scid cells efficiently integrate hairpin and linear dna substrates
TRANSCRIPT

MOLECULAR AND CELLULAR BIOLOGY, June 1994, p. 3876-3883 Vol. 14, No. 60270-7306/94/$04.00+0Copyright X 1994, American Society for Microbiology
scid Cells Efficiently Integrate Hairpin and Linear DNA SubstratesJANE E. STAUNTON' AND DAVID T. WEAVER' 2*
Department of Microbiology and Molecular Genetics, Harvard Medical School, ' and Division ofTumor Immunology, Dana-Farber Cancer Institute,2 Boston, Massachusetts 02115
Received 6 December 1993/Returned for modification 20 January 1994/Accepted 18 March 1994
The scid mouse mutation affects V(D)J rearrangement and double-strand break repair. scid V(D)Jrearrangement is characterized by defective coding joint formation which prevents the development of matureB and T cells. Hairpin DNA has been implicated in the formation of V(D)J coding joints. We found scid cellsto be proficient in hairpin processing in the context of DNA integration. In addition, we found that the sciddefect did not impair integration of linear DNA via nonhomologous recombination. Therefore, hairpinprocessing and integration of DNA into the genome are distinct from hypersensitivity to ionizing radiation andthe defect in V(D)J recombination.
scid (severe combined immune deficiency) mice were firstidentified because of their immunodeficiency (3), now knownto be caused by defective V(D)J rearrangement during lym-phocyte development. Germ line antibody and T-cell receptorloci contain coding elements flanked by recombination signalsequences (heptamer-spacer-nonamer). Productive rearrange-ment yields precise signal joints, whereas coding junctionsusually contain small DNA additions or deletions which lenddiversity to the resulting transcription unit (2). In scid cells,signal sequences are joined with relatively high fidelity butrearrangements are nonproductive because of large deletionsof DNA from coding junctions (13, 19, 20, 28).The scid mutation also impairs double-strand break repair in
a variety of cell types (1, 9, 12). The kinetics of the repair defectindicate that scid cells repair some breaks at a rate comparableto that of wild-type cells but that a subpopulation of the breaksremain unrepaired in scid cells. The end structures created bygamma irradiation have not been well studied, but someevidence exists that the termini are modified (14). Suchmodifications may require processing before DNA ends can bejoined. In both V(D)J joining and DNA repair, the scidmutation affects the joining of a subset of DNA ends. A modelconsistent with both phenotypes is that the wild-type scid geneproduct is involved in the processing of specific DNA endstructures.The observation of palindromic P nucleotides at rearranged
coding junctions prompted a model that involves an interme-diate hairpin structure (15, 18, 21). The model proposes thathairpins arise during coding joint formation but not duringsignal joint formation. Roth and coworkers have providedfurther support for this model with physical evidence of codingjoint hairpin DNA in thymocytes of scid but not wild-type mice(26, 27). If the observed hairpins represent actual intermedi-ates, the scid defect may lie in a failure to effectively resolvehairpin structures.Here we demonstrate that scid mutant cells efficiently re-
solve hairpin DNA substrates during integration into thechromosome. We also examined whether the scid mutationaffects integration of nonhomologous DNA into the chromo-some. We show that integration rates of DNA into thechromosome vary considerably between cell lines but that
* Corresponding author. Phone: (617) 632-3826. Fax: (617) 632-4569. Electronic mail address: [email protected].
reduced integration frequency is not an intrinsic scid pheno-type.
MATERUILS AND METHODS
Cell culture. Fibroblast cell lines were grown in Dulbeccomodified Eagle medium (DMEM; Gibco) with 10% newborncalf serum. ScSV3 cells are simian virus 40-transformed scidfibroblasts, and Sc3T3 cells are spontaneously transformed scidfibroblasts (12). The scid cells used were from CB-17 mice andare thus genetically closely related to BALB/3T3 cells; how-ever, they are more similar to NIH 3T3 cells in morphologyand growth properties. Primary embryonic fibroblasts wereprepared by published methods (29). Briefly, day 16 to 17embryos were aborted from pregnant CB-17 or scid CB-17mice. Individual embryos were incubated in trypsin overnight,and adherent cells were isolated. Cells were passaged inDMEM plus 10% fetal calf serum for at least 20 passages. Cellswere then cultured in DMEM with 5% each fetal calf serumand newborn calf serum. All transfection experiments wereperformed on cells between 20 and 40 passages.
Transfections. Cells were transfected by CaPO4 as previ-ously described (24), with some modifications. Exponentiallygrowing cells were plated 16 to 24 h prior to transfection at105/100-mm-diameter dish for cell lines and at 2 x 105 per dishfor early-passage cells. At 1 to 2 h before transfection, cellswere fed with fresh medium. For each set of transfections, abatch CaPO4 precipitate was made with linearized pPGKNeoor pPGKHygro plasmid DNA (5 ,ug per plate). After 20 to 24h, cells were washed and refed with medium. At 48 h,transfected cells were trypsinized, counted, and plated at 105 to106/100-mm plate for drug selection and at 100 to 500/100-mmplate for determination of cell viability by colony counting after12 to 14 days. Selection with G418 (400 ,ug/ml; Gibco) orhygromycin B (500 ,ug/ml; Calbiochem) was begun 1 day laterand continued for 12 to 14 days. At this time, plates werestained with crystal violet for colony counting by standardmethods (12).
Hairpin transfections were performed similarly to thosedone with linear DNA. From 100 ng to 1 ,ug of hairpin DNAwas used per transfection; cotransfections with linearpPGKHygro (see Table 3) used 100 ng of double-hairpin (dhp)molecules and 5 ,ug of pPGKHygro.For electroporation, cells were trypsinized and resuspended
in DMEM at a concentration of 3 x 106 to 5 x 106/ml in avolume of 0.7 ml per transfection. Cells were electroporated at
3876

scid CELLS INTEGRATE HAIRPINS 3877
300 V and 1,180 ,uF with 5 jig of linear DNA and/or 0.1 to 1 jgof hairpin DNA (Cell-Porator; Bethesda Research Laborato-ries). Immediately after electroporation, each transfection wasdiluted into 10 ml of DMEM plus serum. For colony isolation(see below), cells were immediately plated at several dilutionsand placed under selection 24 h later. For hairpin electropo-ration (see Table 3), cells were replated at various dilutions 24h after transfection and placed under selection on the follow-ing day.
Integration frequencies were calculated for each experimentby dividing the total number of drug-selected colonies by thetotal number of unselected colonies. For each set of transfec-tions, the mean integration frequency was calculated per cellline. Mean frequencies were then compared to those of theothers within the same set (those transfected with the samebatch of transfection precipitate) by arbitrarily setting thevalue of the BALB/3T3 cells to 1.00 to obtain the correctedfrequency (Vc.rr). The corrected frequencies were then aver-aged for each cell line (Iav). The statistical significance of thedifference between cell lines within a given experiment wasderived by using the Student t test and published significance(P) values (16).
Southern blot analysis. pPGKNeo/XbaI or pPGKNeo/AflIIItransfectant clones were selected from 106 cells for eachexpanded colony. Cells were trypsinized and resuspended inTris-EDTA-NaCl solution plus 1% sodium dodecyl sulfate(SDS) and 10 ,ug of proteinase K per ml. Ten micrograms ofgenomic DNA was digested, fractionated on 0.75% agarosegels, and blotted onto a nitrocellulose or Biotrans membrane.DNA was fixed on the membrane via UV cross-linking (Strata-linker; Stratagene). Blots were prehybridized for at least 4 h at42°C in 50% formamide-5X SSPCE (1 x SSPCE is 120 mMNaCl, 15 mM sodium citrate, 13 mM KH2PO4, and 0.1 mMEDTA [pH 7.2])-5 x Denhardt's solution-500 ,ug of denaturedherring sperm DNA per ml-0.1% SDS. Probes for blots werelabeled with [t_-32P]dCTP (Amersham) by using mixed oligo-nucleotide priming and Klenow fragment DNA polymerase(7). Blots were hybridized for at least 16 h at 42°C with 50%formamide-5X SSPCE-1 x Denhardt's solution-100 jLg ofdenatured herring sperm DNA per ml-2.5% dextran sulfate.Blots were washed at 68°C with 2x SSC (1 x SSC is 0.15 MNaCl plus 0.015 M sodium citrate)-0.1% SDS.
Clones were screened for single-copy inserts which could beused for restriction digests to look for deletions at the un-selected end of the plasmid DNA. For Xbal colonies, DNAwas digested with HindlIl and probed with a 2.3-kb PvuIIrestriction fragment to look for single-copy events. DNA wasthen cut with PvuII and probed with the same restrictionfragment to assay for a PvuII site 140 bp from the site oflinearization.To identify hairpin DNAs, exonuclease V reactions were
fractionated on 0.8% agarose gels and blotted with 0.4 NNaOH onto positively charged nylon filters (Biotrans plus).Blots were probed with an [c_-32P]dCTP random-oligonucleo-tide-labeled PGKNeo fragment. Hybridization and washeswere carried out at 68°C with 6x SSC.
Hairpin DNA substrates. From 100 to 500 ,ug of pPGKNeoplasmid DNA was cut with XbaI and HindIII. The 1.9-kbPGKNeo fragment was isolated from a 0.9% Tris-acetate-EDTA gel by using sodium iodide and glass beads. Linearmonomer DNA was self-ligated in 0.2 ml at room temperaturefor 4 h with T4 DNA ligase (New England Biolabs). Concate-merized monomers were digested with Hinidlll, and the result-ing dimer-sized fragments were isolated on a 0.9% Tris-acetate-EDTA gel as described above. DNA was boiled for 5min and cooled on ice in the presence of 50 mM NaCl.
NaCl-dependent conversion to monomer size was monitoredanalytically with a 0.9% agarose gel. Single-hairpin (shp)molecules generated in this manner have a hairpin at one endand a free double-stranded end. The shps were self-ligated asdescribed above to form dhps that were isolated on 0.9%Tris-acetate-EDTA agarose prior to transfection.
Exonuclease V treatment of DNA. A linear monomer anddimer mixture and a solution of shp and dhp molecules (100 to500 ng of each) were each treated with 0 to 1.0 U ofexonuclease V (United States Biochemicals) for 30 min at37°C. The reaction volume was 30 jl and contained the bufferrecommended by the supplier, including 1 mM ATP.
RESULTS
Integration of linear plasmid DNA in scid and wild-typecells. Given its effect on V(D)J rearrangement and double-strand break repair, the scid mutation may be required foradditional recombination processes in the cell. To examine thishypothesis, we looked at another form of nonhomologousrecombination, integration of exogenous DNA into the chro-mosome. We assayed the frequency of stable transfectantsresulting from transfection of linearized plasmid DNA con-taining the neomycin phosphotransferase (neo) gene (encodesG418 resistance) or the hygromycin B phosphotransferasegene (encodes hygromycin B resistance). If scid cells weredefective for this form of nonhomologous recombination, weexpected to see a decreased level of stable drug-resistantclones. Transfected cells were plated for selection with theappropriate drug and separately plated to determine thenumber of viable cells by colony formation. A transfection-and-integration frequency was determined from the number ofresistant colonies per 103 viable (colony-forming) cells.The frequency of integration for a given cell line varied
between experiments, but within an experimental set, thevalues were more consistent. Thus, to compare cell lines ofdifferent origins, we used a normalization procedure. For eachset, the mean integration frequency was determined per cellline and these values were compared within the set by assigningthe BALB/3T3 cell line an arbitrary value of 1.0. The I,crr wascalculated for each cell line per experiment, and the crr valueswere averaged across sets for each cell line to yield the Iav.These refinements allowed us to compare different cell linesdespite individual experimental variation.We prepared primary fibroblast cell lines from CB-17 and
CB-17 scid mice in parallel (see Materials and Methods). ScF4and ScF6 fibroblasts exhibited the scid phenotype, as reflectedin X-ray sensitivity and defective V(D)J recombination capac-ity (4, 12) (data not shown). We transfected linear plasmidDNA and tabulated the integration frequency as describedabove. All transfections were done on cells between passages20 and 40. The ScF4, ScF6, and CB1 fibroblast lines had similarlinear plasmid integration values (Table 1; ScF6 Iav, 1.03; ScF4Iav 0.91; CB1 Iav, 1.0). In each experiment, the integrationfrequencies of ScF4 and ScF6 were not significantly differentfrom those of CB1 (P > 0.1). Thus, the scid gene product is notessential for the nonhomologous integration required forstable transfection. These results are in contrast to those ofHarrington et al. ( 11) who found scid cells to be 10- to 100-foldless efficient than wild-type cells. Our CB17 and scid cellsdisplayed comparable levels of DNA uptake, as verified bylacZ reporter gene transient transfection assays (data notshown).We also compared the transfection frequencies of wild-type
and scid fibroblast cell lines. scid lines integrated linear DNAat a frequency 10-fold lower than that of BALB/3T3 cells
VOL. 14, 1994

3878 STAUNTON AND WEAVER
TABLE 1. Integration of linear DNA into early-passage scid and wild-type fibroblasts
Cell line No. of viable cells No. of resistant Integration frequency Ic SD Ioand expt" (103) colonies (1o-3)b m +ScorrCB1Hi 220, 600, 410 234, 478, 360 1.06, 0.80, 0.88 0.91 + 0.14 1.00 1.00H2 306, 280, 332 169, 281, 425 0.55, 1.00, 1.28 0.95 ± 0.37 1.00Gl 570, 720, 800 820, 860, 924 1.44, 1.20, 1.16 1.26 + 0.15 1.00
ScF6HI 560, 570, 630 338, 818, 598 0.60, 1.44, 0.95 1.00 + 0.42 1.09 1.03H2 425, 370 336, 496 0.79, 1.34 1.07 + 0.39 1.13Gl 2,560, 2,950, 2,320 3,300, 3,040, 2,400 1.29, 1.03, 1.03 1.12 ± 0.15 0.89
ScF4, Gl 750, 650 854, 750 1.14, 1.15 1.15 ± 0.11 0.91 0.91
" G, G418 selection (pPGKNeo-transfected DNA); H, hygromycin B selection (pPGKHygro-transfected DNA).b The integration frequency is the number of resistant colonies per 103 viable cells.' In, mean integration frequency for each cell line per set of transfections.
(Sc3T3 Iav, 0.11; ScSV3 IaV, 0.08; Table 2). These results efficiency of integration seen in the cell lines may thus reflectrepresent several experimental sets; in every case, the scid cell the state of transformation or other cell line-specific factorsIeorr value was less than 20% of that of BALB/3T3 cells. The rather than the scid mutation.efficiency of the other wild-type cell line, NIH 3T3 (lay, 0.43), We also examined whether scid integration events werewas also reduced relative to that of BALB/3T3 cells but was accompanied by large deletions in the plasmid DNA, analo-still greater than that of the scid lines. In one experiment, the gous to the large deletions which accompany scid V(D)J codingNIH 3T3 integration frequencies were very close to those of joint formation (13). Associated deletion was examined by twoBALB/3T3 cells (experiment H1; IaV, 1.32 versus 1.53). In the approaches. First, we reasoned that a scid-specific defect mayother three experiments, NIH 3T3 cells and both scid lines had be manifested in transfection efficiency when plasmid DNA isintegration frequencies statistically significantly different from linearized near the selectable marker. In the NIH 3T3, BALB/those of BALB/3T3 cells (P < 0.1). Except in experiment H2, 3T3, Sc3T3, and ScSV3 cell lines, we found that plasmidthe scid and BALB/3T3 differences were more significant (P < pPGKNeo DNA integrated 1.5 times more efficiently when0.05). Thus, the scid cell lines were less efficient than BALB/ linearized 1.2 kb (AflIlI linearization) rather than 50 bp (XbaI3T3 cells in the integration of linear DNA, but the results linearization) from the phosphoglycerate kinase (PGK) pro-obtained with NIH 3T3 cells indicate that cell line differences moter. Sc3T3 and ScSV3 transfections also showed no in-unrelated to scid influence transfection efficiency. These cell creased positional sensitivity (data not shown), arguing thatlines have been in extended culture and differ somewhat in extended deletions at the plasmid integration site are not moremorphology and growth rate, in contrast to the more closely pronounced for scid cells.related early-passage fibroblasts (Table 1). The differences in The second test was molecular characterization of a set of
TABLE 2. Integration of linear DNA into scid and wild-type cell linesa
Cell line, No. of viable cells No. of resistant I (103) /m + SD Iand expt (103) colonies I, DI:" a
BALB/3T3Gi 146, 214, 160 3,908, 3,624, 3,124 26.77, 16.93, 19.53 21.08 ± 5.1 1.00 1.00Hi 328, 378 532, 540 1.62, 1.43 1.53 ± 0.14 1.00H2 450, 300 1,680, 2,352 3.73, 7.84 5.79 ± 2.9 1.00H3 80, 140 462, 532 5.78, 3.80 4.79 + 1.4 1.00
NIH 3T3Gi 410, 210, 320 1,850, 1,906, 2,006 4.51, 9.08, 6.27 6.62 + 2.3 0.31 0.43Hi 530, 580 700, 832 1.32, 1.43 1.38 ± 0.08 0.90H2 3,500, 1,700 1,375, 750 0.39, 0.44 0.42 + 0.03 0.07H3 670, 430 1,159, 1,103 1.73, 2.57 2.15 + 0.59 0.45
Sc3T3GI 3,000, 1,760, 1,120 1,031, 909, 691 0.34, 0.52, 0.62 0.49 + 0.14 0.02 0.11Hi 1,630, 2,360 668, 321 0.41, 0.14 0.27 + 0.19 0.18H2 4,030, 3,040 2,000, 1,790 0.50, 0.59 0.54 + 0.07 0.09H3 610, 420 488, 300 0.80, 0.71 0.76 + 0.06 0.16
ScSV3G1 2,110, 1,740, 1,700 963, 1,145, 1,096 0.46, 0.66, 0.64 0.59 + 0.11 0.03 0.08Hi 2,090, 2,400 339, 345 0.16, 0.14 0.15 + 0.01 0.01H2 410, 360 32, 188 0.08, 0.52 0.30 ± 0.31 0.05H3 270, 220 90, 182 0.33, 0.83 0.58 + 0.35 0.12
a For abbreviations, see the footnotes to Table 1.
MOL. CELL. BIOL.
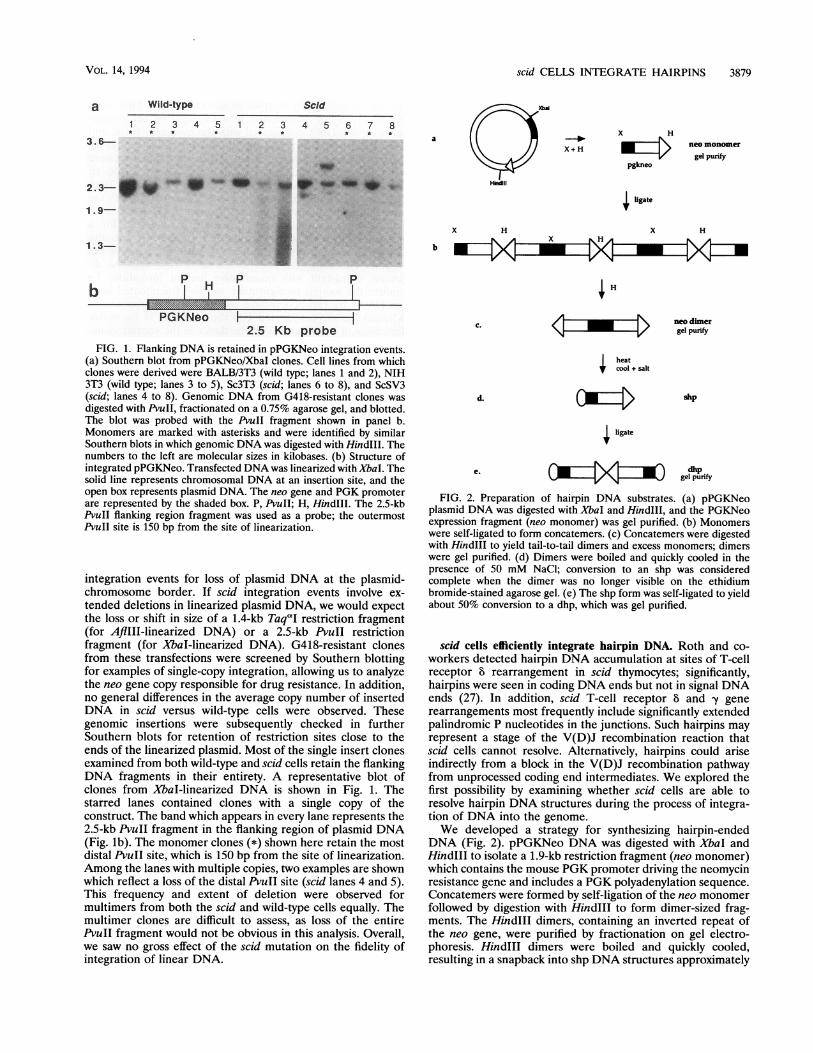
scid CELLS INTEGRATE HAIRPINS 3879
a Wild-type Scid
1 2 3 4 5 1 2 3 4 5 6 7 8* * * * * * * * *
3.6-
2.3- 10 t# ~ fg 'mtt:wIW m &V
1 .9-
1 .3-
bp H pI I l
st.l 4-I.
.x H
X+H
pgkneo
neo monomer
gel purify
ligate
X H X H
b _
Hp
PGKNeo I-2.5 Kb probe
1IFIG. 1. Flanking DNA is retained in pPGKNeo integration events.
(a) Southern blot from pPGKNeo/XbaI clones. Cell lines from whichclones were derived were BALB/3T3 (wild type; lanes 1 and 2), NIH3T3 (wild type; lanes 3 to 5), Sc3T3 (scid; lanes 6 to 8), and ScSV3(scid; lanes 4 to 8). Genomic DNA from G418-resistant clones wasdigested with PvuII, fractionated on a 0.75% agarose gel, and blotted.The blot was probed with the PvuII fragment shown in panel b.Monomers are marked with asterisks and were identified by similarSouthern blots in which genomic DNA was digested with HindIl. Thenumbers to the left are molecular sizes in kilobases. (b) Structure ofintegrated pPGKNeo. Transfected DNA was linearized with XbaI. Thesolid line represents chromosomal DNA at an insertion site, and theopen box represents plasmid DNA. The neo gene and PGK promoterare represented by the shaded box. P, PvuII; H, HindIll. The 2.5-kbPvuII flanking region fragment was used as a probe; the outermostPvuII site is 150 bp from the site of linearization.
integration events for loss of plasmid DNA at the plasmid-chromosome border. If scid integration events involve ex-tended deletions in linearized plasmid DNA, we would expectthe loss or shift in size of a 1.4-kb TaqaI restriction fragment(for AflIII-linearized DNA) or a 2.5-kb PvuII restrictionfragment (for XbaI-linearized DNA). G418-resistant clonesfrom these transfections were screened by Southern blottingfor examples of single-copy integration, allowing us to analyzethe neo gene copy responsible for drug resistance. In addition,no general differences in the average copy number of insertedDNA in scid versus wild-type cells were observed. Thesegenomic insertions were subsequently checked in furtherSouthern blots for retention of restriction sites close to theends of the linearized plasmid. Most of the single insert clonesexamined from both wild-type and scid cells retain the flankingDNA fragments in their entirety. A representative blot ofclones from XbaI-linearized DNA is shown in Fig. 1. Thestarred lanes contained clones with a single copy of theconstruct. The band which appears in every lane represents the2.5-kb PvuII fragment in the flanking region of plasmid DNA(Fig. lb). The monomer clones (*) shown here retain the mostdistal PvuII site, which is 150 bp from the site of linearization.Among the lanes with multiple copies, two examples are shownwhich reflect a loss of the distal PvuII site (scid lanes 4 and 5).This frequency and extent of deletion were observed formultimers from both the scid and wild-type cells equally. Themultimer clones are difficult to assess, as loss of the entirePvuII fragment would not be obvious in this analysis. Overall,we saw no gross effect of the scid mutation on the fidelity ofintegration of linear DNA.
C. <zm
d.
|heatcool + salt
,:4
neo dimergel purify
shp
4gligate
dhp~~~Dgel p-urify
FIG. 2. Preparation of hairpin DNA substrates. (a) pPGKNeoplasmid DNA was digested with XbaI and HindIll, and the PGKNeoexpression fragment (neo monomer) was gel purified. (b) Monomerswere self-ligated to form concatemers. (c) Concatemers were digestedwith HindIll to yield tail-to-tail dimers and excess monomers; dimerswere gel purified. (d) Dimers were boiled and quickly cooled in thepresence of 50 mM NaCl; conversion to an shp was consideredcomplete when the dimer was no longer visible on the ethidiumbromide-stained agarose gel. (e) The shp form was self-ligated to yieldabout 50% conversion to a dhp, which was gel purified.
scid cells efficiently integrate hairpin DNA. Roth and co-workers detected hairpin DNA accumulation at sites of T-cellreceptor 8 rearrangement in scid thymocytes; significantly,hairpins were seen in coding DNA ends but not in signal DNAends (27). In addition, scid T-cell receptor 8 and -y generearrangements most frequently include significantly extendedpalindromic P nucleotides in the junctions. Such hairpins mayrepresent a stage of the V(D)J recombination reaction thatscid cells cannot resolve. Alternatively, hairpins could ariseindirectly from a block in the V(D)J recombination pathwayfrom unprocessed coding end intermediates. We explored thefirst possibility by examining whether scid cells are able toresolve hairpin DNA structures during the process of integra-tion of DNA into the genome.We developed a strategy for synthesizing hairpin-ended
DNA (Fig. 2). pPGKNeo DNA was digested with XbaI andHindIIl to isolate a 1.9-kb restriction fragment (neo monomer)which contains the mouse PGK promoter driving the neomycinresistance gene and includes a PGK polyadenylation sequence.Concatemers were formed by self-ligation of the neo monomerfollowed by digestion with Hindlll to form dimer-sized frag-ments. The HindlIl dimers, containing an inverted repeat ofthe neo gene, were purified by fractionation on gel electro-phoresis. HindlIl dimers were boiled and quickly cooled,resulting in a snapback into shp DNA structures approximately
p-.J..i.r,\.-., ='..n:ME-aa, m 1,
VOL. 14, 1994

3880 STAUNTON AND WEAVER
0a)EA Ec0E
BDE-o
Q)
7.2_
3.6-
2.3-1.9 -1.3-
C
LO o Lf Co0 0
o _: 0
dimer +
monomer + w
units ExoV
* * 4- dhp0
4- shp
FIG. 3. Characterization of hairpin DNAs. (A and B) Mobilities ofmonomer and dimer DNAs and shps and dhps in agarose gel electro-phoresis. The numbers to the left of each panel are molecular sizes inkilobases. (C) Digestion by exonuclease V. Monomer and dimer linearDNAs or shps and dhps were digested with 0, 0.5, or 1.0 U ofexonuclease V for 30 min, fractionated on an agarose gel, and blotted.Blots were probed with a PGKNeo fragment.
the size of the neo monomer. By design, these moleculescontain one hairpin terminus and one restriction fragment(HindIII) end and are termed shp molecules. dhps wereformed by self-ligation of the shp molecules.We confirmed that the ends of the shp and dhp structures
are indeed blocked by hairpins by several criteria. First, bothhairpin types ran as expected in native agarose gel electro-phoresis; shp DNA comigrated with the monomer form, anddhp DNA comigrated with the dimer form (Fig. 3a and b).Second, the neo monomer, the neo dimer, shps, and dhps weretreated with exonuclease V. Exonuclease V digests single- ordouble-stranded DNA but requires an open 3' or 5' end. Boththe linear neo monomer and dimer were digested fully by thelowest enzyme concentration (Fig. 3c). The shp DNA was alsovulnerable to exonuclease V digestion, as expected for amolecule with a single nonhairpin end. In contrast, the dhpDNA was resistant to digestion, even with an excess of theenzyme. Therefore, the ends are blocked from nuclease di-gestion, consistent with quantitative hairpin formation. Toconfirm the hairpin configuration, we also fractionated hair-pin DNAs by two-dimensional agarose gel electrophoresis.Whereas the shp comigrated with the monomer in the nativedimension, it comigrated with the dimer in the second, dena-turing, dimension (data not shown).
Purified hairpin DNA molecules were used in transfectionexperiments similar to those used to measure the integration oflinear DNA. We compared wild-type and scid fibroblasts forthe ability to integrate the shp substrates (Fig. 2). In eachexperiment, the efficiency of hairpin integration was normal-ized relative to a parallel or internal control for integration oflinear DNA, thus eliminating the effects of transfection effi-ciency differences between cell lines. Separate hairpin DNApreparations were used for the two sets of transfections. Wecalculated integration frequencies as discussed above, by usingthe number of resistant colonies divided by the number ofviable colonies for each transfection (Imn and 'shp in Table 3).For each set, duplicate parallel transfections were done on thesame day with neo monomer or shp DNA. The duplicates wereaveraged to derive the mean efficiency for each pair. The ratioof the shp-to-monomer frequency (hp/mn) for each set wasthen calculated from these mean values. The hp/mn valueswithin a set were compared by deriving a corrected ratio(hp/mncorr) relative to a BALB/3T3 value of 1.0. The two setsof corrected ratios were averaged to obtain the averagecorrected ratio (hp/mnav).
Purified shp DNA was effectively integrated in all of the celllines tested (Table 3). NIH 3T3 (hp/mncorr, 1.49), Sc3T3(hp/mncorr, 1.51), and ScSV3 (hp/mncorr, 1.41) cells were
TABLE 3. Integration of shp DNA into scid and wild-type cell lines'
Cell line and set /nb (103) Mean I__ + SD Ishpd (10-3) Mean l,hpc - SD hp/mn hp/mncorr hp/mna,BALB/3T3
1 0.30, 0.20 0.25 10.07 0.23, 0.18 0.21 +0.04 0.82 1.00 1.002 4.30, 5.00 4.65 + 0.05 1.80, 2.00 1.90 0.14 0.41 1.00
NIH 3T31 3.90 3.90 0.87, 2.30 1.59 ± 1.0 0.41 0.49 1.492 5.60, 6.40 6.00 ± 0.57 5.70, 6.50 6.10 +0.57 1.02 2.49
Sc3T31 0.21 0.21 0.09, 0.13 0.11 + 0.03 0.53 0.64 1.512 1.30, 1.70 1.50 +0.28 1.10, 1.80 1.45 ± 0.50 0.97 2.37
ScSV31 0.61, 0.57 0.59 + 0.02 0.58, 0.43 0.51 ± 0.11 0.86 1.03 1.412 3.50, 4.70 4.10 ±0.85 4.00, 2.00 3.00 + 1.40 0.73 1.78
aLinear monomer and shp DNAs are described in Fig. 1. Set 1 was transfected via CaPO4; set 2 transfection was done via electroporation.b I,n, transfection efficiency per 103 viable cells for monomer DNA.c Mean I.n and mean Ishp, mean efficiencies for each cell line for a given set.d Ishp, efficiency for shp DNA.
7.2 _3.6-2.3-1.9-1.3-
MOL. CELL. BIOL.

scid CELLS INTEGRATE HAIRPINS 3881
TABLE 4. Integration of dhp DNA into scid and wild-type cells'
Cell line No. of colonies with:andset DNA ratio dhp/linear (hp/ln) Mean hp/In + SD hp/lncorr hp/ln;,,,and set Linear DNA dhp DNA
BALB/3T31 532, 540 17, 20 0.03, 0.04 0.03 _ 0.004 1.00 1.002 1,680, 2,352 367, 513 0.22, 0.22 0.22 + 0.0 1.00
NIH 3T31 700, 832 14, 36 0.02, 0.04 0.03 + 0.02 0.92 1.092 1,375, 750 322, 240 0.23, 0.32 0.28 + 0.06 1.27
Sc3T31 668, 321 44, 2 0.07, 0.01 0.04 + 0.04 1.05 1.362 2,000, 1,790 620, 748 0.31, 0.42 0.36 + 0.08 1.67
SvSV31 339, 345 10, 12 0.03, 0.03 0.03 + 0.0 0.93 1.892 32, 188 38, 11 1.19, 0.06 0.62 + 0.8 2.85
" hp/In is the ratio of G418-resistant colonies (from dhp) to hygromycin B-resistant colonies (from pPGKHygro) within the same transfection. Mean hp/in is the meanratio for a set of duplicate transfections. hp/nc,,rr is the corrected mean per set, relative to a BALB value of 1.0. hp/ln,, is the average corrected ratio of the two sets.
comparable to the normalizing cell line, BALB/3T3 (hp/mncorr,1.0). This indicates that the presence of one end with a hairpinstructure did not greatly affect DNA integration. These exper-iments indicate that scid cells are able to process hairpin endswith an efficiency similar to that of wild-type cell lines. In all ofthe cell lines, the hairpin and monomer DNAs integrated withsimilar frequencies (hp/mn, 0.41 to 1.02). Thus, all of the cellswere able to process the hairpin end sufficiently to allowintegration.We performed similar experiments with the dhp substrate
which we have shown to be covalently closed at both ends. Inthese experiments, dhp DNA was cotransfected with linearizedDNA containing the hygromycin B phosphotransferase-encod-ing gene, providing an internal control for each transfection.Transfected cells were plated in parallel for selection with eachdrug. The number of hygromycin B-resistant colonies (linearDNA) and the number of G418-resistant colonies (dhp DNA)were quantitated for each transfection (Table 4). The ratio ofdhp colonies to linear colonies (hp/mn) was calculated for eachtransfection, and the mean for each set (mean hp/mn) wasdetermined. We also corrected the values by setting theBALB/3T3 cell line control hp/mncorr value at 1.0, as for linearDNA transfections, and the corrected values of the two setswere averaged for each cell line (hp/mnav).The scid and wild-type cell lines had comparable abilities to
integrate dhp DNAs (Table 4). Two scid cell lines wereproficient at hairpin DNA integration, showing a slightlyhigher hp/mnav than did wild-type cells (Sc3T3 hp/mnav, 1.36;ScSV3 hp/mnav, 1.89). Similarly, in transfections of dhp DNAwithout addition of linear monomer DNA, no scid defect wasseen (data not shown). Thus, hairpin-ended DNA integratesinto either wild-type or scid fibroblast cell lines, indicating thatscid cells are able to resolve hairpin structures. We cannotexclude the possibility that transfection leads to nicked hair-pins prior to integration.
shp DNA was integrated at least as efficiently as the corre-sponding linear DNA neo fragment (Table 2). The absoluterate of dhp integration in Table 3 is more difficult to estimatebecause the cotransfected marker DNA was at -50-foldexcess. If we assume both that integration frequency is linearwith respect to the amount of DNA transfected and that thetwo selective drugs are comparable, the dhps would haveintegrated as efficiently as the linear DNA.
DISCUSSION
Integration of hairpin DNA. Our data indicate that DNAmolecules in which one or both termini are blocked by hairpinsare efficiently integrated into the genome in scid and wild-typecells (Tables 3 and 4). Because these blocked DNA ends mustbe resolved prior to or during the process of integration, it islikely that they convert to an integration-efficient form first.Several previous observations have implicated free double-stranded DNA ends as the most effective integration substrate.First, linearized plasmid DNA integrates more efficiently thancircular plasmid DNA. Second, integration junctions of theintroduced DNAs are most often conserved, suggesting thatthe integration occurs directly through these termini (8, 23).Third, when DNA is linearized with restriction enzymes whichremain bound to terminal nucleotides, integration efficiencydrops, suggesting that protein-free ends are preferred (10).Fourth, dideoxynucleotide addition to double-stranded DNAends reduces the efficiency of end joining (6).
Hairpin-ended DNA could be resolved for integration byeither of two pathways. Single-stranded or double-strandedcuts at or near the termini would be expected to form efficientintegration-joining substrates. On the other hand, strandbreaks further internal in hairpin-ended molecules could alsopromote integration, although not in a simple mechanism. Twoobservations support a pathway by which the hairpins areresolved near their termini. scid and wild-type cells efficientlyrecircularize linear plasmid DNA containing terminal hairpins(17). First, these end-joined junctions frequently contain in-serted palindromic nucleotides consistent with nicking only 1to 5 bp within the hairpins. Second, we showed that theposition of linearization of DNA relative to the PGK promotershows that the integration efficiency can be influenced by theextent of deletion at the ends. Therefore, the proximity of thePGK promoter to hairpins in our assays is expected to limit theterminal deletion required to retain neo expression. Since wedid find efficient hairpin integration (Tables 3 and 4), terminalresolution of these hairpins is also the most likely option.
Implications for V(D)J rearrangement. scid V(D)J recom-bination events demonstrate that coding and signal jointformation are separable events. Productive signal joints areobserved in scid cells at a slightly reduced level, whereasnormal coding joints are difficult to detect by most assays (13,
VOL. 14, 1994

3882 STAUNTON AND WEAVER
19). Also, hairpin-ended 8 coding sequences accumulate in scidthymocytes as if hairpin resolution were blocked during TCR 8recombination (27). The lack of productive coding joints mightbe explained by a slow rate of formation where the cleavedDNA is vulnerable to exonucleases. Alternatively, coding jointformation could be dependent on hairpin resolution. Whenhairpin resolution is blocked, junctions either do not form at allor form by secondary pathways leading to extensive deletions,i.e., the scid defect. A hairpin model also explains the appear-ance of P nucleotides in coding junctions (18, 27). The numberof P nucleotides would be determined by the position at whichthe hairpin is nicked and whether 3' or 5' overhangingnucleotides are chewed back or filled in. Most frequently, Pnucleotides are 1 or 2 bp, suggesting that hairpins are resolvedinternally 1 or 2 bp in their single-stranded DNA loop.P-nucleotide occurrence is variable and dependent on thecomposition of the adjacent coding sequence, suggesting thateither hairpin formation or the position of nicking is sequencedependent (4, 22).Our results do not preclude a scid-specific requirement in
hairpin resolution for V(D)J coding end joining. In both ourassay and the end-joining assay (17), hairpin nicking couldoccur during transfection and bypass the requirement forscid-dependent resolution activity. It is also possible thathairpin processing is sufficient for integration and end joiningbut somehow limiting in lymphocyte V(D)J rearrangement.Our assay may not be sensitive enough to detect quantitativedifferences in hairpin processing between cell lines becausetransfected cells take up many copies of the exogenous DNAbut only a single integrated copy is required for G418 resis-tance. However, hairpin-resolved junctions have been recov-ered at similar levels in scid and wild-type cells (17).
Coding junction formation does not require DNA homologynear the coding ends (4, 5) and is therefore similar tononhomologous reactions, such as random integration and endjoining. However, because we have seen no evidence for a sciddefect in hairpin processing when investigating random DNAintegration, there may be mechanistically separable classes ofnonhomologous recombination events, only some of whichoverlap with the V(D)J recombination pathway.
Integration of linear DNA. Early-passage scid cells efficientlyintegrate linear DNA (Table 1). Several factors led us toconclude that the normal integration frequency is not acompound effect that masks a scid deficiency. First, there wereno significant differences among ScF4, ScF6, and CB1 fibro-blasts in DNA uptake control assays (data not shown). Second,for scid cell lines in which an integration deficiency wasobserved, Southern blot data indicated that the stably inte-grated plasmid copy number was comparable for scid andwild-type cells (Fig. 1 and data not shown). There may be aredundancy in recombination and/or repair such that anotherprotein(s) functionally substitutes for that encoded by scid insome circumstances. scid mice are vulnerable to infection butnot particularly susceptible to tumor formation (25). Possibly,the scid-encoded protein acting in V(D)J recombination isinvolved in highly specific protein-protein interactions thatrestrict the ability of a redundant protein to act. Similarly, scidmay be defective in only a subset of chromosome damagerepair. scid cells repair a fraction of double-stranded breaks ata wild-type rate, but overall repair is less than half of that ofwild-type cells (12). This repair defect may result from areduced repair threshold, or the unrepaired breaks may con-tain end structures that cannot be rejoined by scid cells. TheDNA integration assay may be insensitive to this difference,since it requires integration of only a single plasmid.
Spontaneously or simian virus 40-transformed scid cell lines
reduced the integration efficiency 10-fold (Table 2). However,our results obtained with primary fibroblasts suggest that suchdifferences are likely not due to the scid mutation alone.Wild-type (BALB/3T3 and NIH 3T3) cell lines showed atwofold variation in integration efficiency. Wild-type cell vari-ations could be due to strain differences, as has been observedwith radiation sensitivity (12). Also, we have previously shownthat the X-ray sensitivities of related scid cell lines can differsignificantly (12). It was previously shown that scid fibroblastshad a 10- to 100-fold-reduced ability to integrate linear plasmidDNA (11), whereas scid pre-B cells showed normal DNAintegration (19). The DNA I value of a cell line is probably acomplex function influenced by a number of factors, includingthe growth regulation and transformation status of the cells.
ACKNOWLEDGMENTS
We thank Nikolai Boubnov, John Petrini, and Simon Powell forhelpful discussions and Susanna Lewis for sharing a manuscript priorto publication. We also thank Chuck Stiles for BALB/3T3 cells, and weare very grateful to David Ord for assistance in the preparation ofprimary fibroblasts.
This work was supported by NIH grant CA54326 to D.T.W.
REFERENCES1. Biedermann, K. A., J. Sun, A. J. Giaccia, L. M. Tosto, and J. M.
Brown. 1991. scid mutation in mice confers hypersensitivity toionizing radiation and a deficiency in DNA double-strand breakrepair. Proc. Natl. Acad. Sci. USA 88:1394-1397.
2. Blackwell, T. K., and F. W. Alt. 1988. Immunoglobulin genes, p.1-60. In B. D. Hames and D. M. Glover (ed.), Molecularimmunology. IRL Press, Oxford.
3. Bosma, G. C., R. P. Custer, and M. J. Bosma. 1983. A severecombined immunodeficiency mutation in the mouse. Nature (Lon-don) 301:527-530.
4. Boubnov, N. V., Z. P. Wills, and D. T. Weaver. 1993. V(D)Jrecombination coding junction formation without DNA homol-ogy: processing of coding termini. Mol. Cell. Biol. 13:6957-6968.
5. Boubnov, N. V., Z. P. Wills, and D. T. Weaver. Inhibition of theinitiation of V(D)J recombination by coding DNA flanking signalelements of either spacer length. Submitted for publication.
6. Chang, X.-B., and J. H. Wilson. 1987. Modification of DNA endscan decrease end joining relative to homologous recombination inmammalian cells. Proc. Natl. Acad. Sci. USA 84:4959-4963.
7. Feinberg, A., and B. Vogelstein. 1984. A technique for radiolabel-ing DNA restriction endonuclease fragments to high specificactivity. Annu. Rev. Immunol. 4:529-591.
8. Folger, K. R., E. A. Wong, G. Wahl, and M. R. Capecchi. 1982.Patterns of integration of DNA microinjected into cultured mam-malian cells: evidence for homologous recombination betweeninfected plasmid DNA molecules. Mol. Cell. Biol. 2:1372-1387.
9. Fulop, G. M., and R. A. Phillips. 1990. The scid mutation in micecauses a general defect in DNA repair. Nature (London) 347:479-482.
10. Gesew, N., A. Nepveu, and P. Chartrand. 1987. Linear DNA musthave free ends to transform rat cells efficiently. Mol. Gen. Genet.206:121-125.
11. Harrington, J., C.-L. Hsieh, J. Gerton, G. Bosma, and M. R.Lieber. 1992. Analysis of the defect in DNA end joining in themurine scid mutation. Mol. Cell. Biol. 12:4758-4768.
12. Hendrickson, E. A., X.-Q. Qin, E. A. Bump, D. G. Schatz, M.Oettinger, and D. T. Weaver. 1991. A link between double-strandbreak-related repair and V(D)J recombination: the scid mutation.Proc. Natl. Acad. Sci. USA 88:4061-4065.
13. Hendrickson, E. A., D. G. Schatz, and D. T. Weaver. 1988. The scidgene encodes a trans-acting factor that mediates the rejoiningevent of Ig gene rearrangement. Genes Dev. 2:817-829.
14. Henner, W. D., S. M. Grunberg, and W. A. Haseltine. 1983.Enzyme action at 3' termini of ionizing radiation-induced DNAstrand breaks. J. Biol. Chem. 258:15198-15205.
15. Lafaille, J. J., A. DeCloux, M. Bonneville, Y. Takagaki, and S.Tonegawa. 1989. Junctional sequences of T cell receptor y8 genes:
MOL. CELL. BIOL.

scid CELLS INTEGRATE HAIRPINS 3883
implications for yb T cell lineages and for a novel intermediate ofV-(D)-J joining. Cell 59:859-870.
16. Leaverton, P. E. 1991. A review of biostatistics, 4th ed. Little,Brown, Boston.
17. Lewis, S. M. 1994. P-nucleotide insertions and the resolution ofhairpin DNA structures in mammalian cells. Proc. Natl. Acad. Sci.USA 91:1332-1336.
18. Lieber, M. R. 1991. Site-specific recombination in the immunesystem. FASEB J. 5:2934-2944.
19. Lieber, M. R., J. E. Hesse, S. Lewis, G. C. Bosma, N. Rosenberg,K. Mizuuchi, M. J. Bosma, and M. Gellert. 1988. The defect inmurine severe combined immune deficiency: joining of signalsequences but not coding segments in V(D)J recombination. Cell55:7-16.
20. Malynn, B. A., T. K. Blackwell, G. M. Fulop, G. A. Rathbun,A. J. W. Furley, P. Ferrier, L. B. Heinke, R. A. Phillips, G. D.Yancopoulos, and F. W. Alt. 1988. The scid defect affects the finalstep of the immunoglobulin VDJ recombinase mechanism. Cell54:453-460.
21. McCormack, W. T., L. W. Tjoelker, L. M. Carlson, B. Petryniak,C. F. Barth, E. H. Humphries, and C. B. Thompson. 1989. Chicken1gL gene rearrangement involves deletion of a circular episomeand addition of single nonrandom nucleotides to both codingsegments. Cell 56:785-791.
22. Meier, J. T., and S. M. Lewis. 1993. P nucleotides in V(D)J
recombination: a fine-structure analysis. Mol. Cell. Biol. 13:1078-1092.
23. Murnane, J. P., M. J. Yezze, and B. R. Young. 1990. Recombina-tion events during integration of transfected DNA into normalhuman cells. Nucleic Acids Res. 18:2733-2738.
24. Petrini, J. H. J., J. W. Donovan, C. DiMare, and D. T. Weaver.1994. Normal V(D)J coding junction formation in DNA ligase Ideficiency syndromes. J. Immunol. 152:176-183.
25. Phillips, R. A., and G. M. Fulop. 1989. Pleiotropic effects of thescid mutation: effects on lymphoid differentiation and on repair ofradiation damage. Curr. Top. Microbiol. Immunol. 152:11-17.
26. Roth, D., P. Nakajima, J. Menetski, M. Bosma, and M. Gellert.1992. V(D)J recombination in mouse thymocytes: double-strandbreaks near T cell receptor 8 rearrangement signals. Cell 69:41-53.
27. Roth, D. B., J. P. Menetski, P. B. Nakajima, M. J. Bosma, and M.Gellert. 1992. V(D)J recombination: broken DNA molecules withcovalently sealed (hairpin) coding ends in scid mouse thymocytes.Cell 70:983-991.
28. Schuler, W., I. J. Weiler, A. Schuler, R. A. Phillips, N. Rosenberg,T. W. Mak, J. F. Kearney, R. P. Perry, and M. J. Bosma. 1986.Rearrangement of antigen receptor genes is defective in mice withsevere combined immune deficiency. Cell 46:963-972.
29. Todaro, G. J., and H. Green. 1963. Quantitative studies of thegrowth of mouse embryo cells in culture and their developmentinto established lines. J. Cell Biol. 17:299-313.
VOL. 14, 1994