, a new C–Hγii singlet develops at lowerfields, which corresponds to the two C–Hγ protons of theundeprotonated moieties. The downfield shift reflects thedecrease in the electrostatic repulsion between the C–Hγfragments, following deprotonation.
Thus, it has been demonstrated that in the presence ofDBU, even in excess, 23+ behaves as a monoprotic acid.Stepwise deprotonation of the other two C–Hγ fragmentsmay occur, but would require a stronger base than DBU.
Eur. J. Org. Chem. 2011, 6434–6444 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 6437
A preliminary knowledge of the acidic tendencies of 23+ isimportant when studying its interaction with basic anions,such as fluoride.
The Interaction of 23+ with Fluoride
Figure 6 (a) shows the absorption spectra taken over thecourse of the titration of an MeCN solution of [2](PF6)3
(2.00 �10–4 m) with an MeCN solution of [Bu4N]F·3H2O(2.16�10–2 m).
Figure 6. (a) and (b) Absorption spectra taken over the course ofthe titration of an MeCN solution of [2](PF6)3 (2.00�10–4 m) with[Bu4N]F·3H2O (2.16�10–2 m); (c) Titration profile at 281 nm,which indicates the 1:1 stoichiometry of the interaction.
It is observed that fluoride addition induces well definedspectral modifications, and the presence of seven isosbesticpoints seems to indicate a definite receptor–anion equilib-rium. Titration profiles (Figure 6, c) taken at 281 nm clearlyindicate the 1:1 stoichiometry of the complexation equilib-rium, which can be described by Equation (2).
23+ + F–p [2···F]2+ (2)
The sharp discontinuity at the equivalent point preventssafe determination of the equilibrium constant, K. On 10-fold dilution of the receptor solution (to 2�10–5 m), thetitration profiles kept the same discontinuity at the equiva-lent point. Thus, we can only assess that logK for equilib-rium (2) is � 7. Anion complexation induces a significantblueshift of the bands in the 270–285 nm range. This canbe ascribed to the fact that the interaction of F– at the C–H fragment, which possesses a formal negative charge onthe carbon atom, destabilises the excited state, which makesthe hν2 transition take place at higher energy (and pertinentabsorption bands shift to lower wavelengths). Fluoride-in-duced deprotonation of an imidazolium C–H fragmentseems to be ruled out in view of the substantial differenceof the limiting spectrum with respect to that recorded in thepresence of excess DBU (Figure 4, a).
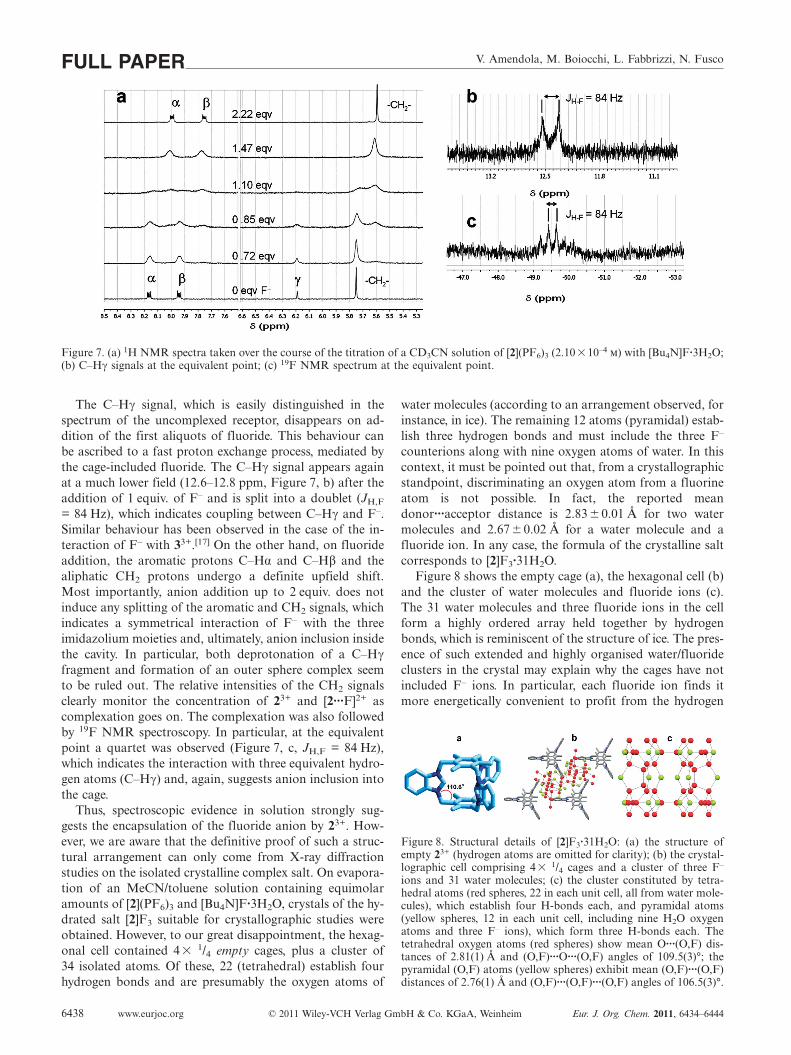
Spectrophotometric titration data provided significantinformation on the formation equilibrium and stoichiome-try of the cage–fluoride complex. However, no informationwas obtained on the nature of the receptor–anion interac-tion, whether involving equivalent or inequivalent C–H···Fhydrogen bonds. A more direct insight came from a 1HNMR titration experiment in CD3CN. Pertinent spectra areshown in Figure 7 (a).
V. Amendola, M. Boiocchi, L. Fabbrizzi, N. FuscoFULL PAPER
Figure 7. (a) 1H NMR spectra taken over the course of the titration of a CD3CN solution of [2](PF6)3 (2.10�10–4 m) with [Bu4N]F·3H2O;(b) C–Hγ signals at the equivalent point; (c) 19F NMR spectrum at the equivalent point.
The C–Hγ signal, which is easily distinguished in thespectrum of the uncomplexed receptor, disappears on ad-dition of the first aliquots of fluoride. This behaviour canbe ascribed to a fast proton exchange process, mediated bythe cage-included fluoride. The C–Hγ signal appears againat a much lower field (12.6–12.8 ppm, Figure 7, b) after theaddition of 1 equiv. of F– and is split into a doublet (JH,F
= 84 Hz), which indicates coupling between C–Hγ and F–.Similar behaviour has been observed in the case of the in-teraction of F– with 33+.[17] On the other hand, on fluorideaddition, the aromatic protons C–Hα and C–Hβ and thealiphatic CH2 protons undergo a definite upfield shift.Most importantly, anion addition up to 2 equiv. does notinduce any splitting of the aromatic and CH2 signals, whichindicates a symmetrical interaction of F– with the threeimidazolium moieties and, ultimately, anion inclusion insidethe cavity. In particular, both deprotonation of a C–Hγfragment and formation of an outer sphere complex seemto be ruled out. The relative intensities of the CH2 signalsclearly monitor the concentration of 23+ and [2···F]2+ ascomplexation goes on. The complexation was also followedby 19F NMR spectroscopy. In particular, at the equivalentpoint a quartet was observed (Figure 7, c, JH,F = 84 Hz),which indicates the interaction with three equivalent hydro-gen atoms (C–Hγ) and, again, suggests anion inclusion intothe cage.
Thus, spectroscopic evidence in solution strongly sug-gests the encapsulation of the fluoride anion by 23+. How-ever, we are aware that the definitive proof of such a struc-tural arrangement can only come from X-ray diffractionstudies on the isolated crystalline complex salt. On evapora-tion of an MeCN/toluene solution containing equimolaramounts of [2](PF6)3 and [Bu4N]F·3H2O, crystals of the hy-drated salt [2]F3 suitable for crystallographic studies wereobtained. However, to our great disappointment, the hexag-onal cell contained 4� 1/4 empty cages, plus a cluster of34 isolated atoms. Of these, 22 (tetrahedral) establish fourhydrogen bonds and are presumably the oxygen atoms of
www.eurjoc.org © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2011, 6434–64446438
water molecules (according to an arrangement observed, forinstance, in ice). The remaining 12 atoms (pyramidal) estab-lish three hydrogen bonds and must include the three F–
counterions along with nine oxygen atoms of water. In thiscontext, it must be pointed out that, from a crystallographicstandpoint, discriminating an oxygen atom from a fluorineatom is not possible. In fact, the reported meandonor···acceptor distance is 2.83 �0.01 Å for two watermolecules and 2.67 �0.02 Å for a water molecule and afluoride ion. In any case, the formula of the crystalline saltcorresponds to [2]F3·31H2O.
Figure 8 shows the empty cage (a), the hexagonal cell (b)and the cluster of water molecules and fluoride ions (c).The 31 water molecules and three fluoride ions in the cellform a highly ordered array held together by hydrogenbonds, which is reminiscent of the structure of ice. The pres-ence of such extended and highly organised water/fluorideclusters in the crystal may explain why the cages have notincluded F– ions. In particular, each fluoride ion finds itmore energetically convenient to profit from the hydrogen
Figure 8. Structural details of [2]F3·31H2O: (a) the structure ofempty 23+ (hydrogen atoms are omitted for clarity); (b) the crystal-lographic cell comprising 4� 1/4 cages and a cluster of three F–
ions and 31 water molecules; (c) the cluster constituted by tetra-hedral atoms (red spheres, 22 in each unit cell, all from water mole-cules), which establish four H-bonds each, and pyramidal atoms(yellow spheres, 12 in each unit cell, including nine H2O oxygenatoms and three F– ions), which form three H-bonds each. Thetetrahedral oxygen atoms (red spheres) show mean O···(O,F) dis-tances of 2.81(1) Å and (O,F)···O···(O,F) angles of 109.5(3)°; thepyramidal (O,F) atoms (yellow spheres) exhibit mean (O,F)···(O,F)distances of 2.76(1) Å and (O,F)···(O,F)···(O,F) angles of 106.5(3)°.

Fluoride Inclusion in the Smallest Trisimidazolium Macrotricycle
bonds donated by the water molecules of the cluster ratherthan from the three imidazolium C–H fragments inside thecage. In this context, it should be considered that, in anaqueous MeCN solution (MeCN/H2O, 80:20 v/v) of 23+,addition of even a large excess of F– does not induce anyspectral modification, thus ruling out fluoride inclusion. Itappears that crystallisation of the caged fluoride complexcould take place only under strictly anhydrous conditions.In any case, [Bu4N]F·3H2O as a source of fluoride shouldbe avoided.
Titration of an MeCN solution of [2](PF6)3 with anykind of anion (halides, inorganic oxoanions, carboxylates)as tetrabutylammonium salts, did not induce any significantmodification in the absorption and 1H NMR spectra, whichruled out the formation of an inclusion complex. Thus, 23+
exerts specific recognition of the fluoride ion, based on theprinciple of size exclusion.
In this context, it may be useful to compare the behav-iour of the trisbenzimidazolium cage 23+ with that of thetrisimidazolidinium cage 53+.[17] This cage selectively in-cludes F– in strongly acidic aqueous solution (pH = 1) withan extremely high association constant (logK = 12.5). Un-der these conditions, the two bridgehead tertiary amine ni-trogen atoms were protonated. The crystal structure of[5H2···F]4+ showed that F– receives two H-bonds from twoN–H fragments of the two tertiary ammonium groups in alinear coordination geometry. The three imidazolidiniumC–H fragments are not involved in H-bonding with theanion because they point outside the cavity. Note that thisorientation of imidazolidinium C–H fragments was ob-served in the uncomplexed, unprotonated 53+. These obser-vations provide a further validation for the high affinity of
Figure 9. (a) Absorption spectra taken over the course of the titration of an MeCN solution of [4](PF6)3 (2.00 �10–4 m) with [Bu4N]-F·3H2O; (b) lines: concentration profiles of the species present at the equilibrium over the course of the titration; symbols: titrationprofiles at selected wavelengths, which satisfactorily superimpose on the concentration profiles of the pertinent species (MH3
3+ = 43+).Vertical axes on the right indicate the absorbances at 284, 270 and 300 nm in sequence.
Eur. J. Org. Chem. 2011, 6434–6444 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 6439
23+ towards fluoride, whose imidazolium C–H fragmentspoint towards the centre of the cavity and emphasise therole of receptor preorganisation in molecular recognition.
Halide Interactions with the Tripodal Receptor 43+ –Fluoride and Chloride
For comparative purposes, the anion binding tendenciesof the parent tripodal receptor 43+ towards anions were in-vestigated through titration experiments in MeCN, follow-ing the procedure described above.
Figure 9 (a) shows spectra measured on addition of upto 4 equiv. of F–. The spectra show three clear isosbesticpoints until the addition of 1 equiv. of F–. Thereafter, theabsorbance increases continuously at 240 nm, and a broaddefined band, centred at 300 nm, forms and develops. Asobserved in the titration of 23+ with DBU (Figure 4, a),development of a band at 300 nm is indicative of the depro-tonation of the imidazolium C–H fragment. Interestingly,the titration profile at 300 nm, shown in part b of Figure 9,begins to increase just after the addition of 1 equiv. of fluo-ride. It is suggested that, on addition of the first equiv. ofF–, the 1:1 complex [4···F]2+ forms, in which the anion isexpected to interact with the three imidazolium C–H frag-ments in a symmetric triangular arrangement, as hypoth-esised for the cage complex, see Equation (3), MH3
3+ = 43+.However, the complex is not stable with respect to the ad-dition of further F–, and the deprotonation of one imid-azolium C–H fragment takes place, with the simultaneousformation of the hydrogendifluoride anion, according toEquation (4).
MH33+ + F–
p [MH3···F]2+ (3)
log K1 = 5.75�0.02
[MH3···F]2+ + F–p MH2
2+ + HF2– (4)
log K2 = 4.01 �0.03
The fitting of spectroscopic data over the 260–320 nminterval, using a nonlinear least-squares procedure,[18] wasobtained by assuming the occurrence of stepwise equilibria
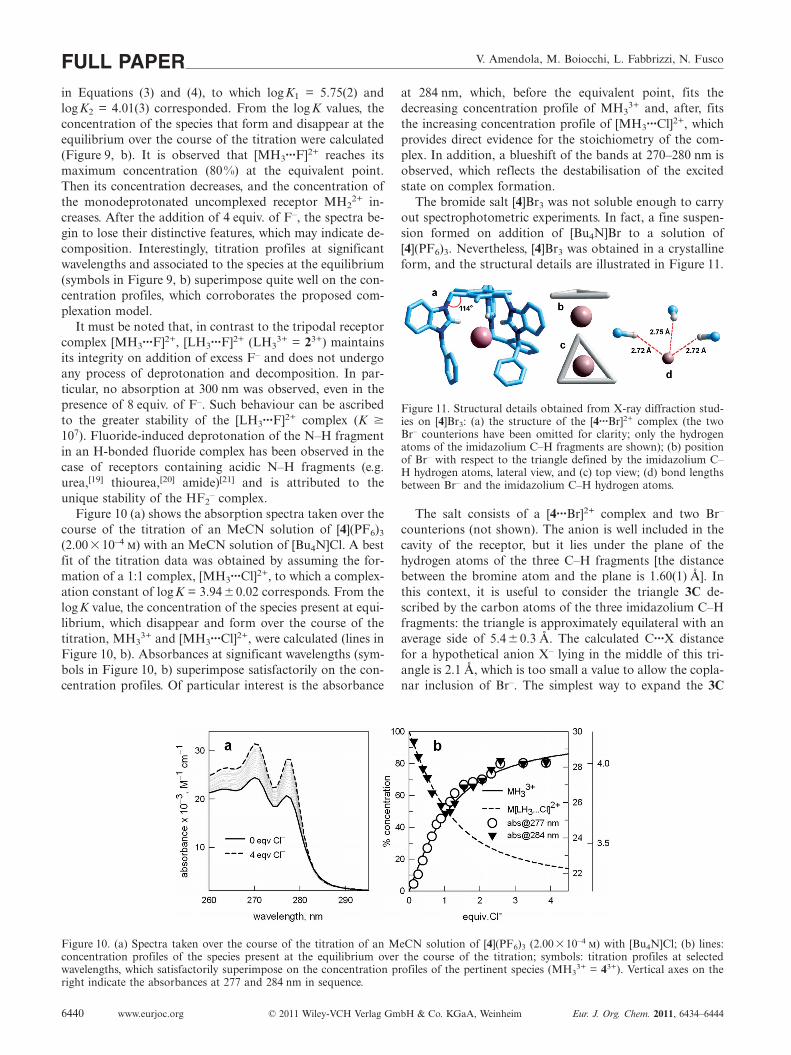
V. Amendola, M. Boiocchi, L. Fabbrizzi, N. FuscoFULL PAPERin Equations (3) and (4), to which logK1 = 5.75(2) andlogK2 = 4.01(3) corresponded. From the log K values, theconcentration of the species that form and disappear at theequilibrium over the course of the titration were calculated(Figure 9, b). It is observed that [MH3···F]2+ reaches itsmaximum concentration (80%) at the equivalent point.Then its concentration decreases, and the concentration ofthe monodeprotonated uncomplexed receptor MH2
2+ in-creases. After the addition of 4 equiv. of F–, the spectra be-gin to lose their distinctive features, which may indicate de-composition. Interestingly, titration profiles at significantwavelengths and associated to the species at the equilibrium(symbols in Figure 9, b) superimpose quite well on the con-centration profiles, which corroborates the proposed com-plexation model.
It must be noted that, in contrast to the tripodal receptorcomplex [MH3···F]2+, [LH3···F]2+ (LH3
3+ = 23+) maintainsits integrity on addition of excess F– and does not undergoany process of deprotonation and decomposition. In par-ticular, no absorption at 300 nm was observed, even in thepresence of 8 equiv. of F–. Such behaviour can be ascribedto the greater stability of the [LH3···F]2+ complex (K �107). Fluoride-induced deprotonation of the N–H fragmentin an H-bonded fluoride complex has been observed in thecase of receptors containing acidic N–H fragments (e.g.urea,[19] thiourea,[20] amide)[21] and is attributed to theunique stability of the HF2
– complex.Figure 10 (a) shows the absorption spectra taken over the
course of the titration of an MeCN solution of [4](PF6)3
(2.00� 10–4 m) with an MeCN solution of [Bu4N]Cl. A bestfit of the titration data was obtained by assuming the for-mation of a 1:1 complex, [MH3···Cl]2+, to which a complex-ation constant of logK = 3.94 �0.02 corresponds. From thelogK value, the concentration of the species present at equi-librium, which disappear and form over the course of thetitration, MH3
3+ and [MH3···Cl]2+, were calculated (lines inFigure 10, b). Absorbances at significant wavelengths (sym-bols in Figure 10, b) superimpose satisfactorily on the con-centration profiles. Of particular interest is the absorbance
Figure 10. (a) Spectra taken over the course of the titration of an MeCN solution of [4](PF6)3 (2.00 �10–4 m) with [Bu4N]Cl; (b) lines:concentration profiles of the species present at the equilibrium over the course of the titration; symbols: titration profiles at selectedwavelengths, which satisfactorily superimpose on the concentration profiles of the pertinent species (MH3
3+ = 43+). Vertical axes on theright indicate the absorbances at 277 and 284 nm in sequence.
www.eurjoc.org © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2011, 6434–64446440
at 284 nm, which, before the equivalent point, fits thedecreasing concentration profile of MH3
3+ and, after, fitsthe increasing concentration profile of [MH3···Cl]2+, whichprovides direct evidence for the stoichiometry of the com-plex. In addition, a blueshift of the bands at 270–280 nm isobserved, which reflects the destabilisation of the excitedstate on complex formation.
The bromide salt [4]Br3 was not soluble enough to carryout spectrophotometric experiments. In fact, a fine suspen-sion formed on addition of [Bu4N]Br to a solution of[4](PF6)3. Nevertheless, [4]Br3 was obtained in a crystallineform, and the structural details are illustrated in Figure 11.
Figure 11. Structural details obtained from X-ray diffraction stud-ies on [4]Br3: (a) the structure of the [4···Br]2+ complex (the twoBr– counterions have been omitted for clarity; only the hydrogenatoms of the imidazolium C–H fragments are shown); (b) positionof Br– with respect to the triangle defined by the imidazolium C–H hydrogen atoms, lateral view, and (c) top view; (d) bond lengthsbetween Br– and the imidazolium C–H hydrogen atoms.
The salt consists of a [4···Br]2+ complex and two Br–
counterions (not shown). The anion is well included in thecavity of the receptor, but it lies under the plane of thehydrogen atoms of the three C–H fragments [the distancebetween the bromine atom and the plane is 1.60(1) Å]. Inthis context, it is useful to consider the triangle 3C de-scribed by the carbon atoms of the three imidazolium C–Hfragments: the triangle is approximately equilateral with anaverage side of 5.4�0.3 Å. The calculated C···X distancefor a hypothetical anion X– lying in the middle of this tri-angle is 2.1 Å, which is too small a value to allow the copla-nar inclusion of Br–. The simplest way to expand the 3C
Fluoride Inclusion in the Smallest Trisimidazolium Macrotricycle
triangle involves increasing the C–C–N angle of the CH2
spacer linking the 1,3,5-trimethylbenzene cap and the imid-azolium subunit. The CH2 carbon atom has an sp3 natureand is prepared to form bond angles of 109.5°. This sizecan be expanded within reasonable limits for steric reasons.In the bromide complex, this angle falls in the range112.3(7)–114.1(6)°, which may represent the impassableboundary in the present ligating framework. Note that inthe highly symmetric empty cage 23+ (Figure 6, a) all six C–C–N angles have a size of 110.6(6)°, which generate a regu-lar equilateral triangle 3C of side 4.54 Å. Thus, for reasonsof size and steric constraints of the receptor, Br– cannotplace itself in the middle of the 3C triangle but must staybelow it to profit from the H-bonds. The C···Cl distanceobserved in the imidazolium complexes is 4.44 Å, whichmakes the coplanar inclusion of chloride difficult.
No significant spectral modifications were observed onaddition of [Bu4N]I to a MeCN solution of [4](PF6)3
(2 �10–4 m).
Oxoanion Interactions with 43+ – Acetate and Nitrate
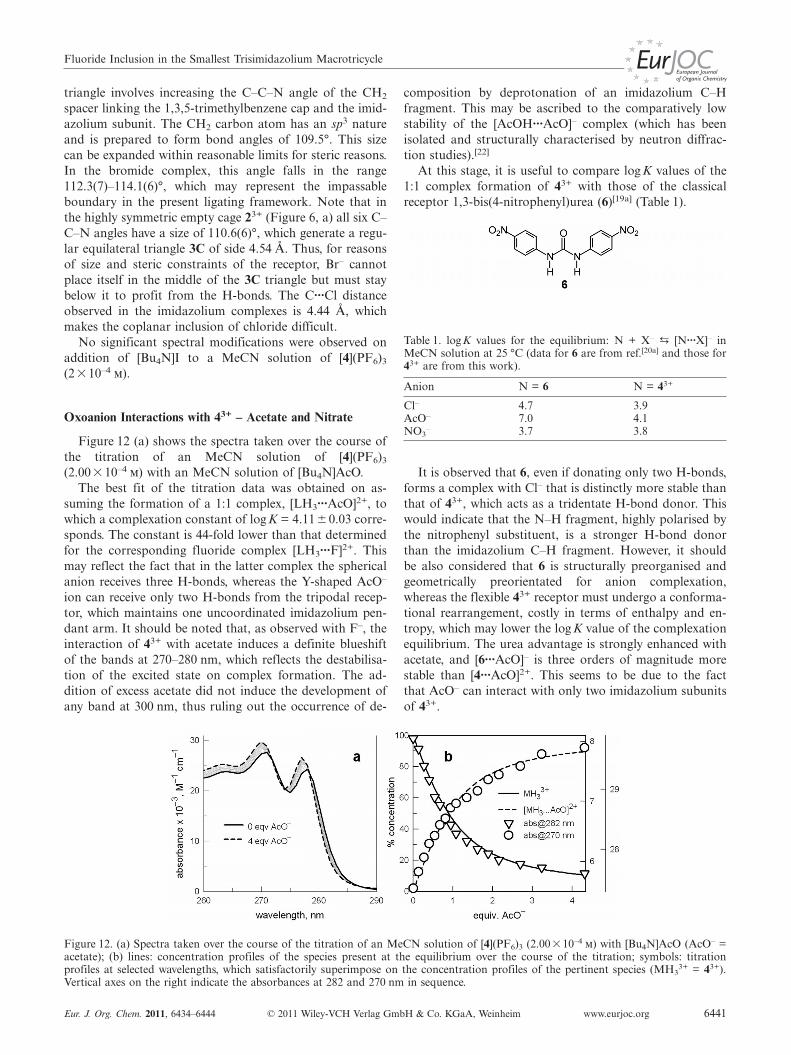
Figure 12 (a) shows the spectra taken over the course ofthe titration of an MeCN solution of [4](PF6)3
(2.00� 10–4 m) with an MeCN solution of [Bu4N]AcO.The best fit of the titration data was obtained on as-
suming the formation of a 1:1 complex, [LH3···AcO]2+, towhich a complexation constant of log K = 4.11�0.03 corre-sponds. The constant is 44-fold lower than that determinedfor the corresponding fluoride complex [LH3···F]2+. Thismay reflect the fact that in the latter complex the sphericalanion receives three H-bonds, whereas the Y-shaped AcO–
ion can receive only two H-bonds from the tripodal recep-tor, which maintains one uncoordinated imidazolium pen-dant arm. It should be noted that, as observed with F–, theinteraction of 43+ with acetate induces a definite blueshiftof the bands at 270–280 nm, which reflects the destabilisa-tion of the excited state on complex formation. The ad-dition of excess acetate did not induce the development ofany band at 300 nm, thus ruling out the occurrence of de-
Figure 12. (a) Spectra taken over the course of the titration of an MeCN solution of [4](PF6)3 (2.00�10–4 m) with [Bu4N]AcO (AcO– =acetate); (b) lines: concentration profiles of the species present at the equilibrium over the course of the titration; symbols: titrationprofiles at selected wavelengths, which satisfactorily superimpose on the concentration profiles of the pertinent species (MH3
3+ = 43+).Vertical axes on the right indicate the absorbances at 282 and 270 nm in sequence.
Eur. J. Org. Chem. 2011, 6434–6444 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 6441
composition by deprotonation of an imidazolium C–Hfragment. This may be ascribed to the comparatively lowstability of the [AcOH···AcO]– complex (which has beenisolated and structurally characterised by neutron diffrac-tion studies).[22]
At this stage, it is useful to compare log K values of the1:1 complex formation of 43+ with those of the classicalreceptor 1,3-bis(4-nitrophenyl)urea (6)[19a] (Table 1).
Table 1. logK values for the equilibrium: N + X–p [N···X]– in
MeCN solution at 25 °C (data for 6 are from ref.[20a] and those for43+ are from this work).
Anion N = 6 N = 43+
Cl– 4.7 3.9AcO– 7.0 4.1NO3
– 3.7 3.8
It is observed that 6, even if donating only two H-bonds,forms a complex with Cl– that is distinctly more stable thanthat of 43+, which acts as a tridentate H-bond donor. Thiswould indicate that the N–H fragment, highly polarised bythe nitrophenyl substituent, is a stronger H-bond donorthan the imidazolium C–H fragment. However, it shouldbe also considered that 6 is structurally preorganised andgeometrically preorientated for anion complexation,whereas the flexible 43+ receptor must undergo a conforma-tional rearrangement, costly in terms of enthalpy and en-tropy, which may lower the log K value of the complexationequilibrium. The urea advantage is strongly enhanced withacetate, and [6···AcO]– is three orders of magnitude morestable than [4···AcO]2+. This seems to be due to the factthat AcO– can interact with only two imidazolium subunitsof 43+.

V. Amendola, M. Boiocchi, L. Fabbrizzi, N. FuscoFULL PAPER
Figure 13. (a) Absorption spectra taken over the course of the titration of an MeCN solution of [4](PF6)3 (2.00�10–4 m) with [Bu4N]NO3; (b) titration profiles at selected wavelengths.
On the other hand, the triangular NO3– ion is expected
to form a stable complex with receptor 43+, which possessestrigonal symmetry. Figure 13 (a) shows the spectra recordedin the titration of an MeCN solutions of [4](PF6)3 with[Bu4N]NO3.
On anion addition, the bands at 270–280 nm undergo thetypical blueshift and their intensity smoothly increases toreach saturation (see the profile at 277 nm in Figure 13, b).On the other hand, the profile at 284 nm is interesting inthat it defines the 1:1 stoichiometry of the process of com-plex formation. On nonlinear least-squares treatment of ti-tration data over the wavelength interval 260–290 nm, alogK value of 3.77� 0.04 for the 1:1 complexation equilib-rium was obtained. Note that this value is the same as thatobserved with the corresponding complex of 6 (Table 1)fully compensating for the urea advantage, which suggeststhat NO3
– receives three H-bonds from 43+.Clear cut information on the bonding mode in the
[4···NO3]2+ complex came from X-ray diffraction studies on[4](NO3)(PF6)2·MeCN, which was obtained from evapora-tion of an MeCN solution containing equimolar amountsof [4](PF6)3 and [Bu4N]NO3. Figure 14 (a) shows the struc-ture of [4···NO3]2+. The nitrate ion is well included in cavityof the receptor, but, like Br–, it lies below the plane definedby the hydrogen atoms of the three imidazolium C–H frag-ments [Figure 14, b; the distance between the nitrogen atom
Figure 14. Structural details obtained through X-ray diffractionstudies on [4](NO3)(PF6)2·MeCN: (a) the structure of [4···NO3]2+
(PF6– counterions and the MeCN molecule have been omitted for
clarity; only the hydrogen atoms of the imidazolium C–H frag-ments are shown); (b) position of NO3
– with respect to the triangledefined by imidazolium C–H hydrogen atoms, lateral view, and (c)top view; (d) bond lengths between NO3
– oxygen atoms and imid-azolium C–H hydrogen atoms: each oxygen atom establishes a bi-furcated H-bond interaction.
www.eurjoc.org © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2011, 6434–64446442
of NO3– and the centroid of the triangle whose vertices are
C–H hydrogen atoms is 1.28(1) Å].Moreover, the NO3
– ion is staggered with respect to thethree C–H hydrogen atoms (Figure 14, c) and each nitrateoxygen atom does not interact with a single C–H fragment,but establishes bifurcated H-bond interactions with two C–H fragments (Figure 14, d). The bifurcated interactions ac-count for the comparatively high stability of the[4···NO3]2+ complex. These interactions, either six bifur-cated or three collinear, would be certainly more intense ifNO3
– lay in the plane of the three C–H fragments. However,such a structural arrangement is prevented by the same geo-metric restrictions experienced by Br– in [4···Br]2+. The sideof the 3C triangle in [4···NO3]2+ is 5.5�0.3 Å and the C–C–N angles of the CH2 spacers fall in the range 113.2(3)–114.0(3)°, which again appears to be the impassable limitfor an aliphatic carbon atom.
Thus, the structural investigation of [4···Br]2+ and[4···NO3]2+ has shown that the supposed flexible and adapt-able trisbenzimidazolium tripodal receptor 43+ presents itsown steric constraints. Nevertheless, it does not exert anydrastic size exclusion selectivity, like cage 23+, because it canaccommodate anions of varying size and shape in its bowl-shaped cavity by modulating the distance between the anionand the 3C triangle.
Conclusions
Spectrophotometric and 1H NMR titration experimentsin MeCN have provided evidence for the encapsulation offluoride by 23+. Anion inclusion could not be supported byX-ray diffraction studies, probably because crystallisationof the [2]F3 salt took place in the presence of water, and acluster of 31 water molecules offered the three F– counteri-ons an alternative, more comfortable shelter. Geometricconsiderations suggest that the included fluoride should liein the middle of the equilateral triangle 3C, whose verticesare the carbon atoms of the three imidazolium C–H frag-ments. The parent tripodal derivative 43+ includes F– inMeCN to give a stable complex, in which the anion proba-bly lies in the middle of the 3C triangle and receives threecoplanar H-bonds from the imidazolium C–H fragments,

Fluoride Inclusion in the Smallest Trisimidazolium Macrotricycle
as hypothesised for the corresponding cage complex. Thistype of coordination is restricted to fluoride, because largeranions such as Br– and NO3
– cannot be accommodated ina coplanar fashion in the 3C triangle, whose size cannot beexpanded at will, and is limited by the sp3 nature of thecarbon atoms of the CH2 spacers linking each benzimidaz-olium subunit to the 1,3,5-trialkyl cap. Thus, Br– and NO3
–
stay below the 3C triangle and profit from H-bonds. Cage23+ is a totally specific receptor for fluoride and puts theprinciple of size exclusion into practice. A thermodynamiccage effect exists because the [2···F]2+ complex (logK � 7)is more stable than the [4···F]2+ tripodal complex (logK =5.75), which probably reflects the high degree of preorganis-ation of 23+. Moreover, whereas the cage complex [2···F]2+
is stable in the presence of excess fluoride, the tripodal com-plex [4···F]2+, on addition of further F–, decomposes due tothe deprotonation of an imidazolium C–H fragment andthe formation of the very stable [HF2]– H-bonded complex.
It remains to compare the results of this study on 23+
(containing two 1,3,5-trimethylbenzene caps) with those re-ported on 3a3+ (benzene caps) and 3b3+ (1,3,5-triethylben-zene).[15] Spring, Kim and Yoon suggested that 3b3+ in-cludes F– by profiting from anion–π interactions with the1,3,5-triethylbenzene caps. Such interactions are establishedbecause the induction effect exerted by the electron-rich1,3,5-triethylbenzene rings is large enough to compensatefor the electrostatic repulsions. These π interactions cannotbe established by the electron-poor benzene caps of 3a3+,which prevents anion inclusion. The 1,3,5-trimethyl caps in23+ seem to be electron rich enough π donors to compensatefor electrostatic repulsions to afford fluoride inclusion. Inany case, steric effects and geometric restrictions remain de-termining factors. Size exclusion specificity is a rare andintriguing phenomenon in molecular recognition. Fluoride,the smallest stable anion, is the perfect guest to challengethe synthetic skill of receptor designers.
Experimental SectionGeneral: Methods and procedures, syntheses and characterisationsof receptors 23+ and 43+, crystal structure determinations of[2]F3·31H2O, [4](NO3)(PF6)2·0.5MeCN and [4]Br3·3H2O are de-scribed in detail in the Supporting Information.
CCDC-818804 (for [2]F3·31H2O), -818805 (for [4](NO3)(PF6)2·0.5MeCN) and -818806 (for [4]Br3·3H2O) contain the supplemen-tary crystallographic data for this paper. These data can be ob-tained free of charge from The Cambridge Crystallographic DataCentre via www.ccdc.cam.ac.uk/data_request/cif.
Supporting Information (see footnote on the first page of this arti-cle): Details on (i) general procedures; (ii) synthesis and characteri-sation of 23+ and 43+; (ii) details on crystallographic studies on[2]F3·31H2O, [4](NO3)(PF6)2·0.5MeCN and [4]Br3·3H2O.
Acknowledgments
The financial support by the Italian Ministry of University andResearch (PRIN-Dispositivi Supramolecolari) is gratefully ack-nowledged.
Eur. J. Org. Chem. 2011, 6434–6444 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 6443
[1] J.-M. Lehn, Supramolecular Chemistry, Concepts and Perspec-tives, VCH, Weinheim, 1995.
[2] a) C. J. Pedersen, J. Am. Chem. Soc. 1967, 89, 7017–7036; b)H. K. Frensdorff, J. Am. Chem. Soc. 1971, 93, 600–606.
[3] a) B. Dietrich, J.-M. Lehn, J.-P. Sauvage, Tetrahedron Lett.1969, 10, 2889–2892; b) J.-M. Lehn, J.-P. Sauvage, J. Am.Chem. Soc. 1975, 97, 6700–6707.
[4] F. P. Schmidtchen, M. Berger, Chem. Rev. 1997, 97, 1609–1646.[5] C. H. Park, H. E. Simmons, J. Am. Chem. Soc. 1968, 90, 2431–
2432.[6] E. Graf, J.-M. Lehn, J. Am. Chem. Soc. 1976, 98, 6403–6405.[7] F. P. Schmidtchen, Angew. Chem. 1977, 89, 751; Angew. Chem.
Int. Ed. Engl. 1977, 16, 720–721.[8] a) J.-M. Lehn, E. Sonveaux, A. K. Willard, J. Am. Chem. Soc.
1978, 100, 4914–4916; b) J.-M. Lehn, R. Méric, J.-P. Vigneron,I. Bkouche-Waksman, C. Pascard, J. Chem. Soc., Chem. Com-mun. 1991, 62–64.
[9] a) A. P. Bisson, V. M. Lynch, M.-C. C. Monahan, E. V. Anslyn,Angew. Chem. 1997, 109, 2435; Angew. Chem. Int. Ed. Engl.1997, 36, 2340–2342; b) S. O. Kang, D. Powell, V. W. Day, K.Bowman-James, Angew. Chem. 2006, 118, 1955; Angew. Chem.Int. Ed. 2006, 45, 1921–1925.
[10] C. A. Ilioudis, D. A. Tocher, J. W. Steed, J. Am. Chem. Soc.2004, 126, 12395–12402.
[11] M. V. Baker, M. J. Bosnich, C. C. Williams, B. W. Skelton,A. H. White, Aust. J. Chem. 1999, 52, 823–826. The crystalstructure of the same salt has been reported also by: Y. Yuan,Z.-L. Jiang, J.-M. Yan, G. Gao, A. S. C. Chan, R.-G. Xie,Synth. Commun. 2000, 30, 4555–4561.
[12] C. E. Willans, K. M. Anderson, P. C. Junk, L. J. Barbour, J.Steed, Chem. Commun. 2007, 3634–3636: [1b][FeIIICl4]Br
[13] a) J. Yoon, S. K. Kim, N. J. Singh, K. S. Kim, Chem. Soc. Rev.2006, 35, 355–360; b) Z. Xu, S. K. Kim, J. Yoon, Chem. Soc.Rev. 2010, 39, 1457–1466.
[14] a) K. Sato, S. Arai, T. Yamagishi, Tetrahedron Lett. 1999, 40,5219–5222; b) J. Howarth, N. A. Al-Hashimy, TetrahedronLett. 2001, 42, 5777–5779; c) H. Ihm, S. Yun, H. G. Kim, J. K.Kim, K. S. Kim, Org. Lett. 2002, 4, 2897–2900; d) S. Yun, H.Ihm, H. G. Kim, C.-W. Lee, B. Indrajit, K. S. Oh, Y. J. Gong,J. W. Lee, J. Yoon, H. C. Lee, K. S. Kim, J. Org. Chem. 2003,68, 2467–2470; e) Y. Bai, B.-G. Zhang, J. Xu, C.-Y. Duan, D.-B. Dang, D.-J. Liu, Q.-J. Meng, New J. Chem. 2005, 29, 777–779; f) D.-B. Qin, F.-B. Xu, X.-J. Wan, Y.-J. Zhao, Z.-Z. Zhang,Tetrahedron Lett. 2006, 47, 5641–5643; g) S. K. Kim, N. J.Singh, J. Kwon, I.-C. Hwang, S. J. Park, K. S. Kim, J. Yoon,Tetrahedron 2006, 62, 6065–6072; h) V. Amendola, M. Boioc-chi, B. Colasson, L. Fabbrizzi, M.-J. Douton-Rodriguez, F.Ugozzoli, Angew. Chem. 2006, 118, 7074; Angew. Chem. Int.Ed. 2006, 45, 6920–6924; i) Z. Xu, S. Kim, K.-H. Lee, J. Yoon,Tetrahedron Lett. 2007, 48, 3797–3800; j) N. J. Singh, E. J. Jun,K. Chellappan, D. Thangadurai, R. P. Chandran, I.-C. Hwang,J. Yoon, K. S. Kim, Org. Lett. 2007, 9, 485–488; k) N. Singh,D. O. Jang, Org. Lett. 2007, 9, 1991–1994; l) N. Al-Hashimy,D. J. Brougham, J. Howarth, A. Farrell, Brid Quilty, K. Nolan,Tetrahedron Lett. 2007, 48, 125–128; m) B. Wannalerse, T. Tun-tulani, B. Tomapatanaget, Tetrahedron 2008, 64, 10619–10624;n) S. K. Kim, D. Seo, S. J. Han, G. Son, I.-J. Lee, C. Lee, K. D.Lee, J. Yoon, Tetrahedron 2008, 64, 6402–6405; o) V.Amendola, M. Boiocchi, B. Colasson, L. Fabbrizzi, E. Mon-zani, M.-J. Douton-Rodriguez, C. Spadini, Inorg. Chem. 2008,47, 4808–4816; p) K. Ghosh, I. Saha, A. Patra, TetrahedronLett. 2009, 50, 2392–2397; q) S. Kumar, S. Kumar, TetrahedronLett. 2009, 50, 4463–4466; r) S. H. Mashraqui, R. Betkara, M.Chandiramania, D. Quinonero, A. Frontera, Tetrahedron Lett.2010, 51, 596–599.
[15] K. Chellappan, N. J. Singh, I.-C. Hwang, J. W. Lee, K. S. Kim,Angew. Chem. 2005, 117, 2959; Angew. Chem. Int. Ed. 2005,44, 2899–2903.

V. Amendola, M. Boiocchi, L. Fabbrizzi, N. FuscoFULL PAPER[16] Z. Xu, N. J. Singh, S. K. Kim, D. R. Spring, K. S. Kim, J.
Yoon, Chem. Eur. J. 2011, 17, 1163–1170.[17] B. Zhang, P. Cai, C. Duan, R. Miao, L. Zhu, T. Niitsu, H.
Inoue, Chem. Commun. 2004, 2206–2207.[18] The Hyperquad® package was used, see: P. Gans, A. Sabatini,
A. Vacca, Talanta 1996, 43, 1739–1753; http://www.hyperquad.co.uk/index.htm; accessed 27th April 2011.
[19] a) M. Boiocchi, L. Del Boca, D. Esteban-Gómez, L. Fabbrizzi,M. Licchelli, E. Monzani, J. Am. Chem. Soc. 2004, 126, 16507–16514; b) M. Boiocchi, L. Del Boca, D. Esteban-Gómez, L.Fabbrizzi, M. Licchelli, E. Monzani, Chem. Eur. J. 2005, 11,3097–3104; c) D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli, J.Org. Chem. 2005, 70, 5717–5720; d) V. Amendola, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli, Acc. Chem. Res. 2006, 39,
www.eurjoc.org © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2011, 6434–64446444
343–353; e) V. Amendola, L. Fabbrizzi, L. Mosca, Chem. Soc.Rev. 2010, 39, 3889–3915.
[20] a) D. Esteban Gómez, L. Fabbrizzi, M. Licchelli, E. Monzani,Org. Biomol. Chem. 2005, 3, 1495–1500; b) M. Bonizzoni, L.Fabbrizzi, A. Taglietti, F. Tiengo, Eur. J. Org. Chem. 2006,3567–3574.
[21] a) S. Camiolo, P. A. Gale, M. B. Hursthouse, M. E. Light, A. J.Shi, Chem. Commun. 2002, 758–759; b) T. Gunnlaugsson, P. E.Kruger, P. Jensen, F. M. Pfeffer, G. M. Hussey, TetrahedronLett. 2003, 44, 8909–8913.
[22] M. J. Barrow, M. Currie, K. W. Muir, J. C. Speakman, D. N. J.White, J. Chem. Soc. Perkin Trans. 2 1975, 15–18.
Received: June 20, 2011Published Online: September 14, 2011