regulatory affairs: how understanding the current fda processes will shape the future of your...
TRANSCRIPT

Angela Kustas and Shivani Pandya CMC Consulting Group
© 2015 CMC Consulting Group. All Rights Reserved.
Regulatory Affairs: How understanding the current FDA processes will shape the future of your medical device
FDA 101
MassMedic’s Medical Device Regulatory Affairs 101: Are you sure you know the best regulatory strategy for your
device? ™ comprehensively covered both current and upcoming regulatory landscapes for Medical Devices in the
United States. The all-day intensive event, led by Dr. Michael Drues, PhD, President of Vascular Sciences and
Adjunct Professor of Regulatory Science, Medicine, Biomedical Engineering, and Biotechnology at several
universities and medical schools, clearly defined and analyzed the seven different pathways to approval and
emphasized the importance of choosing best practices for product approval. FDA regulatory affairs for medical
devices may be somewhat complex, but when approached correctly can be a powerful tool for a company’s
success on the market.
CMC had the pleasure of sitting down with Dr. Drues after the event to further discuss his views on regulatory
strategy and how he anticipates changes in the process of FDA filings.
CMC shares the belief that a comprehensive regulatory strategy, that utilizes
health economics, can be an effective tool in creating a barrier to entry for
competitors. Having a strategic plan in place, whether created internally or
through clinical advisory services, can help firms save time and money. Such
services position companies to “sling-shot” out of the approval process into
market sales. A clear understanding of how and when to use each of the
seven pathways to approval is the key to getting approved by the FDA and
gaining strong adoption for the product once approved.
The FDA will never approve bare minimum requirements because eventually
these devices are going to be used on humans, whether it be one or one
million. Beyond the economic value of regulatory services, working with the
right advisors (such as Vascular Sciences and CMC’s Medical Affairs Division)
can ensure the product is approved with the highest standards for safety and
efficacy.
Michael Drues’s perspective: “Sooner or later, a family member or friend, perhaps even you are going to be on the receiving end of one of these medical devices. When that day occurs, what will it take for us to put our personal stamp of endorsements on this particular product to say that it is okay to use this product, be it in my spouse, in my mother, or in my son? Then and only then should we go to FDA and say this is what we do, and this is why it makes sense. Then you have the discussion... Most people take the approach of what’s the minimal amount of work that I have to do in order to get FDA to sign off. And I just am so troubled by that approach, for a bunch of reasons.”

Angela Kustas, Shivani Pandya
CMC Consulting Group
2
Before considering the best pathway for your device, two questions must be answered:
1. Is the product actually a regulated medical device? a. The FDA defines a regulated medical device as “an instrument, apparatus, implement,
machine, contrivance, implant, in vitro reagent, or other similar or related article…intended to affect the structure or any function of the body of man or other animals and which does not achieve any of its primary intended purposes intended through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achieving of any of its primary intended purposes.”
b. Put simply: The distinction of medical devices lies in its intended use and primary mechanism of action (PMOA) and needs not to alter the body in any way.
2. Is the device Class I, Class II, or Class II?
a. The FDA website has a product classification database to help users classify their devices. b. The table below shows a brief guide to the difference between each of the three classes.
SEVEN PATHWAYS
510(K)
The vast majority of medical devices are brought to market through a 510(K)
From January to July 2015, there were 1,783 cleared devices.
Standard FDA Submission costs $5,228 and, on average, takes around 123 days to complete. These are strictly the costs associated with FDA filings, additional costs may apply for performing the required biocompatibility studies and additional testing. (2014)
Only ~ 70% of devices are cleared upon the first submission. However, the success rate jumps to 84% after resubmission. (2014)
Class I Class II Class III
Controls General controls General controls Special controls
General controls PMA
Description Class I devices present minimal harm to users and have history of safe use. These simple devices have the lowest risk.
Class II devices are slightly more complex making general controls not enough to ensure safety. These require guidance documents to ensure safe and effective use.
Class III devices are typically implantable or invasive and are used to support or sustain human life. With this comes the highest risk of illness or injury.
Examples Tongue depressors, bandages
Wheel chairs, x-rays, hospital beds
Artificial heart valves, pacemakers
Michael Drues on 510(k) submissions: “You can spin the 510K however you want to but at the end of the day you have to show that your device is the same as another device, substantially equivalent, or basically the same as, both in terms of technology as well as in terms of labeling. Sometimes, that’s an advantage and at other times, it is not.”

Angela Kustas, Shivani Pandya
CMC Consulting Group
3
The rate of submissions that were not accepted for filing review has fallen between 2014 to 2015 from 47.1% to 38.4%.
PREMARKET APPROVAL
Standard FDA Submission costs are $261,388 and takes around 297 days on average to complete (2014). Additional costs are related to the performance of clinical trials which are usually requested by a PMA.
Note that the costs of PMA submission for a small business drop by 75% to $65,347.
60% (2015) of PMA submissions were accepted for filing review while 100% were accepted in 2014.
DE NOVO
Experts predict that the de novo will be the fastest growing pathway to market.
The de novo is simpler than a traditional 510k because there is no substantial equivalence required.
The average review time for a de novo submission is 166 days.
HUMANITARIAN DEVICE EXEMPTION (HDE)
The HDE is exclusively for small patient populations (equivalent to the orphan status for pharmaceuticals). The intended use must be applicable to less than 4,000 people.
The HDE requires no proof of efficacy. Instead, the filer must show probable benefit.
If applicable, a company may use the HDE as a strategy to gain approval by narrowing their device to a small population and then later going after a larger market through PMA expansion.
CUSTOM DEVICE EXEMPTION (CDE)
In the past, CDEs were most commonly used for personalized devices such as dental appliances, prescription lenses, and prosthetic limbs.
As personalized medicine and 3D printing technology continue to make waves, CDE popularity will follow, making it the most competitively aggressive choice.
PRODUCT DEVELOPMENT PROTOCOL (PDP)
The PDP is technically a sub-set of the PMA.
The PDP can be viewed as a contract, or agreement, with the FDA.
Michael Drues on the De novo pathway: “One of the huge advantages of the de novo in my opinion is you are starting off with a blank canvas in terms of labeling and you can literally paint onto that canvas anything you want, assuming that you can substantiate it of course. You don’t have as much freedom in the 510K world because you have to use the same or very similar labeling to get the devices cleared.”
Did you know that the majority of medical devices are not required to go through clinical trials?
95% of devices currently on the market have not gone through clinical trials.
Angela Kustas, Shivani Pandya
CMC Consulting Group
4
When a PDP reaches successful completion by the FDA, it can be marketed with an approved PMA status.
EMERGENCY USE PROVISION
Used exclusively in high risk situations where a patient has few alternatives, such as brain trauma from a car crash.
Emergency use devices are a great way to gain feasibility with patients without clinical trials, with potential for expanded access.
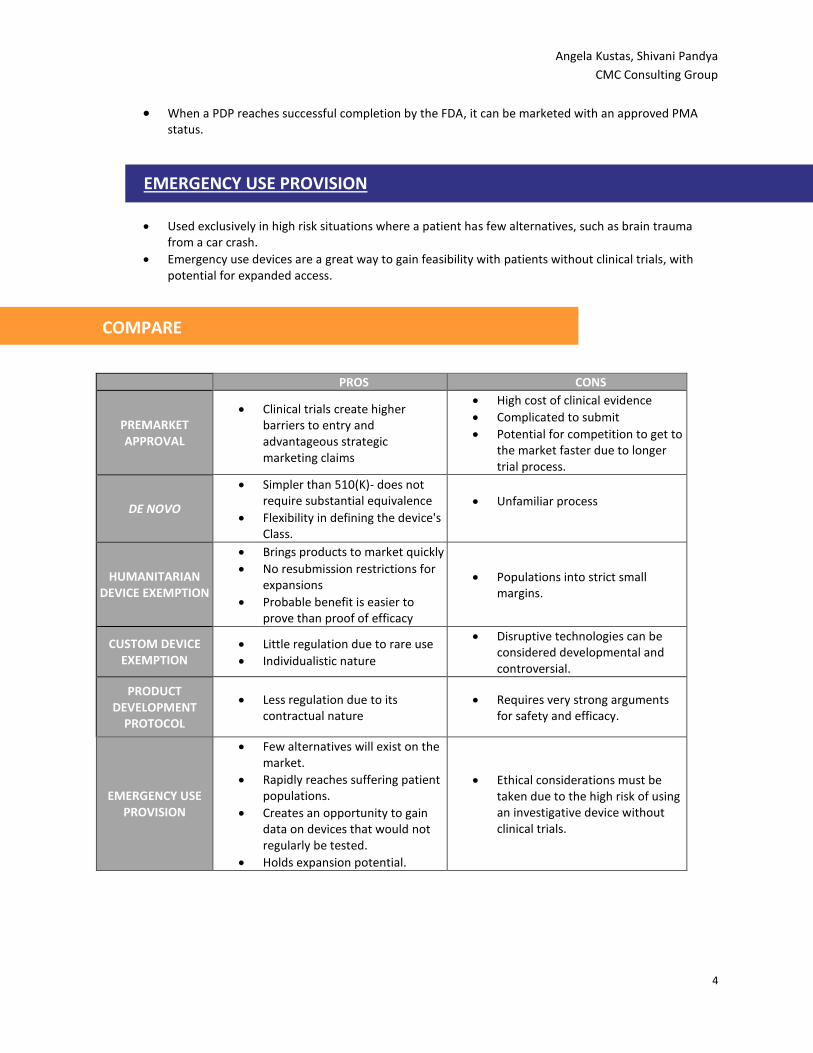
COMPARE
PROS CONS
PREMARKET APPROVAL
Clinical trials create higher barriers to entry and advantageous strategic marketing claims
High cost of clinical evidence
Complicated to submit
Potential for competition to get to the market faster due to longer trial process.
DE NOVO
Simpler than 510(K)- does not require substantial equivalence
Flexibility in defining the device's Class.
Unfamiliar process
HUMANITARIAN DEVICE EXEMPTION
Brings products to market quickly
No resubmission restrictions for expansions
Probable benefit is easier to prove than proof of efficacy
Populations into strict small margins.
CUSTOM DEVICE EXEMPTION
Little regulation due to rare use
Individualistic nature
Disruptive technologies can be considered developmental and controversial.
PRODUCT DEVELOPMENT
PROTOCOL
Less regulation due to its contractual nature
Requires very strong arguments for safety and efficacy.
EMERGENCY USE PROVISION
Few alternatives will exist on the market.
Rapidly reaches suffering patient populations.
Creates an opportunity to gain data on devices that would not regularly be tested.
Holds expansion potential.
Ethical considerations must be taken due to the high risk of using an investigative device without clinical trials.

Angela Kustas, Shivani Pandya
CMC Consulting Group
5
TAKEAWAYS
It’s important to understand and consider all of the possible pathways to market for your medical device. Don’t
neglect your relationship with the FDA as a part of your strategic tool belt. It can be just as powerful as a competitive
pricing, application, or marketing strategy. In fact, choosing the right way to the market can supplement your overall
business strategy.
There is a time and place for each and every FDA filing. Unfortunately, there is no one-size-fits all scenario. Gaining
approval or clearance for a device is empirical, based on its PMOA and its intended use. However, each pathway
has its advantages and disadvantages. For example, consider the de novo submission first for mobile medical apps,
imaging, and IVDs as they are likely to lack substantial equivalents. Strong clinical evidence based medicine lends
itself to a PMA submission that provides the company with stronger barriers to entry from the competition. Far
too many companies are penny wise and pound foolish and try and do the minimal amount of work to get FDA
approval. This minimalistic approach is fraught with danger and often leads to poor patient outcomes and very
expensive recalls for the product. It is our opinion that product quality and patient safety can’t be compromised
and needs to be the company’s first priority so that the product can be sustainable in the market place.
Gaining FDA approval or clearance for a medical device is not an easy feat. Understanding the routes to market is
only the first step. Drues analogized, “to view the whole interaction between a company and the FDA very much
like a poker game. Just because somebody understands the rules of poker, doesn't necessarily mean that they are
going to be good poker players and certainly doesn't mean they are going to win the game.” To add to his example,
sometimes you need to (legally) stack the deck.
Bringing in a regulatory consultant is a key way to ensure you take the correct
necessary actions and get approval your product, the first time. Experts from
Vascular Sciences, and CMC’s Medical Affairs Division (MAD) speak the FDA
language and work directly with clients and FDA to bring new technologies to
market. A strong understanding of health economics shows direct
improvements in quality of care and cost savings to our ever burdened health
care system. Working with regulatory experts can make the process with FDA
more straight-forward and transparent so you don’t have to “gamble” with
the submission process.
All of the information in this document was obtained from the Medical Device Regulatory Affairs 101 seminar and from an exclusive interview with Michael Drues To learn more, visit: http://www.meddeviceonline.com
Michael Drues on FDA Strategy: “I view the whole interaction between the company and FDA very much like a poker game. Just because somebody understands the rules of poker, it doesn't necessarily mean that they are going to be good poker players and certainly doesn't mean they are going to win the game.”

Angela Kustas, Shivani Pandya
CMC Consulting Group
6
REGULATORY EXPERTS
Michael Drues, MS, PhD, is the President of Vascular Sciences, an education, training, &
consulting company offering a broad range of services to medical device, pharmaceutical
& biotechnology companies including (but not limited to): stimulating & innovative
educational programing, brain-storming sessions, prototype design, product
development, benchtop & animal testing , regulatory strategy & clinical trial design, FDA
presentation preparation & defense, reimbursement, clinical acceptance, business
development & technology assessment.
Michael Drues received his degrees in Biomedical Engineering from Iowa State University
and has worked for & consulted with leading medical device, pharmaceutical &
biotechnology companies ranging in size from start-ups to Fortune 100 companies. He
also works on a regular basis for the U.S. Food & Drug Administration (FDA), Health
Canada, the US & European Patent Offices, the Centers for Medicare & Medicaid Services
(CMS) & other regulatory & governmental agencies around the world.
Giacomo Basadonna, MD, PhD, is the Managing Director of the Medical Affairs
Consulting Division (MAD) at CMC. Dr. Basadonna has developed a team of healthcare
advisors who work with our clients throughout the timeline of their projects from proof of
concept to market implementation. He is a member of 23 national committees, 3 hospital
committees and 14 professional societies. Having held offices in these committees
throughout his career; Dr. Basadonna has designed and obtained 16 grants, including
those from large companies and government departments. His previous hospital
appointments include UC Davis Medical Center, Yale New Haven Hospital, and UMass
Memorial Hospital.
Additionally, Dr. Basadonna has held Chief Medical Officer positions for companies,
including Z-Medica. Giacomo holds an MD and PhD from the University of Milan and has
presented at over 80 major conferences and TV appearances during the course of his
career. His publications include over 80 abstracts, 13 limited dissertations, and over 80
publications in peer review journals
Mitchell Sanders MS, PhD, is the Managing Director of the Drug and Device Discovery
Lab (3DL) at CMC. Mitch has 30+ years of experience in studying bacterial biofilms and
chronic wound infections. With ECI Biotech, Mitchell has produced over 12 peer-reviewed
publications and 27 worldwide patents in medical device and in vitro diagnostics.
Mitchell is an expert in clinical and translational research and is a reviewer for the Wound
Healing Society, CIMIT, MassVentures, MIT, WPI, Tech Sandbox, Piranha Pond, SBANE and
the Venture Forum. Mitchell has an MS and PhD from WPI in molecular biology and
biomedical sciences with 2 Postdocs (biochemistry and pathogen genetics) at the
Whitehead Institute/MIT.

Angela Kustas, Shivani Pandya
CMC Consulting Group
7
REFERENCES
1. Drues, Michael. "FDA 101 Follow Up." Personal interview. 13 Aug. 2015.
2. Drues, Michael. "Medical Device Regulatory Affairs 101." Are You Sure You Know the Best Regulatory
Strategy for Your Device? ™. MassMedic, Waltham. Lecture.
3. "U.S. Food and Drug Administration." Classify Your Medical Device. N.p., 29 July 2014. Web.
4. "U.S. Food and Drug Administration." MDUFA Quarterly Performance Reports. N.p., 31 July 2015. Web. 29
Oct. 2015.
5. "U.S. Food and Drug Administration." Medical Devices. N.p., 21 Oct. 2015. Web.

Angela Kustas, Shivani Pandya
CMC Consulting Group
8
ABOUT CMC CONSULTING GROUP
The CMC Group is an international advisory firm providing integrated
transaction services, management and medical affairs consulting and
contract research to companies in the life science industries. This
integration provides clients a seamless interface between strategy and
implementation and incorporates a range of perspectives designed to
optimize engagement outcomes. With offices in the United States, Asia
and throughout the EU, the firm complements global industry knowledge
with rich local market insight.
LOCATIONS Munich, Germany Phone: +49 89 41614220 [email protected]
Boston, USA Phone: +1 508 7209803 [email protected]
Paris, France Phone: +33 1 44549623 [email protected]
Amsterdam, Netherlands Phone: +31 35 6940999 [email protected]
Milan, Italy Phone: +39 0396260093 [email protected]
Barcelona, Spain Phone: +34 93 4067171 [email protected]
Poznan, Poland Phone: +48 61 66001509 [email protected]
Penang, Malaysia Phone: +60 4 2362566 [email protected]
Shanghai, China Phone: +86 21 61721632 [email protected]
Your Global Partner for Growth in Healthcare
LEARN MORE www.cmc-co.net