small-molecule targeting of a diapophytoene desaturase ... · proposed biosynthetic pathway for...
TRANSCRIPT

174 nature chemical biology | vol 12 | march 2016 | www.nature.com/naturechemicalbiology
articlepublished online: 18 January 2016 | doi: 10.1038/nchembio.2003
Staphylococcus aureus is an important human pathogen that causes life-threatening infections1. With the emergence of multidrug resistance, S. aureus poses a serious public health
threat. In particular, MRSA is now responsible for more deaths in the United States than AIDS2,3. In view of these facts, there is an urgent need to develop new agents to target this pathogen4.
Over 90% of all S. aureus human clinical isolates produce the golden carotenoid pigment (staphyloxanthin)5, an important viru-lence factor that shields S. aureus from host oxidant killing6–8. The proposed biosynthetic pathway for staphyloxanthin starts with farnesyl diphosphate (a key intermediate in the isoprenoid biosyn-thetic pathway) and consists of six enzymes: 4,4′-diapophytoene synthase (CrtM), 4,4′-diapophytoene desaturase (CrtN), 4,4′-diaponeurosporene oxidase (CrtP), 4,4′-diaponeurosporen-aldehyde dehydrogenase (AldH), glycosyltransferase (CrtQ) and acyltrans-ferase (CrtO)9,10 (Supplementary Results, Supplementary Fig. 1). In S. aureus, five of the staphyloxanthin biosynthesis genes are orga-nized in a crtOPQMN operon whose expression is dependent on a functional sigB operon11–13 (Supplementary Fig. 1). Recently, it has been reported that a number of S. aureus genes, such as ispA13, SAUSA300_1470 (ref. 14), citZ13, citB15 and ccpE15, as well as the small RNA ssrA16, influence production of carotenoid pigment.
Following increased insight on the important role of staphylox-anthin in S. aureus pathogenesis8, antivirulence drugs that target CrtM have been suggested as alternatives or adjuncts to conven-tional antibiotics, as they limit pathogenesis and increase bacterial susceptibility to host killing6,8. Notably, the compound BPH-652, a human squalene synthase inhibitor developed as a potential cholesterol-lowering drug, inhibits CrtM and thus blocks staphy-loxanthin biosynthesis, with the result that S. aureus is more suscep-tible to the innate host immune system and is relatively avirulent6. Hence, chemically targeting the staphyloxanthin biosynthesis may offer potential ways to treat complicated S. aureus infections.
In this study, we screened existing drugs against S. aureus infection. We identified naftifine (1), an FDA-approved antifungal drug, as an inhibitor of S. aureus infection. We found that the mode
of action of naftifine is mediated by inhibiting the carotenoid pig-ment synthesis of S. aureus, most likely targeting CrtN. We showed that naftifine is a promising candidate for further development as an antivirulence agent for the treatment of S. aureus infection.
RESULTSNaftifine is a staphyloxanthin biosynthesis inhibitorTo probe the possible use of an existing drug as an antivirulence agent against S. aureus, we screened an in-house collection com-posed of commercially available known drugs for inhibitors of staphyloxanthin biosynthesis of S. aureus Newman, a highly virulent clinical isolate of methicillin-sensitive S. aureus (MSSA)17. Of the 412 drugs we tested, three (ibandronate, terbinafine (2) and naftifine) inhibited staphyloxanthin biosynthesis (Fig. 1a). Notably, nafti-fine was an effective staphyloxanthin biosynthesis inhibitor, with a half-maximal inhibitory concentration (IC50) of 296 nM (Fig. 1b). The addition of naftifine to staphylococcal cultures (up to 0.2 mM, corresponding to ~64.8 μg/ml) did not affect the growth of S. aureus Newman (Supplementary Fig. 2a), indicating that naftifine selectively inhibits staphyloxanthin biosynthesis and does not func-tion as an antibiotic against S. aureus.
The minimum inhibitory concentration of naftifine against S. aureus has been reported to be >256 μg/ml18, and the minimum bactericidal concentration, as high as 1.25% (w/v)19. In line with those studies, we observed that the antimicrobial concentration that inhibited growth of 90% of the strain (MIC90) of the naftifine for S. aureus Newman was ~0.5% (w/v) (corresponding to 5 mg/ml) (Supplementary Fig. 2b). Hence, it is likely that naftifine has a very weak antibacterial effect against S. aureus in vitro.
Naftifine blocked staphyloxanthin biosynthesis of several MRSA strains (USA300 LAC, USA400 MW2 and Mu50) at low micromolar concentrations (Supplementary Fig. 2c–e), suggesting that the pigment-inhibitory effect of naftifine is not restricted to the Newman strain. As naftifine was a more potent inhibitor of staphyloxanthin biosynthesis than ibandronate or terbinafine (Fig. 1a), we focused on its mode of action and efficacy in this study.
1State Key laboratory of Drug research, Shanghai Institute of materia medica, chinese academy of Sciences, Shanghai, china. 2Shanghai Key laboratory of New Drug Design, School of Pharmacy, East china University of Science and Technology, Shanghai, china. 3humanwell healthcare (Group) co. ltd., Wuhan, china. 4Shanghai Institute of materia medica, chinese academy of Sciences, Shanghai, china. 5These authors contributed equally to this work. *e-mail: [email protected] or [email protected]
small-molecule targeting of a diapophytoene desaturase inhibits S. aureus virulenceFeifei chen1,5, hongxia di1,5, youxin Wang2,5, Qiao cao1, bin Xu1, Xue Zhang1, nana yang1, guijie liu1, cai-guang yang1, yong Xu3, hualiang Jiang1, Fulin lian4, naixia Zhang4, Jian li2* & lefu lan1*
The surge of antibiotic resistance in Staphylococcus aureus has created a dire need for innovative anti-infective agents that attack new targets, to overcome resistance. In S. aureus, carotenoid pigment is an important virulence factor because it shields the bacterium from host oxidant killing. Here we show that naftifine, a US Food and Drug Administration (FDA)-approved anti-fungal drug, blocks biosynthesis of carotenoid pigment at nanomolar concentrations. This effect is mediated by competitive inhibition of S. aureus diapophytoene desaturase (CrtN), an essential enzyme for carotenoid pigment synthesis. We found that naftifine attenuated the virulence of a variety of clinical S. aureus isolates, including methicillin-resistant S. aureus (MRSA) strains, in mouse infection models. Specifically, we determined that naftifine is a lead compound for potent CrtN inhibitors. In sum, these findings reveal that naftifine could serve as a chemical probe to manipulate CrtN activity, providing proof of concept that CrtN is a druggable target against S. aureus infections.
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

nature chemical biology | vol 12 | march 2016 | www.nature.com/naturechemicalbiology 175
articleNATURE CHEMICAL bIoLogy doi: 10.1038/nchembio.2003
Naftifine sensitizes S. aureus to immune clearanceBecause naftifine inhibited production of golden carotenoid pig-ment in S. aureus (Fig. 1a), and because the pigment shields S. aureus from host oxidant killing6–8, we next examined whether naftifine sensitizes S. aureus to killing by hydrogen peroxide (H2O2). We exposed S. aureus Newman to naftifine (a final concentration of 100 μM) for 24 h. Then we collected the treated cells and incubated them with H2O2. The naftifine-treated S. aureus cells were killed more efficiently by H2O2 as compared to the untreated S. aureus (mock-treated) (survival, 0.07% vs. 9.17%; Fig. 1c). Next, we deter-mined whether naftifine could render S. aureus more susceptible to immune clearance in human blood. We measured survival in naftifine-treated and untreated S. aureus Newman cells mixed with whole blood. Survival of the naftifine-treated S. aureus was ~20 times lower than that of the untreated S. aureus (1.1% vs. 20.2%; Fig. 1d), suggesting that naftifine makes S. aureus more susceptible to immune clearance in human blood.
Naftifine attenuates the virulence of S. aureus NewmanAs naftifine sensitized S. aureus Newman bacteria to immune clearance in an ex vivo assay (Fig. 1d), we next analyzed naftifine’s in vivo efficacy. To evaluate the efficacy of naftifine on the outcome of S. aureus sepsis, mice received intraperitoneal injections with a total dose of 225 mg/kg naftifine hydrochloride in 12-h intervals. When infected with 5 × 107 colony-forming units (CFU) S. aureus Newman bacteria via retro-orbital injection, the mock-treated mice died within 1 d after challenge, whereas naftifine-treated mice survived up to 5 d, and more than 70% (10/14) of the naf-tifine-treated mice were alive at day 12 after infection (Fig. 1e). To examine the role of naftifine in reducing S. aureus virulence, we measured bacterial survival in host organs. Naftifine treat-ment reduced bacterial survival in the kidney, heart and liver by
1.1, 1.4 and 2.0 log10 CFU/organ, respec-tively. These results demonstrate that naftifine treatment in vivo attenuates the virulence of S. aureus Newman.
Naftifine does not affect crtOPQMN expressionTo explore the mechanism by which naftifine inhibits the production of staphyloxanthin, we examined whether the pigment-inhibitory effect of naftifine was caused by the down-regulation of crtOPQMN, the operon encod-ing staphyloxanthin-synthesis enzymes9,20. We cultured S. aureus Newman harboring the promoter-lacZ reporter plasmid pCL-crtO-lacZ15 in tryptone soya broth (TSB) medium at 37 °C for 6 h, then treated the bacterial cultures with naftifine hydrochloride (100 μM) and sam-pled them at different time points thereafter. Naftifine did not inhibit the promoter activ-ity of crtOPQMN (Supplementary Fig. 3a) but blocked production of carotenoid pigment (Supplementary Fig. 3b). Thus, the inhibition of pigment production by naftifine was not due to the inhibition of expression of staphy-loxanthin biosynthesis genes.
Effect of naftifine on isoprenoid biosynthesis pathwayThe first step in staphyloxanthin biosynthe-sis is the head-to-head condensation of two molecules of farnesyl diphosphate by the dehydrosqualene synthase CrtM into 4,4′-diapophytoene9 (Supplementary Fig. 1). The
substrate farnesyl diphosphate is produced by the farnesyl diphos-phate synthase IspA, the final enzyme in the isoprenoid biosynthetic pathway9 (Supplementary Fig. 1). We examined whether naftifine inhibits the isoprenoid biosynthesis. We extracted isoprenoids from wild-type S. aureus Newman (untreated and naftifine-treated), ispA mutant strain, and ispA mutant strain complemented with ispA (ispA-C) (Supplementary Table 1), and analyzed the samples by reverse-phase high-performance liquid chromatography (RP-HPLC) (absorption at 210 nm). Disruption of ispA resulted in colorless bacteria as well as a profound change in the RP-HPLC pro-file of isoprenoids (Supplementary Fig. 3c–e). In contrast, although naftifine treatment rendered S. aureus Newman cells colorless, it had no obvious inhibitory effect on the RP-HPLC profile of the isoprenoid extracts (Supplementary Fig. 3f).
In support of previous studies showing that ibandronate is a farnesyl pyrophosphate synthase (IspA) inhibitor21, we observed that the ligand-induced protein stabilization effect of S. aureus 6His-ispA protein was apparent for ibandronate (Supplementary Fig. 4a). Furthermore, ibandronate treatment resulted in a pro-found change in the RP-HPLC profile of isoprenoids derived from the S. aureus Newman strain (Supplementary Figs. 3c and 4b). Thus, unlike naftifine, ibandronate may inhibit IspA, thereby preventing the biosynthesis of isoprenoid compounds that are essential for staphyloxanthin biosynthesis. Taken together, we concluded that the inhibitory effect of naftifine on carotenoid pigment production was not mediated by the blockade of the isoprenoid biosynthesis pathway.
Naftifine inhibits CrtN function in vivo In light of the reports that crtM and crtN together are sufficient for the production of yellow-colored pigment in Escherichia coli and Staphylococcus carnosus9,20, we reasoned that naftifine may inhibit
a b dc
0 1 2 3 4 5 6 7 8 9 10 11 120
20
40
60
80
100
MockNaftifine
***
Newman
Time (d)
Perc
ent s
urvi
val (
%)
–9 –8 –7 –6 –5 –4
020406080
100120
IC50: 296 nM
Log [Naftifine] (M)
Perc
enta
ge o
fpi
gmen
t pro
duct
ion
05
1015
2025
Mock
Whole blood
***
Surv
ival
(%)
0123456789
10Kidney
* Heart* Liver
**
Mock
Newman
Limit of detection
log 10
CFU
/org
an
0.01
0.1
1
10
100
Surv
ival
(%)
H2O2
***Ibandronate
Terbinafine
Naftifine
Concentration (µM)
0
e f
1 cm
200100101
NaftifineMockNaftifineMockNaftifine
Naftifine
Mock
Naftifine
Figure 1 | Naftifine has antivirulence properties against S. aureus Newman. (a) Effect of ibandronate sodium, terbinafine hydrochloride or naftifine hydrochloride on wild-type S. aureus Newman pigmentation. The images show the spun-down cells. (b) Ic50 of naftifine hydrochloride for pigment production of S. aureus Newman. Data shown represent the mean ± s.d. from triplicate experiments; the level of the pigment production by untreated S. aureus Newman was set to 100%. (c,d) Effect of naftifine hydrochloride on the susceptibility of S. aureus Newman to killing by either h2o2 (c) or human whole blood (d). ***P < 0.001 via two-tailed t-test (n = three biological replicates, each with two technical replicates). (e) Effect of naftifine hydrochloride in protecting mice (n = 14) from lethal S. aureus infection challenged with 5 × 107 cFU Newman bacteria. mock vs. naftifine, ***P < 0.001; log-rank test. (f) Effect of naftifine treatment on S. aureus Newman bacteria survival in the kidneys, hearts and livers of mice (n = 15) challenged with 4 × 106 cFU S. aureus Newman bacteria. *P < 0.05, **P < 0.01; mann-Whitney test, two-tailed. Each symbol represents the value for an individual mouse. horizontal bars indicate observation means, and the dashed line marks limit of detection.
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.
176 nature chemical biology | vol 12 | march 2016 | www.nature.com/naturechemicalbiology
article NATURE CHEMICAL bIoLogy doi: 10.1038/nchembio.2003
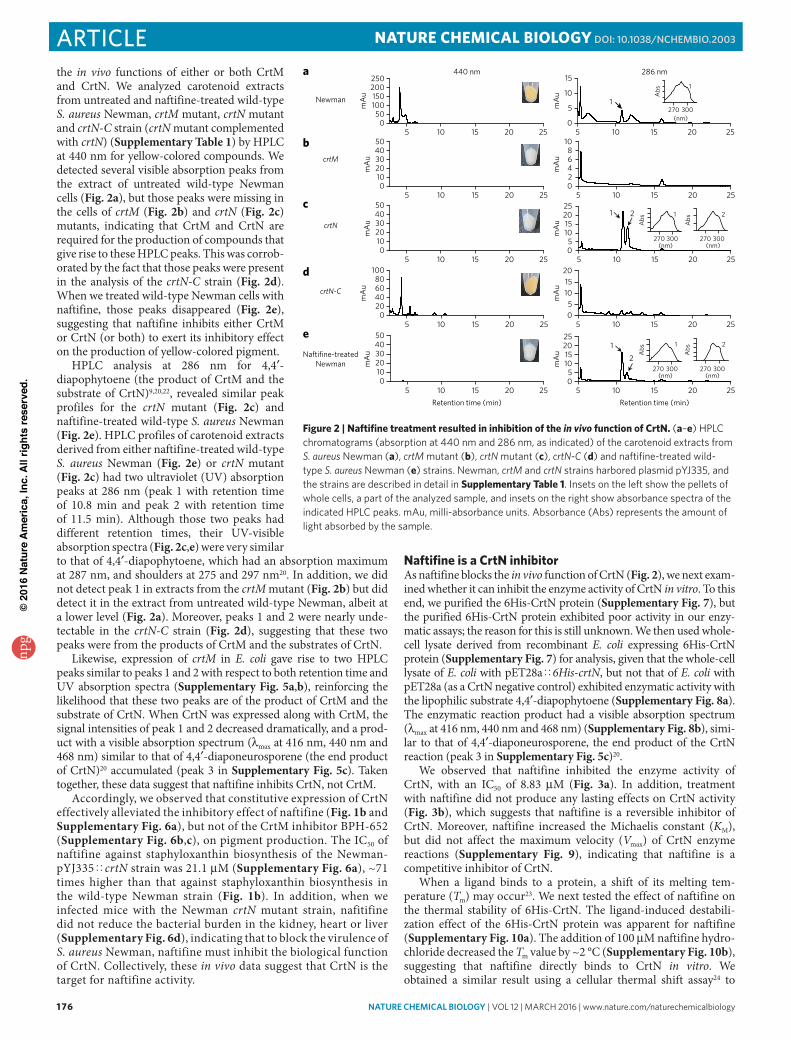
the in vivo functions of either or both CrtM and CrtN. We analyzed carotenoid extracts from untreated and naftifine-treated wild-type S. aureus Newman, crtM mutant, crtN mutant and crtN-C strain (crtN mutant complemented with crtN) (Supplementary Table 1) by HPLC at 440 nm for yellow-colored compounds. We detected several visible absorption peaks from the extract of untreated wild-type Newman cells (Fig. 2a), but those peaks were missing in the cells of crtM (Fig. 2b) and crtN (Fig. 2c) mutants, indicating that CrtM and CrtN are required for the production of compounds that give rise to these HPLC peaks. This was corrob-orated by the fact that those peaks were present in the analysis of the crtN-C strain (Fig. 2d). When we treated wild-type Newman cells with naftifine, those peaks disappeared (Fig. 2e), suggesting that naftifine inhibits either CrtM or CrtN (or both) to exert its inhibitory effect on the production of yellow-colored pigment.
HPLC analysis at 286 nm for 4,4′-diapophytoene (the product of CrtM and the substrate of CrtN)9,20,22, revealed similar peak profiles for the crtN mutant (Fig. 2c) and naftifine-treated wild-type S. aureus Newman (Fig. 2e). HPLC profiles of carotenoid extracts derived from either naftifine-treated wild-type S. aureus Newman (Fig. 2e) or crtN mutant (Fig. 2c) had two ultraviolet (UV) absorption peaks at 286 nm (peak 1 with retention time of 10.8 min and peak 2 with retention time of 11.5 min). Although those two peaks had different retention times, their UV-visible absorption spectra (Fig. 2c,e) were very similar to that of 4,4′-diapophytoene, which had an absorption maximum at 287 nm, and shoulders at 275 and 297 nm20. In addition, we did not detect peak 1 in extracts from the crtM mutant (Fig. 2b) but did detect it in the extract from untreated wild-type Newman, albeit at a lower level (Fig. 2a). Moreover, peaks 1 and 2 were nearly unde-tectable in the crtN-C strain (Fig. 2d), suggesting that these two peaks were from the products of CrtM and the substrates of CrtN.
Likewise, expression of crtM in E. coli gave rise to two HPLC peaks similar to peaks 1 and 2 with respect to both retention time and UV absorption spectra (Supplementary Fig. 5a,b), reinforcing the likelihood that these two peaks are of the product of CrtM and the substrate of CrtN. When CrtN was expressed along with CrtM, the signal intensities of peak 1 and 2 decreased dramatically, and a prod-uct with a visible absorption spectrum (λmax at 416 nm, 440 nm and 468 nm) similar to that of 4,4′-diaponeurosporene (the end product of CrtN)20 accumulated (peak 3 in Supplementary Fig. 5c). Taken together, these data suggest that naftifine inhibits CrtN, not CrtM.
Accordingly, we observed that constitutive expression of CrtN effectively alleviated the inhibitory effect of naftifine (Fig. 1b and Supplementary Fig. 6a), but not of the CrtM inhibitor BPH-652 (Supplementary Fig. 6b,c), on pigment production. The IC50 of naftifine against staphyloxanthin biosynthesis of the Newman-pYJ335<crtN strain was 21.1 μM (Supplementary Fig. 6a), ~71 times higher than that against staphyloxanthin biosynthesis in the wild-type Newman strain (Fig. 1b). In addition, when we infected mice with the Newman crtN mutant strain, nafitifine did not reduce the bacterial burden in the kidney, heart or liver (Supplementary Fig. 6d), indicating that to block the virulence of S. aureus Newman, naftifine must inhibit the biological function of CrtN. Collectively, these in vivo data suggest that CrtN is the target for naftifine activity.
Naftifine is a CrtN inhibitorAs naftifine blocks the in vivo function of CrtN (Fig. 2), we next exam-ined whether it can inhibit the enzyme activity of CrtN in vitro. To this end, we purified the 6His-CrtN protein (Supplementary Fig. 7), but the purified 6His-CrtN protein exhibited poor activity in our enzy-matic assays; the reason for this is still unknown. We then used whole-cell lysate derived from recombinant E. coli expressing 6His-CrtN protein (Supplementary Fig. 7) for analysis, given that the whole-cell lysate of E. coli with pET28a<6His-crtN, but not that of E. coli with pET28a (as a CrtN negative control) exhibited enzymatic activity with the lipophilic substrate 4,4′-diapophytoene (Supplementary Fig. 8a). The enzymatic reaction product had a visible absorption spectrum (λmax at 416 nm, 440 nm and 468 nm) (Supplementary Fig. 8b), simi-lar to that of 4,4′-diaponeurosporene, the end product of the CrtN reaction (peak 3 in Supplementary Fig. 5c)20.
We observed that naftifine inhibited the enzyme activity of CrtN, with an IC50 of 8.83 μM (Fig. 3a). In addition, treatment with naftifine did not produce any lasting effects on CrtN activity (Fig. 3b), which suggests that naftifine is a reversible inhibitor of CrtN. Moreover, naftifine increased the Michaelis constant (KM), but did not affect the maximum velocity (Vmax) of CrtN enzyme reactions (Supplementary Fig. 9), indicating that naftifine is a competitive inhibitor of CrtN.
When a ligand binds to a protein, a shift of its melting tem-perature (Tm) may occur23. We next tested the effect of naftifine on the thermal stability of 6His-CrtN. The ligand-induced destabili-zation effect of the 6His-CrtN protein was apparent for naftifine (Supplementary Fig. 10a). The addition of 100 μM naftifine hydro-chloride decreased the Tm value by ~2 °C (Supplementary Fig. 10b), suggesting that naftifine directly binds to CrtN in vitro. We obtained a similar result using a cellular thermal shift assay24 to
5 10 15 20 2505
1015
2025
Retention time (min)
mA
u
5 10 15 20 2505
1015
2025
mA
u
5 10 15 20 250
5
10
15
mA
u
5 10 15 20 250
1020304050
Retention time (min)
mA
u
5 10 15 20 250
20406080
100
mA
u
5 10 15 20 250
1020304050
mA
u
5 10 15 20 250
1020304050
mA
u
5 10 15 20 250
50100150
200250
mA
u
286 nm
1
1
2
2
1
440 nm
270 300
1
(nm)
Abs
5 10 15 20 2502468
10
mA
u
270 300
1
(nm)
Abs
270 300
2
(nm)
Abs
5 10 15 20 2505
1015
20
mA
u
270 300
1
(nm)
Abs
270 300
2
(nm)
Abs
Newman
crtM
crtN
Naftifine-treatedNewman
crtN-C
a
b
c
d
e
Figure 2 | Naftifine treatment resulted in inhibition of the in vivo function of CrtN. (a–e) hPlc chromatograms (absorption at 440 nm and 286 nm, as indicated) of the carotenoid extracts from S. aureus Newman (a), crtM mutant (b), crtN mutant (c), crtN-C (d) and naftifine-treated wild-type S. aureus Newman (e) strains. Newman, crtM and crtN strains harbored plasmid pYJ335, and the strains are described in detail in Supplementary Table 1. Insets on the left show the pellets of whole cells, a part of the analyzed sample, and insets on the right show absorbance spectra of the indicated hPlc peaks. mau, milli-absorbance units. absorbance (abs) represents the amount of light absorbed by the sample.
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

nature chemical biology | vol 12 | march 2016 | www.nature.com/naturechemicalbiology 177
articleNATURE CHEMICAL bIoLogy doi: 10.1038/nchembio.2003
examine the ligand-induced thermal stabilization of CrtN protein in a Staphylococcus carnosus TM300<pTXcrtMN strain9 (Fig. 3c,d and Supplementary Fig. 11), which indicates that naftifine inter-acts with CrtN in a cellular context. Naftifine also blocked pigment production by S. carnosus TM300<pTXcrtMN (Supplementary Fig. 12), which is consistent with the finding that CrtN is a target for naftifine activity (Fig. 2). Moreover, the direct binding of naftifine to CrtN was further confirmed by the use of T1ρ (Supplementary Figs. 13 and 14 ) and saturation transfer difference NMR spectroscopy experiments (Fig. 3e). Hence, taken together, we concluded that CrtN is a direct target of naftifine.
Naftifine is a well-known inhibitor of squalene epoxidase (SE)19 whereas CrtN is a desaturase that catalyzes the formation of the first yellow-colored carotenoid intermediate product (4,4′-diaponeurosporene), which is formed via successive dehydrogena-tion reactions (Supplementary Fig. 1) (refs. 20,25). Although SE and CrtN catalyze different reactions, their substrates have a similar scaf-fold. The natural substrates of CrtN (dehydrosqualene, diapophyto-fluene and diapo-ζ-carotene) have a very similar scaffold to that of SE (i.e., squalene) (Supplementary Fig. 15a). It has been reported recently that CrtN can desaturate squalene into dehydrosqualene26. Hence, SE and CrtN may have similar mechanisms of substrate
binding. This hypothesis awaits further inves-tigation, however. ClustalW analysis revealed that CrtN has amino acid sequence similarity with SEs, albeit at a low level (~10% identity) (Supplementary Fig. 15b). These observations generally support and are consistent with our finding that naftifine is an inhibitor of CrtN.
Naftifine is a lead compound for potent CrtN inhibitorsAs described above, terbinafine, an analog of naftifine, could block pigment production of S. aureus, albeit less potently than naftifine (Fig. 1a). In addition, we observed that the naftifine analog butenafine (3) had no obvi-ous inhibitory effect on pigment production, even at concentrations of up to 1, 250 μM (Supplementary Fig. 16; also see this supple-mentary figure for structures of numbered compounds). We then sought to develop potent inhibitors of staphyloxanthin biosyn-thesis based on a naftifine scaffold. Removing or changing the N-methyl substituent of naf-tifine (JX04, 4; JX05, 5; and JX06, 6) led to a dramatic loss of pigment-production inhibition activity (Supplementary Fig. 16). In addition, we observed that an allyl moiety was essential
for pigment-production inhibition activity (butenafine vs. JX08, 8) (Supplementary Fig. 16). Generally, alkyl (cyclohexyl, JX10, 10; tert-butyl acetenyl, terbinafine) substituents or hetroaryl (pyri-dyl, JX11, 11) group were not beneficial, but the introduction of a large aromatic substituent (naphthalen-2-yl, JX09, 9) was favorable (Supplementary Fig. 16). In the studied set of the substituents on the phenyl ring, the potency increased in the order methyl > tert- butyl > hydrogen (JX07 > JX08 > nafitine). JX07 (7) and JX09 exhibited approximately ten times more effectiveness than naftifine in inhibit-ing carotenoid pigment production (Supplementary Fig. 16). This effect correlated generally with their CrtN-inhibitory activities. JX07 and JX09 inhibited CrtN enzyme activity with a 10.8- and 82.5-fold higher potency than naftifine, respectively (Supplementary Fig. 16). Hence, in addition to supporting the finding that naftifine is a CrtN inhibitor (Fig. 3), these results suggest that naftifine could serve as a lead for the development of highly potent inhibitors of CrtN.
CrtN is important for S. aureus pathogenesisGiven that CrtN is a target of naftifine (Figs. 2 and 3) and that naf-tifine blocks S. aureus Newman’s virulence (Fig. 1), we next sought to determine the contribution of CrtN to S. aureus pathogenesis. We subjected the test strains, wild-type S. aureus Newman, crtN
12111314
2,16,20
17,19
3,6,18
74,5,8
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.01H (p.p.m.)
260
50
100
48 50 52 54 56 58
CrtN-mockCrtN-naftifine
Cellular thermal shift assay
Rela
tive
band
inte
nsity
(%)
*
****
*****
N
12
345
6
7
89
10
1112
13
1415
1617
1819
20CH3
2121
Mock
Naftifine
Temperature (°C)
2 3 4 5 60
20
40
60
80
100 CrtN: IC50 = 8.83 µMIn
hibi
tory
rate
(%) b
log10 [naftifine] (nM)
0 200 400 600 8000
0.05
0.10
0.15
0.20
100 (µM)0 (µM)
A45
0 nm
Lysate of E. coli pET28a::6His-crtN (µl)
20 (µM)
26.048.0
52.054.4
56.358.0
Temperature (°C)
a c
d e
Figure 3 | Naftifine is a CrtN inhibitor. (a) relative crtN activities, measured in the presence of variable concentrations of naftifine. Data shown represent the mean ± s.d. from triplicate experiments. (b) Effect of naftifine treatment on crtN enzyme activity. (c,d) Western blot image (c) and the cellular thermal shift assay curves (d) showing the effect of naftifine hydrochloride (100 μm) on the Tm of crtN protein in S. carnosus Tm300 cells. The full western blot image is shown in Supplementary Figure 11. In d, data are presented as means ± s.e.m., n = 5 independent experiments (Supplementary Fig. 11); *P < 0.05, **P < 0.01, ***P < 0.001; two-tailed t-test. (e) Saturation transfer difference spectrum recorded for 200 μm naftifine hydrochloride in the presence of crtN (5 μm). Nmr data for protons in the structure of naftifine hydrochloride were generally assigned (Supplementary Fig. 14).
0
2
4
6
8
10
12
14 USA400 MW2
Kidney
**Heart
*
Limit of detection
log 10
CFU
/org
an
0123456789
1011
USA300 crtN USA300 crtN crtN crtN
*** ***Kidney
**
Heart
Limit of detection
USA300 LAC
log 10
CFU
/org
an
Newman crtN crtN-C Newman crtN
Limit of detection
*** ***Kidney
******
Heart
Newman
log 10
CFU
/org
an
0123456789
101112
crtN-C crtN-C crtN-C crtN-C crtN-C
a b c
Figure 4 | CrtN is important for S. aureus pathogenesis. (a) virulence of S. aureus wild-type strain Newman (harboring pcl-lacZ) (n = 14) and crtN mutant complemented with vector (pcl-lacZ) alone (crtN) (n = 15) or pcl<crtN (crtN-C) (n = 14). (b) virulence of S. aureus wild-type strain USa300 lac (harboring pcl-lacZ) (n = 14) and crtN mutant complemented with vector (pcl-lacZ) alone (crtN) (n = 15) or pcl<crtN (crtN-C) (n = 14). (c) virulence of S. aureus USa400 mW2 crtN mutant complemented with vector (pcl-lacZ) alone (crtN) (n = 14) or pcl<crtN (crtN-C) (n = 13). horizontal bars indicate observation means, and dashed lines mark limits of detection. *P < 0.05, **P < 0.01, ***P < 0.001; mann-Whitney test (two-tailed).
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

178 nature chemical biology | vol 12 | march 2016 | www.nature.com/naturechemicalbiology
article NATURE CHEMICAL bIoLogy doi: 10.1038/nchembio.2003
mutant strain and ctrN-C strain (crtN<pCL-crtN; Supplementary Table 1) to a mouse model of abscess formation, and examined bacte-rial survival in the host organs. Inactivation of crtN decreased bacterial survival in both kidney (~1.98 log10 CFU/organ reduction) and heart (~2.60 log10 CFU/organ reduction) (Fig. 4a). Introduction of the crtN gene significantly increased (>2.0 log10 CFU/organ) bacterial survival (Fig. 4a). These results indicate that CrtN is genetically essential for full virulence of S. aureus Newman. Moreover, we obtained similar results when we repeated this exper-iment with the crtN mutant of USA300 LAC strain (Fig. 4b) or USA400 MW2 strain (Fig. 4c), two current clones responsible for the epidemic of community-acquired MRSA (CA-MRSA) infec-tions in the United States27. Hence, it is likely that CrtN is important in promoting the virulence of pigmented S. aureus. These results are also in line with previous studies showing that staphyloxanthin is an important virulence factor of S. aureus6,8. Indeed, CrtN was essential for the carotenoid pigment production in Newman, USA300 LAC and USA400 MW2 strains (Supplementary Fig. 17).
Naftifine is effective against MRSA infectionsAs was the case with S. aureus Newman, staphyloxanthin bio-synthesis of USA300 LAC, USA400 MW2, and Mu50 strains was inhibited by naftifine, with IC50 values of 2.91 μM, 1.96 μM and 1.08 μM, respectively (Supplementary Fig. 2c–e). We next exam-ined whether the inhibitory effect of naftifine on S. aureus virulence is restricted to the Newman strain. We infected mock-treated or naftifine-treated mice with MRSA USA400 MW2. Treatment with naftifine significantly decreased the staphylococcal loads in both kidneys (~0.95 log10 CFU/organ reduction) and hearts (~1.58 log10 CFU/organ reduction) (Fig. 5a). In addition, treatment with nafti-fine also resulted in a reduction (~0.8 log10 CFU/organ) of bacterial loads in the livers of the infected mice, although the decrease was not significant (Fig. 5a). Accordingly, we observed that naftifine significantly protected mice from challenges with two different lethal doses of USA400 MW2 bacteria (Fig. 5b and Supplementary Fig. 18a). These results suggest that the inhibitory effect of naftifine on S. aureus virulence is not restricted to the Newman strain.
To further examine the role of naftifine in blocking the virulence of multidrug-resistant S. aureus, we used a Mu50 strain and a mouse model of abscess formation. S. aureus Mu50 strain is a hospital-acquired MRSA strain with vancomycin-intermediate resistance (MRSA/VISA) isolated in Japan28. Although treatment of mice with naftifine resulted in a slight reduction (~0.23 log10 CFU/organ reduc-tion) in staphylococcal loads in the kidney tissues only, naftifine caused a >1.0 log10 CFU/organ reduction in staphylococcal loads in hearts and a >1.8 log10 CFU/organ reduction in staphylococcal loads in liver tissues (Supplementary Fig. 18b). To our surprise, naftifine was as efficacious as vancomycin in preventing the colonization of Mu50 in the liver of the infected mice (Fig. 5c). Accordingly, naftifine prevented S. aureus Mu50 to form liver abscess (Fig. 5d). These results suggest that CrtN inhibitor naftifine is also efficacious in mouse models infected with clinically relevant multidrug-resistant S. aureus. The naftifine analog JX07 had similar effects to naftifine in vitro (Supplementary Fig. 16) as well as in vivo (Fig. 5c), indicat-ing that CrtN is a common target of these two compounds.
DISCUSSIoNNaftifine is an allylamine derivative19. Although its precise mecha-nism of action is as-yet unclear, naftifine is believed to inhibit SE and thus lead to fungal cell lysis19. In addition, naftifine was highly selective for fungal SE, as opposed to mammalian SE19. Currently, some fungal SE inhibitors, such as terbinafine, naftifine and buten-afine, are in clinical use29. SE catalyzes the epoxidation of the C-C double bond of squalene to yield 2,3-oxidosqualene, the key step of sterol biosynthesis pathways in eukaryotes30. Sterol biosynthesis is viewed primarily as a eukaryotic process31 and is not yet identified
in S. aureus. Using BLASTP algorithms for similarity searching against proteins of S. aureus Newman strain, we observed the first hit with an E value of 0.016 and a bit score of 34.3 for S. cerevisiae SE. Therefore, it is very likely that S. aureus lacks the SE homolog.
The sensitivity to naftifine of staphyloxanthin biosynthesis in S. aureus Newman (IC50, 0.296 μM) is ~30-fold higher than that of in vitro CrtN enzyme (Supplementary Fig. 16). The reason for this difference is currently unknown. It is possible that naftifine accu-mulates in the cytoplasm of S. aureus, leading to higher intracellular drug levels. Meanwhile, in addition to the possible accumulation of naftifine, as-yet unknown additional effects are probably an important factor. Further research is needed to uncover the detailed mechanism of action of naftifine. Nonetheless, it is clear that naf-tifine is a reversible competitive inhibitor of CrtN (Fig. 3 and Supplementary Fig. 9) and a promising lead compound targeting staphyloxanthin biosynthesis (Supplementary Fig. 16).
Reports indicate that both naftifine and terbinafine show very weak antibacterial effects against S. aureus in vitro18,19,32 but have nevertheless proved efficacious in treating S. aureus infections33–35. For instance, terbinafine effectively treated patients with either pyoderma33 or nonbullous impetigo34,35. These previous data suggest that clinically relevant anti-infective properties against S. aureus were also observed for the CrtN inhibitors. Given that CrtN is required for the full virulence of S. aureus strains (Fig. 4), the inhi-bition of CrtN may be responsible, at least in part, for the achieve-ment of these therapeutic efficacies. We should, however, keep in mind that S. aureus virulence is multifactorial1 and that not all S. aureus isolates express staphyloxanthin5, which may limit the therapeutic utility of this molecule to some extent.
0 2 4 6 8 10 12 14 16 180
20
40
60
80
100
Mock 5 × 107 CFUNaftifine
USA400 MW2
*
Time (d)
Perc
ent s
urvi
val (
%)
0123456789
10Kidney
Heart Liver**** n.s.
USA400 MW2
Limit of detection
0123456789
101112
***n.s.
***
Limit of detection
Liver
Mu50
***
log 10
CFU
/org
anlo
g 10 C
FU/o
rgan
1 cm
Uninfected liver
Mu50-infected Naftifine-treated
Mock Naf Mock Naf Mock Naf
Mock Van Naf JX07
a b
c d
Figure 5 | Naftifine blocks the virulence of MRSA strains. (a) Effect of naftifine (Naf) on S. aureus survival in the kidneys, hearts and livers of mice (n = 13 for mock and n = 14 for naftifine-treated) challenged with 7 × 106 cFU USa400 mW2 bacteria. (b) Effect of naftifine in protecting mice (n = 14 for mock and n = 15 for naftifine-treated) from lethal S. aureus infection challenged with 5 × 107 cFU USa400 mW2 bacteria. mock vs. naftifine, *P < 0.05; log-rank test. (c) Effect of vancomycin (van), naftifine (Naf) and naftifine analog JX07 on S. aureus survival in the livers of mice (n = 13 for mock, n = 14 for vancomycin-treated and naftifine-treated, and n = 15 for JX07-treated) challenged with 1.2 × 108 cFU mu50 bacteria. (d) Photographs of the livers of uninfected mice, mu50-infected mice and mu50-infected mice treated with naftifine. **P < 0.01, ***P < 0.001, n.s. indicates no significant difference (in a,c: mann-Whitney two-tailed test); each symbol represents the value for an individual mouse; horizontal bars indicate observation means, and dashed lines mark the limit of detection.
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

nature chemical biology | vol 12 | march 2016 | www.nature.com/naturechemicalbiology 179
articleNATURE CHEMICAL bIoLogy doi: 10.1038/nchembio.2003
To our knowledge, diphenylamine is probably the only known CrtN inhibitor that has been demonstrated so far36,37, although its inhibitory effects on S. aureus virulence in vivo remain to be eluci-dated. Diphenylamine inhibited S. aureus CrtN with an IC50 value of 20 μM37 and inhibited staphylococcal growth above 148 μM36. In line with these previous studies, we observed that diphenylamine blocks staphyloxanthin biosynthesis of S. aureus Newman strain with an IC50 of 55 μM. Hence, it is likely that diphenylamine is much less effective (>150-fold) than naftifine (0.296 μM, Fig. 1b) in inhibiting staphyloxanthin biosynthesis of S. aureus.
In sum, our findings, together with the established safety and efficacy profile of naftifine and its analog in patients with S. aureus infections33–35, make CrtN an attractive and druggable target against infections caused by pigmented S. aureus. This also opens a new area of research for the investigation, optimization and application of naftifine in fighting S. aureus infections.
received 1 July 2015; accepted 13 November 2015; published online 18 January 2016
METHoDSMethods and any associated references are available in the online version of the paper.
references1. Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532
(1998).2. DeLeo, F.R. & Chambers, H.F. Reemergence of antibiotic-resistant
Staphylococcus aureus in the genomics era. J. Clin. Invest. 119, 2464–2474 (2009).
3. Klein, E., Smith, D.L. & Laxminarayan, R. Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999-2005. Emerg. Infect. Dis. 13, 1840–1846 (2007).
4. Arias, C.A. & Murray, B.E. Antibiotic-resistant bugs in the 21st century—a clinical super-challenge. N. Engl. J. Med. 360, 439–443 (2009).
5. Kloos, W.E. & Jorgensen, J.H. Staphylococci. in Manual of Clinical Microbiology edn. 4 (eds. Lennette, E.H., Balows, A., Hausler Jr., W.J. & Shadomy, H.J.) 143–153 (American Society for Microbiology, Washington, DC, 1985).
6. Liu, C.I. et al. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 319, 1391–1394 (2008).
7. Clauditz, A., Resch, A., Wieland, K.P., Peschel, A. & Götz, F. Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect. Immun. 74, 4950–4953 (2006).
8. Liu, G.Y. et al. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J. Exp. Med. 202, 209–215 (2005).
9. Pelz, A. et al. Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J. Biol. Chem. 280, 32493–32498 (2005).
10. Kim, S.H. & Lee, P.C. Functional expression and extension of staphylococcal staphyloxanthin biosynthetic pathway in Escherichia coli. J. Biol. Chem. 287, 21575–21583 (2012).
11. Bischoff, M. & Berger-Bächi, B. Teicoplanin stress-selected mutations increasing sigma(B) activity in Staphylococcus aureus. Antimicrob. Agents Chemother. 45, 1714–1720 (2001).
12. Palma, M. & Cheung, A.L. sigma(B) activity in Staphylococcus aureus is controlled by RsbU and an additional factor(s) during bacterial growth. Infect. Immun. 69, 7858–7865 (2001).
13. Lan, L., Cheng, A., Dunman, P.M., Missiakas, D. & He, C. Golden pigment production and virulence gene expression are affected by metabolisms in Staphylococcus aureus. J. Bacteriol. 192, 3068–3077 (2010).
14. Fey, P.D. et al. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio 4, e00537–12 (2013).
15. Ding, Y. et al. Metabolic sensor governing bacterial virulence in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 111, E4981–E4990 (2014).
16. Liu, Y. et al. SsrA (tmRNA) acts as an antisense RNA to regulate Staphylococcus aureus pigment synthesis by base pairing with crtMN mRNA. FEBS Lett. 584, 4325–4329 (2010).
17. Duthie, E.S. & Lorenz, L.L. Staphylococcal coagulase; mode of action and antigenicity. J. Gen. Microbiol. 6, 95–107 (1952).
18. Porretta, G.C. et al. Antifungal agents, Part 11. Biphenyl analogues of naftifine: synthesis and antifungal activities. Arch. Pharm. (Weinheim) 328, 667–672 (1995).
19. Gupta, A.K., Ryder, J.E. & Cooper, E.A. Naftifine: a review. J. Cutan. Med. Surg. 12, 51–58 (2008).
20. Wieland, B. et al. Genetic and biochemical analyses of the biosynthesis of the yellow carotenoid 4,4′-diaponeurosporene of Staphylococcus aureus. J. Bacteriol. 176, 7719–7726 (1994).
21. Russell, R.G. Ibandronate: pharmacology and preclinical studies. Bone 38 (suppl. 1), S7–S12 (2006).
22. Leejae, S., Hasap, L. & Voravuthikunchai, S.P. Inhibition of staphyloxanthin biosynthesis in Staphylococcus aureus by rhodomyrtone, a novel antibiotic candidate. J. Med. Microbiol. 62, 421–428 (2013).
23. Pantoliano, M.W. et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 6, 429–440 (2001).
24. Martinez Molina, D. et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 (2013).
25. Raisig, A. & Sandmann, G. 4,4′-diapophytoene desaturase: catalytic properties of an enzyme from the C30 carotenoid pathway of Staphylococcus aureus. J. Bacteriol. 181, 6184–6187 (1999).
26. Furubayashi, M., Li, L., Katabami, A., Saito, K. & Umeno, D. Construction of carotenoid biosynthetic pathways using squalene synthase. FEBS Lett. 588, 436–442 (2014).
27. David, M.Z. & Daum, R.S. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23, 616–687 (2010).
28. Kuroda, M. et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240 (2001).
29. El-Gohary, M. et al. Topical antifungal treatments for tinea cruris and tinea corporis. Cochrane Database Syst. Rev. 8, CD009992 (2014).
30. Belter, A. et al. Squalene monooxygenase—a target for hypercholesterolemic therapy. Biol. Chem. 392, 1053–1075 (2011).
31. Volkman, J.K. Sterols in microorganisms. Appl. Microbiol. Biotechnol. 60, 495–506 (2003).
32. Alsterholm, M., Karami, N. & Faergemann, J. Antimicrobial activity of topical skin pharmaceuticals—an in vitro study. Acta Derm. Venereol. 90, 239–245 (2010).
33. Nolting, S. & Bräutigam, M. Clinical relevance of the antibacterial activity of terbinafine: a contralateral comparison between 1% terbinafine cream and 0.1% gentamicin sulphate cream in pyoderma. Br. J. Dermatol. 126 (suppl. 39), 56–60 (1992).
34. Ciftci, E., Guriz, H. & Aysev, A.D. Mupirocin vs terbinafine in impetigo. Indian J. Pediatr. 69, 679–682 (2002).
35. Koning, S. et al. Interventions for impetigo. Cochrane Database Syst. Rev. 1, CD003261 (2012).
36. Hammond, R.K. & White, D.C. Inhibition of vitamin K2 and carotenoid synthesis in Staphylococcus aureus by diphenylamine. J. Bacteriol. 103, 611–615 (1970).
37. Raisig, A. & Sandmann, G. Functional properties of diapophytoene and related desaturases of C30 and C40 carotenoid biosynthetic pathways. Biochim. Biophys. Acta 1533, 164–170 (2001).
acknowledgmentsWe thank M. Wu for helpful discussion, S.F. Reichard for editing the manuscript, and F. Götz (University of Tübingen) for providing the S. carnosus strains TM300<pTX15 and TM300<pTXcrtMN. This work was supported by the National Natural Science Foundation of China (21472207 and 31270126 to L.L.; 21222211 and 91313303 to J.L.), Hundred Talents Program of the Chinese Academy of Sciences (L.L.), Shanghai Institute of Materia Medica Foundation (CASIMM0120152018 to L.L.), Shanghai Municipal Education Commission and Shanghai Education Development Foundation (14SG28 to J.L.) and Foundation of the State Key Laboratory of Drug Research (SIMM1302KF-01 to J.L.). We acknowledge the National Science and Technology Major Project “Key New Drug Creation and Manufacturing Program” (2013ZX09507-004 to C.-G.Y.) and Shanghai Committee of Science and Technology (12ZR1453200 to F.C.).
author contributionsF.C. performed the biological experiments and drafted the biology part of the manu-script. H.D. performed the biological experiments. Y.W. synthesized naftifine derivatives. F.L. and N.Z. performed one-dimensional NMR spectroscopy analysis. Q.C., B.X., X.Z. and N.Y. participated in the mouse infection experiment; G.L. and C.-G.Y. contributed to protein purification; Y.X. and H.J. contributed to data analysis. J.L. designed the chemi-cal synthesis and drafted the chemistry part of the manuscript. L.L. conceived the study, designed the biological experiments and wrote the manuscript. All authors contributed to interpretation of data, and read and approved the final manuscript.
competing financial interestsThe authors declare competing financial interests: details accompany the online version of the paper.
additional informationAny supplementary information, chemical compound information and source data are available in the online version of the paper. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Correspondence and requests for materials should be addressed to L.L. or J.L.
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

nature chemical biology doi:10.1038/nchembio.2003
oNLINE METHoDSEthics statement. Animal experiments were performed in strict accordance with the regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of People’s Republic of China (11-14-1988). The animal study protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the Shanghai Public Health Clinical Center (permit 2013P201) and were performed in accordance with the relevant guidelines and regulations. The laboratory animal usage license number is SYXK-HU-2010-0098, certified by Shanghai Committee of Science and Technology. The animals were randomized, but the investigators were not blinded to the experimental conditions. Experiments with blood from human volunteers involved protocols that were reviewed, approved and per-formed under regulatory supervision of Shanghai Institute of Materia Medica’s Institutional Review Board. Written informed consent was obtained.
Bacterial strains, plasmids and culture conditions. Supplementary Table 1 lists the bacterial strains and plasmids used in this study. Unless otherwise noted, S. aureus strains were grown in TSB (Difco 286220) or on tryptic soy agar (TSA) plates without glucose supplement. E. coli strains were grown in Luria Bertani (LB) broth or on LB agar plates. For plasmid maintenance, antibiotics were used at the following concentrations where appropriate: for S. aureus, erythromycin at 10 μg/ml, chloramphenicol at 10 μg/ml; for S. carnosus, tetracycline at 10 μg/ml; for E. coli, ampicillin at 100 μg/ml and kanamycin at 50 μg/ml. Unless otherwise noted, cultures were incubated at 37 °C in a shaker (IKA KS 4000i Control Orbital Shaker) with 250 r.p.m. of aeration.
Market drug library and screening. 412 market drugs (see in Supplementary Data Set 1) were dissolved in DMSO (dimethyl sulfoxide) and yielded 10 mM solution, respectively. All drugs used for the screening had a purity of at least 98%. Drug screening was performed as follows. Briefly, overnight cultures were diluted 1:100 in TSB medium, and drug was added to each 20-ml tube (with a tube volume to medium volume ratio 10:1) to 200 μM. The S. aureus Newman bacteria were cultured at 37 °C for 24 h, with shaking at 250 r.p.m. 1.5 ml of bacteria cultures were centrifuged, and the pigmentation of S. aureus bacteria was visually analyzed. The control (40 μl DMSO) has no obvious effect on the growth and pigmentation of S. aureus Newman strain.
General information of the chemistry. Naftifine hydrochloride (BF1016, Shanghai Biosundrug Co., Ltd, 99% purity), terbinafine hydrochloride (D2049, Tokyo Chemical Industry Co., Ltd, >98% purity), butenafine hydrochlo-ride (B3293, Tokyo Chemical Industry Co., Ltd, >98% purity) and iband-ronate sodium (CV6199, Shanghai Civi Chemical Technology Co., Ltd, 99% purity) were used in this study except for the screening assay. BPH-652 was synthesized by Shanghai ChemPartner Co. Ltd with a purity of 96.7%. The synthesis of naftifine analogs and spectroscopic data are described in the Supplementary Note.
Constructions of USA300 LAC crtN mutant and USA400 MW2 crtN mutant. USA300 LAC and USA400 MW2 variants containing the crtN<erm allele from Newman crtN mutant13,38 were constructed by phage transduction with φ85 phage. Transductants were selected on TSA containing erythromycin 150 μg/ml (to overcome background resistance). Successful crosses of the crtN<erm insertion were confirmed by PCR on purified genomic DNA using oligonucleotides crtN-F and crtN-R (Supplementary Table 2). A com-plemented version of either the USA300 LAC crtN mutant or the USA400 MW2 crtN mutant was constructed by the introduction of integration vector pCL-crtN (Supplementary Table 1).
Construction of plasmids. To construct the plasmid for the constitutive expres-sion of crtN, a ~1.5 kilobase (kb) DNA covering 30 base pairs (bp) of crtN upstream region, the crtN gene and a 40 bp downstream of crtN was ampli-fied from S. aureus Newman genomic DNA with primers crtN-F and crtN-R (Supplementary Table 2), and then cloned into pYJ335. The construct with crtN in the same orientation as the tetracycline-inducible xyl/tetO promoter was confirmed by DNA sequencing, yielding plasmid pYJ335<crtN. Plasmid pYJ335<ispA was constructed as described above with primers IspA-F and IspA-R (Supplementary Table 2).
To construct the plasmid pCL-crtN, a ~2.3 kb DNA fragment containing the crtN gene and the tetracycline-inducible xyl/tetO promoter of pYJ335 was amplified from pYJ335<crtN plasmid DNA with primers crtN-pCL-F
(with EcoRI site) and crtN-pCL-R (with KpnI site) (Supplementary Table 2). The resulting PCR products were cut with the restriction enzymes EcoRI and KpnI, and ligated with plasmid pCL-LacZ that had been cut with the same enzyme, generating pCL-crtN (Supplementary Table 1). The pCL-crtN plasmid was electroporated into the S. aureus strain RN4220 and then trans-formed into mutants using bacteriophage φ85. As a control, empty pCL-LacZ vector was integrated into the chromosomes of wild-type and crtN mutant with a similar method.
To construct the plasmid pET28a<crtN, the crtN gene was amplified from genomic DNA of S. aureus Newman with primers crtN-PF (with BamHI site) and crtN-PR (with XhoI site) (Supplementary Table 2). The amplified fragment was digested with BamHI and XhoI, and inserted into pET28a digested with the same pair of restriction enzymes to generate pET28a<crtN (Supplementary Table 1). Similarly, pET28a<ispA was con-structed with primers IspA-PF and IspA-PR, pET28a<crtM was constructed with primers CrtM-PF (with BamHI site) and crtM-PR (with XhoI site), and pET28a<crtMN was constructed using primers CrtM-PF (with BamHI site) and crtN-PR (with XhoI site).
b-galactosidase assays. The product (7-hydroxy-4-methylcoumarin, 4MU) was detected using a Synergy 2 (Biotek) plate reader following the manufacturer’s instructions. The reaction was monitored at 460 nm with an excitation wave-length of 365 nm. Each sample was tested in triplicate. Relative β-galactosidase (lacZ) activity was normalized by cell density at 600 nm (OD600).
Protein expression, purification and preparation of anti-CrtN. The proteins were expressed in E. coli strain BL21 (DE3), and purifications were performed as follows. Briefly, the E. coli strain was subcultured into 1,000 ml of LB broth supplemented with 50 μg/ml kanamycin to obtain an optical density at 600 nm (OD600) of ~0.1 and grown to an OD600 of ~0.5. Expression of 6His-crtN was induced with 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) overnight at 16 °C with shaking (200 r.p.m.). Cells were harvested, and the pellets were suspended in 30 ml of buffer A (50 mM Tris-HCl, pH 8.0; 150 mM NaCl and 1 mM DTT), and lysed at 4 °C by sonication. The lysate was centrifuged at 15,000g for 30 min, and the supernatants were loaded onto a nickel-nitrilotriacetic acid col-umn (His Trap; GE Healthcare). After being equilibrated with buffer A, 6His-crtN was eluted with a 0–100% gradient of buffer B (50 mM Tris-HCl, pH 8.0; 150 mM NaCl, 1 mM DTT, 500 mM imidazole). The purified protein was veri-fied by SDS-PAGE followed by Coomassie blue staining. Anti-CrtN polyclo-nal antibody was prepared by immunizing a rabbit with the purified 6His-crtN protein (Shanghai Immune Biotech CO., Ltd); the validation profile of CrtN antibody is shown in the Supplementary Note. Purification of 6His-ispA was performed as CrtN except the concentration of NaCl in buffers was 300 mM.
One-dimensional NMR spectroscopy analysis. 6His-crtN was first purified by Ni affinity chromatography and subsequently purified by size-exclusion chro-matography using a calibrated HiLoad 16/60 Superdex 200 pg (GE Healthcare, 28-9893-35) pre-equilibrated with buffer A. Sample fractions were analyzed for purity by SDS-PAGE, and the pure fractions were pooled and concentrated. Protein concentration was measured according to the Beer-Lambert law.
Naftifine hydrochloride was dissolved with D2O to 10 mM as stock solu-tion, and ligand observed T1ρ and saturation transfer difference NMR spec-troscopy experiments were applied to investigate ligand-protein interactions. All NMR spectra were acquired at 25 °C on a Bruker Avance III-600 MHz (proton frequency) spectrometer equipped with a cryogenically cooled probe (Bruker biospin). Samples containing 200 μM naftifine hydrochloride (dissolved in D2O) in the absence or presence of CrtN were used in NMR spectroscopy data acquisition, as indicated.
Measurement of pigment production. S. aureus bacteria were cultured in TSB (4 ml) medium (with or without inhibitor compounds) at 37 °C for 48 h, with shaking at 250 r.p.m. Prior to the assays, 3 ml bacteria cultures were centrifuged and washed twice with 0.01 M phosphate-buffered saline (PBS) (8g NaCl, 0.2g KCl, 1.44g Na2HPO4, 0.24g KH2PO4, pH 7.4, ddH2O to 1, 000 ml). Pigment was extracted thrice with methanol and added to a total volume of 1 ml. The absorbance value was determined at 450 nm using a NanoDrop 2000c (Thermo Scientific) spectrophotometer.
High-performance liquid chromatography (HPLC). S. aureus bacteria were grown overnight in tubes and diluted in 50 ml fresh TSB medium with or
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

nature chemical biologydoi:10.1038/nchembio.2003
without ibandronate sodium (100 μM) or naftifine hydrochloride (100 μM), as indicated. Cells were further grown with shaking at 37 °C for 24 h, and harvested by centrifugation. The cells were washed twice with PBS buffer and suspended in 500 μl 1× TE (10 mM Tris, pH 8.5, and 1 mM EDTA) buffer. After the addition of lysostaphin (final concentration of 50 μg/ml), suspen-sions were incubated at 37 °C for 30 min. Then, 20 ml methanol/0.3% aqueous NaCl (10:1, v/v) was added, and the mixtures were vortexed vigorously. Subsequently, equal volume of hexane was added for the extraction of isopre-noids. The extraction was repeated twice, and the combined extracts were con-centrated in a rotary evaporator. Finally, 500 μl methanol/2-propanol (1:1, v/v) was used to dissolve the extracts. The dissolved extracts were filtered and 50 μl samples were analyzed by high-performance liquid chromatography (HPLC, Agilent 1260 infinity) on a Shim-pack CLC-ODS reversed-phase column (228-00808-91) (6 mm × 150 mm; eluent, methanol/2-propanol, 1:1 (v/v); flow rate, 1 ml/min). Isoprenoids were monitored at 210 nm.
For analysis of carotenoids, strains were grown overnight in tubes and diluted in 50 ml TSB fresh medium (100 μM naftifine hydrochloride was used where appropriate, as indicated). Cells were further grown with shak-ing at 37 °C for 24 h, and harvested by centrifugation. Briefly, wet cells harvested from 50-ml cultures were extracted with 20 ml of acetone. To this, 10 ml of hexane and 10 ml of aqueous NaCl (10%, w/v) were added, and the mixture was shaken vigorously to remove oily lipids. The upper phase containing the carotenoids was dried with anhydrous MgSO4 and con-centrated in a rotary evaporator. Finally, a 500 μl acetonitrile/isopropanol mixture (85:15, v/v) was used to dissolve the extracts. The dissolved extracts were filtered, and 50-μl samples were analyzed through a Spherisorb ODS2 column (250 mm × 4.6 mm; particle diameter, 5 μm; Waters) and eluted with an acetonitrile-isopropanol mixture (85:15, v/v) at a flow rate of 1 ml/min, using an Alliance high-pressure liquid chromatogra-phy (HPLC) system (Agilent 1260 Infinity) equipped with a photodiode array detector.
In vitro thermal shift assays. For an in vitro thermal shift assay of CrtN, E. coli/ pET28a<crtN bacteria were grown at 37 °C overnight in LB medium. Overnight cultures were sub-cultured into 1 l of LB broth supplemented with 50 μg/ml kanamycin to obtain an optical density at 600 nm (OD600) of ~0.1 and grown to an OD600 of ~0.5. Expression of 6His-crtN protein was induced with 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) overnight at 16 °C with shaking (200 r.p.m.). Cells were harvested, and the pellets were suspended in 40 ml of HEPES buffer (20 mM HEPES, pH 7.5; 500 mM NaCl, 1 mM DTT) and lysed at 4 °C by sonication. After that, a 100 μl substrate 4,4′-diapophytoene emul-sion (see below) and 7 μl FAD stock solutions (10 mM) were added into 600 μl lysate containing the 6His-crtN protein, and mixed by vortexing the solutions. The reaction mixture was diluted with 553 μl HEPES buffer and divided into two aliquots (630 μl each), with one diluent treated with 70 μl naftifine hydro-chloride (final concentration: 100 μM, ~11-fold higher than the IC50 value of naftifine for in vitro CrtN activity) and the other aliquot treated with the mock solution (ddH2O). After a 60-min incubation at 37 °C, the lysates were cen-trifuged at 20,000g for 20 min at 4 °C, and the soluble fraction was collected. The soluble fractions were divided into smaller (50 μl) aliquots and heated individually at different temperatures (26.0, 48.0, 51.9, 55.8, 60.0 and 64.6 °C) for 5 min with a C1000TM thermal cycler (BIO-RAD) followed by cool-ing for 3 min at room temperature. The heated lysates were centrifuged at 20,000g for 20 min at 4 °C to separate the soluble fractions from precipitates. The supernatants were transferred to new microtubes, analyzed by SDS-PAGE, and then visualized by Coomasie Blue G-250 staining. The relative abun-dance was determined by densitometric analysis using ImageQuant software. PageRuler Prestained Protein Ladder SM0672 (Fermentas) was used as a molecular weight reference.
For fluorescence-based thermal shift (FTS) assay of purified 6His-ispA protein, 6His-ispA was appropriately diluted in a buffer containing 50 mM imidazole, pH 8.0, 500 mM NaCl, and 1 mM DTT. All assay experiments used 2 μg proteins per well and 1 μl 100× Sypro Orange (Invitrogen) up to a total volume of 20 μl. 96-well PCR plates were sealed with optical seal, shaken and centrifuged after the protein and the compounds were added. Thermal scanning (25−95 °C at 1 °C/min) was performed using a Fast 7500 Real-Time PCR System (Applied Biosystems), and fluorescence intensity was measured after every 20 s. Curve fitting, melting temperature calculation, and report generation on the raw FTS data were performed using Protein thermal shift
software (Applied Biosystems). All experiments were performed in triplicate. Data analysis was performed with Graphpad 5.0 software. Pre-melt (initial) and post-melt (final) fluorescence signals of all samples were normalized to relative values of 0 and 1, respectively.
Cellular thermal shift assay. For cellular thermal shift assay24, S. carnosus TM300<pTXcrtMN bacteria (Supplementary Table 1) were grown at 37 °C overnight in TSB medium without glucose. The overnight cultures were washed twice and diluted 100-fold in 50 ml of fresh TSB medium contained in a 250-ml flask with or without naftifine hydrochloride (final concentration, 100 μM), and shaken at 250 r.p.m. at 37 °C for 24 h. Xylose was added with a final concentration of 1% (w/v) to induce expression of crtMN. S. carnosus cells were harvested by centrifugation and washed twice with Tris buffer (50 mM Tris, 150 mM NaCl and 10 mM CaCl2, pH 7.9) and resuspended in the same buffer with OD600 ≈ 50. A 1-ml bacterial suspension was crushed with a FastPrep-24 sample preparation system (MP Biomedicals) (6.5 m/s, 40s, three times), the cell debris was centrifuged at 20,000g for 20 min at 4 °C, and the soluble fraction (lysate) was collected.
The respective lysates were divided into smaller (50 μl) aliquots and heated individually at different temperatures (26.0, 48.0, 52.0, 54.4, 56.3 and 58.0 °C) for 10 min with a C1000 thermal cycler (BIO-RAD) followed by cooling for 3 min at room temperature. The heated lysates were centrifuged at 20,000g for 20 min at 4 °C to separate the soluble fractions from precipitates. The supernatants were transferred to new microtubes and analyzed by SDS-PAGE followed by western blot analysis.
For western blot analysis, 35 μl sample was mixed with 10 μl of 5× SDS load-ing buffer (250 mM Tris-HCl (pH 6.8), 10% SDS, 0.5% bromophenol blue, 50% glycerol and 1 mM DTT) and then heated at 100 °C for 10 min. SDS PAGE was carried out using a 8% slab gel with a 5% stacking gel, and then transferred onto PVDF (Bio-Rad) membranes. PageRuler Prestained Protein Ladder SM0672 (Fermentas) was used as a molecular weight reference. For the detection of CrtN protein, anti-CrtN polyclonal antibody (described above, 1:1,000) and anti-rabbit IgG conjugated to horseradish peroxidase (HRP) (NA934, GE 425 Healthcare, 1:5,000) were used. The resulting chemiluminescent light was detected by a Tanon-5200 multi according to the manufacturer’s recommen-dation, and the relative abundance was determined by densitometric analysis using ImageQuant software.
Effect of naftifine on bacterial growth of S. aureus Newman. Naftifine hydro-chloride was dissolved with dimethyl sulfoxide (DMSO) to a concentration of 20 mM as a stock concentration. Naftifine was diluted with fresh TSB medium to 1,000 μl with the final concentration of naftifine hydrochloride at either 0.2 mM or 0.5 mM. 100 μl of each dilution was distributed in 96-well plates along with growth controls (containing equal amount of DMSO). 60 μl of paraffin wax was used to cover the dilutions to avoid evaporation of the medium. The dilu-tions were plated at 37 °C for 4 h to dissolve naftifine sufficiently. Overnight cultured S. aureus Newman strain was washed twice with PBS and diluted with fresh medium to obtain an optical density at 600 nm (OD600) of ~1.0. Test and growth control wells were inoculated with 5 μl of a bacterial suspension (final OD600 ≈ 0.05). The 96-well plates were incubated at 37 °C for 15 h, and OD600 was recorded every half an hour with a Synergy 2 (Biotek) plate reader following the manufacturer’s instructions.
Naftifine hydrochloride has influence on the culture turbidity (OD600) due to its poor solubility in TSB medium, especially at high concentrations. To test the effects of high concentrations of naftifine on the bacterial growth of S. aureus Newman, naftifine hydrochloride (power) was added directly to 4 ml TSB medium in 20-ml tube with final concentrations of 0.5% or 1% (wt/v). Subsequently, overnight cultured strains were transferred to the medium with a ratio of 1:100 (v/v). The tubes were incubated at 37 °C for 24 h with shaking (250 r.p.m.), and then dilutions were plated on a TSA agar plate for enumera-tion of colony forming units (CFU). Analysis of bacterial growth in the TSB medium alone was used as a control.
CrtN enzyme activity assays. Diapohytoene was purified from diapophytoene-producing E. coli BL21 (DE3)/pET28a<crtM (Supplementary Table 1) by extraction with acetone, as described above for the extraction of carotenoids. Diapophytoene emulsion was prepared by dissolving 24 mg of phosphatidyl-choline (Sigma-Aldrich) in 200 μl CHCl3, and mixed with 8 mg diapophy-toene. The dried phosphatidylcholine-diapophytoene mixture was incubated
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

nature chemical biology doi:10.1038/nchembio.2003
for 6 h with shaking (250 r.p.m.), and then dilutions were plated on a TSA agar plate for enumeration of surviving CFU.
Mouse infection models. Overnight cultures of S. aureus were washed and diluted 100-fold in fresh TSB medium in a 250-ml flask with a volume-to- medium volume ratio of 5:1. The liquid culture was grown at 37 °C for about 3 h with shaking (OD600 ≈ 0.6), 250 r.p.m. of aeration, and then the bacteria were harvested and washed twice with ice-cold, phosphate-buffered saline (PBS). CFUs per milliliter was determined before mice were inoculated.
6−8-week-old female BALB/c mice were obtained from SIPPR-BK Lab Animal Ltd. (http://www.slarc.org.cn) and housed under specified pathogen−free conditions. Mice were anaesthetized with pentobarbital sodium (intraperitoneal injection, 80 mg/kg) and were infected by retro-orbital injection with a suspension of staphylococci. For each experimental group, at least eight mice were used, as indicated.
To evaluate the efficacy of compounds (naftifine, vancomycin and JX07) on the outcome of S. aureus pathogenesis, female BALB/c mice received intra-peritoneal injections at a total dose of 225 mg/kg naftifine hydrochloride (or vancomycin) dissolved in sterile ddH2O in 12-h intervals for 108 h (4.5 d). Twelve hours after the first injection of naftifine hydrochloride, vancomycin or mock (ddH2O) control, mice were challenged with S. aureus bacteria.
For the lethal challenge experiments, female BALB/c mice were infected with 5 × 107 CFU of S. aureus Newman, 5 × 107 or 2.5 × 108 CFU of S. aureus MW2, and the log-rank test was used to analyze mortality data. For the mouse model of abscess formation, female BALB/c mice were challenged with either 4 × 106 CFU of S. aureus Newman and its variants, 4 × 106 CFU of S. aureus USA300 LAC and its variants, 7 × 106 CFU of S. aureus USA400 MW2 or 1.2 × 108 CFU of S. aureus Mu50. To test the efficacy of naftifine against Newman crtN mutant infection, female BALB/c mouse was challenged with 4 × 107 CFU. Animals were killed 5 d after infection. Kidneys, hearts or livers were aseptically removed and homogenized in PBS plus 0.1% Triton X-100 to obtain single-cell suspensions, and serial dilutions of each organ were plated on TSA (Difco) plates for the enumeration of CFUs. Statistical significance was determined by the Mann-Whitney test (two-tailed).
Statistical analysis. Student’s t-tests (two-tailed) were used to compare two data sets (Figs. 1c,d and 3d). The log-rank test was used for survival analysis (Figs. 1e and 5b and Supplementary Fig. 18a). The non-parametric Mann-Whitney test (two-tailed) was used to compare two groups of observations (Figs. 1f, 4a–c and 5a,c, and Supplementary Figs. 6d and 18b).
with 2 ml 0.02 M HEPES buffer (pH 7.5), then sonicated in ice water until the emulsion was homogeneous39.
For the assay of diapophytoene desaturase CrtN activity, the reaction mixture contained a total of 700 μl of the following: 0.02 M HEPES buffer (pH 7.5), 50 μl diapophytoene emulsion, 70 μl different concentrations of naftifine or mock (ddH2O), 3.5 μl FAD stock solution (10 mM) and 300 μl CrtN lysate (~1.41 mg CrtN, estimated by western blot using known concen-tration of the purified 6His-crtN protein). The experiments were performed under anaerobic conditions by adding a final concentration of 20 U/ml glucose oxidase (Sigma-Aldrich, G2133), 20,000 U/ml catalase (Sigma-Aldrich, C1345) and 2 mM glucose as an oxygen-trapping system. The reaction mixture was started by adding the lysate and incubated for 14 h at 37 °C with gentle shaking. The reaction was terminated by adding 500 μl methanol. Pigments were extracted twice against 700 μl chloroform. The organic phase was concen-trated, redissolved in 200 μl chloroform, analyzed by reading the absorbance under 450 nm for the relative quantification of diaponeurosporene. IC50 was defined as the concentration of naftifine hydrochloride that is required for 50% inhibition under our experimental conditions, and was determined by constructing a dose-response curve using Graphpad 5.0 software.
To determine whether the inhibitory effect of naftifine on CrtN was reversible or irreversible, the reaction test was carried out as described above, except that a different mount of CrtN lysate (~0.47 mg CrtN/100 μl lysate) was used, and the total reaction volume was scaled to 1,100 μl. The reaction rate was determined by detecting the absorbance under 450 nm for diaponeu-rosporene quantification after starting the reaction for 4 h.
Lineweaver–Burk40 plot was used to determine the CrtN enzyme kinetics. Enzyme reaction was carried out with different volumes of diapophytoene emulsion (0 μl, 67 μl, 80 μl, 100 μl, 133 μl, 200 μl and 400 μl), either in the absence or presence of naftifine (final concentration, 100 μM). The initial reac-tion rate was determined by monitoring the increase in absorbance at 450 nm within the first 1.5 h (the reaction rate was kept steady during the first 3 h in our enzymatic assay of CrtN). A double reciprocal plot (1/V versus 1/[S]) where V is reaction velocity and [S] is substrate concentration was plotted. The kinetic constants (KM and Vmax) were calculated.
Hydrogen peroxide killing and human whole blood killing. S. aureus was cul-tured in TSB with or without naftifine hydrochloride (100 μM). After 24 h, bacteria were washed twice in PBS, and diluted to a concentration of 2.5 × 106 CFU per 250 μl reaction mixture in a 2-ml Eppendorf tube. H2O2 was added to a 1.5% final concentration and the tubes were incubated for 1 h at 37 °C with shaking at 250 r.p.m. The reaction was stopped by the addition of 1,000 U/ml exogenous catalase (Sigma-Aldrich). Bacterial viability assessed by dilutions on TSA plates for enumeration of surviving CFUs.
For whole blood killing assay, overnight cultures of strains with or without naftifine hydrochloride (100 μM) were centrifuged and suspended in sterile PBS to generate a suspension of 1 × 107 CFU/ml. Whole blood from healthy human volunteer was collected using a BD VACUTAINER PT tube. 450 μl of whole blood was transferred into a 20-ml tube and mixed with 50 μl bacterial sample, which resulted in 1 × 106 CFU/ml. The tubes were incubated at 37 °C
38. Bae, T. et al. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc. Natl. Acad. Sci. USA 101, 12312–12317 (2004).
39. Schaub, P. et al. On the structure and function of the phytoene desaturase CRTI from Pantoea ananatis, a membrane-peripheral and FAD-dependent oxidase/isomerase. PLoS One 7, e39550 (2012).
40. Lineweaver, H. & Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 56, 658–666 (1934).
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.