structural analysis of a ternary complex of allantoate amidohydrolase from escherichia coli reveals...
TRANSCRIPT

doi:10.1016/j.jmb.2007.02.028 J. Mol. Biol. (2007) 368, 450–463
Structural Analysis of a Ternary Complex of AllantoateAmidohydrolase from Escherichia coli Revealsits Mechanics
Rakhi Agarwal1, Stephen K. Burley2 and Subramanyam Swaminathan1⁎
1Biology Department,Brookhaven NationalLaboratory, Upton,NY 11973, USA2SGX Pharmaceuticals Inc.,San Diego, CA 92121, USAAbbreviation used: MAD, multiwdispersion.E-mail address of the correspondi
0022-2836/$ - see front matter © 2007 E
Purine metabolism plays a major role in regulating the availability of purinenucleotides destined for nucleic acid synthesis. Allantoate amidohydrolasecatalyzes the conversion of allantoate to (S)-ureidoglycolate, one of thecrucial alternate steps in purine metabolism. The crystal structure of aternary complex of allantoate amidohydrolase with its substrate allantoateand an allosteric effector, a sulfate ion, from Escherichia coli was determinedto understand better the catalytic mechanism and substrate specificity. The2.25 Å resolution X-ray structure reveals an α/β scaffold akin to zincexopeptidases of the peptidase M20 family and lacks the (β/α)8-barrel foldcharacteristic of the amidohydrolases. Arrangement of the substrate and thetwo co-catalytic zinc ions at the active site governs catalytic specificity forhydrolysis of N-carbamyl versus the peptide bond in exopeptidases. In itscrystalline form, allantoate amidohydrolase adopts a relatively openconformation. However, structural analysis reveals the possibility of asignificant movement of domains via rotation about two hinge regions uponallosteric effector and substrate binding resulting in a closed catalyticallycompetent conformation by bringing the substrate allantoate closer to co-catalytic zinc ions. Two cis-prolyl peptide bonds found on either side of thedimerization domain in close proximity to the substrate and ligand-bindingsites may be involved in protein folding and in preserving the integrity ofthe catalytic site.
© 2007 Elsevier Ltd. All rights reserved.
Keywords: allantoate amidohydrolase; crystal structure; di-zinc-dependentexopeptidases; allosteric effector; hinge region
*Corresponding authorIntroduction
The purine catabolic pathway regulates the purinepool in the cell to ensure adequate supplies ofprecursors for nucleic acid synthesis. Unlike purinebiosynthesis, purine catabolism differs both amongand within different plants and animals. Moreover,purine catabolic end products vary between species.In higher primates, including humans, uric acid isthe end product. In other mammals, allantoin isformed.1 In plants, purine nucleotides are catabo-lized via ureides, allantoin, and allantoate to NH3and CO2 through the conventional purine catabolicpathway. However, in specific organs such as the
avelength anomalous
ng author:
lsevier Ltd. All rights reserve
roots of ureide-accumulating plants allantoin and/or allantoate constitute the end products of thepathway and are translocated to different parts ofthe plant, such as shoots and leaves, where degrada-tion is ultimately completed.Allantoin utilization by Escherichia coli as a sole
source of nitrogenunder anaerobic conditionwas firstsuggested by Vogels and van der Drift.2 Allantoinamidohydrolyase (allantoinase) catalyzes hydrolysisof the internal amide bond of allantoin, giving rise toallantoate (Figure 1). This enzyme is found in manyplants.3 Typically, allantoate is hydrolyzed to ureaand ureidoglycolate, which is further degraded toglyoxylate and urea. Urea formed via this pathwaymay be hydrolyzed to ammonia and CO2 by urease.However, several lines of evidence suggest theexistence of an alternate “allantoate amidohydrolase(allantoate deiminase)” pathway in plants andmicroorganisms.3,4 In this pathway, allantoate ishydrolyzed to CO2, NH3, and ureidoglycine, which
d.

Figure 1. The allantoin catabolic pathway in Escher-ichia coli K12.
451Structure of Allantoate Amidohydrolase
is unstable and readily undergoes deamination toureidoglycolate (either spontaneously or enzymati-cally by allantoate amidohydrolase). Two differentenzymes can then act upon ureidoglycolate. Ureido-glycolate hydrolase gives rise to glyoxylate and urea,allowing glyoxylate to enter general glyoxalate anddicarboxylate metabolism. A second enzyme, urei-doglycolate dehydrogenase, oxidizes ureidoglycolateto oxalureate (Figure 1). Subsequently, oxalureate canbe converted into oxamate and carbamylphosphate,which can be further metabolized to CO2, NH4
+, andATP.Allantoate amidohydrolases from various bacteria
have been studied extensively.4–7 The enzyme isthought to be unstable, is readily inactivated, andshows pH-dependent activity.8,9 Here, we presentthe X-ray crystal structure of allantoate amidohy-drolase from E. coli K12 bound to its substrate allan-toate and the anionic ligand sulfate. The enzymeactive center contains two zinc ions. Substrate, i.e.allantoate, and sulfate ligand-binding residues havebeen identified. The basis of substrate selectivity andthe role of anionic ligand sulfate as an allostericeffector are discussed. Two cis-peptide bonds areidentified and their possible role in folding andfunction of allantoate amidohydrolase is discussed.A comparison with structural neighbors, the relativepositioning of substrate, ligand and hinge regionsprovides insights into the mechanism(s) of enzy-matic catalysis.
Results and Discussion
Structure determination
The final refined model for the native protein is adimer with each half consisting of residues 3–412,
two zinc ions, and one sulfate anion. The allantoatesubstrate is present only in protomer A. Both proto-mers are essentially identical in structure, with aroot-mean-square deviation (r.m.s.d.) of 0.670 Å for406 equivalent α-carbon atomic pairs. The entirepolypeptide chain (residues 3–412) fall within theallowed regions of the Ramachandran diagram asshown in Table 1.10
It is remarkable that the active site structures of thenative (PDB code 1Z2L) and Se-Met (PDB code2IMO) forms of the protein differ markedly from oneanother in one important respect. Specifically, thezinc ion pair is absent from the Se-Met structure. Wesuggest that this difference is due to the pH at whichthe crystals were grown (native pH 8.5 versus Se-MetpH 4.6). The His side-chains responsible for zinc ioncoordination in the native structure adopt differentrotomer positions in the Se-Met structure, whichpresumably reflects differences in their protonationstates at acidic pH. The loss of activity of theenzyme at the low pH (<7) is possibly due toprotonation of the bridging hydroxide, protonationof the general base (Glu128) or protonation of aLewis acid (His384), in addition to the loss of metalions. Not surprisingly, the Se-Met active site alsolacks both bound sulfate and allantoate. Allsubsequent discussion here of structure and func-tion of allantoate amidohydrolase will be limited tothe native form of the enzyme.
Overall structure
E. coli allantoate amidohydrolase exists as ahomodimer both in our crystal structure (moleculesA and B are related by a non-crystallographic 2-fold)and in solution as determined by gel-filtrationchromatography (data not shown). The enzymepossesses the M20_dimer motif with substantialsolvent-accessible surface area (∼2278 Å2) buried ondimerization. The three-dimensional structure of theallantoate amidohydrolase is shown schematically inFigure 2. Each polypeptide chain folds into twostructural domains: a large catalytic domain bearingmetal ions, sulfate, and substrate-binding sites and asmaller domain,which supports dimerization (Figure3(a) and (b)). The dimerization domain also con-tributes to substrate binding by the other protomer,suggesting that the dimer visualized by X-ray crystal-lography and detected in solution by analytical gel-filtration chromatography represents the biochemi-cally/biologically active form of the enzyme.
Catalytic domain
This domain is composed of both N-terminal(residues 3–212) and C-terminal residues (residues330–412). It has mixed three-layer α/β/α-sandwicharchitecture. The central mixed polarity β-sheetconsists of eight β-strands (strand order 1, 2, 3, 4,5, 6, 15, 16) and is flanked on one side by four longα-helices and on the other by two short α-helices(Figure 2). Two zinc ions bind at one end of the fourcentral β-strands in the active site, which is typical

Table 1. Data collection and refinement statistics
Native Selenomethionine
A. Cell parameters and data statisticsSpace group P21212 P21212Unit cell parameters
a (Å) 95.53 95.21b (Å) 186.58 184.3c (Å) 49.22 48.9
Native Peak Se-MAD Inflection Remote
Wavelength (Å) 1.1 0.9793 0.9795 0.940Resolution (Å) 50–2.25 50–3.0 50–3.0 50–3.0Molecules/asymmetric unit 2 2 2 2Redundancy 15.5 12.9 13.1 12.8I/σ 21.2 6.8 6.9 5.5Rmerge 0.066 (0.46) 0.12 (0.63) 0.11 (0.60) 0.14 (0.73)No. unique reflections 41,530 17,912 17,947 17,792Phasing power (iso/ano) 0.686FOMa (after SHARP) 0.57FOMa (after DM) 0.93
B. Refinement statistics: NativeResolution range (Å) 50–2.25No. reflections 40,329Completeness (%) 97.2 (96.1)R-factor 0.22Rfree 0.27No. protein atoms 6407No. zinc atoms 4No. allantoate 1No. sulfate ions 2No. water molecules 194r.m.s.d from ideal
Bond lengths (Å) 0.0075Bond angles (deg.) 1.43
Average B-factorAll protein atoms (Å2) 33.4Allantoate (molecule A) 27.0Zinc (Å2) 39.5Sulfate (Å2) 28.2
Ramachandran plotMost favored regions (%) 89Additionally allowed regions (%) 10.3Generously allowed regions (%) 0.4Disallowed regions (%) 0.3
Values in parentheses are for the highest resolution shell.a FOM; figure of merit.
452 Structure of Allantoate Amidohydrolase
of exopeptidases from the peptidase_M20 family(INTERPRO # IPR002933).
Dimerization domain
The dimerization domain of allantoate amido-hydrolase consists of a 117-residue insertion be-tween β-strands 6 and 13 (residues 213–329). Thisinserted domain folds into a four-stranded antipa-rallel β-sheet (strand order 7, 10, 12, 13), flanked onone side by two long α-helices (9 and 10) (Figure 2).The fold of the dimerization domain resembles thoseof domains found in carboxypeptidase G2 ofPseudomonas species strain RA-16,11 and β-alaninesynthase from Saccharomyces kluyveri.12
The active site
Biochemical studies of allantoate amidohydro-lases from various sources revealed the metal-
dependent exopeptidase nature of the enzyme.6,7,9
However, the presence of bi-metal center was notrevealed until our structure determination. Thecatalytic activity of allantoate amidohydrolase fromStreptococcus allantoicus has been tested in thepresence of various divalent cations at pH 6.05, pH7.45, and pH 8.45, of which manganese yielded thehighest activity at pH 8.45.8 There is generalagreement that the optimum pH for activity is ∼8for all allantoate amidohydrolases and they are lessactive under acidic conditions, although the precisepH-dependence activity profiles of enzymes fromdifferent sources vary.6,8
The enzyme active site is located within a deepcleft formed between the dimerization and catalyticdomains. Figure 4(a) illustrates the bi-metal activecenter (metal ion separation=3.4 Å; Table 2). Thetwo zinc ions have subtly different coordinationgeometries. Zinc 1 is penta-coordinated by His83,Asp94, and His192, plus two water molecules

Figure 2. Topological representation of allantoate amidohydrolase (α-helices and β-strands are numberedsequentially; N and C termini are labeled).
453Structure of Allantoate Amidohydrolase
(Wat341 and Wat 344). Zinc 2 displays distortedtetrahedral coordination by Asp94, Glu129, andHis384, plus Wat344. We believe that the bridgingWat344 represents the classic nucleophile requiredfor attacking the carbon-nitrogen bond of the sub-strate upon activation by the side-chain of nearbyGlu128.The structural similarity of allantoate amidohy-
drolase to the di-zinc-dependent exopeptidases andthe observed pattern of side-chain–metal coordi-nation suggest that the most likely identity of thebound metals in our structure is a pair of zincions.13,14 Moreover, the allantoate amidohydrolasesbelong to the peptidase_M20 family, which encom-passes a range of zinc metallopeptidases distributedamong several subfamilies. Metalloproteases are themost diverse of the four main types of proteases,with more than 30 families described to date.15 Inthese enzymes, a divalent cation, usually a zincion, is thought to activate a water molecule duringcatalysis. Typically, the metal ion is held in place by,usually three, amino acid side-chain ligands . Aminoacids known for metal ion coordination are His, Glu,Asp, and Lys, and at least one other residue isrequired for catalysis, which may play the role of anelectrophile. Although a metal analysis for thisparticular enzyme from E. coli has not been carriedout, structural comparisons and the observed coor-dination geometries support the presence of zinc.13
This was confirmed also by scanning near the
absorption edge of zinc at the synchrotron source.The role of a bi-metal center for the activation ofnucleophilic water is well known for hydrolysis ofcarbon–nitrogen bonds.
Bipartite substrate and ligand-binding site:substrate selectivity and allosteric effector
Within the active site cleft of molecule A, twosignificant electron density features were observedin the difference Fourier syntheses that could bemodeled as the substrate allantoate, although itwas not specifically included during crystallization,and a sulfate ion (SO4 415). A composite omit mapcontoured at the 1σ level at the substrate and thesulfate ion is shown in stereo view in Figure 5.Within molecule B, the electron density map did notpermit unambiguous modeling of allantoate. Allan-toate and the sulfate ion (or possibly phosphate)were probably carried through protein expressionand purification steps. The electron density mapallowed them to be modeled unambiguously. Theelectron density for allantoate was well definedand ruled out the possibility of product formation(Figure 5). The probable reason for this is thatthe molecule exists in a partially open conforma-tion, the protein–substrate complex represents half-filled active sites, or because the protein waspurified at ∼pH 7 while the optimum activityrequires pH>8.

Figure 3. Ribbon representation of allantoate amidohydrolase. Each protomer is composed of catalytic anddimerization domains. Molecules A and B are shown in green and dark pink, respectively, with zinc ions (metallic green),allantoate (magenta), and sulfate anion (yellow). Views (a) and (b) are related by a 90° rotation about the vertical.
454 Structure of Allantoate Amidohydrolase
Restricting our analysis of enzyme–substrateinteractions to molecule A, we detected a total ofnine contacts for allantoate (Figure 4(b), Table 2);two with the sulfate anion, three with the samepolypeptide chain (Arg290 and His384), four withthe dimerization domain of the molecule B (His228and Asn277). SO4 415 (molecule A) interacts withthe Gln 215, Arg217, Arg290, Gly359, and allantoate(Figures 4(b) and 6, Table 2). Arg290 and Gly359 areconserved among all allantoate amidohydrolasesfrom different species.Several anions enhance allantoate amidohydrolase
activity.8 Given these observations and our structuralfindings, we suggest that the sulfate (or possiblyphosphate) anion represents an allosteric effector ofthe enzyme that is responsible for stabilizing sub-
strate binding. In addition, this anion effectormay actas a counterion during enzyme-mediated catalysis.Allantoate amidohydrolase has specificity for
allantoate. A comparison of the binding site residueswith those of the four structurally similar proteins(β-alanine synthase, PDB code 1R3N; carboxypepti-dase G2, PDB code 1CG2; putative peptidase T, PDBcode 1VIX; succinyl diaminopimelate desuccinylase,PDB code 1VGY) gives some insights into specificity.Of the four residues involved in binding allantoate(His228, Asn277, Arg290, and His384, all numberingaccording to 1Z2L), only His228 and Arg290 areconserved among all five proteins (Table 3). His384is conserved among four of the five proteins, exceptin 1CG2. Asn277 is conserved among four of the fiveproteins, except in 1VIX. These variations could be

Figure 4. (a) The di-zinc center of allantoate amidohydrolase., showing both zinc cations (red), the nucleophilic watermolecule (Wat344), and the sulfate anion allosteric effector. (b) Allantoate substrate and sulfate ion allosteric effectorbinding sites. Residues originating from the other half of the homodimer are labeled with B.
455Structure of Allantoate Amidohydrolase
responsible for substrate specificity. Gly359 isconserved among four of the five proteins, exceptin 1VGY (Thr). The sulfate-binding residue, Gln215,is not conserved (1R3N, Tyr; ICG2 Ile; IVGY, Ser;1VIX, Ala).
1R3N, β-alanine synthase, represents the onlyone of the related structures that was determinedas an enzyme-product complex with which a directcomparison of the substrate and allosteric site canbe performedwith our structure of allantoate amido-

†http://www.bioinfo3d.cs.tau.ac.il/HingeProt/index.html
Table 2. Active site interaction distances (allantoate isrepresented as LAL)
Het Het atom Residue Atom Distance (Å)
Zn A: 511 ZN His A: 83 NE2 2.26Asp A: 94 OD2 2.24His A: 192 NE2 2.32H2O: 341 O 2.31H2O: 344 O 2.55
Zn A: 512 ZN Asp A: 94 OD1 2.07Glu A: 129 OE1 2.86
OE2 2.40His A: 384 NE2 2.27H2O: 344 O 2.48
SO4 A: 415 O2 Gln A: 215 OE1 2.96NE2 3.05
Arg A: 217 NH2 2.97Arg A: 290 NH1 2.73LAL A: 414 O2 2.77
O3 Gln A: 215 NE2 3.18Gly A: 359 N 2.32
O4 LAL A: 414 N3 2.771Al A: 414 N4 His B: 228 NE2 π–π
Asn B: 277 OD1 3.51N3 Asn B: 277 OD1 3.01
SO4 A: 415 O4 2.79N12 Asn B: 277 O 3.03O2 Arg A: 290 NH1 3.01
NH2 2.15SO4 A: 415 O2 2.77
456 Structure of Allantoate Amidohydrolase
hydrolase. This comparison demonstrated that theallosteric or sulfate binding site is completelydifferent and thus generates a different environ-ment for substrate binding. For β-alanine synthasethe β-aminoisobutyrate binds to the Arg and Asncorresponding to Arg290 and Asn277 (1Z2L),respectively. However, the presence of Tyr andTrp instead of Gln215 and Arg217, respectively in1Z2L, results in a distinct charge distribution that isunlikely to bind either sulfate or phosphate in β-alanine synthase. We believe, therefore, that subtledifferences in allantoate amidohydrolase versus β-alanine synthase creates an allosteric effector bindingsite in the former, which is not present in the latter.Both small molecule (urea and asparagines) and
ionic (borate and fluoride) inhibitors of allantoateamidohydrolases from various sources have beenidentified.16 Given our structural findings, we sug-gest that asparagine, which is the structural analogof allantoate and ureidoglycine, competes for sub-strate binding. The mechanistic basis for borateinhibition of enzyme activity is less clear, but it mayoccur via displacement of the sulfate ion from itsallosteric binding site.
Functional role of hinge regions duringcatalysis: open and closed conformations
Many proteins are organized as functional do-mains connected by exposed flexible regions,17
known also as hinge regions, which support interdomain flexibility.18 In some protein crystal struc-tures,19–21 large-scalemovements about hinge regionsconnecting globular domains have been observed.Hingemotions are typically thought to followbinding
to another molecule, either a ligand or substrate.Figure 6 shows that in our crystalline conformation ofthe enzyme–substrate–allosteric effector complex,allantoate is not sufficiently close to the active sitezinc ions (∼6 Å) to support catalysis. We thereforesought to examine possible hinge bendingmotions ofthe enzyme.HingeProt† identified two putative hinge residues
in allantoate amidohydrolase (Asn210 and Pro331)that may support transitions between open andclosed conformations (Figure 7(a)). A modest hingeclosure of ∼8° could move the allantoate substrate3–3.5 Å closer to the bi-metal center. In the absenceof bound substrate and/or allosteric effector, wesuggest that the enzyme adopts an open conforma-tion, which may be even more splayed than thatobserved in our crystal structure (Figure 7(a)). Iftrue, the crystallization process would appear tohave stabilized the enzyme in a conformation that isneither fully open nor fully closed. In the case ofallantoate amidohydrolase, we propose that in acompletely transient closed conformation the nar-rowed substrate-binding cleft would allow interac-tion of the substrate molecule with the active sitezinc ions and subsequent catalysis.
Cis-peptide bonds: possible roles in folding,stability and function
The planar peptide bond in proteins is predomi-nantly trans, since cis peptide bonds usually causeunfavorable short contacts between adjacent resi-dues, as is evident from an analysis of structures inthe Protein Data Bank.22,23 The rare presence of apeptide bond in the cis conformation is of interestbecause the cis/trans isomerization of peptide bondson the N-terminal side of proline is thought to playan important role in protein folding.24 Weiss andco-workers have even suggested that cis peptidebonds represent functional determinants of proteinfolding.25 Interestingly, the presence of non-prolinecis-peptide linkages near protein functional siteswas observed by Herzberg and Moult.26 The ener-getic cost by such linkages is sustained to achievethe functionally active three-dimensional conforma-tion of proteins.26
In the present structure, we identified two cis-peptide bonds in both molecules comprising thedimer, Asp260-Pro261 (cis 1) and Arg272-Pro273(cis 2) located on opposite surfaces of the dimeriza-tion domain and near the substrate-binding pocket(Figure 7(b)). These cis peptide residues do notinteract directly with the substrate, but are locatedwithin 10–15 Å of bound allantoate. It is possible thatthe isomerization of cis-Pro273, located at the end ofthe bent β-sheet, would effect the interaction ofAsn277 with the substrate.

Figure 5. A stereo-view of the composite omit map at the active site of molecule A. The electron density is shown inmagenta, contoured at the 1σ level at substrate and allosteric anion sulfate/phosphate shown in ball-and-stick. MoleculeB lacks electron density for allantoate.
457Structure of Allantoate Amidohydrolase
Folding of RNase A is known to require the acidpretreatment and shows two acid-catalyzed pro-cesses, suggesting the presence of two essentialproline residues exhibiting slow-folding and fast-folding species by isomerization. As the protonationof imide bonds is responsible for the acid catalysis of
Figure 6. Relative positioning of the active site and allamidohydrolase, with zinc ions (magenta), sulfate ion (yellowand-stick. Molecules A and B are represented as blue and ma
proline isomerization, the slower reaction possiblyinvolves an imide bond with low pK.24 It may wellbe the bond connecting proline to a positivelycharged amino acid, because the positive chargecould make this bond more difficult to protonate.It is remarkable that allantoate amidohydrolase
osteric-binding and substrate-binding sites in allantoate), and allantoate (red). Selected residues are shown in ball-genta ribbons, respectively.

Table 3. Sequence alignment
Sequence alignment of allantoate amidohydrolase with structural neighbors; β-alanine synthase from Saccharomyces kluyveri (PDB ID1R3N), carboxypeptidase G2 from Pseudomonas sp. (PDB ID 1CG2), succinyl diaminopimelate desuccinylase from Neisseria meningitidisMC58 (PDB ID 1VGY), and a putative peptidase T from E. coli (PDB ID 1VIX). Identical residues are shown in red. The zinc-binding residuesare shown against a black background. The cis-peptides are shown against a red background.
458 Structure of Allantoate Amidohydrolase
exhibits instability and inactivation that requiresacid pre-treatment to restore full catalytic activity invitro.27 The inactive form of the enzyme can beconverted to its active form by reducing the pH to<5in the presence of anions such as oxalate or citrate. Aphosphate ion8 can activate the enzyme only in thepresence of EDTA.28 The metal-free form of theenzyme can be activated by addition of substrateand Mn2+.28 In allantoate amidohydrolase, apositively charged residue (Arg272) precedes thecis-proline, suggesting this enzyme may behavelike RNase A during acid-mediated folding.
Structural homology to di-zinc-dependentexopeptidases: an example of divergentevolution and di-zinc center specificity for theN-carbamyl bond versus the peptide bond
A search with DALI and VAST servers revealedseveral di-zinc-dependent exopeptidases of thepeptidase_M20 family and β-alanine synthase thatare structurally similar to allantoate amidohydro-lase. A ClustalW alignment with β-alanine syn-thase (PDB ID 1R3N), carboxypeptidase G2 fromPseudomonas sp. (PDB ID 1CG2), succinyl diamino-
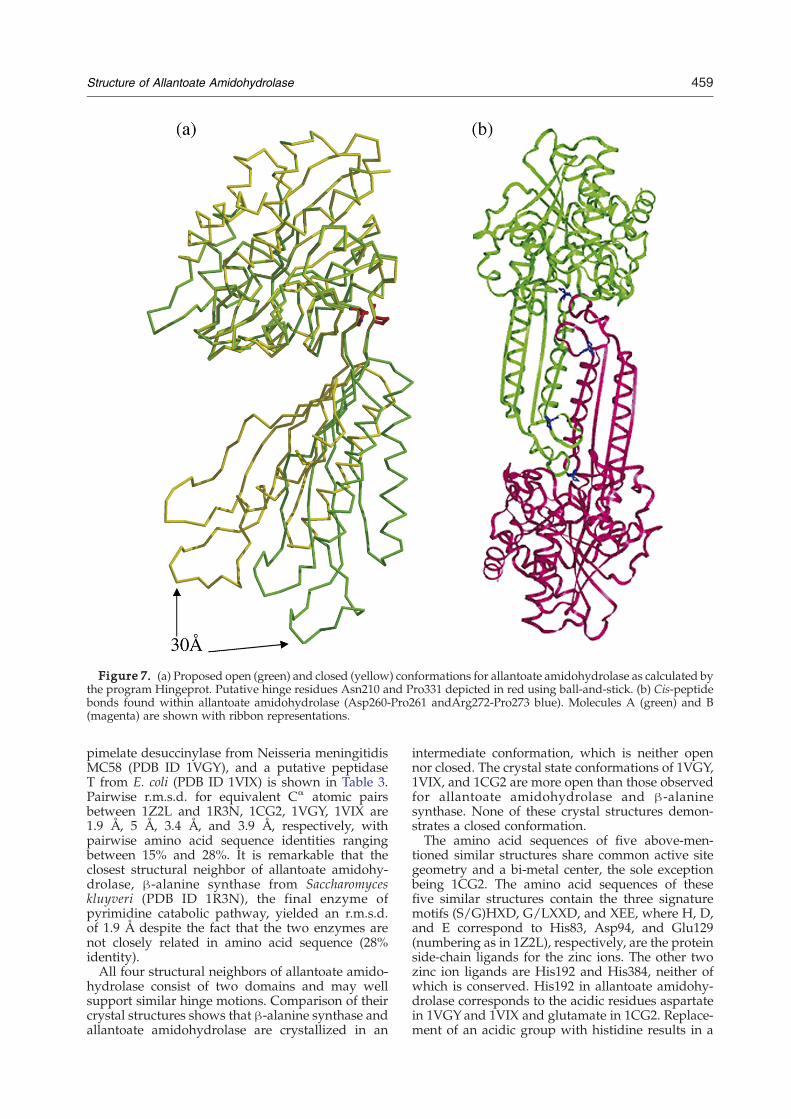
Figure 7. (a) Proposed open (green) and closed (yellow) conformations for allantoate amidohydrolase as calculated bythe program Hingeprot. Putative hinge residues Asn210 and Pro331 depicted in red using ball-and-stick. (b) Cis-peptidebonds found within allantoate amidohydrolase (Asp260-Pro261 andArg272-Pro273 blue). Molecules A (green) and B(magenta) are shown with ribbon representations.
459Structure of Allantoate Amidohydrolase
pimelate desuccinylase from Neisseria meningitidisMC58 (PDB ID 1VGY), and a putative peptidaseT from E. coli (PDB ID 1VIX) is shown in Table 3.Pairwise r.m.s.d. for equivalent Cα atomic pairsbetween 1Z2L and 1R3N, 1CG2, 1VGY, 1VIX are1.9 Å, 5 Å, 3.4 Å, and 3.9 Å, respectively, withpairwise amino acid sequence identities rangingbetween 15% and 28%. It is remarkable that theclosest structural neighbor of allantoate amidohy-drolase, β-alanine synthase from Saccharomyceskluyveri (PDB ID 1R3N), the final enzyme ofpyrimidine catabolic pathway, yielded an r.m.s.d.of 1.9 Å despite the fact that the two enzymes arenot closely related in amino acid sequence (28%identity).All four structural neighbors of allantoate amido-
hydrolase consist of two domains and may wellsupport similar hinge motions. Comparison of theircrystal structures shows that β-alanine synthase andallantoate amidohydrolase are crystallized in an
intermediate conformation, which is neither opennor closed. The crystal state conformations of 1VGY,1VIX, and 1CG2 are more open than those observedfor allantoate amidohydrolase and β-alaninesynthase. None of these crystal structures demon-strates a closed conformation.The amino acid sequences of five above-men-
tioned similar structures share common active sitegeometry and a bi-metal center, the sole exceptionbeing 1CG2. The amino acid sequences of thesefive similar structures contain the three signaturemotifs (S/G)HXD, G/LXXD, and XEE, where H, D,and E correspond to His83, Asp94, and Glu129(numbering as in 1Z2L), respectively, are the proteinside-chain ligands for the zinc ions. The other twozinc ion ligands are His192 and His384, neither ofwhich is conserved. His192 in allantoate amidohy-drolase corresponds to the acidic residues aspartatein 1VGYand 1VIX and glutamate in 1CG2. Replace-ment of an acidic group with histidine results in a

460 Structure of Allantoate Amidohydrolase
decreased negative charge from the protein ligandsthat stabilize the metal-bound hydroxide. There-fore, the zinc-bound water in allantoate amidohy-drolase would be expected to show a lower pKaand higher reactivity than the metal-bound solventin the exopeptidases.29,30 In practice, the hydro-lysis of the N-carbamylamino group does requirean increased nucleophilicity of the attacking wateras compared to that for the hydrolysis of a peptidebond for better resonance stabilization of theureido group and the lower electrophilicity of itscarbonyl carbon.12 Interestingly, a Psi-blast searchwith 1Z2L demonstrated that all known carbamylhydrolases show remarkable identity at these fivepositions (data not shown), including β-alaninesynthase.Although the four structural neighbors described
above have similar polypeptide chain folds andcatalyze similar chemical processes, they have quitedifferent substrate selectivity and specificity. The
Figure 8. The proposed substrate docking at the active sitethe text and only two steps (three stages) are shown here. TheGlu128, which accepts the proton from the zinc-bound water. Tleads to the formation of transition tetrahedral intermediate, stin step 2, cleavage of the scissile bond and the formation of u
observed variation in putative substrate-bindingresidues and structural determinants contributingto substrate specificity is considerable (as discussedearlier). Here, the sulfate ion stabilizes allantoate,and without the sulfate it is likely that dockingof allantoate within the active site during cata-lysis will be affected. The structural similarity ofβ-alanine synthase and allantoate amidohydrolase issurprising, as these two enzymes belong to twodifferent catabolic pathways. We suggest that thesetwo enzymes arose from the same ancestral pepti-dase that evolved into two structurally relatedenzymes with distinct catalytic properties andbiochemical roles within the cell. It is remarkablealso that allantoate amidohydrolase displaysevidence of convergent evolution in that it utilizesthe characteristic bi-metallic center for catalysis,while it lacks the ubiquitous (β/α)8-barrel poly-peptide chain fold typical of the amidohydrolasesuperfamily.
and cleavage of scissile bond. The process is described insubstrate docking at the active site leads to activation ofhe generated hydroxide ion attacks the scissile carbon andage 2 in the Figure via step 1. In further reaction, as shownreidoglycine, NH3 and CO2 takes place.

461Structure of Allantoate Amidohydrolase
Proposed catalytic mechanism
The presence of a previously unrecognized bi-metal center in allantoate amidohydrolase and itsstructural similarity to four enzymes thought toshare a common catalytic mechanism13,31–34 suggeststrongly that substrate (allantoate and ureidogly-cine) cleavage results from activation of a zinc–coordinating water to a hydroxyl ion as a nucleo-phile, which ultimately attacks the scissile bondbetween the carbon and nitrogen of carbamylmoiety and β-amino group, respectively. We, there-fore, propose the following model for the mechan-ism of action of allantoate amidohydrolase. First,ligand and substrate bind to the enzyme. Second,interdomain motion about the Asn210/Pro331hinge permits proper docking of bound substratewithin the active site as shown in Figure 8. Duringcatalysis, the carbonyl oxygen of the N-carbamylgroup replaces Water341 (responsible for coordinat-ing to zinc 1). In the binuclear metal center, the metalion interacting with the substrate is called the β-metal ion.35 Here, zinc 1 is the β-metal ion as itinteracts directly with the substrate. In addition, theside-chain of Gln195 helps to stabilize the P1-carbanyl amide and the carbonyl oxygen atom ofGly359 stabilizes the P2-secondary amide via ahydrogen bond. Conserved Glu128 acts as a generalbase and makes a hydrogen bond with thenucleophilic Wat344, and is thought to act as aproton acceptor leading to formation of hydroxideion (stage1, Figure 8), in addition to helping inbreaking down the tetrahedral intermediate (stage 2,Figure 8) by subsequent shuttling of the proton tothe amino group of the generated ureidoglycine(step 2, Figure 8). In further reaction NH3, CO2 andthe ureidodoglycine are released, making theenzyme free for further catalysis.
Materials and Methods
Gene cloning, expression and protein purification
The target gene allC_ecoli for allantoate amidohydro-lase was cloned from E. coli genomic DNA using primersEc03055cF1 ATTACACATTTCCGTCAAGCTATAG andEc03055cR1 CTTTCTGCCAGGCAAGTTG in psb3 (TC)vector. Native and Se-Met protein expression/purificationwere performed as described.36 Protein yields were∼20 mg/l (native) and ∼18 mg/l (Se-Met).
Crystallization and data collection
Diffraction-quality crystals of native protein wereobtained at room temperature via vapor diffusion againsta reservoir solution containing 30% (w/v) PEG 4000, 0.2 MMgCl2, 0.1 M Tris–HCl (pH 8.5) (1 μl of reservoir solutionplus 1 μl of protein solution at 10 mg/ml) withoutadditional substrate or sulfate. Se-Met crystals of similarquality were obtained via vapor diffusion against areservoir solution containing 20% (w/v) PEG 8000,0.05MKH2PO4 at room temperature. Crystals were frozenby direct immersion in liquid nitrogen using the mother
liquor plus 15% (v/v) glycerol. Both native and Se-Metcrystals were obtained in the orthorhombic space groupP21212 with two molecules per asymmetric unit. X-raydiffraction data (native at 2.25 Å resolution and Se-Met atbetter than 3.0 Å) were collected under standard cryogenicconditions at Beamline X25, National Synchrotron LightSource, Brookhaven National Laboratory, and werereduced and scaled using HKL2000.37 Data collectionand refinement statistics are provided in Table 1.
Structure determination by Se-MAD and refinement
SHELXD38,39 permitted location of 12 of 13 possible Seatoms/monomer with the Se-Met diffraction data. Phaserefinement with SHARP40 followed by density modifica-tion with SOLOMON41 yielded an experimental electrondensity map that was used for manual model buildingusing O.42 The final atomic model was refined againstnative diffraction data with CNS at 2.25 Å resolution.43
Conclusion
This study has, for the first time, identified a di-zincmetal center in allantoate amidohydrolase. Like otheraminopeptidases, the enzyme utilizes a bi-metal-dependent exopeptidase activity but lacks the char-acteristic (β/α)8-barrel polypeptide chain fold of theamidohydrolase superfamily.35 The relative spatialarrangement of the active site residues and the twobound metal ions determines the enzyme specific forhydrolysis of theN-carbamyl bond instead of peptidebonds as seen in structurally related exopeptidases.We believe that enzyme activation via exposure toacidic pH can be explained by the presence of twocis-proline residues flanking an insertion domain,which supports the dimerization necessary forenzyme activity. The allantoate amidohydrolasesmay represent a drug target for pathogenicbacteria growing under anaerobic conditions.
Acknowledgements
We thank Dr S. Eswaramoorthy for helpful discus-sions. The research was supported by NIH grantGM62529 (to S.K.B.) under DOE Prime ContractDEAC02-98CH10886 with the Brookhaven NationalLaboratory. Data for this study were measured atbeamline X25 of the National Synchrotron LightSource, which is supported principally by the Officesof Biological and Environmental Research, and ofBasic Energy Sciences of the US Department ofEnergy, and from the National Center for ResearchResources of the National Institutes of Health.
References
1. Henderson, J. F. & Paterson, A. R. P. (1973). NucleotideMetabolism: An Introduction, New York AcademicPress, pp. 1–304.

462 Structure of Allantoate Amidohydrolase
2. Vogels, G. D. & van der Drift, C. (1976). Degradationof purines and pyrimidines by microorganisms.Bacteriol. Rev. 40, 403–468.
3. Schubert, K. R. & Boland, M. J. (1990). The ureides. InThe Biochemistry of Plants, vol. 16, San Diego AcademicPress, pp. 197–282.
4. Winkler, R. G., Blevins, D. G. & Randall, D. D. (1988).Ureide catabolism in soybeans. III. Ureidoglycolateamidohydrolase and allantoate amidohydrolase areactivities of an allantoate degrading enzyme complex.Plant Physiol. 86, 1084–1088.
5. Xu, Z. W., Zhou, H. X. & Huang, W. N. (2004). Someproperties of the allantoate amidohydrolase fromfrench bean seedlings. Zhi Wu Sheng Li Yu Fen ZiSheng Wu Xue Xue Bao, 30, 460–468.
6. Xu, Z., de Windt, F. E. & van der Drift, C. (1995).Purification and characterization of allantoate amido-hydrolase from Bacillus fastidiosus. Arch. Biochem.Biophys. 324, 99–104.
7. van der Drift, C., deWindt, F. E. & Vogels, G. D. (1970).Allantoate hydrolysis by allantoate amidohydrolase.Arch. Biochem. Biophys. 136, 273–279.
8. van der Drift, C. & Vogels, G. D. (1969). Allantoateamidohydrolase. I. pH- and anion-dependent activa-tion. Enzymologia, 36, 269–277.
9. van der Drift, C. & Vogels, G. D. (1969). Allantoateamidohydrolase. II. Inactivation and instability. Enzy-mologia, 36, 278–286.
10. Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality for assessing theaccuracy of protein structures. J. Appl. Chem. 26,283–291.
11. Rowsell, S., Pauptit, R. A., Tucker, A. D., Melton,R. G., Blow, D. M. & Brick, P. (1997). Crystalstructure of carboxypeptidase G2, a bacterial enzymewith applications in cancer therapy. Structure, 5,337–347.
12. Lundgren, S., Gojkovic, Z., Piskur, J. & Dobritzsch, D.(2003). Yeast beta-alanine synthase shares a structuralscaffold and origin with dizinc-dependent exopepti-dases. J. Biol. Chem. 278, 51851–51862.
13. Auld, D. S. (2001). Zinc coordination sphere in bio-chemical zinc sites. Biometals, 14, 271–313.
14. Lipscomb, W. N. & Strater, N. (1996). Recent advancesin zinc enzymology. Chem. Rev. 96, 2375–2434.
15. Rawlings, N. D. & Barrett, A. J. (1995). Evolutionaryfamilies of metallopeptidases. Methods Enzymol. 248,183–228.
16. Lukaszewski, K. M., Blevins, D. G. & Randall, D. D.(1992). Asparagine and boric acid cause allantoateaccumulation in soybean leaves by inhibiting man-ganese-dependent allantoate amidohydrolase. PlantPhysiol. 99, 1670–1676.
17. Little, J. W. & Hill, S. A. (1985). Deletions within ahinge region of a specific DNA-binding protein. Proc.Natl Acad. Sci. USA, 82, 2301–2305.
18. Dey, D., Oleinikov, A. V. & Traunt, R. R. (1995). Thehinge region of Escherichia coli ribosomal protein L7/L12 is required for factor binding and GTP hydrolysis.Biochimie, 77, 925–930.
19. Sun, Y. J., Rose, J., Wang, B. C. & Hsiao, C. D. (1998).The structure of glutamine-binding protein com-plexed with glutamine at 1.94 Å resolution: compar-isons with other amino acid binding proteins. J. Mol.Biol. 278, 219–229.
20. Spurlino, J. C., Lu, G. Y. & Quiocho, F. A. (1991). The2.3-Å resolution structure of the maltose-or maltodex-trin-binding protein, a primary receptor of bacterial
active transport and chemotaxis. J. Biol. Chem. 266,5202–5219.
21. Sharff, A. J., Rodseth, L. E., Spurlino, J. C. & Quiocho,F. A. (1992). Crystallographic evidence of a largeligand-induced hinge-twist motion between the twodomains of the maltodextrin binding protein involvedin active transport and chemotaxis. Biochemistry, 31,10657–10663.
22. Stewart, D. E., Sarkar, A. & Wampler, J. E. (1990).Occurrence and role ofcis peptide bonds in proteinstructures. J. Mol. Biol. 214, 253–260.
23. Ramachandran, G. N. & Sasisekharan, V. (1968).Conformation of polypeptides and proteins. AdvanProtein Chem. 23, 283–438.
24. Schmid, F. X. & Baldwin, R. L. (1978). Acid catalysis ofthe formation of the slow-folding species of RNase A:evidence that the reaction is proline isomerization.Proc. Natl Acad. Sci. USA, 75, 4764–4768.
25. Weiss, M. S., Metzner, H. J. & Hilgenfeld, R. (1998).Two non-proline cis peptide bonds may be im-portant for factor XIII function. FEBS Letters, 423,291–296.
26. Herzberg, O. & Moult, J. (1991). Analysis of the stericstrain in the polypeptide backbone of protein mole-cules. Proteins: Struct. Funct. Genet. 11, 223–229.
27. Vogels, G. D. (1966). Reversible activation of allanto-ate amidohydrolase by acid-pretreatment and otherproperties of the enzyme. Biochim. Biophys. Acta, 113,277–291.
28. van der Drift, C. & Vogels, G. D. (1967). Activation andinactivation of allantoate amindohydrolase. Biochim.Biophys. Acta, 139, 162–168.
29. Huang, C. C., Lesburg, C. A., Kiefer, L. L., Fierke, C. A.& Christianson, D. W. (1996). Reversal of the hydro-gen bond to zinc ligand histidine-119 dramaticallydiminishes catalysis and enhances metal equilibra-tion kinetics in carbonic anhydrase. Biochemistry, 35,3439–3446.
30. Christianson, D. W. & Fierke, C. A. (1996). Carbonicanhydrase: evolution of the zinc binding site by natureand by design. Accts Chem. Res. 29, 331–339.
31. Vallee, B. L. & Auld, D. S. (1993). Cocatalytic zincmotifs in enzyme catalysis. Proc. Natl Acad. Sci. USA,90, 2715–2718.
32. Vallee, B. L. & Auld, D. S. (1990). Zinc coordination,function, and structure of zinc enzymes and otherproteins. Biochemistry, 29, 5647–5659.
33. Vallee, B. L. & Auld, D. S. (1990). Active-site zincligands and activated H2O of zinc enzymes. Proc. NatlAcad. Sci. USA, 87, 220–224.
34. Matthews, B. W. (1988). Structural basis of the actionof thermolysin and related zinc peptidases. AcctsChem. Res. 21, 333–340.
35. Seibert, C. M. & Raushel, F. M. (2005). Structural andcatalytic diversity within the amidohydrolase super-family. Biochemistry, 44, 6383–6391.
36. Agarwal, R., Bonanno, J. B., Burley, S. K.&Swaminathan,S. (2006). Structure determination of an FMN reductasefrom Pseudomonas aeruginosa PA01 using sulfur anom-alous signal. Acta Crystallog. sect. D, 62, 383–391.
37. Otwinowski, Z. & Minor, W. (1997). Processing ofX-ray diffraction data collected in oscillation mode.Methods Enzymol. 276, 307–326.
38. Sheldrick, G. M. (2002). Macromolecular phasing withSHELXE. Z. Kristallogr. 217, 644–650.
39. Schneider, T. R. & Sheldrick, G. M. (2002). Substruc-ture solution with SHELXD. Acta Crystallog. sect. D,58, 1772–1779.
40. De La Fortelle, E. & Bricogne, G. (1997). Maximum-

463Structure of Allantoate Amidohydrolase
likelihood heavy atom parameter refinement in theMIR and MAD methods. Methods Enzymol. 276,472–493.
41. Collaborative Computational Project, Number 4(1994). The CCP4 suite: programs for protein crystal-lography. Acta Crystallog. sect. D, 50, 760–763.
42. Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard,M. (1991). Improved methods in building protein
models in electron density map and the location oferrors in these models. Acta Crystallog. sect. A, 47,110–119.
43. Brunger, A. T., Adams, P. D., Clore, G. M., Delano,W. L., Gros, P., Grosse-Kunstleve, R. W. et al. (1998).Crystallography and NMR system: a new softwaresuite for macromolecular structure determination.Acta Crystallog. sect. D, 54, 905–921.
Edited by I. Wilson
(Received 8 January 2007; received in revised form 3 February 2007; accepted 5 February 2007)Available online 20 February 2007