review01-natural product reports (2010), 27(8), 1186-1203
TRANSCRIPT

REVIEW www.rsc.org/npr | Natural Product Reports
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
Marine natural products: synthetic aspects†
Jonathan C. Morris*a and Andrew J. Phillipsb
Received 3rd March 2010, Accepted 7th June 2010
DOI: 10.1039/b919366a
Covering: January to December 2008. Previous review: Nat. Prod. Rep., 2009, 26, 245
An overview of marine natural products synthesis during 2008 is provided. As with earlier installments
in this series, the emphasis is on total syntheses of molecules of contemporary interest, new total
syntheses, and syntheses that have resulted in structure confirmation or stereochemical assignments.
1 Introduction
2 Review articles
3 Pinnatoxin
4 Theopederin D
5 Polyketides: callystatin, aburatubolactam, 15G256 (and
related compounds) and bryostatin 16
6 Terpenoids: vannusal B and cortistatin A
7 Largazole and the trichodermamides
8 Acknowledgements
9 References
1 Introduction
This review is designed to provide an overview of key features of
the 2008 literature covering the synthesis of marine natural
products, and should act as a companion to the Marine Natural
Products review published in this journal.1 The emphasis is on
total syntheses of molecules of contemporary interest. Tabulated
data for other syntheses are also provided. While every effort has
been made to be comprehensive within these boundary condi-
tions, we apologize in advance for any oversights.2
2 Review articles
A number of reviews that cover various aspects of marine natural
products synthesis have appeared: ‘‘Synthesis of marine alkaloids
from the oroidin family’’,3 ‘‘Analogues of marine pyrroloimino-
quinone alkaloids: synthesis and antitumor properties’’,4
‘‘Synthetic studies of heterocyclic natural products’’,5 ‘‘Amphi-
dinolides and its related macrolides from marine dinoflagel-
lates’’,6 ‘‘The synthetic challenge of diazonamide A,
a macrocyclic indole bis-oxazole marine natural product’’,7
‘‘Synthesis of marine natural products with antimalarial
activity’’,8 ‘‘The continuing saga of the marine polyether bio-
toxins’’,9 ‘‘Convergent strategies for the total synthesis of poly-
cyclic ether marine metabolites’’,10 ‘‘Natural marine antiviral
aSchool of Chemistry, University of New South Wales, Sydney, Australia2052. E-mail: [email protected]; Fax: +61 2 93856141; Tel:+61 2 93854733bDepartment of Chemistry, Yale University, New Haven, Connecticut,06520, USA.
† Footnote: This paper is part of an NPR themed issue on Synthesis,guest-edited by Andreas Kirschning and Andy Phillips.
1186 | Nat. Prod. Rep., 2010, 27, 1186–1203
products’’,11 ‘‘The biology and chemistry of the zoanthamine
alkaloids’’,12 ‘‘A potential source of anticancer agents: natural
products and their analogs. Extraction, characterization, bio-
logical activity and synthesis’’,13 ‘‘Synthetic efforts toward, and
biological activity of, thyrsiferol and structrurally-related
analogues’’,14 ‘‘Review of cytotoxic cephalostatins and ritter-
azines: isolation and synthesis’’,15 ‘‘The chemistry of marine
furanocembranoids, pseudopteranes, gersolanes, and related
natural products’’,16 and ‘‘The structure activity relationship of
discodermolide analogues’’.17 Other reviews of relevance are
cited in the text.
3 Pinnatoxin
Zakarian’s synthesis of pinnatoxin A 118,19 was based around the
dissection of the target into two key domains, 2 and 3, and
highlighted the power of the Ireland–Claisen rearrangement for
the establishment of quaternary stereocenters (Scheme 1).20 In
this context, when ester 7 (readily prepared by EDCI-mediated
coupling of 4 and 5) was subjected to deprotonation with chiral
amide base 6, the desired (Z)-enolate was formed. Trapping as
the silylketene acetal, followed by warming to room temperature,
resulted in the desired [3,3]-rearrangment to give 9 in an excellent
94% yield. A sequence of 14 steps installed the cyclohexene ring,
and advanced material to 3. Addition of the organolithium
derived from 2 (with t-BuLi) to aldehyde 3 occurred in 75% yield,
and three further steps (desilylation with TBAF, Dess–Martin
oxidation, and vinylmagnesium bromide addition) gave 10.
Ring-closing metathesis with the Hoveyda–Grubbs 2nd-genera-
tion catalyst (25 mol% loading) followed by oxidation gave 11 in
57% yield for the two steps. Diastereoselective conjugate addi-
tion of MeCu(CN)Li installed the final remaining methyl group
(11 / 12, 81%) and spiroketalization with LiBF4 in wet iPrOH
led to 13 in 60% yield. This material was advanced to 14 by
a sequence of 7 steps, and after Staudinger reduction of the azide
with Me3P, the imine was installed using Kishi’s conditions
(triethylammonium 2,4,6-triisopropylbenzoate in xylenes at
85 �C) to yield 15 (70% over two steps). The synthesis was
completed by ester hydrolysis to give (+)-pinnatoxin A.
Nakamura and Hashimoto have also reported a synthesis of
pinnatoxin A (Scheme 2).21 Taking a page from Trost’s recent
successes in complex molecule synthesis [see also the bryostatin
16 synthesis later in this review], the key step was
This journal is ª The Royal Society of Chemistry 2010

Scheme 1 Zakarian’s synthesis of pinnatoxin A. Reagents and conditions: (1) EDCI, DMAP, DMF, 94%; (2) (a) 6, THF, then TMSCl, �78 �C; (b)
25 �C, 6 h, 94%; (3) 2, tBuLi, Et2O,�78 �C, 75%; (4) TBAF, THF; (5) Dess–Martin periodinane, CH2Cl2; (6) H2C]CHMgBr, THF, 75% (over 3 steps);
(7) (a) 25 mol% Hoveyda–Grubbs II, CH2Cl2, 40 �C; (b) Dess–Martin periodinane, pyr, CH2Cl2, 57% (over 2 steps); (8) MeCu(CN)Li, BF3$OEt2, THF,
�78 �C, 81%; (9) LiBF4, 4% aq. iPrOH, 90 �C, 6h, 60%; (10) Me3P, aq. THF; (11) triethylammonium 2,4,6-triisopropylbenzoate, xylenes, 85 �C, 70%
(over 2 steps); (12) LiOH, THF–H2O, 80%.
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
a macrocyclization by Ru-catalyzed enyne isomerization.22
Advanced intermediate 16 was subjected to a five-step sequence
that reorganized protecting groups, and freed the primary
alcohol, which was oxidized with Dess–Martin periodinane to
Jonathan C: Morris
Jonathan C. Morris obtained
his B.Sc. (Hons) degree from
the University of Western
Australia and his Ph.D. degree
from The Australian National
University in Canberra,
Australia. After a postdoctoral
appointment with Phil Magnus
at the University of Texas at
Austin, he joined the faculty at
the University of Canterbury,
New Zealand. In 2004, he
moved to the University of
Adelaide. In late 2009, he was
appointed at the University
of New South Wales. His
research interests focus around the synthesis of biologically
active natural products.
This journal is ª The Royal Society of Chemistry 2010
give the aldehyde 17. Horner–Wadsworth–Emmons reaction
with phosphonate 18 gave the expected enone 19 in 86% yield
(two steps). Wittig methylenation (Ph3P]CH2, 92%) provided
a diene, 20, that was exposed to lactone 21 (10 equivalents) in
Andrew J: Phillips
Andrew J. Phillips obtained his
B.Sc. (Hons) and Ph.D. degrees
from the University of Canter-
bury in Christchurch, New
Zealand. After a postdoctoral
appointment with Peter Wipf at
the University of Pittsburgh, he
joined the faculty at the
University of Colorado. In mid-
2010 he joined the faculty at
Yale University. His research
interests are broadly defined by
the chemistry and biology
of small molecules, including
natural products.
Nat. Prod. Rep., 2010, 27, 1186–1203 | 1187

Scheme 2 The synthesis of pinnatoxin A by Nakamura & Hashimoto. Reagents and conditions: (1) TBAF, THF, reflux, 72 h; (2) TBSCl, imid., DMF,
95% (2 steps); (3) TESOTf, 2,6-lut., CH2Cl2, 94%; (4) TBAF, THF, 0 �C, 91%; (5) Dess–Martin periodinane, pyr, CH2Cl2, (6) 18, LiCl, DIPEA, MeCN,
86% (2 steps); (7) MePPh3Br, nBuLi, THF, 92%; (8) 21 (10 equiv), 4 �A MS, p-xylene, 160 �C, 12 h, 35% of desired (+ 46% of other cycloadducts); (9)
[CpRu(MeCN)3]PF6 (10 mol%), acetone, 50 �C, 15 min, 79%.
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
p-xylenes at 160 �C. Under these conditions the desired endo
product was obtained in 35% yield [3 other diastereomers were
formed, with a total yield in the cycloaddition of 83%]. A seven-
step sequence advanced material to enyne 23, and when treated
with 10 mol% of [CpRu(MeCN)3]PF6, macrocyclization to the
desired product 24 occurred in an impressive 79% yield. This
result underlines the value of this catalyst in complex molecule
synthesis. The synthesis was completed by a further nine
steps that paralleled the general strategy used by Kishi and
Zakarian.23–25
4 Theopederin D
Floreancig’s plan for the synthesis of theopederin D 2526 was
based around connection of known acid chloride 26 with aminal
27 (Scheme 3).27,28 One of the key bond formations en route to 27
was an electron-transfer-initiated cyclization29,30 whereby an
N-acyliminium ion was generated and intercepted by a proximal
acetal. The synthesis of 27 commenced with known homoallylic
alcohol 28, which was converted in six steps and 33% overall
yield to dihydropyran 29. DMDO epoxidation of 29, followed by
addition of trivinylalane to the intermediate alkoxy epoxide and
protection of the alcohol as the 4-(benzyloxy)butyl ether,
produced an intermediate (77% for the three steps) that was
readily ozonized and converted to sulfinylimine 30 by reaction
with (R)-tert-butylsulfonamide in 50% yield. Addition of ben-
zylmagnesium chloride, followed by (i) sulfonamide hydrolysis,
(ii) hydrogenolysis of the benzyl ether and (iii) Cbz group
installation gave alcohol 31. When subjected to iodobenzene
diacetate and I2 under irradiation, Su�arez oxidative ether-
ification31 occurred to give 32 in 80% yield, and set the stage for
the key electron-transfer initiated cyclization. Irradiation of 32
(medium-pressure Hg lamp with Pyrex filtration) in the presence
of 6 mol% of N-methylquinolinium hexafluorophosphate
(NMQPF6) and O2 provided 33 in 76% yield as a 2 : 1 mixture of
diastereomers at C10. The reaction presumably proceeds via
N-acyliminium ion 34, followed by oxononium ion 35. The
1188 | Nat. Prod. Rep., 2010, 27, 1186–1203
ability to generate N-acyliminium ions under such mild condi-
tions is especially noteworthy given the delicate substrate, and
this example should underscore the value of this approach for
complex molecule synthesis. The synthesis was completed by
a four-step sequence that involved oxidation of the lactol-derived
acetal to the lactone with Jones reagent, removal of the Cbz
group, acylation of the aminal with acid chloride 26 and, finally,
removal of the benzoyl group. The overall synthesis provided
theopederin D 25 in 16 steps (longest linear sequence).
5 Polyketides: callystatin, aburatubolactam,15G256 (and related compounds) and bryostatin 16
The Micalizio group has reported a synthesis of callystatin A 36
that features a novel direct reductive coupling of two alkynes
with Ti(II) reagents (Scheme 4).32 The subunit coupling
commenced with a palladium-catalyzed coupling between the
vinylzinc reagent derived from vinyl iodide 37, and (E)-vinyl
iodide 38 and was followed by removal of the terminal TMS
group to give 39. The direct titanium-mediated cross-coupling
between alkyne 40 and diene 39 was accomplished with
ClTi(OiPr)3 and cyclopentylmagnesium chloride in 75% yield
(>5 : 1 regioselectivity) to give 41 (presumably via 42), which
contains the complete callystatin A carbon backbone. TMS
deprotection followed by oxidation and TBS removal concluded
a total synthesis that proceeds in a remarkable 11-step longest
linear sequence.
In a follow-up to their earlier synthesis of the macrolactam
tetramic acid cylindramide A,33 the Phillips group has described
a total synthesis of aburatubolactam A 43 (Scheme 5).34 In the
same vein as cylindramide A, the bicyclo[3.3.0]octane domain
was formed by tandem metathesis of a readily accessible Diels–
Alder adduct. In the case at hand, treatment of 44 with 2.5 mol%
of the Grubbs 1st-generation catalyst under an atmosphere of
ethylene led to fused bicyclic compound 45 in 90% yield. A
15-step sequence advanced this compound to stannyldioxenone
46 in 9% overall and set the stage for an endgame that
This journal is ª The Royal Society of Chemistry 2010

Scheme 4 Micalizio’s Ti-based route to callystatin A. Reagents and
conditions: (1) 37, ZnCl2, tBuLi, Et2O then 38, cat. Pd(PPh3)4; (2) TBAF,
THF 60% (2 steps); (3) nBuLi, ClTi(OiPr)3, c-C5H9MgCl, PhMe, �78 �C
then NH4Cl, �30 �C, 75%; (4) PPTS, H2O, acetone; (5) PCC, AcOH; (6)
HF$pyr, pyr, THF, 24% (3 steps).
Scheme 5 Phillips’ synthesis of aburatubolactam A 43. Reagents and
conditions: (1) 2.5% Grubbs I, CH2]CH2 (1 atm), CH2Cl2, 90%; (2)
PhMe, 110 �C; (3) tert-butyl-b-iodoacrylate, 10 mol% Pd2(dba)3, Ph3As,
NMP; (4) NaOMe, MeOH, 50% (3 steps); (5) TFA, CH2Cl2; (6) DEPC,
Et3N, DMF, rt; (7) HF, MeCN, 46% (3 steps). DEPC ¼ diethy-
lphosphoryl cyanide.
Scheme 3 Floreancig’s synthesis of theopederin D. Reagents and
conditions: (1) DMDO, acetone, then (H2C]CH)3Al, CH2Cl2, 100%; (2)
BBMCl, DIPEA, CH2Cl2, 77%; (3) O3, CH2Cl2, �78 �C; then (R)-tBu-
S(O)NH2, Ti(OiPr)4, CH2Cl2, 50%; (4) BnMgCl, THF, 65%; (5) HCl,
MeOH, 80%; (6) H2, Pd/C, MeOH; then CbzCl, Et3N, CH2Cl2, 70%; (7)
PhI(OAc)2, hn, cyclohexane, 80%; (8) hn, NMQPF6, O2, NaOAc,
Na2S2O3, PhMe, DCE, 76%. BBMCl ¼ 4-(benzyloxy)chlorobutane,
NMQPF6 ¼ N-methylquinolinium hexafluorophosphate.
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
commenced with thermolysis of 46 in the presence of amine 47 to
give b-ketoamide 48. Stille coupling with tert-butyl-b-iodoacry-
late produced the dienoate, and Lacey–Dieckmann cyclization
gave 49 in 50% yield (over three steps). Despite the potential for
subversion of the macrolactamization by stereochemical ques-
tions with respect to the exocyclic olefin of the tetramic acid,
treatment of 49 with TFA to remove the Boc carbamate and tert-
butyl ester, followed by DEPC, smoothly produced the desired
macrocycle. Removal of the TBS group with HF completed the
total synthesis (46% yield over 3 steps).
Barrett and coworkers have developed total syntheses of the
marine antifungal agents 15G256p (50), 15G256i (51), and
15G256b (52), using a biomimetic late-stage aromatization
strategy (Scheme 6).35 This strategy involves the thermolysis of
This journal is ª The Royal Society of Chemistry 2010
dioxinones such as 55 and trapping of the resulting triketoke-
tenes with an alcohol to afford 54. Treatment of 54 with base
generates the aromatic system 55.
To prepare these natural products, an efficient synthesis to the
15G26 monomer unit 60 is required. Using acid chlorides 64 and
65, the readily available dioxinone 56 could be sequentially
C-acylated to provide access to dioxinone 57 in 66% overall yield
Nat. Prod. Rep., 2010, 27, 1186–1203 | 1189

Scheme 6 Resorcylate lactones and a late-stage aromatization strategy.
Scheme 7 Barrett’s synthesis of key monomers 62 and 63. Reagents and
conditions: (1) 64, Bi(OTf)3, �15 / 20 �C, 80%; (2) 65, MgCl2, pyr,
CH2Cl2, 0 to 20 �C, 83%; (3) Pd(PPh3)4 4 mol%, morpholine, CH2Cl2,
0 to 20 �C, 89%; (4) 66, PhMe, 110 �C; (5) K2CO3, iPrOH, CH2Cl2, HCl,
MeOH, 70% (2 steps); (6) Cs2CO3, BnBr, DMF, 0 / 20 �C, 92%; (7) HF,
THF, pyr, 85%; (8) Pd(PPh3)4, morpholine, THF, 93%.
Scheme 8 Completion of Barrett’s synthesis of 15G256p (50), 15G256i
(51), and 15G256b (52). Reagents and conditions: (1) 63, 2,4,6-tri-
chlorobenzoyl chloride, iPr2NEt, then 62, DMAP, PhMe, 86%. (2)
Pd(PPh3)4, morpholine, THF, 87%; (3) H2, Pd/C, EtOAc; (4) HF, THF,
pyr, 86% (2 steps); (5) HF, THF, pyr; (6) 2,4,6-trichlorobenzoyl chloride,
DIPEA, DMAP, PhMe, 76% (2 steps); (7) H2, Pd/C, EtOAc, 75%; (8) 68,
2,4,6-trichlorobenzoyl chloride, iPr2NEt, then 66, DMAP, PhMe, 71%;
(9) Pd(PPh3)4, morpholine, THF; (10) HF, THF, pyr; (11) 2,4,6-tri-
chlorobenzoyl chloride, DIPEA, DMAP, PhMe, 65% (3 steps); (12) H2,
Pd/C, EtOAc, 85%.
Scheme 9 An overview of Trost’s synthesis plan for bryostatin 16.
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
(Scheme 7). The tri-keto ester 58 was obtained in 89% yield after
application of a palladium-catalyzed deallylation–decarboxyl-
ation procedure (4 mol% Pd(PPh3)4, morpholine). Thermolysis
of the dioxinone in the presence of the chiral alcohol 66, followed
by sequential reaction with potassium carbonate and methanolic
hydrogen chloride gave the 15G256 monomer unit 60 in 70%
yield for the two steps. Benzylation (BnBr, Cs2CO3, DMF, 92%)
gave 61, which could then be converted into either alcohol 62 or
acid 63 by the use of selective deprotection protocols.
A Yamaguchi esterification of 62 and 63 gave the tetraester 67
in 86% yield (Scheme 8). Selective deallylation (4 mol%
Pd(PPh3)4, morpholine, 87%) gave the acid 68, which is the key
material for preparing the natural products. 15G256p (50) was
prepared in 86% yield by removal of the benzyl ethers (H2, Pd/C),
then desilylation (HF$pyr).
1190 | Nat. Prod. Rep., 2010, 27, 1186–1203
Conversion of 68 to the symmetrical natural product 15G256i
(51) required a three-step procedure, with the silyl group being
removed first, then a Yamaguchi lactonization, followed by
debenzylation. A 57% yield for the three steps was achieved. The
more complex macrolactone 15G256b (52) was accessed by
firstly converting 68 to macrolactone 69, using a four-step
procedure. Debenzylation of 14 provided the natural product 52
in 85% yield.
The bryostatins are an exciting class of macrolactone natural
products that exhibit exceptional biological activity.36,37 In
particular, it has been found that they have great potential as
anticancer agents when used in combination with other thera-
peutic agents. Due to the scarcity of the natural material and the
need for analogues, there has been much interest in the devel-
opment of efficient syntheses.38
Trost and Dong have reported a highly convergent total
synthesis of bryostatin 16 70 which is a key parent structure that
This journal is ª The Royal Society of Chemistry 2010

Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
could allow access to many other bryostatins.39 Starting from the
readily available aldehyde 75, the synthesis requires 39 steps in
total, with the longest linear sequence being 26 steps. As detailed
in Scheme 9, it was envisaged that the macrocycle could be
formed from 71 using a palladium-catalyzed alkyne–alkyne
coupling, followed by a metal-catalyzed 6-endo-dig cyclization.
The key substrate 3 would be assembled from the fragments 72,
73, and 74.
Lactone 73 was prepared in 11 steps from the readily available
aldehyde 75 (see Scheme 2) (Scheme 10). The same aldehyde was
also used to produce the alkyne coupling partner 72 by firstly
forming the enal, followed by an indium-mediated prop-
argylation. To obtain the enantio-enriched material, the racemic
72 was firstly oxidized (Dess–Martin periodinane) to the ketone,
then a Corey–Bakshi–Shibata reduction gave (R)-72 in 90% ee
and 90% overall yield. A chemoselective ruthenium-catalyzed
tandem alkene–alkyne coupling – Michael addition (13 mol%
CpRu(MeCN)3PF6, DCM) was used to form the cis-tetrahy-
dropyran 76 in 34% yield (80% based on recovered starting
material). Bromination of the vinyl silane (NBS, DMF), followed
by an acid-catalyzed process (10 mol% CSA, MeOH) afforded
the alcohol 77 in 94% overall yield for the two steps. With the
A-ring and B-ring substructures in place, 77 was transformed
into the desired acid 79 in six steps. Esterification of 79 with the
Scheme 10 Trost & Dong’s synthesis of acid 79. Reagents and conditions:
(1) (Z)-1-bromo-2-ethoxyethene, tBuLi, Me2Zn, then 75, Et2O, �78 �C,
97%; (2) (3-bromo-1-propynyl)trimethylsilane, In powder, InF3
(10 mol%), THF, 65 �C, 68%; (3) (i) Dess–Martin periodinane, NaHCO3,
CH2Cl2; (ii) (S)-2-methyl-CBS-oxazaborolidine (5 mol%), catecholbor-
ane, CH2Cl2, �78 �C, 90%, 90% e.e. (2 steps); (4) 1.2 equiv 73,
CpRu(MeCN)3PF6 (13 mol%), CH2Cl2, 34% (80% b.r.s.m.); (5) NBS,
DMF, 98%; (6) CSA (10 mol%), MeOH, 0 �C, 93–96%; (7)
PdCl2(MeCN)2 (10 mol%), dppf, CO, MeOH, NEt3, DMF, 80 �C, 83%
(90% b.r.s.m.); (8) Dess–Martin periodinane, NaHCO3, CH2Cl2, 88%; (9)
Ohira–Bestmann reagent, K2CO3, MeOH, 97%; (10) TBAF, HOAc,
THF, 90% (96% b.r.s.m.); (11) Me3SnOH, 1,2-DCE, 80 �C, 84%; (12)
TESOTf, 2,6-lutidine, CH2Cl2, �10 �C / 0 �C, 76–79%.
This journal is ª The Royal Society of Chemistry 2010
readily available 74 was achieved using Yamaguchi’s conditions
(2,4,6-Cl3C6H2COCl, DMAP, NEt3, PhMe), affording 80 in 92%
yield. Removal of the PMB protecting groups, using oxidative
conditions (DDQ, pH 7 buffer, 2 cycles), gave the macrocycle
precursor 81.
The macrocycle 82 was generated in 56% yield by reaction of
81 with 12 mol% of palladium acetate and 15 mol% tris(2,6-
dimethoxyphenyl)phosphine in PhMe at room temperature
(Scheme 11). It was found that the reaction had to be run at low
concentration (0.002 M) and that the choice of solvent and the
ligand/palladium ratio were critical to the success of the reaction.
Treatment of the resulting alcohol 82 with a cationic gold cata-
lyst (Au(PPh3)SbF6, NaHCO3) initiated a 6-endo-dig cyclization
and afforded the desired ring system 83 in 73% yield. After piv-
alation of the secondary alcohol (Piv2O, DMAP, 62%), efforts
focused on the global deprotection to afford bryostatin 16. After
some experimentation, it was found that treatment of 84 with 5
equivalents of tetrabutylammonium fluoride gave bryostatin 16
70 in 52% yield, after purification using reverse-phase HPLC.
Keck and his group40 have developed a convergent strategy to
highly potent bryostatin analogues, while Wender and coworkers
have reported41 on the development of new highly potent
analogues of the bryostatins, using a Prins-driven macro-
cylization strategy to allow efficient access to the target
molecules.
The isolation of (+)-neopeltolide 85a was reported42 in early
2007, and has attracted a great deal of attention due to the
reported potent biological activity and structural complexity of
the natural product. Indeed, there have been seven total
Scheme 11 Completion of Trost & Dong’s synthesis of bryostatin 16
(70). Reagents and conditions: (1) 79, 2,4,6-trichlorobenzoyl chloride,
NEt3, PhMe, then 74, DMAP, 92%; (2) DDQ, pH 7.0 buffer, DCM, 2
cycles, 75%; (3) Pd(OAc)2 (12 mol%), TDMPP (15 mol%), PhMe, 56%;
(4) AuCl(PPh3) (20 mol%), AgSbF6 (20 mol%), NaHCO3, DCM–MeCN,
0 �C to rt, 73%; (5) Piv2O, DMAP, DCM, 50 �C, 62%; (6) TBAF,
THF, 52%.
Nat. Prod. Rep., 2010, 27, 1186–1203 | 1191

Scheme 12 The original and revised structures of neopeltolide.
Scheme 14 Panek’s synthesis of (+)-neopeltolide (85b). Reagents and
conditions: (1) BH3$SMe2, THF, 0 �C to rt; (2) TBDPSCl, imid., DMF,
0 �C to rt, 80% (over 2 steps); (3) DIBAL-H, diethyl ether, �78 �C; (4)
1,3-propanedithiol, I2, CHCl3, rt, 85% (over 2 steps); (5) t-BuLi, HMPA,
THF, 90, �78 �C, 68%; (6) CaCO3, MeI, MeCN–H2O, rt, 73%; (7)
Zr(OtBu)4, iPrCHO, PhMe, �78 �C; (8) Me3OBF4, Proton Sponge, 4 �A
molecular sieves, CH2Cl2, rt, 90% (over 2 steps); (9) 49% HF in H2O,
MeCN, rt, 91%; (10) (COCl)2, DMSO, CH2Cl2, Et3N,�78 �C to rt, 89%.
(11) 86, TfOH, CH2Cl2–benzene (3 : 1), �78 �C, 75% (d.r. 10 : 1); (12)
NaCN, DMF, 60 �C, 84%; (13) DIBAL-H, Et2O, �78 �C, 96%; (14)
DIBAL-H, CH2Cl2, �78 �C, 60%; (15) NaClO2, 2-methyl-2-butene,
NaH2PO4$H2O, tBuOH, H2O, 85%; (16) 2,4,6-trichlorobenzoyl chloride,
PhMe, DMAP, Et3N, 44%; (17) Hg(O2CCF3)2 then NaBH4, THF–H2O
(1 : 1), 63% (d.r. >20 : 1); (18) (CF3CH2O)2P(O)CH2CO2H, EDCI$HCl,
HOBt$H2O, CH2Cl2, 99%; (19) 94, 18-crown-6, KHMDS, �78 �C, then
95, �85 �C, 62%.
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
syntheses and one formal synthesis of the compound since its
isolation. Importantly, the first total syntheses of the proposed
structure 85a, by the groups of Panek43 and Scheidt44 respec-
tively, lead to the realization that the original stereochemical
assignment was incorrect. Both groups independently assigned
the configuration to structure 85b, and confirmed this by
synthesis (Scheme 12). Their syntheses of the revised structure
are discussed below.
The synthetic plan pursued by the Panek group revolved
around a Yamaguchi macrocyclization and Still–Gennari olefi-
nation (Scheme 13). Key building blocks were allyl silane 86 and
aldehyde 87.
Synthesis of the C7–C16 fragment 87 of neopeltolide was
achieved in 10 steps, starting from commercially available
(R)-(+)-3-methylglutarate (88) (Scheme 14). The dihydropyran
91 was prepared in 75% yield (10 : 1 diasteromeric ratio) by
reacting 87 with the readily available allylsilane 86, using a triflic
acid promoted [4 + 2] annulation. A four-step sequence was then
utilized to prepare the seco acid 92 required for the macro-
cyclization.
After macrocyclization (2,4,6-Cl3C6H2COCl, DMAP, PhMe,
44%), the axial C5 alcohol 93 was obtained by carrying out
a selective oxymercuration (Hg(O2CCF3)2; NaBH4, 63%, >20 : 1
ratio) of the pyran alkene 92. To attach the side-chain, Panek and
coworkers employed a Still–Gennari olefination. Acylation of
alcohol 93 with bis(2,2,2-trifluoroethyl)phosphonoacetic acid
(EDCI$HCl, HOBt$H2O, 99%) afforded phosphonate 94.
Deprotonation (KHMDS, 18-crown-6, �78 �C) of the phos-
phonate 94, followed by addition of the readily available alde-
hyde 95 at �85 �C provide a 7 : 1 mixture of neopeltolide (85b)
and the corresponding (E)-olefin. The longest linear sequence
was 19 steps, with an overall yield of 1.3%.
Scheme 13 Panek’s retrosynthetic analysis of (+)-neopeltolide (85b).
1192 | Nat. Prod. Rep., 2010, 27, 1186–1203
The Scheidt synthesis also focuses on the preparation of the
macrolactone moiety, but the endgame differs in that it was
proposed to attach the side-chain 96 via a Mitsunobu inversion
(Scheme 15). To prepare the macrolactone 97, they planned to
Scheme 15 Scheidt’s retrosynthesis of (+)-neopeltolide (85b)
This journal is ª The Royal Society of Chemistry 2010
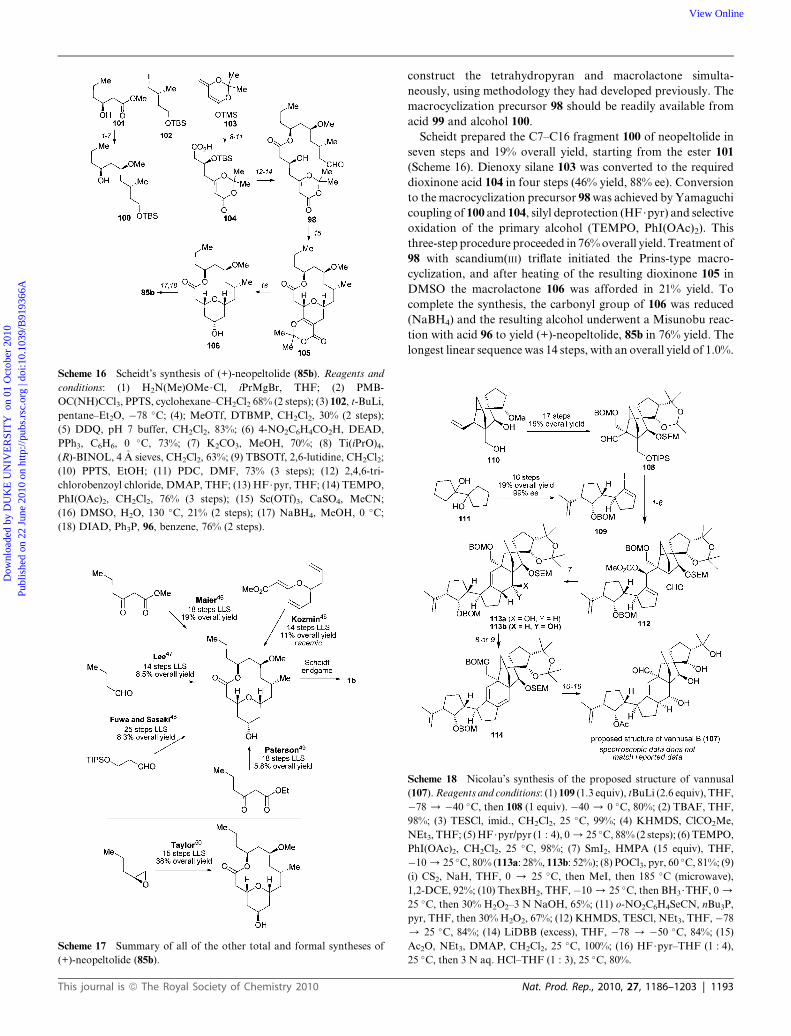
Scheme 16 Scheidt’s synthesis of (+)-neopeltolide (85b). Reagents and
conditions: (1) H2N(Me)OMe$Cl, iPrMgBr, THF; (2) PMB-
OC(NH)CCl3, PPTS, cyclohexane–CH2Cl2 68% (2 steps); (3) 102, t-BuLi,
pentane–Et2O, �78 �C; (4); MeOTf, DTBMP, CH2Cl2, 30% (2 steps);
(5) DDQ, pH 7 buffer, CH2Cl2, 83%; (6) 4-NO2C6H4CO2H, DEAD,
PPh3, C6H6, 0 �C, 73%; (7) K2CO3, MeOH, 70%; (8) Ti(iPrO)4,
(R)-BINOL, 4 �A sieves, CH2Cl2, 63%; (9) TBSOTf, 2,6-lutidine, CH2Cl2;
(10) PPTS, EtOH; (11) PDC, DMF, 73% (3 steps); (12) 2,4,6-tri-
chlorobenzoyl chloride, DMAP, THF; (13) HF$pyr, THF; (14) TEMPO,
PhI(OAc)2, CH2Cl2, 76% (3 steps); (15) Sc(OTf)3, CaSO4, MeCN;
(16) DMSO, H2O, 130 �C, 21% (2 steps); (17) NaBH4, MeOH, 0 �C;
(18) DIAD, Ph3P, 96, benzene, 76% (2 steps).
Scheme 17 Summary of all of the other total and formal syntheses of
(+)-neopeltolide (85b).
This journal is ª The Royal Society of Chemistry 2010
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
construct the tetrahydropyran and macrolactone simulta-
neously, using methodology they had developed previously. The
macrocyclization precursor 98 should be readily available from
acid 99 and alcohol 100.
Scheidt prepared the C7–C16 fragment 100 of neopeltolide in
seven steps and 19% overall yield, starting from the ester 101
(Scheme 16). Dienoxy silane 103 was converted to the required
dioxinone acid 104 in four steps (46% yield, 88% ee). Conversion
to the macrocyclization precursor 98 was achieved by Yamaguchi
coupling of 100 and 104, silyl deprotection (HF$pyr) and selective
oxidation of the primary alcohol (TEMPO, PhI(OAc)2). This
three-step procedure proceeded in 76% overall yield. Treatment of
98 with scandium(III) triflate initiated the Prins-type macro-
cyclization, and after heating of the resulting dioxinone 105 in
DMSO the macrolactone 106 was afforded in 21% yield. To
complete the synthesis, the carbonyl group of 106 was reduced
(NaBH4) and the resulting alcohol underwent a Misunobu reac-
tion with acid 96 to yield (+)-neopeltolide, 85b in 76% yield. The
longest linear sequence was 14 steps, with an overall yield of 1.0%.
Scheme 18 Nicolau’s synthesis of the proposed structure of vannusal
(107). Reagents and conditions: (1) 109 (1.3 equiv), tBuLi (2.6 equiv), THF,
�78 / �40 �C, then 108 (1 equiv). �40 / 0 �C, 80%; (2) TBAF, THF,
98%; (3) TESCl, imid., CH2Cl2, 25 �C, 99%; (4) KHMDS, ClCO2Me,
NEt3, THF; (5) HF$pyr/pyr (1 : 4), 0 / 25 �C, 88% (2 steps); (6) TEMPO,
PhI(OAc)2, CH2Cl2, 25 �C, 98%; (7) SmI2, HMPA (15 equiv), THF,
�10 / 25 �C, 80% (113a: 28%, 113b: 52%); (8) POCl3, pyr, 60 �C, 81%; (9)
(i) CS2, NaH, THF, 0 / 25 �C, then MeI, then 185 �C (microwave),
1,2-DCE, 92%; (10) ThexBH2, THF,�10 / 25 �C, then BH3$THF, 0 /
25 �C, then 30% H2O2–3 N NaOH, 65%; (11) o-NO2C6H4SeCN, nBu3P,
pyr, THF, then 30% H2O2, 67%; (12) KHMDS, TESCl, NEt3, THF,�78
/ 25 �C, 84%; (14) LiDBB (excess), THF, �78 / �50 �C, 84%; (15)
Ac2O, NEt3, DMAP, CH2Cl2, 25 �C, 100%; (16) HF$pyr–THF (1 : 4),
25 �C, then 3 N aq. HCl–THF (1 : 3), 25 �C, 80%.
Nat. Prod. Rep., 2010, 27, 1186–1203 | 1193

Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
The syntheses and subsequent structural revisions of Panek
and Scheidt have triggered a great deal of work, with a further six
new syntheses (Kozmin,45 Maier,46 Lee,47 Fuwa-Sasaki,48 Pater-
son,49 and Taylor50) reported in 2008. Due to space constraints,
these syntheses are not discussed, but a summary of the number
of steps and the overall yield for the sequence is provided in
Scheme 17. It should be noted that all but one (Taylor) have
utilized the Scheidt endgame, whereby they attach the oxazole
sidechain using a Mitsunobu inversion.
Kozmin and coworkers have carried out an investigation of
the mode of action of neopeltolide, 85b and have identified the
cytochrome bc1 complex of the mitochondrial respiratory chain
as the primary cellular target.45
6 Terpenoids: vannusal B and cortistatin A
Vannusal B 107 represents a significant synthetic challenge, with
seven rings and 13 stereogenic centers.51 However, Nicolaou and
Scheme 19 Highlights of the first three total syntheses of cortistatin A by B
conditions: (1) Co(acac)2 (0.2 equiv), PhSiH3 O2, THF, HC(OMe)3, 23 �C, 12
(5 equiv), Br2 (5 equiv), CH2Cl2, �30 �C, 10 h; then TMSCl, imid., 57%; (3
(1.1 equiv), 23 �C, 5 h; (5) LiBr, Li2CO3, DMF, 80 �C, (65% over 2 steps); (6) A
DMAP, CH2Cl2, 89%; (7) MgBr2$Et2O, 2,6-tBu2Py, PhH; then PPTS, butan
50 �C, 6 h, then I2 (2 equiv), Et3N (3 equiv), THF, 23 �C, 5 min; (b) 7-(trim
DMSO, 23 �C, 10 min, 53% (over 2 steps); (9) RANEY� Ni, iPrOH, H2O, 5
Reagents and conditions: (1) 3-oxocyclohex-1-enyl trifluoromethanesulfonat
Pd/BaSO4 (5% wt/wt, 0.24 equiv), H2, MeOH–THF, 64%; (4) K2CO3, dioxane
(dr � 1 : 1); (7) Me2NH, THF, Ti(OiPr)4, 80 �C, 5 h, 45%. (C) Shair’s synthes
70 �C, 1 h (49%, 2 steps); (3) SOCl2, pyr, CH2Cl2; (4) NaHMDS, THF, the
(3 steps); (6) CHBr3, KOtBu, hexane, 0 �C; (7) TASF (1.2 equiv), DMF, 80 �C
50 �C, 40 min, 65% (3 steps).
1194 | Nat. Prod. Rep., 2010, 27, 1186–1203
coworkers have synthesised the proposed structure of vannusal B
using a convergent strategy, starting from aldehyde 108 and
iodide 109 (Scheme 18).52 Unfortunately, the spectroscopic data
for the synthesized material does not match the reported data of
the natural product.
A six-step sequence, starting from 108 and 109, generates the
aldehyde carbonate 112. Formation of the carbon skeleton 113
(as a 1.9 : 1 mixture of alcohols) was achieved in 80% yield by
treatment of 112 with samarium iodide. Each of the diastereo-
mers of 113 could be independently deoxygenated to afford diene
114. This material was transformed to the proposed structure 107
using a seven-step sequence. At the time of writing, the Nicolaou
group has revised the structure on the basis of further synthetic
studies.53,54
The cortistatin family of steroidal alkaloids have generated
significant interest from the synthesis community55 since their
discovery in 2006–2007.56–58 Aside from their interesting
9-(10,19)-abeo-androstane type skeleton, attention has no doubt
aran, Nicolaou & Chen, and Shair. (A) Baran’s synthesis. Reagents and
h; then TsOH$H2O, rt, 2 h; then K2CO3 MeOH, 6 h, 65%; (2) PhI(OAc)2
) DBU, LiCl, THF, 85%; (4) SmI2, DMPU–THF, 23 �C; then TBCHD
lH3, THF, 23 �C, 1 h; then K2CO3, MeOH, 23 �C, 12 h; then Ac2O, Et3N,
one: H2O, 90 �C; then K2CO3, MeOH, 82%; (8) (a) N2H4, Et3N, EtOH,
ethylstannyl)isoquinoline (4 equiv), Pd(PPh3)4 (0.5 equiv), CuCl, LiCl,
0 �C, 1 h, 50% (at �50% conversion). (B) Nicolaou & Chen’s synthesis.
e, 10% Pd(PPh3)4, CuI, Et3N, DMF, 85%; (2) IBX, DMSO, 81%; (3)
, 52%; (5) tBuOOH, DBU, CH2Cl2, 70%; (6) NaBH4, CeCl3, MeOH, 80%
is. Reagents and conditions: (1) PPTS, Me2CO–H2O; (2) NaOMe, MeOH,
n PhNTf2; (5) Me(OiPr)2SiCH2MgCl, 5 mol% Pd(PPh3)4, THF, rt, 62%
, 30 min, 66% (2 steps); (8) Me2NH (3 equiv), ZnBr2 (1.5 equiv), CH3CN,
This journal is ª The Royal Society of Chemistry 2010

Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
been honed by their impressive biological activities, most notably
cortistatin A (115), which has highly selective and potent activity
against HUVEC cells (IC50 ¼ 1.8 nM).
The Baran synthesis of cortistatin A 115 commenced with
readily availabile prednisone (116), which was converted in five
steps and 26% overall yield to 117 (Scheme 19).59 Mukaiyama
hydration of the olefin with Co(acac)2 and PhSiH3, followed by
orthoamide formation and acetate cleavage (65% for the three
operations) gave an intermediate that was subjected to a newly
developed procedure that used in situ-formed acetylhypobromite
for the functionalization of hydrocarbons. Under direction of the
proximal alcohol, the C19 methyl group was dibrominated to
give 118 in 57% yield (with the alcohol being protected as the
TMS ether to prevent intramolecular etherification). Treatment
of 118 with DBU at elevated temperature resulted in enolization
and intramolecular alkylation with the germinal dibromide to
provide bromocyclopropane 119 as a single diastereomer. Ring
expansion mediated by SmI2, followed by treatment with 2,4,4,6-
tetrabromo-2,5-cyclohexadienone (TBCHD), gave 120. Elimi-
nation with LiBr/Li2CO3, removal of the orthoester, and finally
acetylation produced diene 121. Lewis acid-mediated SN20 reac-
tion installed the oxabicylo[3.2.1]octane motif, and removal of
the protecting groups led to 122.
Barton iodination of the ketone 122 produced an intermediate
iodide that was readily coupled with 7-(trimethyl-
stannyl)isoquinoline, leading to 123 in 53% yield (over the two
steps). The synthesis was completed by hydrogenation of the
styryl olefin using RANEY� nickel to give cortistatin A, 115 in
50% yield (at �50% conversion).
The Nicolaou synthesis, which was executed in conjunction
with David Chen at A-Star in Singapore, began with hydrindane
124, a derivative of the Hajos–Parrish ketone (Scheme 19).60
Nine steps converted 124 to alkyne 125, and set the stage for
a key four-step sequence that assembled the polycyclic
Scheme 20 (a) The synthesis of the pentacyclic cortistatin core by
Yamashita & Hirama. Reagents and conditions: (1) cyclohexane-1,3-
dione, piperidine, EtOAc, 87%, d.r. ¼ 5 : 1; (2) HF$pyr, THF, rt; (3) I2,
Ph3P, imid., THF, 87% (2 steps); (4) Et3B, (Me3Si)3SiH, THF, �78 �C.
(b) The key step of the Danishefsky synthesis of the cortistatin core.
Reagents and conditions: (1) TBAF, THF, 88%.
This journal is ª The Royal Society of Chemistry 2010
framework of cortistatin. After Sonagashira coupling of
the alkyne with 3-(triflyloxy)cyclohexenone (125 / 126), the
dithiane was removed under oxidative conditions with IBX, and
reduction of the alkyne with H2 and Pd/BaSO4 gave 127. An
elegant sequence of tandem heteroconjugate addition and aldol
condensation gave 128. After conversion of 128 to 129 (9 steps,
6% overall), functionalization of the cyclohexenone ring
commenced with conjugate epoxidation from the b-face to give
epoxyketone 130. Subsequent reduction with NaBH4/CeCl3 gave
the desired alcohol as a 1 : 1 mixture of separable diastereomers
(the minor could be recycled by quantitative oxidation with
Dess–Martin periodinane), and the synthesis was completed by
Ti(OiPr)4-mediated epoxide opening with Me2NH to yield cor-
tistatin A, 115 in 45% yield (unoptimized).
The departure point for the Shair synthesis was the Hajos–
Parrish ketone 131, which was advanced to 132 by a five-step
sequence of standard transformations (Scheme 19).61 Removal of
Scheme 21 (a) Sarpong’s synthesis of the pentacyclic cortistatin core.
Reagents and conditions: (1) 10–20 mol% PtCl2, PhH, rt / 40 �C; (2)
TsNHNH2, Et3N, 1,2-DCE, 65 �C, 95% (after one recycle); (3)
MgBr2$OEt2, Me2S (20 equiv), CH2Cl2, �78 �C / rt; (4) TESCl, imid.,
DMF; (5) MCPBA, NaHCO3, CH2Cl2, 0 �C, 46% (3 steps); (6) nBuLi,
THF, 0 �C, 1 h; (7) PhI(OAc)2, CH2Cl2–iPrOH–TFE (5 : 3 : 2), 0 �C, 30
min, 60% (2 steps). (b) Corey’s benzylic cyanation and Demjanov rear-
rangement approach to the cortistatin core. Reagents and conditions: (1)
DDQ, TMSCN, LiClO4, CH2Cl2, �10 �C, 95%; (2) LiAlH4, THF, 98%;
(3) NaNO2, AcOH, H2O–THF, 61%.
Nat. Prod. Rep., 2010, 27, 1186–1203 | 1195

Scheme 22 An overview of macrocyclization strategies for largazole, and the syntheses by Luesch and Williams. (a) Solution structure of largazole [as
determined by Phillips and co-workers]. (b) Sites of macrocyclization. (c) Luesch & Hong’s synthesis. Reagents and conditions: (1) Et3N, EtOH, 51%; (2)
TFA, CH2Cl2; (3) 158, DMAP, 94% (2 steps); (4) Boc-L-valine, 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, CH2Cl2, 99%; (5) LiOH, THF–H2O; (6)
TFA, CH2Cl2; (7) HATU, HOAt, DIPEA, CH2Cl2, 64% (3 steps); (8) Grubbs II (30 mol%), PhMe, 41%. (d) Williams’ synthesis. Reagents and conditions:
(1) Fmoc-L-valine, EDCI, DMAP, CH2Cl2; (2) Et2NH, CH3CN; (3) PyBOP, DIPEA, CH2Cl2, 78% (3 steps); (4) TFA, CH2Cl2; (5) HATU, HOBt,
DIPEA, CH2Cl2, 77% (2 steps); (6) iPr3SiH, TFA, CH2Cl2; (7) H3C(CH2)6COCl, Et3N, CH2Cl2, 89% (2 steps).
Scheme 23 (a) The syntheses of largazole by Phillips and Cramer.
Reagents and conditions: (1) Fmoc-L-valine, EDCI, DMAP (Phillips) or
Fmoc-L-valine, DIC, DMAP (Cramer); (2) Et2NH, 62% (2 steps, Phillips)
or piperidine, 93% (2 steps, Cramer); (3) DCC, PFP, 52% (Phillips) or
DCC, DMAP, 97%, (Cramer); (4) TFA, CH2Cl2 (Phillips) or TFA,
Et3SiH, CH2Cl2; (5) PyAOP, MeCN, DMAP, 50% (2 steps, Phillips) or
HATU, DIPEA, THF, 68–78% (2 steps, Cramer); (6) 157, 20% Grubbs
II, 34% (Phillips) or 157, 20% Grela catalyst, 54%. (b) The Ghosh mac-
rolactamization leading directly to largazole. Reagents and conditions: (1)
TFA, CH2Cl2; (2) HATU, HOAt, DIPEA, CH2Cl2, 40% (2 steps).
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
the cyclic ketal with PPTS, followed by intramolecular aldol
reaction, produced 133 in 49% yield. A three-step process of
SOCl2-mediated elimination to the a,b-unsaturated ketone, vinyl
triflate formation and Pd(0)-catalyzed coupling with
Me(OiPr)2SiCH2MgCl provided 134 in 62% overall for the three
steps. Cyclopropanation with dibromocarbene (generated from
CHBr3 with tBuOK as base) gave 135. Warming this compound
in the presence of TASF at 80 �C in DMF produced the desired
ring-expanded compound 136 in 66% yield (over 2 steps). Five
further steps were employed to convert 136 to the precursor 137,
which is required for the key tandem Mannich-etherification
process. In this sequence, treatment of the aldehyde 137 with
Me2NH and ZnBr2 in CH3CN at 50 �C resulted in iminium ion
formation, cyclization of the olefin onto the iminium ion, and
trapping of the carbenium ion with the MEM ether. Termination
of this cascade occurred by scission of the MEM ether to give 138
in an excellent 65% yield (over the proceeding three steps). A
further six steps converted 138 into cortistatin A, 115.
Cortistatin also stimulated a number of studies that resulted
in novel and/or concise approaches to the framework of the
cortistatins. Yamashita and Hirama employed a Knoevenagel
reaction of 139 with cyclohexane-1,3-dione in the presence of
piperidine at room temperature (Scheme 20).62 The intermediate
product, 140, underwent electrocyclization to produce tetracycle
141 in 87% yield and with 5 : 1 diastereoselectivity. Removal of
the TBS ether and conversion of the alcohol to the iodide
(141 / 142, 87%) was followed by radical cyclization with Et3B
and (Me3Si)3SiH to give 143 in 78% yield. A similar strategy
1196 | Nat. Prod. Rep., 2010, 27, 1186–1203 This journal is ª The Royal Society of Chemistry 2010

Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
involving formation of the pentacyclic core by an alkylative
dearomatization was published by Danishefsky. In this case, the
key step was the reaction of 144 with TBAF, which proceeded
to give 145 in an excellent 88% yield.63 Danishefsky has also
published a synthesis of the cortistatin core that employs
a novel a,b-unsaturated nitrone–aryne [3 + 2] cycloaddition
followed by N–O bond reduction, elimination and electro-
cyclization.64
Sarpong and co-workers decribed a concise synthesis of the
pentacyclic cortistatin core that employed enyne cyclization
chemistry developed in their group (Scheme 21).65 When the key
substrate 148 (obtained from 146 and 147 in a four-step
sequence) was subjected to PtCl2, the desired product 149 was
obtained in 95% yield. A three-step sequence of (i) selective
reduction of the least hindered double bond with diiimide, (ii)
exchange of the PMB group for a TES group, and (iii)
Scheme 24 The syntheses of trichodermamide by Zakarian and Joulli�e. (a) Z
(2) LiOH, aq. THF, 90%; (3) TBSCl, imid., DMAP, DMF, 90 �C, 96%; (4) Sm
9-BBN, THF then NaOH, H2O2, 92%; (6) Swern oxidation, 99%; (7) (i) N
CH2Cl2–MeOH, 94%; (8) LDA, Me3SiCl, THF; (9) iC5H11ONO, TiCl4, D
Zn(OTf)2, EtSH, NaHCO3, CH2Cl2, 88%; (12) MsCl, pyr, CH2Cl2, 90%; (1
conditions: (1) TFAA, 90% H2O2, Na2HPO4, CH2Cl2, 95%; (2) TsOH, 2,2-D
EtOH, 95%; (5) Dess–Martin reagent, CH2Cl2, 95%; (6) NH2OH$HCl, NaOA
FeCl3$H2O, CH2Cl2, 85%; (9) 1,10-thiocarbonyldiimidazole, PhMe, reflux, 95
(11) K2CO3, MeOH, 72%; (12) TBAF, THF, 85%; (13) MsCl, NEt3, LiCl, C
This journal is ª The Royal Society of Chemistry 2010
epoxidation led to 150. Eliminative opening of the epoxide 150
gave the allylic alcohol, which upon exposure to PhI(OAc)2 was
oxidized to the dienone and resulted in the formation of the
oxabicylo[3.2.1]octene ring, yielding 151 in 60% yield (over two
steps).
Corey has also described preliminary ring-expansion studies
directed toward the cortistatins (Scheme 21).66 Readily avail-
able estrone derivative 152 was treated with DDQ and
TMSCN (at levels great enough to ensure that [TMSCN] >
3.0 M) to yield the benzylic cyanide 153. Reduction to the
primary amine (LAH, 98%, 153 / 154) was followed by
a Demjanov ring expansion using NaNO2 and AcOH to give
155 in 61% yield. Other reports on the cortistatins include
a CD-ring synthesis by intramolecular Michael-aldol reaction
and an intramolecular [4 + 3] cycloaddition approach to
a tetracyclic model system.67,68
akarian: Reagents and conditions: (1) vinylidene carbonate, 165 �C, 64%;
I2, MeOH, THF, 99%; (5) (i) MePPh3Br, KHMDS, THF, 99%, then (ii)
aClO2, NaH2PO4, 2-methyl-2-butene, tBuOH, H2O, then TMSCHN2,
BMP, CH2Cl2, 82% (2 steps); (10) EDCI, DMAP, CH2Cl2, 62%; (11)
3) LiCl, DMF, 74%; (14) aq. HF, THF, 90%. (b) Joulli�e: Reagents and
MP, acetone, 99%; (3) NaBH4, CeCl3, EtOH, �15 �C, 80%; (4) NaBH4,
c, EtOH–H2O then NaOH, 65%; (7) TBDPSCl, imid., CH2Cl2, quant.; (8)
%; then P(OMe)3, MW, 150 �C, 84%; (10) EDCI, 30% pyr, CH2Cl2, 83%;
H2Cl2, 60%.
Nat. Prod. Rep., 2010, 27, 1186–1203 | 1197

Table 1 First total syntheses of marine natural products reported in 2008
Compound Reference Notes
Gung and Omollo81 � 5 steps from known compound� Resolution� Absolute configuration determined
Giddens et al.82 � Pseudopyrinone A: 3 steps from known compound� Pseudopyrinone B: 5 steps from known compound� Biological activity: good potency and selectivity
against parasitic protozoa
Greshock et al.83 � 18 steps from commercially available 6-hydroxyindole
� Biomimetic synthesis� Racemic synthesis
Kumar and Shaw84 � 16 steps from commercially available materials� Non-racemic synthesis� Biological activity: potent cytotoxicity
Ghosh, Kumar and Shashidhar85 � 21 steps from known compound� Non-racemic synthesis� Determined absolute configuration� Biological activity: cytotoxic against 3YI rat normal
fibroblast cells
Skepper et al.86 � 5 steps from pentadecyne� Non-racemic synthesis� Biological activity: significant antifungal
Cordes et al.87 � 7 steps from 2,6-dimethoxybenzaldehyde
Crimmins and Ellis88 � 8 steps from known intermediate used to prepare11-acetoxy-4-deoxtasbestinin D
� Non-racemic synthesis
Sofiyev, Navarro and Trauner89 � 13 steps from known compounds� Biomimetic synthesis� No protecting groups used
1198 | Nat. Prod. Rep., 2010, 27, 1186–1203 This journal is ª The Royal Society of Chemistry 2010
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online

Table 1 (Contd. )
Compound Reference Notes
Seden et al.90 � 16 steps (longest liner sequence) from3-benzoyloxypropanal
� Non-racemic synthesis
Hamel et al.91 � 24 steps (longest liner sequence) from knowncompound
� Non-racemic synthesis� Biological activity: potent in vitro antiproliferative
activity
Niethe, Fischer and Blechert92 � 19 steps (longest linear sequence) from alanine� Non-racemic synthesis� Established absolute configuration of natural
product� Biological activity: activity against malaria causing
plasmodia and some trypanosomes
Hupp and Tepe93 � 14 steps from known compound� Racemic synthesis� Biological activity: cytotoxicity
Xie et al.94 � 19 steps (longest linear sequence) from alanine� Non-racemic synthesis� Established absolute configuration of natural
product� Biological activity: activity against malaria causing
plasmodia and some trypanosomes
Jiang et al.95 � 16 steps (longest linear sequence) fromcommercially available (+)-dehydro-epiandrosterone
� Non-racemic synthesis� Biological activity: cytotoxic against human solid
tumour cell lines
Ard�a et al.96 � 16 steps (longest linear sequence) from L-glutamicacid
� Non-racemic synthesis
This journal is ª The Royal Society of Chemistry 2010 Nat. Prod. Rep., 2010, 27, 1186–1203 | 1199
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online

Table 1 (Contd. )
Compound Reference Notes
Liu, Cui and Nan97 � 15 steps (longest linear sequence) from knowncompound
� Non-racemic synthesis� revision of structure
Lewis, Daniels and Lindsley98 � 3 steps from known compounds� Non-racemic synthesis� Revision of stereochemistry� Biological activity: antileishmanial
Eade et al.99 � 6 steps from known compound� Racemic synthesis� Biomimetic synthesis
Paterson, Razzak and Anderson100 � 17 steps from known compound� Non-racemic synthesis� Biological activity: ornithine decarboxylase
induction inhibitors
Li et al.101 � 11 steps from known compound� Non-racemic synthesis� Biological activity: antifungal
Smith et al.102 � Data for the synthetic material doesn’t match thatreported for each natural product
� 25 steps from known compound� Non-racemic� Potent cytotoxicity
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
7 Largazole and the trichodermamides
Along with cortistatin A, largazole 156 proved to be one of the
hottest targets of the year. In both cases the high level of interest
was underpinned by some very impressive biological activity – in
1200 | Nat. Prod. Rep., 2010, 27, 1186–1203
the case of largazole it was nanomolar GI50 values against
a number of cancer cell lines, with �15-fold differential activity
for transformed vs. non-transformed cells. The structure eluci-
dation by Luesch69 was quickly followed by several total
syntheses, including those of Luesch,70,71 Williams,72 Phillips,73
This journal is ª The Royal Society of Chemistry 2010

Table 2 New total syntheses of marine natural products previously prepared that were reported in 2008
Compound Reference Compound Reference
Aaptamine Larghi et al.103 (�)-Agelastatin A Yoshimitsu et al.104
Aigialomycin D Chrovian et al.105 Amaminol B Jacobs et al.106
(�)-Amathaspiramide F Sakaguchi et al.107 Amphidinolide J Barbazanges et al.108
Attenols A and B Fuwa et al.109 (-)-Brevenal Ebine et al.110
Chondramide C Eggert et al.111 (�)-Cylindricine C Flick et al.112
Dictyopterene A Hohn et al.113 (�)-Dysibetaine Isaacson et al.114
Eicosanoid Kumaraswamy and Padmaja115 Et-743 Fishlock and Williams116
(�)-Flustramines A and C, (�)-flustramide A, and (�)- and(+)-debromoflustramines A
Kawasaki et al.117 (�)-Hirsutene and (�)-1-desoxyhypnophilin
Jiao et al.118
(+)-Isolaurepan Tripathi and Kumar119
Lamellarins O, P, Q and R Fukuda et al.120
Malyngamide U and its 20-epimer Feng et al.121
Manzacidins A and C Oe et al.122
(+)-Monocerin Kwon et al.123
Pachastrissamine (jaspine B) Passiniemi and Koskinen124
(+)-Phorboxazole A Smith III et al.125
Preclathridines A and C, andisonaamines A and C
Alifanov et al.126
(+)-Psymberin (irciniastatin A) Smith III et al.127 Siphonarienal and siphonarienone Lum et al.128
Solandelactones A, B, E and F White et al.129 (+)-Spongistatin 1 Smith III et al.130
(+)-Tedanolide and (+)-13-deoxytedanolide
Dunetz et al.131 (�)-Tridachiahydropyrone Sharma et al.132
Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
and Cramer.74 Further syntheses continued at a steady rate
through 2008, and by the end of year a further three had been
recorded (by Ghosh,75 Ye,76 and Doi77). Perhaps not surprisingly
given the relative simplicity of the target, key strategic questions
revolved around the timing of the thioester incorporation and the
position of macrocyclization. Most groups arrived at over-
lapping solutions, and the position of macrocyclization is shown
in Scheme 22, along with the almanac of key building blocks
employed in the various approaches (157 / 160).
The Luesch synthesis involved the coupling of thiazolyl nitrile
162 and a-methylcysteine 163 to give thiazolylthiazoline 159 in
51% yield. Removal of the Boc carbamate and acylation of the
amine with acid 158 under standard EDCI conditions produced
164 in 94% yield (over 2 steps) and was followed by coupling with
Boc-valine under Yamaguchi mixed anhydride conditions to
yield the precursor to the macrocyclization, 165. Removal of the
methyl ester (LiOH, aq. THF) and the Boc group (TFA) was
followed by HATU-mediated cyclization to give the desired
macrocycle 166 in 64% yield (over 3 steps). The synthesis was
then completed by appending the thioester side chain by cross-
metathesis between 164 and 157 in the presence of 30 mol%
Grubbs 2nd-generation catalyst, to give largazole in 41% yield.
The Williams synthesis involved acylation of alcohol 167 with
Fmoc-valine to produce 168 (Scheme 22). Removal of the Fmoc
carbamate was followed by coupling to acid 169 in the presence
of PyBOP, to give 170 in 78% yield (over 3 steps). Treatment with
TFA in CH2Cl2 removed the Boc and TMSE protecting groups,
and was followed by macrolactamization with HATU to give 171
in 77% yield (over 2 steps). Liberation of the thiol with TFA–
iPr3SiH, followed by acylation with capryloyl chloride, gave
largazole in 89% yield for these two steps.
The Phillips group and the Cramer group arrived at remark-
ably similar syntheses in an independent fashion (Scheme 23).
Readily accessible allylic alcohol 158 was acylated with Fmoc
valine and after removal of the Fmoc group, thiazolylthiazoline
acid 169 could be coupled to the amine to give 174. Acidic
conditions cleaved the acid-sensitive Boc carbamate and tert-
butyl ester, and macrocyclization was readily achieved with
This journal is ª The Royal Society of Chemistry 2010
either PyAOP (Phillips, 50% over 2 steps) or HATU (Cramer,
68–78% over 2 steps). The syntheses were then completed by
cross-metathesis with olefin 157 in the presence of either Grubbs
2nd-generation catalyst 175 (Phillips, 41%) or the Grela catalyst
176 (Cramer, 54%). An alternative macrocyclization substrate
was reported by Ghosh as a part of their total synthesis of lar-
gazole (see also Scheme 23). Treatment of 177 with TFA
removed the tert-butyl ester and Boc carbamate, and the inter-
mediate amino acid salt could by cyclised with HATU/HOAt to
give largazole directly in 40% yield.
Both the Zakarian and Joulli�e groups have completed total
syntheses of the 4H-5,6-dihydro-1,2-oxazine-containing natural
product trichodermamide B, and the Joulli�e group has also
completed trichodermamide A (Scheme 24).78,79 The Zakarian
synthesis is predicated on the formation of the oxazine by
application of an enolate nitrosation followed by a Lewis acid
mediated hetero-Cope rearrangement as the key reaction, and
the synthesis commenced with a Diels–Alder reaction between
179 and vinylidene carbonate to give 180 in 64% yield. Hydro-
lysis of the carbonate was accompanied by a surprising inversion
of the stereochemistry at the b-hydroxy position to give 181, and
after removal of the methoxy groups and protection of the
alcohols as TBS ethers, 181 was obtained. Wittig olefination,
hydroboration–oxidation and conversion to the methyl ester
gave 184 (via 183) and set the stage for the key transformation.
Deprotonation of the ester and conversion to the silylketene
acetal 185 was followed by treatment with isoamyl nitrite in the
presence of TiCl4 to give 186 in an excellent 82% yield over the
two steps. A sequence of seven steps advanced material to 187,
which was readily reacted with aminocoumarin 188 to yield 189.
Removal of the benzylidene acetal with Zn(OTf)2 and EtSH,
followed by mesylation of the less hindered alcohol, gave 190.
SN2 displacement of the mesylate was readily achieved with LiCl
in DMF, and the synthesis was completed by removal of the
TBDPS protecting group with aqueous HF.
The Joulli�e synthesis employed (�)-quinic acid 192 as a start-
ing material, and a 13-step sequence led to 193. Epoxidation with
in situ-formed CF3CO3H and acetonide formation gave 194.
Nat. Prod. Rep., 2010, 27, 1186–1203 | 1201

Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
Reduction of the lactone in two steps was accompanied by
migration of the TBDPS group to the primary alcohol, which
facilitated the protecting group and redox reorganizations
required to arrive at ketone 195. Upon reaction with hydroxyl-
amine, an intermediate oxime was presumably formed that
underwent intramolecular O-alkylation with the epoxide to
provide oxazine 196 as a single diastereomer in 65% yield. A
three-step sequence of TBDPS protection, acetonide cleavage
and Corey–Winter olefination gave 197. Oxidation-state
manipulations over five steps gave 198, and at this juncture the
synthesis could be completed by a sequence that was analogous
to that described by Zakarian.
A large number of other total syntheses of marine natural
products were reported in the review period, and papers
describing first total syntheses are presented in Table 1. New
total syntheses of compounds previously prepared are summa-
rized in Table 2.
8 Acknowledgements
We would like to thank Professor John Blunt and Professor
Murray Munro (University of Canterbury, Christchurch, New
Zealand) for a copy of the 2008 version of the MarinLit data-
base80 which facilitated data collection for this review. Members
of the Morris group are thanked for their assistance in drawing
structures.
9 References
1 J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote andM. R. Prinsep, Nat. Prod. Rep., 2010, 27, 165.
2 Literature searching was performed by using MarinLit, SciFinder,Web of Science, and a variety of publishing house search engineswith combinations of ‘marine’, ‘natural products’, ‘synthesis’, and‘total synthesis’.
3 H.-D. Arndt and M. Riedrich, Angew. Chem., Int. Ed., 2008, 47,4785–4788.
4 E. Delfourne, Anticancer Agents Med. Chem., 2008, 8, 910–916.5 M. Joullie, S. Berritt and E. M. Forbeck, Curr. Opin. Drug Discovery
Dev., 2008, 11, 829–852.6 J. i. Kobayashi, J. Antibiot., 2008, 61, 271–284.7 M. Lachia and C. J. Moody, Nat. Prod. Rep., 2008, 25, 227–253.8 I. Mancini, G. Guella and A. Defant, Mini-Rev. Med. Chem., 2008,
8, 1265–1284.9 K. C. Nicolaou, M. O. Frederick and R. J. Aversa, Angew. Chem.,
Int. Ed., 2008, 47, 7182–7225.10 Y. Usami, H. Ichikawa and M. Arimoto, Int. J. Mol. Sci., 2008, 9,
401–421.11 M. J. Abad Martinez, L. M. B. Del Olmo and P. B. Benito, Stud.
Nat. Prod. Chem., 2008, 35, 101–134.12 D. C. Behenna, J. L. Stockdill and B. M. Stoltz, Angew. Chem., Int.
Ed., 2008, 47, 2365–2386.13 J. Cossy, C. R. Chim., 2008, 11, 1303–1305.14 R. D. Little and G. A. Nishiguchi, Stud. Nat. Prod. Chem., 2008, 35,
3–56.15 B. R. Moser, J. Nat. Prod., 2008, 71, 487–491.16 P. A. Roethle and D. Trauner, Nat. Prod. Rep., 2008, 25, 298–317.17 S. J. Shaw, Mini-Rev. Med. Chem., 2008, 8, 276–284.18 T. Chou, O. Kamo and D. Uemura, Tetrahedron Lett., 1996, 37,
4023–4026.19 D. Uemura, T. Chou, T. Haino, A. Nagatsu, S. Fukuzawa,
S.-z. Zheng and H.-s. Chen, J. Am. Chem. Soc., 1995, 117, 1155–1156.
20 C. E. Stivala and A. Zakarian, J. Am. Chem. Soc., 2008, 130, 3774–3776.
21 S. Nakamura, F. Kikuchi and S. Hashimoto, Angew. Chem., Int. Ed.,2008, 47, 7091–7094.
22 B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2000, 122, 714–715.
1202 | Nat. Prod. Rep., 2010, 27, 1186–1203
23 J. A. McCauley, K. Nagasawa, P. A. Lander, S. G. Mischke,M. A. Semones and Y. Kishi, J. Am. Chem. Soc., 1998, 120, 7647–7648.
24 F. Matsuura, J. Hao, R. Reents and Y. Kishi, Org. Lett., 2006, 8,3327–3330.
25 F. Matsuura, R. Peters, M. Anada, S. S. Harried, J. Hao andY. Kishi, J. Am. Chem. Soc., 2006, 128, 7463–7465.
26 N. Fusetani, T. Sugawara and S. Matsunaga, J. Org. Chem., 1992,57, 3828–3832.
27 M. E. Green, J. C. Rech and P. E. Floreancig, Angew. Chem., Int.Ed., 2008, 47, 7317–7320.
28 P. J. Kocienski, R. Narquizian, P. Raubo, C. Smith and F. T. Boyle,Synlett, 1998, 1432–1434.
29 P. E. Floreancig, Synlett, 2007, 191–203.30 W. Tu and P. E. Floreancig, Org. Lett., 2007, 9, 2389–2392.31 J. I. C. Pedro de Armas, C. G. Francisco, R. Hern�andez,
J. A. Salazar and E. Su�arez, J. Chem. Soc., Perkin Trans. 1, 1989,405–411.
32 H. A. Reichard, J. C. Rieger and G. C. Micalizio, Angew. Chem., Int.Ed., 2008, 47, 7837–7840.
33 A. C. Hart and A. J. Phillips, J. Am. Chem. Soc., 2006, 128, 1094–1095.
34 J. A. Henderson and A. J. Phillips, Angew. Chem., Int. Ed., 2008, 47,8499–8501.
35 I. Navarro, J.-F. o. Basset, S. v. Hebbe, S. M. Major, T. Werner,C. Howsham, J. BraIckow and A. G. M. Barrett, J. Am. Chem.Soc., 2008, 130, 10293–10298.
36 K. J. Hale, M. G. Hummersone, S. Manaviazar and M. Frigerio,Nat. Prod. Rep., 2002, 19, 413–453.
37 D. J. Newman, Anticancer Agents from Natural Products, 2005, 137–150.
38 P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc.Chem. Res., 2008, 41, 40–49.
39 B. M. Trost and G. Dong, Nature, 2008, 456, 485.40 G. E. Keck, M. B. Kraft, A. P. Truong, W. Li, C. C. Sanchez,
N. Kedei, N. E. Lewin and P. M. Blumberg, J. Am. Chem. Soc.,2008, 130, 6660.
41 P. A. Wender, B. A. DeChristopher and A. J. Schrier, J. Am. Chem.Soc., 2008, 130, 6658.
42 A. E. Wright, J. C. Botelho, E. Guzman, D. Harmody, P. Linley,P. J. McCarthy, T. P. Pitts, S. A. Pomponi and J. K. J. Reed,J. Nat. Prod., 2007, 70, 412.
43 W. Youngsaye, J. T. Lowe, F. Pohlki, P. Ralifo and J. S. Panek,Angew. Chem., Int. Ed., 2007, 46, 9211.
44 D. W. Custar, T. P. Zabawa and K. A. Scheidt, J. Am. Chem. Soc.,2008, 130, 804–805.
45 O. A. Ulanovskaya, J. Janjic, M. Suzuki, S. S. Sabharwal,P. T. Schumacker, S. J. Kron and S. A. Kozmin, Nat. Chem. Biol.,2008, 4, 418–424.
46 V. V. Vintonyak, B. Kunze, F. Sasse and M. E. Maier, Chem. Eur. J.,2008, 14, 11132–11140.
47 S. K. Woo, M. S. Kwon and E. Lee, Angew. Chem., Int. Ed., 2008,47, 3242.
48 H. Fuwa, S. Naito, T. Goto and M. Sasaki, Angew. Chem., Int. Ed.,2008, 47, 4737–4739.
49 I. Paterson and N. A. Miller, Chem. Commun., 2008, 4708.50 R. Kartika, T. R. Gruffi and R. E. Taylor, Org. Lett., 2008, 10, 5047.51 F. Guella, F. Dini and F. Pietra, Angew. Chem., Int. Ed., 1999, 38,
1134–1136.52 K. C. Nicolaou, H. Zhang, A. Ortiz and P. Dagneau, Angew. Chem.,
Int. Ed., 2008, 47, 8605–8610.53 K. C. Nicolaou, A. Ortiz and H. Zhang, Angew. Chem., Int. Ed.,
2009, 48, 5648–5652.54 K. C. Nicolaou, H. Zhang and A. Ortiz, Angew. Chem., Int. Ed.,
2009, 48, 5642–5647.55 C. F. Nising and S. Braese, Angew. Chem., Int. Ed., 2008, 47, 9389–
9391.56 S. Aoki, Y. Watanabe, M. Sanagawa, A. Setiawan, N. Kotoku and
M. Kobayashi, J. Am. Chem. Soc., 2006, 128, 3148–3149.57 S. Aoki, Y. Watanabe, D. Tanabe, A. Setiawan, M. Arai and
M. Kobayashi, Tetrahedron Lett., 2007, 48, 4485–4488.58 Y. Watanabe, S. Aoki, D. Tanabe, A. Setiawan and M. Kobayashi,
Tetrahedron, 2007, 63, 4074–4079.59 R. A. Shenvi, C. A. Guerrero, J. Shi, C.-C. Li and P. S. Baran, J. Am.
Chem. Soc., 2008, 130, 7241–7243.
This journal is ª The Royal Society of Chemistry 2010

Dow
nloa
ded
by D
UK
E U
NIV
ER
SIT
Y o
n 01
Oct
ober
201
0Pu
blis
hed
on 2
2 Ju
ne 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
9366
AView Online
60 K. C. Nicolaou, Y.-P. Sun, X.-S. Peng, D. Polet and D. Y. K. Chen,Angew. Chem., Int. Ed., 2008, 47, 7310–7313.
61 H. M. Lee, C. Nieto-Oberhuber and M. D. Shair, J. Am. Chem. Soc.,2008, 130, 16864–16866.
62 S. Yamashita, K. Iso and M. Hirama, Org. Lett., 2008, 10, 3413–3415.
63 M. Dai and S. J. Danishefsky, Tetrahedron Lett., 2008, 49, 6610–6612.
64 M. Dai, Z. Wang and S. J. Danishefsky, Tetrahedron Lett., 2008, 49,6613–6616.
65 E. M. Simmons, A. R. Hardin, X. Guo and R. Sarpong, Angew.Chem., Int. Ed., 2008, 47, 6650–6653.
66 L. Kurti, B. Czako and E. J. Corey, Org. Lett., 2008, 10, 5247–5250.67 N. Kotoku, Y. Sumii, T. Hayashi and M. Kobayashi, Tetrahedron
Lett., 2008, 49, 7078–7081.68 D. T. Craft and B. W. Gung, Tetrahedron Lett., 2008, 49, 5931–5934.69 K. Taori, V. J. Paul and H. Luesch, J. Am. Chem. Soc., 2008, 130,
1806–1807.70 Y. Ying, K. Taori, H. Kim, J. Hong and H. Luesch, J. Am. Chem.
Soc., 2008, 130, 8455–8459.71 Y. Ying, Y. Liu, S. R. Byeon, H. Kim, H. Luesch and J. Hong, Org.
Lett., 2008, 10, 4021–4024.72 A. Bowers, N. West, J. Taunton, S. L. Schreiber, J. E. Bradner and
R. M. Williams, J. Am. Chem. Soc., 2008, 130, 11219–11222.73 C. G. Nasveschuk, D. Ungermannova, X. Liu and A. J. Phillips,
Org. Lett., 2008, 10, 3595–3598.74 T. Seiser, F. Kamena and N. Cramer, Angew. Chem., Int. Ed., 2008,
47, 6483–6485.75 A. K. Ghosh and S. Kulkarni, Org. Lett., 2008, 10, 3907–3909.76 Q. Ren, L. Dai, H. Zhang, W. F. Tan, Z. S. Xu and T. Ye, Synlett,
2008, 2379–2383.77 Y. Numajiri, T. Takahashi, M. Takagi, K. Shin-ya and T. Doi,
Synlett, 2008, 2483–2486.78 C.-D. Lu and A. Zakarian, Angew. Chem., Int. Ed., 2008, 47, 6829–
6831.79 X. Wan and M. M. Joullie, J. Am. Chem. Soc., 2008, 130, 17236–
17237.80 MarinLit Database, Department of Chemistry, University of
Canterbury: http://www.chem.canterbury.ac.nz/marinlit/marinlit.shtml.
81 B. W. Gung and A. O. Omollo, J. Org. Chem., 2008, 73, 1067–1070.82 A. C. Giddens, L. Nielsen, H. I. Boshoff, D. Tasdemir, R. Perozzo,
M. Kaiser, F. Wang, J. C. Sacchettini and B. R. Copp, Tetrahedron,2008, 64, 1242–1249.
83 T. J. Greshock, A. W. Grubbs, P. Jiao, D. T. Wicklow, J. B. Gloerand R. M. Williams, Angew. Chem., Int. Ed., 2008, 47, 3573–3577.
84 V. Kumar and A. K. Shaw, J. Org. Chem., 2008, 73, 7526–7531.85 S. Ghosh, S. U. Kumar and J. Shashidhar, J. Org. Chem., 2008, 73,
1582–1585.86 C. K. Skepper, D. S. Dalisay and T. F. Molinski, Org. Lett., 2008,
10, 5269–5271.87 J. Cordes, C. Wessel, K. Harms and U. Koert, Synthesis, 2008, 2217–
2220.88 M. T. Crimmins and J. M. Ellis, J. Org. Chem., 2008, 73, 1649–1660.89 V. Sofiyev, G. Navarro and D. Trauner, Org. Lett., 2008, 10, 149–
152.90 P. T. Seden, J. P. H. Charmant and C. L. Willis, Org. Lett., 2008, 10,
1637–1640.91 C. Hamel, E. V. Prusov, J. Gertsch, W. B. Schweizer and
K. H. Altmann, Angew. Chem., Int. Ed., 2008, 47, 10081–10085.92 A. Niethe, D. Fischer and S. Blechert, J. Org. Chem., 2008, 73, 3088–
3093.93 C. D. Hupp and J. J. Tepe, Org. Lett., 2008, 10, 3737–3739.94 W. Xie, D. Ding, G. Li and D. Ma, Angew. Chem., Int. Ed., 2008, 47,
2844–2848.95 B. Jiang, H. P. Shi, M. Xu, W. J. Wang and W. S. Zhou,
Tetrahedron, 2008, 64, 9738–9744.
This journal is ª The Royal Society of Chemistry 2010
96 A. Arda, R. G. Soengas, M. I. Nieto, C. Jimenez and J. Rodriguez,Org. Lett., 2008, 10, 2175–2178.
97 S. Liu, Y.-M. Cui and F.-J. Nan, Org. Lett., 2008, 10, 3765–3768.98 J. A. Lewis, R. N. Daniels and C. W. Lindsley, Org. Lett., 2008, 10,
4545–4548.99 S. J. Eade, M. W. Walter, C. Byrne, B. Odell, R. l. Rodriguez,
J. E. Baldwin, R. M. Adlington and J. E. Moses, J. Org. Chem.,2008, 73, 4830–4839.
100 I. Paterson, M. Razzak and E. A. Anderson, Org. Lett., 2008, 10,3295–3298.
101 S. Li, S. Liang, Z. S. Xu and T. Ye, Synlett, 2008, 569–574.102 A. B. Smith, III, M. O. Duffey, K. Basu, S. P. Walsh,
H. W. Suennemann and M. Frohn, J. Am. Chem. Soc., 2008, 130,420.
103 E. L. Larghi, B. V. Obrist and T. S. Kaufman, Tetrahedron, 2008, 64,5236–5245.
104 T. Yoshimitsu, T. Ino and T. Tanaka, Org. Lett., 2008, 10, 5457–5460.
105 C. C. Chrovian, B. Knapp-Reed and J. Montgomery, Org. Lett.,2008, 10, 811–814.
106 W. C. Jacobs and M. Christmann, Synlett, 2008, 247–251.107 K. Sakaguchi, M. Ayabe, Y. Watanabe, T. Okada, K. Kawamura,
T. Shiada and Y. Ohfune, Org. Lett., 2008, 10, 5449–5452.108 M. Barbazanges, C. Meyer and J. Cossy, Org. Lett., 2008, 10, 4489–
4492.109 H. Fuwa and M. Sasaki, Org. Lett., 2008, 10, 2549–2552.110 M. Ebine, H. Fuwa and M. Sasaki, Org. Lett., 2008, 10, 2275–2278.111 U. Eggert, R. Diestel, F. Sasse, R. Jansen, B. Kunze and M. Kalesse,
Angew. Chem., Int. Ed., 2008, 47, 6478–6482.112 A. C. Flick, M. J. A. Caballero and A. Padwa, Org. Lett., 2008, 10,
1871–1874.113 E. Hohn, J. Palecek and J. Pietruszka, Synlett, 2008, 971–974.114 J. Isaacson, M. Loo and Y. Kobayashi, Org. Lett., 2008, 10, 1461–
1463.115 G. Kumaraswamy and M. Padmaja, J. Org. Chem., 2008, 73, 5198–
5201.116 D. Fishlock and R. M. Williams, J. Org. Chem., 2008, 73, 9594–
9600.117 T. Kawasaki, M. Shinada, M. Ohzono, A. Ogawa, R. Terashima and
M. Sakamoto, J. Org. Chem., 2008, 73, 5959–5964.118 L. Jiao, C. Yuan and Z.-X. Yu, J. Am. Chem. Soc., 2008, 130, 4421–
4430.119 D. Tripathi and P. Kumar, Tetrahedron Lett., 2008, 49, 7012–7014.120 T. Fukuda, E. Sudo, K. Shimokawa and M. Iwao, Tetrahedron,
2008, 64, 328–338.121 J.-P. Feng, Z.-F. Shi, Y. Li, J.-T. Zhang, X.-L. Qi, J. Chen and
X.-P. Cao, J. Org. Chem., 2008, 73, 6873–6876.122 K. Oe, T. Shinada and Y. Ohfune, Tetrahedron Lett., 2008, 49, 7426–
7429.123 H. K. Kwon, Y. E. Lee and E. Lee, Org. Lett., 2008, 10, 2995–2996.124 M. Passiniemi and A. M. P. Koskinen, Tetrahedron Lett., 2008, 49,
980–983.125 A. B. Smith, T. M. Razler, J. P. Ciavarri, T. Hirose, T. Ishikawa and
R. M. Meis, J. Org. Chem., 2008, 73, 1192–1200.126 D. S. Ermolat’ev, V. L. Alifanov, V. B. Rybakov, E. V. Babaev and
E. V. Van der Eycken, Synthesis, 2008, 2083–2088.127 A. B. Smith, J. A. Jurica and S. P. Walsh, Org. Lett., 2008, 10, 5625–5628.128 T.-K. Lum, S.-Y. Wang and T.-P. Loh, Org. Lett., 2008, 10, 761–
764.129 J. D. White, C. M. Lincoln, J. Yang, W. H. C. Martin and
D. B. Chan, J. Org. Chem., 2008, 73, 4139–4150.130 A. B. Smith, T. Tomioka, C. A. Risatti, J. B. Sperry and
C. Sfouggatakis, Org. Lett., 2008, 10, 4359–4362.131 J. R. Dunetz, L. D. Julian, J. S. Newcom and W. R. Roush, J. Am.
Chem. Soc., 2008, 130, 16407–16416.132 P. Sharma, N. Griffiths and J. E. Moses, Org. Lett., 2008, 10, 4025–
4027.
Nat. Prod. Rep., 2010, 27, 1186–1203 | 1203