research article an improved distance matrix...
TRANSCRIPT

Research ArticleAn Improved Distance Matrix Computation Algorithm forMulticore Clusters
Mohammed W Al-Neama12 Naglaa M Reda3 and Fayed F M Ghaleb3
1 Department of Mathematics Faculty of Science Al-Azhar University Cairo Egypt2 Education College for Girls Mosul University Mosul Iraq3 Department of Mathematics Faculty of Science Ain Shams University Cairo Egypt
Correspondence should be addressed to MohammedW Al-Neama mwneamauomosuleduiq
Received 19 January 2014 Revised 11 May 2014 Accepted 18 May 2014 Published 12 June 2014
Academic Editor Horacio Perez-Sanchez
Copyright copy 2014 MohammedW Al-Neama et al This is an open access article distributed under the Creative CommonsAttribution License which permits unrestricted use distribution and reproduction in any medium provided the original work isproperly cited
Distance matrix has diverse usage in different research areas Its computation is typically an essential task in most bioinformaticsapplications especially in multiple sequence alignment The gigantic explosion of biological sequence databases leads to an urgentneed for accelerating these computations DistVect algorithm was introduced in the paper of Al-Neama et al (in press) to presenta recent approach for vectorizing distance matrix computing It showed an efficient performance in both sequential and parallelcomputing However themulticore cluster systems which are available now with their scalability and performancecost ratio meetthe need formore powerful and efficient performanceThis paper proposesDistVect1 as highly efficient parallel vectorized algorithmwith high performance for computing distance matrix addressed to multicore clusters It reformulates DistVect1 vectorizedalgorithm in terms of clusters primitives It deduces an efficient approach of partitioning and scheduling computations convenientto this type of architecture Implementations employ potential of both MPI and OpenMP libraries Experimental results show thatthe proposed method performs improvement of around 3-fold speedup upon SSE2 Further it also achieves speedups more than 9orders of magnitude compared to the publicly available parallel implementation utilized in ClustalW-MPI
1 Introduction
Distance matrix (DM) refers to a two-dimensional arraycontaining the pairwise distances of a set of elements DMhas a broad range of usage in various scientific researchfields It is used intensively in data clustering [1] patternrecognition [2] image analysis [3] information retrieval[4] and bioinformatics In bioinformatics it is mainly usedin constructing the so-called phylogenetic tree which isa diagram that depicts the lines of evolutionary descentof different species organisms or genes from a commonancestor [5]
The explosive growth of genomes makes the ability toalign a huge number of long sequences become more essen-tial For example the Ribosomal Database Project Release
10 [6] consists of more than million sequences This leadsto the massive number of distance calculations As for justaligning 100000 sequences approximately 5 billion distancesneed to be computed to construct a complete DM Even if thesequences are short and pairwise distance calculations canbe done relatively quickly say at a rate of 5 000minus1 sec theiralignment still requires almost 12 days of CPU time Anotherdifficulty is how to store the DM elements as it will take up to40GB of memory This leads to the need of new approachesto accelerate the distance calculations and to handle storageefficiently
On the other hand the rapid development of highperformance computing (HPC) hardware provides high-performance cost ratio computational capability The highprecisions of multicore computers clusters and grids have
Hindawi Publishing CorporationBioMed Research InternationalVolume 2014 Article ID 406178 12 pageshttpdxdoiorg1011552014406178
2 BioMed Research International
become increasingly available and more powerful nowadays[7] Therefore it is of interest to use high-performance tech-nologies to unlock the potential of such systems Meanwhileparallel programming libraries such as OpenMP and MPImade it possible for programmers to take advantage of thegreat computational capability of multicore and clusters forgeneral purpose usage
Many attempts have been produced to efficiently computethe distance matrix As seen in the next section the onesthat used GPUs are really fast but the length of sequencesis limited Other methods that can handle long sequencesand produce accurate alignment are relatively slow Ourmotivation is to provide an efficient method that combinesthe speed and the ability to align long sequences
This paper is an extension to our work in [8] Animproved version of DistVect algorithm is proposed Thecomputations are redistributed efficiently to manage the loadimbalance aiming at enhancing the overall performanceThealgorithm has been upgraded to the hybrid model so as toreap the maximum benefits of fine- and coarse-grainedmod-els The proposed method is devoted to multicore clustersdue to its popularity nowadays and is for achieving higherprocessing speed
The main contributions of this paper are
(i) designing a highly parallel extended DistVect algo-rithm for distance matrix computation on multicoreclusters calledDistVect1 to align huge sequences fast
(ii) implementing the proposed DistVect1 algorithmusing C++ with MPI and OpenMP on BibliothecaAlexandrina platform
(iii) carrying out comprehensive experiments using awidevariety of real dataset sizes and showing that ourdeveloped program outperforms both ClustalW-MPIand SSE2 in terms of execution time
(iv) investigating the impact of increasing both the num-ber and the length of sequences on the speedupand demonstrating that DistVect1 yields significantspeedup when the length of genomes increases
The rest of this paper is organized as follows Section 2summarizes briefly the fundamental methods and algo-rithms concerning the distance matrix Section 3 explainsthe DistVect algorithm Section 4 discusses the improvedDistVect1 Section 5 describes the implementation procedurebriefly and presents results with a detailed analysis FinallySection 6 concludes the paper and suggests future work
2 Related Work
Distance matrix computation is considered as the substantialstage of most multiple sequence alignment tools To align adataset of size 119873 times 119871 where 119873 is the number of sequencesand 119871 is their average length the computation of the DMelements requires lceil119873(119873 minus 1)2rceil pairwise comparisons Eachcomparison uses a matrix of size (119871+1)times(119871+1) to obtain thedistance These computations can become prohibitive when119873 and 119871 are very large (ie in the tens of thousands) There
are few multiple alignment programs that handle datasetsof this size with acceptable accuracy such as MAFFT [9]DIALIGN [10] and Clustal [11] Most accurate methodscould only routinely handle hundreds or few thousands ofsequences like MUSCLE [12] Probcoms [13] and T-Coffee[14]
Promising solutions have been found for parallelizingDM calculations Various parallel algorithms were presentedto overcome speedspace obstacles for different HPC systemssuch as multiprocessor machines and workstation clustersOne category focuses on parallelizing the operations onsmaller data components Typical implementations of thisapproach usingmultithreading are in [15 16]The others con-centrate on distributing each independent pair of sequenceson different processors The most popular parallel methodthat uses this approach is ClustalW-MPI [17] It is targeted forworkstation clusters with distributed memory architectureIts main contribution was providing an efficient distributedmemory implementation of ClustalW that can be run on awide range of distributed memory PC clusters and parallelmulticomputers
Wirawan et al exploit in [18] the use of an intertaskapproach with SIMD model They take advantage of thefact that all elements in the same minor diagonal can becomputed independently in parallelThey used common Intelprocessors with the SSE2 instruction set supporting 16-bitelements and produced a software tool called SSE2 whichwas mainly written in C with p-thread API This approachhas been exploited in some recent methods [15 19ndash21] whereparallelism occurs within a single pair of sequences to avoiddata dependencies within the alignment matrix
GPU has been used in [22] to accelerate sequencealignment It has reformulated dynamic programming-basedalignment algorithms as streaming algorithms in terms ofcomputer graphics primitives Experimental results show thatthe GPU-based approach allows speedups of over one orderof magnitude with respect to optimized CPU implementa-tions Nevertheless this is not severe since 998 percent of thesequences in the database are of length lt4096 Furthermoreit is reasonable to expect that the allowed texture buffer sizeswill increase in next-generation graphics hardware
Also CUDASW++ [20] parallelizes Smith-Watermanalgorithm forCUDAGPU that computes the similarity scoresof a query sequence paired with each sequence in a databasePerformance analysis shows substantial improvement to theoverall performance on the order of three to four giga-cell updates per second The single-GPU version achievesan average performance of 9509 GCUPS with a lowestperformance of 9039 GCUPS and a highest performance of9660 GCUPS and the dual-GPU version achieves an averageperformance of 14484 GCUPS with a lowest performance of10660 GCUPS and a highest performance of 16087 GCUPSBut it supports query sequences of length up to 59K and forquery sequences ranging in length from 144 to 5478
This approach has been further explored by its authorsresulting in optimized SIMT and partitioned vectorized algo-rithmCUDASW++ 20 [19] with an astonishing performance
BioMed Research International 3
of up to 17 GCUPS on a GeForce GTX 280 and 30 GCUPS ona dual-GPU GeForce GTX 295
Likewise CUDASW++ 30 [21] couples CPU and GPUSIMD instructions and carries out concurrent CPU andGPU computations It employs SSE-based vector executionunits as accelerators and employs CUDA PTX SIMD videoinstructions to gain more data parallelism beyond the SIMTexecution model Evaluation shows that CUDASW++ 30gains a performance improvement over CUDASW++ 20 upto 29 and 32 with a maximum performance of 1190 and1856 GCUPS on a single-GPU GeForce GTX 680 and adual-GPU GeForce GTX 690 graphics card respectively Italso has demonstrated significant speedups over SWIPE andBLAST+ However the longest query sequence was of length5478 to search against the Swiss-Prot protein databases thathave the largest sequence length 35213
However most of GPU implementations cannot alignsequences longer than 59000 residues This is due to theintrinsic SIMD characteristics of the GPG where pipeliningallows a great speedup factor yet intense memory usage maylead to bottlenecks This leads to the deployment of otherarchitectures such as many-cores [23] MC64-ClustalWP2has been developed recently as a new implementation of theClustalW algorithm to align long sequences in architectureswith many cores It runs multiple alignments 18 times fasterthan the original ClustalWalgorithmand can align sequencesthat are relatively long (more than 10 kb)
Authors proposed a vectorized distance matrix computa-tion algorithm called DistVect [8] The algorithm addressesthe problem of building a parallel tool for multicores thatproduces the alignment of multiple sequences in a short timewithout using much storage space The main contributionwas the vectorization of all used matrices in computationExperimentally the proposed method achieved good abil-ity of aligning large number of sequences through pow-erful improved storage handling capabilities with efficientimprovement of the overall processing time
3 DistVect Algorithm
DistVect [8] is an accelerated algorithm that computes thedistance matrix for aligning huge datasets It has the advan-tage of exhausting less space It takes as input 119873 sequences1198781
1198782
119878119873
of average length 119871 with their substitutionmatrix sbt and the gab cost 119892 It outputs a distance vector DVcontaining the similarity score (distance) for each of the twosequences It works on vectorizing matrices presented by Liuet al in [22] and used byWirawan et al in [18] It parallelizesthe computations of resolving vectors taking in account theadvantage of the independence of the elements of the minordiagonals of the matrices
To compute the number of exact matches and make itsuitable for a fine-grained parallel implementation Liu et al[22] formulated a recurrence relation for the number of exactmatch computations that is more suitable for implementation
using a linear gap penalty This formula facilitates the calcu-lations without computation of the actual alignment Giventwo sequences 119878
119894
and 119878119895
of lengths 119871119894
and 119871119895
the distancesare computed using the matrices119873119867 as shown below
DM (119878119894
119878119895
) = 1 minus119873 (119894max 119895max)
min (119871119894
119871119895
)
119873 (119909 119910)
=
0 if 119867(119909 119910) = 0
119873 (119909 minus 1 119910 minus 1)
+119898 (119909 119910) if 119867(119909 119910) = 119867 (119909 minus 1 119910 minus 1)
+sbt (119878119894
(119909) 119878119895
(119910))
119873 (119909 119910 minus 1) if 119867(119909 119910) = 119867 (119909 119910 minus 1) + 119892
119873 (119909 minus 1 119910) if 119867(119909 119910) = 119867 (119909 minus 1 119910) + 119892
119867 (119909 119910) = max
0
119867 (119909 minus 1 119910 minus 1) + sbt (119878119894
(119909) 119878119895
(119910))
119867 (119909 minus 1 119910) + 119892
119867 (119909 119910 minus 1) + 119892
119898 (119909 119910) = 1 119878119894
(119909) = 119878119895
(119910)
0 otherwise(1)
where 119867(119909 0) = 119867(0 119910) = 119873(119909 0) = 119873(0 119910) = 0 and 1 le
119909 le 119871119894
1 le 119910 le 119871119895
In [8] the authors proved mathematically that the above
equations can be transformed to the following equationsusing vectors only
DV (ℎ) = 1 minus119896max
min (119871119894
119871119895
)
119873119909
V (119896)
=
0 if 119881119909 (119896) = 0
119873119909
1
(119896 minus 1)
+119898 (119896) if 119881119909 (119896) = 119881119909
1
(119896 minus 1)
+sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119873119909
2
(119896) if 119881119909 (119896) = 119881119909
2
(119896) + 119892
119873119909
2
(119896 minus 1) if 119881119909 (119896) = 119881119909
2
(119896 minus 1) + 119892
119881119909
(119896)
= max
0
119881119909
1
(119896 minus 1) + sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119881119909
2
(119896 minus 1) + 119892
119881119909
2
(119896) + 119892
119898 (119896) = 1 119878119894
(119896) = 119878119895
(119909 minus 119896 + 1)
0 otherwise(2)
4 BioMed Research International
where 119896max is the highest score of 119873 which indicates thenumber of exact matches in the optimal local alignment 1 le
ℎ le lceil119873(119873minus 1)2rceil max(2 119909 minus 119871) le 119896 le min((119909 + 1) (119871 + 1))1 le 119909 le (2119871 minus 1) 119871 is average lengths and 119873
1
1198732
are theexact match values in each iteration
The main idea was based on the fact that any element inan antidiagonal of119867 needs only the values of three elementsfrom the previous two antidiagonalsThis leads to the fact thatonly three vectors could be sufficient One vector119881 is used asa current vector and the previously calculated vectors119881
1
and1198812
as buffers Also three vectors 119873V 1198731 and 1198732
substitutematrix 119873 Finally the distance matrix DM is replaced by adistance vector DV EachDV element is evaluated in terms ofthe highest score 119896max Figure 1 showsDistVectmethodologywhen aligning two sample sequences of length 8
4 Proposed Algorithm
Although DistVect implementation on a multicore usingOpenMp and multithreading has achieved good speedupcompared to ClustalW-MPI [17] it shows load unbalancingTo overcome this problem we propose using a better compu-tation partitioning technique and accompanying extensionsto operate on recent available clusters system that offers morepower to parallelism This is done in order to enhance theperformance In this section an improved version ofDistVectis proposed
DistVect computes the similarity scores using only threevectors instead of the matrix DM using fine-grained paral-lelism and multithreading It distributes all cellsrsquo computa-tions of the antidiagonal vector119881 over the available 119875 coresThe value of each cell is evaluated in terms of its diagonalneighbor stored at119881
1
with its left and upper neighbors storedat 1198812
and the maximum value is selected indicating thehighest score (see Figure 1)
When analyzing the data dependencies between DMcomputations it appears that it may be decomposed throughthree distinct approaches
(i) First the fine-grain approach like multithreadedClustalW [16] is the one in which the computationsof each similarity matrix are decomposedThe imple-mentation of this approach in our work [8] showshigh sensitivity to the length of sequences and animbalanced workload among CPU cores
(ii) Second the coarse-grain approach partitionsDM intoblocks (tiles) and assigns the computation of each oneto a node as in [17 24] However this approach isgood only when the number of sequences is too largecompared to their length and vice versa
(iii) Third the hybrid approach (which is used in theproposed algorithm) combines the two previousapproaches to benefit from the strong points of bothof them
As mentioned above DistVect algorithm faces some per-formance challenges when applying the fine-grain approachThe reason is that the size of the vector 119881 varies during thecomputation and the number of cells to be computed at each
0 0 0 0 0 0 0 0
0
0
0
0
0
0
0
0
V1
V2
V
Deleted
Figure 1 DistVect alignment using fine-grain approach
0
0 0 0 0 0 0 0 0
0
0
0
0
0
0
0
V1
V2
V
Deleted
Figure 2 Distance computation using cyclic partitioning
iteration step is not the same Thus when we distribute thecells on the diagonal to 119875 cores we notice that the workloadis not well balanced among coresThat is because the numberof cells is not always divisible by 119875 Therefore at each stepsome cores have idle time leading to work overhead on othercores Consider Figure 1 at iteration 1 (|119881| = 1) only core 1works at iteration 2 (|119881| = 2) only cores 1 and 2 work Whileat iterations 6 and 9 (|119881| = 6) cores 1 and 2 compute two cellscores 3 and 4 compute one cell only Therefore not all corescompute the same number of cells
On the other hand we think this computation modelrequires a large number of cores for aligning real biologicaldata That is because our main contribution is based onthe acceleration of similarity score computation which is afunction of the sequencesrsquo length 119871 And we concentrate ondealing with long sequences but each core calculates oneelement of119881 at a timeThus as the number of cores increases
BioMed Research International 5
the parallelization factor will increase leading to a higheracceleration
Figure 2 demonstrates our proposed cyclic partitioningtechnique when handling the two sequences considered atFigure 1 with 4 cores The four cores act in parallel on each ofthe first four consecutive rows then they sequentially act onthe second consequent 4 rows
Furthermore all lceil119873(119873minus1)2rceil distance computations arepartitioned into119872 splits and distributed over the119872 nodes inthe cluster system All 119872 nodes work in parallel Each nodewill operate sequentially on aligning the set of lceil119873(119873minus1)2119872rceil
pairs at its own split whose size is 119899split Figure 3 explains howthe alignments of 4 sequences of length 8 are scheduled over acluster system of 2 nodes each node has 4 cores In this casethere are 2 splits each split is of size 3
The pseudocode of this proposed method is depicted inAlgorithm 1 And the distance vector DV is computed usingthe following modified recurrences
119881119909
(119896)
= max
0
119881119909
1
(119896 minus 1) + sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119881119909
2
(119896 minus 1) + 119892
119881119909
2
(119896) + 119892
(3)
119873119909
V (119896)
=
0 if 119881119909 (119896) = 0
119873119909
1
(119896 minus 1) + 119898 (119896) if 119881119909 (119896)= 119881119909
1
(119896 minus 1)
+sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119873119909
2
(119896) if 119881119909 (119896) = 119881119909
2
(119896) + 119892
119873119909
2
(119896 minus 1) if 119881119909 (119896) = 119881119909
2
(119896 minus 1) + 119892
(4)
where 1 le 119896 le 119875
5 Performance Evaluation
The proposed algorithm DistVect1 has been implementedin C++ using OpenMP and MPI libraries The introducedhybrid paradigm matches the characteristics of a cluster ofmulticore system very well The resulting program has theadvantage of coarse-grained parallelism on process level inwhich each MPI process is executed on one multicore nodeAlso it has the advantage of fine-grained parallelismon a looplevel in which each MPI process spawns a team of threads tooccupy the multicore processors when encountering parallelsections of code using OpenMP However the obtainedresults should be carefully analyzed in order to realize the setof items that interferewith the established comparisonmodel
51 Experimental Setup The presented program has beenimplemented on Sun Microsystems cluster provided byLinkSCEEM-2 systems at Bibliotheca Alexandrina Egypt
This platform specification is as follows
(i) 130 nodes and 64GB memory (the allowed numberof nodes for one user is 32 nodes) each node containstwo Intel quad core Xeon 283GHz processors (64-bittechnology)
(ii) 8 Gbyte RAM 80 Gbyte hard disk and a dual port infin band (10Gbps)
(iii) a Giga Ethernet Network port the operating systemis 64-bit Linux
Real protein sequences of different lengths were used forcomputation They are available online at NCBI [25] Theycomprise a subset of variant family of viruses Each usedbenchmark dataset with the average sequence length and thestandard deviation is given in Table 1The standard deviationgives us a numerical measure of the scatter of a dataset Thismeasure is useful for making comparisons between datasetsthat go beyond simple visual impressions
Comprehensive experiments have been conducted onthe specified platform using different groups of sequencesselected from the indicated datasets They evaluated theimplementation of DistVect1 versus that of DistVect and twopopular efficient programs
(i) ClustalW-MPI available at httpwwwmybiosoft-warecomalignment3052
(ii) SSE available at httpssourceforgenetprojectsdist-matcomp
For SSE2 program we have used the SSE approachwithin each MPI process after distributing the overall work-load using the proposed scheduling technique as clarifiedat Figure 3 Thus each node applies its multithreadingapproach on its assigned matrices one at a time Biblio-theca Alexandria used platform support Advanced VectorExtensions (AVX) which is used during implementing SSE2code
52 Execution Time Speedup and Efficiency In this subsec-tion three common performance measurements the execu-tion time the speedup and the efficiency are used Parallelexecution time represents the elapsed time for completecomputation of the distance matrix including all additionscomparisons and maximum operations Parallel speedup isevaluated by the ratio between the parallel execution time ofthe two involved programs Parallel efficiency is computed bydividing the corresponding parallel speedups by the numberof cores Results for variable number of available nodes withtheir analysis are presented below
As a start experiments were executed on a single nodewith 8 cores Since the amount of computations requiredfor a sequence depends on its length multiple lengths ofsequences fromHIV Coronaviridae andHAhaemagglutininwere tested The execution time (in seconds) of the pro-posed DistVect1 implementation against ClustalW-MPI andDistVect was recorded Figure 4(a) shows how the perfor-mance of N(L) is affected by the sequencesrsquo number 119873 and
6 BioMed Research International
Node 1 Node 2
Core 0
Core 1
Core 2
Core 3
Core 0
Core 1
Core 2
Core 3
Node interconnect
0 0 0 0 0 0 0 00 0 0 0 0 0 0 0
00000000
00000000
S1 times S4
S1 times S3
S1 times S2
S2 times S4
S3 times S4
S2 times S3
Figure 3 Proposed scheduling on multicore cluster
Input Number of sequences N average sequencesrsquo length L sequencesrsquo dataset119878 = 119878
1
1198782
119878119873
substitution matrix sbt gab cost g Number of Nodes MNumber of cores POutput Distance Vector DV(1) Let ℎ = 0
(2) Let 119899119878119901119897119894119905
larr [119873(119873 minus 1)2119872]
(3) ⊳ Distributing splits overM nodes(4) for119873119900119889119890119904 larr 1119872 do in parallel(5) Identify Split(6) for119875119886119894119903119904 larr 1 119899
119878119901119897119894119905
do(7) Get 119878
119894
and 119878119895
from Split(8) Initialize 1198811
1
1198812
1
1198731
1
and1198732
1
(9) for119909 larr 1 119871 + 119875 do(10) ⊳ Distributing distance computation over P cores(11) for 119896 larr 1 119875 do in parallel(12) Compute 119881119909(119896) using (3)(13) if 119878
119894
(119896) = 119878119895
(119909 minus 119896 + 1) then Let119898(119896) = 1
(14) else Let119898(119896) = 0
(15) end if(16) Compute119873119909V (119896) using (4)(17) end for 119896(18) Let 119881119909
1
= 119881119909
2
and 119881119909
2
= 119881119909
(19) Let1198731199091
= 119873119909
2
and119873119909
2
= 119873119909
(20) end for119909(21) Identify 119896max(22) Let DV(ℎ) = 1 minus (119896max119871)
(23) Increment ℎ(24) end for Pairs(25) end forNodes(26) return DV
Algorithm 1 DistVect1 Hybrid DistVect Algorithm
length 119871 DistVect1 shows slightly higher performance thanthat of the two other programswhen119871 ranges in the hundredsand119873 ranges in the thousands Further it presented superiorperformance when 119873 is small compared to 119871 and 119871 exceedsa thousand As shown in Figure 4(b) the maximum achievedspeedups when comparing set 500(9200) were (41) and(25)
Next variant subsets of the specified datasetwith differentsizes were tested on 8 nodes Figure 5 shows that ClustalW-MPI could not handle long sequenceswith119871 overriding thirtythousand That is because ClustalW-MPI approach stronglydepends on the number of sequences and focuses on thedistribution of sequences across nodes without regardingtheir length Also DistVect and SSE2 concentrate on the
BioMed Research International 7
Table 1 Used benchmark dataset specifications
Benchmark Number of sequences Average length Standard deviation
Human immunodeficiency virus (HIV) 3000 858 107456000 858 14632
HA hemagglutinin (influenza B virus) 2000 1741 326044000 1728 36661
Several viruses within the Coronaviridae family 200 30488 908534500 29414 1066417
Herpesviridae (large family of DNA viruses) 20 163654 473303250 161000 4289981
20
80
320
1280
5120
20480
81920
ClustalW-MPIDistVect
DistVect1
400(408)
400(856)
800(454)
2000(266)
4000(247)
1000(858)
500
Exec
utio
n (s
)
Number of sequences (length of sequences)
(9200)
(a)
115
225
335
445
ClustalW-MPIDistVect1DistVectDistVect1
Spee
dup
4000(247)
2000(266)
1000(858)
400(408)
800(454)
400(856)
500
Number of sequences (length of sequences)(9200)
(b)
Figure 4 Performance comparisons using one node and 8 cores
thread level parallelism approach which is based on 119871As a result our program performed better than the threeprograms in all cases and operated smoothly with very longsequences This accomplishment is due to the perfect vec-torization and hybrid partitioning approaches For examplefor comparing set 200(30488) ClustalW-MPI did not workDistVect1 consumed 23467 sec SSE2 exhausted 22949 secand DistVect1 achieved it in 8017 sec
Also the same dataset was tested on 16 nodes Resultsgiven in Figure 6 demonstrate that as the number of nodesincreases the performance upgrades For example comparedto the same set 200(30488) ClustalW-MPI works but slowlytakes 26824 sec DistVect consumed better time 12955 secSSE2 exhausted less period 8166 sec and DistVect1 presentsthe fastest achievement 3226 sec
Finally when tests were performed on 32 nodes as seenin Figure 7 DistVect1rsquos overall performance is typically muchbetter when compared to other programs In fact it exhibitsits superiority when 119871 is in tens of thousands The maximumspeedup for set 20(163354) was up to 9 3 and 2 with respectto the ClustalW-MPI SSE2 and DistVect respectively
To ensure that DistVect1 execution time decreases as thenumber of nodes increases Figure 8 illustrates the relationbetween the execution time and the number of nodes As aconsequence it is concluded that DistVect1 implementationacts almost monotonically increasing whenever the numberof available nodes increases Therefore we can state thatDistVect1 execution time is inversely in proportion to thenumber of nodes
To emphasize that the proposed DistVect1 program ismore efficient than other implemented programs the parallelefficiency in terms of the maximum number of availablecores has been evaluated Results are recorded in Table 2 Itis clear that DistVect1 efficiency is monotonically increasingas the length of sequences increases Thus we can state thatDistVect1 efficiency is directly in proportion to the sequencesrsquolength In addition DistVect1rsquos supreme efficiency was up to029 0086 and 0092 with respect to the ClustalW-MPISSE2 and DistVect respectively for the longest sequencelength (163 k)
53 GCUPS A performance measurement commonly usedin computational biology is billion cell updates per second(GCUPS) A GCUPS represents the time for a completecomputation of one entry in the similarity matrix includingall comparisons additions andmaximumoperationsWehavescanned the datasets with their average length as mentionedabove Our implementation on a cluster of multicores allowshandling sequences up to a length of 163 k Table 3 comparesthe corresponding GCUPS performance values
8 BioMed Research International
128000
64000
32000
16000
8000
4000
2000
1000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
1
15
2
25
3
35
4
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
Number of sequences (length of sequences)
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
(b)
Figure 5 Performance comparisons using 8 nodes
5001000200040008000
160003200064000
128000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
200
(30
488
)
20
(163
354
)
6000
(858
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVectDistVectDistVect1
Spee
dup
6000(858)
3000(858)
2000(1741)
4000(1728)
200(30488)
50(161000)
Number of sequences (length of sequences)
(b)
Figure 6 Performance comparisons using 16 nodes
Table 2 Efficiency comparisons using 32 nodes
Number of sequences (Length of sequence) ClustalW-MPI SSE2 DistVect6000 (858) 01899 00401 006423000 (858) 02025 00414 006252000 (1750) 02087 00436 006774000 (1750) 02061 00449 00696200 (30500) 02578 00881 0091050 (161000) 02730 00823 0091920 (163650) 02901 00863 00919
BioMed Research International 9
250500
1000200040008000
160003200064000
3000
(858
)
2000
(1741
)
200
(30
488
)
6000
(858
)
20
(163
354
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
ClustalW-MPISSE2
DistVectDistVect1
Number of sequences (length of sequences)
Exec
utio
n (s
)
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
500
(30
000
)
Number of sequences (length of sequences)
(b)
Figure 7 Performance comparisons using 32 nodes
01000020000300004000050000600007000080000
Excu
tion
time (
s)
8 nodes16 nodes32 nodes
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
Figure 8 DistVect1 performance against number of nodes
54 Space Reduction Thehuge growth of sequence databasesthat exceed current programsrsquo capability and computer sys-tems capacity gives rise to the importance of consideringspace complexity as an intrinsic performance metric In thefollowing we study the exhausted storage space as a functionof 119871 the sequence length and 119873 the number of sequencesDistVect1 has reduced the overall consumed space by otherprograms in two ways
(i) The compensation of the distance matrix DM by thedistance vector DV reduces space from 119873
2 into ℎ =
119873(119873 minus 1)2 that is almost to the half as shown inTable 4
(ii) The substitution of matrices 119867 and 119873 by vector Vrsquosand three 1198731015840V119904 reduces space from 2 times (119871 + 1)
2 to 6119871as shown in Table 5
55 Load Balancing and Occupancy To study the occupancyof processors and memory real snapshots have been takenas seen in Figure 9 They record the history of CPUs mem-ory and network during execution They demonstrate howdifferent proposed versions of the algorithm improve loadbalancing and memory bandwidth The load unbalancingof DistVect is apparent in Figure 9(a) On the contrary theoptimal load balancing of DistVect1 with the full use of allCPUs by 100 is obvious Also the reduction of memoryoccupancy is very clear in Figure 9(b)
As a conclusion a comparison of DistVect1 proposedmethod with studied GPU and cluster implementationsdiscussed in this paper is illustrated in Table 6 For eachprogram it specifies its platform and records the maximumsequence length that may be handled the highest speedup
10 BioMed Research International
Table 3 Performance comparison (in GCUPS) for scanning the datasets
Number of sequences (Length of sequence) DistVect1 DistVect SSE2 ClustalW-MPI20 (163650) 232 079 084 0252000 (1750) 582 268 417 0874000 (1750) 627 281 436 0953000 (858) 666 333 503 1036000 (858) 669 326 522 110200 (30500) 1169 401 415 14250 (161000) 703 239 267 080500 (30000) 1075 392 385 000
Table 4 Storage Comparisons between DV DM (in MW)
N 20 50 200 500 2000 3000 4000 6000DM 00004 00025 004 025 4 9 16 36DV 000019 0001225 00199 012475 1999 449 799 17998
(a) DistVect
(b) DistVect1
Figure 9 CPUs memory and network occupancy
Table 5 Storage comparisons between119867rsquos 119881rsquos (in MW)
L 858 1728 29414 161000Hrsquos + Nrsquos 1475 597 17348 518V rsquos +119873Vrsquos 0005 001 0176 0966
and GCUPS which can be reached Records certify ourpresumed idea that although GPU programs are very fastwith high GCUPS they cannot align long sequences It alsoproves that our proposal is superlative in handling longsequences with good speedup
The above presented results reveal the best performanceof theDistVect1 against other implementationsThis achieve-ment is due to two main reasons (1) the superiority ofDistVect1 in using vectors instead of matrices leading to abetter caching and (2) the optimal use of multicore clustersystem because of the proposed partitioning and schedul-ing techniques especially when the length of sequencesincreases thus obtaining a higher parallelization factor
6 Conclusion and Future Work
In this paper we presented and evaluatedDistVect1 algorithman improved version of the vectorized parallel algorithmDistVect for efficiently computing the distance matrix usingmulticore cluster systems The new suggested partition-ing and scheduling approaches integrated with MPI andOpenMP primitives have achieved a significant improve-ment to the overall performance Implementations wereconducted on Bibliotheca Alexandria cluster system using 32nodes (8 cores)
The resulting program DistVect1 was able to computedistances between very long sequences up to 163 k It outper-forms SSE2 with 3-fold speedup and ClustalW-MPI 013 with
BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

2 BioMed Research International
become increasingly available and more powerful nowadays[7] Therefore it is of interest to use high-performance tech-nologies to unlock the potential of such systems Meanwhileparallel programming libraries such as OpenMP and MPImade it possible for programmers to take advantage of thegreat computational capability of multicore and clusters forgeneral purpose usage
Many attempts have been produced to efficiently computethe distance matrix As seen in the next section the onesthat used GPUs are really fast but the length of sequencesis limited Other methods that can handle long sequencesand produce accurate alignment are relatively slow Ourmotivation is to provide an efficient method that combinesthe speed and the ability to align long sequences
This paper is an extension to our work in [8] Animproved version of DistVect algorithm is proposed Thecomputations are redistributed efficiently to manage the loadimbalance aiming at enhancing the overall performanceThealgorithm has been upgraded to the hybrid model so as toreap the maximum benefits of fine- and coarse-grainedmod-els The proposed method is devoted to multicore clustersdue to its popularity nowadays and is for achieving higherprocessing speed
The main contributions of this paper are
(i) designing a highly parallel extended DistVect algo-rithm for distance matrix computation on multicoreclusters calledDistVect1 to align huge sequences fast
(ii) implementing the proposed DistVect1 algorithmusing C++ with MPI and OpenMP on BibliothecaAlexandrina platform
(iii) carrying out comprehensive experiments using awidevariety of real dataset sizes and showing that ourdeveloped program outperforms both ClustalW-MPIand SSE2 in terms of execution time
(iv) investigating the impact of increasing both the num-ber and the length of sequences on the speedupand demonstrating that DistVect1 yields significantspeedup when the length of genomes increases
The rest of this paper is organized as follows Section 2summarizes briefly the fundamental methods and algo-rithms concerning the distance matrix Section 3 explainsthe DistVect algorithm Section 4 discusses the improvedDistVect1 Section 5 describes the implementation procedurebriefly and presents results with a detailed analysis FinallySection 6 concludes the paper and suggests future work
2 Related Work
Distance matrix computation is considered as the substantialstage of most multiple sequence alignment tools To align adataset of size 119873 times 119871 where 119873 is the number of sequencesand 119871 is their average length the computation of the DMelements requires lceil119873(119873 minus 1)2rceil pairwise comparisons Eachcomparison uses a matrix of size (119871+1)times(119871+1) to obtain thedistance These computations can become prohibitive when119873 and 119871 are very large (ie in the tens of thousands) There
are few multiple alignment programs that handle datasetsof this size with acceptable accuracy such as MAFFT [9]DIALIGN [10] and Clustal [11] Most accurate methodscould only routinely handle hundreds or few thousands ofsequences like MUSCLE [12] Probcoms [13] and T-Coffee[14]
Promising solutions have been found for parallelizingDM calculations Various parallel algorithms were presentedto overcome speedspace obstacles for different HPC systemssuch as multiprocessor machines and workstation clustersOne category focuses on parallelizing the operations onsmaller data components Typical implementations of thisapproach usingmultithreading are in [15 16]The others con-centrate on distributing each independent pair of sequenceson different processors The most popular parallel methodthat uses this approach is ClustalW-MPI [17] It is targeted forworkstation clusters with distributed memory architectureIts main contribution was providing an efficient distributedmemory implementation of ClustalW that can be run on awide range of distributed memory PC clusters and parallelmulticomputers
Wirawan et al exploit in [18] the use of an intertaskapproach with SIMD model They take advantage of thefact that all elements in the same minor diagonal can becomputed independently in parallelThey used common Intelprocessors with the SSE2 instruction set supporting 16-bitelements and produced a software tool called SSE2 whichwas mainly written in C with p-thread API This approachhas been exploited in some recent methods [15 19ndash21] whereparallelism occurs within a single pair of sequences to avoiddata dependencies within the alignment matrix
GPU has been used in [22] to accelerate sequencealignment It has reformulated dynamic programming-basedalignment algorithms as streaming algorithms in terms ofcomputer graphics primitives Experimental results show thatthe GPU-based approach allows speedups of over one orderof magnitude with respect to optimized CPU implementa-tions Nevertheless this is not severe since 998 percent of thesequences in the database are of length lt4096 Furthermoreit is reasonable to expect that the allowed texture buffer sizeswill increase in next-generation graphics hardware
Also CUDASW++ [20] parallelizes Smith-Watermanalgorithm forCUDAGPU that computes the similarity scoresof a query sequence paired with each sequence in a databasePerformance analysis shows substantial improvement to theoverall performance on the order of three to four giga-cell updates per second The single-GPU version achievesan average performance of 9509 GCUPS with a lowestperformance of 9039 GCUPS and a highest performance of9660 GCUPS and the dual-GPU version achieves an averageperformance of 14484 GCUPS with a lowest performance of10660 GCUPS and a highest performance of 16087 GCUPSBut it supports query sequences of length up to 59K and forquery sequences ranging in length from 144 to 5478
This approach has been further explored by its authorsresulting in optimized SIMT and partitioned vectorized algo-rithmCUDASW++ 20 [19] with an astonishing performance
BioMed Research International 3
of up to 17 GCUPS on a GeForce GTX 280 and 30 GCUPS ona dual-GPU GeForce GTX 295
Likewise CUDASW++ 30 [21] couples CPU and GPUSIMD instructions and carries out concurrent CPU andGPU computations It employs SSE-based vector executionunits as accelerators and employs CUDA PTX SIMD videoinstructions to gain more data parallelism beyond the SIMTexecution model Evaluation shows that CUDASW++ 30gains a performance improvement over CUDASW++ 20 upto 29 and 32 with a maximum performance of 1190 and1856 GCUPS on a single-GPU GeForce GTX 680 and adual-GPU GeForce GTX 690 graphics card respectively Italso has demonstrated significant speedups over SWIPE andBLAST+ However the longest query sequence was of length5478 to search against the Swiss-Prot protein databases thathave the largest sequence length 35213
However most of GPU implementations cannot alignsequences longer than 59000 residues This is due to theintrinsic SIMD characteristics of the GPG where pipeliningallows a great speedup factor yet intense memory usage maylead to bottlenecks This leads to the deployment of otherarchitectures such as many-cores [23] MC64-ClustalWP2has been developed recently as a new implementation of theClustalW algorithm to align long sequences in architectureswith many cores It runs multiple alignments 18 times fasterthan the original ClustalWalgorithmand can align sequencesthat are relatively long (more than 10 kb)
Authors proposed a vectorized distance matrix computa-tion algorithm called DistVect [8] The algorithm addressesthe problem of building a parallel tool for multicores thatproduces the alignment of multiple sequences in a short timewithout using much storage space The main contributionwas the vectorization of all used matrices in computationExperimentally the proposed method achieved good abil-ity of aligning large number of sequences through pow-erful improved storage handling capabilities with efficientimprovement of the overall processing time
3 DistVect Algorithm
DistVect [8] is an accelerated algorithm that computes thedistance matrix for aligning huge datasets It has the advan-tage of exhausting less space It takes as input 119873 sequences1198781
1198782
119878119873
of average length 119871 with their substitutionmatrix sbt and the gab cost 119892 It outputs a distance vector DVcontaining the similarity score (distance) for each of the twosequences It works on vectorizing matrices presented by Liuet al in [22] and used byWirawan et al in [18] It parallelizesthe computations of resolving vectors taking in account theadvantage of the independence of the elements of the minordiagonals of the matrices
To compute the number of exact matches and make itsuitable for a fine-grained parallel implementation Liu et al[22] formulated a recurrence relation for the number of exactmatch computations that is more suitable for implementation
using a linear gap penalty This formula facilitates the calcu-lations without computation of the actual alignment Giventwo sequences 119878
119894
and 119878119895
of lengths 119871119894
and 119871119895
the distancesare computed using the matrices119873119867 as shown below
DM (119878119894
119878119895
) = 1 minus119873 (119894max 119895max)
min (119871119894
119871119895
)
119873 (119909 119910)
=
0 if 119867(119909 119910) = 0
119873 (119909 minus 1 119910 minus 1)
+119898 (119909 119910) if 119867(119909 119910) = 119867 (119909 minus 1 119910 minus 1)
+sbt (119878119894
(119909) 119878119895
(119910))
119873 (119909 119910 minus 1) if 119867(119909 119910) = 119867 (119909 119910 minus 1) + 119892
119873 (119909 minus 1 119910) if 119867(119909 119910) = 119867 (119909 minus 1 119910) + 119892
119867 (119909 119910) = max
0
119867 (119909 minus 1 119910 minus 1) + sbt (119878119894
(119909) 119878119895
(119910))
119867 (119909 minus 1 119910) + 119892
119867 (119909 119910 minus 1) + 119892
119898 (119909 119910) = 1 119878119894
(119909) = 119878119895
(119910)
0 otherwise(1)
where 119867(119909 0) = 119867(0 119910) = 119873(119909 0) = 119873(0 119910) = 0 and 1 le
119909 le 119871119894
1 le 119910 le 119871119895
In [8] the authors proved mathematically that the above
equations can be transformed to the following equationsusing vectors only
DV (ℎ) = 1 minus119896max
min (119871119894
119871119895
)
119873119909
V (119896)
=
0 if 119881119909 (119896) = 0
119873119909
1
(119896 minus 1)
+119898 (119896) if 119881119909 (119896) = 119881119909
1
(119896 minus 1)
+sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119873119909
2
(119896) if 119881119909 (119896) = 119881119909
2
(119896) + 119892
119873119909
2
(119896 minus 1) if 119881119909 (119896) = 119881119909
2
(119896 minus 1) + 119892
119881119909
(119896)
= max
0
119881119909
1
(119896 minus 1) + sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119881119909
2
(119896 minus 1) + 119892
119881119909
2
(119896) + 119892
119898 (119896) = 1 119878119894
(119896) = 119878119895
(119909 minus 119896 + 1)
0 otherwise(2)
4 BioMed Research International
where 119896max is the highest score of 119873 which indicates thenumber of exact matches in the optimal local alignment 1 le
ℎ le lceil119873(119873minus 1)2rceil max(2 119909 minus 119871) le 119896 le min((119909 + 1) (119871 + 1))1 le 119909 le (2119871 minus 1) 119871 is average lengths and 119873
1
1198732
are theexact match values in each iteration
The main idea was based on the fact that any element inan antidiagonal of119867 needs only the values of three elementsfrom the previous two antidiagonalsThis leads to the fact thatonly three vectors could be sufficient One vector119881 is used asa current vector and the previously calculated vectors119881
1
and1198812
as buffers Also three vectors 119873V 1198731 and 1198732
substitutematrix 119873 Finally the distance matrix DM is replaced by adistance vector DV EachDV element is evaluated in terms ofthe highest score 119896max Figure 1 showsDistVectmethodologywhen aligning two sample sequences of length 8
4 Proposed Algorithm
Although DistVect implementation on a multicore usingOpenMp and multithreading has achieved good speedupcompared to ClustalW-MPI [17] it shows load unbalancingTo overcome this problem we propose using a better compu-tation partitioning technique and accompanying extensionsto operate on recent available clusters system that offers morepower to parallelism This is done in order to enhance theperformance In this section an improved version ofDistVectis proposed
DistVect computes the similarity scores using only threevectors instead of the matrix DM using fine-grained paral-lelism and multithreading It distributes all cellsrsquo computa-tions of the antidiagonal vector119881 over the available 119875 coresThe value of each cell is evaluated in terms of its diagonalneighbor stored at119881
1
with its left and upper neighbors storedat 1198812
and the maximum value is selected indicating thehighest score (see Figure 1)
When analyzing the data dependencies between DMcomputations it appears that it may be decomposed throughthree distinct approaches
(i) First the fine-grain approach like multithreadedClustalW [16] is the one in which the computationsof each similarity matrix are decomposedThe imple-mentation of this approach in our work [8] showshigh sensitivity to the length of sequences and animbalanced workload among CPU cores
(ii) Second the coarse-grain approach partitionsDM intoblocks (tiles) and assigns the computation of each oneto a node as in [17 24] However this approach isgood only when the number of sequences is too largecompared to their length and vice versa
(iii) Third the hybrid approach (which is used in theproposed algorithm) combines the two previousapproaches to benefit from the strong points of bothof them
As mentioned above DistVect algorithm faces some per-formance challenges when applying the fine-grain approachThe reason is that the size of the vector 119881 varies during thecomputation and the number of cells to be computed at each
0 0 0 0 0 0 0 0
0
0
0
0
0
0
0
0
V1
V2
V
Deleted
Figure 1 DistVect alignment using fine-grain approach
0
0 0 0 0 0 0 0 0
0
0
0
0
0
0
0
V1
V2
V
Deleted
Figure 2 Distance computation using cyclic partitioning
iteration step is not the same Thus when we distribute thecells on the diagonal to 119875 cores we notice that the workloadis not well balanced among coresThat is because the numberof cells is not always divisible by 119875 Therefore at each stepsome cores have idle time leading to work overhead on othercores Consider Figure 1 at iteration 1 (|119881| = 1) only core 1works at iteration 2 (|119881| = 2) only cores 1 and 2 work Whileat iterations 6 and 9 (|119881| = 6) cores 1 and 2 compute two cellscores 3 and 4 compute one cell only Therefore not all corescompute the same number of cells
On the other hand we think this computation modelrequires a large number of cores for aligning real biologicaldata That is because our main contribution is based onthe acceleration of similarity score computation which is afunction of the sequencesrsquo length 119871 And we concentrate ondealing with long sequences but each core calculates oneelement of119881 at a timeThus as the number of cores increases
BioMed Research International 5
the parallelization factor will increase leading to a higheracceleration
Figure 2 demonstrates our proposed cyclic partitioningtechnique when handling the two sequences considered atFigure 1 with 4 cores The four cores act in parallel on each ofthe first four consecutive rows then they sequentially act onthe second consequent 4 rows
Furthermore all lceil119873(119873minus1)2rceil distance computations arepartitioned into119872 splits and distributed over the119872 nodes inthe cluster system All 119872 nodes work in parallel Each nodewill operate sequentially on aligning the set of lceil119873(119873minus1)2119872rceil
pairs at its own split whose size is 119899split Figure 3 explains howthe alignments of 4 sequences of length 8 are scheduled over acluster system of 2 nodes each node has 4 cores In this casethere are 2 splits each split is of size 3
The pseudocode of this proposed method is depicted inAlgorithm 1 And the distance vector DV is computed usingthe following modified recurrences
119881119909
(119896)
= max
0
119881119909
1
(119896 minus 1) + sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119881119909
2
(119896 minus 1) + 119892
119881119909
2
(119896) + 119892
(3)
119873119909
V (119896)
=
0 if 119881119909 (119896) = 0
119873119909
1
(119896 minus 1) + 119898 (119896) if 119881119909 (119896)= 119881119909
1
(119896 minus 1)
+sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119873119909
2
(119896) if 119881119909 (119896) = 119881119909
2
(119896) + 119892
119873119909
2
(119896 minus 1) if 119881119909 (119896) = 119881119909
2
(119896 minus 1) + 119892
(4)
where 1 le 119896 le 119875
5 Performance Evaluation
The proposed algorithm DistVect1 has been implementedin C++ using OpenMP and MPI libraries The introducedhybrid paradigm matches the characteristics of a cluster ofmulticore system very well The resulting program has theadvantage of coarse-grained parallelism on process level inwhich each MPI process is executed on one multicore nodeAlso it has the advantage of fine-grained parallelismon a looplevel in which each MPI process spawns a team of threads tooccupy the multicore processors when encountering parallelsections of code using OpenMP However the obtainedresults should be carefully analyzed in order to realize the setof items that interferewith the established comparisonmodel
51 Experimental Setup The presented program has beenimplemented on Sun Microsystems cluster provided byLinkSCEEM-2 systems at Bibliotheca Alexandrina Egypt
This platform specification is as follows
(i) 130 nodes and 64GB memory (the allowed numberof nodes for one user is 32 nodes) each node containstwo Intel quad core Xeon 283GHz processors (64-bittechnology)
(ii) 8 Gbyte RAM 80 Gbyte hard disk and a dual port infin band (10Gbps)
(iii) a Giga Ethernet Network port the operating systemis 64-bit Linux
Real protein sequences of different lengths were used forcomputation They are available online at NCBI [25] Theycomprise a subset of variant family of viruses Each usedbenchmark dataset with the average sequence length and thestandard deviation is given in Table 1The standard deviationgives us a numerical measure of the scatter of a dataset Thismeasure is useful for making comparisons between datasetsthat go beyond simple visual impressions
Comprehensive experiments have been conducted onthe specified platform using different groups of sequencesselected from the indicated datasets They evaluated theimplementation of DistVect1 versus that of DistVect and twopopular efficient programs
(i) ClustalW-MPI available at httpwwwmybiosoft-warecomalignment3052
(ii) SSE available at httpssourceforgenetprojectsdist-matcomp
For SSE2 program we have used the SSE approachwithin each MPI process after distributing the overall work-load using the proposed scheduling technique as clarifiedat Figure 3 Thus each node applies its multithreadingapproach on its assigned matrices one at a time Biblio-theca Alexandria used platform support Advanced VectorExtensions (AVX) which is used during implementing SSE2code
52 Execution Time Speedup and Efficiency In this subsec-tion three common performance measurements the execu-tion time the speedup and the efficiency are used Parallelexecution time represents the elapsed time for completecomputation of the distance matrix including all additionscomparisons and maximum operations Parallel speedup isevaluated by the ratio between the parallel execution time ofthe two involved programs Parallel efficiency is computed bydividing the corresponding parallel speedups by the numberof cores Results for variable number of available nodes withtheir analysis are presented below
As a start experiments were executed on a single nodewith 8 cores Since the amount of computations requiredfor a sequence depends on its length multiple lengths ofsequences fromHIV Coronaviridae andHAhaemagglutininwere tested The execution time (in seconds) of the pro-posed DistVect1 implementation against ClustalW-MPI andDistVect was recorded Figure 4(a) shows how the perfor-mance of N(L) is affected by the sequencesrsquo number 119873 and
6 BioMed Research International
Node 1 Node 2
Core 0
Core 1
Core 2
Core 3
Core 0
Core 1
Core 2
Core 3
Node interconnect
0 0 0 0 0 0 0 00 0 0 0 0 0 0 0
00000000
00000000
S1 times S4
S1 times S3
S1 times S2
S2 times S4
S3 times S4
S2 times S3
Figure 3 Proposed scheduling on multicore cluster
Input Number of sequences N average sequencesrsquo length L sequencesrsquo dataset119878 = 119878
1
1198782
119878119873
substitution matrix sbt gab cost g Number of Nodes MNumber of cores POutput Distance Vector DV(1) Let ℎ = 0
(2) Let 119899119878119901119897119894119905
larr [119873(119873 minus 1)2119872]
(3) ⊳ Distributing splits overM nodes(4) for119873119900119889119890119904 larr 1119872 do in parallel(5) Identify Split(6) for119875119886119894119903119904 larr 1 119899
119878119901119897119894119905
do(7) Get 119878
119894
and 119878119895
from Split(8) Initialize 1198811
1
1198812
1
1198731
1
and1198732
1
(9) for119909 larr 1 119871 + 119875 do(10) ⊳ Distributing distance computation over P cores(11) for 119896 larr 1 119875 do in parallel(12) Compute 119881119909(119896) using (3)(13) if 119878
119894
(119896) = 119878119895
(119909 minus 119896 + 1) then Let119898(119896) = 1
(14) else Let119898(119896) = 0
(15) end if(16) Compute119873119909V (119896) using (4)(17) end for 119896(18) Let 119881119909
1
= 119881119909
2
and 119881119909
2
= 119881119909
(19) Let1198731199091
= 119873119909
2
and119873119909
2
= 119873119909
(20) end for119909(21) Identify 119896max(22) Let DV(ℎ) = 1 minus (119896max119871)
(23) Increment ℎ(24) end for Pairs(25) end forNodes(26) return DV
Algorithm 1 DistVect1 Hybrid DistVect Algorithm
length 119871 DistVect1 shows slightly higher performance thanthat of the two other programswhen119871 ranges in the hundredsand119873 ranges in the thousands Further it presented superiorperformance when 119873 is small compared to 119871 and 119871 exceedsa thousand As shown in Figure 4(b) the maximum achievedspeedups when comparing set 500(9200) were (41) and(25)
Next variant subsets of the specified datasetwith differentsizes were tested on 8 nodes Figure 5 shows that ClustalW-MPI could not handle long sequenceswith119871 overriding thirtythousand That is because ClustalW-MPI approach stronglydepends on the number of sequences and focuses on thedistribution of sequences across nodes without regardingtheir length Also DistVect and SSE2 concentrate on the
BioMed Research International 7
Table 1 Used benchmark dataset specifications
Benchmark Number of sequences Average length Standard deviation
Human immunodeficiency virus (HIV) 3000 858 107456000 858 14632
HA hemagglutinin (influenza B virus) 2000 1741 326044000 1728 36661
Several viruses within the Coronaviridae family 200 30488 908534500 29414 1066417
Herpesviridae (large family of DNA viruses) 20 163654 473303250 161000 4289981
20
80
320
1280
5120
20480
81920
ClustalW-MPIDistVect
DistVect1
400(408)
400(856)
800(454)
2000(266)
4000(247)
1000(858)
500
Exec
utio
n (s
)
Number of sequences (length of sequences)
(9200)
(a)
115
225
335
445
ClustalW-MPIDistVect1DistVectDistVect1
Spee
dup
4000(247)
2000(266)
1000(858)
400(408)
800(454)
400(856)
500
Number of sequences (length of sequences)(9200)
(b)
Figure 4 Performance comparisons using one node and 8 cores
thread level parallelism approach which is based on 119871As a result our program performed better than the threeprograms in all cases and operated smoothly with very longsequences This accomplishment is due to the perfect vec-torization and hybrid partitioning approaches For examplefor comparing set 200(30488) ClustalW-MPI did not workDistVect1 consumed 23467 sec SSE2 exhausted 22949 secand DistVect1 achieved it in 8017 sec
Also the same dataset was tested on 16 nodes Resultsgiven in Figure 6 demonstrate that as the number of nodesincreases the performance upgrades For example comparedto the same set 200(30488) ClustalW-MPI works but slowlytakes 26824 sec DistVect consumed better time 12955 secSSE2 exhausted less period 8166 sec and DistVect1 presentsthe fastest achievement 3226 sec
Finally when tests were performed on 32 nodes as seenin Figure 7 DistVect1rsquos overall performance is typically muchbetter when compared to other programs In fact it exhibitsits superiority when 119871 is in tens of thousands The maximumspeedup for set 20(163354) was up to 9 3 and 2 with respectto the ClustalW-MPI SSE2 and DistVect respectively
To ensure that DistVect1 execution time decreases as thenumber of nodes increases Figure 8 illustrates the relationbetween the execution time and the number of nodes As aconsequence it is concluded that DistVect1 implementationacts almost monotonically increasing whenever the numberof available nodes increases Therefore we can state thatDistVect1 execution time is inversely in proportion to thenumber of nodes
To emphasize that the proposed DistVect1 program ismore efficient than other implemented programs the parallelefficiency in terms of the maximum number of availablecores has been evaluated Results are recorded in Table 2 Itis clear that DistVect1 efficiency is monotonically increasingas the length of sequences increases Thus we can state thatDistVect1 efficiency is directly in proportion to the sequencesrsquolength In addition DistVect1rsquos supreme efficiency was up to029 0086 and 0092 with respect to the ClustalW-MPISSE2 and DistVect respectively for the longest sequencelength (163 k)
53 GCUPS A performance measurement commonly usedin computational biology is billion cell updates per second(GCUPS) A GCUPS represents the time for a completecomputation of one entry in the similarity matrix includingall comparisons additions andmaximumoperationsWehavescanned the datasets with their average length as mentionedabove Our implementation on a cluster of multicores allowshandling sequences up to a length of 163 k Table 3 comparesthe corresponding GCUPS performance values
8 BioMed Research International
128000
64000
32000
16000
8000
4000
2000
1000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
1
15
2
25
3
35
4
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
Number of sequences (length of sequences)
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
(b)
Figure 5 Performance comparisons using 8 nodes
5001000200040008000
160003200064000
128000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
200
(30
488
)
20
(163
354
)
6000
(858
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVectDistVectDistVect1
Spee
dup
6000(858)
3000(858)
2000(1741)
4000(1728)
200(30488)
50(161000)
Number of sequences (length of sequences)
(b)
Figure 6 Performance comparisons using 16 nodes
Table 2 Efficiency comparisons using 32 nodes
Number of sequences (Length of sequence) ClustalW-MPI SSE2 DistVect6000 (858) 01899 00401 006423000 (858) 02025 00414 006252000 (1750) 02087 00436 006774000 (1750) 02061 00449 00696200 (30500) 02578 00881 0091050 (161000) 02730 00823 0091920 (163650) 02901 00863 00919
BioMed Research International 9
250500
1000200040008000
160003200064000
3000
(858
)
2000
(1741
)
200
(30
488
)
6000
(858
)
20
(163
354
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
ClustalW-MPISSE2
DistVectDistVect1
Number of sequences (length of sequences)
Exec
utio
n (s
)
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
500
(30
000
)
Number of sequences (length of sequences)
(b)
Figure 7 Performance comparisons using 32 nodes
01000020000300004000050000600007000080000
Excu
tion
time (
s)
8 nodes16 nodes32 nodes
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
Figure 8 DistVect1 performance against number of nodes
54 Space Reduction Thehuge growth of sequence databasesthat exceed current programsrsquo capability and computer sys-tems capacity gives rise to the importance of consideringspace complexity as an intrinsic performance metric In thefollowing we study the exhausted storage space as a functionof 119871 the sequence length and 119873 the number of sequencesDistVect1 has reduced the overall consumed space by otherprograms in two ways
(i) The compensation of the distance matrix DM by thedistance vector DV reduces space from 119873
2 into ℎ =
119873(119873 minus 1)2 that is almost to the half as shown inTable 4
(ii) The substitution of matrices 119867 and 119873 by vector Vrsquosand three 1198731015840V119904 reduces space from 2 times (119871 + 1)
2 to 6119871as shown in Table 5
55 Load Balancing and Occupancy To study the occupancyof processors and memory real snapshots have been takenas seen in Figure 9 They record the history of CPUs mem-ory and network during execution They demonstrate howdifferent proposed versions of the algorithm improve loadbalancing and memory bandwidth The load unbalancingof DistVect is apparent in Figure 9(a) On the contrary theoptimal load balancing of DistVect1 with the full use of allCPUs by 100 is obvious Also the reduction of memoryoccupancy is very clear in Figure 9(b)
As a conclusion a comparison of DistVect1 proposedmethod with studied GPU and cluster implementationsdiscussed in this paper is illustrated in Table 6 For eachprogram it specifies its platform and records the maximumsequence length that may be handled the highest speedup
10 BioMed Research International
Table 3 Performance comparison (in GCUPS) for scanning the datasets
Number of sequences (Length of sequence) DistVect1 DistVect SSE2 ClustalW-MPI20 (163650) 232 079 084 0252000 (1750) 582 268 417 0874000 (1750) 627 281 436 0953000 (858) 666 333 503 1036000 (858) 669 326 522 110200 (30500) 1169 401 415 14250 (161000) 703 239 267 080500 (30000) 1075 392 385 000
Table 4 Storage Comparisons between DV DM (in MW)
N 20 50 200 500 2000 3000 4000 6000DM 00004 00025 004 025 4 9 16 36DV 000019 0001225 00199 012475 1999 449 799 17998
(a) DistVect
(b) DistVect1
Figure 9 CPUs memory and network occupancy
Table 5 Storage comparisons between119867rsquos 119881rsquos (in MW)
L 858 1728 29414 161000Hrsquos + Nrsquos 1475 597 17348 518V rsquos +119873Vrsquos 0005 001 0176 0966
and GCUPS which can be reached Records certify ourpresumed idea that although GPU programs are very fastwith high GCUPS they cannot align long sequences It alsoproves that our proposal is superlative in handling longsequences with good speedup
The above presented results reveal the best performanceof theDistVect1 against other implementationsThis achieve-ment is due to two main reasons (1) the superiority ofDistVect1 in using vectors instead of matrices leading to abetter caching and (2) the optimal use of multicore clustersystem because of the proposed partitioning and schedul-ing techniques especially when the length of sequencesincreases thus obtaining a higher parallelization factor
6 Conclusion and Future Work
In this paper we presented and evaluatedDistVect1 algorithman improved version of the vectorized parallel algorithmDistVect for efficiently computing the distance matrix usingmulticore cluster systems The new suggested partition-ing and scheduling approaches integrated with MPI andOpenMP primitives have achieved a significant improve-ment to the overall performance Implementations wereconducted on Bibliotheca Alexandria cluster system using 32nodes (8 cores)
The resulting program DistVect1 was able to computedistances between very long sequences up to 163 k It outper-forms SSE2 with 3-fold speedup and ClustalW-MPI 013 with
BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 3
of up to 17 GCUPS on a GeForce GTX 280 and 30 GCUPS ona dual-GPU GeForce GTX 295
Likewise CUDASW++ 30 [21] couples CPU and GPUSIMD instructions and carries out concurrent CPU andGPU computations It employs SSE-based vector executionunits as accelerators and employs CUDA PTX SIMD videoinstructions to gain more data parallelism beyond the SIMTexecution model Evaluation shows that CUDASW++ 30gains a performance improvement over CUDASW++ 20 upto 29 and 32 with a maximum performance of 1190 and1856 GCUPS on a single-GPU GeForce GTX 680 and adual-GPU GeForce GTX 690 graphics card respectively Italso has demonstrated significant speedups over SWIPE andBLAST+ However the longest query sequence was of length5478 to search against the Swiss-Prot protein databases thathave the largest sequence length 35213
However most of GPU implementations cannot alignsequences longer than 59000 residues This is due to theintrinsic SIMD characteristics of the GPG where pipeliningallows a great speedup factor yet intense memory usage maylead to bottlenecks This leads to the deployment of otherarchitectures such as many-cores [23] MC64-ClustalWP2has been developed recently as a new implementation of theClustalW algorithm to align long sequences in architectureswith many cores It runs multiple alignments 18 times fasterthan the original ClustalWalgorithmand can align sequencesthat are relatively long (more than 10 kb)
Authors proposed a vectorized distance matrix computa-tion algorithm called DistVect [8] The algorithm addressesthe problem of building a parallel tool for multicores thatproduces the alignment of multiple sequences in a short timewithout using much storage space The main contributionwas the vectorization of all used matrices in computationExperimentally the proposed method achieved good abil-ity of aligning large number of sequences through pow-erful improved storage handling capabilities with efficientimprovement of the overall processing time
3 DistVect Algorithm
DistVect [8] is an accelerated algorithm that computes thedistance matrix for aligning huge datasets It has the advan-tage of exhausting less space It takes as input 119873 sequences1198781
1198782
119878119873
of average length 119871 with their substitutionmatrix sbt and the gab cost 119892 It outputs a distance vector DVcontaining the similarity score (distance) for each of the twosequences It works on vectorizing matrices presented by Liuet al in [22] and used byWirawan et al in [18] It parallelizesthe computations of resolving vectors taking in account theadvantage of the independence of the elements of the minordiagonals of the matrices
To compute the number of exact matches and make itsuitable for a fine-grained parallel implementation Liu et al[22] formulated a recurrence relation for the number of exactmatch computations that is more suitable for implementation
using a linear gap penalty This formula facilitates the calcu-lations without computation of the actual alignment Giventwo sequences 119878
119894
and 119878119895
of lengths 119871119894
and 119871119895
the distancesare computed using the matrices119873119867 as shown below
DM (119878119894
119878119895
) = 1 minus119873 (119894max 119895max)
min (119871119894
119871119895
)
119873 (119909 119910)
=
0 if 119867(119909 119910) = 0
119873 (119909 minus 1 119910 minus 1)
+119898 (119909 119910) if 119867(119909 119910) = 119867 (119909 minus 1 119910 minus 1)
+sbt (119878119894
(119909) 119878119895
(119910))
119873 (119909 119910 minus 1) if 119867(119909 119910) = 119867 (119909 119910 minus 1) + 119892
119873 (119909 minus 1 119910) if 119867(119909 119910) = 119867 (119909 minus 1 119910) + 119892
119867 (119909 119910) = max
0
119867 (119909 minus 1 119910 minus 1) + sbt (119878119894
(119909) 119878119895
(119910))
119867 (119909 minus 1 119910) + 119892
119867 (119909 119910 minus 1) + 119892
119898 (119909 119910) = 1 119878119894
(119909) = 119878119895
(119910)
0 otherwise(1)
where 119867(119909 0) = 119867(0 119910) = 119873(119909 0) = 119873(0 119910) = 0 and 1 le
119909 le 119871119894
1 le 119910 le 119871119895
In [8] the authors proved mathematically that the above
equations can be transformed to the following equationsusing vectors only
DV (ℎ) = 1 minus119896max
min (119871119894
119871119895
)
119873119909
V (119896)
=
0 if 119881119909 (119896) = 0
119873119909
1
(119896 minus 1)
+119898 (119896) if 119881119909 (119896) = 119881119909
1
(119896 minus 1)
+sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119873119909
2
(119896) if 119881119909 (119896) = 119881119909
2
(119896) + 119892
119873119909
2
(119896 minus 1) if 119881119909 (119896) = 119881119909
2
(119896 minus 1) + 119892
119881119909
(119896)
= max
0
119881119909
1
(119896 minus 1) + sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119881119909
2
(119896 minus 1) + 119892
119881119909
2
(119896) + 119892
119898 (119896) = 1 119878119894
(119896) = 119878119895
(119909 minus 119896 + 1)
0 otherwise(2)
4 BioMed Research International
where 119896max is the highest score of 119873 which indicates thenumber of exact matches in the optimal local alignment 1 le
ℎ le lceil119873(119873minus 1)2rceil max(2 119909 minus 119871) le 119896 le min((119909 + 1) (119871 + 1))1 le 119909 le (2119871 minus 1) 119871 is average lengths and 119873
1
1198732
are theexact match values in each iteration
The main idea was based on the fact that any element inan antidiagonal of119867 needs only the values of three elementsfrom the previous two antidiagonalsThis leads to the fact thatonly three vectors could be sufficient One vector119881 is used asa current vector and the previously calculated vectors119881
1
and1198812
as buffers Also three vectors 119873V 1198731 and 1198732
substitutematrix 119873 Finally the distance matrix DM is replaced by adistance vector DV EachDV element is evaluated in terms ofthe highest score 119896max Figure 1 showsDistVectmethodologywhen aligning two sample sequences of length 8
4 Proposed Algorithm
Although DistVect implementation on a multicore usingOpenMp and multithreading has achieved good speedupcompared to ClustalW-MPI [17] it shows load unbalancingTo overcome this problem we propose using a better compu-tation partitioning technique and accompanying extensionsto operate on recent available clusters system that offers morepower to parallelism This is done in order to enhance theperformance In this section an improved version ofDistVectis proposed
DistVect computes the similarity scores using only threevectors instead of the matrix DM using fine-grained paral-lelism and multithreading It distributes all cellsrsquo computa-tions of the antidiagonal vector119881 over the available 119875 coresThe value of each cell is evaluated in terms of its diagonalneighbor stored at119881
1
with its left and upper neighbors storedat 1198812
and the maximum value is selected indicating thehighest score (see Figure 1)
When analyzing the data dependencies between DMcomputations it appears that it may be decomposed throughthree distinct approaches
(i) First the fine-grain approach like multithreadedClustalW [16] is the one in which the computationsof each similarity matrix are decomposedThe imple-mentation of this approach in our work [8] showshigh sensitivity to the length of sequences and animbalanced workload among CPU cores
(ii) Second the coarse-grain approach partitionsDM intoblocks (tiles) and assigns the computation of each oneto a node as in [17 24] However this approach isgood only when the number of sequences is too largecompared to their length and vice versa
(iii) Third the hybrid approach (which is used in theproposed algorithm) combines the two previousapproaches to benefit from the strong points of bothof them
As mentioned above DistVect algorithm faces some per-formance challenges when applying the fine-grain approachThe reason is that the size of the vector 119881 varies during thecomputation and the number of cells to be computed at each
0 0 0 0 0 0 0 0
0
0
0
0
0
0
0
0
V1
V2
V
Deleted
Figure 1 DistVect alignment using fine-grain approach
0
0 0 0 0 0 0 0 0
0
0
0
0
0
0
0
V1
V2
V
Deleted
Figure 2 Distance computation using cyclic partitioning
iteration step is not the same Thus when we distribute thecells on the diagonal to 119875 cores we notice that the workloadis not well balanced among coresThat is because the numberof cells is not always divisible by 119875 Therefore at each stepsome cores have idle time leading to work overhead on othercores Consider Figure 1 at iteration 1 (|119881| = 1) only core 1works at iteration 2 (|119881| = 2) only cores 1 and 2 work Whileat iterations 6 and 9 (|119881| = 6) cores 1 and 2 compute two cellscores 3 and 4 compute one cell only Therefore not all corescompute the same number of cells
On the other hand we think this computation modelrequires a large number of cores for aligning real biologicaldata That is because our main contribution is based onthe acceleration of similarity score computation which is afunction of the sequencesrsquo length 119871 And we concentrate ondealing with long sequences but each core calculates oneelement of119881 at a timeThus as the number of cores increases
BioMed Research International 5
the parallelization factor will increase leading to a higheracceleration
Figure 2 demonstrates our proposed cyclic partitioningtechnique when handling the two sequences considered atFigure 1 with 4 cores The four cores act in parallel on each ofthe first four consecutive rows then they sequentially act onthe second consequent 4 rows
Furthermore all lceil119873(119873minus1)2rceil distance computations arepartitioned into119872 splits and distributed over the119872 nodes inthe cluster system All 119872 nodes work in parallel Each nodewill operate sequentially on aligning the set of lceil119873(119873minus1)2119872rceil
pairs at its own split whose size is 119899split Figure 3 explains howthe alignments of 4 sequences of length 8 are scheduled over acluster system of 2 nodes each node has 4 cores In this casethere are 2 splits each split is of size 3
The pseudocode of this proposed method is depicted inAlgorithm 1 And the distance vector DV is computed usingthe following modified recurrences
119881119909
(119896)
= max
0
119881119909
1
(119896 minus 1) + sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119881119909
2
(119896 minus 1) + 119892
119881119909
2
(119896) + 119892
(3)
119873119909
V (119896)
=
0 if 119881119909 (119896) = 0
119873119909
1
(119896 minus 1) + 119898 (119896) if 119881119909 (119896)= 119881119909
1
(119896 minus 1)
+sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119873119909
2
(119896) if 119881119909 (119896) = 119881119909
2
(119896) + 119892
119873119909
2
(119896 minus 1) if 119881119909 (119896) = 119881119909
2
(119896 minus 1) + 119892
(4)
where 1 le 119896 le 119875
5 Performance Evaluation
The proposed algorithm DistVect1 has been implementedin C++ using OpenMP and MPI libraries The introducedhybrid paradigm matches the characteristics of a cluster ofmulticore system very well The resulting program has theadvantage of coarse-grained parallelism on process level inwhich each MPI process is executed on one multicore nodeAlso it has the advantage of fine-grained parallelismon a looplevel in which each MPI process spawns a team of threads tooccupy the multicore processors when encountering parallelsections of code using OpenMP However the obtainedresults should be carefully analyzed in order to realize the setof items that interferewith the established comparisonmodel
51 Experimental Setup The presented program has beenimplemented on Sun Microsystems cluster provided byLinkSCEEM-2 systems at Bibliotheca Alexandrina Egypt
This platform specification is as follows
(i) 130 nodes and 64GB memory (the allowed numberof nodes for one user is 32 nodes) each node containstwo Intel quad core Xeon 283GHz processors (64-bittechnology)
(ii) 8 Gbyte RAM 80 Gbyte hard disk and a dual port infin band (10Gbps)
(iii) a Giga Ethernet Network port the operating systemis 64-bit Linux
Real protein sequences of different lengths were used forcomputation They are available online at NCBI [25] Theycomprise a subset of variant family of viruses Each usedbenchmark dataset with the average sequence length and thestandard deviation is given in Table 1The standard deviationgives us a numerical measure of the scatter of a dataset Thismeasure is useful for making comparisons between datasetsthat go beyond simple visual impressions
Comprehensive experiments have been conducted onthe specified platform using different groups of sequencesselected from the indicated datasets They evaluated theimplementation of DistVect1 versus that of DistVect and twopopular efficient programs
(i) ClustalW-MPI available at httpwwwmybiosoft-warecomalignment3052
(ii) SSE available at httpssourceforgenetprojectsdist-matcomp
For SSE2 program we have used the SSE approachwithin each MPI process after distributing the overall work-load using the proposed scheduling technique as clarifiedat Figure 3 Thus each node applies its multithreadingapproach on its assigned matrices one at a time Biblio-theca Alexandria used platform support Advanced VectorExtensions (AVX) which is used during implementing SSE2code
52 Execution Time Speedup and Efficiency In this subsec-tion three common performance measurements the execu-tion time the speedup and the efficiency are used Parallelexecution time represents the elapsed time for completecomputation of the distance matrix including all additionscomparisons and maximum operations Parallel speedup isevaluated by the ratio between the parallel execution time ofthe two involved programs Parallel efficiency is computed bydividing the corresponding parallel speedups by the numberof cores Results for variable number of available nodes withtheir analysis are presented below
As a start experiments were executed on a single nodewith 8 cores Since the amount of computations requiredfor a sequence depends on its length multiple lengths ofsequences fromHIV Coronaviridae andHAhaemagglutininwere tested The execution time (in seconds) of the pro-posed DistVect1 implementation against ClustalW-MPI andDistVect was recorded Figure 4(a) shows how the perfor-mance of N(L) is affected by the sequencesrsquo number 119873 and
6 BioMed Research International
Node 1 Node 2
Core 0
Core 1
Core 2
Core 3
Core 0
Core 1
Core 2
Core 3
Node interconnect
0 0 0 0 0 0 0 00 0 0 0 0 0 0 0
00000000
00000000
S1 times S4
S1 times S3
S1 times S2
S2 times S4
S3 times S4
S2 times S3
Figure 3 Proposed scheduling on multicore cluster
Input Number of sequences N average sequencesrsquo length L sequencesrsquo dataset119878 = 119878
1
1198782
119878119873
substitution matrix sbt gab cost g Number of Nodes MNumber of cores POutput Distance Vector DV(1) Let ℎ = 0
(2) Let 119899119878119901119897119894119905
larr [119873(119873 minus 1)2119872]
(3) ⊳ Distributing splits overM nodes(4) for119873119900119889119890119904 larr 1119872 do in parallel(5) Identify Split(6) for119875119886119894119903119904 larr 1 119899
119878119901119897119894119905
do(7) Get 119878
119894
and 119878119895
from Split(8) Initialize 1198811
1
1198812
1
1198731
1
and1198732
1
(9) for119909 larr 1 119871 + 119875 do(10) ⊳ Distributing distance computation over P cores(11) for 119896 larr 1 119875 do in parallel(12) Compute 119881119909(119896) using (3)(13) if 119878
119894
(119896) = 119878119895
(119909 minus 119896 + 1) then Let119898(119896) = 1
(14) else Let119898(119896) = 0
(15) end if(16) Compute119873119909V (119896) using (4)(17) end for 119896(18) Let 119881119909
1
= 119881119909
2
and 119881119909
2
= 119881119909
(19) Let1198731199091
= 119873119909
2
and119873119909
2
= 119873119909
(20) end for119909(21) Identify 119896max(22) Let DV(ℎ) = 1 minus (119896max119871)
(23) Increment ℎ(24) end for Pairs(25) end forNodes(26) return DV
Algorithm 1 DistVect1 Hybrid DistVect Algorithm
length 119871 DistVect1 shows slightly higher performance thanthat of the two other programswhen119871 ranges in the hundredsand119873 ranges in the thousands Further it presented superiorperformance when 119873 is small compared to 119871 and 119871 exceedsa thousand As shown in Figure 4(b) the maximum achievedspeedups when comparing set 500(9200) were (41) and(25)
Next variant subsets of the specified datasetwith differentsizes were tested on 8 nodes Figure 5 shows that ClustalW-MPI could not handle long sequenceswith119871 overriding thirtythousand That is because ClustalW-MPI approach stronglydepends on the number of sequences and focuses on thedistribution of sequences across nodes without regardingtheir length Also DistVect and SSE2 concentrate on the
BioMed Research International 7
Table 1 Used benchmark dataset specifications
Benchmark Number of sequences Average length Standard deviation
Human immunodeficiency virus (HIV) 3000 858 107456000 858 14632
HA hemagglutinin (influenza B virus) 2000 1741 326044000 1728 36661
Several viruses within the Coronaviridae family 200 30488 908534500 29414 1066417
Herpesviridae (large family of DNA viruses) 20 163654 473303250 161000 4289981
20
80
320
1280
5120
20480
81920
ClustalW-MPIDistVect
DistVect1
400(408)
400(856)
800(454)
2000(266)
4000(247)
1000(858)
500
Exec
utio
n (s
)
Number of sequences (length of sequences)
(9200)
(a)
115
225
335
445
ClustalW-MPIDistVect1DistVectDistVect1
Spee
dup
4000(247)
2000(266)
1000(858)
400(408)
800(454)
400(856)
500
Number of sequences (length of sequences)(9200)
(b)
Figure 4 Performance comparisons using one node and 8 cores
thread level parallelism approach which is based on 119871As a result our program performed better than the threeprograms in all cases and operated smoothly with very longsequences This accomplishment is due to the perfect vec-torization and hybrid partitioning approaches For examplefor comparing set 200(30488) ClustalW-MPI did not workDistVect1 consumed 23467 sec SSE2 exhausted 22949 secand DistVect1 achieved it in 8017 sec
Also the same dataset was tested on 16 nodes Resultsgiven in Figure 6 demonstrate that as the number of nodesincreases the performance upgrades For example comparedto the same set 200(30488) ClustalW-MPI works but slowlytakes 26824 sec DistVect consumed better time 12955 secSSE2 exhausted less period 8166 sec and DistVect1 presentsthe fastest achievement 3226 sec
Finally when tests were performed on 32 nodes as seenin Figure 7 DistVect1rsquos overall performance is typically muchbetter when compared to other programs In fact it exhibitsits superiority when 119871 is in tens of thousands The maximumspeedup for set 20(163354) was up to 9 3 and 2 with respectto the ClustalW-MPI SSE2 and DistVect respectively
To ensure that DistVect1 execution time decreases as thenumber of nodes increases Figure 8 illustrates the relationbetween the execution time and the number of nodes As aconsequence it is concluded that DistVect1 implementationacts almost monotonically increasing whenever the numberof available nodes increases Therefore we can state thatDistVect1 execution time is inversely in proportion to thenumber of nodes
To emphasize that the proposed DistVect1 program ismore efficient than other implemented programs the parallelefficiency in terms of the maximum number of availablecores has been evaluated Results are recorded in Table 2 Itis clear that DistVect1 efficiency is monotonically increasingas the length of sequences increases Thus we can state thatDistVect1 efficiency is directly in proportion to the sequencesrsquolength In addition DistVect1rsquos supreme efficiency was up to029 0086 and 0092 with respect to the ClustalW-MPISSE2 and DistVect respectively for the longest sequencelength (163 k)
53 GCUPS A performance measurement commonly usedin computational biology is billion cell updates per second(GCUPS) A GCUPS represents the time for a completecomputation of one entry in the similarity matrix includingall comparisons additions andmaximumoperationsWehavescanned the datasets with their average length as mentionedabove Our implementation on a cluster of multicores allowshandling sequences up to a length of 163 k Table 3 comparesthe corresponding GCUPS performance values
8 BioMed Research International
128000
64000
32000
16000
8000
4000
2000
1000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
1
15
2
25
3
35
4
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
Number of sequences (length of sequences)
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
(b)
Figure 5 Performance comparisons using 8 nodes
5001000200040008000
160003200064000
128000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
200
(30
488
)
20
(163
354
)
6000
(858
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVectDistVectDistVect1
Spee
dup
6000(858)
3000(858)
2000(1741)
4000(1728)
200(30488)
50(161000)
Number of sequences (length of sequences)
(b)
Figure 6 Performance comparisons using 16 nodes
Table 2 Efficiency comparisons using 32 nodes
Number of sequences (Length of sequence) ClustalW-MPI SSE2 DistVect6000 (858) 01899 00401 006423000 (858) 02025 00414 006252000 (1750) 02087 00436 006774000 (1750) 02061 00449 00696200 (30500) 02578 00881 0091050 (161000) 02730 00823 0091920 (163650) 02901 00863 00919
BioMed Research International 9
250500
1000200040008000
160003200064000
3000
(858
)
2000
(1741
)
200
(30
488
)
6000
(858
)
20
(163
354
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
ClustalW-MPISSE2
DistVectDistVect1
Number of sequences (length of sequences)
Exec
utio
n (s
)
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
500
(30
000
)
Number of sequences (length of sequences)
(b)
Figure 7 Performance comparisons using 32 nodes
01000020000300004000050000600007000080000
Excu
tion
time (
s)
8 nodes16 nodes32 nodes
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
Figure 8 DistVect1 performance against number of nodes
54 Space Reduction Thehuge growth of sequence databasesthat exceed current programsrsquo capability and computer sys-tems capacity gives rise to the importance of consideringspace complexity as an intrinsic performance metric In thefollowing we study the exhausted storage space as a functionof 119871 the sequence length and 119873 the number of sequencesDistVect1 has reduced the overall consumed space by otherprograms in two ways
(i) The compensation of the distance matrix DM by thedistance vector DV reduces space from 119873
2 into ℎ =
119873(119873 minus 1)2 that is almost to the half as shown inTable 4
(ii) The substitution of matrices 119867 and 119873 by vector Vrsquosand three 1198731015840V119904 reduces space from 2 times (119871 + 1)
2 to 6119871as shown in Table 5
55 Load Balancing and Occupancy To study the occupancyof processors and memory real snapshots have been takenas seen in Figure 9 They record the history of CPUs mem-ory and network during execution They demonstrate howdifferent proposed versions of the algorithm improve loadbalancing and memory bandwidth The load unbalancingof DistVect is apparent in Figure 9(a) On the contrary theoptimal load balancing of DistVect1 with the full use of allCPUs by 100 is obvious Also the reduction of memoryoccupancy is very clear in Figure 9(b)
As a conclusion a comparison of DistVect1 proposedmethod with studied GPU and cluster implementationsdiscussed in this paper is illustrated in Table 6 For eachprogram it specifies its platform and records the maximumsequence length that may be handled the highest speedup
10 BioMed Research International
Table 3 Performance comparison (in GCUPS) for scanning the datasets
Number of sequences (Length of sequence) DistVect1 DistVect SSE2 ClustalW-MPI20 (163650) 232 079 084 0252000 (1750) 582 268 417 0874000 (1750) 627 281 436 0953000 (858) 666 333 503 1036000 (858) 669 326 522 110200 (30500) 1169 401 415 14250 (161000) 703 239 267 080500 (30000) 1075 392 385 000
Table 4 Storage Comparisons between DV DM (in MW)
N 20 50 200 500 2000 3000 4000 6000DM 00004 00025 004 025 4 9 16 36DV 000019 0001225 00199 012475 1999 449 799 17998
(a) DistVect
(b) DistVect1
Figure 9 CPUs memory and network occupancy
Table 5 Storage comparisons between119867rsquos 119881rsquos (in MW)
L 858 1728 29414 161000Hrsquos + Nrsquos 1475 597 17348 518V rsquos +119873Vrsquos 0005 001 0176 0966
and GCUPS which can be reached Records certify ourpresumed idea that although GPU programs are very fastwith high GCUPS they cannot align long sequences It alsoproves that our proposal is superlative in handling longsequences with good speedup
The above presented results reveal the best performanceof theDistVect1 against other implementationsThis achieve-ment is due to two main reasons (1) the superiority ofDistVect1 in using vectors instead of matrices leading to abetter caching and (2) the optimal use of multicore clustersystem because of the proposed partitioning and schedul-ing techniques especially when the length of sequencesincreases thus obtaining a higher parallelization factor
6 Conclusion and Future Work
In this paper we presented and evaluatedDistVect1 algorithman improved version of the vectorized parallel algorithmDistVect for efficiently computing the distance matrix usingmulticore cluster systems The new suggested partition-ing and scheduling approaches integrated with MPI andOpenMP primitives have achieved a significant improve-ment to the overall performance Implementations wereconducted on Bibliotheca Alexandria cluster system using 32nodes (8 cores)
The resulting program DistVect1 was able to computedistances between very long sequences up to 163 k It outper-forms SSE2 with 3-fold speedup and ClustalW-MPI 013 with
BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

4 BioMed Research International
where 119896max is the highest score of 119873 which indicates thenumber of exact matches in the optimal local alignment 1 le
ℎ le lceil119873(119873minus 1)2rceil max(2 119909 minus 119871) le 119896 le min((119909 + 1) (119871 + 1))1 le 119909 le (2119871 minus 1) 119871 is average lengths and 119873
1
1198732
are theexact match values in each iteration
The main idea was based on the fact that any element inan antidiagonal of119867 needs only the values of three elementsfrom the previous two antidiagonalsThis leads to the fact thatonly three vectors could be sufficient One vector119881 is used asa current vector and the previously calculated vectors119881
1
and1198812
as buffers Also three vectors 119873V 1198731 and 1198732
substitutematrix 119873 Finally the distance matrix DM is replaced by adistance vector DV EachDV element is evaluated in terms ofthe highest score 119896max Figure 1 showsDistVectmethodologywhen aligning two sample sequences of length 8
4 Proposed Algorithm
Although DistVect implementation on a multicore usingOpenMp and multithreading has achieved good speedupcompared to ClustalW-MPI [17] it shows load unbalancingTo overcome this problem we propose using a better compu-tation partitioning technique and accompanying extensionsto operate on recent available clusters system that offers morepower to parallelism This is done in order to enhance theperformance In this section an improved version ofDistVectis proposed
DistVect computes the similarity scores using only threevectors instead of the matrix DM using fine-grained paral-lelism and multithreading It distributes all cellsrsquo computa-tions of the antidiagonal vector119881 over the available 119875 coresThe value of each cell is evaluated in terms of its diagonalneighbor stored at119881
1
with its left and upper neighbors storedat 1198812
and the maximum value is selected indicating thehighest score (see Figure 1)
When analyzing the data dependencies between DMcomputations it appears that it may be decomposed throughthree distinct approaches
(i) First the fine-grain approach like multithreadedClustalW [16] is the one in which the computationsof each similarity matrix are decomposedThe imple-mentation of this approach in our work [8] showshigh sensitivity to the length of sequences and animbalanced workload among CPU cores
(ii) Second the coarse-grain approach partitionsDM intoblocks (tiles) and assigns the computation of each oneto a node as in [17 24] However this approach isgood only when the number of sequences is too largecompared to their length and vice versa
(iii) Third the hybrid approach (which is used in theproposed algorithm) combines the two previousapproaches to benefit from the strong points of bothof them
As mentioned above DistVect algorithm faces some per-formance challenges when applying the fine-grain approachThe reason is that the size of the vector 119881 varies during thecomputation and the number of cells to be computed at each
0 0 0 0 0 0 0 0
0
0
0
0
0
0
0
0
V1
V2
V
Deleted
Figure 1 DistVect alignment using fine-grain approach
0
0 0 0 0 0 0 0 0
0
0
0
0
0
0
0
V1
V2
V
Deleted
Figure 2 Distance computation using cyclic partitioning
iteration step is not the same Thus when we distribute thecells on the diagonal to 119875 cores we notice that the workloadis not well balanced among coresThat is because the numberof cells is not always divisible by 119875 Therefore at each stepsome cores have idle time leading to work overhead on othercores Consider Figure 1 at iteration 1 (|119881| = 1) only core 1works at iteration 2 (|119881| = 2) only cores 1 and 2 work Whileat iterations 6 and 9 (|119881| = 6) cores 1 and 2 compute two cellscores 3 and 4 compute one cell only Therefore not all corescompute the same number of cells
On the other hand we think this computation modelrequires a large number of cores for aligning real biologicaldata That is because our main contribution is based onthe acceleration of similarity score computation which is afunction of the sequencesrsquo length 119871 And we concentrate ondealing with long sequences but each core calculates oneelement of119881 at a timeThus as the number of cores increases
BioMed Research International 5
the parallelization factor will increase leading to a higheracceleration
Figure 2 demonstrates our proposed cyclic partitioningtechnique when handling the two sequences considered atFigure 1 with 4 cores The four cores act in parallel on each ofthe first four consecutive rows then they sequentially act onthe second consequent 4 rows
Furthermore all lceil119873(119873minus1)2rceil distance computations arepartitioned into119872 splits and distributed over the119872 nodes inthe cluster system All 119872 nodes work in parallel Each nodewill operate sequentially on aligning the set of lceil119873(119873minus1)2119872rceil
pairs at its own split whose size is 119899split Figure 3 explains howthe alignments of 4 sequences of length 8 are scheduled over acluster system of 2 nodes each node has 4 cores In this casethere are 2 splits each split is of size 3
The pseudocode of this proposed method is depicted inAlgorithm 1 And the distance vector DV is computed usingthe following modified recurrences
119881119909
(119896)
= max
0
119881119909
1
(119896 minus 1) + sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119881119909
2
(119896 minus 1) + 119892
119881119909
2
(119896) + 119892
(3)
119873119909
V (119896)
=
0 if 119881119909 (119896) = 0
119873119909
1
(119896 minus 1) + 119898 (119896) if 119881119909 (119896)= 119881119909
1
(119896 minus 1)
+sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119873119909
2
(119896) if 119881119909 (119896) = 119881119909
2
(119896) + 119892
119873119909
2
(119896 minus 1) if 119881119909 (119896) = 119881119909
2
(119896 minus 1) + 119892
(4)
where 1 le 119896 le 119875
5 Performance Evaluation
The proposed algorithm DistVect1 has been implementedin C++ using OpenMP and MPI libraries The introducedhybrid paradigm matches the characteristics of a cluster ofmulticore system very well The resulting program has theadvantage of coarse-grained parallelism on process level inwhich each MPI process is executed on one multicore nodeAlso it has the advantage of fine-grained parallelismon a looplevel in which each MPI process spawns a team of threads tooccupy the multicore processors when encountering parallelsections of code using OpenMP However the obtainedresults should be carefully analyzed in order to realize the setof items that interferewith the established comparisonmodel
51 Experimental Setup The presented program has beenimplemented on Sun Microsystems cluster provided byLinkSCEEM-2 systems at Bibliotheca Alexandrina Egypt
This platform specification is as follows
(i) 130 nodes and 64GB memory (the allowed numberof nodes for one user is 32 nodes) each node containstwo Intel quad core Xeon 283GHz processors (64-bittechnology)
(ii) 8 Gbyte RAM 80 Gbyte hard disk and a dual port infin band (10Gbps)
(iii) a Giga Ethernet Network port the operating systemis 64-bit Linux
Real protein sequences of different lengths were used forcomputation They are available online at NCBI [25] Theycomprise a subset of variant family of viruses Each usedbenchmark dataset with the average sequence length and thestandard deviation is given in Table 1The standard deviationgives us a numerical measure of the scatter of a dataset Thismeasure is useful for making comparisons between datasetsthat go beyond simple visual impressions
Comprehensive experiments have been conducted onthe specified platform using different groups of sequencesselected from the indicated datasets They evaluated theimplementation of DistVect1 versus that of DistVect and twopopular efficient programs
(i) ClustalW-MPI available at httpwwwmybiosoft-warecomalignment3052
(ii) SSE available at httpssourceforgenetprojectsdist-matcomp
For SSE2 program we have used the SSE approachwithin each MPI process after distributing the overall work-load using the proposed scheduling technique as clarifiedat Figure 3 Thus each node applies its multithreadingapproach on its assigned matrices one at a time Biblio-theca Alexandria used platform support Advanced VectorExtensions (AVX) which is used during implementing SSE2code
52 Execution Time Speedup and Efficiency In this subsec-tion three common performance measurements the execu-tion time the speedup and the efficiency are used Parallelexecution time represents the elapsed time for completecomputation of the distance matrix including all additionscomparisons and maximum operations Parallel speedup isevaluated by the ratio between the parallel execution time ofthe two involved programs Parallel efficiency is computed bydividing the corresponding parallel speedups by the numberof cores Results for variable number of available nodes withtheir analysis are presented below
As a start experiments were executed on a single nodewith 8 cores Since the amount of computations requiredfor a sequence depends on its length multiple lengths ofsequences fromHIV Coronaviridae andHAhaemagglutininwere tested The execution time (in seconds) of the pro-posed DistVect1 implementation against ClustalW-MPI andDistVect was recorded Figure 4(a) shows how the perfor-mance of N(L) is affected by the sequencesrsquo number 119873 and
6 BioMed Research International
Node 1 Node 2
Core 0
Core 1
Core 2
Core 3
Core 0
Core 1
Core 2
Core 3
Node interconnect
0 0 0 0 0 0 0 00 0 0 0 0 0 0 0
00000000
00000000
S1 times S4
S1 times S3
S1 times S2
S2 times S4
S3 times S4
S2 times S3
Figure 3 Proposed scheduling on multicore cluster
Input Number of sequences N average sequencesrsquo length L sequencesrsquo dataset119878 = 119878
1
1198782
119878119873
substitution matrix sbt gab cost g Number of Nodes MNumber of cores POutput Distance Vector DV(1) Let ℎ = 0
(2) Let 119899119878119901119897119894119905
larr [119873(119873 minus 1)2119872]
(3) ⊳ Distributing splits overM nodes(4) for119873119900119889119890119904 larr 1119872 do in parallel(5) Identify Split(6) for119875119886119894119903119904 larr 1 119899
119878119901119897119894119905
do(7) Get 119878
119894
and 119878119895
from Split(8) Initialize 1198811
1
1198812
1
1198731
1
and1198732
1
(9) for119909 larr 1 119871 + 119875 do(10) ⊳ Distributing distance computation over P cores(11) for 119896 larr 1 119875 do in parallel(12) Compute 119881119909(119896) using (3)(13) if 119878
119894
(119896) = 119878119895
(119909 minus 119896 + 1) then Let119898(119896) = 1
(14) else Let119898(119896) = 0
(15) end if(16) Compute119873119909V (119896) using (4)(17) end for 119896(18) Let 119881119909
1
= 119881119909
2
and 119881119909
2
= 119881119909
(19) Let1198731199091
= 119873119909
2
and119873119909
2
= 119873119909
(20) end for119909(21) Identify 119896max(22) Let DV(ℎ) = 1 minus (119896max119871)
(23) Increment ℎ(24) end for Pairs(25) end forNodes(26) return DV
Algorithm 1 DistVect1 Hybrid DistVect Algorithm
length 119871 DistVect1 shows slightly higher performance thanthat of the two other programswhen119871 ranges in the hundredsand119873 ranges in the thousands Further it presented superiorperformance when 119873 is small compared to 119871 and 119871 exceedsa thousand As shown in Figure 4(b) the maximum achievedspeedups when comparing set 500(9200) were (41) and(25)
Next variant subsets of the specified datasetwith differentsizes were tested on 8 nodes Figure 5 shows that ClustalW-MPI could not handle long sequenceswith119871 overriding thirtythousand That is because ClustalW-MPI approach stronglydepends on the number of sequences and focuses on thedistribution of sequences across nodes without regardingtheir length Also DistVect and SSE2 concentrate on the
BioMed Research International 7
Table 1 Used benchmark dataset specifications
Benchmark Number of sequences Average length Standard deviation
Human immunodeficiency virus (HIV) 3000 858 107456000 858 14632
HA hemagglutinin (influenza B virus) 2000 1741 326044000 1728 36661
Several viruses within the Coronaviridae family 200 30488 908534500 29414 1066417
Herpesviridae (large family of DNA viruses) 20 163654 473303250 161000 4289981
20
80
320
1280
5120
20480
81920
ClustalW-MPIDistVect
DistVect1
400(408)
400(856)
800(454)
2000(266)
4000(247)
1000(858)
500
Exec
utio
n (s
)
Number of sequences (length of sequences)
(9200)
(a)
115
225
335
445
ClustalW-MPIDistVect1DistVectDistVect1
Spee
dup
4000(247)
2000(266)
1000(858)
400(408)
800(454)
400(856)
500
Number of sequences (length of sequences)(9200)
(b)
Figure 4 Performance comparisons using one node and 8 cores
thread level parallelism approach which is based on 119871As a result our program performed better than the threeprograms in all cases and operated smoothly with very longsequences This accomplishment is due to the perfect vec-torization and hybrid partitioning approaches For examplefor comparing set 200(30488) ClustalW-MPI did not workDistVect1 consumed 23467 sec SSE2 exhausted 22949 secand DistVect1 achieved it in 8017 sec
Also the same dataset was tested on 16 nodes Resultsgiven in Figure 6 demonstrate that as the number of nodesincreases the performance upgrades For example comparedto the same set 200(30488) ClustalW-MPI works but slowlytakes 26824 sec DistVect consumed better time 12955 secSSE2 exhausted less period 8166 sec and DistVect1 presentsthe fastest achievement 3226 sec
Finally when tests were performed on 32 nodes as seenin Figure 7 DistVect1rsquos overall performance is typically muchbetter when compared to other programs In fact it exhibitsits superiority when 119871 is in tens of thousands The maximumspeedup for set 20(163354) was up to 9 3 and 2 with respectto the ClustalW-MPI SSE2 and DistVect respectively
To ensure that DistVect1 execution time decreases as thenumber of nodes increases Figure 8 illustrates the relationbetween the execution time and the number of nodes As aconsequence it is concluded that DistVect1 implementationacts almost monotonically increasing whenever the numberof available nodes increases Therefore we can state thatDistVect1 execution time is inversely in proportion to thenumber of nodes
To emphasize that the proposed DistVect1 program ismore efficient than other implemented programs the parallelefficiency in terms of the maximum number of availablecores has been evaluated Results are recorded in Table 2 Itis clear that DistVect1 efficiency is monotonically increasingas the length of sequences increases Thus we can state thatDistVect1 efficiency is directly in proportion to the sequencesrsquolength In addition DistVect1rsquos supreme efficiency was up to029 0086 and 0092 with respect to the ClustalW-MPISSE2 and DistVect respectively for the longest sequencelength (163 k)
53 GCUPS A performance measurement commonly usedin computational biology is billion cell updates per second(GCUPS) A GCUPS represents the time for a completecomputation of one entry in the similarity matrix includingall comparisons additions andmaximumoperationsWehavescanned the datasets with their average length as mentionedabove Our implementation on a cluster of multicores allowshandling sequences up to a length of 163 k Table 3 comparesthe corresponding GCUPS performance values
8 BioMed Research International
128000
64000
32000
16000
8000
4000
2000
1000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
1
15
2
25
3
35
4
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
Number of sequences (length of sequences)
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
(b)
Figure 5 Performance comparisons using 8 nodes
5001000200040008000
160003200064000
128000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
200
(30
488
)
20
(163
354
)
6000
(858
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVectDistVectDistVect1
Spee
dup
6000(858)
3000(858)
2000(1741)
4000(1728)
200(30488)
50(161000)
Number of sequences (length of sequences)
(b)
Figure 6 Performance comparisons using 16 nodes
Table 2 Efficiency comparisons using 32 nodes
Number of sequences (Length of sequence) ClustalW-MPI SSE2 DistVect6000 (858) 01899 00401 006423000 (858) 02025 00414 006252000 (1750) 02087 00436 006774000 (1750) 02061 00449 00696200 (30500) 02578 00881 0091050 (161000) 02730 00823 0091920 (163650) 02901 00863 00919
BioMed Research International 9
250500
1000200040008000
160003200064000
3000
(858
)
2000
(1741
)
200
(30
488
)
6000
(858
)
20
(163
354
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
ClustalW-MPISSE2
DistVectDistVect1
Number of sequences (length of sequences)
Exec
utio
n (s
)
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
500
(30
000
)
Number of sequences (length of sequences)
(b)
Figure 7 Performance comparisons using 32 nodes
01000020000300004000050000600007000080000
Excu
tion
time (
s)
8 nodes16 nodes32 nodes
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
Figure 8 DistVect1 performance against number of nodes
54 Space Reduction Thehuge growth of sequence databasesthat exceed current programsrsquo capability and computer sys-tems capacity gives rise to the importance of consideringspace complexity as an intrinsic performance metric In thefollowing we study the exhausted storage space as a functionof 119871 the sequence length and 119873 the number of sequencesDistVect1 has reduced the overall consumed space by otherprograms in two ways
(i) The compensation of the distance matrix DM by thedistance vector DV reduces space from 119873
2 into ℎ =
119873(119873 minus 1)2 that is almost to the half as shown inTable 4
(ii) The substitution of matrices 119867 and 119873 by vector Vrsquosand three 1198731015840V119904 reduces space from 2 times (119871 + 1)
2 to 6119871as shown in Table 5
55 Load Balancing and Occupancy To study the occupancyof processors and memory real snapshots have been takenas seen in Figure 9 They record the history of CPUs mem-ory and network during execution They demonstrate howdifferent proposed versions of the algorithm improve loadbalancing and memory bandwidth The load unbalancingof DistVect is apparent in Figure 9(a) On the contrary theoptimal load balancing of DistVect1 with the full use of allCPUs by 100 is obvious Also the reduction of memoryoccupancy is very clear in Figure 9(b)
As a conclusion a comparison of DistVect1 proposedmethod with studied GPU and cluster implementationsdiscussed in this paper is illustrated in Table 6 For eachprogram it specifies its platform and records the maximumsequence length that may be handled the highest speedup
10 BioMed Research International
Table 3 Performance comparison (in GCUPS) for scanning the datasets
Number of sequences (Length of sequence) DistVect1 DistVect SSE2 ClustalW-MPI20 (163650) 232 079 084 0252000 (1750) 582 268 417 0874000 (1750) 627 281 436 0953000 (858) 666 333 503 1036000 (858) 669 326 522 110200 (30500) 1169 401 415 14250 (161000) 703 239 267 080500 (30000) 1075 392 385 000
Table 4 Storage Comparisons between DV DM (in MW)
N 20 50 200 500 2000 3000 4000 6000DM 00004 00025 004 025 4 9 16 36DV 000019 0001225 00199 012475 1999 449 799 17998
(a) DistVect
(b) DistVect1
Figure 9 CPUs memory and network occupancy
Table 5 Storage comparisons between119867rsquos 119881rsquos (in MW)
L 858 1728 29414 161000Hrsquos + Nrsquos 1475 597 17348 518V rsquos +119873Vrsquos 0005 001 0176 0966
and GCUPS which can be reached Records certify ourpresumed idea that although GPU programs are very fastwith high GCUPS they cannot align long sequences It alsoproves that our proposal is superlative in handling longsequences with good speedup
The above presented results reveal the best performanceof theDistVect1 against other implementationsThis achieve-ment is due to two main reasons (1) the superiority ofDistVect1 in using vectors instead of matrices leading to abetter caching and (2) the optimal use of multicore clustersystem because of the proposed partitioning and schedul-ing techniques especially when the length of sequencesincreases thus obtaining a higher parallelization factor
6 Conclusion and Future Work
In this paper we presented and evaluatedDistVect1 algorithman improved version of the vectorized parallel algorithmDistVect for efficiently computing the distance matrix usingmulticore cluster systems The new suggested partition-ing and scheduling approaches integrated with MPI andOpenMP primitives have achieved a significant improve-ment to the overall performance Implementations wereconducted on Bibliotheca Alexandria cluster system using 32nodes (8 cores)
The resulting program DistVect1 was able to computedistances between very long sequences up to 163 k It outper-forms SSE2 with 3-fold speedup and ClustalW-MPI 013 with
BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 5
the parallelization factor will increase leading to a higheracceleration
Figure 2 demonstrates our proposed cyclic partitioningtechnique when handling the two sequences considered atFigure 1 with 4 cores The four cores act in parallel on each ofthe first four consecutive rows then they sequentially act onthe second consequent 4 rows
Furthermore all lceil119873(119873minus1)2rceil distance computations arepartitioned into119872 splits and distributed over the119872 nodes inthe cluster system All 119872 nodes work in parallel Each nodewill operate sequentially on aligning the set of lceil119873(119873minus1)2119872rceil
pairs at its own split whose size is 119899split Figure 3 explains howthe alignments of 4 sequences of length 8 are scheduled over acluster system of 2 nodes each node has 4 cores In this casethere are 2 splits each split is of size 3
The pseudocode of this proposed method is depicted inAlgorithm 1 And the distance vector DV is computed usingthe following modified recurrences
119881119909
(119896)
= max
0
119881119909
1
(119896 minus 1) + sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119881119909
2
(119896 minus 1) + 119892
119881119909
2
(119896) + 119892
(3)
119873119909
V (119896)
=
0 if 119881119909 (119896) = 0
119873119909
1
(119896 minus 1) + 119898 (119896) if 119881119909 (119896)= 119881119909
1
(119896 minus 1)
+sbt (119878119894
(119896) 119878119895
(119909 minus 119896 + 1))
119873119909
2
(119896) if 119881119909 (119896) = 119881119909
2
(119896) + 119892
119873119909
2
(119896 minus 1) if 119881119909 (119896) = 119881119909
2
(119896 minus 1) + 119892
(4)
where 1 le 119896 le 119875
5 Performance Evaluation
The proposed algorithm DistVect1 has been implementedin C++ using OpenMP and MPI libraries The introducedhybrid paradigm matches the characteristics of a cluster ofmulticore system very well The resulting program has theadvantage of coarse-grained parallelism on process level inwhich each MPI process is executed on one multicore nodeAlso it has the advantage of fine-grained parallelismon a looplevel in which each MPI process spawns a team of threads tooccupy the multicore processors when encountering parallelsections of code using OpenMP However the obtainedresults should be carefully analyzed in order to realize the setof items that interferewith the established comparisonmodel
51 Experimental Setup The presented program has beenimplemented on Sun Microsystems cluster provided byLinkSCEEM-2 systems at Bibliotheca Alexandrina Egypt
This platform specification is as follows
(i) 130 nodes and 64GB memory (the allowed numberof nodes for one user is 32 nodes) each node containstwo Intel quad core Xeon 283GHz processors (64-bittechnology)
(ii) 8 Gbyte RAM 80 Gbyte hard disk and a dual port infin band (10Gbps)
(iii) a Giga Ethernet Network port the operating systemis 64-bit Linux
Real protein sequences of different lengths were used forcomputation They are available online at NCBI [25] Theycomprise a subset of variant family of viruses Each usedbenchmark dataset with the average sequence length and thestandard deviation is given in Table 1The standard deviationgives us a numerical measure of the scatter of a dataset Thismeasure is useful for making comparisons between datasetsthat go beyond simple visual impressions
Comprehensive experiments have been conducted onthe specified platform using different groups of sequencesselected from the indicated datasets They evaluated theimplementation of DistVect1 versus that of DistVect and twopopular efficient programs
(i) ClustalW-MPI available at httpwwwmybiosoft-warecomalignment3052
(ii) SSE available at httpssourceforgenetprojectsdist-matcomp
For SSE2 program we have used the SSE approachwithin each MPI process after distributing the overall work-load using the proposed scheduling technique as clarifiedat Figure 3 Thus each node applies its multithreadingapproach on its assigned matrices one at a time Biblio-theca Alexandria used platform support Advanced VectorExtensions (AVX) which is used during implementing SSE2code
52 Execution Time Speedup and Efficiency In this subsec-tion three common performance measurements the execu-tion time the speedup and the efficiency are used Parallelexecution time represents the elapsed time for completecomputation of the distance matrix including all additionscomparisons and maximum operations Parallel speedup isevaluated by the ratio between the parallel execution time ofthe two involved programs Parallel efficiency is computed bydividing the corresponding parallel speedups by the numberof cores Results for variable number of available nodes withtheir analysis are presented below
As a start experiments were executed on a single nodewith 8 cores Since the amount of computations requiredfor a sequence depends on its length multiple lengths ofsequences fromHIV Coronaviridae andHAhaemagglutininwere tested The execution time (in seconds) of the pro-posed DistVect1 implementation against ClustalW-MPI andDistVect was recorded Figure 4(a) shows how the perfor-mance of N(L) is affected by the sequencesrsquo number 119873 and
6 BioMed Research International
Node 1 Node 2
Core 0
Core 1
Core 2
Core 3
Core 0
Core 1
Core 2
Core 3
Node interconnect
0 0 0 0 0 0 0 00 0 0 0 0 0 0 0
00000000
00000000
S1 times S4
S1 times S3
S1 times S2
S2 times S4
S3 times S4
S2 times S3
Figure 3 Proposed scheduling on multicore cluster
Input Number of sequences N average sequencesrsquo length L sequencesrsquo dataset119878 = 119878
1
1198782
119878119873
substitution matrix sbt gab cost g Number of Nodes MNumber of cores POutput Distance Vector DV(1) Let ℎ = 0
(2) Let 119899119878119901119897119894119905
larr [119873(119873 minus 1)2119872]
(3) ⊳ Distributing splits overM nodes(4) for119873119900119889119890119904 larr 1119872 do in parallel(5) Identify Split(6) for119875119886119894119903119904 larr 1 119899
119878119901119897119894119905
do(7) Get 119878
119894
and 119878119895
from Split(8) Initialize 1198811
1
1198812
1
1198731
1
and1198732
1
(9) for119909 larr 1 119871 + 119875 do(10) ⊳ Distributing distance computation over P cores(11) for 119896 larr 1 119875 do in parallel(12) Compute 119881119909(119896) using (3)(13) if 119878
119894
(119896) = 119878119895
(119909 minus 119896 + 1) then Let119898(119896) = 1
(14) else Let119898(119896) = 0
(15) end if(16) Compute119873119909V (119896) using (4)(17) end for 119896(18) Let 119881119909
1
= 119881119909
2
and 119881119909
2
= 119881119909
(19) Let1198731199091
= 119873119909
2
and119873119909
2
= 119873119909
(20) end for119909(21) Identify 119896max(22) Let DV(ℎ) = 1 minus (119896max119871)
(23) Increment ℎ(24) end for Pairs(25) end forNodes(26) return DV
Algorithm 1 DistVect1 Hybrid DistVect Algorithm
length 119871 DistVect1 shows slightly higher performance thanthat of the two other programswhen119871 ranges in the hundredsand119873 ranges in the thousands Further it presented superiorperformance when 119873 is small compared to 119871 and 119871 exceedsa thousand As shown in Figure 4(b) the maximum achievedspeedups when comparing set 500(9200) were (41) and(25)
Next variant subsets of the specified datasetwith differentsizes were tested on 8 nodes Figure 5 shows that ClustalW-MPI could not handle long sequenceswith119871 overriding thirtythousand That is because ClustalW-MPI approach stronglydepends on the number of sequences and focuses on thedistribution of sequences across nodes without regardingtheir length Also DistVect and SSE2 concentrate on the
BioMed Research International 7
Table 1 Used benchmark dataset specifications
Benchmark Number of sequences Average length Standard deviation
Human immunodeficiency virus (HIV) 3000 858 107456000 858 14632
HA hemagglutinin (influenza B virus) 2000 1741 326044000 1728 36661
Several viruses within the Coronaviridae family 200 30488 908534500 29414 1066417
Herpesviridae (large family of DNA viruses) 20 163654 473303250 161000 4289981
20
80
320
1280
5120
20480
81920
ClustalW-MPIDistVect
DistVect1
400(408)
400(856)
800(454)
2000(266)
4000(247)
1000(858)
500
Exec
utio
n (s
)
Number of sequences (length of sequences)
(9200)
(a)
115
225
335
445
ClustalW-MPIDistVect1DistVectDistVect1
Spee
dup
4000(247)
2000(266)
1000(858)
400(408)
800(454)
400(856)
500
Number of sequences (length of sequences)(9200)
(b)
Figure 4 Performance comparisons using one node and 8 cores
thread level parallelism approach which is based on 119871As a result our program performed better than the threeprograms in all cases and operated smoothly with very longsequences This accomplishment is due to the perfect vec-torization and hybrid partitioning approaches For examplefor comparing set 200(30488) ClustalW-MPI did not workDistVect1 consumed 23467 sec SSE2 exhausted 22949 secand DistVect1 achieved it in 8017 sec
Also the same dataset was tested on 16 nodes Resultsgiven in Figure 6 demonstrate that as the number of nodesincreases the performance upgrades For example comparedto the same set 200(30488) ClustalW-MPI works but slowlytakes 26824 sec DistVect consumed better time 12955 secSSE2 exhausted less period 8166 sec and DistVect1 presentsthe fastest achievement 3226 sec
Finally when tests were performed on 32 nodes as seenin Figure 7 DistVect1rsquos overall performance is typically muchbetter when compared to other programs In fact it exhibitsits superiority when 119871 is in tens of thousands The maximumspeedup for set 20(163354) was up to 9 3 and 2 with respectto the ClustalW-MPI SSE2 and DistVect respectively
To ensure that DistVect1 execution time decreases as thenumber of nodes increases Figure 8 illustrates the relationbetween the execution time and the number of nodes As aconsequence it is concluded that DistVect1 implementationacts almost monotonically increasing whenever the numberof available nodes increases Therefore we can state thatDistVect1 execution time is inversely in proportion to thenumber of nodes
To emphasize that the proposed DistVect1 program ismore efficient than other implemented programs the parallelefficiency in terms of the maximum number of availablecores has been evaluated Results are recorded in Table 2 Itis clear that DistVect1 efficiency is monotonically increasingas the length of sequences increases Thus we can state thatDistVect1 efficiency is directly in proportion to the sequencesrsquolength In addition DistVect1rsquos supreme efficiency was up to029 0086 and 0092 with respect to the ClustalW-MPISSE2 and DistVect respectively for the longest sequencelength (163 k)
53 GCUPS A performance measurement commonly usedin computational biology is billion cell updates per second(GCUPS) A GCUPS represents the time for a completecomputation of one entry in the similarity matrix includingall comparisons additions andmaximumoperationsWehavescanned the datasets with their average length as mentionedabove Our implementation on a cluster of multicores allowshandling sequences up to a length of 163 k Table 3 comparesthe corresponding GCUPS performance values
8 BioMed Research International
128000
64000
32000
16000
8000
4000
2000
1000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
1
15
2
25
3
35
4
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
Number of sequences (length of sequences)
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
(b)
Figure 5 Performance comparisons using 8 nodes
5001000200040008000
160003200064000
128000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
200
(30
488
)
20
(163
354
)
6000
(858
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVectDistVectDistVect1
Spee
dup
6000(858)
3000(858)
2000(1741)
4000(1728)
200(30488)
50(161000)
Number of sequences (length of sequences)
(b)
Figure 6 Performance comparisons using 16 nodes
Table 2 Efficiency comparisons using 32 nodes
Number of sequences (Length of sequence) ClustalW-MPI SSE2 DistVect6000 (858) 01899 00401 006423000 (858) 02025 00414 006252000 (1750) 02087 00436 006774000 (1750) 02061 00449 00696200 (30500) 02578 00881 0091050 (161000) 02730 00823 0091920 (163650) 02901 00863 00919
BioMed Research International 9
250500
1000200040008000
160003200064000
3000
(858
)
2000
(1741
)
200
(30
488
)
6000
(858
)
20
(163
354
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
ClustalW-MPISSE2
DistVectDistVect1
Number of sequences (length of sequences)
Exec
utio
n (s
)
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
500
(30
000
)
Number of sequences (length of sequences)
(b)
Figure 7 Performance comparisons using 32 nodes
01000020000300004000050000600007000080000
Excu
tion
time (
s)
8 nodes16 nodes32 nodes
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
Figure 8 DistVect1 performance against number of nodes
54 Space Reduction Thehuge growth of sequence databasesthat exceed current programsrsquo capability and computer sys-tems capacity gives rise to the importance of consideringspace complexity as an intrinsic performance metric In thefollowing we study the exhausted storage space as a functionof 119871 the sequence length and 119873 the number of sequencesDistVect1 has reduced the overall consumed space by otherprograms in two ways
(i) The compensation of the distance matrix DM by thedistance vector DV reduces space from 119873
2 into ℎ =
119873(119873 minus 1)2 that is almost to the half as shown inTable 4
(ii) The substitution of matrices 119867 and 119873 by vector Vrsquosand three 1198731015840V119904 reduces space from 2 times (119871 + 1)
2 to 6119871as shown in Table 5
55 Load Balancing and Occupancy To study the occupancyof processors and memory real snapshots have been takenas seen in Figure 9 They record the history of CPUs mem-ory and network during execution They demonstrate howdifferent proposed versions of the algorithm improve loadbalancing and memory bandwidth The load unbalancingof DistVect is apparent in Figure 9(a) On the contrary theoptimal load balancing of DistVect1 with the full use of allCPUs by 100 is obvious Also the reduction of memoryoccupancy is very clear in Figure 9(b)
As a conclusion a comparison of DistVect1 proposedmethod with studied GPU and cluster implementationsdiscussed in this paper is illustrated in Table 6 For eachprogram it specifies its platform and records the maximumsequence length that may be handled the highest speedup
10 BioMed Research International
Table 3 Performance comparison (in GCUPS) for scanning the datasets
Number of sequences (Length of sequence) DistVect1 DistVect SSE2 ClustalW-MPI20 (163650) 232 079 084 0252000 (1750) 582 268 417 0874000 (1750) 627 281 436 0953000 (858) 666 333 503 1036000 (858) 669 326 522 110200 (30500) 1169 401 415 14250 (161000) 703 239 267 080500 (30000) 1075 392 385 000
Table 4 Storage Comparisons between DV DM (in MW)
N 20 50 200 500 2000 3000 4000 6000DM 00004 00025 004 025 4 9 16 36DV 000019 0001225 00199 012475 1999 449 799 17998
(a) DistVect
(b) DistVect1
Figure 9 CPUs memory and network occupancy
Table 5 Storage comparisons between119867rsquos 119881rsquos (in MW)
L 858 1728 29414 161000Hrsquos + Nrsquos 1475 597 17348 518V rsquos +119873Vrsquos 0005 001 0176 0966
and GCUPS which can be reached Records certify ourpresumed idea that although GPU programs are very fastwith high GCUPS they cannot align long sequences It alsoproves that our proposal is superlative in handling longsequences with good speedup
The above presented results reveal the best performanceof theDistVect1 against other implementationsThis achieve-ment is due to two main reasons (1) the superiority ofDistVect1 in using vectors instead of matrices leading to abetter caching and (2) the optimal use of multicore clustersystem because of the proposed partitioning and schedul-ing techniques especially when the length of sequencesincreases thus obtaining a higher parallelization factor
6 Conclusion and Future Work
In this paper we presented and evaluatedDistVect1 algorithman improved version of the vectorized parallel algorithmDistVect for efficiently computing the distance matrix usingmulticore cluster systems The new suggested partition-ing and scheduling approaches integrated with MPI andOpenMP primitives have achieved a significant improve-ment to the overall performance Implementations wereconducted on Bibliotheca Alexandria cluster system using 32nodes (8 cores)
The resulting program DistVect1 was able to computedistances between very long sequences up to 163 k It outper-forms SSE2 with 3-fold speedup and ClustalW-MPI 013 with
BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

6 BioMed Research International
Node 1 Node 2
Core 0
Core 1
Core 2
Core 3
Core 0
Core 1
Core 2
Core 3
Node interconnect
0 0 0 0 0 0 0 00 0 0 0 0 0 0 0
00000000
00000000
S1 times S4
S1 times S3
S1 times S2
S2 times S4
S3 times S4
S2 times S3
Figure 3 Proposed scheduling on multicore cluster
Input Number of sequences N average sequencesrsquo length L sequencesrsquo dataset119878 = 119878
1
1198782
119878119873
substitution matrix sbt gab cost g Number of Nodes MNumber of cores POutput Distance Vector DV(1) Let ℎ = 0
(2) Let 119899119878119901119897119894119905
larr [119873(119873 minus 1)2119872]
(3) ⊳ Distributing splits overM nodes(4) for119873119900119889119890119904 larr 1119872 do in parallel(5) Identify Split(6) for119875119886119894119903119904 larr 1 119899
119878119901119897119894119905
do(7) Get 119878
119894
and 119878119895
from Split(8) Initialize 1198811
1
1198812
1
1198731
1
and1198732
1
(9) for119909 larr 1 119871 + 119875 do(10) ⊳ Distributing distance computation over P cores(11) for 119896 larr 1 119875 do in parallel(12) Compute 119881119909(119896) using (3)(13) if 119878
119894
(119896) = 119878119895
(119909 minus 119896 + 1) then Let119898(119896) = 1
(14) else Let119898(119896) = 0
(15) end if(16) Compute119873119909V (119896) using (4)(17) end for 119896(18) Let 119881119909
1
= 119881119909
2
and 119881119909
2
= 119881119909
(19) Let1198731199091
= 119873119909
2
and119873119909
2
= 119873119909
(20) end for119909(21) Identify 119896max(22) Let DV(ℎ) = 1 minus (119896max119871)
(23) Increment ℎ(24) end for Pairs(25) end forNodes(26) return DV
Algorithm 1 DistVect1 Hybrid DistVect Algorithm
length 119871 DistVect1 shows slightly higher performance thanthat of the two other programswhen119871 ranges in the hundredsand119873 ranges in the thousands Further it presented superiorperformance when 119873 is small compared to 119871 and 119871 exceedsa thousand As shown in Figure 4(b) the maximum achievedspeedups when comparing set 500(9200) were (41) and(25)
Next variant subsets of the specified datasetwith differentsizes were tested on 8 nodes Figure 5 shows that ClustalW-MPI could not handle long sequenceswith119871 overriding thirtythousand That is because ClustalW-MPI approach stronglydepends on the number of sequences and focuses on thedistribution of sequences across nodes without regardingtheir length Also DistVect and SSE2 concentrate on the
BioMed Research International 7
Table 1 Used benchmark dataset specifications
Benchmark Number of sequences Average length Standard deviation
Human immunodeficiency virus (HIV) 3000 858 107456000 858 14632
HA hemagglutinin (influenza B virus) 2000 1741 326044000 1728 36661
Several viruses within the Coronaviridae family 200 30488 908534500 29414 1066417
Herpesviridae (large family of DNA viruses) 20 163654 473303250 161000 4289981
20
80
320
1280
5120
20480
81920
ClustalW-MPIDistVect
DistVect1
400(408)
400(856)
800(454)
2000(266)
4000(247)
1000(858)
500
Exec
utio
n (s
)
Number of sequences (length of sequences)
(9200)
(a)
115
225
335
445
ClustalW-MPIDistVect1DistVectDistVect1
Spee
dup
4000(247)
2000(266)
1000(858)
400(408)
800(454)
400(856)
500
Number of sequences (length of sequences)(9200)
(b)
Figure 4 Performance comparisons using one node and 8 cores
thread level parallelism approach which is based on 119871As a result our program performed better than the threeprograms in all cases and operated smoothly with very longsequences This accomplishment is due to the perfect vec-torization and hybrid partitioning approaches For examplefor comparing set 200(30488) ClustalW-MPI did not workDistVect1 consumed 23467 sec SSE2 exhausted 22949 secand DistVect1 achieved it in 8017 sec
Also the same dataset was tested on 16 nodes Resultsgiven in Figure 6 demonstrate that as the number of nodesincreases the performance upgrades For example comparedto the same set 200(30488) ClustalW-MPI works but slowlytakes 26824 sec DistVect consumed better time 12955 secSSE2 exhausted less period 8166 sec and DistVect1 presentsthe fastest achievement 3226 sec
Finally when tests were performed on 32 nodes as seenin Figure 7 DistVect1rsquos overall performance is typically muchbetter when compared to other programs In fact it exhibitsits superiority when 119871 is in tens of thousands The maximumspeedup for set 20(163354) was up to 9 3 and 2 with respectto the ClustalW-MPI SSE2 and DistVect respectively
To ensure that DistVect1 execution time decreases as thenumber of nodes increases Figure 8 illustrates the relationbetween the execution time and the number of nodes As aconsequence it is concluded that DistVect1 implementationacts almost monotonically increasing whenever the numberof available nodes increases Therefore we can state thatDistVect1 execution time is inversely in proportion to thenumber of nodes
To emphasize that the proposed DistVect1 program ismore efficient than other implemented programs the parallelefficiency in terms of the maximum number of availablecores has been evaluated Results are recorded in Table 2 Itis clear that DistVect1 efficiency is monotonically increasingas the length of sequences increases Thus we can state thatDistVect1 efficiency is directly in proportion to the sequencesrsquolength In addition DistVect1rsquos supreme efficiency was up to029 0086 and 0092 with respect to the ClustalW-MPISSE2 and DistVect respectively for the longest sequencelength (163 k)
53 GCUPS A performance measurement commonly usedin computational biology is billion cell updates per second(GCUPS) A GCUPS represents the time for a completecomputation of one entry in the similarity matrix includingall comparisons additions andmaximumoperationsWehavescanned the datasets with their average length as mentionedabove Our implementation on a cluster of multicores allowshandling sequences up to a length of 163 k Table 3 comparesthe corresponding GCUPS performance values
8 BioMed Research International
128000
64000
32000
16000
8000
4000
2000
1000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
1
15
2
25
3
35
4
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
Number of sequences (length of sequences)
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
(b)
Figure 5 Performance comparisons using 8 nodes
5001000200040008000
160003200064000
128000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
200
(30
488
)
20
(163
354
)
6000
(858
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVectDistVectDistVect1
Spee
dup
6000(858)
3000(858)
2000(1741)
4000(1728)
200(30488)
50(161000)
Number of sequences (length of sequences)
(b)
Figure 6 Performance comparisons using 16 nodes
Table 2 Efficiency comparisons using 32 nodes
Number of sequences (Length of sequence) ClustalW-MPI SSE2 DistVect6000 (858) 01899 00401 006423000 (858) 02025 00414 006252000 (1750) 02087 00436 006774000 (1750) 02061 00449 00696200 (30500) 02578 00881 0091050 (161000) 02730 00823 0091920 (163650) 02901 00863 00919
BioMed Research International 9
250500
1000200040008000
160003200064000
3000
(858
)
2000
(1741
)
200
(30
488
)
6000
(858
)
20
(163
354
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
ClustalW-MPISSE2
DistVectDistVect1
Number of sequences (length of sequences)
Exec
utio
n (s
)
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
500
(30
000
)
Number of sequences (length of sequences)
(b)
Figure 7 Performance comparisons using 32 nodes
01000020000300004000050000600007000080000
Excu
tion
time (
s)
8 nodes16 nodes32 nodes
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
Figure 8 DistVect1 performance against number of nodes
54 Space Reduction Thehuge growth of sequence databasesthat exceed current programsrsquo capability and computer sys-tems capacity gives rise to the importance of consideringspace complexity as an intrinsic performance metric In thefollowing we study the exhausted storage space as a functionof 119871 the sequence length and 119873 the number of sequencesDistVect1 has reduced the overall consumed space by otherprograms in two ways
(i) The compensation of the distance matrix DM by thedistance vector DV reduces space from 119873
2 into ℎ =
119873(119873 minus 1)2 that is almost to the half as shown inTable 4
(ii) The substitution of matrices 119867 and 119873 by vector Vrsquosand three 1198731015840V119904 reduces space from 2 times (119871 + 1)
2 to 6119871as shown in Table 5
55 Load Balancing and Occupancy To study the occupancyof processors and memory real snapshots have been takenas seen in Figure 9 They record the history of CPUs mem-ory and network during execution They demonstrate howdifferent proposed versions of the algorithm improve loadbalancing and memory bandwidth The load unbalancingof DistVect is apparent in Figure 9(a) On the contrary theoptimal load balancing of DistVect1 with the full use of allCPUs by 100 is obvious Also the reduction of memoryoccupancy is very clear in Figure 9(b)
As a conclusion a comparison of DistVect1 proposedmethod with studied GPU and cluster implementationsdiscussed in this paper is illustrated in Table 6 For eachprogram it specifies its platform and records the maximumsequence length that may be handled the highest speedup
10 BioMed Research International
Table 3 Performance comparison (in GCUPS) for scanning the datasets
Number of sequences (Length of sequence) DistVect1 DistVect SSE2 ClustalW-MPI20 (163650) 232 079 084 0252000 (1750) 582 268 417 0874000 (1750) 627 281 436 0953000 (858) 666 333 503 1036000 (858) 669 326 522 110200 (30500) 1169 401 415 14250 (161000) 703 239 267 080500 (30000) 1075 392 385 000
Table 4 Storage Comparisons between DV DM (in MW)
N 20 50 200 500 2000 3000 4000 6000DM 00004 00025 004 025 4 9 16 36DV 000019 0001225 00199 012475 1999 449 799 17998
(a) DistVect
(b) DistVect1
Figure 9 CPUs memory and network occupancy
Table 5 Storage comparisons between119867rsquos 119881rsquos (in MW)
L 858 1728 29414 161000Hrsquos + Nrsquos 1475 597 17348 518V rsquos +119873Vrsquos 0005 001 0176 0966
and GCUPS which can be reached Records certify ourpresumed idea that although GPU programs are very fastwith high GCUPS they cannot align long sequences It alsoproves that our proposal is superlative in handling longsequences with good speedup
The above presented results reveal the best performanceof theDistVect1 against other implementationsThis achieve-ment is due to two main reasons (1) the superiority ofDistVect1 in using vectors instead of matrices leading to abetter caching and (2) the optimal use of multicore clustersystem because of the proposed partitioning and schedul-ing techniques especially when the length of sequencesincreases thus obtaining a higher parallelization factor
6 Conclusion and Future Work
In this paper we presented and evaluatedDistVect1 algorithman improved version of the vectorized parallel algorithmDistVect for efficiently computing the distance matrix usingmulticore cluster systems The new suggested partition-ing and scheduling approaches integrated with MPI andOpenMP primitives have achieved a significant improve-ment to the overall performance Implementations wereconducted on Bibliotheca Alexandria cluster system using 32nodes (8 cores)
The resulting program DistVect1 was able to computedistances between very long sequences up to 163 k It outper-forms SSE2 with 3-fold speedup and ClustalW-MPI 013 with
BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 7
Table 1 Used benchmark dataset specifications
Benchmark Number of sequences Average length Standard deviation
Human immunodeficiency virus (HIV) 3000 858 107456000 858 14632
HA hemagglutinin (influenza B virus) 2000 1741 326044000 1728 36661
Several viruses within the Coronaviridae family 200 30488 908534500 29414 1066417
Herpesviridae (large family of DNA viruses) 20 163654 473303250 161000 4289981
20
80
320
1280
5120
20480
81920
ClustalW-MPIDistVect
DistVect1
400(408)
400(856)
800(454)
2000(266)
4000(247)
1000(858)
500
Exec
utio
n (s
)
Number of sequences (length of sequences)
(9200)
(a)
115
225
335
445
ClustalW-MPIDistVect1DistVectDistVect1
Spee
dup
4000(247)
2000(266)
1000(858)
400(408)
800(454)
400(856)
500
Number of sequences (length of sequences)(9200)
(b)
Figure 4 Performance comparisons using one node and 8 cores
thread level parallelism approach which is based on 119871As a result our program performed better than the threeprograms in all cases and operated smoothly with very longsequences This accomplishment is due to the perfect vec-torization and hybrid partitioning approaches For examplefor comparing set 200(30488) ClustalW-MPI did not workDistVect1 consumed 23467 sec SSE2 exhausted 22949 secand DistVect1 achieved it in 8017 sec
Also the same dataset was tested on 16 nodes Resultsgiven in Figure 6 demonstrate that as the number of nodesincreases the performance upgrades For example comparedto the same set 200(30488) ClustalW-MPI works but slowlytakes 26824 sec DistVect consumed better time 12955 secSSE2 exhausted less period 8166 sec and DistVect1 presentsthe fastest achievement 3226 sec
Finally when tests were performed on 32 nodes as seenin Figure 7 DistVect1rsquos overall performance is typically muchbetter when compared to other programs In fact it exhibitsits superiority when 119871 is in tens of thousands The maximumspeedup for set 20(163354) was up to 9 3 and 2 with respectto the ClustalW-MPI SSE2 and DistVect respectively
To ensure that DistVect1 execution time decreases as thenumber of nodes increases Figure 8 illustrates the relationbetween the execution time and the number of nodes As aconsequence it is concluded that DistVect1 implementationacts almost monotonically increasing whenever the numberof available nodes increases Therefore we can state thatDistVect1 execution time is inversely in proportion to thenumber of nodes
To emphasize that the proposed DistVect1 program ismore efficient than other implemented programs the parallelefficiency in terms of the maximum number of availablecores has been evaluated Results are recorded in Table 2 Itis clear that DistVect1 efficiency is monotonically increasingas the length of sequences increases Thus we can state thatDistVect1 efficiency is directly in proportion to the sequencesrsquolength In addition DistVect1rsquos supreme efficiency was up to029 0086 and 0092 with respect to the ClustalW-MPISSE2 and DistVect respectively for the longest sequencelength (163 k)
53 GCUPS A performance measurement commonly usedin computational biology is billion cell updates per second(GCUPS) A GCUPS represents the time for a completecomputation of one entry in the similarity matrix includingall comparisons additions andmaximumoperationsWehavescanned the datasets with their average length as mentionedabove Our implementation on a cluster of multicores allowshandling sequences up to a length of 163 k Table 3 comparesthe corresponding GCUPS performance values
8 BioMed Research International
128000
64000
32000
16000
8000
4000
2000
1000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
1
15
2
25
3
35
4
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
Number of sequences (length of sequences)
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
(b)
Figure 5 Performance comparisons using 8 nodes
5001000200040008000
160003200064000
128000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
200
(30
488
)
20
(163
354
)
6000
(858
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVectDistVectDistVect1
Spee
dup
6000(858)
3000(858)
2000(1741)
4000(1728)
200(30488)
50(161000)
Number of sequences (length of sequences)
(b)
Figure 6 Performance comparisons using 16 nodes
Table 2 Efficiency comparisons using 32 nodes
Number of sequences (Length of sequence) ClustalW-MPI SSE2 DistVect6000 (858) 01899 00401 006423000 (858) 02025 00414 006252000 (1750) 02087 00436 006774000 (1750) 02061 00449 00696200 (30500) 02578 00881 0091050 (161000) 02730 00823 0091920 (163650) 02901 00863 00919
BioMed Research International 9
250500
1000200040008000
160003200064000
3000
(858
)
2000
(1741
)
200
(30
488
)
6000
(858
)
20
(163
354
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
ClustalW-MPISSE2
DistVectDistVect1
Number of sequences (length of sequences)
Exec
utio
n (s
)
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
500
(30
000
)
Number of sequences (length of sequences)
(b)
Figure 7 Performance comparisons using 32 nodes
01000020000300004000050000600007000080000
Excu
tion
time (
s)
8 nodes16 nodes32 nodes
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
Figure 8 DistVect1 performance against number of nodes
54 Space Reduction Thehuge growth of sequence databasesthat exceed current programsrsquo capability and computer sys-tems capacity gives rise to the importance of consideringspace complexity as an intrinsic performance metric In thefollowing we study the exhausted storage space as a functionof 119871 the sequence length and 119873 the number of sequencesDistVect1 has reduced the overall consumed space by otherprograms in two ways
(i) The compensation of the distance matrix DM by thedistance vector DV reduces space from 119873
2 into ℎ =
119873(119873 minus 1)2 that is almost to the half as shown inTable 4
(ii) The substitution of matrices 119867 and 119873 by vector Vrsquosand three 1198731015840V119904 reduces space from 2 times (119871 + 1)
2 to 6119871as shown in Table 5
55 Load Balancing and Occupancy To study the occupancyof processors and memory real snapshots have been takenas seen in Figure 9 They record the history of CPUs mem-ory and network during execution They demonstrate howdifferent proposed versions of the algorithm improve loadbalancing and memory bandwidth The load unbalancingof DistVect is apparent in Figure 9(a) On the contrary theoptimal load balancing of DistVect1 with the full use of allCPUs by 100 is obvious Also the reduction of memoryoccupancy is very clear in Figure 9(b)
As a conclusion a comparison of DistVect1 proposedmethod with studied GPU and cluster implementationsdiscussed in this paper is illustrated in Table 6 For eachprogram it specifies its platform and records the maximumsequence length that may be handled the highest speedup
10 BioMed Research International
Table 3 Performance comparison (in GCUPS) for scanning the datasets
Number of sequences (Length of sequence) DistVect1 DistVect SSE2 ClustalW-MPI20 (163650) 232 079 084 0252000 (1750) 582 268 417 0874000 (1750) 627 281 436 0953000 (858) 666 333 503 1036000 (858) 669 326 522 110200 (30500) 1169 401 415 14250 (161000) 703 239 267 080500 (30000) 1075 392 385 000
Table 4 Storage Comparisons between DV DM (in MW)
N 20 50 200 500 2000 3000 4000 6000DM 00004 00025 004 025 4 9 16 36DV 000019 0001225 00199 012475 1999 449 799 17998
(a) DistVect
(b) DistVect1
Figure 9 CPUs memory and network occupancy
Table 5 Storage comparisons between119867rsquos 119881rsquos (in MW)
L 858 1728 29414 161000Hrsquos + Nrsquos 1475 597 17348 518V rsquos +119873Vrsquos 0005 001 0176 0966
and GCUPS which can be reached Records certify ourpresumed idea that although GPU programs are very fastwith high GCUPS they cannot align long sequences It alsoproves that our proposal is superlative in handling longsequences with good speedup
The above presented results reveal the best performanceof theDistVect1 against other implementationsThis achieve-ment is due to two main reasons (1) the superiority ofDistVect1 in using vectors instead of matrices leading to abetter caching and (2) the optimal use of multicore clustersystem because of the proposed partitioning and schedul-ing techniques especially when the length of sequencesincreases thus obtaining a higher parallelization factor
6 Conclusion and Future Work
In this paper we presented and evaluatedDistVect1 algorithman improved version of the vectorized parallel algorithmDistVect for efficiently computing the distance matrix usingmulticore cluster systems The new suggested partition-ing and scheduling approaches integrated with MPI andOpenMP primitives have achieved a significant improve-ment to the overall performance Implementations wereconducted on Bibliotheca Alexandria cluster system using 32nodes (8 cores)
The resulting program DistVect1 was able to computedistances between very long sequences up to 163 k It outper-forms SSE2 with 3-fold speedup and ClustalW-MPI 013 with
BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

8 BioMed Research International
128000
64000
32000
16000
8000
4000
2000
1000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
1
15
2
25
3
35
4
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
Number of sequences (length of sequences)
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
(b)
Figure 5 Performance comparisons using 8 nodes
5001000200040008000
160003200064000
128000
Exec
utio
n (s
)
3000
(858
)
2000
(1741
)
200
(30
488
)
20
(163
354
)
6000
(858
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
Number of sequences (length of sequences)
ClustalW-MPISSE2
DistVectDistVect1
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVectDistVectDistVect1
Spee
dup
6000(858)
3000(858)
2000(1741)
4000(1728)
200(30488)
50(161000)
Number of sequences (length of sequences)
(b)
Figure 6 Performance comparisons using 16 nodes
Table 2 Efficiency comparisons using 32 nodes
Number of sequences (Length of sequence) ClustalW-MPI SSE2 DistVect6000 (858) 01899 00401 006423000 (858) 02025 00414 006252000 (1750) 02087 00436 006774000 (1750) 02061 00449 00696200 (30500) 02578 00881 0091050 (161000) 02730 00823 0091920 (163650) 02901 00863 00919
BioMed Research International 9
250500
1000200040008000
160003200064000
3000
(858
)
2000
(1741
)
200
(30
488
)
6000
(858
)
20
(163
354
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
ClustalW-MPISSE2
DistVectDistVect1
Number of sequences (length of sequences)
Exec
utio
n (s
)
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
500
(30
000
)
Number of sequences (length of sequences)
(b)
Figure 7 Performance comparisons using 32 nodes
01000020000300004000050000600007000080000
Excu
tion
time (
s)
8 nodes16 nodes32 nodes
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
Figure 8 DistVect1 performance against number of nodes
54 Space Reduction Thehuge growth of sequence databasesthat exceed current programsrsquo capability and computer sys-tems capacity gives rise to the importance of consideringspace complexity as an intrinsic performance metric In thefollowing we study the exhausted storage space as a functionof 119871 the sequence length and 119873 the number of sequencesDistVect1 has reduced the overall consumed space by otherprograms in two ways
(i) The compensation of the distance matrix DM by thedistance vector DV reduces space from 119873
2 into ℎ =
119873(119873 minus 1)2 that is almost to the half as shown inTable 4
(ii) The substitution of matrices 119867 and 119873 by vector Vrsquosand three 1198731015840V119904 reduces space from 2 times (119871 + 1)
2 to 6119871as shown in Table 5
55 Load Balancing and Occupancy To study the occupancyof processors and memory real snapshots have been takenas seen in Figure 9 They record the history of CPUs mem-ory and network during execution They demonstrate howdifferent proposed versions of the algorithm improve loadbalancing and memory bandwidth The load unbalancingof DistVect is apparent in Figure 9(a) On the contrary theoptimal load balancing of DistVect1 with the full use of allCPUs by 100 is obvious Also the reduction of memoryoccupancy is very clear in Figure 9(b)
As a conclusion a comparison of DistVect1 proposedmethod with studied GPU and cluster implementationsdiscussed in this paper is illustrated in Table 6 For eachprogram it specifies its platform and records the maximumsequence length that may be handled the highest speedup
10 BioMed Research International
Table 3 Performance comparison (in GCUPS) for scanning the datasets
Number of sequences (Length of sequence) DistVect1 DistVect SSE2 ClustalW-MPI20 (163650) 232 079 084 0252000 (1750) 582 268 417 0874000 (1750) 627 281 436 0953000 (858) 666 333 503 1036000 (858) 669 326 522 110200 (30500) 1169 401 415 14250 (161000) 703 239 267 080500 (30000) 1075 392 385 000
Table 4 Storage Comparisons between DV DM (in MW)
N 20 50 200 500 2000 3000 4000 6000DM 00004 00025 004 025 4 9 16 36DV 000019 0001225 00199 012475 1999 449 799 17998
(a) DistVect
(b) DistVect1
Figure 9 CPUs memory and network occupancy
Table 5 Storage comparisons between119867rsquos 119881rsquos (in MW)
L 858 1728 29414 161000Hrsquos + Nrsquos 1475 597 17348 518V rsquos +119873Vrsquos 0005 001 0176 0966
and GCUPS which can be reached Records certify ourpresumed idea that although GPU programs are very fastwith high GCUPS they cannot align long sequences It alsoproves that our proposal is superlative in handling longsequences with good speedup
The above presented results reveal the best performanceof theDistVect1 against other implementationsThis achieve-ment is due to two main reasons (1) the superiority ofDistVect1 in using vectors instead of matrices leading to abetter caching and (2) the optimal use of multicore clustersystem because of the proposed partitioning and schedul-ing techniques especially when the length of sequencesincreases thus obtaining a higher parallelization factor
6 Conclusion and Future Work
In this paper we presented and evaluatedDistVect1 algorithman improved version of the vectorized parallel algorithmDistVect for efficiently computing the distance matrix usingmulticore cluster systems The new suggested partition-ing and scheduling approaches integrated with MPI andOpenMP primitives have achieved a significant improve-ment to the overall performance Implementations wereconducted on Bibliotheca Alexandria cluster system using 32nodes (8 cores)
The resulting program DistVect1 was able to computedistances between very long sequences up to 163 k It outper-forms SSE2 with 3-fold speedup and ClustalW-MPI 013 with
BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 9
250500
1000200040008000
160003200064000
3000
(858
)
2000
(1741
)
200
(30
488
)
6000
(858
)
20
(163
354
)
4000
(1728
)
50
(161
000
)
500
(30
000
)
ClustalW-MPISSE2
DistVectDistVect1
Number of sequences (length of sequences)
Exec
utio
n (s
)
(a)
0123456789
10
ClustalW-MPIDistVect1SSE2DistVect1DistVectDistVect1
Spee
dup
6000
(858
)
3000
(858
)
2000
(1741
)
4000
(1728
)
200
(30
488
)
50
(161
000
)
20
(163
354
)
500
(30
000
)
Number of sequences (length of sequences)
(b)
Figure 7 Performance comparisons using 32 nodes
01000020000300004000050000600007000080000
Excu
tion
time (
s)
8 nodes16 nodes32 nodes
3000
(858
)
2000
(1741
)
20
(163
354
)
200
(30
488
)
6000
(858
)
50
(161
000
)
4000
(1728
)
500
(30
000
)
Number of sequences (length of sequences)
Figure 8 DistVect1 performance against number of nodes
54 Space Reduction Thehuge growth of sequence databasesthat exceed current programsrsquo capability and computer sys-tems capacity gives rise to the importance of consideringspace complexity as an intrinsic performance metric In thefollowing we study the exhausted storage space as a functionof 119871 the sequence length and 119873 the number of sequencesDistVect1 has reduced the overall consumed space by otherprograms in two ways
(i) The compensation of the distance matrix DM by thedistance vector DV reduces space from 119873
2 into ℎ =
119873(119873 minus 1)2 that is almost to the half as shown inTable 4
(ii) The substitution of matrices 119867 and 119873 by vector Vrsquosand three 1198731015840V119904 reduces space from 2 times (119871 + 1)
2 to 6119871as shown in Table 5
55 Load Balancing and Occupancy To study the occupancyof processors and memory real snapshots have been takenas seen in Figure 9 They record the history of CPUs mem-ory and network during execution They demonstrate howdifferent proposed versions of the algorithm improve loadbalancing and memory bandwidth The load unbalancingof DistVect is apparent in Figure 9(a) On the contrary theoptimal load balancing of DistVect1 with the full use of allCPUs by 100 is obvious Also the reduction of memoryoccupancy is very clear in Figure 9(b)
As a conclusion a comparison of DistVect1 proposedmethod with studied GPU and cluster implementationsdiscussed in this paper is illustrated in Table 6 For eachprogram it specifies its platform and records the maximumsequence length that may be handled the highest speedup
10 BioMed Research International
Table 3 Performance comparison (in GCUPS) for scanning the datasets
Number of sequences (Length of sequence) DistVect1 DistVect SSE2 ClustalW-MPI20 (163650) 232 079 084 0252000 (1750) 582 268 417 0874000 (1750) 627 281 436 0953000 (858) 666 333 503 1036000 (858) 669 326 522 110200 (30500) 1169 401 415 14250 (161000) 703 239 267 080500 (30000) 1075 392 385 000
Table 4 Storage Comparisons between DV DM (in MW)
N 20 50 200 500 2000 3000 4000 6000DM 00004 00025 004 025 4 9 16 36DV 000019 0001225 00199 012475 1999 449 799 17998
(a) DistVect
(b) DistVect1
Figure 9 CPUs memory and network occupancy
Table 5 Storage comparisons between119867rsquos 119881rsquos (in MW)
L 858 1728 29414 161000Hrsquos + Nrsquos 1475 597 17348 518V rsquos +119873Vrsquos 0005 001 0176 0966
and GCUPS which can be reached Records certify ourpresumed idea that although GPU programs are very fastwith high GCUPS they cannot align long sequences It alsoproves that our proposal is superlative in handling longsequences with good speedup
The above presented results reveal the best performanceof theDistVect1 against other implementationsThis achieve-ment is due to two main reasons (1) the superiority ofDistVect1 in using vectors instead of matrices leading to abetter caching and (2) the optimal use of multicore clustersystem because of the proposed partitioning and schedul-ing techniques especially when the length of sequencesincreases thus obtaining a higher parallelization factor
6 Conclusion and Future Work
In this paper we presented and evaluatedDistVect1 algorithman improved version of the vectorized parallel algorithmDistVect for efficiently computing the distance matrix usingmulticore cluster systems The new suggested partition-ing and scheduling approaches integrated with MPI andOpenMP primitives have achieved a significant improve-ment to the overall performance Implementations wereconducted on Bibliotheca Alexandria cluster system using 32nodes (8 cores)
The resulting program DistVect1 was able to computedistances between very long sequences up to 163 k It outper-forms SSE2 with 3-fold speedup and ClustalW-MPI 013 with
BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology
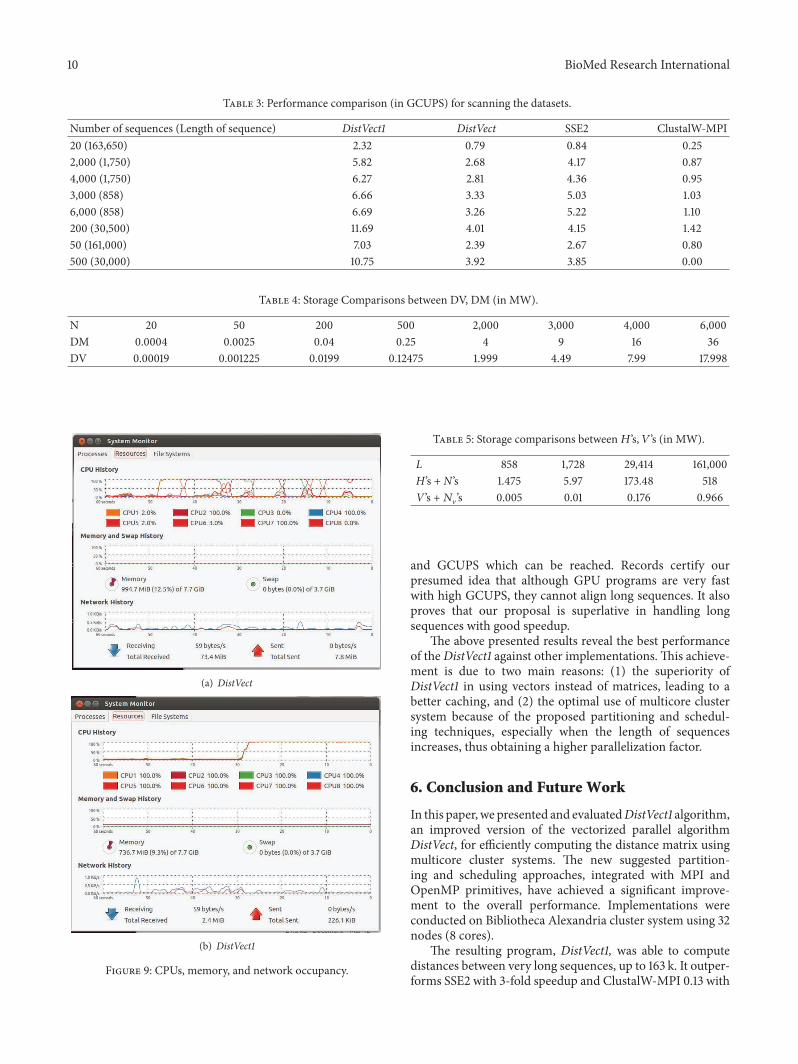
10 BioMed Research International
Table 3 Performance comparison (in GCUPS) for scanning the datasets
Number of sequences (Length of sequence) DistVect1 DistVect SSE2 ClustalW-MPI20 (163650) 232 079 084 0252000 (1750) 582 268 417 0874000 (1750) 627 281 436 0953000 (858) 666 333 503 1036000 (858) 669 326 522 110200 (30500) 1169 401 415 14250 (161000) 703 239 267 080500 (30000) 1075 392 385 000
Table 4 Storage Comparisons between DV DM (in MW)
N 20 50 200 500 2000 3000 4000 6000DM 00004 00025 004 025 4 9 16 36DV 000019 0001225 00199 012475 1999 449 799 17998
(a) DistVect
(b) DistVect1
Figure 9 CPUs memory and network occupancy
Table 5 Storage comparisons between119867rsquos 119881rsquos (in MW)
L 858 1728 29414 161000Hrsquos + Nrsquos 1475 597 17348 518V rsquos +119873Vrsquos 0005 001 0176 0966
and GCUPS which can be reached Records certify ourpresumed idea that although GPU programs are very fastwith high GCUPS they cannot align long sequences It alsoproves that our proposal is superlative in handling longsequences with good speedup
The above presented results reveal the best performanceof theDistVect1 against other implementationsThis achieve-ment is due to two main reasons (1) the superiority ofDistVect1 in using vectors instead of matrices leading to abetter caching and (2) the optimal use of multicore clustersystem because of the proposed partitioning and schedul-ing techniques especially when the length of sequencesincreases thus obtaining a higher parallelization factor
6 Conclusion and Future Work
In this paper we presented and evaluatedDistVect1 algorithman improved version of the vectorized parallel algorithmDistVect for efficiently computing the distance matrix usingmulticore cluster systems The new suggested partition-ing and scheduling approaches integrated with MPI andOpenMP primitives have achieved a significant improve-ment to the overall performance Implementations wereconducted on Bibliotheca Alexandria cluster system using 32nodes (8 cores)
The resulting program DistVect1 was able to computedistances between very long sequences up to 163 k It outper-forms SSE2 with 3-fold speedup and ClustalW-MPI 013 with
BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 11
Table 6 DistVect1 comparison against other programs
Software Platform Maximum sequence length Highest GCUPS Highest speedupCUDASW++ 2 Dual-GPU GeForce GTX 295 59 k 28 (144ndash5478) 178 with (CUDASW++ 10)CUDASW++ 3 Dual-GPU GeForce GTX 690 59 k 18560 (144ndash5478) 32 with (CUDASW++ 20)MC64-ClustalWP2 Intel Xeon Quad Core 300 k 092 776 with (ClustalW-MPI)SSE2 CellBE 0858 k 00058 7620 with ClustalW sequentialClustalW-MPI PC cluster 1100 007 145 with ClustalW sequentialDistVect1 Cluster of multicores 163 k 1169 92 with (ClustalW-MPI)
9-fold speedup Its efficiency reaches 029 0086 and 0092over the ClustalW-MPI SSE2 and DistVect respectivelyMoreover it accomplishes 100 of CPUs occupancy withoptimal load balancing and less memory exhaustion Theperformance figures also vary from a low of 627 GCUPSto a high of 1169 GCUPS as the lengths of the querysequences increase from 1750 to 30500 We believe thatif more cores are provided a better performance will beachieved and a higher speedup with improved efficiency willbe accomplished
For future work it is planned to develop an efficientmultiple sequence alignment tool as an extension to theproposed work Furthermore it is expected to exploit theproposed parallel program for providing better solutions fora class of widely encountered problems in bioinformatics andimage processing
Conflict of Interests
The authors declare that there is no conflict of interestsregarding the publication of this paper
Acknowledgments
The authors would like to thank Bibliotheca Alexandriafor granting the access for running their computations onits platform This work has been accomplished with theaid of proficient discussions and technical support of theSupercomputer laboratory team members at BibliothecaAlexandria Egypt
References
[1] C-E Chin A C-C Shth and K-C Fan ldquoA novel spectralclustering method based on pairwise distance matrixrdquo Journalof Information Science and Engineering vol 26 no 2 pp 649ndash658 2010
[2] R Hu W Jia H Ling and D Huang ldquoMultiscale distancematrix for fast plant leaf recognitionrdquo IEEE Transactions onImage Processing vol 21 no 11 pp 4667ndash4672 2012
[3] A Roman-Gonzalez ldquoCompression techniques for image pro-cessing tasksrdquo International Journal of Advanced Research inComputer Science and Software Engineering vol 3 no 7 pp379ndash388 2013
[4] J Venna J Peltonen K Nybo H Aidos and S Kaski ldquoInforma-tion retrieval perspective to nonlinear dimensionality reductionfor data visualizationrdquo Journal of Machine Learning Researchvol 11 pp 451ndash490 2010
[5] A Y Zomaya Parallel Computing for Bioinformatics and Com-putational Biology Models Enabling Technologies and CaseStudies John Wiley amp Sons 2006
[6] J R ColeQWang E Cardenas et al ldquoTheRibosomalDatabaseProject improved alignments and new tools for rRNA analysisrdquoNucleic Acids Research vol 37 no 1 pp D141ndashD145 2009
[7] B SchmidtBioinformatics High Performance Parallel ComputerArchitectures CRC Press 2011
[8] M W Al-Neama N M Reda and F F M Ghaleb ldquoFastvectorized distance matrixcomputation for multiple sequencealignment on multi-coresrdquo International Journal of Biomathe-matics In press
[9] K Katoh K-I Kuma H Toh and T Miyata ldquoMAFFT version5 improvement in accuracy of multiple sequence alignmentrdquoNucleic Acids Research vol 33 no 2 pp 511ndash518 2005
[10] A R Subramanian M Kaufmann and B MorgensternldquoDIALIGN-TX greedy and progressive approaches forsegment-based multiple sequence alignmentrdquo Algorithms forMolecular Biology vol 3 no 1 article 6 2008
[11] T Kim and H Joo ldquoClustalXeed a GUI-based grid computa-tion version for high performance and terabyte size multiplesequence alignmentrdquo BMC Bioinformatics vol 11 article 4672010
[12] R C Edgar ldquoMUSCLE multiple sequence alignment with highaccuracy and high throughputrdquo Nucleic Acids Research vol 32no 5 pp 1792ndash1797 2004
[13] J D Thompson ldquoThe clustal series of programs for multiplesequence alignmentrdquo inTheProteomics Protocols Handbook pp493ndash502 Humana Press 2005
[14] J Zola X Yang S Rospondek and S Aluru ldquoParallel T-Coffeea parallel multiple sequence alignerrdquo in Proceedings of the20th ISCA International Conference on Parallel and DistributedComputing Systems (ISCA PDCS rsquo07) pp 248ndash253 2007
[15] A Wirawan C K Kwoh and B Schmidt ldquoMulti-threadedvectorized distance matrix computation on the CELLBE andx86SSE2 architecturesrdquoBioinformatics vol 26 no 10 pp 1368ndash1369 2010
[16] K Chaichoompu and S Kittitornkun ldquoMultithreadedClustalWwith improved optimization for intel multi-core processorrdquo inProceedings of the International Symposium on Communicationsand Information Technologies (ISCIT rsquo06) pp 590ndash594 October2006
[17] K-B Li ldquoClustalW-MPI ClustalW analysis using distributedand parallel computingrdquoBioinformatics vol 19 no 12 pp 1585ndash1586 2003
[18] A Wirawan B Schmidt and C K Kwoh ldquoPairwise distancematrix computation formultiple sequence alignment on the cellbroadband enginerdquo in Computational SciencemdashICCS 2009 vol5544 of Lecture Notes in Computer Science pp 954ndash963 2009
12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

12 BioMed Research International
[19] Y Liu B Schmidt and D L Maskell ldquoMSAProbs multiplesequence alignment based on pair hidden Markov models andpartition function posterior probabilitiesrdquo Bioinformatics vol26 no 16 pp 1958ndash1964 2010
[20] D Hains Z Cashero M Ottenberg W Bohm and SRajopadhye ldquoImproving CUDASW a parallelization of smith-waterman for CUDA enabled devicesrdquo in Proceedings of the25th IEEE International Parallel and Distributed ProcessingSymposium Workshops and Phd Forum (IPDPSW rsquo11) pp 490ndash501 May 2011
[21] Y Liu A Wirawan and B Schmidt ldquoCUDASW++ 30 accel-erating Smith-Waterman protein database search by couplingCPU and GPU SIMD instructionsrdquo BMC Bioinformatics vol14 article 117 2013
[22] W Liu B Schmidt G Voss and W Muller-Wittig ldquoStreamingalgorithms for biological sequence alignment on GPUsrdquo IEEETransactions on Parallel and Distributed Systems vol 18 no 9pp 1270ndash1281 2007
[23] D Dıaz F J Esteban P Hernandez et al ldquoMc64-clustalwp2a highly-parallel hybrid strategy to align multiple sequences inmany-core architecturesrdquo PLOS ONE vol 9 no 4 Article IDe94044 2014
[24] J Cheetham FDehne S Pitre A Rau-Chaplin and P J TaillonldquoParallel CLUSTAL W for PC clustersrdquo in ComputationalScience and Its ApplicationsmdashICCSA 2003 vol 2668 of LectureNotes in Computer Science pp 300ndash309 2003
[25] National Center for Biotechnology Information (NCBI)httpwwwncbinlmnihgov
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology