regulation of prostate cancer cell division by glucose
TRANSCRIPT

Regulation of Prostate Cancer Cell Divisionby Glucose
GOPAL SINGH,1,2 CLAIR L. LAKKIS,1,2 RODOLFO LAUCIRICA,3
AND DANIEL E. EPNER1,2*1Houston Veterans Affairs Medical Center, Baylor College of Medicine, Houston, Texas
2Department of Medicine, Baylor College of Medicine, Houston, Texas3Department of Pathology, Baylor College of Medicine, Houston, Texas
Previous studies have shown that rapid cell proliferation is associated withelevated glucose consumption. However, those studies did not establish whetherglucose is required for prostate cancer cell proliferation or define the molecularmechanisms by which glucose regulates cell division. We addressed these issuesby studying two metastatic human prostate cancer cell lines: DU145, which isandrogen independent and highly proliferative; and LNCaP, which is androgendependent and relatively slow growing. We found that proliferation of DU145cells was significantly inhibited by reduction of glucose in the medium to 0.5 g/L,which is half the physiologic concentration, whereas LNCaP cells grew at controlrates even in the presence of only 0.05 g/L glucose. Glucose deprivation ofDU145 cells caused a 90% reduction in DNA synthesis; a 10–20-fold reductionin cyclins D and E and CDK4 levels; and cell cycle arrest in G0-G1. However,glucose deprivation did not cause global inhibition of protein synthesis, sincemutant p53 levels increased in glucose-deprived DU145 cells. This observedincrease in mutant p53 levels was not associated with a rise in p21 levels.Glucose deprivation of DU145 cells also led to apparent dephosphorylation ofmutant retinoblastoma (RB) protein. We conclude that: 1) high levels of glucoseconsumption are required for rapid proliferation of androgen-independent pros-tate cancer cells, 2) glucose may not be required for slow growth of androgen-dependent prostate cancer cells, and 3) glucose promotes passage of cells throughearly G1 by increasing the expression of several key cell cycle regulatory proteinsthat normally inhibit RB function. J. Cell. Physiol. 180:431–438, 1999.© 1999 Wiley-Liss, Inc.
Many common cancers, such as prostate cancer, re-spond poorly to conventional chemotherapy drugs,which generally work by inhibiting rapid DNA synthe-sis. Fortunately, major advances in molecular biologyin recent years have led to the discovery of severalnovel targets for cancer treatment, including angiogen-esis, invasion and metastasis, genetic abnormalities,and resistence to apoptosis. However, recent studieshave essentially ignored one of the earliest known hall-marks of cancer: increased aerobic glycolysis (Warburg,1930).
Glycolysis is the metabolic pathway by which glucoseis converted to pyruvate with the generation of 2 mol ofATP per mol of glucose. Most nondividing normal tis-sues, such as muscle, fully oxidize pyruvate formedfrom glycolysis to carbon dioxide and water in mito-chondria, yielding 38 mol of ATP per mol of glucose.These normal tissues generally do not convert glucoseto lactic acid except when starved of oxygen. In con-trast, tumors convert much of the glucose they con-sume into lactic acid even in the presence of oxygen.The rate at which tumors convert glucose to lactic acidvia glycolysis is generally correlated with their rate ofproliferation and degree of “aggressiveness” (Burk et
al., 1967; Venuta and Rubin, 1973; Rheinwald andGreen, 1974; Fagan and Racker, 1978; Sariban-Sohraby et al., 1983; Racker et al., 1985; Baulida et al.,1992; Casciari et al., 1992; Hue and Rousseau, 1993;Burger et al., 1994). Like cancer cells, some rapidlydividing normal cells also have high rates of aerobicglycolysis. For instance, lymphocytes produce largequantities of lactic acid when stimulated to divide (Pol-gar et al., 1968; Sariban-Sohraby et al., 1983; Mar-janovic et al., 1991; Burger et al., 1994), with peakglycolytic activity during S phase (Wang et al., 1976;Marjanovic et al., 1991; Netzker et al., 1994). Lympho-cytes also fail to divide when deprived of glucose (Wadaet al., 1993; Greiner et al., 1994).
While previous studies have shown a correlation be-tween glucose consumption and cell division for se-
Contract grant sponsor: National Cancer Institute; Contractgrant number: 1R29CA/78355-01.
*Correspondence to: Daniel E. Epner, M.D., VAMC, Medical Ser-vice (111H), 2002 Holcombe Blvd., Houston, TX 77030. E-mail:[email protected]
Received 23 October 1998; Accepted 21 April 1999
JOURNAL OF CELLULAR PHYSIOLOGY 180:431–438 (1999)
© 1999 WILEY-LISS, INC.

lected cell types, they have not defined the molecularmechanisms by which glucose regulates cancer cell di-vision or determined whether glucose is universallyrequired for proliferation of cancer cells. We addressedthese issues by using flow cytometry, Western blotanalysis, measurement of DNA synthesis, and mea-surement of cellular ATP content to compare the re-sponses of two disparate human prostate cancer celllines to glucose deprivation.
MATERIALS AND METHODSCell culture
Cells were routinely passaged at 37°C in a mixture of5% carbon dioxide/ 95% air in medium supplementedwith 10% fetal bovine serum (FBS), penicillin 100U/ml, and streptomycin sulfate 100 mg/ml. DU145 cells(American Type Culture Collection, Manassas, VA)were grown in Eagle’s minimum essential medium andLNCaP cells (American Type Culture Collection) weregrown in RPMI-1640 medium. All growth media werepurchased from Gibco BRL (Grand Island, NY). D-glucose-depleted medium was made by adding the ap-propriate quantities of cell culture grade D-glucoseand/or L-glucose to glucose-free medium and supple-menting with 10% dialyzed serum.
Measurement of DNA synthesis by 5-Bromo-2*-deoxy-uridine labeling
We used the 5-Bromo-29-deoxy-uridine (BrdU) label-ing and detection kit II (Boehringer-Mannheim Bio-chemical, Indianapolis, IN) to measure cellular DNAsynthesis. Briefly, the procedure involved growing cellson coverslips, aspirating growth medium, incubating inthe presence of 10 mmol BrdU in phosphate-bufferedsaline (PBS) for 30 min, washing three times with PBS,and fixing in 70% ethanol in 50 mmol glycine buffer(pH 2.0) at –20°C for 30 min. This was followed bythree PBS washes, incubating with anti-BrdU mousemonoclonal antibody for 30 min at 37°C followed bythree PBS washes, incubating with anti-mouse Ig-al-kaline phospatase from sheep in triethanolaminebuffer for 30 min at 37°C, and followed by three PBSwashes, incubating with nitroblue tetrazolium colorsubstrate solution for 20 min at room temperature.
DNA flow cytometryEach sample consisted of 106 prostate cancer cells/
ml, to which 105 fresh chicken erythrocyte nuclei wereadded as an internal standard. Nuclei were stainedaccording to established procedures (Vindelov et al.,1983). Twenty thousand cells per sample were ana-lyzed on a FACScan flow cytometer (Becton Dickinson,San Jose, CA). Aneuploid cancer cells met the followingcriteria: two individual G0-G1 peaks or an asymmetricG0-G1 peak with two well-defined G2/M peaks. ModFitLT software (Verity Software House, Topsham, ME)was used to quantitate the proportion of cells in eachphase of the cell cycle.
Measurement of intracellular ATP levelsIntracellular ATP levels were measured with the
Bioluminescent Somatic Cell Assay Kit (Sigma Chem-ical Co., St. Louis, MO). This kit employs the mostwidely used and sensitive method for ATP determina-tion, the luciferin-luciferase bioluminescent assay
(Stanley, 1986). Twenty thousand cells were seeded perwell in 12-well plates 1 day before experiments. At thebeginning of experiments, cells were washed twice withPBS and 2 ml of the appropriate test medium wasadded to each well. At the time of the assay, mediumwas aspirated from each well and cells were treated inthe wells with 0.25 ml of a 2:1:1 mixture of somatic cellreleasing agent, water, and cell culture medium con-taining neither glucose nor serum at 37°C for 10 min.Somatic cell releasing reagent increased membranepermeability and released cellular ATP. ATP assay mixwas then added to each sample. ATP assay mix con-tained luciferase, luciferin, and MgSO4. In the pres-ence of magnesium, luciferase converted free ATP andluciferin to adenyl-luciferin and inorganic phosphate.Adenyl-luciferin was then converted to oxyluciferinwith generation of light. This procedure is sensitiveenough to measure ATP released by fewer than 10 cellsand light intensity is linearly related to ATP concen-tration for up to 105 cells (Campbell, 1988). An internalstandard was used for each sample to allow conversionof light units to ATP quantity. Readings were obtainedwith a model TD-20e luminometer (Turner Designs,Sunnyvale, CA) with a 10-sec delay time, a 30-sec in-tegrate time, and no predelay time. All experimentswere repeated at least three times on separate wells.Using this procedure, we found a linear relationshipbetween cell number and ATP content for 103–105 cellsper well when identical growth conditions were usedfor each well. Percent ATP content was calculated bydividing average ATP content of test samples by aver-age ATP content of control samples (grown in mediumcontaining 10% nondialyzed fetal calf serum [FCS] and2 g/L D-glucose).
Measurement of lactic acid productionOne day prior to each experiment, 20,000 cells were
seeded in each well of 12-well tissue culture plates.Cells were grown in standard growth medium with10% FBS until the time of the experiment. At thebeginning of each experiment, medium was aspiratedfrom each well, cells were washed twice with glucose-free medium, and 3 ml of various test media wereadded to each well. Eight hours later, medium wasremoved, allowed to reach room temperature for 15min, and immediately analyzed for lactic acid contentwith a YSI 2700 select biochemistry analyzer (YellowSprings Instrument Co., Inc., Yellow Springs, OH).Lactic acid content was also determined at the begin-ning of the experiment by separately analyzing testmedium momentarily placed on cells and then imme-diately removed. Cells from each well were trypsinizedand counted with a hemacytometer. All measurementswere repeated on at least three separate wells. Lacticacid production (mmol/ h /106 cells) was calculated asfollows: ([lactic acid8 h]-[lactic acid0 h]) 3 0.003 L/ 8 h /0.02 3 106 cells, where [ ] indicates concentration inmM, 0.003 was the volume of cell suspension, and 8 hwas the duration of the experiment.
Western analysis. Western analysis was done ac-cording to established protocols (Scopes and Smith,1995). Antiobodies to cyclin D (catalog number sc-451),CDK4 (catalog number sc-260), p21 (catalog numbersc-397), cyclin E (catalog number sc-481), and retino-blastoma (RB) protein (catalog number sc-102) were
432 SINGH ET AL.
purchased from Santa Cruz Biotechnology (SantaCruz, CA). Antibody to p53 was purchased from Onco-gene Research Products (Cambridge, MA; catalog num-ber OP43).
RESULTSEffect of glucose on proliferation of
prostate cancer cellsTwo prostate cancer cell lines were used for the cur-
rent experiments: LNCaP and DU145. LNCaP cellswere derived from a human lymph node metastasis,are androgen dependent, and have normal p53 and RBgenes (Bookstein et al., 1990; Isaacs et al., 1991; Car-roll et al., 1993). DU145 cells were derived from ahuman brain metastasis, are androgen independent,express truncated and phosphorylation-defective RB(Bookstein et al., 1990), and have two mutant p53alleles (Isaacs et al., 1991; Carroll et al., 1993).
We first studied the effect of glucose deprivation oncell proliferation. Glucose deprivation was achieved byreplacing some or all of the D-glucose in the growthmedium with L-glucose, a stereoisomer of glucose thatis neither actively transported into cells nor metabo-lized by them. We used dialyzed serum for experimentsinvolving D-glucose deprivation, since dialyzed serumcontains only about 1% of the physiologic D-glucoseconcentration. Tumor cells deprived of glucose in thecurrent studies were not completely starved of nutri-ents, but instead relied upon mitochondrial metabo-lism of amino acids for ATP production (Pedersen,1978; Kouvroukoglou et al., 1998).
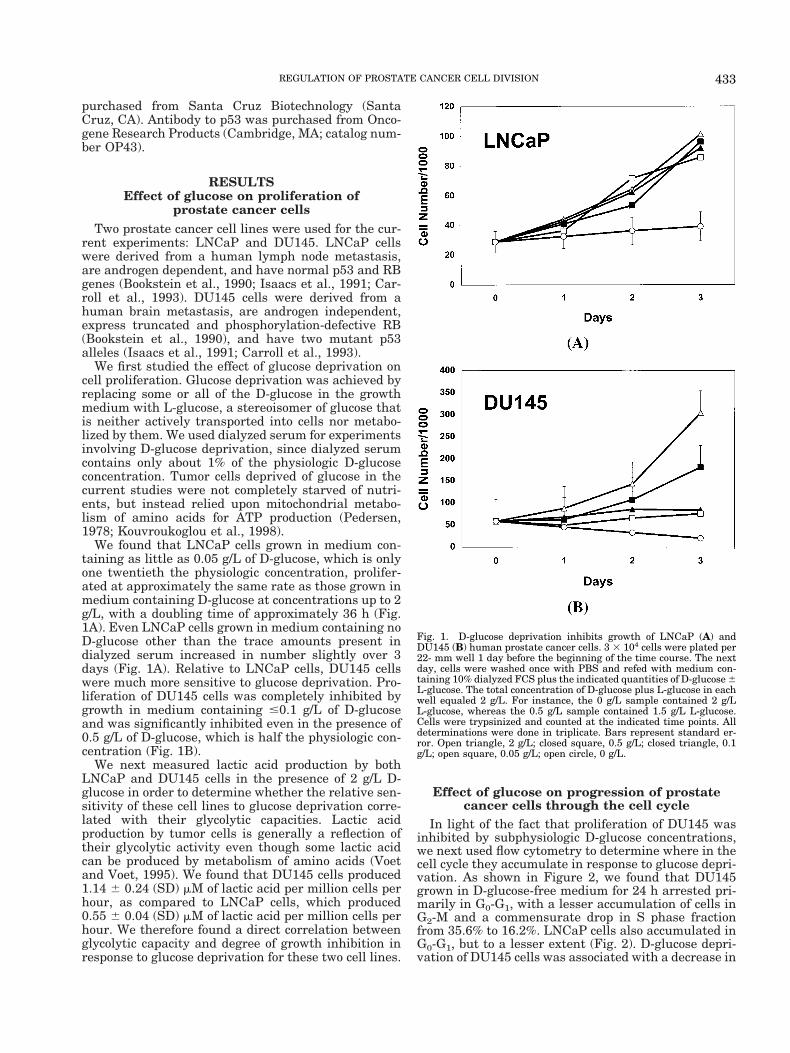
We found that LNCaP cells grown in medium con-taining as little as 0.05 g/L of D-glucose, which is onlyone twentieth the physiologic concentration, prolifer-ated at approximately the same rate as those grown inmedium containing D-glucose at concentrations up to 2g/L, with a doubling time of approximately 36 h (Fig.1A). Even LNCaP cells grown in medium containing noD-glucose other than the trace amounts present indialyzed serum increased in number slightly over 3days (Fig. 1A). Relative to LNCaP cells, DU145 cellswere much more sensitive to glucose deprivation. Pro-liferation of DU145 cells was completely inhibited bygrowth in medium containing #0.1 g/L of D-glucoseand was significantly inhibited even in the presence of0.5 g/L of D-glucose, which is half the physiologic con-centration (Fig. 1B).
We next measured lactic acid production by bothLNCaP and DU145 cells in the presence of 2 g/L D-glucose in order to determine whether the relative sen-sitivity of these cell lines to glucose deprivation corre-lated with their glycolytic capacities. Lactic acidproduction by tumor cells is generally a reflection oftheir glycolytic activity even though some lactic acidcan be produced by metabolism of amino acids (Voetand Voet, 1995). We found that DU145 cells produced1.14 6 0.24 (SD) mM of lactic acid per million cells perhour, as compared to LNCaP cells, which produced0.55 6 0.04 (SD) mM of lactic acid per million cells perhour. We therefore found a direct correlation betweenglycolytic capacity and degree of growth inhibition inresponse to glucose deprivation for these two cell lines.
Effect of glucose on progression of prostatecancer cells through the cell cycle
In light of the fact that proliferation of DU145 wasinhibited by subphysiologic D-glucose concentrations,we next used flow cytometry to determine where in thecell cycle they accumulate in response to glucose depri-vation. As shown in Figure 2, we found that DU145grown in D-glucose-free medium for 24 h arrested pri-marily in G0-G1, with a lesser accumulation of cells inG2-M and a commensurate drop in S phase fractionfrom 35.6% to 16.2%. LNCaP cells also accumulated inG0-G1, but to a lesser extent (Fig. 2). D-glucose depri-vation of DU145 cells was associated with a decrease in
Fig. 1. D-glucose deprivation inhibits growth of LNCaP (A) andDU145 (B) human prostate cancer cells. 3 3 104 cells were plated per22- mm well 1 day before the beginning of the time course. The nextday, cells were washed once with PBS and refed with medium con-taining 10% dialyzed FCS plus the indicated quantities of D-glucose 6L-glucose. The total concentration of D-glucose plus L-glucose in eachwell equaled 2 g/L. For instance, the 0 g/L sample contained 2 g/LL-glucose, whereas the 0.5 g/L sample contained 1.5 g/L L-glucose.Cells were trypsinized and counted at the indicated time points. Alldeterminations were done in triplicate. Bars represent standard er-ror. Open triangle, 2 g/L; closed square, 0.5 g/L; closed triangle, 0.1g/L; open square, 0.05 g/L; open circle, 0 g/L.
433REGULATION OF PROSTATE CANCER CELL DIVISION

nuclear accumulation of BrdU, an indicator of DNAsynthesis. BrdU incorporation by DU145 cells grown inthe absence of D-glucose was reduced by 43% after 24 hand by 90% after 48 h as compared to cells grown undercontrol conditions (Fig. 3).
Effect of glucose on expression of cell cycleregulatory molecules
We next determined whether glucose affected ex-pression and/or posttranslational modification of mol-
ecules that are known to regulate entry of cells into theS phase and thereby play key roles in tumor initiationand progression. These include the tumor suppressorproteins p53 and RB, which are frequently inactivatedin human cancers (Jacks and Weinberg, 1996; Levine,1997; Weinberg, 1991, 1996); p21, an inhibitor of cy-clin-dependent kinases that is transcriptionally acti-vated by p53 (el-Deiry et al., 1993; Elledge, 1996);cyclins D and E, which govern phosphorylation of RBand control exit from G1 (Sherr, 1996); and CDK4, the
Fig. 2. D-glucose deprivation arrests DU145 (A,B) and LNCaP (C,D)human prostate cancer cells in the G0-G1 phase of the cell cycle. 5 3106 cells were plated per 100-mm dish 1 day before the beginning ofthe experiment. The next day, cells were washed once with PBS andrefed with medium containing 10% dialyzed FCS plus either 2 g/L
D-glucose (A,C) or 2 g/L L-glucose (B,D). One day later, cells weretrypsinized and analyzed for DNA content by flow cytometry as de-scribed in Materials and Methods. The vertical axis represents thenumber of cells present in each of the phases of the cell cycle.
434 SINGH ET AL.

catalytic partner of cyclin D (Sherr, 1996). We focusedprimarily on DU145 cells for these experiments, sincethey were more sensitive to glucose deprivation thanwere LNCaP cells.
Figure 4A shows the results of Western analysis ofcyclin D levels in DU145 cells grown in the presence ofeither 2 g/L D-glucose or 2 g/L L-glucose for up to 48 h.Cyclin D levels were reduced by 30% in cells deprivedof D-glucose for 24 h as compared to control cells andreduced 28-fold after 48 h of D-glucose deprivation.D-glucose deprivation had no appreciable effect on cy-clin D levels at earlier time points. CDK4 levels mir-rored cyclin D1 levels, with a 40% reduction for DU145cells deprived of D-glucose for 24 h as compared tocontrol and a 24-fold reduction after 48 h (Fig. 4B).Cyclin E levels were also reduced in DU145 cells to asimilar extent (Fig. 4C).
Since cyclins D and E and CDK4 govern phosphory-lation (inactivation) of RB during cell cycle progressionin early G1, the above results suggest that RB dephos-phorylation may occur in response to glucose depriva-tion. To test that possibility, we analyzed RB by West-ern analysis. We found that DU145 cells grown understandard conditions contained at least two forms of RB:an upper band that presumably represented phosphor-ylated or partially phosphorylated RB, and a moreabundant, lower band that represented hypophosphor-ylated RB (Fig. 4D). The upper band was virtuallyundetectable in growth-arrested DU145 cells grown inthe absence of D-glucose for as little as 4 h (Fig. 4D).We did a similar analysis of LNCaP cells and foundthat glucose deprivation had no appreciable effect oneither the abundance or phosphorylation status ofwild-type RB (data not shown).
We next tested whether the p53 pathway, which
normally inhibits cell cycle progression in late G1, isinvolved in growth inhibition of prostate cancer cells inresponse to glucose deprivation. To do so, we first de-termined whether glucose deprivation altered p53 lev-els in LNCaP and DU145 cells. We found that wild-type p53 was undetectable in LNCaP cells cultured inthe presence of D-glucose and remained undetectableafter glucose deprivation for up to 48 h (Fig. 4E). Incontrast, levels of mutant p53 increased up to fourfoldin DU145 cells grown in the absence of D-glucose for16, 24, or 48 h as compared to cells grown in thepresence of 2 g/L D-glucose (Fig. 4F). One conclusionthat can be drawn from these results is that glucosedeprivation does not cause a global, nonspecific inhibi-tion of protein synthesis. In order to determine thefunctional significance, if any, of the observed accumu-lation of mutant p53 in DU145 cells, we measured theeffect of glucose deprivation on levels of p21, a down-stream target of p53. We found that p21 was undetect-able by Western analysis of DU145 cells under stan-dard growth conditions and that p21 levels did notincrease in response to glucose deprivation for 48 h (notshown). In all Western blot experiments, equal proteinloading per lane was verified by Coumassie staining ofduplicate gels (Fig. 4G). Dose-response relationshipsfor cell cycle regulatory proteins are shown in Figure4H.
We next tested whether an association existed be-tween intracellular ATP levels and growth inhibition ofprostate cancer cells in response to glucose deprivation.To do so, we measured ATP levels in DU145 and LN-CaP cells grown in medium containing either 2 g/LL-glucose or 2 g/L D-glucose for 3 days. Surprisingly,we found that the ATP content per cell was 18% higherin DU145 cells grown in the complete absence of D-glucose, whereas ATP content of LNCaP cells wasequivalent in the presence or absence of D-glucose.These results suggest that growth arrest of humanprostate cancer cells in response to glucose deprivationis not simply due to ATP depletion.
DISCUSSIONThe current studies show that rapidly dividing
DU145 prostate cancer cells depend on high levels ofglucose consumption, whereas relatively slow-growingLNCaP cells are much less dependent on glucose. Wecannot yet conclude whether LNCaP cells can prolifer-ate slowly in the complete absence of glucose, sincetrace amounts of glucose were present in dialyzed se-rum. Nonetheless, our results are consistent with earlystudies that showed a direct relationship between gly-colytic capacity and growth rate for several rat hepa-toma tumors (Burk et al., 1967; Lo et al., 1968; Weberet al., 1971) and lend further support to the hypothesisthat high glucose consumption is required for rapidproliferation of most, if not all, cancer cells (Wang etal., 1976).
Glucose probably facilitates cell division through anumber of mechanisms. First, glucose obviously plays acritical role in ATP production, although one of ourexperiments would seem to suggest otherwise. Wefound that glucose deprivation of DU145 and LNCaPcells for 3 days did not cause ATP depletion. However,we measured ATP levels, not ATP production rate.Prior studies have clearly established that glycolysis
Fig. 3. Glucose deprivation inhibits DNA synthesis in DU145 cells.3 3 104 cells were plated per 22-mm well 1 day before the beginningof the time course. The next day, cells were washed once with PBS andrefed with medium containing 10% dialyzed FCS plus either 2 g/LD-glucose or 2 g/L L-glucose. Stained nuclei actively synthesizingDNA were counted at baseline and after 1 or 2 days by BrdU labelingas described in Materials and Methods. All determinations were donein triplicate. Bars represent standard error. Open square, D-glucose;closed square, L-glucose.
435REGULATION OF PROSTATE CANCER CELL DIVISION

Fig. 4. Glucose regulates the abundance of several cell cycle reg-ulatory proteins in human prostate cancer cells. A-G: For eachsample, 2 3 106 cells were plated in a 60-mm dish 1 day before thebeginning of the experiment. The following day, cells were washedonce with PBS and refed with medium supplemented with 10%dialyzed FCS plus either 2 g/L D-glucose (D) or 2 g/L L-glucose (L).Cells were lysed at the indicated times, and lysates were analyzedby Western analysis as described in Materials and Methods. Rela-tive density of bands as compared to the 0-h sample is shown forcyclin D, CDK4, cyclin E, and p53. A: Cyclin D levels in DU145
cells. B: CDK4 levels in DU145 cells. C: Cyclin E levels in DU145cells. D: RB levels and phosphorylation status in DU145 cells. E:p53 levels in LNCaP and DU145 cells grown in the presence of D-or L-glucose for 48 h. F: p53 levels in DU145 cells. G: Coummasieblue-stained duplicate gel of DU145 cell lysates used for Westernblots, indicating equivalent protein loading per lane. H: Dose-response relationships between D-glucose concentration and levelsof cell cycle regulatory proteins in DU145 cells. Cells were culturedfor 2 days in the presence of the indicated concentrations of D-glucose and then analyzed by Western analysis.
436 SINGH ET AL.

has the capacity to generate ATP much more rapidlythan mitochondrial respiration, even though respira-tion is much more efficient than glycolysis (Voet andVoet, 1995). Cells deprived of glucose in our studiesrelied on mitochondrial metabolism of amino acids forATP production and were therefore probably unable togenerate ATP quickly enough to meet the energy re-quirements of rapid cell division. The fact that steady-state ATP levels did not fall in response to glucosedeprivation suggests that ATP consumption was alsoreduced.
While glucose is undoubtedly an important source ofenergy in rapidly dividing cells, it also has other func-tions. Several studies indicate that glucose serves as aprecursor for DNA synthesis (Pouyssegur et al., 1980;McKeehan, 1982; Newsholme et al., 1985; Medina andNunez de Castro, 1990). In one such study, fibroblastmutants deficient in the glycolytic enzyme phosphoglu-cose isomerase were unable to generate ATP from theglycolytic cascade, as would be expected, but were stillable to use glucose as raw material for DNA synthesisvia the pentose phosphate pathway (Pouyssegur et al.,1980). Other studies have shown that glucose can beconverted to diacylglycerol, a ligand for protein kinaseC, suggesting that glucose plays an additional role insignal transduction (Chiarugi et al., 1990; Rossi et al.,1991; Farese et al., 1994).
Current and previous studies address the potentialrole of RB in growth regulation of DU145 cells. Book-stein et al. (1990) showed that DU145 cells express atruncated form of RB that lacks exon 21. Stable trans-fection of DU145 cells with wild-type RB suppressedtumorigenicity in nude mice but did not affect theirgrowth rate in culture (Bookstein et al., 1990). Whilethose studies do not support a role for endogenous RBin regulation of DU145 cell proliferation, they do notnecessarily exclude such a role. We found that glucosedeprivation of DU145 cells led to arrest in G1; down-regulation of cyclin D, cyclin E, and CDK4; and prob-able dephosphorylation of RB within 4 h. Our resultssuggest that mutant RB is involved in growth inhibi-tion of DU145 cells in response to glucose deprivation.Additional studies will be required to determinewhether RB also plays a role in regulating proliferationof other cell types in response to glucose.
We also addressed the possible role of p53 in regu-lating prostate cancer cell growth. We found that wild-type p53 was undetectable in LNCaP cells at baselineand did not accumulate in response to prolonged glu-cose deprivation despite the fact that proliferation wassignificantly inhibited under those conditions (Fig. 4E).In addition, accumulation of mutant p53 in glucose-deprived DU145 cells was not accompanied by a com-mensurate rise in p21. p53 is therefore probably notinvolved in glucose regulation of prostate cancer cellgrowth.
Our studies suggest that two classical hallmarks ofcancer, namely increased aerobic glycolysis and uncon-trolled proliferation, are closely integrated. Aerobicglycolysis in tumors was a major focus of cancer re-search for 50 years, but has largely been ignored inrecent years in favor of molecular genetic studies. Lackof current interest in the field does not necessarilyreflect a lack of clinical relevance, however. In fact,recent studies strongly suggest that elevated aerobic
glycolysis in tumors can potentially be targeted forcancer treatment (Eskey et al., 1993; Hamilton et al.,1995; Shim et al., 1997). In order for that goal to berealized, however, the molecular mechanisms by whichglucose promotes uncontrolled proliferation of cancercells will need further clarification.
ACKNOWLEDGMENTSSupported by a Merit Review Award from the De-
partment of Veterans Affairs, the Chao Fund (BaylorCollege of Medicine), the Caroline Wiess Law Award(Baylor College of Medicine), and the Joe and JaniceIngram Fund (Baylor College of Medicine).
LITERATURE CITEDBaulida J, Onetti R, Bassols A. 1992. Effects of epidermal growth
factor on glycolysis in A431 cells. Biochem Biophys Res Commun183:1216–1223.
Bookstein R, Shew J, Chen P, Scully P, Lee WH. 1990. Suppression oftumorigenicity of human prostate carcinoma cells by replacing amutated RB gene. Science 247:712–715.
Burger C, Wick M, Brusselbach S, Muller R. 1994. Differential induc-tion of “metabolic genes” after mitogen stimulation and duringnormal cell cycle progression. J Cell Sci 107:241–252.
Burk D, Woods M, Hunter J. 1967. On the significance of glycolysis forcancer growth, with special reference to Morris rat hepatomas.J Natl Cancer Inst 38:839–863.
Campbell AK. 1988. Chemiluminescence. Chichester, UK: Ellis Hor-wood.
Carroll AG, Voeller HJ, Sugars L, Gelmann EP. 1993. p53 oncogenemutations in three human prostate cancer cell lines. Prostate 23:123–134.
Casciari JJ, Sotirchos SV, Sutherland RM. 1992. Variations in tumor cellgrowth rates and metabolism with oxygen concentration, glucose con-centration, and extracellular pH. J Cell Physiol 151:386–394.
Chiarugi V, Basi G, Quattrone A, Micheletti R, Ruggiero M. 1990. Theold and the new in transformed cell signalling: glycolysis, diacyl-glycerol and protein kinase C. Second Messengers Phosphoproteins13:69–85.
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM,Lin D, Mercer WE, Kinzler KW, Vogelstein B. 1993. WAF1, apotential mediator of p53 tumor suppression. Cell 75:817–825.
Elledge SJ. 1996. Cell cycle checkpoints: preventing an identity crisis.Science 274:1664–1672.
Eskey CJ, Koretsky AP, Domach MM, Jain RK. 1993. Role of oxygenvs. glucose in energy metabolism in a mammary carcinoma per-fused ex vivo: direct measurement by 31P NMR. Proc Natl Acad SciUSA 90:2646–2650.
Fagan JB, Racker E. 1978. Determinants of glycolytic rate in normaland transformed chick embryo fibroblasts. Cancer Res 38:749–759.
Farese RV, Standaert ML, Arnold TP, Yamada K, Musunuru K,Hernandez H, Mischak H, Cooper DR. 1994. Preferential activationof microsomal diacylglycerol/protein kinase C signaling during glu-cose treatment (de novo phospholipid synthesis) of rat adipocytes.J Clin Invest 93:1894–1899.
Greiner EF, Guppy M, Brand K. 1994. Glucose is essential for prolif-eration and the glycolytic enzyme induction that provokes a tran-sition to glycolytic energy production. J Biol Chem 269:31484–31490.
Hamilton E, Fennell M, Stafford DM. 1995. Modification of tumorglucose metabolism for therapeutic benefit. Acta Oncol 34:429–433.
Hue L, Rousseau GG. 1993. Fructose 2,6-bisphosphate and the controlof glycolysis by growth factors, tumor promoters and oncogenes.Adv Enzyme Regul 33:97–110.
Isaacs WB, Carter BS, Ewing CM. 1991. Wild-type p53 suppressesgrowth of human prostate cancer cells containing mutant p53 al-leles. Cancer Res 51:4716–4720.
Jacks T, Weinberg RA. 1996. Cell-cycle control and its watchman.Nature 381:643–644.
Kouvroukoglou S, Lakkis CL, Wallace JD, Zygourakis K, Epner DE.1998. Bioenergetics of rat prostate cancer cell migration. Prostate34:137–144.
Levine AJ. 1997. p53, the cellular gatekeeper for growth and division.Cell 88:323–331.
Lo C, Cristofalo VJ, Morris HP, Weinhouse S. 1968. Studies on res-piration and glycolysis in transplanted hepatic tumors of the rat.Cancer Res 28:1–10.
437REGULATION OF PROSTATE CANCER CELL DIVISION

Marjanovic S, Skog S, Heiden T, Tribukait B, Nelson BD. 1991.Expression of glycolytic isoenzymes in activated human peripherallymphocytes: cell cycle analysis using flow cytometry. Exp Cell Res193:425–431.
McKeehan WL. 1982. Glycolysis, glutaminolysis and cell prolifera-tion. Cell Biol Int Rep 6:635–650.
Medina MA, Nunez de Castro I. 1990. Glutaminolysis and glycolysisinteractions in proliferant cells. Int J Biochem 22:681–683.
Netzker R, Hermfisse U, Wein KH, Brand K. 1994. Expression ofglycolytic isozymes in rat thymocytes during cell cycle progression.Biochim Biophys Acta 1224:371–376.
Newsholme EA, Crabtree B, Ardawi MS. 1985. The role of high ratesof glycolysis and glutamine utilization in rapidly dividing cells.Biosci Rep 5:393–400.
Pedersen PL. 1978. Tumor mitochondria and the bioenergetics ofcancer cells. In: Homburger F, editor. Progress in experimentaltumor research. Basel: Karger. p 190–274.
Polgar PR, Foster JM, Cooperband SR. 1968. Glycolysis as an energysource for stimulation of lymphocytes by phytohemagglutinin. ExpCell Res 49:231–237.
Pouyssegur J, Franchi A, Silvestre P. 1980. Relationship betweenincreased aerobic glycolysis and DNA synthesis initiation studiedusing glycolytic mutant fibroblasts. Nature 287:445–447.
Racker E, Resnick RJ, Feldman R. 1985. Glycolysis and methylami-noisobutyrate uptake in rat-1 cells transfected with ras or myconcogenes. Proc Natl Acad Sci USA 82:3535–3538.
Rheinwald JG, Green H. 1974. Growth of cultured mammalian cellson secondary glucose sources. Cell 2:287–293.
Rossi F, Grzeskowiak M, Della Bianca V, Sbarbati A. 1991. De novosynthesis of diacylglycerol from glucose. A new pathway of signaltransduction in human neutrophils stimulated during phagocytosisof beta-glucan particles. J Biol Chem. 266:8034–8038.
Sariban-Sohraby S, Magrath IT, Balaban RS. 1983. Comparison ofenergy metabolism in human normal and neoplastic (Burkitt’s lym-phoma) lymphoid cells. Cancer Res 43:4662–4664.
Scopes RK, Smith JA. 1995. Analysis of proteins. In: Ausubel FM,Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K,editors. Current protocols in molecular biology. New York: Wiley. p.10.0.1–10.20.13
Sherr CJ. 1996. Cancer cell cycles. Science 274:1672–1677.Shim H, Dolde C, Lewis BC, Wu C-S, Dang G, Jungmann RA, Dalla-
Favera R, Dang CV. 1997. c-Myc induction of LDH-A: implicationsfor tumor metabolism and growth. Proc Natl Acad Sci USA 94:6658–6663.
Stanley PE. 1986. Extraction of adenosine triphosphate from micro-bial and somatic cells. Methods Enzymol 133:14–22.
Venuta S, Rubin H. 1973. Sugar transport in normal and Rous sar-coma virus transformed chick embryo fibroblasts. Proc Natl AcadSci USA 70:653–657.
Vindelov LL, Christensen IJ, Nissen NI. 1983. Standardization ofhigh-resolution flow cytometric DNA analysis by the simultaneoususe of chicken and trout red blood cells as internal reference stan-dards. Cytometry 3:328–331.
Voet D, Voet JG. 1995. Glycolysis. In: Biochemistry. New York: Wiley.p. 443–483.
Wada HG, Indelicato SR, Meyer L, Kitamura T, Miyajima A, Kirk G,Muir VC, Parce JW. 1993. GM-CSF triggers a rapid, glucose depen-dent extracellular acidification by TF-1 cells: evidence for sodium/proton antiporter and PKC mediated activation of acid production.J Cell Physiol 154:129–138.
Wang T, Marquardt C, Foker J. 1976. Aerobic glycolysis during lym-phocyte proliferation. Nature 261:702–705.
Warburg OH. 1930. The metabolism of tumors. London: Constable.Weber G, Stubbs M, Morris HP. 1971. Metabolism of hepatomas of
different growth rates in situ and during ischemia. Cancer Res31:2177–2183.
Weinberg RA. 1991. Tumor suppressor genes. Science 254:1138 –1146.
Weinberg RA. 1996. How cancer arises. Sci Am 275:62–70.
438 SINGH ET AL.