read the small print of the 1572
DESCRIPTION
The Essential GCP Document. Read the SMALL PRINT of the 1572. Good Clinical Practice (GCP). A standard for the design, conduct, performance, monitoring, auditing, recording, analyses, and reporting of clinical trials that provides assurance that - PowerPoint PPT PresentationTRANSCRIPT

Read the SMALL PRINT of the 1572
The Essential GCP Document

Good Clinical Practice (GCP)
A standard for the design, conduct, performance, monitoring, auditing, recording, analyses, and reporting of clinical trials that provides assurance that
the Data and Reported Results are Credible, and Accurate,
and that the Rights, Integrity, and Confidentiality of Trial
Subjects are Protected.
= Quality Data
= Ethics
Quality Data + Ethics = GCP

Obligations of Investigators Video
A “GCP Day” in the life of a clinical researcher

GCPs: Discussion
What “ethics” process was shown in the video?– the Rights, Integrity, and Confidentiality of Trial
Subjects are Protected.
What “data quality” concepts were shown?– the Data and Reported Results are Credible, and
Accurate

Form FDA 1572: It’s the Law!
21 Code of Federal Regulations 312.53 (c) (1)– Before permitting an investigator to begin
participation in an investigation, the sponsor shall obtain the following:1) A signed investigator statement (Form FDA-1572)

Form FDA 1572: Location
DHHS’s Program Support Center (PSC): http://forms.psc.gov/ – Click on “FDA” for current version of 1572 (and
instructions)
Note: In last year there have been 3 versions– Sep 30, 2002– Nov 30, 2002– Jan 31, 2006
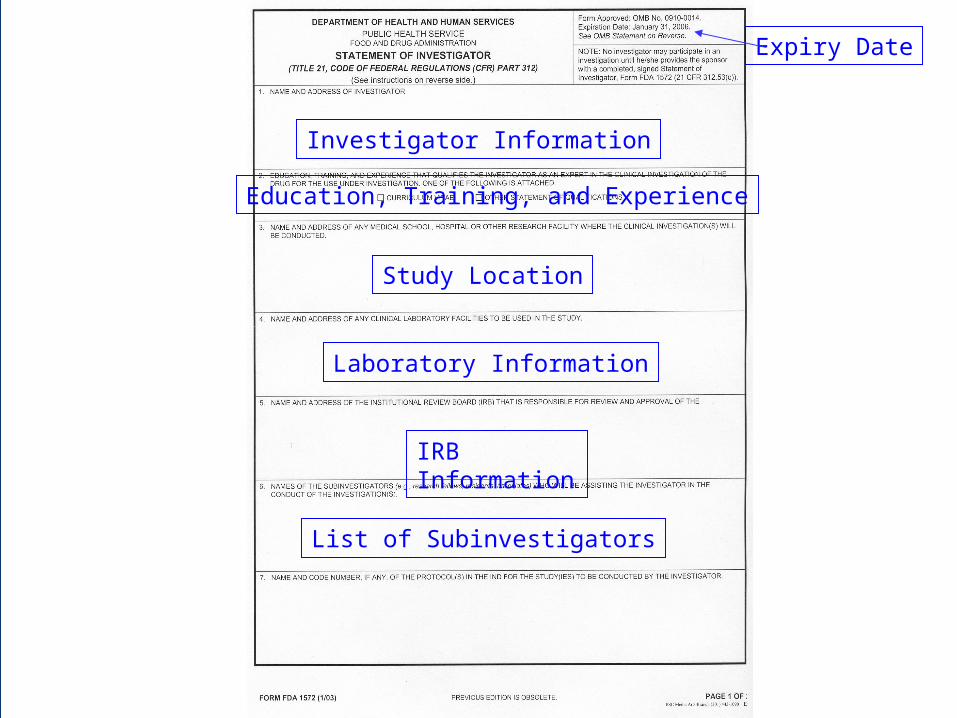
Investigator Information
Education, Training, and Experience
Study Location
Laboratory Information
IRB Information
List of Subinvestigators
Expiry Date

Bioresearch Monitoring Information System File (BMIS)
http://www.fda.gov/oc/gcp/clinenforce.html Abstracts IND study information from 1572 and
other documents Contains information on:
– Investigators – Contract Research Organizations (CROs)– Institutional Review Boards (IRBs)
Lists separate information entry each time a new IND is submitted

Expiry Date
Investigator Information
Education, Training, and Experience
Study Location
Laboratory Information
IRB Information
List of Subinvestigators
Protocol/IND Number (Sponsor Information)

IND Requirement
Investigational New Drug [21CFR312.3]– New drug, or – New biological drug – New biological product used in vitro for diagnostic
purposes
Phase 1, 2, or 3 studies– Administered or dispensed to, or used in, one or
more human subjects

Clinical Phase
Filing Instructions
Investigator Signatureand Date
Commitments• S• M• A• L• L
• P• R• I• N•T

1572 Commitments: Box 9
S _______ M ______ ______ A _____ to _______ L ____ __________ ________ L et FDA Inspect
__P__ _______ _______ R etain Records I _____ _______ N ____ _ _ _ T ____ ______

1572 Commitments: Box 9
S _______ M ______ ______ A _____ to _______ L ____ __________ ________ L et FDA Inspect
__P__ _______ _______ R etain Records I _____ _______ N ____ _ _ _ T ____ ______
upervise
aintain Recordsdhere Protocolearn Investigator’s Brochure
Re ort Adverse Events
nform Subjects
otify I R Brain Staff

1572 Commitments: Box 9
S _______ M ______ ______ A _____ to _______ L ____ __________ ________ L et FDA Inspect
__P__ _______ _______ R etain Records I _____ _______ N ____ _ _ _ T ____ ______
upervise
aintain Recordsdhere Protocolearn Investigator’s Brochure
Re ort Adverse Events
nform Subjects
otify I R Brain Staff
Data Quality
Ethics
PeopleNeeds

1572: Discussion Points
For what studies is the 1572 used?
Who can be the investigator (i.e. signatory)?
Who can be a sub investigator (Box 6)?

1572 Problem Cases

FDA Warning Letters (WL)
A post FDA inspection document An informal advisory to a firm communicating
FDA's position on a matter but does not commit FDA to taking enforcement action
http://www.fda.gov/oc/gcp/clinenforce.html

WL (5 Jun 02): Hassman, MD
The investigator agreement you signed requires you to personally conduct or supervise the clinical investigation (see FDA Form 1572). – FDA’s investigation revealed that you failed to
adequately supervise those aspects of clinical investigations which you did not personally conduct. As described in more detail …, this lack of supervision resulted in submission of false information to the sponsor and failure to maintain adequate and accurate case histories.

WL(17 Apr 02): Yu, MD, PhD
“You failed to obtain a signed investigator statement, Form FDA 1572– from all investigators prior to permitting them to
begin participation in the investigation.”
“You failed to provide a complete list of the sub-investigators – who assisted you in the conduct of the
investigation.”

FDA Problem Investigators Lists
Restricted List– Names of all clinical investigators who have agreed
to certain restrictions with respect to their conduct of clinical investigations
http://www.fda.gov/ora/compliance_ref/bimo/restlist.htm

Restrictions List: Cases
David P. Faxon, MD, Los Angeles, CA
CDER R 19-Jun-2002
By consent agreement. For 3 years: Shall not be principal investigator for more than two (2) FDA regulated clinical investigations at any one time (with additional provisions); shall not be principal clinical investigator for study that enrolls more than 25 subjects at the site where he is the principal investigator; and additional provisions.
Lois Anne Katz, MD, New York, NY
CDER R 23-Aug-2002
For 3 years: Shall not be principal investigator for more than one additional study clinical investigation at any one time; attend at least educational programs on clinical research studies; arrange training and education of staff in conduct of clinical trials; provide annual certification of compliance

“The obligations of the 1572 must be applied to all clinical
research."Anonymous
15 2