soft gelatin capsules (softgels
DESCRIPTION
capsulas molesTRANSCRIPT

REVIEW
Soft Gelatin Capsules (Softgels)
RAMPURNA PRASAD GULLAPALLI
Elan Pharmaceuticals, 800 Gateway Blvd., South San Francisco, California 94080
Received 13 December 2009; revised 15 February 2010; accepted 19 February 2010
Published online 5 April 2010 in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/jps.22151
The contentsauthor in his pesarily representaffiliates.
Corresponden914-316-4935; F
Journal of Pharm
� 2010 Wiley-Liss
ABSTRACT: It is estimated that more than 40% of new chemical entities (NCEs) coming out ofthe current drug discovery process have poor biopharmaceutical properties, such as low aqueoussolubility and/or permeability. These suboptimal properties pose significant challenges for theoral absorption of the compounds and for the development of orally bioavailable dosage forms.Development of soft gelatin capsule (softgel) dosage form is of growing interest for the oraldelivery of poorly water soluble compounds (BCS class II or class IV). The softgel dosage formoffers several advantages over other oral dosage forms, such as delivering a liquid matrixdesigned to solubilize and improve the oral bioavailability of a poorly soluble compound as a unitdose solid dosage form, delivering low and ultra-low doses of a compound, delivering a lowmelting compound, and minimizing potential generation of dust during manufacturing andthereby improving the safety of production personnel. However, due to the very dynamicnature of the softgel dosage form, its development and stability during its shelf-life arefraught with several challenges. The goal of the current review is to provide an in-depthdiscussion on the softgel dosage form to formulation scientists who are considering developingsoftgels for therapeutic compounds. � 2010 Wiley-Liss, Inc. and the American Pharmacists Association
J Pharm Sci 99:4107–4148, 2010
Keywords: softgel; soft gelatin capsule; for
mulation; encapsulation; poorly soluble; bioavail-ability; cross-linking; dissolution; physical stability; chemical stabilityINTRODUCTION
Oral absorption of a compound can be influenced by avariety of factors, such as the physicochemicalproperties, formulation, and dose of the compoundand the physiology and pathology of the gastro-intestinal tract (GIT). Regardless of other factors, it isreasonable to conclude that the compound must be inthe solution form or solubilized form in the GIT todiffuse into and across the enterocytes lining theintestinal lumen.1,2 The advent of combinatorialchemistry and high throughput screening (HTS)has resulted in the identification of many highlypotent new chemical entities (NCEs) that usuallyhave less than desirable physicochemical properties,that is, high molecular weight, high lipophilicity(log P), and low aqueous solubility.3 More than 40% of
of this article represent the efforts and views of thersonal and professional capacity and do not neces-the views of Elan Pharmaceuticals, Inc. and/or its
ce to: Rampurna Prasad Gullapalli (Telephone:ax: 650-877-7461; E-mail: [email protected])
aceutical Sciences, Vol. 99, 4107–4148 (2010)
, Inc. and the American Pharmacists Association
JOURNAL OF
NCEs coming out of the combinatorial chemistry andHTS technologies have been thought to belong to thiscategory.4 Poor aqueous solubility has been identifiedas the single largest physicochemical challenge forthe oral absorption of compounds and almostinevitably leads to their lower oral bioavailabilitiesfrom the conventional dose forms.3
Traditional approaches to enhance the absorptionof a compound relate to improving its solubility andrate of dissolution in the GIT fluids. These approachesinclude using a form of the compound with optimumaqueous solubility, for example, salt form,5–7 amor-phous form,8,9 prodrug form,10 nanosizing,11–13 oremploying a vehicle in which the compound is solubleand remain solubilized upon contact with the GITaqueous environment.8,14
The least complex way to present a compound to theGIT for absorption is to administer the compound as asolution or solubilized form, thereby removing anydissolution rate-limiting step in the absorptionprocess.2 As the compound is already in solution atthe site of absorption, it could yield a faster, uniform,and enhanced absorption. Occasionally, nonaqueous(organic) vehicles are employed to solubilize poorlywater soluble compounds for oral and parenteral
PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010 4107

4108 GULLAPALLI
use.15 Use of the nonaqueous vehicles in oralproducts may be complicated by many factors. First,some nonaqueous vehicles, such as dimethylforma-mide (DMF) and dimethyl sulfoxide (DMSO), mayhave substantial solubilizing capacity for manycompounds. However, such vehicles are not pharma-ceutically acceptable. Second, some vehicles, thoughpharmaceutically acceptable, may not exert sufficientsolubilizing action to be of any practical value forsome compounds unless the dose is low. Otherwise,the volume of the vehicle required cannot be readilycontained in a convenient dose unit. Third, it is alsopossible that precipitation of the compound from thesolution may follow administration when the solubi-lized compound encounters the aqueous environmentin the GIT, resulting in little or no absorptionenhancement.
When a compound demonstrates sufficient solubi-lity in a pharmaceutically acceptable nonaqueousvehicle, soft gelatin capsules may be ideal to deliverthe solution as a solid dosage form. Soft gelatincapsules (SGC), also referred to as softgels orsoft elastic capsules (SEC), have gained popularityin delivering therapeutic compounds solubilized orsuspended in nonaqueous vehicles. A softgel is a one-piece, hermetically sealed soft gelatin shell contain-ing a solution, a suspension, or a semisolid, referred toas fill formulation, fill material, or fill. Softgels offermany advantages over other conventional oral dosageforms, including improving swallowbility, maskingodors and unpleasant taste, protecting the encapsu-lated compound against oxygen and light, and able toreadily dissolve in the gastric juices of the GIT. Theabsorption of poorly soluble compounds encapsulatedin softgels may also be higher compared to that fromother conventional dosage forms not only due to thesolubilization of the compounds in the fill formulationbut also due to the fill excipient induced inhibition ofP-glycoprotein-mediated drug efflux and reducedenzyme-catalyzed degradation of the compound inthe lumen of the GIT.16–24 Softgels also offer theadvantage of accurately delivering therapeuticagents that require ultra-low doses (e.g., cardiacglycosides, vitamin D analogs).
One of the major challenges in the development ofthe softgel dosage form is that the system is verydynamic in terms of (a) the physical migration ofcomponents between the shell and the fill and theshell and the external environment, and (b) theoccurrence of physical and chemical interactionswithin and between the shell and fill components.It is critical to understand these intricacies to developa softgel product that is stable and provides desiredin vitro and in vivo characteristics. This review articledeals with the various aspects of softgel dosage form,including selection of fill and shell compositions andmanufacturing process and the influence these
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
formulation and manufacturing components on thestability, dissolution, and bioavailability of the softgeldosage form. This review will be limited to onlysoftgels prepared from gelatin and will not addresssoftgels prepared from nongelatin material.
SOFTGEL MANUFACTURING PROCESS
The manufacturing process for the production ofsoftgels is the rotary die process, invented byScherer,25–29 in which a molten mass of a gelatinsheath (shell) formulation at 57–608C is fed from areservoir onto two separate rotating cool castingdrums (cooling drums) to form two spaced flat sheetsor ribbons of gelatin in a semi-molten state. Two tonesoftgels may be produced by utilizing two separatereservoirs of different color gelatin masses, eachsupplying one of the two ribbons for the coolingdrums. These flat ribbons are extracted from thecooling drums and are fed around rollers thatlubricate them, usually with fractionated coconutoil (e.g., Miglyol1 812, Sasol; Captex1 355, Abitec)/lecithin blend, and then brought together at aconvergent angle into the nip of a pair of roller diesthat include opposing die cavities. The lubricationstep is necessary to avoid the sticking of ribbonstogether and to other machine parts. The typicalspeed at which a gelatin ribbon is drawn into anencapsulation station is limited to around 2.5 cm/sdue to the limitation of a conventional encapsulationmachine. However, higher speeds may be achievedwith some modifications to the encapsulationmachine.30 A fill formulation, to be encapsulated,flowing from its own reservoir through a tube undergravity, is fed into a positive displacement pump.Accurately metered volumes of the fill formulationare injected from a wedge (heated to 37–408C) into thespace between gelatin ribbons as they pass betweenthe cavities on the die rolls. As the ribbons meet on therim of the opposing die cavities, the bottom lips of thecavities initially seal, forming what is referred to as‘‘lower seam.’’ The fill formulation is then injectedprecisely into the semi formed softgel. The softgelhalves are then sealed together (forming the ‘‘upperseam’’) by the application of heat and pressure. Theheated wedge provides enough heat to the gelatinribbons that aids in the sealing of the two-halves of asoftgel. It is extremely critical to avoid entrapment offill material within the upper seam, a concern duringthe encapsulation of highly viscous, viscoelastic, andstringy materials (e.g., povidone, surfactants). Suchentrapment potentially results in the formation ofweak seams and subsequent leakage of a softgelproduct. This phenomenon is not a concern in the caseof the lower seam, as it has already formed before theinjection of the fill material into the softgel.
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4109
The softgels, severed around each of the diecavities, are ejected by the continuous rotation ofthe dies and are carried on a conveyer into a tumbledryer. The part of the gelatin sheath that is severedfrom the segments forming the softgels (referred to as‘‘net’’) is then collected for recovery and recycling ofgelatin31,32 or for disposal. Photographs of softgelencapsulating machine and tumble dryer are shownin Figure 1. During the encapsulation, a series of in-process checks such as ribbon thickness (VernierCalipers), seam thickness (microscopy), fill weights,and shell weights are performed at regular intervals.The various steps involved in the manufacturing of asoftgel product can be grouped into five components:(a) gel mass, (b) fill formulation, (c) ribbons forencapsulation, (d) drying, and (e) finishing. Thecritical parameters in each manufacturing compo-nent and their effects on a softgel product are listed inTable 1.
Gel Mass
A gel mass (shell formulation) is usually preparedfrom gelatin, plasticizer(s), water, and other minoradditives such as opacifiers, colorants, flavors, sweet-
Figure 1. Photographs of Softgel Manufacturing Equipment.(A) Encapsulating Machine; (B) Tumble Dryer (Courtesy of Pii(Pharmaceutics International, Inc.)).
DOI 10.1002/jps JO
eners, and preservatives. The gel mass is prepared byinitially mixing water and plasticizer(s) with gelatingranules in a suitable reactor (kettle) to form a fullyhydrated fluff at room temperature. The material isthen melted thoroughly at high temperatures (�90–958C) under vacuum (29.5 inch Hg) with slow mixinguntil a clear gel is obtained. The gel mass is thentransferred into electrically heated holding tanks andkept at 57–608C for encapsulation.33 If the gelformulation requires an opacifier, the opacifier isinitially dispersed and wetted thoroughly in glycerinbefore its addition to the molten gel mass. Dispersionand wetting of the opacifier is usually accomplishedin a manufacturing setting by mixing it with glycerinin rotating drums or using drum mixers for extendedperiods. Other ingredients, such as colorants, flavors,and preservatives, may be added and mixed at highspeeds. Gelatin undergoes depolymerization whenthe gel mass is stored at high temperatures, resultingin a reduction of the gel strength and viscosity withtime.34 Thus, the temperature and time, to which thegel mass is exposed, need to be carefully monitoredand controlled throughout the encapsulation process.The gel mass is checked for clarity, color, consistency,and moisture content before encapsulation to insurethat the gel will run properly on the encapsulationmachine.
Fill Formulation
A fill formulation for encapsulation into softgels canbe a solution, liquid-in-liquid dispersion, or a solid-in-liquid suspension. Fill formulations are preparedusing standard procedures employed in pharmaceu-tical solution, suspension, and semisolid manufactur-ing. Fill formulations after compounding aredeaerated thoroughly under vacuum to eliminateany of the entrapped air in the formulation. Thedeaeration is a critical step in the manufacturing of asoftgel product that affects not only the fill viscosity,blend uniformity, fill weight uniformity, and thuscontent uniformity during manufacturing, but alsothe physical and chemical stability of the finishedsoftgel product during its shelf-life. Smaller scalebatches of fill formulations can be deaerated in apressure resistant stainless steel container undervacuum. Larger scale fill formulations are typicallyvacuum transferred into pressure resistant stainlesssteel tanks and further deaerated under vacuum (e.g.,FrymaKoruma Vacuum Deaerator). For highly vis-cous fill formulations, deaeration process can be aidedby moderate mixing with or without the use of heat.The length of deaeration of a fill formulation isinfluenced by a variety of factors, such as thecomposition (e.g., lipophilic vs. hydrophilic, solutionvs. suspension, presence or absence of viscosifiers andsurfactants), amount, and viscosity of the fill mate-
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

Table 1. Critical Parameters in a Softgel Product Manufacturing
Manufacturing Component Parameter Effects
Gel Mass Composition: gelatin type andconcentration, plasticizer(s) type and
concentration, moisture contentProcess: gel aging
temperature and time
Gel rheology, ribbon integrity andstrength, seam cutting and strength,softgel drying time, softgel hardnessand brittleness, oxygen and volatile
solute permeability, physicaland chemical stability
Fill Formulation Composition: type (hydrophilic vs.lipophilic), solution versus suspension,
particle size, morphology(suspensions), viscosifiers, surfactants
Process: mixing, temperature, inertenvironment (nitrogen blanketing,
yellow lights), deaeration
Fill rheology, blend and contentuniformity, fill weight variation, seamintegrity (due to fill trapping), softgel
drying time, softgel hardness andbrittleness, physical and
chemical stability
Encapsulation Cooling drums and dies speeds,cooling drums temperature, wedge
temperature, ribbon thickness,softgel size
Fill weight variation, seamthickness and strength,
production rate, drying time
Drying Primary drying (tumble/rotary)conditions, Secondary drying
(tray) conditions—temperatureand humidity, drying rate
(air flow), drying time
Fill moisture content, shell moisturecontent, softgel hardness and
brittleness, case hardening, shellcross-linking, dissolution
Finishing Sizing, polishing, printing,inspection, packaging
(container-closure)
Product quality, physical andchemical stability
4110 GULLAPALLI
rial, deaeration temperature, and the type of deaera-tion equipment used.
The fill formulation may be maintained at up to amaximum of 35–378C at the time of encapsulation tofacilitate the encapsulation process and highertemperatures should be avoided as they couldinterfere with the sealing of softgels. Fill formulationswhich are viscoelastic (stringy), shear sensitive(shear-thickening or dilatant material), and solidifyduring the encapsulation process, can pose significantchallenges during the manufacture of softgels. Suchfill material may be encapsulated into softgels bycontinuous heating of the material in its reservoir andin the conveying tubing to higher temperatures untilit reaches the dosing pump where it is cooled to lowertemperatures just before it reaches the wedge forencapsulation.35
Ribbons for Encapsulation
During the manufacture of a softgel product usingthe rotary die process, the gel mass is spread ontothe cooling drums to form ribbons with the aid ofmetering devices known as spreader boxes. Aspreader box, as invented by Scherer,36 consists ofa hopper having an elongated slot at its bottom and arotating roll (spindle) spaced between the two edges ofthe slot to provide a gap through which the molten gelmass is extruded by the rotation of the roll. Themechanism allows the volume of the gel mass passing
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
through the gap uniform along the entire length of thegap thereby resulting in the formation of a ribbonwith uniform thickness across its entire width andlength. The thickness of the ribbon can be controlledthrough controlling the flow of gel mass that can bereadily accomplished by increasing or decreasingthe speed of the rotating roll. The rotating roll in thehopper mechanism also helps in removing macro-scopic air bubbles from the viscous gel mass before itpasses through the gap thus enhancing the quality ofthe film deposited on the cooling drum. The spreaderboxes control the flow of gel mass onto the coolingdrums to a ribbon thickness within �10% of setspecification. The wet ribbon thickness may varyfrom 0.022 inches to 0.045 inches, with larger softgelsizes requiring thicker shell to accommodate thehigher structural strength required during theirmanufacture.33
The gel mass is typically at about 57–608C when itcomes into contact with the cooling drums. Thetemperature of the cold air used to cool the drums istypically about 13–148C.33 The air cooling processmay be inefficient in cooling the drums uniformlyacross their entire surface.37 US patent 7,078,05437
proposes that the surface of the cooling drums and thegelatin sheaths spread over them may be better andmore evenly cooled by the cooling drums that use aliquid coolant (e.g., water) than by the ones that useair coolant. The gelatin sheaths containing propylene
DOI 10.1002/jps

Figure 2. Dynamics of Water Migration during Softgel DryingProcess Water migration patterns during drying of a softgel productcontaining a typical (A) PEG 400 fill formulation; (B) mixed mediumchain mono-, di-, and triglycerides fill formulation with or withoutan added surfactant(s); and (C) medium chain triglycerides (MCT)or long chain triglycerides (LCT) fill formulation. Yellow innercore represents fill formulation and brown exterior representsgelatin shell. Numerical values represent approximate percentwater content, w/w. ( ) Water migration from shell to fill, ( ) watermigration from fill to shell, and ( ) water migration from shell toenvironment.
SOFT GELATIN CAPSULES 4111
glycol as the plasticizer are substantially tackier thanthose containing glycerol or sorbitol as the plasticizerand thus difficult to extract from the cooling drums.The use of a liquid coolant in the cooling drums mayresult in such gelatin sheaths containing propyleneglycol sticking less strongly to the cooling drums andthus may be easily removed from them. Anotheradvantage of using a liquid coolant over cold air incooling the drums is that the cooling processtemperature can be altered and controlled moreprecisely with the former to meet the coolingrequirements for various gel mass compositions.For example, the temperature of the coolant watercan be altered precisely from about 20–228C, optimaloperational temperature for a gel mass in the absenceof propylene glycol, to about 18–208C and 16–188C,optimal operational temperatures for gel massescontaining 10% and 21% propylene glycol, respec-tively, as the plasticizer.37
Drying
Softgels formed at the encapsulating machine arehighly flexible due to excessive moisture content ofthe starting gel mass.34,38–41 Softgels originating fromthe encapsulating machine undergo a primary shorttime, low intensity drying process followed by asecondary longer time, high intensity drying process.Softgels are initially tumble dried in a hollow drum(s)with perforated walls (Fig. 1B) (primary or rotarydrying process).38 Dry air is continuously pumpedthrough the rotating drum(s) at an air temperaturelower than 358C. The warm air being blown onto thesoftgels may penetrate the shell and cause it to dryfrom the inside by moving the water outward to thesurface of the softgel. The warm temperature alsohelps to keep the gel in semi-fluid state that promotesfurther sealing of softgels. Sometimes adsorbenttowels are used in the tumble dryer to remove anylubricant carried over during the encapsulationprocess. By the time the softgels exit this tumbledrying process, a significant portion of water fromthe shell has been removed into the fill and/or into theenvironment.
After the softgels exit the tumble dryer (primaryor rotary drying process), they are spread on totrays. The final drying phase (secondary or traydrying process) of the softgels is accomplished byinserting the stacks of trays into a drying tunnelmaintained at controlled temperature (21–248C) andlow relative humidity (20–30%) conditions.42 Thetime for the secondary drying process for softgelsmay vary from few hours to few days, depending onthe nature of the fill formulation (i.e., hydrophilicvs. lipophilic), nature of the shell formulation (e.g.,type and concentration of plasticizer(s)), thicknessof the shell, and size of the softgels. The shellformulation at the time of encapsulation process
DOI 10.1002/jps JO
typically contains water somewhere from 30 to40% w/w.34,39–41 During the encapsulation andsubsequent primary drying processes, dependingon the nature of the fill formulation encapsulated,the fill may pick up water anywhere from zeropercent to 20% w/w. During the secondary dryingprocess, the fill may loose some water leaving anamount anywhere from zero percent to 8% w/w inthe fill. The rate and extent of this water migrationprocesses in both directions are influenced by thenature of the fill formulation, nature of the shellformulation, thickness of the shell, and size of thesoftgels. The dynamics of water migration duringthe softgel drying processes is depicted in Figure 2.
The moisture content of the fill in a softgel istypically measured using a Karl–Fisher apparatus.The softgel is cut open at the seam with a knife andthe fill is collected into a syringe. The syringe iscapped tightly to prevent any moisture transferbetween the fill and the surroundings until measure-ments are completed. The moisture content of theshell is measured using a loss on drying (LOD)apparatus. The softgel is cut open at the seam with aknife and its fill contents are drained. The shell isthen given a quick wash in a suitable organic solvent,
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4112 GULLAPALLI
such as petroleum ether, and wiped clean of anyremaining fill contents with a paper towel before themeasurements are initiated.
The hardness of a softgel is measured using aBareiss hardness tester. Photographs of the Bareissdigital hardness testers are presented in Figure 3.The instrument operates by compressing the softgelunder test for a brief period (e.g., 20 s) between aplunger attached to a load cell and a platform which isautomatically raised. To test a softgel for its hardness,it is placed horizontally on the platform with its seamcontour aligned parallel to the platform and theplatform then raised enough so that the softgel is incontact with both the platform and the plunger.During the test, the platform rises automatically andthe load cell indicator displays the value of theresistance of the softgel to the compressive force.After the test period, the resistance value is displayedin Newtons (N) and represents the hardness of thesoftgel under test.41
Drying is a dynamic process and the processcontinues until the gelatin shell returns to itsequilibrium moisture content, which is in the rangeof 10–15% w/w.43 Softgels containing a lipophilic fillgenerally dry faster than those containing a hydro-philic fill and typically reach equilibrium shellmoisture within 24 h. If water migrates into the fillfrom the shell extensively, more typical of polyethy-lene glycol fills, it needs to migrate back out until thefill moisture content reaches equilibrium with themoisture content of the shell for the optimal physicalstability of a softgel product. The polyethylene glycolbased fills may take from 7 to 10 days to reach
Figure 3. Photographs of the Bareiss Hardness Testers A.Analogue hardness tester (Model HP) with the stand (Model BS61); B. Digital hardness tester (Model HPE II) with the stand(Model BS 61 II). (Courtesy of Heinrich Bareiss PruefgeraetebauGmbH.) http://www.bareiss.de/english/produkte/pruefgeraete_start.html.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
acceptable moisture levels and may still contain up to10% water after drying.
Softgels permitted to come to moisture equilibriumat the controlled temperature (21–248C) and relativehumidity (20–30%) conditions of the secondary dryingprocess are considered ‘‘dry.’’42 The shell material ofsuch ‘‘dry’’ softgels usually contains 8–16% w/w waterdepending on the specific gelatin shell formulationused. The range of water content between 7.6% and14% in gelatin was hypothesized to correspond to thewater sorbed by the polar groups in gelatin or thestructural water, which is bound with the proteins byhydrogen bonding both inside and outside the helicalfragments.44,45 The fill material of the ‘‘dry’’ softgelsusually contains as high as 6–10% w/w water for asimple polyethylene glycol 400 based formulation toless than 0.5% w/w water for a medium chaintriglyceride (MCT) or a long chain triglyceride(LCT) oil based formulation (Fig. 2).
After the softgels are ‘‘dry,’’ they may be subjectedto an additional step, known as stress relieving step ortempering step, to improve the overall quality of asoftgel product.42 The step involves a change in theconditions of temperature and relative humidity fromthe secondary drying step and is accomplished at atemperature range of 32–388C and a relative humid-ity range of 35–60%. The stress relieving step cantake place in the same tunnel as the secondary dryingstep, and thus requires no additional equipment orlabor. The step is intended to remove any dimplespresent in the shell and any bubbles present in the fill.In addition, the step also reduces the dimensionalstandard deviation thereby resulting in more dimen-sionally uniform batches of softgels.42 The dimen-sional uniformity of a softgel product is typicallymeasured by the standard deviation in the lengthwiseand widthwise measurement of an oblong softgel orthe diameter wise of a round softgel.
The rate and extent of softgel drying are the criticalprocessing parameters that should be carefullycontrolled. Removal of too much water may resultin hard, brittle softgels that have a higher propensityto develop cracked shells and/or require a longer timefor dissolution. On the other hand, insufficient dryingmay result in very soft softgels that become tacky and/or tightly stick to each other with time. If the softgelsare subjected to rapid drying conditions, such as highdrying temperature, very low relative humidity, and/or high airflow, the product may under go aphenomenon referred to as ‘‘case hardening.’’46 Casehardening occurs when the exterior surface of thesoftgel dries very rapidly and forms a temporary sealthat prevents further egress of moisture from the filland shell. The hardness of such a softgel increasestemporarily reaching an acceptable value. However,the excess moisture entrapped within the fill andshell migrates slowly during storage, resulting in a
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4113
very soft softgel product. An acceleration of the dryingprocess can also potentially lead to considerablechanges in the structure and properties of the shellmaterial. Though the presence of a plasticizer in theshell formulation may mitigate these deleteriouseffects of the faster drying conditions to some extent,the drying process should be carefully controlled tominimize its effects on the thermal and mechanicalproperties of the shell material.47
Finishing
After drying, the softgels are sorted (sized),48
polished, printed,49 and inspected for their quality.The softgels are then packaged into suitable contain-ers, typically of low density polyethylene (LDPE)bags, high density polyethylene (HDPE) bottles, orblisters. The recommended storage conditions for thesoftgels include a temperature range of 15–308C and arelative humidity of not more than 50%.43,50 Whenstored under these conditions, the equilibriummoisture content of the shell material and oxygenpermeability through the material are minimal, thusimproving the stability of the softgel product.43
The readers are directed to the review by Wilkinsonand Hom51 to obtain an extensive understanding ofthe design of a softgel manufacturing facility andequipment used in the softgel manufacturing.
FILL FORMULATIONS
Types of fill formulations that can be delivered usingsoftgel delivery system include: solutions, suspen-sions, emulsions, microemulsions, self-emulsifyingdrug delivery systems (SEDDS), and self-microemul-sifying drug delivery systems (SMEDDS). The con-sistency of a fill formulation may vary from a freeflowing liquid (e.g., Rocaltrol1 Softgels, RochePharmaceuticals; Hectorol1 Softgels, Genzyme Cor-poration) to a thick suspension (e.g., Zantac1 Soft-gels, GlaxoSmithKline; Prometrium1 Softgels,Solvay Pharmaceuticals). In the case of self-micro-emulsifying (SMEDDS) and self-emulsifying(SEDDS) formulations, a compound dissolved in alipophilic vehicle containing one or more emulsifiersand cosolvents, forms a microemulsion (droplet size�0.15mm) when the ratio of emulsifier to lipid is high(>1) or a fine emulsion (droplet size >0.15mm) whenthe ratio is <1, respectively, upon dilution withaqueous fluids in vitro or in vivo.52
Compounds belonging to class II and class IV ofthe Biopharmaceutics Classification System (BCS)show extremely low aqueous solubility throughoutthe physiological pH range, resulting in low andinconsistent bioavailability.53 However, when such acompound demonstrates enhanced solubility in anonaqueous vehicle, softgel delivery system may be
DOI 10.1002/jps JO
promising in improving its bioavailability. A vehiclefor the development of a softgel fill formulation shouldideally satisfy the following criteria:
� P
URNA
harmaceutically acceptable for oral use.
� S ufficient solubilizing capacity to dissolve agiven dose in a small volume.
� Y ield a fill formulation that is stable and compa-tible with gelatin shell material.
� Y ield a fill formulation that is easier to compoundand encapsulate.
� P revent precipitation of the solubilized com-pound within the softgel product during its man-ufacturing and shelf-life, and upon contact withthe aqueous environment in vitro (dissolution)and in vivo (GIT).
Vehicles suitable for encapsulation into softgels canbe broadly classified into two groups:
A. H
L
ydrophilic vehicles.
B. L ipophilic vehicles (lipid based fill formulations).Hydrophilic Vehicles
Hydrophilic vehicles for softgel fill formulationsinclude polyethylene glycols (e.g., PEG 400, PEG600), methoxypolyethylene glycols (e.g., MPEG 350,MPEG 550), diethyleneglycol monoethyl ether(Transcutol1), tetrahydrofurfurylalcohol polyethy-lene glycol (Glycofurol), propylene carbonate, N-methyl-2-pyrrolidone (NMP), polyoxyethylene–poly-oxypropylene copolymers (Poloxamers), propyleneglycol, glycerin, ethyl alcohol, and water. The use ofpropylene glycol, glycerin, and water is restricted toless than 10% of the total fill formulation, as thesevehicles can also act as plasticizers for the gelatinshell.54 Similarly, use of lower molecular weightpolyethylene glycols (e.g., PEG 200, PEG 300) in thefill formulations is limited due to their ability todiffuse into the shell and thereby act as gelatinplasticizers.55–57 The extent of diffusion of a poly-ethylene glycol from the fill into the shell decreaseswith an increase in its molecular weight.40,41 The useof volatile components, such as ethyl alcohol, in the fillformulations is limited due to their ability to rapidlydiffuse through the shell material, and carrying outother fill components in the process.54,58–60
Pros and Cons of Use of Polyethylene Glycols in Softgels
Polyethylene glycols, due to their ability to bemiscible with aqueous fluids in all proportions anddissolve many pharmaceutical compounds at thesame time make them ideal vehicles for the delivery ofmany poorly soluble compounds in softgels. Com-pounds with poor bioavailability and considerable
OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
4114 GULLAPALLI
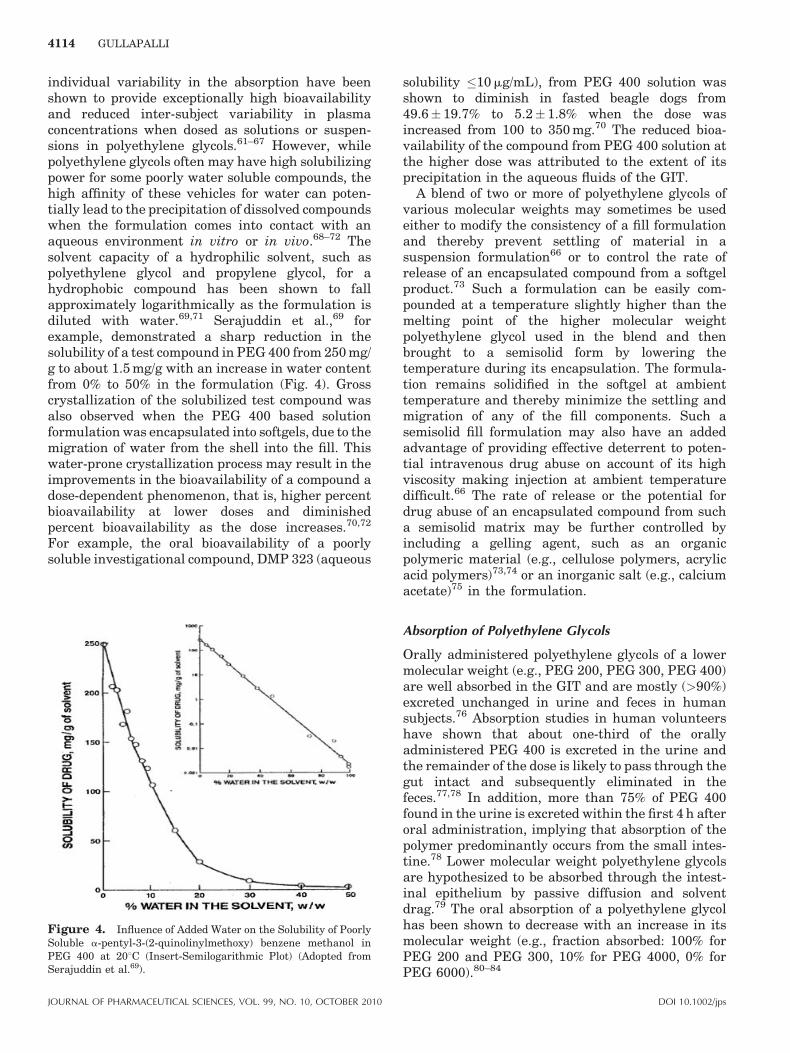
individual variability in the absorption have beenshown to provide exceptionally high bioavailabilityand reduced inter-subject variability in plasmaconcentrations when dosed as solutions or suspen-sions in polyethylene glycols.61–67 However, whilepolyethylene glycols often may have high solubilizingpower for some poorly water soluble compounds, thehigh affinity of these vehicles for water can poten-tially lead to the precipitation of dissolved compoundswhen the formulation comes into contact with anaqueous environment in vitro or in vivo.68–72 Thesolvent capacity of a hydrophilic solvent, such aspolyethylene glycol and propylene glycol, for ahydrophobic compound has been shown to fallapproximately logarithmically as the formulation isdiluted with water.69,71 Serajuddin et al.,69 forexample, demonstrated a sharp reduction in thesolubility of a test compound in PEG 400 from 250 mg/g to about 1.5 mg/g with an increase in water contentfrom 0% to 50% in the formulation (Fig. 4). Grosscrystallization of the solubilized test compound wasalso observed when the PEG 400 based solutionformulation was encapsulated into softgels, due to themigration of water from the shell into the fill. Thiswater-prone crystallization process may result in theimprovements in the bioavailability of a compound adose-dependent phenomenon, that is, higher percentbioavailability at lower doses and diminishedpercent bioavailability as the dose increases.70,72
For example, the oral bioavailability of a poorlysoluble investigational compound, DMP 323 (aqueous
Figure 4. Influence of Added Water on the Solubility of PoorlySoluble a-pentyl-3-(2-quinolinylmethoxy) benzene methanol inPEG 400 at 208C (Insert-Semilogarithmic Plot) (Adopted fromSerajuddin et al.69).
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
solubility �10mg/mL), from PEG 400 solution wasshown to diminish in fasted beagle dogs from49.6� 19.7% to 5.2� 1.8% when the dose wasincreased from 100 to 350 mg.70 The reduced bioa-vailability of the compound from PEG 400 solution atthe higher dose was attributed to the extent of itsprecipitation in the aqueous fluids of the GIT.
A blend of two or more of polyethylene glycols ofvarious molecular weights may sometimes be usedeither to modify the consistency of a fill formulationand thereby prevent settling of material in asuspension formulation66 or to control the rate ofrelease of an encapsulated compound from a softgelproduct.73 Such a formulation can be easily com-pounded at a temperature slightly higher than themelting point of the higher molecular weightpolyethylene glycol used in the blend and thenbrought to a semisolid form by lowering thetemperature during its encapsulation. The formula-tion remains solidified in the softgel at ambienttemperature and thereby minimize the settling andmigration of any of the fill components. Such asemisolid fill formulation may also have an addedadvantage of providing effective deterrent to poten-tial intravenous drug abuse on account of its highviscosity making injection at ambient temperaturedifficult.66 The rate of release or the potential fordrug abuse of an encapsulated compound from sucha semisolid matrix may be further controlled byincluding a gelling agent, such as an organicpolymeric material (e.g., cellulose polymers, acrylicacid polymers)73,74 or an inorganic salt (e.g., calciumacetate)75 in the formulation.
Absorption of Polyethylene Glycols
Orally administered polyethylene glycols of a lowermolecular weight (e.g., PEG 200, PEG 300, PEG 400)are well absorbed in the GIT and are mostly (>90%)excreted unchanged in urine and feces in humansubjects.76 Absorption studies in human volunteershave shown that about one-third of the orallyadministered PEG 400 is excreted in the urine andthe remainder of the dose is likely to pass through thegut intact and subsequently eliminated in thefeces.77,78 In addition, more than 75% of PEG 400found in the urine is excreted within the first 4 h afteroral administration, implying that absorption of thepolymer predominantly occurs from the small intes-tine.78 Lower molecular weight polyethylene glycolsare hypothesized to be absorbed through the intest-inal epithelium by passive diffusion and solventdrag.79 The oral absorption of a polyethylene glycolhas been shown to decrease with an increase in itsmolecular weight (e.g., fraction absorbed: 100% forPEG 200 and PEG 300, 10% for PEG 4000, 0% forPEG 6000).80–84
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4115
Influence of PEG 400 on GIT Motility and Absorption ofCompounds
Mechanistically, PEG 400 was shown to have aconcentration-dependent effect on gastrointestinalmotility and transit.78,85,86 Radiolabeled studies havesuggested that the mean gastric residence time(MGRT) of an aqueous vehicle containing PEG 400was similar to that of an aqueous vehicle free fromPEG 400 (MGRT of about 20 min). The mean smallintestinal transit time (MSITT), in contrast, wasshown to decrease with an increase in the concentra-tion of PEG 400 in the aqueous vehicle (MSITT of153 min at 10 g PEG 400 dose in aqueous vehicleversus 236 min for PEG 400 free aqueous vehicle).85
The mechanism behind the influence of PEG 400 onthe intestinal transit of a coadministered liquidformulation was attributed to the incomplete absorp-tion of the polymer from the intestine and theresulting polymer induced osmotic activity. PEG400 would probably increase the luminal fluid volumevia the retention of water, which in turn stimulatesintestinal motility and hence transit.87 Since thesmall intestine is the primary site for absorption ofmany compounds, a reduction in the contact timewith this region of the GIT could potentially impactthe bioavailability of orally administered compounds.The absorption of low permeability compounds (i.e.,BCS class III and class IV) is likely to be the mostsusceptible to changes in intestinal residence time.88
PEG 400 at higher levels (�2.5 g), for example, wasshown to reduce the small intestinal residence time ofa liquid formulation, resulting in a significantreduction in the oral bioavailability of ranitidine,dissolved within.78,85,86 However, at a lower concen-tration (1 g), PEG 400 was shown to significantlyenhance the absorption of ranitidine, possibly viamodulation of intestinal permeability.86 PEG 400 wasalso thought to have a significant impact on effluxprocess via P-gp transporter inhibition and metabo-lism of some drugs during their migration throughthe GIT.89
Solubility Enhancers for Hydrophilic Vehicles
It is desirable to produce a highly concentratedsolution of a compound as it allows the encapsulationof a unit dose of the compound in a softgel that issmall enough to swallow easily and thus improvingpatient acceptance of the medication. Yu et al.,90
Morton et al.,91 and Shelley et al.92 inventedmethods for enhancing the solubility and producinghighly concentrated solutions for acidic, basic, andamphoteric compounds in hydrophilic vehicles sui-table for filling softgels (referred to as enhancedsolubility system or ESS).93 These inventions allowthe improvement in the solubility of some compoundsin polyethylene glycols by 40–400% using an ionizing
DOI 10.1002/jps JO
agent (i.e., counter-ion; neutralizing agent). Forexample, the solubility of acidic compounds (e.g.,ibuprofen, naproxen, indomethacin, acetaminophen)in polyethylene glycols can be enhanced throughpartial ionization of these compounds with a hydro-xide ion species (e.g., sodium hydroxide, potassiumhydroxide, ammonium hydroxide). Whereas, thesolubility of basic compounds (e.g., thioridazine,cimetidine, ranitidine, nifedipine) can be enhancedthrough partial ionization with a hydronium ionspecies (e.g., hydrochloric acid, hydrobromic acid,sulfuric acid, an organic acid). For amphotericcompounds, either hydroxide ion or hydroniumion sources may be utilized to effect enhancedsolubilization.
The solubility enhancing techniques employedby Yu et al.,90 Morton et al.,91 and Shelley et al.92
result in a softgel fill formulation containing acompound as a mixture of its unionized form (freeacid or base) and its corresponding ionized form (i.e.,salt form). When used these neutralization (counterion) techniques to obtain a highly concentratedsolution of a compound, it is essential to keep theapparent pH of the final fill formulation at leastbetween 2.5 and 7.5. At pH values below 2.5, gelatin ishydrolyzed94 causing leakage of the softgel, whereasat pH values above 7.5, gelatin may be eitherhydrolyzed94 or tanned (i.e., cross-linked) resultingin decreased solubility of the gelatin shell.33 Inaddition, the ionizing agents used as solubilityenhancers contain a highly reactive species whichmay react adversely with other ingredients present inthe softgel.95 The use of a highly reactive species, suchas hydroxide ion, may be substituted with a milderand relatively neutral salt, such as ammoniumacetate,96 an alkali metal acetate,92 or a combinationof alkali metal acetate/lactate,97 in enhancing thesolubility of acidic compounds that is more compatiblewith the other softgel components.
Alternately, the solubility of some compounds(e.g., acetaminophen, danazol, ibuprofen) in hydro-philic vehicles can also be improved significantly byusing povidone (polyvinylpyrrolidone, PVP) as asolubility enhancer.55,56,67,98 Unlike the solubilityenhancing techniques employed by Yu et al.,90
Morton et al.,91 and Shelley et al.,92 the use ofpovidone as a solubility enhancer results in a softgelfill formulation containing a compound in its originalform that is very compatible with the other softgelcomponents. In addition, as povidone is available ina variety of molecular weights ranging from 2500 to3000000,99 the viscosity of a fill formulation can becontrolled through the selection of appropriatemolecular weight and concentration of the polymerwithout adversely affecting the solubility of dis-solved compounds.98 An advantage of using a higheramount of a lower molecular weight povidone as a
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4116 GULLAPALLI
solubility enhancer is the reduction in the amountpolyethylene glycol available in the fill formulationfor any potential reactions with acidic compoundssuch as ibuprofen free acid (e.g., esterificationreactions), a well known disadvantage that resultsin the reduction of available ibuprofen in its freeform.100 Use of povidone of a lower molecular weightalso yields a fill formulation of a lower viscosity andthus improving the product manufacturability anddissolution characteristics.98
Lipophilic Vehicles (Lipid Based Fill Formulations)
Lipophilic vehicles for softgel fill formulations includefree fatty acids (e.g., oleic acid), fatty acid esters ofhydroxyl compounds, such as ethyl alcohol, propyleneglycol, glycerin, sorbitol, sucrose, polyethylene gly-cols, and polyethoxylated fatty acid esters. The fattyacid composition of these esters may vary from shortchain (SC, <C8) to medium chain (MC, C8-C10) tolong chain (LC, �C12).
Classification of Lipid Based Fill Formulations
Lipid based fill formulations generally comprise of acompound dissolved or suspended in one or moreexcipients consisting of triglycerides (TG), mixedglycerides, surfactants, and cosolvents. The simplestlipid formulation is one in which a compound isdissolved in a digestible oil, usually a long chaintriglyceride (LCT, e.g., vegetable oil) or a mediumchain triglyceride (MCT, e.g., fractionated coconutoil). These formulations may be appropriate forpotent compounds and/or highly lipophilic com-pounds (log P> 4) and require digestion of theformulation before absorption.71,101,102 Some com-pounds may have limited solubility in triglyceridesor may yield lower bioavailability when dosed inthese formulations.103,104 The solvent capacity oftriglyceride vehicles for some compounds may beimproved by blending them with other mixedglycerides, that is, diglycerides (DG) and monogly-cerides (MG), and free fatty acids (FA). Theadvantage of using these mixed glycerides is thatthese components are similar to the natural diges-tion products of the triglycerides and do not interferewith the regular lipid digestion and absorptionprocesses. These triglyceride and mixed glyceridesformulations are referred to as type I under lipidformulation classification system (LFCS), proposedby Pouton71,101,102 and Porter et al.105 The solubi-lization of a compound in a type I lipid formulationmay sometimes be improved with the inclusion of alipophilic surfactant (HLB< 12). This approach isused primarily to promote the emulsification of aformulation vehicle in the aqueous fluids of the GITunder gentle agitation. This type of lipid formulationis likely to retain its solvent capacity for the
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
dissolved compound after dispersion in the aqueousfluids. These formulations are referred to as type IIunder LFCS. In contrast, a type I lipid formulationmay include a hydrophilic surfactant (HLB> 12)and/or a water soluble cosolvent (e.g., propyleneglycol, polyethylene glycol, ethyl alcohol) to increasethe solvent capacity of the formulation for acompound. These formulations are referred to astype III under LFCS. A type IV formulationcomprises predominantly of a hydrophilic cosolventand a surfactant, and with or without minimal oilcomponents.
The digestion (lipolysis) of a triglyceride withinthe GIT by the pancreatic lipase/colipase complexinto amphiphilic diglycerides, monoglycerides, andfree fatty acids can enhance the dissolution rate of apoorly soluble compound coadministered with thevehicle (the subject will be discussed in more detailin Lipid Digestion and Absorption and Influence onAbsorption of Compounds Section). However, thepoor miscibility of the undigested triglyceride withthe GIT aqueous environment may lead to highlyvariable gastric emptying and/or dispersion into anemulsion which in turn, can result in variableabsorption of the compound from the GIT.106 Thedispersibility of the triglyceride in the GIT fluids canbe enhanced by including a surfactant in theformulation. Interestingly, Lacy et al.,107 usingin vitro digestion experiments, demonstrated thatthe lipolysis of a triglyceride might be retarded by ahydrophilic surfactant (i.e., HLB >10) (e.g., Cremo-phor RH 40, Cremophor EL, Polyoxyethylenesorbitan monooleate), typically used in the lipidbased fill formulation for a poorly soluble compound(type III formulation). Lacy et al.107 further demon-strated that the inhibitory effect of a hydrophilicsurfactant on the in vitro lipolysis of a triglyceridecould be countered substantially by the inclusion of alipophilic surfactant (i.e., HLB< 10) in the formula-tion. Some examples of such beneficial lipophilicsurfactants include, free fatty acids (e.g., oleic acid,linoleic acid, linolenic acid), mono- and/or di-glycer-ides (e.g., glyceryl mono- and di-caprylate/caprate,distilled acetylated monoglycerides), sorbitan fattyacid esters (e.g., sorbitan monolaurate, sorbitanmonooleate), and polyglycerol esters of fatty acids(e.g., polyglyceryl oleate).
Few examples of currently marketed lipid basedformulations delivered using softgel technology areincluded in Table 2. The list includes such wellknown products as Cyclosporine A (Sandimmune1
[type II] and Neoral1 [type III]; Novartis Pharma-ceuticals, Australia), Ritonavir (Norvir1; AbbottLaboratories), Lopinavir/Ritonavir (Kaletra1; AbbottLaboratories), Saquinavir (Fortovase1; Roche Phar-maceuticals), Amprenavir (Agenerase1 [type IV];GlaxoSmithKline), Calcitriol (Rocaltrol1 [type I];
DOI 10.1002/jps

Table 2. Examples of Softgel Products with Their Fill Compositions and Dissolution Methods
Drug Product Information Fill Excipients Properties, Dissolution Method
Hydrophilic fillsAgenerase1, Amprenavir, 50and 150 mg, GlaxoSmithKline,NDA 21-007
PEG 400, D-a-tocopheryl polyethyleneglycol 1000 succinate (TPGS),
propylene glycol
HIV-protease inhibitor, MW 505.63,Aq. Sol. 40mg/mL, clog P 3.29306,BCS Class II307
Softgel-introductory dosage form.Amprenavir softgel product waslater reformulated into a tabletformulation (Lexiva1, NDA 21-548)containing its phosphate esterprodrug, fosamprenavir calcium,with improved aqueous solubility
Apparatus I at 50 rpm in 900 mLof 0.1 N HCl at 37� 0.58Ca
Targretin1, Bexarotene,75 mg, Eisai/Ligand,
PEG 400, polysorbate 20, povidoneK-90, butylated hydroxyanisole (BHA)
Antineoplastic, MW 348.48,Aq. Sol. insoluble
NDA 21-055 Softgel-introductory dosage formApparatus II at 50 rpm in
900 mL of 0.05 M phosphatebuffer, pH 7.5 containing 0.5%hexadecyltrimethylammoniumbromide (HDTMA) at 37� 0.58Ca
Lanoxicap1, Digoxin, 0.05,0.1, and 0.2 mg, GlaxoSmithKline,
PEG 400, ethyl alcohol,propylene glycol
Heart failure, cardiotonic, MW 780.94,Aq. Sol. 10mg/mL, BCS Class I308,309
NDA 18-118 More complete absorption of digoxinfrom soft capsules and recommendedoral doses are only 80% of those fortablets and elixir
Apparatus I at 120 rpm in 600 mL ofwater (37� 0.58C)b
Zarontin1, Ethosuximide,250 mg, Pfizer, NDA 12-380
PEG 400 Anticonvulsant, MW 141.17, Aq. Sol.freely soluble
Softgel-introductory dosage formApparatus I at 50 rpm in 900 mL of
phosphate buffer, pH 6.8 at 37�0.-0.58Cc
VePesid1, Etoposide, 50 mg,Bristol-Myers-Squibb,
PEG 400, glycerin, water, citric acid Antineoplastic, MW 588.56, Aq. Sol.0.20 mg/mL310
NDA 19-557 Softgel-introductory dosage formApparatus II at 50 rpm in 900 mL
of acetate buffer, pH 4.5 at 37�0.-0.58Cc
Advil1, ibuprofen, 200 mg, PEG 600, potassium hydroxide, Anti-inflammatoryWyeth, NDA 20-402 water, (ibuprofen is present as
free acid/potassium salt)MW 206.28, Aq. Sol. 10mg/mL,
BCS Class II308,309
Initial launch as tablet (NDA 18-989);softgel as line-extension
Apparatus I at 150 rpm in 900 mL ofphosphate buffer, pH 7.2 at 37�0.-0.58Ca
Ibuprofen Capsules, Ibuprofen,200 mg, Banner Pharmacaps,Inc., NDA 21-472
PEG 600, D-a-tocopherylpolyethylene glycol 1000 succinate
(TPGS), povidone, (ibuprofen ispresent as free acid)
Advil PM Liqui-Gels1, Ibuprofen200 mg and, Diphenhydramine HCl25 mg, Wyeth, NDA 21-393
PEG 600, potassium hydroxide,water, (ibuprofen is present as
free acid/potassium salt)
Anti-inflammatory/antihistaminic,sedative, hypnotic, MW 206.28 and291.82, Aq. Sol. 10mg/mL and1 g/mL, BCS Class II308,309
Apparatus I at 100 rpm in 900 mLof 200 mM phosphate buffer,pH 7.2 at 37�0.58Ca
Aleve1, Naproxen sodium, 220 mg,Bayer HealthCare, NDA 21-920
PEG 400, propylene glycol,povidone, lactic acid
Anti-inflammatory,MW 252.24, soluble
(Continued)
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
SOFT GELATIN CAPSULES 4117

Table 2. (Continued )
Drug Product Information Fill Excipients Properties, Dissolution Method
Initial launch as tablet (NDA 17-581;as free acid); softgel as second-line
Apparatus II at 50 rpm in 900 mLof 0.1 M phosphate buffer (pH 7.4),at 37�0.58Cd
Procardia1, Nifedipine, 10 and20 mg, Pfizer, NDA 18-482
PEG 400, glycerin, peppermintoil, sodium saccharin
Antianginal, MW 346.33, Aq. Sol.12mg/mL, BCS Class II308,309,311
Softgel-introductory dosage form,tablet as line-extension(NDA 19-684; higher doses)
Apparatus II at 50 rpm in 900 mLof SGF at 37�0.58Cc
Nimotop1, Nimodipine, 30 mg,Bayer HealthCare, NDA 18-869
PEG 400, glycerin, peppermintoil, water
Vasodilator, MW 418.44, Aq. Sol.4mg/mL, BCS Class II312
Softgel-introductory dosage formApparatus II at 50 rpm in 900 mL
of water, containing 0.5% sodiumdodecyl sulfate at 37� 0.58Ca
Hytrin1, Terazosin HCl,1, 2, 5, and 10 mg, Abbott
PEG 400, glycerin, povidone Benign prostatic hyperplasia (BPH),MW 459.92, Aq. Sol. 24.2 mg/mL
Laboratories, NDA 20-347 Initial launch as tablet (NDA 19-057);softgel as line-extension
Navelbine1, Vinorelbine tartrate,20, 30, and 80 mg, PierreFabre, NDA 20-388
PEG 400, ethanol, glycerol, water NSCLC, breast cancer, multiplemyeloma, MW 1079.11, Aq.Sol. >1000 mg/mL
Lipophilic solution fillsOne Alpha1, Alfacalcidol,0.25mg, 0.5mg, and 1mg,LEO Pharma, Ex-US
Sesame oil, dl-alpha-tocopherol Renal osteodystrophy,hyperparathyroidism,hypocalcaemia, rickets,MW 400.64, Aq. Sol. insoluble
Rocaltrol1, Calcitriol, 0.25mgand 0.5mg, Roche Pharmaceuticals,
Fractionated coconut oil, butylatedhydroxyanisole (BHA), butylated
Calcium regulator, MW 416.64,Aq. Sol. insoluble
NDA 18-044 hydroxytoluene (BHT) Softgel-introductory dosage formUSP rupture testd
Hectorol1, Doxercalciferol, 0.5, 1,and 2.5mg, Bone Care Int.,
Fractionated coconut oil, ethyl alcohol,butylated hydroxyanisole (BHA)
Antihyperparathyroidism, MW 412.65,Aq. Sol. insoluble
Inc., NDA 20-862 Softgel-introductory dosage formUSP rupture testd
Marinol1, Dronabinol, 2.5, 5,and 10 mg, Unimed andRoxane, NDA 18-651
Sesame oil Anorexia, nausea, MW 314.46,log P 3.78
Softgel-introductory dosage formApparatus II at 100/150 rpm in
500 mL of water, containing 10%Labrasol (37�0.58C)a. Plus USPrupture test,c,d
Avodart1, Dutasteride, 0.5 mg,GlaxoSmithKline, NDA 21-319
Medium chain mono- and diglycerides,butylated hydroxytoluene (BHT)
Benign prostatic hyperplasia (BPH),MW 528.53, Aq. Sol.<0.038 ng/mL,log P 5.09
Softgel-introductory dosage formApparatus II at 50 rpm in 900 mL
of 0.1 N HCl containing 2% sodiumdodecyl sulfate at 37� 0.58Ca
Drisdol1, Ergocalciferol, 50000IU, Sanofi-Aventis, NDA 3-444
Soybean oil Parathyroid disease, refractoryrickets, MW 396.65, Aq. Sol. insolu-ble
Softgel-introductory dosage formUSP disintegration testc
Claritin1, Loratadine, 10 mg,Schering-Plough, NDA 21-952
Caprylic/capric glycerides,polysorbate 80, povidone
Antihistamine, MW 382.88, Aq.Sol. insoluble
Initial launch as tablet (NDA 19-658);softgel as line-extension
(Continued)
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010 DOI 10.1002/jps
4118 GULLAPALLI

Table 2. (Continued )
Drug Product Information Fill Excipients Properties, Dissolution Method
Amitiza1, Lubiprostone, 24mg,Sucampo and Takeda,NDA 21-908
Medium chain triglycerides Chronic idiopathic constipation,MW 390.46, Aq. Sol. insoluble
Softgel-introductory dosage formZemplar1, Paricalcitol, 1, 2,and 4mg, Abbott Laboratories,
Medium chain triglycerides,ethyl alcohol, butylated
Antihyperparathyroidism, MW 416.64,Aq. Sol. insoluble, BCS Class IV
NDA 21-606 hydroxytoluene (BHT) Softgel-introductory dosage formFortovase1, Saquinavir, 200 mg,Roche Pharmaceuticals,NDA 20-828
Medium chain mono- and diglycerides,povidone, DL-a-tocopherol
HIV-protease inhibitor, MW 670.84,Aq. Sol. 2.2 mg/mL, log P 2.73, clog P4.73, BCS Class IV305–307
Fortovase1 softgel product wasreplaced with tablet (Invirase1
500 mg) and powder filled HGCcapsule (Invirase1 200 mg)formulations containing itsmesylate salt, with reduceddosing (NDA 21-785 and NDA 20-6-28)
Apparatus II at 50 rpm in 900 mLof citrate buffer containing 0.582%anhydrous dibasic sodium phosphateand 1.67 g citric acid monohydrateat 37�0.58Cc
Andriol1, Testosteroneundecanoate, 40 mg,Organon, EX-US
Castor oil, propylene glycolmonolaurate
Hypogonadism, MW 456.70,Aq. Sol. insoluble
Depakene1, Valproic acid,250 mg, Abbott Laboratories,
Corn oil Antiepileptic, MW 144.21, Aq. Sol.1.3 mg/mL, BCS Class II309
NDA 18-081 Softgel-introductory dosage formApparatus II at 50 rpm in 900 mL
of SIF TS without enzyme and withmonobasic sodium phosphate (inst-ead of monobasic potassium phos-phate)and pH adjusted to 7.5 with 5 M so-dium hydroxide; containing 0.5% s-odiumdodecyl sulfate at 37� 0.58Cc
Lipophilic suspension fillsToctino1, Alitretinoin, 10and 30 mg, BasileaPharmaceutica AG, Ex-US
Refined soya-bean oil, partiallyhydrogenated soya-bean oil, mediumchain triglycerides, yellow beeswax,
All-rac-a-tocopherol
Atopic dermatitis, MW 300.44,Aq. Sol. insoluble
Symmetrel1, AmantadineHCl, 100 mg, NovartisPharmaceuticals, Ex-US
Rape seed oil, soybean lecithin,blend white beeswax and
hydrogenated soya and vegetable oils
Infections, influenza, Parkinson’s,MW 187.71, Aq. Sol. freely soluble
Lamprene1, Clofazimine, 50and 100 mg, NovartisPharmaceuticals,NDA 19-500
Rapeseed oil, hydrogenated soybeanoil, partially hydrogenated vegetable
oils, propylene glycol, beeswax,butylated hydroxytoluene (BHT),
soybean lecithin, p-methoxyacetophenone, sodium ethyl
paraben, sodium propyl paraben
Antileprosy, MW 473.40, Aq. Sol.0.49mg/mL, log P 4.36, BCS ClassII/IV307,313
Softgel-introductory dosage form
USP rupture testd
Accutane1, Isotretinoin, 10, 20, Soybean oil, beeswax, hydrogenated Antiachne, MW 300.44, log P¼ 6.6117
and 40 mg, Roche Pharmaceuticals, soybean oil flakes, hydrogenated Softgel-introductory dosage formNDA 18-662 vegetable oil, butylated hydroxyanisole
(BHA), edetate disodiumApparatus I at 100 rpm in 900 mL
of 0.05 M potassium phosphatedibasic buffer, pH 7.8 containing0.5% lauryldimethylamine-oxide(LDAO) at 37�0.58Ca
(Continued)
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
SOFT GELATIN CAPSULES 4119

Table 2. (Continued )
Drug Product Information Fill Excipients Properties, Dissolution Method
Glakay1, Menatetrenone, 15 mg,Eisai Co., Ex-US
L-Aspartic acid, carnauba wax,hydrogenated oil, ethyl
parahydroxybenzoate, propylparahydroxybenzoate, propylene
glycol esters of fatty acids,glyceryl monooleate
Osteoporosis, MW 444.65, Aq.Sol. insoluble
Prometrium1, Progesterone,100 and 200 mg, Solvay/Schering,NDA 19-781
Peanut oil, lecithin Hormone, MW 314.46, Aq. Sol.insoluble, log P 3.78305
Softgel-introductory dosage formUSP rupture testd
Zantac1, Ranitidine HCl, 150and 300 mg, GlaxoSmithKline,NDA 20-095
Medium chain triglycerides,mixed glycerides of long chainfatty acids (Gelucire1 33/01)
Treatment of ulcers, MW 350.86,Aq. Sol. soluble
Withdrawn from marketApparatus II at 50 rpm in 900 mL
of water at 37�0.58CSelf-emulsifying (SEDDS) and
self-microemulsifying (SMEDDS) fillsSandimmune1, CyclosporinA, 25, 50, and 100 mg, NovartisPharmaceuticals, NDA 50-625
Corn oil, ethyl alcohol, linoleoylmacrogolglycerides
Immunosuppressant MW 1202.61,Aq. Sol. 40mg/mL, LogP 2.92, BCSClass IV305,314,315
Neoral1, Cyclosporin A, 25 and100 mg, Novartis Pharmaceuticals,NDA 50-715
Corn oil-mono- and di-triglycerides,ethyl alcohol, polyoxyl 40
hydrogenated-castor oil, propyleneglycol, DL-a-tocopherol
Apparatus II at 75 rpm in 500 mL (for25 mg dose) or 1000 mL (for 100 mgdose) of 0.1 N HCl containing 2 mg/mL (for 25 mg dose) or 4 mg/mL (for100 mg dose) of N,N-dimethydode-cylamine N-oxide at 37� 0.58Ca, plusUSP rupture test,c,d
Cyclosporine Capsules, Cyclosporin A,25 and 100 mg, Eon Labs Mfg., Inc.,ANDA 65-017
PEG 400, ethyl alcohol, D-a-tocopherylpolyethylene glycol 1000 succinate(TPGS), polyoxyl 40 hydrogenated-
castor oilKaletra1, Lopinavir 133.3 mg,Ritonavir 33.3 mg, AbbottLaboratories, NDA 21-226
Oleic acid, polyoxyl 35 castor oil,propylene glycol
HIV-protease inhibitor, lopinavir-MW628.80, Aq. Sol. 40mg/mL, clog P 6.-09, BCS Class IV306,307,316
Softgel-introductory dosage formKaletra1 softgel product was
replaced with a tablet formulationwith increased drug loading(strength—200 mg/50 mg) and withreduced food effects (NDA 21-906)
Apparatus II at 50 rpm in 900 mLof 10 mM sodium phosphate mono-basic solution containing 0.05 Mpolyoxyethylene 10 lauryl ether,pH 6.8 at 37�0.58Ca
Norvir1, Ritonavir, 100 mg, Oleic acid, ethyl alcohol, polyoxyl 35 HIV-protease inhibitorAbbott Laboratories, NDA 20-659 castor oil, butylated hydroxytoluene
(BHT)MW 720.94, Aq. Sol. 1mg/mL,
clog P 4.94, BCS Class IV306,309,311
Softgel-introductory dosage formApparatus II at 50 rpm in 900 mL
of 0.1 N HCl, containing 0.025 Mpolyoxyethylene 10 lauryl etherat 37�0.58Ca
Information on fill excipients was collected from label, prescription information, or available FDA approval packages; information onindication, molecular weight (MW), and aqueous solubility was collected from The Merck Index, 2006 or otherwise referenced.
aFDA Website—http://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_SearchResults_Dissolutions.cfm?PrintAll¼1.bBritish Pharmacopeia, 1998.cUSP/NF Monographs.dUS Pharmacopeia, 2009.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010 DOI 10.1002/jps
4120 GULLAPALLI

SOFT GELATIN CAPSULES 4121
Roche Pharmaceuticals), and Isotretinoin (Accu-tane1; Roche Pharmaceuticals).
Characteristics of Lipid Based Fill Formulations
A type II formulation yields a broader particle sizedistribution in micron size upon aqueous dilution(SEDDS), whereas a type III formulation yields aclear or almost clear dispersion with micron (SEDDS)or submicron (SMEDDS) size distribution. A distinc-tion between type III and type II lipid formulations isthat the water soluble components in the formerformulation will tend to part from the oil duringdispersion and become dissolved in the aqueousphase. Pouton71 speculated that the forces involvedin this phase separation may be the driving force forthe emulsification of a type III formulation and coinedthe phrase ‘‘diffusion and stranding’’ to describe theprocess. In contrast, the lipophilic surfactant in a typeII formulation would less likely disperse extensivelyinto the aqueous phase and promote the emulsifica-tion of glycerides in the aqueous phase. Theunfavorable consequence of type III and also typeIV formulations is that the dissolved compound maybe partially or completely precipitated when theseformulations come in to contact with an aqueousenvironment in vitro or in vivo.70,103 The extent ofprecipitation of a dissolved compound from theseformulations will ultimately depend on the aqueoussolubility of the compound and hydrophilicity of thelipid formulation. Compounding the problem further,the precipitated compound can take on a variety offorms including the amorphous form, crystallineform, and of varying particle size distributionsdepending upon the nature of the aqueous environ-ment and the kinetic conditions that exist in theGIT.24 It is essential to evaluate these propertiesin vitro during the selection of a softgel fill vehicle asthey could potentially impact the in vivo bioavail-ability of a solubilized compound. For example, theoral bioavailability of a poorly soluble investigationalcompound, WIN 54954, in fasted beagle dogs wasshown to be erratic and inconsistent when it wasadministered as a solution in PEG 600-Polysorbate80 solvent blend (type IV formulation).103 In contrast,the plasma profiles of the compound were consistentwhen it was administrated as a type II formulationconsisting of a MCT-ethoxylated glyceryl trioleateblend. The erratic and inconsistent absorption of thecompound from the hydrophilic PEG formulation wasattributed to its erratic and inconsistent precipitationin the GIT aqueous environment. Whereas, theimprovement in the plasma profiles from the typeII formulation was attributed to the dispersion ofthe triglyceride into fine emulsion droplets (diameter<3mm) with the compound still dissolved within.
DOI 10.1002/jps JO
Lipid Digestion and Absorption and Influence onAbsorption of Compounds
The consumption of a meal, particularly one contain-ing a large quantity of lipids, stimulates a number ofphysiological responses, including a reduction ingastric transit (with no change in small intestinaltransit108), alterations in gastric pH, secretion ofpancreatic enzymes, stimulation of biliary lipidrelease from the gallbladder, and promotion oflymphatic transport.109–111 The resultant release ofbiliary lipids, primarily bile salts, phospholipids, andcholesterol, promotes the formation of a number ofcolloidal species in association with the digestion(lipolysis) products from the triglyceride (e.g., digly-cerides, monoglycerides, and fatty acids) within thesmall intestine.112,113 A prerequisite to the lipidabsorption process is the micellar diffusion of theselipid digestion products across the aqueous boundarylayer and then into the microclimate adjacent to theintestinal membrane. Once in the enterocytes, fattyacids and monoglycerides of carbon chain length morethan 12 are combined with phospholipids/cholesterolto form triglyceride, which packs into chylomicrons(0.05–0.75mm) and enter the lymph system. Chylo-microns are the major lipid transporting lipoproteinsin intestinal lymph and are composed primarily of atriglyceride core which is stabilized in the aqueousenvironment of the lymph by a surface coating ofphospholipids, cholesterol, and proteins.114 Thephospholipids in chylomicrons are mainly of endo-genous origin, the cholesterol derives from manysources (diet, blood, digestive secretions, newlysynthesized in the enterocytes), and the triglyceridescontain both endogenous and exogenous fattyacids.114,115 The lymphatic transport of a lipophiliccompound, via chylomicrons, may eventually bedetermined by its solubility and partitioning intothe triglyceride core of the chylomicrons.116 Lipophi-lic compounds with (a) a high log P (>5), (b)significant solubility in LCT (�50 mg/mL), and (c)administered either in fed state or with an appro-priate lipid source in fasted state, are thought topotentially gain direct access to the systemic circula-tion through the intestinal lymphatic transport,resulting in the improved bioavailability of thesecompounds.116–121 Compounds transported from theintestinal lumen by the intestinal lymph gain accessto the general circulation of the body at the junction ofthe left internal jugular and left subclavian veins,thereby bypassing the hepatic system.116
Food intake, due to its affect on reduced gastrictransit, can increase the solubilization time and thusthe solubility of a poorly soluble compound in thestomach contents.108 The formation of the aforemen-tioned mentioned colloidal species further aids in thesolubilization of poorly soluble compounds in the GIT,
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4122 GULLAPALLI
whereas the stimulation of lymphatic transport aidsin the improved absorption and reduced hepaticmetabolism of these compounds. Even relativelysmall quantities of LCT (e.g., 2 g) are thought to becapable of stimulating the gall bladder contractionand thereby elevating intestinal bile salts, phospho-lipids, and cholesterol levels. The extent of thisstimulatory response appears to increase as thequantity of LCT administered increases.110 Thiseffect is a likely contributor to the ability of LCTbased formulations to reduce food effects and toenhance the oral bioavailability of some poorly solublecompounds. In contrast, administration of similarquantities of MCT was shown to have relativelylimited effects on the gallbladder contraction andwould not stimulate appreciable increase in theintestinal concentration of biliary-derived lipids.110
In addition, fatty acids with carbon chain length lessthan 12 directly enter the portal blood leading to theliver and then into the systemic circulation, thuscircumventing the lymphatic transport mechanism.Thus, fatty acids with medium chain length aretransported primarily by the portal blood, whereasones with longer chain length are incorporatedprimarily into the chylomicrons and transported inthe lymph.114 An understanding of how lipids ofvarying chain lengths of fatty acids could influencethe mode of transportation and absorption of dis-solved compounds across the GIT provides valuableguidance to the formulation scientist in the selectionof an appropriate lipid in the softgel fill formulation.
The absorption of lipophilic compounds in fastedstate was shown to be significantly higher from aformulation containing even a small amount of LCTthan that from a formulation containing either lipidsof MCT or no lipids.121–126 For example, Fischleret al.122 demonstrated a higher relative oral bioavail-ability and more rapid absorption of clomethiazole infasting healthy volunteers when it was administeredas a softgel containing arachis oil based formulationcompared to a tablet formulation. The oral bioavail-ability of clomethiazole was also shown to increasewith increase of coadministered oil amount. Inanother example, the bioavailability of the poorlysoluble anti-malarial compound, halofantrine (aqu-eous solubility <0.1mg/mL; LCT solubility >50 mg/mL; log P 8.5) solubilized in LCT and administeredin fasted state was shown to be similar to that fromof a lipid-free formulation administered in-fedstate.125,127 In lymph cannulated, fasted rats, thecalculated relative systemic exposure of orallyadministered halofantrine (i.e., sum of plasma avail-ability and lymphatic transport) was shown toincrease with an increase in the fatty acid chainlength of the coadministered lipid (C18-LCT, 22.7% ofdose>C8–10-MCT, 19.2%>C4-SCT, 15.2%>C0-tri-glyceride free, 6.4%).121 This was attributed to the
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
relatively increased contribution of the intestinallymphatic transport (C18, 15.8% of dose>C8–10,5.5%>C4, 2.22%>C0, 0.34%) to the overall absorp-tion of the compound. In a concurrent study, after oraladministration of the three types of triglycerides(LCT, MCT, and SCT) to the lymph cannulated,fasted rats, the amount of LCT present in the lymphwas three- and ninefold, respectively, higher afterLCT administration compared to that after MCT andSCT administration. This was thought to be due tothe stimulatory effect of exogenously administeredLCT on the secretion and lymphatic transport ofendogenous LCT, and the effect was hypothesizedto diminish with a decrease in the chain length ofthe exogenously administered triglyceride (i.e.,LCT>MCT> SCT).121,128,129 The lipophilic com-pounds administered in LCT were suggested to betransported in the lymph in association with theresynthesized exogenously provided LCT present inthe core of the lymph lipoproteins (i.e., chylomicrons),as opposed to in association with the more polar,endogenously derived surface components such asphospholipid and protein.121 Though, these and manyother studies demonstrated that LCT vehicles couldenhance lymphatic uptake and yield relatively highconcentrations of a lipophilic compound in the lymph,it should not be overlooked that the overall compounduptake may be some 50–150 times greater than thatobserved to occur via the lymphatics due to thelimited lymph flow.117,118
The relative oral bioavailability of anotherpoorly soluble compound, danazol (aqueous solubility0.42mg/mL at 378C; LCT-soybean oil solubility4.8 mg/mL at 378C; log P 4.5; BCS Class II130), infasted beagle dogs from various formulations was alsoshown to be in the order of LCT solutionffiLCT-SMEDDS>MCT-SMEDDS>micronized powder.126
In a concurrent in vitro digestion studies, danazol wasobserved to precipitate more extensively upon aqu-eous dilution of the MCT-SMEDDS formulationcompared to the LCT-SMEDDS formulation. Thebioavailability of danazol from the micronized powderwas significantly higher in the fed state compared tothat in the fasted state. Importantly, the relativebioavailability of danazol from the LCT solution andLCT-SMEDDS in the fasted state was shown to bestatistically indistinguishable from that of themicronized powder administered in the fed state.These studies clearly demonstrate that an appro-priate lipid formulation may be capable of achievingthe same positive food effect as the postprandialadministration of a poorly soluble compound.
Another extensively studied example of lipidicformulation is of cyclosporin A (aqueous solubility40mg/mL; log P 2.92; BCS Class IV), which is suppliedas a self-emulsifying oil solution (SEDDS) underthe trade name Sandimmune1 (Novartis Pharma-
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4123
ceuticals) and as a microemulsion preconcentrate(SMEDDS) under the trade name Neoral1 (NovartisPharmaceuticals). Sandimmune1 cyclosporine fillformulation comprises of corn oil (LCT), linoleoylmacrogolglycerides (Labrafil1 M-2125-CS), and ethylalcohol (Tab. 2), which forms a coarse emulsion ondispersion into an aqueous media.131 The triglycerideexcipients in Sandimmune1 formulation requirefurther lipolysis in vivo into diglycerides, monogly-cerides, and free fatty acids, for efficient releaseand absorption of cyclosporine.52,132,133 In contrast,Neoral1 cyclosporine fill formulation comprises ofcorn oil mono- and di-glycerides, polyoxyl 40 hydro-genated castor oil (Cremophor1 RH 40), propyleneglycol, ethyl alcohol, and dl-a-tocopherol (Tab. 2),which spontaneously forms a microemulsion with adroplet size below 100 nm when introduced into anaqueous media.131 The improved dispersion charac-teristics and presence of the rapidly absorbable mono-and di-glycerides, which would not require furtherlipolysis in vivo (thus circumventing the lypolyticprocess) have been suggested to be responsible for theincreased bioavailability and reduced inter- andintra-subject variability of cyclosporine from Neoral1
formulation.134,135 The bioavailability of cyclosporinefrom Neoral1formulation, for example, was shown tobe significantly higher (174–239%), dose propor-tional, and free from food effects with reduced inter-and intra-subject variability compared to thatfrom Sandimmune1formulation.134,135 In addition,the potential inhibitory effect of the polyethoxylatedsurfactant (i.e., Cremophor RH 40) in Neoral1
formulation on CYP3A and P-gp efflux functional-ities18,20–22,136 may also contribute to the increasein bioavailability of cyclosporine from Neoral1
formulation.105
It is important to bear in mind that the influence ofthe type of formulation and the type of lipid used inthe formulation on the bioavailability can vary fromone compound to other and with the amount of doseadministered. Grove et al.,137,138 for example, demon-strated that the bioavailability of poorly solubleseocalcitol in rats was similar from a simple oilsolution or a SMEDDS and was not influenced by thechain length of the lipid used in the formulations. Thebioavailability of another poorly soluble investiga-tional compound, LAB687 (aqueous solubility0.17mg/mL; log P 4.7), was shown to be similar froma lipid formulation (corn oil glycerides, CremophorRH 40, ethyl alcohol, and propylene glycol) and a PEG3350-polysorbate 80 formulation in fasted beagledogs.8
Fill Formulation Development
Typically, solubility determinations are carried out byequilibrating a suspension containing an excessamount of a compound in a solvent at a constant
DOI 10.1002/jps JO
temperature (e.g., 258C, 378C) and analyzing thesupernatant, collected after centrifugation and filtra-tion of the suspension, using an appropriate analy-tical method.104,117,130 The method is modified whendeveloping a solution fill formulation for softgelencapsulation to take the water migration processesoccur in a softgel into account.139 The solubility of acompound in a solvent is determined by dissolvingincreasing amounts of the compound in a fixedamount of the solvent. A portion of the solution ateach concentration is mixed with water to mimicwater migration and retention processes occur in asoftgel. The highest concentration of the compound atwhich no precipitation is observed in the presence ofwater at the equilibrium level is assumed to be theequilibrium solubility of the compound in the ‘‘softgelcompatible vehicle.’’ When evaluating the solubility ofa compound in a semisolid or a solid vehicle at roomtemperature, solutions of varying concentrations ofthe compound are prepared at a temperature abovethe melting point of the vehicle. The solutions arethen allowed to solidify at room temperature. Thesolid solutions are observed periodically under apolarized microscope for the presence of any crystalsof the compound.69
A hypothetical example of a softgel solution fillformulation development for a compound is illu-strated in Table 3. In this example, the solubility ofthe compound in a neat vehicle is evaluated atincrements of 10 mg/g (e.g., maximum solubility�50 mg/g). Depending on the type of vehicle (i.e.,PEG, mixed medium chain glycerides, MCT, or LCT),the solutions are mixed with water at two levels thatthe fill solutions are expected to be exposed to duringthe encapsulation, drying phase, and subsequentequilibrium (Fig. 2). These solutions are placed atroom temperature (RT) and accelerated physicalstability conditions (e.g., 48C and �208C) for abouta month. Solutions that do not show any crystal-lization, when observed visually and under a micro-scope, at a water level (higher) representing theprimary drying phase within 24 h and at a water level(lower) representing the equilibrium phase for amonth at all stability conditions are considered forfurther development (e.g., �20 mg/g in the example).It is, however, worth mentioning two aspects of watermigration and its influence on physical stability of adissolved compound in softgels. First, though acompound dissolved in a vehicle for encapsulationmay exhibit precipitation upon exposure to a higherwater level during the primary drying phase, thecompound may re-dissolve in the vehicle uponremoval of the excess water during the secondarydrying phase. Thus, it may be meaningful to fill thesolution formulation into empty soft gelatin capsules(referred to as air-fills) and monitor the precipitation-re-dissolution phenomenon during cycling of the
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

Table 3. Experimental Design for the Development of a Hypothetical Softgel Solution Fill Formulation
Fill Vehicle
WaterContent Before
Encapsulation (%)
Mimicking Primary Drying Mimicking Equilibrium
Amount of Water to be Added (%)
PEG based �1–2a, 317 �16 �8Mixed MC glycerides �0.15a, 318 �6 �4MCT/LCT �0.1a, 253,319 �0.1a, 253 �0.1a, 253
Stability conditionsCompoundConc. (mg/g)
�208C 48C RT �208C 48C RT �208C 48C RT
Physicalstability—initial10 H H H H H H H H H20 H H H H H H H H H30 H H H H H H H H H40 H H H H H H H H H50 H H H Xb Xb Xb H H H60 Insoluble
Physical stability—24 h10 H H H H H H H H H20 H H H H H H H H H30 H H H H H H H H H40 H H H Xb Xb Xb H H H50 H H H Xb Xb Xb H H H60 Insoluble
Physical stability—1 month10 H H H Observations are discontinued
after 24 h as the fill formulationis expected to be exposed to thishigh level of water only during
the first 24 h
H H H
20 H H H H H H30 H H H Xb Xb Xb
40 H H H Xb Xb Xb
50 H H H Xb Xb Xb
60 Insoluble
(X) Precipitation (crashing out) of the compound; (H) no precipitation, but rejected due to precipitation of the compound within 24 h at awater level approximated during the primary drying process; (H) physically stable fill formulation for further chemical stability evaluationand subsequent encapsulation.
aTypical water content initially present in the vehicle; no water addition is required.bMay not be applicable to MCT/LCT based fill formulations as water is unlikely to migrate into these hydrophobic vehicles.
4124 GULLAPALLI
capsules between high humidity and low humidityconditions. A similar cycling procedure has been usedby Raikes et al.,140 though, to study the moistureuptake by softgels under various relative humidityconditions. Secondly, the migration of water from theshell into the fill may be of an advantage whenencapsulating fills formulated using the enhancedsolubility system (ESS) techniques (discussed inSolubility Enhancers for Hydrophilic Vehicles Sec-tion), where a portion of the compound is present inits water soluble salt form. The chemical stability ofthe selected fill formulations is evaluated at 408C orhigher in the presence of added water and gelswatches that represent various potential gelatinshell formulations used for encapsulation. Alter-nately, the selected fill formulations, with no addedwater, may also be filled into air-fills and exposed toelevated temperatures and relative humidities for
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
chemical stability evaluation. It is important to keepin mind that the selected gel swatches and air-fills forstability evaluation have compositions commonlyused at various softgel manufacturers, as it wouldoffer the flexibility of manufacturing the final softgelproduct at more than a single manufacturer.
When a compound is soluble and has demonstratedstability in a softgel compatible vehicle, it can beencapsulated into softgels as a solution with minimalformulation effort. On the other hand, compoundsthat do not have sufficient solubility in softgelcompatible vehicles may require encapsulation assuspensions. The dispersed material in a suspensionshould have a particle size of 180mm or finer (passthrough a #80 mesh) to achieve an acceptable blenduniformity during encapsulation and content uni-formity in the final softgel product.33 When a vehiclefor a suspension fill formulation has any solubility for
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4125
the compound, the dispersed material may undergoOswald ripening and/or secondary nucleation,12
resulting in changes in the particle size distributionsand/or in the polymorphic nature during the shelf-lifeof a softgel product. This aspect of stability asuspension fill formulation is typically investigatedby subjecting the formulation with the added water(shown in Tab. 3) to the stress testing of cyclingbetween 40 and 48C, in addition to other testsdescribed above for a solution fill formulation. Thesamples are observed at the end of each cycle (e.g.,48 h at 408C, followed by another 48 h at 48C may beconsidered as one cycle) for any changes in theparticle size, morphology, and polymorphism. Tem-perature cycling may accelerate the dissolution of thesuspended compound in the formulation vehicle atthe higher temperature and re-crystallization uponcooling, thus provide valuable information on thephysical stability of a softgel product during its shelf-life. However, the transformation may occur rapidlyor slowly and may be reversible (enantiotropic) orirreversible (monotropic).12
A suspension fill formulation may also require asuspending (thickening/viscosifying) agent to preventsettling of the dispersed material and to maintainhomogeneity throughout the encapsulation process.33
The widely used suspending agents for oil basedformulations include beeswax,141 hydrogenated vege-table oils,142 and glycerol esters of fatty acids with lowHLB values (e.g., Gelucire1 33/01, Gelucire1 39/01,Gelucire1 43/01, Compritol1 888, from GattefosseeCorporation).143,144 These suspending agents, due totheir hydrophobic and high viscous nature, could alsominimize the migration of water from the shell intothe fill and the diffusion of water soluble compoundsfrom the fill into the shell and thereby improve thephysical stability of a softgel product. The suspendingagents used for polyethylene glycol based formula-tions include higher molecular weight polyethyleneglycols (e.g., PEG 1500, PEG 4000, PEG 6000),66
cellulose polymers,73,74 colloidal silicon dioxide, poly-vinylpyrrolidone, calcium acetate,75 and mixtures ofmono-, di-, and triglycerides/mono- and di-fatty acidesters of polyethylene glycols (e.g., Gelucire1 44/14,Gelucire1 50/13, from Gattefossee Corporation).
Polymorphism is a common phenomenon amongmany pharmaceutical active ingredients and excipi-ents.145–147 Substantial differences can exist in therate of dissolution and bioavailability of the variouspolymorphic forms of a compound.146,148–151 Theseeffects of polymorphism are more critical especially inthe case of compounds belonging to BCS class II andclass IV. It is critical to start the fill formulationdevelopment with a stable polymorph to prevent anypotential precipitation of the dissolved compound dueto conversion of an unstable or a metastable form intothe stable form. Ritonavir softgel product (Norvir1,
DOI 10.1002/jps JO
Abbott Laboratories) is a classic example of howpolymorphic transformation of a soluble polymorph(I) to a less soluble form (II) could affect the quality ofa softgel product.152,153
SHELL FORMULATIONS
A softgel shell formulation typically consists of a film-forming material, such as gelatin, water dispersibleor soluble plasticizer(s), and water. The formulationmay also contain other minor additives, such asopacifiers, colorants, flavors, sweeteners, and pre-servatives. Softgels may also be coated with a varietyof polymers for certain targeted enteral deliveryapplications.154
Gelatin
The Unites States Pharmacopeia/National Formulary(USP/NF) defines gelatin as a product obtained by thepartial hydrolysis of collagen derived from the skin,white connective tissue, and bones of animals. Gelatincan be derived from many different sources of collagenwith cattle bones, hides, pigskins, and fish being theprinciple commercial sources. It contains a mixture ofwater soluble proteins (84–90%), mineral salts (1–2%),and water (8–15%). The protein fraction containsalmost entirely of amino acids linked by amide bondsforming a linear polymer with a molecular weightranging from 15000 to 250000 Da.155–158 The watercontent in gelatin usually originates from its manu-facturing process.34,159
Gelatin is derived from collagen by thermaldenaturing with the aid of either a dilute acid (typeA gelatin) or a dilute alkali (type B gelatin). Gelatin isusually characterized by the mode of its productionand how it is produced has marked influence on itsproperties. Gelatin is amphoteric in nature with itsisoelectric points (IEP) ranging from 7.0 to 9.0 for typeA gelatin and from 4.7 to 5.3 for type B gelatin,respectively.158 The alkaline hydrolysis causes agreater degree of deamidation of the asparagineand glutamine amino acids in collagen, resulting inthe production of a larger number of free carboxylicacid groups in gelatin than that from acid hydrolysis.The greater degree of deamidation and the resultinglarger number of free carboxylic acid groups from thealkaline hydrolytic process accounts for the relativelylower isoelectric point of type B gelatin compared tothat of type A gelatin.160 Type A gelatin usuallydisplays relatively higher plasticity and elasticitythan type B gelatin, whereas type B gelatin displaysrelatively higher gel strength.
Commercially available gelatin is odorless, taste-less, and free-flowing granular material with a light-amber to light-yellowish tint.158 The most favorableaspect of gelatin for its use in softgels is its ability to
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

Figure 5. Influence of pH and Temperature on the First OrderRate Constant (h�1) for Decay of Gel Strength of Limed OsseinGelatin with Bloom Strength 250 g and Isoelectric pH 4.75 (Dataadopted from Croome94).
4126 GULLAPALLI
form thermoreversible gel in water, that is, ability todissolve in hot water and form a gel upon cooling, a‘‘must have’’ quality for sealing the softgels duringencapsulation. Gelatin is soluble in glycerin, propy-lene glycol, dilute acids, and dilute alkalis, butprecipitates in strong acids and alkalis. The rate ofdissolution of type B gelatin is usually higher thanthat of type A gelatin at a given pH of the aqueousmedium.161 The dissolution of gelatin, irrespective ofits type, was shown to be not influenced significantlyby pH at pH values above 3, but was shown to increaseand reach a plateau at pH values below 3. The higherdissolution of gelatin at lower pH may be attributed tothe protonation of amino groups present in thegelatin. However, the dissolution of gelatin atlower pH was shown to decrease in the presence ofadded salts, such as sodium chloride and potassiumchloride.161 Gelatin is insoluble in most organicsolvents, such as alcohols, acetone, and chloroform.156
It is important to keep these solubility characteristicsof gelatin in mind during the development ofdissolution methods for softgel products.
Gelatin undergoes hydrolytic degradation (depoly-merization) in the aqueous gels, the rate and extent ofwhich depend on the pH, temperature, and time, thereaction is allowed to proceed.34,94,162,163 The depo-lymerization reactions in gelatin may be furtheraccelerated by the lowering of its molecular weight.34
The hydrolytic degradation of gelatin results in thereduction of the viscosity and gel-forming ability of agelatin solution, and in the formation of weak seamsduring softgel encapsulation. Though gel strengthand viscosity are the two parameters commonly usedto measure the extent of gelatin hydrolytic degrada-tion, the gel strength has been known to be moresensitive indicator of the degradation reaction.94,162
The degradation of gelatin (expressed as loss of gelstrength as a function of time) was shown to followfirst order reaction kinetics, with the reaction ratesvarying according to the pH and temperature. Inaddition, the rate constants were shown to beminimum between pH values 4 and 7, with the rateaccelerating on either side of this pH region, as shownin Figure 5.94 The minimum hydrolytic degradation ofgelatin within the pH range of 4–7 was furthercorroborated by the data obtained from Courts’investigations,163 where minimum loss of averagemolecular weight of gelatin was observed withinthis pH range. The pH of a gelatin solution remainsconstant when the degradation reaction takes placeat the isoelectric pH of gelatin.163 In contrast,the pH of the solution shifts slightly towards theisoelectric pH when the pH of the reaction medium isother than its isoelectric pH and the shift becomeslarger at high pH values.162,163 Acid treated gelatins(type A) are more susceptible to alkaline degradationthan to acid degradation, whereas alkaline treated
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
gelatins (type B) are more susceptible to aciddegradation.162 From the studies on the hydrolyticdegradation of gelatin, Courts163 suggested that thepeptide bonds involving the amino groups of serineand threonine were labile to both acidic and basichydrolysis, whereas aspartic acid peptides weresusceptible to acid hydrolysis only. The glutamic acidpeptide bonds were reported to be stable, whileglycine peptide bonds were of intermediate instability.
Gelatin is considered as an inactive ingredient bythe Food and Drug Administration (FDA). Softgelmanufacturers’ quality control testing for gelatin,among other things, includes bloom strength, viscos-ity, iron content, and microbial testing.33,50 Bloomstrength, also known as jelly strength, is expressed asthe weight in grams that, when applied with a12.7 mm diameter plastic plunger, will produce adepression exactly 4 mm deep in a jelly containing6.67% w/w of gelatin in water matured for 16–18 h at108C. Bloom strength of gelatin used in a softgel shellmay vary from 150 to 250 g, with the higher the bloomstrength, the more physically stable is the resultingsoftgel shell. As the cost of a softgel product is relateddirectly to the bloom strength of gelatin used, gelatinof a higher bloom strength is usually reserved onlywhen necessary to improve the physical stability of asoftgel product or for large size softgels which requiregreater structural strength during manufacture.33
Viscosity determination is performed on a 6.67% w/wconcentration of gelatin in water at 608C and usuallyranges between 25 and 45 millipoise. Iron levelspresent in the gelatin raw material is derived mainlyfrom the water used in its production and should not
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4127
exceed 15 ppm as higher levels may potentiallyresult in the color reactions with other softgelcomponents. Gelatin is an excellent growth mediumfor many bacteria and thus requires considerablecare during its manufacture and handling to avoidcontamination.
Plasticizers
The high glass transition temperature of anhydrousgelatin (Tg> 1008C)45,164–169 prevents it from forminga flexible and acceptable film readily during themanufacturing of gelatin capsules. Water is aneffective plasticizer for gelatin and reduces the Tg
of gelatin proportionally to its water con-tent.45,165,167,169,170 Coppola et al.,169 for example,reported a decrease in the Tg of gelatin from 1608C to�208C when the water content in gelatin wasincreased from 2 to 28% w/w. However, due to itsvolatile nature, water will be lost during the dryingprocess resulting in a brittle and fragile shell. Thus,nonvolatile plasticizers are included in the productionof gelatin ribbons for softgels. The nonvolatileplasticizers are hypothesized to substitute for waterin the vicinity of the protein chains and reduce theprotein–protein interactions with a consequentincrease in the mobility of protein chains and adecrease in the Tg of gelatin.169,171–173 In addition, aplasticizer, due to its hygroscopic nature, maypromote absorption of moisture by gelatin that alsocontributes to the reduction of the forces between theadjacent polymer chains.155,174 Vanin et al.,174 ineffect, considered the reduction in the Tg of the gelatinfilms as a consequence of the total number of moles ofall plasticizers (i.e., nonaqueous plasticizers andwater) present in the films. Thus, the extent to whicha plasticizer could impart flexibility to a gelatin film isdetermined by its hygroscopicity and its ability tointeract with protein chains and reduce the protein–protein interactions within gelatin.155,174,175 Thereduction in the protein–protein interactions resultsin improved flexibility and handling of the shellmaterial during its manufacturing and shelf-life.
Typical plasticizers used in the softgel shellformulations include glycerin, sorbitol, partiallydehydrated sorbitol (a blend of D-sorbitol, 1,4-sorbi-tan, mannitol, and water; e.g., Sorbitol Special1, fromSPI Pharma; Anidrisorb1 or Polysorb1, fromRoquette), maltitol (hydrogenated corn syrup; e.g.,Lycasin1, Roquette), mannitol, propylene glycol, lowmolecular weight polyethylene glycols, or a blendthereof. Selection of a plasticizer type and itsconcentration (expressed as the plasticizer-to-gelatinratio, P/G) in a shell formulation is determined bygelatin type, composition of fill formulation,60,161 andcompatibility with the ingredients present in a fillformulation.98,100 Plasticizers are used typically atabout 15–30% w/w of the total wet mass of a shell
DOI 10.1002/jps JO
formulation at the time of encapsulation.39,40–42,176
The addition of increasing amounts of a plasticizeralters the physical properties of a gelatin filmresulting in an increase in its flexibility, elongationat break, water retention, water vapor permeability,oxygen permeability, and volatile solute permeabil-ity, and a decrease in its Tg, tensile strength, andelastic modulus.43,56,57,60,155,174,175,177
Among the various plasticizers studied, glycerinwas reported to be the most effective and practicalplasticizer, irrespective of type of gelatin used in ashell formulation.171,175 The higher plasticizingefficacy of glycerin was attributed to its lowermolecular weight178 and higher hygroscopicity175
than other higher polyols. When compared amongvarious nonvolatile plasticizers, the number of molesof a plasticizer in a given amount would be higher inglycerin, and thus their effect on reducing the numberof interactions between the protein polymeric chainswould be more intense.175,179 It also has beentheorized that a plasticizer with a lower glasstransition temperature (Tg) would have a morepronounced plasticizing effect. In this way, theeffectiveness of glycerin might also be explained byits lower Tg (�938C) as compared to sorbitol(�38C).175,180 Propylene glycol is more effective as agelatin plasticizer compared to glycerin.171 However,due to its higher solvent power for gelatin, propy-lene glycol affects the formation of the gel structureadversely, that is, it acts as more of a gel structurebeaker. In addition, due to its higher volatilitycompared with glycerin, the use of propylene glycolresults in the considerable deterioration in themechanical strength of the shell material withtime.171 Furthermore, gelatin sheaths containingpropylene glycol as the plasticizer are substantiallytackier than those containing glycerin or sorbitol asthe plasticizer and require much lower cooling drumtemperatures to extract the sheaths from the drumsduring softgel manufacture.37 The ability of apolyethylene glycol (PEG) to act as a plasticizer isdetermined by its hydrogen bonding potential withthe protein chains in gelatin that in turn is influencedby factors, such as the number of hydroxyl groups permole, molecular size, solubility, and polarity of thePEG. PEG of a lower molecular weight has a largernumber of hydroxyl groups per mole and a higherhygroscopicity compared to those of a higher mole-cular weight PEG and thus exhibits a more pro-nounced plasticizing effect than that by the latter.Gelatin films plasticized with polyethylene glycolshave also shown to exhibit a tendency for theplasticizer to migrate to the surface of the films, aphenomenon referred to as blooming or blushing.57
This phenomenon is thought to take place when theplasticizer concentration exceeds its compatibilitylimit in the polymer, thus causing phase separation
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4128 GULLAPALLI
and physical exclusion of the plasticizer from thepolymer.181
Glycerin, due to its extreme hygroscopic nature,can influence gelatin to pick up substantial amount ofmoisture quickly,169 leading to a soft, tacky, andbloated softgel sheath that will ultimately breakdown or result in softgels sticking together with time.Sorbitol, on the other hand, may prone to crystal-lization from the films that are stored at low tointermediate relative humidity conditions due to theinsufficient availability of water to keep the plasti-cizer in solution (i.e., blooming or blushing).179,182
The crystallization of sorbitol would decrease theamount of ‘‘plasticizing’’ sorbitol that in turn isexpected to increase the molecular interactionswithin the gelatin network and to change themechanical properties of the films.179 Mannitol hasalso been reported to exhibit a similar tendency tocrystallize from gelatin films.57 It is sometimesadvantageous to blend sorbitol with glycerin thatwould yield a better control of the overall moisturecontent of the softgel sheath. Partially dehydratedsorbitol (a blend of D-sorbitol, 1,4-sorbitan, mannitol,and water), in comparison, tends to pick up lessmoisture than glycerin and also not to prone tocrystallization as regular sorbitol. The effectivenessof partially dehydrated sorbitol as a plasticizer isattributed primarily to its 1,4-sorbtan content andthe interactions of 1,4-sorbitan with the gelatinmatrix.172
Gelatin films produced with glycerin as a plastici-zer are less resistant to moisture and more permeableto oxygen and volatile ingredients than thoseproduced with a higher polyol plasticizer, such asxylitol, sorbitol, maltitol, or a blend of glycerin with ahigher polyol.43,60,175,183 The oxygen permeabilitythrough a gelatin film has been shown to increaseexponentially with an increase in glycerin concentra-tion in the film and with an increase in relative
Table 4. Influence of Plasticizer Type and RH on Moisture Cont150 Bloom Lime Bone Gelatin60,183
Plasticizer Typeand Concentrationa
Exposureto % RHb
Glycerin, 20% w/w 11335475
Sorbitol, 20% w/w 33Xylitol, 20% w/w 33
5475
Lycasin, 20% w/w 75
aBased on total wet mass.bExposed to the selected relative humidity (RH) at ambient tempera
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
humidity to which the film is subjected to.43 Duringconcurrent studies, the moisture content of the filmhas also been shown to increase with an increase inglycerin concentration in the film and with anincrease in relative humidity to which the film issubjected to. Based on these observations, Homet al.43 theorized that the plasticizers and environ-mental relative humidity conditions control theequilibrium water content of a gelatin film, and thisequilibrium water content has the greatest effect onthe oxygen permeability through the film. In acomparative study, the oxygen permeability coeffi-cients of gelatin films containing glycerin/sorbitol(1:1) blend, hexaglycerol, and decaglycerol werereported to be about 38.7%, 20.8%, and 19.5%,respectively, of that of a gelatin film containingglycerin, at 43% plasticizer concentration, 72%relative humidity, and room temperature.43 Thus,the use of a nonglycerin plasticizer or a blend ofglycerin and a higher polyol is recommended whenencapsulating oxygen sensitive compounds in thesoftgels.
Volatile fill components, such as ethyl alcohol, canreadily diffuse through conventional softgel shellsand usually disappear by the end of the dryingprocesses during the softgel manufacture.60,183 Mor-eton and Armstrong60,183 have studied the influenceof plasticizer type and relative humidity conditions onthe film moisture content and diffusion of ethylalcohol through the gelatin films in details. A briefsummary of the results reported by these researchersis presented in Table 4. The diffusion of ethyl alcoholthrough the gelatin films plasticized with glycerinwas shown to accelerate with an increase in the filmmoisture content. A similar trend, though at asignificantly much lower rate, was also seen ingelatin films plasticized with xylitol. It appears fromTable 4 that the relative humidity conditions controlthe equilibrium water concentration in a gelatin film,
ent and Diffusion of Ethyl Alcohol through Films Made from
MoistureContent %w/w Range
Apparent DiffusionCoefficient mm2 min�1 Range
4.56–6.44 3.45–3.8110�5
7.43–9.32 4.60–5.2410�5
13.20–19.80 8.61–46.210�5
30.70–34.10 51.7–68.110�5
8.58–8.82 No diffusion detected8.00–8.14 No diffusion detected
13.10–14.39 0.759–1.6810�5
26.50–33.67 5.67–22.110�5
17.00–23.10 1.16–1.4710�5
ture (21.5–23.78C).
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4129
and this equilibrium water concentration appears tohave the greatest effect on the diffusion of a volatilefill component through the film, that is, for a givenplasticizer type, the higher the moisture content of agelatin film, the more rapid the diffusion of a volatilecomponent through it. However, the diffusion processwas significantly lower through the gelatin filmscontaining a higher polyol plasticizer, such as xylitol,sorbitol, or lycasin, compared to that through thefilms containing glycerin as the plasticizer, at similarfilm moisture contents. Thus, the loss of a volatile fillcomponent due to diffusion can be minimized by theuse of a higher polyol plasticizer, maintaining lowshell moisture content, and protecting the softgelproduct against high humidity conditions.
Colorants and Opacifiers
Coloring agents are included in a softgel shellformulation to provide elegance to a softgel productand also to provide a distinctive appearance that mayserve to differentiate a particular softgel product fromothers that have a similar physical appearance. Thecoloring agents can be dyes (water-soluble sub-stances), lakes (insoluble forms of a dye that resultfrom its irreversible adsorption onto a hydrous metaloxide), inorganic pigments (substances such astitanium dioxide or iron oxides), or natural colorants(colored compounds not considered dyes per se, suchas riboflavin).184 The most important properties of acoloring agent are its depth of color and resistance tofading over time. Coloring agents can be graded ontheir efficiency in reflecting desired colors of visiblelight as well as on their molar absorptivities atcharacteristic wavelengths of absorbance.184 Coloringagents are subject to federal regulations and conse-quently the current regulatory status of a givensubstance must be determined before it is used. It isdesirable that the coloring agents should be physi-cally and chemically nonreactive with other ingre-dients present in the softgel product. Anionic dyes areknown to interact to a greater extent with a cationictype A gelatin than with an anionic type B gelatin.185
These interactions could potentially effect the disin-tegration of the gelatin shell.
An opacifier is included in a shell formulation toprovide light resistance when photosensitive com-pounds are encapsulated into softgels. An opacifiermay also be included in a shell formulation whenencapsulating an unaesthetic fill formulation, such asa disperse system that is prone to phase separation orsedimentation. Titanium dioxide is the most com-monly used opacifier and is typically used at about0.5–1.0% w/w of a shell formulation to impartsufficient opacity to the shell. Titanium dioxide is awhite, odorless, tasteless, inert, nonhygroscopicpowder186 that has been shown to undergo no
DOI 10.1002/jps JO
observable interactions with gelatin and otheringredients used in the softgels.187 Due to its highrefractive index (2.55–2.76), titanium dioxide hasexcellent light-scattering properties and the extent oflight-scattering by titanium dioxide can be altered byvarying its particle size186,188,189 and concentrationused in a shell formulation.190 As Rayleigh’s theoryimplies, the shorter wavelengths of light are moreefficiently scattered by smaller particles, that is,the finer the particle size of the opacifier the moreeffective it is in scattering the UV light below 400 nm.However, as the particle size of the opacifier reachesbelow 50 nm, its scattering power decreases in thevisible range making the film more transparent thanone with a larger particle size.188,189
Titanium dioxide is highly hydrophobic and gen-erally occurs as aggregated particles that should bedispersed and wetted thoroughly before its addition tothe molten gel mass.191 The effectiveness of a shellmaterial to provide light protection to an encapsu-lated compound is critically dependent on how wellthe opacifier is dispersed in the gel mass. Ideally, foran opacifier to be more effective against UV lighttransmittance, it should be dispersed as individualcrystals or small agglomerates of two or threecrystals. Aggregates of many finer crystals behaveas though they are one large particle, thus scatteringvisible light and exhibiting poor effectiveness in theUV range.188
The light transmission through a gelatin filmcan also be reduced substantially either with theincrease of its thickness at a given opacifierconcentration or with the increase of the opacifierconcentration in the film.190 The light transmissionthrough a gelatin film was shown to be diminishedwith the increase of titanium dioxide concentrationup to 1% in the film and plateauing afterwards.190
The addition of an opacifier to a shell formulationmay also potentially increase the tortuosity of thepath that the permeating oxygen and light must flowthrough to reach the fill.43 However, the effect of anopacifier on the permeability of oxygen throughgelatin films was shown to be observable onlyat opacifier concentrations substantially higherthan what is commonly used in a capsule shellformulation.
DISSOLUTION
Dissolution of Softgel Shell
The availability of a compound formulated in a softgelfor absorption depends on the initial dissolution andrupture of the softgel shell and subsequent releaseand dissolution of its fill contents in the GIT fluids.Thus, these two processes need to be monitoredduring the release and shelf-life of a softgel product.
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4130 GULLAPALLI
Problems in the dissolution and rupture of the softgelshell may become apparent upon aging, when exposedto physical conditions, such as heat,192,193 hightemperature and humidity, UV radiation, g-radia-tion,194 and rapid drying,47 and when exposed tochemical substances, such as aldehydes, ketones,imines, and carbodiimides.195,196–202 These problemsare attributed to cross-linking of gelatin (pellicleformation) that causes the gelatin shell to becomeswollen, tough, rubbery, and insoluble in water.Cross-linking of gelatin gives rise to the formation of avery thin film during the dissolution testing of asoftgel product. The film is mechanically weak andcan easily be punctured. However, the film does notdisrupt easily with gentle agitation under normaldissolution conditions.203
Mechanism of Gelatin Cross-Linking
The ultimate effects of exposure to elevatedtemperatures on the physical properties of thegelatin shell are known to be similar to those ofexposure to aldehydes, though the mechanisms of
Figure 6. Possible mechanism of formation of mcross-links formation in gelatin (adopted from Tayl
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
chemical reactions occurring within the gelatin aredistinctly different.204,205 Physically, exposure ofgelatin shell to either elevated temperatures oraldehydes results in a decrease of its disintegra-tion, dissolution, and swelling properties, and anincrease of its gel strength, a clear indication of theformation of three dimensional networks withinthe gelatin.
Chemically, aldehydes are known to form methy-lene bonds between two amino groups on adjacentgelatin chains or within the same chain, asillustrated in Figure 6. The aldehyde inducedcross-linking of gelatin is thought to involve the e-amino functional groups present in the lysinemoieties and the guanidino functional groups pre-sent in the arginine moieties of the gelatinchain.196,197,206–211 Both of these amino acids, lysineand arginine, have long, reactive side chains thatcan extend farther out from the polypeptide chainthan other groups and thus can participate moreeasily in the inter and intra molecular reactions.207
In addition, hydrogen bonding is proposed to providethe added stability to the lysine-arginine cross-link
ethylols of lysine and arginine and subsequentor et al.,207 Albert et al.,209 Gold et al.210).
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4131
and arginine-arginine cross-link (Fig. 6). Evidenceobtained from 13C nuclear magnetic resonance(NMR) studies using 13C labeled formaldehyde asa cross-linking agent strongly suggests the forma-tion of methylols (i.e., hydroxylmethyl) of lysineresidues initially and of arginine residues later,which react together resulting in the formation oflysine-to-arginine and arginine-to-arginine cross-links in gelatin, but no evidence has been found inthe formation of lysine-to-lysine cross-links ingelatin exposed to aldehydes.207–209 As the formationof arginine methylols was identified to coincide withthe initiation of gelatin cross-linking reactions, itsformation was hypothesized to be the rate limitingstep in the cross-linking of gelatin. The rate of cross-linking in gelatin by aldehydes was shown to bestrongly influenced by humidity with maximumcross-linking occurring around 60–70% humidity.209
Due to the high pKa value of the e-amino functionalgroup (pKa� 10.79) in lysine and of the guanidinofunctional group (pKa� 12.48) in arginine, boththese functional groups exist in their protonatedforms at acidic pH and thus are not expected to beavailable to react with aldehydes.210 In contrast,since both of these functional groups exist primarilyin their unprotonated forms at alkaline pH, they canpotentially react with aldehydes, resulting in cross-linking. Gold et al.210 suggested that fill formula-tions, containing excipients suspected to be con-taminated with aldehydes, should be formulated atthe lowest possible pH to prevent gelatin cross-linking.
In contrast to the cross-linking mechanism in thepresence of chemical agents, elevated temperat-ures are known to promote condensation reactionsbetween a carboxylic group on a gelatin chain andan amino group on an adjacent gelatin chain (orwithin the same chain), as illustrated in Eq. (1).192–193
Eq. (1) shows the effect of elevated temperatures ongelatin
Chain 1� COOHþH2N� Chain 2 ! Chain 1
� CO�NH� Chain 2þH2O (1)
Whether it is aldehyde induced or heat induced,cross-linking of gelatin results in the formation ofthree dimensional molecular networks of a highermolecular weight with the loss of ionizable groups(i.e., R-NH2 and R-COOH) than the original mole-cules, leading to the reduced solubility of gelatin.Alternately, the loss of ionizable groups in gelatin andthe resulting decrease in its solubility may alsoarise from the ionic, hydrogen, and van der Waalsinteractions of these groups with other compoundsused concurrently in the shell formulation, such asFD&C red #3 and FD&C red #40 dyes.185,212
DOI 10.1002/jps JO
The aldehyde and other carbonyl impurities insoftgels may originate from the autooxidation ofmaterials containing polyoxyethylene moieties intheir structures (e.g., polyethylene glycols, methox-ypolyethylene glycols, polyoxyethylene fatty acidesters, polyoxyethylene sorbitan fatty acid esters,polyoxyl 40 hydrogenated castor oil).95,213–218 Rayoncoiler, included in the package presentations, isknown to be another source that produces furfural(2-furaldehyde) at the accelerated stability conditionsthat could potentially cross-link gelatin.219,220 On theother hand, drug molecules containing carbonylfunctional groups in their structures (e.g., nimesu-lide, rofecoxib, macrolide antibiotics) may also inducethe cross-linking of gelatin and thereby reduce thedissolution of the shell material.221,222
Minimizing Gelatin Cross-Linking
The cross-linking of gelatin in a softgel shell can bereduced by using excipients with low aldehydecontent in the fill formulation, by using excipientscontaining abundant free amino groups (e.g., glycine,lysine) in the shell formulation that can compete withthe amino groups present in the gelatin chain for theavailable aldehydes originating from the componentsused in the softgel (i.e., aldehyde scavengers), and/orby masking the amount of amino groups availablealong the molecular chain of the gelatin throughcovalent bonds with suitable masking agents.223
Succinic acid is an agent often used to mask theamino groups present in the gelatin chain throughcovalent bonds (succinization). The dicarboxylic acidnature enables succinic acid to both the reaction ofone carboxylic group with an accessible amino groupin the molecular chain of the gelatin while the secondcarboxylic group concurrently provides steric preven-tion of access of the cross-linking agent. However, adisadvantage of the succination approach is thegelatin shell prepared using the succinated gelatinis usually highly permeable to volatile solvents (e.g.,ethyl alcohol) and migratable ingredients (e.g.,propylene glycol).
Incorporation of both an amino acid (e.g., glycine)and a carboxylic acid (e.g., citric acid) into the powder-fill of hydrochlorothiazide hard gelatin capsule wasshown to provide a reduction in the cross-linking ofgelatin.224,225 The carboxylic acid was believed toprovide an acidic environment necessary to minimizethe hydrolytic degradation of hydrochlorothiazideinto its carbonyl impurity, whereas the amino acidwas believed to function by acting as the carbonylscavenger. However, this approach is fraught withthe disadvantage of limited solubility of the stabiliza-tion excipients in the nonaqueous vehicles commonlyused in softgels.226 To circumvent these solubilityissues, the WIPO patent application WO 03/103582176
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4132 GULLAPALLI
proposes the use of an amino acid moiety (e.g.,glycine, lysine) in the shell and a softgel compatibleester of carboxylic acid (e.g., tocopherol acetate,tocopherol succinate, D-alpha tocopheryl polyethyleneglycol succinate-TPGS) in the shell, fill, and/orlubricant to reduce the cross-linking of gelatin. Theextent of gelatin cross-linking in the softgel shell mayalso be influenced by the type of functional group(s)present in the structures of the compounds encapsu-lated. Compounds containing either a primary aminegroup(s) (e.g., isoniazid, acyclovir) or a secondaryamine group(s) (e.g., ethambutol) may interact withthe aldehyde impurities present in the fill formula-tion, that is, act as aldehyde scavengers, and therebyprevent the cross-linking of gelatin.222
Dissolution Testing: Influence of Digestive Enzymes onGelatin Cross-Linking
The USP Chapters <711> and <2040> on Dissolu-tion227 provide guidance and procedures for dissolu-tion testing for dosage forms administered orally. Forgelatin capsules that failed tier 1 dissolution testing(i.e., in a medium with no enzymes) due to gelatincross-linking, the USP recommends the use ofdigestive enzymes (i.e., pepsin or pancreatin) in thedissolution medium during tier two dissolutiontesting. These enzymes digest the cross-linked gelatinand thereby promote the dissolution and rupture ofthe cross-linked gelatin shell. The use of thesedigestive enzymes in the dissolution medium isjustified on the grounds that such enzymes are alsopresent in the GIT.228,229
Softgels that failed the tier 1 dissolution testing butpassed the tier 2 dissolution testing (Fail/Pass) wereshown to be bioequivalent to those which passed thetier 1 dissolution testing, whereas softgels that failedboth tiers of dissolution testing (Fail/Fail) were shownto be bioinequivalent to those passed either one orboth tiers of dissolution testing.230 Severe cross-linking of gelatin (Fail/Fail) was thought to result inthe decreased availability of its peptide bondstowards the proteolytic enzymes (i.e., pepsin andpancreatin), leading to decreased rate and extent ofproteolysis of gelatin by these enzymes. The proteo-lytic enzymes may also cease to recognize the carboxylends of the aldehyde-derivatized lysine and argi-nine.231 In some cases, though the severe cross-linking may not adversely affect the bioavailability(AUC(0–1) and Cmax) of a compound, but it couldpotentially delay the onset of its absorption (Tmax) dueto the delayed shell rupture in vivo.231 Based on adetailed gamma scintigraphy study to identify thetime and location of disintegration of cross-linked andnoncross-linked capsules in vivo, Digenis et al.231
suggested that the aldehyde induced cross-links weremore susceptible to cleavage by the pancreatic
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
enzymes in the small intestine as compared to thegastric enzymes like pepsin. It may be the cause forthe delayed capsule rupture in vivo.
Dissolution of Softgel Fill
Changes in the dissolution of a fill material areusually apparent (a) when there is a change inparticle size distributions (e.g., due to Oswaldripening) and/or polymorphic nature (e.g., due tosecondary nucleation) of the suspended material in asuspension fill formulation,12 (b) when there iscrystallization of a solubilized compound from asolution fill formulation,153 or (c) when a poorlysoluble compound is dissolved in hydrophilic solventsand cosolvents (e.g., type IV formulations underPouton’s LFCS109). In the latter case, upon dilutionwith the GIT fluids in vivo or with the dissolutionmedium in vitro, the hydrophilic fill vehicle maydissolve or disperse in the aqueous fluids and therebyexpose the solubilized water-insoluble compound toaqueous fluids, leading to erratic and inconsistentprecipitating of the compound (crashing out).70
Surfactants are commonly used in the dissolutionmedium during the dissolution testing of poorlysoluble compounds and oily formula-tions.1,144,229,232–236 The use of surfactants in thedissolution medium for the dissolution testing ofpoorly soluble compounds has been proposed to bephysiologically meaningful as these surfactantsmimic those natural surfactants, such as bile acids,bile salts, and lecithin present in the GIT.232,233
Additionally, Buri and Hainbert-Droz237 suggestedthat the natural surfactants could be interchangedwith a synthetic surfactant, such as sodium laurylsulfate, for solubilizing poorly soluble compounds.The selection of a type and concentration of asurfactant and other solutes used in the medium fordissolution testing is based on a variety of factors,such as (a) aqueous solubility, ionic nature (pKa; pH-solubility profile), and dose of the compound; (b)compatibility of the surfactant with the compound;(c) compatibility of the surfactant with the gelatinshell;238–241 (d) critical micellar concentration(CMC) of the surfactant; and (e) degree of partition-ing of the compound into the surfactant micelles(i.e., micellar loading).234 Some commonly usedsurfactants in the dissolution medium includesodium lauryl sulfate (SLS), polyoxyethylene sorbi-tan monolaurate (Polysorbate 20), cetyltrimethy-lammonium bromide (CTAB), polyoxyl castor oil(Cremophor EL), hexadecyltrimethylammoniumbromide (HTAB), polyethylene glycol tert-octylphe-nyl ether (Triton), nonylphenol ethoxylate (Tergi-tol), cyclodextrins, and lecithin.234 Use of aqueous-organic solvent mixtures is strongly discouraged asthe dissolution medium for softgels as these solvents
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4133
could potentially prevent dissolution of gelatin shelleven when it is free from cross-linking, resulting inreaching misleading conclusions. In addition, theuse of these aqueous-organic medium has norelevance to the physiological environment and isnot likely to generate meaningful data for in vivointerpretation.229,232,233
Surfactants, used to improve the dissolution ofpoorly soluble compounds and oily formulations, arealso known for their denaturing effects on thedigestive enzymes used in the dissolution mediumfor cross-linked softgel formulations.242,243 The USPrecommended levels of pepsin or pancreatin may besufficient for the digestion of cross-linked softgels inthe absence of a surfactant. The cross-linked softgelswere shown to pass the tier two dissolution testingonly when the enzyme was introduced into themedium initially followed by the surfactant fewminutes afterward. These softgels failed to meetthe dissolution specification when enzyme andsurfactant were introduced into the medium togetherat the onset of dissolution testing. Thus, it wasrecommended to introduce the surfactant into thedissolution medium after the initial enzyme digestionof cross-linked softgels had occurred.244,245
Sodium lauryl sulfate, an anionic surfactant, couldbind to the cationic charges on gelatin at pH valuesequivalent to gastric pH.239,241,246–248 Due to thehigh pKa values of the amino functional groups ingelatin, these groups exist in their protonated form atgastric pH and thus are expected to undergo ionicinteractions with the negative charges on the anionicsurfactant. The anionic charges of gelatin, on theother hand, may bind to cationic surfactants, such ascetylpyridinium chloride, dodecylammonium chlor-ide,249 and dodecyl amine hydrochloride.246 Theseinteractions may influence the solubility and dissolu-tion of the capsule shell.241,247 The rate of dissolutionof a gelatin shell in acidic solutions (pH< 5), forexample, was shown to decrease as the concentrationof sodium lauryl sulfate in the dissolution mediumwas increased.241 In addition, the rate of dissolutionslowdown caused by the surfactant was shown to bemore pronounced with the increased ionic strength inthe medium. Use of a nonionic surfactant (e.g.,polysorbate 20, polysorbate 80) may be consideredin place of an ionic surfactant during the dissolutiontesting of poorly soluble compounds to minimize thegelatin-surfactant interactions and ensuing slow-down of disintegration of gelatin shells.247
Equally important, the impurities present in asurfactant and other electrolytes used in the dissolu-tion medium could influence the CMC of thesurfactant, and size and loading capacity of micellesfor a compound and thus influence the ultimatesolubility and dissolution rate of the compound in thedissolution medium.234,241,250,251 Therefore, the type
DOI 10.1002/jps JO
and concentrations of solutes in the dissolutionmedium used either to adjust pH, buffer strength,or ionic strength of the dissolution medium, or toenhance the solubility of a poorly soluble compound,could play a critical role in the dissolution of a softgelproduct. It is worthwhile to evaluate all thesevariables during the development of dissolutionmethodology for a softgel product. Examples ofdissolution procedures used for some marketed soft-gel products are presented in Table 2.
STABILITY
Physical Stability
Crystallization of Solubilized Compounds
Due to the high initial water content of theshell formulation at the time of encapsulation(� 30%),39,40,41 water migrates from the shell intothe fill and vice versa during the drying andsubsequent equilibrium processes (Fig. 2). Therate and extent of water migration is influencedby the composition of shell formulation (e.g., typeand concentration of plasticizer, presence of addi-tives),37,43 composition of fill formulation,40,41,69,72,139
and environmental conditions to which the softgelsare subjected to.43 The improved bioavailability of acompound encapsulated in a solubilized form is due tothe presentation of the compound at the site ofabsorption as a solution. The objective of improvingbioavailability of a compound through solubilizationmay be defeated if the solubilized compound crystal-lizes either within the softgel due to water migra-tion68,69 or upon coming into contact with the aqueousfluids in the GIT.70,72 Studies by Serajuddin et al.69
demonstrated gross crystallization of a poorly solubletest compound in the softgels when the compound wasencapsulated as a PEG 400 based solution. However,no such crystallization of the compound was observedin the softgels when the test compound was encapsu-lated as a Gelucire1 44/14—PEG 400 (6:1) basedsolution. The investigators attributed the absence ofcrystallization of the compound in the softgelscontaining Gelucire1 44/14—PEG 400 (6:1) basedfill to minimal migration of water from the shell intothe fill. Indeed, the reported water contents of the filland the shell at equilibrium were 6.4� 0.1% and9.6� 0.2%, respectively for the PEG 400 based fillversus 1.1� 0% and 5.6� 0.1%, respectively, for theGelucire1 44/14—PEG 400 based fill. Propyleneglycol, due to its lower viscosity and higher plasticiz-ing efficacy for gelatin, requires lower water contentin the production the gel mass compared to glycerin orsorbitol as the plasticizer. The initial lower watercontent of the shell formulation containing propyleneglycol as the plasticizer may also have the advantage
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4134 GULLAPALLI
of a comparatively smaller amount of water migrationinto the fill that could potentially minimize theprecipitation of sparingly water soluble compounds inthe fill.37 Thus, the extent of water migration withinthe softgel and ultimate physical stability of a softgelproduct can be controlled through the careful selec-tion of a suitable solubilizing vehicle as the fill and asuitable plasticizer in the shell.
Water migration from the shell into a lipid fill,though in minute amounts compared to polyethyleneglycol based fills, may still have profound conse-quences on the solubility of some compounds in lipidvehicles.252–254 The presence of water in a lipidvehicle may alter the solubility of a compound by oneof three ways: (a) via disruption of hydrogen bondingbetween the molecules of the lipid and those of thesolubilized compound and thereby reducing thesolubility if this hydrogen bonding is a major drivingforce for the solubilization of the compound in thelipid;252 (b) via inducing reorganization of the lipidmolecules within the matrix from the originalmolecular arrangement existed in the absence ofwater;254 and/or (c) via formation of a hydrate of thedissolved compound having reduced lipid solubi-lity.253 Land et al.,253 for example, have shown thatthe presence of even ultra-low water content in thelipids was sufficient to induce hydrate formation andreduce the lipid solubility of compounds, such asanhydrous testosterone.
Deterioration of Mechanical Strength of Shell
Polyethylene glycols of a lower molecular weight(�400D), used as fill vehicles, have a higher affinityfor water and glycerin used in the shell formulation,leading to the migration of these shell componentsinto the polyethylene glycol fill. The migration ofwater from the shell into a polyethylene glycol fill maybe reduced to some extent by the use of a highermolecular weight polyethylene glycol that has a lowerhygroscopicity, for example, substituting PEG 600 forPEG 400. Migration of a plasticizer from the shell intothe fill in a softgel could result in the reducedelasticity (flexibility) and increased brittleness of theshell shortly after production or on storage, especiallywhen exposed to cold temperatures.40,41,255 Theelasticity of the shell in such a case could be markedlyimproved by the inclusion of a small amount ofglycerin in the polyethylene glycol fill.255 Theimprovement in the elasticity of the softgel shellcontaining the encapsulated PEG 400 fill with theadded glycerin (or propylene glycol) was attributed tothe equilibration of glycerin between the shell and thefill, resulting in the reduced migration of theplasticizer from the shell into the fill. Substitutionof a portion of glycerin with partially dehydratedsorbitol (Sorbitol Special) as a plasticizer was also
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
shown to provide some marginal improvement in theelasticity to the shell containing the encapsulatedPEG 400 fill without the added glycerin on aging. Incontrast, it was shown that the incorporation of ormigration of increasing amounts of PEG 400 into theshell containing glycerin or sorbitol as the plasticizerreduced its elasticity and increased its brittleness.255
It was speculated that the increased PEG 400concentration in the shell may induce migration ofglycerin from the shell into the fill when glycerin wasthe plasticizer and incompatibility between PEG 400and sorbitol within the shell due to their immiscibilitywhen sorbitol was the plasticizer.
Chemical Stability
Reactions between Encapsulated Compounds andExcipients
Compounds containing reactive moieties in theirstructures have known to react with hydroxylcompounds used in softgels either as solvents,plasticizers, or to meet other functional requirements(e.g., polyethylene glycols, propylene glycol, glycerin,sorbitol, or their partial esters), resulting in theformation of esters, carbonates, and amides.100,256–260
Though these reaction products may be ultimatelyhydrolyzed back to their parent compounds in theGIT, the bioavailabilities of the parent compoundscould be reduced significantly.259,261,262 The rate andextent of these reactions between an encapsulatedcompound (or its degradation product) and a hydroxylcomponent in a softgel are influenced by (a) intrinsicchemical reactivity of the compound, (b) state andextent of ionization of the compound, (c) hydroxylcontent of the solvent, (d) water content of the fill, (e)manufacturing conditions, and (f) storage conditions.
The ionized form of a carboxylic acid compound,such as ibuprofen, (R-COO�) is relatively less reactivetowards a hydroxyl compound compared to itsunionized form (R-COOH).100 In addition, as ester-ification and hydrolytic reactions are generallyreversible, hydrolysis of the formed esters back tothe parent compounds could be potentially achievedthrough increasing the amount of available water inthe fill formulation. Thus, the rate and extent ofdegradation (i.e., esterification) of a carboxylic acidcompound in the presence of a hydroxyl compoundcan be reduced through partial ionization of thecarboxylic acid groups (complete ionization, thoughfurther reduces the esterification reaction, alsoreduces the enhanced solubilization advantagegained by using a counter ion technique, as discussedin Solubility Enhancers for Hydrophilic VehiclesSection), reduction in the amount of availablehydroxyl content (i.e., use of a lower amount of ahydroxyl solvent or use of a solvent with lowerhydroxyl content, e.g., use of PEG 600 instead of PEG
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4135
400 or MPEG instead of PEG), increase in the amountof available water in the softgel, and use of mildermanufacturing conditions.98,100,258
The decomposition of aspirin in polyethyleneglycols in the absence of water was also attributedto the transesterification reactions, resulting in theformation of salicylic acid and acetylated polyethy-lene glycols.256,257,263 These transesterification reac-tions were shown to be affected by temperature (rateat 608C> 458C> 278C> 48C) and the number ofhydroxyl reactive sites available on the polymerchain (rate in PEG>methoxyPEG>PEG acetate).
Compounds encapsulated in softgels could poten-tially undergo hydrolytic degradation during theshelf-life of a product as a result of migration of waterfrom the shell into the fill. During the softgelformulation development for a poorly water solubleinvestigational compound, VX-497, solubilized inPEG 400, Kochling et al.260 demonstrated thehydrolysis of urea bonds in the compound even atstorage temperatures as low as 58C. The hydrolyticdegradation of the compound in PEG 400 vehicle wasattributed to the moisture content of the fill, reportedto be about 7%. The investigators also demonstratedhow reactions between PEG 400 and the ureacarbonyl groups present in the compound and in itsstructurally related process impurity could result inthe formation of a series of PEGylated compounds.The PEGylation reactions of the compound and itsprocess impurity with PEG 400 were shown to betemperature and time dependent, that is, the higherthe manufacturing process temperature and thelonger the mixing time, the greater the concentra-tions of these PEGylated products. In this case, therate of degradation of the compound (i.e., PEGylation)was successfully reduced and the overall quality ofthe drug product was improved through removal ofthe process impurity during the manufacture of thedrug substance, and lowering of the manufacturingtemperature and shortening of the mixing timeduring the manufacture of the drug product.
Autooxidation of Excipients and Affects on Stability ofEncapsulated Compounds
Polyethylene glycols and materials containing poly-oxyethylene moieties in their structures are known toundergo autooxidation in the presence of oxygen andproduce reactive organic peroxides and hydrogenperoxide.95,213–218,264–268 The organic peroxides arefurther degraded to produce short chain carboxylicacids and aldehydes. It has been postulated thatpolyoxyethylene moieties undergo oxidative decom-position at high temperatures in the presence of waterto ethylene glycol, which may then be oxidized furtherto form formaldehyde.269 The amounts of thesereactive peroxides, carboxylic acids, and aldehydes
DOI 10.1002/jps JO
present in a polyethylene glycol or in a relatedmaterial are dependent upon the molecular weightand age of the polymer and the extent of itsexposure to air during its storage.214,267,270 Thehigher the molecular weight of the polymer, thelower the concentrations of these products andthe older the polymer, the greater the concentrationsof these products. The relatively higher levels of theseproducts observed in a polyoxyethylene polymer of alower molecular weight may be related to the increasein the mobility of the polymer chains allowing for theincreased autooxidation reactions.267
The reactive products generated from the autoox-idation of polyoxyethylene polymers have beenimplicated in the degradation of several compoundsformulated in polyethylene glycols.95,215,218,269,271
The carboxylic acids so formed during the autooxida-tion reactions could lower the apparent pH of anaqueous polyethylene glycol solution, the extent ofwhich is dependent upon how much autooxidation ofpolyethylene glycol might have taken place during itshandling and storage.218 The lowered pH can degradecompounds that are susceptible to hydrolytic degra-dation under acidic conditions.95 On the other hand,the reactive aldehydes can cross-link compoundscontaining amino groups in their structures throughmethylene groups.95,272–274 The mechanism of thesecross-linking reactions is similar to the methylenebonding between two amino groups on adjacentgelatin chains, as described in an earlier section.The autooxidation reactions in polyethylene glycolsand their related materials and the formation ofreactive products resulting from these reactions canbe minimized effectively by the use of an antioxidantin the fill formulation, purging the formulation withnitrogen during its manufacturing, and thoroughlydeaerating the formulation under vacuum before itsencapsulation into softgels.215,218
In addition to polyethylene glycols and theirrelated materials, peroxide and aldehyde impuritiesare also known to present in other excipientsand packaging components used with the softgelproducts.219,220,268,275–278 The peroxide impurities inpovidone, a typically used solubility enhancer andviscosifiers in softgels,55,56,67,98 are known to promotesignificant degradation of oxidatively sensitive com-pounds even in solid-state (e.g., degradation ofraloxifene to its N-oxide in tablet formulationscontaining povidone as an excipient277). These per-oxides are present in povidone initially as processimpurities and are known to increase in content withtime in the presence of atmospheric oxygen.279
Formaldehyde, a known contaminant in packagingmaterial, was shown to degrade ropinirole to itshydroxymethyl adduct.276
Equally important, the minor impurities presentin a gelatin shell, for example, ammonia resi-
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
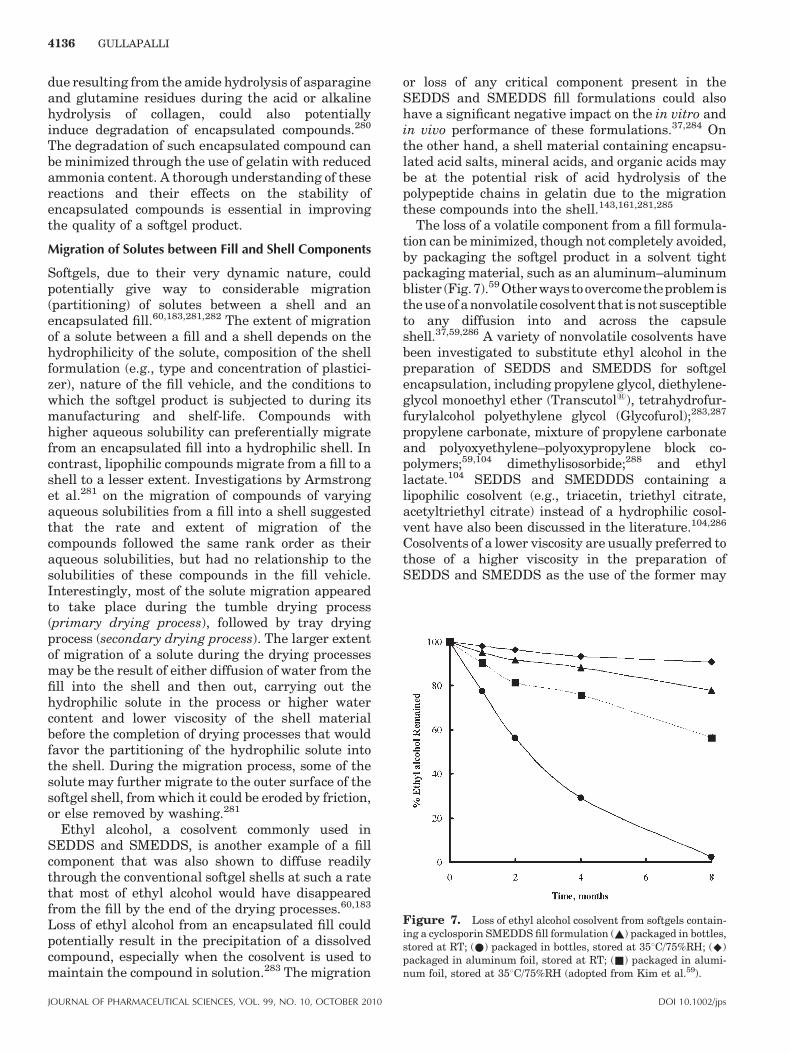
Figure 7. Loss of ethyl alcohol cosolvent from softgels contain-ing a cyclosporin SMEDDS fill formulation (~) packaged in bottles,stored at RT; (*) packaged in bottles, stored at 358C/75%RH; (^)packaged in aluminum foil, stored at RT; (&) packaged in alumi-num foil, stored at 358C/75%RH (adopted from Kim et al.59).
4136 GULLAPALLI
due resulting from the amide hydrolysis of asparagineand glutamine residues during the acid or alkalinehydrolysis of collagen, could also potentiallyinduce degradation of encapsulated compounds.280
The degradation of such encapsulated compound canbe minimized through the use of gelatin with reducedammonia content. A thorough understanding of thesereactions and their effects on the stability ofencapsulated compounds is essential in improvingthe quality of a softgel product.
Migration of Solutes between Fill and Shell Components
Softgels, due to their very dynamic nature, couldpotentially give way to considerable migration(partitioning) of solutes between a shell and anencapsulated fill.60,183,281,282 The extent of migrationof a solute between a fill and a shell depends on thehydrophilicity of the solute, composition of the shellformulation (e.g., type and concentration of plastici-zer), nature of the fill vehicle, and the conditions towhich the softgel product is subjected to during itsmanufacturing and shelf-life. Compounds withhigher aqueous solubility can preferentially migratefrom an encapsulated fill into a hydrophilic shell. Incontrast, lipophilic compounds migrate from a fill to ashell to a lesser extent. Investigations by Armstronget al.281 on the migration of compounds of varyingaqueous solubilities from a fill into a shell suggestedthat the rate and extent of migration of thecompounds followed the same rank order as theiraqueous solubilities, but had no relationship to thesolubilities of these compounds in the fill vehicle.Interestingly, most of the solute migration appearedto take place during the tumble drying process(primary drying process), followed by tray dryingprocess (secondary drying process). The larger extentof migration of a solute during the drying processesmay be the result of either diffusion of water from thefill into the shell and then out, carrying out thehydrophilic solute in the process or higher watercontent and lower viscosity of the shell materialbefore the completion of drying processes that wouldfavor the partitioning of the hydrophilic solute intothe shell. During the migration process, some of thesolute may further migrate to the outer surface of thesoftgel shell, from which it could be eroded by friction,or else removed by washing.281
Ethyl alcohol, a cosolvent commonly used inSEDDS and SMEDDS, is another example of a fillcomponent that was also shown to diffuse readilythrough the conventional softgel shells at such a ratethat most of ethyl alcohol would have disappearedfrom the fill by the end of the drying processes.60,183
Loss of ethyl alcohol from an encapsulated fill couldpotentially result in the precipitation of a dissolvedcompound, especially when the cosolvent is used tomaintain the compound in solution.283 The migration
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
or loss of any critical component present in theSEDDS and SMEDDS fill formulations could alsohave a significant negative impact on the in vitro andin vivo performance of these formulations.37,284 Onthe other hand, a shell material containing encapsu-lated acid salts, mineral acids, and organic acids maybe at the potential risk of acid hydrolysis of thepolypeptide chains in gelatin due to the migrationthese compounds into the shell.143,161,281,285
The loss of a volatile component from a fill formula-tion can be minimized, though not completely avoided,by packaging the softgel product in a solvent tightpackaging material, such as an aluminum–aluminumblister (Fig.7).59 Otherwaystoovercometheproblemisthe use of a nonvolatile cosolvent that is not susceptibleto any diffusion into and across the capsuleshell.37,59,286 A variety of nonvolatile cosolvents havebeen investigated to substitute ethyl alcohol in thepreparation of SEDDS and SMEDDS for softgelencapsulation, including propylene glycol, diethylene-glycol monoethyl ether (Transcutol1), tetrahydrofur-furylalcohol polyethylene glycol (Glycofurol);283,287
propylene carbonate, mixture of propylene carbonateand polyoxyethylene–polyoxypropylene block co-polymers;59,104 dimethylisosorbide;288 and ethyllactate.104 SEDDS and SMEDDDS containing alipophilic cosolvent (e.g., triacetin, triethyl citrate,acetyltriethyl citrate) instead of a hydrophilic cosol-vent have also been discussed in the literature.104,286
Cosolvents of a lower viscosity are usually preferred tothose of a higher viscosity in the preparation ofSEDDS and SMEDDS as the use of the former may
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4137
be better able to promote emulsification with minimaleffort.
ALTERNATE DELIVERY STRATEGIES FORNONAQUEOUS FORMULATIONS
Softgel delivery system offers the advantage ofconveniently delivering a nonaqueous liquid orsemi-solid matrix containing a dissolved or dispersedcompound as a unit dose solid dosage form. However,a variety of factors may influence the decision makingprocess of an organization in the development andintroduction of a compound into commercialization asa softgel product. These factors may be strategic ortechnical and worth further discussion:
(1) M
DOI 10.
anufacture of softgels is inherently a verycomplex, labor-intensive, and time consumingprocess that requires not only acquiring andmaintaining specialized and costly equipment,such as gel reactors, encapsulation machines,dyes, tumble dryers, and drying tunnels butalso in-house technical and operational exper-tise and sourcing high quality gelatin. As aconsequence, organizations are reluctant toset up their own softgel manufacturing opera-tion in-house and prefer to outsource theactivities to an external softgel contract man-ufacturer.
(2) D
ue to the availability of only few contractmanufacturing organizations (CMOs) that spe-cialize in the manufacture of pharmaceuticalquality softgel products (e.g., PharmaceuticsInternational, Inc., Banner Pharmacaps, Inc.,Catalent Pharma Solutions, Accucaps Indus-tries Ltd, Pharmagel Engineering SPA),lengthy product development and manufactur-ing lead timelines are not unusual at theCMOs.(3) A
vailability of limited quantities of a compoundduring the early stages of development maydiscourage an organization from developing asoftgel product for the compound as an earlystage clinical dosage form.(4) I
ntellectual property (IP) rights of a CMO onthe composition of a shell formulation maycomplicate issues related to transferring andmanufacturing the softgel product at an alter-nate CMO, especially when a specialized shellcomposition is used in the product.(5) T
echnically, the high initial water content ofthe shell formulation at the time of encapsula-tion and subsequent migration of any amountof water between the shell and the fill formula-tions could make the softgel dosage form unsui-table for encapsulating compounds that are1002/jps JOURNAL
prone to water induced crystallization or hydro-lytic degradation.
(6) E
ven after an organization chooses to initiatedevelopment and commercialization of a com-pound as a softgel product with improved bioa-vailability as the first-line introductory dosageform, the relatively higher production costscompared to those of some other dosage forms,formulation/excipient related issues, and/orsome other aforementioned issues would encou-rage the organization to pursue an alternatedosage form at a later stage. Some examples ofsuch fruitions include reformulation of Agener-ase1 softgel into Lexiva1 tablet containing aprodrug of the compound (GlaxoSmithKline),reformulation of Fortovase1 softgel intoInvirase1 tablet and capsule containing themesylate salt of the compound (Roche Pharma-ceuticals), and reformulation of Kaletra1softgel into tablet with increased drug loading,improved stability, lowered restrictions on sto-rage conditions, and reduced food effects(Abbott Laboratories). On the other hand, somecompounds that were originally introduced intocommercialization as a conventional tablet orcapsule dosage form were reformulated intosoftgels as a part of an organization’s life-cyclemanagement strategy or some other reason(e.g., Hytrin1, Abbott Laboratories; Claritin1,Schering-Plough). Some softgel products (e.g.,Zantac1, GlaxoSmithKline) were withdrawnfrom the market entirely as they offered noadditional advantage over the tablet formula-tions. Some additional information on the refor-mulation efforts of other softgels are presentedin Table 2.
When formulation of a compound in a nonaqueousvehicle is shown to be the practical option to achieveits acceptable bioavailability (e.g., Neoral1), dictatedby its physical nature (i.e., low melting point, oily,waxy) (e.g., Depakene1), and/or dictated by its ultra-low to low dose requirements (e.g., Rocaltrol1), theprobable solid oral dosage form that has good patientacceptability is the capsule.289 Hard gelatin capsule(HGC) dosage form has been advanced as an alternateto softgel dosage form for encapsulating nonaqueousliquid and semisolid formulations.289–296 There aresubstantial differences between the two types gelatincapsules and each dosage form has been shown tohave its own advantages and challenges. The choice ofsoft gelatin or hard gelatin capsule for encapsulatingliquids will depend on a variety of factors that will bediscussed later. First of all, it is useful to discussbriefly how a formulation scientist can strategizevarious options creatively to overcome some of
OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4138 GULLAPALLI
the challenges posed by the softgel dosage formdevelopment.
It is a reasonable approach to separate the softgeldosage form development into two stages: (1) devel-opment of ‘‘softgel compatible’’ fill formulation in-house using knowledge provided in the currentmanuscript and other literature and (2) encapsula-tion and scale-up manufacturing of the softgelproduct at a CMO using a gel mass of compositioncommonly used across various softgel manufacturers.This approach allows an organization to develop thefill formulation in-house with smaller quantities ofthe compound, keep control of its formulation,process, and IP, and also offers the flexibility ofmanufacturing the final softgel product at more thana single manufacturer. Another advantage of thisapproach is that the fill formulation so developed canbe packaged as a Liquid-In-Bottle (LIB) product andused to advance the early phases of a clinical programby either encapsulating the liquid into two-piece hardgelatin capsule (Liquid-In-Capsule, LIC) or dispen-sing unit doses of the liquid into individual containersfor dilution with a compatible vehicle before use.297
The organization can, in the interim, develop analternate dosage form, if feasible, evaluate the optionof further development of liquid filled hard gelatincapsule (HGC) dosage form, or select softgel CMOsand negotiate contracts, if softgel dosage form is theonly practical option for the compound and to furtheradvance the clinical program. Koon298 has providedsome additional tips on selecting a suitable softgelcontract manufacturer.
When formulation of a compound in a nonaqueousvehicle is shown to be the only practical option, thechoice to continue with liquid filled HGCs or switch tosoftgels for later stage development and subsequentcommercialization is generally determined by twofactors.
Dosage form related: (a) tolerance of capsule shelltowards the fill composition (i.e., hydrophilic orlipophilic, type and amount of PEG and cosolvents,amount of water) and (b) tolerance of fill towards theshell water content (i.e., crystallization and/orhydrolytic degradation of an encapsulated com-pound).
Encapsulation related: (a) existing in-house devel-opment and manufacturing capabilities and person-nel expertise and (b) ease and cost of settingcapabilities and improving personnel expertise.
Empty hard gelatin capsule (HGC) shells aremanufactured separately and supplied for encapsu-lating either a powder fill or a liquid fill. HGC shellsare generally thinner and lighter than those ofsoftgels and are commonly manufactured fromgelatin, water, a colorant(s), and/or an opacifier.HGC shells, unlike those of softgels, do not contain anonvolatile plasticizer (e.g., glycerin, sorbitol) and
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
thus presence of water in the shells is essential tomaintain their integrity. HGC shells must retain amoisture content of 10–18% to maintain theirflexibility.299,300 Below this range, the shells becomebrittle and are prone to breakage, while above thisrange, the shells may deform. Thus, any alteration tothis moisture range, due to migration of moisturebetween the shell and encapsulated fill or between theshell and external environment, could be detrimentalto the integrity of the HGC shells. In contrast, thepresence of plasticizer(s) in the softgel shell impartselasticity to the shell that allows it to accommodate awide range of hydrophilic excipients. For example,polyethylene glycols with molecular weight as low as400 which are hydrophilic have been successfullyencapsulated into softgels (Tab. 2), whereas HGCshells have been reported to be compatible withpolyethylene glycols with molecular weight higherthan 4000.289,296,301 Water and highly hygroscopicexcipients, such as glycerin, sorbitol, propyleneglycol, have also been reported to be incompatiblewith the HGC shells at concentrations as low as5%.291,296 Furthermore, the relatively higher elasti-city of the softgel shell resulting from the presence ofa plasticizer would also provide additional protectionto the softgel product during its handling. On theother hand, due to the substantially lower watercontent of the HGC shells, its migration into the fillwould be insignificant and thus prevent or minimizeany water induced crystallization or hydrolyticdegradation of the dissolved compound encapsulatedin the HGC.
A softgel is a one-piece, hermetically sealed shellcapsule, which is formed, filled entirely with noheadspace, and sealed in one operation. In case ofHGCs, the body of the capsule is filled, capped, andsealed sequentially, leaving substantial headspacewithin�10% of capsule volume.292,302 The presence ofthe headspace within the HGCs may compromise theelegance of a product designed with a transparentshell formulation and the oxidative stability of anencapsulated compound. The loss of fillable volume ina HGC may also result in a relatively larger capsulesize compared to that of a softgel containing a similarfill volume.
As empty HGC shells are supplied ready for usewith various fill volumes292,302 and laboratory scaleequipment is also readily available for filling andsealing HGCs,292,295,302 small scale batches of a liquidfilled HGC product can be easily manufactured in-house using small quantities of a compound forstability and clinical trial purposes during the earlystages of drug development. In fact, based on theauthor’s experience, these small scale batches canalso be manufactured easily using simple equipment,such as a ProFillTM Capsule Filling system (Torpac,Inc. http://www.torpac.com/) and a positive displace-
DOI 10.1002/jps

Table 5. Small Scale Softgel Encapsulation Machines
Machine Designation Supplier Die Roll Specificationsa
R&D 4 inch CapPlus Technologies (Phoenix, AZ) Max. die speed—5 rpm, die rolls—4 inch long2.83 inch diameter
RGY6x15F,RGY6x15S
Pharmaker Machinery (Elmont, NY) Max. die speed—4 rpm, die rolls—3.94 inch long2.52 inch diameter
PSG-100 Pharmaland Technology (Ontario, Canada) Max. die speed—5 rpm, die rolls—3.94 inch long2.56 inch diameter
SS-30 Sky Softgel Co. (Incheon, Korea) Max. die speed—5 rpmRG0.8-110 Zhejiang Fuchang Machinery Co.
(Zhejiang Province, China)Max. die speed—7 rpm, die rolls—4.33 inch long2.83 inch diameter
aOutput depends on softgel size and number of cavities and rpm of die roll.
SOFT GELATIN CAPSULES 4139
ment pipette. Moreover, unlike softgel manufactur-ing, as there is no need for a time consuming gel masspreparation and capsule drying process, liquid filledand sealed HGCs can be manufactured and packagedwithin a short time. The readers are directed to theliterature published by Cole et al.,289 Cole,292
Smith,295 and Rowley303 to obtain any furtherunderstanding of the manufacturing process for theliquid filled HGCs and their characteristics.
R&D scale softgel manufacturing machines havealso been introduced for the production of softgels at asmaller scale. Some of these machines are commer-cially available and offer an organization with theoption to keep its softgel development and manufac-turing activities in-house. Some of the softgelencapsulation machines available commercially arepresented in Table 5. A laboratory scale encapsula-tion machine (Minicap) mimicking the design of astandard softgel encapsulation machine, but utilizinga single pocket die design and requiring as little as50 mL of fill solution has been discussed in theliterature.304 The Minicap has been shown to producesoftgels with comparable qualities as those manu-factured using a standard softgel machine and thuscan be used to produce prototype softgels for fill-shellcompatibility screening and stability evaluation.However, the Minicap machine, developed by Cardi-nal Health (currently Catalent Pharma Solutions),may not be available for purchase.
Though softgel manufacturing machines and aux-iliary equipment are readily available for purchase,due to enormous resources required, for example,designing facilities, sourcing and controlling qualitiesof gelatin, optimizing manufacturing process for gelmass, acquiring and developing in-house talent andso on, an organization has to carefully evaluate theoption of establishing softgel manufacturing capabil-ities in-house against outsourcing and managingthe outsourced activities. It is also essential to bear inmind that the major CMOs that specialize in themanufacture of softgels have their own in-housemachine shop capabilities that can build, replace,
DOI 10.1002/jps JO
and/or repair any existing machine parts right awaywith minimal loss of time.
CONCLUSIONS
Softgels constitute a unique solid dosage form thatcan be used to conveniently encapsulate nonaqueoussolution, suspension, and semisolid formulationsdeveloped to improve the bioavailability of poorlysoluble compounds. In addition to several softgelproducts that are already available commercially, thepatent literature and the number of softgel productscurrently under clinical investigations305 clearlydemonstrate that several pharmaceutical and biotechfirms are actively pursuing the softgel dosage form asa choice to formulate compounds with poor biophar-maceutical properties. The potential of the softgeldosage form in improving the in vitro and in vivoperformance and the physical and chemical stabilityof an encapsulated compound can be maximizedthrough the careful selection of appropriate excipi-ents in the fill and shell formulations. While thedosage form provides immense promise for a varietyof poorly soluble compounds, its dynamic naturecompels the formulation scientist to put in moreefforts than during the development of other conven-tional solid dosage forms, such as tablets andtwo-piece hard gelatin capsules. The potential risksthat may arise during the development and shelf-lifeof a softgel product can be managed through theuse of:
� a
URNA
stable physical form of the drug substance,
� a fill vehicle that minimizes the transfer of waterfrom the shell into the fill and/or lessens theeffect migrated water on the solubilized stateof the drug substance,
� e
xcipients free from interactions with the drugsubstance and/or with each other,� e
xcipients free from impurities (e.g., aldehydes,peroxides) that may adversely effect the gelatinL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

JOURN
4140 GULLAPALLI
shell dissolution process and/or the chemicalstability of the drug substance,
� p
lasticizer(s) that minimizes the transfer of com-ponents from the fill (e.g., ethyl alcohol) or intothe fill (e.g., oxygen, moisture),� in
ert environment during manufacturing, forexample, nitrogen blanketing for oxygen sensi-tive compounds, yellow light for photosensitivecompounds,� t
horough deaeration of the fill formulation toremove any dissolved air (oxygen),� m
oderate drying conditions, and � a ppropriate storage conditions, that is, container/closure, temperature, and relative humidity.
Finally, softgels may not be the appropriate dosageform for some compounds that are highly moisturesensitive or have high therapeutic dose requirements,and alternate dosage forms may be more desirableand cost- and time-effective in such a case.
ACKNOWLEDGMENTS
The author thanks the Library Staff at Elan Phar-maceuticals, Ms. Nancy Phelps, Mr. Cary Cochrell,and Ms. Praveena Raman, for their help in collectingrelevant literature to complete the manuscript. Theauthor also thanks Staff of Elan’s IT department fortheir assistance in the preparation of figures.
REFERENCES
1. Shah VP, Noory A, Noory C, McCullough B, Clarke S, EverettR, Naviasky H, Srinivasan BN, Fortman D, Skelly JP. 1995.In vitro dissolution of sparingly water-soluble drug dosageforms. Int J Pharm 125:99–106.
2. Crowley PJ, Martini LG. 2004. Physicochemical approaches toenhancing oral absorption. Pharm Technol Eur 16:18–27.
3. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 1997.Experimental and computational approaches to estimate solu-bility and permeability in drug discovery and developmentsettings. Adv Drug Deliv Rev 23:3–25.
4. Lipinski C. 2003. Physicochemical properties in drug design/development. 2nd International Drug Discovery and Devel-opment Summit: Novel Concepts and Technologies to Accel-erate Drug Development. Honolulu, HI.
5. Agharkar S, Lindenbaum S, Higuchi T. 1976. Enhancement ofsolubility of drug salts by hydrophilic counter-ions: Properties oforganic salts of an anti-malarial drug. J Pharm Sci 65:747–749.
6. Bastin RJ, Bowker MJ, Slater BJ. 2000. Salt selection andoptimisation procedures for pharmaceutical new chemicalentities. Org Proc Res Dev 4:427–435.
7. Serajuddin ATM. 2007. Salt formation to improve drug solu-bility. Adv Drug Deliv Rev 59:603–616.
8. Dannenfelser R, He H, Joshi Y, Bateman S, SerajuddinATM. 2004. Development of clinical dosage forms for apoorly water soluble drug I: Application of polyethyleneglycol-polysorbate 80 solid dispersion carrier system.J Pharm Sci 93:1165–1175.
AL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
9. Crew MD, Curatolo WJ, Friesen DT, Gumkowski MJ, LorenzDA, Nightlingale JAS, Ruggeri RB, Shanker RM. 2007. Phar-maceutical compositions of cholesteryl ester transfer proteininhibitors. US Patent Application 2007/0282009.
10. Stella VJ, Nti-Addae KW. 2007. Prodrug strategies to over-come poor water solubility. Adv Drug Deliv Rev 59:677–694.
11. Merisko-Liversidge E, Liversidge GG, Cooper ER. 2003. Nano-sizing: A formulation approach for poorly-water-soluble com-pounds. Eur J Pharm Sci 18:113–120.
12. Kipp JE. 2004. The role of solid nanoparticle technology in theparenteral delivery of poorly water-soluble drugs. Int J Pharm284:109–122.
13. Kesisoglou F, Panmai S, Wu Y. 2007. Nanosizing-oral formu-lation development and biopharmaceutical evaluation. AdvDrug Deliv Rev 59:631–644.
14. Hauss DJ. 2007. Oral lipid-based formulations. Adv DrugDeliv Rev 59:667–676.
15. Strickley RG. 2004. Solubilizing excipients in oral and inject-able formulations. Pharm Res 21:201–230.
16. Gumkowski MJ, Fournier LA, Tierney NK, Curatolo WJ.1994. Improved bioavailability through use of soft gelatincapsule formulations of terlakiren, a tripeptide rennin inhi-bitor. Pharm Res 11:S-286.
17. Yu L, Bridgers A, Polli J, Vickers A, Long S, Roy A, Winnike R,Coffin M. 1999. Vitamin E-TPGS increases absorption flux ofan HIV protease inhibitor by enhancing its solubility andpermeability. J Pharm Sci 16:1812–1817.
18. Hugger ED, Novak BL, Burton PS, Audus KL, Borchardt RT.2002. A comparison of commonly used polyethoxylated phar-maceutical excipients on their ability to inhibit P-glycoproteinactivity in vitro. J Pharm Sci 91:1991–2002.
19. Rege BD, Kao JP, Polli JE. 2002. Effects of nonionic surfac-tants on membrane transporters in Caco-2 cell monolayers.Eur J Pharm Sci 16:237–246.
20. Seeballuck F, Ashford MB, O’Driscoll CM. 2003. Theeffects of pluronics block copolymers and cremophor ELon intestinal lipoprotein processing and the potential linkwith P-glycoprotein in Caco-2 cells. Pharm Res 20:1085–1092.
21. Wandel C, Kim RB, Stein M. 2003. ‘‘Inactive’’ excipients suchas Cremophor can affect in vivo drug disposition. Clin Phar-macol Ther 73:394–396.
22. Cornaire G, Woodley J, Hermann P, Cloarec A, Arellano C,Houin G. 2004. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int J Pharm278:119–131.
23. Shono Y, Nishihara H, Matsuda Y, Furukawa S, Okada N,Fujita T, Yamamoto A. 2004. Modulation of intestinal P-glycoprotein function by cremophor EL and other surfactantsby an in vitro diffusion chamber method using the isolated ratintestinal membranes. J Pharm Sci 93:877–885.
24. Pole DL. 2008. Physical and biological considerations for theuse of nonaqueous solvents in oral bioavailability enhance-ment. J Pharm Sci 97:1071–1088.
25. Scherer RP. 1933. Capsule making machine. Canadian PatentCA333007.
26. Scherer RP. 1943. Capsulating mechanism. Canadian PatentCA416849.
27. Scherer RP. 1943. Capsulating mechanism. Canadian PatentCA416850.
28. Scherer RP. 1944. Capsule forming apparatus. CanadianPatent CA422183.
29. Scherer RP. 1948. Capsulating machine. Canadian PatentCA453243.
30. Holland NJ, Tidy GB. 2004. Gelatin encapsulation techniques.US Patent 6,769,226.
31. Carlson GH, Scherer RP. 1960. Process for improvingadhesivity of gelatin used in preparing capsules by con-
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4141
tinuously agitating the gelatin until casting. US Patent2,928,128.
32. Schmidt W. 1999. Method for the purification and recovery ofwaste gelatin using diafilters. US Patent 5,945,001.
33. Stanley JP. 1986. Soft gelatin capsules. In: Lachman L, Lie-berman HA, Kanig JL, editors. The theory and practice ofindustrial pharmacy. 3rd edition. Philadelphia: Lea and Febi-ger. pp. 398–412.
34. Ling WC. 1978. Thermal degradation of gelatin as applied toprocessing of gel mass. J Pharm Sci 67:218–223.
35. Alex R, Gerhards J, Kraemer-Pittrof I, Oeschger R, Rades T.2000. Process for the manufacture of liquid filled capsules.WO/2000/028942.
36. Scherer RP. 1958. Film forming device. US Patent 2,842,797.37. Brox W, Meinzer A, Zande H. 2006. Soft gelatin capsule
manufacture. US Patent 7,078,054.38. SchererRP. 1958. Method for drying gelatin capsules. US
Patent 2,851,786.39. Seager H. 1985. Soft gelatin capsules: A solution to many
tableting problems. Pharm Technol 9:84–104.40. Brox W. 1988. Soft gelatin capsules and methods for their
production. US Patent 4,744,988.41. Brox W. 1988. Gelatin capsule. US Patent 4,780,316.42. Steele D, Dietel G. 1993. Softgel manufacturing process. US
Patent 5,200,191.43. Hom FS, Veresh SA, Ebert WR. 1975. Soft gelatin capsules II.
Oxygen permeability study of capsule shells. J Pharm Sci64:851–857.
44. Kozlov PV, Burdygina GI. 1983. The structure and propertiesof solid gelatin and the principles of their modification. Poly-mer 24:651–666.
45. YakimetsI,WellnerN,SmithAC,WilsonRH,FarhatI,MitchellJ. 2005. Mechanical properties with respect to water content ofgelatin films in glassy state. Polymer 46:12577–12585.
46. Marques MRC, Cole E, Kruep D, Gray V, Murachanian D,Brown WE, Giancaspro GI. 2009. Liquid-filled gelatin cap-sules. Pharmacopeial Forum 35:1029–1041.
47. Reich VG. 1995. Effect of drying conditions on structure andproperties of gelatin capsules: Studies with gelatin films.Pharm Ind 57:63–67.
48. Julius K, Bagusche C. 1998. Method and apparatus for sortingcapsules. US Patent 5,819,953.
49. Benford S, Scherer RP, Moreland ST. 1956. Machine forbranding gelatine capsules. Canadian Patent CA530590.
50. Jimerson RF. 1986. Soft gelatin capsule update. Drug Dev IndPharm 12:1133–1144.
51. Wilkinson PK, Hom FS. 1990. Softgels: Manufacturing con-siderations. In: Tyle P, editor. Specialized drug delivery sys-tems—Manufacturing and production technology. New York:Marcel Dekker, Inc. pp. 409–449.
52. Gao P, Charton M, Morzowich W. 2006. Speeding develop-ment of poorly soluble/poorly permeable drugs by SEDDS/S-SEDDS formulations and prodrugs (Part II). Am Pharm Rev9:16–23.
53. Amidon GL, Lennernas H, Shah VP, Crison JR. 1995.A theoretical basis for a biopharmaceutic drug classification:The correlation of in vitro drug product dissolution and in vivobioavailability. Pharm Res 12:413–420.
54. Nazzal S, Wang Y. 2001. Characterization of soft gelatincapsules by thermal analysis. Int J Pharm 230:35–45.
55. Dhabhar DJ. 1996. Process for reducing the precipitationof difficulty soluble pharmaceutical actives. US Patent5,484,606.
56. Dhabhar DJ. 1996. Concentrated acetaminophen solutioncompositions. US Patent 5,510,389.
57. Cao N, Yang X, Fu Y. 2009. Effects of various plasticizers onmechanical and water vapor barrier properties of gelatinfilms. Food Hydrocolloids 23:729–735.
DOI 10.1002/jps JO
58. Tralhao AML, Watkinson AC, Brain KR, Hadgraft J, Arm-strong NA. 1995. Use of ATR-FTIR spectroscopy to study thediffusion of ethanol through glycerogelatin films. Pharm Res12:572–575.
59. Kim JW, Shin HJ, Yang SG. 1997. Cyclosporine-containingpharmaceutical composition. WO/1997/036610.
60. Moreton RC, Armstrong NA. 1998. The effect of film composi-tion on the diffusion of ethanol through soft gelatin films. Int JPharm 161:123–131.
61. Mallis GI, Schmidt DH, Lindenbaum J. 1975. Superior bioa-vailability of digoxin solution in capsules. Clin Pharnac Ther18:761–768.
62. Gardella LA, Kesler H. 1977. Gelatin-encapsulated digoxin solu-tions and method of preparing the same. US Patent 4,002,718.
63. Ghirardi P, Catenazzo G, Mantero O, Merotti GC, Marzo A.1977. Bioavailability of digoxin in a new soluble pharmaceu-tical formulation in capsules. J Pharm Sci 66:267–269.
64. Johnson BF, Smith G, French J. 1977. The comparability ofdosage regimens of lanoxin tablets and lanoxicaps. Br J ClinPharmacol 4:209–211.
65. Grainger N. 1980. Pharmaceutical compositions. US Patent4,198,391.
66. Launchbury AP, Morton FSS, Rowe D. 1991. PharmaceuticalCompositions. WO/1991/007950.
67. Erlich L, Yu D, Pallister DA, Levinson RS, Gole DG, Wilk-inson PA, Erlich RE, Reeve LE, Viegas TX. 1999. Relativebioavailability of danazol in dogs from liquid-filled hard gela-tin capsules. Int J Pharm 179:49–53.
68. Groves MJ, Bassett B, Sheth V. 1984. The solubility of 17 b-oestradiol in aqueous polyethylene glycol 400. J Pharm Phar-macol 36:799–802.
69. Serajuddin ATM, Sheen PC, Augustine MA. 1986. Watermigration from soft gelatin capsule shell to fill material andits effect on drug solubility. J Pharm Sci 75:62–64.
70. Aungst BJ, Nguyen NH, Rogers NJ, Rowe SM, Hussain MA,White SJ, Shum L. 1997. Amphiphilic vehicles improve theoral bioavailability of a poorly soluble HIV protease inhibitorat high doses. Int J Pharm 156:79–88.
71. Pouton CW. 2000. Lipid formulations for oral administrationof drugs: Non-emulsifying, self-emulsifying and ‘self-microe-mulsifying’ drug delivery systems. Eur J Pharm Sci 11:S93–S98.
72. Roy A, Winnike R, Long S, Rhodes C, Bao Z, Robertson G. 2001.Development of a soft gelatin capsule formulation of a poorlysoluble compound X using a self emulsifying delivery system. In:Oral Drug Absorption: A Renaissance, B.T. Saint-Priest, Cedex,France: Gattefosse N8 94. Gattefosse Corporation. pp. 141–149.
73. Sukuru K. 2007. Hydrophilic vehicle-based dual controlledrelease matrix system. WO/2007/050975.
74. Cody SL, Devlin BT, Dubek J, Hoy MR. 1999. Fill material forsoft gelatin pharmaceutical dosage form. US Patent 5,919,481.
75. Cody SL, Hoy MR, Walter EJ. 1997. Gelling agent for poly-ethylene glycol. US Patent 5,660,859.
76. Chadwick VS, Phillips SF, Hofmann AF. 1977. Measurementsof intestinal permeability using low molecular weight poly-ethylene glycols (PEG-400). 2. Application to normal andabnormal permeability states in man and animals. Gastro-enterology 73:247–251.
77. Chadwick VS, Phillips SF, Hofmann AF. 1977. Measurementsof intestinal permeability using low molecular weight poly-ethylene glycols (PEG 400). I. Chemical analysis and biologicproperties of PEG 400. Gastroenterology 73:241–246.
78. Basit AW, Podczeck F, Newton JM, Waddington WA, Ell PJ,Lacey LF. 2002. Influence of polyethylene glycol 400 on thegastrointestinal absorption of ranitidine. Pharm Res 19:1368–1374.
79. Krugliak P, Hollander D, Ma TY, Tran D, Dadufalza VD, KatzKD, Le K. 1989. Mechanisms of polyethylene glycol 400 per-
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4142 GULLAPALLI
meability of perfused rat intestine. Gastroenterology 97:1164–1170.
80. Shaffer CB, Critchfield FH. 1947. The absorption and excre-tion of the solid polyethylene glycols (carbowax compounds).J Am Pharm Assoc Sci Ed 36:152–157.
81. Shaffer CB, Critchfield FH, Nair JH III. 1950. The absorptionand excretion of a liquid polyethylene glycol. J Am PharmAssoc Sci Ed 39:340–344.
82. DiPiro JT, Michael KA, Clark BA, Dickson P, Tedesco FJ.1986. Absorption of polyethylene glycol after administra-tion of a PEG-electrolyte lavage solution. Clin Pharm 5:153–155.
83. Leung HW, Ballantyne B, Hermansky SJ, Frantz SW. 2000.Peroral subchronic, chronic toxicity and pharmacokineticstudies of a 100-kDa polymer of ethylene oxide (Polyox N-10) in the Fischer 344 rat. Int J Toxicol 19:305–312.
84. Fruijtier-Polloth C. 2005. Safety assessment on polyethyleneglycols (PEGs) and their derivatives as used in cosmeticproducts. Toxicology 214:1–38.
85. Basit AW, Newton JM, Short MD, Waddington WA, Ell PJ,Lacey LF. 2001. The effect of polyethylene glycol 400 ongastrointestinal transit: Implications for the formulation ofpoorly-water soluble drugs. Pharm Res 18:1146–1150.
86. Schulze JDR, Waddington WA, Ell PJ, Parsons GE, CoffinMD, Basit AW. 2003. Concentration-dependent effects of poly-ethylene glycol 400 on gastrointestinal transit and drugabsorption. Pharm Res 20:1984–1988.
87. Schulze JDR, Peters EE, Vickers AW, Staton JS, Coffin MD,Parsons GE, Basit AW. 2005. Excipient effects on gastroin-testinal transit and drug absorption in beagle dogs. Int JPharm 300:67–75.
88. Haruta S, Iwasaki N, Ogawara K, Higaki K, Kimura T. 1998.Absorption behaviour of orally administered drugs in ratstreated with propantheline. J Pharm Sci 87:1081–1085.
89. Johnson BM, Charman WN, Porter CJH. 2002. An in vitroexamination of the impact of polyethylene glycol 400, PluronicP85, and vitamin E d-a-tocopheryl polyethylene glycol 1000succinate on P-glycoprotein efflux and enterocyte-based meta-bolism in excised rat intestine. AAPS PharmSci 4:1–13.
90. Yu MS, Hom FS, Chakrabarti S, Huang C, Patel M. 1991.Solvent system enhancing the solubility of pharmaceuticalsfor encapsulation. US Patent 5,071,643.
91. Morton FSS, Shelley R, Patel MS. 1994. Enhanced solubilitypharmaceutical solutions. US Patent 5,376,688.
92. Shelley RS, Wei Y, Linkin D. 1996. Gelatin capsules contain-ing a highly concentrated acetaminophen solution. US Patent5,505,961.
93. Seager H. 1993. Soft gelatin capsule technology—A route toimproved drug delivery. Pharm Mfg Rev 5:9–10.
94. Croome RJ. 1953. Acid and alkaline hydrolysis of gelatin.J Appl Chem 3:280–286.
95. Bindra DS, Williams TD, Stella VJ. 1994. Degradation of O6-benzylguanine in aqueous polyethylene glycol 400 (PEG 400)solutions: Concerns with formaldehyde in PEG 400. PharmRes 11:1060–1064.
96. Argiriadi AA, Blank RG, Giamalva DH. 1995. Ibuprofenenhancing solvent system. US Patent 5,468,502.
97. Li G, Zheng X, Shelley R, Waranis RP. 2003. Acetaminophen(APAP) solubilization for softgels. AAPS Annual Meeting,Poster 2641.
98. Gullapalli RP. 2001. Ibuprofen-containing softgels. US Patent6,251,426.
99. Kibbe AH. 2006. Povidone. In: Rowe RC, Sheskey PJ, OwenSC, editors. Handbook of pharmaceutical excipients. 5th edi-tion. London: Pharmaceutical Press and Washington, DC:American Pharmacists Association. pp. 611–616.
100. Patel MS, Morton FSS, Seager H, Howard D. 1992. Factorsaffecting the chemical stability of carboxylic acid drugs in
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
enhanced solubility system (ESS) softgel formulations basedon polyethylene glycol (PEG). Drug Dev Ind Pharm 18:1–19.
101. Pouton CW. 2005. Formulation of poorly water-soluble drugsfor oral administration. In: Drug Candidate Optimization,Formulation and Early Development, B.T. GattefosseN8 98. Saint-Priest, Cedex, France: Gattefosse Corporation.pp. 39–51.
102. Pouton CW. 2006. Formulation of poorly water-soluble drugsfor oral administration: Physicochemical and physiologicalissues and the lipid formulation classification system. Eur JPharm Sci 29:278–287.
103. Charman SA, Charman WN, Rogge MC, Wilson TD, Dutko FJ,Pouton CW. 1992. Self-emulsifying drug delivery systems:Formulation and biopharmaceutic evaluation of an investiga-tional lipophilic compound. Pharm Res 9:87–93.
104. Perlman ME, Murdande SB, Gumkowski MJ, Shah TS,Rodricks CM, Thornton-Manning J, Freel D, Erhart LC.2008. Development of a self-emulsifying formulation thatreduces the food effect for torcetrapib. Int J Pharm 351:15–22.
105. Porter CJH, Pouton CW, Cuine JF, Charman WN. 2008.Enhancing intestinal drug solubilization using lipid-baseddelivery systems. Adv Drug Deliver Rev 60:673–691.
106. MacGregor KJ, Embleton JK, Lacy JE, Perry EA, Solomon LJ,Seager H, Pouton CW. 1997. Influence of lipolysis on drugabsorption from the gastro-intestinal tract. Adv Drug Deliv-ery Rev 25:33–46.
107. Lacy JE, Embleton JE, Perry EA. 2000. Delivery systems forhydrophobic drugs. US Patent 6,096,338.
108. Sunesen VH, Vedelsdal R, Kristensen HG, Christrup L, Mul-lertz A. 2005. Effect of liquid volume and food intake on theabsolute bioavailability of danazol, a poorly soluble drug. EurJ Pharm Sci 24:297–303.
109. Fleisher D, Li C, Zhou Y, Pao LH, Karim A. 1999. Drug, mealand formulation interactions influencing drug absorptionafter oral administration. Clinical implications. Clin Pharma-cokinet 36:233–254.
110. Kossena GA, Charman WN, Wilson CG, O’Mahony B, LindsayB, Hempenstall JM, Davison CL, Crowley PJ, Porter CJH.2007. Low dose lipid formulations: Effects on gastric emptyingand biliary Secretion. Pharm Res 24:2084–2096.
111. Lentz KA. 2008. Current methods for predicting human foodeffect. AAPS J 10:282–288.
112. Hernell O, Staggers JE, Carey MC. 1990. Physical-chemicalbehavior of dietary and biliary lipids during intestinal diges-tion and absorption. 2. Phase analysis and aggregation statesof luminal lipids during duodenal fat digestion in healthyadult human beings. Biochemistry 29:2041–2056.
113. Staggers JE, Hernell O, Stafford RJ, Carey MC. 1990. Phy-sical-chemical behavior of dietary and biliary lipids duringintestinal digestion and absorption. 1. Phase behavior andaggregation states of model lipid systems patterned afteraqueous duodenal contents of healthy adult human beings.Biochemistry 29:2028–2040.
114. Clement J. 1980. Intestinal absorption of triglycerols. ReprodNutr Dev 20:1285–1307.
115. Nordskog BK, Phan CT, Nutting DF, Tso P. 2001.An examination of the factors affecting intestinal lymphatictransport of dietary lipids. Adv Drug Deliv Rev 50:21–44.
116. Charman WN, Stella VJ. 1986. Estimating the maximalpotential for intestinal lymphatic transport of lipophilic drugmolecules. Int J Pharm 34:175–178.
117. Nankervis R, Davis SS, Day NH, Shaw PN. 1995. Effect oflipid vehicle on the intestinal lymphatic transport of isotre-tinoin in the rat. Int J Pharm 119:173–181.
118. Nankervis R, Davis SS, Day NH, Shaw PN. 1996. Intestinallymphatic transport of three retinoids in the rat after oraladministration: Effect of lipophilicity and lipid vehicle. Int JPharm 130:57–64.
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4143
119. Porter CJH. 1997. Drug delivery to the lymphatic system. CritRev Ther Drug Carrier Syst 14:333–393.
120. Porter CJH, Charman WN. 1997. Uptake of drugs into theintestinal lymphatics after oral administration. Adv DrugDeliv Rev 25:71–89.
121. Caliph SM, Charman WN, Porter CJH. 2000. Effect of short-,medium-, and long-chain fatty acid-based vehicles on theabsolute oral bioavailability and intestinal lymphatic trans-port of halofantrine and assessment of mass balance in lymph-cannulated and non-cannulated rats. J Pharm Sci 89:1073–1084.
122. Fischler M, Frisch EP, Ortengren B. 1973. Plasma concentra-tions after oral administration of different pharmaceuticalpreparations of clomethiazole. Acta Pharm Suec 10:483–492.
123. Palin KJ, Bell GD, Wilson CG. 1984. The effect of oil on theabsorption of probucol in the rat. J Pharm Pharmacol 36:85P.
124. Palin KJ, Wilson CG. 1984. The effects of different oils on theabsorption of probucol in the rat. J Pharm Pharmacol 36:641–643.
125. Khoo SM, Shackleford DM, Porter CJH, Edwards GA, Char-man WN. 2003. Intestinal lymphatic transport of halofantrineoccurs after oral administration of a unit-dose lipid-basedformulation to fasted dogs. Pharm Res 20:1460–1465.
126. Porter CJH, Kaukonen AM, Boyd BJ, Edwards GA, CharmanWN. 2004. Susceptibility to lipase-mediated digestion reducesthe oral bioavailability of danazol after administration as amedium-chain lipid-based microemulsion formulation. PharmRes 21:1405–1412.
127. Khoo SM, Edwards GA, Porter CJH, Charman WN. 2001.A conscious dog model for assessing the absorption, enterocytebased metabolism, and intestinal lymphatic transport of halo-fantrine. J Pharm Sci 90:1599–1607.
128. Shiau YF, Popper DA, Reed M, Umstetter C, Capuzzi D,Levine GM. 1985. Intestinal triglycerides are derived fromboth endogenous and exogenous sources. Am J Physiol Gas-trointest Liver Physiol 248:G164–G169.
129. Holm R, Porsgaard T, Porter CJH, Hoy CE, Edwards GA,Mullertz A, Kristensen HG, Charman WN. 2006. Lymphaticfatty acids in canines dosed with pharmaceutical formulationscontaining structured triacylglycerols. Eur J Lipid Sci Tech-nol 108:714–722.
130. Zangenberg NH, Mullertz A, Kristensen HG, Hovgaard L.2001. A dynamic in vitro lipolysis model II: Evaluation of themodel. Eur J Pharm Sci 14:237–244.
131. Vonderscher J, Meinzer A. 1994. Rationale for the develop-ment of Sandimmune Neoral. Transplant Proc 26:2925–2927.
132. Reymond J, Sucker H. 1988. In vitro model for ciclosporinintestinal absorption in lipid vehicles. Pharm Res 5:673–676.
133. Reymond J, Sucker H, Vonderscher J. 1988. In vivo model forciclosporin intestinal absorption in lipid vehicles. Pharm Res5:677–679.
134. Kovarik JM, Mueller EA, Van Bree JB, Tetzloff W, Kutz K.1994. Reduced inter- and intraindividual variability in cyclos-porine pharmacokinetics from a microemulsion formulation.J Pharm Sci 83:444–446.
135. Mueller EA, Kovarik JM, Van Bree JB, Tetzloff W, Grevel J,Kutz K. 1994. Improved dose linearity of cyclosporine phar-macokinetics from a microemulsion formulation. Pharm Res11:301–304.
136. Mountfield RJ, Senepin S, Schleimer M, Walter I, Bittner B.2000. Potential inhibitory effects of formulation ingredientson intestinal cytochrome P450. Int J Pharm 211:89–92.
137. Grove M, Pedersen GP, Nielsen JL, Mullertz A. 2005. Bioa-vailability of seocalcitol. I. Relating solubility in biorelevantmedia with oral bioavailability in rats-effect of medium andlong chain triglycerides. J Pharm Sci 94:1830–1838.
138. Grove M, Mullertz A, Nielsen JL, Pedersen GP. 2006. Bioa-vailability of seocalcitol II: Development and characterization
DOI 10.1002/jps JO
of self-microemulsifying drug delivery systems (SMEDDS) fororal administration containing medium and long chain trigly-cerides. Eur J Pharm Sci 28:233–242.
139. Shan Y, Post N, Speer J, Flynn J, Gullapalli R, Agarwal R,Muffuid P. 2005. Investigation of non-aqueous vehicles for apoorly soluble compound intended for softgel dosage form devel-opment. AAPS Annual Meeting and Exposition, Nashville, TN.www.aapsj.org/abstracts/AM_2005/AAPS2005-000990.pdf.
140. Raikes M, Taylor N, Field C. 2009. Moisture sorption experi-ment on soft gel capsules containing a self-emulsifying drugdelivery system. Am Pharm Rev 12:12–17.
141. Neisingh SE, Sam AP, de Nijs H. 1986. A dissolution methodfor hard and soft gelatin capsules containing testosteroneundecanoate in oleic acid. Drug Dev Ind Pharm 12:651–663.
142. Ahmed HA, Infeld MH, Shah NH. 2000. Compositions of andprocess for producing isotretinoin having low particle size.WO/2000/25772.
143. Chopra SK, Makadia TT. 1991. Pharmaceutical capsulescontaining panetidine. US Patent 5,028,432.
144. Anderson NR, Gullapalli RP. 2005. b-Carboline pharmaceu-tical compositions. US Patent 6,841,167.
145. Craig DQM. 1996. The physical characterization of Gelucire50/13. In: Semi-solid Formulations and Oral AbsorptionEnhancement, B.T. Gattefosse N8 89. Gattefosse Corporation,Saint-Priest, Cedex, France. pp. 39–51.
146. Singhal D, Curatolo W. 2004. Drug polymorphism and dosageform design: A practical perspective. Adv Drug Deliv Rev56:335–347.
147. Stahly GP. 2007. Diversity in single- and multiple-componentcrystals. The search for and prevalence of polymorphs andcocrystals. Cryst Growth Des 7:1007–1026 .
148. Aguiar AJ, Krc J, Kinkel AW, Samyn JC. 1967. Effect ofpolymorphism on the absorption of chloramphenicol fromchloramphenicol palmitate. J Pharm Sci 56:847–853.
149. Aguiar AJ, Zelmer JE. 1969. Dissolution behavior of poly-morphs of chloramphenicol palmitate and mefanamic acid.J Pharm Sci 58:983–987.
150. Brice GW, Hammer HF. 1969. Therapeutic nonequivalence ofoxytetracycline capsules. J Am Med Assoc 208:1189–1190.
151. Liebenberg W, de Villiers M, Wurster DE, Swanepoel E,Dekker TG, Lotter AP. 1999. The effect of polymorphism onpowder compaction and dissolution properties of chemicallyequivalent oxytetracycline hydrochloride powders. Drug DevInd Pharm 25:1027–1033.
152. Chemburkar SR, Bauer J, Deming K, Spiwek H, Patel K,Morris J, Henry R, Spanton S, Dziki W, Porter W, Quick J,Bauer P, Donaubauer J, Narayanan BA, Soldani M, Riley D,McFarland K. 2000. Dealing with the impact of ritonavirpolymorphs on the late stages of bulk drug process develop-ment. Org Process Res Dev 4:413–417.
153. Bauer J, Spanton S, Henry R, Quick J, Dziki W, Porter W,Morris J. 2001. Ritonavir: An extraordinary example of con-formational polymorphism. Pharm Res 18:859–866.
154. Davis JD, Touitou E, Rubinstein A. 1990. Targeted enternaldelivery system. US Patent 4,910,021.
155. Sobral PJA, Menegalli FC, Hubinger MD, Roques MA. 2001.Mechanical, water vapor barrier and thermal properties ofgelatin based edible films. Food Hydrocolloids 15:423–432.
156. Singh S, Rama Rao KV, Venugopal K, Manikandan R. 2002.Alteration in dissolution characteristics of gelatin-containingformulations. A review of the problem, test methods, andsolutions. Pharm Technol 26:36–58.
157. Chiou BS, Avena-Bustillos RJ, Shey J, Yee E, Bechtel PG,Imam SH, Glenn GM, Orts WJ. 2006. Rheological andmechanical properties of cross-linked fish gelatins. Polymer47:6379–6386.
158. Price JC. 2006a. Gelatin. In: Rowe RC, Sheskey PJ, Owen SC,editors. Handbook of pharmaceutical excipients. 5th edition.
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4144 GULLAPALLI
London: Pharmaceutical Press and Washington, DC: Amer-ican Pharmacists Association. pp. 295–298.
159. Eastoe JE, Leach AA. 1977. Chemical constitution ofgelatin. In: Ward AG, Courts A, editors. The science andtechnology of gelatin. New York: Academic Press. pp. 91–92.
160. Bowman BJ, Ofner CM. 2002. Hard gelatin capsules. In:Gennadios A, editor. Protein-based films and coatings. Flor-ida: CRC Press. pp. 367–392.
161. Hom FS, Veresh SA, Miskel JJ. 1973. Soft gelatin capsules I:Factors affecting capsule-shell dissolution rate. J Pharm Sci62:1001–1006.
162. Ames WM. 1947. Heat degradation of gelatin. J Soc Chem Ind66:279–284.
163. Courts A. 1954. The N-terminal amino acid residues of gelatin.2. Thermal degradation. Biochem J 58:74–79.
164. Yannas JB, Tobolsky AV. 1964. Viscoelastic properties ofplasticized gelatin films. J Phys Chem 68:3880–3882.
165. Marshall AS, Petrie SEB. 1980. Thermal transitions ingelatin and aqueous gelatin solutions. J Photographic Sci28:128–134.
166. Fraga AN, Williams RJJ. 1985. Thermal properties of gelatinfilms. Polymer 26:113–118.
167. Pinhas MF, Blanshard JMV, Derbyshire W, Mitchell JR.1996. The effect of water on the physicochemical andmechanical properties of gelatin. J Therm Anal 47:1499–1511.
168. Martucci JF, Ruseckaite RA, Vazquez A. 2006. Creep ofglutaraldehyde-crosslinked gelatin films. Mater Sci Eng A435–436: 681–686.
169. Coppola M, Djabourov M, Ferrand M. 2008. Phase diagram ofgelatin plasticized by water and glycerol. Macromol Symp273:56–65.
170. Ni BY, LeFaou A. 1993. Crystalline structure and moistureeffects on deformation mechanisms of gelatin films undermode I stress field. Mater Res Soc Symp Proc (Biomol Mater)292:229–234.
171. Reich VG. 1994. Action and optimization of plasticizers in softgelatin capsules. Pharm Ind 56:915–920.
172. Reich G. 1996. Effect of sorbitol specification on structureand properties of soft gelatin capsules. Pharm Ind 58:941–946.
173. Sanchez AC, Popineau Y, Mangavel C, Larre C, Gueguen J.1998. Effect of different plasticizers on the mechanical andsurface properties of wheat gliadin films. J Agric Food Chem46:4539–4544.
174. Vanin FM, Sobral PJA, Menegalli FC, Carvalho RA, Habi-tante AMQB. 2005. Effects of plasticizers and their concen-trations on thermal and functional properties of gelatin-basedfilms. Food Hydrocolloids 19:899–907.
175. Thomazine M, Carvalho RA, Sobral PJA. 2005. Physicalproperties of gelatin films plasticized by blends of glycerinand sorbitol. J Food Sci 70:172–176.
176. Vrana A. 2003. Reduction of gelatin cross-linking. WO/2003/103582.
177. Bergo P, Sobral PJA. 2007. Effects of plasticizer on physicalproperties of pigskin gelatin films. Food Hydrocolloids 21:1285–1289.
178. Cuq B, Gontard N, Cuq JL, Guilbert S. 1997. Selected func-tional properties of fish myofibrillar protein-based films asaffected by hydrophilic plasticizers. J Agric Food Chem45:622–626.
179. Oses J, Fernandez-Pan I, Mendoza M, Mate JI. 2009. Stabilityof the mechanical properties of edible films based on wheyprotein isolate during storage at different relative humidity.Food Hydrocolloids 23:125–131.
180. Cherian G, Gennadios A, Weller C, Chinachoti P. 1995. Ther-momechanical behavior of wheat gluten films: Effect ofsucrose, glycerin, and sorbitol. Cereal Chem 72:1–6.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
181. Laohakunjit N, Noomhorm A. 2004. Effect of plasticizers onmechanical and barrier properties of rice starch film. Starch/Starke 56:348–356.
182. Sakanaka LS, Sobral PJA, Menegalli FC. 2001. Proceeding ofa meeting on cross-linked gelatin biofilms mechanical andpermeability properties as affected by storage conditions. IIICongreso Ibero-Americano de Ingenieria de Alimentos. Valen-cia, Espana: Press of the Politechnique University. pp. 197–202.
183. Moreton RC, Armstrong NA. 1995. Design and use of anapparatus for measuring diffusion through glycerogelatinfilms. Int J Pharm 122:79–89.
184. Amidon GE, Peck GE, Block LH, Moreton RC, Katdare A,Lafaver R, Sheehan C. 2007. Proposed new USP generalinformation chapter, excipient performance. PharmacopeialForum 33:1311–1323.
185. Cooper JW, Ansel HC, Cadwallader DE. 1973. Liquid andsolid interactions of primary certified colorants with pharma-ceutical gelatins. J Pharm Sci 62:1156–1164.
186. Weller PJ. 2006. Titanium dioxide. In: Rowe RC, Sheskey PJ,Owen SC, editors. Handbook of pharmaceutical excipients.5th edition. London: Pharmaceutical Press and Washington,DC: American Pharmacists Association. pp. 782–784.
187. Samura K, Kato Y, Morita Y, Kawamura M, Koyama N,Osawa S. 1993. Effect of water-insoluble powder additionon physical properties of gelatin gel. Drug Dev Ind Pharm19:2579–2594.
188. Hewitt JP. 1992. Titanium dioxide: A different kind of sun-shield. Drug Cosmet Ind 151:26–32.
189. Allen NS, Edge M, Ortega A, Liauw CM, Stratton J, McIntyreRB. 2002. Behaviour of nanoparticle (ultrafine) titaniumdioxide pigments and stabilisers on the photooxidative stabi-lity of water based acrylic and isocyanate based acrylic coat-ings. Polym Degrad Stab 78:467–478.
190. Matsuda Y, Itooka T, Mitsuhashi Y. 1980. Photostabilityof indomethacin in model gelatin capsules: Effects of filmthickness and concentration of titanium dioxide on the colora-tion and photolytic degradation. Chem Pharm Bull 28:2665–2671.
191. Su KSE, Snyder RR, Scott RR. 1976. Pharmaceutical suspen-sion for opaqing empty gelatin capsules. US Patent 3,992,215.
192. Yannas IV, Tobolsky AV. 1967. Cross-linking of gelatine bydehydration. Nature 215:509–510.
193. Welz MM, Ofner CM. 1992. Examination of self-crosslinkedgelatin as a hydrogel for controlled release. J Pharm Sci81:85–90.
194. Bessho M, Kojima T, Okuda S, Hara M. 2007. Radiation-induced cross-linking of gelatin by using g-rays:Insoluble gelatin hydrogel formation. Bull Chem Soc Jpn80:979–985.
195. Sheehan JC, Hlavka JJ. 1957. The cross-linking of gelatinusing a water-soluble carbodiimide. J Am Chem Soc 79:4528–4529.
196. Davis P, Tabor BE. 1963. Kinetic study of the crosslinking ofgelatin by formaldehyde and glyoxal. J Polymer Sci Part AGeneral Papers 1:799–815.
197. Digenis GA, Gold TB, Shah VP. 1994. Cross-linking of gelatincapsules and its relevance to their in vitro–in vivo perfor-mance. J Pharm Sci 83:915–921.
198. Tomihata K, Ikada Y. 1996. Cross-linking of gelatin withcarbodiimides. Tissue Eng 2:307–313.
199. Bottom CB, Clark M, Carstensen JT. 1997. Dissolution testingof soft shell capsules-acetaminophen and nifedipine. J PharmSci 86:1057–1061.
200. Gold TB, Buice RG, Lodder RA, Digenis GA. 1998. Detection offormaldehyde-induced crosslinking in soft elastic gelatin cap-sules using near-infrared spectrophotometry. Pharm DevTechnol 3:209–214.
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4145
201. Fan H, Dash AK. 2001. Effect of cross-linking on the in vitrorelease kinetics of doxorubicin from gelatin implants. Int JPharm 213:103–116.
202. Liang HC, Chang WH, Liang HF, Lee MH, Sung HW. 2004.Crosslinking structures of gelatin hydrogels crosslinked withgenipin or a water-soluble carbodiimide. J Appl Polymer Sci91:4017–4026.
203. Carstensen JT, Rhodes CT. 1993. Pellicule formation in gela-tin capsules. Drug Dev Ind Pharm 19:2709–2712.
204. Hakata T, Sato H, Watanabe Y, Matsumoto M. 1994.Effect of formaldehyde on the physicochemical propertiesof soft gelatin capsule shells. Chem Pharm Bull 42:1138–1142.
205. Hakata T, Sato H, Watanabe Y, Matsumoto M. 1994.Effect of storage temperature on the physicochemical proper-ties of soft gelatin capsule shells. Chem Pharm Bull 42:1496–1500.
206. Fraenkel-Conrat H, Cooper M, Olcott HS. 1945. Reaction offormaldehyde with proteins. J Am Chem Soc 67:950–954.
207. Taylor SK, Davidson F, Ovenall DW. 1978. Carbon-13 nuclearmagnetic resonance studies on gelatin crosslinking by for-maldehyde. Photogr Sci Eng 22:134–138.
208. Albert K, Peters B, Bayer E, Treiber U, Zwilling M. 1986.Crosslinking of gelatin with formaldehyde: A 13C NMR study.Z Naturforsch 41:351–358.
209. Albert K, Bayer E, Worsching A, Vogele H. 1991. Investigationof the hardening reaction of gelatin with 13C labeled formal-dehyde by solution and solid state 13C NMR spectroscopy.Z Naturforsch 46:385–389.
210. Gold TB, Smith SL, Digenis GA. 1996. Studies on the influ-ence of pH and pancreatin on 13C-formaldehyde inducedgelatin crosslinks using nuclear magnetic resonance. PharmDev Technol 1:21–26.
211. Ofner CM, Zhang Y, Jobeck VC, Bowman BJ. 2001. Cross-linking studies in gelatin capsules treated with formaldehydeand in capsules exposed to elevated temperature and humid-ity. J Pharm Sci 90:79–88.
212. Gautam J, Schott H. 1994. Interaction of anionic compoundswith gelatin I: Binding studies. J Pharm Sci 83:922–930.
213. Azaz E, Donbrow M, Hamburger R. 1973. Incompatibility ofnon-ionic surfactants with oxidisable drugs. Pharm J 211:15.
214. Hamburger R, Azaz E, Donbrow M. 1975. Autoxidation ofpolyoxyethylenic non-ionic surfactants and of polyethyleneglycols. Pharm Acta Helv 50:10–17.
215. McGinity JW, Hill JA, La Via AL. 1975. Influence of peroxideimpurities in polyethylene glycols on drug stability. J PharmSci 64:356–357.
216. Donbrow M, Azaz E, Pillersdorf A. 1978. Autoxidation ofpolysorbates. J Pharm Sci 67:1676–1681.
217. Chafetz L, Hong W, Tsilifonis DC, Taylor AK, Philip J. 1984.Decrease in the rate of capsule dissolution due to formalde-hyde from Polysorbate 80 autooxidation. J Pharm Sci73:1186–1187.
218. Johnson DM, Taylor WF. 1984. Degradation of fenprostalenein Polyethylene glycol 400 solution. J Pharm Sci 73:1414–1417.
219. Hartauer KJ, Bucko JH, Cooke GG, Mayer RF, Schwier JR,Sullivan GR. 1993. The effect of rayon coiler on the dissolutionstability of hard-shell gelatin capsules. Pharm Technol 17:76–83.
220. Schwier JR, Cooke GG, Hartauer KJ, Yu L. 1993. Rayon:Source of furfural—A reactive aldehyde capable of insolubliz-ing gelatin capsules. Pharm Technol 17:78–80.
221. Yamamoto T, Kobayashi M, Matsuura S. 1995. Gelatin coat-ing composition and hard gelatin capsule. US Patent5,419,916.
222. Gholap D, Singh S. 2004. The influence of drugs on gelatincross-linking. Pharm Technol 28:94–102.
DOI 10.1002/jps JO
223. Hwan YJ. 2003. Method of manufacturing succinylated gela-tin. Patent Publication KR20030077501.
224. Adesunloye TA, Stach PE. 1998. Effect of glycine/citric acid onthe dissolution stability of hard gelatin capsules. Drug DevInd Pharm 24:494–500.
225. Adesunloye TA, Stach PE. 1999. Filled gelatin capsules. USPatent 5,874,106.
226. Rama Rao KV, Pakhale SP, Singh S. 2003. A film approach forthe stabilization of gelatin preparations against cross-linking.Pharm Technol 27:54–63.
227. US Pharmacopeia 32. 2009. General Chapter <711> Dissolu-tion and General Chapter <2040> Disintegration and dissolu-tion of dietary supplements. Rockville, MD: US PharmacopeialConvention. pp. 263–264, 783.
228. Piper DW, Fenton BH. 1965. pH stability and activity curvesof pepsin with special reference to their clinical importance.Gut 6:506–508.
229. Shiu GK. 1996. Pharmaceutical product quality research: Anoverview for solid oral dosage forms. J Food Drug Analysis4:293–300.
230. Meyer MC, Straughn AB, Mhatre RM, Hussain A, Shah VP,Bottom CB, Cole ET, Lesko LL, Mallinowski H, Williams RL.2000. The effect of gelatin cross-linking on the bioequivalenceof hard and soft gelatin acetaminophen capsules. Pharm Res17:962–966.
231. Digenis GA, Sandefer EP, Page RC, Doll WJ, Gold TB, Dar-wazeh NB. 2000. Bioequivalence study of stressed and non-stressed hard gelatin capsules using amoxicillin as a drugmarker and gamma scintigraphy to confirm time and GIlocation of in vivo capsules rupture. Pharm Res 17:572–582.
232. Shah VP, Konecny JJ, Everett RL, McCullough B, NoorizadehAC, Skelly JP. 1989. In vitro dissolution profile of water-insoluble drug dosage forms in the presence of surfactants.Pharm Res 6:612–618.
233. Noory C, Tran N, Ouderkirk L, Shah V. 2002. Steps fordevelopment of a dissolution test for sparingly water-solubledrug products. Am Pharm Rev 5:16–21.
234. Brown CK, Chokshi HP, Nickerson B, Reed RA, Rohrs BR,Shah PA. 2004. Acceptable analytical practices for dissolutiontesting of poorly soluble compounds. Pharm Technol 28:56–65.
235. Nishimura H, Hayashi C, Aiba T, Okamoto I, Miyamoto Y,Nakade S, Takeda K, Kurosaki Y. 2007. Application of thecorrelation of in vitro dissolution behavior and in vivo plasmaconcentration profile (IVIVC) for soft-gel capsules-a pointlesspursuit? Biol Pharm Bull 30:2221–2225.
236. Rossi RC, Dias CL, Donato EM, Martins LA, Bergold AM,Froehlich PE. 2007. Development and validation of dissolu-tion test for ritonavir soft gelatin capsules based on in vivodata. Int J Pharm 338:119–124.
237. Buri P, Humbert-Droz J. 1983. Solubilisation de principesactifs insolubles par des constituants des sucs digestifs. 3rdInt Cong Pharm Technol Paris 4:136–143.
238. Larson CE, Greenberg DM. 1933. A paradoxical solubilityphenomenon with gelatin. J Am Chem Soc 55:2798–2799.
239. Pillay V, Fassihi R. 1999. Unconventional dissolution meth-odologies. J Pharm Sci 88:843–851.
240. Pillay V, Fassihi R. 1999. A new method for dissolution studiesof liquid-filled capsules employing nifedipine as a model drug.Pharm Res 16:333–337.
241. Zhao F, Malayev V, Rao V, Hussain M. 2004. Effect of sodiumlauryl sulfate in dissolution media on dissolution of hardgelatin capsule shells. Pharm Res 21:144–148.
242. Bartnik FG. 1992. Interaction of anionic surfactants withproteins, enzymes and membranes. In: Gloxhuber C, KunstlerK, editors. Anionic surfactants: Biochemistry, toxicology, der-matology. 2nd edition (Surfactant Science Series; Vol. 43).New York: Marcel Dekker, Inc. pp. 1–42.
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4146 GULLAPALLI
243. Marchais H, Cayzeele G, Legendre JV, Skiba M, Arnaud P.2003. Cross-linking of hard gelatin carbamazepine capsules:Effect of dissolution conditions on in vitro drug release. Eur JPharm Sci 19:129–132.
244. Joseph M, Mayberry C, Schiermeyer C. 2005. A 2-step dis-solution method for the elimination of crosslinking in nimo-dipine soft gelatin capsules while providing solubility of awater-insoluble drug. AAPS Annual Meeting and Exposition,Nashville, TN. www.aapsj.org/abstracts/AM_2005/AAPS2005-001000.pdf.
245. Speer J, Flynn J, Gullapalli R, Agarwal R, Muffuid P. 2005.Influence of digestive enzymes on dissolution of a poorly watersoluble compound from cross-linked capsules in sodium laurylsulfate medium. AAPS Annual Meeting and Exposition,Nashville, TN. www.aapsj.org/abstracts/AM_2005/AAPS2005-000998.pdf.
246. Tamaki K, Tamamushi B. 1955. The interactions of gelatinmolecule with surface active ions. Bull Chem Soc Jpn 28:555–559.
247. Pennings FH, Kwee BLS, Vromans H. 2006. Influence ofenzymes and surfactants on the disintegration behavior ofcross-linked hard gelatin capsules during dissolution. DrugDev Ind Pharm 32:33–37.
248. Onesippe C, Lagerge S. 2009. Study of the complex formationbetween sodium dodecyl sulphate and gelatin. Colloids Surf A:Physicochem Eng Aspects 337:61–66.
249. Ofner CM, Schott H. 1987. Swelling studies of gelatin II:Effect of additives. J Pharm Sci 76:715–723.
250. Woolfrey SG, Banzon GM, Groves MJ. 1986. The effect ofsodium chloride on the dynamic surface tension of sodiumdodecyl sulfate solutions. J Colloid Interface Sci 112:583–587.
251. Crison JR, Weiner ND, Amidon GL. 1997. Dissolution mediafor in vitro testing of water-insoluble drugs: Effect of surfac-tant purity and electrolyte on in vitro dissolution of carbama-zepine in aqueous solutions of sodium lauryl sulfate. J PharmSci 86:384–388.
252. Cao Y, Marra M, Anderson BD. 2004. Predictive relationshipsfor the effects of triglyceride ester concentration and wateruptake on solubility and partitioning of small molecules intolipid vehicles. J Pharm Sci 93:2768–2779.
253. Land LM, Li P, Bummer PM. 2005. The influence of watercontent of triglyceride oils on the solubility of steroids. PharmRes 22:784–788.
254. Rane SS, Anderson BD. 2008. What determines drug solubi-lity in lipid vehicles: Is it predictable? Adv Drug Deliv Rev60:638–656.
255. Shah NH, Stiel D, Infeld MH, Railkar AS, Malick AW, Patra-wala M. 1992. Elasticity of soft gelatin capsules containingpolyethylene glycol 400-quantitation and resolution. PharmTechnol 3:126–131.
256. Jun HW, Whitworth CW, Luzzi LA. 1972. Decomposition ofaspirin in polyethylene glycols. J Pharm Sci 61:1160–1162.
257. Whitworth CW, Jun HW, Luzzi LA. 1973. Stability of aspirinin liquid and semisolid bases I. Substituted and nonsubsti-tuted polyethylene glycols. J Pharm Sci 62:1184–1185.
258. Wei Y, Shelley RS. 1998. Comparison of lipophilic and hydro-philic ketoprofen solution softgel formulations. AAPS AnnualMeeting, Poster 2134.
259. Greenwald RB, Conover CD, Choe YH. 2000. Poly(ethyleneglycol) conjugated drugs and prodrugs: A comprehensivereview. Crit Rev Ther Drug Carrier Syst 17:101–161.
260. Kochling JD, Miao H, Young CR, Looker AR, Shannon M,Montgomery ER. 2007. Understanding the degradation path-way of a poorly water-soluble drug formulated in PEG-400.J Pharm Biomed Anal 43:1638–1646.
261. Ranucci E, Sartore L, Peroni I, Latini R, Bernasconi R, FerrutiP. 1994. Pharmacokinetic results on naproxen prodrugs based
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
on poly(ethyleneglycol)s. J Biomat Sci Polymer Edn 6:141–147.
262. Sartore L, Perone I, Ferruti P, Latini R, Barnasconi R.1997. Synthesis and pharmacokinetic behaviour of esterderivatives of 4-isobutylphenyl-2-propionic acid (Ibuprofen)with end-hydroxylated poly(N-vinyl pyrrolidinone) andpoly(N-acryloyl morpholine) oligomers. J Biomater SciPolym Ed 8:741–754.
263. Whitworth CW, Asker AF. 1974. Stability of aspirin in liquidand semisolid bases IV: Polyethylene glycol 400 diacetate andtriethylene glycol diacetate. J Pharm Sci 63:1790–1792.
264. Bergh M, Shao LP, Hagelthorn G, Gafvert E, Lars J, NilssonG, Karlberg AT. 1998. Contact allergens from surfactants.Atmospheric oxidation of polyethylene alcohols, formation ofethoxylated aldehydes, and their allergenic activity. J PharmSci 87:276–282.
265. Ha E, Wang W, Wang YJ. 2002. Peroxide formation in poly-sorbate 80 and protein stability. J Pharm Sci 91:2252–2264.
266. Kumar V, Kalonia DS. 2006. Removal of peroxides in poly-ethylene glycols by vacuum drying: Implications in the sta-bility of biotech and pharmaceutical formulations. AAPSPharmSciTech 7:E1–E7.
267. Li Z, Kozlowski BM, Chang EP. 2007. Analysis of aldehydes inexcipients used in liquid/semi-solid formulations by gas chro-matography—Negative chemical ionization mass spectrome-try. J Chromatography A 1160:299–305.
268. Wasylaschuk WR, Harmon PA, Wagner G, Harman AB, Tem-pleton AC, Xu H, Reed RA. 2007. Evaluation of hydroperox-ides in common pharmaceutical excipients. J Pharm Sci96:106.
269. Frontini R, Mielck JB. 1995. Formation of formaldehyde inpolyethylene glycol and in poloxamer under stress conditions.Int J Pharm 114:121–123.
270. Barrio MD, Hu J, Zhou P, Cauchon N. 2006. Simultaneousdetermination of formic acid and formaldehyde in pharma-ceutical excipients using headspace GC/MS. J Pharm BiomedAnal 41:738–743.
271. Nassar MN, Nesarikar VN, Lozano R, Parker WL, Huang Y,Palaniswamy V, Xu W, Khaselev N. 2004. Influence of for-maldehyde impurity in Polysorbate 80 and PEG-300 on thestability of a parenteral formulation of BMS-204352: Identi-fication and control of the degradation product. Pharm DevTech 9:189–195.
272. McGhee JD, Von Hippel PH. 1975. Formaldehyde as a probe ofDNA structure. I. Reaction with exocyclic amino groups ofDNA bases. Biochemistry 14:1281–1296.
273. Chaw YFM, Crane LE, Lange P, Shapiro R. 1980. Isolationand identification of cross-links from formaldehyde-treatednucleic acids. Biochemistry 19:5525–5531.
274. Huang H, Solomon MS, Hopkins PB. 1992. Formaldehydepreferentially interstrand cross-links duplex DNA throughdeoxyadenosine residues at the sequence 50-d(AT). J AmChem Soc 114:9240–9241.
275. Laicher A, Junger H, Klemm FH. 1997. Stability behaviour ofmolsidomine-containing pellet formulations. J Pharm Phar-macol 49:131–134.
276. Stephenson SA, Bryant DK, Thomson CM, Walsgrove TC,Webb ML. 1998. An analytical study of the interaction of lowlevels of formaldehyde with active pharmaceuticals. J PharmPharmacol 50:122.
277. Hartauer KJ, Arbuthnot GN, Baertschi SW, Johnson RA,Luke WD, Pearson NG, Rickard EC, Tingle CA, Tsang PK,Wiens RE. 2000. Influence of peroxide impurities in povidoneand crospovidone on the stability of raloxifene hydrochloridein tablets: Identification and control of an oxidative degrada-tion product. Pharm Dev Technol 5:303–310.
278. Huang T, Garceau ME, Gao P. 2003. Liquid chromatographicdetermination of residual hydrogen peroxide in pharmaceu-
DOI 10.1002/jps

SOFT GELATIN CAPSULES 4147
tical excipients using platinum and wired enzyme electrodes.J Pharm Bio Anal 31:1203–1210.
279. Buhler V. 2008. Kollidon1—Polyvinylpyrrolidone excipientsfor the pharmaceutical industry. 9th edition. Ludwigshafen,Germany: BASF SE Pharma Ingredients & Services. pp. 45–48.
280. Patel K, Kearney AS, Palepu NR. 1997. Investigation of newdegradation products arising from the encapsulation of an oil-based suspension formulation of topotecan. Int J Pharm151:7–13.
281. Armstrong NA, James KC, Pugh KL. 1984. Drug migrationinto soft gelatin capsule shells and its effect on in-vitro avail-ability. J Pharm Pharmacol 36:361–365.
282. Armstrong NA, James KC, Collett D, Thomas M. 1985. Solutemigration from oily solutions into glycerin-gelatin mixtures.Drug Dev Ind Pharm 11:1859–1868.
283. Hauer B, Meinzer A, Posanski U, Richter F. 1994. Pharma-ceutical compositions comprising cyclosporins. US Patent5,342,625.
284. Gursoy RN, Benita S. 2004. Self-emulsifying drug deliverysystems (SEDDS) for improved oral delivery of lipophilicdrugs. Biomed Pharmacother 58:173–182.
285. Marriott C, Kellaway IW. 1978. The release of antibacterialagents from glycerolgelatin gels. Drug Dev Ind Pharm 4:195–208.
286. Hong CI, Kim JW, Choi NH, Shin HJ, Yang SG. 2000. Cyclos-porin-containing microemulsion preconcentrate composition.US Patent 6,063,762.
287. Hauer B, Meinzer A, Posanski U, Richter F. 2007. Pharma-ceutical compositions comprising cyclosporins. US Patent7,235,248.
288. Woo JS. 1997. Cyclosporin-containing soft capsule composi-tions. US Patent 5,603,951.
289. Cole ET, Cade D, Benameur H. 2008. Challenges and oppor-tunities in the encapsulation of liquid and semi-solid formula-tions into capsules for oral administration. Adv Drug DelivRev 60:747–756.
290. Walker SE, Ganley JA, Bedford K, Eaves T. 1980. The filling ofmolten and thixotropic formulations into hard gelatin cap-sules. J Pharm Pharmacol 32:389–393.
291. Cade D, Cole ET, Mayer JPH, Wittwer F. 1986. Liquid filledand sealed hard gelatin capsules. Drug Dev Ind Pharm 12:2289–2300.
292. Cole ET. 1999. Liquid filled and sealed hard gelatincapsules. In: Recent advances in the formulation & develop-ment of poorly-soluble drugs, B.T. Gattefosse N8 92.Saint-Priest, Cedex, France: Gattefosse Corporation. pp.67–77.
293. El Massik MA, Abdallah OY, Galal S, Daabis NA. 2003.Semisolid matrix filled capsules: An approach to improvedissolution stability of phenytoin sodium formulation. DrugDev Ind Pharm 29:531–543.
294. Barakat NS. 2006. Etodolac-liquid-filled dispersion into hardgelatin capsules: An approach to improve dissolution andstability of etodolac formulation. Drug Dev Ind Pharm32:865–876.
295. Smith T. 2006. The hard capsule with the soft center: The re-emergence of liquid-filled hard capsules. Tablets & CapsulesMay: 5–9.
296. Stein D, Bindra DS. 2007. Stabilization of hard gelatin capsuleshells filled with polyethylene glycol matrices. Pharm DevTech 12:71–77.
297. Hariharan M, Ganorkar LD, Amidon GE, Cavallo A, Gatti P,Hageman MJ, Choo I, Miller JL, Shah UJ. 2003. Reducing thetime to develop and manufacture formulations for first oraldose in humans. Pharm Technol 27: 68–84.
298. Koon RC. 2006. How softgels are made and how to select amanufacturer. Tablets & Capsules May: 42–46.
DOI 10.1002/jps JO
299. Kontny MJ, Mulski CA. 1989. Gelatin capsule brittleness as afunction of relative humidity at room temperature. Int JPharm 54:79–85.
300. Jones BE. 2004. Manufacture and properties of two-piece hardcapsules. In: Podczeck F, Jones BE, editors. Pharmaceuticalcapsules. 2nd edition. London: Pharmaceutical Press. pp. 93–94.
301. Fulper D, Wang H, Downey D. 2010. Influence of mechanicalstress on the formation of cracks in two-piece capsules.Tablets & Capsules January: 8–15.
302. Richardson M, Stegemann S. 2007. Filling two-piece hardgelatin capsules with liquids. Tablets & Capsules January:97–110.
303. Rowley G. 2004. Filling of liquids and semi-solids into hardtwo-piece capsules. In: Podczeck F, Jones BE, editors. Phar-maceutical capsules. 2nd edition. London: PharmaceuticalPress. pp. 169–194.
304. Tindal S, Asgarzadeh F. 2006. Laboratory scale softgel encap-sulation (Minicap). AAPS Annual Meeting and Exposition,San Antonio, TX. http://www.aapsj.org/abstracts/AM_2006/AAPS2006-001526.pdf
305. Fatouros DG, Karpf DM, Nielsen FS, Mullertz A. 2007. Clin-ical studies with oral lipid based formulations of poorly solublecompounds. Ther Clin Risk Manage 3:591–604.
306. Garg R, Bhhatarai B. 2004. A mechanistic study of 3-ami-noindazole cyclic urea HIV-1 protease inhibitors using com-parative QSAR. Bioorg Med Chem 12:5819–5831.
307. Heimbach T, Fleisher D, Kaddoumi A. 2007. Overcoming pooraqueous solubility of drugs for oral delivery. In: Stella VJ,Borchardt RT, Hageman MJ, Oliyai R, Maag H, Tilley JW,editors. Prodrugs: Challenges and rewards, part 1. New York:AAPS Press, Springer. p. 175.
308. Kasim NA, Whitehouse M, Ramachandran C, Bermejo M,Lennerna1s H, Hussain AS, Junginger HE, StavchanskySA, Midha KK, Shah VP, Amidon GL. 2004. Molecular proper-ties of WHO essential drugs and provisional biopharmaceu-tical classification. Mol Pharm 1:85–96.
309. Lindenberg M, Kopp S, Dressman JB. 2004. Classification oforally administered drugs on the World Health Organizationmodel list of essential medicines according to the biopharma-ceutics classification system. Eur J Pharm Biopharm 58:265–278.
310. Darwish IA, Florence AT, Saleh AM. 1989. Effects of hydro-tropic agents on the solubility, precipitation, and proteinbinding of etoposide. J Pharm Sci 78:577–581.
311. Branchu S, Rogueda PG, Plumb AP, Cook WG. 2007.A decision-support tool for the formulation of orally active,poorly soluble compounds. Eur J Pharm Sci 32:128–139.
312. Yunzhe S, Rui Y, Wenliang Z, Xing T. 2008. Nimodipine semi-solid capsules containing solid dispersion for improving dis-solution. Int J Pharm 359:144–149.
313. O’Driscoll CM, Griffin BT. 2008. Biopharmaceutical chal-lenges associated with drugs with low aqueous solubility—The potential impact of lipid-based formulations. Adv DrugDeliv Rev 60:617–624.
314. Akhlaghi F, Trull AK. 2002. Distribution of cyclosporinin organ transplant recipients. Clin Pharmacokinet 41:615– 637.
315. Sharma P, Varma MVS, Chawla HPS, Panchagnula R. 2005.Absorption enhancement, mechanistic and toxicity studiesof medium chain fatty acids, cyclodextrins and bile saltsas peroral absorption enhancers. Il Farmaco 60:884–893.
316. Agarwal S, Boddu SHS, Jain R, Samanta S, Pal D, Mitra AK.2008. Peptide prodrugs: Improved oral absorption of lopinavir,a HIV protease inhibitor. Int J Pharm 359:7–14.
317. Price JC. 2006. Polyethylene glycol. In: Rowe RC,Sheskey PJ, Owen SC, editors. Handbook of pharmaceutical
URNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010

4148 GULLAPALLI
excipients. 5th edition. London: Pharmaceutical Press andWashington, DC: American Pharmacists Association.pp. 545–550.
318. Lawrence MJ. 2006. Medium-chain triglycerides. In: Rowe RC,Sheskey PJ, Owen SC, editors. Handbook of pharmaceuticalexcipients. 5th edition. London: Pharmaceutical Press and
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 10, OCTOBER 2010
Washington, DC: American Pharmacists Association.pp. 454–456.
319. Cable CG. 2006. Soybean oil. In: Rowe RC, Sheskey PJ, OwenSC, editors. Handbook of pharmaceutical excipients. 5th edi-tion. London: Pharmaceutical Press and Washington, DC:American Pharmacists Association. pp. 722–724.
DOI 10.1002/jps