quantum chemical calculations on solvation effects for selected photoreactive aromatic organic...
TRANSCRIPT

Computational and Theoretical Chemistry 965 (2011) 346–352
Contents lists available at ScienceDirect
Computational and Theoretical Chemistry
journal homepage: www.elsevier .com/locate /comptc
Quantum chemical calculations on solvation effects for selectedphotoreactive aromatic organic molecules of atmospheric relevance
Gregory Casey, Gregory R. Wentworth, I.P. Hamilton, Hind A. Al-Abadleh ⇑Chemistry Department, Wilfrid Laurier University, Waterloo, ON, Canada N2L 3C5
a r t i c l e i n f o a b s t r a c t
Article history:Received 5 September 2010Received in revised form 30 September2010Accepted 1 October 2010Available online 12 October 2010
Keywords:TD-DFTUV–visible spectraSolvation effectsElectronic propertiesAtmospheric aerosolsPhotochemistry
2210-271X/$ - see front matter � 2010 Elsevier B.V.doi:10.1016/j.comptc.2010.10.001
⇑ Corresponding author. Tel.: +1 519 884 0710x287E-mail address: [email protected] (H.A. Al-Abadl
The application of computational methods to studies in atmospheric chemistry is instrumental in under-standing reaction mechanisms, dynamics, kinetics, particle nucleation and growth, and heterogeneousreactivity. There are a limited number of theoretical and experimental studies on the impact of surfacephotochemical reactions on water solubility and electronic properties of atmospheric aerosols, particu-larly those containing organic matter. We report computational results using density functional theory(DFT/B3LYP) on geometrical and electronic properties of selected aromatic organic molecules of interestto heterogeneous photochemistry in atmospheric aerosols. The impact of simulated explicit and implicitsolvation on the aforementioned properties was examined for a number of organic compounds. Elec-tronic absorption spectra calculated using time-dependent DFT with the B3LYP functional and 6-31G*basis set showed excellent agreement with experimental UV–visible spectra collected for these organiccompounds. Results are discussed in light of the relative importance of implicit and explicit solvationfor calculating the energies of electronic transitions, correlation between water solubility and hydrogenbonding between explicit water molecules and organic molecules.
� 2010 Elsevier B.V. All rights reserved.
1. Introduction
Atmospheric aerosols impact the climate by their ability to ab-sorb and scatter solar radiation. They also affect the formation andlifetime of clouds, and influence the concentration of gas phasespecies by providing surfaces for them to react on [1]. Atmosphericaerosol representation in climate models is still problematic due totheir highly complex physicochemical properties [2]. These models– used by policy makers at the national and international levels –require rates of heterogeneous reactions of aerosols. In particular,photochemistry in bulk aqueous aerosols (e.g. fog) provides impor-tant pathways for degrading water soluble organic carbon [3–6].These reactions are also important sources for secondary organicaerosols (SOA) such as chlorinated organics [7] and organic sulfates[8,9] and nitrates [10–12]. However, the contribution of heteroge-neous photochemical reactions to the budget of gas phase speciesand production of SOA is still largely unexplored [3–6].
Because the molecular environment of the liquid/air and solid/air interfaces is different from that in the bulk, studies that high-light molecular-level differences between bulk and interfacialenvironments were recently the subject of thematic special issuesof J. Phys. Chem. C [13] and Phys. Chem. Chem. Phys. [14]. Infraredspectroscopic analysis on the nature of surface water on organic
All rights reserved.
3.eh).
monolayers [15] and lipids [16] is found to be ‘‘structured” com-pared to bulk liquid water. We recently studied the structure ofwater adsorbed on solid tannic acid (TA) as a model for humic-likesubstances in aerosols (HULIS) under humid conditions (5–30% RH)[17]. Our results show that the structure of water adsorbed on TAresembles that of water at the interface with polar organic solvents[18] in the sense that surface water molecules have fewer hydro-gen bonds than in bulk water suggesting their ability to act ashydrogen bond donors for incoming gas phase molecules.
Little is known about the impact that surface photochemicalreactions have on water solubility and electronic properties ofatmospheric aerosols, particularly those containing organic matter.Based on results from bulk studies, ‘‘photochemically aged” aero-sols might exhibit a shift in their activation properties as cloudcondensation nuclei (CCN), and their overall chemical reactivity(including photoreactivity) depending on the type of products thatform. Finlayson-Pitts and co-workers [19] investigated the photo-oxidation of a fatty acid adsorbed on NaCl mixed with solid NaNO2
and reported the formation of nitrated organic products consistentwith either O� or �OH radical attack depending on the amount ofwater. Kleffmann and co-workers measured fast photosensitizedformation of HONO from the reaction of NO2 on humic acids [20]and mineral dust [21] under humid conditions. Donaldson andco-workers reported photoenhanced uptake of NO2 and O3, andoxidation of halides by chlorophyll at the salt water/air interface[22–24]. We recently reported a proposed mechanism for the

G. Casey et al. / Computational and Theoretical Chemistry 965 (2011) 346–352 347
formation of surface aryl aldehydes from TA photodegradation un-der humid conditions using DRIFTS [17]. In bulk aqueous solutions,UV–vis spectra of aryl aldehydes have better overlap with solarspectrum than TA. Also, it is well-documented that complete min-eralization of dissolved organic matter (DOM) undergoing photo-transformation in aquatic systems results in formation ofproducts that do not absorb simulated solar light [25]. This resultsin the loss of color of DOM (referred to as photobleaching), whichhas consequences for the penetration depth of UV–B radiation inwater bodies, bioavailability of DOM, and its UV-mediated trans-formation. It is not yet clear if photobleaching is important foraerosols containing HULIS, which warrants further investigation.
The application of computational methods to studies in atmo-spheric chemistry is instrumental in understanding reaction mech-anisms, dynamics, kinetics, particle nucleation and growth, andheterogeneous reactivity [6,26]. Density functional theory (DFT)has been the method of choice of a number studies for calculatinggeometrical parameters, binding energies, electronic and vibra-tional spectra of small gas-phase organic clusters of dicarboxylicacids [27,28] and gas phase O3, HNO3, SO2, and NH3 with watermolecules [29,30]. In the present study, we report results fromtime-dependent DFT calculations (TD-DFT) [31] on the geometriesand electronic transitions of selected organic molecules for com-parison with those observed experimentally from solution UV–vis spectra. The organic molecules shown in Scheme 1 were chosenbecause they are potential phototransformation surface productsof gallic acid and TA in the presence of chlorine radicals. Calcula-tions of molecular orbitals (MOs) and electronic transitions wereperformed in vacuum and by simulating solvation effects implicitlyusing the integral equation formalism-polarizable continuummodel (IEFPCM) [32]. For each medium, calculations were per-formed with water molecules (1–8) added around the organic mol-ecules, one at a time, to simulate explicit solvation [33]. The resultsare discussed in light of the relative importance of implicit and ex-plicit solvation for calculating the energies of electronic transitions,correlation between water solubility and hydrogen bonding be-tween explicit water molecules and the organic molecules.
2. Computational and experimental methods
Computations were performed for the organic molecules shownin Scheme 1 using Gaussian ’09 [34] with the B3LYP [35,36] func-
OH
OH
OH
O
OH O
O
Cl
Cl
(A) Gallic acid (B) Tetrachloro-1 (6×104 )elbulosni()L/gm
O
CH3
Cl OH
O H
(D) Ethanone, 1-(2-chlorophenyl) (E) 2,3,4-Trih ydr (1.5×103 mg/L) (5×104 mg/L
Numbers in parentheses represent estimates of water s
Scheme 1. (See above-mentioned re
tional and the double-zeta 6-31G� basis set [37]. In some casesthese were followed by calculations using the triple-zeta 6-311+G�� basis set [38]. However, since only small differences wereseen for the larger basis set (even for molecule (B) which containschlorine atoms [39]), results are reported for the smaller basis setonly. All the molecules are water soluble except (B), which is solu-ble in ethanol. Vibrational frequencies were calculated for all opti-mized structures to confirm that calculated geometries correspondto local minima. Solvation effects were simulated implicitly usingthe IEFPCM solvation model [32] simulating bulk aqueous and[for molecule (B)] ethanol solvation. Explicit addition of up to eightmolecules around each organic molecule was also done for calcu-lations performed in vacuum and in IEFPCM. UV–vis spectra werecalculated using TD-DFT, using the B3LYP functional and 6-31G�basis set in vacuum and using the IEFPCM solvation model [32].TD-DFT has become one of the most widely used methods for cal-culating the properties of electronically excited states [40].Homogenous peak broadening was modeled with a Lorentzianfunction (HWHM = 10 nm) on each calculated excitation, usingthe normalized calculated oscillator strength (f).
Experimental UV–vis spectra were collected using a fiber opticsUV–vis spectrometer (Ocean Optics USB 4000) for the moleculesshown in Scheme 1: gallic acid (3,4,5-trihydroxybenzoic acid,97–102.5%, CAS: 149-91-7, Sigma), chloranil (tetrachloro-1,4-benzoquinone, 99%, CAS: 118-75-2, Sigma), 1,4-benzoquinone(99%, CAS: 106-51-4, Fisher), ethanone-1-(2-chlorophenyl) (20-chloroacetophenone, 97%, CAS: 2142-68-9, Sigma), 2,3,4-trihydr-oxybenzaldehyde (98%, CAS: 2144-08-3, Aldrich), and tannic acid(1,2,3,4,6-pentagalloyl-O-glucose, ACS grade, CAS: 1401-55-4, AlfaAesar). All chemicals were dissolved in milli-Q water (18.5 MX)except chloranil, which was dissolved in ethanol. The concentra-tion of each solution is reported in the caption of Fig. 2.
3. Results and discussion
3.1. Geometric and electronic structure analysis
In order to investigate the effect of solvation on the electronictransitions of the organic molecules listed in Scheme 1, geometryoptimizations were carried out in vacuum and in IEFPCM as a func-tion of explicit water molecules (added one at a time up to eight).Fig. 1 shows the optimized structures calculated in IEFPCM and
Cl
ClO
O,4-benzoquinone (C) 1,4-benzoquinone
6( ×104 mg/L)
OH
OH
H3C O O
HO
OH
O
O
OH
OH
OHoxybenzaldehyde (F) Tannic acid aromatic ) fragment
olubility at 25°C [50].
ference for further information.)
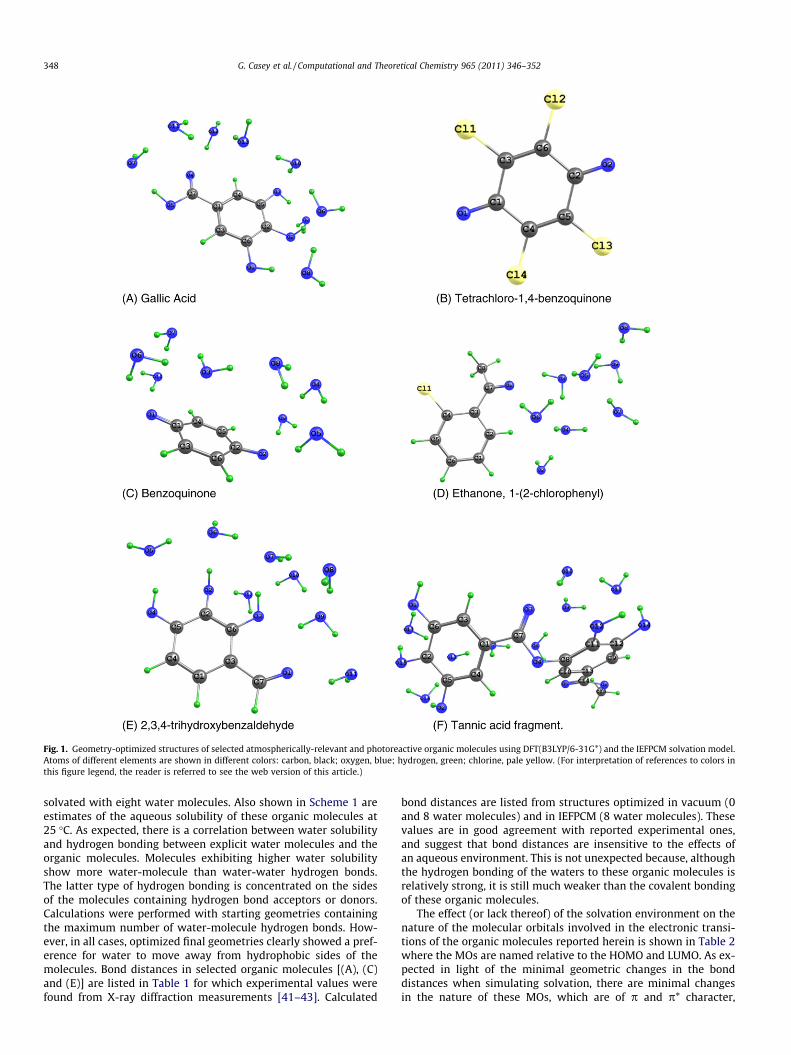
Fig. 1. Geometry-optimized structures of selected atmospherically-relevant and photoreactive organic molecules using DFT(B3LYP/6-31G*) and the IEFPCM solvation model.Atoms of different elements are shown in different colors: carbon, black; oxygen, blue; hydrogen, green; chlorine, pale yellow. (For interpretation of references to colors inthis figure legend, the reader is referred to see the web version of this article.)
348 G. Casey et al. / Computational and Theoretical Chemistry 965 (2011) 346–352
solvated with eight water molecules. Also shown in Scheme 1 areestimates of the aqueous solubility of these organic molecules at25 �C. As expected, there is a correlation between water solubilityand hydrogen bonding between explicit water molecules and theorganic molecules. Molecules exhibiting higher water solubilityshow more water-molecule than water-water hydrogen bonds.The latter type of hydrogen bonding is concentrated on the sidesof the molecules containing hydrogen bond acceptors or donors.Calculations were performed with starting geometries containingthe maximum number of water-molecule hydrogen bonds. How-ever, in all cases, optimized final geometries clearly showed a pref-erence for water to move away from hydrophobic sides of themolecules. Bond distances in selected organic molecules [(A), (C)and (E)] are listed in Table 1 for which experimental values werefound from X-ray diffraction measurements [41–43]. Calculated
bond distances are listed from structures optimized in vacuum (0and 8 water molecules) and in IEFPCM (8 water molecules). Thesevalues are in good agreement with reported experimental ones,and suggest that bond distances are insensitive to the effects ofan aqueous environment. This is not unexpected because, althoughthe hydrogen bonding of the waters to these organic molecules isrelatively strong, it is still much weaker than the covalent bondingof these organic molecules.
The effect (or lack thereof) of the solvation environment on thenature of the molecular orbitals involved in the electronic transi-tions of the organic molecules reported herein is shown in Table 2where the MOs are named relative to the HOMO and LUMO. As ex-pected in light of the minimal geometric changes in the bonddistances when simulating solvation, there are minimal changesin the nature of these MOs, which are of p and p* character,

Table 1Calculated bond distances (DFT/B3LYP-6-31G*) of selected molecules listed in Scheme 1 in vacuum and in IEFPCM, without explicit water molecules and surrounded with 8 watermolecules.
Molecule (Scheme 1) Bonds Bond distances (Å)
In vacuum (0 water) In vacuum (8 water) In IEFPCM (8 water) Experimental Ref.
(A) C1C4 1.404 1.401 1.400 1.392 [41]C1C7 1.482 1.484 1.485 1.480C7O4 1.217 1.242 1.237 1.213C7O5 1.359 1.320 1.327 1.320C1C3 1.400 1.398 1.400 1.394C3C6 1.393 1.400 1.397 1.383C6C2 1.400 1.410 1.410 1.390C6O1 1.362 1.348 1.355 1.371C2C5 1.399 1.407 1.407 1.390C2O2 1.368 1.389 1.383 1.372C5C4 1.387 1.391 1.393 1.382C5O3 1.376 1.375 1.370 1.374
(C) C1C4 1.487 1.484 1.483 1.470 [42]C1O1 1.225 1.235 1.231 1.223C1C3 1.487 1.484 1.482 1.477C3C6 1.343 1.342 1.342 1.334C6C2 1.487 1.475 1.477 1.470C2O2 1.225 1.244 1.240 1.223C2C5 1.487 1.474 1.477 1.477C5C4 1.343 1.344 1.342 1.334C4O1 2.368 2.377 2.370 2.346
(E) C1C3 1.412 1.413 1.415 1.400 [43]C1C4 1.383 1.38 1.377 1.372C3C7 1.446 1.451 1.436 1.426C7O1 1.238 1.233 1.248 1.247C3C6 1.418 1.419 1.422 1.419C6O3 1.351 1.354 1.356 1.344C6C2 1.393 1.399 1.391 1.382C2O2 1.372 1.383 1.373 1.364C2C5 1.398 1.412 1.411 1.395C5O4 1.354 1.336 1.328 1.360C5C4 1.407 1.411 1.418 1.394
G. Casey et al. / Computational and Theoretical Chemistry 965 (2011) 346–352 349
respectively. However, molecular orbital diagrams (not shownhere) of the organic molecules solvated with eight waters showeda number of MOs centred on the water molecules to be degeneratewith the p (below HOMO) molecular orbitals. This complicated theassignment of the electronic vertical transitions. A similar studyfound that when BF�4 weakly binds to a phenyl substituents of apolycyclic chromophore (2.2 Å), the MOs of this charged counterion are degenerate with the p HOMOs of the organic molecule [44].
Fig. 2 shows experimental and calculated UV–vis absorptionspectra using TD-DFT(B3LYP/6-31G�) of the organic moleculeslisted in Scheme 1. The experimental spectra are superimposedwith those calculated in vacuum without explicit water molecules[Calc.(1)] and in IEFPCM with eight explicit water molecules[Calc.(2)]. The vertical line at 290 nm is the solar cutoff and clearlyshows that the organic molecules studied herein will be photoreac-tive under atmospheric conditions. In general, calculated spectraare in excellent agreement with experimental ones collected inaqueous solution. Electronic transitions observed in the TD-DFTspectra calculated using the IEFPCM solvation model are redshifted by less than 20 nm relative to those calculated in vacuum.This suggests that implicit and explicit solvation causes a smallerenergy gap between the MOs involved in these vertical electronictransitions. A theoretical study by Peltier et al. on a polycyclic or-ganic compound also found that simulating bulk solvation usingPCMs does not significantly alter the energy and the intensity ofthe computed transitions [44]. Again, this is not unexpected be-cause, although the hydrogen bonding of the waters to these or-ganic molecules is relatively strong, it is still much weaker thanthe covalent bonding of these organic molecules. However, spectracalculated for molecule (B), in vacuum and simulated solvation byethanol, are red shifted by about 40 nm relative to the experimen-
tal spectrum collected in ethanol. We currently have no explana-tion for this relatively large discrepancy. Table 3 listswavelengths of electronic transitions giving rise to absorptions inthe experimental and calculated spectra (in IEFPCM with eight ex-plicit water molecules) along with their assignments. Experimentalkmax values are in good agreement with those reported in the liter-ature, and primarily correspond to p ? p* transitions with someweak n ? p* transitions. The assignment of the MOs involved inthe calculated transitions is also shown. Fig. 3 shows the variationin the values of kmax as a function of the number of explicit watermolecules obtained from calculated TD-DFT spectra of the watersoluble organic molecules in vacuum and in the IEFPCM solvationmodel. With the exception of molecule (A), the difference betweenvacuum and IEFPCM in the values of kmax for a given cluster isinsignificant. This means that, at the level of simulating the UV–vis spectra for comparison with experimental ones, excluding sol-vation effects will not result in major deviations and will be morecost-effective in terms of computational time and resources.
4. Conclusions and atmospheric implications
In this study, we report TD-DFT calculations on geometries andelectronic properties of selected photoreactive aromatic organicmolecules relevant to HULIS in atmospheric aerosols. Calculationswere performed in vacuum and using the IEFPCM solvation model.For water soluble molecules, calculations were also performed byadding explicit water molecules. Results show that inclusion of sol-vent effects does not significantly shift structural parameters, shapesof molecular orbitals involved in electronic transitions, or energiesof electronic transitions. The results also show that changes to
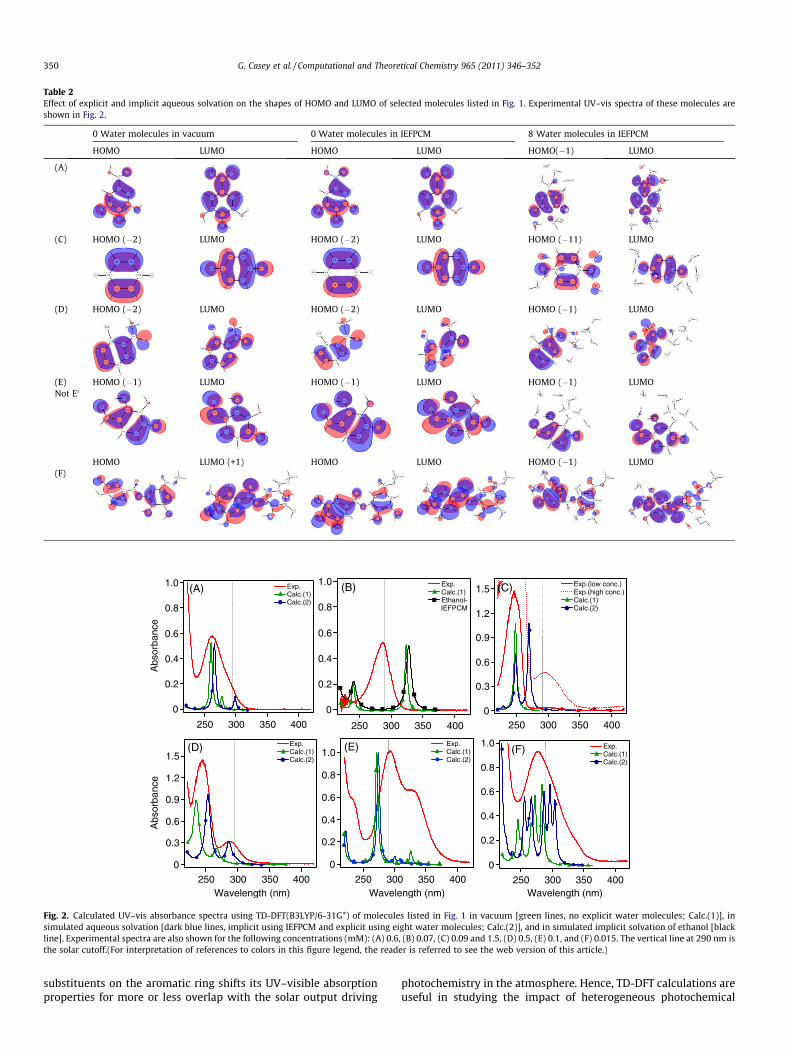
Table 2Effect of explicit and implicit aqueous solvation on the shapes of HOMO and LUMO of selected molecules listed in Fig. 1. Experimental UV–vis spectra of these molecules areshown in Fig. 2.
0 Water molecules in vacuum 0 Water molecules in IEFPCM 8 Water molecules in IEFPCM
HOMO LUMO HOMO LUMO HOMO(�1) LUMO
(A)
(C) HOMO (�2) LUMO HOMO (�2) LUMO HOMO (�11) LUMO
(D) HOMO (�2) LUMO HOMO (�2) LUMO HOMO (�1) LUMO
(E) HOMO (�1) LUMO HOMO (�1) LUMO HOMO (�1) LUMONot E0
HOMO LUMO (+1) HOMO LUMO HOMO (�1) LUMO(F)
1.0
0.8
0.6
0.4
0.2
0
Abs
orba
nce
400350300250
(A) Exp.Calc.(1)Calc.(2)
1.0
0.8
0.6
0.4
0.2
0
400350300250
(B) Exp.Calc.(1)Ethanol-
IEFPCM
400350300250
1.5
1.2
0.9
0.6
0.3
0
(C) Exp.(low conc.)Exp.(high conc.)Calc.(1)Calc.(2)
1.5
1.2
0.9
0.6
0.3
0
Abs
orba
nce
400350300250 Wavelength (nm)
(D) Exp.Calc.(1)Calc.(2)
1.0
0.8
0.6
0.4
0.2
0
400350300250Wavelength (nm)
(E) Exp. Calc.(1) Calc.(2)
1.0
0.8
0.6
0.4
0.2
0
400350300250Wavelength (nm)
(F) Exp.Calc.(1)Calc.(2)
Fig. 2. Calculated UV–vis absorbance spectra using TD-DFT(B3LYP/6-31G*) of molecules listed in Fig. 1 in vacuum [green lines, no explicit water molecules; Calc.(1)], insimulated aqueous solvation [dark blue lines, implicit using IEFPCM and explicit using eight water molecules; Calc.(2)], and in simulated implicit solvation of ethanol [blackline]. Experimental spectra are also shown for the following concentrations (mM): (A) 0.6, (B) 0.07, (C) 0.09 and 1.5, (D) 0.5, (E) 0.1, and (F) 0.015. The vertical line at 290 nm isthe solar cutoff.(For interpretation of references to colors in this figure legend, the reader is referred to see the web version of this article.)
350 G. Casey et al. / Computational and Theoretical Chemistry 965 (2011) 346–352
substituents on the aromatic ring shifts its UV–visible absorptionproperties for more or less overlap with the solar output driving
photochemistry in the atmosphere. Hence, TD-DFT calculations areuseful in studying the impact of heterogeneous photochemical
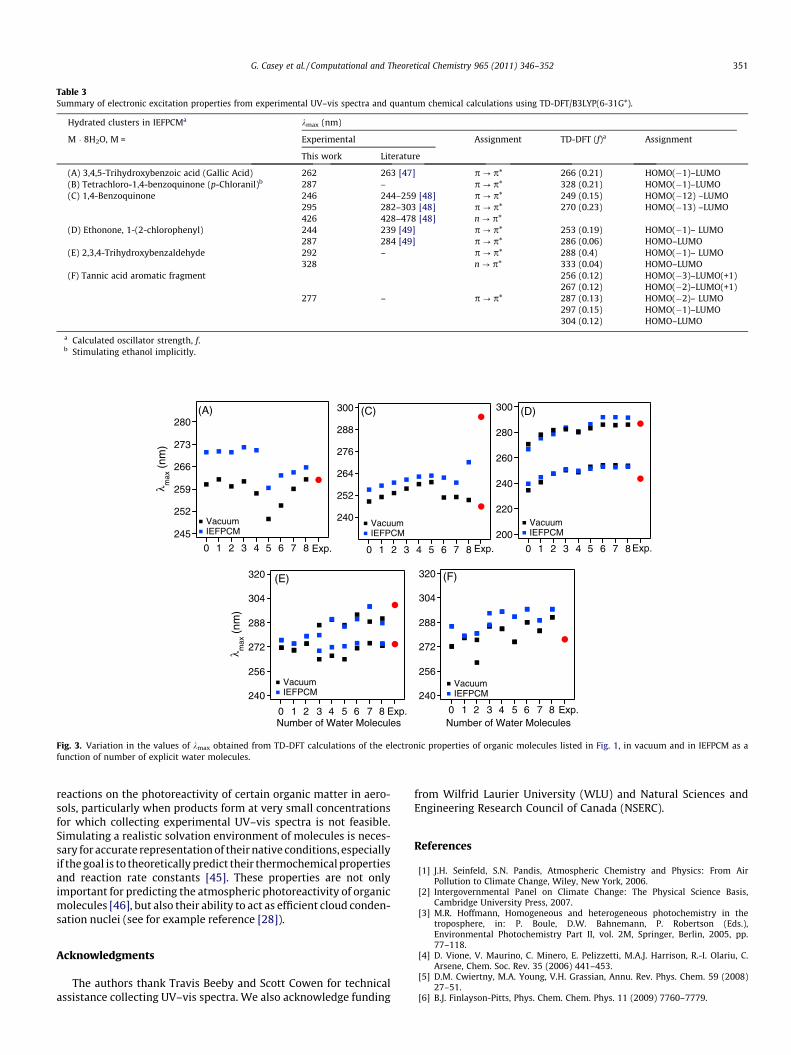
Table 3Summary of electronic excitation properties from experimental UV–vis spectra and quantum chemical calculations using TD-DFT/B3LYP(6-31G*).
Hydrated clusters in IEFPCMa kmax (nm)
M � 8H2O, M = Experimental Assignment TD-DFT (f)a Assignment
This work Literature
(A) 3,4,5-Trihydroxybenzoic acid (Gallic Acid) 262 263 [47] p ? p* 266 (0.21) HOMO(�1)–LUMO(B) Tetrachloro-1,4-benzoquinone (p-Chloranil)b 287 – p ? p* 328 (0.21) HOMO(�1)–LUMO(C) 1,4-Benzoquinone 246 244–259 [48] p ? p* 249 (0.15) HOMO(�12) –LUMO
295 282–303 [48] p ? p* 270 (0.23) HOMO(�13) –LUMO426 428–478 [48] n ? p*
(D) Ethonone, 1-(2-chlorophenyl) 244 239 [49] p ? p* 253 (0.19) HOMO(�1)– LUMO287 284 [49] p ? p* 286 (0.06) HOMO–LUMO
(E) 2,3,4-Trihydroxybenzaldehyde 292 – p ? p* 288 (0.4) HOMO(�1)– LUMO328 n ? p* 333 (0.04) HOMO–LUMO
(F) Tannic acid aromatic fragment 256 (0.12) HOMO(�3)–LUMO(+1)267 (0.12) HOMO(�2)–LUMO(+1)
277 – p ? p* 287 (0.13) HOMO(�2)– LUMO297 (0.15) HOMO(�1)–LUMO304 (0.12) HOMO–LUMO
a Calculated oscillator strength, f.b Stimulating ethanol implicitly.
280
273
266
259
252
245
λ max
(nm
)
9876543210
(A)
Exp.
VacuumIEFPCM
300
288
276
264
252
240
9876543210
(C)
Exp.
VacuumIEFPCM
300
280
260
240
220
2009876543210
(D)
Exp.
VacuumIEFPCM
320
304
288
272
256
240
λ max
(nm
)
9876543210Number of Water Molecules
(E)
Exp.
VacuumIEFPCM
320
304
288
272
256
2409876543210
Number of Water Molecules
(F)
Exp.
VacuumIEFPCM
Fig. 3. Variation in the values of kmax obtained from TD-DFT calculations of the electronic properties of organic molecules listed in Fig. 1, in vacuum and in IEFPCM as afunction of number of explicit water molecules.
G. Casey et al. / Computational and Theoretical Chemistry 965 (2011) 346–352 351
reactions on the photoreactivity of certain organic matter in aero-sols, particularly when products form at very small concentrationsfor which collecting experimental UV–vis spectra is not feasible.Simulating a realistic solvation environment of molecules is neces-sary for accurate representation of their native conditions, especiallyif the goal is to theoretically predict their thermochemical propertiesand reaction rate constants [45]. These properties are not onlyimportant for predicting the atmospheric photoreactivity of organicmolecules [46], but also their ability to act as efficient cloud conden-sation nuclei (see for example reference [28]).
Acknowledgments
The authors thank Travis Beeby and Scott Cowen for technicalassistance collecting UV–vis spectra. We also acknowledge funding
from Wilfrid Laurier University (WLU) and Natural Sciences andEngineering Research Council of Canada (NSERC).
References
[1] J.H. Seinfeld, S.N. Pandis, Atmospheric Chemistry and Physics: From AirPollution to Climate Change, Wiley, New York, 2006.
[2] Intergovernmental Panel on Climate Change: The Physical Science Basis,Cambridge University Press, 2007.
[3] M.R. Hoffmann, Homogeneous and heterogeneous photochemistry in thetroposphere, in: P. Boule, D.W. Bahnemann, P. Robertson (Eds.),Environmental Photochemistry Part II, vol. 2M, Springer, Berlin, 2005, pp.77–118.
[4] D. Vione, V. Maurino, C. Minero, E. Pelizzetti, M.A.J. Harrison, R.-I. Olariu, C.Arsene, Chem. Soc. Rev. 35 (2006) 441–453.
[5] D.M. Cwiertny, M.A. Young, V.H. Grassian, Annu. Rev. Phys. Chem. 59 (2008)27–51.
[6] B.J. Finlayson-Pitts, Phys. Chem. Chem. Phys. 11 (2009) 7760–7779.

352 G. Casey et al. / Computational and Theoretical Chemistry 965 (2011) 346–352
[7] D. Vione, Photochemical transformation processes of environmentalsignificance, in: B. Pignataro (Ed.), Tomorrow’s Chemistry Today, Wiley-VCH,Weinheim, 2008, pp. 429–453.
[8] M.J. Perri, Y.B. Lim, S.P. Seitzinger, B.J. Turpin, Atmos. Environ. 44 (2010) 2658–2664.
[9] L.N. Hawkins, L.M. Russell, D.S. Covert, P.K. Quinn, T.S. Bates, J. Geophys. Res-Atmos. 115 (2010) D13201.
[10] Y. Iinuma, O. Boge, A. Kahnt, H. Herrmann, Phys. Chem. Chem Phys. 11 (2009)7985–7997.
[11] R.A. Zaveri, C.M. Berkowitz, F.J. Brechtel, M.K. Gilles, J.M. Hubbe, J.T. Jayne, L.I.Kleinman, A. Laskin, S. Madronich, T.B. Onasch, M.S. Pekour, S.R. Springston,J.A. Thornton, A.V. Tivanski, D.R. Worsnop, J. Geophys. Res-Atmos. 115 (2010)D12304.
[12] A.W. Rollins, J.D. Smith, K.R. Wilson, R.C. Cohen, Environ. Sci. Technol. 44(2010) 5540–5545.
[13] H. Fairbrother, F.M. Geiger, V.H. Grassian, J.C. Hemminger (Eds.), Physicalchemistry of environmental interfaces, J. Phys. Chem. C, 113 (6) (2009) 2035–2646.
[14] R. Signorell, A.K. Bertram (Eds.), Physical chemistry of aerosols, Phys. Chem.Chem. Phys. 11, 2009, 7741–8104.
[15] S.G. Moussa, T.M. McIntire, M. Szori, M. Roeselová, D.J. Tobias, R.L. Grimm, J.C.Hemminger, B.J. Finlayson-Pitts, J. Phys. Chem. A 113 (2009) 2060–2069.
[16] H.C. Allen, N.N. Casillas-Ituarte, M.R. Sierra-Hernandez, X. Chen, C.Y. Tang,Phys. Chem. Chem. Phys. 11 (2009) 5538–5549.
[17] S. Cowen, H.A. Al-Abadleh, Phys. Chem. Chem. Phys. 11 (2009) 7838–7847.[18] D.K. Hore, D.S. Walker, G.L. Richmond, J. Am. Chem. Soc. 130 (2008) 1800–
1801.[19] F. Karagulian, C.W. Dilbeck, B.J. Finlayson-Pitts, J. Phys. Chem. A 113 (2009)
7205–7212.[20] J. Kleffmann, Chem. Phys. Chem. 8 (2007) 1137–1144.[21] M. Ndour, B. D’Anna, C. George, O. Ka, Y. Balkanski, J. Kleffmann, K. Stemmler,
M. Ammann, Geophys. Res. Lett. 35 (2008) L05812.[22] M. Brigante, D. Cazoir, B. D’Anna, C. George, D.J. Donaldson, J. Phys. Chem. A
112 (2008) 9503–9508.[23] D.I. Resser, A. Jammoul, D. Clifford, M. Brigante, B. D’Anna, C. George, D.J.
Donaldson, J. Phys. Chem. A 113 (2009) 2071–2077.[24] D.I. Resser, C. George, D.J. Donaldson, J. Phys, J. Phys. Chem. A 113 (2009)
8591–8595.[25] B. Sulzberger, E. Durisch-Kaiser, Aquat. Sci. 71 (2009) 104–126.
[26] J.R. Sabin, E. Brandas, M.E. Goodsite, M.S. Johnson (Eds.), Advances in QuantumChemistry: Applications of Theoretical Methods to Atmospheric Science, vol.55, Elsevier, Amsterdam, 2008.
[27] H.I. Kim, W.A. Goddard III, J.L. Beauchamp, J. Phys. Chem. A 110 (2006) 7777–7786.[28] Y. Xu, A.B. Nadykto, F. Yu, L. Jiang, W. Wang, J. Molec. Struc.: THEOCHEM 951
(2010) 28–33.[29] M. Staikova, D.J. Donaldson, Phys. Chem. Earth (C) 26 (2001) 473–478.[30] H. Tachikawa, S. Abe, Inorg. Chim. Acta 358 (2005) 288–294.[31] M.E. Casida, J. Molec. Struc.: THEOCHEM 914 (2009) 3–18.[32] B. Mennucci, J. Tomasi, R. Cammi, J.R. Cheeseman, M.J. Frisch, F.J. Devlin, S.
Gabriel, P.J. Stephens, J. Phys. Chem. A 106 (2002) 6102–6113.[33] C.J. Cramer, Essentials of Computational Chemistry, second ed., Wiley, West
Sussex, England, 2004.[34] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson et al., Gaussian, Inc.,Wallingford, CT, 2009.
[35] A.D. Becke, J. Chem. Phys. 98 (1993) 5648–5652.[36] C.T. Lee, W.T. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785–789.[37] R.S. Mulliken, J. Chem. Phys. 23 (1955) 1833–1840.[38] G.I. Csonka, J. Molec. Struc. 584 (2002) 1–4.[39] R.L. Prasad, A. Kushwaha, S.M. Kumar, R.A. Yadav, Spectrochim. Acta A 69
(2008) 304–311.[40] A. Dreuw, M. Head-Gordan, Chem. Rev. 105 (2005) 4009–4037.[41] F. Billes, I. Mohammed-Ziegler, P. Bombicz, Vib. Spectrosc. 43 (2007) 193–202.[42] F. Van Bolhuis, C.T. Kiers, Acta Cryst. B 34 (1978) 1015–1016.[43] S.W. Ng, Acta Cryst. E 64 (2008) 1565.[44] C. Peltier, P.P. Laine, G. Scalmani, M.J. Frisch, C. Adamo, I. Ciofini, J. Molec.
Struc.: THEOCHEM 914 (2009) 94–99.[45] J. Raul Alvarez-Idaboy, A. Galano, Atmospheric reactions of oxygenated volatile
organic compounds + OH radicals: role of hydrogen-bonded intermediates andtransition states. In: J.R. Sabin, E. Brandas, M.E. Goodsite, M.S. Johnson (Eds.),Advances in Quantum Chemistry: Applications of Theoretical Methods ofAtmospheric Science, vol. 55, Elsevier, Amsterdam, 2008, pp. 245–274.
[46] J.M. Andino, J.N. Smith, R.C. Flagan, W.A. Goddard III, J.H. Seinfeld, J. Phys.Chem. 100 (1996) 10967–10980.
[47] L. Ku, Y. Li, X. Lu, Spectrochim. Acta A 74 (2009) 829–834.[48] R.W. Bigelow, J. Chem. Phys. 68 (1978) 5086–5096.[49] W.J. Horton, D.E. Robertson, J. Org. Chem. 25 (1960) 1016–1020.[50] S. Banerjee, Water Res. 30 (1996) 1047–1061.