research-repository.uwa.edu.auresearch-repository.uwa.edu.au/files/3238276/mukai_shin_2011.pdf ·...
TRANSCRIPT

Stable Analogues of Phosphohistidine
Shin Mukai
B. Eng. (Master’s)
Chemistry
School of Biomedical, Biomolecular and Chemical Sciences
This thesis is presented for the degree of Doctor of
Philosophy to The University of Western Australia

ii

iii
Candidate Declaration
The work described in this thesis was carried out by the author during the period of
April 2007 to January 2011 in the School of Biomedical, Biomolecular and Chemical
Sciences at The University of Western Australia under the supervision of Associate
Professor Matthew J. Piggott. Unless duly described, this work is original.
………………………..
Shin Mukai
April 2011

iv
DECLARATION FOR THESES CONTAINING PUBLISHED WORK AND/OR WORK PREPARED FOR
PUBLICATION
The examination of the thesis is an examination of the work of the student. The work must have
been substantially conducted by the student during enrolment in the degree.
Where the thesis includes work to which others have contributed, the thesis must include a
statement that makes the student’s contribution clear to the examiners. This may be in the form
of a description of the precise contribution of the student to the work presented for examination
and/or a statement of the percentage of the work that was done by the student.
In addition, in the case of co-authored publications included in the thesis, each author must give
their signed permission for the work to be included. If signatures from all the authors cannot be
obtained, the statement detailing the student’s contribution to the work must be signed by the
coordinating supervisor.
Please sign one of the statements below.
1. This thesis does not contain work that I have published, nor work under review for publication.
Student Signature
.........................................................................................................................................................
2. This thesis contains only sole-authored work, some of which has been published and/or
prepared for publication under sole authorship. The bibliographical details of the work and where
it appears in the thesis are outlined below.
Student Signature
.........................................................................................................................................................
3. This thesis contains published work and/or work prepared for publication, some of which has
been co-authored. The bibliographical details of the work and where it appears in the thesis are
outlined below.
The student must attach to this declaration a statement for each publication that clarifies the
contribution of the student to the work. This may be in the form of a description of the precise
contributions of the student to the published work and/or a statement of percent contribution by
the student. This statement must be signed by all authors. If signatures from all the authors cannot
be obtained, the statement detailing the student’s contribution to the published work must be
signed by the coordinating supervisor.
Student Signature ………………………………………………………………………………………….
Coordinating Supervisor Signature. ..……………………………………………………………………

v
Summary
Phosphorylation is a crucial post-translational modification of proteins. This chemical
modification acts as an on-off switch for protein function; for example, the catalytic
activity of many enzymes, and most aspects of cell signalling, are controlled by
phosphorylation. Most commonly, the hydroxyl group of serine, threonine and tyrosine
residues is phosphorylated. There is a mass of information about proteins that contain
phosphohydroxyamino acid residues and the kinases that catalyse their phosphorylation.
Phosphorylation of one of the imidazole nitrogens of histidine residues in proteins also
plays a very important role in nature. However, information about phosphohistidine-
containing proteins is limited owing to the chemical lability of the phosphoramidate
functional group and the lack of commercially available phosphohistidine antibodies.
Attempts to raise antibodies using phosphohistidine have failed due to the instability of
the latter towards hydrolysis.
The primary aim of the research described in this thesis was, therefore, to synthesise
stable triazolylphosphonate analogues of N1- and N3-phosphohistidine for the purpose
of generating antibodies, which can recognise phosphohistidine residues in a protein
context.
N
N
HN
NH
O
PO
OO
N
N
HN
NH
O
POO
O3
11
3
or
proteinprotein
protein
protein
N
NH
HN
NH
O
3
1
proteinprotein
histidine kinase
N N
NPO
O
O
HN
O
ORR
N NN
HN
OR
O
PO
OO
R
Figure S1. Histidine phosphorylation and phosphohistidine analogues
Chapter 1 of this thesis provides an introduction to protein phosphorylation and the
history of detection of phosphohistidine in both prokaryotes and eukaryotes. The
chapter also includes a summary of similar work by other groups published during the
completion of the research described in this thesis.
vi
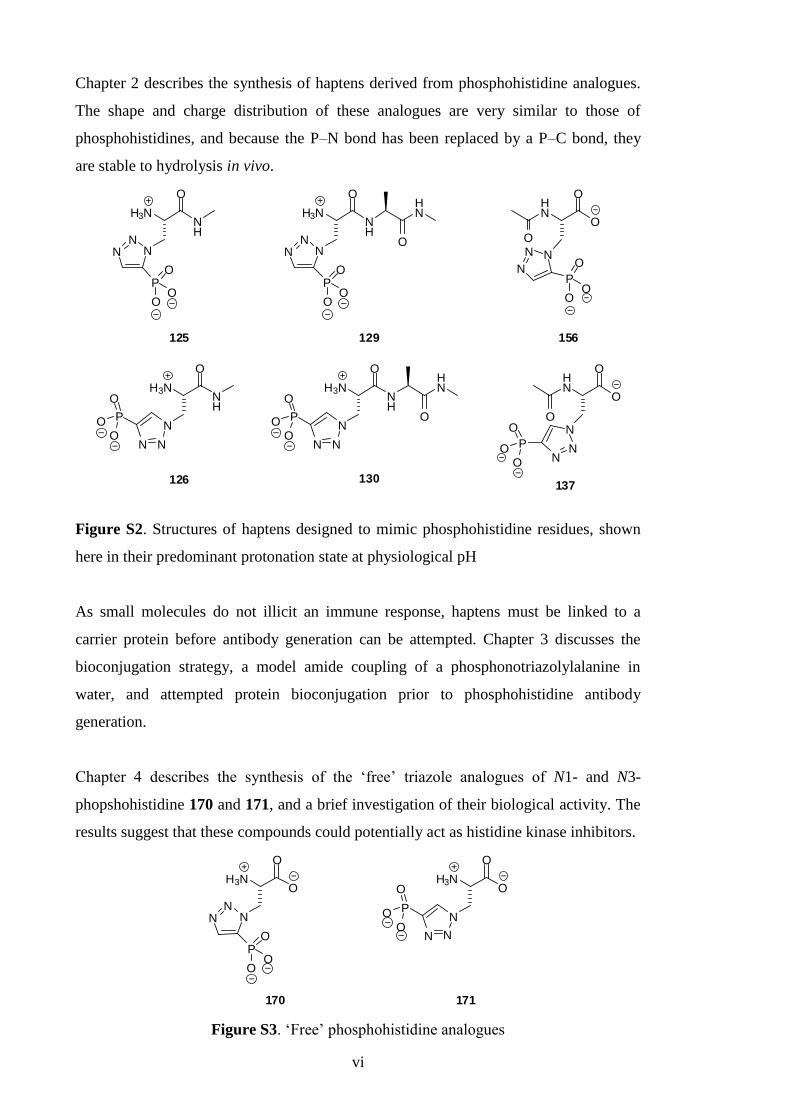
Chapter 2 describes the synthesis of haptens derived from phosphohistidine analogues.
The shape and charge distribution of these analogues are very similar to those of
phosphohistidines, and because the P–N bond has been replaced by a P–C bond, they
are stable to hydrolysis in vivo.
N N
NPO
O
OH3N
O
NH
N NN
H3NNH
O
PO
OO
N NN
H3NNH
O
PO
OO
HN
O
HN
O
O
NN
NP
O
OO
O
N N
NPO
O
OH3N
O
NH
HN
O
HN
O
O
ON
NNP
O
OO
125 129 156
137130126
Figure S2. Structures of haptens designed to mimic phosphohistidine residues, shown
here in their predominant protonation state at physiological pH
As small molecules do not illicit an immune response, haptens must be linked to a
carrier protein before antibody generation can be attempted. Chapter 3 discusses the
bioconjugation strategy, a model amide coupling of a phosphonotriazolylalanine in
water, and attempted protein bioconjugation prior to phosphohistidine antibody
generation.
Chapter 4 describes the synthesis of the „free‟ triazole analogues of N1- and N3-
phopshohistidine 170 and 171, and a brief investigation of their biological activity. The
results suggest that these compounds could potentially act as histidine kinase inhibitors.
N N
NPO
O
OH3N
O
O
N NN
H3NO
O
PO
OO
170 171
Figure S3. „Free‟ phosphohistidine analogues

vii
Chapter 5 details the preparation of Fmoc-protected phosphohistidine analogues 180
and 181 for potential use in (automated) solid-phase peptide synthesis. In addition, this
chapter discusses the potential utility of some of the compounds described in this thesis
for preparing affinity chromatography media for the purpose of isolating histidine
kinases and signalling molecules that recognise the phosphohistidine motif.
N N
NPEtO
EtO
OFmocN
O
O
N NN
FmocNO
O
PO
OEtOEt
H H
180 181
Figure S4. Fmoc derivatives of phosphohistidine analogues
Finally, as the extra nitrogen atom in the triazoles above alters the basicity of the
heterocycle, and therefore might affect phosphohistidine antibody generation, an
imidazole analogue 231 of N1-phosphohistidine and a pyrazole analogue 209 of N3-
phosphohistidine were targeted. Chapter 6 describes the attempted synthesis of these
compounds.
N
NPEtO
EtO
OBocN
O
OR'
N N
BocNOR'
O
PO
OEt
H H
231 209
OEt
Figure S5. Imidazole analogue and pyrazole analogues
The thesis ends with a conclusion, summarising the achievements and outstanding
goals, and possible future work in this area.

viii
Acknowledgements
I would like to extend my sincere appreciation and gratitude to my supervisor, A/Prof.
Matthew Piggott for his advice, support and encouragement. I am also grateful to my
co-supervisor, Prof. Paul Attwood for his guidance and dedication.
I would like to thank Dr Paul Besant for giving me several skills in biochemistry and
encouraging me to challenge a full marathon. It was one of the greatest achievements in
my life to complete the Perth Marathon 2010.
Thanks must go to the Piggott research group members, in particular, Michael Gandy,
Katie Punch and Blake Nguyen for their friendship and assistance.
I would like to thank Dr Lindsay Byrne for his invaluable NMR lessons, Dr Gavin
Flematti for his assistance with HPLC analyses and Dr Anthony Reeder for conducting
mass spectrometry on my samples.
An expression of gratitude is extended to Mr. Kim Foo (First year laboratory), Mr.
Graeme Cuffe (First year laboratory), Mr. Oscar Del Borrello (Second year laboratory),
Dr Sato Juniper (Graduate Research School) and Dr Joanne Edmondston (Student
Services) for their friendly assistance and expertise.
I am grateful to Prof. Allan McKinley for his support throughout the course of my PhD.
I wish to thank the supervisors in my Master‟s research, Prof. Sei-ichi Nishimoto
(Kyoto University) and A/Prof. Kazuhito Tanabe (Kyoto University) for their
encouragement.

ix
The financial assistance of a SIRF scholarship and a UIS scholarship from The
University of Western Australia is gratefully acknowledged.
I would like to extend a big thank-you to my family and friends.
Thankfully, I am alive and healthy, which enables me to pursue my dreams.
One Life, No Regret.

x
Abbreviations
The following abbreviations are used in this thesis.
Abbreviation Full Name
Ac
ADDP
ADP
All
ATP
aq.
B
Bn
Boc
Boc2O
BSA
But
CAN
Cbz
COD
conc.
Cp*
DCC
DCM
DEAD
DIAD
DIPEA
DMF
DMSO
DNA
EDCI
ESI
Fmoc
FmocCl
HATU
acetyl
azodicarboxylic acid dipiperidide
adenosine 5'-diphosphate
allyl
adenosine 5'-triphosphate
aqueous
base
benzyl
tert-butoxycarbonyl
di-tert-butyl dicarbonate
bovine serum albumin
tert-butyl
ceric ammonium nitrate
carbobenzyloxy
1,5-cyclooctadiene
concentrated
pentamethylcyclopentadienylbis(triphenylphosphine)
N,N′-dicyclohexylcarbodiimide
dichloromethane
diethyl azodicarboxylate
diisopropyl azodicarboxylate
N,N-diisopropylethylamine
N,N-dimethylformamide
dimethyl sulfoxide
deoxyribonucleic acid
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide
electrospray ionisation
fluorenylmethoxycarbonyl
fluorenylmethoxycarbonyl chloride
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium
hexafluorophosphate

xi
HCTU
HMBC
HPLC
HRMS
IR
KLH
MALDI
NBD
Me
MES
Ms
Nu
OVA
Ph
iPr
r.t.
sat.
SDS-PAGE
TFA
TLC
THF
TIPS
TMAD
TMS
TMSBr
Tris
Ts
TsCl
Tza
O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium
hexafluorophosphate
heteronuclear multiple bond coherence
high performance liquid chromatography
high resolution mass spectrometry
infrared (absorption spectrometry)
keyhole limpet hemocyanin
matrix-assisted laser desorption ionisation
norbornadiene
methyl
2-(N-morpholino)ethanesulfonic acid
mesyl (methanesulfonyl)
nucleophile
ovalbumin
phenyl
isopropyl
room tempetature
saturated
sodium dodecyl sulfate polyacrylamide gel electrophoresis
trifluoroacetic acid
thin layer chromatography
tetrahydrofuran
triisopropylsilyl
N,N,N′,N′-tetramethylazodicarboxamide
trimethylsilyl
trimethylsilyl bromide
tris(hydroxymethyl)aminomethane
tosyl (4-toluenesulfonyl)
tosyl (4-toluenesulfonyl) chloride
triazolylalanine

xii
Table of Contents
Candidate declaration iii
Summary v
Acknowledgements viii
Abbreviations x
Chapter 1
Introduction 1
1.1 Protein phosphorylation 2
1.2 Phosphohistidine 3
1.2.1 Synthesis of phosphohistidine 4
1.2.2 The two-component system in prokaryotic signalling 5
1.2.3 Histidine phosphorylation in mammalian cells 7
1.2.4 Instability of phosphohistidine 8
1.3. Detection of phosphohistidine 11
1.3.1 Detection using [32
P]ATP 11
1.3.2 Mass spectrometric detection 12
1.4. Phosphohistidine antibodies 15
1.4.1 Antibodies 17
1.4.2 Polyclonal and monoclonal antibodies 18
1.4.3 Haptens 20
1.4.4 Stable analogues of phosphohistidine 20
1.5 Stable phosphohistidine analogue targets: Aims 24
1.6.1 Similar work published by the Muir group 27
1.6.2 Similar work published by the Webb group 30

xiii
Chapter 2
Synthesis of Triazolylphosphonate Analogues of Phosphohistidine 33
2.1 Strategy 34
2.2 Synthesis of a protected azidoalanine 34
2.2.1 Mitsunobu reaction 35
2.3. Synthesis of diethyl ethynylphosphonate 37
2.4 Azide-Alkyne Cycloadditions 42
2.4.1 Background 42
2.4.2 Copper-catalysed azide-alkyne cycloadditions 43
2.4.3 Ruthenium-catalysed azide-alkyne cycloadditions 45
2.4.4 Synthesis of protected triazolylalaninephosphonates
(NBoc-Tza(POEt2)-OMe) 46
2.5 Synthesis of haptens 51
2.5.1 N-methylamide haptens 52
2.5.2 Dipeptide haptens 54
2.6 Synthesis of haptens ready for bioconjugation through
the carboxyl group 60
2.6.1 Attempts to convert NBoc derivatives to NAc derivatives 60
2.6.2 Synthesis of NAc-Tza(POEt2)-OMe 63
2.6.3 Synthesis of N-acetyl haptens 65
2.6.4 Synthesis of NAc-Tza(POEt2)-OAll 67
2.6.5 Attempted deallylation 70
2.6.6 Synthesis of NAc-Tza(POEt2)-OBn 71
2.6.7 Synthesis of N-acetyl haptens-2 73
Chapter 3
Hapten-Carrier Protein Conjugation Strategies 77
3.1 Introduction to bioconjugation methods 78

xiv
3.2 Trial conjugation-1 80
3.3 Trial conjugation-2 82
Chapter 4
Synthesis of Free Phosphosphohistidine Analogues and Evaluation of
their Ability to Inhibit a Mammalian Histidine Kinase 87
4.1 Possible biological activity of the phosphohistidine analogues 88
4.2 Competitive and non-competitive inhibitors of histidine kinases 88
4.3 Synthesis of free triazolylalaninephosphonates 90
4.4 Inhibition of a mammalian histidine kinase by
the free phosphohistidine analogues 96
Chapter 5
Preparation for Solid-Phase Peptide Synthesis and Affinity Chromatography 101
5.1 Introduction to solid-phase peptide synthesis 102
5.2 SPPS by the Muir and Webb groups 104
5.3 Synthesis of Fmoc derivatives of N1- and
N3-phosphohistidine analogues 104
5.4 Preparation for affinity chromatography 109
Chapter 6
Attempted Synthesis of Pyrazole and Imidazole Analogues of Phosphohistidine 111
6.1 Attempted synthesis of a phosphonopyrazole analogue
of N3-phosphohistidine 112
6.1.1 Synthesis of pyrazolylphosphonates 112

xv
6.1.2 Attempted synthesis of protected pyrazolylalanines 116
6.2 Imidazole analogus of N1-phosphohistidine 121
Conclusion 124
Chapter 7
Experimental 125
7.1 General Details 126
7.2 Experimental for Chapter 2 127
7.3 Experimental for Chapter 3 163
7.4 Experimental for Chapter 4 165
7.5 Experimental for Chapter 5 172
7.6 Experimental for Chapter 6 176
References 184


1
Chapter 1
Introduction

2
1.1 Protein phosphorylation
Protein phosphorylation plays a very important role in the regulation of cellular function
in both prokaryotes and eukaryotes, and has been intensively studied since it was
discovered more than 50 years ago.1 Indeed, investigation of intracellular signalling
almost always involves the study of protein phosphorylation.2 In signalling cascades,
proteins are often phosphorylated, in reactions catalysed by protein kinases, and
dephosphorylated by phosphatases (Scheme 1).1 Protein phosphorylation, in its simplest
form, involves the transfer of the -phosphoryl group from adenosine 5'-triphosphate
(ATP) to a side chain of an amino acid residue, most usually a hydroxyamino acid
residue, in the substrate protein, thereby causing conformational changes or protein-
protein interactions that alter its biological activity.3 For example, phosphorylation of
transcription factors is central to the regulation of gene expression.2 There is increasing
evidence that the activation of nuclear steroid hormone receptors, which are
transcription factors, is also controlled by their phosphorylation status.4
NO
OHOH
OP
OP
OP
N
N
N
NH2
ATP
NO
OHOH
OP
OP
N
N
N
NH2
Protein OH Protein O P
O
O
O
O
O
OOO
OO
ADP
O
OO
O O
Kinase
Phosphatase
Scheme 1. Protein hydroxyphosphorylation
Most commonly, it is the hydroxyl of serine, threonine and tyrosine residues that is
phosphorylated to form a phosphoester.3 There is a mass of information about proteins
that contain phosphoesters, and on the associated kinases.3 The free phosphoamino
acids phosphoserine (2), phosphothreonine (3) and phosphotyrosine (4) are illustrated in
Figure 1.

3
O
H3NO
O
2 3 4
POO
H3NO
O
POO
OP
O
OO
H3NO
O
O
OO
Figure 1. Structures of phosphohydroxyamino acids
There are, however, protein phosphorylation events that occur on other amino acid
residues; perhaps the most important of these is at histidine residues. Although
phosphohydroxyamino acids dominate the protein phosphorylation literature,
phosphohistidine has been reported to occur more abundantly than phosphotyrosine, but
less so than phopshoserine.5
1.2 Phosphohistidine
Histidine kinases catalyse the phosphorylation of either N3 (5) or N1 (6) of the side-
chain imidazole of histidine residues (Scheme 2).5
1,3-Diphosphohistidine can be
chemically synthesised; however, this has never been found to occur naturally.5
N
N
HN
NH
O
PO
OO
N
N
HN
NH
O
POO
O
5 6
3
1 1
3
or
proteinprotein
proteinprotein
N
NH
HN
NH
O
3
1
proteinprotein
histidine kinase
Scheme 2. Protein histidine residue phosphorylation
As mentioned above, two isomers of phosphohistidine exist (Figure 2).5 Isomer 8 is
referred to as N1-phosphohistidine by IUPAC and Chemical Abstracts, whereas 7 is
regarded as N1-phosphohistidine by the majority of the research community.6-8
Both of
these numbering systems have been accepted in the literature; however, to avoid
confusion, the more widely used numbering system has been chosen in this thesis, that

4
is, isomer 7 is referred to as N1-phosphohistidine, and the other, isomer 8, as N3-
phosphohistdine.
H3N
O
O
NN P
O
OO
H3N
O
O
NN
PO
OO
Widely used numbering system
IUPAC numbering system
N1-phosphohistidine
N3-phosphohistidine
N3-phosphohistidine
N1-phosphohistidine
7 8
Figure 2. Two numbering systems for phosphohistidine
1.2.1 Synthesis of phosphohistidine
The first report on synthetic phosphorylation of histidine appeared in 1947, when
phosphorus oxychloride was used as an electrophile (Scheme 3).9 An alternative
employing phosphoramidate was published in 1956 (Scheme 3).10
31P NMR studies on the synthesis using phosphoramidate elucidated that N1-
phosphohistidine (7) is formed more rapidly than N3-phosphohistidine (9), which can
be formed directly from histidine or via hydrolysis of the 1-phosphoramidyl group of
1,3-diphosphohistidine (8).5

5
N
N
H3NO
O
PO
OO
8
3
1
+N
NH
H3NO
O
3
1
P
O
ClCl
Cl
Et3N H2O
or
P
O
HOO
NH3 H2O
(A)
(B)
N
N
H3NO
O
PO
OO
3
1
H2O
P
O
OO
7
N
N
H3NO
O
POO
O
9
1
3
Histidine
Scheme 3. Synthetic phosphorylation of histidine
1.2.2 The two-component system in prokaryotic signalling
Histidine phosphorylation is best understood in prokaryotic signalling, in the context of
the „two-component histidine kinase system‟.11,12
This signal transduction system
frequently occurs to connect external stimuli such as temperature, osmolarity, chemo
attractants and pH to various crucial intracellular events such as gene regulation.5,11
In
general, two component systems consist of a protein histidine kinase and a response-
regulator protein. The histidine kinases are the intracellular parts of membranous sensor
proteins that detect environmental changes. Activation of the sensor usually results in
dimerisation of the sensor proteins and autophosphorylation of a conserved histidine
residue in the kinase domain, that is, a self-catalysed phosphorylation reaction involving
ATP. A phosphoryl transfer reaction then ensues between the phosphohistidine in the
histidine kinase domain and a conserved aspartate residue in the response regulator

6
protein (RRP) (Figure 3).5,11,13,14
This results in the activation of the response-regulator
protein, which most often is a transcription factor that interacts with DNA to control
gene expression.5,11,13,14
The phosphorylated response-regulator protein can be
inactivated by specific phosphatases.15
Thus, the activation level of response-regulator
proteins is controlled by their phosphorylation via histidine kinases and their
dephosphorylation by phosphatases.15
Figure 3. Typical two-component histidine kinase signalling pathway. P = Phosphoryl
group, RRP = Response Regulator Protein
Some two-component systems are associated with the virulence of pathogenic bacteria.
For example, stimulation of the VanS-VanR system by vancomycin, the antibiotic of
last resort, leads to the expression of vancomycin-reisistance genes in some strains of
bacteria.11
The emergence of methicillin-resistant Staphylococcus aureus (MRSA)
bacterial strains resistant to vancomycin is a major concern for humanity.13

7
Consequently, effective methods to deal with those antibiotic-resistant bacterial
infections are being investigated, and inhibitors of bacterial two-component histidine
kinases systems could prove useful as novel antibiotics.13
1.2.3 Histidine phosphorylation in mammalian cells
Protein phosphorylation of histidine residues in mammalian cells, although not as well
studied as two-component systems, is also known, and there is evidence that suggests a
correlation between histidine phosphorylation and various cellular functions and
dysfunctions such as cancer.8,16
While, mechanistically, histidine phosphorylation in
bacterial proteins is reasonably well understood, by comparison there is sparse
information about histidine phosphorylation in mammalian cells. The only well-
characterised mammalian histidine kinase, nucleoside diphosphate kinase (NDPK),
catalyses phosphorylation of ribonucleosides via a phosphohistidyl-enzyme
intermediate, in addition to phosphorylating various proteins such as the beta subunit of
some trimeric G-proteins. 5,11,17
Although there are several reports regarding the
importance of histidine phosphorylation in eukaryotic systems, elucidation of signalling
pathways involving phosphohistidine has been painfully slow.5,11
This is mainly
because phosphohistidine is chemically unstable,5,11
as explained in the following
section.
A mammalian histidine kinase of particular interest is histone H4 histidine kinase
(HHK). Histidine phosphorylation on histone H4 is a vital process in regenerating liver,
and HHK activity is upregulated in liver cancer (Figure 4).18
Although elucidation of
the roles and functions of HHK is not complete, HHK could be a potential therapeutic
target for liver cancer.18

8
Figure 4. Histone H4 kinase (HHK) activity in human foetal liver (Fetal), human liver
cancer (HCCT), normal tissues surrounding the cancer (HCCN) and normal adult liver
(Normal)18
1.2.4 Instability of phosphohistidine
Phosphohistidine has a phosphoramidate bond that differs significantly from the
phosphoester in the phosphohydroxyamino acids. Phosphoramidates are significantly
less hydrolytically stable than phosphoesters.5 The ΔG
o for hydrolysis of the
phosphoester group in the phosphohydroxyamino acids in proteins is in the range 6.5
to 9.5 kcal mol–1
, whereas the ΔGo
for hydrolysis of the phosphoramidate group in
phosphohistidine residues is of the order 12 to 14 kcal mol–1
.19
A detailed review on
phosphohistidine by Attwood et al. discusses the thermodynamic stability of
phosphoramidates in relation to carboxylic amides (Figure 5).5 To briefly summarise,
amides 10 are dramatically more thermodynamically stable (with respect to hydrolysis)
than phosphoramidates as, in the former, the lone pair of electrons on the nitrogen is
delocalised onto the carbonyl, and this interaction makes the hydrolytically susceptible
C–N bond stronger. As a result of the delocalisation, the carbonyl carbon is also less
electrophilic, which hampers the attack of nucleophiles such as water. It follows that the

9
charge-separated canonical form 10a greatly contributes to the stability of amides. The
electron delocalisation characteristic of amides also explains the lack of basicity of the
amide nitrogen, and the restricted rotation around the CN bond.
N
O
N
O
PN
O
O
O
PN
O
O
O
10
11
10a
11a
Figure 5. Resonance structures of amides 10 and phosphoramidate 11
Although phosphoramidates 11 are notionally similar to amides, the nitrogen lone pair
electron in phosphoramidates 11 is not delocalised onto the phosphoryl group and the
canonical form 11a is not a significant resonance contributor. Presumably this is due to
poor overlap between the orbital containing the nitrogen lone pair electrons and the
phosphoryl π-bond, although there is some debate about whether the bond commonly
represented as P=O has any -bond character.20
Phosphohistidine is rapidly hydrolysed under acidic conditions, whereas the
phosphoester amino acids can survive low pH. The half-lives of phosphoserine 2 and
phosphothreonine 4 are approximately 18 h in 1 M HCl at 100 °C, and that of
phosphotyrosine 3 is approximately 5 h (Scheme 4).21
In contrast, the half-lives of N1-
phosphohistidine 7 and N3-phosphohistidine 8 are 18 s and 25 s, respectively, in 1M
HCl at 49 °C (Scheme 4).22
It has been proposed that N3-phosphohistidine is kinetically
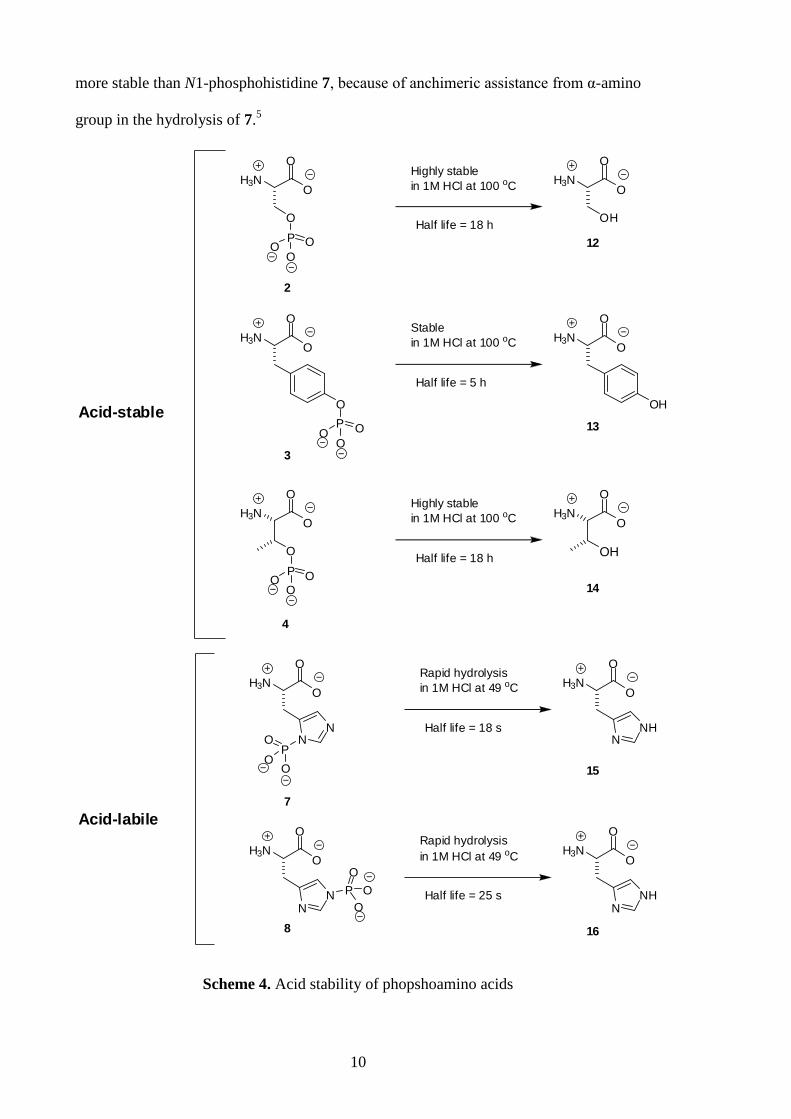
10
more stable than N1-phosphohistidine 7, because of anchimeric assistance from α-amino
group in the hydrolysis of 7.5
H3NO
O
NN
Rapid hydrolysis
in 1M HCl at 49 oC
Half life = 18 s
H3NO
O
NNH
PO
OO
H3NO
O
O
Highly stable
in 1M HCl at 100 oC
Half life = 18 h
H3NO
O
OH
P O
OO
H3NO
OStable
in 1M HCl at 100 oC
Half life = 5 h
H3NO
O
O
P
OO O
OH
H3NO
OHighly stable
in 1M HCl at 100 oC
Half life = 18 h
H3NO
O
O
P OO
O
OH
2
3
4
H3NO
O
NN P
O
O
O
Rapid hydrolysis
in 1M HCl at 49 oC
Half life = 25 s
H3NO
O
NNH
8
7
12
13
14
15
16
Acid-stable
Acid-labile
Scheme 4. Acid stability of phopshoamino acids

11
1.3 Detection of phosphohistidine
The acid-lability of phosphohistidine poses some issues when attempting to undertake
biochemical characterisation of phosphorylated proteins. For example, standard
phosphoamino acid analysis involves hydrolysis of the peptide bonds of a protein with 6
M hydrochloric acid.14
The phosphoester amino acids survive this treatment method but
this process results in rapid hydrolysis of the P–N bond of phosphohistidine.14
Additionally, radioactive peptide mapping and Edman degradation are useful for
detection of phosphorylated serine, tyrosine or threonine residues in a protein.23
However, since these methods also require acidic conditions,23
they cannot be utilised to
analyse phosphohistidine. For these reasons, insights into the role of phosphohistidines
have been slow to surface,24
and several alternative techniques have been developed in
order to measure histidine phosphorylation:8
1.3.1 Detection using [32
P]ATP
Using this methodology, a substrate protein subjected to [32
P]ATP, in the presence of
a histidine kinase, is digested to the component amino acids by the protease cocktail,
pronase E.18
Qualitative identification of the resultant phosphoamino acids, is achieved
by comparison with authentic phosphoamino acid standards, by reversed-phase TLC.
The TLC plate is subsequently analysed with a phosphorimager (Figure 6).18

12
Figure 6. Phosphoamino acid analysis of histidine phosphorylation.18
The left panel is a
ninhydrin-stained reversed-phase TLC plate. A digested phosphohistidine-containing
protein was developed on the TLC alongside with free histidine (left lane),
phosphohistidine (PH), phosphotyrosine (PY), phosphoserine (PS), phosphothreonine
(PT) and phosphoarginine (PR) standards. The right panel is a phosphoimage of the
same TLC plate, showing 32
P-labelled phosphohistidine.
1.3.2 Mass spectrometric detection
Mass analysis of phosphohistidine-containing proteins has made detection of
phosphohistidine possible without radioactive materials.5 Solvent conditions and
matrixes for ionisation and duration of exposure to the solvents or matrixes need to be
carefully selected.5 For example, proteolytic digests of phosphohistidine-containing
proteins were performed, and then the purified phosphohistidine was detected by
positive ion electrospray mass spectrometry (Figure 7).25
However, a relatively large

13
amount of phosphoprotein is required so that this detection can be achieved, and
therefore this method has yet to be applied to proteins in cellular extracts.25
Figure 7. Positive ion ESI-MS spectrum of a proteolysed phosphoprotein showing a
peaks corresponding to phosphohistidine (pH), ( [M+H]+, m/z = 236)
25
A combination of enrichment techniques and mass spectrometry analyses enables
researchers to identify phosphorylated proteins, determine the site of phosphorylation
and characterise conditions under which these phosphorylation occurred.26
For example,
enrichment using immobilised metal-ion affinity chromatography (IMAC) has been
developed for phosphoproteins containing phosphohydroxyamino acids i.e.
phosphoserine, phosphotyrosine and phosphothreonine.26
Napper and co-workers
reported a novel combination of IMAC and positive ion matrix-assisted laser desorption
ionisation time of flight mass spectrometry (MALDI-TOF MS).26
Various resin-
immobilised divalent metal cations such as gallium, nickel, iron, zinc, cobalt, copper
have been immobilised on a resin of IMAC to purify phosphoproteins, and copper(II)
ions were adopted to isolate a phosphohistidine-containing protein26
but a significant

14
amount of nonphosphorylated (HPr and HPr-H2O) was observed in the positive ion
MALDI TOF-MS owing to the instability of phosphohistidine (Figure 8).26
Figure 8. Positive ion MALDI TOF-MS of a sample enriched by IMAC. HPr :
histidine-containing protein, P-HPr : phosphorylated histidine-containing protein, HPr-
H2O : histidine-containing protein which lost water.
The negative ionisation mode also caused dephosphorylation of the intact
phosphohistidine-containing protein (Figure 9).26
These results indicate that detection of
histidine phosphorylated proteins utilising a combination of IMAC and mass
spectrometry has proven challenging.

15
Figure 9. Negative ion MALDI TOF-MS of a sample enriched by IMAC.
Abbreviations as in Figure 8
1.4 Phosphohistidine antibodies
As mentioned above, phosphorylation of proteins on hydroxyamino acid residues is
well understood, and most progress in protein phosphorylation research has been made
in this area.3 Phosphohydroxyamino acids are detectable using conventional
methods.3,14
In addition, there are commercially available antibodies that can selectively
recognise phosphotyrosine and both phosphoserine and phosphothreonine residues.5
An example of the convenience that antibodies afford is depicted in Figure 10. If the
task is to identify phosphoproteins and no antibodies are available, a mixture of proteins
in a cell extract has to be separated by denaturing polyacrylamide gel electrophoresis
(SDS-PAGE), and then each protein is hydrolysed to its component amino acids. These
amino acids can be analysed by mass spectrometry, HPLC and so on, as described
above. Where antibodies are available, the detection of phosphoamino acids is
accomplished by a Western blot, which is vastly more concise than the standard amino
acid analysis. In this analytical technique, proteins are transferred to a membrane after

16
their separation by SDS-PAGE, and then the membrane is immersed in a solution of
antibodies. The antibodies that bind to the phosphoamino acid residues can be detected
by commercially available secondary antibodies, which are either tagged with a
fluorophore or an enzyme that catalyses a chromogenic reaction.
HN
NH
proteinprotein
O
H3O+
POO
O
H3NO
O
POO
O
Separation bySDS-PAGE
Hydrolysis of peptidebonds in each protein
Analysis of amino acids
C
C Hydrolysis of peptide bonds isNOT required
Detection by antibodiesfor phosphoamino acids
O O
Figure 10. Conventional phosphoamino acid analysis and detection of phosphoamino
acids using antibodies. Red antibody = anti-phosphohistidine antibody. Black antibody
= secondary antibody. Yellow sphere C = chromophore.
The significant progress in understanding protein phosphorylation and the associated
signalling pathways, in recent times, has been possible due largely to the use of
phosphoester-recognising antibodies. Conversely, progress on phosphohistidine
research has lagged behind due to the lack of such antibodies. Despite significant
investment of time and effort, efforts to generate phosphohistidine antibodies (see
below) have been unsuccessful.

17
1.4.1 Antibodies
Antibodies are glycoproteins referred to as immunoglobulins, produced by specialised B
lymphocytes in response to a large foreign molecule: the antigen. Antibodies comprise
four polypeptides: two identical heavy chains and two identical light chains held
together to form a „Y-shaped‟ protein structure (Figure 11).27,28
The amino acid
sequence of the amino termini of both the the light and heavy chains differs between
individual antibodies, and these parts are thus referred to as „the variable regions‟.27,28
The remaining parts of the polypeptide chains are referred to as „the constant regions‟,
and are common in each class of immunogloblins.27,28
The variable regions of the light
chain and of the heavy chains consist of 110-130 amino acid residues, and form an
antigen binding site that enables each antibody molecule to recognise a specific antigen
(Figure 11).27,28
. As antibodies differ in the binding site, they can function in a variety
of immune responses and at particular stages of the immune response.27,28
Figure 11. Structure of an antibody

18
1.4.2 Polyclonal and monoclonal antibodies
There are two forms in which antibodies for immunoassays are obtained: „polyclonal
antibodies‟ and „monoclonal antibodies‟. Polyclonal antibodies are a mixture of
different antibodies generated by multiple B lymphocytes.27,28
Antigens are generally
complex and possesses numerous surface features, or epitopes, that are specifically
recognised by the antibody. Many different cell lines of B lymphocytes produce
antibodies to the same antigen, and each antibody clone recognises different epitopes on
the same antigen.27,28
A large number of antigen-specific antibody clones are observed
in serum obtained from an immunised animal, and antibodies purified from these
samples are referred to as polyclonal antibodies.27,28
In contrast, monoclonal antibodies are pure antibodies generated by a single clone of B
lymphocytes and recognising a single epitope on an antigen.27,28
The first discovery of
monoclonal antibodies was in the sera of patients suffering from multiple myeloma,
where high levels of an identical antibody were produced by expansion of malignant
plasma cells.29
A hybridoma technology to produce monoclonal antibodies was invented
by Köhler and Milstein in the mid-1970s, and this achievement earnt them the Nobel
Prize in Physiology or Medicine in 1984.30
In this method, isolated splenic B cells are
fused with myeloma cells from the same species, generating immortal hybridomas,
which retain their abilities to produce unique monoclonal antibodies.30
Table 1. Differences between polyclonal antibodies and monoclonal antibodies
Polyclonal antibodies Monoclonal antibodies
from multiple B lymphocytes from a single clone of B lymphocytes
detect multiple epitopes on an antigen detect a single epitope on an antigen

19
Production of polyclonal antibodies does not require complex technology, and
therefore the production time scale is short.29
On the other hand, the production of a
hybridoma, and hence monoclonal antibodies, is complex and time-consuming.29
While
polyclonal antibodies can be expected to be obtained within several months after
commencing immunisation, it can take more than a year to generate hybridomas and
subsequently produce monoclonal antibodies.29
However, they have the considerable
advantage of being a constant and renewable source of antibodies that, more so than
polyclonal antibodies, provide highly reproducible results.29
In addition, monoclonal
antibodies are monospecific, which enables them to efficiently detect target antigens
among various related molecules in tissues or affinity purification.
The high specificity of monoclonal antibodies can, at times, be a disadvantage.29
In the
case where minor structural transformation of an epitope caused by genetic
polymorphism, glycosylation, denaturation and so on occurs, a monoclonal antibody
may not bind to the antigen.29
In contrast, as polyclonal antibodies are heterogeneous
and recognise multiple epitopes on an antigen, they are less likely to be affected even if
a single or small number of epitopes are structurally transformed.29
Polyclonal
antibodies are also less susceptible to changes in pH and salt concentration changes than
monoclonal antibodies.29
Because both polyclonal antibodies and monoclonal
antibodies have advantages and disadvantages, several factors need to be considered in
deciding which antibodies to be used.29
1.4.3 Haptens
Unlike large molecules like proteins, small molecules do not induce an immune
response, that is, although they are antigenic, they are not immunogenic.31
Thus, in the
case where small molecules are utilised as antigens, they are called „haptens‟ and must
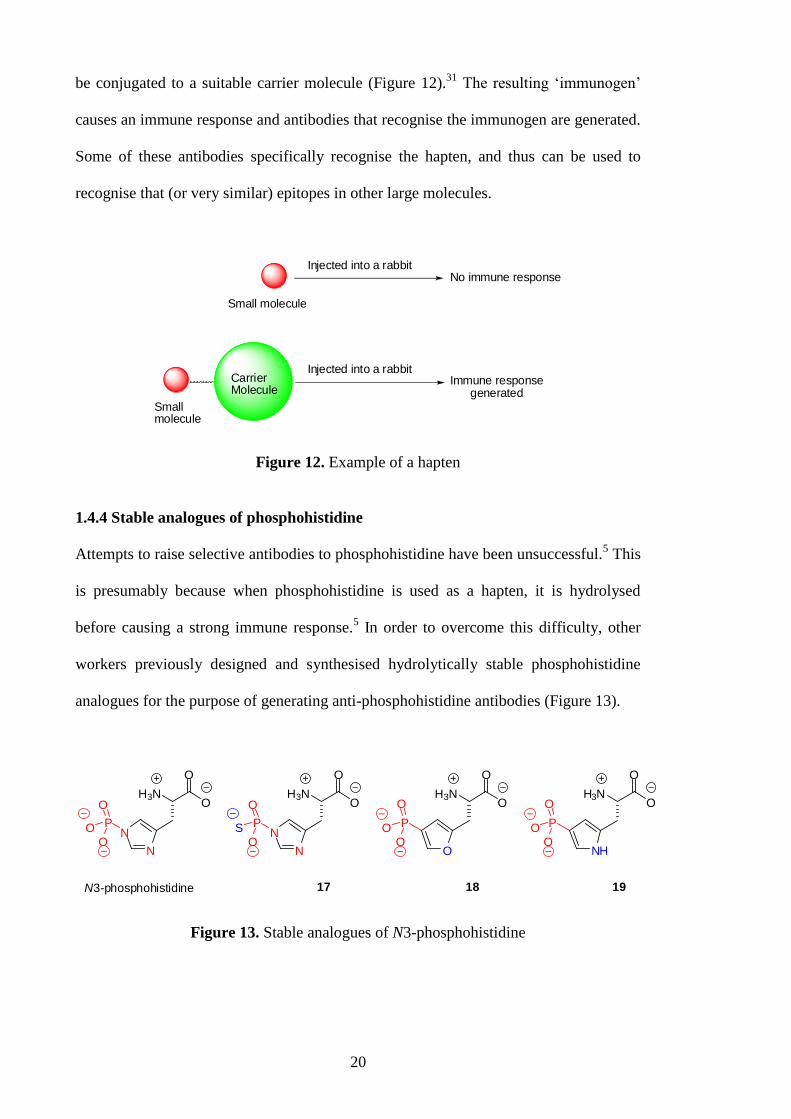
20
be conjugated to a suitable carrier molecule (Figure 12).31
The resulting „immunogen‟
causes an immune response and antibodies that recognise the immunogen are generated.
Some of these antibodies specifically recognise the hapten, and thus can be used to
recognise that (or very similar) epitopes in other large molecules.
CarrierMolecule
Small molecule
No immune responseInjected into a rabbit
Smallmolecule
Immune responsegenerated
Injected into a rabbit
Figure 12. Example of a hapten
1.4.4 Stable analogues of phosphohistidine
Attempts to raise selective antibodies to phosphohistidine have been unsuccessful.5 This
is presumably because when phosphohistidine is used as a hapten, it is hydrolysed
before causing a strong immune response.5 In order to overcome this difficulty, other
workers previously designed and synthesised hydrolytically stable phosphohistidine
analogues for the purpose of generating anti-phosphohistidine antibodies (Figure 13).
N
N
H3NO
O
PSO
O
O
H3NO
O
POO
O
NH
H3NO
O
POO
O
N
N
H3NO
O
POO
O
N3-phosphohistidine 17 18 19
Figure 13. Stable analogues of N3-phosphohistidine

21
The Turck group prepared a synthetic peptide containing thiophosphohistidine 17
(Scheme 5).32
In their strategy, the peptide 20 was treated with thiophosphoryl chloride,
followed by RP-HPLC purification to afford the peptide with a thiophosphohistidine
residue, the identity of which was confirmed by ESI-MS.32
LAAAGGGG NH2NH
O
NNH
PSCl3 LAAAGGGG NH2NH
O
NN P
O
SO
Et3N
H2O
20 21
Scheme 5. Synthesis of a thiophosphohistidine-containing peptide by Turck‟s group
The Pirrung group also synthesised thiophosphohistidine using potassium
thiophosphoramidate 22 (Scheme 6).33
H3NO
O
NNH
H3NO
O
NN P
O
OS
P
O
HOS
NH2
H2O
16 23
22
Scheme 6. Synthesis of thiophosphohistidine 23 by Pirrung‟s group
Thiophosphohistidine is hydrolytically more stable than phosphohistidine, possibly
because sulfur has lower electronegativity than oxygen, thereby imparting an increase in
the stability of the thiophosphoramidate P–N bond.5,33
However, attempts to produce
antibodies utilising the thiophosphohistidine analogue as a hapten were unsuccessful.34
The inference for this failure is that thiophosphohistidine is still not sufficiently
hydrolytically stable in vivo to allow an immune response to develop.
The other method of creating a more stable analogue involves replacing the
phosphoramidate P–N bond with a P–C bond (giving a phosphonate), which is

22
hydrolytically inert. Reymond and co-workers made the furylphosphonate analogue 18
of N3-phosphohistidine (8) in five steps from the furan 2435
using a variation of the
amidomalonate synthesis (Scheme 7).36
Radical bromination of the furan 24 using N-
bromosuccinimide gave the bromide 25 in excellent yield.37
α-Alkylation of 26 with 25
provided the diester 27.37
Hydrolysis and decarboxylation of 27 gave the racemic amino
acid derivative 28.37
Enzymatic resolution using papain,38
by which only S-configured
NBoc-protected amino acid esters are hydrolysed, furnished the enantiopure carboxylic
acid 29. The free amino and phosphonic acid groups were revealed by heating the
carboxylic acid in 1M HCl under reflux to afford 30 as a hydrochloride.36
OP OEt
OEt
ONBS
AIBN
CCl4
Reflux
OP OEt
OEt
OBr
BocNH
OEtO
OEt
O
NaH
EtOH
BocNH CO2Et
CO2Et
OP
O
OEt
OEt
1) NaOH, EtOH2) HCl3) Dioxane, reflux
BocNH
OP
O
OEt
OEt
OEt
O
BocNH
OP
O
OEt
OEt
OH
O
Reflux
HClClH3N
OP
O
OH
OH
OH
OPapain
96% 80%
76%
95%
40%
24 25 27
26
30 29 28
Scheme 7. Synthesis of a furylphosphonate analogue of N3-phosphoihistidine by the
Reymond group36
However, successful production of anti-phosphohistidine antibodies using the N3-furan
analogue 30 has not been reported. This is possibly because the furan oxygen is a
weaker hydrogen bond acceptor than the nitrogen of phosphohistidine. Alternatively, as
there is no record in the literature, it may be that it was not attempted.

23
The Reymond group also synthesised the pyrrole analogue 19, and the Attwood
research group attempted to raise antibodies utilising a short peptide containing this
analogue (Scheme 8).39
Although antibodies were raised, they only recognised the
peptide containing 19 and were not cross-reactive with phosphohistidine-containing
peptides.39
H3NNH
HN
NH
O O
O
HN
O
O
O
NHPO
OO
N
O
O
HS
Hapten
H3NNH
HN
NH
O O
O
HN
O
O
O
NHPO
OO
S
N
O
O
KLH
KLH Immunogen
Carrier protein
NH
H3NO
O
PO
O
O
19
Scheme 8. Carrier protein-hapten conjugation strategy used by the Attwood group
There are several possible reasons for this failure:
1. While phosphorylated histidine residues in proteins are generally internal, the
analogue contains a terminal phosphohistidine analogue residue. The negatively charged
carboxylate is very different from the neutral peptide linkage occurring in native
histidine-phosphorylated proteins.
2. The non-phosphorylated ring nitrogen in phosphohistidine residues is a good
hydrogen bond acceptor, whereas the pyrrole nitrogen is a hydrogen bond donor.
Hydrogen bonding to the nitrogen of phosphohistidine is likely to be critical in antibody
recognition.

24
3. The pyrrole analogue only mimics N3-phosphohistidine, and therefore if there are
N1-phosphohistidine residues in a protein of interest, these would probably not be
recognised by the antibodies due to the different spatial arrangement of phosphoryl
group relative to the rest of the hapten.
1.5 Stable phosphohistidine analogue targets: Aims
At the outset, the primary aim of the research described in this thesis was to
1. Synthesise hydrolytically stable phosphohistidine analogues.
2. Produce generic phosphohistidine antibodies (i.e., that recognise the phosphohistidine
epitope in all, or most histidine-phosphorylated proteins).
3. Investigate the intrinsic biological activity of the analogues.
4. Undertake solid-phase peptide synthesis to incorporate the analogues into short
peptides, for the purpose of generating antibodies to specific histidine-phosphorylated
proteins.
Several analogues that addressed all of the deficiencies of the previous phosphohistidine
analogues described above were designed. A subset comprising a triazole (31) and
imidazole (33) analogue of N1-phosphohistidine and a triazole (32) and pyrazole (34)
analogue of N3-phosphohistidine is shown in Figure 14.

25
N
N
H3NO
O
PO
OO
N N
HN
OR'
O
PO
OO
N NN
HN
OR'
O
PO
OO
N
N
POO
OH3N
O
O
RR
N
NPO
O
O
HN
O
OR'
N N
NPO
O
O
HN
O
OR'R R
N3-phosphohistidine
N1-phosphohistidine
32
31
34
33
Figure 14. Primary N1- and N3-phosphohistidine analogue targets
In these compounds, R and R' represent an appropriate protecting group, which can be
removed selectively.
Like the previous phosphohistidine analogues 18 and 19, these compounds are
phosphonates and, instead of the phosphoramidate P–N bond, contain a C–P bond,
which makes them non-hydrolysable. The other reasons why these analogues were
chosen for the potential generation of antibodies are detailed below:
1. The phosphoryl group and the unsubstituted heterocyclic nitrogen in N1- and N3-
phosphohistidines, illustrated in red in Figure 14, would play an important role in the
binding interactions with antibodies. In the analogues, both of these features are
retained.
2. The planar, aromatic nature and sp2-hybridisation of all atoms in the imidazole ring
of phosphohistidine are maintained by the introduction of another one or two

26
heteroatoms, marked in blue, into the heterocycle. The shapes of these analogues are as
similar to the phosphohistidines as possible and any non-covalent antibody-antigen
interactions with the heteroaromatic ring should therefore be transferable.
3. By using the chiral pool, the analogues can be prepared without any need for
enantioselective reactions or resolutions, which simplifies the synthesis and purification
and avoids waste.
With respect to the last statement, the proposed analogues could be obtained from L-
serine, which is an inexpensive and enantiopure starting material (Scheme 9).
Importantly, throughout the synthesis, the reaction conditions have to be chosen
carefully to avoid racemisation.
H3N
O
O
L-serine
HO
N
NPO
O
O
HN
O
ORR
N NN
HN
OR
O
PO
OO
R
N N
NPO
O
O
HN
O
ORR
N N
HN
OR
O
PO
OO
R
Scheme 9. Simplified retrosynthetic analysis for the target phosphohistidine analogues

27
In 2010, two independent groups published work very similar to aspects of the research
described in this thesis. To simplify comparison of our work with theirs, that work is
summarised in the next two sections.
1.6.1 Similar work published by the Muir group
In 2010 the Muir research group, at Rockerfeller University, reported the NBoc-
protected triazolylalaninephosphonate esters 35 and 36 (Figure 15), analogues of N1-
and N3-phosphohistidine, respectively.40
BocNOH
OH
N
N N
P
O
OEt
OEt
BocNOH
OH
N NN
PO
OEtEtO
35 36
Figure 15. Proteted triazolylphosphonate analogues of phosphohistidines synthesised
by the Muir group
The synthesis of 35 and 36 is shown in Scheme 10. A copper(I)-catalysed cycloaddition
of the azide 37 and alkyne 38 gave the 1,4-triazole 36 exclusively.41
In contrast, the
ruthenium(II)-catalysed reaction of 37 and 38 did not proceed as expected, which
required them to protect the carboxylic acid by reaction with benzyl bromide, providing
the benzyl ester 40. The ruthenium(II)-catalysed reaction42
of the azide 40 and alkyne
38 provided the 1,5-triazole 41, and the crude product was subsequently subjected to
hydrogenolysis to afford the free carboxylic acid 35.

28
BocNOH
OH
N
N N
P
O
OEt
OEt
BocNOBn
OH
N NN
PO
OEtEtO
36
BocNOH
OH
N3
37
BocNOBn
OH
N3
BocNOH
OH
N NN
PO
OEtEtO
35
40
41
P H
O
EtO
EtO
38
CuI, DIPEA
DMF, 72%
Br
DIPEA, DMF
63%
Cp*RuCl(COD)
P H
O
EtO
EtO
38
PhMe, r.t.
Pd/C
H2
THF
68% (Two steps)
39
Scheme 10. Synthesis of peptide precursors 35 and 36 by Muir‟s group
Compounds 35 and 36 were used in the solid-phase synthesis of peptides containing the
phosphohistidine analogues, specifically, the histone H4 tail peptides 45 and 46. The
phosphonic acid was revealed during the cleavage of the synthesised peptides from the
resin by hydrofluoric acid (Scheme 11).40
Furthermore, Muir and co-workers extended
the histone H4 tail peptide 46 to full-length histone H4 utilising native chemical
ligation.40

29
H2NHN
RKVLR
O
H2N
Boc SPPS
HATU DIPEA
DMF
HATU DIPEA
DMF
HNRKVLR
O
HN
OBocNH
N
N
N
PO
EtOOEt
NH2
RKVLR
O
HN
ONH
N
N
N
PO
HOOH
CGAKR
O
Ac
HNRKVLR
O
HN
OBocNH
N
N
NPEtO
EtO
NH2
RKVLR
O
HN
ONH
N
N
NPHO
HO
CGAKR
O
Ac
O
O
Boc SPPSCleavage from the resin
Boc SPPSCleavage from the resin
42
36 35
44 43
46 45
Scheme 11. Solid-Phase Peptide Synthesis (SPPS) of histone H4 Tail Peptides
containing stable triazolylphosphonate analogues of phosphohistidine by the Muir
group40
Polyclonal antibodies were raised using peptide 46 as an immunogen, and the antibodies
generated specifically recognised phosphorylated histone H4, but not the non-
phosphorylated protein.40
Although these antibodies do not recognise phosphohistidine
residues in other proteins (i.e. they are not generic), this is the first example of
antibodies that specifically recognise the histidine-phosphorylated form of a protein.
This result showed that the triazolylphosphonate analogues are good mimics of
phosphohistidines and, to some extent, validated the approach to produce generic
phosphohistidine antibodies described in this thesis.

30
1.6.2 Similar work published by the Webb group
Most recently, Webb and co-workers, from the University of Leeds, reported solid-
phase peptide synthesis using an Fmoc-protected triazolylphosphonate analogue 47 of
N3-phosphohistidine (Figure 16).43
FmocNOH
O
N
N N
P
O
OEt
OEt
H
47
Figure 16. SPPS-ready N3-phosphohistidine analogue synthesised by Webb‟s group.43
The Webb group‟s synthesis of 47 is depicted in Scheme 12. A Cu(II)-catalysed diazo-
transfer reaction converted N--Boc-diaminopropionic acid 48 to the azide 37, which
was subjected to a cycloaddition with the alkyne 38 in the presence of a copper(I)
catalyst, affording the triazole 36, reported by the Muir group. The Boc group was
removed by trifluoroacetic acid (TFA) to produce, after neutralisation, the free amino
acid 49. Treatment with FmocCl furnished the Fmoc-protected amino acid 47.
Alternatively, a copper(I)-catalysed cycloaddition of commercially available Fmoc-
azidoalanine 50 and the alkyne 38 afforded 47 in excellent yield.

31
BocNOH
OH
N3
BocNOH
OH
NH2
BocNOH
OH
N
N N
P
O
OEt
OEt
H2NOH
O
N
N N
P
O
OEt
OEt
FmocNOH
O
N
N N
P
O
OEt
OEt
H
FmocNOH
OH
N3
48 37
47
36
4950
TfN3, CuSO4
K2CO3, MeOH
78% CuSO4
sodium ascorbate
P H
O
EtO
EtO
38
tBuOH/H2O
88%
(1) TFA(2) H2O
92%
68%CuSO4
sodium ascorbate
P H
O
EtO
EtO
38
tBuOH/H2O
96%
FmocCl
Dioxane/H2O
Na2CO3
Scheme 12. Synthesis of the SPPS-ready N3-phosphohistidine analogue 47 by Webb‟s
group43
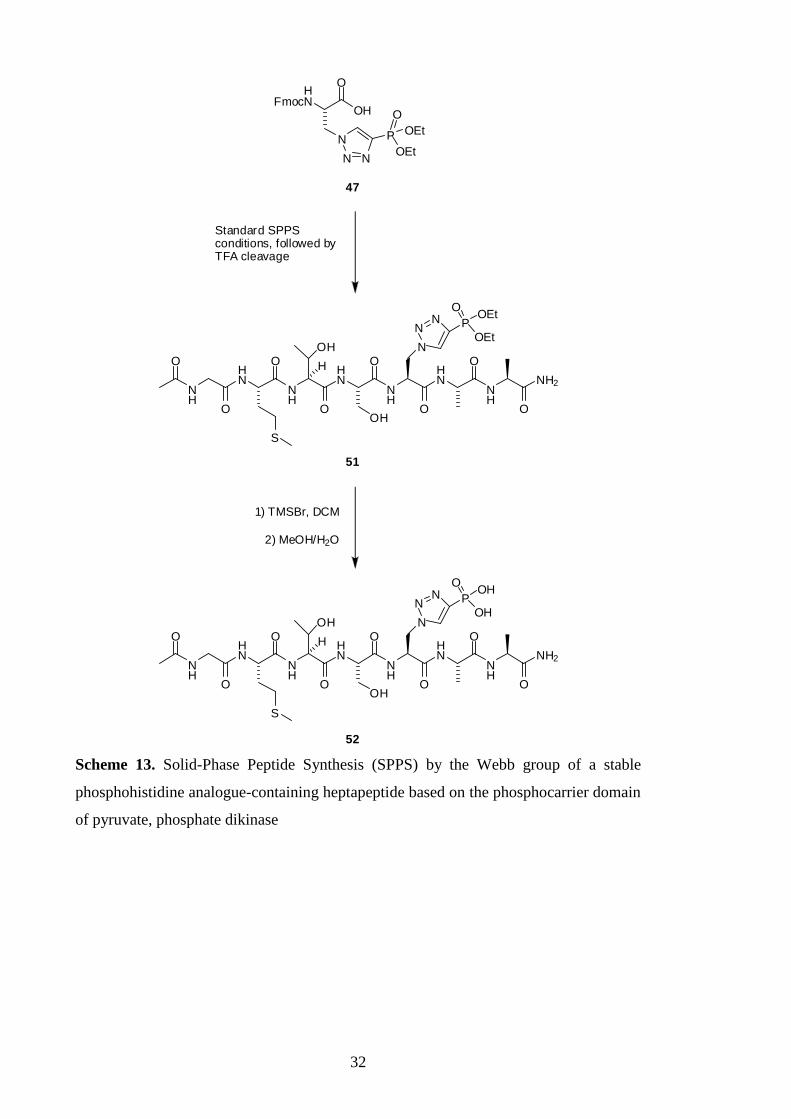
A standard Fmoc SPPS protocol was applied to incorporate 47 into the heptapeptide 51,
based on the phosphocarrier domain of pyruvate, orthophosphate dikinase (Scheme 13).
In contrast to the Muir group‟s approach, the phosphonic acid of the heptapeptide 51
was not revealed during cleavage of 51 from the resin with TFA. However, the
deprotection was achieved using trimethylsilyl bromide (TMSBr), providing the
heptapeptide 52 as a mixture with the mono- and diethyl phosphoesters.43
Purification
of the heptapeptide 52 was accomplished by HPLC.
32
FmocNOH
OH
N
N N
P
O
OEt
OEt
NH
HN
NH
HN
NH
HN
NH
O
O
O
O
O
O
O
NH2
O
S
H
OH
OH
N
NN P
OEt
OEtO
NH
HN
NH
HN
NH
HN
NH
O
O
O
O
O
O
O
NH2
O
S
H
OH
OH
N
NN P
OH
OHO
Standard SPPSconditions, followed byTFA cleavage
1) TMSBr, DCM
2) MeOH/H2O
47
51
52
Scheme 13. Solid-Phase Peptide Synthesis (SPPS) by the Webb group of a stable
phosphohistidine analogue-containing heptapeptide based on the phosphocarrier domain
of pyruvate, phosphate dikinase

33
Chapter 2
Synthesis of Triazolylphosphonate
Analogues of Phosphohistidine

34
2.1 Strategy
The plan to prepare the triazolylalanine (Tza) analogues of phosphohistidine involved
azide-alkyne cycloadditions (Scheme 14). It was envisioned that this reliable reaction
could provide access to both N1- (55) and N3- (56) phosphohistidine analogues.
HN
O
O
N3
R1 R2
P
O
O
O
H
R3
HN
O
O
N
R1 R2HN
O
O
N
R1 R2
N
N
N NP
P
O
O
OO
O
+
R3
R3
R3R3
OR353
54
55 56
Scheme 14. Plan to synthesise the Tza analogues of phosphohistidine. R1–R
3 are
protecting groups that can potentially be removed orthogonally
Thus, a protected azidoalanine 53 and an ethynylphosphonate 54 were initially required.
2.2 Synthesis of a protected azidoalanine
The synthesis of the protected triazole analogues of phosphohistidine began with the
literature preparation of the azide 6044
from the inexpensive, enantiopure starting
material L-serine 57 (Scheme 15). Fischer esterification of 57 gave the methyl ester
hydrochloride 58,45
and the amino group was protected as a t-butyl carbamate, affording
59.45

35
H3N
O
O
OH
BocN
O
OMe
OH
HClH3N
O
OMe
OH
BocN
O
OMe
N3
H
HN3
PPh3
DEAD
THF
SOCl2
MeOH
Boc2O
Et3N
Dioxane
MeCN74%
90%
72%
57 58 59
60
Scheme 15. Synthesis of N-Boc-azidoalanine methyl ester 60
2.2.1 Mitsunobu reaction
The substitution reaction between an alcohol and a weak acid, which was later named
for its inventor, was first reported by Oyo Mitsunobu in 1967. It is utilised to convert an
alcohol to the corresponding ester, nitrile and so on, with inversion of the
configuration46
and has been widely used due to its reliability, effectiveness and
versatility.47
With respect to the mechanism (Scheme 16),46
the reaction commences
with formation of the adduct 63 between triphenylphosphine 61 and an
azodicarboxylate, most commonly diethyl azodicarboxylate (DEAD) 62. The proton of
the acid 64 (the conjugate base of which is the nucleophile) is abstracted by the
generated betaine 63. Due to a strong affinity between oxygen and phosphorus, the
positively charged phosphorus in the phosphonium intermediate 65 is attacked by the
oxygen in the alcohol, (most probably with general base catalysis, as shown in Scheme
16), providing the anion 68 and O-alkylated phosphine oxide 69. The nucleophile 67
undergoes an SN2 reaction with the electrophile 69 to afford the substation product 71
and triphenylphosphine oxide 72.

36
N NEtO OEt
O O
Ph3P
N N OEt
O
EtO
O
PPh3
NuH
N NH
OEt
O
EtO
O
PPh3
R1
R2
O
61
62 63
64
65 66
H
Nu
N NH
OEt
O
EtO
O
R1
R2
OPh3P
NH
NH
OEt
O
EtO
ONu
R1
R2
OPh3P
R1
R2
Nu Ph3P O
68 6970
64
69
67
7271
67
HNu
Scheme 16. Mechanism for the Mitsunobu reaction
In the case where a very weak acid participates in the reaction, the nitrogen anion 68
can attack the electrophile 69, furnishing the undesired side-product 75 (Scheme 17).
N N OEt
O
EtO
O
PPh3
R3
R4
OH
N NH
OEt
O
EtO
O
PPh3
R3
R4
O
N NH
OEt
O
EtO
O
63
65 74
68
R3
R4
OPh3P
R3
R4
NEtO
O
NHEtO
O
Ph3P O69
72
75
73
Scheme 17. Formation of the undesired side-product 75

37
The Mitsunobu reaction of 59 with hydrazoic acid converted the alcohol to the
corresponding azide 6044
in acceptable yield (Scheme 15). The optical rotation of the
azide 60 was close to the published value.48
As mentioned above, DEAD 62 is the most common azo compound used for Mitsunobu
reactions, and the reagent used in the literature synthesis of 60.44
Although it is
commercially available, it is a sea freight item and hard to acquire in Australia.
Accordingly, DEAD was substituted with diisopropyl azodicarboxylate (DIAD), which
is more readily available. However, the azide 60 and the hydrazine by-product derived
from DIAD had similar mobility on silica gel, which hampered the purification of the
azide. It was therefore necessary to synthesise DEAD 62. Fortunately this was achieved
easily, in two steps from hydrazine 76 (Scheme 18). Diethyl hydrazodicarboxylate 7049
was obtained by the reaction of hydrazine 76 and ethyl chloroformate 77. Oxidation of
70 with concentrated nitric acid produced DEAD 62.50
NH2H2N
Cl
O
O
Na2CO3
EtOH/H2O
NH
NH
O
O O
Oconc. HNO3
CHCl3
NN
O
O O
O
60%70%
76 70 62
77
Scheme 18. Synthesis of DEAD
2.3 Synthesis of diethyl ethynylphosphonate
The ethynylphosphonate 8243,51
required for the Huisgen cycloaddition reactions and
their metal-catalysed variants was synthesised as depicted in Scheme 19. TIPS-
acetylene 78 was treated with ethylmagnesium bromide to form the magnesium
acetylide, which was quenched with diethyl chlorophosphate 80,52
affording the TIPS-
protected ethynylphosphonate 81 in good yield (Method A). Although 80 is
commercially available, it is also readily prepared from diethyl phosphite 79 (Scheme

38
19). Deprotection of 81 was achieved with potassium fluoride to furnish the
ethynylphosphonate 82 in acceptable yield.53
TIPS-actylene was used because with
TMS-acetylene 83 the intermediate 84 was not able to be isolated in pure form; the silyl
group was spontaneously cleaved during normal purification techniques. However,
subsequently, it was found that the target alkyne 82 was obtained in the best overall
yield when the deprotection of the crude intermediate 84 was deliberately completed by
treatment with weak aqueous base (Method B).51
Method A
H TIPS
EtMgBr
THF
P
O
EtO
EtOCl
P
O
EtO
EtOH
Et3N
CCl4
P TIPS
O
EtO
EtO
P H
O
EtO
EtO
KF
MeOH80%
61%
85%
78 81
80
79
82
Method B
H TMS
EtMgBr
THF
P
O
EtO
EtOCl
P TMS
O
EtO
EtO
P H
O
EtO
EtO
K2CO3
H2O
75% (overall)80
84 8283
Scheme 19. Synthesis of the ethynylphosphonate 82
Webb and co-workers have recently reported that the ethynylphosphonate 82 can be
synthesised by direct addition of the ethynylmagnesium bromide 85 to diethyl

39
chlorophosphate 80 (Scheme 20).43
Their approach reduces the number of steps but the
alkyne 82 is produced in better yield by the methods described above.
H MgBr
THF
P
O
EtO
EtOCl
P H
O
EtO
EtO
24%
80
85 82
Scheme 20. Synthesis of the alkyne 82 by the Webb group43
Because of the expense of the ethynylsilanes above, alternative syntheses of
ethynylphosphonate 82 were investigated (Scheme 21). The Grignard reagent formed by
treatment of the much cheaper 2-methyl-3-butyn-1-ol 86 with an excess of
ethylmagnesium bromide reacted smoothly with diethyl chlorophosphate 80, affording
the protected ethynylphosphonate 87.
H
EtMgBr
THF
P
O
EtO
EtOCl
P
O
EtO
EtO
OHOH
77%
Cl
P(OEt)3
NiCl2 OH
47%
80
86 87 88
Scheme 21. Synthesis of the protected ethynylphosphonate 87
Compound 87 was also formed by a nickel(II)-catalysed Arbuzov reaction of
triethylphosphite and the chloride 88.54
The Arbuzov reaction is, traditionally, the
reaction of a trialkylphosphite 89 and an alkyl halide 90 to provide the corresponding
phosphonate 93 (Scheme 22).55,56

40
(EtO)3PRX P
O
EtO
OEtR X P
O
EtO
OEtR
+ EtX
8990
91 92 93 94
Scheme 22. Mechanism for the Arbuzov reaction
The Arbuzov reaction can also be applied to aryl or alkynyl halides (Scheme 23), but
requires transition metal catalysis; nickel(II) chloride (NiCl2) 95 is used in many
cases.55-57
In the proposed reaction mechanism,55,56
NiCl2 95 is reduced in situ to a
tetrakis(trialkylphosphite) nickel(0) complex 96. Oxidative addition of 96 to an alkynyl
halide (97 in this case) gives 98, and reductive elimination subsequently furnishes the
desired phosphonate 102, regenerating Ni(0).

41
Ni
PEtO
OEt
OEt
PEtOOEt
OEt
X Ni
PEtO
OEt
OEt
PEtOOEt
OEt
X-
+
Ni
PEtO
OEt
O
PEtO
OEt
OEt
P
OEt
OEtOEt
EtX
P(OEt)3
NiCl2
P(OEt)3
X
P
O
OEt
OEt
[(EtO)3P]2Ni
95
[(EtO)3P]4Ni
96
97
98 99
100
102
101
89
R R R
RR
89
Scheme 23. Mechanism for the Nickel(II)-catalysed Arbuzov reaction
Unfortunately, the deprotection of 87 did not proceed as expected (Scheme 24). Under
each of the conditions attempted,58-60
complete consumption of the starting material 87
was observed, but the desired ethynylphosphonate 82 was not produced. These
alternative routes to the alkyne 82 had to be abandoned at this point, but fortunately
there was sufficient material to proceed with.

42
P
O
EtO
EtO
P H
O
EtO
EtO
OH
(1) KOH, i-PrOH, 70 oC
(2) NaH, toluene, 110 oC
(3) K2CO3, 18-crown-6, toluene, 110 oC
Attempted conditions
87 82
Scheme 24. Attempted deprotection of 82
2.4 Azide-Alkyne Cycloadditions
2.4.1 Background
1,3-Dipolar cycloadditions are pericyclic reactions of a dipolarophile, for example an
alkene or an alkyne, and a 1,3-dipolar compound such as a nitrile oxide or an azide,
producing the corresponding 5-membered heterocyclic compounds.61,62
A particularly
popular example42,63
is the 1,3-dipolar cycloaddition between an azide and an alkyne to
furnish a 1,2,3-triazole, known as a Huisgen cycloaddition after Rolf Huisgen
established the general application of this reaction in the 1960s.61,62
In the thermal
reaction of an azide 103 and an alkyne 104, a mixture of the two regioisomers 105 and
106 is afforded, and electronic and steric factors affect the regioselectivity of the
reaction (Scheme 25).42,63
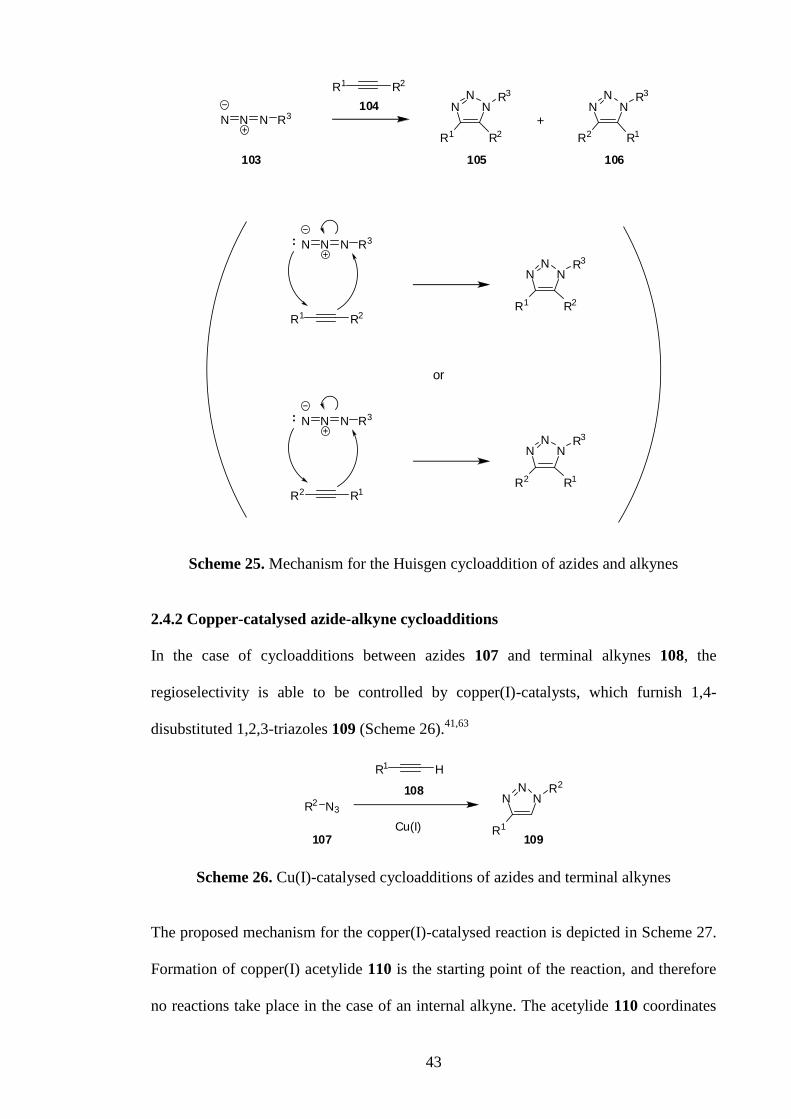
43
R1 R2
N N N R3
NN
NR3
R2R1
R2 R1
N N N R3
NN
NR3
R1R2
or
NN
NR3
R2R1
NN
NR3
R1R2
+
R1 R2
N N N R3
103 105 106
104
Scheme 25. Mechanism for the Huisgen cycloaddition of azides and alkynes
2.4.2 Copper-catalysed azide-alkyne cycloadditions
In the case of cycloadditions between azides 107 and terminal alkynes 108, the
regioselectivity is able to be controlled by copper(I)-catalysts, which furnish 1,4-
disubstituted 1,2,3-triazoles 109 (Scheme 26).41,63
R1 H
R2 N3N
NN
R2
R1Cu(I)107
108
109
Scheme 26. Cu(I)-catalysed cycloadditions of azides and terminal alkynes
The proposed mechanism for the copper(I)-catalysed reaction is depicted in Scheme 27.
Formation of copper(I) acetylide 110 is the starting point of the reaction, and therefore
no reactions take place in the case of an internal alkyne. The acetylide 110 coordinates

44
to an azide 107 to regiospecifically form a copper(III) metallacycle 112, and subsequent
reductive elimination, followed by hydrolysis of the copper-carbon bond in 113, gives a
1,4-substituted triazole 109.63
NN
N
R1
R2
[LnCu]+
CuLn
NN
N
R1
R2
R1 H
R1 CuLn
N N N R2
R1 CuLn
N N N R2
N N NR2
107
108
110
111
112
113 109
CuLn
R1
C
Acetylideformation
Reductiveelimination
Productrelease
Coordination betweenazide and acetylide
Regiospecific formationof Cu(III) metallacylce
Scheme 27. Proposed mechanism for copper(I)-catalysed cycloadditions of azides and
terminal alkynes63
There are several sources of copper(I),63
including the iodide, bromide and triflate, that
can be used to catalyse these reactions. A nitrogen base, for example, 2,6-lutidine,
triethylamine, diisopropylethylamine or pyridine, is required in these cases in order to
impede the formation of undesired by-products such as diacetylenes, bis-triazoles, and
5-hydroxytriazoles, although this is not always successful.41,63
Accordingly, it is
generally more efficient to generate copper(I) in situ by reducing copper(II) salts.63
The
combination of copper(II) sulfate and sodium ascorbate is particularly popular.63
Under
these conditions, the reaction can be conducted at room temperature and in a wide range
of solvents, such as aqueous tert-butyl alcohol, ethanol, dimethylformamide and water

45
without any organic co-solvent.63
The modular nature, high and reliable yields, atom
economy, wide scope and simple purification of copper(I)-catalysed azide-alkyne
cycloadditions has led them to become the prototypical „click‟ reaction.64,65
In
particular, copper(I)-catalysed cycloadditions are extremely powerful tools for
bioconjugation, both in vitro and in vivo, because of the perfect regioselectivity and
chemoselectivity of the reactants, as well as the high reliability.65
Azide-alkyne
cycloadditions have had a significant impact on a variety of research fields, including
medicinal and materials chemistry.65
2.4.3 Ruthenium-catalysed azide-alkyne cycloadditions
In contrast to copper catalysts, the cycloadditions of azides 114 and terminal alkynes
115 in the presence of ruthenium(II)-catalysts, give 1,5-substituted triazoles 116
predominantly (Scheme 28).42
R1 H
R2 N3N
NN
R2
Ru(II)
114
115
116
R1
Scheme 28. Ruthenium(II)-catalysed cycloadditions of azides and terminal alkynes
A mechanistic proposal for the ruthenium(II)-catalysed reactions is described in Scheme
29.66
Unlike the copper catalysed reaction, the Ru(II)-catalysed variant does not involve
formation of a metal acetylide and can be applied to both terminal and internal
alkynes.42,66
The displacement of the spectator ligands by an azide 114 and alkyne 115
initiates the reaction and produces 118. Subsequent oxidative coupling gives 119,
followed by reductive elimination of the ruthenium and the release of the 1,5-substituted
triazoles 116. Several commercially available ruthenium(II) catalysts, including
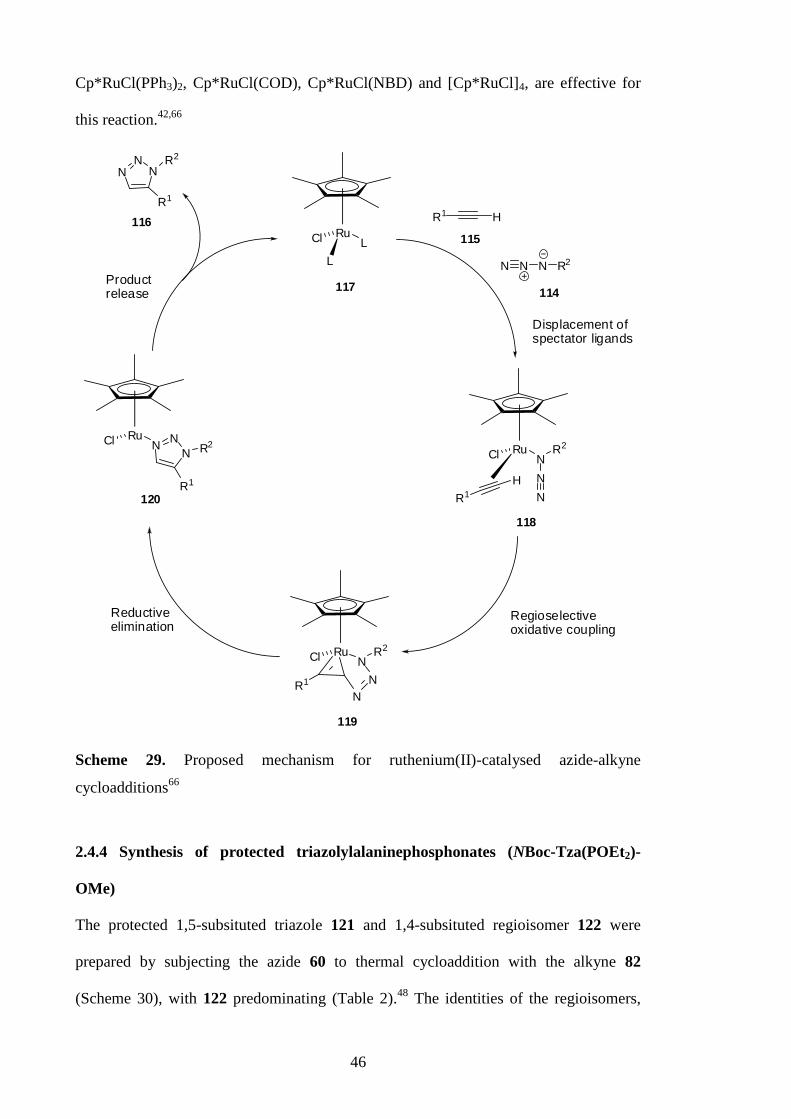
46
Cp*RuCl(PPh3)2, Cp*RuCl(COD), Cp*RuCl(NBD) and [Cp*RuCl]4, are effective for
this reaction.42,66
RuCl
L
L
R1
H
N N N R2
RuCl N
N
N
R2
RuCl N
N
NR1
R2
R1 H
RuCl NN
NR2
R1
N NN R2
R1
Displacement ofspectator ligands
Regioselectiveoxidative coupling
Reductiveelimination
Productrelease
116
115
114117
118
119
120
Scheme 29. Proposed mechanism for ruthenium(II)-catalysed azide-alkyne
cycloadditions66
2.4.4 Synthesis of protected triazolylalaninephosphonates (NBoc-Tza(POEt2)-
OMe)
The protected 1,5-subsituted triazole 121 and 1,4-subsituted regioisomer 122 were
prepared by subjecting the azide 60 to thermal cycloaddition with the alkyne 82
(Scheme 30), with 122 predominating (Table 2).48
The identities of the regioisomers,

47
which were easily separated by column chromatography, were confirmed by an HMBC
experiment (Figure 17). Specifically, the 1,4-substituted triazole 122 was identified by a
three-bond correlation between the -protons and the triazolyl carbon. In the 13
C NMR
spectra of both regioisomers the signals arising from the two triazolyl carbons are split
into doublets, due to coupling with the phosphorus nucleus (Figures 18 and 19). In
particular, the C–P coupling constant is very large (220 Hz). In contrast, H–P coupling
was not observed with the triazoles‟ protons in the 1H NMR spectra.
The formation of both isomers was both expected and useful, giving access to both
protected N1-PHis (121) and N3-PHis (122) analogues. However, from the perspective
of preparative efficiency, it was of interest to control the regioselectivity of the
cycloaddition reaction and, accordingly, catalysis was explored. The copper(I)-catalysed
reaction63
furnished the triazole analogue 122 exclusively and in excellent yield (Table
2). The exclusive formation of the isomeric analogue 121 is far more challenging.
Ruthenium(II) catalysis successfully reversed the regioselectivity under these conditions
(Table 2).42
However, the reaction was not regiospecific and it would be preferable to
optimise the reaction conditions to accomplish the exclusive formation of the 1,5-
substituted triazole 121. A possible solution for this matter would be to increase the
amount of the ruthenium catalyst to 10 mol% and conduct the catalysed cycloaddition at
room temperature.40
However, as discussed later in Chapter 4, changing the protecting
groups on the azide substrate also achieved the desired regiospecificity.

48
BocN
O
OMe
N3
HBocN
OMe
OH
N
N N
P
O
OEt
OEt
BocNOMe
OH
N NN
PO
OEtEtO
P H
O
EtO
EtO
+conditions
60
82
121 122
Scheme 30. Azide-alkyne cycloadditions. Conditions: (a) PhMe, 110 ºC (b) CuSO4 (10
mol%), sodium ascorbate, 1:1 t-BuOH-H2O (c) Cp*RuCl(PPh3)2 (1.5 mol%), PhMe, 60
ºC. See table 2 for yields.
Table 2. Yields of azide-alkyne cycloadditions under various conditions.
Product Thermal Cu(I)-catalysed Ru(II)-catalysed
121 21 % – 56 %
122 58 % 95% 21 %
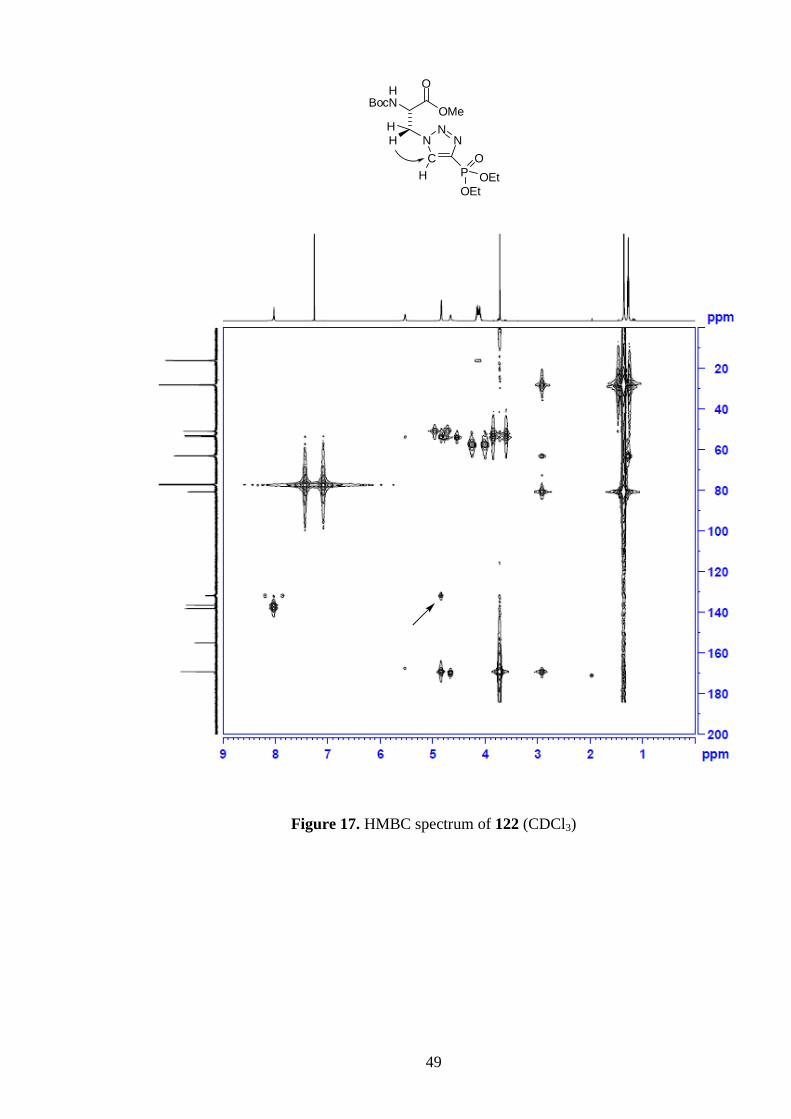
49
BocNOMe
OH
N
C
NN
P
OEt
O
OEt
HH
H
Figure 17. HMBC spectrum of 122 (CDCl3)

50
Figure 18. 1H and
13C NMR spectra of 121 (CDCl3)

51
Figure 19. 1H and
13C NMR spectra of 122 (CDCl3)

52
2.5 Synthesis of haptens
As discussed in Chapter 1, small molecules do not elicit an immune response, and must
be conjugated to a carrier protein before being used to raise antibodies.67
To allow this
conjugation, modification of the phosphohistidine analogues to give a series of haptens
was carried out. It was intended to conjugate through amide linkages so that the haptens
better mimic the naturally occurring phosphohistidine residues in proteins (Figure 20).
To maximise the chances of generating a generic phosphohistidine antibody, for each
regioisomer, three haptens were targeted, with the intention of preparing six different
immunogens for use in antibody generation in the same animal.
H3NNH
O
NN
N
P
O
OO
conjugated to a carrier proteinthrough the amino group
amide mimics peptide bondsin native P-His residues
Figure 20. Example of a target hapten
2.5.1 N-methylamide haptens
The methyl esters 121 and 122 were converted to the amides 123 and 124 simply by
mild treatment with methylamine (Scheme 31).68
Concomitant deprotection69
of the
amine and phosphonic acid, in the presence of the amide, was conducted using
hydrogen bromide in acetic acid, providing the haptens 125 and 126 in good yield.
These haptens can be conjugated through the amino group by coupling with aspartate
and glutamate side chains on the surface of the carrier protein.

53
BocNOMe
OH
N
N N
P
O
OEt
OEt
BocNNH
OH
N
N N
P
O
OEt
OEt
BrH3NNH
O
N
N N
P
O
OH
OH
HBr
AcOH
73 %90%
BocNOMe
OH
N NN
PO
OEtEtO
BocNNH
OH
HBr
AcOH
71 %
MeNH2
MeOH,H2O
85%
121
122
123
124
125
126
NN
N
P
O
EtOOEt
BrH3NNH
O
NN
N
P
O
HOOH
MeNH2
MeOH,H2O
Scheme 31. Synthesis of N-methylamide haptens
Figure 21. 1H NMR spectrum of 125 (D2O)

54
Figure 22. 1H NMR spectrum of 126 (D2O)
2.5.2 Dipeptide haptens
The dipeptide haptens 129 and 130 were also prepared, as depicted in Scheme 32 and
33. Careful saponification70
of 121 and 122 gave the carboxylic acids 35 and 36,
reported by Muir40
(See section 1.6.1 for an account of very similar work published
during the completion of this research). The NMR spectra of 35 and 36 were compared
with those published.40
The chemical shifts in both the 1H and
13C NMR spectra
acquired in CDCl3 were in accord with those reported, but the signals were considerably
more broadened in the present case, possibly because of some kind of dynamic
phenomenon (Figures 23 and 24). On the other hand, sharp peaks were seen in the NMR
spectra acquired in CD3OD (Figures 23 and 24).

55
BocNOMe
OH
N
N N
P
O
OEt
OEt
BocNOH
OH
N
N N
P
O
OEt
OEt
LiOH
MeOH/H2O
88%122 36
BocNOMe
OH
N NN
PO
OEtEtO
BocNOH
OH
N NN
PO
OEtEtO
LiOH
MeOH/H2O
86%
121 35
0 oC
0oC
Scheme 32. Careful saponification of the methyl esters 121 and 122
(A)
(B)
Figure 23. 1H NMR of 35. (A) CD3OD, (B) CDCl3

56
(A)
(B)
Figure 24. 1H NMR of 36. (A) CD3OD, (B) CDCl3
The specific rotations of the carboxylic acids 35 and 36 were lower in magnitude than
those reported by Muir40
(Table 3), which suggested that the compounds had been
partially racemised in the hydrolysis step, and this was confirmed by enantioselective
HPLC. The enantiomeric excess determined were almost consistent with those
determined from the specific rotations (Table 3). Although this partial racemisation was
unfortunate, it should be noted that even racemic compounds would suffice for antibody
production. Nevertheless, this problem was later overcome (see Chapter 4).

57
Table 3. Specific rotations and enantiomeric excesses for the carboxylic acids 35 and
36.a
[α]D ee
Muir40
This work [α]D HPLC
35 +1.8° (1.0) +1.2° (0.5) 67% 70%
36 +60° (1.0) +40.5° (0.7) 68% 74%
a [α]D values were recorded at 21 C (Muir) and ambient temperature (this work).
Concentrations in g.100 mL–1
in CHCl3 are given in brackets. The ee values were
determined by comparison with the optical rotations reported by Muir and coworkers40
([α]D) and relative integrals of the peaks corresponding to the enantiomers by
enantioselective HPLC.
O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
(HCTU)-mediated peptide coupling reactions of the enantiomerically enriched
carboxylic acids 35 and 36 and N-methyl-L-alaninamide 133, prepared by treatment of
L-serine methyl ester 13271
with methylamine,72
afforded the dipeptides 127 and 128 in
acceptable yields (Scheme 33).73
The 1H and
13C NMR spectra of 127 and 128
contained several minor peaks in addition to those expected for the desired compounds.
These were originally ascribed to the presence of rotamers (a common occurrence with
amides). But, given that the carboxylic acids 35 and 36 were partially racemised, the
additional peaks may actually have been due to diastereomers. Indeed, the
diastereomeric ratio calculated from the 1H NMR spectrum of the dipeptide 127 was in
accord with the enantiomeric ratio of the carboxylic acid 35 (Table 4). On the other
hand, the diastereomeric ratio of the dipeptide 128 did not match the enantiomeric ratio
of the carboxylic acid 35. These data indicate that further epimersation of the Tza
stereocentre may have occurred in the peptide coupling reaction.
58
Table 4. Enantiomeric ratio of 35 and 36 and diastereomeric ratio of 127 and 128
Phosphonoamino acid Enantiomeric ratio Dipeptide Diastereomeric ratioa
35 17:3 127 17:3
36 87:13 128 17:8
a As judged by comparison of relative integrals for the sets of signals in the
1H NMR
spectrum of the column chromatography-purified product.
Removal of the remaining protecting groups was achieved with hydrogen bromide in
acetic acid,69
giving the dipeptide haptens 129 and 130, retaining the diastereomeric
ratio (as judged from the 1H NMR spectra), in good yields (Scheme 33).
BocN
O
NH
H
O
HN
DIPEA
H2NNH
O
HCTU
DMF
60%
BocNOH
OH
N NN
PO
OEtEtO
35
BrH3N
O
NH
O
HN
HBr
AcOH
BocNOH
OH
N
N N
P
O
OEt
OEt
BocNNH
OH
N
N N
O
HN
PO
OEtOEt
BrH3NNH
O
N
N N
O
HN
PO
OHOH
36
DIPEA
H2NNH
O
HCTU
DMF
61%
HBr
AcOH
127
128
129
130
H2NNH
O
ClH3NOEt
O
H3NO
OSOCl2
EtOH MeNH2
131 132 133
133
133N N
N
N
N
N
PPEtOEtO
O O
HOHO
68% over 2 steps
Scheme 33. Synthesis of dipeptide haptens and N-methyl-L-alaninamide

59
Figure 25. 1H NMR spectrum of 127 (CD3OD)
Figure 26. 1H NMR spectrum of 129 (D2O)

60
Figure 27. 1H NMR spectrum of 128 (CD3OD)
Figure 28. 1H NMR spectrum of 130 (D2O)

61
2.6 Synthesis of haptens ready for bioconjugation through the carboxyl group
Other haptens, which could be linked through the carboxyl group by coupling to lysine
residues on the surface of the carrier protein, were also targeted (Figure 29).
HN
OH
O
NN
N
P
O
OO
conjugated to a carrier proteinthrough the carboxyl group
O
amide mimics peptide bondsin native P-His residues
Figure 29. Example of a hapten
2.6.1 Attempts to convert NBoc derivatives to NAc derivatives
The first strategy to obtain these bioconjugation precursors is shown in Scheme 34. The
Boc-amino protecting group in 122 was removed by TFA, giving the amine 134 in good
yield after neutralisation.74
However, neither acylation of the amine 134 by acetic
anhydride nor peptide coupling with N-acetyl-L-alanine 13875
was successful, judging
by ESI-MS and 1H NMR spectra of the crude products, although the starting materials
seemed to be completely consumed. The reasons for this are unclear, but it is thought
that some inherent property of 134 was responsible.

62
BocNOMe
OH
N
N N
P
O
OEt
OEt
H2NOMe
O
N
N N
P
O
OEt
OEt
TFA
DCM
80%
Ac2O HN
OMe
O
N
N N
P
O
OEt
OEt
O
LiOH
MeOH/H2O
HN
OH
O
N
N N
P
O
OEt
OEt
O
HBr
AcOH
HN
OH
O
N
N N
P
O
OH
OH
O
H2NOMe
O
N
N N
P
O
OEt
OEt
HN
OMe
O
N
N N
P
O
OEt
OEt
O
LiOH
MeOH/H2O
HBr
AcOH
NH
O
HN
OH
O
O
HCTU
DIPEA
DMF
HN
OH
O
N
N N
P
O
OEt
OEt
O
NH
OHN
OH
O
N
N N
P
O
OH
OH
O
NH
O
122 134 135
136137
134 139
140141
138
Et3N
DCM
Scheme 34. Attempted conversion of NBoc derivatives to NAc derivatives

63
Figure 30. 1H NMR of 134 (CDCl3)
2.6.2 Synthesis of NAc-Tza(POEt2)-OMe
Due to the difficulties in replacing the NBoc group with an NAc group, it was decided
to incorporate the NAc group earlier on in the synthesis. The known azide 14376
was
synthesised in two steps from L-serine methyl ester 58 (Scheme 35). An amide coupling
reaction using N,N′-dicyclohexylcarbodiimide (DCC) furnished N-acetyl-L-serine
methyl ester 14277
in excellent yield. The alcohol 142 was converted to the azide 143 by
a Mitsunobu reaction in acceptable yield.
ClH3N
O
OMe
OH
HN
O
OMe
OHO
HN
O
OMe
N3
O
AcOH
DCC
DCM
HN3
DIAD
PPh3
THF90%
60%58 142 143
Scheme 35. Synthesis of the azide 143
Various conditions were applied to azide-alkyne cycloadditions of the azide 143 and the
ethynylphosphonate 82 (Scheme 36), with similar results to those observed with the
NBoc derivative. In the absence of a catalyst, the isomeric triazole analogues 144 and
145 were produced, with the 1,4-substituted triazole 145 predominating (Table 5). The
copper(I)-catalysed reaction furnished the 1,4-substituted triazole 145 exclusively in

64
excellent yield (Table 5). The regioselectivity was successfully reversed in the presence
of the ruthenium(II) catalyst, affording the 1,5-substituted triazole 144 as the major
product (Table 5).
HN
O
OMe
N3
HN
OMe
O
N
N N
P
O
OEt
OEt
HN
OMe
O
N NN
PO
OEtEtO
P H
O
EtO
EtO
+conditions
O O O
143
82
144 145
Scheme 36. Azide-alkyne cycloadditions. Conditions: (a) PhMe, 110 ºC (b) CuSO4 (10
mol%), sodium ascorbate, 1:1 t-BuOH/H2O (c) Cp*RuCl(PPh3)2 (1.5 mol%), PhMe, 60
ºC
Table 5. Yields of Huisgen cycloadditions under various conditions
Product Thermal Cu(I)-catalysed Ru(II)-catalysed
144 26% – 57%
145 60% 91% 19%
Figure 31. 1H NMR spectrum of 144 (CDCl3)

65
Figure 32. 1H NMR spectrum of 145 (CDCl3)
2.6.3 Synthesis of N-acetyl haptens
The next task was to transform the methyl esters 144 and 145 to the corresponding
carboxylic acids, followed by deprotection of the phosphonic acid to provide the target
haptens. The ester 145 was carefully saponified with lithium hydroxide (Scheme 37).
78
The target carboxylic acid 146 was obtained but the yield was extremely poor. It should
be noted that the saponification using lithium hydroxide needed to be conducted at 0 ºC.
When it was carried out at room temperature, only the elimination product 147 was
observed.
Owing to the poor yield, other conditions for the hydrolysis were investigated. When
potassium carbonate was used as a base,79
the desired carboxylic acid 146 was obtained
in better yield. Under these conditions, the reaction did not proceed at 0 ºC, and
therefore it was performed at room temperature. The optical rotation of the carboxylic
acid 146 obtained under these conditions was greater in magnitude than that using
lithium hydroxide (Table 6), indicating that as expected, enolate formation and
At the time this experiment was conducted, it was not known that these conditions caused partial
racemisation.

66
racemisation accompanied the elimination of the triazole substrate, which presumably
occurred by an E1cB mechanism.
AcNOMe
OH
N
N N
P
O
OEt
OEt
AcNOH
OH
N
N N
P
O
OEt
OEt
+
AcNOH
OH
conditions
145 146 147
Scheme 37. Hydrolysis of the methyl ester 145 under various conditions
Table 6. Results of the hydrolysis of 145.
Ester Conditions 146 (Yield) 147 Specific Rotation
145 LiOH, MeOH, H2O, 0 ºC 31% – +5.1°
145 LiOH, MeOH, H2O, RT – Observed –
145 K2CO3, MeOH, H2O, RT 47% – +8.8°
145 K2CO3, MeOH, H2O, 0 ºC no reaction –
Figure 33. 1H NMR spectrum of 146 (CD3OD)
The carboxylic acid 146 synthesised by hydrolysis of the methyl ester 145 using K2CO3
was subjected to hydrogen bromide in acetic acid to reveal the phosphonic acid,69
giving the hapten 137 in good yield (Scheme 38).

67
HN
OH
O
N
N N
P
O
OEt
OEt
O
HBr
AcOH
HN
OH
O
N
N N
P
O
OH
OH
O
70%146 137
Scheme 38. Deprotection of 146
The same strategies were applied to 1,5-substituted triazole 144 (Scheme 39). However,
a mixture of the desired carboxylic acid 148 and elimination product 147, which were
not separable by normal-phase chromatography, was obtained. Accordingly, the poor
yield, difficult purification and assumed racemisation required another approach to be
taken.
AcNOMe
OH
N NN
PO
OEtEtO
Conditions
AcNOH
OH
N NN
PO
OEtEtO
AcNOH
OH
+
144 148
147
Scheme 39. Hydrolysis of the methyl ester 140. Conditions (a) K2CO3, MeOH/H2O, r.t.
(b) LiOH, MeOH/H2O, 0 ºC.
2.6.4 Synthesis of NAc-Tza(POEt2)-OAll
The alternative route was designed to circumvent the problems of racemisation and
elimination during the saponification of methyl esters. An allyl ester was initially
chosen as it can be removed by deprotection using palladium(II)-catalysis.80
Thus, L-
serine 57 was treated with acetic anhydride in acetic acid,75
providing a mixture of N-
acetyl-L-serine 149 and N-acetyl-O-acetyl-L-serine 150 (Scheme 40). The mixture was
esterified with allyl bromide to give the desired ester 151 and undesired ester 152, with

68
the latter dominating.81
The alcohol 151 was converted to the novel azide 153 by a
Mitsunobu reaction in acceptable yield.
AcOH
Ac2OHN
O
OH
OH
H3N
O
O
OHO
HN
O
OH
OO
O
HN
O
O
OHO
HN
O
O
OO
O
DMF
DIPEA
21% (over two steps)
45% (over two steps)
+
Br
57 149 150
151
152
HN
O
O
N3
O
153
HN3
DIAD
PPh3
THF
61%
Scheme 40. Synthesis of N-acetyl azidoalanine allyl ester 153
The azide 153 was subjected to cycloaddition reactions with the ethynylphosphonate 82
under various conditions (Scheme 41). Interestingly, the thermal reaction furnished the
1,5-substituted triazole 154 and 1,4-substituted triazole 155 in the ratio of 1:1 (Table 7),
whereas the 1,4-substituted triazole 145 was a major product in the case of the thermal
reaction of the NAc protected methyl ester 143 (2:1). The reasons for the altered
regioselectivity brought about by the allyl ester are not obvious. As expected, the 1,4-

69
substituted triazole 155 was formed exclusively in the copper(I)-catalysed reaction
(Table 7). The innate tendency of the allyl group to favour more of the 1,5-triazole was
further enhanced with Ru(II) catalysis (Table 7), with the regioselectivity now
significantly better than previously observed.
AcN
O
OAll
N3
HAcN
OAll
OH
N
N N
P
O
OEt
OEt
AcNOAll
OH
N NN
PO
OEtEtO
P H
O
EtO
EtO
+conditions
153
154 155
82
Scheme 41. Azide-alkyne cycloadditions. Conditions: (a) PhMe, 110 ºC (b) CuSO4 (10
mol%), sodium ascorbate, 1:1 t-BuOH-H2O (c) Cp*RuCl(PPh3)2 (1.5 mol%), PhMe, 60
ºC
Table 7. Yields of azide-alkyne cycloadditions under various conditions
Product Thermal Cu(I)-catalysed Ru(II)-catalysed
154 41 % – 73 %
155 44 % 84% 10 %
Figure 34. 1H NMR spectrum of 154 (CDCl3)

70
Figure 35. 1H NMR spectrum of 155 (CDCl3)
2.6.5 Attempted deallylation
Despite similar precedents,80
the target carboxylic acids 148 and 146 were not obtained
by palladium(0)-catalysed cleavage of the allylesters 154 or 155 (Scheme 42). Although
what occurred in the reactions are unclear, the starting materials were not completely
consumed and there was no trace of the target compounds, judging by the ESI-MS and
1H NMR spectra of the crude products.
Pd(PPh3)4
Morpholine
THF
HBr
AcOH
AcNOAll
OH
N
N N
P
O
OEt
OEt
Pd(PPh3)4
Morpholine
THF
AcNOH
OH
N
N N
P
O
OEt
OEt
HBr
AcOH
AcNOH
OH
N
N N
P
O
OH
OH
AcNOAll
OH
N NN
PO
OEtEtO
154
AcNOH
OH
N NN
PO
OEtEtO
148
AcNOH
OH
N NN
PO
OHHO
156
155 146 137
Scheme 42. Attempted deallylation

71
In the proposed mechanism for removal of allyl protecting group by a palladium
catalyst,80
palladium(0) species coordinate to the double bond of the allyl protecting
group, followed by oxidative addition to form a -allyl complex (Scheme 43).
R O
O
R OH
OPd+ (II)
L
L
R O
O
Pd (0)
L
L
coordination
Oxidative addition
+
Pd (II)
L
L
B
Pd (0)
L
L +B
Pd (0)
L
L
Scheme 43. Proposed mechanism for removal of the allyl protecting group
As to why this normally reliable deprotection failed in the current case, it is possible
that the Pd formed a stable complex with the substrate, which has many groups capable
of coordinating Pd, shutting down the catalytic cycle.
2.6.6 Synthesis of NAc-Tza(POEt2)-OBn
Due to the difficulties with the deallylation, an alternative route was designed to
circumvent the saponification step, and a benzyl group was chosen as an acid-labile
carboxyl protecting group, which would be cleaved by treatment with hydrogen
bromide in acetic acid, used for deprotection of the phosphonate, thereby also avoiding
one step. The synthesis was similar to that of the allyl esters. The ester 160 was
synthesised in 2 steps75,81
from L-serine 57 with the undesired ester 161 being a by-
product (Scheme 44). The alcohol 160 was transformed to the azide 162 by a Mitsunobu
reaction.

72
AcOH
Ac2OHN
O
OH
OH
H3N
O
O
OHO
HN
O
OH
OO
O
HN
O
O
OHO
HN
O
O
OO
O
DMF
DIPEA
31% (over two steps)
45% (over two steps)
+
Br
HN3
DIAD
PPh3
THF
HN
O
O
N3
O
61%
57
157 158
159
161
160
162
Scheme 44. Synthesis of the azide 162
The azide 162 was subjected to azide-alkyne cycloadditions as before (Scheme 45). The
thermal reaction furnished the 1,5-substituted triazole 163 and 1,4-substituted triazole
164 in the ratio of 1:1 (Table 8). As anticipated, the copper(I)-catalysed reaction gave
164 exclusively (Table 8). A remarkable difference observed in the case of the benzyl
esters was the exclusive formation of the 1,5-substituted triazole 163 using the
ruthenium catalyst (Table 8). The findings thus far suggest that bulky carboxyl
protecting groups assist the formation of the 1,5-substituted triazole in the click
reaction, although the exact mechanisms are still being elucidated.

73
HN
O
OBn
N3
HN
OBn
O
N
N N
P
O
OEt
OEt
HN
OBn
O
N NN
PO
OEtEtO
P H
O
EtO
EtO
+conditions
O O O
162
163 164
82
Scheme 45. Azide-alkyne cycloadditions. Conditions: (a) PhMe, 110 ºC (b) CuSO4 (10
mol%), sodium ascorbate, 1:1 t-BuOH-H2O (c) Cp*RuCl(PPh3)2 (1.5 mol%), PhMe, 60
ºC
Table 8. Yields of azide-alkyne cycloadditions under various conditions.
Product Thermal Cu(I)-catalysed Ru(II)-catalysed
163 43% – 81%
164 45% 87% –
2.6.7 Synthesis of N-acetyl haptens-2
The protected carboxylic acid and phosphonic acid of 163 and 164 were revealed
concomitantly utilising hydrogen bromide in acetic acid,69
giving the haptens 156 and
137 in good yields.. The specific rotation of the hapten 137 obtained in this way was
greater in magnitude than that obtained via saponification, suggesting that the partial
racemisation observed under basic conditions was avoided by using protonolysis (Table
9).
74
HN
OBn
O
N
N N
P
O
OEt
OEt
HN
OBn
O
N NN
PO
OEtEtO
O
O
HBr
AcOH
HN
OH
O
N
N N
P
O
OH
OH
O
67%
HBr
AcOH
69%
HN
OH
O
N NN
PO
OHHO
O
163
164 137
156
Scheme 46. Synthesis of the conjugation precursors
Table 9. Specific rotations of the hapten 137
Hapten 137 1 step from 164 2 steps from 145
[α]D +10.0º (c 1.0, H2O) +6.1º
(c 1.0, H2O)
Figure 36. 1H NMR spectrum of 163 (CDCl3)

75
Figure 37. 1H NMR spectrum of 156 (D2O)
Figure 38. 1H NMR spectrum of 164 (CDCl3)
Figure 39. 1H NMR spectrum of 137 (D2O)

76

77
Chapter 3
Hapten-Carrier Protein Conjugation Strategies

78
3.1 Introduction to bioconjugation methods
As mentioned in the previous chapter, small molecules must be conjugated to a large
molecule, usually a „carrier‟ protein, in order to cause an immune response.67
Small
molecules can be coupled to carrier proteins by various reactions, which involve
existing functional groups or added reactive groups.82
Among several conjugation
methods, one of the most common procedures is coupling of haptens and carrier
proteins through amide linkages using water-soluble carbodiimides such as N-(3-
dimethylaminopropyl)-N′-ethylcarbodiimide (EDCI) (Scheme 47).83
O
O
aspartate/glutamateresidue
carrierprotein
H2N Hapten
O
NH
carrierprotein
Hapten
EDCI
Scheme 47. An example of a coupling of carrier protein and hapten using EDCI
Reactive esters, for example N-hydroxysuccinimide (NHS) esters, are often used to
allow conjugation through amide linkages (Scheme 48).82
These esters can selectively
react with alkyl amines in the presence of anilines, alcohols, phenols, and histidine.82
The optimum pH for reactions of the alkyl amines and reactive esters in aqueous
systems is 8–9.82
O
O
carrierprotein
N
O
O
H2N Hapten
O
HN
carrierprotein
Hapten
Scheme 48. Coupling of carrier protein and amino-bearing hapten using an activated
ester

79
Commercially available maleimide-activated carrier proteins are also frequently utilised
for the bioconjugation of haptens containing thiol groups, which undergo conjugate
addition with the maleimide to form stable thioethers (Scheme 49).82
The optimum pH
for the reactions is about 7.0, as maleimide can be hydrolysed to a maleamic acid
derivative above pH 8.0.82
Ncarrierprotein
O
O
HS Hapten
Ncarrierprotein
O
O
S Hapten
Scheme 49. Coupling of maleimide-activated carrier protein and a thiol-bearing hapten
Although other carrier proteins such as ovalbumin (OVA) and bovine serum albumin
(BSA) are utilised as carriers, keyhole limpet hemocyanin (KLH) is the most common
immunostimulator.84
KLH is more immunogenic compared with other carrier proteins
because of its high molecular weight.84
KLH also possesses a large number of lysine
and aspartic acid residues to which haptens can be attached.84
The 1,4-triazolylalanine
derivatives 126, 130 and 137 will be conjugated to KLH as depicted in Scheme 50, and
the same strategies will be applied to the 1,5-triazolylalanine derivatives 125, 129 and
156.
The resulting immunogens (i.e., the six different bioconjugates) will be co-administered
to rabbits to maximise the chances of producing phosphohistidine-recognising
antibodies.

80
KLH
O
O
aspartate/glutamateresidue
EDCI
BrH3N
O
NH
N
N N
P
O
OO
126 (Hapten 1)
KLH
O
HN
NH
O
N
N N
P
O
OO
carrier protein
Amides mimic peptide bondsin native P-His residues
KLH
O
O
aspartate/glutamateresidue
EDCI
BrH3N
O
NH
N
N N
130 (Hapten 2)
KLH
O
carrier protein
HN
O
PO
OO
HN
O
NH
N
N N
HN
O
PO
OO
KLH
carrier protein
H3N
Lysine residue
EDCI
HN
O
OH
N
N N
P
O
OO
137 (Hapten 3)
O
KLHNH
OHN
Immunogen 1
Immunogen 2
N
N N
P
O
O
O
O
Immunogen 3
Scheme 50. Hapten-Carrier protein Conjugation Strategies
3.2 Trial Conjugation-1
A trial conjugation in water was performed using the hapten 126 and azidoacetic acid
166, prepared by the reaction of bromoacetic acid 165 and sodium azide (Scheme 51).85
After purification by Dowex anion exchange chromatography, the slightly impure
desired amide 167 was obtained, as judged by the 1H NMR,
13C NMR and ESI-MS

81
spectra of the product. No trace of undesired phosphoramidates 168 (Scheme 52),
resulting from self-condensation of the phosphonic acid, was observed. This result
indicated that the haptens possessing a phosphonic acid could be linked to a carrier
protein by an EDCI-mediated amide coupling in water.
OHN3
O
EDCI
NaHCO3
H2O
60%
OHBr
O
OHN3
O
NaN3
H2O
61%
BrH3N
O
NH
N
N N
P
O
OHOH
HN
O
NH
N
N N
P
O
OHOH
O
N3
165 166
126
166
167
Scheme 51. Trial conjugation of the hapten 126 and azidoacetic acid 166
R1 NH2
EDCI
P
O
OH
OHR2
P
O
OH
NHR2
R1
168
Scheme 52. Potential, undesired phosphoramidate formation resulting from activation
of a phosphonic acid by EDCI

82
Figure 40. 1H and
13C NMR of 167 (D2O)
3.3 Trial Conjugation-2
Conjugation of the hapten 126 and a protein was then attempted. OVA, which can also
be used as a carrier protein and is more affordable than KLH, was chosen for model
studies. Although several attempts were made (Scheme 53) based on literature
precedents86,87
for the coupling of amino-bearing haptens, none of them were
successful, as judged by MALDI-TOF mass spectra of the products after dialysis, which
was virtually the same as that of unreacted OVA.

83
O
O
aspartate/glutamateresidue
EDCI
BrH3N
O
NH
N
N N
P
O
OHOH
126 (Hapten 1)
O
HN
NH
O
N
N N
P
O
OHOH
carrier protein
Amides mimic peptide bondsin native P-His residues
OVA OVA
Attempted conditions
1) EDCI, NaHCO3, H2O
2) EDCI, H2O
3) EDCI, 0.1M MES buf fer (pH 4.7)
Scheme 53. Attempted conjugation of the hapten 126 and OVA
At this time, we have no explanations for the failure of this coupling, and there was
insufficient time to conduct any further work in this area, but several control
experiments are possible in order to investigate the causes of the failure and search for
suitable bioconjugation conditions.
For example, N-methylalaninamide 133 and N-acetylalanine 138 would be useful for
the control experiments (Scheme 54). Success in these couplings will validate the
bioconjugation procedure and point to an inherent difficulty in amide couplings of the
hapten(s). Given the previously observed inability to acylate 134 with small molecules
(see section 2.6.1), this is a distinct possibility.

84
O
O
carrier protein
OVA
H2NNH
O
EDCI
O
HN
NH
OOVA
OVA
HN
OH
O
EDCI
O
H3N OVANH
OHN
O
133
138
Scheme 54. Possible control experiments for the bioconjugation
In the case where EDCI-mediated peptide coupling reaction is not suitable for the
bioconjugation of the haptens and a carrier protein, other methods without using EDCI
will need to be found. The first candidate is using the activated ester, which allows
conjugation through amide linkages.82
O
O
KLH
N
O
O
BrH3N
O
NH
N
N N
P
O
OO
126 (Hapten 1)
KLH
O
HN
NH
O
N
N N
P
O
OO
Immunogen 4
Scheme 55. Conjugation of the hapten and a carrier protein using an activated ester
Alternatively, now that the azide 167 has been produced in the trial conjugation of the
hapten 126 and azidoacetic acid 166 (Scheme 51), another approach utilising a
copper(I)-catalysed azide-alkyne cycloaddition („click‟) reaction can be proposed
(Scheme 56). KLH possessing alkynes on the surface could be synthesised by the

85
reaction of the lysine residues with the succinimidyl ester 169.88
As the „click‟ reaction
can be conducted in water with no organic co-solvents,63
the cycloaddition of the azide
167 alkyne 170 should give immunogen 5.
KLH NH3
lysine residue
KLH NH
Carrier protein
O
KLH NH
O
N N
N
HN
O
NH
O
N
NN
P
O
O
O
H
N3
HN
O
NH
O
N
NN
P
O
O
O
CuSO4
Sodium ascorbate
Immunogen 5
NO
O
HO
O
H2O
169
167
170
Scheme 56. Proposed conjugation strategy using a copper(I)-catalysed azide-alkyne
cycloaddition (click) reaction

86

87
Chapter 4
Synthesis of Free Phosphohistidine
Analogues and Evaluation of their Ability to Inhibit a
Mammalian Histidine Kinase

88
4.1 Possible biological activity of phosphohistidine analogues
The possible intrinsic biological activity of the phosphohistidine analogues captured our
interest. For example, they may exhibit antibacterial activity by interfering with protein-
protein recognition in bacterial two-component systems. In addition, as product-mimics,
they could potentially inhibit histidine kinase activity, and therefore antiproliferative
activity in mammalian cells. The roles of histidine kinases in mammalian cells are
poorly understood, but inhibition of their activity could provide some insight into their
functions and lead to the invention of useful medicines, including anticancer drugs.8,18
4.2 Competitive and non-competitive inhibitors of histidine kinases
As with any enzyme, there are two common types of inhibition of histidine kinases:
competitive and non-competitive. In competitive inhibition, an inhibitor prevents a
histidine kinase from binding the substrate protein and/or ATP by blocking the binding
site of the histidine kinase, and thereby, the transfer of a phosphoryl group does not
occur (Figure 41).89
For instance, the Winkler group reported that the novel
thienopyridine TEP inhibited autophosphorylation of a bacterial histidine kinase, His-
HpkA77, competitively with respect to ATP.89

89
Competitive inhibition
Histidine kinase
ATP
Inhibitor
Inhibition by blockingthe binding site
N S
HN Br
N
H2N
TEP
O
NH2
Figure 41. Mechanism of competitive inhibition and the structure of TEP
On the other hand, in non-competitive inhibition, an inhibitor binds to a histidine kinase
on a site different from the active site, which causes structural transformation of the
histidine kinase, thereby preventing binding of the substrates (ATP or the substrate
protein) (Figure 42).15,90
For example, it was found by Hock‟s group that RWJ-49815
and Closantel interfered with autophosphorylation of a bacterial two-component
histidine kinase, Kin A, due to non-specific structural transformation caused by the
inhibitor intercalating in the hydrophobic core of the enzyme.13,15,90

90
Non-competitive inhibition
Histidine kinase
ATP
Inhibitor
Inhibition by causing structuraltranformation of the kinase
O
HN NH2
NH
RWJ-49815
NH
O
CN
Cl
I
OH
I
Cl
Closantel
Figure 42. Mechanism of non-competitive inhibition and the structures of RWJ-49815
and Closantel
4.3 Synthesis of free triazolylalaninephosphonates
In order to evaluate the biological activities, the free triazolylalaninephosphonates 170
and 171 (Figure 43) were synthesised.
H3NO
O
N NN
PO
OO
H3NO
O
N
N N
P
O
O
O
170 171
Figure 43. Structures of ‘free‟ N1-(170) and N3-phosphohistidine (171) analogues
Initial attempts at global deprotection of 121 and 122 using hydrochloric acid or
hydrobromic acid were unsuccessful (Scheme 57).36
Although what occurred was not
completely elucidated, the methyl esters seemed to be stable under these conditions,
judging by 1H NMR spectra of the crude materials.
91
BocNOMe
OH
N NN
PO
OEtEtO
BocNOMe
OH
N
N N
P
O
OEt
OEt
XH3NOH
O
N NN
PO
OHHO
XH3NOH
O
N
N N
P
O
OH
OH
121
122
(X=Cl or Br)
(X=Cl or Br)
H3O Cl or H3O Br
H3O Cl or H3O Br
170
171
Scheme 57. Attempted global deprotections using mineral acids
Therefore, the carboxylic acids 35 and 36, which were synthesised by the hydrolysis of
the methyl esters 121 and 122 as described in the previous section, were utilised for the
synthesis of the free phosphonoamino acids. The amine and phosphonic acid were
concomitantly deprotected by hydrogen bromide in acetic acid to afford the free
analogues 170 and 171 as hydrobromides in acceptable yields (Scheme 58).69
However,
it should be remembered that the saponification to provide the carboxylic acids caused
partial racemisation and, accordingly, the free phosphonoamino acids 170 and 171,
prepared in this way, were not enantiopure but enantiomerically enriched.
BocNOMe
OH
N NN
PO
OEtEtO
BocNOH
OH
N NN
PO
OEtEtO
BocNOMe
OH
N
N N
P
O
OEt
OEt
BocNOH
OH
N
N N
P
O
OEt
OEt
LiOH
MeOH/H2O
88%
LiOH
MeOH/H2O
86%
HBr
AcOH
72%
HBr
AcOH
70%
BrH3NOH
O
N NN
PO
OHHO
BrH3NOH
O
N
N N
P
O
OH
OH
36
35
122
121 170
171
Scheme 58. Synthesis of phosphonoamino acids 170 and 171

92
To avoid racemisation, a strategy similar to that used for the synthesis of the haptens
137 and 156 (section 2.6.7) was used; the methyl ester was replace with a benzyl ester,
which is cleavable by protonolysis or hydrogenolysis (Scheme 59). The amino group of
L-serine 57 was protected to give the t-butyl carbamate 172,91
followed by alkylation
with benzyl bromide 159 to give the benzyl ester 173.81
A Mitsunobu reaction
converted the alcohol 173 to the corresponding azide 17440
in acceptable yield.
H3N
O
O
OH
BocN
O
O
N3
H
BocN
O
OH
OH
HBocN
O
O
OH
HBoc2O
NaOH
Dioxane
H2O
DIPEA
DMF
HN3
DEAD
PPh3
THF
72%
85%
75%
Br
57 172 173
159
174
Scheme 59. Synthesis of the azide 174
The azide 174 was subjected to azide-alkyne cycloadditions to furnish the protected
triazolylalanines 175 and 176 (Scheme 60). The thermal reaction gave a 1:1 mixture of
175 and 176, which were separable by flash chromatography (Table 10). In the same
reaction of the analogous methyl ester 60 (Scheme 30, page 48), the 1,5- and 1,4-
triazoles 121 and 122 were produced in the ratio of 1:2. Judging by these results, the
formation of the 1,5-triazole 175 was assisted by the benzyl protecting group. As
anticipated, the copper(I)-catalysed reaction gave the 1,4-triazole 176 exclusively in
good yield (Table 10). As with the N-acetyl analogue 163 (Scheme 45, page 73),
93
exclusive production of the regioisomer 175 was observed in the presence of the
ruthenium(II)-catalyst (Table 10).
BocN
O
O
N
H
NN
P O
OEtEtO
BocN
O
O
N
H
NN
PO
OEtEtO
P H
O
EtO
EtO
BocN
O
O
N3
H
conditions +
174
82
175 176
Scheme 60. Azide-alkyne cycloadditions of benzyl esters. Conditions: (a) PhMe, 110 ºC
(b) CuSO4 (10 mol%), sodium ascorbate, 1:1 t-BuOH-H2O (c) Cp*RuCl(PPh3)2 (1.5
mol%), PhMe, 60 ºC.
Table 10. Yields of Azide-Alkyne cycloadditions under various conditions
Product Thermal Cu(I)-catalysed Ru(II)-catalysed
175 41% – 80%
176 42% 88% –
Figure 44. 1H NMR spectrum of 175 (CDCl3)
94
Figure 45. 1H NMR spectrum of 176 (CDCl3)
Gratifyingly, all protecting groups were successfully removed in one step utilising
hydrogen bromide in acetic acid, producing the free phosphonamino acids 170 and 171
as the hydrobromides in acceptable yield (Scheme 61). Their specific rotations were
greater in magnitude than those of the same compounds produced via the methyl esters
(121 and 122). These results suggested that the more enantiopure free analogues were
obtained by employing a benzyl protecting group for the carboxylic acid in place of a
methyl protecting group (Table 11).
BocN
O
OBn
N
H
N
N
PO
EtOOEt
BrH3N
O
OH
NN
N
PO
HOOH
HBr
AcOH
65%
BocN
O
OBn
N
H
N N
P
O
OEt
OEt
BrH3N
O
OH
N
N N
P
O
OH
OH
HBr
AcOH
67%
175
176
170
171
Scheme 61. Improved and simplified methods to synthesise the free phosphonoamino
acids 170 and 171

95
Table 11. Specific rotations of 170 and 171
One step from the benzyl ester Two steps from the methyl ester
[α]D of 170 7.7° (c 0.5 H2O) 5.6° (c 0.8 H2O)
[α]D of 171 25.0° (c 0.5 H2O) 18.8° (c 0.5 H2O)
Figure 46. 1H and
13C NMR of 170 (D2O)

96
Figure 47. 1H and
13C NMR of 171 (D2O)
4.4 Inhibition of a mammalian histidine kinase by the free phosphohistidine
analogues
The roles of histidine kinases and histidine-phosphorylated proteins in mammalian cells
are poorly understood, except for one well characterised mammalian histidine kinase,
NDPK.17
Although the phosphorylation of histone H4 is associated with regeneration in
liver following injury and with hepatocellular carcinoma, it has not been established
whether cause or effect defines this relationship.18
However, inhibition of histidine
kinases‟ activity could provide some insight into their functions, and, in particular,
inhibition of the histone H4 histidine kinase associated with hepatocellular carcinoma18
could be of great clinical importance.8 Accordingly, the inhibitory activity of the free

97
phosphohistidine analogues 170 and 171 against transfer of a phosphoryl group from
[32
P]ATP to histone H4 by a guinea pig liver histidine kinase was measured. The
“kinase” has not been fully characterised but its ability to phosphorylate a histone H4
histidine has been reasonably well investigated.34
Histones are proteins that are
intimately associated with DNA, helping to store it in compact form as chromatin, and
are also involved in the regulation of gene expression.
In these experiments, solutions containing histone as substrate, the guinea pig liver
histidine kinase and [32
P]ATP were incubated with various concentrations of the
analogues. The positive control did not contain the phosphohistidine analogue, and the
phosphorylation rate should be the highest in these samples. The negative controls had
either no histone or no histidine kinase, and no phosphorylation should be observed
(Table 12).
Table 12
Component Assay NaBr control Positive control Negative controls
170 or 171 + – – –
histone + + + + [–]
kinase + + + – [+]
Following incubation, the phosphorylated histone was separated by SDS-PAGE and
quantified with a phosphoimager. As shown in Figures 48 and 49, both of the free
phosphohistidine analogues 170 and 171 inhibited the phosphorylation relative to the
positive control. Unexpectedly, the phosphorylation was also inhibited by sodium
bromide (NaBr), which was included as another control to account for the fact that the
inhibitors were hydrobromides. However, the inhibitory effects of the inhibitors are
greater than that of NaBr.
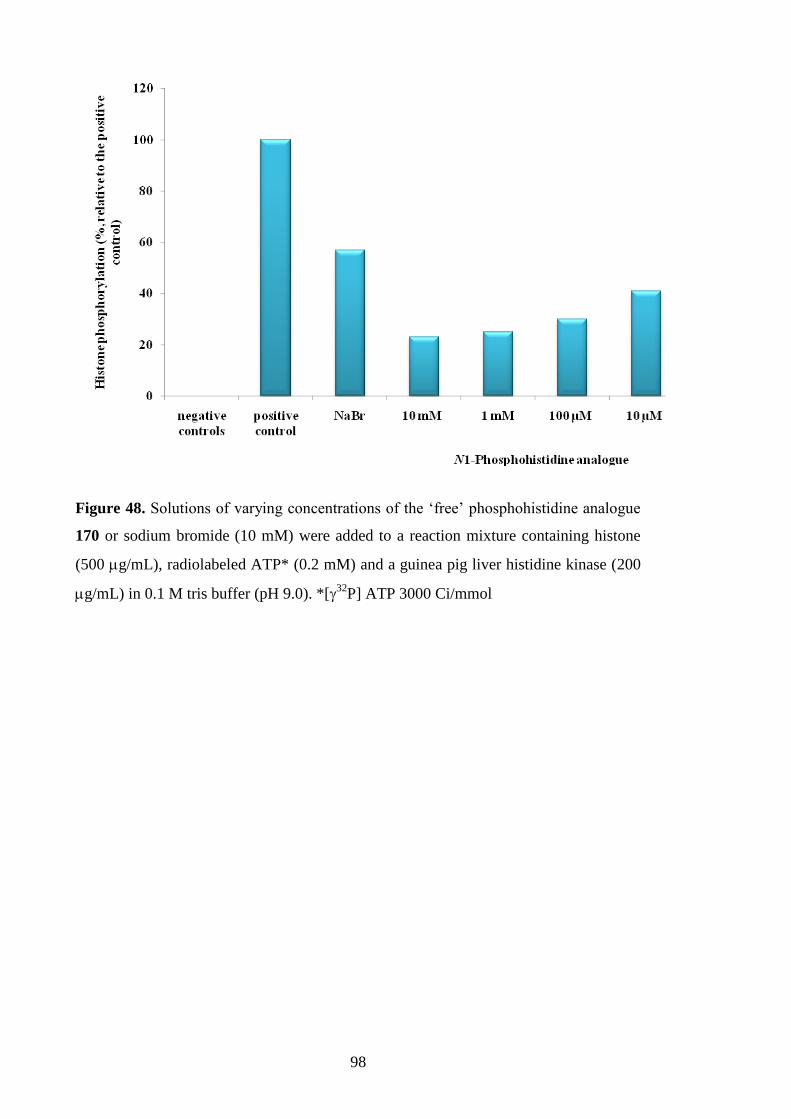
98
Figure 48. Solutions of varying concentrations of the „free‟ phosphohistidine analogue
170 or sodium bromide (10 mM) were added to a reaction mixture containing histone
(500 g/mL), radiolabeled ATP* (0.2 mM) and a guinea pig liver histidine kinase (200
g/mL) in 0.1 M tris buffer (pH 9.0). *[32
P] ATP 3000 Ci/mmol
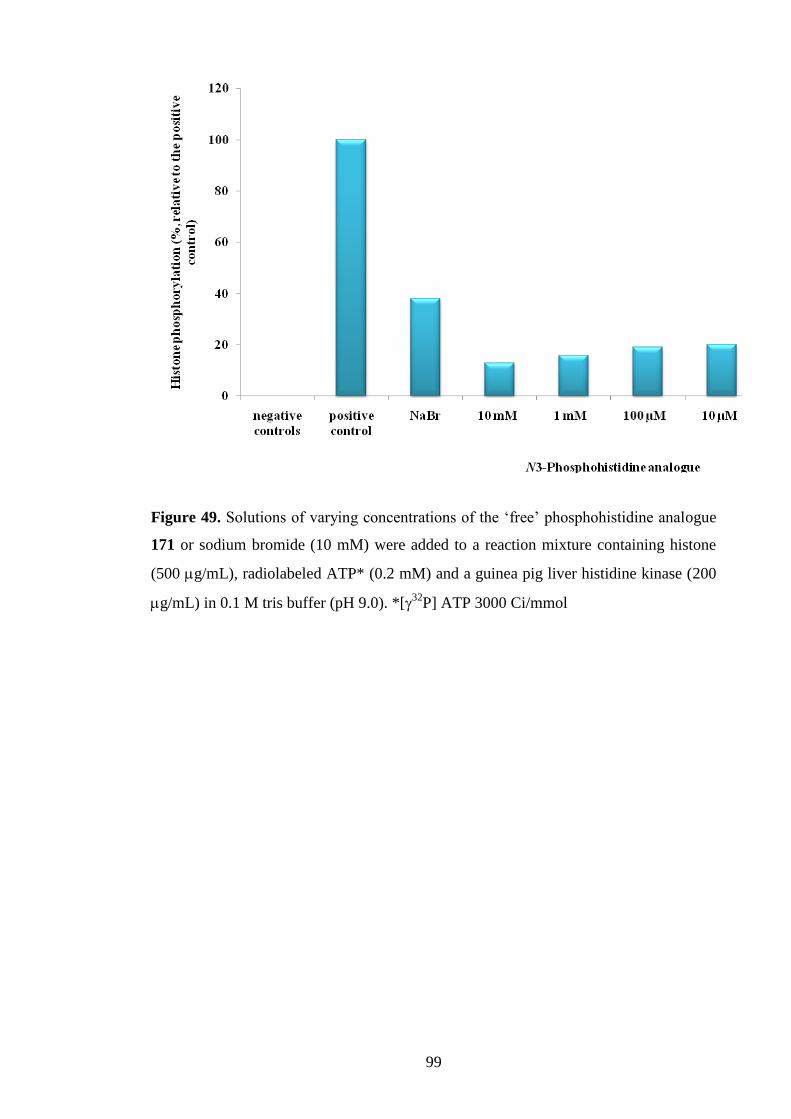
99
Figure 49. Solutions of varying concentrations of the „free‟ phosphohistidine analogue
171 or sodium bromide (10 mM) were added to a reaction mixture containing histone
(500 g/mL), radiolabeled ATP* (0.2 mM) and a guinea pig liver histidine kinase (200
g/mL) in 0.1 M tris buffer (pH 9.0). *[32
P] ATP 3000 Ci/mmol

100
These results indicate that the free phosphohistidine analogues 170 and 171 could be
mammalian histidine kinase inhibitors. Owing to time constrains, it remains to repeat
these experiments to determine the IC50 values and to determine what type of inhibition
is occurring, i.e., competitive or non-competitive, with respect to both of the substrates.
Although these results are promising, it may be better to conduct similar experiments on
a well-characterised histidine kinase, such as NDPK or a two-component histidine
kinase like EnvZ or CheA, as the identity of the histone H4 kinase has not yet been
confirmed. Indeed, compounds related to 170 and 171 may assist in the conclusive
confirmation that the guinea pig enzyme used in this study is a histidine kinase. The last
and crucial step in this identification is the purification of this putative histidine kinase.
The commercial sample is not pure, and purification has been challenging.34
As it seems
apparent that 170 and 171 bind to the enzyme, their derivatives may prove useful for
purification by affinity chromatography, as detailed in the next section.

101
Chapter 5
Preparation for Solid-Phase Peptide Synthesis
and Affinity Chromatography

102
5.1 Introduction to solid-phase peptide synthesis
Solid-phase peptide synthesis (SPPS) has been highly useful for producing peptides
with specific sequences since it was first reported a few decades ago.92
The synthetic
peptides can be used for a variety of purposes such as haptens and synthetic vaccines.92
Merrifield and co-workers developed SPPS using NBoc-protected amino acids, as
outlined in Scheme 62.93
An NBoc-protected amino acid is usually attached to a
crosslinked polystyrene resin.92
The Boc protecting group is removed by TFA, followed
by neutralisation of the resultant ammonium group with triethylamine.92
Peptide
coupling of a second Boc-protected amino acid and the resin-bound amino acid is
accomplished using a carbodiimide activating agent such as dicyclohexylcarbodiimide
(DCC).92
The cycle of reactions is repeated as required, and then the synthesised peptide
is usually cleaved from the resin with anhydrous HF.92
ResinO
O
BocN
R1
H 1. TFA
2. Et3N
ResinO
O
H2N
R1
OH
O
BocN
R2
H
CN N
ResinO
OHN
R1O
R2
BocNH
OH
OHN
R1O
R2
FH3N
HF
R3 R4
Scheme 62. Boc SPPS
Although Boc SPPS is fast, and cost of the reagents is relatively low, it does have
several disadvantages.92
The final side-chain functional group deprotection and cleavage
from the resin uses anhydrous HF, requiring special equipment and making the process

103
potentially hazardous.92
In addition, both deprotection and cleavage from the resin can
be achieved using acids, and it follows that the Boc SPPS lacks orthogonality.92
In order to overcome these problems, Fmoc SPPS has been developed (Scheme 63).92
An Fmoc-protected amino acid is usually attached to a resin with 4-
hydroxymethylphenoxy substitution.92
The Fmoc protecting group is base-labile, which
enables the N-terminal α-amino group to be revealed by piperidine.92
Most importantly,
the final cleavage from the resin and deprotection of side-chain functional groups can be
accomplished using TFA, which is vastly safer than hydrofluoric acid.92
Fmoc SPPS
utilises a base for the deprotection and an acid for the final cleavage and side-chain
deprotection, and therefore it possesses orthogonality.92
O ResinO
O
FmocN
R1
H piperidineO Resin
O
O
H2N
R1
OH
O
FmocN
R2
H
O ResinO
OHN
R1O
R2
FmocNH
OH
OHN
R1O
R2
H2N
O ResinO
OHN
R1O
R2
H2N
TFA
piperidine
N
N
N
ON
PF6
Scheme 63. Fmoc SPPS

104
5.2 SPPS by the Muir and Webb groups
As described in the introduction, during the completion of the research described in this
thesis, the Boc-protected N1- and N3-phosphohistidine analogues 35 and 36, and the
Fmoc derivative of the N3-analogue 47 were synthesised and applied the to SPPS by the
Muir40
and Webb43
research groups, respectively (Figure 50).
BocNOH
OH
N
N N
P
O
OEt
OEt
BocNOH
OH
N NN
PO
OEtEtO
FmocNOH
OH
N
N N
P
O
OEt
OEt
35 36 47
Figure 50. Peptide precursors synthesised by the Muir and Webb groups
Comparing these two methods, Muir‟s strategy revealed the phosphonic acid during
cleavage of the synthesised peptide from a resin using anhydrous HF,40
whereas Webb‟s
strategy required an extra step to deprotect the phosphonic acid using trimethylsilyl
bromide after the synthesised peptide had been detached from a resin.43
However, the
latter method is safer and more convenient than the former in that usage of anhydrous
HF can be avoided.
5.3 Synthesis of Fmoc derivatives of N1- and N3-phosphohistidine analogues
The plan in the current project was to synthesise Fmoc derivatives of N1- and N3-
phosphohistidine analogues and incorporate them into peptides using Fmoc SPPS. The
synthesis began with commercially available Fmoc-L-serine monohydrate 177, and the
carboxylic acid was protected as the benzyl ester 17894
in excellent yield (Scheme 64).81
A Mitsunobu reaction converted the alcohol 178 to the novel azide 179 in acceptable
yield.
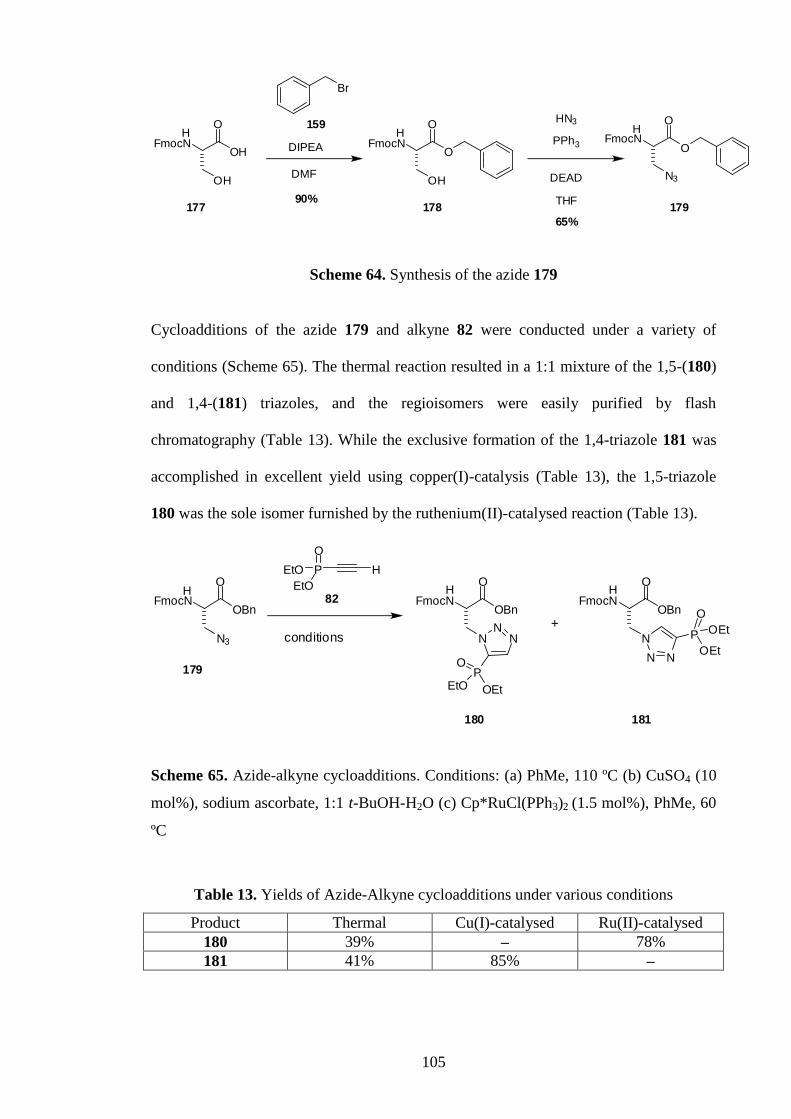
105
FmocN
O
O
N3
H
FmocN
O
OH
OH
HFmocN
O
O
OH
H
DIPEA
DMF
HN3
DEAD
PPh3
THF
Br
177
159
178 17990%
65%
Scheme 64. Synthesis of the azide 179
Cycloadditions of the azide 179 and alkyne 82 were conducted under a variety of
conditions (Scheme 65). The thermal reaction resulted in a 1:1 mixture of the 1,5-(180)
and 1,4-(181) triazoles, and the regioisomers were easily purified by flash
chromatography (Table 13). While the exclusive formation of the 1,4-triazole 181 was
accomplished in excellent yield using copper(I)-catalysis (Table 13), the 1,5-triazole
180 was the sole isomer furnished by the ruthenium(II)-catalysed reaction (Table 13).
FmocN
O
OBn
N3
HFmocN
OBn
OH
N
N N
P
O
OEt
OEt
FmocNOBn
OH
N NN
PO
OEtEtO
P H
O
EtO
EtO
+conditions
179
82
180 181
Scheme 65. Azide-alkyne cycloadditions. Conditions: (a) PhMe, 110 ºC (b) CuSO4 (10
mol%), sodium ascorbate, 1:1 t-BuOH-H2O (c) Cp*RuCl(PPh3)2 (1.5 mol%), PhMe, 60
ºC
Table 13. Yields of Azide-Alkyne cycloadditions under various conditions
Product Thermal Cu(I)-catalysed Ru(II)-catalysed
180 39% – 78%
181 41% 85% –

106
Figure 51. 1H NMR spectrum of 180
Figure 52. 1H NMR spectrum of 181
Although time did not permit any further synthetic work in this area, the peptide
precursors 182 and 47 for Fmoc SPPS can be obtained by hydrogenolysis of 180 and
181, respectively (Scheme 66). It should be noted that Fmoc chemistry of the N1-
phosphohistidine analogue 182 has not been reported.

107
FmocN
O
OBn
NN
N
PO
OEtEtO
H
H2/Pd
MeOH
FmocN
O
OH
NN
N
PO
OEtEtO
H
180 182
FmocNOBn
OH
N
N N
P
O
OEt
OEt
181
H2/Pd
MeOH
FmocNOH
OH
N
N N
P
O
OEt
OEt
47
Scheme 66. Proposed synthesis of SPPS precursors 182 and 47.
Although Webb and co-workers published the synthesis of a short peptide using the N3-
phosphohistidine analogue 47,43
there is still scope for improvement. The ethyl
phosphonate was not able to be cleanly cleaved under the deprotection conditions,
giving a mixture of free phosphonic acid, ethyl phosphonate and diethyl phosphonate,
which reduced the yield of the target peptide.43
In SPPS of peptides containing
phosphoamino acids, phosphonic and phosphoric acids are usually protected as dibenzyl
or di-tert-butyl phosphonates, because they can be removed easily by treatment with
TFA during cleavage of the synthesised peptides from a resin. In order to apply this
strategy to the current problem, the novel ethynylphosphonate 185 will need to be
synthesised (Scheme 67).

108
H TMS
EtMgBr
THF
P
O
O
OCl
P TMS
O
ButO
ButO
P H
O
ButO
ButO
K2CO3
H2O
184 18583
183
Scheme 67. Proposed synthesis of the novel ethynylphosphonate 185
The Grignard reagent formed by treatment of TMS-acetylene 83 with an excess of
ethylmagnesium bromide should react with di-tert-butyl chlorophosphate 183,95
affording the protected ethynylphosphonate 185 after treatment of the intermediate 184
with a weak base. The phosphonate 185 would be subjected to cycloadditions with the
azide 179 to afford the triazoles 186 and 187, hydrogenolysis of which would provide
the SPPS precursors 188 and 189, respectively.
FmocNOBn
O
N3
HFmocN
OBn
O
N
H
N
N
FmocNOBn
O
N
H
N N
P
O
OBut
OBut
PO
OButButO
+
Conditions
185
186 187
179
H2/Pd
FmocNOH
O
N
H
N
N
FmocNOH
O
N
H
N N
P
O
OBut
OBut
PO
OButButO
+
188 189
Scheme 68. Proposed cycloadditions to furnish SPPS precursors 188 and 189

109
5.4 Preparation for affinity chromatography
There are commercially available affinity chromatography resins carrying immobilised
phosphoserine, phosphotyrosine or phosphothreonine, which can be utilised for
convenient and rapid purification of particular proteins that bind to the phosphoamino
acids. The experiments described in Chapter 4 suggested that there is an affinity
between the free phosphohistidine analogues 170 and 171 and the histidine kinase,
implying that an affinity resin with the covalently bound phosphonotriazololylalanines
could be of great value for purification/isolation of this and other histidine kinases and,
potentially, signaling proteins that recognise phosphohistidine residues.
The Fmoc derivatives 180 and 181 described in this chapter may prove useful in
preparing affinity chromatography resins. It remains to remove the benzyl and ethyl
protecting groups to obtain the precursors 190 and 191 for affinity chromatography
carrying the phosphohistidine analogues (Scheme 69). Deprotection using hydrogen
bromide in acetic acid has been dependable throughout this project, and there is
literature precedent suggesting that fluorenylmethyl carbamates are stable under these
conditions.96
FmocN
O
OBn
N
N N
P
O
OEtOEt
H
FmocN
O
OBn
NN
N
PO
OEtEtO
H
HBr
AcOH
FmocN
O
OH
N
N N
P
O
OHOH
H
FmocN
O
OH
NN
N
PO
OHHO
H
HBr
AcOH
180
181
190
191
Scheme 69. Proposed synthesis of the precursors for affinity chromatography
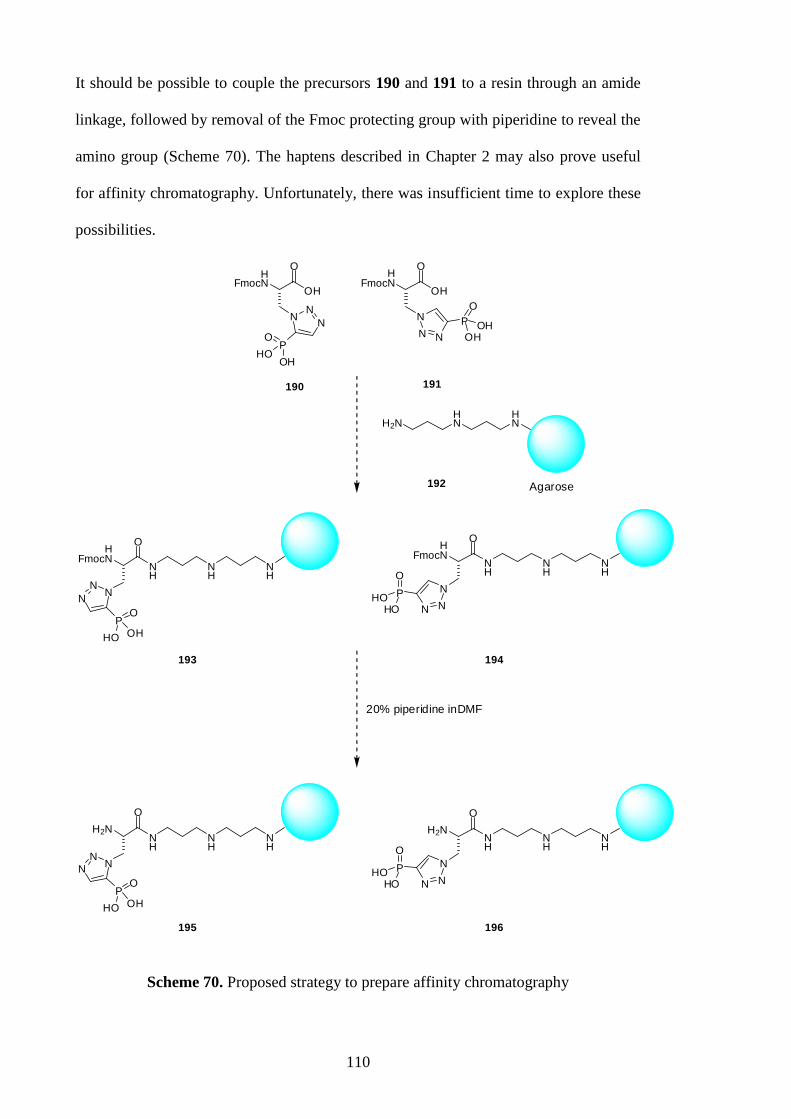
110
It should be possible to couple the precursors 190 and 191 to a resin through an amide
linkage, followed by removal of the Fmoc protecting group with piperidine to reveal the
amino group (Scheme 70). The haptens described in Chapter 2 may also prove useful
for affinity chromatography. Unfortunately, there was insufficient time to explore these
possibilities.
HN
HNH2N
Agarose
FmocN
O
OH
N
N N
P
O
OHOH
HFmocN
O
OH
NN
N
PO
OHHO
H
FmocN
O
NH
H
NH
NH
FmocN
O
NH
H
NH
NH
N
N NP
O
HOHO
NN
N
PO
OHHO
H2N
O
NH
NH
NH
H2N
O
NH
NH
NH
N
N NP
O
HOHO
NN
N
PO
OHHO
20% piperidine inDMF
190 191
192
194193
196195
Scheme 70. Proposed strategy to prepare affinity chromatography

111
Chapter 6
Attempted Synthesis of Pyrazole and Imidazole Analogues of
Phosphohistidine

112
6.1 Attempted synthesis of a phosphonopyrazole analogue of N3-phosphohistidine
The extra nitrogen atom in the triazoles described in this thesis does lower the basicity
of the heterocycle relative to histidine. Thus, in an attempt to more closely match the
pKa of phosphohistidine, which may be important in antibody recognition, a synthesis of
phosphonopyrazole and phosphonoimidazole analogues has been pursued.
The initial plan to reach the target analogue 199 involved synthesising the
phosphonopyrazole 197 and subjecting it to a displacement reaction with a serine-
derived tosylate 198 (Scheme 71).
N
NHPEtO
EtO
O
RNOR'
O
OTs
H
RNOR'
O
N
H
N
P
O
OEt
OEt
197 199
198
Scheme 71. Plan to synthesise a pyrazole N3-phosphohistidine analogue
6.1.1 Synthesis of pyrazolylphosphonates
The starting point for the synthesis was iodination of pyrazole 200 to give 4-
iodopyrazole 20197
in excellent yield. An initial attempt to install the phosphono group
used a Nickel(II)-catalysed Arbuzov reaction55
of the unprotected pyrazole 201 with
triethyl phosphite (Scheme 72). Unfortunately, no reaction was observed, which
prompted us to protect the pyrazole nitrogen as the p-toluenesulfonamide 203.98
Nevertheless, as judged by the 1H NMR and ESI-MS spectra of the crude product, the
phosphorylation reaction of 203 was unsuccessful, although 203 was completely
consumed (Scheme 72).

113
N
NH
N
NHI
CAN
I2
MeCN
N
NHP
O
EtO
EtO
P(OEt)3
NiCl2
N
NTsI
TsCl
pyridine
DCM
N
NTsP
O
EtO
EtO
P(OEt)3
200 201 202
203 204
93%
63%
150 oC
NiCl2 150 oC
Scheme 72. Attempted Arbuzov reactions to furnish phosphonopyrazoles
The third attempt to synthesise the phosphonate 204 used iodine-magnesium exchange
of the 203 with isopropylmagnesium bromide,99
followed by quenching with diethyl
chlorophosphate 80. 1H NMR and ESI-MS spectra of the crude product indicated the
perfect consumption of 203, but there was no trace of the desired phosphonate 204
(Scheme 73).
N
NTsI
N
NTsP
O
EtO
EtO
iPrMgBr
THF
ClP
O
EtO
EtO
203 204
80
Scheme 73. Attempted iodine-magnesium exchange to give the phosphonopyrazole 204
Because of the difficulties above, another strategy to obtain the phosphonate 204 was
investigated. There is literature precedent in which copper(I)-catalysed coupling of aryl
halides 205 and dibutyl phosphate 206 proceeded in excellent yields (Scheme 74).100

114
I
R CuI
Cs2CO3
H2NNH2
HP
O
BuO
BuO
206 P
R
O
OBu
OBu
PhMe
110C
205 207
R Yield
OMe 85%
NH2 86%
Scheme 74. Precedent for Cu(I)-catalysed C-P bond formation100
According to the procedures,100
Cu(I)-catalysed coupling of the iodides 201 or 203 and
diethyl phosphite 79 was attempted; however, no reaction was observed in the case of
iodopyrazole 201, and only the reduced compound was produced with the N-tosyl
derivative 203 (Scheme 75).
N
NHI
N
NHP
O
EtO
EtO
CuI
HP
O
EtO
EtO
Cs2CO3
H2NNH2
N
NTsI
HP
O
EtO
EtO
CuI
Cs2CO3
H2NNH2
201
N
NTsP
O
EtO
EtO
202
204203
79
79
110 oC
110 oC
Scheme 75. Attempted Cu(I)-catalysed coupling of iodopyrazoles and diethyl phosphite

115
Finally, palladium-catalysed coupling101
of the iodide 203 and diethyl phosphite 79
gave the target phosphonate 204 in acceptable yield (Scheme 76). Historically, N-
detosylation of aromatic heterocycles has been conducted by using a base, such as
potassium hydroxide, in methanol.102
However, the formation of toxic methyl p-
toluenesulfonate is problematic under these conditions.103
The use of aqueous THF with
a phase transfer catalyst in place of methanol can prevent toxic alkyl p-
toluenesulfonates from being generated.102
Detosylation was achieved, in this case, by
heating with potassium hydroxide in aqueous dioxane, providing the target
phosphonopyrazole 202 in good yield, without the need for the phase transfer catalyst.
The unprotected pyrazole 201 also underwent the palladium(II)-catalysed coupling101
with diethyl phosphate 79, furnishing the phosphonopyrazole 202 in acceptable yield
(Scheme 76). Despite more troublesome purification using this strategy, the yield of the
phosphonate was ameliorated compared with the two step procedure.
N
NTsI
N
NTsP
O
EtO
EtO
PPh3
Et3N
HP
O
EtO
EtO
Pd(OAc)2
EtOH
N
NHP
O
EtO
EtO
KOH
Dioxane
H2O
N
NHI
N
NHP
O
EtO
EtO
HP
O
EtO
EtO
Pd(OAc)2
PPh3
Et3N
EtOH
203 204 202
202201
79
79
50%
53%
47%
Scheme 76. Synthesis of the phosphonopyrazole 202
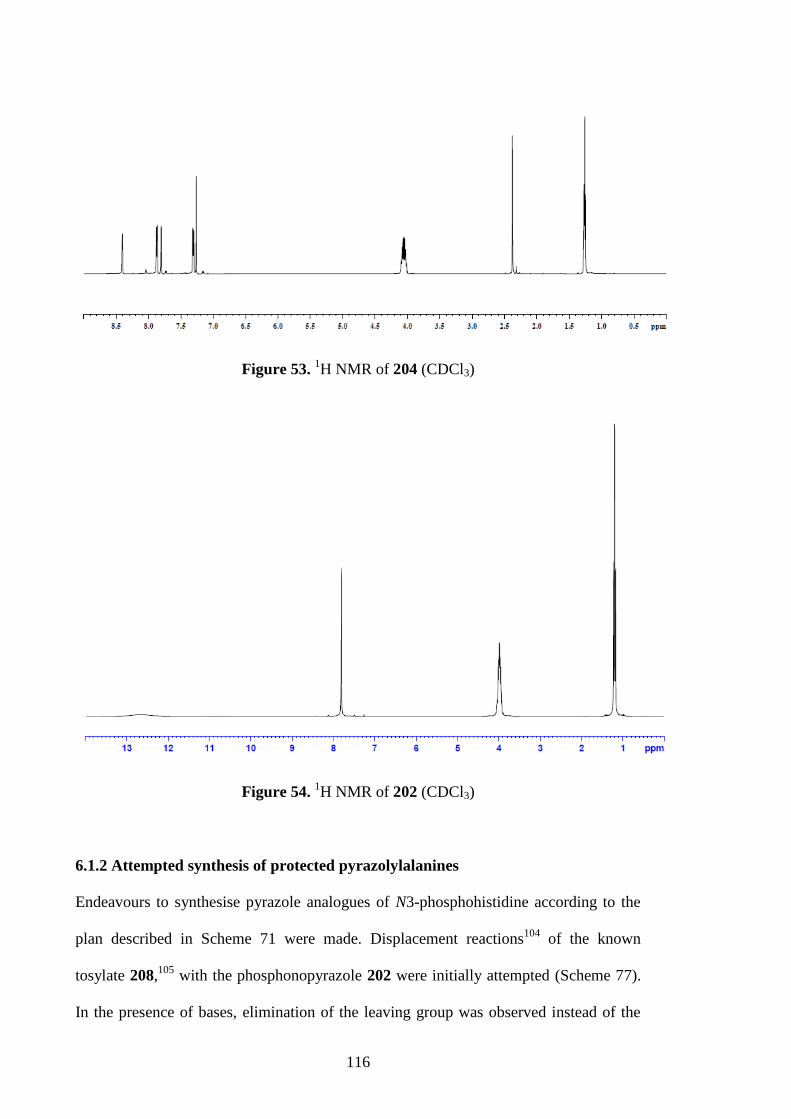
116
Figure 53. 1H NMR of 204 (CDCl3)
Figure 54. 1H NMR of 202 (CDCl3)
6.1.2 Attempted synthesis of protected pyrazolylalanines
Endeavours to synthesise pyrazole analogues of N3-phosphohistidine according to the
plan described in Scheme 71 were made. Displacement reactions104
of the known
tosylate 208,105
with the phosphonopyrazole 202 were initially attempted (Scheme 77).
In the presence of bases, elimination of the leaving group was observed instead of the
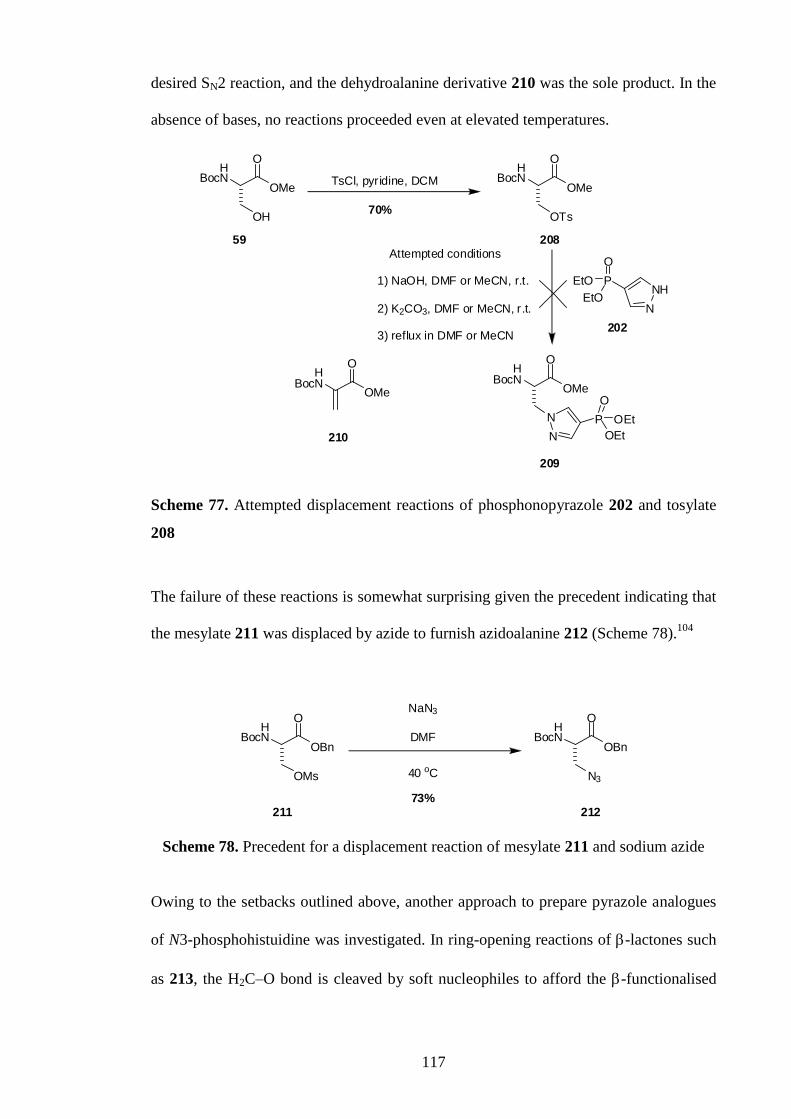
117
desired SN2 reaction, and the dehydroalanine derivative 210 was the sole product. In the
absence of bases, no reactions proceeded even at elevated temperatures.
N
NHP
O
EtO
EtO
BocNOMe
OH
N
N
P
O
OEt
OEt
Attempted conditions
1) NaOH, DMF or MeCN, r.t.
2) K2CO3, DMF or MeCN, r.t.
3) reflux in DMF or MeCN
BocNOMe
OH
BocNOMe
OH
OH
BocNOMe
OH
OTs
TsCl, pyridine, DCM
70%
59 208
209
210
202
Scheme 77. Attempted displacement reactions of phosphonopyrazole 202 and tosylate
208
The failure of these reactions is somewhat surprising given the precedent indicating that
the mesylate 211 was displaced by azide to furnish azidoalanine 212 (Scheme 78).104
BocNOBn
OH
OMs
BocNOBn
OH
212211
NaN3
DMF
40 oC N3
73%
Scheme 78. Precedent for a displacement reaction of mesylate 211 and sodium azide
Owing to the setbacks outlined above, another approach to prepare pyrazole analogues
of N3-phosphohistuidine was investigated. In ring-opening reactions of -lactones such
as 213, the H2C–O bond is cleaved by soft nucleophiles to afford the -functionalised
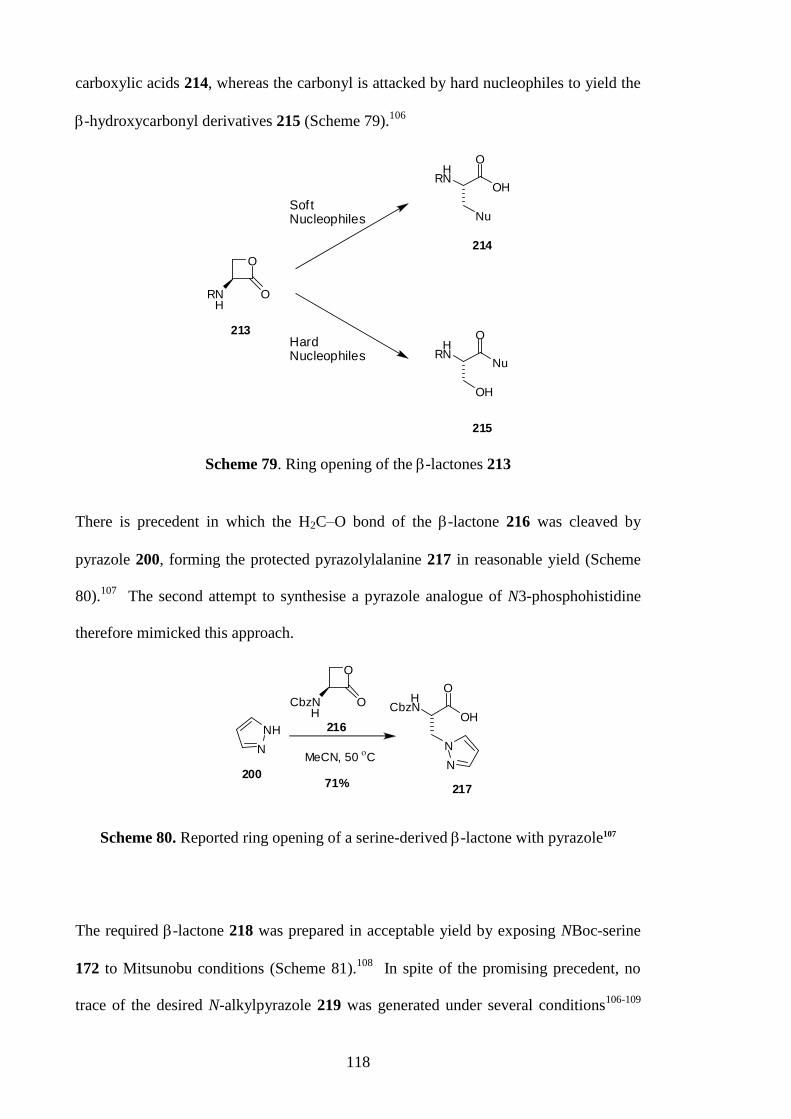
118
carboxylic acids 214, whereas the carbonyl is attacked by hard nucleophiles to yield the
-hydroxycarbonyl derivatives 215 (Scheme 79).106
O
RNH
O
SoftNucleophiles
HardNucleophiles
RNOH
OH
Nu
RNNu
OH
OH
213
214
215
Scheme 79. Ring opening of the -lactones 213
There is precedent in which the H2C–O bond of the -lactone 216 was cleaved by
pyrazole 200, forming the protected pyrazolylalanine 217 in reasonable yield (Scheme
80).107
The second attempt to synthesise a pyrazole analogue of N3-phosphohistidine
therefore mimicked this approach.
O
CbzNH
O
N
NH
CbzNOH
O
N
H
NMeCN, 50
C
71%200
217
216
Scheme 80. Reported ring opening of a serine-derived -lactone with pyrazole107
The required -lactone 218 was prepared in acceptable yield by exposing NBoc-serine
172 to Mitsunobu conditions (Scheme 81).108
In spite of the promising precedent, no
trace of the desired N-alkylpyrazole 219 was generated under several conditions106-109

119
(Scheme 81). Although the -lactone 218 was completely consumed, what occurred is
still unclear. It is possible that the reduced nucleophilicity of 202 relative to pyrazole
200, owing to the electron-withdrawing phosphoryl group, contributed to the
unexpected result, with decomposition of -lactone 218 occurring more rapidly than the
desired reaction.
BocNOH
OH
OH
DEAD
PPh3
THF
O
OBocNH
BocNOH
OH
N
N
P
O
OEt
OEt
N
NHP
O
EtO
EtO
Attempted conditions
1) MeCN or DMF, 50 oC
2) K2CO3, MeCN or DMF, 50 oC
3) Cs2CO3, MeCN or DMF, 50 oC
4) NaH, MeCN or DMF, 50 oC
202
45%172
218
219
Scheme 81. Attempted ring opening reactions of the -lactone
Most recently, a Mitsunobu reaction was chosen for the purpose of making the pyrazole
analogue, because of the report of a conventional Mitsunobu reaction of 4-nitropyrazole
220 and the alcohol 221 (Scheme 82).110
Presumably, this reaction is only possible
because of the enhanced acidity of the pyrazole NH due to the electron-withdrawing
nitro group. It was felt that the phosphoryl group in 202 might similarly lower the pKa
sufficiently for a Mitsunobu reaction to be viable.

120
NNH
O O
OH
DEAD
PPh3
THF
69%
O O
NN
NO2
NO2
220
221
222
Scheme 82. An example of a Mitsunobu reaction of a pyrazole.110
When conventional Mitsunobu conditions were applied to the alcohol 59 and
phosphonopyrazole 202, the reaction did not proceed as anticipated, probably because
the pKa of the phosphonopyrazole is not low enough (Scheme 83).
N
NHP
O
EtO
EtO
BocNOMe
OH
OH
BocNOMe
OH
N
N
P
O
OEt
OEtDEAD
PPh3
THF
202
209
59
Scheme 83. Unsuccessful Mitsunobu reaction of diethyl 4-pyrazolylphosphonate
Tsunoda and co-workers proved that a combination of either azodicarboxylic acid
dipiperidide (ADDP, 223)111
or N,N,N′,N′-tetramethylazodicarboxamide (TMAD)112
and
tributylphosphine (PBu3) is a powerful tool for Mitsunobu reactions of alcohols and
weak acids with pKa values higher than 11.111,112
Therefore, in the current work, ADDP
223 was prepared by treating DEAD 62 with piperidine (Scheme 84).113
Due to time
constraints, the Mitsunobu reaction employing ADDP 223 and PBu3 (Scheme 85) was
not attempted, but is planned for future work.111

121
N N O
OO
O N N N
OO
N
62 223
NH
70%
Et2O
Scheme 84. Synthesis of ADDP
N
NHP
O
EtO
EtO
BocNOMe
OH
OH
BocNOMe
OH
N
N
P
O
OEt
OEt
ADDP
PBu3
THF
202
209
59
Scheme 85. Proposed Mitsunobu reaction using ADDP and PBu3
6.2 Imidazole analogues of N1-phosphohistidine
Imidazole analogues of N1-phosphohistidine have also been pursued. The synthesis
began with the preparation of 4(5)-iodoimidazole 226, via the triiodide 225, as reported
previously (Scheme 86).114
Surprisingly, the palladium-catalysed phosphonation101
that
worked well in the synthesis of the phophonopyrazoles 202 and 204 was ineffective for
the preparation of the target phosphonoimidazoles 228 and 230, although ESI-MS
spectra of the crude products indicated the perfect consumption of the starting materials
(Scheme 87).

122
NHN NHN
I
I I
NHN
I
I2
KI
NaOH
H2O
Na2SO3
EtOH
H2O
60% over two steps
NHN
I
224 225 226 227
Scheme 86. Synthesis of 4(5)-iodoimidazole
HP
O
EtO
EtO
Pd(OAc)2
PPh3
Et3N
EtOH
NHN
I
NHN
PO
OEtOEt
226
79
228
NTsNNTsN
229 230
I
TsCl
Et3N
THF
70%
PO
EtOOEt
HP
O
EtO
EtO
Pd(OAc)2
PPh3
Et3N
EtOH
79
Scheme 87. Attempted phosphonation of 4(5)-iodoimidazole
Although time did not permit any further synthetic work in this area, alternative
strategies to synthesise the novel phosphonoimidazole have been considered. Several
articles reported that iodine-magnesium exchange of N-protected iodoimidazoles
proceeded smoothly in DCM despite Grignard reactions normally being performed in
etheral solvents.115,116
It is proposed that 4-iodo-1-p-toluenesulfonylimidazole 229114
may undergo such iodine-magnesium exchange, and quenching the reaction with diethyl

123
chlorophosphate 80 should provide the protected phosphonoimidazole 230 (Scheme
88). The tosyl protecting group could then be removed by potassium hydroxide in
aqueous dioxane to afford the desired phosphonoimidazole 228.
NTsN
I
ClP
O
EtO
EtO
iPrMgBr
DCMNTsN
PO
EtOOEt
NHN
PO
EtOOEt
KOH
Dioxane
H2O229
230 228
80
Scheme 88. Proposed route to the imidazolylphosphonate 228
A Mitsunobu reaction utilising a combination of ADDP and PBu3 could potentially
furnish an imidazole analogue of N1-phosphohistidine 231; however, it is likely to be
complicated by competing formation of the 1,5-substituted imidazole 232 (Scheme 89).
More likely, an alternative approach will be required.
NHN
PO
EtOOEt
BocNOMe
OH
OH
BocNOMe
OH
NN
ADDP
PBu3
THF
BocNOMe
OH
NN
PP
O
EtOOEt
O
OEtEtO
+
228
231 232
55
Scheme 89. Challenging Mitsunobu reaction for the imidazole analogue 231

124
Conclusion
The primary aim of the research described in this thesis was to synthesise a series of
haptens, based on stable N1- and N3-phosphohistidine analogues, to be utilised for
generation of generic phosphohistidine antibodies. The first goal, the synthesis of
haptens incorporating triazolylalaninephosphonates, was achieved. Several strategies to
conjugate the haptens to carrier proteins were attempted, but none have been successful
to date. However, it is likely that with further experimentation, bioconjugation will be
achieved and antibody generation will ensue soon after.
Towards the conclusion of the work described herein, virtually identical research was
published by other groups,40,43
including the generation of polyclonal antibodies that
specifically recognise histidine-phosphorylated histone H4.40
However, the antibodies
do not recognise other histidine-phosphorylated proteins,40
and therefore we are still in
pursuit of generic phosphohistidine antibodies.
The biological activities of the phosphohistidine analogues synthesised in this project
were of particular interest, and accordingly, the free triazolylalaninephosphonate
analogues of N1- and N3-phosphohistidine have been synthesised. In preliminary
experiments, the analogues were found to inhibit a mammalian histidine kinase. This
observation may be the starting point for the discovery of more potent histidine kinase
inhibitors with potential therapeutic use. In addition, these inhibitors may be useful for
affinity chromatography to allow purification/isolation of histidine kinases and,
potentially, other signaling proteins that recognise histidine-phosphorylated proteins.

125
Chapter 7
Experimental

126
General details
All solvents were distilled prior to use; anhydrous solvents and reagents were distilled
under N2. THF for organometallic reactions was distilled from sodium benzophenone
ketyl. All reaction temperatures refer to bath temperatures. Organic extracts were dried
over anhydrous MgSO4 and then filtered. Solvents were evaporated at vacuum or
under a stream of N2.
Flash chromatography was conducted with Merck silica gel 60. Analytical TLC was
performed on Whatman flexible plates (250 m layer, silica gel 60 F254). Spots were
visualised under UV light and by staining with ammonium molybdate or ninhydrin.
Melting points were measured on a Kofler hot stage melting point apparatus and are
uncorrected. IR spectra were acquired using a Perkin-Elmer Spectrum One FTIR
spectrometer. Mass spectra were acquired on an Applied Biosystems QSTAR pulsar I
quadrupole time-of-flight instrument or a Waters LCT Premier XE spectrometer.
Histone phosphorylation was quantified using a Fuji Photo Film BAS-III
phosphoimager. NMR spectra were acquired on Varian Inova 300 (300 MHz, 1H; 75.5
MHz, 13
C; 120 MHz, 31
P), Varian 400 (400 MHz, 1H; 100 MHz,
13C), Bruker Avance
500 (500 MHz, 1H; 125 MHz,
13C) or Bruker AV600 (600 MHz,
1H; 150 MHz,
13C;
240 MHz, 31
P) spectrometers, as indicated. Chemical shifts are expressed in ppm,
relative to CHCl3 (1H, 7.26 ppm), CDCl3 (
13C, 77.0 ppm), CD3SOCD2H (
1H, 2.50
ppm), (CD3)2SO (13
C, 39.5 ppm), CHD2OD (1H, 3.30 ppm), CD3OD (
13C, 49.0 ppm),
and 85% H3PO4 (external capillary, 31
P, 0 ppm), as appropriate. Routine assignments
of 13
C NMR spectra were made with the assistance of DEPT 135 and DEPT 90
experiments.

127
HPLC
HPLC was conducted using a Hewlett Packard 1050 HPLC system equipped with a
multiple wavelength detector (MWD) and a 250 × 10 mm i.d., 5 µm, Apollo C18
reversed-phase column (Grace-Davison), with a 33 mm × 7 mm guard column of the
same material. The samples were eluted at 4 mL/min with 30% (v/v) MeCN-water.
UV absorbance was measured at 220 nm.
Enantioselective chromatography of the carboxylic acids 35 and 36 was carried out
using a 250 × 4.6 mm i.d., 5 µm Chiracel OD-H column (Diacel), eluted at 1.0 mL/min
with 8% isopropanol/hexanes, and detection at 220 nm.
~Chapter 2~
ClH3NOMe
O
OH
(S)-3-Hydroxy-1-methoxy-1-oxopropan-2-aminium chloride (58)
This known compound was synthesised according to the method of Rudd.45
Thionyl chloride (8.70 mL, 121 mmol) was added dropwise to a suspension of L-serine
(12.5 g, 120 mmol) in MeOH (240 mL) at 0 ºC. The resulting mixture was allowed to
warm to room temperature and stirred for 18 h. The MeOH was evaporated, and the
residue was washed with Et2O. The solid was recrystallised from MeOH to afford the
title hydrochloride 58 as a white solid (13.5 g, 74 %). 1H NMR (500 MHz, CD3OD):
4.10-4.15 (m, 1H, -H), 3.90-4.00 (m, 2H, -H), 3.81 (s, 3H, OCH3). 13
C NMR (125
MHz, CDCl3): δ 169.3 (CO), 60.6 (), 56.1 (OCH3), 53.8 (). MS (ESI) m/z: 120
[M+H]+
(where M = free base). []D = +4.1 (c 1.0, MeOH) [lit.45
+3.9 (c 4.05,
MeOH)].

128
BocNOMe
OH
OH
(S)-Methyl-2-(tert-butoxycarbonylamino)-3-hydroxypropanoate (59)
This known compound was synthesised according to the method of Rudd with slight
modifications.45
Boc anhydride (7.00 g, 32.1 mmol) was added to a suspension of the hydrochloride 58
(5.00 g, 32.1 mmol) and triethylamine (13.4 mL, 96.1 mmol) in MeCN (100 mL) at 0
ºC. The resulting mixture was allowed to warm to room temperature and stirred for 6 h,
and then the MeCN was evaporated. The residue was diluted with sat. aq. KHSO4 and
then extracted with DCM (3 × 50 mL). The organic extract was washed with sat. aq.
NaHCO3 (50 mL) and brine (50 mL), dried and concentrated under reduced pressure.
The crude product was subjected to flash chromatography. Elution with 2:3
EtOAc/hexanes gave 59 as a pale yellow oil (6.37 g, 90%). 1H NMR (500 MHz,
CDCl3): 5.62 (br d, J = 7.0 Hz, 1H, NH), 4.28 (m, 1H, -H), 3.88 (m [appearent br d],
1H, -Ha), 3.77 (dd, 1H, J = 11.0, 3.5 Hz, -Hb), 3.69 (s, 3H, OCH3), 1.37 (s, 9H, t-
Bu). 13
C NMR (125 MHz, CDCl3): δ 171.4 (CO) 155.7 (NCO), 80.0 [OCCH3)3], 62.9
(), 55.6 (OCH3), 52.3 (), 28.1 [CCH3)3]. MS (ESI) m/z: 242 [M+Na]+. []D = +10.1
(c 1.0, CH3Cl) [lit.117
+9.7 (c 4.29, CH3Cl)].
Preparation of a 2.5 M hydrazoic acid (HN3) solution.118
Concentrated sulfuric acid (3.60 mL) was added dropwise to a suspension of sodium
azide (8.45 g, 130 mmol) in toluene (51.5 mL) and water (4.20 mL) at 0 ºC. The
resulting mixture was stirred for 1 h at 0 ºC, and then the precipitated Na2SO4 was
filtered off. The obtained HN3 solution was immediately used for a Mitsunobu reaction.

129
NH
NH
O
OO
O
Ethyl hydrazodicarboxylate (70)
This known compound was synthesised according to the methods of Wang.119
Ethyl chloroformate (4.60 mL, 48.4 mmol) was added dropwise to a solution of
hydrazine monohydrate (1.80 mL, 37.2 mmol) at 0 ºC. After the addition, a solution of
Na2CO3 (5.30 g, 50.0 mmol) in water (40 mL) was added to the mixture simultaneously
with ethyl chloroformate (4.60 mL, 48.4 mmol) at 0 ºC. The resulting mixture was stirrd
at 0 ºC for 1 h, and then the precipitate was filtered, washed with cold water (30 mL),
EtOH (30 mL) and Et2O (30 mL) and dried under vacuum, affording 70 as a white solid
(3.93 g, 60%). 1H NMR (300 MHz, d4-DMSO): 9.00 (br s, 2H, NHCH2CH3), 4.06 (q,
J = 6.9 Hz, 4H, NHCH2CH3), 1.20 (t, J = 6.9 Hz, 6H, NHCH2CH3). 13
C NMR (75 MHz,
d4-DMSO): δ 156.9 (NCO), 60.9 (CH2), 14.8 (CH3).
N N O
OO
O
Diethylazodicarboxylate (62)
This known compound was synthesised according to the methods of Steel.120
Concentrated nitric acid (20 mL) was added dropwise to a suspension of 70 (3.00 g,
17.0 mmol) in CHCl3 (20 mL) at 0 ºC. The resulting solution was stirred at 0 ºC for 3 h
and then diluted with water (50 mL). The layers were separated, and the aqueous phase
was extracted with CHCl3 (3 × 60 mL). The organic extract was washed with water (50
mL), sat. aq. NaHCO3 (2 × 50 mL) and brine (50 mL), dried and concentreated under a
stream of N2, affording 62 as an orange liquid (2.07 g, 70%) in a sufficiently pure state.
1H NMR (300 MHz, CDCl3): 4.10 (q, J = 7.2 Hz, 4H, NCH2CH3), 1.01 (t, J = 7.2 Hz,
6H, NCH2CH3) 13
C NMR (75 MHz, CDCl3): δ 159.9 (NCO), 65.0 (CH2), 13.2 (CH3).

130
BocN
O
OMe
N3
H
(S)-Methyl-3-azido-2-(tert-butoxycarbonylamino)propanoate (60)
This compound was synthesised according to the method of Boger44
with slight
modifications.
A stirred solution of the alcohol 59 (3.61 g, 16.5 mmol) and triphenylphosphine (6.49 g,
24.7 mmol) in anhydrous THF (20 mL) at –78 ºC under N2 was treated with a 2.5 M
solution of HN3 in toluene (9.85 mL, 24.7 mmol) and DEAD (3.90 mL, 24.7 mmol).
The resulting solution was allowed to warm to room temperature. After 5 h the solution
was diluted with water (100 mL), the layers were separated and the aqueous phase was
extracted with Et2O (3 × 80 mL). The combined organic phase was washed with brine
(50 mL), dried and concentrated under reduced pressure. The residue was subjected to
flash chromatography. Elution with 1:4 Et2O/hexanes gave 7 as a colourless oil (2.90 g,
72 %). 1H NMR (600 MHz, CDCl3): 5.61 (br d, 1H, NH), 4.28 (m, 1H, -H), 3.55 (s,
3H, COOCH3), 3.49 (m [apparent br d], 2H, -H), 1.23 (s, 9H, t-butyl). 13
C NMR (150
MHz, CDCl3) 169.7, 154.6, 79.5, 77.2, 53.0, 51.9, 27.6. MS (ESI) m/z: 343 [M+Na]+.
[]D = +36.4 (c 2.0, CHCl3) [lit.48
+36.0 (c 0.6, CHCl3)]. The 1H NMR data are
similar to those reported.48
P
O
EtO
EtOCl
Diethyl chlorophosphate (80)
This known compound was synthesised according to the method of Steinberg.52
Triethylamine (1.10 mL, 7.90 mmol) was added to a solution of diethyl phosphite (8.60
mL, 67.0 mmol) in carbon tetrachloride (13 mL) at 0 ºC. The resulting solution was
allowed to warm up to room temperature, stirred for 18 h and then volatile components

131
were removed under reduced pressure. The residue was distilled under vacuum at 50 ºC,
giving 80 as a pale yellow liquid (9.77 g, 85%). 1H NMR (400 MHz, CDCl3): 4.10 (m,
4H, OCH2CH3), 1.33 (t, J = 5.2 Hz, 6H, OCH2CH3) 13
C NMR (100 MHz, CDCl3): δ
65.7 (d, JC,P = 7.4 Hz, CH2), 15.5 (d, JC,P = 8.2 Hz, CH3). 31
P NMR (120 MHz, CDCl3):
δ 5.35.
P TIPS
O
EtO
EtO
Diethyl (triisopropylsilyl)ethynylphosphonate (81)
Ethyl bromide (1.00 mL, 13.7 mmol) was added dropwise to a suspension of stirred Mg
turnings (416 mg, 17.0 mmol) in THF (10 mL) at 0 ºC under N2 and the resulting
mixture was heated at reflux for 30 min. The ethynylmagnesium bromide solution thus
prepared was added dropwise via canula to a solution of TIPS-acetylene (3.00 mL, 13.7
mmol) in THF (10 mL) at 0 ºC under N2. The reaction mixture was allowed to warm to
room temperature and stirring was continued for 1 h before being cooled to 0 ºC and
treated dropwise with diethyl chlorophosphate (2.20 mL, 15.2 mmol). The resulting
solution was allowed to warm to room temperature and after 1 h was diluted with sat.
aq. NH4Cl (30 mL) and extracted with Et2O (3 × 50 mL). The organic extract was
washed with brine (30 mL), dried and concentrated under reduced pressure, and the
residue was subjected to flash chromatography. Elution with 1:4 EtOAc/hexanes gave
81 as a pale yellow oil (3.49 g, 80%). Rf = 0.80 (1:1 EtOAc/hexanes). 1H NMR (600
MHz, CDCl3): 4.11 (m, 4H, OCH2CH3), 1.32 (dt, JH,P = 0.6, J = 7.1 Hz, 6H,
OCH2CH3), 1.03–1.10 (m, 21H, i-Pr). 13
C NMR (150 MHz, CDCl3): δ 106.4 (d, JC,P =
37.6 Hz, C2), 96.3 (d, JC,P = 269.4 Hz, C1), 63.0 (d JC,P = 5.4 Hz), 18.3 (i-Pr CH3), 15.9
(d, JC,P = 6.8 Hz, OCH2CH3), 10.7 (CSi). 31
P NMR (120 MHz, CDCl3): δ –7.38; MS
(ESI) m/z: 319 [M+H]+, 341 [M+Na]
+. This compound has been reported but not
characterised.121

132
P H
O
EtO
EtO
Diethyl ethynylphosphonate (82)
(Method A; Removal of TIPS)
A solution of 81 (1.00 g, 3.14 mmol) and KF (0.38 g, 6.5 mmol) in MeOH (10 mL) was
stirred for 2 h and then concentrated under reduced pressure. The residue was diluted
with water (50 mL) and extracted with Et2O (3 × 40 mL). The organic extract was
washed with brine (30 mL), dried and concentrated under reduced pressure. The residue
was subjected to flash chromatography. Elution with 1:1 Et2O/hexanes gave 82 as a pale
yellow oil (310 mg, 61%). 1H NMR (500 MHz, CDCl3): 4.04 (m, 4H, CH2), 3.00 (d,
JH,P = 13.5 Hz 1H, C), 1.24 (dt, JH,P = 0.5 J = 7.0 Hz, 6H, 2 × CH3). 13
C NMR (125
MHz, CDCl3): δ 88.1 (d, J = 50.6 Hz, CH), 73.8 (d, J = 289 Hz, CP), 63.1 (d, J = 5.5
Hz, OCH2CH3), 15.7 (d, J = 7.0 Hz, OCH2CH3). MS (ESI) m/z: 163 [M+H]+. The
1H
and 13
C NMR data are similar to those reported.43
(Method B; Grignard reaction using TMS acetylene)
The procedure reported by Mignani51
was slightly modified.
Ethyl bromide (1.20 mL, 16.1 mmol) was added dropwise to a suspension of stirred Mg
turnings (487 mg, 20.0 mmol) in THF (20 mL) at 0 ºC under N2 and the resulting
mixture was heated at reflux for 30 min. The ethynylmagnesium bromide solution thus
prepared was added dropwise via canula to a solution of TMS-acetylene (2.30 mL, 16.3
mmol) in THF (10 mL) at 0 ºC under N2. The reaction mixture was allowed to warm to
room temperature and stirring was continued for 1 h before being cooled to 0 ºC and
treated dropwise with diethyl chlorophosphate (2.50 mL, 17.2 mmol). The resulting
solution was allowed to warm to room temperature and after 1 h was diluted with sat.
aq. NH4Cl (30 mL) and extracted with Et2O (3 × 50 mL). The organic extract was
washed with brine (30 mL), dried and concentrated under reduced pressure. The

133
obtained residue was dissolved in 10% aq. Na2CO3 (50 mL), stirred for 18 h, then
diluted with sat. aq. NH4Cl (30 mL) and extracted with Et2O (3 × 50 mL). The organic
extract was washed with brine (30 mL), dried and concentrated under reduced pressure.
The residue was subjected to flash chromatography. Elution with 1:3 EtOAc/hexanes
gave 82 as a pale yellow oil (2.01 g, 77%), identical with the material described above.
P
O
EtO
EtO
OH
Diethyl 3-hydroxy-3-methylbut-1-ynylphosphonate (87)
(Method A: Grignard reaction)
Ethyl bromide (1.00 mL, 13.7 mmol) was added dropwise to a suspension of stirred Mg
turnings (416 mg, 17.0 mmol) in THF (10 mL) at 0 °C under N2 and the resulting
mixture was heated at reflux for 30 min. The ethynylmagnesium bromide solution thus
prepared was added dropwise via canula to a solution of the alkyne 86 (1.13 g, 13.4
mmol) in THF (10 mL) at 0 ºC under N2. The reaction mixture was allowed to warm to
room temperature and stirring was continued for 1 h before being cooled to 0 ºC and
treated dropwise with diethyl chlorophosphate (2.20 mL, 15.2 mmol). The resulting
solution was allowed to warm to room temperature and after 1 h was diluted with sat.
aq. NH4Cl (30 mL) and extracted with Et2O (3 × 50 mL). The organic extract was
washed with brine (30 mL), dried and concentrated under reduced pressure, and the
residue was subjected to flash chromatography. Elution with 2:3 EtOAc/hexanes gave
87 as a pale yellow oil (2.27 g, 77%). Rf = 0.20 (1:1 EtOAc/hexanes). 1H NMR (500
MHz, CDCl3): 3.44 (m, 4H, OCH2CH3), 2.33 (s, 1H, OH), 1.07 (s, 6H, CCH3), 0.69
(dt, JH,P = 1.0, J = 7.0 Hz, 6H, OCH2CH3). 13
C NMR (125 MHz, CDCl3): δ 83.1 (d, JC,P
= 4.5 Hz, C2), 72.7 [C(CH3)2], 72.4 (d, JC,P = 7.0 Hz, C1), 62.0 (d, JC,P = 5.9 Hz,
OCH2CH3), 29.2 (d, JC,P = 4.7 Hz, [C(CH3)2], 14.6 (d, JC,P = 6.8 Hz, OCH2CH3). 31
P
NMR (120 MHz, CDCl3): δ –5.22. MS (ESI) m/z: 243 [M+Na]+.

134
(Method B: Ni(II)-catalysed Arbuzov reaction)
A solution of the chloride 88 (1.00 g, 8.43 mmol) and NiCl2.5H2O (340 mg, 1.55
mmol) in triethylphosphite (5.00 mL, 29.2 mmol) was heated at reflux for 6 h, and
then the excess triethylphosphite was removed under reduced pressure. The residue
was diluted with water (50 mL) and extracted with EtOAc (3 × 50 mL). The organic
extract was washed with brine (50 mL), dried and concentrated under reduced
pressuere. The residue was subjected to flash chromatography. Elution with 2:3
EtOAc/hexanes gave 87 as a pale yellow oil (874 mg, 47%), identical in every respect
with the material described above.
BocNOMe
OH
N
N N
P
O
OEt
OEt
BocNOMe
OH
N NN
PO
OEtEtO
121 122
Cycloadditions of the azide 60 and alkyne 82
Thermal
A solution of the azide 60 (150 mg, 0.614 mmol) and the alkyne 82 (100 mg, 0.614
mmol) in toluene (5 mL) under N2 was heated at reflux for 5 h. The toluene was
evaporated and the residue was subjected to flash chromatography. Elution with 1:1
EtOAc/hexanes gave 121 as a pale yellow oil (53 mg, 21%), identical with the material
described below. Further elution with EtOAc gave 122 as a pale yellow oil (144 mg,
58%), identical with the material described below.

135
Ru(II)-catalysed
(S)-Methyl-2-(tert-butoxycarbonylamino)-3-(5-(diethoxyphosphoryl)-1H-1,2,3-
triazol-1-yl)propanoate (121)
Cp*RuCl(PPh3)2 (5 mg, 1.5 mol%) was added to a solution of the azide 60 (100 mg,
0.41 mmol) and alkyne 82 (67 mg, 4.1 mmol) in toluene (5 mL) under N2. The resulting
solution was stirred at 60 ºC for 24 h, then diluted with water (10 mL) and extracted
with EtOAc (3 × 30 mL). The extract was washed with brine (10 mL), dried and
concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:1 EtOAc/hexanes gave 121 as a pale yellow oil (93 mg,
56%). Rf = 0.15 (1:1 EtOAc/hexanes). IR (thin film) cm–1
: 3306 (NH), 1747 (m, C=O),
1715 (s, NC=O). 1H NMR (600 MHz, CDCl3): 7.95 (s, 1H, triazolyl), 5.69 (br d, J =
6.0 Hz, 1H, NH), 4.88–5.00 (m, 3H, -CH & -CH2), 4.17 (m, 4H, 2 × OCH2CH3),
3.76 (s, 3H, OCH3), 1.36 (s, 9H, t-Bu), 1.35 (m, 6H, 2 × OCH2CH3). 13
C NMR (125
MHz, CDCl3): δ 169.5 (CO2), 154.8 (NCO2), 140.0 (d, JC,P = 20.4 Hz, triazolyl CH),
126.7 (d, JC,P = 219.6 Hz, CP), 79.8 [OC(CH3)3], 63.4 (d, JC,P = 5.9 Hz, OCH2CH3a),
63.3 (d, JC,P = 5.8 Hz, OCH2CH3b), 53.2 (), 52.5 (OCH3), 50.4 (), 27.8 [C(CH3)3],
15.9 (d, JC,P = 4.4 Hz, OCH2CH3a), 15.8 (d, JC,P = 4.5 Hz, OCH2CH3b). 31
P NMR (240
MHz, CDCl3): δ 4.03. MS (ESI) m/z: 407 [M+H]+, 429 [M+Na]
+. HRMS (ESI):
observed, 407.1688, [C15H27N4O7P+H]+ requires, 407.1690. []D = –31.0º (c 1.7,
EtOAc). Further elution with EtOAc gave 122 as a pale yellow oil (35 mg, 21%),
identical with the material described below.

136
Cu(I)-catalysed
(S)-Methyl-2-(tert-butoxycarbonylamino)-3-(4-(diethoxyphosphoryl)-1H-1,2,3-
triazol-1-yl)propanoate (122)
CuSO4.5H2O (30 mg, 0.120 mmol) was added to a solution of the azide 60 (300 mg,
1.23 mmol), the alkyne 82 (200 mg, 1.23 mmol) and sodium ascorbate (24 mg, 0.12
mmol) in 1:1 t-BuOH/water (6 mL). The resulting solution was stirred for 24 h, then
diluted with water (10 mL) and extracted with DCM (3 × 30 mL). The organic extract
was washed with brine (10 mL), dried and concentrated under reduced pressure. The
residue was subjected to flash chromatography. Elution with EtOAc gave 122 as a pale
yellow oil (429 mg, 88%). Rf = 0.05 (1:1 EtOAc/hexanes). IR (thin film) cm–1
: 3293
(NH), 1744 (m, C=O), 1716 (s, NC=O).1H NMR (600 MHz, CDCl3): 8.03 (s, 1H,
triazolyl), 5.78 (br d, J = 7.7 Hz, 1H, NH), 4.76 (dd, J = 13.5, 4.5 Hz, 1H, -Ha), 4.70
(dd, J = 14.0, 6.5 Hz, 1H, -Hb), 4.55 (m, 1H, -H), 3.99 (m, 4H, 2 × OCH2CH3), 3.59
(s, 3H, OCH3), 1.21 (s, 9H, t-Bu), 1.144 (t, J = 7.2 Hz, 3H, OCH2CH3a), 1.140 (t, J =
7.2 Hz, 3H, OCH2CH3b). 13
C NMR (150 MHz, CDCl3): δ 169.1 (CO2), 154.8 (NCO2),
136.7 (d, JC,P = 239 Hz, CP), 131.6 (d, JC,P = 32 Hz, triazolyl CH), 80.0 [OC(CH3)3],
62.59 (d, JC,P = 5.7 Hz, OCH2CH3a), 62.58 (d, JC,P = 5.9 Hz, OCH2CH3b), 53.3 (α),
52.5 (OMe), 50.1 (), 27.7 [C(CH3)3], 15.8 (d, JC,P = 6.5 Hz, OCH2CH3). 31
P NMR (240
MHz, CDCl3): δ 6.97. MS (ESI) m/z: 407 [M+H]+. HRMS (ESI): observed, 407.1696,
[C15H27N4O7P+H]+ requires, 407.1690. [D = –15.9º (c 2.2, EtOAc).

137
BocNNH
OH
NN
N
P
O
EtOOEt
(S)-tert-Butyl-3-(5-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-yl)-1-(methylamino)-1-
oxopropan-2-ylcarbamate (123)
33% Aqueous methylamine (2.40 mL, 25 mmol) was added to a solution of 121 (240
mg, 0.60 mmol) in MeOH (5 mL). The solution was stirred at room temperature for 4 h,
and then concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:20 MeOH/DCM gave 123 as a pale yellow oil (207 mg,
85%). Rf = 0.20 (1:20 MeOH/DCM). IR (thin film) cm–1
: 3307 (NH), 1716 (ester C=O),
1672 (amide C=O). 1H NMR (500 MHz, CD3OD): 8.11 (s, 1H, triazolyl), 5.12 (br d,
1H, J = 11.0 Hz, -H), 4.74 (m, 2H, -H), 4.25 (m, 4H, 2 × OCH2CH3), 2.75 (s, 1H,
NHCH3), 1.38, (t, 6H, J = 7.5 Hz, 2 × OCH2CH3), 1.33 (s, 9H, t-Bu). 13
C NMR
(125MHz, CD3OD): δ 171.5, 157.2, 141.6 (d, JCP = 20.8 Hz, triazolyl CH), 127.9 (d, JCP
= 222 Hz, CP), 80.9, 65.5 (d, JC,P = 5.8 Hz, OCH2CH3a), 65.4 (d, JC,P = 5.9 Hz,
OCH2CH3b), 55.7, 52.3, 28.6, 26.5, 16.54 (d, JC,P = 2.9 Hz, OCH2CH3a), 16.49 (d, JC,P
= 3.0 Hz, OCH2CH3b). 31
P NMR (120 MHz, CD3OD): δ 4.74. MS (ESI) m/z: 406
[M+H]+, 428 [M+Na]
+. HRMS (ESI): observed, 428.1673, [C15H28N5O6P+Na]
+
requires, 428.1669. [D = –33.8o (c 1.0, MeOH).

138
BocNNH
OH
N
N N
P
O
OEt
OEt
(S)-tert-Butyl-3-(4-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-yl)-1-(methylamino)-1-
oxopropan-2-ylcarbamate (124)
33% aqueous methylamine (3.00 mL, 31 mmol) was added to a solution of triazole 122
(300 mg, 0.74 mmol) in MeOH (5 mL). The solution was stirred at room temperature
for 4 h, and then concentrated under reduced pressure. The residue was subjected to
flash chromatography. Elution with 1:10 MeOH/DCM gave 124 as a pale yellow oil
(270 mg, 90%). Rf = 0.20 (1:9 MeOH/DCM). IR (thin film) cm–1
: 3305 (NH), 1712
(ester C=O), 1666 (amide C=O). 1H NMR (500 MHz, CD3OD): 8.39 (s, 1H, triazolyl),
4.92 (dd, 1H, J = 12.5 Hz, 3.0 Hz, -H), 4.63 (m, 2H, -H), 4.14 (m, 4H, 2 ×
OCH2CH3), 2.70 (s, 1H, CONHCH3), 1.32 (s, 9H, t-Bu), 1.288 (t, 3H, J = 7.0 Hz,
OCH2CH3a ), 1.287 (t, 3H, J = 7.0 Hz, OCH2CH3b). 13
C NMR (125 MHz, CD3OD): δ
171.3, 157.1, 137.4 (d, JC,P = 242.2 Hz,CP), 133.5 (d, JCP = 33.2 Hz, triazolyl CH),
81.0, 64.45 (d, JC,P = 4.2 Hz, OCH2CH3a), 64.50 (d, JC,P = 4.2 Hz, OCH2CH3b), 55.7,
52.3, 28.6, 26.5, 16.6 (d, JC,P = 6.5 Hz, OCH2CH3). 31
P NMR (120 MHz, CD3OD): δ
8.73. MS (ESI) m/z: 406 [M+H]+, 428 [M+Na]
+. HRMS (ESI): observed, 406.1855,
[C15H28N5O6P+H]+ requires, 406.1850. [D = 5.4º (c 1.0, MeOH).

139
BrH3NNH
O
NN
N
P
O
HOOH
(S)-1-(Methylamino)-1-oxo-3-(5-phosphono-1H-1,2,3-triazol-1-yl)propan-2-
aminium bromide (125)
A solution of 123 (100 mg, 0.25 mmol) in 33% HBr in acetic acid (2.00 mL, 8.1 mmol)
was stirred at room temperature for 48 h. The reaction mixture was concentrated under
vacuum and the residue was purified by Dowex cation exchange chromatography.
Elution with water gave the hapten 125 as a white solid (54 mg, 71%), mp 141–143 C.
Rf = 1.0 on reverse-phase TLC (H2O). IR (KBr disk) cm–1
: 300–3500 (br, NH3 + OHs),
1685 (C=O). 1H NMR (600 MHz, D2O): 7.73 (s, 1H, triazolyl), 4.89 (dd, J = 6.6 Hz,
2.4, 2H, -H), 4.37 (t, J = 6.0 Hz, 1H, -H), 2.51 (s, 1H, NHCH3). 13
C NMR (125 MHz,
D2O): δ 166.4, 138.4 (br s, triazolyl CH), 134.6 (d, JC,P = 192.1 Hz, CP), 52.5, 49.0,
25.9. 31
P NMR (120 MHz, D2O): δ 3.48. MS (ESI) m/z: 250 [M+H]+
(where M = free
base). HRMS (ESI): observed, 250.0703, [C6H11N5NaO4P+H]+ requires, 250.0700. [D
= +38.4º (c 1.0, H2O).
BrH3NNH
O
N
N N
P
O
OH
OH
(S)-1-(Methylamino)-1-oxo-3-(4-phosphono-1H-1,2,3-triazol-1-yl)propan-2-
aminium bromide (126)
A solution of 124 (100 mg, 0.25 mmol) in 33% HBr in acetic acid (2.00 mL, 8.1 mmol)
was stirred at room temperature for 48 h. The reaction mixture was concentrated under
vacuum and the residue was purified by Dowex cation exchange chromatography.
Elution with water gave 126 as a white solid (56 mg, 73%), mp 145–146 C. Rf = 1.0 on

140
reverse-phase TLC (H2O). IR (KBr disk) cm–1
: 3100–3500 (br, NH3 + OHs), 1646
(C=O). 1H NMR (500 MHz, D2O): 8.14 (s, 1H, triazolyl), 4.97 (d, J = 5.5 Hz, 2H, -
H), 4.54 (t, J = 5.0 Hz, 1H, -H), 2.69 (s, 1H, NHCH3). 13
C NMR (125 MHz, D2O): δ
167.0, 144.5 (d, JC,P = 214.7 Hz, CP), 129.7 (d, JC,P = 28.9 Hz, triazolyl CH), 52.9, 49.8,
26.3. 31
P NMR (120 MHz, D2O): δ 1.55 MS (ESI) m/z: 272 [M+Na]+, 294 [M+2NaH]
+
(where M = free base). HRMS (ESI): observed, 272.0522, [C6H12N5NaO4P+Na]+
requires, 2720519. [D = +27.2º (c 1.0, H2O).
BocNOH
OH
N NN
PO
OEtEtO
(S)-2-(tert-Butoxycarbonylamino)-3-(5-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-
yl)propanoic acid (35)
LiOH.H2O (5 mg, 0.1 mmol) was added to a stirred solution of 121 (51 mg, 0.13 mmol)
in 3:1 MeOH/water (4 mL) at 0 ºC. After 2 h, the reaction mixture was acidified to pH 4
with sat. aq. KHSO4 and then extracted with DCM (3 × 40 mL). The organic extract
was washed with brine (10 mL), dried and concentrated under reduced pressure. The
residue was subjected to flash chromatography. Elution with 1:9 MeOH/DCM gave 35
as a colourless oil (41 mg, 86 %). Rf = 0.10 (1:9 MeOH/DCM). 1
H NMR (500 MHz,
CD3OD): 8.09 (s, 1H, triazolyl), 5.15 (m, -CH), 4.74 (m, 2H, -CH2), 4.25 (m, 4H, 2
× OCH2CH3), 1.36 (t, J = 7.0 Hz, 6H, 2 × OCH2CH3), 1.30 (s, 9H, t-Bu). 13
C NMR (125
MHz, CD3OD): 175.5 (CO2), 158.0 (NCO), 142.3 (d, JC,P = 20.6 Hz, triazolyl CH),
128.4 (d, JC,P = 222.5 Hz, CP), 81.1 [OC(CH3)3], 66.3 (d, JC,P = 5.5 Hz, OCH2CH3a),
66.2 (d, JC,P = 5.5 Hz, OCH2CH3b), 57.1 (), 54.5 (), 29.5 [C(CH3)3], 17.4 (d, JC,P =
6.3 Hz, OCH2CH3a), 17.3 (d, JC,P = 5.9 Hz, OCH2CH3b). MS (ESI) m/z: 393 [M+H]+,

141
415 [M+Na]+. A sample that had partially degraded was further purified by HPLC in
order to compare to the reported spectra in CDCl3. 1H NMR (500 MHz, CDCl3): 8.00
(br s, 1H, triazolyl), 5.90 (br s, 1H, NH) 5.07 (br m, 1H, -CH), 4.91 (br m, 2H, -
CH2), 4.22 (m, 4H, 2 × OCH2CH3), 1.35–1.40 (m, 6H, 2 × OCH2CH3), 1.36 (s, 9H, t-
Bu). 13
C NMR (125 MHz, CDCl3): δ 171.1 (br, CO2), 155.8 (NCO), 140.5 (br s,
triazolyl CH), 126.7 (br d, JC,P = 231.5 Hz, CP), 80.8 [OC(CH3)3], 64.3 (br s,
OCH2CH3), 53.8 (), 51.1 (), 28.2 [C(CH3)3], 16.1 (br s, OCH2CH3). [D = +1.2o (c
0.5, CDCl3). The NMR data are similar to those reported.40
BocNOH
OH
N
N N
P
O
OEt
OEt
(S)-2-(tert-Butoxycarbonylamino)-3-(4-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-
yl)propanoic acid (36)
LiOH.H2O (5 mg, 0.1 mmol) was added to a stirred solution of 122 (51 mg, 0.13 mmol)
in 3:1 MeOH/water (4 mL) at 0 ºC. After 2 h, the reaction mixture was acidified to pH 4
with sat. aq. KHSO4, and then extracted with DCM (3 × 40 mL). The extract was
washed with brine (10 mL), dried and concentrated under reduced pressure. The residue
was subjected to flash chromatography. Elution with 1:4 MeOH/DCM gave 36 as a
colourless oil (42 mg, 88 %). Rf = 0.10 (1:4 MeOH/DCM). 1H NMR (500 MHz,
CD3OD): 8.39 (s, 1H, triazolyl), 5.01 (br dd, J = 17.5, 10.2 Hz, 1H, -CH2a), 4.71 (m
[apparent dd], 1H, -CH2b), 4.51 (m, 1H, -CH), 4.17 (m, 4H, 2 × OCH2CH3), 1.37 (s,
9H, t-Bu), 1.32 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3). 13
C NMR (125 MHz, CD3OD): δ
175.2 (CO2), 158.2 (NCO), 138.0 (d, JC,P = 242.2 Hz, CP), 134.2 (d, JC,P = 31.8 Hz,
triazolyl CH), 81.4 [OC(CH3)3], 65.4 (d, JC,P = 3.4 Hz, OCH2CH3a), 65.3 (d, JC,P = 3.3
Hz, OCH2CH3b), 57.5 (), 54.0 (), 29.5 [C(CH3)3], 17.4 (d, JC,P = 6.5 Hz, OCH2CH3).

142
MS (ESI) m/z: 415 [M+Na]+. A sample that had partially degraded was further purified
by HPLC in order to compare to the reported spectra in CDCl3. 1H NMR (600 MHz,
CDCl3): 8.28 (br s, 1H, triazolyl), 6.4 (v br s, OH), 5.55 (br s, 1H, NH), 4.98 (m, 2H,
-CH2), 4.75 (br s, 1H, -H), 4.20 (br m, 4H, 2 × OCH2CH3), 1.43 (s, 9H, t-Bu), 1.34 (2
× overlapping t at 1.34 and 1.33 [apparent q], J = 6.0 Hz, 6H, 2 × OCH2CH3). 13
C NMR
(150 MHz, CDCl3): δ 170.5 (br CO2), 155.4 (NCO), 136.4 (br d, JC,P = 231.6 Hz, CP),
132.5 (br s, triazolyl CH), 80.7 [OC(CH3)3], 63.85 (br s, OCH2CH3a), 63.76 (br s,
OCH2CH3b), 53.7 (br, ), 51.2 (), 28.2 [C(CH3)3], 16.1 (br s, OCH2CH3). [D =
+40.5o (c 0.7, CHCl3). The NMR data are similar to those reported.
40
BocN
O
N
NN
N
HH
O
HN
PO
OEtEtO
tert-Butyl-(S)-3-(5-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-yl)-1-((S)-1-
(methylamino)-1-oxopropan-2-ylamino)-1-oxopropan-2-ylcarbamate (127)
HCTU (635 mg, 1.53 mmol) was added to a solution of 35 (500 mg, 1.27 mmol), N-
methyl-L-alaninamide 133 (196 mg, 1.91 mmol) and diisopropylethylamine (DIPEA)
(450 mL, 2.55 mmol) in DMF (10 mL) at 0 ºC. The resulting solution was stirred at
room temperature for 18 h, then diluted with sat. aq. NH4Cl (20 mL) and extracted with
EtOAc (3 × 40 mL). The organic extract was washed with brine (20 mL), dried and
concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:50 MeOH/DCM gave a 17:3 ratio of 127 and the R,S
diastereomer as a colourless oil (363 mg, 60%). Rf = 0.15 (1:50 MeOH/DCM). IR (thin
film) cm–1
: 3307 (NH), 1600-1750 (broad peak, C=Os). 1H NMR (600 MHz, CD3OD):
8.16 (s, 1H, triazolyl), 5.14 (m [apparent d], 1H, -Ha), 4.80-4.87 (m [under water
signal], 2H, -Hb & -H), 4.39 (q, 1H, J = 7.2 Hz, -Hala), 4.29 (m, 4H, 2 ×

143
OCH2CH3), [2.78, 2.77* (2 s, 3H, NCH3)], 1.36-1.44 (m, 18 H, t-Bu & 2 × OCH2CH3 &
CCH3). 13
C NMR (150 MHz, CD3OD): δ [175.9*, 175.0 (CCO)], 170.7 (CCO), [158.4*,
157.3 (NCO2)], 141.5 (d, JC,P = 21.1 Hz, triazolyl CH), 128.0 (d, JC,P = 221.5 Hz, CP),
[82.1*, 81.0 (OC(CH3)3)], 65.5 (d, JC,P = 5.6 Hz, OCH2CH3a), 65.4 (d, JC,P = 5.4 Hz,
OCH2CH3b), 55.7, 52.4, 50.5, 28.6 [C(CH3)3], 26.4 (NCH3), 18.2 (CCH3) 16.55 (d, JC,P
= 2.9 Hz, OCH2CH3a), 16.51 (d, JC,P = 2.7 Hz, OCH2CH3b). 31
P NMR (120 MHz,
CD3OD): δ 4.64. MS (ESI) m/z: 477 [M+H]+, 499 [M+Na]
+. HRMS (ESI): observed,
477.2227, [C18H33N6O7P+H]+ requires, 477.2221. [D = –19.1º (c 1.0, MeOH).
BocNNH
OH
N
N N
O
HN
PO
OEtOEt
tert-Butyl-(S)-3-(4-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-yl)-1-((S)-1-
(methylamino)-1-oxopropan-2-ylamino)-1-oxopropan-2-ylcarbamate (128)
HCTU (1.27 g, 3.06 mmol) was added to a solution of 36 (1.00 g, 2.55 mmol), N-
methyl-L-alaninamide 133 (391 mg, 3.82 mmol) and DIPEA (900 mL, 5.10 mmol) in
DMF (20 mL) at 0 ºC. The resulting solution was stirred at room temperature for 18 h,
then diluted with sat. aq. NH4Cl (30 mL) and extracted with EtOAc (3 × 50 mL). The
organic extract was washed with brine (30 mL), dried and concentrated under reduced
pressure. The residue was subjected to flash chromatography. Elution with 1:20
MeOH/DCM gave a 17:8 ratio of 128 and the R,S diastereomer as a colourless oil (741
mg, 61%). Rf = 0.15 (1:20 MeOH/DCM). IR (thin film) cm–1
: 3293 (NH), 1600-1750
(broad peak, C=Os). 1H NMR (500 MHz, CD3OD): [8.46, 8.45* (2 s, 1H, triazolyl)],
[5.00, 4.98* (2 dd, J = 13.5, 4.0 Hz, 1H, -Ha)], 4.72-4.79 (m, 1H, -Hb), 4.58-4.70 (m,
1H, -H), 4.36 (m, 1H, -Hala), 4.21 (m, 4H, 2 × OCH2CH3), [2.78, 2.76* (2 s, 3H,
* Peak corresponding to the minor diastereomer

144
NCH3)], 1.42 (s, 9H, t-Bu), 1.35-1.40 (m, 9H, 2 × OCH2CH3 & CCH3). 13
C NMR (125
MHz, CD3OD): δ [176.05*, 175.96 (2 s, CCO)], [171.7*, 171.4 (2 s, CCO)], [158.4*,
158.2 (2 s, NCO2)], 138.2 (d, JC,P = 242.2 Hz, CP), [134.53, 134.47* (2 d, JC,P = 33.2
Hz, triazolyl CH)], [82.2*, 82.1 (2 s, OC(CH3)3)], 65.44 (d, JC,P = 2.8 Hz, OCH2CH3a),
65.39 (d, JC,P = 2.8 Hz, OCH2CH3b), [56.7*, 56.4 (2 s)], [53.1, 52.7* (2 s)], 51.4,
[29.50*, 29.47 (2 s, C(CH3)3)], 27.3 (NCH3), [19.0, 18.8* (2 s, CCH3)], 17.4 (d, JC,P =
6.4 Hz, OCH2CH3). 31
P NMR (120 MHz, CD3OD): δ 8.73. MS (ESI) m/z: 477 [M+H]+,
499 [M+Na]+. HRMS (ESI): observed, 477.2227, [C18H33N6O7P+H]
+ requires,
477.2221. [D = –5.0° (c 1.0, MeOH).
BrH3N
O
N
NN
N
H
O
HN
PO
OHHO
(S)-1-((S)-1-(Methylamino)-1-oxopropan-2-ylamino)-1-oxo-3-(5-phosphono-1H-
1,2,3-triazol-1-yl)propan-2-aminium bromide (129)
A solution of 127 (100 mg, 0.210 mmol) in 33% HBr in acetic acid (1.8 mL, 7.34
mmol) was stirred at room temperature for 48 h. The reaction mixture was concentrated
under vacuum and the residue was purified by Dowex cation exchange chromatography.
Elution with water gave a 17:3 ratio of the hapten 129 and the R, S diastereomer as a
white solid (55 mg, 66%), mp 137–138 C. Rf = 1.0 on reverse-phase TLC (H2O). IR
(KBr disk) cm–1
: 3000–3500 (br, NH3 + OHs), 1685 (br, C=Os). 1H NMR (500 MHz,
D2O): [7.69*, 7.67 (s, 1H, triazolyl)], [4.86*, 4.75 (2 dd, J = 14.5, 4.5 Hz, 2H, -H)],
4.46 (m, 1H, -H), 4.16 (q, J = 7.0 Hz, 1H, -Hala), [2.62*, 2.60 (2 s, 3H, CH3)], 1.20
(d, 3H, J = 7.5 Hz, CCH3). 13
C NMR (125 MHz, D2O): δ [174.5, 172.8* (2 s, CO)],
162.7 (CO), [138.4, 138.1* (2 d, JC,P = 172.3 Hz, CP), [136.8*, 133.68 (2 d, JC,P = 15.3
* Peak corresponding to the minor diastereomer

145
Hz, triazolyl CH)], 56.1, [49.2*, 50.2 (2 s)], 48.5, [25.23, 25.19* (2 s, CCH3)], [15.8*,
15.6 (2 s, NCH3)]. 31
P NMR (120 MHz, D2O): δ –3.58. MS (ESI) m/z: 321 [M+H]+, 343
[M+Na]+ (where M = free base). HRMS (ESI): observed, 321.1070, [C9H17N6O5P + H]
+
requires, 321.1076. [D = –28.8º (c 1.0, H2O).
BrH3NNH
O
N
N N
O
HN
PO
OHOH
(S)-1-((S)-1-(Methylamino)-1-oxopropan-2-ylamino)-1-oxo-3-(4-phosphono-1H-
1,2,3-triazol-1-yl)propan-2-aminium bromide (130)
128 (100 mg, 0.210 mmol) in 33% HBr in acetic acid (1.8 mL, 7.34 mmol) was stirred
at room temperature for 48 h. The reaction mixture was concentrated under vacuum and
the residue was purified by Dowex cation exchange chromatography. Elution with water
gave a 17:8 ratio of the hapten 130 and the R,S diastereomer as a white solid (53 mg,
63%), mp 145–147 C. Rf = 1.0 on reverse-phase TLC (H2O). IR (KBr disk) cm–1
:
3000–3500 (br, NH3 + OHs), 1685 (C=Os). 1
H NMR (500 MHz, D2O): [8.06, 8.05* (2
s, 1H, triazolyl)], 4.83-4.96 (m, 2H, -H), [4.51, 4.45 (2 m [apparent t], 1H, -H)],
[4.14, 4.04* (2 q, J = 6.0 Hz, 1H, -Hala)], [2.60, 2.54* (2 s, 3H, CH3)], [1.21, 1.12*
(2 d, 3H, J = 6.0 Hz, CCH3)]. 13
C NMR (125 MHz, D2O): δ [174.5*, 174.3 (2 s, CO)],
[166.1*, 166.0 (2 s, CO)], 144.0 (d, JC,P = 220.9 Hz, CP), 130.0 (br s, triazolyl CH),
[52.4*, 52.3 (2 s)], [50.0, 49.8* (2 s)], [49.3, 49.2* (2 s)], [25.9, 25.8* (2 s, CCH3)],
16.4 (NCH3). 31
P NMR (120 MHz, D2O): δ 1.32. MS (ESI) m/z: 321 [M+H]+, 343
[M+Na]+ (where M = free base). HRMS (ESI): observed, 321.1075, [C9H17N6O5P + H]
+
requires, 321.1076. [D = –63.5º (c 1.0, H2O).
* Peak corresponding to the minor diastereomer

146
H2NNH
O
N-Methyl-L-alaninamide (129)
This known compound was synthesised according to the method of Shimizu122
and
Ottenheqm72
with slight modifications.
Thionyl chloride (12.5 mL, 172 mmol) was added dropwise to L-alanine (4.46 g, 50
mmol) in EtOH (50 mL) at 0 ºC. The resulting solution was stirred at room temperature
for 18 h, and then the solvent was removed under reduced pressure. The product was
used for the next step without further purification. The crude hydrochloride was
dissolved in 40% aq. MeNH2 (6.0 mL, 77 mmol) at 0 ºC. The resulting solution was
stirred at room temperatrure for 1 h, and the solvent was removed under reduced
pressure. The residue was subjected to flash chromatography. Elution with 1:4
MeOH/DCM gave 129 as a white solid (3.47 g, 68%), mp 210–211 ºC. Rf = 0.10 (1:4
MeOH/DCM). 1H NMR (500 MHz, CD3OD): 3.95 (m, 1H, -H), 2.77 (s, 3H,
NHCH3), 1.50 (d, J = 7.0 Hz, 3H, CHCH3). 13
C NMR (125 MHz, CD3OD): δ 172.6
(CO), 50.4 (), 26.4 (NHCH3), 18.2 (CHCH3). MS (ESI) m/z: 103 [M+H]+, 125
[M+H]+. [D = +8.0 (c 1.0, EtOH) [lit.
123 +7.7 (c 0.7, EtOH)].
H2NOMe
O
N
N N
P
O
OEt
OEt
(S)-Methyl-2-amino-3-(4-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-yl)propanoate
(134)
TFA (1.00 mL, 13.1 mmol) was added to a solution of 122 in DCM (10 mL) at 0 ºC.
The solution was allowed to warm to room temperature and stirred for 5 h, and then
volatile components were removed under reduced pressure. The residue was diluted
with sat. aq. NaHCO3 (30 mL) and extracted with EtOAc (3 × 50 mL). The organic

147
extract was washed with brine (30 mL), dried and concentrated under reduced pressure.
The residue was subjected to flash chromatography. Elution with 1:10 MeOH/DCM
gave 134 as a pale yellow oil (302 mg, 80%). Rf = 0.35 (1:9 MeOH/DCM). IR (thin
film) cm–1
: 3392 (NH), 1741 (C=O). 1H NMR (500 MHz, CDCl3): 8.19 (s, 1H,
triazolyl), 4.75 (dd, J = 13.5, 4.0 Hz, 1H, -Ha), 4.54 (dd, J = 13.5, 7.0 Hz, 1H, -Hb),
4.17 (m, 4H, 2 × OCH2CH3), 3.97 (s, 1H, br s), 3.74 (s, 3H, OCH3), 1.312, (t, J = 7.0
Hz, 3H, OCH2CH3a), 1.311, (t, J = 7.0 Hz, 3H, OCH2CH3b). 13
C NMR (150 MHz,
CDCl3): δ 172.3 (CO2), 136.9 (d, JC,P = 239.6 Hz, CP), 132.0 (d, JC,P = 33.3 Hz,
triazolyl CH), 63.0 (d, JC,P = 5.8 Hz, OCH2CH3), 54.2 (α), 53.3 (OMe), 52.6 (), 16.0
(d, JC,P = 6.5 Hz, OCH2CH3). 31
P NMR (120 MHz, CDCl3): δ 7.73. MS (ESI) m/z: 307
[M+H]+. HRMS (ESI): observed, 307.1160, [C10H19N4O5P+H]
+ requires, 307.1166.
[D = –4.9º (c 1.0, CHCl3).
HN
OH
O
O
N-Acetyl-L-alanine (138)
This known compound was synthesised according to the method of Schmidt with slight
modifications.75
Acetic anhydride (6.30 mL, 67.3 mmol) was added to a suspension of L-alanine (5.00 g,
56.1 mmol) in acetic acid (25 mL). The suspension was stirred at room temperature for
18 h, resulting in a clear solution. The acetic acid and excess acetic anhydride were
removed under vacuum, and the residue was subjected to a C18 reverse-phase
chromatography. Elution with water gave 138 as a colourless oil (5.30 g, 72%). 1H
NMR (500 MHz, CD3OD): 4.33 (m, 1H, -H), 1.93, 1.92 (rotamers, 3H, s, COCH3),
1.33, 1.31 (rotamers, d, J = 7.0 Hz, 3H, CCH3). 13
C NMR (125 MHz, CD3OD): δ 174.7,

148
173.0 52.7, 22.3, 17.4. MS (ESI) m/z: 132 [M+H]+, 154 [M+H]
+. []D = 62.7 (c 1.0,
H2O) [lit.124
62.0 (c 3.0, H2O)].
HN
O
OMe
OHO
(S)-Methyl-2-acetamido-3-hydroxypropanoate (142)
This known compound was synthesised according to the method of Maruyama77
with
slight modifications.
DCC (3.90 g, 18.9 mmol) was added to a suspension of 58 (2.85 g, 18.3 mmol), acetic
acid (1.10 mL, 19.3 mmol) and triethylamine (2.60 mL, 18.7 mmol) in DCM (60 mL) at
0 ºC. The reaction mixture was allowed to warm to room temperature and stirred for 18
h, and then dicyclohexylurea was filtered off. The filtrate was concentrated under
reduced pressure, and the residue was subjected to flash chromatography. Elution with
EtOAc gave 142 as a pale yellow oil (2.66 g, 90%). 1H NMR (500 MHz, CDCl3): 7.28
(br d, 1H, J = 8.0 Hz, NH), 4.34 (m, 1H, -H), 3.71 (dd, 1H, J = 11.0, 4.5 Hz, -Ha),
3.58 (dd, J = 11.5, 3.5 Hz, -Hb), 1.80 (s, 3H, NHCOCH3). 13
C NMR (125 MHz,
CDCl3): δ 171.0, 170.6, 61.7 (COOCH3), 54.2 (-C), 51.8 (-C), 22.0 (NHCOCH3) MS
(ESI) m/z: 162 [M+H]+. [D = +10.5º (c 1.0, MeOH). The
1H NMR data are similar to
those reported.77
HN
O
OMe
N3
O
(S)-Methyl-2-acetamido-3-azidopropanoate (143)
A solution of the alcohol 142 (2.00 g, 12.4 mmol) and triphenylphosphine (3.90 g, 14.9
mmol) in anhydrous THF (50 mL) at 78 ºC under N2 was treated with a 2.5 M HN3
solution in toluene (6.00 mL, 15.1 mmol) and DIAD (3.00 mL, 15.3 mmol). The
resulting solution was allowed to warm to room temperature, stirred for 5 h, and then

149
diluted with water (100 mL). The layers were separated and the aqueous phase was
extracted with Et2O (3 × 100 mL). The organic extract was washed with brine (50 mL),
dried and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:1 Et2O/hexanes gave 143 as a yallow oil (1.39 g, 60
%). 1H NMR (300 MHz, CDCl3): 6.51 (br d, 1H, J = 5.4 Hz, NH), 4.74 (m, 1H, -H),
3.79 (s, 1H, OCH3, 3H), 3.73 (dd, 2H, J = 3.6, 1.2 Hz, -H), 2.05 (s, 3H, NCH3). 13
C
NMR (75 MHz, CDCl3): δ 170.1, 169.5, 53.8, 52.9, 52.2, 22.9. MS (ESI) m/z: 187
[M+H]+, 209 [M+H]
+. [D = +79.3
o (c 1.0, CHCl3). [lit.
125 +74.2 (c 1.4, CHCl3)]. The
1H NMR data are similar to those reported.
125
HN
OMe
O
N
N N
P
O
OEt
OEt
HN
OMe
O
N NN
PO
OEtEtO
O O
144 145
Cycloadditions of the azide 143 and alkyne 82
Thermal
A solution of the azide 143 (1.00 g, 5.37 mmol) and the alkyne 82 (871 mg, 5.37 mmol)
in toluene (10 mL) under N2 was heated at reflux for 5 h. The toluene was evaporated,
and the residue was subjected to flash chromatography. Elution with (7:3
EtOAc/hexanes) gave 144 as a pale yellow oil (486 mg, 26%), identical with the
material described below. Further elution with EtOAc gave 145 as a pale yellow oil
(1.12 g, 60%), identical with the material described below.

150
Ru(II)-catalysed
(S)-Methyl-2-acetamido-3-(5-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-
yl)propanoate (144)
Cp*RuCl(PPh3)2 (13 mg, 1.5 mol%) was added to a solution of the azide 143 (200 mg,
1.07 mmol) and the alkyne 82 (173 mg, 1.07 mmol) in toluene (20 mL) under N2. The
resulting solution was stirred at 60 ºC for 24 h, then diluted with water (20 mL) and
extracted with EtOAc (3 × 100 mL). The extract was washed with brine (20 mL), dried
and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 7:3 EtOAc/hexanes gave 144 as a pale yellow oil (213
mg, 57%). Rf = 0.35 (EtOAc). IR (thin film) cm–1
: 3283 (NH), 1748 (ester C=O), 1671
(amide C=O). 1H NMR (500 MHz, CDCl3): 7.81 (s, 1H, triazolyl), 7.51 (br d, 1H, J =
8.0 Hz, NH), 4.99 (dd, 1H, J = 8.0, 4.0 Hz, -H), 4.88 (dd, 1H, J = 14.0, 4.5 Hz, -Ha),
4.75 (dd, 1H, J = 14.0, 8.0 Hz, -Ha), 4.05 (m, 4H, 2 × OCH2CH3), 3.58 (s, 3H,
OCOCH3) 1.79 (s, 3H, NHCOCH3), 1.20 (m, 6H, 2 × OCH2CH3). 13
C NMR (125 MHz,
CDCl3): δ 170.1, 169.1, 139.9 (d, JC,P = 20.4 Hz, triazolyl CH), 126.7 (JC,P = 220.4 Hz,
CP), 63.6 (d, JC,P = 5.9 Hz, OCH2CH3), 52.5, 51.8, 50.0, 22.2, 15.81 (JC,P = 3.6 Hz,
OCH2CH3a), 15.76 (JC,P = 3.8 Hz, OCH2CH3b). 31
P NMR (120 MHz, CDCl3): δ 4.36.
MS (ESI) m/z: 349 [M+H]+, 371 [M+Na]
+. HRMS (ESI): observed, 371.1097,
[C12H21N4O6P+Na]+ requires, 371.1091. [D = +15.7
o (c 1.0, CH3Cl). Further elution
with EtOAc gave 145 as a pale yellow oil (71 mg, 19%), identical with the material
described below.

151
Cu(I)-catalysed
(S)-Methyl-2-acetamido-3-(4-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-
yl)propanoate (145)
CuSO4.5H2O (34 mg, 0.136 mmol) was added to a solution of the azide 143 (250 mg,
1.34 mmol), the alkyne 82 (217 mg, 1.34 mmol) and sodium ascorbate (27 mg, 0.136
mmol) in 1:1 t-BuOH/water (15 mL). The resulting solution was stirred for 24 h, then
diluted with water (10 mL) and extracted with EtOAc (3 × 100 mL). The organic extract
was washed with brine (20 mL), dried and concentrated under reduced pressure. The
residue was subjected to flash chromatography. Elution with EtOAc gave 145 as a pale
yellow oil (425 mg, 91%). Rf = 0.10 (EtOAc). IR (thin film) cm–1
: 3272 (NH), 1747
(ester C=O), 1673 (amide C=O). 1H NMR (500 MHz, CDCl3): 8.11 (s, 1H, triazolyl),
7.57 (br d, 1H, J = 7.5 Hz, NH), 4.78 (m, 2H, -H), 4.69 (dd, 1H, J = 14.0, 7.0 Hz, -
H), 3.96 (m, 4H, 2 × OCH2CH3), 3.55 (s, 3H, OCOCH3) 1.78 (s, 3H, NHCOCH3), 1.121
(t, 3H, J = 7.0 Hz, OCH2CH3a), 1.118 (t, 3H, J = 7.0 Hz, OCH2CH3b). 13
C NMR (125
MHz, CDCl3): δ 170.5, 168.9, 136.3 (d, JC,P = 240.2 Hz, CP), 131.6 (d, JC,P = 33.1 Hz,
triazolyl CH), 62.7 (d, JC,P = 5.8 Hz, OCH2CH3), 52.4, 52.0, 49.8, 22.0, 15.70 (d, JC,P =
0.6 Hz, OCH2CH3a), 15.65 (JC,P = 0.75 Hz, OCH2CH3b). 31
P NMR (120 MHz, CDCl3):
δ 7.71. MS (ESI) m/z: 349 [M+H]+, 371 [M+Na]
+. HRMS (ESI): observed, 371.1086,
[C12H21N4O6P+Na]+ requires, 371.1091. [D = +62.0
o (c 1.0, CH3Cl).

152
HN
O
OH
NO
N N
P
O
OEtOEt
(S)-2-Acetamido-3-(4-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-yl)propanoic acid
(Method A; saponification using LiOH)
LiOH.H2O (60 mg, 1.44 mmol) was added to a solution of 145 (500 mg, 1.44 mmol) in
3:1 MeOH/water (15 mL) at 0 ºC. After 2 h, the reaction mixture was acidified to pH 4
~ 5 with sat. aq. KHSO4 and then extracted with EtOAc (3 × 50 mL). The organic
extract was washed with brine (10 mL), dried and concentrated under reduced
pressure. The residue was subjected to flash chromatography. Elution with 1:4
MeOH/DCM gave 146 as a colourless oil (144 mg, 30 %). [D = +5.1º (c 1.0,
MeOH). Identical in every respect except the optical activity with the material
described below.
(Method B; saponification using K2CO3)
K2CO3 (200 mg, 1.44 mmol) was added to a solution of 145 (500 mg, 1.44 mmol) in 3:1
MeOH/water (15 mL) at 0 ºC. The solution was allowed to warm to room temperature
and stirred for 2 h, and the reaction mixture was acidified to pH 4 with sat. aq. KHSO4
and extracted with EtOAc (3 × 50 mL). The organic extract was washed with brine (10
mL), dried and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:4 MeOH/DCM gave 146 as a colourless oil (226 mg,
47 %). Rf = 0.10 (1:4 MeOH/DCM). IR (thin film) cm–1
: 3293 (NH), 1720 (C=O), 1618
(NC=O).1H NMR (500 MHz, CD3OD): 8.47 (s, 1H, triazolyl), 5.06 (m, 1H, -Ha),
4.78-4.86 (m, 2H, -Hb + -H), 4.20 (m, 4H, 2 × OCH2CH3), 1.96 (s, 3H, NCH3),
1.361 (t, J = 7.0 Hz, 3H, OCH2CH3a), 1.360 (t, J = 7.2 Hz, 3H, OCH2CH3b). 13
C NMR
(125 MHz, CD3OD): δ 175.7 (CO), 173.9 (CO), 138.0 (d, JC,P = 242.3 Hz, CP), 134.3

153
(d, JC,P = 33.3 Hz, triazolyl CH), 65.47 (d, JC,P = 0.9 Hz, OCH2CH3a), 65.43 (d, JC,P =
0.7 Hz, OCH2CH3b), 57.0 (α), 53.8 (), 23.5 (NHCH3), 17.4 (d, JC,P = 6.5 Hz,
OCH2CH3). 31
P NMR (120 MHz, CD3OD): δ 8.90. MS (ESI) m/z: 335 [M+H]+, 357
[M+Na]. HRMS (ESI): observed, 357.1046, [C11H19N4O6P+Na]+ requires, 357.0934.
[D = +8.8º (c 1.0, MeOH).
HN
O
O
OHO
HN
O
O
OAcO
(S)-Allyl-2-acetamido-3-hydroxypropanoate (151)
(S)-Allyl-2-acetamido-3-acetoxypropanoate (152)
Acetic anhydride (6.30 mL, 67.0 mmol) was added to a suspension of L-serine (5.90 g,
56.1 mmol) in acetic acid (25 mL) at room temperature. The mixture was stirred at
room temperature for 18 h, and then concentrated under reduced pressure. The residue
was dissolved in DMF (25 mL), and allyl bromide (4.9 mL 56 mmol) and DIPEA (9.8
mL, 56 mmol) were added to the solution. The resulting solution was stirred at room
temperature for 18 h, then diluted with sat. aq. NH4Cl and extracted with EtOAc (3 × 50
mL). The organic extract was washed with brine, dried and concentrated under reduced
pressure. The residue was subjected to flash chromatography. Elution with 1:1
EtOAc/hexanes gave 152 as a white solid (5.79 g, 45%), mp 81–82 C. Rf = 0.40
(EtOAc). IR (thin film) cm–1
: 3294 (NH), 1747 (ester C=O), 1661 (amide C=O). 1H
NMR (500 MHz, CDCl3): 7.27 (d, 1H, J = 8.0 Hz), 5.56 (m, 1H, OCH2CHCH2), 4.98
(dd, 1H, J = 17.0, 1.5 Hz, OCH2CHCH2a), 4.89 (dd, 1H, J = 10.0, 1.0 Hz,
OCH2CHCH2b), 4.53 (1H, m, -H), 4.30 (m, 2H, OCH2CHCH2), 4.04 (dd, 2H, J = 6.5,
5.0 Hz, -H), 1.69 (s, 3H, OCOCH3), 1.68 (s, 3H, NHCOCH3). 13
C NMR (125 MHz,
CDCl3): δ 169.9, 169.6, 168.5, 130.8, 117.7, 65.2, 62.9, 50.9, 21.7, 20.1. MS (ESI) m/z:
230 [M+H]+, 252 [M+Na]
+. HRMS (ESI): observed, 230.1020, [C10H15NO5+H]
+

154
requires, 230.1023. [D = +58.8o (c 1.0, CH3Cl). Further elution with EtOAc gave 151
as a brown oil (2.21 g, 21%). Rf = 0.15 (EtOAc). IR (thin film) cm–1
: 3366 (NH), 1741
(ester C=O), 1655 (amide C=O). 1H NMR (400 MHz, CDCl3): 7.28 (d, 1H, J = 7.6
Hz, NH), 5.77 (m, 1H, OCH2CHCH2), 5.20 (ddd, 1H, J = 17.6, 3.2, 1.6 Hz,
OCH2CHCH2a), 5.11 (ddd, 1H, J = 10.4, 2.4, 1.2 Hz, OCH2CHCH2b), 4.51 (2H, dt, J =
6.0 Hz, 1.2 Hz, OCH2CHCH2), 4.47 (dd, 1H, J = 8.0, 4.0 Hz, -H), 3.83 (dd, 1H, J =
11.2, 4.0 Hz, -Ha), 3.71 (dd, 1H, J = 11.2, 3.2 Hz, -Hb), 1.90 (s, 1H, NHCOCH3). 13
C
NMR (100 MHz, CDCl3): δ 171.2, 170.1, 131.2, 118.3, 65.8, 62.0, 54.5, 22.3. MS (ESI)
m/z: 188 [M+H]+, 210 [M+Na]
+. HRMS (ESI): observed, 188.0914, [C8H13NO4+H]
+
requires, 188.0917. [D = +33.6o (c 1.0, CH3Cl).
HN
O
O
N3
O
(S)-Allyl-2-acetamido-3-azidopropanoate (153)
A solution of the alcohol 151 (2.00 g, 10.7 mmol) and triphenylphosphine (3.36 g, 12.8
mmol) in anhydrous THF (40 mL) at 78 ºC under N2 was treated with a 2.5 M HN3
solution in toluene (5.20 mL, 12.8 mmol) and DEAD (2.00 mL, 12.7 mmol). The
resulting solution was allowed to warm to room temperature, stirred for 5 h and then
diluted with water (100 mL). The layers were separated and the aqueous phase was
extracted with Et2O (3 × 100 mL). The organic extract was washed with brine (50 mL),
dried and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:1 Et2O/hexanes gave 153 as a brown oil (1.38 g, 61 %).
Rf = 0.40 (Et2O). IR (thin film) cm–1
: 3274 (NH), 2107 (N3) 1743 (ester C=O), 1660
(amide C=O). 1H NMR (500 MHz, CDCl3): 6.84 (d, 1H, J = 7.5 Hz, NH), 5.83 (m,
1H, OCH2CHCH2), 5.27 (dd, 1H, J = 17.0, 1.5 Hz, OCH2CHCH2a), 5.20 (dd, 1H, J =
10.5, 1.0 Hz, CH2CHCH2b), 4.72 (m, 1H, -H), 4.60 (dd, 2H, J = 4.5, 1.5 Hz,

155
OCH2CHCH2), 3.68 (d, 2H, -H), 1.99 (s, 3H, NHCOCH3). 13
C NMR (125 MHz,
CDCl3): δ 170.2, 169.2, 130.9, 119.0, 66.3, 52.1, 52.0, 22.6. MS (ESI) m/z: 235
[M+Na]+. HRMS (ESI): observed, 235.0808, [C8H13NO4+H]
+ requires, 235.0802. [D
= +16.3o (c 1.0, CH3Cl).
HN
O
O
NO
N N
P
O
OEtOEt
HN
O
O
NN
N
PO
EtO
O
OEt
154 155
Cycloadditions of the azide 153 and alkyne 82
Thermal
A solution of the azide 153 (1.00 g, 4.71 mmol) and the alkyne 82 (764 mg, 4.71 mmol)
in toluene (10 mL) under N2 was heated at reflux for 5 h. The toluene was evaporated,
and the residue was subjected to flash chromatography. Elution with 7:3
EtOAc/hexanes gave 154 as a pale yellow oil (722 mg, 41%), identical with the material
described below. Further elution with EtOAc gave 155 as a pale yellow oil (774 mg,
44%), identical with the material described below.
Ru(II)-catalysed
(S)-Allyl-2-acetamido-3-(5-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-yl)propanoate
(154)
Cp*RuCl(PPh3)2 (15 mg, 19 mmol) was added to a solution of the azide 153 (250 mg,
1.2 mmol) and the alkyne 82 (191 mg, 1.2 mmol) in toluene (10 mL) under N2. The
resulting solution was stirred at 60 ºC for 24 h, then diluted with water (10 mL) and
extracted with EtOAc (3 × 30 mL). The extract was washed with brine (10 mL), dried
and concentrated under reduced pressure. The residue was subjected to flash

156
chromatography. Elution with 1:1 EtOAc/hexanes gave 154 as a pale yellow oil (323
mg, 73%). Rf = 0.30 (EtOAc). IR (thin film) cm–1
: 3284 (NH), 1747 (ester C=O), 1682
(amide C=O). 1H NMR (500 MHz, CDCl3): 7.88 (s, 1H, triazolyl), 7.19 (br d, 1H, J =
8.0 Hz, NH), 5.78 (m, 1H, OCH2CHCH2), 5.23 (ddd, 1H, J = 17.0, 2.5, 1.0 Hz
OCH2CHCH2a), 5.15 (ddd, 1H, J = 10.5, 2.5, 1.0 Hz, OCH2CHCH2b), 5.11 (m, 1H, -
H) 4.95 (dd, 1H, J = 14.0, 4.5 Hz, -Ha), 4.86 (dd, 1H, J = 14.5, 8.0 Hz, -Hb), 4.56
(m, 2H, OCH2CHCH2), 4.13 (m, 4H, 2 × OCH2CH3), 1.88 (s, 1H, NHCOCH3), 1.29 (m,
6H, 2 × OCH2CH3). 13
C NMR (125 MHz, CDCl3): δ 170.1, 168.5, 140.1 (d, JC,P = 20.4
Hz, triazolyl CH), 131.0, 126.9 (d, JC,P = 220.4 Hz, CP), 118.8, 66.3, 63.73 (JC,P = 4.5
Hz, OCH2CH3a), 63.69 (JC,P = 4.7 Hz, OCH2CH3b), 52.1, 50.2, 22.5, 16.01 (d, JC,P =
3.6 Hz, OCH2CH3a), 15.96 (d, JC,P = 3.8 Hz, OCH2CH3b). 31
P NMR (120 MHz,
CDCl3): δ 4.48. MS (ESI) m/z: 375 [M+H]+, 397 [M+Na]
+. HRMS (ESI): observed,
375.1422, [C14H23N4O6P+H]+ requires, 375.1428. [D = +21.0
o (c 1.0, CH3Cl). Further
elution with EtOAc gave 155 as a pale yellow oil (44 mg, 10%). Identical with the
material described below.
Cu(I)-catalysed
(S)-Allyl-2-acetamido-3-(4-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-yl)propanoate
(155)
CuSO4.5H2O (30 mg, 0.12 mmol) was added to a solution of the azide 153 (250 mg, 1.2
mmol), the alkyne 82 (191 mg, 1.2 mmol) and sodium ascorbate (23 mg, 0.12 mmol) in
1:1 t-BuOH/water (6 mL). The resulting solution was stirred at room temperature for 24
h, then diluted with water (10 mL) and extracted with DCM (3 × 30 mL). The organic
extract was washed with brine (10 mL), dried and concentrated under reduced pressure.
The residue was subjected to flash chromatography. Elution with EtOAc gave 155 as a
pale yellow oil (371 mg, 84%). Rf = 0.15 (EtOAc). IR (thin film) cm–1
: 3271 (NH),

157
1745 (ester C=O), 1674 (amide C=O). 1H NMR (500 MHz, CDCl3): 8.10 (s, 1H
triazolyl), 7.21 (br d, 1H, J = 7.5 Hz, NH), 5.80 (m, 1H, OCH2CHCH2), 5.25 (ddd, 1H,
J = 17.5, 3.0, 1.5 Hz, OCH2CHCH2a), 5.19 (ddd, 1H, J = 10.5, 2.5, 1.5 Hz,
OCH2CHCH2b), 4.93 (m, 1H, -H), 4.85 (m, 2H, -H), 4.57 (dt, 2H, J = 5.5 Hz, 1.0
Hz, OCH2CHCH2), 4.09 (m, 4H, 2 × OCH2CH3), 1.92 (s, 1H, NHCOCH3), 1.251 (t, 3H,
J = 7.5 Hz, OCH2CH3a), 1.248 (t, 3H, J = 7.5 Hz, OCH2CH3b). 13
C NMR (125 MHz,
CDCl3): δ 170.6, 168.3, 136.9 (d, JC,P = 239.8 Hz, CH), 131.8 (d, JC,P = 33.1 Hz,
triazolyl CH), 130.9, 119.2, 66.6, 63.0 (d, JC,P = 0.9 Hz, OCH2CH3a), 62.9 (d, JC,P = 0.8
Hz, OCH2CH3b), 52.4, 50.2, 22.5, 16.02 (d, JC,P = 1.3 Hz, OCH2CH3a), 15.96 (d, JC,P =
1.1 Hz, OCH2CH3b). 31
P NMR (120 MHz, CDCl3): δ 7.52. MS (ESI) m/z: 375 [M+H]+.
HRMS (ESI): observed, 375.1433, [C14H23N4O6P+H]+ requires, 375.1428. [D =
+25.3o (c 1.0, CH3Cl).
HN
O
O
OHO
HN
O
O
OO
O
(S)-Benzyl-2-acetamido-3-hydroxypropanoate (160)
(S)-Benzyl-2-acetamido-3-acetoxypropanoate (161)
Acetic anhydride (6.30 mL, 67.0 mmol) was added to a suspension of L-serine (5.90 g,
56.1 mmol) in acetic acid (25 mL) at room temperature. The mixture was stirred at
room temperature for 18 h, and then concentrated under reduced pressure. The residue
was dissolved in DMF (25 mL), and benzyl bromide (6.70 mL 56.4 mmol) and DIPEA
(9.80 mL, 56.3 mmol) were added to the solution. The resulting solution was stirred at
room temperature for 18 h, then diluted with sat. aq. NH4Cl and extracted with EtOAc
(3 × 50 mL). The organic extract was washed with brine (30 mL), dried and
concentrated under reduced pressure. The residue was subjected to flash

158
chromatography. Elution with 1:1 EtOAc/hexanes gave 161 as a pale yellow oil (7.05 g,
45%). Rf = 0.50 (EtOAc). IR (thin film) cm–1
: 3293 (NH), 1746 (ester C=O), 1661
(amide C=O). 1H NMR (400 MHz, CDCl3): 7.28 (m, 5H, aromatic), 7.00 (br d, 1H, J
= 8.0 Hz), 5.13 (dd, 2H, J = 31.6 Hz, 12.4 Hz, OCH2Ph), 4.87 (m, 1H, -H), 4.42 (dd,
1H, J = 11.2 Hz, 4.0 Hz, -Ha), 4.29 (dd, 1H, J = 11.2 Hz, 3.6 Hz, -Hb), 1.97 (s, 3H,
NHCOCH3), 1.87 (s, 3H, OCOCH3). 13
C NMR (100 MHz, CDCl3): δ 170.1, 169.9,
169.1, 134.7, 128.2, 128.1, 127.9, 67.0, 63.5, 51.3, 22.4, 20.0. MS (ESI) m/z: 280
[M+H]+, 302 [M+Na]
+. HRMS (ESI): observed, 302.1005, [C14H17NO5+Na]
+ requires,
302.0999. [D = +24.5o (c 1.0, CH3Cl). Further elution with EtOAc gave 160 as a white
solid (4.15 g, 31%), mp 60–62 C. Rf = 0.20 (EtOAc). IR (thin film) cm–1
: 3313 (NH),
1742 (ester C=O), 1657 (amide C=O). 1H NMR (400 MHz, CDCl3): ; 7.30 (m, 5H,
aromatic), 7.14 (br d, 1H, J = 7.6 Hz, NH), 5.14 (d, 2H, J = 1.2 Hz, OCH2Ph), 4.63 (m,
1H, -H), 3.95 (dd, 1H, J = 11.6 Hz, 4.0 Hz, -Ha), 3.81 (dd, 1H, J = 11.2 Hz, 3.2 Hz,
-Hb), 1.97 (s, 3H, NHCOCH3). 13
C NMR (100 MHz, CDCl3): δ 171.1, 170.3, 135.0,
128.4, 128.2, 127.8, 67.1, 62.4, 54.7, 22.6. MS (ESI) m/z: 238 [M+H]
+, 260 [M+Na]
+.
HRMS (ESI): observed, 238.1077, [C12H16NO4+H]+ requires, 238.1074. [D = +14.3°
(c 1.0, CH3Cl).
HN
O
O
N3
O
(S)-Benzyl-2-acetamido-3-azidopropanoate (162)
A solution of the alcohol 160 (2.00 g, 12.4 mmol) and triphenylphosphine (3.90 g, 14.9
mmol) in anhydrous THF (50 mL) at 78 °C under N2 was treated with a 2.5 M HN3
solution in toluene (4.10 mL, 10.3 mmol) and DIAD (2.00 mL, 10.2 mmol). The
resulting solution was allowed to warm to room temperature, stirred for 5 h and then

159
diluted with water (100 mL). The layers were separated and the aqueous phase was
extracted with Et2O (3 × 100 mL). The organic extract was washed with brine (50 mL),
dried and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:1 Et2O/hexanes gave 162 as a yellow oil (1.35 g, 61
%). Rf = 0.45 (Et2O). IR (thin film) cm–1
: 3293 (NH), 2106 (azide), 1743 (ester C=O),
1655 (amide C=O). 1H NMR (500 MHz, CDCl3): 7.26 (m, 5H, aromatic), 7.15 (br d, J
= 7.5 Hz, NH), 5.11 (d, 2H, OCH2Ph), 4.76 (m, 1H, -H), 3.63 (dd, 2H, J = 4.0 Hz, 1.5
Hz), 1.95 (s, 3H, NHCOCH3). 13
C NMR (125 MHz, CDCl3): δ 170.1, 169.2, 134.5,
128.2, 128.1, 127.8, 67.2, 51.9, 51.8, 22.2. MS (ESI) m/z: 263 [M+H]+, 285 [M+Na]
+.
HRMS (ESI): observed, 263.1133, [C12H14N4O3+H]+ requires, 263.1139. [D = +22.8º
(c 1.0, CHCl3).
AcNOBn
OH
N
N N
P
O
OEt
OEt
AcNOBn
OH
N NN
PO
OEtEtO
163 164
Cycloadditions of the azide 162 and alkyne 82
Thermal
A solution of the azide 162 (1.00 g, 3.81 mmol) and the alkyne 82 (618 mg, 3.81 mmol)
in toluene (10 mL) under N2 was heated at reflux for 5 h. The toluene was evaporated,
and the residue was subjected to flash chromatography. Elution with 1:1
EtOAc/hexanes gave 163 as a pale yellow oil (697 mg, 43%), identical with the material
described below. Further elution with EtOAc gave 164 as a pale yellow oil (729 mg,
45%), identical with the material below.

160
Ru(II)-catalysed
(S)-Benzyl-2-acetamido-3-(5-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-
yl)propanoate (163)
Cp*RuCl(PPh3)2 (9 mg, 1.5 mol%) was added to a solution of the azide 162 (200 mg,
0.763 mmol) and the alkyne 82 (124 mg, 0.763 mmol) in toluene (20 mL) under N2.
The resulting solution was stirred at 60 ºC for 24 h, then diluted with water (10 mL) and
extracted with EtOAc (3 × 100 mL). The extract was washed with brine (20 mL), dried
and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 3:2 EtOAc/hexanes gave 163 as a pale yellow oil (262
mg, 81%). Rf = 0.40 (EtOAc). IR (thin film) cm–1
: 3290 (NH), 1747 (ester C=O), 1683
(amide C=O). 1H NMR (500 MHz, CDCl3): 7.83 (s, 1H, triazolyl), 7.44 (br d, 1H, J =
8.5 Hz, NH), 7.16 (m, 5H, aromatic), 5.09 (dd, 1H, J = 8.0, 4.5 Hz, -H), 5.02 (d, 2H, J
= 6.5 Hz, OCH2Ph), 4.90 (dd, 1H, J = 14.0, 4.5 Hz, -Ha), 4.81 (dd, 1H, J = 14.0, 8.0
Hz, -Hb), 4.00 (m, 4H, 2 × OCH2CH3), 1.81 (s, 3H, NHCOCH3), 1.18 (m, 6H, 2 ×
OCH2CH3). 13
C NMR (125 MHz, CDCl3): δ 170.1, 168.5, 140.0 (d, JC,P = 20.6 Hz,
triazolyl CH), 134.5, 128.1, 128.0, 127.8, 126.6 (d, JC,P = 220.4 Hz, CP), 67.2, 63.45 (d,
JC,P = 6.2 Hz, OCH2CH3a), 63.40 (d, JC,P = 6.0 Hz, OCH2CH3b), 52.8, 50.0, 22.1, 15.70
(d, JC,P = 4.2 Hz, OCH2CH3a), 15.65 (d, JC,P = 4.2 Hz, OCH2CH3b). 31
P NMR (120
MHz, CDCl3): δ 4.35. MS (ESI) m/z: 425 [M+H]
+, 447 [M+Na]
+. HRMS (ESI):
observed, 425.1590, [C18H25N4O6P+H]+ requires, 425.1584. [D = +14.0° (c 1.0,
CH3Cl).

161
Cu(I)-catalysed
(S)-Benzyl-2-acetamido-3-(4-(diethoxyphosphoryl)-1H-1,2,3-triazol-1-
yl)propanoate (164)
CuSO4.5H2O (20 mg, 0.0801 mmol) was added to a solution of the azide 162 (200 mg,
0.763 mmol), the alkyne 82 (124 mg, 0.763 mmol) and sodium ascorbate (16 mg,
0.0808 mmol) in 1:1 t-BuOH/water (10 mL). The resulting solution was stirred at room
temperature for 24 h, then diluted with water (10 mL) and extracted with EtOAc (3 ×
100 mL). The organic extract was washed with brine (10 mL), dried and concentrated
under reduced pressure. The residue was subjected to flash chromatography. Elution
with EtOAc gave 164 as a pale yellow oil (282 mg, 87%). Rf = 0.20 (EtOAc). IR (thin
film) cm–1
: 3271 (NH), 1744 (ester C=O), 1675 (amide C=O). 1H NMR (500 MHz,
CDCl3): 8.10 (s, 1H, triazolyl), 7.67 (br d, 1H, J = 8.0 Hz, NH), 7.15 (m, 5H,
aromatic), 5.00 (s, 2H, OCH2Ph), 4.88 (dd, 1H, J = 7.0, 4.5 Hz, -H), 4.81 (dd, 1H, J =
14.0, 4.5 Hz, -Ha), 4.71 (dd, 1H, J = 14.0, 7.5 Hz, -Hb), 3.98 (m, 4H, 2 × OCH2CH3)
1.79 (s, 3H, NHCOCH3), 1.13 (t, 6H, J = 7.0 Hz, 2 × OCH2CH3). 13
C NMR (125 MHz,
CDCl3): δ 170.3, 168.3, 136.3 (d, JC,P = 239.8 Hz, CP), 134.4, 131.5 (d, JC,P = 33.1 Hz,
triazolyl CH), 128.0, 127.9, 127.7 67.1, 62.5, (d, JC,P = 5.7 Hz, OCH2CH3) 52.1, 49.7,
22.0, 15.6 (d, JC,P = 6.4 Hz, OCH2CH3). 31
P NMR (120 MHz, CDCl3): δ 7.73. MS (ESI)
m/z: 425 [M+H]+, 447 [M+Na]
+. HRMS (ESI): observed, 447.1410,
[C18H25N4O6P+Na]+ requires, 447.1404. [D = +13.9° (c 1.0, CH3Cl).

162
HN
O
OH
NN
N
PO
OHHO
O
(S)-2-Acetamido-3-(5-phosphono-1H-1,2,3-triazol-1-yl)propanoic acid (156)
A solution of 163 (200 mg, 0.47 mmol) in 33% HBr in acetic acid (4.0 mL, 16 mmol)
was stirred at room temperature for 48 h. The reaction mixture was concentrated under
vacuum and the residue was purified by Dowex anion exchange chromatography.
Elution with water gave 156 as a white solid (90 mg, 69%), mp 146–148 C. IR (KBr
disk) cm–1
: 3000–3500 (br, NH3 + OHs), 1717 (C=O), 1650 (NC=O). 1H NMR (500
MHz, D2O): 7.83 (s, 1H, triazolyl), 4.71 (dd, 1H, J = 14.0, 3.5 Hz, -H), 4.54 (dd, 1H,
J = 9.0, 3.5 Hz, -Ha), 4.46 (dd, 1H, J = 14.0, 9.5 Hz, -Hb), 1.38 (s, 3H, CH3). 13
C
NMR (125 MHz, D2O): δ 173.8, 170.5, 136.1 (d, JC,P = 197.9 Hz, CP), 134.5 (br s,
triazolyl), 51.6, 51.1, 21.5. 31
P NMR (120 MHz, D2O): δ –2.65. MS (ESI) m/z: 279
[M+H]+, 301 [M+Na]
+. HRMS (ESI): observed, 301.0305 [C7H11N4O6P+Na]
+ requires,
301.0308. [D = –23.8° (c 1.0, H2O).
HN
O
OH
N
N N
P
O
OHOH
O
(S)-2-Acetamido-3-(4-phosphono-1H-1,2,3-triazol-1-yl)propanoic acid (137)
(Method A: From the benzylester 164)
A solution of 164 (200 mg, 0.47 mmol) in 33% HBr in acetic acid (4.0 mL, 16 mmol)
was stirred at room temperature for 48 h. The reaction mixture was concentrated under
vacuum and the residue was subjected to Dowex anion exchange chromatography.
Elution with water gave 137 as a white solid (88 mg, 67%), mp 135–137 C. IR (KBr
disk) cm–1
: 3100–3500 (br, NH3 + OHs), 1732 (C=O), 1637 (NC=O). 1H NMR (500

163
MHz, D2O): 8.10 (s, 1H, triazolyl), 4.71 (dd, 2H, J = 12.5, 5.5 Hz, -H), 4.60 (dd, 1H,
J = 13.5, 7.0 Hz, -H), 1.67 (s, 3H, CH3). 13
C NMR (125 MHz, D2O): δ 173.9, 171.1,
139.9 (d, JC,P = 223.7 Hz, CP), 131.5 (d, JC,P = 30.2 Hz, triazolyl), 52.2, 50.5, 21.5
(CH3). 31
P NMR (120 MHz, D2O): δ 2.68. MS (ESI) m/z: 279 [M+H]+, 301 [M+Na]
+.
HRMS (ESI): observed, 279.0491 [C7H11N4O6P+H]+ requires, 279.0489. [D = +10.0°
(c 1.0, H2O).
(Method B: From the carboxylic acid 146)
A solution of 146 (200 mg, 0.60 mmol) in 33% HBr in acetic acid (5.0 mL, 20 mmol)
was stirred at room temperature for 48 h. The reaction mixture was concentrated under
vacuum and the residue was subjected to Dowex anion exchange chromatography.
Elution with water gave 137 (117 mg, 70%) as a white solid. [D = +6.1º (c 1.0, H2O).
Identical in every respect except the optical activity with the material described above.
~Chapter 3~
OHN3
O
Azidoacetic acid (166)
This compound was synthesised according to the method of Taylor126
with slight
modifications.
Sodium azide (1.87 g, 28.8 mmol) was added to a solution of bromoacetic acid (1.00 g,
7.20 mmol) in water (10 mL) at 0 ºC. The solution was allowed to warm to room
temperature, stirred for 18 h, then diluted with 1 M HCl (20 mL) and extracted with
EtOAc (3 × 50 mL). The organic extract was washed with brine (50 mL), dried and
concentrated under reduced pressure, affording 166 as a pale yellow oil (371mg, 51%).
1H NMR (500 MHz, CDCl3): 11.55 (br s, 1H, COOH), 3.95 (2H, s, 2H).
13C NMR

164
(125 MHz, CDCl3): δ 173.9 (CO), 49.8 (CH2). MS (ESI) m/z: 102 [M+H]+, 124
[M+Na]+. The
1H NMR data are similar to those reported.
126
HN
O
N
NN
N
P O
OHHO
H
O
N3
(S)-1-(2-(2-Azidoacetamido)-3-(methylamino)-3-oxopropyl)-1H-1,2,3-triazol-4-
ylphosphonic acid (167)
EDCI (37 mg, 0.20 mmol) was added to a solution of 126 (54 mg, 0.16 mmol),
azidoacetic acid 166 (16 mg, 0.16 mmol) and NaHCO3 (19 mg, 0.23 mmol) in water (5
mL). The solution was stirred at room temperature for 18 h and then subjected to
Dowex anion exchange chromatography. Elution with water gave 167 as a white solid
(32 mg, 60%), mp 125–126 C. Rf = 1.0 on reverse-phase TLC (H2O). IR (KBr disk)
cm–1
: 3000–3500 (br, NH3 + OHs), 2119 (N3) 1644 (C=O). 1H NMR (500 MHz, D2O):
8.04 (s, 1H, triazolyl), 4.83-4.90 (m, 2H, -H), 4.72-4.77 (m, 1H, -H), 3.96 (s, 2H,
CH2N3), 2.65 (s, 3H, NCH3). 13
C NMR (125 MHz, D2O): δ 170.6, 169.9, 144.3 (d, JC,P
= 213.6 Hz, CP), 129.3 (d, JC,P = 28.8 Hz, triazolyl CH), 53.2, 51.5, 50.0, 25.9. 31
P
NMR (120 MHz, D2O): δ 1.67. MS (ESI) m/z: 377 [M+2NaH]+. HRMS (ESI):
observed, 377.0464 [C8H12N8O5P+2NaH]+ requires, 377.0458. [D = 1.8
o (c 1.0,
H2O).

165
~Chapter 4~
BocN
O
OH
OH
H
(S)-2-(tert-Butoxycarbonylamino)-3-hydroxypropanoic acid (172)
This known compound was synthesised according to the Method of Sinou with slight
modifications.91
Boc anhydride (7.20 g, 33 mmol) was added to a solution of L-serine (3.00 g, 29 mmol)
in 1:1 dioxane/1 M aq. NaOH (50 mL) at 0 ºC. The resulting solution was stirred at 0 ºC
for 3 h, then acidified to pH 4 ~ 5 with sat. aq. KHSO4 and extracted with EtOAc (3 ×
100 mL). The organic extract was washed with brine (50 mL), dried and concentrated
under reduced pressure, giving 172 as a colourless oil (4.97 g, 85%). 1H NMR (500
MHz, CDCl3): 5.88 (br d, J = 7.5 Hz, 1H, NH), 4.34 (m, 1H, -H), 3.99 (m, 1H, -
Ha), 3.83(m, 1H, -Hb). 13
C NMR (125 MHz, CDCl3): δ 173.9 (CO), 156.1 (NCO),
80.4 [OCCH3)3], 62.8 (), 55.3 (), 27.5 [CCH3)3]. MS (ESI) m/z: 206 [M+H]+, 228
[M+Na]+. [D = 9.1
o (c 1.0, MeOH) [lit.
127 +8.7 (c 2.8, CHCl3)]. The
1H NMR data
are similar to those reported.91
BocN
O
O
OH
H
(S)-Benzyl 2-(tert-butoxycarbonylamino)-3-hydroxypropanoate (173)
Benzyl bromide (1.80 mL, 15.2 mmol) was added to a solution of 172 (3.00 g, 14.6
mmol) and DIPEA (1.30 mL, 15.2 mmol) in DMF (20 mL) under N2 at 0 ºC. The
resulting solution was allowed to warm up to room temperature, stirred for 18 h, then
diluted with sat. aq. NH4Cl (30 mL) and extracted with EtOAc (3 × 50 mL). The
organic extract was washed with brine (20 mL), dried and concentrated under reduced

166
pressure. The residue was subjected to flash chromatography. Elution with 1:4
EtOAc/hexanes gave 173 as a colourless oil (3.66 g, 85%). 1H NMR (500 MHz,
CDCl3): 7.27-7.27 (m, 5H, aromatic), 5.71 (d, J = 8.0 Hz, 1H, NH), 5.15 (d, J = 2.5
Hz, 2H, OCH2Ph), 4.39 (m, 1H, -H), 3.95 (m [apparent br d], 1H, -Ha), 3.84 (dd, J =
11.0, 3.5 Hz, 1H, -Hb), 1.42 (s, 9H, t-Bu). 13
C NMR (125 MHz, CDCl3): δ 170.8
(CO2), 155.7 (NCO2), 135.1 (ArC), 128.4 (ArH), 128.2 (ArH), 127.9 (ArH), 80.0
[C(CH3)3], 67.1 (OCH2Ph), 62.9 (), 55.7 (), 28.1 [C(CH3)3]. MS (ESI) m/z: 296
[M+H]+, 318 [M+Na]
+. [D = 19.6 (c 1.0, MeOH) [lit.
128 19.0 (c 1.0, CHCl3)]. The
NMR data are similar to those reported.129
BocN
O
O
N3
H
(S)-Benzyl-3-azido-2-(tert-butoxycarbonylamino)propanoate (174)
A solution of the alcohol 173 (1.50 g, 5.08 mmol) and triphenylphosphine (1.60 g, 6.10
mmol) in anhydrous THF (20 mL) at 78 ºC under N2 was treated with a 2.5 M HN3
solution in toluene (2.50 mL, 6.27 mmol) and DEAD (1.00 mL, 6.35 mmol). The
resulting solution was allowed to warm to room temperature, stirred for 5 h and then
diluted with water (100 mL). The layers were separated and the aqueous phase was
extracted with Et2O (3 × 80 mL). The organic extract was washed with brine (50 mL),
dried and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:4 Et2O/hexanes gave 174 as a colourless oil (1.18 g, 72
%). Rf = 0.20 (1:4 Et2O/hexanes). 1H NMR (400 MHz, CDCl3): 7.34 (m, 5H,
aromatic), 5.53 (br d, J = 7.6 Hz, 1H, NH), 5.19 (d, J = 2.8 Hz, 2H, OCH2Ph), 4.50 (m,
1H, -H), 3.71 (d, J = 3.6 Hz, 2H, -H), 1.44 (s, 9H, t-Bu). 13
C NMR (100 MHz,
CDCl3): δ 169.3 (CO2), 154.7 (NCO2), 134.6 (ArC), 128.24 (ArH), 128.17 (ArH), 127.9
(ArH), 80.0 [OC(CH3)3], 67.2 (OCH2Ph), 53.3 (α), 52.2 (), 27.8 [C(CH3)3]. MS (ESI)

167
m/z: 267 [M+Na]+. []D = +8.2 (c 1.0, CHCl3) [lit.
40 +8.0 (c 1.0, CHCl3)]. The
1H and
13C NMR data are similar to those reported.
40
BocNOBn
OH
N
N N
P
O
OEt
OEt
BocNOBn
OH
N NN
PO
OEtEtO
175 176
Cycloadditions of the azide 174 and alkyne 82
Thermal
A solution of the azide 174 (200 mg, 0.624 mmol) and the alkyne 82 (101 mg, 0.624
mmol) in toluene (10 mL) under N2 was heated at reflux for 5 h. The toluene was
evaporated and the residue was subjected to flash chromatography. Elution with 1:1
EtOAc/hexanes gave 175 as a pale yellow oil (123 mg, 41%), identical with the material
described below. Further elution with EtOAc gave 176 as a pale yellow oil (129 mg,
42%), identical with the material described below.
Ru(II)-catalysed
(S)-Benzyl-2-(tert-butoxycarbonylamino)-3-(5-(diethoxyphosphoryl)-1H-1,2,3-
triazol-1-yl)propanoate (175)
Cp*RuCl(PPh3)2 (8 mg, 1.6 mol%) was added to a solution of the azide 174 (200 mg,
0.624 mmol) and the alkyne 82 (101 mg, 0.624 mmol) in toluene (20 mL) under N2.
The resulting solution was stirred at 60 ºC for 24 h, then diluted with water (20 mL) and
extracted with EtOAc (3 × 100 mL). The extract was washed with brine (20 mL), dried
and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 3:2 EtOAc/hexanes gave 175 as a pale yellow oil (241
mg, 80%). Rf = 0.20 (1:1 EtOAc/hexanes). IR (thin film) cm–1
: 3305 (NH), 1747 (m,

168
C=O), 1716 (s, NC=O). 1H NMR (500 MHz, CDCl3): 7.94 (s, 1H, triazolyl), 7.29 (m,
5H, aromatic), 5.75 (br s, 1H, NH), 5.15 (s, 2H, OCH2Ph), 4.88-5.00 (m, 3H, -H + -
H), 4.14 (m, 4H, 2 × OCH2CH3), 1.34 (s, 9H, t-Bu), 1.27-1.36, (m, 6H, 2 × OCH2CH3).
13C NMR (125 MHz, CDCl3): δ 169.1 (CO2), 155.0 (NCO2), 140.3 (d, JC,P = 20.4 Hz,
triazolyl CH), 134.8 (ArC), 128.5 (ArH), 128.4 (ArH), 128.2 (ArH), 127.0 (d, JC,P =
219.6 Hz, CP), 80.1 [OCCH3)3], 67.6 (OCH2Ph), 63.64 (d, JC,P = 5.7 Hz, OCH2CH3a),
62.58 (d, JC,P = 5.8 Hz, OCH2CH3b), 53.5 (α), 50.6 (), 28.1 [C(CH3)3], 16.09 (d, JC,P =
4.8 Hz, OCH2CH3a), 16.09 (d, JC,P = 4.9 Hz, OCH2CH3b). 31
P NMR (120 MHz,
CDCl3): δ 4.60. MS (ESI) m/z: 483 [M+H]+, 505 [M+Na]
+. HRMS (ESI): observed,
505.1820, [C21H31N4O7P+Na]+ requires, 505.1823. [D = +9.2º (c 1.0, CHCl3).
Cu(I)-catalysed
(S)-Benzyl-2-(tert-butoxycarbonylamino)-3-(4-(diethoxyphosphoryl)-1H-1,2,3-
triazol-1-yl)propanoate (176)
CuSO4.5H2O (16 mg, 0.0641 mmol) was added to a solution of the azide 174 (200 mg,
0.624 mmol) and alkyne 82 (101 mg, 0.624 mmol) and sodium ascorbate (13 mg,
0.0656 mmol) in 1:1 t-BuOH/water (10 mL). The resulting solution was stirred at room
temperature for 24 h, then diluted with water (20 mL) and extracted with EtOAc (3 ×
100 mL). The organic extract was washed with brine (20 mL), dried and concentrated
under reduced pressure. The residue was subjected to flash chromatography. Elution
with EtOAc gave 176 as a pale yellow oil (265 mg, 88%). Rf = 0.10 (1:1
EtOAc/hexanes). IR (thin film) cm–1
: 3293 (NH), 1744 (m, C=O), 1716 (s, NC=O).1H
NMR (500 MHz, CDCl3): 8.03 (s, 1H, triazolyl), 7.27 (5H, m, aromatic) 5.71 (br d, J
= 7.5 Hz, 1H, NH), 5.12 (d, J = 2.0 Hz, 2H, OCH2Ph), 4.84 (m, 2H, -H), 4.68 (m, 1H,
-H), 4.10 (m, 4H, 2 × OCH2CH3), 1.33 (s, 9H, t-Bu), 1.261, (t, J = 7.0 Hz, 3H,
OCH2CH3a), 1.259, (t, J = 7.0 Hz, 3H, OCH2CH3b). 13
C NMR (125 MHz, CDCl3): δ

169
168.6 (CO2), 154.9 (NCO2), 137.0 (d, JC,P = 239.4 Hz, CP), 134.4 (ArC) 131.2 (d, JC,P =
33.1 Hz, triazolyl CH), 128.4 (ArH), 128.4 (ArH), 128.2 (ArH), 80.4 [OC(CH3)3], 67.8
(OCH2Ph), 62.8 (d, JC,P = 5.8 Hz, OCH2CH3), 53.5 (α), 50.5 (), 27.9 [CCH3)3], 15.9 (d,
JC,P = 6.5 Hz, OCH2CH3). 31
P NMR (120 MHz, CDCl3): δ 7.52. MS (ESI) m/z: 483
[M+H]+, 505 [M+Na]
+. HRMS (ESI): observed, 505.1825, [C21H31N4O7P+Na]
+
requires, 505.1823. [D = +11.1º (c 1.0, CHCl3).
BrH3NOH
O
N NN
PO
OHHO
(S)-1-Carboxy-2-(5-phosphono-1H-1,2,3-triazol-1-yl)ethanaminium bromide (170)
(Method A: From the benzyl ester 175)
A solution of 175 (100 mg, 0.207 mmol) in 33% HBr in acetic acid (2.0 mL, 8.1 mmol)
was stirred at room temperature for 48 h. The reaction mixture was concentrated under
vacuum and the residue was subjected to Dowex cation exchange chromatography.
Elution with water gave 170 as a white solid (43 mg, 65%), mp 138–140 C. Rf = 1.0 on
reverse-phase TLC (H2O). IR (KBr disk) cm–1
: 3200–3500 (br, NH3 + OHs), 1600–
1750 (br, obscures C=O). 1
H NMR (600 MHz, D2O): 7.86 (s, 1H, triazolyl), 5.11 (dd,
J = 15.0, 3.6 Hz, 1H,-CH) 4.98 (dd, J = 15.6, 7.2 Hz, 1H, -CH), 4.27 (dd, J = 7.8, 3.6
Hz, 1H, -CH). 13
C NMR (125 MHz, D2O): δ 169.6 (CO), 139.9 (d, JC,P = 18.4 Hz,
triazolyl CH), 135.6 (d, JC,P = 191.8 Hz, CP), 53.2 (α), 49.0 (). 31
P NMR (120 MHz,
D2O): δ –3.14. MS (ESI) m/z: 237 [M+H]+ 259 [M+Na]
+, 281 [M+2NaH]
+ (where M =
free base). HRMS (ESI): observed, 259.0201, [C5H10N4O5P+Na]+ requires, 259.0203.
[]D = 7.7° (c 0.5 H2O).

170
Method B (From the carboxylic acid 35)
A solution of 35 (90 mg, 0.23 mmol) in 33% HBr in acetic acid (2.0 mL, 8.1 mmol) was
stirred at room temperature for 48 h. The reaction mixture was concentrated under
vacuum, and the residue was subjected to Dowex cation exchange chromatography.
Elution with water gave 170 as a white solid (51 mg, 70%). [D = –5.6º (c 0.8, H2O).
Identical in every respect except the optical activity with the material described above.
BrH3N
O
OH
N
N N
P
O
OH
OH
(S)-1-Carboxy-2-(4-phosphono-1H-1,2,3-triazol-1-yl)ethanaminium bromide (171)
(Method A: From the benzyl ester 176)
A solution of 176 (100 mg, 0.207 mmol) in 33% HBr in acetic acid (2.0 mL, 8.1 mmol)
was stirred at room temperature for 48 h. The reaction mixture was concentrated under
vacuum and the residue was purified by Dowex cation exchange chromatography.
Elution with water gave 171 as a white solid (44 mg, 67%), mp 133–135 C. Rf = 1.0 on
reverse-phase TLC (H2O). IR (KBr disk) cm
–1: 3300–3500 (br, NH3 + OHs), 1716
(C=O). 1H NMR (600 MHz, D2O): 8.12 (s, 1H, triazolyl), 4.98–5.00 (m, 2H, -CH2),
4.47 (dd, J = 5.4, 4.8 Hz, 1H, α-CH). 13
C NMR (125 MHz, D2O): δ 170.7 (CO), 143.7
(d, JC,P = 217.6 Hz, CP), 130.2 (d, JC,P = 31.7 Hz, triazolyl CH), 54.5 (α), 50.0 (). 31
P
NMR (120 MHz, D2O): δ 1.95; MS (ESI) m/z: 237 [M + H]+
(where M = free base).
HRMS (ESI): observed, 237.0385, [C5H10N4O5P]+ requires, 237.0383. []D = 25.0° (c
0.5 H2O).

171
(Method B: From the carboxylic acid 36)
A solution of 36 (90 mg, 0.23 mmol) in 33% HBr in acetic acid (2.0 mL, 8.1 mmol) was
stirred at room temperature for 48 h. The reaction mixture was concentrated under
vacuum and the residue was purified by Dowex cation exchange chromatography.
Elution with water gave 171 as a white solid (52 mg, 72%). [α]D = –18.8° (c 0.5, H2O).
Identical in every respect except the optical activity with the material descrived above.
Inhibition assay
Solutions of varying concentrations of the „free‟ phosphohistidine analogue 170 or 171
(10 mM, 1 mM, 100 M and 10 M) or sodium bromide (10 mM) were added to a
reaction mixture containing histone (500 g/mL), radiolabeled ATP* (0.2 mM) and
transglutaminase from guinea pig liver (200 g/mL) in 0.1 M tris buffer (pH 9.0) at
room temperature. Alongside these samples, separate control reactions were prepared
that contained either no inhibitor (positive control), no kinase or no histone (negative
controls). Each sample was incubated at 37 ºC for 1 h, and the reaction was quenched
by the addition of 2 × SDS sample buffer (12 L). The phophorylated histone was
subsequently separated by polyacrylamide (15%) gel electrophoresis, and the
phosphorylation of the histone was quantified with a phosphoimager.
*[32
P] ATP 3000Ci/mmol

172
~Chapter 5~
FmocN
O
O
OH
H
(S)-Benzyl-2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-3-hydroxypropanoate
(178)
Benzyl bromide (1.40 mL, 11.8 mmol) was added to a solution of Fmoc-L-serine
monohydrate (2.00 g, 5.79 mmol) and DIPEA (1.00 mL, 11.6 mmol) in DMF (20 mL)
at 0 °C. The solution was allowed to warm to room temperature, stirred for 18 h, then
diluted with sat. aq. NH4Cl (30 mL) and extracted with EtOAc (3 × 50 mL). The
organic extract was washed with brine (50 mL), dried and concentrated under reduced
pressure. The residue was subjected to flash chromatography. Elution with 2:3
EtOAc/hexanes gave 178 as a white solid (2.18 g, 90%). 1H NMR (500 MHz, CDCl3):
7.81 (d, J = 7.5 Hz, 2H), 7.70 (br d, J = 5.5 Hz, 2H), 7.46 (t, J = 7.5 Hz, 4H), 6.47 (br d,
J = 5.0 Hz, 1H, NH), 5.27 (s, 2H, OCH2Ph), 4.66 (m, 1H, -H), 4.53 (m[apparent t],
1H, H-10'a), 4.43 (m [apparent t], 1H, H-10'b), 4.66 (t, J = 7.0 Hz, 1H, H-9'), 4.12
(m[apparent br d], 1H, -Ha), 3.99 (m[apparent br d], 1H, -Hb). 13
C NMR (125 MHz,
CDCl3): δ 170.4, 156.3, 143.5, 143.3, 140.9, 134.8, 128.2, 128.0, 127.7, 127.3, 126.7,
119.6, 67.0, 66.9, 62.4, 56.0, 46.6. MS (ESI) m/z: 440 [M+Na]+. []D = +0.7 (c 1.0,
DCM) [lit.94
+0.6 (c 1.0, DCM)]. The 1H and
13C NMR data are similar to those
reported.94
FmocN
O
OBn
N3
H
(S)-Benzyl-2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-3-azidopropanoate (179)
A solution of the alcohol 178 (2.00 g, 4.8 mmol) and triphenylphosphine (1.51 g, 5.76
mmol) in anhydrous THF (40 mL) at –78 ºC under N2 was treated with a 2.5 M HN3

173
solution in toluene (2.30 mL, 5.75 mmol) and DEAD (910 mL, 5.78 mmol). The
resulting solution was allowed to warm to room temperature, stirred for 5 h and then
diluted with water (100 mL). The layers were separated and the aqueous phase was
extracted with Et2O (3 × 100 mL). The organic extract was washed with brine (50 mL),
dried and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:4 Et2O/hexanes gave 179 as a white solid (1.38 g, 65
%). mp 70–72 ºC. Rf = 0.15 (1:1 Et2O/hexanes). IR (thin film) cm–1
: 3337 (NH), 2107
(N3) 1650-1750 (br, C=O + NC=O). 1H NMR (500 MHz, CDCl3): 7.80 (d, J = 7.5 Hz,
2H), 7.64 (br d, J = 7.0 Hz, 2H), 7.33-7.46 (m, 9H), 5.85 (br d, J = 7.5 Hz, 1H, NH),
5.26 (d, J = 4.5 Hz, 2H, OCH2Ph), 4.64 (m, 1H, -H), 4.45 (m, 2H, H-10'), 4.26 (t, J =
7.5Hz, 1H, H-9'), 3.79 (br s, 2H, -H). 13
C NMR (125 MHz, CDCl3): δ 169.2 (CO2),
155.6 (NCO2), 143.6, 143.5, 141.2, 134.8 (ArC), 128.6 (ArH), 128.2 (ArH), 127.6
(ArH), 127.0, 125.0, 119.9, 67.8, 67.2, 53.9 (α), 52.4 (), 46.9 (C-9'). MS (ESI) m/z:
443 [M+H]+, 465 [M+Na]
+. HRMS (ESI): observed, 465.1539, [C25H22N4O4+Na]
+
requires 465.1533. [D = +25.7º (c 1.0, CHCl3).
FmocN
O
OBn
N
N N
P
O
OEtOEt
HFmocN
O
OBn
NN
N
PO
EtO
H
180 181
OEt
Cycloadditions of the azide 179 and alkyne 82
Thermal
A solution of the azide 179 (100 mg, 0.226 mmol) and the alkyne 82 (37 mg, 0.228
mmol) in toluene (10 mL) was heated at reflux for 5 h. The toluene was evaporated
under vacuum, and the residue was subjected to flash chromatography. Elution with 2:3

174
EtOAc/hexanes gave 180 as a pale yellow oil (53 mg, 39%). Further elution with
EtOAc gave 181 as a pale yellow oil (56 mg, 41%).
Ru(II)-catalysed
(S)-Benzyl-2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-3-(5-
(diethoxyphosphoryl)-1H-1,2,3-triazol-1-yl)propanoate (180)
Cp*RuCl(PPh3)2 (3 mg, 1.7 mol%) was added to a solution of the azide 179 (100 mg,
0.226 mmol) and the alkyne 82 (37 mg, 0.228 mmol) in toluene (20 mL) under N2. The
resulting solution was stirred at 60 °C for 24 h, then diluted with water (10 mL) and
extracted with EtOAc (3 × 100 mL). The organic extract was washed with brine (10
mL), dried and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 2:3 EtOAc/hexanes gave 180 as a pale yellow oil (107
mg, 78%). Rf = 0.20 (1:1 EtOAc/hexanes). IR (thin film) cm–1
: 3272 (NH), 1700-1750
(br, C=O + NC=O). The NMR spectra showed the presence of the rotamers. 1H NMR
(500 MHz, CDCl3): [7.99, 7.98 (2 s, 1H, triazolyl)], 7.74 (d, J = 7.5 Hz, 2H), 7.54 (d,
J = 7.5 Hz, 2H), 7.23-7.43 (m, 9H), [6.42, 6.26 (2 br d, J = 7.5 Hz, 1H, NH)], 5.25 (d, J
= 5.0 Hz, 2H, OCH2Ph), 4.98–5.21 (m, 3H, -H + -H), 4.07-4.70 (m, 7H, H-10' + H-9'
+ 2 × OCH2CH3), 1.33 (t, J = 7.0 Hz, 3H, OCH2CH3a), 1.31 (t, J = 9.5 Hz, 3H,
OCH2CH3b). 13
C NMR (125 MHz, CDCl3): δ [168.7, 168.4 (2 s, CO2)], [156.9, 155.8,
(2 s, NCO2)], [143.7, 143.5, 141.44, 141.39 (4 s)], [141.15, 141.14, 140.59, 140.56 (4
s)], [140.34, 140.27 (2 d, JC,P = 20.3 Hz, triazolyl CH)], [134.7, 134.6 (2 s, ArC)],
[130.3, 128.3 (2 s)], 128.6 (ArH), 128.53 (ArH), 128.47 (ArH), [127.7, 126.63, 126.62
(3 s)], [127.3, 127.2 (2 d, JC,P = 221.4 Hz, CP)], [127.01, 126.99 (2 s)], [126.5, 126.4 (2
s)], [120.0, 119.9 (2 s)], [67.9, 67.4 (2 s)], [65.0, 64.0 (2 s)], [63.99, 63.92 (2 d, JC,P =
5.9 Hz, OCH2CH3a)], [63.8, 63.7 (2 d, JC,P = 5.5 Hz, OCH2CH3b)], [54.3, 54.0 (2 s, α)],
[50.5, 50.4 (2 s, )], 46.9 (C-9'), 16.11 (d, JC,P = 6.0 Hz, OCH2CH3a), 16.07 (d, JC,P =

175
5.2 Hz, OCH2CH3b). 31
P NMR (120 MHz, CDCl3): δ 4.54. MS (ESI) m/z: 605 [M+H]+,
627 [M+Na]+. HRMS (ESI): observed, 605.2168, [C31H33N4O7P+H]
+ requires,
605.2160. [D = +2.3° (c 1.0, CHCl3).
Cu(I)-catalysed
(S)-Benzyl-2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-3-(4-
(diethoxyphosphoryl)-1H-1,2,3-triazol-1-yl)propanoate (181)
CuSO4.5H2O (6 mg, 0.0240 mmol) was added to a solution of the azide 179 (100 mg,
0.226 mmol), the alkyne 82 (37 mg, 0.228 mmol) and sodium ascorbate (5 mg, 0.0252
mmol) in 1:1 t-BuOH/water (15 mL). The resulting solution was stirred for 24 h, then
diluted with water (20 mL) and extracted with EtOAc (3 × 100 mL). The organic extract
was washed with brine (20 mL), dried and concentrated under reduced pressure. The
residue was subjected to flash chromatography. Elution with EtOAc gave 181 as a white
solid (116 mg, 85%), mp 100–101 C. Rf = 0.10 (1:1 EtOAc/hexanes). IR (thin film)
cm–1
: 3271 (NH), 1700-1750 (br, C=O + NC=O). 1H NMR (500 MHz, CDCl3): 8.10
(s, 1H, triazolyl), 7.73 (d, J = 7.5 Hz, 2H), 7.54 (d, J = 7.0 Hz, 2H), 7.22-7.39 (m, 9H),
6.31 (br d, J = 7.5 Hz, 1H, NH), 5.17 (br s, 2H, OCH2Ph), 4.90 (m[apparent br d], 2H,
-H), 4.83 (m, 1H, -H), 4.33 (m[apparent t], 2H, H-10'), 4.09-4.22 (m, 5H, H-9' + 2 ×
OCH2CH3), 1.26-1.32 (m, 6H, 2 × OCH2CH3). The 13
C NMR spectra showed the
presence of the rotamers. 13
C NMR (125 MHz, CDCl3): δ 168.3 (CO2), 155.7 (NCO2),
143.3, 141.0, 137.2 (d, JC,P = 239.1 Hz, CP), 134.4 (ArC), 131.8 (d, JC,P = 33.5 Hz,
triazolyl CH), 128.5 (ArH), 128.46 (ArH), 128.3 (ArH), 127.6, [126.92, 126.88 (2 s)],
[124.93, 124.85 (2 s)], 119.8, 68.0, 67.2, 62.9 (d, JC,P = 5.7 Hz, OCH2CH3), 54.0 (α),
50.4 (), 46.8 (C-9') 16.03 (br s, OCH2CH3a), 15.98 (d, JC,P = 1.0 Hz, OCH2CH3b). 31
P
NMR (120 MHz, CDCl3): δ 7.56. MS (ESI) m/z: 605 [M+H]+, 627 [M+Na]
+. HRMS

176
(ESI): observed, 627.1975, [C31H33N4O7P+Na]+ requires, 627.1979. [D = +10.4º (c
1.0, CHCl3).
~Chapter 6~
N
NHI
4-Iodopyrazole (201)
This known compound was synthesised according to the method of Casida with slight
modification.97
Ceric ammonium nitrate (CAN) (5.65 g, 10.3 mmol) was added to a solution of
pyrazole (1.17 g, 17.2 mmol) and iodine (2.61 g, 10.3 mmol) in MeCN (50 mL) at room
temperature. The solution was stirred at room temperature for 5 h, then diluted with
10% aq. Na2SO3 (50 mL) and extracted with EtOAc (3 × 50 mL). The organic extract
was washed with brine (50 mL), dried and concentrated under reduced pressure. The
residue was subjected to flash chromatography. Elution with 2:3 EtOAc/hexanes gave
201 as a white solid (3.11 g, 93%). 1H NMR (500 MHz, CDCl3): 11.95 (br s, 1H,
NH), 7.61 (br s, pyrazolyl, 2H). 13
C NMR (125 MHz, CDCl3): δ 138.7 (pyrazolyl CH),
56.4 (CI). MS (ESI) m/z: 194 [M+H]+. The
1H and
13C NMR data are similar to those
reported.97
N
NTsI
4-Iodo-1-tosyl-1H-pyrazole (203)
This known compound was synthesised according to the method of Obala with slight
modification.98
Tosyl chloride (3.33 g, 17.5 mmol) was added to a solution of 201 (3.05 g, 15.7 mmol)
and pyridine (4.80 mL, 59.5 mmol) in DCM (10 mL) at room temperature. The solution
was stirred at room temperature for 3 h, then diluted with 1M HCl (20 mL) and

177
extracted with DCM (3 x 40 mL). The organic extract was washed with brine (20 mL),
dried and concentrated under reduced pressure. The residue was recrystallised from
EtOH (50 mL), and 203 was obtained as a white solid (3.44 g, 63%). 1H NMR (500
MHz, CD3OD): ; 8.15 (s, 2H, pyrazolyl), 7.70 (d, J = 6.5 Hz, 2H, aromatic), 7.23 (d, J
= 8.0 Hz, 2H, aromatic), 2.36 (s, 3H, CH3). 13
C NMR (125 MHz, CD3OD): δ 143.3,
141.9, 140.3, 129.9, 126.9, 58.9, 21.3. MS (ESI) m/z: 348 [M+H]+. The
1H NMR data
are similar to those reported.98
N
NTsP
O
EtO
EtO
Diethyl-1-tosyl-1H-pyrazol-4-ylphosphonate (204)
Palladium acetate (6 mg, 10 mol%) was added to a solution of 203 (100 mg, 0.29
mmol), diethyl phosphite (80 L, 0.63 mmol), triethylamine (80 L, 0.58 mmol) and
triphenyl phosphine (23 mg, 30 mol%) in EtOH (10 mL) under N2. The resulting
solution was heated at reflux for 6 h, then diluted with sat. aq. NH4Cl (10 mL) and
extracted with EtOAc (3 × 30 mL). The organic extract was washed with brine (20 mL),
dried and concentrated under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:1 EtOAc/hexanes gave 204 as a pale yellow oil (52 mg,
50%). Rf = 0.15 (1:1 EtOAc/hexanes). IR (thin film) cm–1
: 1386 (S=O). 1H NMR (600
MHz, CDCl3): 8.40 (br s, 1H, pyrazolyl), 7.87 (d, J = 5.5 Hz, aromatic), 7.80 (d, J =
1.0 Hz 1H, pyrazolyl), 7.31 (d, J = 5.5 Hz 2H, aromatic), 4.06 (m, 4H, 2 × OCH2CH3),
2.37 (s, 3H, CH3), 1.26 (dt, J = 5.5, 2.0 Hz, 6H, 2 × OCH2CH3). 13
C NMR (150 MHz,
CDCl3): δ 146.6 (ArC), 145.8 (d, JC,P = 13.4 Hz, pyrazolyl), 135.9 (d, JC,P = 23.4 Hz,
pyrazolyl), 132.8 (ArC), 130.1 (ArH), 128.4 (ArH), 111.40 (d, JC,P = 217.0 Hz, CP),
62.4 (d, JC,P = 5.6 Hz, OCH2CH3), 21.6 (CH3), 16.1 (d, JC,P = 6.5 Hz, OCH2CH3). 31
P

178
NMR (120 MHz, CDCl3): δ 11.74. MS (ESI) m/z: 359 [M+H]+. HRMS (ESI): observed,
359.0828, [C14H19N2O5PS+H]+ requires, 359.0825.
N
NHP
O
EtO
EtO
Diethyl-1H-pyrazol-4-ylphosphonate (202)
(Method A: Removal of the tosyl group)
KOH (160 mg, 2.9 mmol) was added to a solution of 204 (200 mg, 0.56 mmol) in 1:1
dioxane/H2O (10 mL) at room temperature. The solution was heated at reflux for 6 h,
and then the solvents were evaporated. The residue was diluted with sat. aq. NH4Cl (30
mL) and extracted with EtOAc (3 × 50 mL). The residue was subjected to flash
chromatography. Elution with EtOAc gave 202 as a white solid (61 mg, 53%), mp 85–
87 C. Rf = 0.10 (EtOAc). IR (thin film) cm–1
: 3152 (NH); 1H NMR (300 MHz, CDCl3):
7.81 (s, 2H, pyrazolyl), 3.97 (m, 4H, CH2), 1.20 (t, J = 6.9 Hz, 6H, CH3). 13
C NMR
(75 MHz, CDCl3): δ 138.2 (d, JC,P = 17.1 Hz, CP), 107.6 (pyrazolyl CH), 104.6
(pyrazolyl CH), 62.5 (d, JC,P = 5.5 Hz, CH2), 16.4 (d, JC,P = 6.5 Hz, CH3). 31
P NMR
(120 MHz, CDCl3): δ 16.49. MS (ESI) m/z: 205 [M+H]+, 227 [M+Na]
+. HRMS (ESI):
observed, 205.0735, [C7H13N2O3P+H]+ requires, 205.0737.
(Method B: Direct phosphonation of 4-iodopyrazole)
Palladium acetate (6 mg, 10 mol%) was added to a solution of 201 (56 mg, 0.29 mmol),
diethyl phosphite (80 L, 0.63 mmol), triethylamine (80 L, 0.58 mmol) and triphenyl
phosphine (23 mg, 30 mol%) in EtOH (10 mL) under N2. The resulting solution was
heated at reflux for 6 h, then diluted with sat. aq. NH4Cl (10 mL) and extracted with
EtOAc (3 × 30 mL). The organic extract was washed with brine (20 mL), dried and
concentrated under reduced pressure. The residue was subjected to flash

179
chromatography. Elution with EtOAc gave 202 as a white solid (49 mg, 47%). Identical
in every respect with the material described above.
BocNOMe
O
OTs
H
(S)-Methyl-2-(tert-butoxycarbonylamino)-3-(tosyloxy)propanoate (208)
This known compound was synthesised according to the method of Silks105
with slight
modifications.
Tosyl chloride (3.93 g, 20.6 mmol) was added to a solution of 59 (3.00 g, 13.7 mmol)
and pyridine (4.50 mL, 55.6 mmol) in DCM (20 mL) at 0 C. The solution was allowed
to warm to room temperature, stirred for 40 h and then diluted with sat. aq. NH4Cl (50
mL). The layers were separated, and the aqueous phase was extracted with DCM (3 ×
50 mL). The organic extract was washed with brine (50 mL), dried and concentrated
under reduced pressure. The residue was subjected to flash chromatography. Elution
with 1:9 EtOAc/hexanes gave 208 as a white solid (4.45 g, 87%). 1H NMR (500 MHz,
CDCl3): 7.72 (d, 2H, J = 8.5 Hz, ), 7.32 (d, 2H, J = 8.0 Hz, ), 5.3 (br d, 1H, J = 8.0
Hz, NH), 4.45-4.50 (m, 1H, -H), 4.35 (dd, 1H, J = 10.5, 3.0 Hz, -Ha), 4.25 (dd, 1H, J
= 10.0, 3.0 Hz, -Hb), 3.65 (s, 3H, OCH3), 2.41 (s, 3H, CCH3), 1.38 (s, 9H, t-Bu). 13
C
NMR (125 MHz, CDCl3): δ 168.9 (CO2), 154.9 (NCO2), 145.1, 132.2, 129.8, 127.9,
80.3 [OCCH3)3], 69.4 (-H), 52.8 (OCH3), 52.7 (-H), 28.1 [C(CH3)3], 21.5 (CCH3).
MS (ESI) m/z: 396 [M+Na]+. []D = +3.3 (c 1.0, MeOH) [lit.
130 +3.0 (c 2.0, MeOH)].
The 1H and
13C NMR data are similar to those reported.
105

180
O
OBocNH
(S)-tert-Butyl 2-oxooxetan-3-ylcarbamate (218)
This known compound was synthesised according to the method of Levesque with
slight modifications.131
DEAD (0.950 mL, 6.02 mmol) was added to a solution of 172 (1.00 g, 4.87 mmol) and
triphenylphosphine (1.55 g, 5.92 mmol) in THF (20 mL) at –78 ºC. The solution was
allowed to warm to room temperature and stirred for 5 h, and then volatile components
were removed under reduced pressure. The residue was subjected to flash
chromatography. Elution with 1:4 EtOAc/hexanes gave the title lactone in a slightly
impure state. Further purification was achieved by recrystallisation from CHCl3,
affording 218 as a white solid (410 mg, 45 %). 1H NMR (500 MHz, CDCl3): ; 5.63 (br
d, 1H, J = 6.5 Hz, NH), 5.06 (m [apparent q], 1H, -H), 4.45-4.43 (m, 2H, -H), 1.42
(s, 9H, t-Bu). 13
C NMR (125 MHz, CDCl3): δ; 169.7 (CO2), 154.6 (NCO2), 81.2
[OC(CH3)3], 66.5 (-H), 59.3 (-H), 28.1 [CCH3)3]. MS (ESI) m/z: 188 [M+H]+, 210
[M+Na]+. []D = 27.1 (c 1.0, MeCN) [lit.
132 27.0 (c 1.0, MeCN)]. The
1H NMR
data are similar to those reported.131
N N N
OO
N
Azodicarboxylic acid dipiperidide (223)
This known compound was synthesised according to the method of Kanety-Londner.113
Piperidine (1.96 g, 23.0 mmol) was added to a solution of DEAD (1.00 g, 5.74 mmol) in
Et2O (10 mL), and the resulting mixture was stirred at room temperature for 18 h. The
precipitate was filtered, washed with Et2O and then recrystallised from CHCl3,
providing 223 as an orange solid (1.01 g, 70%). 1H NMR (500 MHz, CD3OD): 3.65

181
(m, 4H), 3.46 (m, 4H), 1.71 (m, 8H), 1.58 (m, 4H). 13
C NMR (125 MHz, CD3OD): δ
162.1 (CO), 46.7, 45.6, 27.0, 26.5, 25.0. MS (ESI) m/z: 275 [M+Na]+.
NHN
I
4(5)-Iodoimidazole (226)
This known compound was synthesised according to the method of Still with slight
modifications.114
A solution of iodine (6.71 g, 26.5 mmol) in 20 % aqueous potassium iodide (50 mL)
was added to a solution of imidazole (1.00 g, 14.7 mmol) in 2.0 M aqueous NaOH (100
mL). The resulting mixture was stirred at room temperature for 18 h and then
neutralised by acetic acid. The produced precipitate was filtered and then washed with
water. The product was dried and used for the next step without further purification. A
mixture of the crude product and sodium sulfate (8.40 g, 66.7 mmol) was heated at
reflux in 30 % aqueous EtOH (100 mL) for 18 h, and then the EtOH was removed under
reduced pressure. The remaining mixture was filtered, and the filtrate was extracted
with Et2O (3 × 100 mL). The organic extract was washed with brine (50 mL), dried and
concentrated under reduced pressure. The residue was recystallised from EtOH,
providing 226 as a white solid (1.71 g, 60%). 1H NMR (500 MHz, CD3OD): 7.62 (d, J
= 1.0 Hz, 1H, imidazolyl), 7.19 (d, J = 1.5 Hz, 1H, imidazolyl). 13
C NMR (125 MHz,
CD3OD): δ 138.9 (pyrazolyl), 124.8 (CI). MS (ESI) m/z: 194 [M+H]+. The
1H NMR
data are similar to those reported.114

182
NTsN
I
4-Iodo-1-p-toluenesulfonylimidazole (229)
This known compound was synthesised according to the method of Still with slight
modifications.114
Tosyl chloride (2.00 g, 10.5 mmol) was added to a solution of 226 (2.00 g, 10.3 mmol)
and triethylamine (1.50 mL, 10.8 mmol) in THF (20 mL). The resulting solution was
stirred at room temperature for 18 h, then diluted with water and extracted with EtOAc
(3 × 50 mL). The organic extract was washed with brine (50 mL), dried and
concentrated under reduced pressure. The residue was recrystallised from EtOH,
providing 229 as a white solid (2.51 g, 70%). 1H NMR (500 MHz, CD3OD): 8.17 (d, J
= 1.5 Hz, 1H, imidazolyl), 7.95 (d, J = 8.5 Hz, 2H, aromatic), 7.73 (d, J = 1.5 Hz, 1H,
imidazolyl), 7.47 (d, J = 8.5 Hz, 2H, aromatic), 2.44 (s, 3H, CH3). 13
C NMR (125 MHz,
CD3OD): δ 148.6, 140.0, 135.6, 131.8, 130.1, 128.9, 124.5, 21.7. MS (ESI) m/z: 348
[M+H]+. The
1H NMR data are similar to those reported.
114

183

184
References
1. Burnett, G.; Kennedy, E. P. J Biol Chem 1954, 211, 969-980.
2. Mandell, J. W. Am. J. Pathol 2000, 163, 1687-1698.
3. Besant, P. G.; Tan, E.; Attwood, P. Int J Biochem Cell Biol 2003, 35, 297-309.
4. Weigel, N. L.; Moore, N. L. Nucl. Recept. Signaling 2007, 5, 1-13.
5. Attwood, P. V.; Besant, P. G.; Zu, X. L.; Piggott, M. J. Amino Acids 2007, 32, 145-
156.
6. Hultquist, D. E.; Moyer, R. W.; Boyer, P. D. Biochemistry. 1966, 5, 322-331.
7. Pirrung, M. C. Chem. Biol. 1999, 6, 167–175.
8. Besant, P. G.; Attwood, P. V. Biochim. Biophys. Acta 2005, 1754, 281-290.
9. Severin, S. E.; Yudelovich, R. Y. Biokhimiya 1947, 12, 105-110.
10. Muller, T.; Rathlev, T.; Rosenberg, T. Biochim. Biophys. Acta 1956, 19, 563-564.
11. Klumpp, S.; Krieglstein, J. Eur. J. Biochem. 2002, 269, 1067-1071.
12. Stock, A. M.; Rovinson, V. L.; Goudreau, P. N. Annu. Rev. Biochem 2000, 69, 183-
215.
13. Matsushita, M.; Janda, K. D. Biorg. Med. Chem. 2002, 10, 855-867.
14. Saito, H. Chem. Rev. 2001, 101, 2497-2509.
15. Barrett, J. F.; Goldschmidt, R. M.; Lawrence, L. E.; Foleno, B.; Chen, R.; Demers,
J. P.; Johnson, S.; Kanojia, R.; Fernandez, J.; Bernstein, J.; Licata, L.; Donetz, A.;
Huang, S.; Hlasta, D. J.; Macielag, M. J.; Ohemeng, K.; Frechette, R.; Frosco, M. B.;
Klaubert, D. H.; Whiteley, J. M.; Wang, L.; Hoch, J. A. Proc. Natl. Acad. Sci. USA.
1998, 95, 5317–5322.
16. Steeg, P. S.; Palmieri, D.; Ouatas, T.; Salerno, M. Cancer Lett. 2001, 190, 1-12.
17. Schneider, B.; Norda, A.; Karlsson, A.; Veron, M.; Deville-Bonne, D. Protein Sci.
2002, 11, 1648-1656.
18. Tan, E.; Besant, P. G.; Zu, X. L.; Turck, C. W.; Bogoyevitch, M. A.; Lim, S. G.;
Attwood, P. V.; Yeoh, G. C. Carcinogenesis 2004, 11, 2083-2088.
19. Stock, J. B.; Stock, A. M.; Mottonen, J. M. Nature. 1990, 344, 395-400.
20. Modro, T. A. ACS Symp. Ser. 1981, 171, 619–622.
21. Duclos, B.; Marcandier, S.; Cozzone, A. J. Methods Enzymol. 1991, 210, 10-21.
22. Hultquist, D. E. Biochim. Biophys. Acta 1968, 153, 329-340.
23. Boyle, W. J.; Geer, P. V. D.; Hunter, T. Methods Enzymol. 1991, 201, 110-148.
24. Besant, P. G.; Attwood, P. V. Int J Biochem Cell Biol 2000, 32, 243-253.
25. Zu, X. L.; Besant, P. G.; Imhof, A.; Attwood, P. V. Amino Acids 2007, 32, 347–357.
26. Napper, S.; Kindrachuk, J.; Olson, D. J. H.; Ambrose, S. J.; Dereniwsky, C.; Ross,
A. R. S. Anal. Chem. 2003, 75, 1741-1747.
27. Sites, D. P. Basic & Clinical Immunology 1976.
28. Alberts, D. Molecular Biology of the Cell 1983.
29. Lipman, N. S.; Jackson, L. R.; Trudel, L. J.; Weis-Garcia, F. ILAR Journal 2005, 46,
258-268.
30. Köhler, G.; Milstein, C. Nature. 1975, 256, 495-497.
31. Sell, S. Immunology, Immunopathology and Immunity 1987.
32. Lasker, M.; Bui, C. D.; Besant, P. G.; Sugawara, K.; Thai, P.; Medzihradszky, G.;
Turck, C. W. Protein Sci. 1999, 8, 2177-2185.
33. Pirrung, M. C.; James, K. D.; Rana, V. S. J. Org. Chem. 2000, 65, 8448-8453.
34. Dr Paul Besant. Personal Communication. School of Biomedical, Biomolecular and
Chemical Sciences, The University of Western Australia.
35. Truel, I.; Mohamed-Hachi, A.; About-Jaudet, E.; Colligoron, N. Synth. Commun.
1997, 27, 1165.
36. Schenkles, C.; Emi, B.; Reymond, J. L. Bioorg. Med. Chem. Lett. 1999, 9, 1443-
1446.

185
37. Schneider, H.; Sigmund, G.; Schricker, B.; Thirring, K.; Bemer, H. J. Org. Chem.
1993, 58, 683.
38. Schricker, B.; Thirring, K.; Berner, H. Bioorg. Med. Chem. Lett. 1992, 5, 387.
39. Prof. Paul Attwood. Personal Communication. School of Biomedical, Biomolecular
and Chemical Sciences, The University of Western Australia.
40. Kee, J.-M.; Villani, B.; Carpenter, L. R.; Muir, T. W. J. Am. Chem. Soc. 2010, 132,
14327–14329.
41. Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057-3064.
42. Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.;
Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998-15999.
43. McAllister, T. E.; Nix, M. G.; Webb, M. E. Chemical Communications (Cambridge)
2010.
44. Boger, D. L.; Honda, T.; Menezes, R. F.; Colletti, S. L.; Dang, Q.; Yang, W. J. Am.
Chem. Soc. 1994, 116, 82-92.
45. Trost, B. M.; Rudd, M. T. Org. Lett. 2003, 5, 4599-4602.
46. Mitsunobu, O. Synthesis 1981, 1-28.
47. Lipshutz, B. H.; Chung, D. W.; Rich, B.; Corral, R. Org. Lett. 2006, 8, 5069-5072.
48. Dondoni, A.; Giovannini, P. P.; Massi, A. Org. Lett. 2004, 6, 2929-2931.
49. Xun, S.; LeClair, G.; Zhang, J.; Chen, X.; Gao, J. P.; Wang, Z. Y. Org. Lett. 2006,
8, 1697-1700.
50. Cohen, S. G.; Zand, R.; Steel, C. J. Am. Ceram. Soc. 1961, 83, 2895-2900.
51. Vuilhorgne, M.; Malpart, J.; Mutti, S.; Mignani, S. J. Heterocycl. Chem. 2003, 40,
159-162.
52. Steinberg, G. M. J. Org. Chem. 1950, 15, 637-647.
53. Yamauchi, Y.; Yuki, M.; Tanabe, Y.; Miyake, Y.; Inada, Y.; Uemura, S.;
Nishibayashi, Y. J. Am. Chem. Soc. 2008, 130, 2908-2909.
54. Saalfrank, R. W.; Welch, A.; Haubner, M.; Bauer, U. Liebigs Annalen 1996, 2, 171-
181.
55. Balthazor, T. M.; Grabia, R. C. J. Org. Chem. 1980, 45, 5425-5426.
56. Yao, Q.; Levchik, S. Tetrahedron Lett. 2006, 47, 277-281.
57. Hägele, G.; Goudetsidis, S.; Wilke, E.; Seega, J.; Blum, H.; Murray, M.
Phosphorus, Sulfur Silicon Relat. Elem. 1990, 48, 131-140.
58. Jacobsen, M. F.; Andersen, C. S.; Knudsen, M. M.; Gothelf, K. V. Org. Lett. 2007,
9, 2851-2854.
59. Patrick, D. A.; Bakunov, S. A.; Bakunova, S. M.; Kumar, E. V. K. S.; Lombardy, R.
J.; Jones, S. K.; Bridges, A. S.; Zhirnov, O.; Hall, J. E.; Wenzler, T.; Brun, R.; Tidwell,
R. R. J. Med. Chem. 2007, 50, 2468-2485.
60. Boyall, D.; Lopez, F.; Sasaki, H.; Frantz, D.; Carreira, E. M. Org. Lett. 2000, 2,
4233-4236.
61. Huisgen, R. Angew. Chem. 1963, 75, 604.
62. Gothelf, K. V.; Jørgensen, K. A. Chem. Rev. 1998, 98, 863-909.
63. Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem. Int.
Ed. 2002, 41, 2596-2599.
64. Johnson, J. A. Org. Lett.
65. Kolb, H. C.; Sharpless, K. B. Drug Discovery Today 2003, 8, 1128-1137.
66. Boren, B. C.; Narayan, S.; Rasmussen, L. K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.;
Fokin, V. V. J. Am. Chem. Soc. 2008, 130, 14900.
67. Kirkley, J. E.; Goldstein, A. L.; Naylor, P. H. Immunobiology 2001, 203, 601-615.
68. Lesma, G.; Meschini, E.; Recca, T.; Sacchetti, A.; Silvani, A. Tetrahedron 2007, 53,
5567-5578.
69. Coeffard, V.; Beaudet, I.; Evain, M.; Grognec, E. L.; Quintard, J. P. Eur. J. Org.
Chem. 2008, 19, 3344-3351.

186
70. Angione, M. C.; Miller, S. J. Tetrahedron 2006, 62, 5254-5261.
71. Lee, S. J.; Kim, E.; Seo, M. L.; Do, Y.; Lee, Y.-A.; Lee, S. S.; Jung, J. H.; Kogiso,
M.; Shimizu, T. Tetrahedron 2008, 64, 1301-1308.
72. Feenstral, R. W.; Stokkmgreef, E. H. M.; Relchwem, A. M.; Lousberg, W. B. H.;
Ottenheqm, H. C. J. Tetrahedron 1990, 46, 1745-1756.
73. Mak, C. C.; Brik, A.; Lerner, D. L.; Elder, J. H.; Morris, G. M.; Olsonb, A. J.;
Wong, C. H. Biorg. Med. Chem. 2003, 11, 2025–2040
74. Stocking, E. M.; Schwarz, J. N.; Senn, H.; Salzmann, M.; Silks, L. A. Journal of the
chemical society, Perkin Transactions 1997, 1, 2443-2447.
75. Sims, J. W.; Schmidt, E. W. J. Am. Chem. Soc. 2008, 130, 11149-11155.
76. Davoli, P.; Forni, A.; Moretti, I.; Prati, F. Tetrahedron: Asymmetry 1995, 6, 2011-
2016.
77. Maruyama, K.; Hashimoto, M.; Tamiaki, H. J. Org. Chem. 1992, 57, 6143-6150.
78. Angione, M. C.; Miller, S. J. Tetrahedron 2006, 62, 5254-5261.
79. Conchillo, A.; Camps, F.; Messeguer, A. J. Org. Chem. 1990, 55, 1728-1735.
80. Schmittberger, T.; Cotte, A.; Waldmann, H. Chemical Communications
(Cambridge) 1998.
81. Levengood, M. L.; Donk, W. A. Nat. Protoc. 2006, 6, 3001-3010.
82. Brinkley, M. Bioconjugate Chem. 1992, 3, 2-13.
83. Brandon, D. L.; Binder, R. G. Food and Agricultural Immunology 2006, 17, 53-61.
84. Helling, S. A. F.; Zhang, S. O. S.; Lloyd, S. H. I. K. O.; Livingston, P. O. Cancer
Immunol Immunother 1995, 41, 185-192.
85. Lai, J. H.; Marsilje, T. H.; Choi, S.; Nair, S. A.; Hangauer, D. G. J.Pept.Res. 1998,
51, 271-281.
86. Zhang, H.; Duan, Z.; Wang, L.; Zhang, Y.; Wang, S. J. Agric. Food. Chem. 2006,
54, 4499-4505.
87. Hermanson, G. T. Bioconjugate Techniques 1996.
88. Galibert, M.; Dumy, P.; Boturyn, D. Angew. Chem. Int. Ed. 2009, 48, 2576 –2579.
89. Gilmour, R.; Foster, E.; Sheng, Q.; McClain, J. R.; Riley, A.; Sun, P.-M.; Ng, W.-
L.; Yan, D.; Nicas, T. I.; Henry, K.; Winkler, M. E. J. Bacteriol., 187, 8196–8200.
90. Stephenson, K.; Yamaguchi, Y.; Hoch, J. A. J. Biol. Chem. 2000, 275, 38900.
91. Bayardon, J.; Sinou, D. Tetrahedron: Asymmetry 2005, 16, 2965-2972.
92. Fields, G. B.; Noble, R. L. Int. J. Pept. Protein Res. 1990, 35, 161-214.
93. Merrifield, R. B. J. Am. Chem. Soc. 1963, 85, 2149-2154.
94. Huang, Y.; Dey, S.; Zhang, X.; nnichsen, F. S.; Garner, P. J. Am. Chem. Soc. 2004,
126, 4626-4640.
95. Gajda, T.; Andrzej Zwierzak Synthesis 1976, 4, 243-244.
96. Brown, Z. Z.; Schafmeister, C. E. Org. Lett. 2010, 12, 1436-1439.
97. Sammelson, R. E.; Casida, J. E. J. Org. Chem. 2003, 68, 8075-8079.
98. Heinisch, G.; Holzer, W.; Obala, C. Monatsh. Chem. 1988, 119, 253--262.
99. Felding, J.; Kristensen, J.; Bjerregaard, T.; Sander, L.; Vedsø, P.; Begtrup, M. J.
Org. Chem. 1999, 64, 4196-4198.
100. Gelman, D.; Jiang, L.; Buchwald, S. L. Org. Lett. 2003, 5, 2315-2318.
101. Bessmertnykh, A.; Douaihy, C. M.; Muniappan, S.; Guilard, R. Synthesis 2008, 10,
1575-1579.
102. Liu, Y.; Shen, L.; Prashad, M.; Tibbatts, J.; Repic, O.; Blacklock, T. J. Org.
Process Res. Dev. 2008, 12, 778-780.
103. Shirley, D. A.; Roussel, P. A. J. Am. Chem. Soc. 1953, 75, 375.
104. Roth, S.; Thomas, N. R. Synthesis 2010, 4, 607-609.
105. Stocking, E. M.; Schwarz, J. N.; Senn, H.; Salzmann, M.; Silks, L. A. Journal of
the chemical society, Perkin Transactions 1997, 1, 2443-2447.
106. Spetzler, J. C.; Jensen, T. H. J. Pept. Sci. 1999, 5, 582-592.

187
107. Amold, L. D.; Kalantar, T. H.; Vederas, J. C. J. Am. Chem. Soc. 1985, 107, 7105-
7109.
108. Paradis-Bleau, C.; Beaumont, M.; Sanschagrin, F.; Voyerb, N.; Levesqu, R. C.
Biorg. Med. Chem. 2007, 15, 1330-1340.
109. Nakatani, S.; Hidaka, K.; Ami, E. i.; Nakahara, K.; Sato, A.; Nguyen, J.-T.;
Hamada, Y.; Hori, Y.; Ohnishi, N.; Nagai, A.; Kimura, T.; Hayashi, Y.; Kiso, Y. J.
Med. Chem. 2008 51, 2992-3004.
110. Walczak, K.; Wamberg, M.; Pedersen, E. B. Helv. Chim. Acta 2004, 87, 469-478.
111. Tsunoda, T.; Yamamiya, Y.; Ito, S. Tetrahedron Lett. 1993, 34, 1639.
112. Tsunoda, T.; Yamamiya, Y.; Kawamura, Y.; Ito, S. Tetrahedron Lett. 1995, 36,
2529.
113. Kosower, E. M.; Londnerle, H. K. J. Am. Chem. Soc. 1976, 98, 3001-3007.
114. Panosyan, F. B.; Still, I. W. J. Can. J. Chem. 2001, 79, 1110-1114.
115. Turner, R. M.; Lindell, S. D. J. Org. Chem. 1991, 56, 5739-5740.
116. Carver, D. S.; Lindell, S. D.; Saville-Stones, E. A. Tetrahedron 1997, 53, 14481-
14496.
117. Vaswani, R. G.; Chamberlin, A. R. J. Org. Chem. 2008, 73, 1661-1681.
118. Yeager, A. R.; Finney, N. S. J. Org. Chem. 2004, 69, 613-618.
119. Xun, S.; LeClair, G.; Zhang, J.; Chen, X.; Gaoa, J. P.; Wang, Z. Y. Org. Lett. 2006,
8, 1697–1700.
120. Cohen, S. G.; Zand, R.; Steel, C. J. Am. Chem. Soc. 1961, 83, 2895-2900.
121. Lecercle´, D.; Sawicki, M.; Taran, F. d. r. Org. Lett. 2006, 8, 4283-4285.
122. Lee, S. J.; Kim, E.; Seo, M. J.; Do, Y.; Lee, Y. A.; Lee, S. S.; Jung, J. H.; Kogiso,
M.; Shimizu, T. Tetrahedron 2008, 2008, 1301-1308.
123. Bergel, F.; Peutherer, M. A. J. Chem. Soc. 1964, 3973-3981.
124. Wolfrom, M. L.; Remieux, R. U.; Olin, S. M. J. Am. Chem. Soc. 1949, 71, 2870-
2873.
125. Davoli, P.; Forni, A.; Moretti, I.; Prati, F. Tetrahedron 1995, 6, 2011-2016.
126. Cai, J.; Li, X.; Yue, X.; Taylor, J. S. J. Am. Chem. Soc. 2004, 126, 16324-16325.
127. Marcantoni, E.; Massaccesi, M.; Torregiani, E. J. Org. Chem. 2001, 66, 4430-
4432.
128. Nakata, T.; Nakatani, M.; Takahashi, M.; Okai, J.; Kawaoka, Y.; Kouge, K.; Okai,
H. Bull. Chem. Soc. Jpn. 1996, 69, 1099-1106.
129. Tummatorn, J.; Albiniak, P. A.; Dudley, G. B. J. Org. Chem. 2007, 72, 8962-8964.
130. Jackson, R. F. W.; Perez-Gonzalez, M. Organic Syntheses. 2005, 81, 77-88.
131. Paradis-Bleau, C.; Beaumont, M.; Sanchagrin, F.; Voyer, N.; Levesque, R. C.
Biorg. Med. Chem. 2007, 15, 1330-1340.
132. Solorzano, C.; Antonietti, F.; Duranti, A.; Tontini, A.; Rivara, S.; Lodola, A.;
Vacondio, F.; Tarzia, G.; Piomelli, D.; Mor, M. J. Med. Chem. 2010, 53, 5770-5781.