signal transduction, calcium and acute pancreatitis
TRANSCRIPT

Invited Review
Pancreatology 2003;3:497–505DOI: 10.1159/000075581
Signal Transduction, Calcium and AcutePancreatitis
Robert Suttona David Criddlea,b Michael G.T. Raratya Alexei Tepikinb
John P. Neoptolemosa Ole H. Petersenb
Departments of aSurgery and bPhysiology, University of Liverpool, Liverpool, UK
Published online: December 11, 2003
Prof. Robert Sutton, DPhil, FRCSDepartment of Surgery, University of Liverpool5th Floor UCD Block, Royal Liverpool University HospitalDaulby Street, Liverpool L69 3GA (UK)Tel. +44 151 7064170, Fax +44 151 7065826, E-Mail [email protected]
ABCFax + 41 61 306 12 34E-Mail [email protected]
© 2003 S. Karger AG, Basel1424–3903/03/0036–0497$19.50/0
Accessible online at:www.karger.com/pan
Key WordsCell signalling W Calcium W Acute pancreatitis W
Pathogenesis
AbstractEvidence consistently suggests that the earliest changesof acute pancreatitis are intracellular, the hallmark ofwhich is premature intracellular activation of digestivezymogens, accompanied by disruption of normal signaltransduction and secretion. Principal components ofphysiological signal transduction include secretagogue-induced activation of G-protein-linked receptors, fol-lowed by generation of inositol 1,4,5-trisphosphate, nico-tinic acid adenine dinucleotide phosphate and cyclicADP-ribose. In response, calcium is released from endo-plasmic reticulum terminals within the apical, granularpole of the cell, where calcium signals are usually con-tained by perigranular mitochondria, in turn respondingby increased metabolism. When all three intracellularmessengers are administered together, even at thresh-old concentrations, dramatic potentiation results in sus-tained, global, cytosolic calcium elevation. Prolonged,global elevation of cytosolic calcium is also induced byhyperstimulation, bile salts, alcohol and fatty acid ethylesters, and depends on continued calcium entry into thecell. Such abnormal calcium signals induce intracellularactivation of digestive enzymes, and of nuclear factor ÎB,
as well as the morphological changes of acute pancreati-tis. Depletion of endoplasmic reticulum calcium and mi-tochondrial membrane potential may contribute to fur-ther cell injury. This review outlines current understand-ing of signal transduction in the pancreas, and its appli-cation to the pathophysiology of acute pancreatitis.
Copyright © 2003 S. Karger AG, Basel
Premature Enzyme Activation Is the Hallmarkof Acute Pancreatitis
The incidence of acute pancreatitis is of the order of 40per 100,000 per year and is increasing [1]. Severe compli-cations that require major intervention and intensive caresupport develop in 20–30% of patients, and carry a mor-tality of 20–50% [1]. Despite recognition of precipitatingfactors, understanding of the pathogenesis of acute pan-creatitis has been limited, and randomised trials havelargely failed to show consistent benefits in favour of anytreatment [1].
Acute pancreatitis is an autolytic process, with acti-vated pancreatic enzymes identifiable within [2–4] andsubsequently around acinar cells, precipitated by ductalhypertension, bile salts, ethanol, ischaemia, hypercal-caemia, hyperlipidaemia, viral infections, drugs or toxins[1, 5]. Evidence consistently suggests that the initialchanges are intracellular: apical enzyme activation, cyto-

498 Pancreatology 2003;3:497–505 Sutton/Criddle/Raraty/Tepikin/Neoptolemos/Petersen
skeletal disruption, loss of secretory polarity, vacuolisa-tion, co-localisation of zymogen granules and lysosomes,and expression of pro-inflammatory cytokines all occur[6–10]. Co-localisation of zymogen granules and lyso-somes may be an initiating event [4]; alternatively, suchco-localisation may be protective [8]. It is also uncertainwhich of the many acinar cell pro-enzymes can initiateacute pancreatitis; substantial evidence favours trypsino-gen as the critical mediator [11–14]; pro-cathepsin B andgranular pro-elastase also have important early effects[15, 16].
Despite a series of safeguards, the central role of tryp-sinogen as a one-way master switch for digestive enzymeactivation carries a liability: this molecule, and thus thepancreas, is vulnerable to premature activation. Muta-tions of the PRSS1 cationic trypsinogen gene (includingR122H and N29I) render prematurely activated intracel-lular trypsinogen resistant to normal, protective inactiva-tion, causing hereditary pancreatitis [11, 12]. Hereditarypancreatitis is a rare autosomal dominant disorder result-ing in recurrent acute pancreatitis, leading to chronic pan-creatitis and a lifetime risk of pancreatic cancer of 40–50%. In addition, mutations of the pancreatic secretorytrypsin inhibitor contribute to progression of the disease[13]. The importance of trypsin is underlined by ourrecent demonstration that on admission to hospital, uri-nary trypsinogen activation peptide levels are highly pre-dictive of both the presence and severity of acute pancre-atitis [14].
Abnormal Elevation in Cytosolic CalciumInitiates Enzyme Activation
Stimulation of acinar cells with a physiological concen-tration of cholecystokinin (up to 10 pM) elicits repetitivecytosolic calcium spikes, primarily due to release of cal-cium from intracellular stores [17, 18]. Such calcium spik-ing evokes normal secretion, but not intracellular trypsinactivation [19]. On the other hand, cholecystokinin hyper-stimulation (10 nM) evokes a sustained elevation of thecytosolic calcium level that induces trypsinogen activa-tion [19]; in vivo, cholecystokinin hyperstimulation hasbeen found to induce acute pancreatitis in all species thathave been examined [2–10, 19].
Abnormally high levels of cytosolic calcium are instru-mental in damage to other cell types, including from abu-sive stimulation, ischaemia-reperfusion, toxins, viruses,in immune cell killing and apoptosis [5, 17, 20, 21]. Pro-longed elevations of cytosolic calcium can activate de-
gradative calcium-dependent enzymes, leading to necro-sis or apoptosis, depending on the degree of damage [21].We have proposed [5] and tested [19] the hypothesis thatacute pancreatitis is caused by pathological increases inacinar cytosolic calcium. Our results [19] agree with thoseof others [22, 23] in support of this, as discussed below(‘Abnormal Elevation of Cytosolic Calcium TriggersAcute Pancreatitis’). However, before considering the evi-dence further, it is appropriate to review normal pancreat-ic acinar cell signalling, discussed in the next section [andmore extensively in 17, 18 and 24].
Signal Transduction Has Been Defined inNormal Acinar Cells
The normal calcium-signalling process within the pan-creatic acinar cell has been worked out in some detail [17,18, 24–39]. Acetylcholine binding to muscarinic receptorsactivates phospholipase C, generating inositol 1,4,5-tris-phosphate. This messenger binds to specific receptors(calcium channels) in the endoplasmic reticulum mem-brane, causing release of calcium stored inside the endo-plasmic reticulum. Since acetylcholine-induced calciumspikes are blocked by ryanodine as well as inositol 1,4,5-receptor antagonists, the spiking process is probably dueto calcium-mediated actions on both sets of receptors (seefig. 1) [25].
In contrast to the actions of acetylcholine, physiologi-cal concentrations of cholecystokinin (up to 10 pM) gener-ate very little or no inositol trisphosphate [25, 26]. Chole-cystokinin-induced calcium spiking is totally dependenton functional nicotinic acid adenine dinucleotide phos-phate receptors, and normally requires operational cyclicADP-ribose receptors [25]. Nevertheless, cholecystoki-nin-induced calcium spiking is also dependent on func-tional inositol 1,4,5-trisphosphate receptors [25]. In addi-tion, apical calcium spikes induced by cholecystokinin aredramatically potentiated (globalised) by inositol 1,4,5-trisphosphate, but not by cyclic ADP-ribose nor nicotinicacid adenine dinucleotide phosphate, whereas calciumresponses to acetylcholine are globalised by intracellularcyclic ADP-ribose and nicotinic acid adenine dinucleo-tide phosphate, but not inositol 1,4,5-trisphosphate [27].When all three intracellular messengers are administeredtogether at threshold concentrations, dramatic potentia-tion results in sustained, global, cytosolic calcium eleva-tion [27]. Thus, two different intracellular signal path-ways converge on a common oscillator unit (see fig. 1)[25].

Signal Transduction, Calcium and AcutePancreatitis
Pancreatology 2003;3:497–505 499
Fig. 1. Physiological calcium release mecha-nisms in normal pancreatic acinar cells[from 25]. CCK = Cholecystokinin; ACh =acetylcholine; NAADP = nicotinic acid ade-nine dinucleotide phosphate; cADPR =cyclic adenosine diphosphate ribose;Ins(1,4,5)P3 = inositol 1,4,5-trisphosphate;Ca2+ = calcium; Ca-Cam = calcium-calmo-dulin complex; NAADPR = NAADP recep-tor; RyR = ryanodine receptor; InsP3R =InsP3 receptor.
Signal transduction in the pancreatic acinar cell ishighly polarised, which is suited to its principal functionof digestive enzyme secretion. As described, physiologicalcytosolic calcium signals occur principally in the apical,granular area of the cell, regulating exocytotic secretion[17, 18]. The functionally important calcium releasechannels are clustered in thin endoplasmic reticulum ele-ments extending into the apical pole [28]; followingrelease, most apical, cytosolic calcium signals are prevent-ed from spreading to the basolateral part of the cell by amitochondrial buffer barrier that surrounds the granularregion [29]. Such apical signals do not result in significantdepletion of the endoplasmic reticulum calcium store andcan go on for some time, in the complete absence of exter-nal calcium [30]. However, to restore normal apical cyto-solic levels, calcium is extruded by calcium-ATPase, athigh concentration within the apical plasma membrane[31]; calcium is also extruded by granular exocytosis [32].These small calcium fluxes prompt re-entry of small quan-tities of calcium through store-operated channels in thebasal membrane, independently of ATP, but driven byendoplasmic reticulum uptake via calcium-ATPase [33],and through endocytosis [34].
In contrast, maximal or supramaximal stimulationcauses substantial calcium depletion of the endoplasmicreticulum [30]; under these circumstances, the cytosoliccalcium signals spread from the apical region throughout
the cell [35]. Calcium-induced calcium release is themajor force behind such global transients, the mapping ofwhich (with calcium uncaging experiments) has shownthe extreme sensitivity of the apical region, in contrast tothe basolateral region [36]. With global calcium release,sustained elevations of cytosolic calcium become depen-dent on external calcium [19]. The entry of such externalcalcium is again through store-operated channels in thebasal membrane, with calcium-ATPase-mediated uptakeinto the endoplasmic reticulum, within which calciummoves rapidly across the cell [28, 30]. Whilst basal cyto-solic levels are re-established by calcium extrusion withinseconds of the cessation of stimulation, endoplasmicreticulum calcium-ATPase refilling takes several minutes[37]. Also, the endoplasmic reticulum calcium reuptakerate declines until equilibrium is reached with the passiveendoplasmic reticulum leak rate, at a calcium concentra-tion (100–300 ÌM) far higher than within the cytosol(!1 ÌM) [37].
Perigranular mitochondria take up calcium during api-cal cytosolic calcium spiking [38], not only restrictingcytosolic calcium release to the apical pole [29], but alsotriggering increased mitochondrial metabolism during ex-ocytosis [39]. There are also peripheral mitochondriapositioned close to the basolateral plasma membrane thattake up calcium during store-operated calcium entry, andperinuclear mitochondria that prevent or delay calcium

500 Pancreatology 2003;3:497–505 Sutton/Criddle/Raraty/Tepikin/Neoptolemos/Petersen
entry into the nucleoplasm; calcium uptake by all groupsof mitochondria are linked to specific subcellular meta-bolic demands [38, 39].
Calcium Signalling Is Progressively Disruptedin Acute Pancreatitis
We have shown progressive disruption of acinar cellcalcium signalling to be an integral feature of early caeru-lein-induced acute pancreatitis in mice [9]. Followinginduction of acute pancreatitis in vivo, acinar cells andcell clusters were harvested by collagenase digestion invitro, and calcium signalling examined by digital imagingmicrofluorimetry with Fura-2. In isolated acinar cells fol-lowing only a single dose of caerulein in vivo, typical high-frequency oscillations of 2–3 per min followed expo-sure to quasi-physiological doses of acetylcholine (25–100 nmol/l) and slower transients every 2–3 min followedcholecystokinin (10–20 pmol/l). With cells isolated afteran increasing number of doses of caerulein in vivo,responses became more frequently abnormal, and in-cluded transient single increases without further re-sponses, as well as slow initiation of responses, minimal oreven absent responses. Thus, normal oscillatory calciumsignals evoked by low agonist concentrations were selec-tively abolished in acute pancreatitis, with the apical poleof the acinar cell as the site of primary abnormality [9].This was especially clear in experiments that showed aprogressive loss of the normal pattern of global calciumrelease following supramaximal stimulation. In healthycells, such stimulation initiates cytosolic calcium releasewithin the apical pole, that spreads to the basolateralregions within 200–300 ms [35]. With increasing doses ofcaerulein in vivo, this sequence was progressively defi-cient, such that after 7 injections, more than 50% of cellshad lost this pattern, displaying simultaneous apical andbasal calcium release. Furthermore, the basolateral re-gions showed relatively sparing of calcium-signalling ab-normalities, since both calcium reuptake and extrusionwere found to be intact. Thus, all groups of experimentaland control cells demonstrated typical patterns of cyto-solic calcium release following inhibition of the endoplas-mic reticulum calcium reuptake pump with thapsigargin.Recently, similar overall changes in calcium signallinghave been found in isolated acini following pancreaticduct ligation-induced acute pancreatitis in the rat [40].
The demonstration of a primary signal transductionabnormality at the apical pole provides an explanation asto why digestive enzyme secretion is diminished in acute
pancreatitis [6, 9]. These data also point to a pathogeneticprocess that may be initiated within the apical pole, sinceearly in the course of the disease other areas of the cellremain intact.
Abnormal Elevation of Cytosolic CalciumTriggers Acute Pancreatitis
We have examined the role of abnormal calcium sig-nals in the initiation of acute pancreatitis, through investi-gation of acinar cell changes induced in vitro by hyper-stimulation of isolated acinar cells and cell clusters iso-lated from normal mouse acini [19]. Trypsinogen activa-tion and the occurrence of vacuoles were seen in the apicalpole of cells hyperstimulated with cholecystokinin orexposed to thapsigargin for 60 min [19], treatments thatproduce prolonged elevations in cytosolic calcium. Theresulting changes could be prevented by preincubationwith a membrane-permeable specific calcium chelator(BAPTA-AM, see fig. 2) or by simply removing extracel-lular calcium [19]. Since chelation of cytosolic calciumwith BAPTA does not inhibit agonist-evoked loss of cal-cium from the endoplasmic reticulum [37], it wouldappear to be the sustained rise in cytosolic calcium thatcauses the intracellular changes, as reported by others [22,23]. Nor were changes in mitochondrial membrane poten-tial implicated, as the inner mitochondrial membranemaintained its potential throughout the 60 min of hyper-stimulation [19]. After the peak of calcium release, a smallelevation of calcium persists with continuing hyperstimu-lation, that as indicated depends on the continued entry ofcalcium from the external medium, and which results intrypsinogen activation [19, 22, 23]. Higher concentrationsof external calcium result in greater amounts of trypsino-gen activation [41]; indeed hypercalcaemia without hy-perstimulation can cause acute pancreatitis [42]. Further-more, abnormal cytosolic calcium release has now beenfound to induce trypsinogen activation, vacuolisation andacinar cell damage by a number of investigators [19, 22,23, 41]. Together, these data suggest a key role for abnor-mal, sustained elevations of cytosolic calcium in thepathogenesis of acute pancreatitis, that contrast with thenormal oscillatory changes, and again implicate the gran-ule-rich apical pole as the site of primary injury [19, 20].
Experiments with fluorescent dyes coupled to enzyme-specific amino acid moieties (without cleavage of whichfluorescence does not occur) and visualised by confocalmicroscopy showed that within 300 s of the onset ofhyperstimulation, fluorescence occurred within multiple,

Signal Transduction, Calcium and AcutePancreatitis
Pancreatology 2003;3:497–505 501
discrete, rounded compartments, each up to 1 Ìm diame-ter and situated throughout the granular region of the cell[19, 43]. These enlarged modestly during hyperstimula-tion and became less distinct as fluorescence appeared inthe surrounding apical cytosol. By 60 min, typical vac-uoles were demonstrable by electron microscopy. Whenthe abnormal calcium signals were attenuated with thecalcium chelator BAPTA, no trypsinogen or cathepsin Bactivation occurred, and no morphological abnormalitiesdeveloped, again confirming their dependence on abnor-mal calcium signalling. The spatio-temporal characteris-tics of intracellular enzyme activation seen in these exper-iments suggested that activation first occurs within thezymogen granules themselves, and that activated granulesbecome vacuoles, which then leak activated enzymes intothe surrounding cytosol [43].
Abnormal, prolonged cytosolic calcium may provokeacinar cell enzyme activation through excessive depletionof calcium from and potassium entry into zymogen gran-ules [19]. Zymogen granules are calcium stores [32], with-in which calcium maintains granular integrity, and anacidic pH maintains the condensed, inactive state of theenclosed digestive enzymes [44]. Zymogen and othersecretory granules release calcium through inositol 1,4,5-trisphosphate and cyclic ADP-ribose triggered channels[32, 45, 46], contributing to the apical cytosolic calciumrelease that leads to exocytosis [19]. Zymogen granule cal-cium release is followed by potassium entry [45], account-ing for a modest pH increase within maturing granulesand preparing enzymes for secretion. If granular calciumdepletion is excessive before exocytosis is possible, potas-sium entry may induce not only unfolding but also prema-ture activation of digestive enzymes [47].
Recent experiments have examined the potential roleof factors that precipitate acute pancreatitis in producingabnormal signal transduction. Thus, we [48] and others[49] have found various naturally occurring bile salts toinduce abnormal, prolonged elevations in cytosolic cal-cium that are similar to those induced by hyperstimula-tion, and are likely to be associated with acute pancreati-tis. We found bile salts to induce global calcium oscilla-tions and extended calcium transients that spread fromthe apical pole, unaffected by atropine, but abolished bycaffeine or by prior depletion of calcium stores [48]. Suchcalcium release may thus occur through activation of ino-sitol 1,4,5-trisphosphate receptors, although blockade ofthe endoplasmic reticulum calcium reuptake pump hasalso been suggested [49]. Even though the debate stillrages over the clinical importance of retrograde passage ofbile into the pancreatic duct, put forward by Opie [50]
Fig. 2. Effect of cholecystokinin on cytosolic calcium (thin line) andtrypsinogen activation (thick line) in the pancreatic acinar cell[adapted from 19]. A Physiological stimulation producing normalcalcium oscillations and no trypsinogen activation (arbitrary units).B Hyperstimulation producing abnormal, prolonged elevation of cal-cium and increasing trypsinogen activation, reduced by a membrane-permeant trypsin inhibitor, benzamidine (rapid washout of rhodam-ine 110). C After loading with the calcium chelator BAPTA, hyper-stimulation producing reduced elevation of calcium and no trypsino-gen activation.
500
400
300
200
100
0
250
200
150
100
50
0
Cal
ciu
m (
n)
M
Tryp
sin
(u
nit
s)
CCK 10pM
0 200 400 600 800
Time (s)
A
500
400
300
200
100
0
250
200
150
100
50
0
Cal
ciu
m (
n)
M
Tryp
sin
(u
nit
s)
CCK 10nM
0 200 400 600 800
Time (s)
B
Benzamidine
500
400
300
200
100
0
250
200
150
100
50
0
Cal
ciu
m (
n)
M
Tryp
sin
(u
nit
s)
BAPTA + CCK 10nM
0 200 400 600 800
Time (s)
C
Benzamidine
Calcium
Trypsin

502 Pancreatology 2003;3:497–505 Sutton/Criddle/Raraty/Tepikin/Neoptolemos/Petersen
100 years ago to account for fatal acute pancreatitis in 2patients who were found at post mortem to have gall-stones impacted at the sphincter of Oddi, bile salts perfu-sion of the pancreatic duct is an established experimentalmodel of acute pancreatitis [6]. Thus, signal transductionexperiments with bile salts provide further evidence insupport of an important role for abnormal, prolonged ele-vations of cytosolic calcium in the pathogenesis of experi-mental acute pancreatitis.
Further work has demonstrated global pancreatic aci-nar cell cytosolic calcium release in response to ethanoland free fatty acid ethyl esters [51], important pancreaticmetabolites of ethanol, which also induce acute pancreati-tis [52]. In addition, oxidative stress, associated withknown precipitants of acute pancreatitis, causes sustainedacinar cell free ionised cytosolic calcium release [53].Water immersion stress, which induces heat-shock pro-teins and prevents hyperstimulation-induced acute pan-creatitis, attenuates the calcium release normally conse-quent upon caerulein hyperstimulation [54]. It is also ofnote that nuclear factor ÎB activation, which leads to aci-nar cell cytokine expression independently of trypsinogenactivation, has been found to depend on abnormal acinarcell cytosolic calcium release [54–56].
Cytokine Expression Is Dependent on CalciumSignalling
Cells produce cytokines in response to injury, to enrolvital reparative responses [57]. Acute pancreatitis inducesacinar cell expression of cytokines (e.g., tumour necrosisfactor ·) [10, 57], including chemokines (e.g., monocytechemotactic protein (1) that signal to leukocytes [55, 58].We and others have found isolated acinar cell chemokineexpression to occur by a nuclear factor ÎB-dependentmechanism within 30 min of cholecystokinin hyperstimu-lation [55, 58, 59], through prolonged elevation of cyto-solic calcium [55, 59]. However, the mechanisms induc-ing acinar cell cytokine expression are not fully under-stood, as the isolation process per se can induce cytokineexpression, again via nuclear factor ÎB [60]. Nuclear fac-tor ÎB appears to be essential to the pathogenesis of acutepancreatitis [61], although of itself does not cause trypsin-ogen activation [62]. It is thus of considerable interest thatactivation of nuclear factor ÎB requires not only proteinkinase C activation, but also an elevation of cytosolic cal-cium [54–56].
Subcellular Organelle Stress Contributes toPathogenesis
We have shown that the initial phase (within the first30 min) of acinar cell enzyme activation is criticallydependent on the prolonged elevation of cytosolic cal-cium, and not depletion of calcium within the endoplas-mic reticulum [19]. However, calcium depletion fromwithin the endoplasmic reticulum is a recognised sourceof cell stress, the end result of which may include misfold-ing of proteins and impaired export through the Golgiapparatus [63]. Excessive accumulation of misfolded pro-teins in the endoplasmic reticulum may in turn exacer-bate calcium release, causing the production of reactiveoxygen species and activation of nuclear factor ÎB [63,64]. The action of store-operated calcium, which may notonly activate but also control enzyme secretion by meansof a switch between phospholipase-Cß or phosphoinosi-tide 3-kinase signalling, may be central in this [65]. Thus,wortmannin, which inhibits phosphoinositide 3-kinases,also protects against experimentally-induced acute pan-creatitis [66]. Further evidence of the importance of cal-cium depletion of the endoplasmic reticulum comes fromexperiments with thapsigargin [19]. This substance,which blocks calcium-ATPase and depletes endoplasmicreticulum calcium, produced more dramatic acinar cellvacuolisation than cholecystokinin hyperstimulation,which does deplete endoplasmic reticulum calcium to thesame extent [19]. Furthermore, the calcium chelatorBAPTA was less effective in preventing vacuolisationproduced by thapsigargin than by cholecystokinin hyper-stimulation [19].
Lysosomes have previously been implicated in thepathogenesis of acute pancreatitis, although our recentevidence [43] brings an initiating role [4] into question(see above: ‘Abnormal Elevation of Cytosolic CalciumTriggers Acute Pancreatitis’). Lysosomes are a potentialsource of calcium release, but whether their calcium con-tributes to pathogenesis remains undetermined.
Mitochondrial metabolism is necessary for the provi-sion of energy throughout the cell. We have previouslyshown that administration of menadione, an oxidant,activates the permeability transition pore, leading to lossof the ability of perinuclear mitochondria to delay cal-cium entry into the nucleoplasm [67]. Also, the combina-tion of induction of the permeability transition pore,which causes partial mitochondrial depolarisation, andcytosolic calcium release results in activation of caspases,following which the pancreatic acinar cell proceeds downan apoptotic pathway [67]. With more marked interfer-
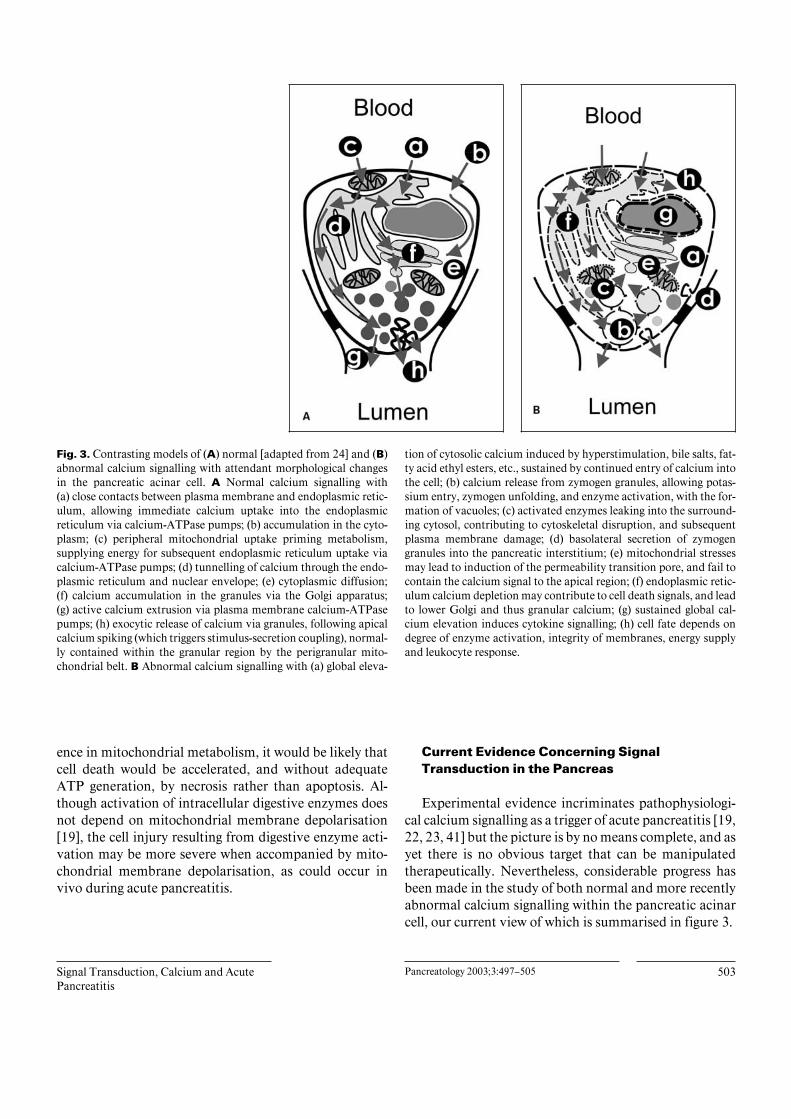
Signal Transduction, Calcium and AcutePancreatitis
Pancreatology 2003;3:497–505 503
Fig. 3. Contrasting models of (A) normal [adapted from 24] and (B)abnormal calcium signalling with attendant morphological changesin the pancreatic acinar cell. A Normal calcium signalling with(a) close contacts between plasma membrane and endoplasmic retic-ulum, allowing immediate calcium uptake into the endoplasmicreticulum via calcium-ATPase pumps; (b) accumulation in the cyto-plasm; (c) peripheral mitochondrial uptake priming metabolism,supplying energy for subsequent endoplasmic reticulum uptake viacalcium-ATPase pumps; (d) tunnelling of calcium through the endo-plasmic reticulum and nuclear envelope; (e) cytoplasmic diffusion;(f) calcium accumulation in the granules via the Golgi apparatus;(g) active calcium extrusion via plasma membrane calcium-ATPasepumps; (h) exocytic release of calcium via granules, following apicalcalcium spiking (which triggers stimulus-secretion coupling), normal-ly contained within the granular region by the perigranular mito-chondrial belt. B Abnormal calcium signalling with (a) global eleva-
tion of cytosolic calcium induced by hyperstimulation, bile salts, fat-ty acid ethyl esters, etc., sustained by continued entry of calcium intothe cell; (b) calcium release from zymogen granules, allowing potas-sium entry, zymogen unfolding, and enzyme activation, with the for-mation of vacuoles; (c) activated enzymes leaking into the surround-ing cytosol, contributing to cytoskeletal disruption, and subsequentplasma membrane damage; (d) basolateral secretion of zymogengranules into the pancreatic interstitium; (e) mitochondrial stressesmay lead to induction of the permeability transition pore, and fail tocontain the calcium signal to the apical region; (f) endoplasmic retic-ulum calcium depletion may contribute to cell death signals, and leadto lower Golgi and thus granular calcium; (g) sustained global cal-cium elevation induces cytokine signalling; (h) cell fate depends ondegree of enzyme activation, integrity of membranes, energy supplyand leukocyte response.
ence in mitochondrial metabolism, it would be likely thatcell death would be accelerated, and without adequateATP generation, by necrosis rather than apoptosis. Al-though activation of intracellular digestive enzymes doesnot depend on mitochondrial membrane depolarisation[19], the cell injury resulting from digestive enzyme acti-vation may be more severe when accompanied by mito-chondrial membrane depolarisation, as could occur invivo during acute pancreatitis.
Current Evidence Concerning SignalTransduction in the Pancreas
Experimental evidence incriminates pathophysiologi-cal calcium signalling as a trigger of acute pancreatitis [19,22, 23, 41] but the picture is by no means complete, and asyet there is no obvious target that can be manipulatedtherapeutically. Nevertheless, considerable progress hasbeen made in the study of both normal and more recentlyabnormal calcium signalling within the pancreatic acinarcell, our current view of which is summarised in figure 3.

504 Pancreatology 2003;3:497–505 Sutton/Criddle/Raraty/Tepikin/Neoptolemos/Petersen
Future research perspectives must address the generalrelevance of calcium signal transduction abnormalities,and determine whether the majority if not all precipitantsof acute pancreatitis initiate the disease by these means.In this regard, extension of these studies to other in vitroand in vivo models of acute pancreatitis will be useful [68,69]. The calcium-signalling abnormalities themselves re-quire further characterisation, to distinguish principalfeatures that are disease initiators, including amplitude,
timing and subcellular location. In turn, the effects ofthese abnormalities on several of the subcellular organelles may be revealing, including the endoplasmic reticu-lum, mitochondria and zymogen granules. As the treat-ment of acute pancreatitis is one of the most importantareas of pancreatology, it is to be hoped that continuedresearch of these phenomena will eventually lead to thedevelopment of more effective treatments.
References
1 Neoptolemos JP, Raraty M, Finch M, SuttonR: Acute pancreatitis: The substantial humanand financial costs. Gut 1998;42:886–891.
2 Leach SD, Modlin IM, Scheele GA, GorelickFS: Intracellular activation of digestive zymo-gens in rat pancreatic acini. Stimulation byhigh doses of cholecystokinin. J Clin Invest1991;87:362–366.
3 Mithofer K, Fernandez-del Castillo C, RattnerD, Warshaw AL: Subcellular kinetics of earlytrypsinogen activation in acute rodent pancre-atitis. Am J Physiol 1998;274:G71–G79.
4 Hofbauer B, Saluja AK, Lerch MM, Bhagat L,Bhatia M, Lee HS, Frossard JL, Adler G, SteerML: Intra-acinar cell activation of trypsinogenduring caerulein-induced pancreatitis in rats.Am J Physiol 1998;275:G352–G362.
5 Ward JB, Petersen OH, Jenkins SA, Sutton R:Is an elevated concentration of acinar cytosolicfree ionised calcium the trigger for acute pan-creatitis? Lancet 1995;346:1016–1019.
6 Niederau C, Niederau M, Luthen R, Stroh-meyer G, Ferrell LD, Grendell JH: Pancreaticexocrine secretion in acute experimental pan-creatitis. Gastroenterology 1990;99:1120–1127.
7 O’Konski MS, Pandol SJ: Effects of caeruleinon the apical cytoskeleton of the pancreatic aci-nar cell. J Clin Invest 1990;86:1649–1657.
8 Gorelick FS, Matovcik LM: Lysosomal en-zymes and pancreatitis. Gastroenterology1995;109:620–625.
9 Ward JB, Sutton R, Jenkins SA, Petersen OH:Progressive disruption of acinar cell calciumsignalling is an early feature of caerulein-induced pancreatitis in mice. Gastroenterology1996;111:481–491.
10 Gukovskaya AS, Gukovsky I, Zaninovic V,Song M, Sandoval D, Gukovsky S, Pandol SJ:Pancreatic acinar cells produce, release, andrespond to tumor necrosis factor-·. Role in reg-ulating cell death and pancreatitis. J Clin In-vest 1997;100:1853–1862.
11 Whitcomb DC, Gorry MC, Preston RA, FureyW, Sossenheimer MJ, Ulrich CD, Martin SP,Gates LK Jr, Amann ST, Toskes PP, Liddle R,McGrath K, Uomo G, Post JC, Ehrlich GD:Hereditary pancreatitis is caused by a mutationin the cationic trypsinogen gene. Nat Genet1996;14:141–145.
12 Sahin-Toth M: The pathobiochemistry of he-reditary pancreatitis: Studies on recombinanthuman cationic trypsinogen. Pancreatology2001;1:461–465.
13 Pfutzer RH, Barmada MM, Brunskill AP,Finch R, Hart PS, Neoptolemos J, Furey WF,Whitcomb DC: SPINK1/PSTI polymorphismsact as disease modifiers in familial and idio-pathic chronic pancreatitis. Gastroenterology2000;119:615–623.
14 Neoptolemos JP, Kemppainen EA, Mayer JM,Fitzpatrick J, Raraty MGT, Slavin J, BegerHG, Hietaranta AJ, Puolakkainen PA: Earlyprediction of severity in acute pancreatitis byurinary trypsinogen activation peptide: A mul-ticentre study. Lancet 2000;355:1955–1960.
15 Halangk W, Lerch MM, Brandt-Nedelev B,Roth W, Ruthenbuerger M, Reinheckel T,Domschke W, Lippert H, Peters C, Deussing J:Role of cathepsin B in intracellular trypsinogenactivation and the onset of acute pancreatitis. JClin Invest 2000;106:773–781.
16 Halangk W, Kruger B, Ruthenburger M, Stur-zebecher J, Albrecht E, Lippert H, Lerch MM:Trypsin activity is not involved in premature,intrapancreatic trypsinogen activation. Am JPhysiol 2002;282:G367–G374.
17 Berridge MJ, Lipp P, Bootman MD: The versa-tility and universality of calcium signalling.Nat Rev Mol Cell Biol 2000;1:11–21.
18 Williams JA: Intracellular signaling mecha-nisms activated by cholecystokinin-regulatingsynthesis and secretion of digestive enzymes inpancreatic acinar cells. Annu Rev Physiol2001;63:77–97.
19 Raraty M, Ward J, Erdemli G, Vaillant C,Neoptolemos JP, Sutton R, Petersen OH: Cal-cium-dependent enzyme activation and va-cuole formation in the apical granular region ofpancreatic acinar cells. Proc Natl Acad SciUSA 2000;97:13126–13131.
20 Parekh AB: Calcium signaling and acute pan-creatitis: Specific response to a promiscuousmessenger. Proc Natl Acad Sci USA 2000;97:12933–12934.
21 Nicotera P, Bellomo G, Orrenius S: Calcium-mediated mechanisms in chemically inducedcell death. Annu Rev Pharmacol Toxicol 1992;32:449–470.
22 Saluja AK, Bhagat L, Lee HS, Bhatia M, Fros-sard JL, Steer ML: Secretagogue-induced diges-tive enzyme activation and cell injury in ratpancreatic acini. Am J Physiol 1999;276:G835–G842.
23 Kruger B, Albrecht E, Lerch MM: The role ofintracellular calcium signaling in prematureprotease activation and the onset of pancreati-tis. Am J Pathol 2000;157:43–50.
24 Ashby MC, Tepikin AV: Polarized calcium andcalmodulin signaling in secretory epithelia.Physiol Rev 2002;82:701–734.
25 Cancela JM, Gerasimenko OV, GeramisenkoJV, Tepikin AV, Petersen OH: Two differentbut converging messenger pathways to intracel-lular Ca2+ release: The roles of nicotinic acidadenine dinucleotide phosphate, cyclic ADP-ribose and inositol trisphosphate. EMBO J2000;19:2549–2557.
26 Matozaki T, Goke B, Tsunoda Y, RodriguezM, Martinez J, Williams JA: Two functionallydistinct cholecystokinin receptors show differ-ent modes of action on Ca2+ mobilisation andphospholipid hydrolysis in isolated rat pan-creatic acini. Studies using a new cholecystoki-nin analog, JMV-180. J Biol Chem 1990;265:6247–6254.
27 Cancela JM, Van Coppenolle F, Galione A,Tepikin AV, Petersen OH: Transformation oflocal Ca2+ spikes to global Ca2+ transients: Thecombinatorial roles of multiple Ca2+ releasingmessengers. EMBO J 2002;21:909–919.
28 Mogami H, Nakano K, Tepikin AV, PetersenOH: Ca2+ flow via tunnels in polarized cells:Recharging of apical Ca2+ stores by focal Ca2+
entry through basal membrane patch. Cell1997;88:49–55.
29 Tinel H, Cancela J, Mogami H, GerasimenkoJV, Gerasimenko OV, Tepikin AV, PetersenOH: Active mitochondria surrounding the pan-creatic acinar granule region prevent spreadingof inositol trisphosphate-evoked local cytosolicCa2+ signals. EMBO J 1999;18:4999–5008.
30 Park MK, Petersen OH, Tepikin AV: The en-doplasmic reticulum as one continuous Ca2+
pool: Visualization of rapid Ca2+ movementsand equilibration. EMBO J 2000;19:5729–5739.

Signal Transduction, Calcium and AcutePancreatitis
Pancreatology 2003;3:497–505 505
31 Belan PV, Gerasimenko OV, Tepikin AV, Pe-tersen OH: Localization of Ca2+ extrusion sitesin pancreatic acinar cells. J Biol Chem 1996;271:7615–7619.
32 Gerasimenko OV, Gerasimenko JV, Belan PV,Petersen OH: Inositol trisphosphate and cyclicADP-ribose-mediated release of Ca2+ from sin-gle isolated pancreatic zymogen granules. Cell1996;84:473–480.
33 Scott SR, Kiessling K, Parekh AB: An examina-tion of the role of intracellular ATP in the acti-vation of store-operated Ca2+ influx and Ca2+-dependent capacitance increases in rat baso-philic leukaemia cells. Pflügers Arch 1998;436:928–933.
34 Gerasimenko JV, Tepikin AV, Petersen OH,Gerasimenko OV: Calcium uptake via endocy-tosis with rapid release from acidifying endo-somes. Curr Biol 1998;8:1335–1338.
35 Thorn P, Lawrie AM, Smith PM, GallacherDV, Petersen OH: Local and global cytosolicCa2+ oscillations in exocrine cells evoked byagonists and inositol trisphosphate. Cell 1993;74:661–668.
36 Ashby MC, Craske M, Park MK, GerasimenkoOV, Burgoyne RD, Petersen OH, Tepikin AV:Localized Ca2+ uncaging reveals polarized dis-tribution of Ca2+-sensitive Ca2+ release sites:Mechanism of unidirectional Ca2+ waves. JCell Biol 2002;158:283–292.
37 Mogami H, Tepikin AV, Petersen OH: Termi-nation of cytosolic Ca2+ signals: Ca2+ reuptakeinto intracellular stores is regulated by the freeCa2+ concentration in the store lumen. EMBOJ 1998;17:435–442.
38 Park MK, Ashby MC, Erdemli G, PetersenOH, Tepikin AV: Perinuclear, perigranularand sub-plasmalemmal mitochondria have dis-tinct functions in the regulation of cellular cal-cium transport. EMBO J 2001;20:1863–1874.
39 Voronina S, Sukhomlin T, Johnson PR, Er-demli G, Petersen OH, Tepikin A: Correlationof NADH and Ca2+ signals in mouse pancreaticacinar cells. J Physiol 2002;539:41–52.
40 Mooren FC, Hlouschek V, Finkes T, Turi S,Weber IA, Singh J, Domschke W, Schneken-burger J, Kruger B, Lerch MM: Early changesin pancreatic acinar cell calcium signaling afterpancreatic duct obstruction. J Biol Chem 2003;278:9361–9369.
41 Frick TW, Fernandez-del Castillo C, BimmlerD, Warshaw AL: Elevated calcium and activa-tion of trypsinogen in rat pancreatic acini. Gut1997;41:339–343.
42 Mithofer K, Fernandez-del Castillo C, FrickTW, Lewandrowski KB, Rattner DW, War-shaw AL: Acute hypercalcemia causes acutepancreatitis and ectopic trypsinogen activationin the rat. Gastroenterology 1995;109:239–246.
43 Raraty MGT, Neoptolemos JP, Petersen OH,Sutton R: Initiation, site and sequence of intra-cellular enzyme activation in early experimen-tal acute pancreatitis (abstract). Pancreas 2002;25:446.
44 Laine J, Lebel D: Efficient binding of regulatedsecretory protein aggregates to membranephospholipids at acidic pH. Biochem J 1999;338:289–294.
45 Nguyen T, Chin WC, Verdugo P: Role of Ca2+/K+ ion exchange in intracellular storage andrelease of Ca2+. Nature 1998;395:908–192.
46 Yoo SH, So SH, Kweon HS, Lee JS, Kang MK,Jeon CJ: Coupling of the inositol 1,4,5-tris-phosphate receptor and chromogranins A andB in secretory granules. J Biol Chem 2000;275:12553–12559.
47 Reeves EP, Lu H, Jacobs HL, Messina CG,Bolsover S, Gabella G, Potma EO, Warley A,Roes J, Segal AW: Killing activity of neutro-phils is mediated through activation of pro-teases by K+ flux. Nature 2002;416:291–297.
48 Voronina S, Longbottom R, Sutton R, PetersenOH, Tepikin A: Bile acids induce calcium sig-nals in mouse pancreatic acinar cells: Implica-tions for bile-induced pancreatic pathology. JPhysiol 2002;540:49–55.
49 Kim JY, Kim KH, Lee JA, Namkung W, SunAQ, Ananthanarayanan M, Suchy FJ, ShinDM, Muallem S, Lee MG: Transporter-me-diated bile acid uptake causes Ca2+-dependentcell death in rat pancreatic acinar cells. Gastro-enterology 2002;122:1941–1953.
50 Opie EL: The etiology of acute hemorrhagicpancreatitis. Johns Hopkins Hosp Bull 1901;12:182–188.
51 Raraty MGT, Lloyd Mills C, Ward JB, Neopto-lemos JP, Sutton R, Petersen OH: Ethanol andfatty acid ethyl esters, but not acetaldehyde,induce elevations in cytosolic calcium in mousepancreatic acinar cells typical of acute pancre-atitis (abstract). Digestion 1998;59:249.
52 Werner J, Laposata M, Fernandez-del CastilloC, Saghir M, Iozzo RV, Lewandrowski KB,Warshaw AL: Pancreatic injury in rats inducedby fatty acid ethyl ester, a nonoxidative metab-olite of alcohol. Gastroenterology 1997;113:286–294.
53 Klonowski-Stumpe H, Schreiber R, Grolik M,Schulz HU, Haussinger D, Niederau C: Effectof oxidative stress on cellular functions andcytosolic free calcium of rat pancreatic acinarcells. Am J Physiol 1997;272:G1489–G1498.
54 Hietaranta AJ, Singh VP, Bhagat L, van AckerGJ, Song AM, Mykoniatis A, Steer ML, SalujaAK: Water immersion stress prevents caeru-lein-induced pancreatic acinar cell NF-ÎB acti-vation by attenuating caerulein-induced intra-cellular Ca2+ changes. J Biol Chem 2001;276:18742–18747.
55 Han B, Logsdon CD: CCK stimulates mob-1expression and NF-ÎB activation via proteinkinase C and intracellular Ca2+. Am J Physiol2000;278:C344–C351.
56 Hietaranta AJ, Saluja AK, Bhagat L, Singh VP,Song AM, Steer ML: Relationship between NF-ÎB and trypsinogen activation in rat pancreasafter supramaximal caerulein stimulation. Bio-chem Biophys Res Commun 2001;280:388–395.
57 Bhatia M, Brady M, Zagorski J, Christmas SE,Campbell F, Neoptolemos JP, Slavin J: Treat-ment with neutralising antibody against cyto-kine induced neutrophil chemoattractant(CINC) protects rats against acute pancreatitisassociated lung injury. Gut 2000;47:838–844.
58 Bhatia M, Brady M, Kang YK, Costello E,Newton DJ, Christmas SE, Neoptolemos JP,Slavin J: MCP-1 but not CINC synthesis isincreased in rat pancreatic acini in response tocerulein hyperstimulation. Am J Physiol 2002;282:G77–G85.
59 Tando Y, Algul H, Wagner M, Weidenbach H,Adler G, Schmid RM: Caerulein-induced NF-ÎB/Rel activation requires both Ca2+ and pro-tein kinase C as messengers. Am J Physiol1999;277:G678–G686.
60 Blinman TA, Gukovsky I, Mouria M, Zaninov-ic V, Livingston E, Pandol SJ, Gukovskaya AS:Activation of pancreatic acinar cells on isola-tion from tissue: Cytokine upregulation via p38MAP kinase. Am J Physiol 2000;279:C1993–C2003.
61 Algul H, Tando Y, Schneider G, WeidenbachH, Adler G, Schmid RM: Acute experimentalpancreatitis and NF-ÎB/Rel activation. Pan-creatology 2002;2:503–509.
62 Chen X, Ji B, Han B, Ernst SA, Simeone D,Logsdon CD: NF-ÎB activation in pancreasinduces pancreatic and systemic inflammatoryresponse. Gastroenterology 2002;122:448–457.
63 Paschen W: Dependence of vital cell functionon endoplasmic reticulum calcium levels: Im-plications for the mechanisms underlying neu-ronal cell injury in different pathological states.Cell Calcium 2001;29:1–11.
64 Pahl HL: Signal transduction from the endo-plasmic reticulum to the cell nucleus. PhysiolRev 1999;79:683–701.
65 Campos-Toimil M, Bagrij T, Edwardson JM,Thomas P: Two modes of secretion in pan-creatic acinar cells: Involvement of phosphati-dylinositol 3-kinase and regulation by capacita-tive Ca2+ entry. Curr Biol 2002;12:211–215.
66 Singh VP, Saluja AK, Bhagat L, van Acker GJ,Song AM, Soltoff SP, Cantley LC, Steer ML:Phosphatidylinositol 3-kinase-dependent acti-vation of trypsinogen modulates the severity ofacute pancreatitis. J Clin Invest 2001;108:1387–1395.
67 Gerasimenko JV, Gerasimenko OV, PalejwalaA, Tepikin AV, Petersen OH, Watson AJ: Men-adione-induced apoptosis: Roles of cytosolicCa2+ elevations and the mitochondrial perme-ability transition pore. J Cell Sci 2002;115:485–497.
68 Schneider A, Whitcomb DC, Singer MV: Ani-mal models in alcoholic pancreatitis – Whatcan we learn? Pancreatology 2002;2:189–203.
69 Keck T, Campo-Ruiz V, Warshaw AL, Ander-son RR, Fernandez-del Castillo C, Gonzalez S:Evaluation of morphology and microcircula-tion of the pancreas by ex vivo and in vivoreflectance confocal microscopy. Pancreatolo-gy 2001;1:48–57.