role of erk1/2 in the crosstalk between the pdgf- and...
TRANSCRIPT

Role of ERK1/2 in the Crosstalk between the PDGF- and Estrogen-Signalling Pathways in
Neonatal Testicular Gonocyte Proliferation
Monty Mazer
Experimental Medicine
McGill University, Montreal
August 2010
A Thesis submitted to McGill University in partial fulfillment of the requirements of the degree
of M.Sc.
© Monty Mazer, 2010

Abstract
Gonocytes are the precursors of spermatogonial stem cells from which spermatozoa
originate. We have shown that neonatal rat gonocytes proliferate in response to the combined
action of PDGF and 17β-estradiol (E2). The xenoestrogens Bisphenol A and genistein, previously
shown to alter the male reproductive system, stimulated gonocyte proliferation through
crosstalk with the PDGF pathway in a manner similar to E2, while testosterone and
progesterone did not affect gonocyte proliferation. Gonocytes expressed Raf1, MEK1, ERK1/2
and PI3K, and proliferated through ERK1/2 activation. PDGF and estrogen induced rapid ERK2
phosphorylation and their combination maintained activated ERK2 for 60 minutes, localized
mainly in the cytosol. E2 induced a rapid increase of estrogen receptor β immunoreactivity in
gonocyte cytosol. PDGF increased a cytosolic PDGFRβ signal, suggesting a role for the variant
V1-PDGFRβ previously identified in gonocytes. These data suggest that PDGF and estrogen are
required to maintain ERK2 activation which mediates gonocyte proliferation, and that estrogen
exerts rapid non-genomic effects in gonocytes.

Resumé
Les gonocytes sont les précurseurs des cellules souche spermatogoniales dont sont issus
les spermatozoa. Nous avons montré que les gonocytes néonataux de rat prolifèrent en
réponse à la combinaison de PDGF et d' estradiol (E2). Les xenoestrogenes Bisphenol A et
genistein, connus pour leurs effets perturbateurs sur le système reproductif masculin,
stimulaient la proliferation des gonocytes par un mécanisme similaire à celui de l' estradiol,
tandis que ni la testosterone ni la progesterone n' avaient d' effet sur la proliferation. Les
gonocytes exprimaient Raf1, MEK1, ERK1/2 et PI3K, et proliferaient via activation de ERk1/2.
PDGF et estrogen induisaient une rapide phosphorylation de ERK2, leur action combinée
maintenant cette phosphorylation pour 60 min, principalement dans le cytosol. E2 induisait
aussi l' augmentation rapide de l'immunoreactivité de l'estrogène receptor β dans le cytosol.
PDGf augmentait le signal cytosolic de PDGFRβ, suggerant un role du variant V1-PDGFRβ,
identifié préalablement dans les gonocytes. Ces résultats suggèrent que le PDGF et l'estrogène
sont nécessaires à la maintenance de l'activation de ERK2, médiateur de la prolifération, et que
l'estrogene exerce des effets rapides et non-génomiques dans les gonocytes.

Acknowledgments
I would like to graciously thank my Master’s supervisor, Dr. Martine Culty for all of her guidance and assistance with my research. Dr. Culty was instrumental in setting up my project and afforded me the tools to plan experiments independently while always providing insights and direction when necessary. Dr. Culty has been incredibly understanding, patient and helpful in providing me an excellent education during my years working in her laboratory, while ensuring that the research lab was a comfortable environment for everyone to be optimally productive. I am especially grateful to her for all the time that she spent helping me with papers, presentations and especially with writing this thesis, and as busy as she might have been, she always found time to meet to discuss results or to help me with anything I needed.
I would also like to acknowledge my coworkers, Annie Boisvert and Gurpreet Manku who have been incredibly helpful in assisting me throughout the course of my studies and in the process; we have also become very good friends. They were always there to help with anything I needed, and were reliable people to turn to for guidance when it was necessary. It was a true pleasure working with them.
As well, I have to thank the rest of the Culty / Papadopoulos labs for all their help and insights throughout. My two years working in the lab were truly enjoyable, and the people who worked with me created a friendly and warm working atmosphere.
The most important thank you must go to my endlessly supportive and helpful family, and especially to my loving wife Daniella, who stood by me through many late nights and weeks with countless hours of work as I balanced numerous things at once in order to finish my thesis. She showed her support every single day and was always understanding of the pressures of a Master’s student. No matter what, she was always there when I needed her and eager to help in any way possible to relieve some of the load. She definitely deserves a vacation as much as I do.
This thesis is dedicated in memory of my grandfather, Sid Mazer ל"ז , who passed away less than two weeks ago. He never stopped encouraging me to reach for the sky, and until the last day that I spoke with him was asking when I would finish my thesis. I have learned so much from him throughout my life, and his determination and his “never give up” attitude are lessons that I have always admired and continue to personify in everything I do.

Abbreviations
- 4OHT: 4-hydroxytamoxifen
- 3βAdiol: 5α Androstane-3β, 17β-diol
- As: Spermatogonia ASingle
- Apr: Spermatogonia Apaired
- Aal: Spermatogonia Aaligned
- AdDP: 4,4’-(1,3-adamantanediyl)diphenol
- AdP: Adamantly substituted phenol (4-(1-anamantyl)phenol)
- AdMP: 2-(1-adamantyl)-4-methylphenol
- AF-(1/2):activation factor 1/2
- AP-1: Activator protein 1
- APP: amyloid precursor peptide
- AR: androgen receptor
- ArKO: aromatase knock out
- BMP: Bone morphogenic protein
- BPA: Bisphenol-A
- cAMP: cyclic AMP (adenosine monophosphate)
- CIS: Carcinoma in situ
- CREB: cAMP response element binding protein
- CV: Cardiovascular
- CYP19: cytochrome P-450 aromatase
- Cyp40: cyclophilin 40
- DAG: 1,2-diacylglycerol
- DBD: DNA binding domain
- DES: Diethylstilbestrol
- DHT: 5α-dihydrotestosterone
- DNA: deoxyribonucleic acid
- dpc: Days post coitum
- E2: 17β-estradiol

- EGF: epidermal growth factor
- ER: estrogen receptor
- ERE: estrogen response element
- ERK: estracellular signal regulated protein kinase
- ERKO: estrogen receptor knock out mouse
- ERR: estrogen related receptor
- FBS: Foetal bovine serum
- FSH: Follicle-stimulating hormone
- GDNF: Glial cell derived neurotrophic factor
- GPCR: G-protein coupled receptor
- Grb2/7: Growth factor receptor bound protein 2/7
- GTPase: enzyme to hydrolyze guanosine triphosphate
- HGF: hepatocyte growth factor
- HSP: Heat shock protein
- ICC: Immunocytochemistry
- IHC: Immunohistochemistry
- IGF-1: Insulin-like growth factor 1
- IGF-1R: Insulin-like growth factor 1 receptor
- Il-1β: interleukin 1β
- IP3: inositol triphosphate
- JAK: Janus Kinase
- JNK: c-jun N-terminal kinase
- kDa: Kilodalton
- KSR: kinase suppressor of ras
- LBD: ligand binding domain
- LH: Luteinizing hormone
- MAPK: Mitogen activated protein kinase
- MAPKK: MAPK kinase (MEK)
- MAPKKK: MAPK kinase kinase (MEK kinase)

- MEK: MAPK/ERK kinase
- mER: membrane-bound estrogen receptor
- MKP: MAPK phosphatase
- MNAR: modulator of non-genomic activity of estrogen receptor
- MP1: MEK partner 1
- MTA1-S: metastatic tumour antigen 1 short form
- NES: nuclear export signal
- NF-κB: Nuclear factor- kappa B
- Ngn3: Neurogenin 3
- NR: nuclear receptor
- Oct-3/4: Octamer-4
- p75NTP: p75 neurotrophin marker
- PDGF: Platelet-derived growth factor
- PDGFR: PDGF receptor
- PDK1: pyruvate dehydrogenase kinase isoenzyme 1
- PDPN: Podoplanin
- PGC: Primordial germ cell
- PI3K: phosphatidylinositol 3-kinase
- PIP2: phosphatidylinositol-4,5-bisphosphate (PI 4,5 P2)
- PIP3: phosphatidylinositol-3,4,5-triphosphate (PtdIns (3,4,5) P3)
- PKA: Protein kinase A
- PKC: Protein kinase C
- PLC: Phospholipase C
- PMC: peritubular myoid cell
- PND: postnatal day
- PP2A: Protein phosphatase 2A
- ptch1: Protein patched homologue 1
- PTP-SL: Phosphotyrosine-specific phosphatase-SL
- RA: Retinoic acid

- Rap1: Ras proximate 1
- RAR-RXR: Retinoic acid receptor – Retinoid X receptor complex
- ROS: reactive oxygen species
- RTK: Receptor Tyrosine kinase
- SAPK: stress activated protein kinase
- SCF: Stem cell factor
- SERM: selective ER modulator
- SH2/3: Src homology 2/3
- SSC: Spermatogonial stem cell
- SOS: Son of sevenless
- Sry: Sex determining region Y gene
- STAT: Signal transducer and activator of transcription protein
- TDS: Testicular dysgenesis syndrome
- VEGF: vascular endothelial growth factor
- TFAP2C: Transcription factor AP-2 gamma
- TGCT: Testicular germ cell tumour
- TGF: transforming growth factor
- TNF-α: tumour necrosis factor α
- V1-PDGFRβ: Variant form of the PDGFRβ

Publications
Thuillier R, Mazer M, Manku G, Boisvert A, Wang Y, Culty, M (2010). Interdependence of platelet-derived growth factor and estrogen-signaling pathways in inducing neonatal rat testicular gonocytes proliferation. Biology of Reproduction, 82(5): 825-836
*A portion of the work for this thesis was published in this paper.

Table of Contents
1. Introduction 1
2. Germ Cells and Foetal Testis Development 1
2.1. Testis Structure 1
2.2. Germ Cell Origin 2
2.3. Foetal Testis Formation and Development 3
2.4. Neonatal Gonocyte Development 5
3. Spermatogenesis 9
3.1. Stem cell Renewal mechanisms 10
3.2. Hormonal Control of Spermatogenesis 13
4. The Study of Gonocytes
4.1. Scientific Models Appropriate for the Study of Neonatal Germ Cells 14
5. Testicular Dysgenesis Syndrome 15
6. Platelet Derived Growth Factor Signalling Pathway
6.1. PDGF signalling molecule 17
6.2. PDGF Receptors 18
6.3. PDGF in Testis Development and Function 21
6.4. Effect of PDGF on gonocytes 23
6.5. V1-Variant form of PDGFRβ 24
6.6. Pathologies Involving the PDGF Signalling Pathway 24
7. Extracellular-Stimulated Downstream Signalling Pathways 25
7.1. Mitogen Activated Protein Kinase Pathway 25
7.2. Phosphatidylinositol 3-Kinase 28
8. Estrogen
8.1. Endogenous Estrogens 29
8.2. Exogenous Estrogens 31
8.3. Estrogen Receptors
8.3.1. Genomic Function of the Estrogen Receptor 32
8.3.2. Estrogen Receptor Structure 33

8.3.3. Non-Genomic effects of ERs 34
8.4. Reproductive Effects of Estrogens
8.4.1. Estrogen signalling in Females 34
8.4.2. Estrogen Signalling in Male Reproductive System 35
8.5. Estrogen in Male Reproductive Development 36
8.6. The Estrogen Hypothesis 38
8.7. Approaches Used to Study the Role of Estrogen/ERs in Males
8.7.1. Laboratory results of estrogen exposure in vivo 40
8.7.2. Knockout Mice 42
9. Cell Signalling Cross Talk Mechanisms 45
9.1. Intercommunication of separate downstream pathways 46
9.2. Crosstalk between Estrogen Receptor and Growth
Factor Receptors / MAPK Pathway 47
10. Summary 51
11. Materials and Methods
11.1. Gonocyte Isolation 53
11.2. Cell Culture for Short Term Molecular Profile 56
11.3. PDGF-depleted FBS 57
11.4. Protein Analysis – Western Blot 57
11.5. Nuclear Isolation 59
11.6. Immunocytochemistry 59
11.7. Proliferation Assay 60
11.8. Immunohistochemistry 61
11.9. V1-PDGFRβ Vector Transfection and Live Cell Imaging 61
12. Results
12.1. Charcoal Stripped FBS and PDGF-Depleted Serum 63
12.2. Gonocyte Expression of Downstream Molecules
of the PDGF Signalling Pathway 63
12.3. In Vitro Exposure to Xenoestrogens and Phytoestrogens

Induce Proliferation in Neonatal Gonocytes 65
12.4. Treatment of Gonocytes with Other Steroid Hormones 68
12.5. ERK2 Activation via PDGF-BB and 17β-estradiol 72
12.6. PDGF and Estrogen Increase Expression of PDGFRβ
and ERβ Immunoreactivity In Vitro 77
12.7. Live Cell Imaging of Gonocytes Transfected with an
EGFP-V1-PDGFRβ Construct, Preliminarly Observations 78
13. Discussion 90
14. Conclusion 97

1. Introduction
Spermatogenesis is the male germ cell pathway necessary for procreation and
regeneration of the species. For viable fertilization to occur in any species, a high percentage of
healthy haploid gametes must be produced by the reproductive center of the organism. This
pathway in mammals is comprised of numerous cell divisions, regulatory mechanisms, positive
and negative feedback, and a host of other processes, all necessary for successful production of
fertile spermatozoa. While every step of germ cell progression between fertilization of a new
zygote and the organism’s subsequent production of sperm at the onset of puberty is crucial,
many of the intermediate stages are rarely discussed and not often investigated. Of particular
note, many scientists overlook the entire gonocyte stage. Gonocytes are cells that differentiate
from primordial germ cells and, through multiple mitotic phases and a lengthy quiescent
period, progress to spermatogonial stem cells, the first cells of the spermatogenetic pathway.
More detailed study of this area is necessary, given the evidence that the precursor to
Testicular Germ Cell Tumours (TGCT), Carcinoma in Situ (CIS), originates from this stage of
development.
2. Germ Cells and Foetal Testis Development
2.1 Testis Structure
The male reproductive system is made up of a number of organs which produce and
harbour the cells necessary for reproduction. The male testes, or gonads, are the organs where
spermatogenesis takes place. They are surrounded by a fibrous enveloping capsule and are
split into two separate compartments, the interstitium and the seminiferous tubules. The
interstitium is comprised of the Leydig cells as well as abundant vasculature, lymphatic vessels
and macrophages. The seminiferous tubules are a system of well organized convoluted tubules
which connects at the end to the rete testis. The seminiferous tubules are bound in place by
endothelial cells and are formed by an outer basement membrane covered with peritubular
myoid cells. The inside of the tubules consist of the germ cells surrounded by Sertoli cells.
Peritubular myoid cells (PMCs) are contractile cells which among other functions are used for
motility of the sperm through the tubules to the rete testis. Important for the growth and
1

hormonal regulation of germ cell development, the Leydig cells are the major source of the
androgen testosterone as well as many other hormones in the adult testis (Russell et al, 1990).
Sertoli cells are the somatic cells of the seminiferous tubules, bound to the germ cells by
intercellular gap junctions (Orth and Boehm, 1990) and they are responsible for providing
nutrition and structural support to the germ cells (Gnessi et al, 1995). Sertoli cells align their
nuclei along the basal membrane and extend their cytoplasm and extracellular matrices into
the middle of the tubule in immature testes, and retract to form a lumen in mature testes.
They function in regulating spermatogenesis, secrete liquid to fill the lumen and deliver
nutrition to the germ cells. They cease their mitotic divisions at puberty and Sertoli cell
numbers remain stable throughout the remainder of the man’s life.
2.2 Germ Cell Origin
Germ cells are the only cells of multicellular organisms that can undergo meiosis. They
are the cells that are capable of combining with a germ cell of the opposite sex to create new
organisms of a given species (Alberts et al, 2002). The germ cell lineage in both males and
females before gender specification and differentiation, begin as Primordial Germ Cells (PGCs),
pluripotent cells in the embryonal ectoderm, originating as part of the epiblast (Rouiller-Fabre
et al, 2003). Germ cells are first seen in mice at 7.5 days post coitum (dpc) and are
characterized by their alkaline phosphatase activity, as well as their retention of the
transcription factor Oct-4, a protein originally expressed in all totipotent embryonic cells that
becomes restricted to PGC. They total nearly 100 cells (Ohmura et al, 2004;De Rooij,
1998;Ohbo et al, 2003). It is of interest that there is no direct germ cell lineage from
fertilization. The germ cells originate as normal pluripotent embryonal cells that are induced to
PGC specification under the control of the bone morphogenic proteins BMP-4 and 8b at their
specific location in the extraembryonic region posterior to the primitive streak (Ying et al,
2000;Lawson et al, 1999). This group of cells migrates through the primitive streak towards the
exoderm where some cells remain in the genital ridge as primordial germ cells, while others
continue onwards to form the mesoderm (McLaren, 1999). PGC migration is dependent on
their expression of the receptor c-KIT (Sutton, 2000) a protein also found in a number of
2

embryonic stem cells (Ashman et al, 1991). PGC migration in humans begins at the stalk of the
allantois during the 4th week of gestation until week 5 when they reach the gonad. Testis cord
formation is then initiated over the next two weeks until week 7 (Lambrot et al, 2006). At this
point, the genital ridge, or gonad, is bipotent and can develop into either an ovary or testis. Sex
determination is decided based on the expression of the Sry gene on the Y chromosome in the
somatic cells destined to become foetal Sertoli cells. The expression of the Sry gene begins on
gestational day 10.5 and continues until day 12.5, inducing expression of the transcription
factor Sox9. The expression of both these factors is essential for development of the male
gonad, and failure to express either Sry or Sox9 will default the gonad into forming an ovary
(Basciani et al, 2010). Epigenetic changes as well as the majority of genetic imprinting in the
germ cell lineage takes place around 10.5 dpc. More imprints are set in place prior to birth
(Chuma et al, 2005). The testis begins to be formed on 11.5 dpc in mice and 12.5 dpc in rats
when the PGCs arrive at the genital ridge and are surrounded by Sertoli cells, which are
differentiating from their embryological precursor into the gonadal somatic cells, thereby
creating the seminiferous tubules (Lambrot et al, 2006).
2.3 Foetal Testis Formation and Development
Testis formation requires a tightly regulated and coordinated sequence of proliferation,
migration, apoptosis and differentiation affecting several somatic cell types and germ cells
(Puglianiello et al, 2004). Any changes or abnormalities to this process can lead to infertility or
cancer (Wang and Culty, 2007). By 13 dpc there are approximately 10,000 PGCs in each of the
two forming gonads (Chuma et al, 2005;De Rooij, 1998). Once embedded in the Sertoli cell
matrix, the PGCs are designated as gonocytes, although some authors refer to them as
prespermatogonia, prospermatogonia or postmigratory PGCs (Olaso and Habert, 2000).
Although there are not many morphological differences between PGCs and gonocytes, which
appear so far to present similar gene expression profiles (Culty, 2009;Gaskell et al, 2004) one
functional difference is that gonocytes can only be cultured in vitro in the presence of Sertoli
cells, while PGCs can be cultured with any somatic cell type (De Rooij, 1998). Germinative cells
are generally identified by high levels of alkaline phosphatase activity (Puglianiello et al, 2004).
3

Mesenchymal cells migrate from the mesonephros into the gonad and differentiate into
myoid cells, pericytes and endothelium, which are critical steps in organized testis formation
(Puglianiello et al, 2004). Foetal Leydig cells appear in the interstitium on 12.5 dpc (Schmahl et
al, 2008) and begin to mature starting at 14.5 dpc with the production of testosterone, which
contributes to the development of male sex characteristics. Although mature adult Leydig cells
are stimulated by luteinizing hormone (LH) to produce testosterone, at this point until
approximately 20 dpc, Leydig cells function independent of gonadotropins. There are two
different Leydig cell populations found in the testis interstitial tissue at different developmental
periods. Although both cell types serve similar roles as the main steroidogenic cells of the
testis, they are morphologically and functionally quite different, supporting the idea that these
two types of Leydig cells arise from dissimilar precursor cells lineages. While foetal Leydig cells
reach their functional peak around gestational day 19, adult Leydig cells become fully matured
by 56 days after birth in rodents. Foetal Ledig cells are smaller and more sporadically located in
the interstitium and begin to atrophy and disperse over the first two weeks after birth. At this
point, Leydig stem cells begin to develop and produce progenitor Leydig cells which are spindle
shaped cells expressing a receptor for LH as well as steroidogenic enzyme activity, though they
produce very little testosterone. The cells enlarge and decrease their proliferative abilities until
day 56, when they are immature Leydig cells producing mostly 5α-reduced androgens. A final
proliferation and differentiation step results in mature adult Leydig cells producing testosterone
for testis function (Dong et al, 2007).
Gonocytes undergo two active periods of proliferation separated by a quiescent period
spanning from 17.5 dpc until neonatal day 3. Although in rats and mice the second proliferative
stage takes place postnatally, in humans, gonocyte proliferation and migration all occur in the
gestational period and spermatogonia remain quiescent from birth until pre-puberty. During
the first and second active periods, gonocytes proliferate and simultaneously undergo
apoptosis (Lambrot et al, 2006). Indeed germ cell apoptosis occurs mainly after the first
postnatal week and during the second week, when the cells have differentiated into
spermatogonia (Jahnukainen et al, 2004). Cells that failed to migrate and become
spermatogonia are then eliminated by apoptosis (Tres and Kierszenbaum, 2005). Proliferation
4

in rat gonocytes continues until day 6 when the first spermatogonia can be identified (Boulogne
et al, 2003). Proliferation and apoptosis can regulate the Sertoli cell / gonocyte ratio.
Alternatively, it is proposed that the importance of building this germinitive pool through both
proliferation and apoptosis is to negatively select the abnormally developed gonocytes to
prevent serious fertility problems or defects in future offspring (Olaso and Habert, 2000).
Sertoli cells continue to grow until the 3rd postnatal week, enlarging the diameter and length of
the seminiferous cords (Boulogne et al, 2003).
2.4 Neonatal Gonocyte Development
Beginning on gestational day 17.5 following the first proliferative stage, gonocytes arrest
their mitotic cell cycle at the G0/G1 phase until after birth. Termed “reproliferation”,
gonocytes end their quiescent period and activate proliferation on neonatal day 3 in rats and
around day 1.5 in mice. Simultaneously, gonocytes migrate to the basement membrane in a
process identified by some as prespermatogenesis. To be consistent with their function,
Hilscher uses different nomenclature to describe gonocytes. He coined the terms ‘multiplying’
and ‘transitional-prospermatogonia’ (Hilscher, 1991). Independent of all factors outside of the
testis, gonocytes will continue to proliferate and migrate in vitro in organ culture or coculture
with Sertoli cells. It was first hypothesized by McGuinness and Orth (McGuinness and Orth,
1992), and subsequently proven by studies in many different species and strains of mice, that
migration occurs independently of proliferation, and both can take place independent of the
other process. Studies showed that although most gonocytes proliferated before they
migrated, there were also cells which migrated to the basement membrane prior to
proliferation. Nagano et al. showed that gonocyte migration began in mice on day 18.5 post
coitum and continued without proliferation until neonatal day 1.5. From then on, migrated
cells were seen to proliferate prior to, as well as after migration. These two major events in
prespermatogenesis must be regulated by completely different mechanisms. In order to
migrate, gonocytes extend pseudopods from their cytoplasm to move around the Sertoli cell
matrix. By neonatal day 5 or 6 in rats, all normal gonocytes have reached the basement
membrane (Nagano et al, 2000). Any abnormal gonocytes or poorly formed spermatogonia
5

that either had problems with migration or with other functions will ultimately degenerate and
be eliminated by apoptosis, which is seen at low levels in rat germ cells until postnatal day
(PND) 20 (Basciani et al, 2008;Roosen-Runge and Leik, 1968). As part of this systematic process,
following the proliferation and migratory stages, gonocytes begin to differentiate into more
mature germ cells (Boulogne et al, 2003).
The next logical step in the germ cell lineage is formation of spermatogonial stem cells
by gonocytes, which will then differentiate to give rise to type A spermatogonia as the first
official step of spermatogenesis at the onset of puberty. Although this appears the reasonable
pathway, it is most likely the case that the gonocytes are in fact a heterogeneous population
where a portion of cells are already committed in foetal or early neonatal life to differentiate
directly into spermatogonia of the first spermatogenic wave and not the adult stem cell type.
This fact was supported by the work of Yoshida et al. (2006) who showed a subset of neonatal
gonocytes led to the formation of differentiating spermatogonia negative for the transcription
factor neurogenin 3 (Ngn3), rather than to the generation of Ngn3-positive spermatogonial
stem cells. Although there are currently no specific markers that designate each discreet
subset of gonocytes, the simultaneous investigation of several gene sets has clearly shown that
gonocytes are not all uniform in their profiles and some gonocytes might express proteins more
similar to stem cells, while other are closer to spermatogonia (Culty, 2009;Yoshida et al,
2006;De Rooij, 1998).
In order to investigate the foetal germ cell commitment to spermatogenesis and the
ability for germ cells to populate an infertile environment, three important studies were
conducted that would eventually help mould our understanding of germ cell development and
function. Brinster and Zimmermann showed that transplantation of spermatogonial stem cells
and spermatogonia populations into an infertile mouse induced normal spermatogenesis and
fertile gametes (1994). In another study, Ohta et al. (2004) transplanted foetal gonocytes into
infertile mice to explore when gonocytes become committed to spermatogenesis. They
observed that although the 14.5 dpc gonocytes did produce viable spermatozoa, germ cells
from 12.5 dpc mice did not induce spermatogenesis. This time period correlates well with the
6

Mig
rato
ry (m
ouse
)
Prim
ordi
al G
erm
Cel
lG
onoc
yte/
Pres
perm
atog
onia
SSC/
Sper
mat
ogon
ia
Mou
se(d
ays)
Rat
(day
s)
Hum
an(w
eeks
)
7.5
13.5
1619
813
1821
1.5
Prem
igra
tory
Post
mig
rato
ry
11.5
37
5
Mito
ticQ
uies
cent B I R T H
3
4-5 5
Mito
tic
Mig
rato
ry
(rat
)
Apop
tosis
Apop
tosis
Cord
form
atio
n
820
A s>A
pr>
A al>
A1-
4>
In >
B 10 10
36 Birt
h
Sper
mat
ogen
esis
Gen
erat
ion
of sp
erm
atog
onia
l ste
m ce
lls
TGCT
sCI
S ce
lls
Mito
tic16
8-12
SSC
A pal
e/A d
ark/
B
Qui
esce
nt
P U B E R T Y
30-4
0
11-1
3 ye
ars
7

Figure 1: Early Male Germ cell Timeline. Timeline depicting the majorprocesses in the prespermatogenesis phase of male germ cell developmentuntil the onset of puberty in the human, rat and mouse. The first stagepresented in the diagram is the undifferentiated embryonic primordialgerm cell (PGC), and the last stage is the differentiated type Bspermatogonia. The picture under the timeline, beginning from the left,represents a cross section of a foetal seminiferous cord as it develops intoa seminiferous tubule. PGCs and gonocytes are found in the center of thecord, gonocytes migrate out to the basement membrane and the Sertolicell cytoplasm eventually retracts forming the lumen of the tubule. Onceadjacent to the basement membrane, gonocytes will differentiate intospermatogonial stem cells which will eventually produce sperm whichenters the lumen of the tubules. In the event of an error in one of theregulation mechanisms, gonocytes may not properly migrate anddifferentiate and may fail to be eliminated by apoptosis, leading tocarcinoma in situ (CIS), which can progress to form testicular germ celltumours (TGCT).
8

first proliferation stage of foetal gonocytes, and highlighted a critical functional difference
between PGC and their descendents the gonocytes. It is important to realize though, that germ
cells will only continue to differentiate and eventually undergo meiosis when in direct
communication with Sertoli cells. Finally, Chuma et al. transplanted epiblast cells and PGCs into
infertile mice, and showed that they were able to continue normal development in the
presence of mature Sertoli and Leydig cells to eventually become spermatogonial stem cells
and produce viable spermatozoa (Chuma et al, 2005).
3. Spermatogenesis
There are three major steps in spermatogenesis; proliferation of the germ cells,
separation of genetic material via meiosis, and spermatozoa development (Russell et al, 1990).
There are many different models for how spermatogenesis in rodents occurs, most of which are
quite similar, most often differing in where the germ cells irreversibly differentiate downstream
in the pathway. I will discuss briefly the Huckins and Oakberg’s (Huckins, 1971;Oakberg, 1971)
As model. Gonocyte differentiation, whether or not through extra mitotic divisions, results in
type A spermatogonia at the basement membrane of the seminiferous tubules. These first
spermatogonia are called Asingle (As) and function as the spermatogonial stem cell (SSC). SSCs
can divide to replenish the SSC population by creating two separate cells, which both act as
stem cells, or they can divide into two daughter cells that remain attached together through
intercellular bridges to become Apair (Apr) (De Rooij, 1998). Alternatively, it is possible that the
SSC will differentiate asymmetrically into one stem cell and another cell that will immediately
progress to spermatogenesis (De Rooij, 2001). Under normal conditions, SSCs renew
themselves at a 1:1 ratio with Apr cells. The Apr cells divide into 4, 8, 16 and rarely 32 Aaligned (Aal)
cells. Until this point, the cells are considered undifferentiated spermatogonia (De Rooij, 1998).
The first differentiation occurs as the Aal cells become A1, a process that is regulated by the
active metabolite of vitamin A, retinoic acid (De Rooij, 2001), as well as cyclin D2 (Beumer et al,
2000). The A1 cells subsequently divide six times, becoming A2, A3, A4, A-intermediate cells
and spermatogonia B (De Rooij, 1998). The next division defines the transformation to primary
spermatocytes, called preleptotene. Before meiosis can take place, the cells remain in
9

prophase for three weeks, during which they go through many phases, characterized by
increases in cellular and nuclear size as well as changes in chromatin conformation in
preparation for division. The designations given to the different phases are leptotene,
zygotene, pachytene and diplotene. The first division, meiosis I, creates two secondary
spermatocytes, and meiosis II splits these cells into spermatids (Russell et al, 1990). The final
process, which contains nineteen steps in rats and sixteen in mice, is called spermiogenesis.
The spermatids develop a flagellum, concentrate their nuclear material in the head which is
surrounded by an acrosome, and remove the remaining cytoplasm, creating the spermatozoa.
The spermatozoa are then released into the lumen of the tubules and travel to the rete testis.
They are stored in the epididymis for final maturation (chapter 6). Unlike in the rodent, human
spermatogenesis undergoes fewer divisions and only has one intermediate stage between the
SSC and type B spermatogonia, termed Apale Spermatogonia. In total, a single mouse SSC will go
through approximately 13 divisions yielding 8192 sperm, while human SSCs only go through 4
divisions and therefore yield only 16 sperm (Schlatt, 2010).
3.1 Stem Cell Renewal Mechanisms
Under normal circumstances, the 1:1 ratio of stem cells to Apr spermatogonia is
sufficient to maintain a large stem cell pool. In a case where the stem cell pool had been
depleted due to toxic substances or irradiation, the remaining stem cells are able to begin
renewing themselves at a higher than normal rate until the stem cell pool is returned to
normal. SSCs have been shown to preferentially occupy specific areas of the tubule periphery
which are in proximity of the blood vessels, defining a stem cell “niche” (Yoshida et al, 2007).
The stem cells occupy open areas on the Sertoli matrix, and when there are low numbers of
stem cells, Sertoli cells secrete high amounts of GDNF to stimulate stem cell renewal. It is
generally accepted that spermatogonia Apr and Aal are undifferentiated. The question arises
whether or not they maintain their stem cell capabilities. It has been noted that in some
mutant mice, there were some aligned spermatogonia with odd numbers of cells instead of
even numbers. It is conceivable that this is an emergency mechanism to replenish the stem cell
10

Spermatogonia
Dark
Pale B
Prim
ary
Sper
mat
ocyt
e (4
n)
Seco
ndar
y Sp
erm
atoc
yte
(2n)
Mei
osis
1 Mei
osis
2
SpermatogoniaA s A p
r
A al
A 1 A 2 A 3 A 4
B
Prim
ary
Sper
mat
ocyt
e (4
n)
Seco
ndar
y Sp
erm
atoc
yte
(2n)
Mei
osis
1
Mei
osis
2
Sper
mat
id(1
n)
Sper
mat
ozoa
Undifferentiated
DifferentiatingIn
term
edia
te
11

Figure 2: Spermatogenesis. Representative diagram of thespermatogenic cycle in human and rodents. The diagram shows themain steps of male germ cell development starting at thespermatogonial stem cell that take place throughout life. The cycleincludes 3 phases, the proliferative phase (spermatogonia), meioticphase (spermatocytes) and spermiogenesis (spermatidmetamorphose into spermatozoa). The first spermatogonial stepsare initiated before puberty in rodents. The whole cycle takesaround 50 days in rat and 64 days in human, leading to theformation of 100-200 millions spermatozoa in men and billions inrodents.
12

pool. Once the aligned cells differentiate to A1, the cells are then irreversibly directed towards
becoming spermatozoa (De Rooij, 2001).
3.2 Hormonal Control of Spermatogenesis
Testis function and embryonic development are regulated by the pituitary gonadotropic
hormones luteinizing hormone (LH) and follicle-stimulating hormone (FSH). Both FSH and LH
are secreted in cycles, a number of times each day, followed by a surge of testosterone. LH
stimulates Leydig cell testosterone production, which is secreted in the testes to regulate
spermatogenesis. Testosterone and FSH act indirectly on the germ cells though the Sertoli cells
to trigger spermatogenetic processes. FSH stimulates the Sertoli cells to secrete androgen
binding protein and inhibin which both act directly on the germ cells. All of the necessary
hypothalamic hormones are regulated negatively by testosterone levels (Strauss and Barbieri,
2004). Although the pituitary gonadotropins oversee complete testes development and
function, they cannot precisely regulate the detailed activities of individual cells in all of these
processes. For precise and accurate organization of the developing testis, there is a host of
paracrine and autocrine factors acting on the various cells (Gnessi et al, 1995). Included in
these factors are hepatocyte growth factors (HGFs), transforming growth factors (TGFs),
neurotrophins, and platelet-derived growth factors (PDGFs), which will be discussed later in
depth. HGF is critical for seminiferous cord formation, activating cell proliferation, and
migration (Ricci et al, 2004). Studies have shown that Retinoid acid (RA) has a small
proliferative effect on organ cultures of PND3 rat gonocytes (Livera et al, 2000) and in
gonocyte-Sertoli cell co-cultures (Boulogne et al, 2003), as well as in PND2 mice (Zhou et al,
2008a). However, RA treatment was shown to have the opposite effect on foetal and neonatal
gonocytes co-cultured for 3 to 6 days with a mixture of testicular somatic cells (Boulogne et al,
2003). It appears that retinoids, acting via specific RAR-RXR receptor dimers, affect both
mitosis and apoptosis in neonatal germ cells and these effects differ in function depending on
the conditions and cell types present in the cultures. Alternatively, Retinoic acid plays an active
role in gonocyte differentiation and maturation at the neonatal day 3 active stage in rat (Wang
and Culty, 2007) and in mouse (Zhou et al, 2008a;Zhou et al, 2008b).
13

4. The Study of Gonocytes
4.1 Scientific Models Appropriate for the Study of Neonatal Germ Cells
Investigation of germ cells in general, and more specifically, PGCs, Gonocytes and SSCs,
is extremely difficult for a number of reasons. PGCs and gonocytes are quite fragile in foetal life
and are not easily isolated nor identified due to lack of adequate specific markers. Studying
spermatogonial stem cells has the added difficulty of being one of a host of morphologically
similar cells in the testis. In 1994, Brinster’s lab discovered a functional test for SSCs, but low
cell numbers impairs the ability to study them. In addition, the study of most germ cell
functions require primary culture, as immortalizing germ cells would certainly change their
ability to proliferate and differentiate compared to in vivo (De Rooij, 2001). It is therefore
imperative that there be many different methods of gonocyte study, each of which examining
gonocyte function from a unique angle, enabling researchers to extract invaluable information
about a specific dimension about gonocytes. Of course, none of the methods are perfect, and
all of them must be employed in order to fully understand the big picture of foetal reproductive
development. In vivo studies commonly use transgenic mice or knockout genes to observe
testicular development following manipulation of specific factors or receptors. Studying murine
embryonic exposure to external factors such as estrogenic compounds or various
environmental toxins can also be useful in understanding early reproductive development.
Methods have been devised to study different actions of germ cells in vitro. Feeder cultures
and organ cultures are often used, which allow the tissue to continue its normal foetal and
neonatal growth in medium without serum or external nutrients (Lambrot et al, 2006;Livera et
al, 2006;Olaso and Habert, 2000). Organ culture, or organotypic culture, was developed to
create an in vivo-like environment for the germ cells in vitro (Rouiller-Fabre et al, 2003).
Gonocytes can also be purified and then cocultured with Sertoli cells or grown in medium alone
in order to dilute the testicular hormonal factors which can affect the development of these
cells. Long term gonocyte cultures require somatic cells in order for them to properly survive.
Using organ culture methods or cocultures with different somatic cells has a serious
shortcoming in that one cannot study the direct effects of a treatment of the germ cells without
14

accounting for an indirect effect of the treatment via somatic cells as well as the paracrine
relationship between germ cells and the surrounding somatic cells. Any contact with other cells
means that treatments will not only affect the germ cells, but will cause the somatic cells to
play a role in the reaction as well. Somatic cells are known to regulate germ cell growth by
secreted factors or directly through gap junctions. Alternatively, culturing germ cells alone
means that they are not in their native environment and might not function exactly as they
would in vivo. It is therefore crucial that one carefully decides the purpose of a study, before
choosing a method (Olaso and Habert, 2000).
5. Testicular Dysgenesis Syndrome
Over the past half century, there has been a steady increase in male reproductive
system disorders. These disorders include hypospadia, cryptorchidism, impaired
spermatogenesis, infertility and testicular cancer, all of which fall under the broader title of
testicular dysgenesis syndrome (TDS) (Sharpe and Skakkebaek, 2008). The syndrome
encompassing all of these developmental disorders describes how reproductive disorders are
all interconnected and ultimately can lead to infertility and testicular cancer. There is a direct
correlation between the number and severity of minor disorders and the development of some
type of reproductive cancer. The outcomes of TDS range from very few problems and almost
flawless fertility, to more severe developmental or pathogenic problems. Although there are
genetic abnormalities that can explain many of these disorders, it is generally accepted that the
main cause of the majority of TDS cases is environmentally related. There is a clear correlation
between the risks of TDS and the geographical location and environmental exposures of
pregnant mothers, suggesting that even testicular dysgenesis that is not apparent or that is
asymptomatic until much later in life, very often is predisposed by events during the life of the
developing foetus (Sonne et al, 2008). One of the main proposed causes of TDS is in utero
estrogen exposure which will be discussed in detail in a later section.
The incidence of testicular cancer has doubled since 1960, and germ cell testicular
cancer is the most common cancer in men aged 15 to 35 (Holmes, Jr. et al, 2008). Germ cell
tumours are almost completely curable if detected early and generally have a very positive
15

prognosis, although consequences of intense chemotherapy and radiotherapy can be harmful.
Effects include secondary malignancy and reduced fertility. If detected too late, testicular germ
cell tumours (TGCTs) can be lethal (Hoei-Hansen et al, 2007). Almost all germ cell tumours,
whether they develop into seminomas, or the more aggressive non-seminomas and embryomal
carcinomas, originate as Carcinoma in Situ (CIS), as defined by Skakkebaek in 1972 (Joensen et
al, 2007). Seminomas appear from primordial germ cells or gonocytes, and non-seminomas are
made up of neoplastic tissue, which usually consists of somatic or embryonal tissue (McIntyre
et al, 2005). The non-seminomas contain pluripotent cells which make up a heterogeneous
cancer population. Embryonal carcinomas can differentiate into different types of tissues,
producing a teratoma (Sonne et al, 2008). CIS can remain latent and symptom free for five to
fifteen years before becoming invasive (Hoei-Hansen et al, 2007). CIS cells are bigger than
spermatogonia and have a very large nucleus and well-defined nucleolus reminiscent of
gonocytes (Joensen et al, 2007). CIS testicular tissue has smaller and less developed
seminiferous tubules along with impaired spermatogenesis. It is very clear that CIS contains
distinct morphological similarities to primordial germ cells and gonocytes, and are believed to
originate directly from these lines of spermatogonial precursors in utero (Rajpert-De Meyts E.
and Hoei-Hansen, 2007;Sonne et al, 2008). These CIS cells which are derived from gonocytes or
their predecessor primordial germ cells and spermatogonial precursor cells, are inhibited from
differentiation and therefore always maintain immature germ cell morphology (Horwich et al,
2006). Not only do they have common morphological similarities to foetal/neonatal germ cells,
they have practically identical protein profiles, staining positive for stem cell markers such as c-
KIT, Oct3/4, NANOG, PDPN and TFAP2C (Joensen et al, 2007). Usually CIS cells remain in the
seminiferous tubules until puberty when they start to proliferate. These cells are able to enter
the lumen and can often be detected in the semen (Hoei-Hansen et al, 2007). With the onset of
pubertal proliferation, CIS cells undergo mitosis without differentiation, accumulating further
genetic mutations, en route towards genetic instability. This can eventually lead to invasive
testicular cancer (Sonne et al, 2008).
Given the prevalence of germ cell cancers and the increasing evidence of their gonocyte
origin, there is a growing need for investigative study in the field of foetal and neonatal
16

gonocyte growth and development. Unfortunately, the principal factor impeding CIS research
is that rodents do not appear to express a CIS or TGCT phenotype. For this reason, other
species and cell lines are necessary to complement rat and mouse studies. Nevertheless,
before experimenting on larger animals with more similar developmental patterns as humans,
one must gain a profound understanding of testis development in established model systems.
Ultimately, understanding normal testis developmental progression and mechanisms of
differentiation can lead to a better comprehension of what may go wrong to cause testicular
cancer (Joensen et al, 2007).
6. Platelet Derived Growth Factor Signalling Pathway
6.1 PDGF Signalling Molecule
Platelet derived growth factor (PDGF) is a growth factor that was first discovered in
blood plasma and, as the name suggests, was assumed to have originated in platelets. PDGF
was discovered as a molecule secreted into the plasma that could stimulate the proliferative
effect of fibroblasts. It is now known that PDGF is expressed in a large number of different
tissues and plays a role in many important activities in development (Basciani et al, 2010). The
paracrine activity of growth factors is a critical step in the communication and organization of
developing tissues (Ricci et al, 2004). Until roughly twenty years ago, there were only two
known PDGF isoforms; A and B. More recently C and D isoforms have been identified. Each
PDGF molecule, although expressing various degrees of homology, is transcribed from a unique
gene, on chromosomes 7, 22, 4 and 11 respectively in human. All of the PDGF hormones
contain six exons except for PDGF-D which is coded by seven. PDGF molecules are active in
their dimerized form, and dimerize, immediately after secretion, into homodimers or
heterodimers depending on the degree of expression of each of the molecules in a given tissue.
The only two PDGFs that are known to form heterodimers thus far are the A and B homologues.
Each PDGF contains eight cysteine residues in its polypeptide chain, forming the cysteine knot
which is responsible for intermolecular binding and dimerization. PDGF molecules have specific
sequence homology to another family of growth factors called the vascular endothelial growth
factor (VEGF) family. These two groups of growth factors share a conserved 80 – 90 amino acid
17

sequence which codes for the growth receptor binding domain. The propeptides of PDGF-A
and B are activated by proteolytic cleavage in the endoplasmic reticulum, while the C and D
isoform propeptides are only activated extracellularly. PDGF acts directly on specific receptors
to induce a wide variety of cellular functions such as proliferation, survival, angiogenesis,
migration, cytoskeletal rearrangements, as well as glycosaminoglycan, proteoglycan and
collagen production and secretion. It is also a promoter of tissue remodelling and embryonic
development of the kidneys, brain, lungs, heart and testis. Generally, PDGF molecules act upon
fibroblasts, neurons, endothelial cells and epithelium (Basciani et al, 2010).
6.2 PDGF Receptors
There are two specific PDGF receptors, alpha and beta, which are encoded on human
chromosomes four and five. The PDGF receptor (PDGFR) is a tyrosine kinase receptor
containing five extracellular immunoglobulin repeats, a single transmembrane and
juxtamembrane domain, two tyrosine kinase domains and a C-terminal domain. Binding PDGF
dimers causes receptor dimerization, and each receptor dimer has a unique affinity for specific
ligand isoforms. The αα homodimer binds to PDGF-AA, BB, AB and CC with preferential affinity
for AA and AB, the ββ homodimer binds to PDGF-BB and DD with highest affinity for BB, and the
αβ heterodimer binds to PDGF-AB, BB, CC and DD (Mariani et al, 2002). Upon ligand-binding,
the receptors are activated and autophosphorylated on tyrosine residues opening docking sites
for Src homology 2 (Sh2) and Sh3 domains. Activated PDGFRs can bind to Grb2, Shc, Grb7, Nck
and Crk which activate effectors such as PI3 kinase (PI3K), phospholipase C γ, STATs, Ras and
JAK/STAT. These effectors will trigger various downstream pathways of second messengers
such as; 1,2-diacylglycerol, inositol triphosphate, GTPase and MAP kinase, and downstream
transcription factors such as c-jun and c-fos. Generally, different receptors and ligands are
found to affect different tissues, and often different receptor/ligand patterns can generate
contradictory actions (Basciani et al, 2010). In cases where both types of PDGF receptors in a
single tissue or cell induce opposing actions on cell activity, it is very likely that the α-β
heterodimer will be involved in mediating the response (Mariani et al, 2002). The study of
PDGF receptor knockout mice have shown that PDGF-A is important for the development of
18

Grb
2
PV1
-PDG
FRβ
Ras
Raf
MEK
ERK
Surv
ival
Prol
ifera
tion
PP
PP
PP
PDG
FRβ/
βPD
GFR
α/α
DB
B B
A A
A D
C C
PDG
FRα/
β
C- Jun
C- Fos
JAK
P STAT
PP
STAT
STAT
PP
STAT
PI3K
AKT
Angi
ogen
esis
Mig
ratio
n
Colla
gen
and
GAG
prod
uctio
n
19

Figure 3: PDGF / PDGFR, receptor - ligand induced pathways. Representativediagram depicting the preferential affinity binding of PDGF A, B, C and D to thetwo receptors, alpha and beta. Three of the most common and well knownpathways are described in the schematic, all of which can be, and are found tobe, induced by each of the three PDGFR dimers. JAK/STAT: JAK is recruited toactivated autophosphorylated tyrosine kinase sites on the PDGFR and isphosphorylated itself, changing into its active form. It induces phosphorylationof STAT which dimerizes and enters the nucleus. STAT acts as a transcriptionfactor to promote transcription of genes. PI3K: Activated PDGFR canphosphorylate PI3K and activate it which will then travel downstream, andactivates a host of different molecules, most notably AKT (PKB). MAPK: PDGFRrecruits a Grb-SOS complex and activates it which will localize Ras to the plamamembrane in close proximity to the receptor. Activated Ras will then activatethe classic MAPK downstream pathway of Raf, MEK and ERK. ERK cantranslocate into the nucleus and modulate transcription.Gonocytes have been shown to express a variant form of PDGFRβ called V1-PDGFRβ. Its exact function is not yet known.
20

alveolar smooth muscle and oligodendrocytes, while PDGF-B is necessary for microvascular
pericytes, and kidney messangial cell migration and organization (Basciani et al, 2002). PDGF
function is regulated in the cells via specific mechanisms. SHP-2 binds to a phosphorylated
tyrosine near the C-terminal of PDGFRs and acts as a phosphatase, causing dephosphorylation
of the receptor (Lu et al, 1998). Activated PDGFRs are also often internalized into the cell and
degraded in lysosomes (Dai, 2010).
6.3 PDGF in Testis Development and Function
From early in embryogenesis, beginning at 12.5 dpc, Sertoli cells produce and secrete
PDGF in higher doses than observed in the developing female gonad. PDGF in the Sertoli cells is
negatively regulated by the gonadotropin FSH from the pituitary (Gnessi et al, 1995). Until 17.5
dpc, PDGFRα is only sporadically expressed throughout the testis and PDGFRβ is expressed
mainly in the interstitial tissue. By 18.5 dpc, gonocytes are also expressing PDGFRβ. There is
high expression of PDGF ligands and their receptors until PND 5 when the expression levels
drop drastically (Basciani et al, 2010). In the adult testis, the only significantly expressed PDGF
receptor/ligand combination is the alpha receptor and PDGF-AA isoform, expressed and
secreted primarily by the Leydig cells. Only in mice do Sertoli cells continue to produce PDGF in
adults (Mariani et al, 2002). PDGFRα and β are both found in the prenatal PMCs. In growing
and functional testis, PDGF-BB stimulates contraction of PMCs as well as cell proliferation.
PDGFR-BB also inhibits 5α-reductase and 3β-hydroxysteroid dehydrogenase activity in the
Leydig cells, stimulating increased testosterone production (Basciani et al, 2010). Postnatally,
PDGF is responsible for a number of actions in the testis such as increased testosterone
production, chemotaxis, proliferation and migration in PMCs, and germ cell proliferation and
differentiation. PDGF levels in the human testis are highest during the active foetal periods
between weeks 16 to 20 and significantly decreased during weeks 24 to 28. During the high
expression periods there is proliferation of gonocytes and foetal Leydig cells as well as
migration of the PMCs. At the end of the second trimester gonocytes begin their quiescent
period. PDGF levels again rise in the adult testis, evidence that it is also involved in
21

spermatogenesis. PDGF profiles were especially high in Leydig cell tumours (Basciani et al,
2002).
Testis development requires the communication and activation of different cell types
mediated through PDGF secretion from the Sertoli cells. Essentially Sertoli cells, through PDGF
production, are the conductors orchestrating the entire process of testis cord formation, and
without them, the testis would develop as an unorganized heterogeneous collection of cells.
Stimulation of organ cultures of 11.5 dpc urogenital ridge sections with PDGF-BB induces testis
cord formation through the MAPK and PI3K pathways. Alternatively, PDGF inhibitors block this
effect and repress the formation of seminiferous cords. Mesenchymal cells migrate from the
mesonephros to the gonad and develop into myoid cells, which play an integral role in testis
organization by surrounding the seminiferous cords and secrete contributions to the basal
lamina. Migratory mesonephric cells, myoid cell precursors, which also express p75
neurotrophin marker (p75NTP), are the only gonadal cells expressing PDGFRβ before 12.5 dpc.
PDGF-BB induces mesonephric cell proliferation, migration and chemotaxis through the
formation of cellular lamellipodia, a process that is vital for testis formation (Puglianiello et al,
2004). These cells in vivo migrate towards the testis around 13.5 dpc and enter the
interstitium. During this embryonic stage, PDGF-BB induces proliferation which causes
testicular growth and development. Migration of the testicular cells during development is a
“male specific event” which is regulated by the second messenger PI3K downstream to the
PDGF receptor (Ricci et al, 2004). PDGFRβ knockout mice die prenatally but generally develop
normally until 16-19 dpc. This drastic change appears to be due to defective PDGF-induced
migration of mesenchymal cells to their proper destinations. Without migration, vascular
smooth muscle cells and pericytes cannot develop appropriately (Puglianiello et al, 2004).
PDGFRα is also very important in testis development. Without PDGFRα, adult Leydig cell
production and function is impaired. Studies with PDGFRα KO mice showed that foetal Leydig
cells were practically normal while the adult Leydig cell population was almost non-existent.
This led to a significant decrease in testis size and spermatogenic arrest early in puberty.
Because foetal Leydig cells developed normally, embryonic testosterone levels were relatively
22

normal allowing masculinisation and testicular descent to proceed predictably. The PDGFRα KO
mice also caused a decreased expression in various other genes necessary for Leydig cell
development such as ptch1 (Brennan et al, 2003).
Imatinib mesylate is a tyrosine kinase receptor inhibitor that is used in cancer treatment
to block PDGFR and c-kit, a specific receptor for stem cell factor (SCF), attenuating the
proliferative activity of the tumour cells. C-kit, similar to PDGFR, is very important in testicular
development and is expressed on PMCs, Leydig cells, PGCs and migratory postnatal gonocytes,
while SCF is produced and secreted by the Sertoli cells. This factor is involved in the maturation
of spermatogonia and Leydig cells and migration of the gonocytes to the basement membrane.
While migrating gonocytes express c-kit, they lose the receptor after migration and only
express it again at the onset of spermatogenesis. In vivo treatment of PND 5 mice with imatinib
for three days caused a decrease in migration and proliferation of the gonocytes and decreased
induction of the stem cell pool, increased apoptosis in germ cells, and caused development of
shorter seminiferous tubules and lower testis weight. In adult mice that were treated
neonatally, there are increased levels of LH and FSH as a compensation for testis size and stem
cell count, but spermatozoa production is normal in the treated mice (Nurmio et al,
2007;Nurmio et al, 2008).
6.4 Effect of PDGF on Gonocytes
PDGF is not required for foetal gonocyte proliferation as germ cells do not express the
PDGF receptors before gestational day 13.5. PDGFRβ knockout studies show that in early
embryogenesis Knockout mice maintain a healthy germ cell population at this early stage of
development. It is hypothesized that Sertoli cells regulate the proliferatory and migratory stage
of the postnatal rat gonocytes, and later embryological foetal human gonocytes. It is well
known that PDGF is responsible for similar actions in many developing tissues and therefore it
was proposed that PDGF could be involved in this stage of testes development as well (Basciani
et al, 2008). Our lab has previously shown that rat gonocytes express PDGFRα and PDGFRβ
postnatally. At PND3 gonocytes also express a variant form of PDGFRβ in the cytosol which
contains no ligand binding domains but maintains tyrosine kinsase autophosphorylation
23

domains (Wang and Culty, 2007;Thuillier et al, 2003). Previous data from our lab proved that
PND3 gonocytes are stimulated to proliferate by both PDGF and 17β-estradiol, both of which
are produced and secreted by neonatal Sertoli cells (Li et al, 1997). These experiments were
conducted with pure gonocytes cultures in order to prevent any intercellular communication
with other testicular cell types. The proliferative effect of 17β-estradiol was inhibited by an
antagonist to estrogen receptors (Li et al, 1997). Foetal exposure to various estrogenic
compounds caused up-regulation of the gonocyte expression of PDGF receptor β (Thuillier et al,
2003;Wang and Culty, 2007), further suggesting an interaction between PDGF and estrogen
pathways in gonocytes. Treatment of mice between PND1 and 5 with imatinib confirmed the
involvement of PDGF-BB in gonocyte proliferation that our laboratory had previously described
in rats (Basciani et al, 2008). The inhibition of the PDGFR caused delayed gonocyte maturation
with a decrease in numbers as well. There was also an observed decrease in the migration of
gonocytes to the basal lamina of the tubules. Not only was there reduced proliferation but the
treated gonocytes also experienced an increase in apoptosis (Basciani et al, 2008).
6.5 V1-Variant Form of PDGFRβ
There are many known variant transcripts of the PDGFRs found in normal and cancerous
tissues, although their roles and functions remain puzzling (Mosselman et al, 1996;Mosselman
et al, 1994;Palumbo et al, 2002;Vu et al, 1989;Heinrich et al, 2003). We identified a variant
form of PDGFRβ in PND3 gonocytes named V1-PDGFRβ that is a cytosolic molecule missing part
of the extracellular ligand binding domain. It has active tyrosine kinase activity and is only
expressed at specific stages of testis development. V1-PDGFRβ transcripts are comprised of
intron 6 until exon 23. In the F9 teratocarcinoma cell line, V1-PDGFRβ was seen to play a role in
retinoic acid induced differentiation (Wang and Culty, 2007).
6.6 Pathologies Involving the PDGF Signalling Pathway
Increased PDGFR activity is linked to various pathologies, most importantly cancer.
PDGFs play a role in a number of different cancers including lung, prostate and renal. These
cancers can either use PDGF as an autocrine factor or through paracrine stimulation from other
secretory tissues. Most known gliomas express high levels of PDGF ligands as well as their
24

receptors. Another role for PDGF in cancer is stabilization of the vasculature through pericyte
recruitment, thus playing a role in angiogenesis of tumour blood vessels (Dai, 2010).
7. Extracellular-Stimulated Downstream Signalling Pathways
Extracellular stimulation of cells to elicit a critical change or specific cellular activity can
be initiated through two major pathways. Steroids can enter the cell and bind to their specific
receptor which will then activate one of hundreds of various cellular pathways. Most
extracellular signalling molecules or hormones cannot enter the cell due to their hydrophilic
properties and therefore must affect cellular functions from the outside. Substrate binding to
membrane bound receptors causes a conformational change in the receptor which elicits either
autophosphorylation of the intracellular domains of the receptor or stimulates specific GTPase
activity through G-protein coupled receptors. This enzyme activation recruits other proteins
which will then stimulate a cascade of secondary messengers to be phosphorylated or
dephosphorylated in order to direct a very precise response. Large scale amplification of
second messengers such as cyclic AMP or MAPK allow for strong responses to very minute
extracellular stimulations. Alternatively, scaffolding proteins function to maintain close
proximity between downstream enzymes or distinct communicating pathways to increase the
rate and specificity of the response. The nuclear receptor family response to steroid
stimulation will be discussed in depth in the following sections dealing with estrogenic cellular
activity. We will not be discussing G-protein coupled receptors, although they are equally
important in cell signalling. We will restrict our discussion of downstream pathways to the two
major pathways stimulated by growth factor tyrosine kinase receptors, and more specifically
the two pathways reported in PDGF induced testicular development (Ricci et al, 2004), the
mitogen activated protein kinase (MAPK) pathway and phosphatidylinositol 3-kinase (PI3K)
pathway.
7.1 Mitogen Activated Protein Kinase Pathway
The MAPK pathway is a linear downstream pathway cascade of kinase molecules
ranging from three to five molecules, each of which is phosphorylated by the previous member.
This cascade is involved in proliferation, gene transcription, migration, differentiation,
25

development, learning, survival as well as apoptosis (Robinson and Cobb, 1997). This is a very
highly conserved system of molecules and is present in the majority of eukaryotic cell types in
very high sequence conservation (Schaeffer and Weber, 1999;Kolch, 2000). The MAPK
molecule is the last enzyme in the cascade which carries out the necessary action, and as such
is phosphorylated by MAPK kinase, also called the MAPK/ERK kinase (MEK) which is in turn
phosphorylated by MAPKKK (or MEK kinase) such as Raf (Robinson and Cobb, 1997). MAPKs
are inactivated by the MAPK phosphatases (MKP) (Rumora and Grubisic, 2009). There are four
major known classes of MAPKs; extracellular signal regulated protein kinase (ERK), p38, c-jun N-
terminal kinase (JNK) (also known as stress activated protein kinase (SAPK) (Kim and Choi,
2010)) and BMK, each of which has a number of different isoforms or related pathways. The
ERK pathway is often involved in proliferation and is activated by growth factors (Rumora and
Grubisic, 2009) while JNK and p38 are commonly activated by stress factors such as tumour
necrosis factor α (TNFα), interleukin 1β (Il-1β) or cellular stress (Kim and Choi, 2010). It is
commonly observed that JNK and ERK have opposing actions in a single cell, one promoting
apoptosis and the other functioning in cell survival (Robinson and Cobb, 1997). The first
described MAPK, and the most researched pathway to date is ERK1/2 which is transcribed by
the erk1 and erk2 genes and are 42 and 44 kDa respectively (Shaul and Seger, 2007).
The first step of ERK activation is receptor ligand binding which triggers
autophosphorylation of the receptor on tyrosine residues. Ras, a small G-protein, is activated
and attaches to the receptor on SH2 (phospho-tyrosine) domains (Kim and Choi, 2010). Ras
activation is regulated by the RTK-Grb2-SOS complex coming to the membrane and docking to
the receptor. ERK can induce a negative feedback mechanism by phosphorylating SOS and
disassembling the whole complex (Kolch, 2000). This activation is also regulated by the
scaffolding proteins kinase suppressor of ras (KSR) or MEK partner 1 (MP1) (Kim and Choi,
2010). The specific enzymes that are activated for the phosphorylation of ERK1/2 in the
pathway are Raf and MEK1/2. Ras and Raf are very well known oncogenes, overexpression or
autonomous expression of which is responsible in part for a large number of cancers. Raf is a
cytosolic protein that is recruited to the phospholipid membrane by activated Ras where it is
activated through phosphorylation (Shaul and Seger, 2007;Kolch, 2000). For the process to
26

continue, MEK is required to bind to an active Raf molecule (Robinson and Cobb, 1997). MEKs
are very specific for Raf due to their proline rich domain, unlike all other MAPKKs. Upon
binding, MEKs are subsequently phosphorylated by Raf on two serine residues in the activation
loop (Shaul and Seger, 2007). Without this sequence, MEK cannot bind and therefore cannot
be activated by Raf (Schaeffer and Weber, 1999). MEK has many regulatory domains and
phosphorylation sites and can be controlled through a host of different enzymes including ERK
via a feedback mechanism which will either downregulate or upregulate MEK’s signal. MEK is
deactivated by the serine/threonine phosphatase PP2A. In the final tier of the MAPK cascade,
MEK phosphorylates ERK on a tyrosine and threonine residues in its activation domain. MEK
contains a docking site for ERK, enabling more precise and quick activation. Deactivation of ERK
is through dephosphorylation by PP2A, PTP-SL and MKPs. ERK is a serine / threonine kinase and
will phosphorylate substrates in the cytosol or the nucleus for activation. In the nucleus ERK
often activates the transcription factors Elk1m, c-fos, p53, Ets1/2 and c-jun (Shaul and Seger,
2007).
Both MEK and ERK are capable of translocation into the nucleus at rest, and at higher
frequencies when activated. Unlike ERK, MEK has a nuclear export signal (NES) and is exported
out of the nucleus via the exportin system upon entering. This is hypothesized to function in
activating nuclear-localized ERK molecules, or in recruiting ERK molecules that generally reside
in the nucleus. When inactive, MEK and ERK molecules are bound to docking proteins, ensuring
that they are, for the most part, found in the cytoplasm. When MEK or ERK is activated,
conformational changes occur, releasing them from their docking proteins, allowing for passive
or active (in the case of ERK, only as a homodimer) translocation into the nucleus. ERK is able
to dock itself to nuclear proteins when it is activated. In the nucleus, ERK is able to activate
transcription, proliferation and cell survival, while if activated ERK is recruited to the cell
membrane it can become a pro-apoptotic signal.
Specificity is a very important aspect of extracellular signalling. It is the objective of
rigorous research to understand exactly how this specificity and regulation occurs with such
precision. Additionally, it is important to understand exactly how it is possible that we get a
27

very specific response from the combination of various stimuli, activating pathways that often
can share similar molecules. Many molecules are ubiquitous among different pathways, but can
still be signal-specific in individual cells (Schaeffer and Weber, 1999). In PC12 cells, long-term
ERK phosphorylation led to cell differentiation, while shorter activation times led to
proliferation. Scaffolding proteins are very important as well to allow for proper cascade
activation. Scaffolds are integral in defining the cellular location of the ERK molecules and
forming multi-protein complexes, as well as coupling the MAPK signalling molecules with other
downstream pathways in a form of signalling crosstalk (Shaul and Seger, 2007).
The MAPK pathway is an active component of various pathologies. Its role in cancer, as
discussed above, is very important to our understanding of the disease and is the focus of a
considerable amount of ongoing therapeutic research. In addition, MAPK can function in
neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease. In
Alzheimer’s disease, there is build up of amyloid-β plaque in the brain causing significant
memory loss and mental deterioration. One proposed mechanism is triggered by oxidative
stress forming reactive oxygen species (ROS) which activates JNK and p38. This triggers
neuronal destruction as well as stabilization of amyloid precursor peptide (APP) (Kim and Choi,
2010).
7.2 Phosphatidylinositol 3-Kinase
PI3K is a downstream pathway that also can induce proliferation, migration and
apoptosis. The PI3K pathway is based on the conversion of inositol lipids into signalling
molecules through phosphorylation. PI3K, which is made up of a p85 regulatory subunit and a
p110 catalytic subunit, phosphorylates phosphatidylinositol-4,5-bisphosphate (PI 4,5 P2 (PIP2))
to phosphatidylinositol-3,4,5-triphosphate (PtdIns (3,4,5) P3 (PIP3)) which then binds to either
phosphoinositide dependant protein kinase 1 (PDK1) or Akt via a PH (pleckstrin homology)
domain (domain that binds to triphosphorylated lipid inositols). This pathway can either be
activated by tyrosine kinase receptors or G-protein coupled receptors. There are other classes
of PI3K that activate different pathways as well. When Akt binds to the PtdIns (3,4,5) P3, it is
phosphorylated in its activation domain by PDK and by mTOR complex 2. Akt can now migrate
28

to the rest of the cytosol or the nucleus. There are over 100 substrates that Akt can affect in
the cell (Martelli et al, 2010). In certain cell types, PI3K actually functions as an activator for the
MAPK pathway. In mesangial cell proliferation and migration, PI3K induces MAPK response, and
ERK is turned off when the cells are inhibited by a PI3K inhibitor (Choudhury et al, 1997). As
part of my thesis research, our lab has recently showed that PDGF-induced PND3 gonocyte
proliferation is only through the MAPK pathway and not via the PI3K downstream cascade
(Thuillier et al, 2010). (See Results and discussion sections)
8. Estrogen
8.1 Endogenous Estrogens
Estrogens (oestrogens) are classically known as the “female sex hormones”, as their
function in the female reproductive system well preceded knowledge of their function in other
systems. Estrogens are crucial for the development, maintenance and function of the female
secondary sex characteristics. Found in their highest concentrations in females of reproductive
age, they function in developing breasts and the endometrium, and play a role in regulating the
menstrual cycle. In addition to their sexual functions, estrogens regulate growth, bone
mineralization, brain masculinisation and cardiovascular functions in males as well as females
(Luconi et al, 2002); and they also play a role in the immune system and the central nervous
system (Heldring et al, 2007). In the absence of estrogen, bones cannot mineralize and the
epiphyseal plate cannot close. For this reason, estrogen deficiencies often lead to very tall
stature and continued growth with a high incidence of osteoporosis. Estrogens play a
protective role in the cardiovascular system by inhibiting vascular smooth muscle cells from
proliferating, thereby preventing vascular injury response as well as preventing the
development of atherosclerosis, a disorder due to lipid deposits in the large arteries (Luconi et
al, 2002). Three main types of endogenous estrogens are found in the endocrine system; 17β
estradiol (E2), estrone and estriol, E2 being the strongest and most potent of the three.
Estrogens are steroid hormones, and therefore are derived from cholesterol, a molecule
combining four five-carbon and six-carbon rings. They are secreted as endocrine messengers
and therefore circulate through the bloodstream until reaching their target tissue where they
29

will bind the estrogen receptors (ERs), triggering the expression of specific genes regulated by
estrogen (Heldring et al, 2007). The enzyme aromatase, generally found in the endoplasmic
reticulum of estrogen secreting tissues, catalyzes the irreversible conversion of androgens such
as testosterone to estrogens. Aromatase is an enzymatic complex containing a ubiquitous
reductase and a cytochrome P450 aromatase which harbours a heme and a steroid binding
pocket (Carreau et al, 2003).
What defines a molecule as an estrogen? Originally scientists used to test possible
estrogen candidate molecules by implanting them into the uterus of female rats or rabbits to
see if there was a change in cell proliferation. If growth occurred, this suggested that the
molecule was estrogenic, therefore estrogens were considered uterotropic molecules (Koehler
et al, 2005). Today there are two in vitro methods performed at the molecular level using
either recombinant ERs or gene reporter assays, which are more precise than the latter in
defining estrogenic molecules. These new techniques have provided scientists with a new
means of defining estrogenic molecules. The first method is a competitive binding assay testing
the new compounds for their ability to displace the binding of E2, the most common estrogen,
from ERs. There are two main types of ERs, ERα and ERβ that will be discussed below in greater
detail, and both types of recombinant receptors are used for testing the possible estrogenic
activity of compounds. One can use fluorescently conjugated molecules to view binding of the
unknown chemical compared to E2 to find out how strong their relative affinity is to the
receptors. If they do bind, they are thought to have estrogen-like properties. The second and
more definitive method uses a luciferase-reporter-gene assay. The substance to be tested is
incubated with cells from a cell line that contains ER and an estrogen-dependent luciferase
gene containing an ER binding site on its promoter sequence. If this gene is transcribed in the
presence of the chemical, then the molecule can be considered estrogenic. If the chemical
binds to the ER, but does not induce gene transcription, it is regarded as an antiestrogen
(Gutendorf and Westendorf, 2001).
30

8.2 Exogenous Estrogens
There are many different molecules called xenoestrogens, created by humans or
originating from other natural sources such as plants that are present in the environment that
can bind and affect the activity of ERs, (Nikov et al, 2001). They are usually ingested or
absorbed into the body and are often found in our water sources. Although they act like
estrogens, their chemical structure doesn’t always appear similar (Nadal et al, 2000). These
chemicals are found in nature in a very diverse array of structures and can bind to the ERs with
different affinities, displaying estrogen-like functions (Nikov et al, 2001). Natural or synthetic
exogenous compounds that interact with the normal ERs are called endocrine disrupters and
display either estrogenic or antiestrogenic properties, either functioning similarly to natural
estrogens or inhibiting the transcriptional function of ERs (antiestrogens) (Gutendorf and
Westendorf, 2001). Some xenoestrogens are produced naturally by plants such as soy
(phytoestrogens), while others are synthesized as industrial chemicals, for example; pesticides
and herbicides, which become environmental pollutants. Generally, these molecules will have
a common structural motif containing a phenol attached to a bulky hydrophobic structure. Due
to different structural properties, some of these substances will have higher affinities to the
ligand binding domain (LBD) of the receptors than others. AdDP1 appears to bind more strongly
to both ERs than AdP2, and AdMP3 does not bind to either protein. These molecules have
reduced binding affinity compared with DES4 and 4OHT5
1 4,4’-(1,3-adamantanediyl)diphenol
, although AdDP will bind to ERβ with
higher affinity than 4OHT (Nikov et al, 2001). DES is one of the most potent xenoestrogens that
was used to prevent miscarriages in the 1940’s to 1970’s, until it was associated with birth
defects in males and females alike. Another extremely well-known xenoestrogen is bisphenol-A
(BPA) which is usually found in canned food, plastic bottles and dental sealants (Nadal et al,
2000). A well known ER antagonist is ICI 182780 which is often used in research to examine
whether or not ER binding is required in the specific effect of an estrogenic compound. It
2 Adamantly substituted phenol (4-(1-anamantyl)phenol) 3 2-(1-adamantyl)-4-methylphenol 4 diethylstilbestrol 5 4-hydroxytamoxifen
31

competitively blocks the receptors, and allows scientist to see if the estrogenic effects continue
in the absence of a functioning ER (Delbes et al, 2006). ICI 182780 is also called Faslodex or
Fulvestrant and is used clinically to treat cancer (AstraZeneca, 2004). Another family of
compounds interacting with ER are the pharmacological modulators called Selective ER
Modulators (SERM), such as raloxifen and tamoxifen that have differential affinity for the two
ERs. SERMs can be agonists for one receptor and antagonists for the other (Luconi et al, 2002).
Many of these molecules disrupt the endocrine signalling of the receptors, and depending on
the dosages and period of exposure can either cause or prevent different diseases including
cancer. Alternatively, other exogenous estrogen compounds might also be able to prevent
cancer in the right doses. One of the xenoestrogens believed to function this way is the most
common phytoestrogen in soy, genistein, which has a very high affinity to ERβ and therefore
slows down proliferation in some tissues. Other synthetic estrogens are used clinically for the
same purpose (Heldring et al, 2007).
8.3 Estrogen Receptors
8.3.1 Genomic Function of the Estrogen Receptor
In order for the Estrogens to perform their biological functions, they must travel to the
cells in the targeted tissues and subsequently bind to their specific receptors. There are two
known estrogen receptors (ERs), each transcribed from different chromosomes (10 and 12 in
mouse, 1 and 6 in rat and 6 and 14 in humans (Delbes et al, 2006)), namely ERα and ERβ (Segars
and Driggers, 2002). Estrogen receptors were first discovered in the 1950’s by Elwood V.
Jensen who identified the ERα protein (Jensen, 2004). ERβ was discovered much later in 1996
by Jan-Åke Gustafsson while looking for an androgen receptor in prostate cells (Maher, 2006).
ERs function as transcription factors (Segars and Driggers, 2002) and are part of the steroid
nuclear receptor superfamily, which functions in regulating gene expression (Delbes et al,
2006). After ER binds directly to its specific ligand, it releases associated receptor inactivating
heat shock proteins (usually HSP90). Binding to estrogen causes the ER to change its
conformation allowing it to form a stable dimer with another activated receptor of the same
type. The dimer then translocates through the nuclear membrane into the nucleus (Luconi et
32

al, 2002). ERs can bind directly to DNA on selected estrogen response elements (ERE) found
near or in the promoter region of the targeted gene. In order to fully activate the transcription
of their target proteins, the ERs must also come in contact with coregulatory proteins that can
either enhance or repress their functions (Segars and Driggers, 2002). It is the ER dimer that
interacts with the ERE and attracts other molecules to the promoter to either suppress or
activate gene transcription (Nikov et al, 2001). ERs can also be modified by phosphorylation
(Segars and Driggers, 2002). Because transcription is regulated by hormones, the pattern of the
genes that are modulated depends on all of the signalling pathways that are active at the time
of ER activation in the targeted gene (Heldring et al, 2007).
8.3.2 Estrogen Receptor Structure
Each of the two ERs has several isoforms, which are truncated versions of the wild-type
protein, some due to differential splicing of the C-terminal. There are no reported functions of
the ERα variant isoforms, although there is a lot of research being conducted concerning
different functions of truncated versions of ERβ (Delbes et al, 2006). Human ERα is a protein
encoded by 9 exons, and is built up of 595 amino acids and a molecular weight of 66kDa. ERβ is
smaller at 530 amino acids and 54kDa, but is also encoded by 9 exons. Both receptors have the
structure of a common nuclear receptor (NR) with six functional domains. The N-terminal is the
least conserved domain (Luconi et al, 2002). The central domain or C domain contains two zinc-
fingers creating the DNA binding domain (DBD). The zinc finger motifs are coordinated by eight
cysteine residues (Pettersson and Gustafsson, 2001). This domain is the most evolutionarily
conserved and is very important for the binding of the receptor to the DNA (Heldring et al,
2007). The DBD is connected to the ligand binding domain (LBD) through the D domain, which
acts as a hinge between the two domains. The hinge domain is not very conserved between
different NRs and associates itself with HSP90. The LBD, or E/F domain, is multifunctional as it
binds to ER agonists or antagonists. It also plays a role in dimerization, transactivation, cofactor
binding, and can bind to a second HSP90 molecule (Pettersson and Gustafsson, 2001).
Transcription is activated in the receptor through two activation functions (AF-1 and AF-2) on
either end of the receptor molecule, both of which accepts and binds to coregulatory proteins
33

(Heldring et al, 2007). While both ERs share similar affinities for E2, they have varying affinities
for other natural and synthetic estrogen molecules as well as other various antagonistic ligands
(Luconi et al, 2002). There are two common isoforms of ERβ; ERβ1 and ERβ2. ERβ2 has 54 extra
nucleotides, 18 of which are inserted into the LBD (Pettersson and Gustafsson, 2001). It
appears that ERβ2 is a suppressor of ERβ1 and ERα (Luconi et al, 2002) and interestingly it is
found in human foetal gonocytes (Gaskell et al, 2004).
8.3.3 Non-Genomic Effects of ERs
Recent research has shown that in addition to the transcriptional function of ERs,
estrogens are capable of triggering a very rapid cascade of responses. These effects are much
faster than those produced by ER working on its own accord and can happen within seconds to
minutes following stimulation. These responses are activated through second messengers such
as calcium, activated kinases and tyrosine kinases, PKA6 and PKC7
8.4 Reproductive Effects of Estrogens
as well as the ERK pathway.
These effects, unlike those generally activated by estrogens through the classical ER
mechanism, occur in the cytoplasm or on the cellular membrane. These pathways are possibly
triggered by different functions of the known ERs or perhaps a completely new type of ER
(Luconi et al, 2002). An estrogen-activating membrane bound G protein-coupled receptor has
been found in breast cancer as well as on adult testis (Delbes et al, 2006). Nongenomic effects
of estrogens will be discussed in more depth with more recent research and clinically relevant
examples in the following sections on signalling crosstalk mechanisms.
8.4.1 Estrogen Signalling in Females
In the female reproductive system E2 is synthesized in the ovaries by granulosa cells via
aromatization of testosterone. Estrogens are also made in adipose tissue, skeletal muscles,
skin, hair and bone. Women of reproductive age produce the highest concentrations of
estrogens; post-menopausal women are more susceptible to heart disease and bone fracture,
6 Protein Kinase A 7 Protein Kinase C
34

due to their low levels of estrogen production. Hormone replacement therapy was thought to
be very reliable in decreasing these risks, but has recently been shown to pose other health
risks such as increased risk of stroke in postmenopausal women and is now recommended only
in specific cases for treatment of menopause (Anderson et al, 2004). In adult females, ERβ is
found in the granulosa cells where its levels of expression fluctuate throughout the menstrual
cycle. It is also present in the glandular epithelial cells. ERα is abundant in the theca cells as
well as luminal, glandular epithelial cells and stroma. Women deficient in estrogens develop
normally until puberty, but then show failure in growth of the breasts, enlargement of clitoris
and unfused epiphyses as well as amenorrhea. Such patients, when treated with estrogens and
progesterone begin normal puberty cycles (Pettersson and Gustafsson, 2001). Estrogens play a
crucial role in the early development of the female reproductive organs as they directly
stimulate secondary sex characteristics, breast development, uterine development and
maturation of the fallopian tubes (Waterloo, 2007).
8.4.2 Estrogen Signalling in Male Reproductive System
ERs were first located in the male reproductive system over thirty years ago in the
epididymis, almost twenty years after Jensen’s discovery of the estrogen receptor. It was then
apparent that estrogen played a role in male foetal organ development, but not much else was
known. Even as late as the 1990’s some scientists continued to think that the ERs present in
the male reproductive system were simply the residue of embryological differentiation (Hess
and Carnes, 2004). Due to differences in laboratory methods of research, there are differing
opinions as to which receptors and isoforms are found in which tissues and cells. It appears
that ERβ is more prevalent in the accessory organs, and is expressed mainly in the prostate,
bladder, seminal vesicles and testis (Luconi et al, 2002), while ERα is mainly in the efferent
ductules and Leydig cells (Hess and Carnes, 2004). Both receptors are found in the epididymis
and spermatogonia as well as elongated spermatids (Luconi et al, 2002). Our own studies
identified ERβ in germ cells. Including late foetal to neonatal gonocytes, and in older germ cell
stages (Wang et al, 2004). There are many ligands other than the common endogenous
estrogens that are able to bind to ERs, as they are, unlike many other nuclear receptors, not
35

highly specific receptors, and may accept a large number of different molecules with a
surprising diversity in structure (Heldring et al, 2007).
8.5 Estrogen in Male Reproductive Development
ERs are found in the foetal testis from a very early stage of development. ERα is found
in the mouse gonad 10.5 days postconception and is prominent in Leydig cells only until birth.
ERβ is seen in the foetus a few days after ERα and is generally located in the gonocytes, Sertoli
and Leydig cells. With the knowledge of estrogen production in the foetal and neonatal testis
in addition to the presence of ERs, it was anticipated that estrogens played an important role in
foetal development. It is now known that estrogen is important in the regulation of the adult
male reproductive system as well. By inactivating specific estrogen receptors, scientists
showed how estrogens functioned in the developing testis. In the foetus, excess estrogens
inhibit the proper development of the testis. ERβ is involved in gametogenesis since mice
expressing a mutant inactive ERβ presented increased gonocyte numbers from late foetal to
neonatal periods, due in part to decreased germ cell apoptosis. However, estrogens appear to
exert either transient proliferative effects in vivo (Vigueras-Villasenor et al, 2006) and on
isolated gonocytes (Li et al, 1997;Thuillier et al, 2010), no effect on gonocytes used in organ
cultures or co-cultures, and negative effects on foetal gonocyte numbers (Delbes et al, 2007).
However, ERβ knockout male mice are fertile (Gould et al, 2007;Krege et al, 1998) suggesting
that ERβ is not essential for male germ cell development or that redundancy occurs in the germ
cells of male transgenic mice due to the presence of another yet uncharacterized estrogen-
dependent receptor. In this context, PGCs were shown to express the estrogen related
receptor beta (ERR-β) which was found to play a role in their proliferation (Mitsunaga et al,
2004). Studies in our lab have shown that estrogen combined with PDGF enhances gonocyte
proliferation, most probably via ERβ which is strongly expressed in gonocytes (Li et al,
1997;Thuillier et al, 2010). It has also been shown that ERα inhibits foetal germ cell growth,
indicating a delicate balance between the two functions in fetal development (Carreau et al,
2006). Another possible function of estrogens in the developing testis might be to establish the
adhesion between Sertoli cells and germ cells (Hess and Carnes, 2004;MacCalman et al, 1997).
36

ERα was proposed to mediate the negative effects of estrogens on foetal Leydig cells (Delbes et
al, 2004;Delbes et al, 2006). After puberty, estrogens have been shown to play a role in the
regulation of spermatogenesis, spermatid maturation, germ cell number and viability (Carreau
et al, 2003). By injecting small doses of estrogens into animals, scientists were able to
accelerate the onset of spermatogenesis.
In rats, plasma estrogen levels rise throughout gestation and peak at 17.5 days, while
the mother’s plasma estrogen levels are highest at 18 days gestation. Also located in the foetal
testis is the estrogen producing enzyme aromatase indicating that estrogens can be
manufactured in the testicular tissue. Aromatase is most prominent in foetal Sertoli cells. Due
to this, they are probably the cells producing the most estrogens in the foetal testis. However,
Leydig cells were demonstrated to be the primary estrogen producer cells in adult testis (Delbes
et al, 2006). The major aromatase molecule found in the testis is cytochrome P-450 aromatase
(CYP19) (Wahlgren et al, 2008). Testicular estrogen levels are generally much higher than
plasma levels, indicating that the majority of estrogens produced in males are synthesized by
the testicular tissue (Hess and Carnes, 2004). Aromatase has been found in germ cells, which
appears to be another large producer of estrogens. Aromatase also plays a role in elongated
spermatid mobility (Nitta et al, 1993;Berensztein et al, 2006;Carreau et al, 2003). A study
conducted by Wahlgren et al (2008), demonstrated that estrogen can induce DNA synthesis and
a proliferatory effect not only in developing male germ cells, but in adult spermatogonia as
well. 5α Androstane-3β, 17β-diol (3βAdiol) is a specific ERβ agonist derived from 5α-
dihydrotestosterone (DHT) secreted by Leydig cells, and is found in elevated levels in the rat
testis. In vitro studies show that 3βAdiol was able to induce DNA production in premitotic
spermatogonia, evidence that estrogens are important in spermatogenesis (Wahlgren et al,
2008). Estrogens are crucial for the development and maintenance of the male reproductive
system and must be in perfect balance with testosterone in order for normal development to
take place. Too much or too little estrogen can be harmful to development; therefore it is clear
that “endogenous estrogens are essential for maintenance of male fertility” (Delbes et al,
2006).
37

8.6 The Estrogen Hypothesis
Increasing concern has been generated in the medical community throughout the past
fifty years regarding research documenting a gradual increase in severe male reproductive
disorders. As society is becoming more industrialized, it is hard not to notice the detrimental
effects that it has on the well-being of many different organisms in the environment. Studies
have shown that over this period of time there has been an unprecedented upsurge in
testicular cancer, cryptorchidism and hypospadia (Delbes et al, 2004) as well as a large
reduction in average male sperm counts from 170 to 70 million spermatozoa per millilitre
(Delbes et al, 2006). Both cryptorchidism, a malfunction in testicular descent, and hypospadia,
the abnormal positioning of the urethra on the penis, are associated with increased likelihood
of cancer. The cause of these findings was first hypothesized by Sharpe and Skakkebaek in 1993
to be environmental, when they stated that the current situation is due to an escalating
quantity of xenobiotics and xenoestrogens in our environment, making the risk of exposure
very likely. They stated that “estrogen-like molecules could alter adult male fertility by acting
early in gonad development and that inappropriate exposure to estrogen during foetal or
neonatal life could affect adult reproductive function”. Irrefutable evidence for this statement
can be observed in the reproductive development of males exposed to DES in their foetal life.
This group of male offspring, most of who were born between the 1940’s and 1970’s, had an
abnormally high incidence of reproductive disorders in their adult life (Delbes et al, 2004).
Many of these changes to the endocrine and reproductive system have been shown to be
completely irreversible. Exposure to exogenous estrogens appears to affect females as
negatively as it does in males (Iguchi et al, 2001).
There are many other occurrences in nature that strengthen this hypothesis and have
prompted focused research in this field. Alligators in a chemically polluted lake in Florida
showed a very large upsurge in abnormalities to their reproductive organs during development
(Gutendorf and Westendorf, 2001) compared with those alligators in a nearby lake unpolluted
by agricultural waste containing xenoestrogens. The affected alligators had low testosterone
levels and micropenis with abnormally developed testicular tissue (Delbes et al, 2006). Fish in a
38

lake near a sewage disposal site also displayed a decrease in sperm quality. Trout in Lake
Ontario died due to the dioxin-like compounds in the water (Gutendorf and Westendorf, 2001).
The most extreme example observed in human society is apparent in the Aamjiwnaang First
Nation community in Canada, where over the course of ten years the gender ratio of viable
offspring saw drastic changes whereby only 30% of the children born to this community were
male. This phenomenon was suggested to be due to the nearby industrial and environmental
chemicals (Delbes et al, 2006).
In light of the given data and the numerous examples of estrogen-induced disorders, it is
still very unclear how these estrogenic compounds induce their negative effects on the male
reproductive system, and whether or not there actually is a direct estrogenic effect at all.
Certainly, it is well documented that estrogen plays an important role in foetal and neonatal
testicular development and an imbalance of estrogens is a cause for concern, and similarly it is
important to address the fact that unrealistically high dosages of in utero estrogenic exposure
can cause serious mal-developed reproductive organs, however the majority of studies
experimenting with normal, potential daily exposure levels of exogenous estrogens have only
observed minimal, benign changes in the reproductive system. Nevertheless, there is a concern
surrounding potentially harmful estrogenic compounds found in everyday life such as the
phytoestrogen genistein in soy products and BPA, which can be found in plastic containers.
Testicular estrogen exposure is even further complicated by the fact that it has been shown
that at different stages, estrogens can be pro-proliferative or pro-apoptotic and each of these
functions have been linked directly to the estrogen receptor pathway. In order to further
understand the mechanisms behind changes caused by foetal estrogen exposure, our lab
conducted various experiments, observing estrogen-induced changes in the PDGFR, estrogen
receptor and MAPK pathways in foetal, neonatal, prepubescent and adult rats.
Estrogen receptor β in gonocytes are complexed together by a host of chaperone
proteins including Hsp90, the cochaperone p23 and the immunophilin cyclophilin 40 (Cyp40),
which stabilize the receptor in the cytosol, protecting it from degradation and exposing it for
ligand binding. When estrogen binds to its receptor, Hsp90 is dissociated but p23 remains
39

attached as ERβ translocates to the nucleus. In utero exposure to BPA, genistein, coumestrol
and DES increase mRNA and protein levels, at 21dpc and PND3, of Hsp90, but not those of p23.
DES is the only estrogenic compound that increases Cyp40 levels in the neonatal testis. Unlike
the other estrogens that increase Hsp90 in the gonocytes, BPA increased Hsp90 levels in the
whole testis. These changes were only found in neonatal rat testis, as Hsp90 levels were
normal in PND21 prepubescent rats. The only notable change at PND21 was a significant
increase in spermatogonia (Wang et al, 2004), although by PND60 in adult rats, spermatogonial
numbers were back to normal (Thuillier et al, 2009). Exogenous estrogen exposure to foetal
rats induced an increase in PDGFRα and β mRNA as well as protein in PND3 rats compared to
controls. There was a notable increase in PDGFRβ in gonocytes. DES exposure gave the most
intriguing result, as it increased PDGFRβ in the seminiferous cords at lower doses, but then
actually decreased it when exposed to a higher dosage. Foetal rats showed a significant
increase in PDGFβ in exposed testis (Thuillier et al, 2003). These results are very important,
leading to our understanding that estrogen-induced proliferation is somehow modulated
through the PDGF pathway (Thuillier et al, 2010). In addition to this, our lab has recently
observed that rats exposed to BPA and genistein in utero caused increased levels of ERK1/2 and
Raf1 in PND3 rat testis. Similar to previous findings, these changes were no longer present in
PND60 rats, although it was noted that PND60 rats had an increased number of Leydig cells,
although testosterone levels in the testis were found to be normal. At experimental levels of
exogenous estrogens similar to potential environmental exposures, there was no significant
decrease in fertility observed in treated adult rats (Thuillier et al, 2009).
8.7 Approaches Used to Study the Role of Estrogen/ERs in Males
8.7.1 Laboratory Results of Estrogen Exposure in Vivo
In view of the natural models mentioned above, scientists set out to understand how
exogenous estrogens affected organisms in vivo. In order to gain an understanding of the
mechanisms used by estrogens in the male reproductive system, scientists studied animals at
different stages of development following exposure to large amounts of estrogens as compared
to unexposed animals. It appears that most of the testicular disorders originate from exposure
40

during foetal development. For instance, an irradiation-induced decrease in the number of
gonocytes during development was shown to lower sperm count later in life (Moreno et al,
2001). When viewing data on patients exposed to DES as a foetus, it was interesting to find
that some patients were unharmed, while others had highly deformed reproductive organs. It
was understood that the differences in intensities of reproductive deficiency were based on the
stage of pregnancy in which they were exposed. DES appears to have a greater negative effect
during the first semester of pregnancy (Delbes et al, 2006). Some of the malformations
observed while exposing foetal mice to exogenous estrogens were low testicular weight, low
germ cell count, increased apoptosis in germ cells, and a high rate of tumours (Delbes et al,
2004). In vitro studies showed that DES exposure in early development disrupted cord
formation as well as establishment of many of the reproductive cells. Pregnant mice were
given exogenous estrogens either by gavage, injection or ingestion via their drinking water in
order to expose foetal mice. The results of these studies displayed both short term and long
term repercussions in the affected mice (Delbes et al, 2006). Some of the short term effects
included accelerated testicular development and abnormal gonocyte differentiation (Delbes et
al, 2004). It is conclusive though, that foetal development is very sensitive to exogenous
estrogens (Delbes et al, 2006). Although it is now clearer that estrogens can play a role in
testicular development, this is not proof that the small amounts of estrogens that we are
exposed to chronically actually contribute to testicular diseases. Whereas most experimental
effects of estrogen are detrimental to the reproductive organs, there have also been studies
describing positive effects of specific estrogens as shown in germ cell development. Indeed,
one study showed that exposure to mild doses of genistein actually increased sperm’s fertility
(Delbes et al, 2004). Some of the natural or synthetic non-proliferative or antiestrogen
molecules are now used to treat breast cancer as they prevent the cells from continuously
multiplying. The first attempt to use anti-estrogens to prevent cancer was through the use of
an anti-ERα molecule. However over time the tumour grew resistance to this molecule. For
this reason scientists have attempted to create an ERβ agonist to treat cancer instead, in view
of the pro-differentiation and/or anti-proliferation role of ERβ in prostate and breast tissue
(Heldring et al, 2007). Indeed, ERβ ligands were shown to slow prostate tumour growth in nude
41

mice and promise to lead to the development of novel and efficient therapeutic approaches
(Koehler et al, 2005).
8.7.2 Knockout Mice
One method of understanding how a specific gene or protein works in the body is to
study the animal in the complete absence of its expression and/or function. By inactivating a
target gene, we can see the functioning differences between the wild-type animal and its
mutant. The most common technique used in laboratories is the knockout mouse; a mouse
that is created with the specific gene encoding the target protein deleted from its genome.
Many studies have been performed using ER knockout (ERKO) mice to better understand the
role of estrogen in males and females. The first completely ER inactive ERKO mouse was
produced in 1996 by Korach et al. in order to better understand ER’s function in the male
reproductive system. This breakthrough preceded the discovery of ERβ, and therefore in
actuality was only an ERαKO mouse. At this point in time it was assumed that the whole ER
function was removed from these mice, however with the discovery of ERβ, it was soon
understood that only ERα was affected, leaving ERβ fully functional in the original ERKO mouse.
Korach and his team made their ERKO mouse by disrupting the ERα gene by the use of
homologous recombination. The heterozygous mice were mated in order to obtain progeny
homozygous for the deletion of the ER gene. In order to understand the function of ER, they
performed long and short term mating assays with wild-type, heterozygous and ERαKO mice
and then euthanized them to see the anatomical differences between the different clones. In a
two month mating period the wild-type and heterozygous male mice produced equal numbers
of viable offspring, while the ERαKO mice did not produce a single litter. It was also noted that
in the short term mating (one day per week for three weeks), the ERαKO mice produced almost
no copulatory plugs, while the males in the other two categories seamed to mount the females
at almost every opportunity. After euthanasia, testis weight was much lower in ERαKO mice,
while the other organs did not differ much from the wild-type. ERαKO mice also had higher
testosterone levels. The epididymides of ERαKO mice contained low numbers of sperm after 20
weeks of life and the testis contained atrophic and degenerating seminiferous tubules. At birth
42

the ERKO testis appeard quite normal but as the mice grew older, more degeneration occurred
in their tubules, and their lumens became increasingly dilated. Furthermore, ERαKO mice
presented a much higher percentage of immotile sperm, which again increased with age. In
vitro fertilization showed that even the motile sperm were less fertile than wild-type
spermatids. This study clearly showed that ERαKO mice were completely infertile and the lack
of ERα affected sperm number and function, as well as a suggested behavioural change in the
mouse’s sex drive. Estrogen appears to be involved in regulating luminal fluid production in the
Sertoli cells, which is why ERKO mice produced an overabundance of the fluid and the
seminiferous tubules were dilated. Another cause might be due to inhibited reabsorption in
the efferent ducts of the testis due to loss of estrogen function causing fluid accumulation.
Fluid accumulation might be an important explanation as to why the seminiferous tubules
degenerate over time. As fluid accumulates in the testis, intratesticular pressure increases,
blocking off regular blood flow to the surrounding tissues. As the pressure rises, the less
vascularised tissues degenerate first, followed by the sections with more vasculature (Eddy et
al, 1996). There is also an increase in cryptorchidism in ERαKO mice, similar to the observations
made following high levels of exogenous estrogen exposure (Luconi et al, 2002).
Although ERα is important for testicular function it is not responsible for
spermatogenesis. By implanting ERαKO germs cells into an empty wild-type testis, normal
sperm is produced and the cells can generate live offspring. ERβ probably does influence
spermatogenesis as diets high in soy increase sperm production in adults and genistein has a
much higher affinity to ERβ than ERα. However, studies in rats exposed neonatally to genistein
showed that small numbers of these rats were sterile in adulthood compared to none in control
animals, suggesting that neonatal exposure can have deleterious effects in a subset of animals
(Atanassova et al, 2000). Additionally, long term exposures to ICI 182,780 cause gradual
testicular atrophy. This effect was thought to have to do with fluid accumulation, but that was
only the case in rats and not in mice. Thus, estrogens may have a direct effect on the ERβ
molecules on germ cells, where ICI 182,780 functions as an antiestrogen (Hess and Carnes,
2004).
43

ERβKO mice were created similarly to ERαKO mice by inserting a neomycin resistance
gene into the third exon of the ERβ gene using homologous recombination. The results were
quite different than those of the ERαKO mice. Unlike the female ERαKO mice that were
completely infertile, these females were fertile and breast development was normal. The only
effect on females was that their litter size and number was smaller than for the wild-type. Male
ERβKO mice were fertile as well, and their testicular tissue showed no abnormalities, although
older males had epithelial hyperplasia in the prostatic ducts and bladder wall (Krege et al,
1998). A later study showed that homozygous and heterozygous mice carrying an inactivating
mutation to ERβ expressed an increase in gonocyte number and density, although the actual
testis size remained the same. There was also a decrease in gonocyte apoptosis. From this
study, it was concluded that endogenous estrogens normally directly inhibit germ cell growth
during foetal development. Estrogens are crucial for testis development and germ cell
proliferation, although ERβ does not affect Leydig and Sertoli cells (Delbes et al, 2004). It has
now also been shown in the male prostate that there is an abundance of 3βAdiol, the second
most prominent estrogen in the body, which is a very good ligand for ERβ, allowing it to prevent
proliferation in the prostate. ERβKO mice also showed that many cells in the prostate do not
differentiate completely and are therefore still capable of proliferation (Koehler et al, 2005).
A third strain of knockout mice was created to further understand the function of
estrogen in males. These mice were deficient in the gene that produces the aromatase
enzyme, rendering them unable to produce estrogens. Male mice of this breed developed
normally and reproduced until they reached five months of age, at which point they
progressively lost their spermatogenesis function to become deficient of round spermatids and
completely infertile by one year of age (Carreau et al, 2003). At this point, the testis also
became deficient of round spermatids (Hess and Carnes, 2004). The reason for the inhibition of
spermatogenesis in ArKO mice is different than in ERαKO mice, and is due to a complication in
germ cell differentiation. Estrogen is used as a survival factor for normal spermatids, and when
this hormone is not present, they cannot differentiate and therefore begin to apoptose.
44

Experiments on mice defective for both ER molecules (ERαβKO mice), showed results
similar to those of the ERαKO mouse. Although from these results it appears that the only truly
crucial receptor is ERα, it is important to note that we are still not perfectly clear on the
function of ERβ and therefore it is hard to see what it does based on knockout models. It also
might be the case that, although we have knocked out the target receptor or protein that we
believe is important, there may be alternative molecules that perform the functions of the ERs
or of the aromatase enzyme in their absence (Luconi et al, 2002). Female ERαβKO mice
displayed a distinct phenotypical difference from those only deficient of ERα. Whereas it is
clear that in females there is a specific function for ERβ, it has yet to be completely defined in
males (Couse et al, 1999). From the results using the different knockout mice, it appears that
the two estrogen receptors can often play opposite roles in various experimental situations. It
may be crucial for the body to have the proper balance between the two of them in order to
develop and function correctly (Heldring et al, 2007). Although a distinct functional role for ERβ
has yet to be defined in male testicular development, and more specifically in the
differentiation of the male germ cells and reproductive organs, it is clear that due to their high
expression in testicular cells at this developmental period there must be a crucial
developmental role for ERβ.
9. Cell Signalling Cross Talk Mechanisms
Studies in our lab have demonstrated that unlike PND2 gonocytes, PND3 gonocytes
respond to PDGF as well as 17β-estradiol by proliferating in a non-additive manner, suggesting
the existence of crosstalk between the two signalling pathways. PDGF and 17β-estradiol are
likely activators for the regulation of the second proliferative phase in vivo given that both
factors are found at high levels in rats until PND5 or 6. Testosterone, the male reproductive
hormone, which is synthesized by Leydig cells in early neonatal life, could also play a role in
germ cell development and differentiation, although these cells are generally considered to be
androgen receptor deficient (Johnston et al, 2001). Because our initial study suggested that
PDGF and 17β-estradiol might crosstalk to stimulate gonocyte proliferation, we decided to
determine which downstream cascade of PDGF was involved in this process by using inhibitors
45

and antagonists specific for either PDGF-activated signalling molecules or ER. My master’s
thesis work is part of this study, which will be presented in details in the results and discussion
sections.
There are a number of models in the literature of different downstream pathways
working together in order to activate a single cellular function. As mentioned above, there are
many novel estrogen-induced functions, above and beyond transcription of genes expressing
the ERE in their promoter region. Roles in proliferation and rapid effects of estrogens have
stimulated a new field of research in secondary and non-genomic mechanisms of estrogen/ER
binding. Similarly, as it has become clear that one of the most important regulatory
mechanisms of the MAPK pathway is its interaction with other downstream pathways, it is
important to study where these interactions take place and how to manage them in
pathological situations. It is becoming increasingly evident that many known cellular actions
actually require some degree of physical or chemical crosstalk between different pathways,
including nuclear receptor pathways. The models in place are valuable tools in our
investigation of PND3 gonocyte proliferation and, using our experimental data, we can begin to
hypothesize how this crosstalk works and at which point in the respective downstream
pathways it takes place.
9.1 Intercommunication of Separate Downstream Pathways
Analogous to computer programming, where if the right buttons are not pressed and
the settings are not in complete coordination the program will not run, cell signalling requires
all of the right inputs and stimuli to be activated simultaneously or in the correct sequence of
events to elicit the proper response. Increasing research has shown that each cellular function
is actually the result of many factors all working in communication. In specific cells, high levels
of cyclic AMP (cAMP) can turn on or turn off Raf molecules, to direct a specific response upon
tyrosine kinase receptor stimulation. There is a small GTPase designated as Rap1 that responds
directly to cAMP that can activate or inhibit specific Rafs in a dose dependant, cell specific
manner (Schaeffer and Weber, 1999). In this way, the same growth factor will have a different
response based on the levels of cAMP in the cell at the time of stimulation. There are small
46

molecules such as small G-proteins and others that can communicate between different MAPK
pathways to increase or attenuate a response to an extracellular stimulation. There are
transcription factors and other important cellular enzymes that are affected either positively or
negatively by more than one MAPK. This allows for different stimulations to have additive
affects on the specific factors (Schaeffer and Weber, 1999). Raf or MEK have been shown to be
activated by cyclins or cAMP which then stimulates a normal MAPK cascade. P38, a MAPK, can
activate phospholipase A2 in platelets upon stimulation by collagen (Robinson and Cobb, 1997).
9.2 Crosstalk between Estrogen Receptor and Growth Factor Receptors / MAPK
Pathway
Nongenomic estrogen activity generally works alongside numerous different pathways.
In contrast to the classical localization and function of estrogen receptors, ERs have been
detected on cell membranes, denoted as membrane estrogen receptors (mERs), and activate
downsteam pathways. There are numerous different hypotheses as to what these receptors
are and how they work. It is assumed via immunofluorescent cytology that there are some
systems and cell types expressing the ordinary ERα and ERβ docked to the membranes, while
there have also been novel ERs discovered as well, most notably, the ER-X receptor found in the
brain, uterus and lungs (Hirahara et al, 2009) and GPR30 which is an estrogen binding G-protein
coupled receptor (Fox et al, 2009). Very often, these systems express different splice variants
of ER that help with the cellular response to estrogen (Hirahara et al, 2009). mER behaviour
resembles that of G-protein coupled receptors, activating G-protein GTPase activity (Simoncini
et al, 2006), and mERs are usually found interacting with growth factor receptors or activating
MAPK cascades on their own. Interaction with growth factor receptors can be either on the
same cell as the mER or on a neighbouring cell in a paracrine or juxtacrine crosstalk (Osborne et
al, 2005). Through this mechanism, ERs can activate pathways including MAPK, PI3K, GSK-3β,
Src, STATs nitric oxide synthesis and intercellular calcium flux (Collins and Webb, 1999;Hirahara
et al, 2009). Src is an important molecule, as ER has no kinase activity; most estrogen induced
phosphorylation of other pathways is modulated by this kinase (Fox et al, 2009). ERα has an
Sh2 domain and can bind directly to Scr to activate MAPK (Geffroy et al, 2005).
47

There are a number of proposed mechanisms for how ERs can dock to the plasma
membrane given that they have no transmembrane domains in their amino acid sequence. ERs
have been shown to be recruited to the membrane and dock to growth factor receptors via
activation of docking proteins. Alternatively, ERs can attach to the end of the cytoskeleton near
the plasma membrane using caveolin-1 as a docking protein. This complex can directly target
MAPK for activation (Hirahara et al, 2009). Caveolin has also been shown to be upregulated
upon estrogen receptor activation (Park et al, 2009). Wong et al. published a paper in 2002
describing a protein called modulator of non-genomic activity of estrogen receptor (MNAR)
which complexes with ERs and Src as a scaffolding protein to regulate estrogen-induced
proliferation. Although the author retracted this paper in 2009, the scaffolding complex is still a
valid mechanism of mER localization and activation (Wong et al, 2002), and other groups have
reported similar results in their studies (Osborne et al, 2005). It was reported by Cheskis et al.
that MNAR is integral in ER’s stimulation of the MAPK pathway via phosphorylation of MNAR by
c-Src. c-Src is activated by dephosphorylation of its inhibitory phosphotyrosine sites or by
binding to receptors via its Sh2 and Sh3 domains. It is not yet known whether or not ER
requires a scaffolding protein to activate PI3K (Cheskis et al, 2008) although it is known that ERs
somehow bind to PI3K to activate Akt (also called protein kinase B, PKB) (Geffroy et al, 2005).
Other scaffolding proteins are also reported to form complexes with ER to enable the non-
genomic estrogen responses are Shc, IGF-1R (Fox et al, 2009) and MTA1-S (Osborne et al, 2005).
Fu et al. showed in bovine arterial endothelial cells that estrogen-induced proliferation
was caused by expression of the cell cycle modulator cyclin D1 through the ERK pathway. This
ER was found to be bound to the membrane and is likely to be a novel ER. The promoter region
of cyclin D1 contains binding sites for NF-κB, AP-1 and Sp1, all of which can be activated by ERK.
This study has important implications for cardiovascular (CV) disease, as postmenopausal
women tend to be at a higher risk for suffering from CV disease due to estrogen deficiency. The
finding that estrogen induces endothelial cell proliferation is an important revelation in
understanding this phenomenon and possible remedies (Fu et al, 2007). Oligodendrocytes and
myelin cells also express mERs, and estrogen induces proliferation of these cells as well.
Estrogen deficiency is a common risk factor for neuron demyelination and multiple sclerosis,
48

and treatment with estrogen has been shown to produce some positive results (Hirahara et al,
2009). Osteoporosis is another disease that is frequent in estrogen deficient postmenopausal
women. Bone, which is in constant remodelling, requires estrogenic activity for proper strength
and structure. An imbalance of estrogen can result in destruction of calcified bone. Although it
has been proposed that estrogen hormone therapy could reverse osteoporotic changes,
estradiol therapy is not recommended (Sehmisch et al, 2008). Instead, due to the increased risk
of breast cancer with estrogen supplementation, it was proposed to use phytoestrogens
instead. The phytoestrogen resveratrol, which comes from such plants as nuts, berries and
grapes, appears to be a promising treatment for osteoporosis by stimulating estrogen-induced
proliferation and maturation of osteoblasts through the ERK and p38 pathways (Dai et al, 2007).
Resveratrol has been shown to prevent development of osteoclasts through RANK-L, and
induces apoptosis in these cells (He et al, 2010). There are groups working on resveratrol
analogues with more potent effects on osteoclast and osteoblast differentiation (Kupisiewicz et
al, 2010), as well, the phytohormone 8-prenylnaringenin has been shown to have a much
stronger anti-osteoporotic effect than resveratrol (Sehmisch et al, 2008).
Bouskine et al. demonstrates how estrogens can stimulate proliferation in the JKT-1
testicular germ cell tumour cell line through ERK and protein kinase A (PKA) activation. ERK and
PKA then activate the transcription factor cAMP response element binding protein (CREB). It
was proposed that this does not go through the classical ERs as its function is inhibited by G-
protein inhibitors as opposed to estrogen antagonists. Although this study concluded that
inappropriate foetal exposure to estrogen can cause germ cell hyperplasia leading to testicular
cancer, it was also noted that in these cells ERβ acts as a tumour suppressor gene and prevents
cancer progression (Bouskine et al, 2008). Crosstalk-induced proliferation in cancer cells has
commonly been observed between estrogen receptors and growth factors. Normal mammary
growth requires communication between estrogen and epidermal growth factor (EGF) while
both estrogen and IGF-1 are necessary for uterine tissue proliferation. When any of these
factors are overexpressed, this leads to hyperplasia of the tissue. Proliferation in many breast
cancer models is stimulated by a rapid estrogenic response through MAPK. Proliferation is
often through the STAT5 molecule which is activated by Src or EGF receptor (Fox et al, 2009). In
49

endometrial carcinoma cells, treatment with estrogen induces phosphorylation of ERK, and an
influx of calcium into the cell. When estrogen binds to its membrane receptor, mER stimulates
the opening of L-type calcium channels generating an influx of calcium into the cell. A
conceivable mechanism for this action is ER stimulation of phospholypase C through a G-
protein. In this model, ERK activation is indirectly associated to estrogen receptor activation
through changes in the calcium electrochemical gradient (Zhang et al, 2009).
Growth factors can also be used in eliciting an estrogenic response. In endometrial
adenocarcinoma cells, EGF activates nuclear ER-induced transcription of genes through specific
EREs. EGF ligand binding caused a downstream pathway that activates ER in the nucleus to
induce transcription (Ignar-Trowbridge et al, 1995;Ignar-Trowbridge et al, 1992). Ginsenosides,
a potent phytoestrogen derived from ginseng root is proposed as another possible treatment
for postmenopausal effects such as cardiovascular and central nervous system diseases.
Unfortunately, ginsenoside also induces breast cancer growth and development. This
phytoestrogen stimulates a normal genomic response as well as a non-genomic estrogenic
response. It induces proliferation of MCF7 breast cancer cells via stimulation of a tyrosine
kinase receptor through the MAPK pathway which activates ERK. ERK is required to
phosphorylate ERα on serine 118 in the nucleus in order to fully activate it (Lau et al, 2009).
Activation of ERK is involved in a very intricate crosstalk mechanism whereby reactive
oxygen species, secreted in very low concentrations by spermatozoa, self-stimulate themselves
activating a number of different signalling pathways which are all required to activate and
upregulate protein tyrosine phosphorylation for spermatozoa capacitation prior to fertilization
of the oocyte. The main crosstalk involves the ERK signalling cascade in parallel with the PKA
downstream pathway through phosphorylation of downstream molecules. The mechanism
proposed is that ERK activation works in parallel with adenylyl cyclase, cAMP, PKA and its
downstream molecules, and they converge together for the phosphorylation of protein
tyrosine. This crosstalk is set in place to ensure the coordinated precise timing of spermatozoa
capacitation directly before fertilization (Awda and Buhr, 2010;de and O'Flaherty,
2008;O'Flaherty et al, 2006).
50

Estrogens have been shown to induce increased PDGF and PDGFR expression in uterus
and vagina, demonstrating that estrogens can affect PDGF-dependent responses by increasing
PDGFRs and their ligands in specific tissues (Gray et al, 1995). An observation made in the
MCF7 breast cancer cell line is that progesterone, through its specific receptor, induces PDGF-
AA production and secretion. Although the PDGFR in many breast cancer cells is down
regulated, the PDGF-AA was shown to play a paracrine role by increasing proliferation and
viability of vascular smooth muscle cells in vitro in a crosstalk through PDGFRα. Progesterone
stimulation of the cancers cells induces communication with the endothelial cells to promote
vascularisation for the tumour to grow. This is important, given that tumour progression
requires the continued growth of blood vessels through vascular smooth muscle cells.
Targeting the progesterone pathway could provide a successful treatment for these types of
cancers (Soares et al, 2007). Testosterone and DHT are other critical steroid hormones in male
reproductive tissues that have been shown to crosstalk with growth factors or other steroid
hormone pathways. For example, AR was found to interact with ERs via Src in a human
prostate carcinoma cell line (Migliaccio et al, 2000) and it was shown to crosstalk with a number
of growth factor pathways such as that of IGF-1 (Kaarbo et al, 2007;Culig et al, 1994). Because
testosterone is produced by the foetal Leydig cells still present in neonatal testis in rodents, one
could consider the possibility that it might also act on gonocyte functions (Habert et al, 2001).
Although most germ cell stages do not express androgen receptor (AR) and are therefore not
considered as direct androgen targets (Vornberger et al, 1994), a recent study of the Habert lab
suggested that AR might be transiently expressed in foetal gonocytes where androgens were
found to inhibit proliferation (Merlet et al, 2007). This led to experiments I performed in which
the effects of testosterone and progesterone on gonocyte proliferation were examined (see
results and discussion sections, Thuillier et al. 2010).
10. Summary
Previous findings in our lab have shown that PDGF-BB and 17β-estradiol are able to
stimulate PND3 gonocyte proliferation, through activation of their respective receptors. This
was confirmed by in vitro experiments where AG370, a specific inhibitor of the kinase activity of
51

PDGFR, attenuated the PDGF-BB-stimulated proliferation, and ICI 182780, an ER antagonist,
arrested the estrogen-induced proliferative effect. The results obtained with ICI 182780
confirmed those observed earlier with a different ER antagonist, ICI 164384, showing that the
17β-estradiol effect requires binding on an estrogen receptor (Li et al, 1997). These earlier
experiments also showed that adding AG370 together with 17β-estradiol on gonocytes blocked
the proliferative effect on estrogen, while adding ICI 182780 with PDGF-BB prevented PDGF to
stimulate gonocyte proliferation.
In view of the knowledge listed above and the earlier findings in our laboratory, the goal
of my research was to extend and clarify our understanding of the mechanisms involved in the
stimulation of gonocyte proliferation by PDGF and 17β-estradiol, focusing on identifying the
downstream molecules mediating this process. My hypothesis was that PDGF and estrogen are
working in concert via crosstalk to activate gonocyte proliferation, and are both in some way
involved in ERK1/2 activation to transform the cell from the quiescent state to the proliferative
phase. My goal was to identify which signalling pathway was involved in the PDGF effect and
whether it was also under the control of estrogen.
52

11. Materials and Methods
11.1 Gonocyte Isolation
Male albino Sprague Dawley rats were purchased at post natal day 2 (PND2) from
Charles Rivers Laboratories. The pups were euthanized by inducing hypothermia and
decapitation at PND3 according to the guide for the care and use of experimental animals from
the Canadian Council on Animal Care and McGill University. Neonatal rats are resistant to CO2
asphyxia and were therefore euthanized by decapitation. Dissection was performed by
abdominal incision to remove and isolate the undescended testes from 30 pups. Testes were
kept in a 50mL tube of RPMI 1640 supplemented with 100 U/ml penicillin, and 100 mg/ml
streptomycin, on ice for the duration of the dissection. The testes were manually decapsulated,
and then processed for tissue dissociation. The cell isolation procedure was executed according
to the protocol described by Li et al. (1997), which is a modified version of that used by Van
Dissel-Emiliani et al. (1989). Under sterile conditions, the decapsulated testes were incubated
with 3.8mL RPMI, 2.5mL type IV Collagenase (stock 2mg/mL), 1mL Hyaluronidase (stock
1mg/mL) and 0.8 mL DNase I (stock 1mg/mL) in a shaking water bath at 37℃ for 30min in order
to cause the seminiferous tubules to dissociate from the interstitium. After decantation of the
digest, the pellet of remaining tissues containing the seminiferous cords was processed for
further tissue digestion, while the supernatant, including a mixture of mainly dissociated Leydig
cells, vascular endothelial and blood cells was discarded. The tubule fragments were
dissociated by adding 3 ml of 0.25% trypsin + 1 mM EDTA and 0.1 ml of DNAse I for 15 min at
37℃ to dissociate the Sertoli cells, peritubular myoid cells (PMCs) and gonocytes. The
digestion was arrested by adding 5 ml of 10% foetal bovine serum (FBS) (which neutralizes
trypsin) in RPMI. After collection of the supernatant, the remaining undigested tissue was
incubated a second time with trypsin-EDTA and DNase I, and stopped by addition of 10% FBS.
The final cell suspensions were filtered through a 40 μm nylon filter to remove any cell
aggregates or tissue fragments. The gonocytes were quantified with a hemacytometer,
differentiated from the other cell types by their larger size and round, smooth morphology.
The cells were collected by centrifugation at 800g, the pellet was resuspended in RPMI and the
53

X 30
Hya
luro
nida
se
Colla
gena
seSe
min
ifero
usTu
bule
s
Deca
psul
atio
n
Tryp
sin, E
DTA
Tubu
les a
nd se
para
ted
cells
Filtr
atio
n
Ove
rnig
ht in
cuba
tion
Gono
cyte
s, S
erto
li an
d M
yoid
cells
BSA
grad
ient
m
ixer
4%2%
BSA
Gra
dien
t
Frac
tion
Colle
ctio
n
Purif
ied
Gono
cyte
s
2:15
hr
Sedi
men
tatio
n
54

Figure 4: Gonocyte Isolation Protocol. Schematic representation of thedissection and digestion of rat testis for the purpose of isolating gonocytes, theprespermatogonial germ cell pool in neonates. Testis are collected in ice-coldRPMI and then manually decapsulated to allow the tissue to be exposed to thedissociation enzymes. Collagenase and hyaluronidase are used to dissociatethe interstitial tissue from the seminiferous cords, while maintaining the cordsintact. This digestion removes the Leydig cell population from the sample, aswell as the vasculature and other interstitial cell types. The next digestion iswith trypsin-EDTA, which breaks down the cords and dissociates the cells fromtheir tight intercellular adhesions. We now have a cell suspension of somaticcells and 0.5-1% gonocytes. Overnight incubation allows Sertoli cells and myoidcells to adhere to the culture dish, removing 50% of the somatic cells from thesuspension. Sedimentation by gravity on a BSA gradient allows the cells tofraction out by size. Fractions of 2mL are collected and observed on thehemacytometer to determine the numbers and purity of the gonocytes in eachfraction. Fractions with the highest purity of gonocytes are pooled togetherand used for our experiments. Other sets of gonocytes at lower purities aresometimes also kept and used for different investigations.
55

cells were cultured overnight in 150 mm large culture plates with 5% FBS in RPMI in 5% CO2 at
37℃. This incubation was crucial to gonocyte isolation as Sertoli and myoid cells are adherent
to culture dishes but gonocytes are not. This allowed us to separate out 50% of the
Sertoli/myoid cells before applying the cells to a BSA gradient. The cells were collected after 20
hours in culture, centrifuged and resuspended in 2mL RPMI, and then fractionated through a 2-
4% BSA gradient. The cells were allowed to sediment by gravity for 2 hours and fifteen
minutes, approximately 30x 2mL fractions were collected, and the number of gonocytes
determined on a hematocytometer. The fractions with the highest number and purity of
gonocytes compared with somatic cells were pooled together and centrifuged. Fractions of
lower purity that still contained a high yield of gonocytes were pooled separately. The total
yield of the high purity fraction was between 2 X 105 and 4 X 105 gonocytes at 45 to 70% purity.
The lower purity fraction often yielded similar numbers of gonocytes at purities ranging from
20-40%. Different purities were selected depending on the quantity of cells yielded from a
single dissection as well as the type of experiment being performed, with western blotting
requiring higher percentages of gonocytes and cytochemistry allowing for lower purities. The
final cell suspensions were resuspended in RPMI without FBS and incubated with or without
treatments for 30 and 60 minutes at 37℃.
11.2 Cell Culture for Short Term Molecular Profiles
For cytological studies, between 8,000–15,000 cells per well were treated for short term
assays of 30 and 60 minute cultures in RPMI, without FBS. FBS contains many hormones and
growth factors in it and therefore for short term assays, it was important to have no external
factors acting upon the cells other than the treatment itself. Cells were also cultured with FBS
for 4 and 8 hours to observe translational and early transcriptional changes in protein
expression as well as 20 - 24 hours for proliferative and gene transcription studies.
Alternatively, for some proliferative assays, cells were also cultured with charcoal stripped
(Biomeda) or PDGF-depleted serum. When conducting cultures for western blot analysis,
25,000–30,000 cells were treated in each well or tube. Because these studies were short term
analyses, most of the experiments were conducted in microcentrofuge tubes and the cells were
56

incubated in a 37℃ shaking water bath as opposed to using cell culture plates. Treatments
included 10ng/mL human PDGF-BB, 10-6M 17β-estradiol, 100µM AG370 (Biomol Research
Laboratories), 100µM ICI 182780 (Tocris) and 10µM UO126 (Calbiochem), between 10-6M and
10-15M of Genistein (synthetic) and Bisphenol A (4,4’-isopropylidenediphenol; BPA;
ICN/Millipore Biomedicals). Cells were also treated with 10-6M testosterone and 10-6M
progesterone. Although cells were in sterile tubes, the water bath was not sterile, thus cells
were monitored for viability and contamination. Cell viability was excellent, and no cultures
showed any sign of contamination.
11.3 PDGF-depleted FBS
FBS was prepared overnight at 4℃ with specific antibodies against PDGF-AA, BB and AB
(Biodesign International and Oncogene research products). In order to remove antibody-PDGF
complexes, the FBS was run through a protein A HiTrap column that binds antibodies together
with their antigens. Removal of most of the PDGF was documented with Western blot analysis.
11.4 Protein Analysis – Western Blot
Western blot analysis was carried out in order to a) determine gonocyte expression of
cell signalling molecules, growth factors and hormone receptors as well as germ cell markers,
and b) investigate changes in expression of these factors when gonocytes were treated with
PDGF and estrogen in the short term. Expression of these molecules provides clues for the
mechanisms behind gonocyte proliferation as well as the commitment of gonocytes to
differentiation. Changes in expression can illustrate which factors are activated or expressed
after short term exposure to PDGF and estrogen, leading to an understanding of the crosstalk
between the two distinct pathways.
The cell cultures were stopped after 30-60 minutes by addition of 10µM Protease
inhibitor cocktail (Sigma P8340), 10µM phosphatase inhibitor (Roche, PhosSTOP – 1 tablet for
10mL) and 4µM okadaic acid (Calbiochem, 495609) and centrifugation at 4℃ for 2 minutes at
5000g. The supernatant was removed and the cells were lysed and stored in a Laemmli buffer
containing 1X DTT including protease and phosphatase inhibitors as well as bromophenol blue
57

as a loading dye. Proteins were quantified using a BCA assay and spectrophotometer. Protein
samples were loaded onto a 4-20% or 10% tris-glycine (Invitrogen) gel and proteins were
separated via SDS-page electrophoresis. 20-30μg of protein or protein from 20,000-30,000
cells was applied to each well in the electrophoresis gels. Following electrophoresis, the
samples were transferred onto a nitrocellulose membrane using 1X Novex Tris-Glycine Transfer
Buffer (Invitrogen LC3675, 25X stock) in 20% methanol. Membranes were blocked with 5%
powdered milk or 5% BSA and incubated overnight with specific primary antibodies at
concentrations of 1:250 to 1:1000 in 1X TTBS at 4℃. The primary antibodies included
antibodies specific for ERK and phosphorylated-ERK (p-ERK) (PhosphoPlus MAPK kit from Cell
Signalling #9100), Raf1, PDGFRβ, MEK1, PI3K (from Santa Cruz #SC227, SC432, SC219, SC7189),
MIS/AMH (R&D MAB1146), ERβ (ABR PA1-313), ERα (Upstate #06-935), and PDGFRα (Upstate
#07-276). Secondary antibodies were incubated at 1:10,000 dilutions and were coupled with
streptavidin-horseradish-peroxidase (HRP) (Zymed Invitrogen Corp.). Loading controls for these
samples were histone-1 and Tubulin (Chemicon MAB052 and Abcam ab0742-300 HRP bound).
Negative controls were performed by incubation with rabbit and mouse IgG instead of primary
antibody or by using a secondary antibody alone, prior to reprobing the membranes with
specific primary antibodies. ERβ antibody signal was effectively blocked by incubation with the
specific peptide used to produce the antibody. The immunoreactive bands were visualized
using ECL-Plus detection reagents (Amersham). The images were collected and densitometry
analysis performed on a FujiFilm Luminescent Image Analyzer (LAS-4000) and Multi Guage V3.0
software. Using normalization against tubulin and histone gave quialitativly similar results but
higher variability due to the inconstancy in the Histone and Tubulin immunoreactions between
different experiments. Within each experiment, the same amount of total protein per sample
was loaded onto the polyacrylamide gels. In some of the experiments we verified, by using two
different protein concentrations on the same gel, that the immunoreactions of the proteins of
interest were proportional to the amount of protein loaded on the gel. Therefore we chose to
directly compare the immunoreactive bands of treated samples with the control and expressed
the data as a fold change for each protein analyzed. This allowed us to better express the
average changes observed between numerous experiments.
58

11.5 Nuclear Isolation
Nuclear extractions were performed to provide insight into which of the downstream
signalling molecules were activated inside the nucleus or translocated into the nucleus at the
time of activation following PDGF and estrogen treatments.
The cells were incubated for 30 or 60 minutes at 37℃ with the different reagents, then
protease and phosphatase inhibitors were added and the cells were centrifuged for 2 minutes
at 5000g and the supernatants were removed. The samples were kept on ice for the duration
of the cell fractionation and the cell pellets were resuspended with tissue homogenizing buffer
(with added protease and phosphatase inhibitors) and a cell fractionation buffer (0.25M
sucrose, 25mM KCl, 50mM Triethanolamine, 5mM MgCl2, 0.5mM PMSF, 1mM DTT). Cells were
transferred to a manual glass cell homogenizer tube and were lysed with a Teflon coated tissue
grinder. The cell lysates were centrifuged at 800g in order to pellet the nuclei, while cytosolic
proteins and small organelles (mitochondria, ER…) remained in the supernatant. The nuclear
fraction was resuspended in Laemmli buffer. The supernatant was transferred to a 10kDa
microcon filter (Millipore, YM-10) to concentrate the proteins in the supernatant. Most of the
remaining buffer was removed by centrifugation at 4℃ for 1 hour at 14,000g, and the
remaining protein concentrates were collected in a microcentrofuge tube and further
resuspended in Laemmli buffer.
11.6 Immunocytochemistry
Immunocytochemistry was used to localize the various signalling molecules expressed in
the gonocytes that could be activated during proliferation. These experiments helped localize
important signalling players and possible crosstalk mechanisms that could be involved in
proliferation.
Isolated gonocytes were kept for 30 or 60 minutes in microcentrofuge tubes in a 37℃
water bath. The reaction was stopped with 4% paraformaldehyde combined with phosphatase
inhibitors, protease inhibitors and okadaic acid for 7 minutes. The cells were then centrifuged
for 3 minutes at 5000g and resuspended in PBS. Cytological slides of the gonocytes were
59

prepared by cytospin for 10 minutes at 300RPM. The slides were fixed in a 60:40% acetone
methanol mixture for 7 minutes and stored at −20℃. Immunocytochemistry was performed
by exposing slides to 10% Dako-Cytomation Target Retrieval solution (Dako North America Inc.)
at 95℃, then blocking with PBS with 10% BSA and 10% goat serum. Primary antibodies were
applied overnight at 4℃ in 1:25 or 1:100 dilutions. The secondary antibodies were
fluorescently-coupled rabbit and mouse Alexa 488 and 546 antibodies from Invitrogen, and
were incubated with the slides for 1 hour in the dark at 1:300 dilutions. A nuclear (4’,6’-
Diamidino-2-phenylindole, DAPI, Invitrogen) fluorescent stain was applied and the slides were
mounted with Permafluor aqueous mounting medium (Thermo Scientific) and covered with a
glass coverslip. The slides were observed via fluorescent or confocal microscopy. Negative
controls were performed using mouse and rabbit IgG as well as secondary antibody alone.
Immunocytochemistry was used to investigate protein expression of Raf1, MEK1, ERK1/2, p-
ERK1/2, MIS/AMH, PI3K, ERα, ERβ, PDGFRα, and PDGFRβ. Studies were conducted to observe
the expression of certain proteins in control gonocytes as well as to investigate the short term
effects of protein expression and localization following the given treatments.
Photomicrographs were taken of the treated slides focusing the objective to an optimal DAPI
staining to ensure that the picture is taken in the middle of the cell, and intensity of the protein
expression was quantified using ImagePro 6.3 analysis software.
11.7 Proliferation Assay
Cells were cultured with one or more of the following reagents: PDGF-BB, 17β-estradiol,
xenoestrogens, testosterone and progesterone, with or without the specific inhibitors AG370,
UO126 and ICI 182780. The cells were incubated overnight together with 30µg/mL 5’-Bromo-
2’-deoxyuridine (BrdU) and 3µg/mL 5-fluoro-2’-deoxyuridine as described in the cell
proliferation kit from Amersham. Following cell culture and cytospin, the slides were fixed and
stained with a biotinylated anti-BrdU antibody (Neomarkers or Exalpha). The cells were
counted and positive staining was calculated as a percent of the total number of cells. PCNA
was used in some experiments to compare to BrdU in determining the percentage of
proliferating cells. PCNA gave similar results to BrdU.
60

11.8 Immunohistochemistry
Important for understanding the whole picture of PND3 gonocytes, expression of
important factors and receptors were investigating using 5 μm testes paraffin tissue sections.
Paraffin sections of PND3 testes were stained for protein expression of Raf1, MEK1,
ERK1/2, PI3K and MIS/AMH. Two methods were used for deparaffination and antigen retrieval.
Trilogy solution was used in an electric pressure cooker according to the company’s
instructions. Alternatively, slides were rehydrated in xylene and different dilutions of alcohol
and were then heated to 95℃ with DAKO antigen retrieval solution and blocked with 1% BSA,
10% goat serum and 0.02% Triton X100 to prevent nonspecific antibody binding. Primary
antibodies were diluted in PBS with 1% BSA, 0.02% Triton X100 and were incubated at a 1:100
dilution overnight at 4℃. Fluorescent secondary antibodies were used and the nucleus was
stained with either Hoechst 33342 (Invitrogen) or DAPI. The sections were mounted with
ProLong Antifade solution and were viewed under a fluorescent microscope. The same
negative controls were applied on the tissue sections as for the cytospin slides.
11.9 V1-PDGFRβ Vector Transfection and Live Cell Imaging
Green fluorescent protein (GFP)-tagged V1-PDGFRβ variant was transfected into
gonocytes to a) see if we could transiently overexpress a GFP-labelled protein in gonocytes and
b) to monitor V1-PDGFRβ expression and localization in treated cells to observe if V1 plays a
role in short term induction of gonocyte proliferation. Rats were dissected at PND2 and
incubated overnight with a V1-PDGFRβ constructed plasmid. The plasmid was constructed by a
previous student (Wang and Culty (2007) who previously characterized V1-PDGFRβ. The coding
sequence included the complete coding region between exon7 and exon23 of the PDGFRβ
gene. The plasmid was prepared in opti-MEM with a 1:1 ratio in Lipofectamine. Sertoli cells
from the dissection were also plated and incubated on glass confocal plates to prepare a Sertoli
cell matrix for the gonocytes. Following incubation, gonocytes were further purified on a BSA
gradient. They were plated on the Sertoli cell matrices and incubated overnight again in RPMI,
without serum, with V1-PDGFRβ plasmid. This allowed the gonocytes to settle into the matrix
of Sertoli cells and adhere to the somatic cells, also providing more time for the plasmid to be
61

transfected into the cells. After overnight incubation, gonocytes were treated with PDGF-BB
and 17β-estradiol in different combinations with and without inhibitors, and were monitored
for 60 minutes using live cell imaging by confocal microscopy. Short term profiles of the V1-
PDGFRβ expression was observed and photomicrographs were taken.
11.10 Statistical Analysis
The statistical analysis for this study was performed using Microsoft Excel’s
mathematical functions unpaired, two-tailed t-test. Each of the treatments was compared
directly to the control subject, as well as treatments were paired up against each other for
statistical analysis as well. The histograms presented in this study were assembled using
GraphPad Prism (version 5).
62

12. Results
12.1 Charcoal Stripped FBS and PDGF-Depleted Serum
Our first step was to verify that normal FBS was able to provide a steady basal level of
both PDGF and estrogen that would explain why each individual agent was able to stimulate
gonocyte proliferation without addition of the other one. For this, gonocytes were incubated
overnight with PDGF or 17β-estradiol in the presence of either charcoal stripped FBS (void of all
steroids), or PDGF-depleted FBS (figure 5). Gonocytes in culture with charcoal stripped FBS did
not display PDGF-induced proliferation, while cells treated in PDGF-depleted serum also could
not induce estrogen-stimulated proliferation. When 10-6M 17β-estradiol was added to the
charcoal stripped serum, treatment with PDGF was again able to induce proliferation. Similarly,
when PDGF was added to the PDGF depleted serum, estrogen-induced proliferation was re-
established.
12.2 Gonocyte Expression of Downstream Molecules of the PDGF Signalling
Pathway
Studies performed earlier in our lab identified the mRNA expression of Erk1, Raf1, Mek1
and PI3K through in situ hybridization in PND3 rat gonocytes. Erk1 had the highest mRNA
expression while PI3K mRNA was in much lower quantity. To confirm that gonocytes express
the corresponding proteins of these transcripts, we investigated their protein expression using
immunohistochemistry of PND3 normal rat testes. Raf1, Mek1, ERK1/2 and PI3K were all
expressed in the seminiferous cords, specifically located in the cytoplasm of Sertoli cells and
gonocytes, and once again PI3K signal was faint (figure 6A). The Sertoli cell marker AMH/MIS
was also present in the seminiferous cords of the testis sections. We then performed
immunocytochemical analysis to confirm these findings in isolated gonocytes using cells
collected and immunostained immediately after gonocyte isolation from PND3 rats. These
experiments confirm that ERK1/2, Raf1 and Mek1 were expressed in gonocytes (figure 6B). It
was also noticeable that of the morphologically smaller somatic cells present with the
gonocytes, some stained positively for all these factors, while others did not. There were also
AMH positive and AMH negative cells, which most probably distinguished between Sertoli cells
63

*
** **
FBSCharcoal-striped FBS
PDGF17β-estradiol
+---
-+--
+-+-
-++-
+-++
-+++
50
40
30
20
10
0Pr
olife
ratio
n ra
te (%
)
***
***
FBSPDGF-depleted FBS
PDGF17β-Estradiol
*** ***
+---
-+--
+--+
-+-+
+-++
-+++
50
40
30
20
10
0
Prol
ifera
tion
rate
(%)
**
Figure 5: PDGF- and estrogen-induced proliferation in the presence of serum devoid ofsteroids or PDGF. Gonocytes isolated at PND3 were treated with PDGF and estrogen ina medium containing either normal serum, PDGF- or steroid-depleted serum. The cellswere incubated overnight with BrdU, and the proliferating BrdU-positive cells werecounted to calculate their percentage against the total gonocyte numbers. Each valuerepresents proliferation rates ± SEM and was calculated from 3 experiments. For all ofthe histograms in this paper, statistical analysis was performed using a two tailed T-testand the significance values are given as follows: *: P<0.05, **: P<0.01, ***: P<0.001.
64

and PMCs. It is interesting to note that there were some gonocytes expressing AMH, most
likely due to AMH uptake by gonocytes following Sertoli cell secretion. Gonocytes were
examined after one day in culture following isolation, and we were able to confirm the
continued expression of these downstream molecules. The strongest signals were from ERK1/2
and Mek1. Finally, Western blot analysis also confirmed the presence of these downstream
proteins, and we showed that the more prominent ERK isoform in PND3 gonocytes is ERK1
which is expressed ten times higher than ERK2 (figure 6C). Somatic cells had a higher
expression of the proteins examined in total protein extracts as compared to gonocytes (figure
6C).
Given the results above, our lab set out to determine which of the two downstream
pathways was involved in PDGF-induced proliferation. It was observed that gonocyte
treatment with specific inhibitors for Raf1 (iRAF1) and for MEK1/2 (PD98059 and UO126)
significantly attenuated the proliferative response to PDGF-BB following overnight treatment,
while the PI3K inhibitor, wortmannin, did not have a negative effect on PDGF-induced
proliferation of gonocytes at PND3 (figure 7). In order to address the hypothesis that the
effects of PDGF and estrogen are interdependent and crosstalk to stimulate proliferation,
studies were performed where inhibitors for one pathway were incubated with the stimulator
of the other pathway. ICI 182780 was able to negatively affect the proliferative effects of
PDGF-stimulated gonocytes, while AG370 reduced the response to gonocytes to estrogen
stimulation. Similarly, the inhibition of Raf1 and MEK1/2 in the MAPK cascade blocked the
estrogen-induced proliferation (figure 7).
12.3 In Vitro exposure to xenoestrogens and phytoestrogens induce proliferation in
neonatal gonocytes
Earlier experiments in the lab demonstrated that, in the same manner as 17β-estradiol
induced proliferation in PND3 gonocytes, exogenous estrogenic compounds commonly found in
the environment such as genistein and BPA as well as the potent xenoestrogen DES caused
gonocyte proliferation in a dose dependant manner, although the phytoestrogen coumestrol
did not. Addition of estrogen receptor antagonists and PDGF inhibitors showed that these
65
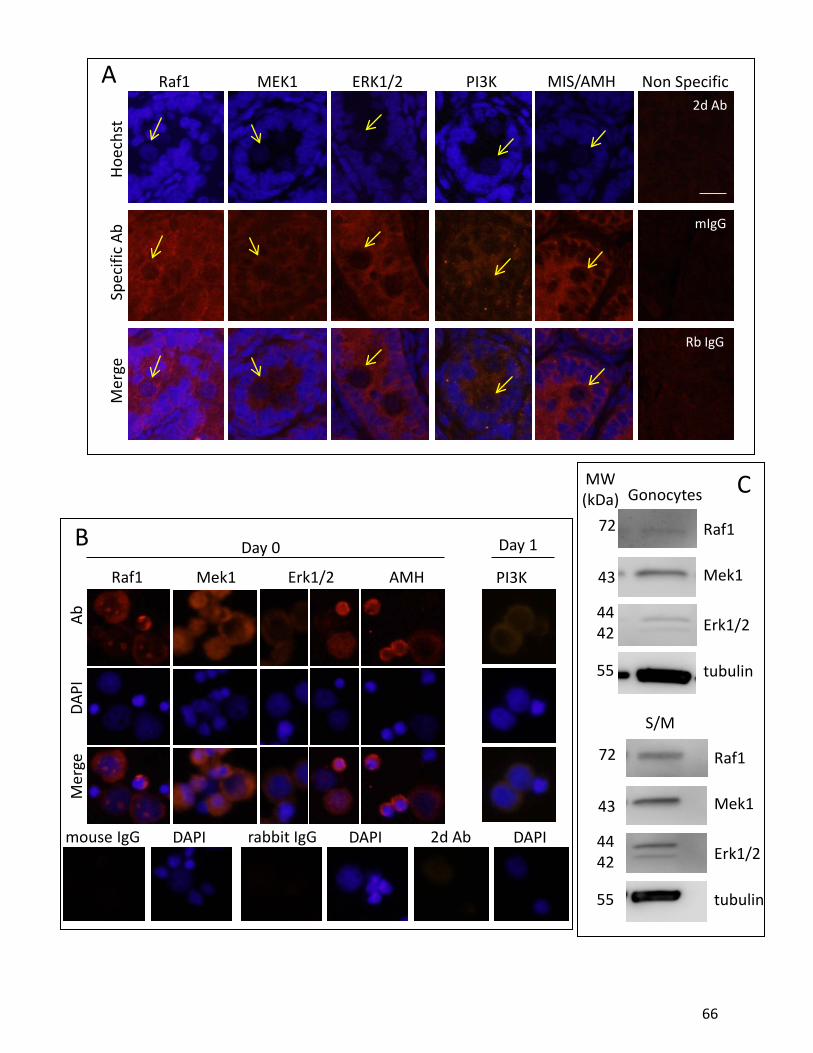
PI3K
DAPI 2d Ab DAPI
Mek1 Erk1/2Raf1 AMH
mouse IgG
DAPI
Mer
geAb
Day 0 Day 1
rabbit IgG DAPI
Raf1 MEK1 ERK1/2 PI3K Non Specific
Hoec
hst
Spec
ific
AbM
erge
MIS/AMH2d Ab
mIgG
Rb IgG
tubulin
S/M
Gonocytes
Raf1
Mek1
Erk1/2
MW(kDa)
4442
72
55
43
4442
72
55
43
tubulin
Raf1
Mek1
Erk1/2
A
B
C
66

Figure 6: Identification of the expression of MAPK downstreamsignalling pathways, PI3K and MIS/AMH in PND3 isolatedgonocytes and testis sections. Previous studies in our lab identifiedmRNA levels of MAPK molecules. (A) Immunohistochemistry wasperformed on PND3 rat testis sections to observe protein profiles ofmolecules that are generally associated with PDGF signalling in thewhole testis. Specific fluorescent antibodies were used to detectprotein expression and a Hoechst nuclear dye was used to identifysingle cells in the tissue. Negative controls shown for non-specificIgG and secondary antibodies alone. (B) Isolated gonocytes werecollected on cytospin slides and the cells treated forimmunocytochemistry at day 0, right after the BSA gradient, andafter one day in culture. Negative controls are shown for rabbit andmouse non-specific IgG and secondary antibody alone. Nuclearstaining was performed using DAPI. (C) Western blot analysisconfirmed protein profiles, and was used to compare proteinexpression in gonocytes to Sertoli and Myoid cells (S/M). Thegonocytes were lysed on day 0, directly after the BSA gradient.
67

compounds behaved similarly to 17β-estradiol. However, in these experiments performed in
the presence of 2.5% serum, genistein and BPA acted at concentrations much lower than
anticipated from their affinity for ERs. Indeed, 17β-estradiol induced proliferation at
concentrations of 10-9M and above, while genistein, BPA and DES were all able to stimulate
proliferation starting at 10-12M. Thus we conducted further experimentation to explore
whether the presence of serum could explain these data. Dose response studies were
performed with genistein and BPA in media deficient of FBS, PDGF-depleted serum or with
charcoal-stripped FBS to see how the xenoestrogens respond in the absence of steroids or
growth factors from the serum. When no FBS was used, PDGF treatment required the addition
of an estrogenic compound such as 17β-estradiol, genistein or BPA to induce proliferation,
reaching a 2-3 fold increase at 10-6M for all of the estrogens (figure 8A). Gonocytes treated in
the presence of PDGF-depleted serum required both PDGF and 10-9M estrogenic compound in
order to induce the same 2-3 fold increase in proliferation. Significant proliferation was also
observed using 10-9M genistein, BPA or 17β-estradiol in the charcoal stripped FBS, which should
still contain other growth factors. These experiments showed that, under these conditions,
exogenous estrogens work in the same manner as 17β-estradiol with similar dose-response
curves (figure 8B). Given that there is little potential to be exposed to only one exogenous
estrogenic compound at a time, we also exposed gonocytes simultaneously to both genistein
and BPA, each in minute, fentomolar concentrations. We had observed that at these
concentrations, neither of the two molecules induced gonocyte proliferation. However, the
two molecules added together indeed did result in a significant increase in gonocyte
proliferation (figure 8C).
12.4 Treatment of Gonocytes with Other Steroid Hormones
As it is well documented that testosterone is the “male sex hormone”, we investigated
whether or not testosterone and another steroid hormone important in sexual differentiation
and function, progesterone, played a role in the proliferative stage of PND3 gonocytes.
Gonocytes were treated with 10-6M testosterone or 10-6M progesterone overnight with or
without FBS. Increased proliferation above the basal rate was not observed in these
68

***
**
PDGF
AG-3
70+ -
- ++ +
- -
Proliferation rate (%)40 30 20 10 0
**
PDGF
iRaf
1- -
+ -- +
+ +
Proliferation rate (%)40 30 20 10 0
**
**
PDGF
UO
126
PD98
059
- - -
+ - -
- + -
+ + -
- - +
+ - +
Proliferation rate (%) 40 30 20 10 0PD
GFW
ortm
anni
n
***
ns
- -+ -
- ++ +
Proliferation rate (%)40 30 20 10 0
*
E2iR
af1
D**
**
- -+ -
- ++ +
Proliferation rate (%)40 30 20 10 0E2
UO
126
PD98
059
***
**
- - -
+ - -
- + -
+ + -
- - +
+ - +
40 30 20 10 0
E
Proliferation rate (%)
***
***
PDGF E2
AG-3
70
- - -
+ - -
- + -
+ + -
- - +
- + +
***
40 30 20 10 050 Proliferation rate (%)
***
***
***
E2IC
I 182
780
- -+ -
- ++ +
Proliferation rate (%) 40 30 20 10 050*
***
***
PDGF
ICI 1
8278
0- -
+ -- +
Proliferation rate (%)40 30 20 10 0
+ +
***
Figu
re7:
PDG
Fan
des
trog
en-in
duce
dpr
olife
ratio
nof
gono
cyte
sin
the
pres
ence
ofsp
ecifi
cin
hibi
tors
.PN
D3go
nocy
tes
wer
etr
eate
dov
erni
ghtw
ithth
efo
llow
ing
trea
tmen
tsin
the
pres
ence
of2.
5%FB
San
dBr
dUin
cultu
reto
mea
sure
the
prol
ifera
tion
rate
ofth
ece
llsin
each
trea
tmen
t.Go
nocy
tes
wer
etr
eate
dw
ithPD
GFw
ithan
dw
ithou
tsp
ecifi
cin
hibi
tors
for
itsty
rosin
eki
nase
rece
ptor
(AG3
70)
and
dow
nstr
eam
path
way
,bl
ocki
ngRa
f1(iR
af1)
and
MEK
1(U
O12
6an
dPD
9805
9).
Incu
batio
nw
ith17
beta
-es
trad
iolw
aspe
rform
edw
ithor
with
outI
CI18
2780
.sim
ilarly
PDGF
trea
tmen
twas
incu
bate
dw
ithth
eER
anta
goni
stan
dce
llsw
ere
trea
ted
with
estr
ogen
alon
gsid
ein
hibi
tors
ofth
ePD
GFR
and
dow
nstr
eam
path
way
.The
sere
sults
indi
cate
acr
osst
alk
betw
een
the
two
path
way
s.Ce
llsw
ere
colle
cted
oncy
tosp
insli
des
and
coun
ted
forp
ositi
vepr
olife
ratio
nst
aini
ng.T
hepe
rcen
tofp
rolif
erat
ing
cells
show
nar
eth
em
eans
±se
mof
seve
rale
xper
imen
ts.
69
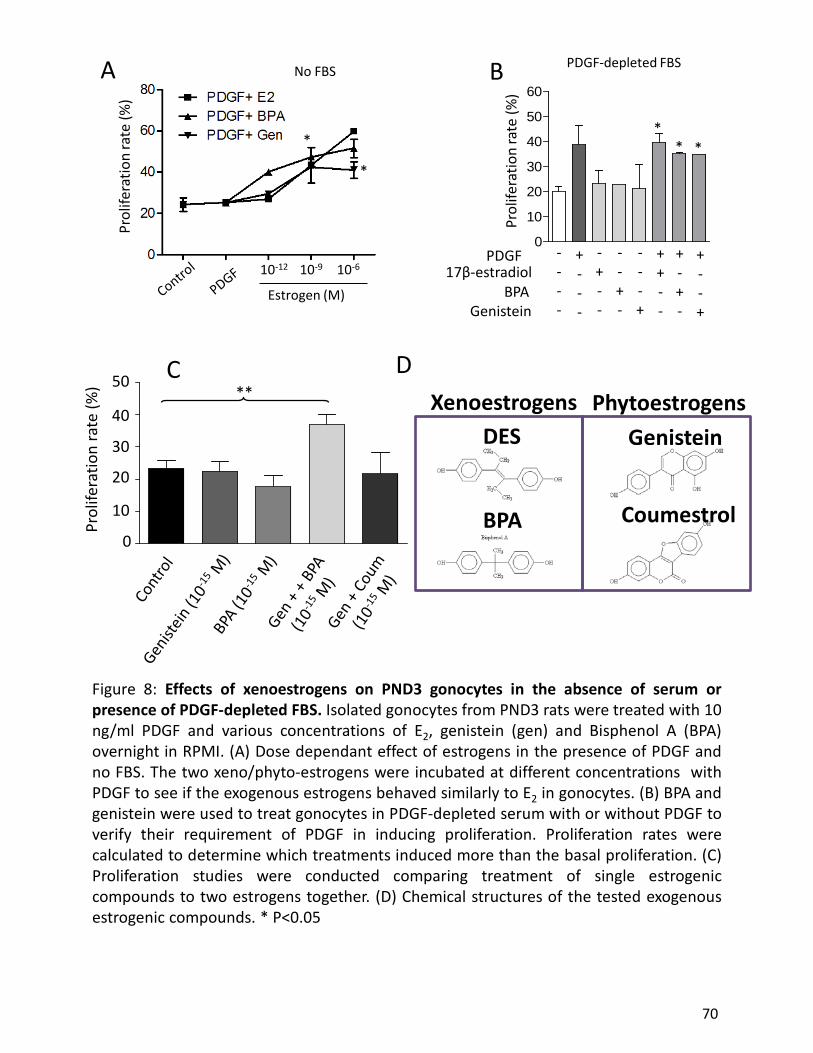
Figure 8: Effects of xenoestrogens on PND3 gonocytes in the absence of serum orpresence of PDGF-depleted FBS. Isolated gonocytes from PND3 rats were treated with 10ng/ml PDGF and various concentrations of E2, genistein (gen) and Bisphenol A (BPA)overnight in RPMI. (A) Dose dependant effect of estrogens in the presence of PDGF andno FBS. The two xeno/phyto-estrogens were incubated at different concentrations withPDGF to see if the exogenous estrogens behaved similarly to E2 in gonocytes. (B) BPA andgenistein were used to treat gonocytes in PDGF-depleted serum with or without PDGF toverify their requirement of PDGF in inducing proliferation. Proliferation rates werecalculated to determine which treatments induced more than the basal proliferation. (C)Proliferation studies were conducted comparing treatment of single estrogeniccompounds to two estrogens together. (D) Chemical structures of the tested exogenousestrogenic compounds. * P<0.05
B
Prol
ifera
tion
rate
(%)
10-12 10-9
Estrogen (M)
10-6
*
*
0
10
20
30
40
50
60
PDGF
Prol
ifera
tion
rate
(%)
17β-estradiol
GenisteinBPA
+---
----
---+
+-+-
--+-
+--+
-+--
++--
** *
PDGF-depleted FBSNo FBSA BPr
olife
ratio
n ra
te (%
) 50
40
30
20
10
0
**C
XenoestrogensDES
BPA
PhytoestrogensGenistein
Coumestrol
D
70

2.5% FBS No FBS
Control
PDGF+E2
Progesterone
TestosteronePr
olife
ratio
n ra
te (%
)
***
Figure 9: Steroid hormone proliferative effect on PND3 gonocytes with and without serum. PND3 gonocytes were treated with 10-6 M testosterone and progesterone and examined for proliferative effect using BrdU in the presence and absence of FBS.
71

experiments, which confirms that the effect of estrogenic compounds on gonocytes is specific,
and that gonocytes are not stimulated to proliferate by the other main steroids produced in the
testis (figure 9).
12.5 ERK2 Activation via PDGF-BB and 17β-estradiol
In order to decipher the molecules involved in the crosstalk between PDGF and estrogen
pathways in proliferative gonocytes, we performed short term time course experiments. We
investigated the phosphorylation of ERK1/2, a molecule that we had found abundantly
expressed in gonocytes, to examine whether activation of the signalling cascade leading to
transcription regulated by the ERK1/2 signaling molecule could serve as the common molecular
element and common target system of the PDGF-estrogen crosstalk. It should be noted that the
extremely low number of cells isolated in each experiment limited the numbers of conditions
that could be tested simultaneously, making it hard to compare each condition with its specific
inhibitor with the same isolation of cells. To appreciate the short term progression of the
pathway, cells were cultured for 15, 30 and 60 minutes in order to make isolated observations
at various short term time points, looking at the expression and phosphorylation (activation) of
ERK. Using Western blot analysis, we were able to distinguish between the two isoforms, ERK1
and ERK2, which are found at 44 and 42 kDa respectively. While the stimulation of gonocytes
with PDGF or estrogen for 15 min showed increased ERK2 phosphorylation in some
experiments, a consistent and overall significant increase of phosphorylated ERK2 was observed
only with the combination of PDGF and estrogen together in four independent experiments
(figure 10B). After 30 minutes of incubation, all 3 conditions caused a strong phosphorylation
of ERK2, while ERK1 phosphorylation remained significantly closer to the basal level (2
experiments). Interestingly, 15 min treatment with genistein, and to a lesser extent BPA,
induced a small increase in ERK2 phosphorylation, yet much lower than the combination of
PDGF + E2 (figure 10D). The fact that ERK2 is the main phosphorylated form of ERK is
interesting given our observation that PND3 gonocytes in vehicle alone appear to express a
much higher concentration of total ERK1 protein, rather than ERK2, and gonocytes treated with
PDGF-BB and 17β-estradiol for 60 minutes and 4 to 8 hours appear to increase their expression
72

of ERK1 protein, which remains in abundance, compared with ERK2 (figure 10B). A significant
increase in phosphorylated ERK2 was observed after 60 minutes stimulation by PDGF and
estrogen added together, but not by any of the single agents (4 independent experiments).
While the presence of UO126 inhibited the effect of PDGF + E2 on ERK2 phosphorylation, the
addition of AG370 did not block ERK2 activation in gonocytes (figure 10A).
Using immunocytochemistry, which could not distinguish between ERK1 and 2, one
could not clearly see the maintenance of phosphorylated ERK2 after 60 min that we had
measured by western blot analysis (figure 10F). However, manual counting of the number of
cells immunopositive for phospho-ERK versus negative cells, showed a clear increase at 60
minutes in the percentage of cells expressing high levels of phosphorylated ERK when treated
with PDGF, as well as a smaller increase observed when gonocytes were treated with PDGF and
E2 together (figure 10E).
The determination by western blot analysis of ERK1 and 2 activation levels in gonocyte
nuclear and cytosolic fractions showed that the majority of the increase in phospho-ERK2 was
localized in the cytosolic fraction of the cells treated with PDGF + E2, while there was no clear
change from the basal level observed in the nuclear fractions after 15 min treatment (figure
10C). This result was confirmed by immunocytochemistry and confocal microscopy, where the
majority of cells treated with PDGF + E2 presented increased phospho-ERK in their cytosol after
30 min incubation, although some cells expressed high levels of phospho-ERK1/2 in their nuclei
figure 10G). It should be noted that PND3 gonocytes are not synchronized and that any given
preparation of gonocytes may include different proportions of quiescent, mitotic and migratory
cells in function of the exact ages of the pups at the time of sacrifice (Culty, 2009). An
interesting observation is that under confocal microscopy it appears that some of the cells
expressing high levels of nuclear phospho-ERK were in the process of proliferation, being
observed in one of the later stages of mitosis (figure 14). Thus, it is possible that gonocyte
proliferation requires ERK translocation into the nucleus during one or more of the phases of
mitosis.
73

30 M
inut
e G
onoc
yte
Trea
tmen
tPh
osph
oryl
ated
ERK
CP
EP+E
P+E+A
G P+E+U
O
P+E+IC
I0246810
Trea
tmen
ts
pERK expression(fold change / Control)
15 M
inut
e G
onoc
yte
Trea
tmen
tPh
osph
oryl
ated
ERK
CP
EP+E
P+E+A
G+UO+IC
I01234
Trea
tmen
ts
pERK expression(fold change / Control)
**
60 M
inut
e G
onoc
yte
Trea
tmen
tPh
osph
oryl
ated
ERK
CP
EP+E P+E
+AG P+E
+UO P+E
+ICI
P+E+A
G+UO+IC
I012351015
Trea
tmen
ts
pERK expression(fold change / Control)
*
*
4 ho
ur tr
eatm
ent
Cont
rol
P+E
PDGF
Estr
ogen
Cont
rol
P+E
PDGF
Estr
ogen
Post
Nat
al d
ay 3
8 ho
ur tr
eatm
ent
Hist
one
P-ER
K
ERK
Phos
phor
ylat
ed E
RK1/
2N
ucle
ar E
xtra
ctio
n Ex
perim
ent
44 42kDa
Nuc
leus
Cyto
sol
CC
CC
P+E
P+E
P+E
P+E
15 m
inut
es in
trea
tmen
t60
min
utes
in tr
eatm
ent
ERK
pERK
Tubu
lin
Cont
rol
PDGF
Estr
ogen
P+E
Cont
rol
PDGF
Estr
ogen
P+E
Post
Nat
al D
ay 3
CP+
EG
BPA
pERK ERK
Tubu
lin
PND3
15
min
ute
trea
tmen
t
A BC D
74

Imm
unoc
ytoc
hem
istr
y30
Min
ute
Gon
ocyt
e Tr
eatm
ent
phos
phor
ylat
ed E
RK
CP
EP+E
AG+UO+IC
I P+AG P+U
O E+ICI
P+E+A
G P+E+U
O P+E+IC
I
P+E+A
LL020406080100
Trea
tmen
ts
Percentage of cells expressing phosphorylated ERK
Imm
unoc
ytoc
hem
istr
y60
Min
ute
Gon
ocyt
e Tr
eatm
ent
phos
phor
ylat
ed E
RK
CP
EP+E
AG+UO+IC
I P+AG P+U
O E+ICI
P+E+A
G P+E+U
O P+E+IC
I
P+E+A
LL020406080
Trea
tmen
ts
Percentage of cells expressing phosphorylated ERK
*
Imm
unoc
ytoc
hem
istr
y30
Min
ute
Gon
ocyt
e Tr
eatm
ent
phos
phor
ylat
ed E
RK in
the
nucl
eus
CP
EP+E
AG+UO+IC
I P+AG P+U
O E+ICI
P+E+A
G P+E+U
O P+E+IC
I
P+E+A
G+UO+IC
I0.
0
0.5
1.0
1.5
2.0
Trea
tmen
ts
Expression of pERK / control
Imm
unoc
ytoc
hem
istr
y30
Min
ute
Gon
ocyt
e Tr
eatm
ent
phos
phor
ylat
ed E
RK in
the
cyto
sol
CP
EP+E
AG+UO+IC
I P+AG P+U
O E+ICI
P+E+A
G P+E+U
O P+E+IC
I
P+E+A
G+UO+IC
I0.
0
0.5
1.0
1.5
2.0
2.5
Trea
tmen
ts
Expression of pERK / control
P+EP EC
pERK
30pE
RK60
pERK
30C
P
EP+
E
cytosolicstaining
E F
G
75

Figure 10: PDGF and estrogen-induced Activation of ERK2 in short term assay.PND3 isolated gonocytes were treated with PDGF with or without estrogen andinhibitors for 15, 30 and 60 minutes. Cells were stopped by either centrifugationor paraformaldehyde and then lysed for western blot analysis or fixed to cells viacytospin centrifugation for immunocytochemistry experiments. (A) western blotprofiles of phosphorylated ERK2. the gels were evenly loaded with the samequantities of protein per well, and therefore we normalized the densitometry ofthe protein bands on the membrane only to the control value of eachexperiment. The results are expressed as the changes in ERK activation betweencontrol (1) and treatment. (B) western blot representations of ERK, phospho-ERKand tubulin/Histone for PND3 gonocytes treated for 15 and 60 minutes as well as4 and 8 hours. (C) Representative membrane expressing phosphorylated ERKfrom a nuclear extraction experiment where the nuclear and cytosolic fractionswere separated and investigated for differences in protein profiles. ERK activationappears to be more prominent in the cytosol than the nucleus. (D) As we havealready observed that genistein and BPA modulate gonocyte proliferation in asimilar manner to 17β-estradiol, this western blot representation shows howgenistein also activates ERK in short term cultures. (E) immunocytochemistryexperiments were performed for short term ERK activation. Pictures of the cellswere captured and cells were counted for activation or inactivation. The data isrepresented as a percentage of the total population of activated cells. (F) Totaland nuclear intensity of immunoreactivity of ICC were calculated. Subtraction ofthe nuclear fraction from the total cell gave the intensity of the given protein inthe cytosolic ring. These histograms compare changes in the control and treatedcell expression of phospho-ERK between the nucleus and cytosol. (G)representative ICC fluorescent pictures of control and treated cells colocalizedwith DAPI nuclear stain. The bottom four pictures are the primary anti phospho-ERK immunoreactivity without the DAPI showing that most of the cells have adistinct cytosolic rings.
76

As expected, UO126 inhibited the ERK1/2 activation in gonocytes treated with PDGF.
Surprisingly, ICI together with E2 or added to the combination of PDGF + E2 induced a stronger
phospho-ERK immunoreactive signal in gonocytes and a higher ratio of positive cells than when
E2 or PDGF were added alone, despite the long term inhibitory effect exerted by ICI on E2- and
PDGF-induced proliferation (figure 10E, figure 11). The same observation was made by western
blot analysis of cells exposed to ICI (figure 10A).
12.6 PDGF and Estrogen Increase Expression of PDGFRβ and ERβ Immunoreactivity
In Vitro
Treatment of PND3 gonocytes with PDGF-BB induced a significant increase in PDGFRβ
immunoreactivity in the cells in general and specifically in the cytosol, as seen via
immunocytochemistry. This expression was seen through the quantification of
immunofluorescent signals in treated cells using an anti PDGFRβ antibody that showed an
increase in PDGFRβ after 30 minutes of treatment with PDGF, estrogen and the two in
combination (figure 12A). Confocal microscopy as well as immunocytochemistry confirmed the
quantitative data, showing that a majority of cells expressed PDGFRβ in the cytosol (figure 12B).
PDGFRβ immunofluorescence was present at high amounts in several dividing cells, and
confocal microscopy analysis showed a number of cells expressing tremendous amounts of
PDGFRβ in the nucleus (figure 14). This is a clear example of the heterogeneity of the isolated
gonocyte population. The clarification of the different PDGFR identities and profiles could be
instrumental in understanding this critical stage of gonocyte development. The nuclear
PDGFRβ is very likely to be the V1 isoform as we observed in live cell imaging experiments (see
section on live cell imaging and discussion). Another important observation is that not all
gonocytes at this stage express PDGFRβ and some cells were seen by confocal microscopy to be
negative for PDGFRβ expression.
Unlike PDGFRβ and activated ERK, which are most commonly found in the cytosol of
gonocytes, ERβ has a number of different expression profiles, all of which are expressed
throughout early neonatal life. Confocal microscopic imaging of PND3 gonocytes stained with
an ERβ antibody presented up to four different protein profiles in a single section. Gonocytes
77

were found to either be completely negative for ERβ, expressed ERβ mostly in the cytosol or
mostly in the nucleus, and on rare occasions, expressed ERβ in the nucleoli of the cell (figure
14). However, ICC analysis showed that the majority of ERβ-expressing gonocytes exhibited
simultaneously cytosolic and nuclear signals (figure 13). Gonocytes with pseudopod-like
projections often expressed high levels of ERβ in the pseudopod (figure 14). An interesting
observation could be made by examining protein expression in dividing cells. Noticeably, some
dividing cells expressed high ERβ levels while others did not. Understanding at which phases
ERβ is or is not expressed can be very important to our understanding of its role in gonocyte
proliferation.
Treatment with PDGF-BB or 17β-estradiol for 60 minutes induced an increase in ERβ
immunoreactivity in the cytosolic ring. These data were supported by western blot analysis of
nuclear and cytosolic fractions of gonocytes in which 60 minutes treatment with either PDGF-
BB or 17β-estradiol caused an apparent increase in cytosolic ERβ compared to the control
(figure 13).
12.7 Live Cell Imaging of Gonocytes Transfected with an EGFP-V1-PDGFRβ
Construct, Preliminary Observations
Because the ICC data demonstrated a relatively strong signal for PDGFRβ in the cytosol
and nucleus that most likely corresponded to the V1-PDGFRβ variant previously found in
gonocytes, preliminary experiments were conducted to investigate its expression and
subcellular localization in control versus PDGF and E2 treated cells. Our laboratory previously
showed that V1-PDGFRβ is missing part of the transmembrane domain, and it was found to be
expressed in the cytosol of transfected cell lines (Wang and Culty, 2007). A first attempt to
transfect PND3 gonocytes for 24 hours without serum yielded only 10-20% positive cells.
Because gonocytes represent roughly 1% of the total cell population isolated from the
seminiferous tubules of PND3 testes and thus the number of cells is always limiting, it was
critical to optimize the method and improve the yield of EGFP-V1-PDGFRβ transfected
gonocytes. We decided to dissect one day earlier, at PND2, in order to allow an extra day of
transfection before the BSA gradient, which separates gonocytes from the Sertoli cells, and to
78

ImmunocytochemistryGonocyte 30 minute treatment
phosphorylated ERKICI 182780 dose response study
CICI50
ICI100
E+ICI50
E+ICI100
0
20
40
60
Treatments
Perc
enta
ge o
f cel
ls e
xpre
ssin
g p
hosp
hory
late
d ER
K
Figure 11: Dose response curve for ICI treatment of gonocytes. A singleexperiment was performed to see if there was a dose dependent effect of ICI onthe cells. Immunocytocytochemistry was performed and fluorescent pictures weretaken of the cells. Cells expressing high levels of pERK were counted versus thecells that had barely any immunoreactive signal.Nearly a doubling of cells expressing phosphorylated ERK was seen when the ICIconcentration in gonocyte treatment for 30 minutes was doubled, reflecting ashort-term agonistic effect of ICI 182780 leading to ERK activation in gonocytes.
79
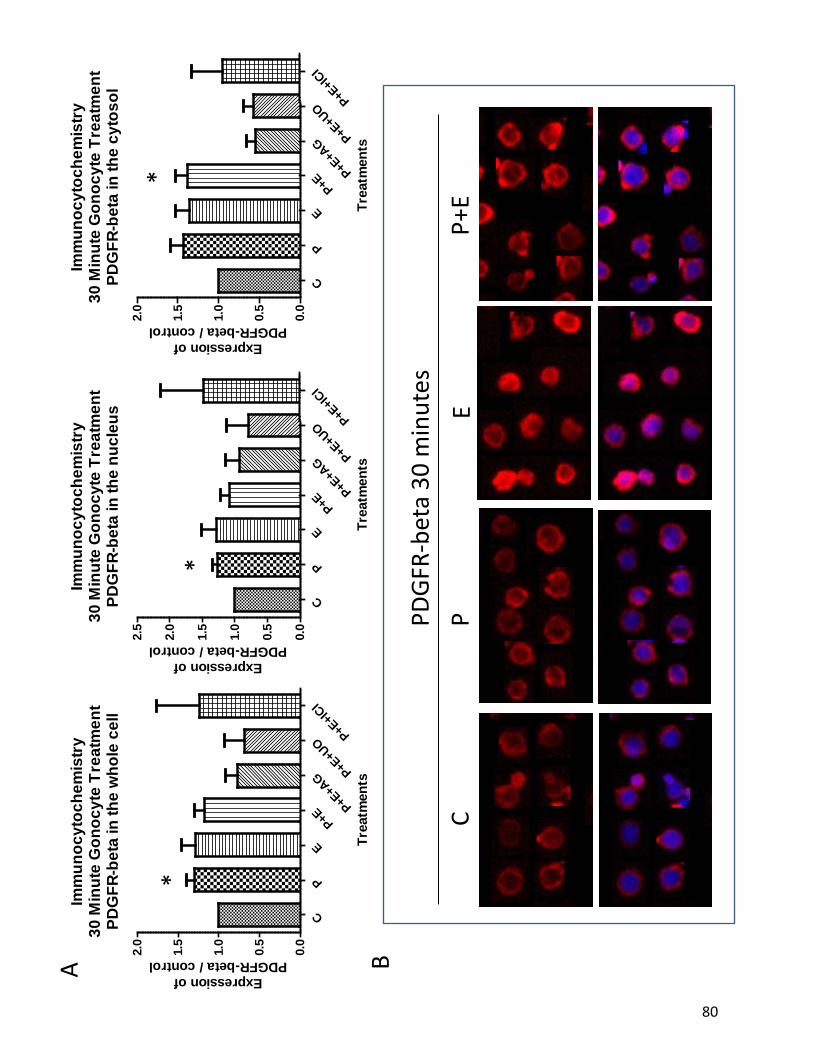
CP
EP+
EPD
GFR-
beta
30
min
utes
Imm
unoc
ytoc
hem
istr
y30
Min
ute
Gon
ocyt
e Tr
eatm
ent
PDG
FR-b
eta
in th
e nu
cleu
s
CP
EP+E
P+E+A
G P+E+U
O
P+E+IC
I0.
0
0.5
1.0
1.5
2.0
2.5
Trea
tmen
tsExpression of
PDGFR-beta / control
Imm
unoc
ytoc
hem
istr
y30
Min
ute
Gon
ocyt
e Tr
eatm
ent
PDG
FR-b
eta
in th
e w
hole
cel
l
CP
EP+E
P+E+A
G P+E+U
O
P+E+IC
I0.
0
0.5
1.0
1.5
2.0
Trea
tmen
ts
Expression ofPDGFR-beta / control
Imm
unoc
ytoc
hem
istr
y30
Min
ute
Gon
ocyt
e Tr
eatm
ent
PDG
FR-b
eta
in th
e cy
toso
l
CP
EP+E
P+E+A
G P+E+U
O
P+E+IC
I0.
0
0.5
1.0
1.5
2.0
Trea
tmen
ts
Expression ofPDGFR-beta / control
**
*
A B
80

Figure 12: Effects of PDGF and estrogen treatment on PDGFR-betain short term PND3 gonocyte assays. (A) ICC quantification ofimmunofluorescent intensity was calculated as a total intensity ofthe signal for each cell. Quantifications were taken for the nucleararea and the whole cell and subtraction of the two provided theintensity in the cytosolic ring. Histograms show the changes in thetreatments against the control values for the nucleus, cytosol andtotal cell intensities. Changes in the immunofluorescence profiles ofPDGFR-beta could be seen at 30 minutes in culture. (B)Representative ICC pictures are shown for control and treated cellsfor 30 minutes in treatment. The top row shows the PDGFR-betaprotein in each treatment, while the bottom row colocalizes theprotein with the nuclear DAPI stain. * p< 0.05
81

Imm
unoc
ytoc
hem
istr
y60
Min
ute
Gon
ocyt
e Tr
eatm
ent
estr
ogen
rece
ptor
bet
a in
the
cyto
sol
CP
EP+E
E+ICI P+E
+AG P+E
+UO P+E
+ICI
P+E+A
G+UO+IC
I0.
0
0.5
1.0
1.5
2.0
2.5
Trea
tmen
ts
Expression of ER-beta / control
Imm
unoc
ytoc
hem
istr
y60
Min
ute
Gon
ocyt
e Tr
eatm
ent
estr
ogen
rece
ptor
bet
a in
the
nucl
eus
CP
EP+E
E+ICI P+E
+AG P+E
+UO P+E
+ICI
P+E+A
G+UO+IC
I0.
0
0.5
1.0
1.5
2.0
Trea
tmen
tsExpression of ER-beta / control
Imm
unoc
ytoc
hem
istr
y60
Min
ute
Gon
ocyt
e Tr
eatm
ent
estr
ogen
rece
ptor
bet
a in
the
who
le c
ell
CP
EP+E
E+ICI P+E
+AG P+E
+UO P+E
+ICI
P+E+A
G+UO+IC
I0.
0
0.5
1.0
1.5
2.0
Trea
tmen
ts
Expression ofER-beta / control
*
CP
EP+
EER
-bet
a 60
min
utes
ER-b
eta
spec
ific
pept
ide
A B
82

Figure 13: Effects of PDGF and estrogen treatment on estrogen receptor-beta in short term PND3 gonocyte assays. (A) Histograms represented in thesame fashion as in the previous figure. Changes in the immunofluorescenceprofiles of ER-beta could be seen at 60 minutes in culture. (B) RepresentativeICC pictures are shown for control and treated cells for 60 minutes intreatment. The top row shows the ER-beta protein in each treatment, whilethe bottom row colocalizes the protein with the nuclear DAPI stain. Negativecontrols were conducted with primary antibody being pre-incubated with itsspecific peptide. * p< 0.05
83

p-ER
KPD
GFR
βER
β
84

ERβ
PDGFRβ p-ERK
Proliferating gonocytes
85

Figure 14: Representative gonocyte images captured with confocal microscopy forlocalization of phosphorylated ERK (p-ERK), PDGFRβ and ERβ. (A) PND3 gonocyteswere incubated for 30 to 60 minutes with various combinations of PDGF and 17β-estradiol. Due to the time constraints and low number of cells when dealing withprimary cultures and confocal microscopy, at this point we have not observedsteady trends between the different treatments. However, by carefully analyzing alarge population of cells under the confocal microscope, we can make a number ofimportant observations that can provide some insights on possible mechanismsinvolved in the different phases of neonatal gonocyte development. ERβ is found inmany different patterns in gonocytes and there is not one protein profile thatstands out as the most common. Some cells have a very distinct cytosolic ring whileothers express ERβ only in the nucleus and others only in the nucleoli. PDGFRβ ispreferentially expressed in the cytosol with most cells presenting a very distinct ringaround the nucleus, while a smaller sub-population of gonocytes do not expressPDGFRβ at all. However, there is also a distinct subset of gonocytes that expressPDGFRβ in the nucleus, including dividing gonocytes, suggesting that a nuclear formof PDGFRβ might be involved in mitosis. p-ERK is also localized mainly in the cytosolof p-ERK positive gonocytes, while other cells were completely negative, showingabsence of activated ERK in these gonocytes. (B) Thanks to a few cells that werestopped during proliferation, we were able to observe potential differences inprotein profiles of actively dividing cells versus non-proliferating cells. Again ERβshowed different profiles which could be due to different phases of mitosis. Most ofthe cells did express ERβ in the nucleus, and only one cell had very spotty staining.PDGFRβ clearly localized in distinct areas of the nucleus during proliferation, butalso in the cytosol. p-ERK was also highly expressed in dividing cells and probablylocated in the nucleus.
86

Cont
rol
PDGF
17β-
Estr
adio
l
87

P+E
AG+U
O+I
CIP+
E+AG
+OU
+ICI
88

Figure 15: Live Cell Imaging of GFP bound V1-PDGFRβ transfected into PND3gonocytes. Cells were transfected for 2 days with a mamalian expression vectorcontaining a GFP-V1-PDGFRβ insert and they were then treated for 60 minuteswith PDGF, estrogen and/or inhibitors. Protein expression profiles wereobserved in order to examine whether PDGF or estrogen induce changes inexpression or subcellular localization of the variant receptor. As we onlyperformed this experiment one time in full, we did not obtain alarge enough number of cells per treatment that would allow finding specifictrends. The V1 variant appeared to be expressed in the nuclei of most cells,although it is interesting that with P+E+AG+UO+ICI, it was seen in a ring aroundthe nucleus, localized in the cytoplasm only.
89

get V1-PDGFRβ transcript and protein expression in the gonocytes at the time of their
maturation to a mitotic phenotype (corresponding to PND3 and 4). After a second night of
transfection, the gonocytes were treated with different combinations of PDGF, Estradiol and
the inhibitors AG370, UO126 and ICI 182780 and observed live using the confocal microscope
for up to 60 minutes. Although these experiments were preliminary, a few observations could
be made from the positive gonocytes in culture. V1-PDGFRβ was primarily expressed in the
nucleus or in the whole cell, but more of the cells appeared to present a nuclear signal for V1-
PDGFRβ. However, in gonocytes incubated with inhibitors, V1-PDGFRβ appeared to be
localized in many cells in a visible cytosolic ring. We also noted that treatment with PDGF and
PDGF + E2 showed a higher expression of EGFP-V1-PDGFRβ than in the control and cells treated
with inhibitors. These preliminary results suggest that PDGF and estrogen may rapidly induce
the translation of V1-PDGFRβ mRNA and may be involved in its nuclear localization (figure 15).
13. Discussion
Because they are the precursor cells of the spermatogonial stem cells, gonocytes represent
a critical developmental stage of the germ line for spermatogenesis and male fertility.
Moreover, it is believed that they are a potential origin of testicular germ cell tumours (Rajpert-
De Meyts E. and Hoei-Hansen, 2007). Thus, understanding gonocyte development can provide
invaluable information in the battle against testicular cancer and its prevention. The goal of my
study was to gain insight on the mechanisms regulating gonocyte proliferation. Through in vitro
studies of purified PND3 rat gonocytes, we concluded that gonocyte proliferation is not only
stimulated by both PDGF and estrogen through their specific receptors, but that they are both
working interdependently. This was shown in studies where gonocytes were treated with PDGF
in the presence of the ER antagonist ICI 182780, and PDGF-induced proliferation was
attenuated. Similar findings were shown for estrogen-induced proliferation in the presence of
PDGFR inhibitors. In order to understand why treatment with only one of the two factors did
induce proliferation, gonocytes were treated with PDGF in charcoal-stripped, steroid depleted,
FBS, or with 17β-estradiol in serum devoid of PDGF. In both cases, induction of proliferation
90

was no longer observed, confirming the hypothesis that proliferation with either PDGF or 17β-
estradiol alone was supported by small amounts of the other factor present in FBS. In addition,
when molecules of the MAPK pathway were inhibited, estrogen-induced proliferation was no
longer detected. We therefore concluded that gonocyte proliferation at PND3 is induced
through crosstalk between estrogen and PDGF, acting through their specific receptors, although
the mechanism by which these two pathways communicated was yet to be defined. It was our
goal to investigate at which point of the receptor signalling and downstream cascades this
crosstalk takes place.
Our first step in investigating the direct effect of PDGF and estrogen on gonocyte
proliferation was to ensure that no external growth factors or steroids found in culture medium
or serum were interfering with our treatments, thereby providing stimulation above the desired
treatments. Experiments employing charcoal-stripped serum and PDGF-depleted serum
confirmed that both PDGF and estrogen together are required, in the absence of other factors,
to induce proliferation of PND3 gonocytes.
Interestingly, we found that exogenous estrogens such as BPA and genistein were able
to stimulate gonocyte proliferation in the absence of FBS but required the presence of PDGF
with a similar dose response curve to E2. This is in contrast to results obtained using normal FBS
where the exogenous estrogens acted at lower concentrations than E2. As well, exogenous
estrogens behaved similarly to E2 as their responses were also inhibited by the PDGFR inhibitor
AG370. The differences between the observations with and without serum could be due to the
presence of low PDGF and E2 levels where the background PDGF and estrogen might have
placed the cells in a more responsive state to the effects of exogenous estrogens. Moreover,
the amount of E2 bound to the steroid binding proteins from serum should be higher than the
amount of exogenous estrogens due to lower affinity of these compounds for steroid binding
proteins, leading to lower levels of free E2 as compared to the levels of exogenous estrogens.
This would result in higher levels of free exogenous estrogens available to stimulate ER in
comparison to E2 for a given concentration of added steroids. However, in the absence of
binding proteins from serum, all of the estrogenic compounds would present the same
91

amounts of free compound and thus the same proliferation efficacy. It is also possible that
different estrogenic compounds possess distinct binding affinities to ER and are able to
stimulate or be stimulated by other cofactors in their immediate environment. The presence of
serum could provide the necessary cofactors for exogenous estrogenic compounds to provide a
stronger response than E2 while without serum they both behave in the same way.
We found that testosterone and progesterone do not induce gonocyte proliferation.
This is a very important observation as the majority of reproductive development in males is
regulated in some way by testosterone, while progesterone and estrogen pathways frequently
interact in cells. Moreover, a recent study reported that foetal gonocytes express the androgen
receptor and that their numbers were decreased by androgen treatment. Our results suggest
that this is not the case in neonatal gonocytes and that the direct effect of androgen on germ
cells might be limited to the foetal period. Although testosterone levels are not as high in
neonatal testis as during foetal life or after puberty, neonatal Leydig cells secrete measurable
amount of testosterone (Culty et al, 2008). However, part of the testosterone produced at this
time is converted to estrogen by the aromatase present in Sertoli cells and to
dihydrotestosterone by the 5α-reductase to support the development of male reproductive
system. Testosterone may play an indirect role in gonocyte development via its action on
somatic cells. Nevertheless, through these observations, it appears that gonocyte proliferation
is highly specific and requires both PDGF and an estrogen for activation.
Gonocyte proliferation is a highly regulated process which is activated at around PND3
in rats and PND1.5 in mice, simultaneously with migration of the seminiferous cords to the
basement membrane. Only gonocytes that reach the basement membrane will further
differentiate into spermatogonia. Although one could assume that such precisely regulated
events would require specific sets of activators unique to either proliferation, migration or
differentiation, studies have shown that some pathways may be involved in more than one
function, such as shown by the dual role of PDGFRs in gonocyte proliferation and migration (Li
et al, 1997;Basciani et al, 2008). Not only do gonocytes depend on the Sertoli cells to produce
the factors regulating their development, but their responsiveness to these molecules is likely
92

regulated by Sertoli cells too, as shown by our laboratory’s earlier studies where quiescent
PND2 rat gonocytes became responsive to mitotic agents after an additional day of maturation
with Sertoli cells (Li et al, 1997). The identification of the MAPK and PI3K signalling pathways in
PND3 gonocytes was an important observation leading to a better understanding of normal
gonocyte development. Since ERK1/2, Raf1, Mek1, as well as PI3K were present in gonocytes,
we investigated which of the two pathways was involved in proliferation through the use of
specific inhibitors for Raf1 (iRAF1) and Mek1/2 (PD98059, UO126), as well as PI3K
(Wortmannin). These experiments showed that gonocyte proliferation is induced primarily
through the MAPK pathway. While PI3K was not required for proliferation, its inhibition led to
small increases in proliferation above the basal level, suggesting that PI3K might be required to
maintain quiescence in neonatal rats (Thuillier et al, 2010).
These experiments were conducted in enriched gonocyte cultures in order to observe
direct effects of PDGF and estrogen on gonocyte proliferation, as opposed to co-cultures or
organ cultures where the gonocytes are in communication with all of the somatic cells and
respond to the factors produced by the Sertoli cells, PMCs and Leydig cells. Although these
latter types of cultures are more similar to the in vivo conditions of gonocyte development,
they would not allow one to study the direct effects of PDGF, estrogen, MAPK and PI3K
inhibitors on gonocytes since most of these proteins are also present in Sertoli and myoid cells.
A critical observation of our study was that MAPK inhibitors were able to attenuate the
proliferative response of gonocytes to estrogen stimulation. This data suggested that ERK1/2 is
involved in the crosstalk mechanism between PDGF and estrogen pathways and its activation is
necessary for gonocyte proliferation. With these data in hand, we were able to continue to the
next stage of the study, which was to examine the kinetics of ERK1/2 activation and whether it
required only PDGF or both PDGF and E2. To this end, we performed short term treatments of
gonocytes and followed ERK1/2 activation by immunoblot analysis and immunocytochemistry
(ICC) analysis.
Western blot analysis showed that ERK2 but not ERK1 was phosphorylated rapidly by
both PDGF and estrogen although ERK1 was the more abundant of the two ERK forms, and that
93

long term ERK2 phosphorylation could only be sustained in the presence of both PDGF and
estrogen. Moreover, the fact that AG370 alone could not block ERK2 activation in cells treated
with PDGF and E2 suggested that ERK2 phosphorylation is not the direct result of PDGFR
activation, but rather it is phosphorylated via a mechanism that is common to both the
estrogenic and PDGF pathways, requiring the activation of both the PDGF and estrogen
pathways for this phosphorylation to take place. Indeed, the finding that xenoestrogens were
able to induce short-term ERK2 phosphorylation but at much lower levels than the combination
of PDGF and E2, while they mimicked the E2 proliferative effect when combined with PDGF,
further supported the idea that estrogens act simultaneously with PDGF to activate ERK2. The
same conclusion could be reached from our ICC data where UO126 but not AG370 was found to
block ERK phosphorylation by PDGF and estrogen. Since the presence of both factors is also
necessary for gonocyte proliferation, this identifies ERK2 phosphorylation as a central event in
the crosstalk of PDGF and estrogen.
It is interesting to note that the addition of ICI 182780 together with E2 or PDGF + E2
surprisingly increased phosphorylated ERK in short term assays. This was unpredicted given the
fact that the same inhibitor was shown to decrease the proliferative capabilities of PDGF and
estrogen in PND3 gonocytes cultured for 1 day in medium containing 2.5% FBS. Although ICI is
a well known estrogen antagonist, there is sufficient documentation indicating its agonistic
effect in specific tissues. ICI 182780 stimulation actually induces MAPK signalling in specific
cells (Zhao et al, 2006;Mercier et al, 2003). Our results suggest that ICI 182780 can induce an
agonistic rapid non-genomic response, while behaving as an estrogen receptor antagonist in
long term, “classic” nuclear effects of ERs. While the existence of non-classic membrane bound
ERs such as GPR30 (Maggiolini and Picard, 2010) or alternative estrogen receptors such as the
ERRs have been reported in male reproductive tissues (Lazari et al, 2009), we have not yet
investigated their presence in PND3 gonocytes. A recent article showed that ERα might play an
important role in early germ cell development, and that ERα actually has a non-genomic effect
in PGCs, as well in the development of foetal germ cells between 11.5 and 12.5 dpc in mice (La
Sala et al, 2010). Indeed, our own studies had shown that besides a very large levels of ERβ,
94

one could detect a weak immunoreactive band of ERα in PND3 gonocytes (Wang et al, 2004),
suggesting that ERα might play a role in neonatal gonocytes.
This study also revealed changes in the levels and sub-cellular localization of ERβ and
PDGFRβ occur within an hour of stimulation with PDGF and E2, suggesting a complex and
dynamic interplay between the receptors in gonocytes. The sub-cellular localization of PDGFRβ
beyond its classical plasma membrane location suggests that the increased expression
corresponds to the induction of V1-PDGFRβ mRNA translation in gonocytes, although one
cannot exclude that it could reflect the internalization and turnover of full length PDGFRs after
their activation.
In view of these results, I propose that this crosstalk is more complicated than a simple dual
activation of MAPK, and that the proliferation process might be regulated at numerous levels.
While ERK activation is important, it appears that it is not the only function of PDGFR activation,
which is required but not sufficient to induce proliferation. Similarly, ERK does not exclusively
require PDGFR for its activation, as it is possible that the rapid, non-genomic response of
estrogen activates ERK, as it was reported for other cell types (Bouskine et al, 2008). There are
many possible mechanisms for this crosstalk given our results. A) PDGF via PDGFRs and E2 via a
non-classical ER could both activate different pools of ERK2 which would then activate the
expression of distinct genes required for proliferation. B) While only one, either PDGF or E2
could induce ERK2 phosphorylation, the other would protect phospho-ERK2 from
dephosphorylation by activating an unknown protein supporting the long term maintenance of
phospho-ERK. C) E2 effects could involve the activation and /or delocalization of both classical
and non-classical ERs. D) One of PDGF’s roles may be to activate V1-PDGFRβ expression and/or
delocalization via ERK activation as a negative feedback signal to cease proliferation. E) AG370
could inhibit V1-PDGFRβ besides the full length receptors, complicating the interpretation of its
effects. F) It is also possible that PDGFR and ERK are required for activation of nuclear ERs and
although with AG370 or ICI 182780 ERK can be phosphorylated by either mER or another
mechanism, PDGFR will not be available to activate nuclear ER (with AG370) or the ER will be
inhibited (by ICI 182780).
95

In support of these hypotheses, we know that gonocytes treated with PDGF have an
increased expression of PDGFRβ, while 30 minute treatment with estrogen increases the
protein expression of ERβ in the cytosol. The PDGFRβ increased in this system is most probably
that of the V1-PDGFRβ variant form. Indeed, live cell imaging data of EGFP-V1-PDGFRβ
expressing cells showed that the presence of PDGFR, ER and MEK inhibitors with PDGF and
estrogen led to V1-PDGFRβ being localized in the cytosol, in contrast with its nuclear expression
in the absence of inhibitors. This suggests that PDGF and estrogen are required for
maintenance of the functional activity and localization of V1-PDGFRβ, although their role in
neonatal gonocytes has yet to be defined. It is less probable that it reflects a recycling
mechanism of the membrane bound receptor, given that the confocal localization of PDGFRβ
varies; in some cells it is found in the nucleus, and other cells it is expressed at high levels of in
pseudopods. Observations of dividing gonocytes by confocal microscopy could possibly lead us
closer to achieving our goal of understanding the progression from quiescent to proliferative
and migratory gonocytes and then to SSCs and differentiating spermatogonia. If we can
understand which proteins must be expressed or activated during the different phases of
mitosis, we should gain a greater appreciation of the dynamic changes occurring in PND3
gonocytes. Given the heterogeneity of the cell population at PND3, as the timeframe between
different periods in neonatal gonocytes are not synchronized and can be as short as a few
hours, finding a way to identify the different sub-sets of gonocytes could prove to be very
important. From our experiments, it appears that gonocytes express high levels of phospho-
ERK during mitosis, either in their nuclei or in the cytosol. They also express ERβ in the nucleus
in some stages and in the cytosol at other times, and express intracellular forms of PDGFRβ,
most probably, V1- PDGFRβ.
In future experiments, it will be interesting to label gonocytes with BrdU or to measure
PCNA expression in order to identify proliferating cells, simultaneously with the determination
of ERK activation. In this way, one could know whether or not a cell is pre- or post-mitotic in
relation to the activation of signalling molecules in short term experiments, including but not
limited to ERK. It is clear that there are likely more than two distinct populations of gonocytes
at PND3, but it is important to at least understand what happens to the molecules that are
96

necessary for proliferation to occur. Moreover, it is unclear whether or not the cells undergo
only one mitosis and then arrest in preparation for differentiation, or if they continue to
proliferate until they are stimulated for differentiation. Looking at single cells is very
complicated in that so far there is no way to tell which stage the cells are in, except when clear
mitotic figures are observed. However, confocal microscopy using multiple antibodies
recognizing ERK, ERβ, PDGFRβ, alternative estrogen receptors and other signalling molecules
could provide important data by revealing simultaneous or successive changes in specific
protein levels and sub-cellular localizations in response to various treatments and time-courses.
In conclusion, my work uncovered new aspects of the regulation of gonocyte
proliferation, highlighting the critical role of ERK2 activation in this process, but also the fact
that ERK phosphorylation is under the dual control of PDGF and estrogen. Although the short-
time studies did not entirely identified the sequence of events occurring downstream of PDGF
and estrogen stimulation, it revealed the existence of a complex interplay between ERK
activation and the levels and sub-cellular localization of PDGF and estrogen receptors.
Moreover, this study showed that estrogen exerts rapid non-genomic effects on gonocytes and
that ICI 182780 behaved as an ER antagonist for long term incubations but as an agonist in
short-term stimulation, suggesting that there may be more than one species of estrogen
receptors in gonocytes. Finally, taken together with earlier studies in our laboratory, changes in
the sub-cellular localization of PDGFRβ suggested that the variant V1- PDGFRβ might play a role
in gonocyte proliferation or in its interruption to allow the further development of the cells.
97

Gon
ocyt
e
PRO
LIFE
RATI
ON
PDG
F
EE
R
E
Gen
e ex
pres
sion
?
Raf
ME
K
EE
RE
ER
PDG
F
ER
K
ER
K
p
Em
ER R
af
ME
K
ER
Kp
V1
UO
126
AG37
0
Tran
slatio
n Ac
tivat
ion
Expr
essio
n
Expr
essio
n
ICI
??
pPD
GF
?
98

Figure 16: Proposed Mechanisms of PDGF and Estrogen Crosstalk in GonocyteProliferation. In light of our observations that UO126 attenuates the phosphorylatedERK signal but AG370 does not and ICI 182780 actually increases ERK phosphorylationin short term treatments, we proposed possible mechanisms in our discussion as tohow PDGF and estrogen-induced proliferation through MAPK might occur. We proposethat short term rapid ER signalling is through a membrane receptor or another type ofnon-genomic mechanism, and this is most likely an important factor in ERKphosphorylation. It is possible that while ICI blocks the nuclear receptor, it works as anagonist for the membrane receptor. We also observed that P and E treatmentsincrease PDGFRβ and ERβ immunofluorescence. We proposed that the increasedPDGFR β could be an induction of translation of the variant V1 form that could play arole in the desensitization of the cells to PDGF to allow differentiation to proceed. Thiscould work through ERK activation via ER or membrane bound PDGFR. However, in theabsence of corresponding western blot analysis, we cannot exclude that the changesin intensity might reflect unmasking of an existing receptor, either by conformationalchange or by change in sub-cellular localization. Similarly, ERK activation might berequired to come from both ER and PDGFR to induce proliferation, but shutting off oneof these receptors might not be enough to attenuate ERK signalling.
99

Literature Cited
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002) Molecular Biology of the Cell. New York:Garland Science. 1463 p.
Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R, Johnson KC, Judd H, Kotchen JM, Kuller L, LaCroix AZ, Lane D, Langer RD, Lasser N, Lewis CE, Manson J, Margolis K, Ockene J, O'Sullivan MJ, Phillips L, Prentice RL, Ritenbaugh C, Robbins J, Rossouw JE, Sarto G, Stefanick ML, Van HL, Wactawski-Wende J, Wallace R, Wassertheil-Smoller S (2004). Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA 291:1701-1712.
Ashman LK, Cambareri AC, To LB, Levinsky RJ, Juttner CA (1991). Expression of the YB5.B8 antigen (c-kit proto-oncogene product) in normal human bone marrow. Blood 78:30-37.
AstraZeneca (2004). Faslodex, Fulvestrant Injection.
Atanassova N, McKinnell C, Turner KJ, Walker M, Fisher JS, Morley M, Millar MR, Groome NP, Sharpe RM (2000). Comparative effects of neonatal exposure of male rats to potent and weak (environmental) estrogens on spermatogenesis at puberty and the relationship to adult testis size and fertility: evidence for stimulatory effects of low estrogen levels. Endocrinology 141:3898-3907.
Awda BJ, Buhr MM (2010). Extracellular signal-regulated kinases (ERKs) pathway and reactive oxygen species regulate tyrosine phosphorylation in capacitating boar spermatozoa. Biol Reprod 83:750-758.
Basciani S, De LG, Dolci S, Brama M, Arizzi M, Mariani S, Rosano G, Spera G, Gnessi L (2008). Platelet-derived growth factor receptor beta-subtype regulates proliferation and migration of gonocytes. Endocrinology 149:6226-6235.
Basciani S, Mariani S, Arizzi M, Ulisse S, Rucci N, Jannini EA, Della RC, Manicone A, Carani C, Spera G, Gnessi L (2002). Expression of platelet-derived growth factor-A (PDGF-A), PDGF-B, and PDGF receptor-alpha and -beta during human testicular development and disease. J Clin Endocrinol Metab 87:2310-2319.
Basciani S, Mariani S, Spera G, Gnessi L (2010). Role of Platelet-Derived Growth Factors in the Testis. Endocr Rev.
Berensztein EB, Baquedano MS, Gonzalez CR, Saraco NI, Rodriguez J, Ponzio R, Rivarola MA, Belgorosky A (2006). Expression of aromatase, estrogen receptor alpha and beta, androgen receptor, and cytochrome P-450scc in the human early prepubertal testis. Pediatr Res 60:740-744.
Beumer TL, Roepers-Gajadien HL, Gademan IS, Kal HB, De Rooij DG (2000). Involvement of the D-type cyclins in germ cell proliferation and differentiation in the mouse. Biol Reprod 63:1893-1898.
100

Boulogne B, Habert R, Levacher C (2003). Regulation of the proliferation of cocultured gonocytes and Sertoli cells by retinoids, triiodothyronine, and intracellular signaling factors: differences between fetal and neonatal cells. Mol Reprod Dev 65:194-203.
Bouskine A, Nebout M, Mograbi B, Brucker-Davis F, Roger C, Fenichel P (2008). Estrogens promote human testicular germ cell cancer through a membrane-mediated activation of extracellular regulated kinase and protein kinase A. Endocrinology 149:565-573.
Brennan J, Tilmann C, Capel B (2003). Pdgfr-alpha mediates testis cord organization and fetal Leydig cell development in the XY gonad. Genes Dev 17:800-810.
Brinster RL, Zimmermann JW (1994). Spermatogenesis following male germ-cell transplantation. Proc Natl Acad Sci U S A 91:11298-11302.
Carreau S, Delalande C, Silandre D, Bourguiba S, Lambard S (2006). Aromatase and estrogen receptors in male reproduction. Mol Cell Endocrinol 246:65-68.
Carreau S, Lambard S, Delalande C, is-Galeraud I, Bilinska B, Bourguiba S (2003). Aromatase expression and role of estrogens in male gonad : a review. Reprod Biol Endocrinol 1:35.
Cheskis BJ, Greger J, Cooch N, McNally C, Mclarney S, Lam HS, Rutledge S, Mekonnen B, Hauze D, Nagpal S, Freedman LP (2008). MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids 73:901-905.
Choudhury GG, Karamitsos C, Hernandez J, Gentilini A, Bardgette J, Abboud HE (1997). PI-3-kinase and MAPK regulate mesangial cell proliferation and migration in response to PDGF. Am J Physiol 273:F931-F938.
Chuma S, Kanatsu-Shinohara M, Inoue K, Ogonuki N, Miki H, Toyokuni S, Hosokawa M, Nakatsuji N, Ogura A, Shinohara T (2005). Spermatogenesis from epiblast and primordial germ cells following transplantation into postnatal mouse testis. Development 132:117-122.
Collins P, Webb C (1999). Estrogen hits the surface. Nat Med 5:1130-1131.
Couse JF, Hewitt SC, Bunch DO, Sar M, Walker VR, Davis BJ, Korach KS (1999). Postnatal sex reversal of the ovaries in mice lacking estrogen receptors alpha and beta. Science 286:2328-2331.
Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H (1994). Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res 54:5474-5478.
Culty M (2009). Gonocytes, the forgotten cells of the germ cell lineage. Birth Defects Res C Embryo Today 87:1-26.
Culty M, Thuillier R, Li W, Wang Y, Martinez-Arguelles DB, Benjamin CG, Triantafilou KM, Zirkin BR, Papadopoulos V (2008). In utero exposure to di-(2-ethylhexyl) phthalate exerts both short-term and long-lasting suppressive effects on testosterone production in the rat. Biol Reprod 78:1018-1028.
101

Dai Y (2010). Platelet-derived growth factor receptor tyrosine kinase inhibitors: a review of the recent patent literature. Expert Opin Ther Pat 20:885-897.
Dai Z, Li Y, Quarles LD, Song T, Pan W, Zhou H, Xiao Z (2007). Resveratrol enhances proliferation and osteoblastic differentiation in human mesenchymal stem cells via ER-dependent ERK1/2 activation. Phytomedicine 14:806-814.
De Rooij DG (1998). Stem cells in the testis. Int J Exp Pathol 79:67-80.
De Rooij DG (2001). Proliferation and differentiation of spermatogonial stem cells. Reproduction 121:347-354.
de LE, O'Flaherty C (2008). Sperm activation: role of reactive oxygen species and kinases. Biochim Biophys Acta 1784:106-115.
Delbes G, Duquenne C, Szenker J, Taccoen J, Habert R, Levacher C (2007). Developmental changes in testicular sensitivity to estrogens throughout fetal and neonatal life. Toxicol Sci 99:234-243.
Delbes G, Levacher C, Habert R (2006). Estrogen effects on fetal and neonatal testicular development. Reproduction 132:527-538.
Delbes G, Levacher C, Pairault C, Racine C, Duquenne C, Krust A, Habert R (2004). Estrogen receptor beta-mediated inhibition of male germ cell line development in mice by endogenous estrogens during perinatal life. Endocrinology 145:3395-3403.
Dong L, Jelinsky SA, Finger JN, Johnston DS, Kopf GS, Sottas CM, Hardy MP, Ge RS (2007). Gene expression during development of fetal and adult Leydig cells. Ann N Y Acad Sci 1120:16-35.
Eddy EM, Washburn TF, Bunch DO, Goulding EH, Gladen BC, Lubahn DB, Korach KS (1996). Targeted disruption of the estrogen receptor gene in male mice causes alteration of spermatogenesis and infertility. Endocrinology 137:4796-4805.
Fox EM, Andrade J, Shupnik MA (2009). Novel actions of estrogen to promote proliferation: integration of cytoplasmic and nuclear pathways. Steroids 74:622-627.
Fu XD, Cui YH, Lin GP, Wang TH (2007). Non-genomic effects of 17beta-estradiol in activation of the ERK1/ERK2 pathway induces cell proliferation through upregulation of cyclin D1 expression in bovine artery endothelial cells. Gynecol Endocrinol 23:131-137.
Gaskell TL, Esnal A, Robinson LL, Anderson RA, Saunders PT (2004). Immunohistochemical profiling of germ cells within the human fetal testis: identification of three subpopulations. Biol Reprod 71:2012-2021.
Geffroy N, Guedin A, Dacquet C, Lefebvre P (2005). Cell cycle regulation of breast cancer cells through estrogen-induced activities of ERK and Akt protein kinases. Mol Cell Endocrinol 237:11-23.
Gnessi L, Emidi A, Jannini EA, Carosa E, Maroder M, Arizzi M, Ulisse S, Spera G (1995). Testicular development involves the spatiotemporal control of PDGFs and PDGF receptors gene expression and action. J Cell Biol 131:1105-1121.
102

Gould ML, Hurst PR, Nicholson HD (2007). The effects of oestrogen receptors alpha and beta on testicular cell number and steroidogenesis in mice. Reproduction 134:271-279.
Gray K, Eitzman B, Raszmann K, Steed T, Geboff A, McLachlan J, Bidwell M (1995). Coordinate regulation by diethylstilbestrol of the platelet-derived growth factor-A (PDGF-A) and -B chains and the PDGF receptor alpha- and beta-subunits in the mouse uterus and vagina: potential mediators of estrogen action. Endocrinology 136:2325-2340.
Gutendorf B, Westendorf J (2001). Comparison of an array of in vitro assays for the assessment of the estrogenic potential of natural and synthetic estrogens, phytoestrogens and xenoestrogens. Toxicology 166:79-89.
Habert R, Lejeune H, Saez JM (2001). Origin, differentiation and regulation of fetal and adult Leydig cells. Mol Cell Endocrinol 179:47-74.
He X, Andersson G, Lindgren U, Li Y (2010). Resveratrol prevents RANKL-induced osteoclast differentiation of murine osteoclast progenitor RAW 264.7 cells through inhibition of ROS production. Biochem Biophys Res Commun 401:356-362.
Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA (2003). PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299:708-710.
Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA (2007). Estrogen receptors: how do they signal and what are their targets. Physiol Rev 87:905-931.
Hess RA, Carnes K (2004). The Role of Estrogen in Testis and the Male Reproductive Tract: a Review and Species Comparison. Animal Reproduction 1:5-30.
Hilscher W (1991). The genetic control and germ cell kinetics of the female and male germ line in mammals including man. Hum Reprod 6:1416-1425.
Hirahara Y, Matsuda K, Gao W, Arvanitis DN, Kawata M, Boggs JM (2009). The localization and non-genomic function of the membrane-associated estrogen receptor in oligodendrocytes. Glia 57:153-165.
Hoei-Hansen CE, Olesen IA, Jorgensen N, Carlsen E, Holm M, Almstrup K, Leffers H, Rajpert-De ME (2007). Current approaches for detection of carcinoma in situ testis. Int J Androl 30:398-404.
Holmes L, Jr., Escalante C, Garrison O, Foldi BX, Ogungbade GO, Essien EJ, Ward D (2008). Testicular cancer incidence trends in the USA (1975-2004): plateau or shifting racial paradigm? Public Health 122:862-872.
Horwich A, Shipley J, Huddart R (2006). Testicular germ-cell cancer. Lancet 367:754-765.
Huckins C (1971). The spermatogonial stem cell population in adult rats. I. Their morphology, proliferation and maturation. Anat Rec 169:533-557.
103

Ignar-Trowbridge DM, Nelson KG, Bidwell MC, Curtis SW, Washburn TF, McLachlan JA, Korach KS (1992). Coupling of dual signaling pathways: epidermal growth factor action involves the estrogen receptor. Proc Natl Acad Sci U S A 89:4658-4662.
Ignar-Trowbridge DM, Pimentel M, Teng CT, Korach KS, McLachlan JA (1995). Cross talk between peptide growth factor and estrogen receptor signaling systems. Environ Health Perspect 103 Suppl 7:35-38.
Iguchi T, Watanabe H, Katsu Y (2001). Developmental effects of estrogenic agents on mice, fish, and frogs: a mini-review. Horm Behav 40:248-251.
Jahnukainen K, Chrysis D, Hou M, Parvinen M, Eksborg S, Soder O (2004). Increased apoptosis occurring during the first wave of spermatogenesis is stage-specific and primarily affects midpachytene spermatocytes in the rat testis. Biol Reprod 70:290-296.
Jensen EV (2004). From Chemical Warfare to Breast Cancer Management. p. 1018-1021.
Joensen UN, Jorgensen N, Skakkebaek NE (2007). Testicular dysgenesis syndrome and carcinoma in situ of the testes. Nat Clin Pract Urol 4:402-403.
Johnston DS, Russell LD, Friel PJ, Griswold MD (2001). Murine germ cells do not require functional androgen receptors to complete spermatogenesis following spermatogonial stem cell transplantation. Endocrinology 142:2405-2408.
Kaarbo M, Klokk TI, Saatcioglu F (2007). Androgen signaling and its interactions with other signaling pathways in prostate cancer. Bioessays 29:1227-1238.
Kim EK, Choi EJ (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802:396-405.
Koehler KF, Helguero LA, Haldosen LA, Warner M, Gustafsson JA (2005). Reflections on the discovery and significance of estrogen receptor beta. Endocr Rev 26:465-478.
Kolch W (2000). Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J 351 Pt 2:289-305.
Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O (1998). Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci U S A 95:15677-15682.
Kupisiewicz K, Boissy P, Abdallah BM, Hansen FD, Erben RG, Savouret JF, Soe K, Andersen TL, Plesner T, Delaisse JM (2010). Potential of resveratrol analogues as antagonists of osteoclasts and promoters of osteoblasts. Calcif Tissue Int 87:437-449.
La Sala G, Farini D, De FM (2010). Rapid estrogen signalling in mouse primordial germ cells. Exp Cell Res 316:1716-1727.
Lambrot R, Coffigny H, Pairault C, Donnadieu AC, Frydman R, Habert R, Rouiller-Fabre V (2006). Use of organ culture to study the human fetal testis development: effect of retinoic acid. J Clin Endocrinol Metab 91:2696-2703.
104

Lau WS, Chen WF, Chan RY, Guo DA, Wong MS (2009). Mitogen-activated protein kinase (MAPK) pathway mediates the oestrogen-like activities of ginsenoside Rg1 in human breast cancer (MCF-7) cells. Br J Pharmacol 156:1136-1146.
Lawson KA, Dunn NR, Roelen BA, Zeinstra LM, Davis AM, Wright CV, Korving JP, Hogan BL (1999). Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev 13:424-436.
Lazari MF, Lucas TF, Yasuhara F, Gomes GR, Siu ER, Royer C, Fernandes SA, Porto CS (2009). Estrogen receptors and function in the male reproductive system. Arq Bras Endocrinol Metabol 53:923-933.
Li H, Papadopoulos V, Vidic B, Dym M, Culty M (1997). Regulation of rat testis gonocyte proliferation by platelet-derived growth factor and estradiol: identification of signaling mechanisms involved. Endocrinology 138:1289-1298.
Livera G, Delbes G, Pairault C, Rouiller-Fabre V, Habert R (2006). Organotypic culture, a powerful model for studying rat and mouse fetal testis development. Cell Tissue Res 324:507-521.
Livera G, Rouiller-Fabre V, Durand P, Habert R (2000). Multiple effects of retinoids on the development of Sertoli, germ, and Leydig cells of fetal and neonatal rat testis in culture. Biol Reprod 62:1303-1314.
Lu X, Qu CK, Shi ZQ, Feng GS (1998). Downregulation of platelet-derived growth factor receptor-beta in Shp-2 mutant fibroblast cell lines. Oncogene 17:441-448.
Luconi M, Forti G, Baldi E (2002). Genomic and nongenomic effects of estrogens: molecular mechanisms of action and clinical implications for male reproduction. J Steroid Biochem Mol Biol 80:369-381.
MacCalman CD, Getsios S, Farookhi R, Blaschuk OW (1997). Estrogens potentiate the stimulatory effects of follicle-stimulating hormone on N-cadherin messenger ribonucleic acid levels in cultured mouse Sertoli cells. Endocrinology 138:41-48.
Maggiolini M, Picard D (2010). The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J Endocrinol 204:105-114.
Maher B (2006). The Discovery of Estrogen Receptor Beta. p. 92.
Mariani S, Basciani S, Arizzi M, Spera G, Gnessi L (2002). PDGF and the testis. Trends Endocrinol Metab 13:11-17.
Martelli AM, Evangelisti C, Chiarini F, McCubrey JA (2010). The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 1:89-103.
McGuinness MP, Orth JM (1992). Reinitiation of gonocyte mitosis and movement of gonocytes to the basement membrane in testes of newborn rats in vivo and in vitro. Anat Rec 233:527-537.
McIntyre A, Summersgill B, Grygalewicz B, Gillis AJ, Stoop J, van Gurp RJ, Dennis N, Fisher C, Huddart R, Cooper C, Clark J, Oosterhuis JW, Looijenga LH, Shipley J (2005). Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res 65:8085-8089.
105

McLaren A (1999). Signaling for germ cells. Genes Dev 13:373-376.
Mercier I, Mader S, Calderone A (2003). Tamoxifen and ICI 182,780 negatively influenced cardiac cell growth via an estrogen receptor-independent mechanism. Cardiovasc Res 59:883-892.
Merlet J, Racine C, Moreau E, Moreno SG, Habert R (2007). Male fetal germ cells are targets for androgens that physiologically inhibit their proliferation. Proc Natl Acad Sci U S A 104:3615-3620.
Migliaccio A, Castoria G, Di DM, de FA, Bilancio A, Lombardi M, Barone MV, Ametrano D, Zannini MS, Abbondanza C, Auricchio F (2000). Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J 19:5406-5417.
Mitsunaga K, Araki K, Mizusaki H, Morohashi K, Haruna K, Nakagata N, Giguere V, Yamamura K, Abe K (2004). Loss of PGC-specific expression of the orphan nuclear receptor ERR-beta results in reduction of germ cell number in mouse embryos. Mech Dev 121:237-246.
Moreno SG, Dutrillaux B, Coffigny H (2001). Status of p53, p21, mdm2, pRb proteins, and DNA methylation in gonocytes of control and gamma-irradiated rats during testicular development. Biol Reprod 64:1422-1431.
Mosselman S, Claesson-Welsh L, Kamphuis JS, van Zoelen EJ (1994). Developmentally regulated expression of two novel platelet-derived growth factor alpha-receptor transcripts in human teratocarcinoma cells. Cancer Res 54:220-225.
Mosselman S, Looijenga LH, Gillis AJ, van Rooijen MA, Kraft HJ, van Zoelen EJ, Oosterhuis JW (1996). Aberrant platelet-derived growth factor alpha-receptor transcript as a diagnostic marker for early human germ cell tumors of the adult testis. Proc Natl Acad Sci U S A 93:2884-2888.
Nadal A, Ropero AB, Laribi O, Maillet M, Fuentes E, Soria B (2000). Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor alpha and estrogen receptor beta. Proc Natl Acad Sci U S A 97:11603-11608.
Nagano R, Tabata S, Nakanishi Y, Ohsako S, Kurohmaru M, Hayashi Y (2000). Reproliferation and relocation of mouse male germ cells (gonocytes) during prespermatogenesis. Anat Rec 258:210-220.
Nikov GN, Eshete M, Rajnarayanan RV, Alworth WL (2001). Interactions of synthetic estrogens with human estrogen receptors. J Endocrinol 170:137-145.
Nitta H, Bunick D, Hess RA, Janulis L, Newton SC, Millette CF, Osawa Y, Shizuta Y, Toda K, Bahr JM (1993). Germ cells of the mouse testis express P450 aromatase. Endocrinology 132:1396-1401.
Nurmio M, Kallio J, Toppari J, Jahnukainen K (2008). Adult reproductive functions after early postnatal inhibition by imatinib of the two receptor tyrosine kinases, c-kit and PDGFR, in the rat testis. Reprod Toxicol 25:442-446.
Nurmio M, Toppari J, Zaman F, Andersson AM, Paranko J, Soder O, Jahnukainen K (2007). Inhibition of tyrosine kinases PDGFR and C-Kit by imatinib mesylate interferes with postnatal testicular development in the rat. Int J Androl 30:366-376.
106

O'Flaherty C, de LE, Gagnon C (2006). Positive role of reactive oxygen species in mammalian sperm capacitation: triggering and modulation of phosphorylation events. Free Radic Biol Med 41:528-540.
Oakberg EF (1971). Spermatogonial stem-cell renewal in the mouse. Anat Rec 169:515-531.
Ohbo K, Yoshida S, Ohmura M, Ohneda O, Ogawa T, Tsuchiya H, Kuwana T, Kehler J, Abe K, Scholer HR, Suda T (2003). Identification and characterization of stem cells in prepubertal spermatogenesis in mice small star, filled. Dev Biol 258:209-225.
Ohmura M, Yoshida S, Ide Y, Nagamatsu G, Suda T, Ohbo K (2004). Spatial analysis of germ stem cell development in Oct-4/EGFP transgenic mice. Arch Histol Cytol 67:285-296.
Ohta H, Wakayama T, Nishimune Y (2004). Commitment of fetal male germ cells to spermatogonial stem cells during mouse embryonic development. Biol Reprod 70:1286-1291.
Olaso R, Habert R (2000). Genetic and cellular analysis of male germ cell development. J Androl 21:497-511.
Orth JM, Boehm R (1990). Functional coupling of neonatal rat Sertoli cells and gonocytes in coculture. Endocrinology 127:2812-2820.
Osborne CK, Shou J, Massarweh S, Schiff R (2005). Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res 11:865s-870s.
Palumbo C, van RK, Gillis AJ, van Gurp RH, de MH, Oosterhuis JW, van Zoelen EJ, Looijenga LH (2002). Expression of the PDGF alpha-receptor 1.5 kb transcript, OCT-4, and c-KIT in human normal and malignant tissues. Implications for the early diagnosis of testicular germ cell tumours and for our understanding of regulatory mechanisms. J Pathol 196:467-477.
Park JH, Lee MY, Han HJ (2009). A potential role for caveolin-1 in estradiol-17beta-induced proliferation of mouse embryonic stem cells: involvement of Src, PI3K/Akt, and MAPKs pathways. Int J Biochem Cell Biol 41:659-665.
Pettersson K, Gustafsson JA (2001). Role of estrogen receptor beta in estrogen action. Annu Rev Physiol 63:165-192.
Puglianiello A, Campagnolo L, Farini D, Cipollone D, Russo MA, Siracusa G (2004). Expression and role of PDGF-BB and PDGFR-beta during testis morphogenesis in the mouse embryo. J Cell Sci 117:1151-1160.
Rajpert-De Meyts E., Hoei-Hansen CE (2007). From gonocytes to testicular cancer: the role of impaired gonadal development. Ann N Y Acad Sci 1120:168-180.
Ricci G, Catizone A, Galdieri M (2004). Embryonic mouse testis development: role of platelet derived growth factor (PDGF-BB). J Cell Physiol 200:458-467.
Robinson MJ, Cobb MH (1997). Mitogen-activated protein kinase pathways. Curr Opin Cell Biol 9:180-186.
107

Roosen-Runge EC, Leik J (1968). Gonocyte degeneration in the postnatal male rat. Am J Anat 122:275-299.
Rouiller-Fabre V, Levacher C, Pairault C, Racine C, Moreau E, Olaso R, Livera G, Migrenne S, Delbes G, Habert R (2003). Development of the foetal and neonatal testis. Andrologia 35:79-83.
Rumora L, Grubisic TZ (2009). A journey through mitogen-activated protein kinase and ochratoxin A interactions. Arh Hig Rada Toksikol 60:449-456.
Russell LD, Ettlin R, Sinha Hikim A, Clegg E (1990) Histological and Histopathological Evaluation of the Testis.Cache River Press.
Schaeffer HJ, Weber MJ (1999). Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol 19:2435-2444.
Schlatt S (2010) Schlatt Laboratory.
Schmahl J, Rizzolo K, Soriano P (2008). The PDGF signaling pathway controls multiple steroid-producing lineages. Genes Dev 22:3255-3267.
Segars JH, Driggers PH (2002). Estrogen action and cytoplasmic signaling cascades. Part I: membrane-associated signaling complexes. Trends Endocrinol Metab 13:349-354.
Sehmisch S, Hammer F, Christoffel J, Seidlova-Wuttke D, Tezval M, Wuttke W, Stuermer KM, Stuermer EK (2008). Comparison of the phytohormones genistein, resveratrol and 8-prenylnaringenin as agents for preventing osteoporosis. Planta Med 74:794-801.
Sharpe RM, Skakkebaek NE (2008). Testicular dysgenesis syndrome: mechanistic insights and potential new downstream effects. Fertil Steril 89:e33-e38.
Shaul YD, Seger R (2007). The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta 1773:1213-1226.
Simoncini T, Scorticati C, Mannella P, Fadiel A, Giretti MS, Fu XD, Baldacci C, Garibaldi S, Caruso A, Fornari L, Naftolin F, Genazzani AR (2006). Estrogen receptor alpha interacts with Galpha13 to drive actin remodeling and endothelial cell migration via the RhoA/Rho kinase/moesin pathway. Mol Endocrinol 20:1756-1771.
Soares R, Guerreiro S, Botelho M (2007). Elucidating progesterone effects in breast cancer: cross talk with PDGF signaling pathway in smooth muscle cell. J Cell Biochem 100:174-183.
Sonne SB, Kristensen DM, Novotny GW, Olesen IA, Nielsen JE, Skakkebaek NE, Rajpert-De ME, Leffers H (2008). Testicular dysgenesis syndrome and the origin of carcinoma in situ testis. Int J Androl 31:275-287.
Strauss J, Barbieri R (2004) Reproductive Endocrinology.Elsevier Saunders.
Sutton KA (2000). Molecular mechanisms involved in the differentiation of spermatogenic stem cells. Rev Reprod 5:93-98.
108

Thuillier R, Manku G, Wang Y, Culty M (2009). Changes in MAPK pathway in neonatal and adult testis following fetal estrogen exposure and effects on rat testicular cells. Microsc Res Tech 72:773-786.
Thuillier R, Mazer M, Manku G, Boisvert A, Wang Y, Culty M (2010). Interdependence of platelet-derived growth factor and estrogen-signaling pathways in inducing neonatal rat testicular gonocytes proliferation. Biol Reprod 82:825-836.
Thuillier R, Wang Y, Culty M (2003). Prenatal exposure to estrogenic compounds alters the expression pattern of platelet-derived growth factor receptors alpha and beta in neonatal rat testis: identification of gonocytes as targets of estrogen exposure. Biol Reprod 68:867-880.
Tres LL, Kierszenbaum AL (2005). The ADAM-integrin-tetraspanin complex in fetal and postnatal testicular cords. Birth Defects Res C Embryo Today 75:130-141.
van Dissel-Emiliani FM, De Rooij DG, Meistrich ML (1989). Isolation of rat gonocytes by velocity sedimentation at unit gravity. J Reprod Fertil 86:759-766.
Vigueras-Villasenor RM, Moreno-Mendoza NA, Reyes-Torres G, Molina-Ortiz D, Leon MC, Rojas-Castaneda JC (2006). The effect of estrogen on testicular gonocyte maturation. Reprod Toxicol 22:513-520.
Vornberger W, Prins G, Musto NA, Suarez-Quian CA (1994). Androgen receptor distribution in rat testis: new implications for androgen regulation of spermatogenesis. Endocrinology 134:2307-2316.
Vu TH, Martin GR, Lee P, Mark D, Wang A, Williams LT (1989). Developmentally regulated use of alternative promoters creates a novel platelet-derived growth factor receptor transcript in mouse teratocarcinoma and embryonic stem cells. Mol Cell Biol 9:4563-4567.
Wahlgren A, Svechnikov K, Strand ML, Jahnukainen K, Parvinen M, Gustafsson JA, Soder O (2008). Estrogen receptor beta selective ligand 5alpha-Androstane-3beta, 17beta-diol stimulates spermatogonial deoxyribonucleic acid synthesis in rat seminiferous epithelium in vitro. Endocrinology 149:2917-2922.
Wang Y, Culty M (2007). Identification and distribution of a novel platelet-derived growth factor receptor beta variant: effect of retinoic acid and involvement in cell differentiation. Endocrinology 148:2233-2250.
Wang Y, Thuillier R, Culty M (2004). Prenatal estrogen exposure differentially affects estrogen receptor-associated proteins in rat testis gonocytes. Biol Reprod 71:1652-1664.
Waterloo (2007). Biology of the Female Reproductive system.
Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ (2002). Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci U S A 99:14783-14788.
Ying Y, Liu XM, Marble A, Lawson KA, Zhao GQ (2000). Requirement of Bmp8b for the generation of primordial germ cells in the mouse. Mol Endocrinol 14:1053-1063.
109

Yoshida S, Sukeno M, Nabeshima Y (2007). A vasculature-associated niche for undifferentiated spermatogonia in the mouse testis. Science 317:1722-1726.
Yoshida S, Sukeno M, Nakagawa T, Ohbo K, Nagamatsu G, Suda T, Nabeshima Y (2006). The first round of mouse spermatogenesis is a distinctive program that lacks the self-renewing spermatogonia stage. Development 133:1495-1505.
Zhang L, Li X, Zhao L, Zhang L, Zhang G, Wang J, Wei L (2009). Nongenomic effect of estrogen on the MAPK signaling pathway and calcium influx in endometrial carcinoma cells. J Cell Biochem 106:553-562.
Zhao L, O'Neill K, Brinton RD (2006). Estrogenic agonist activity of ICI 182,780 (Faslodex) in hippocampal neurons: implications for basic science understanding of estrogen signaling and development of estrogen modulators with a dual therapeutic profile. J Pharmacol Exp Ther 319:1124-1132.
Zhou Q, Li Y, Nie R, Friel P, Mitchell D, Evanoff RM, Pouchnik D, Banasik B, McCarrey JR, Small C, Griswold MD (2008a). Expression of stimulated by retinoic acid gene 8 (Stra8) and maturation of murine gonocytes and spermatogonia induced by retinoic acid in vitro. Biol Reprod 78:537-545.
Zhou Q, Nie R, Li Y, Friel P, Mitchell D, Hess RA, Small C, Griswold MD (2008b). Expression of stimulated by retinoic acid gene 8 (Stra8) in spermatogenic cells induced by retinoic acid: an in vivo study in vitamin A-sufficient postnatal murine testes. Biol Reprod 79:35-42.
110