statistical analysis plan - clinicaltrials.gov · 1 at the screening, the investigator also...
TRANSCRIPT

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 1 of 24
STATISTICAL ANALYSIS PLAN
Protocol Title: A Multicenter, Double-Blind, Randomized,
Comparative, Parallel-Group Clinical Study of the
Efficacy and Safety of BCD-066 (JSC BIOCAD, Russia)
and Aranesp® (Amgen Europe B. V., the Netherlands)
in the Treatment of Anemia in Hemodialysis Patients
with Impaired Kidney Function
Protocol Number:: BCD-066-2
Protocol version 2.2
Version Date: October 10, 2016
SAP version 1.0
SAP version date October 10, 2016
Name and address of the
clinical study sponsor
JSC BIOCAD, Russia
Legal address: 34 A, Ulitsa Svyazi, Strelna,
Petrodvortsoviy District, Saint Petersburg, Russian
Federation, 198515
Postal Address: Petrovo Dalneye, Krasnogorskiy District,
Moscow Region, Russian Federation, 143422.
Tel.: +7 (495) 992-66-28 Fax: +7 (495) 992-82-98
Name and function of
person(s), authorized to sign
the protocol and its
amendments for the sponsor:
Roman Alexeevich Ivanov, Vice President, Research &
Development
Address: Petrovo Dalneye, Krasnogorskiy District,
Moscow Region,
Russian Federation, 143422
Email: [email protected]
Tel.: + 7 (495) 992-66-28 (ext. 154)
Name, position, address and
telephone number of the
Medical Expert assigned by
the Sponsor for this Study
Aleksandr Andreevich Isaev, Medical Advisor, Category II
JSC BIOCAD, 34 A, Ulitsa Svyazi, Strelna,
Petrodvortsoviy District, Saint Petersburg, Russian
Federation, 198515
Email: [email protected]
Tel.: +7 (812) 380 49 33 (ext. 946); +7 (981) 788-04-11
All information contained in this document is strictly confidential and intended for use by investigators, members of Ethics
Committees and Health Authority personnel. This information cannot be disclosed to any other persons or entity, submitted
for publication or used for any purpose other than contemplated herein without the Sponsor`s prior written authorization,
unless it is necessary to get patient's consent for the participation in the study.
The above requirements are effective upon the signing of this protocol.

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 2 of 24
TABLE OF CONTENTS
1. INTRODUCTION ................................................................................................................................................... 3
2. GOALS AND OBJECTIVES ................................................................................................................................. 3
2.1. PURPOSE ......................................................................................................................................................... 3
2.2. OBJECTIVES .................................................................................................................................................. 3
3. STUDY DESIGN ..................................................................................................................................................... 4
3.1. DESIGN ............................................................................................................................................................ 4
3.2. STUDY FLOW CHART/TIME AND EVENTS SCHEDULE ..................................................................... 4
3.3. SELECTION OF STUDY POPULATION .................................................................................................. 11
3.3.1. INCLUSION CRITERIA ........................................................................................................ 11
3.3.2. EXCLUSION CRITERIA ....................................................................................................... 11
4. EFFICACY ASSESSMENT ................................................................................................................................. 13
4.1 PRIMARY ENDPOINT ......................................................................................................................................... 13
5. THE PLANNED ANALYSIS ............................................................................................................................... 14
6. SAMPLE SIZE CALCULATION ....................................................................................................................... 15
7. STATISTICAL ANALYSIS OF POPULATION ............................................................................................... 20
8. ANALYSIS PLAN AND STATISTICAL METHODS ...................................................................................... 21
8.1. THE SOFTWARE ............................................................................................................................................... 21
8.2. DESCRIPTION OF THE STATISTICAL METHODS TO BE EMPLOYED................................................................ 21
8.3. ACCOUNTING FOR MISSING, UNAVAILABLE OR DOUBTFUL DATA, OUTLIERS ............................................... 23
8.6. MULTIVARIATE COMPARISON AND MULTIPLICITY ...................................................................................... 24
8.7. SUBGROUP ANALYSIS, INTERACTION AND RELATED VARIABLES ................................................................... 24
9. OTHER PLANNED ANALYSES ........................................................................................................................ 24
10. DEVIATIONS FROM ANALYSIS METHODS DESCRIBED IN STUDY PROTOCOL ........................... 24

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 3 of 24
1. INTRODUCTION
The Statistical Analysis Plan (SAP) provides a detailed analysis plan and steps of study
report preparation for the clinical trial BCD-066-2.
2. GOALS AND OBJECTIVES
2.1. Purpose
To demonstrate the equivalent efficacy and safety of BCD-066 and Aranesp® in hemodialysis
patients with renal anemia.
2.2. Objectives
Purpose:
To demonstrate the equivalent efficacy and safety of BCD-066 and Aranesp® in hemodialysis
patients with renal anemia.
Study objectives:
1. To evaluate and compare the change in hemoglobin concentration (g/L) from baseline to
the evaluation period (weeks 21 to 24) in the treatment groups;
2. To evaluate and compare the proportion (%) of patients achieving the target hemoglobin
level (100–120 g/L) in the treatment groups at weeks 21 to 24;
3. To evaluate and compare the mean doses of the test drug and the reference drug (µg/kg)
at weeks 21 to 24;
4. To evaluate and compare the proportion of patients receiving blood transfusions
(weeks 1–24);
5. To evaluate and compare the mean hemoglobin levels in the treatment groups at
weeks 21 to 24;
6. To evaluate and compare the proportion (%) of patients in both treatment groups who
maintained hemoglobin at 90–100 g/L for at least the last four weeks of treatment
(weeks 21 to 24);
7. To evaluate and compare the mean hemoglobin levels in the treatment groups during the
main study period (weeks 1 to 24);
8. To evaluate and compare the mean doses of the test drug and the reference drug (µg/kg)
during the main study period (weeks 1 to 24);
9. To evaluate and compare the mean hematocrit levels in the treatment groups during the
main study period (weeks 1 to 24);
10. To evaluate and compare the proportion of patients who required dose titration at the
beginning of the study (weeks 1 to 20);
11. To evaluate and compare the AE/SAE rate and severity, the rate of discontinuation due to
an AE/SAE in both treatment groups at weeks 1 to 24 and 1 to 52;

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 4 of 24
12. To evaluate and compare the rate of arterial and venous thrombotic events (e.g., impaired
cerebral circulation, arteriovenous fistula thrombosis) in both treatment groups at
weeks 1 to 24 and 1 to 52;
13. To evaluate and compare the proportion (%) of patients who developed binding and
neutralizing antibodies to darbepoetin alfa (at weeks 1 to 24 and 1 to 52).
3. STUDY DESIGN
3.1. Design
This clinical study comparing the efficacy and safety of BCD-066 (JSC BIOCAD,
Russia) and Aranesp®
(Amgen Europe B.V., the Netherlands) in the treatment of anemia in
hemodialysis patients with impaired kidney function has been designed as a multicenter, double-
blind, parallel-group randomized study.
This study will include 196 adult patients with renal anemia due to late-stage CKD,
receiving hemodialysis (CKD 5D), and whose dialysis is effective. Eligible patients have
previously received ESA therapy and achieved target Hb levels. The study will not include
patients with non-renal anemia (e.g., anemia due to vitamin B12, folate, or iron deficiency). This
is a maintenance phase study. This means it will include patients previously treated with
erythropoietins. Eligible patients have received rHuEPO (epoetin alfa or epoetin beta
q1w/q2w/q3w, darbepoetin alfa q1w) for at least three months before randomization. During this
period, stable doses of rHuEPO have successfully maintained target Hb concentration (100–
120 g/L).
3.2. Study Flow Chart/Time and Events Schedule

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 5 of 24
Figure 1. Study flow chart
Screening
(28 days)
Test
Group
98 patients
Reference
Group
98 patients
Week
1 Week
1
Week
24
Week
24
B
C
D
0
6
6 s.c.
q1w
А
R
A
N
Е
S
P s.c.
q1w
Final efficacy evaluation, main safety and
immunogenicity assessment
Randomization
1:1
B
C
D
0
6
6 s.c. q1w
maintenance dose
А
R
A
N
Е
S
P
s.c. q1w
maintenance dose
Week
52
Additional safety and immunogenicity assessment
Week
25
Week
25
Week
52

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 6 of 24
Table 1. Schedule of Study Visits and Assessments
Study period Screening Main study period Additional study
period
Visit Screening 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Week 0 1 2 3 4 8 12 16 20 21 22 23 24 32 40 52
Day -28 – 0 1 8 15 22 29 57 85 11
3
14
1
148 155 16
2
169 225 281 365
Informed consent +
Medical history and
complaints
+
Concomitant therapy +1 + + + + + + + + + + + + + + + +
Blood pressure, heart rate,
and body temperature
+ + + + + + + + + + + + + + + + +
Physical examination,
body weight and height
+ + + + + + + + +
CBC (full panel) + + + + + + + +
CBC (“red blood”)2 + + + + + + + + +
Blood chemistry panel + + + + +
Iron metabolism + +
Intact PTH +
Coagulation tests +
Markers for HIV, HCV,
HBV, syphilis3
+ + +
Blood sampling for
BAB/NAB
+ + + + + + +
ECG + + + +
Chest X-ray +

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 7 of 24
Study period Screening Main study period Additional study
period
Visit Screening 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Week 0 1 2 3 4 8 12 16 20 21 22 23 24 32 40 52
Day -28 – 0 1 8 15 22 29 57 85 11
3
14
1
148 155 16
2
169 225 281 365
Echocardiography +
Pregnancy test
+
Eligibility +
Enrollment (randomization
and stratification)
+
IP injection (at the end of
the hemodialysis session)
+ + + + + + + + + + + + + + + +
AE/SAE Recording + + + + + + + + + + + + + + + + + 1 At the screening, the investigator also collects information about the concomitant medications that the subject received within 3 months prior to the enrollment.
2Hb, RBCs, reticulocytes, Hct.
3Syphilis tests will be performed only at screening.

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 8 of 24
Table 2. Clinical and Laboratory Tests Planned for the Study
Examination/
Test Control parameters Number
Physical
examination
Height
Body weight
Skin and mucous membranes
(visual examination)
Lymph nodes (visual
examination, palpation)
Ear/nose/throat, respiratory
organs (examination, lung
auscultation)
Cardiovascular system (heart
auscultation, visual
examination of the area of
major vessels)
GI tract (examination,
abdominal palpation)
Liver span (palpation,
percussion)
Spleen size (palpation,
percussion)
Urogenital system (kidney
punch, palpation of the kidney
and bladder area)
Nervous system (meningeal
signs, focal neurological signs)
Mental health (signs of a
depressive disorder, suicidal
ideation, an acute psychotic
disorder)
×9: at screening, on
days 1, 29, 85, 141,
169, 225, 281, 365
Vital signs BP
HR
body temperature
×17: at screening
and each visit
CBC
Hb
Hct
RBCs
reticulocytes
platelets
WBCs
×8: at screening, on
days 8, 15, 22, 29,
85, 169, 365

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 9 of 24
Examination/
Test Control parameters Number
neutrophils
lymphocytes
monocytes
eosinophils
basophils
ESR
(blood volume: 3 mL)
CBC (“red
blood”)
Hb
Hct
RBCs
reticulocytes
(blood volume: 3 mL)
×9: at screening, on
days 1, 57, 113, 141,
148, 155, 162, 225,
281
Blood chemistry
panel
total protein
glucose
ALT
AST
ferritin
TSAT
C-reactive protein
potassium
(blood volume: 5 mL)
×5: at screening, on
days 29, 85, 169,
365
Iron metabolism ferritin
TSAT
× 2: on days 57 and
113
Blood
coagulation
INR
APTT
fibrinogen
×1: at screening
Blood for intact
PTH
Intact PTH (blood volume:
4 mL)
×1: at screening
Serological tests
for viral
infections1
anti-HIV
anti-HCV
HBsAg
(blood volume: 10 mL)
×3: at screening, on
day 169, day 365
Serological tests
for syphilis1
anticardiolipin antibody
test/RW
(blood volume: 10 mL; at the
×1: at screening

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 10 of 24
Examination/
Test Control parameters Number
screening, blood is sampled to
be tested for all infections)
Pregnancy test2 serum HCG (blood volume:
3 mL)
×1: at screening
Instrumental
examinations
12-lead ECG ×4: at screening, on
day 85, day 169,
day 365
Echocardiography ×1: at screening
Chest X-ray3 ×1: at screening
Immunogenicity
assessment
BABs and NABs in the serum
(blood volume: 2 mL)
×7: at screening,
then once every 2
months until
day 281, and on
day 365

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 11 of 24
3.3. Selection of Study Population
3.3.1. Inclusion criteria
1. Signed Informed Consent Form.
2. Age: 18 to 75 years old (inclusive).
3. Late-stage renal failure.
4. Eligible patients have needed hemodialysis for at least three months before the
enrollment (randomization) in the study.
5. Eligible patients should require in-hospital hemodialysis for at least 12 hours per week.
6. Regular treatment with rHuEPO (epoetin alfa, epoetin beta, or darbepoetin alfa) within
at least three months before the enrollment (randomization) in the study. Epoetins have
been used at a stable dose with the same frequency (q1w/q2w/q3w).
7. Eligible patients have had a target Hb level of 100 to 120 g/L within three months
before the enrollment (randomization) in the study.
8. Effective hemodialysis: dialysis dose (Kt/V) ≥1.21.
9. TSAT ≥20%, ferritin >100 ng/mL.
10. Eligible patients with reproductive potential have agreed to practice acceptable
methods of birth control throughout the entire study period, starting from the screening
and up to four weeks after the last dose of the IP. This requirement does not apply to
patients after surgical sterilization. A combination of at least two birth control methods,
including one barrier contraceptive (e.g., spermicide plus condoms or oral
contraceptives plus condoms), is considered effective.
11. Patient’s ability (in the investigator’s opinion) to follow the protocol procedures.
3.3.2. Exclusion criteria
1 Kt/V = -Ln (R - 0.008 × t) + (4 - 3.5 × R) × UF/W
Where Ln is natural logarithm, R is a ratio between predialysis BUN and postdialysis BUN (or urea), t is dialysis
duration (hrs), UF is ultrafiltration volume (L), W is patient’s weight after dialysis.

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 12 of 24
1. Non-renal anemia: e.g., anemia due to vitamin B12 or folate deficiency, chronic blood
loss, aluminum poisoning, or a chronic disease (C-reactive protein >20 mg/L); sickle-cell
anemia; refractory anemia with excess blasts.
2. Lupus nephritis, CKD due to systemic vasculitis.
3. Platelets <100×109/L.
4. Hemoglobin <100 g/L and >120 g/L.
5. Scheduled kidney transplantation within the study period.
6. The presence of neutralizing/binding antibodies to erythropoietin/darbepoetin in the
blood.
7. History of severe hypersensitivity reactions (anaphylaxis or multiple drug allergy).
8. Vaccination within 8 weeks before randomization.
9. Cirrhosis of the liver with portal hypertension and/or splenomegaly and/or ascites.
10. HIV, active HBV or HCV infection2.
11. ALT, AST > 3×ULN.
12. Documented myelofibrosis.
13. Decompensated heart disease (NYHA Class IV CHF).
14. Resistant hypertension3.
15. Unstable angina.
16. Documented hemoglobinopathy, myelodysplastic syndrome, hematological malignancy.
17. Pure red cell aplasia.
18. Severe secondary hyperparathyroidism (intact PTH >9×ULN).
19. Gastrointestinal hemorrhage within less than three months before randomization.
20. A thrombotic event (e.g., myocardial infarction, a stroke, a transient ischemic attack,
deep vein thrombosis, or pulmonary embolism) within less than six months before
enrollment in the active phase of the study.
2 For eligible patients who tested positive for HBV/HCV markers at the screening, an infectious disease specialist should report the absence of active infection. To confirm the diagnosis of viral hepatitis, additional tests can be performed (e.g., serological examination or qualitative PCR test for viral DNA/RNA) [for details, please refer to section 4.7.4.4.].
3 Resistant hypertension includes all cases of hypertension that are not controlled by the combination of three anti-hypertensive drugs after the patient has achieved his/her dry weight.

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 13 of 24
21. History of acute hemolysis.
22. Seizures, including epilepsy.
23. Major surgery within one month before randomization.
24. Blood transfusion within three months before randomization.
25. An acute or active chronic infection/inflammation, for example, a septic lesion or aseptic
inflammation (hematomas, except for asymptomatic bruising around the venipuncture
site).
26. Any psychiatric disorder, including a history of major depression and/or suicidal
ideation/attempts that can, in the investigator’s opinion, put the patient at risk or affect
patient’s ability to follow the study protocol (including limited capacity).
27. Malignancy, except for cured basal-cell carcinoma and/or cervical carcinoma in situ.
28. Alcohol or drug abuse.
29. Documented hypersensitivity to darbepoetin alfa or any component of the investigational
products.
30. Documented hypersensitivity to iron(III)-hydroxide sucrose complex.
31. Simultaneous participation in another clinical study, previous participation in other
clinical studies within three months prior to the enrollment in this study.
32. Pregnancy and breastfeeding.
4. EFFICACY ASSESSMENT
4.1 Primary endpoint
Change in hemoglobin concentration from baseline (the mean Hb concentration for
screening and visit 1) to the evaluation period (the mean of at least two Hb measurements
between weeks 21 and 24). Change in hemoglobin concentration is calculate as difference
between evaluaton period level and baseline.
4.2. Secondary endpoints
Key secondary endpoints:

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 14 of 24
Proportion (%) of patients achieving the target hemoglobin level (100–120 g/L) at
weeks 21 to 24;
Mean dose of the investigational product (µg/kg) during the last four weeks of treatment
(weeks 21 to 24);
Proportion (%) of patients receiving blood transfusions during the entire treatment period
(weeks 1–24);
Proportion of patients who required dose titration at the beginning of the study (weeks 1
to 20).
Additional secondary endpoints:
Mean hemoglobin during the last four weeks of treatment (weeks 21 to 24);
Proportion (%) of patients who maintained hemoglobin at 90–100 g/L for at least the last
four weeks of treatment (weeks 21 to 24);
Hemoglobin concentration over time during the entire study period (weeks 1 to 24);
Mean dose of the investigational product (µg/kg) during every four weeks of treatment
(weeks 21 to 24);
Mean hemoglobin during every four weeks of treatment (weeks 21 to 24);
Mean hematocrit during every four weeks of treatment (weeks 1 to 24);
Safety assessment:
AE/SAE rate;
Rate of grade 3 to 4 AEs and SAEs;
Rate of study discontinuation due to an AE/SAE;
Rate of arterial and venous thrombotic events (e.g., impaired cerebral circulation,
arteriovenous fistula thrombosis).
Immunogenicity Endpoint
Proportion of BAB-/NAB-positive patients.
5. THE PLANNED ANALYSIS
The statistical analysis will be performed in one step, which will include the final analysis
for the primary endpoint.

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 15 of 24
Final Report
The Final Report will include the analysis of data obtained for both study groups after 24
weeks of blinded treatment with darbepoetin alfa (BCD-066 or Aranesp®) administered at a dose
adjusted with respect to the Hb level (weeks 1 to 24).
Supplementary Report
The Supplementary Report will include the analysis of data obtained for both study
groups after 52 weeks of blinded treatment with darbepoetin alfa (BCD-066 or Aranesp®
)
administered at a dose adjusted with respect to the Hb level (weeks 1 to 52).
6. SAMPLE SIZE CALCULATION
The study aims at demonstrating the equivalent efficacy of the BCD-066 and the
reference drug, Aranesp®
.
Unlike the equality hypothesis testing, testing for the equivalence allows a difference in
the efficacy variable (ε) between the test drug and the reference drugs. This difference should not
exceed a pre-specified equivalence margin (δ) for the test and reference drugs.
The mathematical statement for the null hypothesis (equivalence) is as follows:
𝐻0: |휀| ≥ 𝛿, (1)
which is equivalent to considering the following one-sided hypotheses:
𝐻01: 휀 ≥ 𝛿 and 𝐻02: 휀 ≤ (−𝛿) (2)
The mathematical statement for the alternative hypothesis is as follows:
𝐻1: |휀| < 𝛿, (3)
which is equivalent to considering the following one-sided hypotheses:
𝐻11: 휀 < 𝛿 and 𝐻12: 휀 > (−𝛿) (4)
The null hypothesis (1) is rejected with the significance level α if the following two
inequalities are true:
휀 − 𝛿
𝜎√1
𝑛𝑇+
1𝑛𝑅
< −𝑧𝛼 and 휀 + 𝛿
𝜎√1
𝑛𝑇+
1𝑛𝑅
> 𝑧𝛼,
Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 16 of 24
where 𝑛𝑇 и 𝑛𝑅 is the sample size of the test/reference group, is standard deviation, α is type I
error, ε is the true difference between mean values of the efficacy variable in the groups, δ is the
equivalence margin between the test and reference drugs.
Change in hemoglobin concentration from baseline (the mean Hb concentration for
screening and visit 1) to the evaluation period (the mean of at least two Hb measurements
between weeks 21 and 24) has been chosen as the primary efficacy endpoint.
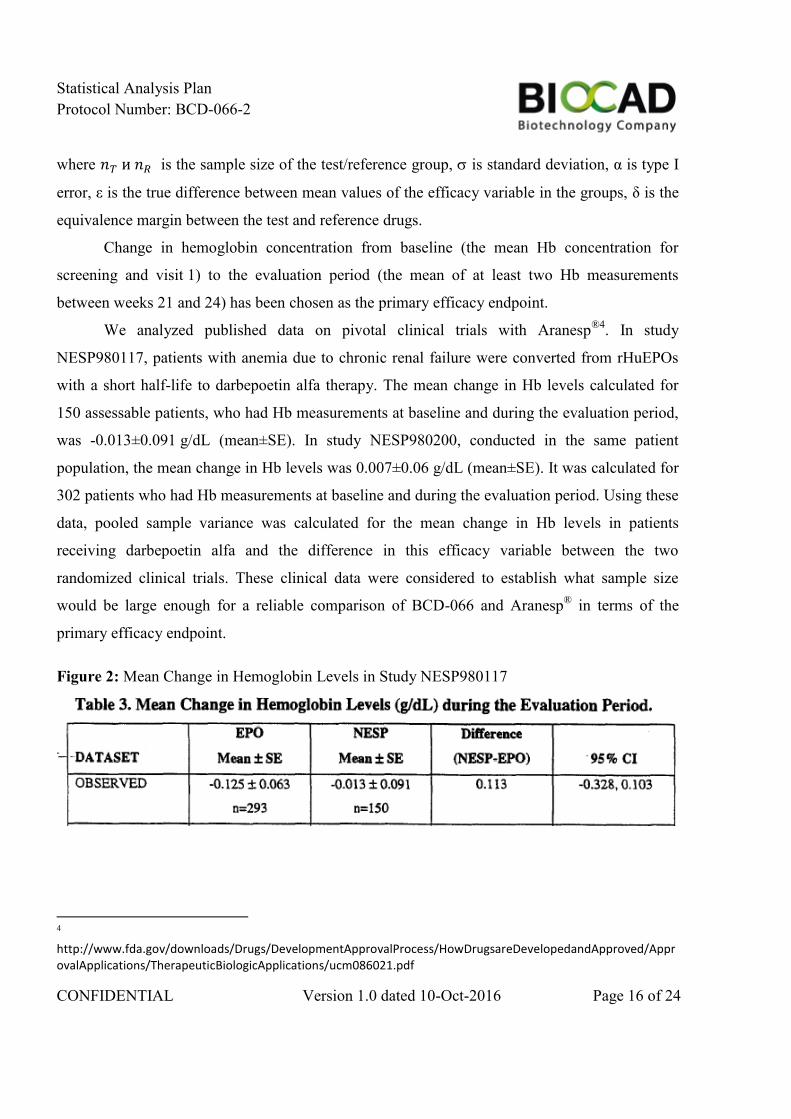
We analyzed published data on pivotal clinical trials with Aranesp®4
. In study
NESP980117, patients with anemia due to chronic renal failure were converted from rHuEPOs
with a short half-life to darbepoetin alfa therapy. The mean change in Hb levels calculated for
150 assessable patients, who had Hb measurements at baseline and during the evaluation period,
was -0.013±0.091 g/dL (mean±SE). In study NESP980200, conducted in the same patient
population, the mean change in Hb levels was 0.007±0.06 g/dL (mean±SE). It was calculated for
302 patients who had Hb measurements at baseline and during the evaluation period. Using these
data, pooled sample variance was calculated for the mean change in Hb levels in patients
receiving darbepoetin alfa and the difference in this efficacy variable between the two
randomized clinical trials. These clinical data were considered to establish what sample size
would be large enough for a reliable comparison of BCD-066 and Aranesp® in terms of the
primary efficacy endpoint.
Figure 2: Mean Change in Hemoglobin Levels in Study NESP980117
4 http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm086021.pdf

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 17 of 24
Figure 3: Mean Change in Hemoglobin Levels in Study NESP980117
A difference of ±0.5 g/dL in Hb level was considered a clinically insignificant difference
for the treatment response between the originator and the biosimilar darbepoetin alfa. The EMA
Guideline on non-clinical and clinical development of similar biological medicinal products
containing recombinant erythropoietins [EMEA/CHMP/BMWP/301636/2008, 18 March 2010]
recommends this equivalence margin (δ) for the chosen primary efficacy endpoint: “If change
from baseline in haemoglobin is used as the primary endpoint, an equivalence margin of
±0.5 g/dL is recommended”.
The sample size was calculated using the formula developed for parallel-group
equivalence studies with equal allocation (Chow S.C., 2008, p.91)5:
𝑛𝑇 =(1 +
1𝑘
) ∗ (𝑧𝛼 + 𝑧𝛽/2)2 ∗ 𝜎2
(𝛿 − |휀|)2=
2 ∗ 1.138506 ∗ (−1.64 − 1.28)2
0.52≈ 78 subjects/group
nC ≈ 78 subjects/group
nт = nc is the sample size of each of the active treatment groups,
zα, zβ/2 are quantiles of normal distribution N(0,1) (mean=0, SD=1). The following values are
used in these calculations: type I error α=0.05, study power should be at least 80%, i.e. type II
error β=0.2.
ε is the true difference in mean values of the efficacy variable (for the biosimilar, ε=0),
δ is the equivalence margin (δ=0.5),
5 Shein-Chung Chow et al. Sample Size in Clinical Research, p.91
Lehana Thabane Sample Size Determination in Clinical Trials, p.10

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 18 of 24
k is the ratio between sample sizes of the test and reference groups: 𝑘 =𝑛𝑇
𝑛𝐶= 1.
σ2 is pooled sample variance calculated as follows:
𝜎2 =𝑠𝑇
2 ∗ (𝑛𝑇 − 1) + 𝑠𝐶2 ∗ (𝑛𝐶 − 1)
𝑛𝑇 + 𝑛𝐶 − 2= 1.138506,
where sT is SD for the test drug calculated as follows:
SET =𝑠𝑇
√nT
, where the sample size 𝑛𝑇 = 150, standard error 𝑆𝐸𝑇 = 0.091
𝑠𝑇 = 𝑆𝐸𝑇 ∗ √𝑛𝑇 = 0.091 ∗ √150 = 1.114518
where sC is SD for the reference drug calculated as follows:
SEC =sC
√nC
, where the sample size 𝑛𝐶 = 302, standard error 𝑆𝐸𝐶 = 0.06
𝑠𝐶 = 𝑆𝐸𝐶 ∗ √𝑛𝐶 = 0.06 ∗ √302 = 1.042689
Figures 4 and 5 present results of sample size calculations in Statistica 10.

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 19 of 24
Figure 4: Sample Size Versus Type I Error Rate
0,00 0,05 0,10 0,15 0,20 0,25 0,30
Type I Error Rate (Alpha)
30
40
50
60
70
80
90
100
110
120
130
Required S
am
ple
Siz
e (
N)

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 20 of 24
Figure 5: Sample Size Versus Study Power
0,6 0,7 0,8 0,9 1,0
Power Goal
50
60
70
80
90
100
110
120
130
140
Re
qu
ire
d S
am
ple
Siz
e (
N)
A further adjustment was made to account for an estimated 25% rate of dropout, resulting
in total planned enrollment of 98 patients per group.
With two groups and a randomization ratio of 1:1, the study should involve 196 patients.
This sample size suffices to show statistically significant differences (or their absence).
7. STATISTICAL ANALYSIS OF POPULATION
Efficacy Analysis
The efficacy will be analyzed in the intent-to-treat (ITT) and per protocol (PP)
populations.
Modified Intent-to-Treat Population

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 21 of 24
The total efficacy population is defined as the ITT population and will include all
randomized patients who have received at least one dose of the test drug or the reference drug.
According to the ITT principle, the patients will be analyzed by treatment groups to which they
have been randomly allocated.
Per Protocol Population
The per protocol (PP) population will include those ITT patients who have completed the
study without major protocol deviations and are assessable for the efficacy at weeks 21 to 24 of
the main study period.
Subjects can be withdrawn from the PP population for the following reasons:
• The subject discontinued his/her participation in the study before day 169 visit;
• Study therapy differs from that assigned by randomization or there were
significant deviations in the treatment regimen (for example, the subject received the test drug
instead of the reference drug, or vice versa);
• There is evidence for subject non-compliance.
Safety Analysis
The safety assessment population will include all subjects who have received at least one
dose of the investigational product and who attended at least one safety assessment visit after the
baseline. The analysis will be performed according to the actually received treatment.
8. ANALYSIS PLAN AND STATISTICAL METHODS
8.1. The software
For statistical analysis and to construct the tables and diagrams would be used the
software STATISTICA 10 and statistical programming language R.
8.2. Description of the Statistical Methods to be Employed
Quantitative Data
The following quantitative data will be analyzed in the study:
Efficacy:
Hemoglobin concentration,

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 22 of 24
Dose of the investigational product,
Hematocrit level
Safety:
CBC results,
Blood chemistry results,
Vital signs
The statistical analysis will include two-tailed hypothesis testing; the chosen significance
level is 0.05.
Quantitative variables will be tested for normality using the Shapiro-Wilk test.
Normally distributed quantitative variables will be tested using the two-sample Student’s
t-test, Welch’s t-test, and ANOVA.
Non-normally distributed quantitative variables will be tested using the Mann-Whitney
U-test, the Wilcoxon test, the Kruskal-Wallis test, and the Friedman test.
Normally distributed quantitative data will be described using the following descriptive
statistics: mean, SD, CV, min, and max. Non-normally distributed quantitative data will be
described using the following descriptive statistics: median, quartiles, CV, min, and max.
Categorical Data
The following categorical data will be analyzed in the study:
Efficacy:
Proportion of patients maintaining the target hemoglobin level,
Proportion of patients receiving blood transfusion,
Proportion of patients who required dose titration at the beginning of the study
(weeks 1 to 20).
Safety:
ECG findings,
Rate and severity grades of AEs/SAEs,
Rate of Grade 3-4 AEs,
The frequency of early withdrawals due to AEs/SAEs,

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 23 of 24
Rate of arterial and venous thrombotic events (e.g., impaired cerebral circulation,
arteriovenous fistula thrombosis).
Immunogenicity:
The percentage of BAB- and NAB-positive patients.
Categorical data will be processed using frequency tables, contingency tables, the
Fisher’s exact test, the test of equal frequencies, Pearson’s 𝜒2 test, and the Cochran-Mantel-
Haenszel test. Percentages or proportions will be used to describe categorical data.
The equivalence hypothesis in terms of the primary endpoint (change in Hb
concentration) will be tested by comparing 95% CIs calculated for the arithmetic means of
changes in the Hb concentration in the study groups with the pre-specified equivalence margin.
The change in the Hb concentration will be calculated for each patient in both study groups. It is
defined as the difference between the mean Hb during the last four weeks of treatment and the
mean Hb at baseline. The former variable is calculated as the arithmetic mean of all Hb values
(at least two) measured between weeks 21 and 24 of the mean study period. The latter one is
calculated as the arithmetic mean of the Hb values measured at the screening and visit 1.
The null hypothesis (𝐻0: |휀| ≥ 𝛿, where δ is the equivalence margin) will be rejected and
the drugs will be considered equivalent if the boundaries of the 95% CI for the difference in
mean change in the Hb level fall within the equivalence margin (δ = 0.5 g/dL).
The Benjamini-Yekutieli correction for multiple testing will be used.
Statistical methods will be chosen according to the type of initial data and their
distribution. Appropriate statistical tests will be established after the data collection has been
completed because the type of data distribution, sample homogeneity, etc. are unknown before
the study start. For appropriate data processing, the list of statistical methods used may expand
during the analysis.
8.3. Accounting for missing, unavailable or doubtful data, outliers
All information specified in the e-CRFs should be supported by relevant data in source
documents.
After entering all data into the electronic database, a database specialist checks it for
inconsistencies, errors, and missing data points. The Clinical Study Database Manager or

Statistical Analysis Plan
Protocol Number: BCD-066-2
CONFIDENTIAL Version 1.0 dated 10-Oct-2016 Page 24 of 24
Medical Expert of JSC BIOCAD generates queries to correct error data or to request missing
data; the queries are site-specific and subject-specific (i.e., individual queries are generated for
each subject). The Clinical Study Monitor will send queries to the study site by fax or email. The
queries should be resolved by the investigator within five business days from the date they have
been submitted to the study site. Copies of responses to queries must be kept at the study site;
original responses must be stored at JSC BIOCAD.
When responses to queries are received from investigators, the database specialist checks
it for inconsistencies, errors, and missing data points. When all data at all study sites have been
collected and entered, the database is closed. Afterwards, statistical processing can be started.
Missing, unused, and spurious data will not be substituted.
Spurious and unevaluable data are revealed during the outlier analysis by examination of
Mahalanobis or Cook distance, visual analysis of scatter plots and box plots.
All actions taken to handle missing, unevaluable, spurious data and outliers before/during
the statistical analysis will be described in the Clinical Study Report.
8.6. Multivariate Comparison and Multiplicity
Refer to Section 8.2.
8.7. Subgroup analysis, interaction and related variables
Not available.
9. OTHER PLANNED ANALYSES
No additional analyses are planned in this study.
10. DEVIATIONS FROM ANALYSIS METHODS DESCRIBED IN STUDY PROTOCOL
This Statistical Analysis Plan has no deviations from methods described in Study
Protocol.