quantitative functional measurement of a protein …
TRANSCRIPT

The Pennsylvania State University
The Graduate School
Integrative Biosciences
QUANTITATIVE FUNCTIONAL MEASUREMENT OF A PROTEIN
USING PHYLOGENETIC PROFILES
A Dissertation in
Integrative Biosciences
by
Kyung Dae Ko
2009 Kyung Dae Ko
Submitted in Partial Fulfillment of the Requirements
for the Degree of
Doctor of Philosophy
August 2009

The dissertation of Kyung Dae Ko was reviewed and approved* by the following:
Randen L. Patterson Assistant Professor of Biology Dissertation Advisor Chair of Committee
Réka Albert Professor of Physics and Biology
Anton Nekrutenko Associate Professor of Biochemistry and Molecular Biology
Michael N. Teng Assistant Professor of Biochemistry and Molecular Biology
Damian van Rossum Assistant Professor, Research of Biology
Peter Hudson Willaman Professor of Biology Director, Huck Institutes of the Life Sciences
*Signatures are on file in the Graduate School

iii
ABSTRACT
In principle, the amino acid sequence of a protein contains structural, functional,
and evolutionary characteristics. Investigating these characteristics using computational
methods provides a powerful resource. However, these methods have limitations in their
ability to annotate the characteristics of proteins accurately. In an attempt to overcome
this drawback, I have developed a unified computational pipeline, called the Gestalt
Domain Detection Algorithm Basic Local Alignment Tool (GDDA-BLAST), for
measuring the structural, functional and evolutionary characteristics of a protein using
phylogenetic profiles. The performance of GDDA-BLAST is better than those of other
method such as SAM and psi-BLAST in homology detection.
Using GDDA-BLAST, I also implemented a classification library to find
quantitative thresholds capable of inferring protein function using phylogenetic profiles.
Using this library, I identified RNA-binding Proteins (RBPs) containing structural unique
motifs by 2695 expanded Position Specific Scoring Metric (PSSM) profiles in a testing
dataset with 37 positive and 118 negative sequences. We achieved 100% specificity,
96.8% accuracy, and 86.5% sensitivity. For the specific nucleotide binding folds (dsRNA
vs. dsDNA, dsRNA vs. dsDNA, and ssRNA vs. ssDNA), our results exceeded those of
obtained using Support Vector Machine (SVM) learning algorithms. Using this method, I
also identified 29 and 168 novel RBPs in yeast and human proteomes. These results
suggest that this method can be used to create PSSM databases for the quantitative
measurement and classification of any protein function.

iv
TABLE OF CONTENTS
LIST OF FIGURES .....................................................................................................vi
LIST OF TABLES.......................................................................................................viii
ACKNOWLEDGEMENTS......................................................................................... ix
Chapter 1 Introduction .................................................................................................1
1.1 Current computational methods for the prediction of protein characteristics ................................................................................................2
1.2 Motivation and Objective ...............................................................................10
Chapter 2 GDDA (Gestalt Domain Detection Algorithm) – BLAST (Basic Local Alignment Tool) with Phylogenetic Profiles........................................................12
2.1 Backgrounds and Motives ..............................................................................12 2.2 GDDA-BLAST with phylogenetic profiles....................................................14 2.3 The prediction of functional characteristics of proteins by GDDA-BLAST..17 2.4 The investigation of evolutionary relations among proteins using GDDA-
BLAST ..........................................................................................................20 2.5 The prediction of structural boundaries of ion-channels using GDDA-
BLAST ..........................................................................................................22 2.6 The discovery of novel lipid-binding domains in vitro ..................................24 2.7 Summary and discussion ................................................................................26
Chapter 3 The Performance of GDDA-BLAST in homology detection .....................29
3.1 The backgrounds and Motives........................................................................29 3.2 Results and Discussion ...................................................................................33
3.2.1 Datasets for the performance evaluation ..............................................33 3.3 Homology detection methods for the performance evaluation.......................35 3.4 The performance evaluation ...........................................................................37 3.5 Summary and discussion ................................................................................41
Chapter 4 The identification of RNA binding proteins using the quantitative functional measurement........................................................................................44
4.1 A classification library for RNA binding proteins .........................................46 4.2 The identification of RNA binding proteins...................................................54 4.3 The investigation of functional relations among RRM containing proteins...64 4.4 Summary.........................................................................................................67
Chapter 5 Summary and Discussion ............................................................................70

v
5.1 Summary.........................................................................................................70 5.2 Discussion.......................................................................................................74
Chapter 6 Future Perspectives .....................................................................................77
Bibliography ................................................................................................................83

vi
LIST OF FIGURES
Figure 1-1: Homology-based methods.........................................................................3
Figure 1-2: The schemes of machine learning methods. .............................................8
Figure 1-3: The schematic diagram of a phylogenetic profile method for function inferences..............................................................................................................9
Figure 2.1: The workflow of GDDA-BLAST. ............................................................16
Figure 2-2: GDDA-BLAST model of the ATP-binding Ankyrin Repeat in TRPV1...............................................................................................................................19
Figure 2-3: Water Channel (Aquaporin) Phylogeny....................................................21
Figure 2-4: GDDA-BLAST models of the ion transport domain in TRPC channels...............................................................................................................................23
Figure 2-5: Functional Information via GDDA-BLAST analysis. ..............................25
Figure 3-1: The statistical information of protein families.. ........................................32
Figure 3-2: Five hierarchical levels of SCOP classification........................................34
Figure 3-3: The schemes of homology-based methods. ..............................................36
Figure 3-4: The ROC graphs for the performance evaluation of GDDA-BLAST.. ....39
Figure 4-1: The structures of RNA binding proteins. ..................................................45
Figure 4-2: The overview of EMSA. ...........................................................................47
Figure 4-3: The problems of functional annotations in conventional programs..........48
Figure 4-4: The pipeline of GDDA-BLAST for the identification of RNA binding proteins. ................................................................................................................49
Figure 4-5: The false positive sequences in phylogenetic profiles from GDDA-BLAST..................................................................................................................50
Figure 4-6: A classification library for the identification of RNA binding proteins. ..50
Figure 4-7: A residue-based phylogenetic profile. ......................................................53

vii
Figure 4-8: The sequence of a hypothetic protein for describing derivation of the feature vector of a protein.....................................................................................54
Figure 4-9: Thresholds for the positive sequences in training sets. .............................55
Figure 4-10: The identification of RRM containing proteins in a testing dataset containing 20 positive and 137 negative sequences. ............................................56
Figure 4-11: The threshold for the classification of single-stranded RNA binding proteins. ................................................................................................................60
Figure 4-12: The classification between double-stranded DNA and single-stranded RNA binding proteins. ...........................................................................62
Figure 4-13: The classification among other DNA and RNA binding proteins ..........62
Figure 4-14: The dendrogram of control sequences. ...................................................65
Figure 4-15: The proteins with U2AF-homology motif (UHM). ................................66
Figure 6-1: The functional dendrogram for the prediction of UHM proteins. Orange boxes indicate the UHM control sequences.............................................79
Figure 6-2: The prediction of UHM candidate proteins. .............................................80
Figure 6-3: The inference of new annotations from reference annotation of NP_005869. ..........................................................................................................82

viii
LIST OF TABLES
Table 4-1: The comparison of the performances between Interproscan and SVM .....48
Table 4-2: Comparison with the sensitivities of other methods for the identification of RRM containing proteins in training and testing sets. ...............57
Table 4-3: Comparison with the sensitivities of other methods for the identification of the four single-fold RNA binding protein groups in training sets. .......................................................................................................................58
Table 4-4: The results of identification for five RNA binding protein groups in yeast and human proteomes..................................................................................58
Table 4-5: The comparison with other methods. .........................................................60
Table 4-6: The classification among six types of RNA binding proteins such as double-stranded RNA binding vs. double stranded DNA binding proteins, single-stranded RNA binding vs. double-stranded DNA binding proteins, and single-stranded RNA binding vs. single-stranded DNA binding proteins. ..........61

ix
ACKNOWLEDGEMENTS
Most of all, I really appreciate God to guide and support me in my graduation, and I am
indebted to many people who have significantly helped me shaping my dissertation.
It is difficult to overstate my gratitude to my advisor, Dr. Randen Patterson. With his
enthusiasm and his inspiration, I was able to endure a long journey at Penn State
University. In my research, he gave me encouragement, advice, and lots of good ideas,
and I could not finish my dissertation without him. I would like to extend my gratitude to
Dr. Damian van Rossum for many things. Whenever I lost my path in my research, he
always helps guided me back to the light. I am also grateful to my thesis committee
members, Dr.Réka Albert, Dr.Anton Nekrutenko and Dr. Michael N. Teng. They
provided me with insightful comments and enthusiastic support during my thesis.
I would like to express my appreciation for my colleagues in the lab, including YooJin
Hong and Gaurav Bhardwaj. I will never forget their support during my research.
I also express my thanks to my friends: Bob and Marin Ford, and Ken and Joyce Layton.
I cannot thank you enough for your support of me and my family.
Finally, I owe everything to my family: my father, my mother, my brother, my father-in-
law, and my mother-in-law for their love and support. I would like to express my deepest
gratitude to my wife for her patience, love, and support. My daughter, Grace, you always
give me happy smile whenever I am exhausted. To them I dedicate my thesis.

Chapter 1
Introduction
Proteins in a cell are involved in the development and process of a cell. Therefore,
the analysis of structures and functions for proteins is important to understand the
pathways of cell interactions. The study of protein evolution also provides clues to
genetic historical information, which shows various combinations during the molecular
evolution. Therefore, the identification and classification of proteins are important in the
analyses of structural and functional characteristics and investigation of molecular
evolutionary history for proteins
A protein usually consists of domains, which can be independent of the rest of the
protein chains. Domains fold autonomously and can bind to ligands or other domains [1].
Domains are components of the protein structure, and can work in the protein as
functional units. Domains may also exist in various evolutionary related proteins among
species. Therefore, the detection of domains plays a very important role in the
identification and classification of proteins.
However, overwhelmed with predicted proteins from genomes, we face several
obstacles to annotate structural, functional and evolutionary properties of proteins. First,
even though experimental methods identify many uncharacterized proteins in proteomes,
the annotations of these proteins take longer time than the identification, and the existing
erroneous annotation can generate the false annotation of a new protein in some case.
Second, the annotation requires the accurate subjective and contextual definition of a

2
protein function because the protein may have multiple functions. Because of these two
problems, the accurate structural and functional annotations of a protein are the
challenging tasks in all biological fields [2]. In this chapter, we start to review current
computational methods for the prediction of protein characteristics and describe their
deficiencies, which lead to our motivation for the development of GDDA (Gestalt
Domain Detection Algorithm)-BLAST. Then, we conclude this chapter by discussing
motivations and objectives of our research in more detail.
1.1 Current computational methods for the prediction of protein characteristics
In principle, the amino acid sequence of a protein can contain structural,
functional, and evolutionary characteristics, and the characteristics have been
investigated using many computational methods such as homology detection, machine
learning method, and phylogenetic profile.
Among these methods, the simplest and fastest algorithm is homology detection.
Homologous proteins generally have high similarities in their structures and functions
from the literatures [53,54]. As establishing a homology between new and reference
proteins, we can infer assorted information such as function, structure, and evolution of
the new protein. Many algorithms for homology detection are classified into three
categories; sequence-sequence comparison, sequence-profile comparison, and profile-
profile comparison [3].
First, shown in Figure 1-1(a), sequence-sequence comparison measures the
similarity between new and reference sequences. If their identity is high, they have

3
structural and functional relationships. Based on these relationships, we can infer the
characteristics of a new protein. [2].
However, if their sequence identity is not high enough to find their relationship,
sequence-sequence comparison algorithms lose the sensitivity to detect the functional or
structural relationship of these sequences. Even though they cannot detect their
relationship, the empirical analyses prove that some sequences with low identity still
have functional and/or structural relationships because these sequences are distantly
related in their evolution [4].
To increase the sensitivity for the detection of remote homologues, instead of
comparing two proteins directly by aligning their sequences, the test sequence is
compared with profiles, which contain common information from known protein
sequences in the same families [4]. Indeed, after building the multiple alignments of
related sequences in the same family, PSSM (Position Specific Scoring Matrices) or
a
EQLAK
E A K Q A A
EAKQ
3.0 0.0 3.0 1.0 0.0 0.0
0.0 4.0 0.0 0.0 3.0 4.0
0.0 0.5 3.0 0.0 0.0 0.0
1.0 0.0 0.0 2.0 0.0 0.0
E A K Q A A
EAKQ
3.0 0.0 3.0 1.0 0.0 0.0
0.0 4.0 0.0 0.0 3.0 4.0
0.0 0.5 3.0 0.0 0.0 0.0
1.0 0.0 0.0 2.0 0.0 0.0
b
E A K Q A A
EAKQ
3.0 0.0 3.0 1.0 0.0 0.0
0.0 4.0 0.0 0.0 3.0 4.0
0.0 0.5 3.0 0.0 0.0 0.0
1.0 0.0 0.0 2.0 0.0 0.0
E A K Q A A
EAKQ
3.0 0.0 3.0 1.0 0.0 0.0
0.0 4.0 0.0 0.0 3.0 4.0
0.0 0.5 3.0 0.0 0.0 0.0
1.0 0.0 0.0 2.0 0.0 0.0
E A K Q A A
EAKQ
3.0 0.0 3.0 1.0 0.0 0.0
0.0 4.0 0.0 0.0 3.0 4.0
0.0 0.5 3.0 0.0 0.0 0.0
1.0 0.0 0.0 2.0 0.0 0.0
E A K Q A A
EAKQ
3.0 0.0 3.0 1.0 0.0 0.0
0.0 4.0 0.0 0.0 3.0 4.0
0.0 0.5 3.0 0.0 0.0 0.0
1.0 0.0 0.0 2.0 0.0 0.0
c
Figure 1-1: Homology-based methods. (a) Sequence-sequence comparison. (b) Sequence-
profile comparison. (C) Profile-profile comparison.

4
HMM (Hidden Markov Model) profile is generated on the basis of the common
information from their multiple alignments. Using PSSM or HMM, sequence-profile
comparison methods such as PSI-BLAST and SAM can increase the sensitivity to detect
the distant homologous sequences with low sequence identities [5,6].
However, if an unknown protein is even distant from the related protein family,
the profile is not sensitive to recognize that this protein belongs to the same family.
Therefore, profile-profile comparison methods such as FFAS[7] and Prof_sim[3] were
developed to solve this problem. Shown in Figure 1-1(b), it first generates the profile
from multiple alignments of sequences related to an unknown sequence. Then, comparing
the profile of the unknown sequence with the profiles of reference sequences, it can
discover the homologous pairs between two profiles.
Even though homology-based method improves the ability to detect functional
and structural relations among proteins, it still has problems to predict the properties of
proteins. First, it is still not sensitive to detect distant homologous sequences below 10%
sequence identity [8]. In fact, two sequences which have very low identity are generally
determined to be unrelated sequences in homology-based method because the possibility
to align them by chance is statistically high. However, Sander and Schneider [55] have
shown that the sequences below 10% sequence identity still have high secondary
structural similarity. Russ et al. [56] have also concluded that a small number of
conserved residues with 8% identity can build 3D folds with similar functions in proteins.
Second, the homology-based method cannot predict the properties of specific
proteins such as enzymes from their homologous pairs because the important residues of
these proteins are not conserved well among sequences even with high sequence

5
similarity [2]. For example, in several researches [57,58], enzymes over 40% sequence
identity can generally establish catalytic functional relationships among them [59].
However, due to high false-negative rate, the information about these functional
relationships is sometimes lost in the sequence over 60% sequence identity. Thus, even
though sequence similarity is generally correlated to functional or structural similarity,
this correlation can be affected by some evolutionary event such as domain shuffling,
which contains the addition, deletion and redistribution of domains [60,61].
Finally, if the existing annotations in databases contain errors, homology-based
method allows these erroneous annotations to amplify and propagate the errors through
the databases [2]. In principle, the addition of more reference sequences to the databases
supports homology-based method to predict the properties of a protein more accurately.
However, if one of these sequences contains erroneous annotations, the new prediction
contains erroneous information. In addition, if iterative computational methods such as
PSI-BLAST and SAM use these databases for the detection of homologous pairs, the
error may propagate an entire PSSM or HMM.
Machine learning method predicts the functional properties of proteins on the
basis of sequence-derived features. Since machine learning method uses physical or
chemical features extracted from the sequences of proteins, it is independent of sequence
similarity. Among many machine learning algorithms, SVM (Support Vector Machine)
and ANN (Artificial Neural Networks) are popularly used for the functional classification
of proteins [9].
SVMs are classified into two groups such non-linear and linear SVM. While non-
linear SVM has the better performance for classifying proteins with diverse sequences or

6
structures than linear SVM, linear SVM is popularly used for general protein
classification because linear SVM is easy to implement. Figure 1-2(a) explains the
procedures to build a SVM. Using feature vectors, SVM first creates a hyper-plane to
divide these feature vectors into two classes with a maximum margin. Eq. (1.1) and
Eq. (1.2) are used for linear and non-linear classification [9]. Then, projecting their
feature vectors into a multi-dimensional space, members and non-members of a
functional class are separated by a hyper-plane in the space. Finally, a new protein can be
classified into a member or non-member class by its feature vector close to the side of the
hyper-plane to which other proteins with similar features are located [9].
where w is vector normal to a hyper-space, xi is a feature vector, b is a parameter, and γi
is group index.
where xi and xj are feature vectors, and σ is standard deviation.
Shown in Figure 1-2(b), ANN has three layers such as input, hidden, and output
layers, and each layer consists of nodes and connections. Each node contains a
classification function, which determines whether each input feature belongs to the
member class or not. Based on the output from each node, the weights of connections
among all nodes are changed using Eq. (1.3). After ANN trains its own network for two-
class classification using training datasets, the trained classifier can predict the functions
of proteins [9].
w x 1 f o r 1, p o s i t i v e c la s s
w x 1 f o r 1, n e g a t i v e c la s si i
i i
b
b
1.1
2
22,j ix x
i jK x x e
1.2

7
where w0j is the output weight of a hidden node j to an output node, g is the output
function, hj is the value of a hidden layer node, xi is the feature vector of a protein whose
components are their computed descriptors, wji is the input weight from an input node i to
a hidden node j, wj is the threshold weight from an input node of value 1 to a hidden node
j, and σ is an active function.
As machine learning method uses physical or chemical features without sequence
similarity for the functional prediction of proteins, it identifies functional or structural
properties of proteins such as enzymes. However, the biased results can be produced by
the number of sequences and properties of features from the datasets because the
accuracy of prediction depends on training sets and feature extracting methods [9]. In fact,
since the training datasets for machine learning models cannot be fully representative of
the members and non-members for particular functional classes of proteins, inadequate
sampling for training and testing datasets can affect the accuracy of prediction for them.
Due to this problem, machine learning method is not applied to classify proteins with
insufficient knowledge about their specific functions. In addition, it is very important to
develop efficient feature extracting methods from sequences for machine learning method
because feature descriptors provide an impact to their performance directly.
0 , j j j i j jj j
g w h h w x w
1.3

8
a b
Figure 1-2: The schemes of machine learning methods. (a) Schematic diagram illustrating
the process of the training and prediction of the functional class of proteins using SVM
[9]. (b) Schematic diagram illustrating the process of the prediction of functional class of
proteins using ANN [9].

9
A phylogenetic profile method encodes the presences or absences of proteins
across genomes for inferring functional relationships among proteins. The basic idea of
the phylogenetic profile method is that functionally related proteins tend to co-evolve in
their organisms because of evolutionary constraints [10]. Thus, if similar proteins are
discovered between two organisms, their phylogenetic profiles are also similar because
they may have functional relationships each other. Figure 1-3 describes the procedures of
the phylogenetic profile method for the functional prediction of proteins. [11].
However, the phylogenetic profiles from genomes are often not informative
because they do not offer information of proteins themselves. Moreover, while the
phylogenetic profiles from prokaryotic genomes describe the functional relationships of
Figure 1-3: The schematic diagram of a phylogenetic profile method for function inferences [11].

10
proteins clearly, the phylogenetic profiles from eukaryotic genomes are less informative
to predict the functional relationship, despite some successful researches for specific
protein function predictions [62]. In addition, the accuracy of the analysis is low due to
the limitation of genome and genome sequences.
1.2 Motivation and Objective
This thesis is motivated by two purposes for the prediction of protein
characteristics to overcome the drawbacks as discussed above. First, we can, in principle,
infer functional, structural and evolutionary properties of a protein on the basis of only its
sequence because its primary amino acid sequence contains information about its
characteristics. However, there is no accurate method to predict these three properties of
a protein together only using its sequence.
To solve this problem, we have developed a unified computational pipeline,
called GDDA-BLAST, for measuring the structural, functional and evolutionary
characteristics of a protein using phylogenetic profiles. Indeed, GDDA-BLAST can
identify structural and functional domain boundaries in TRPC ion channels, and generate
a phylogenetic tree of evolutional related RT sequences which approximates their
evolutionary relationships in our previous studies [12,13]
Based on these previous researches, the objectives of this dissertation are to
improve the performance of GDDA-BLAST in homology detection, and to develop a
method for functional quantitative measurement of a protein. To achieve these objectives,

11
we will investigate the thresholds for the identification of RNA binding proteins and
design a new pylogenetic profile for their functional annotations.
. In this thesis, Chapter 2 describes the background and pipeline of GDDA-
BLAST, and introduces our previous researches using GDDA-BLAST which are
validated by literatures and wet experiments. Chapter 3 explains the background of the
performance evaluation, and compares the performance of GDDA-BLAST with those of
other methods. Chapter 4 reviews computational classifiers for the identification of RNA
binding proteins, and suggests a new method to identify RNA binding proteins by the
quantitative measurement of GDDA-BLAST. In chapter 5, we summarize our results of
evaluations, and discuss the implications of GDDA-BLAST. Finally, in Chapter 6, the
conclusions and recommendations for future research are discussed.

Chapter 2 GDDA (Gestalt Domain Detection Algorithm) – BLAST (Basic Local Alignment
Tool) with Phylogenetic Profiles
2.1 Backgrounds and Motives
Despite decades of researches, it is still unsolved to identify structure, function,
and evolutionary characteristics of a protein from the amino acid sequence. For example,
homolog detection to infer function and structure of an unknown protein has limitation to
identify homologous pairs among highly divergent protein sequences [3]. Indeed, if
pairwise sequence alignments between protein sequences drop down below 25%, the
sequence alignments cannot be reliable for matching two sequences and their alignments
are treated as random events [14]. However, a small number of conserved residues with
8% identity can coordinate the 3-D fold and/or function of proteins, whereas two proteins
with 88% identity can still preserve independent structure and function [15].
Therefore, the abovementioned studies raise fundamental questions about the
structure, sequence and function of a protein. Which residues within amino acid
sequences are important to determine the function and/or structure of a protein? Do
proteins with similar sequence and structure have a common ancestor? Furthermore, if
sequence and structure similarity suggest an evolutionary history, do weak similarities
mean they have different evolutionary history? All of these questions are essentially
connected to the relation among the sequence, structure and function of a protein.

13
However, all these questions have not been clearly solved either experimentally or
theoretically.
For example, common computational alignment programs such as BLAST and
FASTA fail to detect remote homologous sequences with sufficient statistical
significance [16]. To improve the performance of the sequence alignment, Blake and
Cohen [17] built amino acid substation matrices to measure properties of amino acid
residues in a sequence. More recently, advanced sequence comparison methods have
been developed using the shared features from related sequences in the same protein
families. Based on these approaches such as templates [18,19], profiles[20,21] and HMM
(Hidden Markov Models) [22,23],several popular programs such as PSI-BLAST [5] and
SAM [24] have improved the sensitivity to detect the distant homologues. In addition,
threading algorithms are also developed to improve detection of homologous pairs in the
twilight zone [25]. Despite of these improvements, these methods still cannot annotate
the relationships between function and structure of a protein.
The purpose of all these methods is basically to explore information encoded in
sequences. Due to the resent advance of computer technology for knowledge bases and
the analysis of complex data, invaluable information can be teased out from protein
sequences more accurately. Therefore, integrating several advanced methods such as
phylogenetic profiles, RPS(Reverse specific position)-BLAST, and profile databases for
the analysis of biological data, we proposed a unified framework, called GDDA-BLAST,
for inferring structural, functional, and evolutionary information from sequences. In this
chapter, we will introduce the concept and backgrounds of GDDA-BLAST. Then, we
will describe several researches and their results using this computational assay.

14
2.2 GDDA-BLAST with phylogenetic profiles
A phylogenetic profile is a vector that encodes the existence of the protein across
different genomes to predict functional relations and physical interactions between
proteins [26,27]. This approach has applied to one entire sequence with one protein
(single profile method) or separate segments of a sequence with different proteins
(multiple profile method). In principle, when proteins have the similar patterns in their
sequences, the proteins may interact with each other directly or share a common
functional role in their pathways. Thus, the underlying hypothesis of phylogenetic profile
is that functionally linked proteins tend to be inherited or eliminated in a correlated
manner, and, the homologues of the proteins may exist in the same subset of organisms.
Similarly, GDDA-BLAST creates a matrix that encodes the existence of the alignments
of a domain profile across different proteins [12].
The basic idea of GDDA-BLAST is to collect a set of profiles that align to the
query sequence. These profiles can be attained from various knowledge-base sources
such as PDB (Protein Data Bank), Pfam, and SMART, CDD (Conserved Domain
Database) from NCBI (National Center for Biotechnology Information) and/or actual
sequence of a representative protein domain. Then, RPS-BLAST is utilized to compare
query sequences with these profiles. RPS-BLAST generally search protein sequences
against a database of PSSM (position specific scoring matrices) to identify the sequences
with fast speed, and it is informative for the identification of the possible function(s) the
query protein may have. However it is not sensitive to identify divergent sequences. For

15
overcoming this limitation, GDDA-BLAST employed innovative methods to align the
query sequence to the profiles by RPS-BLAST.
First, we utilize a single domain profile database for pairwise comparisons. Since
RPS-BLAST searches aligned profiles in a whole profile database, the searching speed
becomes very slow if a thousand of sequences are used to search the profiles. As dividing
a whole profile database into a number of single domain profile databases, we increase
the speed of profile searches. Next, we record and quantify non-seeded alignments from
unmodified query sequence and “seeded” alignments from modified query sequence. The
modified query sequences are generated with a “seed” from the profile to create a
consistent initiation site. This consistent site assists rps-BLAST to extend an alignment
between highly divergent sequence segments. This approach is designed to amplify and
encode the alignments to hit for any given query sequence. Seeds can be obtained at
multiple proportions (e.g. 3-50% “seed” size) from any region of the profile sequence
(e.g. N-terminal, middle, C-terminal). These seeds are inserted at each position of the
query once at a time. Therefore, a query of N amino acids generates 2*N distinct test
sequences for each seed. Each of these test sequences is aligned by rps-BLAST against
the parent profile.
Based on these innovations, we developed GDDA-BLAST to improve the
performance of RPS-BLAST. Shown in Figure 2-1, the computational pipeline consists
of five procedures. First, we obtain domain profiles from multiple knowledge-based
sources such as Pfam, SMART and CDD or from real sequences. Then we modify the
query sequence with a seed from the profile to create a consistent initiation site. Next,
each of these modified sequences is aligned against the parent profile by rps-BLAST.

16
In the forth procedure, the results are filtered by thresholds such as % identity and %
coverage using Eq. (2.1) and Eq. (2.2).
Where lenalignment = the alignment length = qend – qstart +1
qstart = The start position of a modified query sequence in the alignment
qend = The end position of a modified query sequence in the alignment
Seeding rps-BLAST
Signal collection
Phylogenetic profiles
Figure 2.1: The workflow of GDDA-BLAST. (i and ii) The algorithm begins with a
modification of the query amino acid sequence at each amino acid position via the
insertion of a seed sequence from the profile of interest. These seeds are obtained from
the profile consensus sequences from Conserved Domain Database (CDD). (iii–v) Signals
are collected from optimal alignments between the ‘‘seeded’’ sequences and profiles by
using rps-BLAST and are incorporated as a composite score into an N by M data matrix
[13].
( % ) 1 0 0a l i g n m e n t
p r o f i l e
C o v e r a g el e nl e n
2.1
( % ) 1 0 01
i d e n t i c a l s e e d
a l i g n m e n t s e e d
I d e n t i t y n u m l e nl e n l e n
2.2

17
lenprofile = The length of a consensus sequence of a given profile
lenseed = The sequence length of a seed inserted into the query
numidentical = The number of identical residues in the alignment
The phygenetic profile is finally generated from the filtered sequence alignments
by representing an M (# of profiles) by N (# of queries) matrix. Then, the dedrogram is
produced from this profile on the basis of Pearson’s correlation between query sequences
using equation Eq. (2.3). This dedrogram is used to predict the functional relationships
among query sequences. If a phylogenetic tree is built on the basis of Euclidian distances
between the phylogenetic profiles from Eq. (2.4), we also measure the evolution
distances among sequences. In next chapters, we will introduce our studies, which
discovered experimental results to support our functional and evolutionary predictions
using GDDA-BLAST.
where X and Y are the averages of values in X and Y. X and Y are the standard
deviations of these values.
2.3 The prediction of functional characteristics of proteins by GDDA-BLAST
Since the seeding allows RPS-BLAST to extend the alignment between highly
divergent sequences, we identified divergent domains in proteins using GDDA-BLAST
[12]. Especially, if we use multiple domain profiles as the parent profiles, we detected
1,
1 i ii N
X Y
X X Y Yr
N
2.3
2
1 ,
( , )i M
D X Y X Y i iyx
2.4

18
multiple functional properties of a protein by GDDA-BLAST. For example, ankyrin
repeats can perform a number of functions such as ATP-binding, lipid-binding and
calmodulin-binding [28,29]. However, there are no current domain-detection algorithms
which can resolve their multi-functional nature. Thus, to detect their multi-functional
characteristics, we generated multiple phylogenetic profiles for vanilloid TRP (TRPV)
family using multiple domain profiles such as 131 peripheral lipid-binding (PLB), 98
Integral lipid-binding (ILB), 58 Trafficking (TRFK), 10 Calmodulin-binding (CBD), 4
Ankyrin Repeat (ANK), and 574 ATP (ATP) profiles. Shown in Figure 2-2 (a), we
observed the signals for all of these profiles within the ankyrin repeats of TRPV1 channel
at varying levels of intensity. To validate our predictions, we focused on the signal of
ATP binding domains among these signals.
Lishko et al. recently crystallized the ankyrin repeats of TRPV1 and TRPV2, and
they found their structures to be highly similar [28]. They also discovered both ankyrin
repeats bound to calmodulin, while only TRPV1 was capable of binding ATP in their
assays [28]. Indeed, when we obtain phylogenetic profiles for TRPV1 and TRPV2 using
GDDA-BLAST, we observe calmodulin signals in the ankyrin repeats of both TRPV1
and TRPV2. Comparing the ATP binding signals between two proteins, TRPV1 has a
robust ATP signal within its ankyrin repeats, while the ATP signal of TRPV2 is only
18% of TRPV1 in Figure 2-2 (b). This result suggested that TRPV1 may bind ATP but
TPRV2 may not.
In addition, we predicted the conserved residues from the alignments of ATP
binding domain profiles by GDDA-BLAST. Shown in Figure 2-2 (c), top scoring residue

19
in TRPV1 is E211, which coordinates the N6 amine binding of ATP in the active pocket.
Therefore, all of these results propose that GDDA-BLAST can predict the functional
properties of a protein, which matched the experimental results from the literatures.
a
b c
Figure 2-2: GDDA-BLAST model of the ATP-binding Ankyrin Repeat in TRPV1 [12].
(a) GDDA-BLAST results for human TRPV1 channel using131 peripheral lipid-binding
(PLB), 98 Integral lipid-binding (ILB), 58 Trafficking (TRFK, n=58), 10 Calmodulin-
binding (CBD), 4 Ankyrin Repeat (ANK), and 574 ATP profiles. (b) GDDA-BLAST
results for the screen of 574 ATP profiles in the ankyrin repeat domain of various TRP
channels was integrated to quantify the area under the curve and plotted in a bar graph.
(c) Left: Quantification of amino acid positions in human TRPV1 ankyrin which are
identical or similar in alignments with ATP profiles. Right: Crystal structure of the rat
TRPV1 ankyrin repeat complexed with ATP (PDB: 2PNN). Residues depicted in yellow
are homologous to those derived in human TRPV1

20
2.4 The investigation of evolutionary relations among proteins using GDDA-BLAST
To determine evolutionary relationships between homologous proteins, we should
measure evolutionary rates among the proteins. We assumed that the rate information can
be measured using a phylogenetic profile from GDDA-BLAST. Shown in Figure 2-1,
phylogenetic profiles from GDDA-BLAST are encoded as vectors. As each “seeded”
query can return either no alignment, or an alignment that ranges over %identity
and %coverage using RPS-BLAST; we encode this information into the N X M matrix
with these vectors. Then, an euclidian distance are generated from this N X M vector
matrix on the basis of the simple hypothesis that the distance between each N [query] in
the matrix is proportional to the rate of evolutionary divergence.
Indeed, Figure 2-3 represents the results of our characterization of 20 water-
channel (aquaporin) proteins with 23,605 profiles from the NCBI-CDD database [12]. In
this result, we discover that there are four distinct families with rates that accord with
previous studies employing multiple sequence alignment [30]. From random
considerations, the probability of organizing these twenty sequences correctly into 4
families is 9X10-13. Therefore, these results demonstrate that phylogenetic profiles
derived by GDDABLAST can contain evolutionary rate information, which is
independent of multiple sequence alignment based methods. We believe that rigorous
analyses on benchmark training sets will enable us to make more refined and statistically
robust measurements among distantly related and/or rapidly evolving proteins.

21
Figure 2-3: Water Channel (Aquaporin) Phylogeny [12]. Twenty Zea Maize aquaporin
channels (plasma membrane intrinsic proteins (PIPs), tonoplast intrinsic proteins (TIPs),
Nod26-like intrinsic proteins (NIPs), and small and basic intrinsic proteins (SIPs)) were
screened with GDDA-BLAST. The Euclidian distance is generated from the composite
scores and plotted in an unrooted tree using the MEGA3 minimum evolution algorithm
[31]. Scale bar reflects the Euclidian distance between sequences and color coding
reflects the distinct and known classes of aquaporins. Our results are in excellent accord
with the findings of Chaumontet al [32]

22
2.5 The prediction of structural boundaries of ion-channels using GDDA-BLAST
A recent study by Mio et al. obtained a cryo-EM structure of TRPC3(Transient
Receptor Potential Channel 3) and modeled the six transmembrane helices with the
atomic structure of the potassium channels KcsA and Kv1.2 [33]. Interestingly, these
authors also determined that TRPC3 contains a globular, and presumably hydrophobic,
inner-core surrounded by signal sensing antenna derived from the cytosolic N and C-
termini in Figure 2-4(a). We wondered whether these channel constituents could be
computationally modeled with GDDA-BLAST, by generating phylogenetic profiles from
sequences that comprise the appropriate structural elements/biological functions of
interest.
Initially, we queried human TRPC channels with a curated set of 98
transmembrane domain containing profiles to generate our GDDA-BLAST phylogenetic
profiles. The distribution of the alignments which are above threshold is plotted in
Figure 2-4(b). The results from this experiment accurately model the channel domain in
human TRPC channels when compared with transmembrane predictions by the hidden
Markov model TMHMM and the domain detection algorithm SMART [34,35].
We tested whether key-word searches of the NCBI CDD database (CDD) could
be used to collect additional points of information to our phylogenetic profiles. We
collected 536 profiles in CDD which have the following key words such as channel,
transmembrane, integral membrane, pump and performed our analysis repeatedly in
Figure 2-4(b).

23
a
b
Figure 2-4: GDDA-BLAST models of the ion transport domain in TRPC channels.
(a) 3D reconstruction of TRPC3 channel derived by Mio et al.[33]. Blue lines depict the
plasma-membrane. The scale on the left depicts the cryo-electron microscopic images of
horizontal slices parallel to the plasma-membrane (images 6-9) progressing into the
cytosol (images 10-15). The globular inner shell can be seen as a circular density in the
center of the images. (b) GDDA-BLAST results for human TRPC channels using 98
curated integral lipid-binding (ILB) profiles and 576 profiles parsed with key words for
(channel, transmembrane, integral membrane, and/or pump). The latter were also
analyzed with different % coverage thresholds. Ion transport boundaries in TRPC
channels predicted by SMART (default settings) are noted with the N-terminal boundary
denoted by an arrow. GDDA-BLAST results predict that the globular inner shell domain
is located to the left of the arrow.

24
We observe that alignments against these profiles also model the channel domain
boundaries. In addition, a pronounced peak is evident in TRPC3/6/7 that significantly
differs in TRPC1/4/5. This signal likely represents the hydrophobic globular inner-core
domain in TRPC3 identified by Mio et al.[33], and suggests that the channel domains in
TRPC1/4/5 are likely different structurally and/or functionally from TRPC3/6/7.
To determine whether these signals are robust, we recalculated the data using % coverage
thresholds ranging between 60% and 100% in Figure 2-4(b). Surprisingly, a 60%
threshold does not significantly alter the domain boundaries, but does increase the signal
in our results. Overall, the GDDA-BLAST model of TRPC ion-channel domains is in
excellent accord with other computational models and experimental evidence.
2.6 The discovery of novel lipid-binding domains in vitro
Using lipid-binding profiles, we also predicted the regions of lipid binding in
proteins whose functions are not annotated by any conventional algorithm using GDDA-
BLAST. Then, we designed an assay to validate our prediction for these proteins. Shown
in Figure 2-5 (a), we observe multiple peaks in the histograms generated from these
alignments. Next, we cloned the representative regions from each of these proteins and
prepared bacterially purified protein. These purified proteins were subjected to liposomal
assays containing lipids which mimic the plasma-membrane of animal cells.
Strikingly, each of the fragments containing GDDA-BLAST signals was positive
for lipid-binding in Figure 2-5 (b), whereas our negative controls were not. Although the
physiological relevance of these lipid binding domains remains to be determined, these

25
results clearly demonstrate that phylogenetic profiles generated using ontological
relationships are effective for identifying putative functions within protein domains.
a b
Figure 2-5: Functional Information via GDDA-BLAST analysis (a) GDDA-BLAST
results for three human proteins of unknown function (AAH33897, NP_872401, and
CAB45695.2) using 131 peripheral lipid-binding (PLB) profiles. The white bars depict
regions that we cloned for liposomal experiments in (b). (b) Western analysis of purified
CAB45695, AAH33897, NP_872401, fragments cloned into His vector (1 mg load).
These fragments were tested for binding to liposomes containing phosphatidylcholine
(PC), phosphatidylethanolamine (PE), phosphatidyl serine, and phosphatidylinositol (PI).
All fragments bound to liposomes except fragment 1 (CAB45695: aa 70-180) and the
HIS-tag in perfect accord with the predictions of GDDA-BLAST.

26
2.7 Summary and discussion
In summary, we introduced a new tool for using phylogenetic profiles to infer
structural, functional and evolutionary information from the amino acid sequence of a
protein in these chapters. GDDA-BLAST is a unified computational pipeline for
measuring the structural, functional and evolutionary characteristics of a protein using
phylogenetic profiles with a carefully selected set of profiles. There are two hypotheses to
implement GDDA-BLAST. First, the primary amino acid sequence contains information
of structure, function and evolution of a protein, and, second, the SF&E information can
be inferred from the sequence by a unified method, even if the pair-wise identity of
sequences is below 25%.
Based on these hypotheses, GDDA-BLAST consists of five procedures. First, we
utilize a single domain profile database for pair-wise comparisons. Then, we modify the
query with a “seed”. This seed can be generated from a profile by taking any fraction of
the profile such as N-terminus or C-terminus. This seed is inserted into every position of
the query at a time, creating a consistent initiation site. This site allows rps-BLAST to
extend an alignment even between highly divergent sequences. This resampling strategy
is designed to amplify and encode the alignments possible for any given query sequence.
Next, the results are filtered using thresholds such as % identity and % coverage. The
phylogenetic profiles are finally generated by representing each sequence as a vector of
non-negative numbers. These profiles can be used to create a dendrogram of functional
relationships among proteins using pearson correlations or a phylogenetic tree using
euclidian distances.

27
In our previous studies, GDDA-BLAST can accurately model structural and
functional relationships in TRP channels through these procedures. This is supported by
our findings that GDDABLAST predicts: (i) the ion-channel domains of TRP channels,
(ii) lipid-binding and trafficking function within the previously uncharacterized TRP_2
domain, and (iii) the multi-functional (lipid-, calmodulin-, and ATP-binding) natures of
ankyrin repeats within TRP channels. Our experimental evidences demonstrate that
TRPC3 with TRP_2 is a lipid/trafficking domain that contributes to DAG-sensitive
vesicle fusion. The models of TRPC channels by GDDA-BLAST also recapitulate
experimental evidences from other laboratories. For example, the homologous C-terminal
domain of TRPC6, recently reported to bind both PIP3 and calmodulin in various ion
channels, yet is undetectable by conventional methods [12].
GDDA-BLAST readily predicts this domain and its functions. GDDA-BLAST
also accurately models the ATP-binding activity contained in the ankyrin repeats of the
structurally resolved TRPV channels [12]. We also observe a segmented signal in
TRPC3/6/7 when tested by GDDA-BLAST with transmembrane domain profiles, which
likely represents the globular inner-core domain observed in the cryo-EM structure
obtained by Mio et al. [33]. In addition, GDDA-BLAST predicts that all plasma-
membrane resident ion channels likely contain peripheral-lipid binding and trafficking
domains, based on multiple lipid-binding domains that we also observed in all channels
tested (e.g. aquaporins, and Na+, K+, Cl-, Ca2+channels). All of these channels have
been demonstrated, empirically, to interact with lipids [63].
From these results, we concluded that GDDA-BLAST measurements can be
treated as “fingerprints” of structural, functional and evolutionary information. Through

28
the careful choice of knowledge-base profiles related for either structural or functional
qualities, GDDA-BLAST provides results which can be used to infer evolutionary rate
information, create functional models and identify structural boundaries for protein
sequences, even if no prior information exists. Perhaps most important, GDDA-BLAST
has the capacity to inform laboratory experiments of key amino acids essential to protein
function, thus speeding the discovery process. Our studies here demonstrate one way of
using phylogenetic profiles to quantitatively probe knowledge-bases to obtain structural,
functional and evolutionary information within the same unified framework. Future
works aimed at determining the data points collected by GDDABLAST which are
informative for structural, functional and evolutionary annotation, and which ones are
sufficiently noisy such that they are detrimental to the total information content will
enable us to understand and harness the underlying mechanisms of our algorithm
optimizing and refining our approach. For these purposes, we will suggest the methods to
improve the performance of GDDA-BLAST in next chapter.

Chapter 3
The Performance of GDDA-BLAST in homology detection
3.1 The backgrounds and Motives
Since proteins with similar sequences can share similar structures, the homology
between a know protein and unknown protein is used for investigating the structure and
function prediction of a new protein. In the modeling procedure, a new sequence is
usually compared against all the known sequences in a database. If the homology is
created, the structure and function of the new protein can be inferred from the
homologous protein.
For the identification of the relation, the similarity between the sequences is
calculated from the sequence alignments. If the similarity between two sequences is over
a threshold such as 25%, a literature proposed that the new and known sequences are
closely related [4]. If their sequence identity is not high enough to discover the
relationships, we need to decide whether they are related or not. Sequence-sequence
comparison algorithms generally cut off pair-wise alignments below 25% identity.
However, empirical analyses proved that some sequences with low identity still have
functional and/or structural relationships because these sequences are distantly related in
their evolution [14].
A main reason of this problem is the influence of evolution. Even though the
sequences can be changed significantly due to the mutations and insertions, many

30
proteins still have the same folds and close functional relationships with low sequence
similarity. However, the sensitivity to detect homologous proteins in homology-based
methods suddenly drops below 25 % sequence identity because homology-based methods
discriminate the alignments below 25%.
To detect homologous sequences with weak identities, one of possible solutions is
to increase the sensitivity of sequence comparison. For increasing its sensitivity, we need
to modify a calculating process of a sequence similarity. For example, instead of
comparing two sequences directly, many programs use statistical information of protein
families such as PSSM (Position Specific Scoring Matrix) and HMM (hidden Markov
model)s in Figure 3-1 [6]. While PSSM contains the frequencies of the residues in
specific positions of the sequence, HMMs have the probabilities of the residues which
exist in the positions.
Even though conventional homology-based methods such as PSI (Position
Specific Iterrative)-BLAST and SAM (Sequence Alignment and Modeling system)
increase to the sensitivity to detect the distant homologues on the basis of PSSM or
HMM, they still miss to detect sequences with very weak similarities such as below 10%
because of stringent thresholds for defining significant sequence similarity [3]. In an
attempt to rectify the shortcomings of the methods stated above, the GDDA (Gestalt
Domain Detection Algorithm)-BLAST was developed to increase the sensitivity of RPS-
BLAST by amplifying alignments with low identities. As increasing the sensitivity,
GDDA-BLAST detects the signals of the divergent alignments, which other
computational algorithms cannot detect, between domain profiles and the protein

31
sequence. Based on the signals, GDDA-BLAST can search homologous pairs among a
huge amount of proteins more sensitively. In addition, using multiple domain profiles
from various knowledge-base sources such as PDB, Pfam, SMART, CDD and/or real
sequences, GDDA-BLAST can also generate the phylogenetic profiles from which we
are able to derive biological information related to structures, functions and evolution
from the sequences.
To evaluate the performance of GDDA-BLAST, we need the objective
measurement for functional, structural and evolutionary predictions. Among all these
predictions, we will first evaluate the performance for structural homology detection.
Thus, we select PDB40D-J dataset which contains 935 sequences from SCOP for the
measurement its performance. Using these sequences, we compared the performances of
two methods such as PSI-BLAST and SAM-T21K to detect homologous pairs in pdb40d-
j dataset with that of GDDA-BLAST. We will explains the procedures and dataset for the
performance evaluation, and suggest methods to improve the performance of GDDA-
BLAST in these chapters.

32
a
b
Figure 3-1: The statistical information of protein families. (a) An example of a 49 residue
sample profile, generated from the four-probe sequences located at the left position [52].
(b) The model of HMM, modeling sequences of as and as two regions of potentially
different residue composition [6].

33
3.2 Results and Discussion
3.2.1 Datasets for the performance evaluation
For the evaluation, we used a structural benchmark dataset from Structural
Classification of Proteins (SCOP) database. SCOP database usually provides detailed and
comprehensive information of the structural and evolutionary relationships of proteins
whose structures are already proven in wet-lab experiments. Based on a protein domain
as a unit of classification in SCOP, small proteins with a single domain are treated as a
whole, and the domains within large proteins are classified individually. Thus, Figure 3-2
depicts that the classification in the database consists of five hierarchical levels on the
basis of the evolutionary and structural relationships [36].
In the classification, if the sequence identities between proteins are over 30% or
the functions and structures of proteins, even in low identities, are very similar each other,
these proteins are clustered into the same family which has a common evolutionary origin.
Proteins, whose identities are low and whose common evolutionary origin is probable,
are catagorized into superfamilies. If proteins in different superfamilies and families have
the same major secondary structures, these proteins belong to a common fold. Finally, the
different folds are divided into classes for user convenience. Based on the secondary
structures of which the folds composed, they are assigned to one of these five classes
such as i) all alpha, ii) all beta, iii) alpha and beta, iv) alpha plus beta, and v) multi-
domains [36].

34
Among these hierarchies, we use the sequences in superfamilies to evaluate the
performances of homology detection algorithms because the proteins in superfamilies can
represent the boundaries of groups which share the same structural and functional
features or have the common evolutionary origins [16]. Among many datasets to include
superfamilies, we selected PDB40-J dataset containing 935 sequences, whose sequence
identities are less than 40%, from the literatures [16]. In addition, we extracted 289
sequences in twilight zone, where the sequence identities are below 25%, from PDB40-J
because most of homology-based algorithms lose their sensitivity to detect homologous
sequences in this region.
1086 Folds
1777 Superfamilies
3464 Protein domains
97178 Protein domains from different species
1086 Folds
1777 Superfamilies
3464 Protein domains
97178 Protein domains from different species
Figure 3-2: Five hierarchical levels of SCOP classification [36]. The unit of classification
in SCOP is the protein domain. Small proteins with a single domain are treated as a
whole, and the domains within large proteins are classified individually.

35
After we calculated the sensitivity and specificity using the number of true and
false homology pairs which PSI-BLAST, SAM and GDDA-BLAST detected using these
two datasets, we compared their performances each other on the basis of the sensitivity
and specificity of these methods. The measuring procedures will be discussed in the
following chapters.
3.3 Homology detection methods for the performance evaluation
To evaluate the performance of GDDA-BLAST, we compared its performance to
those of PSI-BLAST and SAM because they are representative methods among many
homology-based methods. Shown in Figure 3-3(a), PSI-BLAST iteratively searches a set
of sequences which may be homologues for the fixed iterations or until it cannot find new
homologues. In the procedures of PSI-BLAST, GAP-BLAST first collects an initial set of
homologues from the sequence database such as NR (Non-Redundant protein database)
for a given query sequence. Then, weighted multiple alignments are generated using the
query sequence and the homologues whose scores are over a specified cut-off value. Next,
a new PSSM is constructed on the basis of the multiple alignments. Using this PSSM, it
searches the database for new homologues. These procedures are repeated until the
results satisfy the conditions given by users [5].
Using HMM instead of PSSM, SAM follows the similar procedures of PSI-
BLAST in Figure 3-3(b). First, SAM creates an initial HMM from a given query
sequence. After searching potential homologues from a sequence database with the initial
HMM, it selects new sequences, which have reliable local alignment scores with the

36
HMM, among potential homologues. After multiple alignments are generated using these
new sequences, a new HMM is created from the multiple alignments. These procedures
repeat for the fixed iterations [22]. For the performance evaluations with our datasets, we
used the default parameters of PSI-BLAST and SAM such as e-value (0.001) and three
iterations.
a
b
Figure 3-3: The schemes of homology-based methods. (a) The scheme of PSI-BLAST
with sequence profiles. (b) The scheme of SAM with HMM

37
3.4 The performance evaluation
After PSI-BLAST, SAM and GDDA-BLAST collect potential homologues in our
dataset, we evaluate their performances following these steps. First, we calculate the
similarity scores between test and reference sequences. Then, we rank test sequences in
ascending order on the basis of similarity scores. After counting the number of true and
false positives and negatives within a sliding window, we draw Receiver Operating
Characteristic (ROC) curve.
For the similarity score of PSI-BLAST and SAM, we calculated E-value which
represents the number of hits that can be shown by chance when searching a database of a
particular size using Eq. (3.1). For the similarity of GDDA-BLAST, we used Hybrid
LogWeighted scoring scheme using Eq. (3.2). This scoring scheme consists of two steps.
First, we calculate the scores of three phylogenetic profiles such as # of hits, % of max.
coverage, and % of avg. identity. Then, we adjust their scores on the basis of the
frequency of the domains aligned with queries.
where K and λ are parameters, m is the length of a domain sequence, n is the length of a
query sequence, and S is bit score.
where H is the number of hit alignments, I is the average of identity, and V is maximum
coverage.
λS
E = K m n e
3.1
( , ) ( ( ) , ( ) ) , , ,x yT
S i m x y P C a d j a d j T H I V
3.2

38
In detail, we rank potential homologues in ascending order on the basis of E-
values after calculating the E-values of all potential homologues in PSI-BLAST and
SAM. Then, changing window size, we count the number of true and false positive and
negative homologous pairs in the potential homologues. For GDDA-BLAST, after
calculating pearson correlation among three phylgenetic profiles, we adjust the value of
each phylgenetic profiles on the basis of the frequency of the domains aligned with
queries. Then we multiplied each scores for total scores together. Based on these scores,
we count the number of true and false positive and negative homologous pairs in the
potential homologues with a sliding window.
Since ROC curve is one of simple methods to represent the relationship between
the FPR (False Positive Rate), which is 1-sepcificity, and sensitivity, we should calculate
sensitivity and specificity for the detection of true homology pairs using the number of
true and false positive and negative homologous pairs. The sensitivity measures the
proportion of true positives using Eq. (3.3), and the specificity measures the proportion of
true negatives using Eq. (3.4).
where TP is the number of true positives, TN is the number of true negatives, and FP is
the number of false positive.
T PS e n s i t i v i t y
T P + F N 3.3
T NS e n s i t i v i t y
T N + F P 3.4

39
Based on the sensitivity and specificity from these equations, we first plotted the
performances among three methods with PDB40-J dataset. Shown Figure 3-4 (a), the X-
axis represents the false positive rate, and Y-axis represents sensitivity. Even though we
could measure the performance of PSI-BLAST by 0.3 in false positive rate because of the
data measuring limitation in PSI-BLAST, the total performance of GDDA-BLAST is
better than those of PSI-BLAST and SAM. When we especially focus on the
a
b
Figure 3-4: The ROC graphs for the performance evaluation of GDDA-BLAST (a) The
comparison of the performances among GDDA-BLAST, PSI-BLAST and SAM using the
dataset of superfamily. (b) The comparison of the performances among GDDA-BLAST,
PSI-BLAST and SAM using the dataset of twilight zone.

40
performances below 0.05 in false positive rate (the red circle in the left of Figure 3-4 (a)),
GDDA-BLAST is superior to other methods in the sensitivity to detect homologous pairs.
Based these results, we concluded that GDDA-BLAST would have the better
performance that those of other methods for the detection of the structural homologues in
a dataset whose sequence identities are over 40%.
Since many homology-based methods lose their sensitivities for the detection of
potential homologues in twilight zone, we also measured the performances of these
methods with sequences in this zone. Although all three methods lose their sensitivities to
detect homologous pairs, the total performance of GDDA-BLAST is still better than
those of others. In the range below 0.05 in false positive rate (the red circle in the left of
Figure 3-4 (b)), while the performance of SAM is better than that of GDDA-BLAST
below 0.02, GDDA-BLAST surpass SAM in the sensitivity of detection. Therefore, these
two ROC curves show that GDDA-BLAST outperforms SAM and PSI-BLAST for the
detection of homologous sequences in superfamilies and twilight zone.

41
3.5 Summary and discussion
We evaluated the performance of GDDA-BLAST for the homology detection in
this chapter. For the evaluation, we selected PDB40D-J to measure the number of true
homologous pairs detected by GDDA-BLAST. PDB40D-J contains 935 sequences which
have pair-wise identities of less 40% in the superfamilies from the structural
classification of proteins (SCOP) database. We also extracted 289 sequences below 25%
pair-wise identity from PDB40D-J to evaluate the performance in twilight zone. 26374
domain profiles from CDD and PDB are used as profiles for GDDA-BLAST.
First, we calculated the similarity scores between each test and reference sequence
to evaluate the performances of GDDA-BLAST, PSI-BLAST, and SAM after aligning
them. For the similarity score of PSI-BLAST and SAM, we use E-value, which
represents the expectation value of hits shown by chance when searching a database of a
particular size. For the similarity score of GDDA-BLAST, Hybrid LogWeighted scoring
scheme is used. Hybrid LogWeighted scoring scheme consists of two steps. First, we
calculate the scores of three phylogenetic profiles such as # of hits, % of maximum
coverage, and % of average identity. Then, their scores are adjusted on the basis of the
frequency of the domains aligned with queries. Next, test sequences are ranked in
ascending order on the basis of similarity scores. Based on the number of true and false
positives and negatives within a sliding window, receiver operating characteristic (ROC)
curve of each method is drawn.
Shown in Figure 3-4, the performance of GDDA-BLAST is better than those of
PSI-BLAST and SAM with datasets in superfamilies and twilight zone. In very low false

42
positive rate (<0.05), the sensitivity of GDDA-BLAST is higher than those of PSI-
BLAST and SAM. This means that GDDA-BLAST is more sensitive to detect
homologous pairs than other methods.
Even though GDDA-BLAST outperforms SAM and PSI-BLAST for the detection
of structural homologues in superfamilies and twilight zone with PDB40D-J dataset,
GDDA-BLAST still have disadvantages which we need to improve. First, we should
develop a method to build domain profiles for the generation of the best phylogenetic
profiles to predict specific functional or structural proteins. Generally, we used domain
profiles selected from CDD and PDB to generate the phylogenetic profiles for the
analysis of proteins. Despite being useful for the functional prediction of some proteins,
these profiles are not enough to predict functions of many proteins because some
domains in the profiles cause to generate noises in the phylogenetic profiles. For example,
if we use domain profiles from CDD to predict evolutionary relationships among RT
sequences, the phylogenetic tree using total domain profiles is worse than a phylogenetic
tree using domain profiles from RT sequences themselves [13].
Second, we need to develop the best scoring scheme for the comparison of the
performances in homology detection because the performance for homology detection
depends on the score for each sequence. Homology-based methods generally represent
the potential homologues with their scores after searching them in the reference database.
Among many scores, e-value and hit score are popular standards to detect homologous
pairs. Since e-value depends on the size of the database and hit score is decided by the
number of identical residues, these methods sometimes miss to detect remote

43
homologous sequences with low sequence identities. To overcome these problems,
GDDA-BLAST uses pearson correlation value to measure the similarity among
phylogenetic profiles from sequences. Although pearson correlation is independent of
sequence identities and the size of a database, pearson correlation itself is not enough to
measure the similarity between sequences because it is too sensitive for noises in the
phylogenetic profiles. Therefore, we need to implement the score system to measure the
similarity of phylogenetic profiles.
Finally, we have to design residue-based phylogenetic profiles for the collection
of accurate information from sequences. In several studies [15,65], a small number of
conserved residues in sequences with 8% sequence identity can coordinate the 3D fold
and/or function of proteins, with large portions of these proteins comprising
heteromorphic pairs. Therefore, if we can extract features of key residues to determine
the functional and structural characteristics of a protein from a sequence, we would
accurately measure the similarity among residue-based phylogenetic profiles from the
sequences.

Chapter 4
The identification of RNA binding proteins using the quantitative functional measurement
RNAs in a cell generally have many functions such as a carrier of genetic
information, a catalyst of biochemical reactions, an adapter molecule in protein synthesis,
and a regulator of RNA splicing/maintenance of telomeres [37]. If we would identify the
functions of RNAs, we should understand the functions of RNA binding proteins because
RNA interacts with a diversity of proteins to regulate a multitude of additional cellular
functions such as pre-mRNA processing, splicing, and translation [38]. Therefore, if we
identify RNA binding proteins related to a specific biological process, we are able to
discover the functions of RNAs in the biological process. However, since RNA structures
are various, the structures of proteins to interact with the RNAs can be very diverse.
Indeed, RNA binding proteins can be classified into six families on the basis of their
basic binding motifs [39], and the proteins in the same family do not share common
structures in Figure 4-1.
For example, while the structure of an arginine-rch motif is unstructured
secondary motif [40,41], the structure of a motif in an αβ protein domain family consists
of several antiparallel β sheets and α helices [42]. In addition, multimeric motif is
composed of multiple proteins or the repeats of the same structural motif [43,44], but
zinc-finger motif contains several zinc-finger peptides and α helices [45,46].

45
a b
c d
e f
Figure 4-1: The structures of RNA binding proteins. (a) The structure of arginine-rich
protein family [40,41]. (b) The structure of all-helical protein family [47]. (c) The
structure of αβ protein [42]. (d) The structure of zinc finger protein family [45,46]. (e)
The structure of multimeric protein family [43,44]. (f) The structure of RNA-targeting
enzyme [48]

46
In addition, even within the same RBP family, the RNA interaction sites need not
to be conserved. Taken together, it is difficult to identify RNA binding proteins in silico
and in vitro. In this chapter, we first start to review existing methods for the identification
of RNA binding proteins. Then, we introduce a computational assay to overcome their
disadvantages. Finally, we analyze and discuss the results in more detail.
4.1 A classification library for RNA binding proteins
We generally use the RNA electrophoretic mobility shift assay for the
identification of RNA binding protein in vitro. In principle, nucleic acid probes which the
protein binds move slowly because the speed of different molecules through the gel is
determined by their size and charge [49]. Based on this property, we can
electrophoretically separate a protein-DNA or protein-RNA mixture from other probes in
Figure 4-2. However, it takes long time to identify RNA binding proteins despite the
accuracy of the identification.
To increase the speed of the analysis, multiple algorithms such as homology-
based methods, support vector machine (SVM) and phylogenetic methods have been
developed for the identification of RNA binding proteins in silco. Among these methods,
SVM is very popular because it can be easily implemented. Bock and Gough [66] have
first shown that SVM is applicable for predicting RNA-binding proteins from protein
primary sequence. In recent studies, Yu et al. [67] have predicted functional classes on
the basis of targets such as rRNA, mRNA, tRNA and viral RNA of RNA binding proteins
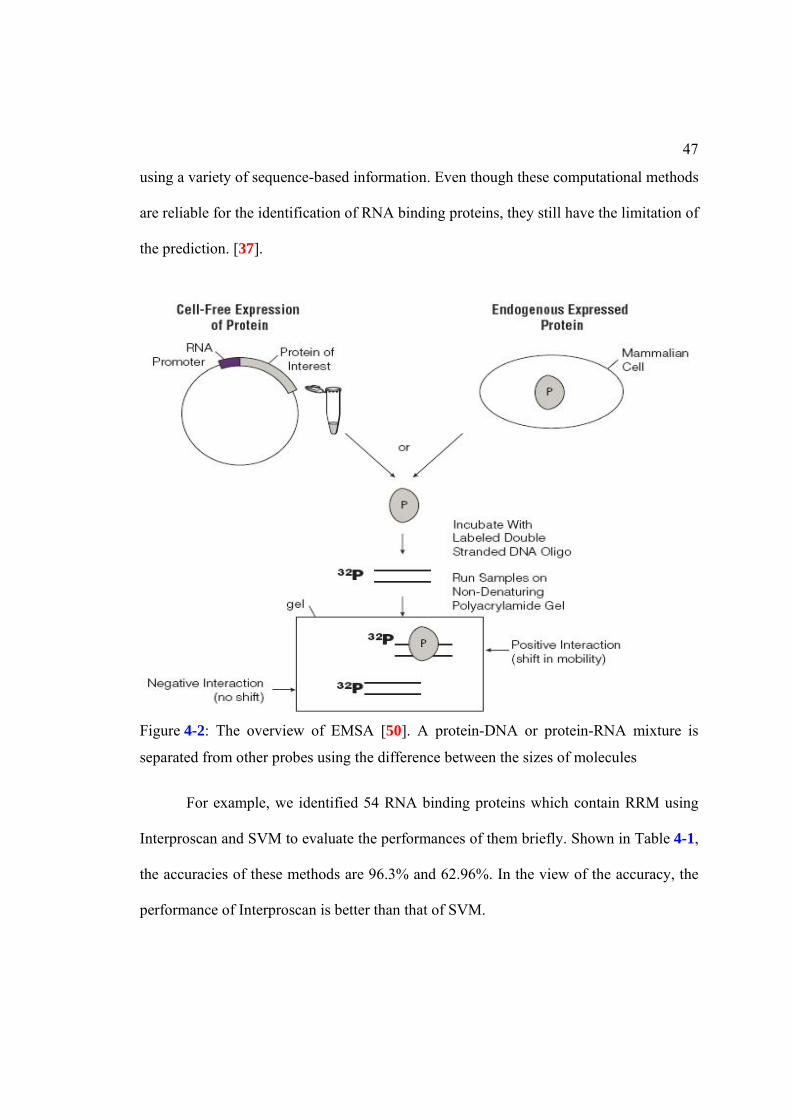
47
using a variety of sequence-based information. Even though these computational methods
are reliable for the identification of RNA binding proteins, they still have the limitation of
the prediction. [37].
For example, we identified 54 RNA binding proteins which contain RRM using
Interproscan and SVM to evaluate the performances of them briefly. Shown in Table 4-1,
the accuracies of these methods are 96.3% and 62.96%. In the view of the accuracy, the
performance of Interproscan is better than that of SVM.
Figure 4-2: The overview of EMSA [50]. A protein-DNA or protein-RNA mixture is
separated from other probes using the difference between the sizes of molecules

48
However, while Interproscan can only detect the regions of RRMs, SVM can
predict the functions of these proteins in Figure 4-3. To overcome this limitation, we need
to develop a new method which can predict the regions of RNA binding domains and
annotate the functions of a protein together using quantitative measurements.
To resolve this question, we applied GDDA-BLAST to identify RNA binding
proteins. Following the procedures in Figure 4-4, we clustered 16 positive sequences with
RRM and 25 negative sequences. After drawing the dendrogram of the sequences, we
found a problem to identify RNA binding using GDDA-BLAST.
Shown in Figure 4-5, the dendrogram of the sequences analyzed by GDDA-
BLAST contains false positive sequences. Because of these sequences, we cannot predict
the function of RNA binding proteins accurately. Thus, we need to develop new
Table 4-1: The comparison of the performances between Interproscan and SVM
Interproscan SVM True positive 52 34 False negative 2 20 The accuracy 96.3% 62.96%
Figure 4-3: The problems of functional annotations in conventional programs (a) The
functional prediction using Interproscan. (b) The functional annotation from NCBI.

49
strategies to eliminate these sequences. The first is to define a threshold to filter the
sequences, and the second is to design a new phylogenetic profile.
To apply these strategies to GDDA-BLAST, we implemented a classification
library for the identification of RNA binding proteins. Shown in Figure 4-6, we first
collect real sequences from a biological database (DB) such as NCBI. Then, we generate
domain profiles from the real sequences. After aligning the sequences against domain
profiles by GDDA-BLAST, we calculate normalized scores of all residues in a query, the
average scores of each query and norms of average scores on the basis of the positive
alignments from GDDA-BLAST. Next, the false positive sequences can be filtered
Figure 4-4: The pipeline of GDDA-BLAST for the identification of RNA binding
proteins. After generating the phylogenetic profiles from the positive alignments, the
pearson correlations among these profiles are calculated. Based on these values, the
sequences are clustered by hierarchical clustering.

50
among queries by a threshold derived from the norms (average scores) for all queries.
After filtering them, we generate a residue distribution matrix of the positive sequences to
investigate the functional or structural relationships of these sequences. Finally, we
cluster the positive sequences using Hierarchical clustering and Pearson’s correlation.
To define a threshold for the elimination of false positive sequences, the
normalized scores of residues are first calculated on the basis of total scores of residues
from the positive alignments using eq. (4.1). In fact, in the alignment between a query
and a domain profile, if two resides are identical, the score for the residue in the query is
Figure 4-5: The false positive sequences in phylogenetic profiles from GDDA-BLAST.
Read boxes represent the negative sequences. The group of positive sequences contains
several negative sequences.

51
assigned 2, and, if two residues are similar, the score is assigned 1. After assigning the
scores to all residues in the query, we add all scores to calculate the total score for the
query. We then normalize the score of each residue by the average of total score.
Then, we calculate the average score of each sequence for filtering the sequences
using eq. (4.2). We finally calculate the norms of average scores to reduce the effect of
the length with eq. (4.3) because these scores are proportional to the length of the query.
Figure 4-6: A classification library for the identification of RNA binding proteins. (i-ii)
Domain profiles are generated on the basis of real sequences from NCBI database. (iii-iv)
The modified sequences are aligned against the parent profile by rps-BLAST to collect
positive alignments. (v-vi) The sequences are divided into positive and negative groups
using a threshold calculated from the average residue scores of queries. (vii-viii) The
functional dendrogram is built using a residue distribution matrix generated from the
positive sequences.

52
To investigate the functional or structural relations among the sequences, we also
designed a new residue-based phylogenetic profile. Shown in Figure 4-7, the matrix
contains the compositions of 20 amino acids and 3 descriptors of chemical features. The
composition of an amino acid is the number of an amino acid divided by the number of
total amino acids.
The 3 descriptors represent global composition of specific chemical groups, and
they consist of composition (C), transition (T), and distribution (D). The composition (C)
is the number of amino acids with a particular group divided by total number of amino
acid in all chemical groups. Transition (T) is the frequency of transition from one
chemical group to another chemical group in a sequence. Distribution (D) is the chain
length within the first, 25%, 50%, 75% and 100% of the amino acid in a specific
chemical group [51].
Figure 4-8 represents an example of the hypothetical protein sequence which has
10 As and 16 Bs. The compositions for these two amino acids are
10*100/(10+16)=38.5% for A and 16*100/(10+16)=61.5% for B. The transition of A is
(10/26)*100=38.46% and the transition of B is (16/26)*100=61.54%. The first, 25%,
50%, 75% and 100% of As are located within the first 1, 4, 12, 17, and 26 residues
respectively. Thus, the D descriptor for As is (1/26)*100=3.8%, (4/26)*100=15.4%,
the sum. of total scoresNormalized score of a residue=Raw score- The length of a query 4.1
the sum of positive normalized scoresAverage score of a query = the num. of residues with the positive scores 4.2
the average scores of a query*100N orm of average score = the length of a query*2 4.3

53
(12/26)*100=46.1%, (17/26)*100=65.4, and 100. In the same way, the D descriptor for
Bs is 7.5%, 23.1%, 53.8%, 79.9%, and 92.3% [64].
Figure 4-7: A residue-based phylogenetic profile. It consists of 20 amino acid
compositions and 19 chemical features such as composition (C), transition (T), and
distribution (D)

54
4.2 The identification of RNA binding proteins
For the first training and testing proteins, we selected RRM containing proteins
because these proteins are abundant in different species and organisms. To discover a
threshold for the identification of the RRM containing proteins, we collected 15 positive
sequences with RRM and 24 negative sequences from PDB (Protein Database) and Swiss
database as a training set. Then, we generated RRM domain profiles on the basis of real
sequences from NCBI database. These domain profiles from real sequences can support
GDDA-BLAST to amplify the weak positive alignments strongly. After calculating norm
of the average score of each query, we drew the distribution graph of the norms of the
average scores for all sequences.
Shown in Figure 4-9(a), the sequences in a training dataset are completely
separated into positive and negative groups. The minimum of the positive group is
Figure 4-8: The sequence of a hypothetic protein for describing derivation of the feature
vector of a protein. Sequence index indicates the position of an amino acid in the
sequence. The index for each type of amino acids in the sequence (A or B) indicates the
position of the first, second, third, … of that type of amino acid (The position of the first,
second, third, …, A is at 1, 3, 4, …). A/B transition indicates the position of AB or BA
pairs in the sequence [64].

55
263.4171 and the maximum of negative group is 127.2489 from the measurement for the
boundary of each group. We finally selected the minimum of the positive group as a
threshold for RRM containing proteins.
Figure 4-9: Thresholds for the positive sequences in training sets (a) The thresholds of
two groups in the training set containing 15 positive and 24 negative sequences. (b) The
thresholds of two groups in the expanded training set containing 55 positive and 151
negative sequences

56
Next, we used 55 positive sequences and 151 negative sequences from a yeast
database and PDB to extend the training dataset. We especially added the 127 sequences
which are proven not to bind nucleic acids from PDB for the measurement of an accurate
threshold [38]. Since the sequences are completely separated and the minimum and
maximum are not changed after the analysis in Figure 4-9(b), we selected the same value
of the first training dataset as a threshold. Based on this threshold, we tried to identify
RRM containing proteins in a testing dataset, which contains 20 positive and 137
negative sequences, and we calculated the accuracy of the identification using Eq. (4.4).
Shown in Figure 4-10, we identified 20 positive sequences in the testing set, and the
accuracy is 100%.
Figure 4-10: The identification of RRM containing proteins in a testing dataset containing
20 positive and 137 negative sequences.

57
Where TP is the number of true positives, TN is the number of true negatives, FP is the
number of false positive, and FN is the number of false negative.
Then, we compared our performance to two other popular algorithms such as
Interproscan and SVM. We observe that, while SVM does not perform well in either the
training or the testing dataset, phylogenetic profiles and Interproscan provide robust
measures in Table 4-2.
To extend upon these discoveries, we performed similar analyses for single-fold
RBP classes. These classes include KH1, double-stranded RNA, and zinc fingers. The
results from these experiments are provided in Table 4-3. We observe that phylogenetic
profiles have 100% accuracy for all single-fold RBPs tested. In comparison, SVM
performs poorly in all of these datasets, while Interproscan performs well.
T P + T NA c c u r a c y
T P + T N + F P + F N 4.4
Table 4-2: Comparison with the sensitivities of other methods for the identification of
RRM containing proteins in training and testing sets.
Method TP FN Sensitivity (%) Pylogenetic classifier 55 0 100
Interproscan 54 1 98.18 Training set
SVM 35 20 63.63 Pylogenetic classifier 20 0 100
Interproscan 20 0 100 Testing set
SVM 12 8 60

58
This methodology is also scalable. Thus, we screened the yeast and human
proteome for RRM, double-stranded RNA binding, KH1, and zinc-finger domains. As
shown in Table 4-4, our methods detect both known and unknown members of the
Table 4-3: Comparison with the sensitivities of other methods for the identification of the
four single-fold RNA binding protein groups in training sets.
Group Method TP FN Sensitivity (%) Phylogenetic classifier 17 0 100
Interproscan 16 1 94.11 Double-
stranded RNA binding SVM 11 6 64.71
Phylogenetic classifier 11 0 100 Interproscan 11 0 100
KH I
SVM 5 6 45.46 Phylogenetic classifier 16 0 100
Interproscan 16 0 100 zf-ccch
SVM 9 7 56.25 Phylogenetic classifier 17 0 100
Interproscan 17 0 100 zf-cchc
SVM 9 8 52.94
Table 4-4: The results of identification for five RNA binding protein groups in yeast and
human proteomes. 29 and 168 novel potential proteins for these groups are identified in
yeast and human proteomes
yeast human RRM 54 23 372 67
Double-stranded RNA binding domain
2 2 39 12
KH1 6 2 56 39 zf-ccch 8 0 41 10 zf-cchc 11 2 11 40
total 81 29 519 168

59
classes of RBPs in both proteomes. In the case of the yeast proteome, we determined that
our method detects all of the previously identified RBPs. These results predict that the
number of RBPs in both proteomes is underestimated by 31% and 34% for the yeast and
human proteome respectively.
In 2008, Shazman et al. demonstrated that SVM methods could be improved by
incorporating electrostatic surface patch information into their analyses [38]. This
provided an excellent benchmark dataset for our study, as well as another algorithm with
which to compare our performance. First, our preliminary experiment using this dataset
as a testing set was performed with class-specific domain profiles. However, these class-
specific profiles were insensitive in this dataset.
Therefore, we wondered whether merely increasing the number of domain
profiles would improve our results. To accomplish this task, we used the PROSITE
database and used a key-word search for “RNA-binding”. The results from this search
were then manually confirmed to ensure the specificity of these sequences. Importantly,
the structure of these sequences was not taken into account. Following, additional
sequences were identified and domain profiles were generated from the non-redundant
NCBI database using PSI-BLAST.

60
Using this expanded PSSM library (2695 profiles), we then analyzed a training set
containing 100 single-stranded RBPs and 127 negative sequences in Figure 4-12. Under
these conditions, we see a clear separation of positive and negative sequences. In our
* 2695 single-stranded RNA binding profiles* Training set
- 100 positive sequences- 127 negative sequences
High threshold : 228.1489
Low threshold : 196.8542
Figure 4-11: The threshold for the classification of single-stranded RNA binding
proteins. 100 positive and 127 negative sequences in a training set are classified using
2695 single-stranded RNA binding profiles. The threshold is 228.1489.
Table 4-5: The comparison with other methods. The table summarizes the results of
identification for single-stranded RNA binding proteins using three methods such as
GDDA-classifier, Interproscan, and support vector machine (SVM). The number of
single-stranded RNA binding proteins is 37, and the number of non-nucleotide binding
proteins is 118.
Sensitivity (%) Specificity (%) Accuracy (%) Pylogenetic classifier 86.5 100 96.8
SVM with features from electrostatic surface patches
80 90 88
Interproscan 78.4 100 94.8 SVM with features from
amino acid sequences 78.4 96.6 92.3

61
testing dataset, which is comprised of 37 positive and 118 negative sequences from the
Shazman et al. study, we achieve 100% specificity, 96.8% accuracy, and 86.5%
sensitivity. In comparison, they reported 90% specificity, 88% accuracy, and 80%
sensitivity in Table 4-5. Thus, with the expansion of the PSSM library, our results rival
those previously obtained.
Using the same paradigm, we generated additional profiles for double-stranded
RNA binding (dsRNA), single-stranded DNA binding (ssDNA), and double-stranded
DNA binding (dsDNA) in Table 4-6. We then compared our results to those from the
Shazman study for the classification of ssRNA and dsDNA binding domains [38]. They
obtained 50% specificity, 51% accuracy, and 53% sensitivity, for this dataset. Our results
using either our ssRNA or dsDNA PSSM libraries are much improved: 97%/83%
specificity, 91%/80% accuracy, and 86%/76% sensitivity respectively.
Table 4-6: The classification among six types of RNA binding proteins such as double-
stranded RNA binding vs. double stranded DNA binding proteins, single-stranded RNA
binding vs. double-stranded DNA binding proteins, and single-stranded RNA binding vs.
single-stranded DNA binding proteins.
profiles Sensitivity(%) Specificity(%) Accuracy (%) dsRNA binding 100 100 100 dsRNAvs.dsDNA dsDNA binding 76.47 85 79.6 ssRNA binding 86.47 97.06 91.55 ssRNAvs.dsDNA dsDNA binding 76.47 83.78 80.28 ssRNA binding 86.47 94.12 88.89 ssRNAvs.ssDNA ssDNA binding 94.12 94.59 94.44

62
We also compared our results in additional testing sets curated from PROSITE,
for proper classification of dsRNA vs. dsDNA and ssRNA vs. ssDNA. Although
attempted, Shazman et al. concluded that to accomplish these comparisons, further
refinement of their method was needed [38]. Conversely, we obtain robust measurements
for these comparisons in Figure 4-13, in particular, for dsRNA binding, where we achieve
100% accuracy.
0
50
100
150
200
250
0 200 400 600 800
The no
rm. of average
score
A query length
dsDNA binding
ssRNA binding
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
0 200 400 600 800
The no
rm. of average
score
A query length
dsDNA binding
ssRNA binding
Accuracy=(32+33) *100/71=91.55%
* 2695 single-stranded RNA binding profiles* Testing set
- double-stranded DNA binding : 34 sequences- single-stranded RNA binding : 37 sequences
Threshold : 228.1489
a
b
0
50
100
150
200
250
0 200 400 600 800
The no
rm. of average
score
A query length
dsDNA binding
ssRNA binding
0
500
1000
1500
2000
2500
3000
0 200 400 600 800
The no
rm. of average
score
A query length
dsDNA binding
ssRNA binding
* 2275 double-stranded DNA binding profiles* Testing set
- double-stranded DNA binding : 34 sequences- single-stranded RNA binding : 37 sequences
Threshold : 207.7592
Accuracy=(26+31) *100/71= 80.28 %
Figure 4-12: The classification between double-stranded DNA and single-stranded RNA
binding proteins. (a) The accuracy of double-stranded DNA binding proteins is 91.55%,
and (b) the accuracy of single-stranded RNA binding is 80.28%.

63
a
b
0
50
100
150
200
250
0 500 1000 1500 2000 2500
The no
rm. o
f average score
A query length
dsDNA binding
dsRNA binding
0
1000
2000
3000
4000
5000
6000
0 500 1000 1500 2000 2500
The no
rm. o
f average score
A query length
dsDNA binding
dsRNA bindingThreshold : 207.7592
* 2275 double-stranded DNA binding profiles* Testing set
- double-stranded DNA binding : 34 sequences- double-stranded RNA binding : 20 sequences
Accuracy=(26+17) *100/54=79.6%
Threshold : 366.2998
* 101 double-stranded RNA binding profiles* Testing set
- double-stranded DNA binding : 34 sequences- double-stranded RNA binding : 20 sequences
Accuracy=(34+20) *100/54=100%
0
50
100
150
200
250
0 200 400 600 800
The n
orm. of average
score
A query length
ssDNA binding
ssRNA binding
0
1000
2000
3000
4000
5000
0 200 400 600 800
The n
orm. of average
score
A query length
ssDNA binding
ssRNA binding
* 2695 single-stranded RNA binding profiles* Testing set
- single-stranded DNA binding : 17 sequences- single-stranded RNA binding : 37 sequences
Threshold : 228.1489
Accuracy=(26+31) *100/71= 80.28 %
c
d
0
200
400
600
800
1000
1200
1400
1600
1800
2000
0 200 400 600 800
The n
orm. of average
score
A query length
ssDNA binding
ssRNA binding
0
1000
2000
3000
4000
5000
6000
7000
0 200 400 600 800
The n
orm. of average
score
A query length
ssDNA binding
ssRNA binding
* 1753 single-stranded DNA binding profiles* Testing set
- single-stranded DNA binding : 17 sequences- single-stranded RNA binding : 37 sequences
Threshold : 1756.5251
Accuracy=(35+16) *100/54=94.44% Figure 4-13: The classification among other DNA and RNA binding proteins. (a) The
accuracy of double-stranded RNA binding proteins is 100%. (b) The accuracy of double-
stranded DNA binding is 79.6%. (c) The accuracy of single-stranded DNA binding
proteins is 88.89% (d) The accuracy of single-stranded RNA binding is 94.44%

64
4.3 The investigation of functional relations among RRM containing proteins
After filtering false positive sequences by the threads, we can investigate the
functional relations among true positive sequences using hierarchical clustering and
pearson correlation. Since residue-based features are more informative than domain-
based features, we generated a phylogenetic profile on the basis of features derived from
amino acid residues. These features represent structural information of a protein from the
amino acid sequence. They consist of the compositions of 20 amino acid, 3 compositions,
1 transition, and 15 distributions of special chemical groups such as hydrophobic,
positive, and negative-charged group. Using this phylogenetic profile, we first clustered
14 control sequences of RRM containing proteins. Shown in Figure 4-14, these sequences
are separately clustered into RNA binding and non-RNA binding groups.
Next, we added sequences from U2AF-homology motif (UHM) group into the
control sequences. UHM is non-canonical type of RRM, which is involved in constitutive
or alternative pre-mRNA splicing [68]. They may also bind ULMs in splicing factors [68].
6 proteins with RRM in Figure 4-15(a) are discovered from the literatures [69]. Since
these proteins bind other proteins instead of RNAs, we assume that they would be
clustered into non-RNA binding group in the dendrogram of control sequences.
Shown in Figure 4-15(b), the UHM sequences are clustered together into non-
RNA binding group. This result suggests that we can predict the functions of unknown
sequences on the basis of this dedrogram using residue-based phylogenetic profiles, if we
add unknown RRM-positive sequences into these control sequences.

65
Figure 4-14: The dendrogram of control sequences. Nucleolin, SXL, PABP, HUD,
hnRNPA1, and PTB bind RNAs, but 2J8A, 1JMT, 1RK8, 2I2Y, 1OPI and 2PE8 bind
other proteins instead of RNAs from the literature [38]. These proteins are divided into
RNA bind and non-RNA bind groups.
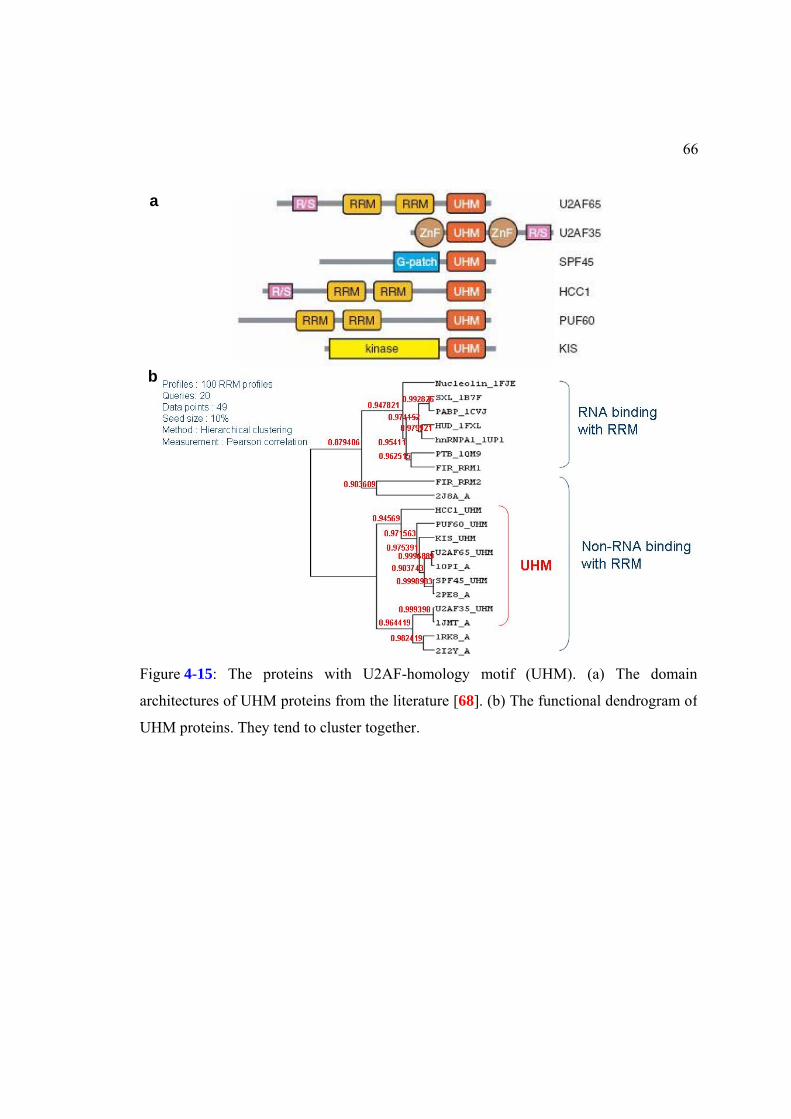
66
a
b
Figure 4-15: The proteins with U2AF-homology motif (UHM). (a) The domain
architectures of UHM proteins from the literature [68]. (b) The functional dendrogram of
UHM proteins. They tend to cluster together.

67
4.4 Summary
For the quantitative functional evaluation, GDDA-BLAST is applied to
investigate functional information in RNA binding proteins. However, false positive
sequences should be filtered in phylogenetic profiles generated from GDDA-BLAST to
investigate the information. To eliminate these sequences, we designed two strategies.
One is the threshold for the filtering, and the other is a new phylogenetic profile
containing accurate information of proteins. For the discovery of the threshold to filter
false positives, we proposed a computational classification assay for the identification of
RNA binding proteins. This assay contains eight procedures, and we found the thresholds
for each protein family using five RNA binding protein families, which contain: (1) RRM,
(2) double-stranded RNA binding domain, (3) K-homology domain, (4) zf-CCCH and (5)
zf-CCHC domain.
In fact, we first calculate the normalized scores of all residues in each sequence to
find the threshold to filter the false positive sequences. After calculating the norms from
the average scores for all sequences, we selected the threshold from the norms. For
example, using 55 positive and 151 negative sequences with RRMs as a training dataset, I
drew the distributions graph on the basis of the norms. Shown in Figure 4-9, the
sequences are divided into positive and negative groups. The minimum of the norm in the
positive group is 263.4171 and the maximum in the negative group is 127.2489. Among
these values, the minimum in the positive group is chosen as a threshold. Based on this
threshold, I identified 20 positive sequences from 157 sequences of a test dataset, and the
accuracy is 100%.

68
Following the same procedures, we found the thresholds for other proteins
containing dsRNA, KH, zf-CCCH, and zf-CCHC domains. Using these thresholds, we
succeeded to classify positive sequences from each testing dataset with 100% accuracy.
These results show that the false positive sequences can be filtered accurately using these
thresholds. I finally identified 82 known and 26 unknown RNA binding proteins in yeast
proteome using the classification library with the same thresholds.
In addition, we identified RBPs containing structural unique motifs by 2695
expanded PSSM profiles in a testing dataset with 37 positive and 118 negative sequences.
We achieved 100% specificity, 96.8% accuracy, and 86.5% sensitivity. For the specific
folds (dsRNA vs. dsDNA, dsRNA vs. dsDNA and ssRNA vs. ssDNA), we also
accomplished the higher accuracies than the SVM using structure features.
To implement a new phylogenetic profile, we used a variety of sequence-based
information from proteins. The new phylogenetic profile contains the compositions of 20
amino acids and 3 descriptors of specific chemical groups such as hydrophobic, negative
and positive charged groups. The composition of an amino acid is the number of an
amino acid divided by the number of total amino acids. The 3 descriptors represent global
composition of specific chemical groups, and consist of composition (C), transition (T),
and distribution (D). The composition (C) is the number of amino acids with a particular
group divided by total number of amino acid in all chemical groups. Transition (T) is the
frequency of transition from one chemical group to another chemical group in a sequence.
Distribution (D) is the chain length within the first, 25%, 50%, 75% and 100% of the
amino acid in a specific chemical group.

69
Using this new residue-based phylogenetic profile, we first clustered the control
sequences containing RRM using hierarchical clustering and Pearson correlation.
Figure 4- shows that the sequences are clustered into RNA binding and non-RNA binding
groups. To investigate functions of this phylogenetic profile, we added UHM sequences,
which bind proteins, into the control sequences. After clustering all of these sequences,
they were clustered into RNA binding and non-RNA binding groups, and the UHM
sequences were located in the non-RNA binding group. Based on these results, we
conclude that the new phylogenetic profile would be helpful to infer the functional
relationships among RNA binding proteins.
To apply this assay to annotate functional characteristics of a protein, we should
consider three issues in the development. First, we need define a format of the annotation
because the means of functions are changed by the view of the annotation. Second, we
need to develop methods to extract accurate features from a sequence for the
phylogenetic profiles. Finally, we need a statistical standard to decide the functional
relationships between proteins.

Chapter 5
Summary and Discussion
5.1 Summary
This thesis described the procedures to develop a unified computational method
for measuring the structural, functional and evolutionary characteristics of a protein from
the amino acid sequence simultaneously. As the computational and biological techniques
are advanced, a huge amount of probable proteins are recently predicted from genomes.
Despite many researches to annotate these proteins accurately, we face several obstacles
to annotate structural, functional and evolutionary properties of the proteins. First, even
though experimental methods identify many uncharacterized proteins in proteomes, the
annotation of these proteins takes longer time than the identification, and existing
erroneous annotation can generate a false annotation of a new protein in some case.
Second, the annotation requires the accurate subjective and contextual definition of
protein function because lots of proteins have multiple functions. Because of these
problems, the accurate structural and functional annotation of a protein is the challenging
task in all biological fields.
In spite of these obstacles, the structural, functional, and evolutionary
characteristics of a protein can be determined by its amino acid sequence because the
protein consists of the amino acid sequence. Many computational methods such as
homology detection, machine learning, and phylogenetic method have investigated these

71
characteristics only using the amino acid sequence. These methods are powerful for the
annotation of some proteins, but they are not enough to annotate all proteins accurately.
For example, homology-based methods usually predict the functions of proteins with
high sequence similarity accurately. However, if the pair-wise sequence similarity
between sequences is lower than 25%, they are not sensitive to identify these distant
homologous sequences. In addition, even though the similarity of some proteins such as
some enzymes is very high, the methods cannot detect their homologous relations
because some residues in the proteins are not reserved among sequences. Finally, if the
existing annotations in databases contain errors, homology-based methods allow these
erroneous annotations to amplify and propagate the errors through the databases.
Since machine learning methods can predict functional properties of proteins on
the basis of sequence derived features, they are independent of sequence similarity.
However, the biased results can be produced by the number of datasets and the sequence-
derived features because their accuracy depends on training sets and feature extracting
methods from sequences. Phylogenetic method infers functional relationships among
proteins on the basis of the presence or absence of the protein across genomes. While the
phylogenetic profiles from prokaryotic genomes describe the functional relationships of
proteins clearly, the phylogenetic profiles from eukaryotic genomes are less informative
to predict the functional relationship, despite some successful researches for the specific
functional prediction of a protein. In addition, the accuracy of the analysis is low due to
the limitation of genome and genome sequences.
In an attempt to overcome these drawbacks, we have developed a unified
computational pipeline, called GDDA-BLAST, for measuring the structural, functional

72
and evolutionary characteristics of a protein using phylogenetic profiles. Our central
hypothesis for the development is that the structural, functional and evolutionary
information can be inferred from the sequence by a unified method, even if the pair-wise
identity of sequences is below 25%. “Seeding” and “pylogenetic profile” are important
innovative processes among five procedures of GDDA-BLAST.
“Seeding” is the resampling strategy designed to amplify and encode the
alignments possible for any given query sequence. This seed can be generated from a
profile by taking any fraction of the profile from N-terminus or C-terminus. Then, it is
inserted into every position of the query at a time, creating a consistent initiation site.
This site allows rps-BLAST to extend an alignment even between highly divergent
sequences.
While a phylogenetic profile generally encodes the presence or absence of a
protein in known genomes, the phylogenetic profile from GDDA-BLAST is a vector
where each entry quantifies the existence of alignments with a domain profile. This
profile represents M (# of profiles) by N (# of queries) matrix. Based on this matrix, we
create a dendrogram of functional relationships among proteins calculating pearson
correlation or a phylogenetic tree measuring Euclidian distance.
To evaluate the performance of this computational pipeline, we measure the
number of true homologous pairs detected by the pipeline. For the performance
evaluation, we selected PDB40D-J containing 935 sequences whose pair-wise identities
are less than 40% in the superfamilies from the structural classification of proteins
(SCOP) database. Then, we extracted 289 sequences below 25% pair-wise identity from

73
PDB40D-J to evaluate the performance in twilight zone. 26374 domain profiles from
CDD and PDB are used as profiles for GDDA-BLAST.
First, we calculated the similarity scores between each test and reference sequence
for potential homologues predicted by GDDA-BLAST, PSI-BLAST, and SAM. For the
similarity score of PSI-BLAST and SAM, we used E-value, which represents the
expectation value of hits shown by chance when searching a particular size of a database.
For the similarity score of GDDA-BLAST, we used Hybrid LogWeighted scoring
scheme. Hybrid LogWeighted scoring scheme consists of two steps. First, we calculate
the scores of three phylogenetic profiles such as # of hits, % of maximum coverage,
and % of average identity. Then, their scores are adjusted on the basis of the frequency
for the domains aligned with queries. Next, test sequences are ranked in ascending order
following similarity scores. Counting the number of true and false positives and negatives
within a sliding window, we plot receiver operating characteristic (ROC) curve of each
method. Shown in Figure 3-4, the performance of GDDA-BLAST is comparable to those
of SAM and PSI-BLAST with datasets in superfamilies and twilight zone.
For the quantitative functional evaluation, GDDA-BLAST is applied to
investigate functional information in RNA binding proteins. When GDDA-BLAST is
applied to identify RNA binding proteins in a quantitative manner, false positive
sequences should be filtered in phylogenetic profiles generated from GDDA-BLAST. To
achieve this purpose, we contrived two strategies: the quantitative threshold and a
residue-based phylogenetic profile. First, we implemented the classification library to
find the quantitative thresholds for RNA binding proteins. Using this library and their
theresholds, we identified RNA binding proteins containing RRM, dsRNA, KH, zf-

74
CCCH, and zf-CCHC domains in their testing datasets with 100% accuracy. Then, we
also identified 82 known and 26 unknown RNA binding proteins in yeast proteome with
the same thresholds and classification library.
After filtering the false positive sequences, we built new phylogenetic profiles
from the true positive sequences to investigate functional relationships among the
sequences. This new phylogenetic profile consists of the compositions of 20 amino acids
and 3 descriptors of chemical features from amino acid residues. The composition of an
amino acid is the number of an amino acid divided by the number of total amino acids.
The 3 descriptors represent global composition of specific chemical groups, and consist
of composition (C), transition (T), and distribution (D). Using this new phylogenetic
profiles, we clustered RRM containing sequences by hierarchical clustering and pearson
correlation. Shown in Figure 4-12, the sequences are divided into RNA binding and non-
RNA binding classes accurately. This functional dendrogram would be good reference to
predict the functions of unknown proteins.
5.2 Discussion
Using a resampling technique and phylogentic genetic profile, we have
successfully developed a unified framework which can quantitatively measure functional,
structural, evolutionary relations among proteins. Through experiments in our researches,
this computational assay has a potential power to resolve challenging problems in
homology detection and functional prediction. However, this assay still has some
drawbacks to improve.

75
In the homology detection, we should first develop a method which can build domain
profiles to generate the best phylogenetic profiles for functional or structural
characterized proteins. We generally use domain profiles from CDD and PDB for the
analysis of proteins. Despite being useful for the functional prediction of some proteins,
these profiles are not satisfied with all requirements for the analysis of protein
characteristics because some domains in the profiles cause to generate noises in the
phylogenetic profiles.
Second, we should implement the best scoring scheme for the comparison of
sequences because the performance for homology detection depends on the score of each
sequence. Even though many homology-based methods generally use e-value and hit
score to detect homology, these methods sometimes miss to detect remote homologous
sequences with low sequence identities because e-value depends on the size of the
database and hit score is decided by the number of identical residues,. To overcome these
problems, GDDA-BLAST uses pearson correlation value to measure the similarity
among phylogenetic profiles from sequences. In spite of the independency of sequence
identities and the size of a database, the pearson correlation itself is not enough to
measure the similarity between sequences because it is too sensitive for noises in the
phylogenetic profiles. Therefore, we need to implement the score system which is
independent of noise to measure the similarity of phylogenetic profiles. Finally, we need
to design residue-based phylogenetic profiles which contain accurate information from
sequences. If we can discover key residues to determine the functional and structural
characteristics of a protein from the sequence, we would extract the unique features only
from the sequences to generate accurate phylogenetic profiles.

76
Especially, to apply this assay to investigate the functional characteristics of a
protein, we should resolve three issues in the development. First, we need to define a
format reliable for multiple contents because some proteins have multiple functions in
different organisms. Second, we should develop methods to extract accurate features
from the amino acid sequence of a protein to generate the phylogenetic profiles reliable
for the purpose of the analyses such as function, structure and evolution. Finally, we have
to develop a statistical measurement to support biological means of our results. Even
though GDDA-BLAST still has many obstacles to overcome, we expect that this pipeline
would be one of the innovative tools to approach undiscovered information in a protein
sequence.

Chapter 6
Future Perspectives
Currently, many researches are devoted to the development of the annotating
methods for the functional or structural characteristics of proteins. Even though advanced
computational technology allows researchers to analyze a huge amount of proteins
automatically with high speed, there are still many problems for accurate annotation of
proteins. One of main problems is that the definition of biological function is ambiguous
and various on the basis of the context in which the function is used [70]. For example,
the function of a protein kinase is the phosphorylation of a hydroxyl group of a specific
substrate in the aspect of biochemistry. However, when protein kinases perform their
functions in different organisms, the function of each kinases are changed following the
organisms [2]. In addition, the functions of kinases also depend on signaling pathways
because the kninases may be part of the signal pathways in a physiological aspect.
Therefore, we should define the aspects of functions before annotating functions of
proteins.
Therefore, we need to define a format of a functional annotation which is satisfied
with a variety of biological aspects. This format is also reliable for an automated
computational and human readable annotation together. Among many type annotations,
The GO (Gene Ontology) annotation serves as one of the most dominant machine-legible
annotations [2]. GO annotation contains the terms representative of three aspects such as
molecular function, biological process and cellular location [71]. Each annotation is

78
connected using DAG (Directed Acyclic Graph). Nodes represent the terms of
annotations and these nodes are assigned from the general means to the specific means in
the graph. As the nodes are connected by following this rule, this graph can describe
functions that are involved in more than a single biological process, cellular compartment
and molecular function because each node may have more than one parent [71].
Based on the concept of GO, we can develop automated functional annotation
system using phylogenetic profiles for proteins instead of gen products. In fact, we would
implement a new annotation system that can annotate proteins with multiple functions in
organisms on the basis of my computational assay. To test our idea, we first collected
6760 yeast proteins 38540 human proteins in proteome databases. With 100 RRM
domain profiles, we discovered 525 candidate proteins containing RRM over the
threshold using my computational assay. After adding these proteins into the RRM
control sequences, we generated residue-based phylogenetic profiles from them. Based
on their phylogenetic profiles, we built a functional dendrogram using hierarchical
clustering and pearson correlation values. Among the clusters in the dendrogram, we first
investigated the proteins which may contain UHM.
Shown in Figure 6-1, orange boxes represent the UHM control sequences, and the
correlation values between the control sequences and new sequences tend to be high.
Then, we annotate the properties of the sequences in the three aspects such as molecular
function, biological process and cellular location on the basis of the annotations from the
NCBI database.

79
Comparing these annotations and correlations of these proteins, we selected 13
potential UHM proteins among the proteins. After calculating the pair-wise sequence
identities between these candidate proteins and control sequences using a local alignment
tool, we finally classified them into closely related and distantly related groups in
Figure 6-2 (a). To prove our annotations, we checked the annotation of each protein in
NCBI, and we found that two sequences such as NP_061862 and NP_060316 bind other
proteins in addition to RNAs. Using the same methodology for other functional
annotations, we can predict novel functional properties of many proteins.
Figure 6-1: The functional dendrogram for the prediction of UHM proteins. Orange
boxes indicate the UHM control sequences.

80
The next example is the functional annotation of a protein, NP_005769. A general
database usually annotates exact functional property of a whole protein but do not
annotate the function of each domain in the protein. Using the dendrogram from GDDA-
BLAST, we can predict the tendency of a function for each RRM in this protein on the
a
b
Figure 6-2: The prediction of UHM candidate proteins (a) the domain architectures of 13
UHM candidate proteins. They are classified into closely related and distantly classes. (b)
The proofs of functional predictions of two proteins from NCBI.

81
basis of functional annotations of adjoining sequences. Shown in Figure 6-3, we can
calculate % of these annotations after counting the number of the same annotation of
adjoining sequences. Then, we can infer the functions of each RRM from statistical
distributions. For example, shown in Figure 6-3 (a), RRM1 might bind RNAs, be
involved in splicing, and belong to nucleus in the aspect of a biological function, process,
and component. Based on these inferences, we can predict new functional characteristics
of each RRM in the protein (Figure 6-3 (b)). To prove these predictions, we searched the
existing annotations of this protein from NCBI. In the annotations from NCBI,
NP_005769 is RNA binding motif protein and it is produced from human RBM5 gene.
This protein binds DNAs, RNAs, nucleotides and proteins with metal ion or zinc ion. It
would be involved in RNA processing, negative regulation of cell cycle and nuclear
mRNA splicing, via spliceosome. In addition, it would be component of intracellular or
nucleus. Comparing these annotations with our new annotations, all of them matched the
annotations of NCBI, and some of them were proven by the literatures. If this annotation
method is applied to study the properties of an unknown protein, we may predict new
functional characteristics of the protein.
From these results, the new functions of a protein may be predicted on the basis of
the annotations of adjoining sequences in the functional dendrogram generated by a
quantitative functional measurement. We need to develop methods to extract reliable
features from a sequence, and statistical methods to prove new annotations for the
inference of the accurate functional annotations from the adjoining-sequence annotations.

82
a
b
Figure 6-3: The inference of new annotations from reference annotation of NP_005869.
(a) The statistical distribution of functional annotations from proteins closely related to
NP_005869. (b) The domain architecture of NP_005869 and new functional annotations
of RRM domains in the protein.

Bibliography
1. Marchler-Baurer A., Panchenko A.R., Benjamin A.S., Thiessen P.A., Geer Y.G.
and Bryant, S.H. CDD: a database of conserved domain alignments with links to
domain three-dimensional structure Nucleic Acids Research, vol.30. no.1. 281-
283 2002
2. Iddo Friedberg, Automated protein function prediction—the genomic challenge
Briefing in Bioinformatics 7(3), 225-242 2006.
3. Yona G., Levitt M., Within the twilight zone: a sensitive profile-profile
comparison tool based on information theory. J Mol Biol. 315(5): 1257-1275
2002.
4. Rychlewski L., Jaroszewski L., Li W. and Godzik A., Comparison of sequence
profiles. Strategies for structural predictions using sequence information. Protein
Sci., 9: 232–241 2000.
5. Altschul S.F., et al , Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs. Nucleic Acids Res. 25(17): 3389-3402 1997.
6. Karplus K., et al Predicting protein structure using hidden Markov models.
Proteins: Struct. Funct. Genet. 1: 134-139 1997.
7. Jaroszewski L., Rychlewski L., Li Z., Li W., Godzik A., FFAS03: a server for
profile-profile sequence alignments. Nucleic Acids Res. 33(Web Server issue):
W284-8 2005.

84
8. Burkhard Rost , Sean I. O'Donoghue , and Chris Sander, Midnight zone of protein
structure evolution, CUBIC(Web Server issue) 1998.
9. Lianyi Han, Juan Cui, Honghuang Lin, Zhiliang Ji, Zhiwei Cao, Yixue Li and
Yuzong Chen, Recent progresses in the application of machine learning approach
for predicting protein functional class independent of sequence similarity,
Proteomics 6: 4023–4037 2006.
10. Pazos, F., Ranea, J. A., Juan, D., and Sternberg, M. J., Assessing protein co-
evolution in the context of the tree of life assists in the prediction of the
interactome, J. Mol. Biol. 352(4): 1002–1015 2005.
11. Zhenran Jiang, Protein Function Predictions Based on the Phylogenetic Profile
Method, Critical Reviews in Biotechnology, 28:233–238 2008.
12. Ko K.D., Hong Y.H., Chang G.S., Bhardwaj G., Rossum D., and Patterson R.L.,
Phylogenetic Profiles as a Unified Framework for Measuring Protein Structure,
Function and Evolution, Phys Arch arXiv:0806.239 2008.
13. Chang G.S, Hong Y.H, Ko K.D., Bhardwaj G., Holmes E.C., Patterson R.L. and
Rossum D., Phylogenetic profiles reveal evolutionary relationships within the
“twilight zone” of sequence similarity, Pro Natl Acad Sci USA 105(36): 13474-
13479 2008.
14. Su Yun Chung and S. Subbiah, A structural explanation for the twilight zone of
protein sequence homology, Structure, 15(4): 1123–1127 1996.

85
15. Russ W.P., Lowery D.M., Mishra P.,. Yaffe M.B, and Ranganathan R., Natural-
like function in artificial WW domains, Nature 437: 579-583 2005.
16. Park J., Karplus K., Barrett C., Hughey R., Haussler D., Hubbard T., and Chothia
C., Sequence comparisons using multiple sequences detect three times as many
remote homologues as pairwise methods, J Mol Biol, 284: 1201-1210 1998
17. Blake J.D. and Cohen F.E., Pairwise sequence alignment below the twilight zone,
J Mol Biol, 307:721–735 2001.
18. Taylor W.R., Identification of protein sequence homology by consensus template
alignment, J Mol Biol, 188:233–258 1986.
19. Yi T.M. and Lander E.S., Recognition of related proteins by iterative template
refinement (ITR). Protein Sci, 3:1315–1328 1994.
20. Gribskov M., McLachlan A.D., Eisenberg D., Profile analysis: Detection of
distantly related proteins. Proc Natl Acad Sci USA, 84:4355–4358 1987.
21. Luthy R., Xenarios I., and Bucher P., Improving the sensitivity of the sequence
profile method, Protein Sci, 3:139–146 1994.
22. Baldi P., Chauvin Y., Hunkapiller T., and. McClure M.A, Hidden Markov models
of biological primary sequence information. Proc Natl Acad Sci USA, 91:1059–
1063 1994.

86
23. Sonnhammer E.L., Eddy S.R., Durbin R., Pfam: A comprehensive database of
protein domain families based on seed alignments, Proteins, 28:405–420 1997.
24. Karplus K., Barrett C., and Hughey R., Hidden Markov models for detecting
remote protein homologies, Bioinformatics, 14(10):846-856 1998.
25. David T. Jones, GenTHREADER: An Efficient and Reliable Protein Fold
Recognition Method for Genomic Sequences, J. Mol. Biol., 287; 797-815 1999.
26. Kim Y. and Subramaniam S., Locally defined protein phylogenetic profiles reveal
previously missed protein interactions and functional relationships, Proteins, 62:
1115-1124 2006.
27. Kim Y., Koyuturk M., Topkara U., Grama A., and Subramaniam S., Inferring
functional information from domain co-evolution, Bioinformatics, 22: 40-49 2006.
28. Lishko P.V., Procko E., Jin X., Phelps C.B., and Gaudet R., The ankyrin repeats
of TRPV1 bind multiple ligands and modulate channel sensitivity, Neuron, 54:
905-918 2007.
29. Batrukova M.A., Betin V.L., Rubtsov A.M., Lopina O.D., Ankyrin: structure,
properties, and functions, Biochemistry (Mosc ), 65: 395-408 2000.
30. Marchler-Bauer A et al , (2005) CDD: a Conserved Domain Database for protein
classification. Nucleic Acids Res, 33 Database Issue: D192-D196.
31. Tamura K, Dudley J, Nei M, and Kumar S (2007) MEGA4: Molecular
Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:
1596-1599 2007.

87
32. Chaumont F., Barrieu F., Wojcik E., Chrispeels M.J., and Jung R., Aquaporins
constitute a large and highly divergent protein family in maize. Plant Physiol 125:
1206-1215 2001.
33. Mio K., Ogura T., Kiyonaka S., Hiroaki Y., Tanimura Y., Fujiyoshi Y., Mori Y.,
and Sato C., The TRPC3 channel has a large internal chamber surrounded by
signal sensing antennas. J Mol Biol 367: 373-383 2007.
34. Letunic I., Copley R.R., Schmidt S., Ciccarelli F.D., Doerks T., Schultz J.,
Ponting C.P., Bork P., SMART 4.0: towards genomic data integration, Nucleic
Acids, Res 32 Database issue: D142-D144 2004.
35. Sonnhammer E.L., von H.G., Krogh A., A hidden Markov model for predicting
transmembrane helices in protein sequences, Proc Int Conf Intell Syst Mol Biol, 6:
175-182 1998.
36. Murzin A. G., Brenner S. E., Hubbard T., and Chothia C., SCOP: a structural
classification of proteins database for the investigation of sequences and
structures, J. Mol. Biol. 247, 536-540, 1995.
37. Chen, Y.C. and Lim, C., Predicting RNA-binding sites from the protein structure
based on electrostatics, evolution and geometry, Nucleic Acids Research, 36(5),
e29 2008.
38. Shazman, S. and Mandel-Gutfreund, Y., Classifying RNA-Binding Proteins
Based on Electrostatic Properties, 4(8), PLOS computational biology e1000146
2008.

88
39. Chen Y. and Varani G., Protein families and RNA recognition, FEBS Journal,
272:2088–2097 2005.
40. Puglisi J.D., Chen L., Blanchard S. and Frankel A.D., Solution structure of a
bovine immunodeficiency virus Tat-TAR peptide-RNA complex, Science, 270:
1200–1203 1995.
41. Ye X., Kumar R.A. and Patel D.J., Molecular recognition in the bovine
immunodeficiency virus Tat peptide-TAR RNA complex, Chem Biol, 2:827–840
1995.
42. Varani G. and Nagai K., RNA recognition by RNP proteins during RNA
processing and maturation, Ann Rev Biophys Biomol Struct, 27:407–445 1998.
43. Antson A.A., Dodson E.J., Dodson G., Greaves R.B., Chen X.P. and Gollnick P.,
Structure of the trp RNA binding attenuation protein, TRAP, bound to RNA,
Nature 401:235–242 1999.
44. Wang X., McLachlan J., Zamore P.D. and Tanaka-Hall T.M. , Modular
recognition of RNA by a human Pumilio-homology domain, Cell 110, 501–512
2002.
45. Lu D., Searles M.A. and Klug A., Crystal structure of a zinc-finger-RNA complex
reveals two modes of molecular recognition, Nature, 426:96–100 2003.

89
46. Hudson B.P., Martinez-Yamout M.A., Dyson H.J. and Wright P.E., Recognition
of the mRNA AU-rich element by the zinc finger domain of TIS11d, Nat Struc
Mol Biol, 11:257–264 2004.
47. Predki P.F., Nayak L.M., Gottlieb M.B.C. & Regan L., Dissecting RNA–protein
interactions: RNARNA Recognition by Rop, Cell 80:41–50 1995.
48. Blaszczyk J., Tropea J.E., Bubunenko M., Routzahn K.M., Waugh D.S., Court
D.L. and Ji X., Crystallographic and modeling studies of RNase III suggest a
mechanism for double-stranded RNA cleavage, Structure, 9:1225–1236 2001.
49. Garner M.M. and Revzin A., A gel electrophoresis method for quantifying the
binding of proteins to specific DNA regions: application to components of the
Escherichia coli lactose operon regulatory system, Nuc. Acids. Res., 9:3047–60
1981.
50. Promega, Protein interaction guide, 24-26.
51. Dubchak I., Muchnik I., Holbrook S.R., and Kim S.H., Prediction of protein
folding class using global description of amino acid sequence, Proc Natl Acad Sci
USA, 92:8700-8704 1995.
52. Gribskov, M., Mclachlan, A.D, and David, E. Profile analysis: Detection of
distantly related proteins Proc. Natl. Acad. Sci. USA, vol. 84. 4355-4358 1987

90
53. Sander, C. and Schneider, R., Database of homology-derived protein structures
and the structural meaning of sequence alignment, Proteins: Struct. Funct. Genet.,
9:56-68 1991.
54. Hilbert, M., Bohm, G. & Jaenicke, R., Structural relationships of homologous
proteins as a fundamental principle in homology modeling, Proteins: Struct.
Funct. Genet., 17:138-151 1993.
55. Chris Sander and Reinhard Schneider, Database of Homology-Derived Structures
and the Structural Meaning of Sequence Alignment, PROTEINS: Structure,
Function, and Genetics, 9:56-68 1991.
56. Russ, W.P., Lowery, D.M., Mishra, P., Yaffe, M.B. and Ranganathan, R.,
Natural-like function in artificial WW domains, Nature, 437: 579-583 2005.
57. Shah, I. and Hunter, L., Predicting enzyme function from sequence: a systematic
appraisal, Proc Int Conf Intell SystMol Biol;5:276–83 1997.
58. Shah, I. and Hunter, L., Identification of divergent functions in homologous
proteins by induction over conserved modules, Proc Int Conf Intell SystMolBiol;
6:157–64 1998.
59. Tian, W. and Skolnick, J., How well is enzyme function conserved as a function
of pairwise sequence identity?, JMol Biol, 333:863–82 2003.
60. Doolittle, R.F. and Bork, P., Evolutionarily mobile modules in proteins, Sci Am,
269:50–6 1993.

91
61. Doolittle RF. The multiplicity of domains in proteins, Annu Rev Biochem,
64:287–314 1995.
62. Ran, J. A., Yeats, C., Grant, A., and Orengo, C. A., Predicting protein function
with hierarchical phylogenetic profiles: the Gene3D phylotuner method applied to
eukaryotic genomes, PLoS. Comput. Biol., doi:10.1371/journal.pcbi.0030237
2007.
63. Suh, B.C. and Hille, B., Regulation of ion channels by phosphatidylinositol 4,5-
bisphosphate, Curr Opin Neurobiol, 15: 370-378 2005.
64. Inna Dubchak, Ilya muchnikt, Stephen R. Holbrook, and Sung-hou Kim,
Prediction of protein folding class using global description of amino acid
sequence, Proc. Natl. Acad. Sci. USA, 92:8700-8704, 1995.
65 Alexande,r P.A., He, Y., Chen, Y., and Orban, J., Bryan PN, The design and
characterization of two proteins with 88% sequence identity but different structure
and function. Proc Natl Acad Sci U S A, 104: 11963-11968 2007.
66 Bock, J.R.and Gough, D.A., Predicting protein–protein interactions from primary
structure. Bioinformatics 17: 455–460 2001.
67 Yu, X., Cao, J., Cai, Y., Shi, T., and Li, Y., Predicting rRNA-, RNA-, and DNA-
binding proteins from primary structure with support vector machines, J. Theor.
Biol. 240: 175–184 2006.

92
68. Corsini, L., Bonnal, S., Basquin, J., Hothorn, M., Scheffzek, K., Valca´rcel, J. and
Sattler, M., U2AF-homology motif interactions are required for alternative
splicing regulation by SPF45, Nature structural & Molecular biology, 14:260-269
2007.
69. Kielkopf C. L., Lücke S., and Green M. R., U2AF homology motifs: protein
recognition in the RRM world Genes Dev.; 18(13): 1513–1526 2004.
70. Rost, B., Liu, J., Nair, R., et al., Automatic prediction of protein function, Cell
Mol Life Sci 60:2637.50 2003.
71. Ashburner, M., Ball, C.A., Blake, J.A., et al., Gene ontology: tool for the
unification of biology. The gene ontology consortium, Nat Genet, 25:25–9 2000.

VITA
Kyung Dae Ko
EDUCATION Penn state University, University Park, PA Ph.D in Bioinformatics & Genomics of IBIOS program(August, 2009) Dissertation topic: “Quantitative functional measurement of a protein using phylogenetic profiles” Master in Computer Science and Engineering, Spring, 2005 Thesis topic: “Designing the Gestalt Detection Domain Algorithm (GDDA) for Detection of Hidden Domains” Master in Electrical Engineering, 2003 Thesis topic: “A design of Directive Photonic-Band-Gap Antennas for a Dual Band operation using CFDTD” PUBLICATION *Kyung Dae Ko, Gaurav Bhardwaj, Yoojin Hong, Gue Su Chang, Kirill Kiselyov, Damian B. van Rossum and Randen L. Patterson, “Phylogenetic profiles reveal structural/functional determinants of TRPC3 signal-sensing antennae”, Communicative & Integrative Biology, Vol. 2, issue 2, March/April 2009 *G.S Chang, *Y.H Hong, K.D. Ko, G. Bhardwaj, E.C. Holmes, R.L. Patterson and D. Rossum, “Phylogenetic profiles reveal evolutionary relationships within the twilight zone of sequence similarilty”, Pro Natl Acad Sci USA, Sept. 2008. *K.D. Ko, *Y.H. Hong, *G.S. Chang, G. Bhardwaj, D. Rossum, and R.L. Patterson, “Phylogenetic Profiles as a Unified Framework for Measuring Protein Structure, Function and Evolution,” Physics Archives, June 2008. Young Ju Lee, Junho Yeo, Kyoung Dae Ko, Raj Mittra, Yoonjae Lee, and Wee Sang Park, “A Novel Design Techinique For Control of Defect Frequencies of An Electromagnetic Bandgap(EBG) Superstrate For Dual-Band Directivity Enhancement,” Microwave and Optical Technology Letters, Vol. 42, No. 1, July 5 2004. Y.J. Lee, J. Yeo, K.D. Ko, R. Mittra, Y. Lee, and S. Park, “Techniques for Controlling the Defect Frequencies of Electromagnetic Bandgap (EBG) Superstrates for Dual-band Directivity Enhancement of a Patch Antenna,” IEEE Antennas & Propagation Society International Symposium/URSI, Monterey, California, Volume: 2 , 20-25 June 2004.