recovery of infectious murine norovirus using pol ii … of infectious murine norovirus using pol...
TRANSCRIPT

Recovery of infectious murine norovirus usingpol II-driven expression of full-length cDNAVernon K. Ward†‡§, Christopher J. McCormick‡, Ian N. Clarke‡, Omar Salim‡, Christiane E. Wobus¶, Larissa B. Thackray¶,Herbert W. Virgin IV¶�, and Paul R. Lambden‡
†Department of Microbiology and Immunology, University of Otago, P.O. Box 56, Dunedin 9054, New Zealand; ‡Molecular Microbiology Group, Universityof Southampton Medical School, Southampton General Hospital, Southampton SO16 6YD, United Kingdom; and Departments of ¶Pathologyand Immunology and �Molecular Microbiology, Washington University School of Medicine, St. Louis, MO 63110
Edited by Mary K. Estes, Baylor College of Medicine, Houston, TX, and accepted by the Editorial Board May 16, 2007 (received for review January 12, 2007)
Noroviruses are the major cause of nonbacterial gastroenteritis inhumans. These viruses have remained refractory to detailed mo-lecular studies because of the lack of a reverse genetics systemcoupled to a permissive cell line for targeted genetic manipulation.There is no permissive cell line in which to grow infectious humannoroviruses nor an authentic animal model that supports theirreplication. In contrast, murine norovirus (MNV) offers a tractablesystem for the study of noroviruses with the recent discovery ofpermissive cells and a mouse model. The lack of a reverse geneticsystem for MNV has been a significant block to understanding thebiology of noroviruses. We report recovery of infectious MNV afterbaculovirus delivery of viral cDNA to human hepatoma cells underthe control of an inducible DNA polymerase (pol) II promoter.Recovered virus replicated in murine macrophage (RAW264.7) cells,and the recovery of MNV from DNA was confirmed throughrecovery of virus containing a marker mutation. This pol II pro-moter driven expression of viral cDNA also generated infectiousvirus after transfection of HEK293T cells, thus providing bothtransduction and transfection systems for norovirus reverse ge-netics. We used norovirus reverse genetics to demonstrate bymutagenesis of the protease–polymerase (pro–pol) cleavage sitethat processing of pro–pol is essential for the recovery of infectiousMNV. This represents the first infectious reverse genetics systemfor a norovirus, and should provide approaches to address funda-mental questions in norovirus molecular biology and replication.
calicivirus � infectious clone � reverse genetics
Noroviruses (Caliciviridae) are the most common cause ofnonbacterial acute gastroenteritis with an estimated 23
million cases and 50,000 hospitalizations per year in the USAalone (1). They are a global problem causing significant mor-bidity and are of particular concern in semiclosed environments,such as the military, hospitals, schools, hotels, and cruise ships(2–5). Significant efforts are being made to prevent, or developtreatments for, this major food-borne disease. To date, noantiviral drugs or vaccines are available.
Human noroviruses are noncultivable, and despite infectiousclone systems being developed for other Caliciviridae genera(6–9), a reverse genetics system for noroviruses has remainedunsolved, apart from Norwalk virus (10), for which the lack ofpermissive cell lines or a small animal model limits options fordirect study (11). An alternative is the recently described murinenorovirus (MNV) (12). This virus has been proposed as a modelnorovirus system, because it can be replicated in mice, grown incultured primary dendritic cells, macrophages, and continuousmacrophage cells, and a range of susceptible mouse strains areavailable for pathogenicity studies (13).
MNV was first identified in RAG2/STAT1�/� mice (12), withhistopathological investigation of MNV infected STAT1�/� miceshowing virus replication in cells resembling macrophages or den-dritic cells (14). Subsequently, MNV has been identified around theworld, with up to 22% of serum from research colonies in theUnited States and Canada proving positive for this virus, often
presenting as a subclinical infection in mice with a functioninginnate immune response (15) (data not shown). The MNV genomeconsists of 7.4 kb of positive-sense single-stranded RNA with a 3�polyA tail. The genome encodes three ORFs comprising a 187.5-kDa replicase polyprotein, the 60-kDa capsid ORF, and a low-abundance 22-kDa basic protein. The autocatalytic replicasepolyprotein is expressed from genomic RNA and includes a proteinof unknown function called N-term and the better defined NTPase,3A-like, VPg, 3C-like, and RNA-dependent RNA polymeraseproteins (16). The capsid protein, VP1, is expressed from a subgenomic RNA fragment. Overall, the genome and expressed pro-teins of MNV correspond well to the genome and expressedproteins of human noroviruses (17–21).
This article reports the development of the first reversegenetics system for MNV that generates infectious virus from agenomic cDNA clone. Two approaches generated infectiousMNV; transduction-based delivery of cDNA under control of apol II promoter into human hepatocellular carcinoma cell lineG2 (HepG2) cells, or transfection of the pol II promoterconstruct into HEK293T cells. Recovered MNV replicated insusceptible RAW 264.7 cells. We show that the MNV genomecan be manipulated and that a genetically modified infectiousvirus can be recovered. This study describes an importantadvance for the field of noroviruses, because it will now bepossible to perform a full genetic analysis of norovirus genefunction. We demonstrate this principle through mutagenesis ofthe pro–pol cleavage site to show that pro–pol cleavage isessential for recovery of infectious virus. The availability of bothtransduction and transfection reverse genetic systems will allowreverse genetic experiments in a range of cell types and mayprovide methods relevant to other noroviruses.
ResultsRecombinant MNV Expression and Recovery. A two-componentbaculovirus expression system used to produce recombinantMNV is shown schematically in supporting information (SI) Fig.4. This system allows the use of a regulated pol II promoter by
Author contributions: V.K.W. and C.J.M. contributed equally to this work; V.K.W., C.J.M.,I.N.C., O.S., L.B.T., and P.R.L. designed research; V.K.W., C.J.M., O.S., C.E.W., L.B.T., and P.R.L.performed research; C.J.M. and H.W.V. contributed new reagents/analytic tools; V.K.W.,C.J.M., I.N.C., O.S., C.E.W., L.B.T., H.W.V., and P.R.L. analyzed data; and V.K.W., C.J.M., I.N.C.,O.S., C.E.W., L.B.T., H.W.V., and P.R.L. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. M.K.E. is a guest editor invited by the EditorialBoard.
Abbreviations: CPE, cytopathic effect; H�V, hepatitis delta virus; HepG2, human hepato-cellular carcinoma cell line G2; MNV, murine norovirus; pol, DNA polymerase; pro, viralprotease; pol, viral polymerase; RACE, rapid amplification of cDNA ends; RAW264.7,Murine macrophage cell line 264.7; Sf9, Spodoptera frugiperda cell line Sf9; VPg, MNV viralprotein genomic.
§Towhomcorrespondenceshouldbeaddressed.E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0700336104/DC1.
© 2007 by The National Academy of Sciences of the USA
11050–11055 � PNAS � June 26, 2007 � vol. 104 � no. 26 www.pnas.org�cgi�doi�10.1073�pnas.0700336104

separate delivery of the encoded MNV genome and a transac-tivator. The baculovirus clone BACTETtTA expressing the tet-racycline repressor/VP16 transactivator from a CAG mamma-lian promoter has been described (22). Recombinant MNVbaculovirus BACTETMNV was propagated in Sf9 cells andconcentrated to a titer of 109 pfu/ml. Transduction of 106 HepG2cells with 108 pfu of BACTETtTA or BACTETMNV alone causedno cytopathic effect (CPE) in the HepG2 cells (Fig. 1 A and B).However, simultaneous cotransduction with both baculovirusesled to rapid CPE within 24 h (Fig. 1C), indicative of theexpression of MNV proteins within transduced cells.
Passage of the double-transduced HepG2 cell lysates inRAW264.7 cells resulted in the development of typical MNVCPE (Fig. 1 E–H), however, no CPE was observed when eitherthe transactivator or MNV baculovirus transduced HepG2 cellmaterial were passaged individually onto RAW264.7 cells (datanot shown). CPE in HepG2 cells was a reliable indicator of MNVproduction. We recovered infectious MNV with two separate
batches of each recombinant baculovirus and six separate co-transduction reactions. The recovery of infectious MNV wasindependently observed in two separate laboratories(Southampton and St. Louis).
The recovered virus could be plaqued on RAW264.7 cells(Fig. 1 D) and HepG2 yields of �2 � 102 pfu/ml were obtained.The recovered MNV could be passaged in RAW264.7 cells,reaching titers of 5 � 107 per ml. Passage of HepG2 lysates uponHepG2 cells did not lead to CPE, confirming the requirement ofthe permissive RAW264.7 cell line for virus propagation. Directtransduction of RAW264.7 cells did not yield a recoverablevirus; however, RAW264.7 cells are not transduced efficiently bybaculovirus (23).
Transfection of HepG2, BHK-21, COS-7, or HEK293T cellswith the pFBTETtTA and pFBTETMNV plasmids produced re-coverable virus. Virus was not recovered from RAW cellstransfected in a similar manner. Of note, transfection of thepFBTETMNV plasmid or the MNV genome in plasmid pSP73
Fig. 1. Recovery of recombinant MNV in HepG2 and RAW 264.7 cells. (A–C) HepG2 cells were transduced with 100 pfu of BACtTA baculovirus (A), BACTET-MNVbaculovirus (B), or both baculoviruses simultaneously (C) and observed 24 h after transduction. (D) Viral plaques of passaged recMNV (105-fold dilution) inRAW264.7 cells are shown at 24 h after infection. (E and F) Mock-infected RAW264.7 cells at 0 (E) and 24 h (F). (G and H) recMNV-infected RAW264.7 cells (MOI �0.25) at 0 (G) and 24 h (H) after infection. (I–N) Expression of nonstructural and structural proteins in 20 h postinfection RAW 264.7 cells was tested byimmunofluorescence of mock or infected cells with capsid (I and J), VPg (K and L), and N-term (M and N) monoclonal antibodies. (O and P) No primary antibodyRAW264.7 or HEK293T cell controls. (Q–T) Expression of nonstructural and structural proteins in mock or pMNV* transfected HEK293T cells was tested byimmunofluorescence with VPg (Q and R) and N-term (S and T) monoclonal antibodies. (U and V) No primary antibody pMNV* and pMNV*/pro–pol transfectioncontrols. (W and X) Expression of VPg (W) and N-term (X) in HEK293T cells transfected with pro–pol cleavage mutation in pMNV*. Cell nuclei were stained withDAPI (I–X) and primary antibodies detected by fluorescence of goat anti-mouse FITC.
Ward et al. PNAS � June 26, 2007 � vol. 104 � no. 26 � 11051
MIC
ROBI
OLO
GY
under the control of the minimal CMV promoter producedrecoverable virus in the absence of the transactivator inHEK293T cells. There was no observable CPE in HEK293Tcells; however, immunofluorescence with monoclonal antibodiesto the VPg and N-term proteins indicated a high number of cellsexpressing the N-term protein (Fig. 1 Q–T). The recovery ofvirus was confirmed by direct plaque assay of the recoveredHEK293T cell lysates on RAW264.7 cells with a yield of 5 � 103
pfu/ml.
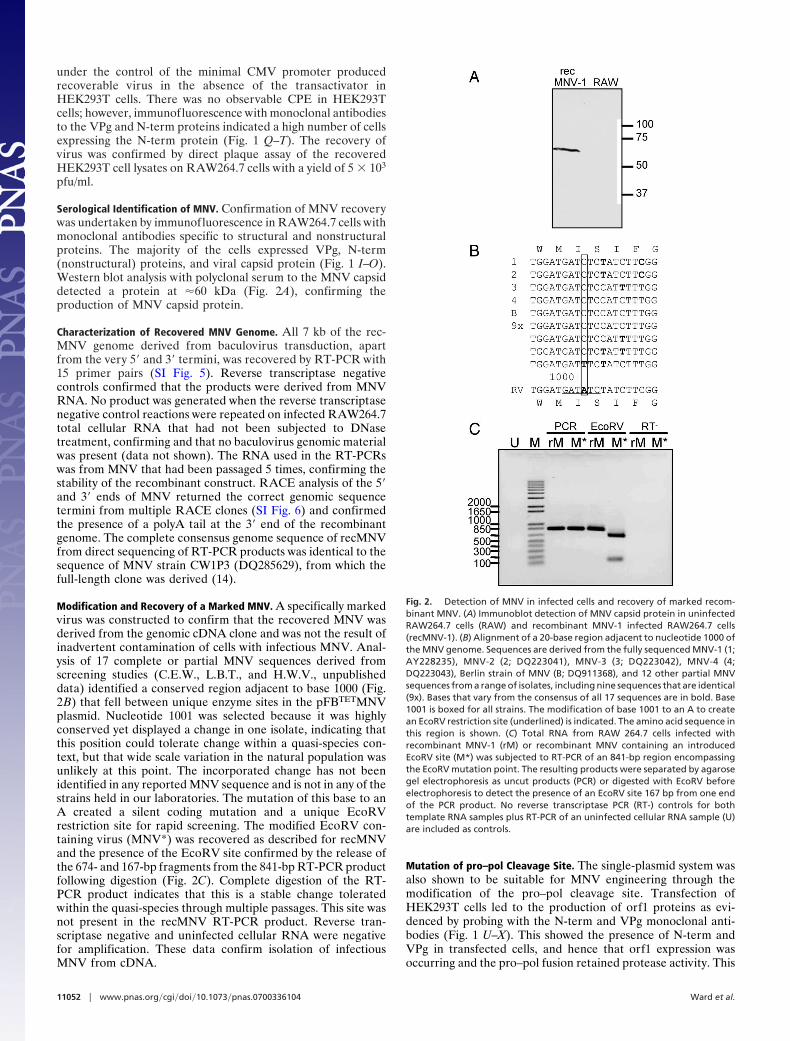
Serological Identification of MNV. Confirmation of MNV recoverywas undertaken by immunofluorescence in RAW264.7 cells withmonoclonal antibodies specific to structural and nonstructuralproteins. The majority of the cells expressed VPg, N-term(nonstructural) proteins, and viral capsid protein (Fig. 1 I–O).Western blot analysis with polyclonal serum to the MNV capsiddetected a protein at �60 kDa (Fig. 2A), confirming theproduction of MNV capsid protein.
Characterization of Recovered MNV Genome. All 7 kb of the rec-MNV genome derived from baculovirus transduction, apartfrom the very 5� and 3� termini, was recovered by RT-PCR with15 primer pairs (SI Fig. 5). Reverse transcriptase negativecontrols confirmed that the products were derived from MNVRNA. No product was generated when the reverse transcriptasenegative control reactions were repeated on infected RAW264.7total cellular RNA that had not been subjected to DNasetreatment, confirming and that no baculovirus genomic materialwas present (data not shown). The RNA used in the RT-PCRswas from MNV that had been passaged 5 times, confirming thestability of the recombinant construct. RACE analysis of the 5�and 3� ends of MNV returned the correct genomic sequencetermini from multiple RACE clones (SI Fig. 6) and confirmedthe presence of a polyA tail at the 3� end of the recombinantgenome. The complete consensus genome sequence of recMNVfrom direct sequencing of RT-PCR products was identical to thesequence of MNV strain CW1P3 (DQ285629), from which thefull-length clone was derived (14).
Modification and Recovery of a Marked MNV. A specifically markedvirus was constructed to confirm that the recovered MNV wasderived from the genomic cDNA clone and was not the result ofinadvertent contamination of cells with infectious MNV. Anal-ysis of 17 complete or partial MNV sequences derived fromscreening studies (C.E.W., L.B.T., and H.W.V., unpublisheddata) identified a conserved region adjacent to base 1000 (Fig.2B) that fell between unique enzyme sites in the pFBTETMNVplasmid. Nucleotide 1001 was selected because it was highlyconserved yet displayed a change in one isolate, indicating thatthis position could tolerate change within a quasi-species con-text, but that wide scale variation in the natural population wasunlikely at this point. The incorporated change has not beenidentified in any reported MNV sequence and is not in any of thestrains held in our laboratories. The mutation of this base to anA created a silent coding mutation and a unique EcoRVrestriction site for rapid screening. The modified EcoRV con-taining virus (MNV*) was recovered as described for recMNVand the presence of the EcoRV site confirmed by the release ofthe 674- and 167-bp fragments from the 841-bp RT-PCR productfollowing digestion (Fig. 2C). Complete digestion of the RT-PCR product indicates that this is a stable change toleratedwithin the quasi-species through multiple passages. This site wasnot present in the recMNV RT-PCR product. Reverse tran-scriptase negative and uninfected cellular RNA were negativefor amplification. These data confirm isolation of infectiousMNV from cDNA.
Mutation of pro–pol Cleavage Site. The single-plasmid system wasalso shown to be suitable for MNV engineering through themodification of the pro–pol cleavage site. Transfection ofHEK293T cells led to the production of orf1 proteins as evi-denced by probing with the N-term and VPg monoclonal anti-bodies (Fig. 1 U–X). This showed the presence of N-term andVPg in transfected cells, and hence that orf1 expression wasoccurring and the pro–pol fusion retained protease activity. This
Fig. 2. Detection of MNV in infected cells and recovery of marked recom-binant MNV. (A) Immunoblot detection of MNV capsid protein in uninfectedRAW264.7 cells (RAW) and recombinant MNV-1 infected RAW264.7 cells(recMNV-1). (B) Alignment of a 20-base region adjacent to nucleotide 1000 ofthe MNV genome. Sequences are derived from the fully sequenced MNV-1 (1;AY228235), MNV-2 (2; DQ223041), MNV-3 (3; DQ223042), MNV-4 (4;DQ223043), Berlin strain of MNV (B; DQ911368), and 12 other partial MNVsequences from a range of isolates, including nine sequences that are identical(9x). Bases that vary from the consensus of all 17 sequences are in bold. Base1001 is boxed for all strains. The modification of base 1001 to an A to createan EcoRV restriction site (underlined) is indicated. The amino acid sequence inthis region is shown. (C) Total RNA from RAW 264.7 cells infected withrecombinant MNV-1 (rM) or recombinant MNV containing an introducedEcoRV site (M*) was subjected to RT-PCR of an 841-bp region encompassingthe EcoRV mutation point. The resulting products were separated by agarosegel electrophoresis as uncut products (PCR) or digested with EcoRV beforeelectrophoresis to detect the presence of an EcoRV site 167 bp from one endof the PCR product. No reverse transcriptase PCR (RT-) controls for bothtemplate RNA samples plus RT-PCR of an uninfected cellular RNA sample (U)are included as controls.
11052 � www.pnas.org�cgi�doi�10.1073�pnas.0700336104 Ward et al.

was confirmed by Western blot analysis of transfected HEK293Tcells with monoclonal antibody to N-term (Fig. 3). Both MNV*and the pro–pol cleavage mutant in pSP73 showed production,and hence cleavage, of N-term from the orf1 polyprotein. It wasalso evident that the pSP vector was more efficient than pFBvectors (Fig. 3). However, no infectious virus could be recoveredfrom cells transfected with the pro–pol cleavage mutant, indi-cating that in contrast to feline calicivirus (24), pro–pol cleavageis essential for the recovery of infectious virus.
DiscussionDespite the existence of a feline calicivirus reverse geneticssystem for over 10 years and systems available for other generaof caliciviruses (6–9), the noroviruses have remained recalcitrantto reverse genetics despite intensive efforts to develop suchsystems by many laboratories over a number of years. The onlyprevious success has been the generation of Norwalk virusparticles in a T7-based system (10). Despite this advance, NVdoes not have a permissive cell line or small animal model inwhich to study the biology and pathology of this genus inconjunction with application of a reverse genetics system. Thisreport provides the essential missing link in the range of toolsnecessary to dissect the molecular and cellular biology of MNVand thus shed light on noroviruses, major contributors to humandisease worldwide. This is complemented by the availability ofmouse strains with variable susceptibility to MNV, known strainsof MNV with a range of pathogenic phenotypes, permissive celllines and perhaps most importantly, the wide range of immu-nological tools available using mice.
Other calicivirus reverse genetic systems have focused on theuse of cytoplasmic T7 promoters in various forms and thecytoplasmic capping function of poxviruses to recover virus (8).The use of poxviruses can cause CPE in host cells and can affectviral recovery. We reasoned that efficient delivery of cDNAcoupled with the use of a pol II promoter would allow nuclearprocessing and export of capped viral transcripts to the cellcytoplasm. This viral transcript would both facilitate expressionof viral nonstructural proteins and act as a template for gener-ation of the negative sense genome, leading to the subsequentproduction of VPg-linked viral RNA.
The HepG2 cell line was selected because, unlike RAW264.7cells, it is highly susceptible to baculovirus transduction (25).Further, Asanaka et al. (10) showed that NV particles could berecovered in a nonpermissive cell line. Although we were unableto recover MNV from transduced RAW264.7 cells, recovery of
MNV from HepG2 cell lysates in RAW264.7 cells was entirelyrepeatable. Although coculture of HepG2 and RAW264.7 cellswas not undertaken, it is reasonable to assume that such a systemcould further streamline recMNV production. The growth ofrecMNV in RAW264.7 cells reached comparable titers to thosereported for wild type virus.
Although transduction of HepG2 cells was successful, a single-plasmid system would be more facile under many circumstances.In addition, it is likely that other noroviruses may requirealternate delivery systems or cells. To address this, we investi-gated the use of the HEK293T cell line, which is highly suscep-tible to transfection. This was also successful and reinforced theuse of a pol II promoter for the production of this norovirusgenome. It is interesting to note that unlike baculovirus trans-duction of HepG2 cells, no transactivator is required inHEK293T cells, and no CPE is observed in these cells.
The use of the two-component baculovirus system was anessential aspect of our experimental design, providing internalcontrols that allowed the individual nonexpressing constructs tobe tested separately. Using mammalian promoters in a baculo-virus system minimizes potential toxicity effects during baculo-virus production, and the separation of transactivator from theexpression construct essentially eliminated any background ex-pression from these promoters in an insect cell. The tTAexpressing baculovirus is a constant in the system, meaning thatall of the viral genomic information is on a single pFastbacplasmid, simplifying future manipulation of the genome.
Comparison of the two systems suggests that the criticalcommon aspect allowing recovery of MNV was the use of a polII promoter for generation of a capped transcript. If thistechnology can be transferred to human noroviruses, then theoption of two delivery systems may increase the range of cells towhich constructs can be delivered efficiently.
Sequence analysis of the recMNV showed that the correct 5�terminus was generated and that the presence of the AgeI siteused to place precisely the 5� terminus of the MNV genome didnot preclude correct transcription initiation. The recovery of thecorrect 5� end is important for authenticity of the recovered virusas future manipulations of the genome will not be biased by 5�terminal variations causing unknown effects. The virus is clearlyable to resolve any capping or processing problems associatedwith the primary transcript. The recovery of a stable markedvirus confirms that MNV is regenerated through reverse genet-ics and, most importantly, confirms that the MNV genome canbe manipulated and that viable virus can be recovered.
Proof of principle that reverse genetics can supply new infor-mation on noroviruses was shown through elimination of thepro–pol consensus cleavage sequence. This mutation in felinecalicivirus (Vesivirus genus) failed to eliminate viral replicationand recovery (24), indicating that processing of pro–pol is notessential for viral recovery in this calicivirus genus. Our datashow that the pro–pol fusion retains protease activity but that novirus can be recovered from this mutant viral genome. This issimilar to poliovirus where the pro–pol fusion (3CD) retainsprotease processing of structural and nonstructural proteins, butno polymerase activity is detectable in the absence of processingto release 3D (polymerase) (26). These data highlight thepotential for differences between vesiviruses and norovirusesand the need for detailed studies of norovirus replication.
In summary, we have developed a robust system that exploitspol II expression of a norovirus genome. The pol II promoter isregulatable through the baculovirus delivery system. The systemexploits the baculovirus transduction of mammalian cells cou-pled with facile production in a two-component expressionsystem where regulated control is desired. In addition a rapid andsimple single-plasmid transfection through HEK293T cells al-lows for the efficient and facile recovery of recombinant MNV.Virus recovery is highly repeatable, the systems can be trans-
Fig. 3. Expression and processing of N-term in transfected HEK293T cells.HEK293T cells were transfected with pMNV* (A) or pro–pol cleavage muta-tions (PPKO) in pFBTETMNV* or pMNV* (B) and subjected to Western blotanalysis with N-term monoclonal antibody. Mock transfections were includedas controls.
Ward et al. PNAS � June 26, 2007 � vol. 104 � no. 26 � 11053
MIC
ROBI
OLO
GY

ferred successfully to other laboratories, and an authentic ge-nome is recovered. This system should allow substantial insightsinto the pathology and biology of the Norovirus genus of theCaliciviridae.
Materials and MethodsCells and Media. Spodoptera frugiperda (Sf9) cells (Invitrogen, Carls-bad, CA) were grown as adherent or suspension cultures in TC100medium supplemented with 10% FBS (Invitrogen). Human hep-atocellular liver carcinoma cells (HepG2) were grown on collagen(Sigma–Aldrich, St. Louis, MO) coated culture ware in DMEMsupplemented with 10% FBS, nonessential amino acids, 25 mMHepes, and glutamax-1 (Invitrogen). RAW 264.7 and HEK293Tcells were grown in DMEM supplemented with 10% FBS, 25 mMHepes, and glutamax-1 (Invitrogen). For immunofluorescence,cells were grown on polylysine-coated cover slips as per manufac-turers instructions (Sigma–Aldrich). BHK-21 and COS-7 cells weregrown in DMEM supplemented with 10% FBS.
Antisera. Monoclonal antibodies to MNV VPg and N-termproteins were generated as described by Oliver et al. (27).Confirmation of monoclonal antibody specificity is provided inSI Fig. 7. Monoclonal antibody A6.2 and rabbit polyclonalantibody to MNV capsid are described in ref. 14.
TET-Activated Expression System. The expression system used was atwo baculovirus system involving transduction of mammalian cellswith one baculovirus expressing a TET-R/VP16 transactivator froma pol II CAG promoter to activate a TET-O/minCMV promoter ona second baculovirus carrying a full length clone of the MNVgenome as described in ref. 22. The parental vector used in thisstudy was designated pFBTET and was constructed from the Fast-Bac vector pFBrep5.1neo(�3�U) (28) containing the TET-O/minCMV promoter, which had been modified to include an AgeIrestriction site immediately before the identified transcription ini-tiation point of the minimal CMV promoter (25).
MNV Genome Cloning. A full length clone of MNV-1 strain CW1P3in the plasmid pSport-1 was used as the template (16). The 5� endof the MNV genome was amplified by PCR, using Accuzymepolymerase (Bioline, Randolph, MA) with primer AgeI-21 (5�AACTTGGGATCCACCGGTGTGAAATGAGGATGGC-AACGC) containing an AgeI site (underlined) followed imme-diately by the first 21 bases of the MNV genome (italics) andprimer MNV1085–1056 (5�CATCCCGATCCCGCCCAA-CAGG), which binds 3� of a unique ApaI site at nucleotideposition 1034 in the MNV genome cDNA. The resulting PCRproduct was cloned into pCRBlunt (Invitrogen), sequenced onan ABI 377 sequencer, and isolated as an AgeI-ApaI fragmentby gel extraction (Sigma–Aldrich; GenElute kit).
The 3� end of the genome was modified to contain a polyA tail,hepatitis delta virus (H�V) ribozyme and a NotI restriction site.Primer BspF (5� CCTCCTAAGCTTCCGGA-CCTACAT-GCGTCAGA), spanning a unique BspEI site in the MNVgenome at nucleotide position 6351, and primer H�VpolyA (5�AGGCTGGGACCATGCCGGCCTTTTTTTTTTTTTTTT-TTTTTTTTTTTTTTTAAAATGCATCTAACT), matching the3� end of the MNV genome (italics), polyA tail, and part of theH�V ribozyme (underlined) were used to amplify the 3� end ofthe MNV genome. The plasmid pFBrep5.1neoT7/NotI (25) wasused as a template with primers H�V (5� AAAGGCCGGCAT-GGTCCCAGC) encoding the 5� end of the H�V ribozyme(underlined) and CAGDOWN (5� CATATGTCCTTC-CGAGTG) to amplify the H�V ribozyme–NotI region from thisplasmid. The two PCR products were gel-purified and used in afusion PCR with primers CAGDOWN and BspF then clonedinto pCRBlunt and sequenced, to create a 1,158-bp BspI-NotIfragment encoding bases 6351–7382 of the MNV genome, a
30-bp polyA-tail, the H�V ribozyme immediately after thepolyA-tail, and a unique NotI site.
The ApaI-BspEI region of the MNV genome (bases 1034–6350) was isolated from the pSport-1 full-length clone andcloned with the 3� BspEI-NotI genome PCR fragment intoApaI-NotI digested pBluescriptIISK� (Invitrogen). The result-ing clone was isolated as an Apa-NotI fragment and along withthe 5� AgeI-ApaI PCR fragment, cloned into AgeI-NotI digestedpFBTET to create the full length MNV clone pFBTET-MNV.
Recombinant Baculovirus Construction. The pFBTET-MNV plasmidwas used to create the Autographa californica nucleopolyhedro-virus bacmid (AcMNPV), pBacTET-MNV, in DH10Bac Esche-richia coli as per manufacturers instructions (Invitrogen). Thebacmid was purified by alkaline lysis extraction, transfected intoSf9 cells with lipofectin (Invitrogen) and grown at 27°C in TC100medium supplemented with 10% FBS (Invitrogen). The result-ing recombinant virus (BACTET-MNV) was propagated in Sf9suspension culture, then concentrated by centrifugation in amicrofuge for 20 min and resuspended in PBS. The viral titer wasdetermined by plaque assay.
Infectious MNV Recovery. HepG2 cells were seeded on collagen-coated 35-mm six-well trays and grown overnight. The cells weretransduced with 100 pfu of BACTET-MNV and 100 pfu ofBACTET-tTA per cell for 4 h then washed and incubated in freshmedium for 24 h. The HepG2 cells were freeze-thawed, and 1 mlof crude lysate used as inoculum for amplification of recombi-nant MNV (recMNV or MNV*) on RAW264.7 cells in 35-mmsix-well trays. Passage and plaque assay of the recovered viruswas undertaken by freeze–thaw and infection with crude lysate.HepG2, BHK-21, Cos-7, and HEK293T cells were seeded onto35-mm six-well trays and grown overnight. FuGene HD (Roche,Basil, Switzerland) was used to transfect 1 �g of plasmid DNAper well and virus recovery on RAW264.7 cells undertaken asabove.
Detection of Recombinant MNV by Immunofluorescence. RAW264.7,HepG2, or HEK293T cells were seeded onto polylysine-coatedcover slips and infected with recombinant MNV then incubatedfor 20 h. The cells were methanol-fixed then probed withmonoclonal antibodies to MNV VPg, N-term, or capsid at1/1,000. Goat anti-mouse FITC secondary antibody was used fordetection, and the cover slips were treated with vectashield(Vector Laboratories, Burlingame, CA) containing DAPI.Background fluorescence was suppressed with Evans blue. Flu-orescent images were observed on a Leica (Wetzlar, Germany)Leitz DMRB fluorescence microscope and captured with a LeicaDFC300FX camera. Representative regions were cropped andoverlays undertaken within Adobe Photoshop CS2 (AdobeSystems, Mountain View, CA).
Western Blot Detection of MNV Proteins. RAW 264.7 or HEK293Tcells were harvested and washed with PBS then subjected toSDS/PAGE analysis. The separated proteins were transferredto Immobilon membrane (Millipore, Billerica, MA) andprobed with rabbit anti-MNV capsid polyclonal antibody (14)or anti-MNV N-term monoclonal antibody. Goat anti-rabbit oranti-mouse alkaline phosphatase antibodies (BioRad, Her-cules, CA) were used as the secondary antibodies, and the blotswere developed with NBT/BCIP or by ECL. The image wasscanned with an HP scanner into Adobe Photoshop CS2(Adobe Systems).
Sequencing the Recombinant MNV Genome. A series of 15 overlap-ping RT-PCRs were performed to sequence all except theextreme termini of the recovered MNV genome (SI Fig. 5 andSI Table 1). The 3� and 5� termini were amplified by RACE
11054 � www.pnas.org�cgi�doi�10.1073�pnas.0700336104 Ward et al.

(SI Fig. 6), the resulting clones were sequenced by using aBeckman CEQ sequencer (Beckman Coulter, Fullerton, CA),and sequences were analyzed by using the Lasergene suite ofDNA analysis programmes (DNAStar, Madison, WI).
Recovery of a Marked Virus. Base 1001 in the 191-bp regionbetween the FseI and ApaI sites of the MNV genomic cDNA wasselected for mutation from C to A to create an EcoRV restrictionsite. Primers MNV382–407 (5� CGGAGGACGCTATGGAT-GCCAAGGAG) and primer ApaRV* (5� TCGAAGGGCCCT-TCGGCCTGCCATTCCCCGAAGATAGATaTCATCCA-GTTGGTC) containing the ApaI restriction site (underlined)and a mismatched base to create an EcoRV site (double under-lined) via an A change at base 1001 (lowercase), were used toamplify the mutated region from the MNV genome. The PCRproduct was digested with ApaI and FseI and ligated directly intothe pFBTETMNV clone to replace the parental sequence and createclone pFBTETMNV*. The clone was used to create a recombinantbacmid and recombinant baculovirus and perform HepG2 trans-duction and RAW cell recovery as described above, to createMNV*. RNA extraction, reverse transcription, and PCR withprimers 3F and #R were performed as described above. Unin-fected cells and reverse transcriptase negative controls were in-cluded. The resulting products for recMNV and MNV* weredigested with EcoRV and analyzed by gel electrophoresis.
pMNV* Construction. The pFBTETMNV* expression cassette, in-cluding minCMVpromoter, full-length genome, polyA tail, andribozyme, was transferred into the plasmid pSP73. The 3� end ofthe genome was isolated as a 3,474-bp fragment from the XhoI
site at position 4596 on the MNV* genome to a HindIII siteoutside the ribozyme on pFBTETMNV* and ligated into pSP73.This clone was digested with XhoI and a 4,967-bp XhoI fragmentcontaining the 5�end of the genome and minCMV promoterfrom pFBTETMNV* ligated into this site. Insert orientation wasconfirmed by digestion with SspI and BssHII.
pro–pol Cleavage Site Mutation. The pro–pol region was modifiedby PCR mutagenesis to alter the glutamine residue in therecognition site to glycine and include an RsrII site for cloneconstruction. Primers MNV5f (SI Table 1) and PPKOr 5�GGAAGCATGGGcGGTCCgccGAACTCCAGAGCCTC-AAGTGTG 3� and primers MNV8r (SI Table 1) and PPKOf5�CTGGAGTTCggcGGACCgCCCATGCTTCCCCGCCCC-TCAGG 3� were used to create two overlapping PCR products(Phusion DNA polymerase; New England Biolabs, Ipswich,MA) with RsrII cleavage sites (underlined). Sequences modifiedfrom the parental sequence are in lowercase. The PCR productswere digested with RsrII, ligated together, gel-purified, andcloned into pCRBlunt. The clone was sequenced, digested withAatII, which flanks the pro–pol boundary, and inserted intoAatII digested pFBTETMNV, and orientation of the insertconfirmed by SfiI digestion. The modified region of the clone wastransferred into the pMNV* vector as an XhoI fragment asdescribed above.
We thank Rachel Skilton for fluorescence microscopy assistance andSarah Garner for providing the VPg and N-term monoclonal antibodies.This work was supported in part by the New Zealand Marsden Fund, theUniversity of Otago (V.K.W.), and Wellcome Trust Grant 069233.
1. Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, Griffin PM,Tauxe RV (1999) Emerg Infect Dis 5:607–625.
2. Clarke IN, Lambden PR (2005) in Topley and Wilson’s Microbiology andMicrobial Infections: Virology, Vol 2, eds Mahy BWJ, ter Meulen W (HodderArnold, London), pp 911–931.
3. Fankhauser RL, Noel JS, Monroe SS, Ando T, Glass RIM (1998) J Infect Dis178:1571–1578.
4. Green KY, Chanock RM, Kapikian AZ (2001) in Fields Virology, eds KnipeDM, Howley PM (Lippincott Williams & Wilkins, Philadelphia), pp 841–874.
5. Hutson AM, Atmar RL, Estes MK (2004) Trends Microbiol 12:279–287.6. Liu GQ, Zhang YY, Ni Z, Yun T, Sheng ZT, Liang HL, Hua JG, Li SM, Du
QY, Chen JP (2006) J Virol 80:6597–6602.7. Chang KO, Sosnovtsev S, Wang Q, Saif LJ, Green KY (2005) J Virol
79:1409–1416.8. Sosnovtsev S, Sosnovtseva S, Green KY (1997) Proceedings of the First
International Symposium on Caliciviruses (Eur Soc Vet Virol Cent Vet Lab,Weybridge, UK), pp 125–130.
9. Sosnovtsev S, Green KY (1995) Virology 210:383–390.10. Asanaka M, Atmar RL, Ruvolo V, Crawford SE, Neill FH, Estes MK (2005)
Proc Natl Acad Sci USA 102:10327–10332.11. Duizer E, Schwab KJ, Neill FH, Atmar RL, Koopmans MPG, Estes MK (2004)
J Gen Virol 85:79–87.12. Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW (2003) Science
299:1575–1578.
13. Wobus CE, Thackray LB, Virgin HW (2006) J Virol 80:5104–5112.14. Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G,
Krug A, Green KY, Virgin HW (2004) PLOS Biology 2:2076–2084.15. Hsu CC, Riley LK, Wills HM, Livingston RS (2006) Comp Med 56:247–251.16. Sosnovtsev SV, Belliot G, Chang KO, Prikhodko VG, Thackray LB, Wobus CE,
Karst SM, Virgin HW, Green KY (2006) J Virol 80:7816–7831.17. Belliot G, Sosnovtsev SV, Mitra T, Hammer C, Garfield M, Green KY (2003)
J Virol 77:10957–10974.18. Blakeney SJ, Cahill A, Reilly PA (2003) Virology 308:216–224.19. Hardy ME (2005) FEMS Microbiol Lett 253:1–8.20. Liu BL, Viljoen GJ, Clarke IN, Lambden PR (1999) J Gen Virol 80:291–296.21. Liu BL, Clarke IN, Lambden PR (1996) J Virol 70:2605–2610.22. McCormick CJ, Rowlands DJ, Harris M (2002) J Gen Virol 83:383–394.23. Condreay JP, Witherspoon SM, Clay WC, Kost TA (1999) Proc Natl Acad Sci
USA 96:127–132.24. Sosnovtsev SV, Garfield M, Green KY (2002) J Virol 76:7060–7072.25. McCormick CJ, Challinor L, Macdonald A, Rowlands DJ, Harris M (2004)
J Gen Virol 85:429–439.26. Harris KS, Reddigari SR, Nicklin MJH, Hammerle T, Wimmer E (1992) J Virol
66:7481–7489.27. Oliver SL, Batten CA, Deng Y, Elschner M, Otto P, Charpilienne A, Clarke
IN, Bridger JC, Lambden PR (2006) J Clin Microbiol 44:992–998.28. McCormick CJ, Brown D, Griffin S, Challinor L, Rowlands DJ, Harris M
(2006) J Gen Virol 87:93–102.
Ward et al. PNAS � June 26, 2007 � vol. 104 � no. 26 � 11055
MIC
ROBI
OLO
GY