role of a tumor-suppressor gene in the negative control of - national
TRANSCRIPT

Proc. Nati. Acad. Sci. USAVol. 86, pp. 8773-8777, November 1989Cell Biology
Role of a tumor-suppressor gene in the negative control ofanchorage-independent growth of Syrian hamster cells
(antioncogenes/signal transduction/cell-shape control of proliferation/chemical carcinogenesis/oncogenes)
MINORu Koi, CYNTHIA A. AFSHARI, Lois A. ANNAB, AND J. CARL BARRETT*Laboratory of Molecular Carcinogenesis, National Institute of Environmental Health Sciences, National Institutes of Health, P.O. Box 12233, ResearchTriangle Park, NC 27709
Communicated by James A. Miller, August 2, 1989 (received for review May 2, 1989)
ABSTRACT Tumor-suppressor genes control the neoplas-tic phenotype of tumor cells, but the function of these genes innormal cells is unknown. In this report we show that the lossof a tumor-suppressor gene function releases negative controlson the growth of cells in agar. This conclusion is based onobservations of cell hybrids and studies of cell variants thathave retained or lost a tumor-suppressor gene function. Non-tumorigenic cell hybrids between normal Syrian hamster em-bryo cells and a benzolalpyrene-transformed tumor-cell line(BP6T) continued to secrete autocrine and/or paracrinegrowth factors produced by the tumor cells but failed torespond to these factors by growing in agar. Normal diploidcells also failed to grow in agar in response to the growth factorsproduced by the tumor cells. Clonal variants of nontumori-genic, immortal Syrian hamster cell lines were isolated thateither retained (termed supB+) or had lost (termed supB-) theability to suppress tumorigenicity ofBP6T tumor cells after cellhybridization. Neither supB+ nor supB- variants grew in agarunder conditions that allowed efficient growth of the tumorcells. However, supB- cells were reversibly induced to grow inagar with high colony-forming effilciencies in the presence oftumor cell-conditioned medium or by supplementation of themedium with a combination of growth factors. Under the sameconditions, the supBI cells failed to grow in agar. This en-hanced growth-factor responsiveness in agar was used to selectfor supB- variants existing at a low frequency in the supB+population. These two phenotypes, loss of tumor-suppressorfunction and enhanced growth-factor responsiveness in agar,were seen to cosegregate. These results indicate the tumor-suppressor gene function in these cells negatively regulates thegrowth response of cells in agar to mitogenic stimuli. Thisgrowth regulation may depend on cell shape or adhesionbecause supB+ and supB- cells grown attached to plasticresponded similarly to growth factors.
Tumor-suppressor genes are a family of normal cellular genesthat are frequently lost or inactivated in neoplastic cells (1-4).The existence of tumor-suppressor genes is supported byseveral lines of evidence, which include suppression oftumorigenicity in cell hybrids (5-7), studies of genetic pre-disposition to cancer in humans and animals (1, 2, 8, 9), andnonrandom chromosome deletions or losses of heterozygos-ity of specific chromosomal regions in tumors (8-12). Al-though progress in cloning putative tumor-suppressor geneshas been made (13-17), the normal functions of these genesat the cellular or biochemical level remain obscure.Our laboratory has previously shown that the loss of
tumor-suppressor gene function is essential in the multistepprocess of neoplastic progression of Syrian hamster embryo(SHE) cells transformed by chemical carcinogens or byoncogene transfection (18-20). After carcinogen treatment,
immortal cell lines were established, which only after mul-tiple passages became tumorigenic. The long preneoplasticphase of neoplastic progression allowed the dissection ofdifferent stages in this process (18, 19). Like normal cells,early-passage preneoplastic cell lines retained the ability tosuppress tumorigenicity oftumor cells in cell-cell hybrids. Atlater passages, this tumor-suppressor phenotype was lost bysome of the cells in the population. By subcloning the non-tumorigenic parental cell lines, subclonal variants were iso-lated that either retained the ability to suppress tumorigenic-ity (termed supB+) or had lost this ability (termed supB-)(18). The tumor-suppressor phenotype is designated supB+because it is operationally defined by the ability to suppressthe tumorigenicity of a particular tumor cell (BP6T). This cellline represents one of at least three different complementa-tion groups we have shown to exist for tumor suppression ofhamster sarcomas (21).BP6T cells have a non-ras transforming gene (22) and, like
other tumor cells (23-27), secrete autocrine (and/or para-crine) growth factors in their conditioned medium as detectedby the stimulation of rat NRK or mouse AKR cells to growin agar. Normal SHE cells do not produce these growthfactors. In this report, we show that a cellular change asso-ciated with the loss of the BP6T tumor-suppressor genefunction is enhanced responsiveness of supB- cells in agar tothe autocrine growth factors produced by tumor cells and topurified growth factors. Therefore, the function of this tumor-suppressor gene appears to involve the negative regulation ofgrowth-factor action under restrictive growth conditions.
MATERIALS AND METHODSCell Lines. Cell medium and growth conditions have been
described (18, 28). SHE cells are normal diploid cells that arenontumorigenic and senesce after '15 passages (18, 28).BP6T tumor cells were derived from a morphologicallytransformed colony of SHE cells after treatment with thechemical carcinogen benzo[alpyrene. These cells are highlytumorigenic and grow efficiently in soft agar (18, 28). Hybridsbetween normal SHE cells and BP6T tumor cells (SHE xBP6T) have been described (18); these hybrid cells aresuppressed for tumorigenicity and anchorage-independentgrowth (18). Two independent, clonally derived cell lineswere established after treatment of normal SHE cells witheither asbestos (1OW) or diethylstilbestrol (DES4). The cellsbecame tumorigenic after multiple passages-i.e., approxi-mately passage 50 for 1OW cells and passage 70 for DES4cells. Like normal SHE cells, early-passage 1OW and DES4cells retained the ability to suppress the tumorigenicity andanchorage-independent growth of BP6T cells in cell hybrids
Abbreviations: PDGF, platelet-derived growth factor; SHE, Syrianhamster embryo; TGF, transforming growth factor; CFE, colony-forming efficiency; EGF, epidermal growth factor.*To whom reprint requests should be addressed.
8773
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.

Proc. Natl. Acad. Sci. USA 86 (1989)
(18). At later passages, this tumor-suppressor phenotype waslost by some of the cells in the population. When isolated,some subclones retained the ability to suppress the tumori-genicity of BP6T cells in cell hybrids (termed supB+ cells),whereas other subclones had lost this ability (termed supB-).Rat NRK and mouse AKR cells were gifts from A. Jetten,
(National Institute of Environmental Health Sciences).Cell Hybridization. The cells were analyzed for their ability
to suppress the tumorigenicity and anchorage-independentgrowth of the BP6T cell line. The ability to suppress tumor-igenicity was determined by fusion of the indicated cells withBP6T cells that were preselected for resistance to both6-thioguanine and ouabain, which allows selection of hybridsformed between BP6T cells and other cells by growth in
hypoxanthine/aminopterin/thymidine (HAT)/ouabain se-lection medium (18). The number of total hybrids was as-sayed at 24 hr after fusion by growth of the cells on plastictissue-culture dishes in HAT/ouabain selection medium (18).At the same time, the number of hybrid cells capable ofgrowth in agar was assayed by suspension of the cells insemisolid agar containing HAT/ouabain-selection medium.The ratio of hybrid colonies growing in agar to total hybrids(tumor-suppression ratio) is used as a quantitative measure ofthe fraction of cells in a culture at a given passage that retainsthe tumor-suppressor phenotype (18).
Soft Agar Assay and Tumorigenicity. Cells (104-105) were
suspended in 0.3% Difco bactoagar in normal growth mediumcontaining 10% fetal bovine serum and 0.1% bactopeptone.All experiments were performed two to four times, and atleast five replicate dishes per condition were used. Forcertain experiments, the agar cultures were supplementedwith the following growth factors: insulin at 1 /.tg/ml (Sigma);epidermal growth factor (EGF) at 50 ng/ml (culture grade,Collaborative Research); and platelet-derived growth factor(PDGF) at 5 ng/ml (Collaborative Research). The suspendedcells were plated in 0.3% agar in 60-mm dishes above a layerof 0.6% agar containing normal medium with 10% fetalbovine serum and 0.1% bactopeptone. Dishes were incubatedat 370C in 10% CO2 for 10-14 days and then scored for colonygrowth. Colonies >60 gm (>30 cells) in diameter werescored. For tumorigenicity assays, 2 x 106 cells were injecteds.c. into weanling nude mice and observed for up to 120 days.
Preparation of Conditioned Medium. BP6T cells or SHE x
BP6T hybrid cells were grown to near confluence in 150-cm2flasks. The cells were washed three times with phosphate-buffered saline. Thirty milliliters of serum-free medium wasadded, and the cultures were incubated at 37°C for 2 days.The medium was then collected, centrifuged at 8000 X g for3 hr, filtered through a 0.22-gm filter, and stored at 4°C. Inthe agar assays, conditioned medium was added 1:1 withnormal serum-free medium to both the base layer of agar aswell as the top layer of agar containing the cells being assayedfor agar growth.
Subcloning. 1oW (passage 17) or DES4 (passage 60) cellswere subcloned by one of two methods: Colonies growing innormal medium on plastic tissue-culture dishes were isolatedby using cloning cylinders and trypsinization or coloniesgrowing in agar supplemented with BP6T-conditioned me-dium were isolated with a Pasteur pipette. The isolated cloneswere grown in 35-mm dishes, subcultured onto 100-mmdishes, and then tested for tumorigenicity, growth in agarwith and without BP6T-conditioned medium, and the abilityto suppress BP6T cells in cell hybrids.
Autoradiography. To assess the growth of cells on plastic,cells were seeded at 5 x 104 cells per well in 8-well Lab Tekslides (Miles Scientific) and grown for 3 days to confluence.On the fourth day, the medium was removed and replacedwith serum-free medium containing 5% platelet-poor plasmaprepared as described (29) and growth factors. [3H]thymidineat 1 ,ACi/ml (Amersham) was added to each well, and the cells
were fixed for autoradiography 48 hr later. Slides weredipped in NTB 2 autoradiographic emulsion (Kodak), ex-posed for 3 days, and developed. Five hundred to onethousand total nuclei were counted in random fields per well.
RESULTSFor our studies, we used a highly tumorigenic Syrian hamstercell line (BP6T), the tumorigenicity and anchorage-indepen-dent growth of which was suppressed by fusion with normaldiploid SHE cells (18). Thus, the normal cells have a tumor-suppressor gene function for BP6T cells, which we havetermed supB+. Two independently derived, carcinogen-in-duced, immortal cell lines (DES4 and 10W) at early nontu-morigenic passages also suppressed BP6T cells in cell hybrids(18). From the DES4 and 1OW parental cell lines, we previ-ously isolated by random subcloning supB+ and supB- cellvariants that differ 20- to 50-fold in their ability to suppress
BP6T cells in cell hybrids (18).As shown in Table 1, the conditioned medium from BP6T
cells stimulated the growth in agar of rat NRK, mouse AKR,and two Syrian hamster supB- cell lines. Based on thebiological activity with NRK and AKR cells (24-26), theBP6T-conditioned medium appeared to contain autocrinegrowth factors with transforming growth factor (TGF) a-likeand P-like activities. Conditioned medium from a nontumor-igenic hybrid between BP6T cells and SHE cells (BP6T X
SHE) stimulated the growth in agar of AKR, NRK,DES4supB-, and 1OWsupB- cells but not of the hybrid cellsthemselves (Table 1). BP6T x SHE hybrid cells also failed togrow in agar in the presence or absence of BP6T-conditionedmedium (Table 1). These results indicate that the hybrid cellsproduce autocrine growth factors but fail to respond to thesegrowth factors.The data presented in Table 1 also show that cells lacking
the tumor-suppressor gene function (supB- cells) failed togrow in agar in normal medium containing only 10% fetalbovine-serum but grew in the presence of BP6T-conditionedmedium. This finding indicates that supB- cells in agar canrespond to the autocrine growth factors produced by thetumor cells. Next, we addressed whether normal SHE cellsand other immortal cell lines (particularly supB+ cells) wouldrespond similarly. As shown in Table 2, normal SHE cells didnot grow significantly in agar in response to BP6T-condi-tioned medium. DES4 and 1OW cells were also examined at
Table 1. Suppressed cell hybrids produce but fail to respond toautocrine growth factors
CFE in agar, %
SHE x BP6TBP6T tumor hybrid-
Normal cell-conditioned conditionedCells assayed medium medium medium
NRK <0.01 36 34AKR <0.01 33 311OW(supB-)2 <0.01 33 NDDES4(supB-)1 <0.01 31 16SHE x BP6T hybrid 0.04 0.02 0.03BP6T 78 80 ND
Suppressed SHE x BP6T cell hybrids were selected as described(18), and conditioned medium was prepared from the parental BP6Ttumor cells and the suppressed SHE x BP6T hybrid cells asdescribed. Cells (104 per 60 mm) were grown in 0.3% agar in eithernormal medium containing 10% fetal bovine serum and 0.1% bac-topeptone (18) or a 1:1 mixture with normal medium and serum-freemedium conditioned by the indicated cells, and colonies in agar werescored after 2 weeks. The % colony-forming efficiency (CFE) is thenumber of colonies formed divided by the number of cells plated x100%. ND, not done.
8774 Cell Biology: Koi et al.

Proc. Natl. Acad. Sci. USA 86 (1989) 8775
Table 2. Correlation of tumor-suppression ability with growthresponsiveness in agar to tumor cell-conditioned medium
CFE in agar, % TumorNormal BP6T tumor cell- suppression
Cell medium conditioned medium ratio
Normal SHE cells <0.001 0.02 0.0011OW cell line
Passage 6 <0.001 <0.03 0.003Passage 17 <0.001 28 0.80
DES4 cell linePassage 35 <0.001 0.3 0.0021Passage 60 <0.001 6.7 0.10
Normal SHE cells were assayed at passage 5. At passage 3, thecells were exposed to chemical carcinogens, and transformed colo-nies (1OW and DES4) were isolated. At the passages examined, thecells were nontumorigenic and failed to grow in agar. The cells wereanalyzed for their ability to suppress tumorigenicity of BP6T cellsand to grow in agar in normal medium or medium supplemented withBP6T-conditioned medium prepared as described in Table 1. Theability to suppress tumorigenicity was determined by fusion of theindicated cells with BP6T cells and selection of the hybrid cells (18).The number of total hybrids was assayed at 24 hr after fusion bygrowth of the cells in hypoxanthine/aminopterin/thymidine (HAT)/ouabain-selective medium on plastic. At the same time the numberof hybrid cells capable of growth in agar was assayed by growth ofthe cells in HAT/oubain-selective medium in agar. The ratio ofhybrid colonies growing in agar to total hybrids (tumor-suppressionratio) is used as a quantitative measure of the fraction of cells in aculture at a given passage that retain or have lost the tumor-suppressive ability for BP6T cells.
different passages, and both failed to grow in agar withnormal growth medium (CFE <0.001%) and were nontu-morigenic at all passages examined. At passage 6, 1OW cellsfailed to grow in agar in the presence of BP6T-conditionedmedium (CFE < 0.03%), but at pasage 17 these cells grewvery efficiently in agar with BP6T-conditioned medium (CFE= 28%). DES4 cells at passage 35 were stimulated to grow inagar with BP6T-conditioned medium to a low level (CFE =0.3%), but at passage 60 DES4 cells were stimulated byBP6T-conditioned medium to grow in agar at a higher level(CFE = 6.7%).The abilities of the cells at different passages to grow in
agar in response to BP6T-conditioned medium were com-pared with their abilities to suppress tumorigenicity of BP6Tcells in cell hybrids. Because an excellent correlation be-tween tumorigenicity and anchorage-independent growthwith Syrian hamster cells has been shown (18, 28), suppres-
sion of growth in agar was used as a quantitative measure oftumor suppression. After fusion of different cells with BP6Tcells, a tumor-suppression ratio was calculated from thefraction of total hybrid colonies on plastic that also grew inagar (18). A low tumor-suppression ratio indicates that thecells retain the tumor-suppressor phenotype (supB+), where-as a ratio of =1.0 indicates that the cells have lost the tumor-suppressor phenotype (supB-). A low tumor-suppressionratio (0.001-0.003) was seen for the normal SHE cells andearly-passage immortal 1OW or DES4 cell lines (Table 2). Atlater passages the tumor-suppression ratio of these originallyclonal cell lines had increased significantly (0.1-0.8). Thetumor-suppression ratios for the 1OW and DES4 cells at differ-ent passages correlated with their responses to growth inagar in the presence of BP6T-conditioned medium (Table 2).To determine the significance of this correlation, we se-
lected clonal variants from the 1OW and DES4 cell lines fortheir ability to form colonies in agar in the presence ofBP6T-conditioned medium. In parallel, colonies growing onplastic dishes were also isolated. The clones isolated bygrowth in agar in the presence of BP6T-conditioned mediumfailed to grow in agar with normal growth medium but grewefficiently in the presence of BP6T-conditioned media (Table3). This result indicates that the growth ofthe cells in agar wascompletely reversible and dependent on the autocrine growthfactors in BP6T-conditioned medium. These cells were stillnontumorigenic when injected into nude mice at 2 x 106 cellsper site. However, when tested for their ability to suppressthe tumorigenicity of BP6T cells in cell hybrids (Table 3),these cells did not have this ability (tumor-suppression ratio,0.95-1.1). In contrast, the clones that were isolated fromcolonies growing on plastic failed to respond to BP6T-conditioned medium in agar and had a significantly lowertumor-suppression ratio (0.03-0.06) (Table 3). Thus, thesupB+ variants were resistant to autocrine growth factorstimulation of growth in agar, whereas the supB- variantsresponded and formed colonies in agar with a high efficiency(CFE = 19-33%). Importantly, when we selected for cellswith one of the two phenotypes (growth factor responsive-ness in agar or tumor suppression), cosegregation of the otherphenotype was seen, strongly suggesting that these twophenotypes are interrelated.The ability of a variety of known growth factors (TGFa,
TGF/3, EGF, PDGF, and insulin), alone or in combination, toreplace BP6T-conditioned medium was examined. No singlegrowth factor alone was as effective as tumor cell-condi-tioned medium in stimulating growth in agar. TGFa weaklystimulated growth in agar of supB- variants (CFE = 1%), but
Table 3. Cosegregation of loss of tumor-suppression ability (supB-) and growth-factor responsiveness in agar
CFE in agar, % Tumor TumorParental Growth condition Normal BP6T tumor cell- suppression suppression
cells Subclone at subcloning medium conditioned medium ratio phenotype
DES4 CT-1 Agar + BP6T CM <0.01 28 0.90 supB-DES4 CT-2 Agar + BP6T CM <0.01 33 1.05 supB-DES4 PL-1 Plastic <0.01 <0.01 0.039 supB+DES4 PL-2 Plastic <0.01 <0.01 0.031 supB+loW CT-1 Agar + BP6T CM 0.05 24 0.95 supB-loW CT-2 Agar + BP6T CM 0.03 30 1.11 supB-loW CT-3 Agar + BP6T CM 0.03 19 1.07 supB-loW PL-1 Plastic <0.01 0.02 0.037 supB+loW PL-2 Plastic <0.01 0.01 0.066 supB+The immortal cell lines (lOW and DES4) were heterogenous for their ability to suppress tumorigenicity of BP6T cells and
to grow in agar in the presence of BP6T-conditioned media (Table 2). Therefore, lOW (passage 17) or DES4 (passage 60)cells were subcloned by one of two methods-colonies growing in agar supplemented with BP6T tumor cell-conditionedmedium (conditionally transformed or CT clones as described in Table 1), or colonies growing in normal medium on plastictissue-culture dishes (PL clones) were isolated. The isolated clones were tested for growth in agar with and withoutBP6T-conditioned medium and their ability to suppress the BP6T cells in cell hybrids, as described in Tables 1 and 2. BP6TCM, BP6T cell-conditioned medium.
Cell Biology: Koi et al.
Proc. Natl. Acad. Sci. USA 86 (1989)
Table 4. Growth factor requirements for conditional growth inagar of supB- cells
CFE in agar, %
loW DES4 loW DES4Addition to agar supB- supB- supBl supB+
None <0.01 <0.01 <0.01 <0.01Insulin <0.01 <0.01 <0.01 <0.01EGF 1.6 <0.01 <0.01 <0.01PDGF 1.0 <0.01 <0.01 <0.01Insulin + PDGF 17 13 <0.01 <0.01EGF + PDGF 5.5 4.7 <0.01 <0.01Insulin + EGF 30 10 <0.01 <0.01Insulin + EGF + PDGF 41 39 <0.01 <0.01
lOW and DES4 are immortal, nontumorigenic SHE cell lines thateither retain (supB+) or have lost (supB-) the ability to suppress thetumorigenicity of BP6T cells in cell hybrids. Cells {103_104) weresuspended in 0.3% agar in normal medium containing 10% fetal calfserum, 0.1% bactopeptone, and designated growth factors as fol-lows: insulin at 1 Ag/ml; EGF at 50 ng/ml; and PDGF at 5 ng/ml.
no increase was seen with TGF8 alone or in combination withTGFa (data not shown). Both EGF and PDGF individuallystimulated growth in agar of 1OWsupB- cells, but the CFEwas <2% compared with >30% with BP6T-conditionedmedium. Next, we tested combinations of growth factors(Table 4), and the most efficient growth in agar of 10WsupB-and DES4supB- cells was seen with three growth factors(EGF, PDGF, and insulin). Under these conditions thesupB- variants formed colonies in agar with a 40% CFE,which is comparable to their CFE on plastic. In contrast,under the same conditions supB+ variants did not grow inagar at a detectable level (CFE < 0.01%). Thus, the supB+variants in agar failed to respond to the mitogenic effects ofmultiple growth factors under conditions where the supB-cells formed colonies with a high frequency (Table 4). Thisstimulation of growth in agar was also reversible (i.e., colo-nies isolated from agar failed to grow in agar unless supple-mented with growth factors), and these clones were nontu-morigenic. When supB + or supB - cells grown on plastic werestimulated with EGF, PDGF, and insulin, no differences in
100 -
80-
.5
C
-0
0IR
60 -
40 -
20 -
0oSHE 1 OWsupB. 1 OWsupB-DES4supB.DES4supB- BP6T
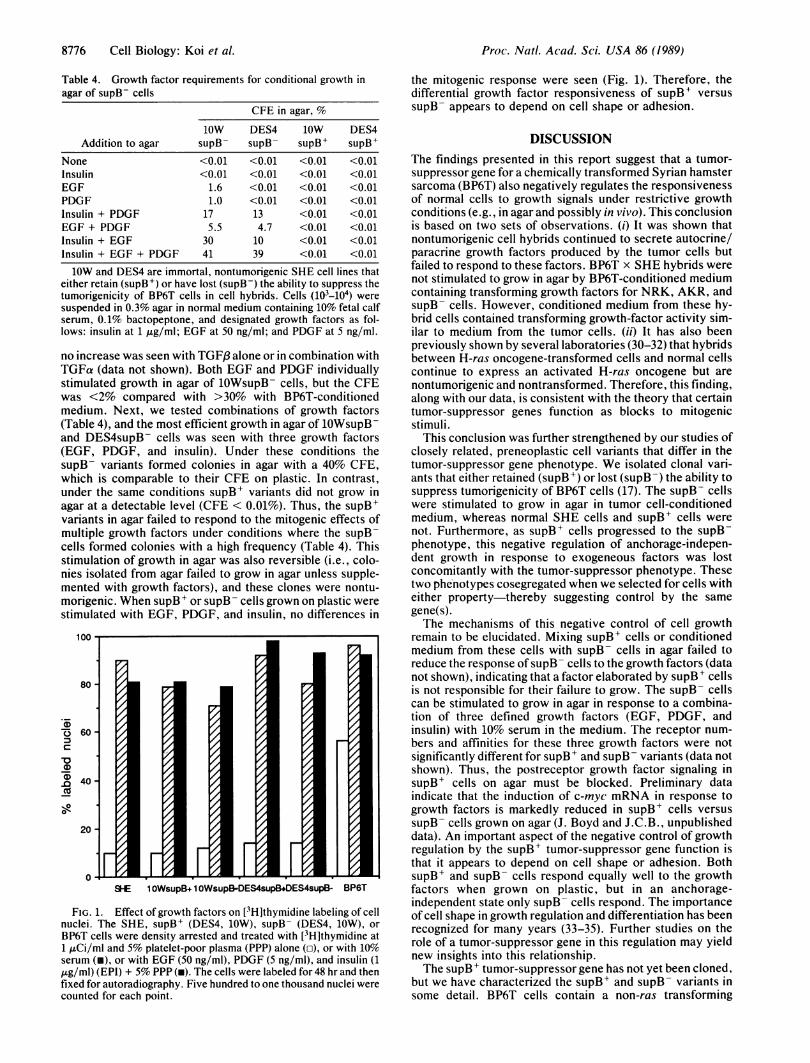
FIG. 1. Effect of growth factors on [3H]thymidine labeling of cellnuclei. The SHE, supB+ (DES4, 10W), supB- (DES4, 10W), orBP6T cells were density arrested and treated with [3H]thymidine at1 ,uCi/ml and 5% platelet-poor plasma (PPP) alone (o), or with 10%serum (i), or with EGF (50 ng/ml), PDGF (5 ng/ml), and insulin (1,ug/ml) (EPI) + 5% PPP (m). The cells were labeled for 48 hr and thenfixed for autoradiography. Five hundred to one thousand nuclei werecounted for each point.
the mitogenic response were seen (Fig. 1). Therefore, thedifferential growth factor responsiveness of supB+ versussupB- appears to depend on cell shape or adhesion.
DISCUSSIONThe findings presented in this report suggest that a tumor-suppressor gene for a chemically transformed Syrian hamstersarcoma (BP6T) also negatively regulates the responsivenessof normal cells to growth signals under restrictive growthconditions (e.g., in agar and possibly in vivo). This conclusionis based on two sets of observations. (i) It was shown thatnontumorigenic cell hybrids continued to secrete autocrine/paracrine growth factors produced by the tumor cells butfailed to respond to these factors. BP6T x SHE hybrids werenot stimulated to grow in agar by BP6T-conditioned mediumcontaining transforming growth factors for NRK, AKR, andsupB- cells. However, conditioned medium from these hy-brid cells contained transforming growth-factor activity sim-ilar to medium from the tumor cells. (ii) It has also beenpreviously shown by several laboratories (30-32) that hybridsbetween H-ras oncogene-transformed cells and normal cellscontinue to express an activated H-ras oncogene but arenontumorigenic and nontransformed. Therefore, this finding,along with our data, is consistent with the theory that certaintumor-suppressor genes function as blocks to mitogenicstimuli.
This conclusion was further strengthened by our studies ofclosely related, preneoplastic cell variants that differ in thetumor-suppressor gene phenotype. We isolated clonal vari-ants that either retained (supB+) or lost (supB-) the ability tosuppress tumorigenicity of BP6T cells (17). The supB- cellswere stimulated to grow in agar in tumor cell-conditionedmedium, whereas normal SHE cells and supB+ cells werenot. Furthermore, as supBI cells progressed to the supB-phenotype, this negative regulation of anchorage-indepen-dent growth in response to exogeneous factors was lostconcomitantly with the tumor-suppressor phenotype. Thesetwo phenotypes cosegregated when we selected for cells witheither property-thereby suggesting control by the samegene(s).The mechanisms of this negative control of cell growth
remain to be elucidated. Mixing supB+ cells or conditionedmedium from these cells with supB- cells in agar failed toreduce the response of supB- cells to the growth factors (datanot shown), indicating that a factor elaborated by supB+ cellsis not responsible for their failure to grow. The supB- cellscan be stimulated to grow in agar in response to a combina-tion of three defined growth factors (EGF, PDGF, andinsulin) with 10% serum in the medium. The receptor num-bers and affinities for these three growth factors were notsignificantly different for supB+ and supB- variants (data notshown). Thus, the postreceptor growth factor signaling insupB+ cells on agar must be blocked. Preliminary dataindicate that the induction of c-myc mRNA in response togrowth factors is markedly reduced in supB+ cells versussupB- cells grown on agar (J. Boyd and J.C.B., unpublisheddata). An important aspect of the negative control of growthregulation by the supB+ tumor-suppressor gene function isthat it appears to depend on cell shape or adhesion. BothsupB+ and supB- cells respond equally well to the growthfactors when grown on plastic, but in an anchorage-independent state only supB- cells respond. The importanceof cell shape in growth regulation and differentiation has beenrecognized for many years (33-35). Further studies on therole of a tumor-suppressor gene in this regulation may yieldnew insights into this relationship.The supB+ tumor-suppressor gene has not yet been cloned,
but we have characterized the supB' and supB- variants insome detail. BP6T cells contain a non-ras transforming
80~ ~ ~ 7-
-
8776 Cell Biology: Koi et A

Proc. Natl. Acad. Sci. USA 86 (1989) 8777
oncogene (22), and BP6T and ras-transformed cells belong todifferent complementation groups for tumor suppression(21). Thus, the supB+ suppressor gene(s) appears to differfrom ras suppressor gene(s). Consistent with this conclusion,the mRNA levels of the K-rev-1, ras suppressor gene (27) arecomparable in supB+ and supB- variants (J. Montgomery,R. W. Wiseman, and J.C.B., unpublished work). Severalgenes are differentially expressed in supB+ and supB- cells,including two differentiation markers (36) and a cytoskeletalprotein, tropomyosin I (37). The supB- cells have a de-creased amount of tropomyosin I, an actin-binding protein,and correspondingly, the actin cables in supB- cells are lessorganized than in supB+ cells (38). It is intriguing to proposethat this change in cytoskeletal organization plays a role inthe growth-factor response differences between supB+ andsupB- cells.
Certain oncogenes may also enhance the responsiveness ofcells to individual growth factors (39-42), but these observa-tions are different from the effects reported here. Oncogenescan replace the action of specific growth factors. Cells withactivated oncogenes (39-45) can proliferate in the absence ofgrowth factors required for normal cells, which generallyrequire multiple growth factors (46-48). In contrast, ourfindings indicate that supB- cells still require multiple growthfactors for efficient growth in agar and that supB+ cells cannotgrow in agar even in the presence of these growth factors.Thus, loss ofthe tumor-suppressor gene function in the supB-cells does not replace the cell's growth factor requirements butreleases cell-shape-dependent restraints to allow the responseof the cells in agar to growth factors.Kaplan and Ozanne (49) reported that subclones of rat
fibroblasts (e.g., F2408 and rat-1 cells) differ in their inherentresponsiveness to growth factor-induced, anchorage-inde-pendent growth. This is similar to our observations withsupB+ and supB- variants. Furthermore, Kaplan andOzanne observed that rat cells resistant to growth factor-induced, anchorage-independent growth are also resistant totransformation by a variety of tumor viruses. Therefore, it ispossible that the rat cell lines described by Kaplan andOzanne also vary in their tumor-suppressor gene activity,which, if correct, would indicate that the differential growthfactor responsiveness of cells in agar is possibly a generalproperty controlled by tumor-suppressor genes.Tumor-suppressor genes are a family of genes that sup-
press the growth of tumor cells. The normal functions ofthese genes are unknown, but it is likely that diverse growthand differentiation pathways are controlled by these genes.Our findings suggest that the tumor-suppressor phenotypelost in the supB- cells is involved in growth-factor respon-siveness of cells in agar. This gene function could directlyinterfere with mitogen-induced signal transduction path-ways, preventing the signal from reaching the nucleus or,alternatively, this gene function may regulate the transcrip-tional or posttranscriptional activation of key mitogen-stimulated genes. The cells described in this report should beextremely useful in understanding the negative regulation ofgrowth-factor responses and the role of tumor-suppressorgenes in modulation of this control.1. Klein, G. (1987) Science 238, 1539-1545.2. Cavenee, W. K., Koufos, A. & Hansen, M. F. (1986) Mutat. Res.
168, 3-143. Sager, R. (1986) Cancer Res. 46, 1573-1580.4. Barrett, J. C. (1987) Cancer Res. 47, 2514-2520.5. Harris, H. (1988) Cancer Res. 48, 3302-3306.6. Stanbridge, E. J., Der, C. J., Doersen, C. J., Nishimi, R. Y., Peehl,
D. M., Weissman, B. E. & Wilkinson, J. (1982) Science 215,252-259.
7. Weissman, B. E., Saxon, P. J., Pasquale, S. R., Jones, G. R.,Geiser, A. G. & Stanbridge, E. J. (1987) Science 236, 175-180.
8. Knudson, A. G., Jr. (1985) Cancer Res. 45, 1437-1443.
9. Cavenee, W. K., Dryja, T. P., Phillips, R. A., Benedict, W. F.,Godbout, R., Gallie, B. L., Murphree, A. L., Strong, L. C. &White, R. L. (1983) Nature (London) 305, 779-784.
10. Murphree, A. L. & Benedict, W. F. (1984) Science 223, 1028-1033.11. Soloman, E., Voss, R., Hall, V., Bodmer, W. F., Jass, J. R.,
Jeffreys, A. J., Lucibello, F. C. & Patel, I. (1987) Nature (London)328, 616-619.
12. Sandberg, A. A. (1987) in Mechanisms of Environmental Carcino-genesis, Role ofGenetic and Epigenetic Changes, ed. Barrett, J. C.(CRC, Boca Raton, FL), Vol. 1, pp. 97-111.
13. Friend, S. H., Bernards, R., Rogelj, S., Weinberg, R. A., Rapaport,J. M., Albert, D. M. & Dryja, T. P. (1986) Nature (London) 328,643-646.
14. Lee, W.-H., Bookstein, R., Hong, F., Young, L.-J., Shew, J.-Y. &Lee, E. Y.-H. P. (1987) Science 235, 1394-1399.
15. Fung, Y.-K. T., Murphree, A. L., T'Ang, A., Qian, J., Hinrichs,S. H. & Benedict, W. F. (1987) Science 236, 1657-1661.
16. Schaefer, R., Iyer, J., Iten, E. & Nirkko, A. C. (1988) Proc. NatIl.Acad. Sci. USA 85,1590-1594.
17. Kitayama, H., Sugimoto, Y., Matsuzaki, T., Ikawa, Y., Noda, M.(1989) Cell 56, 77-84.
18. Koi, M. & Barrett, J. C. (1986) Proc. Natl. Acad. Sci. USA 83,5992-5996.
19. Thomassen, D. G., Gilmer, T. M., Annab, L. A. & Barrett, J. C.(1985) Cancer Res. 45, 726-732.
20. Oshimura, M., Gilmer, T. M. & Barrett, J. C. (1985) Nature (Lon-don) 316, 636-639.
21. Whitehead, R. E., Sugawara, 0. & Barrett, J. C. (1988) Proc. Am.Assoc. Cancer Res. 29, 456 (abstr.).
22. Gilmer, T. M., Annab, L. A. & Barrett, J. C. (1988) Mol. Carcin-ogen. 1, 180-188.
23. Sporn, M. B. & Roberts, A. B. (1985) Nature (London) 313, 745-747.
24. Bowen-Pope, D. F., Vogel, A. & Ross, R. (1984) Proc. Natl. Acad.Sci. USA 81, 2396-2400.
25. Minuto, F., Del Monte, P., Barreca, A., Alama, A., Cariola, G. &Giordano, G. (1988) Cancer Res. 48, 3716-3719.
26. Goustin, A. S., Leof, E. B., Shipley, G. D. & Moses, H. L. (1986)Cancer Res. 46, 1015-1029.
27. Tucker, R. F., Volkenant, M. E., Branum, E. L. & Moses, H. L.(1983) Cancer Res. 43, 1581-1586.
28. Barrett, J. C. & Ts'o, P. 0. P. (1978) Proc. Natl. Acad. Sci. USA75, 3761-3765.
29. Pledger, W. J., Stiles, C. D., Antoniades, H. N. & Scher, C. D.(1977) Proc. Natl. Acad. Sci. USA 74, 4481-4485.
30. Craig, R. & Sager, R. (1985) Proc. Natl. Acad. Sci. USA 82,2062-2066.
31. Geiser, A. C. T., Der, C. J., Marshall, C. J. & Stanbridge, E. J.(1986) Proc. Natl. Acad. Sci. USA 83, 5209-5213.
32. Oshimura, M., Koi, M., Ozawa, N., Sugawara, O., Lamb, P. W. &Barrett, J. C. (1988) Cancer Res. 48, 1623-1632.
33. Folkman, J. & Moscona, A. (1978) Nature (London) 273, 345-349.34. Hay, E. D. (1985) J. Cell. Biochem. 27, 143-156.35. Watt, F. M., Jordan, P. W. & O'Neill, C. H. (1988) Proc. Natl.
Acad. Sci. USA 85, 5576-5580.36. Montgomery, J. C., Hosoi, J., Cizdziel, P. E., Stowers-Hoffman,
J., Wiseman, R. W. & Barrett, J. C. (1989) J. Cell. Biochem. 13B,D234 (abstr.).
37. Wiseman, R. W., Lambert, M. E., Lamb, P. W., Garrels, J. I. &Barrett, J. C. (1988) J. Cell. Biochem. 12A, D325 (abstr.).
38. Boyd, J. & Barrett, J. C. (1989) in Boundaries Between TumorPromotion and Progression During Carcinogenesis, eds. Sudi-lovsky, 0. & Liotta, L. (Plenum, New York), in press.
39. Stem, D. F., Roberts, A. B., Roche, N. S., Sporn, M. B. & Wein-berg, R. A. (1986) Mol. Cell. Biol. 6, 870-877.
40. Sorrentino, V., Drozdoff, V., McKinney, M. D., Zeitz, L. &Fleissner, E. (1986) Proc. Natl. Acad. Sci. USA 83, 8167-8171.
41. Vennstrom, B. & Bravo, R. (1987) Oncogene 1, 271-276.42. Leof, E. B., Proper, J. A. & Moses, H. L. (1987) Mol. Cell. Biol.
7, 2649-2652.43. Armelin, H. A., Armelin, M. C. S., Kelly, J., Stewart, T., Leder,
P., Cochran, B. H. & Stiles, C. D. (1984) Nature (London) 310,655-660.
44. Powers, S., Fisher, P. B. & Pollack, R. (1984) Mol. Cell. Biol. 4,1572-1576.
45. Weissman, B. & Aaronson, S. A. (1985) Mol. Cell. Biol. 5, 3386-3396.
46. Rozengurt, E. (1986) Science 224, 161-166.47. de Asua, L. J., O'Farrell, M. K., Clingan, D. & Rudland, P. R.
(1977) Proc. Natl. Acad. Sci. USA 74, 3845-3899.48. Stiles, C. D. (1983) Cell 33, 653-655.49. Kaplan, P. & Ozanne, B. (1983) Cell 33, 931-938.
Cell Biology: Koi et al.