q. julia aguilar pliego, para la obtencion …148.206.53.84/tesiuami/uam7484.pdf · determinadas...
TRANSCRIPT

"" I
'c
TESIS QUE
HIDROCRACKING DE Pt/ZEOLITA
PRESENTA LA
EN Y "
'Q. JULIA AGUILAR PLIEGO, +
PARA LA OBTENCION DEL GRADO DE
"AESTRO EN QUIMICA
Noviembre, 1985 / ."

i O Q.
ESTA TESIS FUE REALIZADA EN EL LABORATORIO
DE CATALISIS DE LA Urn-IZTAPALAPA, BAJO LA DIRECCION DEL DR. ALBERTO ALARCON DIAZ.

A MIS PADRES, DAVID Y DOLORES
Y
HERMANOS, JUDITH Y JULIAN

AGRADECIMIENTOS
Al Dr. Ricardo G6mez, Jefe del Departamento de Qufmica de la UAM-Iztapalapa.
A CONACYT por su apoyo econ6mico para la realizaci6n de este trabajo.
A Gilbert0 Cbrdova,por su ayuda en la determinaci6n vo- lumétrica.
A Francisco Zubillaga, por la realizacien de los dibu- jos.
A todos los compañeros del Area de Catblisis, por su ayuda y apoyo.
A Fernando Romo, por su gran ayuda durante los cursos recibidos durante la Maestrfa.
Y muy especialmente a Francisco Pedraza, por su cola- boraci6n.
A la Dra. Gabriela Dfaz, por sus consejos.

I N D . 1 C E pdgina
INTRODUCCION 1
CAPITULO I ANTECEDENTES
1.1 Catalizadores monofuncionales
1.2 Reacciones de hidrogenaci6n
1.3 Reacciones de deshidrogenaci6n
1.4 Reacciones de oxidaci6n
1.5 Reacciones de hidrodealquilaci6n
1.6 Otras reacciones
1.7 Catdlisis con catalizadores-metal-zeolita
que presentan cardcter polifuncional.
1.8 Reacciones de isomerizaci6n
1.9 Reacciones de hidroisomerizaci6n
1.10 Otras reacciones
1.11 Reacciones de hidrocracking
CAPITULO I1
DESARROLLO EXPERIMENTAL
3
9
10
11
12
13
18
11.1 Metodo de preparaci6n de catalizadores soportados l8
11.2 Métodos de caracterizacidn 19

11.3
11.4
11.5
M6todos de adsorci6n
a) Mediciones volum6tricas
b) Mediciones gravimgtricas
Difracci6n de Rayos-X
a) Ensanchamiento de las llneas de difracci6n
b) ,Método de desviaci6n a pequeño dngulo
Slntesis de los catalizadores
1) Caracterizaci6n de los catalizadores
2) Resultados
3) Discusi6n
CAPITULO I11
DETERMINACION DE LA ACTIVIDAD CATALITICA EN LA REACCION
DE DEHIDROGENACION DE BENCENO
III. 1 Reaccidn
1) Cdlculos
2) Resultados
3) Discusidn
CAPITULO IV
DETERMINACION DE LA ACTIVIDAD CATALITICA EN LA REACCION
DE HIDROCRACKING DE n-DECANO, DECALINA,Y TETRALINA
21
21
21
22
22
23
24
25
30
31
33
36
38
39
40
4 3

IV.l Hidrocracking de alcanos
a) Catalizadores con propiedades
fuertemente dcidas y con actividad
hidrogenante media.
b) Catalizadores con alta actividad hidro-
genante,
c) Hidrocracking de alquilbencenos.
IV. 2 Reaccidn
1) Cdlculos
2 ) Resultados
3 ) Discusidn
CAPITULO V
CONCLUSIONES
45
45
45
46
52
53
55
55
68
BIBLIOGRAFIA 6 9

INTRODUCCION
Las zeolitas conteniendo metales son eficientes catali- zadores para un gran ntímero de reacciones orgdnicas e inor- gdnicas, muchas de las cuales forman la base de la refinaci6n del petrdleo y de los procesos petroqulmicos. Otros catali- zadores recientemente desarrollados son de uso potencial en el futuro (1).
La actividad, selectividad y estabilidad de los catali- zadores de metales soportados en zeolitas son en gran medida, determinadas por la composici6n qulmica y la estructura del componente zeolltico, la cual es responsable de sus propieda- des Einicas.
Las funciones catallticas de los sistemas metal-zeolita dependen de su preparacibn, modificaci6n y condiciones de aplicaci6n; el mismo catalizador puede ser mono y polifuncio-. nal dependiendo del cardcter de la reacci6n y condiciones de uso. Ademds los avances en este campo dependen de la -aplica- ci6n de nuevos métodos de investigacibn, de la slntesis de nuevas zeolitas y del desarrollo de nuevas t6cnicas que se emplean para la modificaci6n de sus propiedades.
Los sistemas polimetdlicos obtenidos por intercambio ibnico, impregnaci6n (simultdnea o subsecuente), y la combi- nacidn de estos métodos representan nuevas rutas sintgticas de sistemas catallticos.
Otro aspecto interesante de estos materiales radica en la posibilidad de llegar a la preparaci6n de sistemas en los cuales se combinen la actividad catalltica del metal so-
portado como la actividad intrlnseca del soporte. -

2
Una de las reacciones en las cuales los catalizadores del metal soportado en zeolita actdan bifuncionalmente, es la reacci6n de hidrocracki.ng, cuya importancia en los Iílti- mos años se ha ido incrementando en la refinacidn del petr6- leo, en la producci6n de combustibles ligeros y en algunas otras aplicaciones.
Con el fin de estudiar algunos de estos factores se prepararon catalizadores de platino soportado en zeolita NH4-Y, por el método de intercambio i6nico.
Estos catalizadores se caracterizaron qulmica y flisica- mente, asli tambien se determind su actividad en reacciones de hidrogenaci6n de benceno e hidrocracking de hidrocarburos lineales y clclicos, con e1 fin de analizar comparativamente la actividad y selectividad de estos materiales.
En este trabajo se hace primeramente una revisi6n de los estudios realizados sobre este tipo de catalizadores, y los tipos de- reacciones en que son utilizados como cataliza- dores monofuncionales y polifuncionales, posteriormente se presenta el desarrollo experimental en el cual se explica la forma de slntesis de las muestras y su caracterizaci6n qulimica y flsica, el estudio de actividad con las diferen- tes reacciones antes menci.onadas y finalmente se muestran los resultados, discusidn y conclusiones a las que se llega- ron.

3
CAPITULO I
ANTECEDENTES
Los catalizadores metdlicos soportados son ampliamente usados en la refinacidn del petrdleo y en las industrias qulmicas y petroqulmicas (1).
Esos catalizadores son importantes en la slntesis de amonlaco, conversidn de hidrocarburos con vapor de agua para gas de slntesis, reforming, hidrocracking, hidrorefinacidn, hidrodealquilacidn, dehidrociclizacidn, isomerizacidn de pa- rafinas y cicloalcanos, etc.
Las propiedades de esos catalizadores dependen en gran medida del estado y dispersidn del componente metdlico ( 2 , 3 ,
4 , 5 , 6 ) . El soporte por 10 tanto juega un pape1,muy importan- te en este aspecto.
Las zeolitas, debido a SUS propiedades espec€ficas (ha- bilidad de intercambio idnico, estructura cristalina con po- ros regulares de tamaño molecular) se utilizaron como sopor- te para preparar catalizadores conteniendo metales altamente dispersos.
El intergs de los sistemas metal-zeolita surge del estu- dio de la introduccidn de platino y la alta dispersidn obte- nida del metal dentro de las estructuras porosas presentando alta resistencia al envenenamiento con azufre (7).
Las propiedades parti.culares de estos catalizadores han permitido el desarrollo de teorlas en Sreas como: interaccidn metal-soporte, propiedades de partlculas muy pequeñas de va-

rios metales en poros de sdlidos cristalinos, asl como el de- sarrollo de algunos catalizadores eficientes para reacciones importantes practicadas comdnmente en el dmbito comercial, tal como lo es el selectoreformado; un proceso de hidrocracking para n-parafinas con isoparafinas e hidrocarburos aromdticos para reformar gasolinas ( 8 ) .
1.1 Catalizadores monofuncionales
Bajo ciertas condiciones y dependiendo del tipo de reac- ci6n, los sistemas metal-zeolita pueden actuar como cataliza- dor monofuncional y en este caso la zeolita puede ser consi- derada como un soporte inerte. Sin embargo, dado que el es- tado del metal depende de la composici6n y estructura del .cristal de la zeolita, las propiedades de esos catalizadores dependen significativamente del tipo de zeolita usada.
Entre las reacciones en que se utilizan este tipo de catalizadores se mencionan: hidrogenaci6n, oxidaci6n, etc.
A continuaci6n se discuten algunos aspectos de esas' reacciones.
1.2 Reacci6n de hidrogenaci6n
La propiedad hidrogenante de los metales soportados en zeolitas ha sido ampliamente estudiada en reacciones de ole- finas (9, 10, 11, 12), ciclos con dobles enlaces, hidrocar- buros aromdticos y compuestos conteniendo oxlgeno.
Las actividades de los metales del grupo VIIIB soporta- dos en zeolitas del tipo X o Y sodadas,son comparables a la actividad que presentaron 'estos metales en forma mdsica, ju-
-

5
gando estas zeolitas un papel exclusivo de soporte inerte.
Sin embargo una diferencia en actividad es observada cuando se depositan metales en zeolitas previamente decatio- nizadas; las propiedades hidrogenantes del metal se aumentan notablemente, ya que en este caso el metal penetra en las ca- vidades zeollticas.
En las reacciones de hidrogenaci6n la actividad catallE- tica del metal muestra el siguiente orden: Pt-NaY>Pd-NaY > >
Ni-Nay, similar al de los catalizadores mdsicos. Por lo tan- to para estos sistemas, las diferencias en actividad’gara el mismo tipo de metal, serdn debidas a diferencias en la dis- persi6n metdlica (13).
Una propiedad caracterlstica de los catalizadores me- tal-zeolita, es su habilidad para hidrogenar selectivamente mezclas de olefinas.
-Weisz, et.al. (14,15,16,17) mostraron que los sistemas meta¡-zeolita son selectivos para la hidrogenacidn de eti- leno, en presencia de isopropileno o bien de propileno en presencia de isobutileno.
En el caso de catalizadores Pt-zeolita se han encontra- do que los centros activos de Pt no son accesibles para las isoolefinas, por lo tanto estos sustratos no son hidrogena- dos por este tipo de catalizadores.
Las propiedades de las zeolitas conteniendo metales han sido estudiadas con detalle en la reacci6n de hidrogenaci6n de benceno, debido a que e,sta reacci6n es de gran importan- cia tgcnica y es un modelo conveniente para estudiar la de- ._

6
pendencia de la actividad catalItica del metal, del tipo de soporte, de la composici6n de la zeolita, tamaño del cristal metdlico, interaccidn entre metal-zeolita y otros factores (18)
Con mol6culas mds voluminosas como el mesitileno, a pre- sidn atmosf6rica y temperatura de 2OO0C, un catalizador al 53% de Pt-CaX no presenta actividad en dicha reaccibn. En este catalizador todo el platino estd aparentemente dentro de las cavidades de la zeolita y debido al mayor tamaño de la mol6cula de mesitileno, 6sta no alcanza a penetrar en las cavidades de la zeolita. En comparaci6n, si se tiene un ca- takizador al 1.8% Pt-CaX, la conversi6n a hidrocarburos es de 10% en peso, debido a que la hidrogenaci6n se realiza sobre las partlculas de platino localizadas en la superficie de la zeolita.
1.3 Reacci6n de deshidrogenaci6n
Gutyrya, Galich, et.al. (19,20,21) fueron l o s primeros en estudiar los catalizadores de zeolfta Y en reacciones de deshidrogenaci6n de n-parafinas. Logrando la produccidn de olefinas, a partir de n-hexano y n-heptano.
Sin embargo, se encont.r6 que los metales soportados en zeolitas X y A muestran baja actividad y selectividad en la deshidrogenaci6n de n-alcanos del tipo C6 y C7.
La actividad deshidrogenante de estos catalizadores de- ,pende tambien de la dispersi6n. Las muestras con partlculas de platino m6s pequeñas que 20 presentan baja actividad en la reacci6n de deshidrogenacfdn ( 2 2 ) .

7
1.4 Reacci6n de oxidacidn
En 1961, se realiz6 la reaccidn de oxidacidn de etileno con catalizadores de Cu, Ag o. Au soportados en zeolitas ( 2 3 )
y recientemente se ha reportado que los catalizadores'de pla- ta soportada en zeolitas X y A, mostraron actividad en este tipo de reaccibn.
También se observ6,cuando el metal se encuentra en la cavidad (catalizadores preparados por intercambio ibnico), es activo para la reacci6n de oxidaci6n, pero no selectivo en la formaci6n de 6xido de etileno, sin embargo las muestras preparadas por impregnaci6n mostraron gran selectividad y al- .ita actividad.
El orden de selectivid.ad en estos catalizadores varia de la siguiente manera: Ag-CaA>Ag-KA>Ag-NaA>>Ag-NaX. En ge- neral en estos sistemas se encuentra que la actividad y se- lectividad se incrementa linealmente con el aumento de conte- nido de Ag soportada en la zeolita.
Los resultados de estos estudios .han dado origen a in- vestigaciones en las propiedades y especfficidad de los ca- talizadores de metales soportados en zeolitas.
I. 5 Reacci6n de hidrodeal.quilaci6n
Para esta reaccibn, con Tolueno, Cumeno y Etilbenceno, se ha observado que con un catalizador de Ni-CaA al 5% pre- parado por impregnaci6n y reducido, los productos gaseosos consisten principalmente de. CH4, indicando que la dealquila- ci6n proceda por medio de un desdoblamiento sucesivo de cpu- pos CH3. Entonces el orden de demetilaci6n decrece en la si- "

8
guiente forma: Cumeno>Etilbenceno>Tolueno.
Los catalizadores met5licos soportados en zeolitas fau- jasitas muestran alta actividad en hidrodemetilacidn con To- lueno ( 2 4 , 2 5 ) y en la dealquilacidn de aromdticos, conteni- dos en destilados de petr6:Leo ligero.
1.6 Otras reacciones
Los catalizadores de metales soportados en zeolitas pre- sentan actividad en otro tipo de reacciones, a continuaci6n mencionaremos algunos de los resultados de estos estudios.
Hall,et.al. (79) demostraron la eficiencia de la zeoli- ta SK-400 (1% de Ni-NaY y AW-300) como catalizador en la des- composici6n de 3-cloro-3-metilbutileter,produciendo isopreno.
En la reacci6n de n-hexano con vapor de agua para la pro- ducci6n de gas de shtesis se ha observado que los cataliza- dores de Ni y Co soportados en zeolitas Y y mordenitas pre- sentan una actividad apreciable.
Tambign han sido estudiadas las propiedades del catali- zador de Pt-zeolita en la descomposici6n de H202, a 20 y 40°C.
Minachev, et.al. (26), observaron que el catalizador de Ni-NaY acelera la reacci6n de intercambio:
Se ha observado que con zeolitas conteniendo metales como Na, Li o una mezcla de ellos, pueden ser catalizadores -

selectivos en la reaccidn de polimerizaci6n de olefinas. Tambien se encuentra en la literatura que los metales del grupo VIIIB soportados en zeolita Y han sido propuestos co- mo catalizadores para la slntesis de amoniaco.
1.7 Catdlisis con catalizadores metal-zeolita que presen- tan carácter polifuncional.
Estos catalizadores so:n aquellos en que el soporte y la fase soportada activan distintos pasos'elementales de la reacci6n qufmica, asf por ejemplo se mencionan algunas re- acciones qulmicas que son aceleradas por estos catalizado- res: la isomerización de hidrocarburos saturados, monoiso- merización de aromdticos y olefinas, dehidroisomerizaci6n de naftalenos para produci:r arombticos, hidrocracking y
otras reacciones las cuales tienen una gran importancia a nivel industrial.
Los catalizadores de metales soportados en zeolitas han alcanzado un gran desarrollo en varios procesos y se consu- men grandes volhenes de estos materiales anualmente. Se han descubierto nuevas reacciones en estos materiales por lo que el campo de la catblisis heterogenea se ha ampliado considerablemente.
Algunos datos interesantes del mecanismo de las reaccio- nes sobre estos catalizadoxes sirve como base para la pre- sentacidn de nuevas teorfas acerca de la actividad catali- tica de zeolitas y otros sdlidos cristalinos. Sin embargo, no se hablard detalladamente de estos estudios en este cam- po sino de los problemas m6s importantes y los resultados serdn revisados brevemente.

A continuaci6n comentaremos algunos t6picos de estos estudios haciendo 6nfasis en los aspectos mas interesantes.
1.8 Reacciones de isomerizaci6n
Rabo, et.al. (27,28) fueron los primeros en observar la alta actividad de los catalizadores de platino y paladio so- portado en zeolita Yen las reacciones de isonerizaci6n de n-pentano y n-hexano. Para estas reacciones enconetraron que los catalizadores de metales soportados en zeolitas presentan las siguientes caracterlsticas:
a) Las zeolitas conteniendo cationes univalentes son prdcticamente inactivos.
b) Las formas en cationes multivalentes, decationiza- das, hidrogenadas de zeolitas soportando metales, muestran alta actividad (27,2.9,30,31).
c) Los cationes de la zeolita que tengan la carga mbs grande y el radio mds pequeño, presentan la actividad mds alta,
d) Cuando la zeolita es intercambiada en mayor grado, se observa un incremento en la actividad del catalizador sintetizado por intercarbio ibnico.
e) Con un incemento en la relaci6n Si02/A1203 en zeo-
litas tipo faujasita o mordenita, la actividad del catali- zador se incrementa considerablemente lo cual permite rea- lizar la reacci6n a temperaturas menores. Por ejemplo en un catalizador de Pd-Hmordenita se observ6 una alta activi- dad en la isomerizaci6n cuando el soporte present6 una re- lación de Si02/A1203 de 16 a 18, y el valor m6s conveniente

11
en la relacidn de sflice-aldmina en zeolitas faujasita no se ha establecido todavla.
f) La actividad, selectividad y estabilidad del catali- zador, depende de la naturaleza y concentraci6n del componen- te metdlico.
La actividad del catalizador soportado en la zeolita NH4NaY (Si02/A1203 = 5.0) se incrementa linealmente con el aumento de la concentraci6n de Pt y Pd en la reaccidn de iso- merizaci6n (32).
Tambi6n se ha observado que en la reacci6n de isomeriza- cibn, al aumentar el peso molecular de n-parafinas, se in- crementa la conversi6n en esta reaccibn.
1.9 Reacciones de hidroisomerizaci6n
Los hidrocarburos arom=iticos, olefinas y cicloolefinas se hidroisomerizan con cat,alizadores de metal soportado en zeolita, acutando en forma bifuncional. La relaci6n entre los productos de hidrogenaci6n e hidroisomerizaci6n depen- den de la composicidn del catalizador y las condiciones del proceso.
Por ejemplo, sobre el catalizador de CaY(Si02/A1203=3.4) al 5% de Pd a 32OOC y con la relaci6n molar de H2:C6H6=5, dando como producto metilciclopentano en un 20.1% de con- versión y de ciclohexano de 63.5% de conversión. El in- cremento en la relacidn de Si02/A1203 en la zeolita CaY por arriba de 4.5 da como resultado un aumento en la produccidn de metilciclopentano (67.3% de conversi6n).

12
Una alta activi.dad de hidrocracking se observ6 a 32OoC con catalizadores de Rh e Ir, en el catalizador de Rh la conversi6n de los productos fue de 16.9% y de un 60% con el catalizador de Ir y la conversi6n de productos en la hi- droisomerizaci6n fue de 12 y 5.8% respectivamente.
1.10 Otras reacciones
Las zeolitas NaX, NaY y Na-mordenita. conteniendo meta- les del grupo VI11 y fB muestran solamente una funci6n como catalizador en reacciones de hidrocarburos (oxidacitjn, hi- drogenacibn, dehidrogenaci6n). Sin embargo, en la conver- si6n de n-butilaldehido en presencia de Hz, estos sistemas act6an como catalizadores tlpicos de funcitjn bifuncional ( 3 3 )
De esta manera, se tiene un catalizador de Ni-NaY al 4 % preparado por impregnaci6n de la zeolita NaY con una soluci6n de acetilacetonato de Ni en CHC13, con la descom- posicidn subsecuente de la sal en aire corriente a 5OOOC seguida por una reducci6n en presencia de hidr6geno a 45OoC, se produce butanol, isooctanol, 2-etilhexanal y 2-etilhexenal.
As€, las propiedades de los catalizadores son modifi- cadas considerablemente durante la reacci6n; con una dis- minuci6n en la actividad hidrogenante del aldehido a buta- nol, los productos de la condensaci6n se incrementan, la conversien de butilaldehido a 2-etilhexanol y 2-etilhexa- nal alcanza su msximo antes de 2.5 y 5 horas de reaccitjn respectivamente y el producto de 2-etilhexenal se incremen- ta al termino de 9 horas y por lo tanto no es afectada. La

13
conversi6n total del aldehido decrece. Esto es probablemen- te causado por los diferentes coeficientes de adsorci6n del n-butilaldehido y sus productos de conversi6n. A diferen- tes temperaturas y presiones, la reaccidn puede variar. SO- bre zeolita NaY sin nPquel, ocurre s610 condensaci6n prot6- nica de n-butilaldehido a 2-etilhexenal.
La hidrocondensaci6n de pequeños aldehidos puede ser realizada sobre catalizadores de La, Ce, Cd, Ba, Sr, Zn, Ca o zeolitas X y Y conteniendo Pt o metales del grupo del Ni.
Los catalizadores de metal soportado en zeolitas pueden tambi6n acelerar la desproporci6n de alcanos e isoalcanos, C3-C7, a isoalcanos con altos y bajos pesos moleculares asl como la oligomerizacl6n de olefinas C2-C4.
1.11 Reacciones de hidrocracking
En presencia de un metal soportado en una zeolita tipo bcida, como puede ser la tipo X, Y mordenita o erionita, la reacci6n de hidrocracking es'acompañada de varias reac- ciones de hidrocarburos. La medida de una reacci6n de hi- drocracking depende de la composici6n del catalizador del meta1,y su concentración. de la acidez de la zeolita y de los parsmetros de la reacci6n.
Por ejemplo, sobre un catalizador de Rh-CaY al . S % de Rh (Si02/A1203=3.4) a 39OoC, presidn de 30 atm y una rela- cien molar de H2:nC6H14=3.2 la conversi6n a is6meros de hexano y productos de hidrocracking fueron de 25.8% y 21.9%, respectivamente mientras que en un catalizador similar de Ir a 31OOC los valores de conversien fueron de 6.3% y 26.2% respectivamente (34,351 .

14
En las mismas condiciones mencionadas anteriormente, un catalizador de Pt y Pd soportados en una zeolita Cay, el hi- drocracking no excede el 4 % de conversien y la producci6n de isómeros fue de 58.Oa 5 9 . 4 % .
Casos similares a estos fueron observados durante la isomerización de ciclohexano y la hidroisomerizaci6n de ben- ceno ( 3 6 ) .
El hidrocracking de hidrocarburos superiores y fraccio- nes de petrdleo son de inter& particular y de gran impor- tancia practica. La reacci6n de hidrocracking es usada pa- ra obtener hidrocarburos gaseosos como material de alimenta- ci6n para las industrias petroquimicas, para gasolinas y com- bustibles de jets, para combustible diesel, etc.
Muchos, catalizadores,principalmente los que contienen metales del grupo VIIIB (Pt, Pd, Ir, Ni) soportados en zeo- litas y decationizadas y tierras raras en zeolitas tipo Y, ( 3 7 , 3 8 , 3 9 ) se han empleado en la reaccidn de hidrocracking y tambien han resultado eficientes catalizadores de tipo po- limetdlico ( 4 0 ) .
Los catalizadores de 1.r al .1% y Re al . 2 % sobre zeoli- ta decationizada muestran 'alta actividad en la reacci6n de hidrocraking de hidrocarburos al compararlos con los cata- lizadores de Pd y Pt sin soportar.
El uso de las zeolitas como catalizadores en la reacci6n de hidrocracking en la refinaci6n de materiales crudos, evi- ta el envenenamiento debido al N2, S2 o hidrocarburos aromb- ticos policiclicos y tanbign desfavorecen el incremento de productos no deseados.

15
El hidrocracking de hidrocarburos individuales (41,421 y sus mezclas han sido muy estudiadas (43,44,45,46), como ejemplo tenemos'la mezcla de propano y butano que resulta- ron ser los productos predominantes en el hidrocracking de n-heptano con un catalizador de Pt-NaY en un intervalo de 340-400°C, presi6n de 40 atm y una relaci6n molar de H *C H =4.5. La conversi6n total de los productos a 4OOOC fue de.un 65% (42).
2 ' 7 16
Al aumentar el poder hidrogenante del catalizador Pt- NaY con el n-heptano, se increment6 en los productos el contenido de isoheptano y la conversidn total de n-C7H14 y
la actividad de hidrocracking no var€a apreciablemente.
Con catalizadores de poco poder hidrogenante, la can- tidad de hidrocarburos, C5 y C6 aumentd pero esto no co- rresponde a la cantidad de CH4 y C2H6 que se obtiene.
En esta forma la conversidn y la composici6n de los - productos del hidrocracking de n-alcanos se puede controlar
al variar la relaci6n de las funciones de hidrodehidroge- naci6n y cracking del catalizador. Se ha observado que la reactividad de las parafinas, en esta reacci6n, se incre- menta con el aumento de la cadena.
En el hidrocracking de n-decano en un catalizador de Pd-CaY al .5% ocurre a una temperatura por debajo de 250°C y a 3OOOC la conversidn es de 100% (47). La reaccidn pro- cede por un mecanismo dual (48).
Un papel importante realizan los factores de adsorcidn y difusidn en los catalizadores de metales soportados en zeolitas y esto puede comprobarse observando los resulta-

16
dos obtenidos del hidrocracking de n-decano, decalina y la mezcla de estos en catalizadores de mordenita (49). El estudio se realiz6 en dos muestras: una de Pd-HK al . 5 % ( A )
obtenido por impregnaci6n de la zeolita NH4-mordenita con un compuesto de Pd y tratado t6rmicamente en corriente de aire a 538OC y la otra muestra de Pd-HEI al . 5 % preparado con NH4-mordenita dealuminada(B). En la reacci6n de n-de- cano a 26OoC, el catalizador B fue 4.4 veces más activo que el catalizador A , la energla de activacien aparente es de 44 y 33 Kcal/mol respectivamente.
Una relacien similar entre las actividades de estos catalizadores fueron observadas en la reaccidn de hidro- cracking de decalina y bajo las mismas condiciones en que se realiz6 la reaccidn de n-decano, la conversidn de la de- calina result6 ser mbs diflcil. Sin embargo, de la mezcla C10H22 + C10H18 con ambos'catalizadores la decalina pre- senta-un rompimiento m6s rbpido.
Lo anterior puede ser debido a que la decalina es un. hidrocarburo clclico e impide el acceso de las moleculas .de n-decano a los centros activos del catalizador.
Los coeficientes de difusidn de la decalina en A y B a 93OC fueron de 4 y 2 . 5 veces respectivamente, por lo tanto estos valores son mbs bajos que con el n-octano.
Con mordenita dealuminada, la adsorci6n y desorción del hidrocarburo es mds rápida. Por lo tanto el efecto inhibi- dor del catalizador B
Como se puede
de la decalina en la reaccidn con n-decano en presencia es menos predominante.
observar, las reacciones de hidrocracking -

17
tienen gran intergs y su campo de investigaci6n es todavla no agotado por lo cual esta reaccidn forma parte de nuestro estudio con catalizadores de Pt soportado en zeolita NH4Y a diferentes concentraciones de metal.
Esta reaccidn de hidrocracking se efectud con tres reac- tivos diferentes (n-decano, decalina y tetralina) y se estu- dia la actividad de los catalizadores, la distribucidn de los productos y la selectividad.
. I

18
CAPITULO I I
DESARROLLO EXPERIMENTAL
11.1 Método de preparacih de catalizadores soportados
Como se ha mencionado previamente,el metodo de prepara- ci6n de un catalizador soportado, influye directamente en la dispersi6n y localizaci6n del metal en las zeolitas.
El Pt y el Pd son generalmente soportados en zeolitas faujasita por el método de intercambio ibnico, intercambian- do el i6n Pt(NH3)4 ciones acuosas de Pt(NH3),$C12 o PtC12 (PdC12) en solucidn amoniacal (50).
+2 y Pd(NH3) 4 +2 provenientes de las solu-
Tambign pueden emplearse otros mgtodos, como el de im- pregnaci6n, co-precipitaclbn y deposici6n, siendo utiliza- des preferentemente cuando se desea depositar al metal en la superficie externa del soporte (51, 52).
E l metodo de intercambio i6nico en soluciones es una ru- ta.usua1 para introducir al metal en una estructura porosa como en el caso de las zeolitas. El intercambio del i6n se realiza fácilmente en estos materiales, por lo que el cata- lizador alcanza una alta dispersidn metblica. Rabo, et.al. ( 5 3 ) obtuvieron platino atbmico altamente disperso por re- ducci6n de un catalizador PtY al .S%.
E l tamaño y localizaci6n del metal depende de las condi- ciones de los tratamientos t6rmicos y de las condiciones es- tablecidas para la reducci6n. Por ejemplo, se tiene un cata-

19
lizador de Pt/NaY con .24% de Pt obtenido por intercambio i6nico en una solucidn Pt(NH3I4Cl2, resultando partlculas de Pt de 15 a 55 A. Este catalizador se trat6 tgrmicamen- te a 3OOOC en aire y posteriormente se redujo a 5OOOC por 3 horas.(50). A una temperatura menor de reducci6n se ori- gina un tamaño de partxcula menor en zeolitas intercambia- das idnicamente (50).
O
El uso del platino como catalizador es muy extenso ya que es utilizado en una gran variedad de procesos, que van desde la refinacidn petroqulmica, hasta su uso en los esca- pes de automdviles para la purificaci6n de los gases cie com- busti6n (52).
Debido a los recursos limitados y al alto precio de es- te metal, el uso eficiente de cada dtomo utilizado es esen- cial para la produccidn a gran escala. Por lo que este me- tal soportado en diversos :materiales ha sido objeto de mu- chos estudios en l o s Cltimos años. Y por lo tanto su con- secuente interes en e.1 grado de dispersi6n obtenido.
11.2 Metodos de caracterizaci6n
A los 5toraos metdlicos del catalizador que toman parte en la reaccidn se les da el nombre de sitios activos. En el caso de la catálisis heteroggnea encontramos que cuando se tienen partlculas metdlicas pequeñas se dispone de una drea superficial mayor y por lo tanto se encontrardn mayor cantidad de sitios activos.
Es por esto que se hace necesario conocer el grado de dispersi6n de un catalizador soportado y en general, al gra-
-

20
do de dispersi6n se le define como el porcentaje de atomos o molgculas de fase activa, situados en la superficie del catalizador respecto al total presente en toda la masa cata- lctica y para su determinaci6n existen diferentes metodos flsico-qulmicos entre los cuales tenemos: nedicibn del drea superficial por adsorcidn flsica y quimisorci6n, andlisis de rayos-X, microscopla electr6nica, etc.
Las medidas de adsorci6n en catalizadores soportados proporcionan valores de $rea superficial (dispersi6n) pero no dan informacidn de la microestructura de ias partfculas.
Las tecnicas de rayos-X son de dos tipos, dependiendo del dngulo de incidencia del haz estudiado.
A dngulos de incidencia menores de 2Ogrados el rayo efec- ttía un barrido contlnuo en lamuestra llamdndose a esta t6c- I
nica desviaci6n de rayos-X a pequeño 6ngulo.
Los estudios efectuados a dngulos mayores caen dentro de las t6cnicas normales de rayos-X y permiten, ademds de conocer las caracteristicas estructurales de los catalizado- res, el tamaño de partlculn del metal por medio de la medida de ensanchamiento de las llineas de difracci6n.
El uso del microscopio electr6nico complementa y propor- ciona informaci6n de la geometrfa de las partfculas.

21
11.3 Metodos de adsorci6n
Las técnicas mds comGnes para medir drea superficial de catalizadores son: adsorcidn flsica, en donde presentan fuer- zas de Van der Waals y qui.misorci6n de gases, en la cual existen interacciones adsorbato-adsorbente, presentsndose formacT6n de enlaces qulmicos entre la molEcula adsorbida y la superficie metblica.
A continuaci6n se describirdn diferentes metodos para la deteminacidn de dispersibn, basados en la adsorcidn de ga- ses.
a) Mediciones volum6tricas
El aparato bdsico para medidas de gas consiste de un do- sificador de gas,. un aparato para medir su presibn, una cel- da y los aditamentos necesarios para el manejo del.gas y el sistema de vacfo.
La cantidad de gas adsorbido es determinada al tomar la diferencia entre la cantidad de gas dosificada en el volumen que contiene a la muestra y la cantidad registrada en el vo- lumen muerto (52).
b) Mediciones gravimétricas
Por este metodo la masa del material adsorbido es medi- do directamente por medio de una microbalanza.
La aplicabilidad de las medidas de .adsorci6n en la mi- crobalanza necesitan ser analizadas en relacidn al incremen- to de masa esperado y la sensibiiidad disponible.

2 2
Para efectuar un estudio de adsorción de este tipo se requiere que la superficie metslica se encuentre limpia para que se permita suponer que las fuerzas que existen en el sistema se deban a la interacci6n adsorbato-adsorbente.
Para obtener la superficie metdlica limpia del cataliza- dor se realiza una desgasificaci6n a altas temperaturas (52).
11.4 Difracci6n de rayos-X
a) Ensanchamiento de las llneas de difracci6n
El andlisis de difraccidn de rayos-X da informaci6n de la estructura y composici6n cristalina.
Una disminuci6n del tamaño de cristal o de los materia- les policristaltnos causan un ensanchamiento en los perfiles de difracci6n. El tamaño promedio del cristallto se calcula con la ecuaci6n de Scherrer:
donde k es una constante dependiente de la forma del cristal y h es la- longitud de onda del rayo, B es l a anchura de la 11- nea y O es el dngulo de Bragg.
dSe: puede observar el tamaño del cristal con valores me- nores de 100nm, pero la tEcmica es particularmente aprecia- ble a tamaños de cristal de 3 a 50 nm; con valores de 3 nm la linea de difracci6n es t.an ancha que se lleya a perder, mientras que con el tamaño de cristal de 50 nm el cambio en
-

23
la forma del pico es pequeña y el mgtodo por consiguiente no es sensible.
Los lfmites de esta tecnica dependen de la cantidad de metal soportado y algunas dificultades son encontradas con cantidades de metal menores de .S%.
b) Mgtodo de desviacidn a pequeño dngulo
En el método de desviaci6n a pequeño 5ngulo se utiliza la informacidn proporcionada por la desviacidn del haz con valores menores de 5'grados del rayo primario.
La intensidad de barrido I(h) de la radiaci6n depende del ntímero de distribucidn y tipo de 6tomos en la partlcu- la. La intensidad se expresa por la siguiente ecuacidn(54):
I (hl = Ie (h)F2 (h)
en donde h es el 5ngulo de barrido20/h, donde el numerador en radianes, es el dngulo y h la longitud de onda de la ra- diacidn.
Ie(h) es la intensidad del haz de electrones que alcan- zan a la partfcula metslica en el catalizador y F(h) es el factor de estructura de la partfcula.
Esta tgcnica fue utilizada por Somorjai (55) para se- guir el crecimiento de partlculas de Pt soportado en altí- mina durante el tratamiento tgnnico a temperaturas eleva- das.
Tambign en los Gltimos años ha sido de gran inter& la

caracterizaci6n de catalizadores por medio del microscopio electrdnico y este se utiliza frecuentemente para comple- mentar otras tgcnicas analfticas.
El microscopio electr6nico permite observar el tamaño, forma y distribucidn de partlculas.
11.5 Sfntesis de los catalizadores
Se prepararon dos tipos de catalizadores, con un conte- nido metdlico de platino, de 1% y 5 % respectivamente.
Ambos se prepararon de la siguiente manera: se pes6 una cantidad determinada de zeolita NH4Y de fdrmula (NH4)56H5- (A1021 61 (sio2) 131, y se de,shidratd en una estufa durante una hora a 15OOC. . Posteriormente se hizo el intercambio i6nico con una soluci6n acuosa de la concentraci6n requerida de Pt(NH3)2 C12; la cantidad pesada de zeolita se coloca en un vaso de precipitado y se le añade la solucidn acuosa del com- plejo de platino y se mant:iene en agitaci6n durante 24 Hrs.
Al finalizar el proceso de intercambio se filtra la muestra y se lava 3 6 5 veces con agua tridestilada, poste- riormente se seca en una estufa durante una hora a 150OC.
Las zeolitas intercambiadas con Pt2+ fueron pretratadas "insitu", primero en una corriente de O2 a 18OOC durante
' 2 horas, posteriormente se increment6 la temperatura lenta- nente a una velocidad de 2.OoC por minuto hasta 45OoC, man- teniendo este temperatura durante 12 horas.
A continuaci6n se introdujo una corriente de Ni y se disminuye la temperatura hasta 300"C, en estas condiciones
-

se introdujo una corriente de H2 durante 2 horas, al termi- no de este tiempo, se increment6 nuevamente la temperatura hasta 45OOC durante 2 horas.
En la figura 1 se indica la secuencia de los tratamien- tos tgrmicos.
1) Caracterizacih de los catalizadores
Posteriormente se reali'zb la caracterizaci6n de los ca- talizadores por Difracci6n de Rayos-X, por el metodo de Debye-Scherrer y desviacidn a pequeño dngulo, tambi6n por adsorci6n de gases (H2).
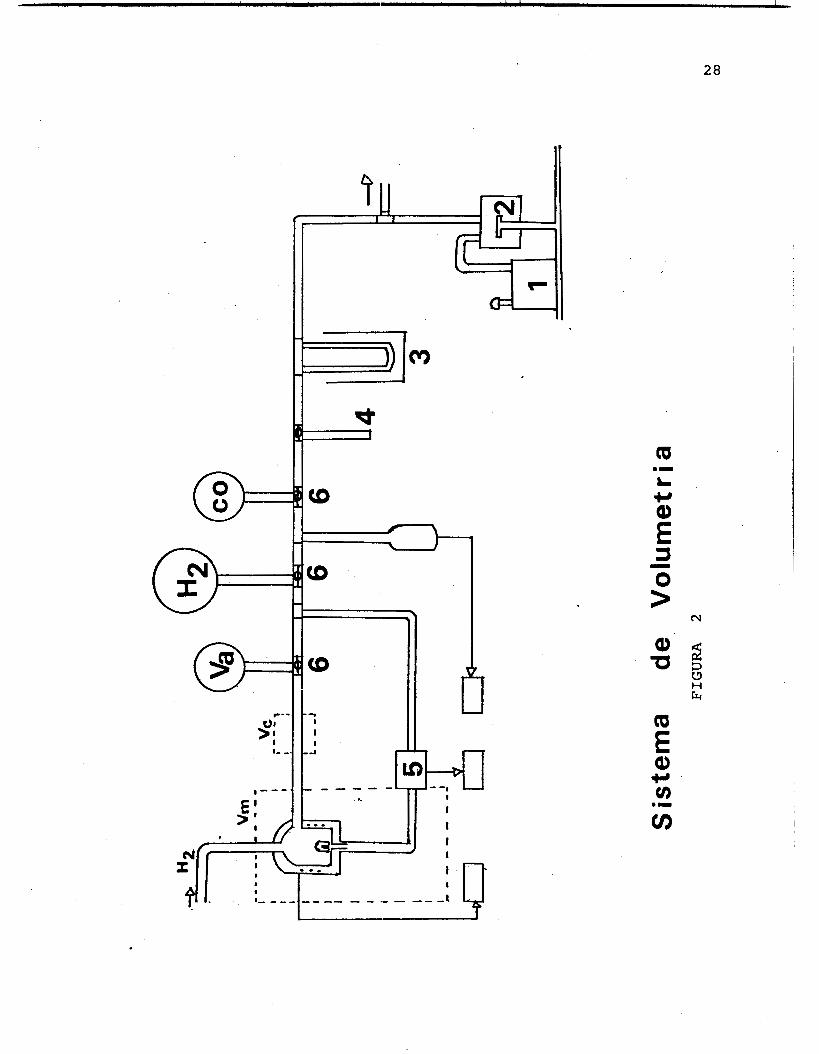
sistema de volumetrZa, est6 construido en vidrio Py- igual que el sistema de vado el cual consta de:
Una bomba mecdnica Una bomba de nitr6geno lfquido Una trampa de nitrtigeno lfquido Un medidor de vacfo tipo Mccleod con intervalo de d 5 a 10-1 Torrs. Un medidor de presiones Valadyne modelo CD-23. VSlvulas para alto vacfo
experimento se realiz6 de la siguiente manera:
reactiva la muestra a 38OOC durante una hora y media, a continuaci6n se hace vacfo en el sistema de vidrio (1 x torrs) , a la misma temperatura; posteriormente se disminuye la temperatura hasta condicones ambiente (25OC)
para efectuar las quimisorciones de hidr6geno.

I
T "C
500
2 50
Diagrama de Tratamientos FIGURA 1
N cn

27
Se efectfia la introduccidn de una determinada cantidad de gas hasta condiciones de equi l ibr io , repi t iendo la opera- c i 6 n incrementando la cantidad de gas introducida hasta ob- tener e l nGmero de puntos necesarios para la construcci6n de l a isote.rma. Toda l a secuencia anterior se efectud con cada muestra.
Con las diferencias de presidn obtenidas es posible cal- cular e l nfimero de moles adsorbidas, representdndose estos en e l e j e de l a s Y y la presi6n en mHg en e l e j e de l a s X
para obtener l a curva (isoterma tipo Langmuir). La cantidad de gas adsorbido se calcula de acuerdo a l o s i g u i e n t e (56):
Para l a primera experiencia:
Nal = N i - NR1
Para l a segunda experiencia:
Na2 = N2T-NR2
N2T = 2Mi-PflVc
Para n experiencias:
Nan = NnT-NRn
NnT=Nni-Pfn-1Vc
NR1 - - Pf 1 (VC+rn) RT
Pf tVc+Vm) NR2 - - RT
2 8
8-
W
El I - "

2 9
En donde:
Naj =
Ni - - NjT =
niunero de moles adsorbidas n€imero de moles suministradas nfimero de moles suministradas totales nhero de moles residuales presidn inici.al presi6n residual volumen calibrado volumen muerto
Para este sistema se calcula previamente:
vc /-
I
vm = 11.13 x lo-',
NGmero de moles aasorbidos: 4 . 3 2 x = Ntimero total de moles adsorbidos
Como el catalizadar se tiene con un contenido met6lico de 1% en Pt, esto correspo.nde a 10 mg de Pt.
g/mol ''meta1 = 5.1 x moles/g de platino
x g de catalizador = 9.69 x moles/g de cata-
lizador g de Pt

30
%D - - nhero. de moles adsorbidos g/catalfzador x 100 = 44.58%
y para obtener el tamaño de partfcula,
g = - 1000 = O
%D 22.43 A
para obtener el ntimero de sitios metdlicos activos,
NO = - - 100 %D x ntimero de moles de Pt totales =
= 2.2735 X Nol de sitios activos/g de cat. "
2) Resultados
TABLA I
O Catalizador No.de moles adsor. 9 A . %D No. Sitios activos/g cat.
PtY 1% 4.32 x 22 44 2.273 x
PtY 5% 6.8 x 74 14 3.417 x

31
Del andlisis por difraccidn de rayos-X, se encontrd que los iones de Ptt2 se encuentran preferentemente dentro de la gran cavidad de la zeolita y las muestras reducidas pre- sentan los siguientes tamaños de partlcula.
Las muestras previamente tratadas con flujo de O2 y N2
(y a temperaturas de 180, 450 y 300OC) fueron reducidas a diferentes temperaturas, desde 200 hasta 45OOC con flujo de H2 y las condiciones en las que .se encontrd una reduccidn total fue con la muestra que se~redujo a 450OC.
. I
TABLA I1
Catalizador p i % D
PtY 1% 1 !j 66
PtY 5% 4 5 2 2
No se encontr6 destruccidn de la cristalinidad de la zeolita.
3 ) Discusidn
Como se muestra en las Tablas I y 11, los valores encon- trados del tamaño de partlcula y dispersidn son los espera- dos segfin' el contenido metslico de cada catalizador (78 ) .
0 9 3 0 4 8

3 2
En el catalizador al 1.8 de Pt se observa una dispersidn superior con respecto al catalizador al 5 % de Pt y la dife- rencia de los resultados de dl’spersidn y tamaño de partlcu- la, entre los dos mgtodos son aceptables (R-X y adsorci6n de gases).
Observando los resulta.dos obtenidos en el andllsis de difraccldn de R-X, para estas muestras reducfdas, se encon- tr6 que se forman aglomerados de dtomos de platino, los cua- les tendersn a megrar a la gran cavidad en funcidn de la temperatura de reducci6n. . TambS6n se encontrd una variacidn en el tamaño de estos aglomerados de acuerdo a las condicio- nes de calentamiento.

I 4
33
CAPITULO I11
DETERMTNACI'ON DE LA ACTIVI'DAD CATALITICA EN LA RGACCION DE HIDROGENACION DE BENCENO
Despues de realizar la caracterizaci6n de los cataliza- dores por los m6todos mencionados anterTormente, se proce- de primeramente a efectuar la reaccf6n de hfdrogenaci6n de Benceno para estudiar la actividad catalftica de estos ca- talizadores.
La hidrogenaci6n de Benceno es uno de los procesos mbs importantes de hidrogenaci.dn de hfdrocarburos aromdticos y
ha sido una reaccrdn efectuada en presencia de diferentes metales soportados.
Se ha encontrado que est8 reacci6n es insensible a la estructura, es decir, es del tipo clasificada por Boudart ( 5 5 ) como reacci6n fbcil.
TambiQn la importancia de esta reaccidn radica en que puede emplearse como prueba catalltica para nuevos materia- les catalfticos, dado que se tiene un solo producto de reac- ci6n (el ciclohexano), est.0 facilita la determinaci6n de la actividad catalltica ( 5 7 ) .
Se ha acordado que dentro del mecanismo de esta reaccibn, el benceno es adsorbido por el s6lido y al encontrarse adsor- bid0 pierde su energfa de resonancia y resulta un valor pe- queño del calor de adsorci6n.

3 4
Selwood (58) mostr6 que cuando el benceno es quimisor- bid0 a l5OoC por el s2stema Ni soportado, la magnetizaci6n relativa del catalizador decrece hasta determinada cantidad, lo cual se expli’carla considerando que el benceno se ha uni- do sobre la superficie por: los 6 enlaces.
Y
6M + C6H6 4)”M
Trabajos recientes han demostrado que la adsorci6n del benceno en un metal, se efectGa por medio de la nube de electrones n del benceno sobre un sitio metdlico (55).
En cuanto a la cingtica de esta reaccidn se ha encontra- do que es de primer orden con respecto al benceno y de or- den cero con respecto al hidrbgeno.
5OoC Ft/zeoli.ta ‘ 0 + calor
Sin embargo, se ha observado que en la reaccidn en fase gaseosa, la velocidad es independiente de la presi6n de ben- ceno y proporcional a la presi6n de H y algunas veces el ex- ponente (en la expresidn cingtica) de la pres36n de hidr6geno
se incrementa con la temperatura, adquiriendo un valor cerca-
2

35
no a la unidad.
'c a P' PY 'gH6 H2
Esta reacci-6n es efect.uada bajo diferentes condiciones des'detemperatura.ambfente hasta 50OOC y a presiones de 0.01 '
a 100 atm6sferas, dependlendo de la selectividad del catali- zador y a temperaturas altas se presenta una disminucf6n de la actividad catalltica (57).
Gallezot, et.al. (59) encontr6 que la velocidad de hi- drogenaci6n de benceno en partlculas de 10 A de platino en muestras de PtNaHY (No - de turnover de 80 a. 100 hr-' a 25OC) son comparables a las medidas en catalizadores de Pt/A1203
O
o Pt/Si02.
Naccache, et.al, (60) encontraron que la velocidad de hidrogenaci6n,a.29S°K es 100 veces nds rdpida en partkulas de 10 Aro en las de 15 a 20 A de platino en PtNaY, que en los catalizadores de Pt/A1,03 o Pt/Si02.
O O
Figueras, et.a1.(61) observaron que las actividades de los catalizadores de PdY para la hidrogenaci6n de benceno se incrementan cuando los sitios de Br'dnsted o cationes multivalentes estdn presentes. Por lo tanto las frecuencias del nbero de turnover en PdHY, PdLaY y PdCeY son 2.5 veces más grandes que en el catalizador de PdNaY y 4 veces mds grandes que en los catalizadores de Pd/A1203 y Pd/Si02.

36
111.1 Reacci6n
Las muestras'de cataltzador reducidas a 45OOC se estu- diaron en esta reacci6n de acuerdo al procedimiento siguien- te: se coloca una cantidad determinada de muestra (reducida) en el reactor diferenc2al que se utiliza en la reducci6n y
en los tratamientos termicos, s6l0 que esta ocasi6n se co- nect6 a un sistema de reaccfdn el cual est5 conectado a un cromatdgrafo de gases. Dlcho sistema de reacci6n se presen- ta en la Figura 3 .
Sistema catalltfco
a) saturadores de benceno b) reactor diferencial c) horno vertical el6ctrico d) . term6metro digital e) cromat6grafo de gases f) registrador g) vaso Deward
El cromat6grafo de gase.s utilizado es un aparato Per- kin-Elmer modelo Zigna 4B,. cuyas caracterSsticas son las siguientes: la columna est5 empacada con Carbowax 20N al 8 % Cromosorb W y es de acero inoxidable de un octavo de. pul- gada de diámetro y 2 mts. de longitud, la columna se mantie- .ne a una temperatura de 70OC. El cromat6grafo se encuentra conectado a un integrador Shimadzu C-R1A.
Ya fijadas las condiciones del cromat6grafo se permite el paso de hidrdgeno ( 2 . 4 ml/s) por el sistema de reaccidn hasta el saturador que contiene benceno, cuya temperatura es de 14OC con el fin de mantener constante la presi6n de va- -

37
O 0 O 0
I

l 1 " 1
38
por del reactivo (-56 torr) y asegurandode este modo una mez- cla benceno-hidr6gen0, con una relaci6n molar constante. Se verificar5 que las vblvulas que aislan el catalizador co- locado en el reactor diferencial est& cerradas para poder efectuar una corrida estsndar, esto es, se introduce la mez- cla gaseosa al cromat6graf0, por medio de una vdlvula de in- yecci6n para efectuar la separacidn de los reactivos. POS- teriormente se efectfia la reaccibn haciendo pasar hidr6geno por el saturador de benceno y por el reactor diferencial, efectuando la reacci6n desde temperatura ambiente hasta 6OOC. Los reactivos y productos son separados por la colum- na del cromat6grafo y determtnadas sus concentracZones. Pa- ra los diferentes catalizadares se sigui6 el mismo procedi- miento.
1) Cdlculos
Para el cdlculo de la ve1ocida.d de reacc26n en moles de benceno transformados por gramo de catalizador se utiliz6 la siguiente expresidn (62)
D X p x 2 7 3 x 1000 %C = 22400 760 T líl x 100
donde
D = flujo de hidrdgeno en ml/s P = presi6n parcia.1 del benceno en el saturador T = temperatura ambiente m = masa del catalizador %C = porcentaje de conversi6n

1 1 1 1 I
39
El cdlculo de la actividad por sitio, T.N,, que indica la relaci6n de mol6culas de benceno transformadas en la u- nidad de tiempo y por sitio activo de metal, se realiz6 con la siguiente expresf6n:
T.N. = . . . ,Y
nGmero de sftios actfvos/g de catalizador
V = velocidad de la reacci6n (moles/g S )
De acuerdo con la ecuacidn de Arrhenius es posible co- nocer la energJa de activacZ6n Ea para esta reaccibn.
k = constante de velocidad A = factor de frecuencia T = temperatura E = energla de activaci6n -a
Al representar grdficamente Ink contra el inverso de la temperatura absoluta, la pendiente de la recta obtenida nos permite determinar correctamente el valor de la energla de activacidn.
2) Resultados
. Los valores obtenidos de .energSas de activacidn son: - PtY 1% de 7 .40 Kcal/mol°K y para PtY 5% de 5.00 Kcal/molOK.

En la Tabla T I X se muestrqn los porcentajes de la conver- si&, las velocrdades y actividades por sitio para la reac- cfdn efectuada a la temperatura de 5OoC (325OK).
En ¡a Figura 4 se representa la velocidad espeeffica en funci6n de la temperatura para los diferentes catalizadores PtY al 1% y PtY al 5% en esta reacci6n.
. . . . .
Catalizador %C v ( ~ 1 0 ' ~ )moles/g s T . N . x
PtY 1% 1.35 1.02
PtY 5% 2.90 2.19
4 4
6 4
3 ) Discusidn
En primer lugar, se observa de la Figura 4 , que ambos catalizadores presentan ac:tividad para laihidrogenaci6n de benceno. Dicha actividad varfa increment6ndose en.el in- tervalo de temperaturas de 30 a 5OoC, para los catalizado- res con diferente contenido metblico.
Con el catalizador PtY al 5 % se observe una mayor con- versibn a ciclohexano en comparaci6n con el catalizador al

093048

1% de Pt, hasta una temperaturq de 5OoC, ya que a partir de esta temperatura se observa con el catalizador al 1% un in- cremento notable de la conversk6n a cfclohexano debido a la exotermicidad de la reaccidn.
Con el catalizador al 5% se observa que a partir de 4OoC
la conversi6n a ciclohexano permanece constante, lo cual nos lleva a pensar en una desactlvacidn de la muestra.

4 3
CAPITULO IY
DETERMINACION DE LA ACTIVIDAD CATALITICA EN LA REACCION DE HIDROCRACKING DE n-DECANO, DECALINA Y TETRALINA
La reaccrdn de hidrocrackfng es el mayor proceso de re- finado que se reali-za en presencfa 'de hidr6geno a presi6n. De esta manera el hidrocrackfng consiste en la conversi6n de hidrocarburos de alto punto de ebullicidn a productos de bajo punto de ebullfcibn (63) .
Las reacciones que ocurren y los productos formados de- Penden del balance entre las contribuciones de cada compo- nente.
Por ejemplo, cuando se efectQa la reacci.6n de hAdro- craking para la producci.6n de gasolinas, se requieren cata- lizadores con propiedades fuertemente acidas, tales como los silico-aluminatos. amorfos: y las zeoiitas ademds del com- ponente hidrogenante.
La presencia de estos materiales en los cataltzadores aceleran reacciones generalmente asociadas al cracking ca- talftico. Estas reacciones producen un alto porcentaje de productos ramificados y productos de cadena lineal, con- servdndose los compuestos de anillos monoclclicos, ademds
-

se efectfia una gran produccidn de is6meros debido a la rup- tura @, formando cati;ones terciaxios como intermediarios.
La funcf6n del componente hidrogenante (el metal) es la de evitar la formaci6n de oleftnas a partir de los cationes o para reducir compuestos de dobles enlaces que se encuen- tren como intermediarios, para obtener posteriormente los alcanos correspondientes.
El tratar de evitar al. mbximo la formaci6n de olefinas en los productos previene la polfmerizaci6n y en consecuen- cia tambi6n evita la formacidn de coque, el cual generalmen- te se deposita sobre los catalizadores y los desactiva.
Se tiene que cuando se hidrogena gas de aceite para producir combustibles de jets, se utilizan catalizadores con una acidez media y con una fuerte actividad hidrogenan- te.
En una categorla especial se estudian los catalizado- res de zeolitas selectivas, en las cuales su estructura porosa permite el acceso de determinados reactivos y la salida de productos y estd limitada a mol6culas de una for- ma y tamaño particular.
Entre los.metales con mayor fuerza hidrogenante se tie- ne al platino; nfquel, paladio, molibdeno y cobalto. Y entre los catalizadores con propfeda,des fuertEmente dcidas y actividad hidrogenante media se tiene a los sulfuros de metales, tales como nfquel, cobalto o tungsteno soportados en sflice-albnlna.

4 5
IV.1 Hidrocracking de alcanos
a) Catalizadores con propiedades fuertemente dcidas y con activrdad hidrogenante -media.
El sulfuro de nfquel sobre Si02-A1203 es uno de los ca- talizadores utilizados frecuentemente para el hidrocracking de gas oil comercial. Se han analizado los resultados de la distribuci6n de productos obtenidos en el hidrocracking de n-decano (63) con los siguientes catalizadores: catali- zador para cracking Si02-A1203 comercial, Ni depositado en Si02-A1203 y sulfuro de Nj, depositado en Si02-A1203.
El catalizador que contiene sulfuro de Ni presenta una mayor conversidn de n-decano que cualquiera de los otros dos catalizadores. La distribuci6n de productos en la reac- ci6n que se emplea el catalizador de sulfuro de Ni es simi- lar a la obtenida con el catalizador de Ni/Si02-A1203.
b) Catalizadores de alta actividad hidrogenante
Los catalizadores de alta actividad en la hidrogenaci6n 'que presentan una acidez media presentan distribuci6n de productos diferente a la obtenida con un catalizador de gran acidez.
i
Las reacciones que se rea1iza.n con este tipo de catali- zadores incluyen isomerizaci6n del reactivo sin efectuar el cracking e hidrogendlisis y en presencia de metales no- bles la cfclizaci6n.

I *
46
En soportes no bci.dos, la hidxogen6lisis es la Gnica reacci6n que presentqn los a,lca,nos con metales no sulfura- dos.
Algunos catalizadores de metales nobles frecuentemente son mencionados como catalizadores de hidrocracking y estbn constitufdos de Pt y Pd sobre Si03-A1203.
ASf 'mismo, se ha observado que en este tipo de catali- zadores con un incremento en el contenido de Pt; se efectGa una adsorci6n selectiva del metal por cada sitio bcido, el cual causa una reducclldn en la acidez de Si02-A1203.
c) Hidrocracking de alquilbencenos
LOS productos de la rea.ccf6n de hi.drocracking de alquil- bencenos, cuando se tiene como reactivo al cicloalcano, pre- sentan una amplia variación- y gran dependencia de la estruc- tura especlf ica dt21 re'activo.
E n la . . rea.cci.611 de t-pentilbenceno se obtienen como productos benceno e isopentano, en el caso de utilizar butil- benceno se obtienen benceno e hidrocarburos de C4 los cuales son una mezcla de butano e isobutano. Cuando los grupos al- quilo son mbs grandes el (anillo de benceno es atacado y los productos de reacci6n llegan a ser mbs complejos, observbndo- se una cicloalquilacibn.
Con catalizadores no 6cidos se efectGa la reacci6n de hidrocracking de a.lquilbencenos, y la reaccidn de hidrogen6- lisis, pero la reacci6n de isomerizaci6n presenta baja con-
-

4 7
versi6n.
Tambtgn es de importancta mencionar que otra caracterls- tica de los catali-zadores para hidrocracking, es que conten- gan al metal Bien dfsperso en los poros de la zeolita (64). Cuando un fuerte materfal hidrogenante como el Pt est6 pre- sente, se observa una alta selectividad hacia la hidroisome- rizaci6n asf como productos primarios .puros de cracking (me- tano, etano).
En este caso se puede tener un conocimiento considera- ble del mecanfsmo de la reacc26n,por esta raz6n Weitkamp(65) introduce el t6rmino de hidrocracking ideal. Tambign se ha reportado que el catalizador Pt/US-Y mostrd un comportamien- to ideal con el hidrocracking de n-decano como reaccidn prue- ba (66). Otra caracterfstlca de este catalizador es la ausen- cia de la desactivaci6n.
La hidroisomerizaci6n y el hidrocracking proceden prin- cipalmente por medio de lones carbonio (67). Los alcanos pueden ser convertidos en iones carbonio directamente por abstracci6n de un hidruro (-H-) o por protonaci6n (+H ) de intermediarios oleflnicos por deshidrogenaci6n en el metal.
+
El patr6n de reaccidn vla olefinas es todavla aceptado para reforming, hidrolsmerlzaci6n e hidrocracking de al- canos, sin embargo, este modelo est5 sujeto a discusi6n (67).
Los iones carbonio pueden reordenarse o bien seguir por una ruptura beta. Esta ruptura da origen a pequeños iones carbonio y a un fragmento oleffnico, el cual es inmediata- mente hidrogenado. La rdpida hidrogenaci6n de hidrocarburos insaturados es una raz6n superior para la ausencia de desac-

tivaci6n del catalfzador por coquificaci6n. Muchas reaccio- nes parqlelas y consecut$.yas de hidrogenacl6n, isomerizaci6n y crackl’ng toman lugar en la hfdroPsomerizaci6n y el hidro- crackhg.
Entonces se puede considerar al n-decano como el modelo ideal de hidrocarburo y un gran ntímero de productos son for- mados. El modelo cinetgco resulta ser como el de una reac- cZ6n compleja y es diffcil de tratar. La aproximaci6n comtín que se le da, es consfderar s6l0 la velocidad.de desapari- cidn global del hidrocarburo (n-decano) reactivo (68,691 .
Una investigacP6n detalla.da real.izada por WeitkamP, et. al. (65, 70, 71) sobre la distrPbuci6n de 10s productos en la hidroisomerizacf6n e hidrocracking de alcanos de cadena larga sobre zeolitas proporciona una importante informaci6n de las reacciones de reordenamiento y rompimiento de iones alquilcarbonio.
Tambfen los estudios realizados con n-decano y n-dodeca- no mostraron que la isomerizacf6n es una funcfdn Gnica de la conversidn total y la selectfvidad hacia isomerizaci6n es cercana al 100% a niveles de conversidn bajos, y esta obser- vaci6n hace suponer; la posibilidad de que un cracking direc- to del n-decano se realice sin que Este sufra un reordena- miento estructural.
En cuanto a la distribuci6n de productos se observa que se forman estructuras de alcanos monoramificados lo cual indica, que estas espectes son predominantes. La cantidad relativa de isomeros diramfficados y multiramificados en los productos de la reacci6n de cracking, se incrementan a altos niveles de conversi6n.
-

4 9
Se obsery6 que el hAdrocracking de n-alcanos es predeci- do por una hldroisomexAzqci6n y este reordenami-ento de la estructura de los n-alcanos ramificados, proviene .de una transfcidn de un 26n carbon.io secundario hacia un i6n car- bonio terciarfo , asi estructuras altamente ramificadas son formadas por reacciones consecutivas, por lo tanto el nhe- ro total de isdmeros que se producen es mayor cuando se tie- nen alcanos de cadena larga.
Por ejemplo, se reporta que de los productos de alcanos de cadena larga [como el n-decano) han sido fdentiffcados 20 isodecanos, de un total de 70 isdmeros posfbles,
Ass mismo, es importante hacer notar que estructuras ra- mificadas pueden presenta,r romp2mientos cuando son adsorbi- das como iones terciarios en lq superficie de la zeolita.
Ademds algunas mol6cul.as de varias ramiffcaciones que forman iones carbonio pueden romperse de acuerdo a 2 6 3
rutas alternas. De la existencia de diferentes rutas depen- de la obtenci6n de productos cra.queados.
Tambih Pniak y Rodomyski. ( 7 2 ) reportaron que para pro- cesos de transformacl6n de naftas pesadas en gasolinas, la actividad de los catalizadores de Pt soportado en zeoltta Y mezclada con gama-alGmi.na, en la reacci6n de hidrocrackhg de n-decano y la distribuci6n de productos fue: productos de isomerizacidn Cisodecanos), productos de hidrocracking y
de dehidrockclizaci6n (arom6ticos de punto de ebullicidn de temperaturas mayores a. las de el n-decano). No se detect6 la presencia de me.tano, benceno, tolueno y xilenos,

50
Las selectividades p’ara esta reacci6n muestran que la posibilidad de romper enlaces centrales C-C en la mol6cula de n-decano, es la mfsma con catalizadores a los cuales se les varfa el contenido de gama-altímina.
Entre los productos detectados en el cracking se en- cuentran pentanos, hexa.nos e isopentanos.
Analizando el mecanl’smo de conversidn de n-decano, co- mo un mecanismo bifuncional cldsico ( 7 3 ) se observan los siguientes pasos:
Decano + H+ -H2 $. Pt + +H2 > iones decilcarbonio
(decanos)
isodecanos S H 2 $. Pt + -H2 < + ioñes decilcarbonio
ramificados. B2 .I -H isodecanos -H + B,
J.
productos craqueados
+ H2 Pt - H2
productos saturados craqueados.

En este mecanismo observamos que los decanos son adsor- bidos en la superficie del platino sopartado en la zeoli- ta-H.
La velocidad de reaccE6n esta determinada por el reorde- namlento de los iones carboni-o del n-decano.
La selecthidad entre el de cracking (B2) y la isomeri- zaci6n (.B1) es determinada (en la ruta A todos los factores permanecen iguales), por la vida media de los iones carbo- nio; los tiempos de vtda media podrían ser mayores como re- sultado de la presencia de fuertes sitios dcidos y por lo tanto existir6 una alta conversidn de productos craqueados.
El valor mdximo para la selectividad de isomerizacidn es tambien determinado por la fuerza de los sitios de Briinsted involucrados en la formaci6n de iones carbonio. Si se efecttia un mecanismo bifuncional, la selectividad hacia isomerizaci6n ser6 menor en presencia de sitios mas fuertes de Bronsted ( 7 3 , 7 4 ) . -
Cuando se tiene la mayor parte del metal presente, can tamaños de partícula de .20 nm de didmetro fuera de las ca- vidades de la zeolita., el criterio de catdlisis bifuncional no es aplicable, ya que la distancia entre las partículas metdlicas y los sitios de Br'dnsted es muy grande ( 7 0 ) .
Beecher, et.al. (-75) estudiaron la reacci6n de hidro- cracking de n-decano y decalina con un catalizador de Pd-so- portado en mordenita-H a una temperatura de reaccf6n de 4 5 0
a 610OV. El n-decano es hidrocraqueado con mayor facilidad que la decalha. La distribuci6n de productos obtenida pa- ra el n-decano es menor en C3-C4 y mayor en C 4 , C5 y C6.
-
093048

1 L 1 I
52
En el caso del hidrocracking de decalina se observa tambien produccfdn de C3-C4 en menor relaci6n y una mayor relac26n de C4-C4, as$ como la producci6n de C5 y c6 y com- puestos de un solo anillo.
Como se ha observado, en los Clltimos años la reaccidn de hidrocracking con cataLizadores de pt soportados en zeo- litas ZSM-5, son de gran i:nteres por su utilidad en una amplia variedad de reacciones (,76, 7 7 ) .
W.2 Reaccfh catalft2ca de h.2drocracking
Para la determTnaci6n de la actividad en la reacci6n de hidrocracking con n-decana, decalina y tetralina se uti- liza un sistema constituldo por un reactor diferencial, hor- no electrico, saturador de arrastre, un cromat6grafo de ga- ses y un integrador.
El cromat6grafo consta de las siguientes caracterfsti- cas; es un aparato Perkin-Elmer 3920-B con una columna ca- pilar de acero inoxidable de 15Q pies de longitud y .O10 pulgadas de didmetro, empacada con escualeno, La temperatu- ra de la columna es de 100OC. El integrador utilizado es marca Shimadzu modelo C-RlA,
En el reactor se coloca una determinada cantidad del ca- talizador tratado de una manera similar a los catalizadores utilizados en la hidrogenaci6n de benceno, Se eleva la tem- peratura hasta 45OOC manteniendo el catalizador bajo corrien- te de hidr6geno.
Se reactiva a esta temperatura durante una hora y enton--

53
. ces se enfrsa hasta 25OOC repitiendose la reacci6n con tem- peraturas de 275, 300, 35Q y 4OOOC.
Se efectca una corrida est6ndar antes de realizar la reacci6n, posterformente se realiza esta con flujo de H2 a las temperaturas antes descrftas.
El saturador que contiene al reactt.vo se mantfene a temperatura ambiente, 1a.s presiones de vapor para cada reac- tivo son: pv. n-decano, 1.7321 torr, PY cis-decaina 1.169 torr y para la trans-decaina 422 torr.
1) C6lculos
Para calcular la yeloct.dad de xeacci,6n, en moles de n-de- .
cano, transformadas por segundo'y por gramo. de catalizador, se utfltza la misma expresi6n que para la hkdrogenaci6n de benceno :
D P 273 x 1000 x '% C " = 22400 x 760 T m 100 x --
en el caso del n-decano:
D = lml/s P F; 1,7321 torr m = 40 mg

I ” , , -
5 4
Habiendo calculado 1s velocidad de reacci6n se realizan los cdlculos de la actji.vidad por sitio, T.N.
‘ V = No - de sttios activos/g de cat
Y = velocidad de reaccidn (moleslg S)
Nota: el nhero de sitios acttvos conszderado fue en base a la superffcie metbl.i.ca.
Para el cdlculo de la selectivfdad, de acuerdo con los valores obtenidos de % de conversidn en el cromatograma, se relacionan con un factor llamado F, defintendose como la relaci6n del peso molecular del producto obtenido entre el peso molecular del reactivo.
P.M. del Producto = P.M. del reactfvo
Este factor calculado se multfplica por la conversidn registrada del cromat6grafo y la suma de estas nuevas con- centraciones representan el 100% de la conversi6n y poste- riormente s610 se necesita relacionar cada concentracidn a esta suma para conacer la abundancia:
%-Kg - - ,7408
%-Cs. = 11.4162
%-iCl0 = 87 .8443

".
55
De una manera similar se efectuaron los cSlculos de selectividad para las reacciones de hidrocracking de deca- lina y de tetralina.
2) Resultados
En la Figura 5 se representa la variaci6n de la velo- cidad especlfica por sitio activo en la reaccidn de hidro- cracking de n-decano efectuada entre 250 y 400OC.
En la Figura 6 se muestra la variacidn de la veloci- dad especffica por sitio en funci6n de la temperatura para
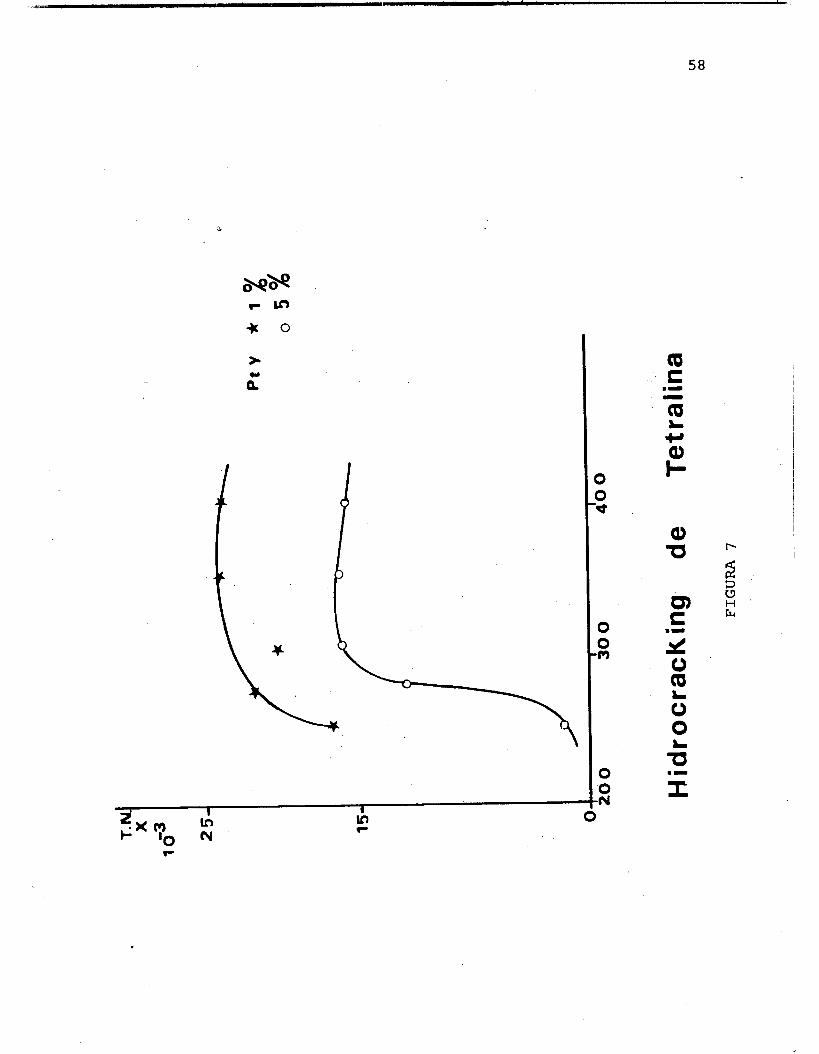
:.. la reaccidn de hidrocracking de decalina y en la Figura 7 se representan los resultados de velocidad especffica para la reacci6n de tetralina.
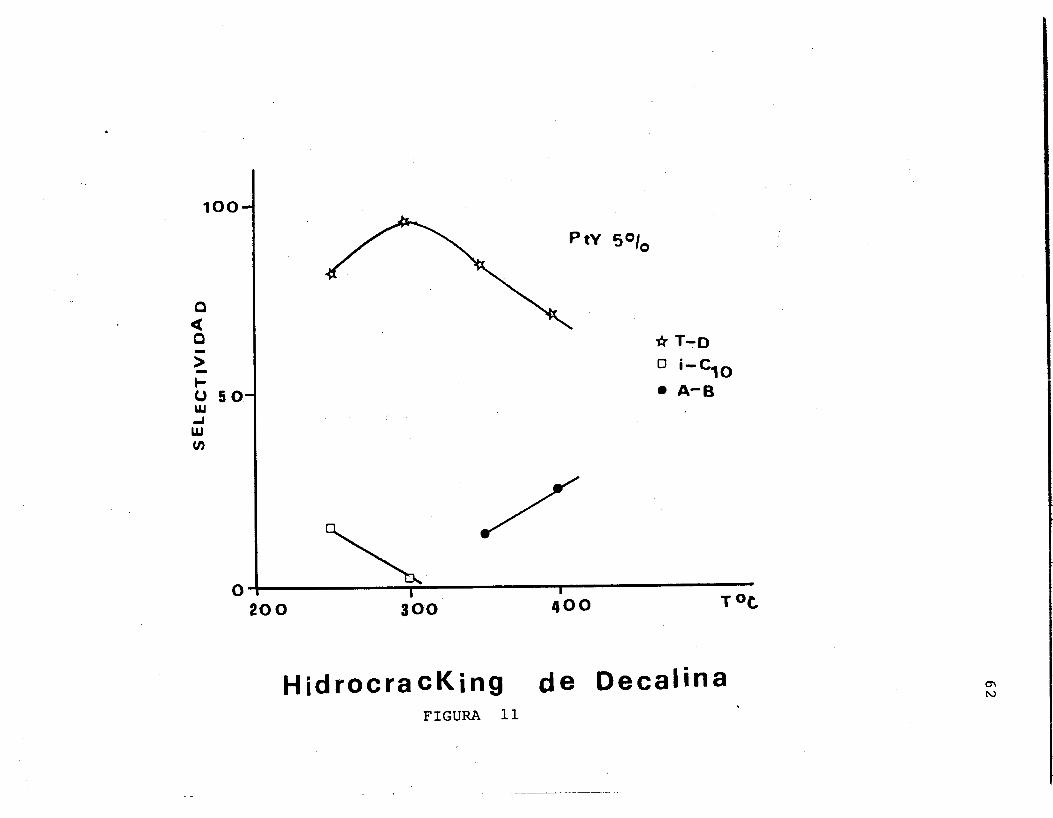
En las Figuras 8 , 9 , 10, 11, 12 y 13 se representan los valores de selectividades de los diferentes productos obte- nidos en las pruebas catallticas realizadas.
3 ) Discusidn
De los resultados presentados en las Figuras 5, 6 y 7
-

t . , " "
56

1 . .
57
2 1
I
O ,o d
O 'O -m
O O
-cy
58
c
o co o O
a S
L
L
.I

O
I G .-
d I o .-
I
o O c
O m
O u c
1 O O
-e
1
O ,o o
O I ,o
(Y
O
59

60

L "
61
\
I o I=
O O 'd
O O
O O 4
o O
'L3
I
L
.I
100
5 0
C 20 o

63
o O
+
L

64
I O m
t
OVQIAl113313S

,
65
los dzferentes ca,talizadores estudiados son activos en las reacciones de hidrocra.cking de n-decano, decalina y tetra- lina.
En el caso de los catalizadores al 1% y 5% de Pt en la reacc26n de n-decano, estos presentan un comportamiento similar en su velocidad especsfica por sitio metblico. Es- te valor es similar al encontrado por Steijins (66) y es posPble explicarlo en base a la facilidad de acceso de las mol6culas lineales a los sitios activos localizados en el interior de la zeolita.
En relacf6n a la velocidad en la reacci6n de hidro- cracking de decalina, se encuentra tarnbien un comportamien- to similar en ambos catalizadores hasta una temperatura de 35OOC y a partir de esta temperatura el catalizador al 1% de Pt presenta una actividad superior (10 veces) con rela- cidn al catalizador al 5% de Pt. Esto podrPa explicarse debido a la variaci6n del car3cter 5cido de los sitios ac- tivós ya que al incrementar la temperatura se favorece la fuer.za bcida en el caso de los catalizadores m& disper- sos (73).
En el caso de los catalizadores al 5% de Pt utilizados en la reacci6n de hidrocracking de tetralina,se encuentra una menor actividad.icatalPtica que para los catalizadores al 1%. A partir de 300°C ¡a actividad catalltica permane- ce constante, dado que la reacci6n se realiza sobre la su- perficie externade la zeolita.
Al analizar los resultados de selectividad encontramos que para la reaccidn de hidrocracking de n-decano, Figuras 9 y 10, la distribuci6n de productos es similar a la obser- "

1 ” ””
66
vada por Steijins (661, en donde se presenta una mayor iS0-
merizaci6n y una menor cantrdad de hidrocracking a tempera- turas menores de 25OOC y a partir de esta temperatura la reaccidn de hidrocrackkng es superior a la de isomerizaci6n.
Con el catalizador de PtY al 1% encontramos que a par- tir de 25OOC el producto princ2pa.l de reaccidn es nC5.
A partir de 3OOOC se empiezan a producir reacciones Se- cundarias, tales como la isomerizaci6n de los productos cra- queados y se llega a detectar la presencia de iCl0.
En el caso del catalizador al 5 % de Pt a la temperatura de 25OOC se presentan reacciones consecutivas de hidro- cracking e isomerizaci6n del n-decano.
Es interesante notar la presencia de una pequeña frac- cidn de iC6 en un intervalo de temperatura de 300 a 400OC.
En el caso de la reaccidn de decalina con el cataliza- dor al 1% de Pt se encuentra, que a partir de 3OOOC la produccidn de nC4 e iC4 es favorable y en el intervalo de temperatura de 300 a 4OOOC se encuentra un incremento gra- dual de la Trans-decalina, asf mismo, a partir de 35OOC este producto presenta una disminuci6n y empieza el rompi- miento y la dehidrogenaci6n de uno de los anillos de la mo- lgcula, siendo corroborado por la presencia de compuestos alquilbencenos.
Con respecto al cataltzador al 5% de Pt no se presenta la formacidn de hidrocarburos ligeros, la reacci6n esta orientada preferentemente a la formaci6n de iClo y a Trans- decalina, con esta molEcula, Trans-decalina se presenta un
-

67
mdximo de selectividad a 300.OC y a partir de esta tempera- tura la selectividad empieza a di-sminuir, nuevamente se encuentra la producci6n de alqutl-bencenos.
Esto podrIa atribuirse a que la reacci6n de hidrocracking se efectGa en la superficie externa de la zeolita debido a que es una molecula ciclica y el acceso al interior de la zeolita resulta diflcil.
Para la reacci6n de htdrocracking de tetralina, los ca- talizadores al l% y al 5% de Pt presentan un comportamiento muy similar, a partir de 25OOC la selectividad est5 orien- tada a la formaci6n de iC5 e ' i C 4 principalmente y a partir de esta temperatura se encuentra una disminución en la se- lectividad de estos productos, entonces la reacci6n se di- rige a la producci6n de Trans-decalina. Esto puede expli- 'carse tambien, a que la tetralina es una mol6cula cfclica y su acceso a las cavidades de la zeolita es poco probable como en el caso de la decalina,

CAPITULO V
CONCLUSIONES
1. Ambos catalizadores, PtY al 1% y PtY al 5% presen- tan actividad catalftica en la reacci6n de hidrogenacidn de beceno y en la reaccfdn de hidrocracking de n-decano, deca- lina y tetralina.
2 . En la reacci6n de hidrocracklng de n-decano, deca- lina y tetralina, la dfstribuci6n de productos resulta di- ferente con cada uno de los reactfvos, debido a que la mo- lgcula de n-decano es un hfdrocarburo lineal y su difusldn y acceso al interior de las cavidades de la zeolita resulta mbs factible que con las mol6culas de hidrocarburos cPcli- cos, como lo son la decalina y la tetralina.
3 . En la reacc$6n de hi.drocracking con n-decano, se en- contr6 preferentemente la formacidn de isheros de C5, C4 y
Cl0. En el caso de la reacci6n con decalina, es notoria una alta producci6n de Trans-decalina en el intervalo de
. temperatura de 300 a 4OOOC y a partir de 35OOC se observa la produccidn de alquil-bencenos, lo cual es trascendente debido a la importancia de estos productos en la industria petroqulmica.

69
BIBLIOGRAFIA
1. Thomas, Ch. L., "Catalytic Processes and Proven Cata- lysts", Academic Press, New York and London, 1970.
-2. Weiz, P.B., Advan. Catal. (1962) 13, 137.
3. Slenkin, A.A., Federovskaya, E.A., USP. Khim (1971140, 1857.
4 . Ciapetta, F.G., Wallace, D.N., Catal. Rev. (1971)5,67.
5. Cinneide, A.D.O., Clarke, J.K.A., Catal. Rev. (197217, 213.
6. Whyte, T.E., Catal. Rev. (1973)8, 117.
7. Rabo, J.A., Schomaker, U., Pickert, P.S., Proc. Intern. Congr. Catal., 3rd. Amsterdam. 1964 (1965)2, 1264.
8. Rabo, J.A., Pickert, P.E., Mays, R.L., Inds. Eng. Chem. (1961) 53, 733.
9. Weisz, P.B., Frielitte, V.J., J. Phys. Chem. (1960)64, 382.
10. Wiesz, P.B.,Fi?ielitte, V.J. Maatman, R.M., Monier, E.B., J. Catal. (1962) 1, 307.
11. Minachev, Rh. M., Garanin, V.I., Knarlamov, V.V., Isa- kova, T. A., Kinet. Catal. (1972) 13, 1104.
12. Minachev, Kh. M., Garanin, V.I., NOVUSOV,T-A-,- Izv.Akad- Nauk, SSSR, Ser. Khim. (19731330.
I

70
13. Aganof, A.V., Maslyanskii, G.N., Rogov, S.P., Havkin, V.A., Shipikin, U..V., Pannikova, R.F., Weltepererabetka Neftekhim. (1971)4, 1.
14. Weisz, P.B., Frieletke, V.J., J. Phys. Chem. (1960164, 382.
15. Weisz, P.B., et.al., J. Catal. (1962)1, 307.
16. Chen, N.Y., Weiz, P.B., Chem. Eng. Progr. Sympos. Ser. (1963) 63.81.
17. .Weisz, P.B., Erdol Kahle (1965) 18, 525.
18. Penchev, V., et.al., Advan. Chem. Ser. (1973)121, 461.
19. Galich, P.N., et.al., Sb. "Sintety Cheskiye Zeolita, izd. An. N. SSSR.M., (1962).p. 260.
21. Galich, P.N., Gutyrya, V.S., Neimark, J.E., et. al., SB. Neftekhimiya, Naukova, Dumka, Riev, (1964), p.13.
22. Kubo, T., Arai, H., Tominaga, H., Kumugi, T., Bull. Chem. Soc. Japan (1972)45, 613.
23. Breck, D.W., Milton, R.N., U.S.Patent 3, 013082; Rzn. Khim, (1963), 8 12111.
24. Pickert, P.E., U.S. Patent 3, 236, 904; C.A. (1966164, * ' 17478a. -

71
25. Ryashententseva, M.A., Minachev, Kh.M., Neftekhimia (1971) 11, 198.-
26. Minachev, Kh. M., Antoshin, G.V., Shpiro,, E'.S., Isakov, Ya, I., ,Izv. mad. Nauk. SSSR, Ser. Khim (197312131.
27. Rabo, J,A., Pickert, P.E., Stamires, D.'N,, Boyle, J. E., Actes Deuxieme. Congr. Internat. Catalyse, Paris, (1960)1961, 2, 2055.
28. Rabo, J-A., Pickert, P.E., Mays, R.L., Ind. Eng. Chem. (1961) 53, 733. I
29. Minachev, Kh.M., Garanin, V.I., Kharlamov, V.V., Isako- va, T.A., Kinet. Catal. (1972)13,1104.
30. Yamamoto, N., et.al. Bull, Japan.Petrol.Inst.(l966)3.13.
31. Minachev, Kh.M., Garanin V.I., Isakov, Ya.I., et-al., Ibid., p. 374.
32. Okruzhnov, A.M., Izmailov, R.I., Verobyanz, R.A., Neftek- himiya (1964)4, 850.
3 3 . Isakov, Ya.I., Minachev, Kh.M., Usachev, N.Ya., Izv. mad. Nauk. SSSR. Ser. Khim (1972)1175.
34. Minachev, Kh.M., Garanin, V.I., Isakov, Ya.I., "Zeolity, iKh sintez, svoistvai primenenie, Nauka, M.L., (19651, p. 374.
35. Minachev, Kh. M., Garanin, V.I., Piguzova, L.I., Vitukhi- na, A.S.., Izv. Akad. Nauk. SSSR, Ser. Khim (1966)129.
-
Y

72
36. Minachev, Kh. M., Garanin, V.I., Piguzova, L.I., Vitikhi- na, A. S . , Izv, Akad. Nauk. SSSR, Ser. Khim (1966)llOl.
37. Richler, H., Schulz, H., Restemeyer, H.O., Weitkamp, V., ErdB1 Khole-Erdas-Petrochem.Ver.Breunst-Chem.(l972)25, 494.
38. Kittrell, J.R., U.S. Patent 3, 558, 472, R.Zh.Khim, (1971), 19, 25711.
39. Kluksdahl, H.E., U.S. Patent 3,660, 310; C.A. (1972)17, 51003a.
40. Pollitzer, E.L., U.S. Patent 3, 594, 310; R.Zh. Khim., (1972), 81111811.
41. Penchev, V., Koleav, M., Izv. m a d . Nauk. SSSR Ser- Khim. (1966) 580.
43. Cole, J . E . , Konmenhoven, H.W., Advan. Chem. Ser.. (1973) 121, 583.
4 4 . Volsfon, I.s., Okruzhnov, A.M., Neftepereraboka. Neftek- him. (1971)8,22.
45. Voorhies, A., Hatcher, W.J., Ind. Eng. Chem. Prod. Res. Develop. (1969)8, 361.
46 . Yan, T . Y . , J. Catal. (1972)25, 204.
-

73
..- . ,
47. Weitkamp, J., Schulz, H., J. Catal. (1973)29, 361.
48. Weiz, P.B., 'Advan. Catal. (1962113, 137.
49. Beecher, R., Voorhies, A., Eberly, P., Ind. Eng. Chem. Prod. Res. Develop (196837, 207.
50. Gallezot, P., Catal. Rev. Sci. Eng., 20, 1, 121-154(1979).
51. Thomas, S.J. and Webb, G. Heterogeneous Catalysis. Oliver and Boyd. London. (1969).
52. Anderson, J.R., Structure of Metallic Catalysts. Acade- mic Press. London (1975)
53. Rabo, J.A., Schomaker, V. and Pickert, E.P., in Procee- dings of the 3rd International Congress on Catalysis, Amsterdam, (1964) 2, North Hollañd.
56. Marchand, I., Tesis Maestrla (1984), U.A.P.
57. Thomas, J.M. and Thomas, W.J., Introduction to the prin- cipes of heterogeneous catalysis. Academic. Press. Lon- don (1975).
58. Selwood, P.W., J. Am. Chem. 79. 4637 (1957).

59.
60.
61.
62.
63.
64.
65.
66 .
67.
68.
69.
Gallezot, J., Datka, J., Massadier, M., Primet and B. Imelik, in Proceedings of the 6th. International Con- gress on Catalysis, London, (1976)2, Chemical Society, London, p. 696.
Naccache, C., Kaufherr, N., Dafaux, M., Pandiera, J. and B.Imelik, in Molecular Sieves-I1 (J.R. Katzer, ea), Am. Chem. Soc., Wash. D.C. (1977), p . 538.
Figueras, F., Gbmez, R., and 'Primet, M., Adv. Chem. Ser., 121, 4 8 0 (1973).
Villamil, A.R.P., Tesis Maestrfa (1983)U.A.M.-I.
Pines, H. The Chemistry of Catalytic Hydrocarbon Conver- sion, Academic. Press.N.Y. (1981).
Jacobs, P.A., Carboniogenic, Activity of Zeolites, Elsevier Scientific Publ. Co. Amsterdam, Oxford, N.Y. (1977).
Weitkamp, J. ACS Symp. Ser. (1975) .20.1.
Steijins, M., Froment, G.F., Jacobs, P.A., Uytterhoeven, J.B., Weitkamp, J. Erdol, Kohle, Erdgas, Petrochem. (1978), 31. 538.
Poutsma, M.L. A.C.S., Monogr. (1976)171, 583.
Beecher, R., Voorhies, A., Eberly, P. Ind. Eng. Chem. Prod. Res. Dev. (1968) 7 , 203.
Engler, B. PhD. Tesis, Technical, University, Aachem Federal Republic'of Germany (1976).

75
70. Weitkamp, J.., Erd61, Kohle, Erdgas, Petrochem. (1978) 31,13.
71. Weitkamp, J. Farag. H. Act. Phys. Chem. (1978124, 327.
72. Pniak, B., and Radomyski, B., React. Kin&. Catal. Lett- (1983)23, 3-4, 387-391.
73. Rudham, R., and Stochwell, S . , in Catalysis by Zeolites vol. t (B. Imelik, et.al. edc.) Elsevier, Amsterdam (1980) , p. 113.
74. Coormadt, H.L., and Garwood, W.E., Ind. Eng. Chem. Prod. Res. dev. (1964) 3.38.
75. Beecher, R., and Voorhies, A. Jr., Eberly, P. Jr. Iand Ec. Product. Research and Development (196817, 3'sep.
0
76. Jacobs, P., Catal. Rev.-Sci. Eng. (1982) 24 (3) .415-440.
77. Bizreh, Y.W., Gates, B.C., J. of Catal. (1984)88, 240-243.
78. Gallezot, P., Catal. Rev. Sci. Eng. 20, (1) , 121-154(1979).