structure-based design of achbp ligands, - tipharma.com · structure-based design of achbp ligands,...
TRANSCRIPT


Structure-based design of AChBP ligands,
new insights and applications
Ewald Edink

Structure-based design of AChBP ligands, new insights and applications Ewald Edink PerkinElmer and Shimadzu Benelux B.V. are greatly acknowledged for their financial support for the printing of this thesis Printed by Wöhrmann Print Service, Zutphen, The Netherlands Cover design: by Ewald Edink Copyright © 2011 Ewald Edink

VRIJE UNIVERSITEIT
Structure-based design of AChBP ligands, new insights and applications
ACADEMISCH PROEFSCHRIFT
ter verkrijging van de graad Doctor aan de Vrije Universiteit Amsterdam,
op gezag van de rector magnificus prof.dr. L.M. Bouter,
in het openbaar te verdedigen ten overstaan van de promotiecommissie
van de faculteit der Exacte Wetenschappen op vrijdag 23 december om 11.45 uur
in de aula van de universiteit, De Boelelaan 1105
door
Ewald Siegfried Edink
geboren te Delft

promotor: prof.dr. R. Leurs copromotor: dr. I.J.P. de Esch

"Als je de beperkingen kent, kun je daarbinnen onbeperkt te werk gaan."
Jules A. Deelder

Leescommissie: prof.dr. C.. Abell prof.dr. A.B. Smit prof.dr. T.K. Sixma
prof.dr. M.J. Smit The investigations described in this thesis were carried out in the Division of Medicinal Chemistry and Pharmaceutical Sciences, Faculty of Sciences, Vrije Universiteit, De Boelelaan 1083, 1081 HV Amsterdam, The Netherlands. The work was performed within the framework of the Dutch Top Institute Pharma, project “New approaches for Ligang-Gated Ion Channel (LGIC) drug discovery (D2-103)”.

Table of Contents
Chapter 1: An Introduction to nicotinic acetylcholine receptors and
acetylcholine-binding protein ........................................................... 9 Chapter 2: Aim and scope of the thesis .......................................................... 35
Chapter 3: Thermodynamic analysis in fragment-based drug discovery ........ 39
Chapter 4: Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis ................................................................ 63
Chapter 5: Structure-based design, synthesis and structure-activity
relationships of dibenzosuberyl- and benzoate substituted tropines as ligands for acetylcholine-binding protein .................... 95 Chapter 6: Structure-based design of novel NSAID ester prodrugs:
Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors ........................................................................ 129 Chapter 7: Summary and conclusions .......................................................... 159 Nederlandse samenvatting .......................................................... 185 Dankwoord................................................................................... 191 List of publications ....................................................................... 195 Curriculum Vitae .......................................................................... 196


Chapter 1
An Introduction to nicotinic acetylcholine receptors
and acetylcholine-binding protein
Ewald Edink, Rob Leurs and Iwan J.P. de Esch

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
10
Synaptic transmission The brain is a highly complex organ and is the center of the human nervous system. It controls movement, memory, learning, thought, consciousness and regulates the body‟s actions and reactions. In addition, the brain is important for processing sensory information enabling us to see, hear, touch, taste and smell. Essential to proper functioning of the brain is the immense network of neuronal cells. The human brain is estimated to contain 100 billion neurons (10
11),
1 with an
average of 7,000 synaptic connections each.2 The synaptic connections, essential
to neuronal function, permit the transmission of electrical or chemical signals from one neuronal cell to another. The cerebral cortex, a part of the brain that plays a key role in memory, attention, language and consciousness, contains 1 billion (10
9)
synapses per cubic millimeter.3 There are two different types of synapses, the
electrical and the chemical synapse. The electrical synapse consists of a cluster of channels that can conduct electric currents mostly by transfer of K
+ ions from one
neuronal cell to another.4 In contrast, at the chemical synapse, small chemical
messengers, i.e., neurotransmitters are released from the presynaptic site of one neuronal cell to interact with receptor proteins on the postsynaptic site of a neighboring neuronal cell (Figure 1). A key role in central nervous system (CNS) signaling via chemical synapses is performed by ligand-gated ion channels (LGICs). LGICs are classified into three superfamilies; the pentameric Cys-loop receptors, the tetrameric glutamate receptors and the dimeric ATP-gated channels. The Cys-loop receptors are ion channels that consist of five protein subunits surrounding an aqueous pore that traverses the cell membrane (Figure 2). When a neurotransmitter binds to the extracellular binding site, a conformational change is induced that results in opening of the ion channel. The opening of the channel allows the influx of ions, thereby directly affecting the membrane potential of the receiving neuronal cell. Certain Cys-loop receptors such as nicotinic acetylcholine and serotonin receptors (nAChRs and 5-HT3R) selectively gate the flux of cations (Na
+, K
+ and Ca
2+) while
others such as γ-aminobutyric acid (GABA) and Glycine receptors are selective for anions (Cl
- and HCO3
-). The influx of cations may result in depolarization of the
post-synaptic cell having an excitatory effect, and allowing further downstream CNS signaling. On the contrary, the influx of anions results in hyperpolarization of the neuronal cell and has an inhibitory effect on the generation of an action potential. Nicotinic Acetylcholine receptor subtypes The most extensively studied prototype of the LGICs is the nAChR. The ionotropic nAChR is activated by its endogenous ligand acetylcholine as well as by nicotine, the addictive constituent of tobacco (Figure 2). To date, seventeen nAChR subunits have been identified which have been divided into muscle-type (α1, β1, γ, δ and ε) and neuronal subunits (α2-α10 and β2-β4).
5 Only two different muscle-
type nAChRs exist; one that contains a subunit composition of two α1 subunits and β1, γ, and δ and a receptor subtype with the same composition except that δ is replaced with ε. The subunits are organized in a clockwise α1γα1β1δ(or ε) arrangement. An α subunit is required for formation of an orthosteric nAChR

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
11
binding site and as such, the muscle-type receptor contains two orthosteric binding sites that are located at the α1-γ and α1-δ subunit interface.
6
Figure 1: Schematic image of interneuronal signaling via chemical synapses. An electric signal travels along a dendrite of one neuronal cell (upper left corner) to another neuronal cell (down right corner). The electric impulse stimulates the release of neurotransmitter molecules from the first neuron, the neurotransmitters subsequently diffuse across the synaptic cleft to interact with postsynaptic receptors of the receiving neuron. The activation of these postsynaptic receptors may stimulate (in case of excitatory receptors) or inhibit further downstream CNS signaling (in case of inhibitory receptors). Adapted from Wikipedia.
7

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
12
Figure 2: Cartoon backbone representation of the 4Å Torpedo nAChR electron microscopy (EM) structure (PDB: 2BG9). A) side view B) view from the extracellular side C) Chemical structures of the endogenous ligand acetylcholine and the potent nAChR agonist nicotine.
Figure 3: nAChR subtypes: A) The muscle-type nAChR is a heteropentamer consisting of four different subunits; α1, β1, γ and δ. The muscle type nAChR contains two orthosteric binding sites (grey squares). B) The neuronal α7 nAChR is an example of a homopentamer and consists only of α7 subunits. This subtype contains five orthosteric binding sites C) The neuronal α4β2 nAChR is one of the many heteropentameric nAChR subtypes. Similar to the muscle-type, this subtype contains two orthosteric binding sites.

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
13
The 12 neuronal nAChR subunits can assemble into a homo- or heteropentameric structures giving rise to a wide variety of possible receptor subunit combinations (Figure 3).
6 By an extensive amount of heterologous expression experiments,
combination rules for nAChR subunits have been identified.6,8,9
It has been discovered that the α(2-6) and β(2-4) subunits can form heteropentameric
complexes, e.g., 42. Separate co-expression of α2, α3 or α4 nAChR subunits in combination with β2 or β4 in Xenopus oocytes or mammalian cell lines results in the formation of heteropentameric functional receptors. Similar to the muscle-type, the orthosteric binding site of neuronal heteromeric nAChRs is located at the interface between an α and a β subunit, allowing the formation of two orthosteric binding sites. The α5 and β3 require co-expression with pairs of α(2-6) and β(2,4) to form more complex pentameric complexes such as (α4)2(β2)2α5. The α5 and β3 nAChR subunits are not involved in the formation of an orthosteric binding site. Instead, they function as a fifth structural subunit influencing the receptor characteristics. For example, incorporation of the α5 subunit increases the conductance of the ion channel and the rate of desensitization.
6,10 Evidence for
even more complex nAChRs exists as the α6 and β4 subunits only form functional receptors when co-expressed with both α5 and β3, indicative of a (α6)2β4β3α5 subtype.
11
Homopentameric nAChRs are assembled by a combination of five α7, α8 or α9 subunits. It should be noted that the α8 subunit has only been identified in birds and is not present in mammals.
12 Co-expression of α7 with β3 or β2 in Xenopus
oocytes yields functional heteropentamers and electrophysiology experiments on neuronal cultures have provided indications of the in vivo existence of heteromeric α7-containing nAChRs.
13-15 Coexpression of α7 with other subunits, such as α5,
influences the desensitization kinetics.16
The α10 subunit is not capable of forming homopentamers but can co-assemble with the α9 to yield heteromeric receptors that exhibit pharmacological profiles quite distinct from other nAChRs.
17,18
In addition to subunit composition, the stochiometry of heteropentameric nAChRs has been shown to be of influence on the receptor‟s characteristics as well, e.g., sensitivity towards acetylcholine. For α4β2 receptors, it is believed that the most prevalent stochiometry consists of (α)2(β)3, although in vitro evidence has been obtained for the existence for an (α)3(β)2 stochiometry as well.
19,20 Receptors with
the (α4)3(β2)2 stochiometry are 20-fold less sensitive to acetylcholine compared to nAChRs with the (α4)2(β2)3 stochiometry.
19
Native expression and functional roles of nAChRs The combination of nAChR subunits in a homo- or heteropentameric manner results in a wide variety of receptor subtypes with distinct characteristics, enabling unique properties and specialized functions.
12 For example, the α7
homopentameric receptor is associated with rapid desensitization and exhibits high Ca
2+ permeability.
21,22 As a result, α7 receptors have been linked to several Ca
2+-
dependant mechanisms, such as neurotransmitter release and stimulation of Ca2+
-dependant kinases.
23-25 The various nicotinic receptor subtypes are differentially
expressed throughout the CNS and the peripheral nervous system (PNS) and specific tissues may be targeted by subtype-selective ligands. Despite their name,

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
14
neuronal nAChRs are also present in non-neuronal tissues such as lymphocytes, macrophages, intestines, lungs, vascular endothelium and other tissues.
26-29 The
name “neuronal” is historically based on the origin of the DNA libraries, the brain, from which receptors were first cloned.
29
The most prevalent nAChR subtypes in the CNS are the α4β2* (~90%) and α7* (~10%).
6 The asterisks used in the nAChR nomenclature indicate that additional
nAChR subunits may be present in the pentameric complexes. The α4β2* subtype is characterized by high affinity for acetylcholine and nicotine but no affinity for the
snake toxin -Bungarotoxin (-Bgt). This receptor subtype has a widespread distribution throughout the CNS, with the highest concentration in the hippocampus, thalamus and cortex.
6 The other major nAChR subtype, the α7*,
binds acetylcholine and nicotine with M affinity and -Bgt with nM affinity. Compared to the α4β2* subtype, the α7* nAChR is characterized by fast desensitization rates and high permeability for Ca
2+.30,31
Similar to the α4β2*, the α7* subtype is also widely distributed throughout the brain with the highest expression in the cortex and hippocampus.
6 The other nAChR subunits have more
localized distributions throughout the CNS, and the interested reader is referred to recent reviews.
6,9,32
The majority of nicotinic receptors in the CNS are found on presynaptic and preterminal locations, the α7*, α4β2* and α3β4* have also been identified to function as postsynaptic receptors.
6,25 The presynaptic and preterminally located
nAChRs modulate the release of acetylcholine and several other neurotransmitters, such as dopamine (DA), norepinephrine (NE), serotonin (5-HT3), glutamate (Glu) and GABA. Because of this modulatory input to several neurotransmitter systems, the nAChRs have been proposed as potential therapeutic targets for a wide variety of CNS-related disorders such as Alzheimer‟s disease, Parkinson‟s disease, Tourette‟s syndrome, schizophrenia, anxiety and depression. Furthermore, nAChR may also be targeted for the treatment of pain, epilepsy, smoking cessation, drug addiction and inflammatory disorders.
6,33-37
The recent discovery that cholinergic pathways are involved in the inhibition of inflammatory responses in the periphery by activating α7 nAChRs, has also opened novel possibilities for the treatment of inflammatory disorders. Using knock-out mice, Wang et al. have shown that the α7 nAChR is an essential mediator in the anti-inflammatory cholinergic pathway and its activation inhibits the release of pro-inflammatory mediators such as TNF-α from macrophages.
38 As such, α7
selective (partial) agonists may therefore have therapeutic utility as novel anti-inflammatory agents. For example, GTS-21 (DMXBA, see below and Figure 6), a selective α7 partial agonist, improves survival in murine endotoxemia and severe sepsis.
39 In Chapter 5, we describe a dual-action approach , in which marketed
non-steroidal anti-inflammatory drugs (NSAIDs) are derivatized into ester prodrugs capable of activating α7 nicotinic receptors. The α7 nicotinic activity of the prodrug and subsequent release of NSAID may result in a synergistic dual activity to efficiently treat inflammations.

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
15
Allosteric Receptor Activation Model The nAChR is considered an allosteric protein. Ligand binding to the extracellular domain induces conformational changes that are transmitted to the transmembrane domain, resulting in receptor activation by opening of the ion channel.
40 The three
most important states of ion channel receptor are the resting, active and desensitized state. When the receptor is in the resting state, the ion channel is closed and no ions can permeate the channel pore. In the active state, the ion channel is open and ions can flow across the cell membrane. The desensitized state refers to a state with a closed ion channel, that cannot be activated despite the presence of an agonist.
41 According to the Monod-Wyman-Changeux (MWC)
model, the receptor protein can spontaneously undergo transitions between the three states and these transitions are in equilibrium, see Figure 4.
42 The model
proposes that agonists and antagonists select and stabilize structurally different conformations. In the absence of a ligand, the equilibrium is strongly shifted to the resting state. In contrast, addition of an agonist results in a shift of the equilibrium towards the active, open channel state. Prolonged exposure to an agonist may shift the equilibrium towards the desensitized state. After removal of the agonist, the equilibrium can shift back to the resting state. Competitive antagonists may stabilize the resting or desensitized state that are both associated with a closed ion channel.
43,44
Figure 4: According to the Monod-Wyman-Changeux (WMC) model, the receptor protein can spontaneously undergo transitions between the resting (A), active conducting (B) and desensitized state (C). These three states are in equilibrium that can be shifted towards the active state by the addition of an agonist. Prolonged exposure may shift the equilibrium towards the desensitized state. The resting or desensitized state are stabilized by competitive antagonists.

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
16

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
17
Figure 5: A) Cartoon and surface representation of 4 Å EM structure of nAChR from Torpedo mamorata (PDB: 2BG9). B) Cartoon and surface representation of 2.2 Å X-ray structure of AChBP from Lymnaea stagnalis in complex with nicotine (PDB: 1UW6). AChBP shares a similar architecture to the extracellular domain of nAChRs but lacks the trans-membrane and intracellular domains. C) Sequence
topology of the Torpedo mamorata subunit. An N-terminal extracellular domain that contains the orthosteric binding site, is followed by 4 transmembrane domains, two intracellular loops, an extracellular loop and an extracellular located C-terminus. The Cys-loop originates from two disulfide-linked cysteines that are separated by 13 amino acids. Besides the Cys-loop cysteines, the residues that make up the principal part of the orthosteric binding site are indicated by circles whereas the residues that form the complementary part are indicated by stars. D) Sequence topology of the Ls-AChBP subunit. Note the conserved Cys-loop and the conservation of principal orthosteric binding site residues. The principal site residues are indicated by small circles whereas complementary site residues are indicated by star-shaped symbols. AChBP sheds light on molecular structure of nAChRS As shown in Figure 5C, all nAChR subunits share a common topology: 1) a conserved extracellular NH2-terminal domain of ~200 amino acids that contains the orthosteric ligand binding site 2) three conserved transmembrane (TM) domains 3) a highly variable cytoplasmic loop 4) a fourth TM domain, and 5) a relatively short variable extracellular COOH-terminal sequence. In addition, the cysteine-loop (Cys-loop) that is established by disulfide bond formation between cysteines 128
and 142 (Torpedo 1 numbering) is a common feature of all nAChR subunits. These Cys residues are separated by 13 amino acids, and the Cys-loop is the hallmark of Cys-loop receptors. The presence of another Cys-Cys pair (residues 192-193 in the Torpedo α1 subunit) differentiates α subunits from non-α subunits. These two adjacent located cysteines form a disulfide bond with each other and their presence is required for binding of agonists.
12
It is notoriously difficult to obtain detailed structural information of membrane-bound proteins. Nevertheless, extensive efforts have been put into elucidating the structure of nAChRs. The high-resolution structure of acetylcholine-binding protein (AChBP) that was obtained in 2001 by Sixma and coworkers, has brought tremendous insight into the structure of the ligand-binding domain (LBD) of nAChRS and other LGICs.
45 The AChBP which is secreted from glial cells of the
freshwater snail Lymnaea stagnalis, displays low but significant amino acid sequence homology with the LBDs of all members of the Cys-loop receptors but lacks their transmembrane- and intracellular domains (Figure 5). AChBP shares the highest sequence identity with the corresponding LBD region in the α7 nAChR (24%).
46 Moreover, AChBP shares many structural hallmark features with the
nAChR LBD, e.g., 1) a stable pentameric complex, 2) conservation of the residues that align the binding pocket, and 3) conservation of the characteristic Cys-loop. However, in AChBP, the cysteines that form the Cys-loop are spaced by 12 hydrophilic instead of 13 hydrophobic amino acid residues.
6

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
18
Due to its structural resemblance to the Cys-loop receptor LBD, the AChBP crystal structure has been shown to be of great use in refining a cryoelectron microscopy structure to 4 Ǻ resolution structure of nAChRs from the electric organs of the Torpedo mamorata (PDB: 1OED and 2BG9).
47,48 Unfortunately, the structural
details are limited by the relative low resolution of these structures. These studies of Unwin and co-workers have, however, provided valuable structural information of nAChRs, such as the pentameric arrangement and location of the orthosteric binding site, as well as the location of the gate within the channel pore. In line with the observed similarities between AChBP and nAChRs, AChBP has been shown to bind a variety of nicotinic ligands with comparable affinities as the
7 nAChR (Table 1, Figure 6).49-51
Furthermore, the ability to signal to a TM domain is conserved, as experiments with AChBP/5-HT3 chimeras have shown that AChBP can be functionally linked to a TM domain and opening of the chimera‟s ion pore can be triggered by addition of acetylcholine.
52 It should be
noted that to obtain a functional AChBP/5-HT3 chimera, some of the AChBP loops at the interface between the binding and channel domain had to be replaced by their 5-HT3 counterparts. The structural resemblance between AChBP and the nAChR extracellular domain (ECD) has recently been confirmed by the crystal
structure of the monomeric mouse nAChR 1 ECD in complex with -bungarotoxin.
53 These findings indicate that AChBP can be used as a structural
model for the nAChR LBD and represents an important step forward in the structural understanding of nAChRs.
Table 1. Binding affinities of reference ligands for Ls-AChBP and the human α7 nAChR.
Ls-AChBP pKi, pKd or pIC50
α7 nAChR pKi, pKd or pIC50
Acetylcholine 5.2 ± 0.1
54 5.1 ± 0.1
55
Nicotine 6.4 ± 0.1 54
6.0 ± 0.1 55
Epibatidine 9.8 ± 0.1 56
9.2 ± 0.1 57
d-tubocurarine 7.0 ± 0.1 49
5.5 ± 0.1 58
α-cobratoxin 8.5 56
8.2 59
α-bungarotoxin 8.7 ± 0.1 56
8.9 60

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
19
Figure 6: Chemical structures of a selection of nAChR ligands Prokaryotic ligand-gated ion channels Next to AChBP X-ray structures, another recent breakthrough has increased understanding on the molecular structure of nAChRs and possible gating mechanisms: the high resolution X-ray crystal structures of two prokaryotic pentameric LGICs, ELIC
61 and GLIC
62,63. These bacterial cation-selective

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
20
channels, display an overall structure that is close to the Torpedo nAChR extracellular domain but lack an N-terminal α-helix and the characteristic Cys-loop. Nevertheless, comparison of the structures of ELIC (closed channel) and GLIC (open channel) provides insight into the gating mechanism of bacterial LGICs, and may be extended to eukaryotic LGICs. AChBP X-ray structures provide insight into ligand-nAChR interactions Currently, more than thirty X-ray structures of AChBPs from three different species i.e., Lymnaea stagnalis (Ls-AChBP)
45,64,65, Aplysia californica (Ac-AChBP)
46,51,66-75
and Bulinus truncatus (Bt-AChBP)50
have been obtained that include structures of the apo form, and of complexes that contain co-crystallized small molecules, buffer molecules or polypeptides (toxins). Analysis of these AChBP crystal structures show that five identical subunits form a stable complex with ~80 Å in diameter and ~60 Å in height. The ligand binding site is located at the interface of two subunits and due to its homopentameric nature, AChBP contains five ligand-binding sites. Similar to nAChRs, the ligand binding site is constructed from loops A, B and C of the principal site which are well-conserved and loops D, E and F from the complementary site that are more variable (Figure 7). Together, these loops form an aromatic cage that consists of Tyr89 (loop A), Trp143 (loopB), Tyr185 and Tyr192 (loop C) and Trp53 (loop D) (unless indicated otherwise, the Ls-AChBP amino acid numbering will be used in this chapter). This aromatic cage has been shown to be very important in ligand-recognition for AChBP
51,64,71 as well as for the
Cys-loop receptors.76
AChBP co-crystal structures with nicotinic ligands exemplify
that Trp143 is an important contributor in terms of cation- interactions in binding of cationic ligands to AChBP. These noncovalent stabilizing interactions originate from electrostatic interactions between the positively charged nitrogen atom of the ligand and the negative electrostatic potential on the face of the fused aromatic rings of the tryptophan residue.
77,78 Evidence for a similar important role of Trp143
in complex formation of acetylcholine and nicotine to α4β2 nAChRs has been obtained by the elegant use of unnatural amino acids.
79 Incorporation of (poly-)
fluorinated tryptophan analogs at this specific position, significantly decreased acetylcholine and nicotine and acetycholine‟s potency, indicative of specific cation-π interactions with Trp143. In addition, AChBP crystal structures show that several other non-aromatic amino acid residues contribute to the ligand binding site, including two vicinal cysteines (Cys187 and 188) that are engaged in a disulfide bond. These two cysteines are located at the top of loop C. This flexible loop closes upon the ligand binding site and forms van der Waals interactions with the ligand (see below). A selection of residues that form the Ls-AChBP orthosteric ligand binding site is depicted in Figure 7C-F and a comparison to their putative counterparts in the human nAChR subunits is depicted in Table 2 and 3. Although differences at the complementary site can be observed, the comparison exemplifies the high homology of the ligand binding interface between AChBP and nAChRs.

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
21
Figure 7: Cartoon and surface representation of 2.2 Å X-ray structure of AChBP from Lymnaea stagnalis in complex with nicotine (PDB: 1UW6) shown in side view (A) and top view (B). The binding site is located at the interface of two subunits (indicated by a black circle) and consists of a principal (C) and a complementary side (D). As a consequence, the AChBP homopentamer has five orthosteric ligand binding sites. A schematic representation showing the loops A-F that shape the orthosteric binding site (E). Close up on the binding pose of nicotine bound to Ls-AChBP (F).

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
22
Ta
ble
2: D
iffe
rence
s in
th
e p
rin
cip
al b
ind
ing
site
re
sid
ues b
etw
een
th
e A
Ch
BP
s a
nd
a s
ele
ctio
n o
f th
e h
um
an
ne
uro
na
l n
AC
hR
su
bun
its. R
esid
ue
nu
mb
ers
acco
rdin
g to
un
ipro
t80 a
re s
how
n in
su
pe
rscrip
t.
4
x
x
x
x
x
x
x)
The
se
re
sid
ues a
re n
ot p
art
of th
e o
rth
oste
ric b
ind
ing
pocke
t. O
nly
th
e c
om
ple
me
nta
ry s
ide
of β
su
bu
nits f
orm
th
e
ort
hoste
ric b
ind
ing
po
cke
t.
2
x
x
x
x
x
x
1
0
Y95
W151
Y192
C194
C195
Y200
9
Y95
W151
Y192
C194
C195
Y200
7
Y93
W149
Y188
C190
C191
Y195
4
Y128
W154
Y195
C197
C198
Y202
Bt
Y88
W142
Y184
C186
C187
Y191
Ac
Y93
W147
Y188
C190
C191
Y195
Ls
Y89
W143
Y185
C187
C188
Y192
Lo
op
A
B
C
C
C
C

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
23
T
ab
le 3
: D
iffe
rence
s in
th
e c
om
ple
me
nta
ry b
ind
ing
site
re
sid
ues b
etw
een
th
e A
Ch
BP
s a
nd
a s
ele
ctio
n o
f th
e h
um
an
ne
uro
na
l n
AC
hR
su
bun
its. R
esid
ue
nu
mb
ers
acco
rdin
g to
un
ipro
t80 a
re s
how
n in
su
pe
rscrip
t.
4
W59
N111
I113
L121
L123
D173
x)
The
se
re
sid
ues a
re n
ot p
art
of th
e o
rth
oste
ric b
ind
ing
pocke
t. O
nly
th
e p
rin
cip
al sid
e o
f α
su
bu
nits fo
rm th
e
ort
hoste
ric b
ind
ing
po
cke
t.
2
W57
N109
V111
F119
L121
D171
1
0
W57
N109
V111
R119
D116
D171
9
W57
N109
V111
T119
D116
D171
7
W55
N107
L109
Q117
L119
G167
4
x
x
x
x
x
x
Bt
W51
R101
V103
I111
V113
Y163
Ac
Y55
I106
V108
M116
I118
S167
Ls
W53
L102
R104
L112
M114
Y164
Lo
op
D
E
E
E
E
F

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
24
Next to the cation-π interactions, an additional strong interaction that is often encountered in co-crystal complexes of nicotinic receptor ligands with AChBPs is the formation of a charged hydrogen bond between the cationic ammonium moiety of ligands with the backbone carbonyl of Trp143 (Figure 8). The work of Dougherty et al. provides strong evidence that the charged hydrogen bond is very important for binding of nicotine to the human α4β2 nicotinic receptor.
79 By converting the
backbone amide to a backbone ester, which is a substantially poorer hydrogen bond acceptor, nicotine‟s potency is decreased 19-fold. The potency of acetylcholine. which contains a quaternary nitrogen atom and cannot engage in a similar hydrogen bond, is not affected by this subtle mutation. In several AChBP X-ray structures, water-mediated hydrogen bonds between a conserved water molecule and ligand hydrogen bond acceptor atoms can also be observed. The conserved water molecule is stabilized by hydrogen bonding to the main chains of Leu102 and Met114 (loop E, complementary site, Figure 8). Dougherty and co-workers have obtained results that indicate that the water-mediated hydrogen bond is of significant importance for binding of nicotine to the
42 nAChR, as well.81
Double mutant cycle analysis on the 42 nAChR in which backbone peptide bonds are replaced by ester bonds and a nicotine analog in which the pyridine moiety is replaced by phenyl, provides evidence for the
formation of the water-mediated hydrogen bond between nicotine and the 42 receptor. These studies illustrate how combining AChBP co-crystal structures with validation experiments on the actual human nicotinic receptors can contribute to our detailed understanding of the structure of nAChRs and reveal the molecular interactions that drive complex formation between small molecular ligands and nAChRs. Conformational changes in loop C By co-crystallizing AChBP in the absence of ligands (apo) and in the presence of agonists and antagonists, the dynamic nature of this nAChR LBD mimic has become apparent. Distinctive conformations of loop C are found in the AChBP X-ray co-crystal structures. When co-crystallized with AChBP, nicotinic agonists such as nicotine
64, carbamylcholine
64, epibatidine
51, reveal a closed conformation of loop
C in which the loop wraps around the ligand and closes upon the binding site. In
contrast, when co-crystallized with antagonists such as -conotoxins46,51,66
and the phycotoxin 13-desmethyl spirolide C
75, loop C is forced in a more open
conformation (8-10 Å compared to agonist bound structures; measured as the
distance between the C atoms of Cys188). The nonpeptidic antagonists methyllycaconitine (MLA) and gymnodimine A stabilize loop C in an intermediate
conformation (5-6 Å). On the other hand, 7 partial agonists display an intermediate positioning of loop C in between antagonist and agonist structures, although the exact positioning of loop C varies between the partial agonists and even between ligand-occupied binding sites on the same homopentameric crystal structure.
73 The conformation of loop C in the apo structure takes an intermediate
conformation and resembles the MLA-bound structure.7 However, loop C could not
be completely refined due to lack of electron density at the tip of loop, indicative that this part of the binding site is very dynamic in the crystal complex in the absence of ligands. In addition, evidence for similar movements of loop C in

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
25
solution has been obtained using fluorescence spectroscopy56,85
, deuterium-hydrogen exchange
86, NMR
87 and surface plasmon resonance (SPR) biosensor
analysis88
. These findings indicate that loop C closure may play an essential role in eliciting the conformational change that is required for opening of the nAChR ion channel. It should be noted, however, that the loop C conformation in AChBP co-crystal structures does not always correspond to the functional profile of the respective ligand on nAChRs. For example, the AChBP co-crystal complex of lobeline, which has been reported to act as a partial agonist
82 as well as an
antagonist83,84
on the α4β2 nAChR, shows a fully closed loop C (PDB: 2BYS).51
Figure 8: X-ray structure of Ls-AChBP in complex with nicotine (PDB: 1UW6) shows that the formation of a cation-π interaction between the charged pyrolidine nitrogen atom and the aromatic side chain of Trp143 is a major contributor to complex formation. In addition, two hydrogen bonds (black dashed lines) with the AChBP binding site are made upon binding. A charged hydrogen bond is established between the positively charged pyrolidine nitrogen atom of nicotine and the backbone carbonyl oxygen atom of Trp143 and the pyridine nitrogen atom is involved in hydrogen bond formation with a conserved water molecule. The residues Ser147 and Ala191 are engaged in a backbone mediated hydrogen bond (black dashed line indicated by the white arrow) between Ser147 and Ala191. Combining the results of molecular dynamics and site-directed mutagenesis experiments indicates that compared to the muscle-type (Ser147 = Gly175) and α7 nAChR (Ser147 = Gly148), this hydrogen bond is much stronger in the α4β2 nAChR (Ser147 = Lys150), shaping the binding pocket in such a way that nicotine establishes a very strong cation-π interaction with Trp143.

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
26
AChBP as template for in silico screening procedures Since AChBP is considered a structural homolog of the extracellular domain of nAChRs, our research group
71,89 and others
90,91 have developed in silico screening
procedures using AChBP X-ray structures to identify novel nAChR ligands. Although, our methods result in high AChBP hit rates and succeeded in identification of novel nAChR ligands, the studies also exemplified some limitations in the use of AChBP X-ray structures. We found that a limited number of AChBP screening hits have affinity for the human nicotinic receptors. Furthermore, no nAChR agonists were identified (only antagonists) when using AChBP as a bait, despite the fact that a nicotine-bound AChBP structure was used. In addition, due to changes in the conformation of loop C, experimentally determined binding modes differed from the predicted in silico docking binding modes. In the high throughput GOLD docking simulations
92 that were used, only local optimization of
protein hydroxyl hydrogen atoms was included, but full side chain or backbone flexibility of binding site residues may be required to account for ligand-induced conformational changes in AChBP. Interestingly, Taylor and co-workers have combined molecular dynamics simulations with molecular docking allowing protein flexibility in a virtual screening protocol. Unfortunately, binding data of the hits on AChBP and nAChRs or structural validation of the predicted binding modes were not reported.
91 Recently, Balle and co-workers have successfully incorporated loop
C flexibility in a retrospective AChBP docking study, resulting in the correct
prediction of 12 out of 15 binding modes (rmsd 2.0 Å).93
Geitmann and co-workers have also performed loop C flexibility simulations and their results were in line with ligand-induced conformational changes that were observed in SPR biosensor measurements on AChBP.
94 These studies underline the importance of
taking protein binding site flexibility into account when performing docking studies. However, inclusion of (full) protein flexibility in protein-ligand docking is computationally demanding, and its application in silico screenings of large compound databases often comes at the cost of screening speed. AChBP X-ray structures provide insight into nAChR subtype selectivity determinants Since the first AChBP crystal structure was solved, AChBP has been widely used as a template to investigate the binding of nicotinic ligands to the nAChR binding sites. Most of the amino acid residues that align the principal part of the binding site are conserved between the nAChR and AChBP (Table 2). Nevertheless, besides Trp53, the residues that shape the complementary part of the binding site are less conserved in the nicotinic receptors (Table 3). Interestingly, differences between AChBP species variants have been identified in the complementary site, as well. For example, comparison between Ls-AChBP and Bt-AChBP reveals that three complementary binding site residues differ; the Lymnaea R104, L112 and M114 are a valine, isoleucine and valine, respectively in Bt-AChBP. Nicotine binds to Ls-AChBP and Bt-AChBP with a binding affinity of 45 nM and 8 nM, respectively. Site-directed mutagenesis of the three residues in Ls-AChBP to their Bt-AChBP counterparts (i.e., Ls-R104V/L112I/M114V), shifts nicotine‟s binding affinity very close to the value of the wildtype Bt-AChBP (Kd = 12 nM). In contrast, these mutations show an opposite trend for acetylcholine; the endogenous neurotransmitter exhibits selectivity for Bt-AChBP as well; Kd = 823 nM and 153 nM

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
27
for Ls-AChBP and Bt-AChBP, respectively. Surprisingly, the triple Ls-AChBP mutant, suffers from a complete loss of binding affinity for acetylcholine (Kd > 10 µM).
50 These mutagenesis studies illustrate that in certain cases (nicotine), the
selectivity of a ligand for one AChBP species variant can be perfectly explained by species differences in the first shell of binding site residues. On the other hand, the example of acetylcholine shows that amino acid residues located outside the first shell of binding site residues may play a role in species/subtype selectivity, as well. A striking example that underlines the important role that residues outside the first shell of the binding site may play in nAChR subtype selectivity is provided by work performed in Dougherty‟s lab.
79 A discrepancy was found between the effect of
replacing the backbone amide carbonyl of Trp143 by a backbone ester between the muscle-type and α4β2 nAChR. As already mentioned above, in the case of the α4β2 nAChR, the potency of nicotine was decreased 19-fold upon this unnatural amino acid mutation, whereas hardly any effect was observed for the muscle-type nAChR. In addition, for muscle-type nicotinic receptors, nicotine‟s potency was unaffected by incorporation of (poly-)fluorinated tryptophans at the position of Trp143. These observations are in line with the substantial lower affinity of nicotine for the muscle-type nAChR compared to the α4β2 and suggest that nicotine has
stronger interactions (cation- and a charged hydrogen bond), with the neuronal nAChR subtype. The first AChBP X-ray structure (PDB: 1I9B) revealed a backbone mediated hydrogen bond between loop B (Ser147) and loop C (Ala191) (Figure 8).
45,95 It is important to note that both residues are located outside the first shell of
amino acid residues that align the binding pocket. In the α4β2 nAChR, the equivalent of Ser147 is a lysine whereas in the muscle-type and α7 receptors which both exhibit substantial lower affinity for nicotine, this residue is a glycine. Molecular dynamics experiments on an α7 homology model suggest that Lys at this specific position in loop B, promotes the formation of the hydrogen bond between loop B and C. On the other hand, substitution for Gly discourages hydrogen bond formation. Substituting the Gly in the α7 or muscle-type for the α4β2 counterpart (Lys) using site-directed mutagenesis, results in significant increases of nicotine‟s potency.
95 Furthermore, in contrast with the muscle nAChR
wildtype, nicotine‟s potency is decreased by incorporation of (poly-)fluorinated tryptophans at the Trp143 position of the G147K mutant, indicative of a strong cation-pi interaction with this aromatic residue.
79 Altogether, these data indicate
that the backbone mediated hydrogen bond between loop B and C that is present in the α4β2, shapes the binding site so that nicotine is in close contact to Trp143. In the muscle-type or α7 receptor, this hydrogen bond is weaker or absent resulting in a more flexible binding site and lower affinity for nicotine resulting from weaker interactions with Trp143. Methyllycaconitine (MLA), a diterpene alkaloid from seeds of the Aconitum or
Delphinium families, is a highly selective 7 nAChR antagonist (Figure 6). The co-crystal structure of MLA in complex with Ac-AChBP shows that MLA is involved in a water-mediated hydrogen bond with Ser165. This complementary binding site residue is located on loop F, which is a highly variable region in the amino acid sequences of nAChRs and as a consequence, loop F may therefore play an

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
28
important role in nAChR subtype selectivity.51
Interestingly, the co-crystal structures of tropisetron, DMXBA (GTS-21) and 4-OH-DMXBA with Ac-AChBP
show that all three 7 partial antagonists interact with Ser165 (chemical structures of DMXBA and tropisetron are depicted in Figure 6).
73 Illustrative of the importance
of loop F for activation of 7 nAChRs, is that alanine substitution at position 165 in
the human and rat 7, has pronounced effect on the potency of DMXBA.96
These findings strongly suggest that residues in loop F are important for the activation of alpha7 nAChRs and may be an important determinant of nAChR subtype selectivity. Lobeline, a piperidine alkaloid found in the Lobelia family, has been claimed to act as a partial agonist
82 as well as an antagonist
83,84 on the α4β2 nAChR and exhibits
substantial selectivity over the α7 subtype (chemical structure of lobeline is depicted in Figure 6).
82,97 The X-ray structure of lobeline in complex with Ac-
AChBP reveals a unique conformational change leading to opening of a subpocket, enabling the binding of the α-hydroxyphenetyl moiety of the ligand.
51 The
subpocket becomes accessible after a change in the rotameric state of gatekeeper residue Tyr91 (tyrosine-flip), which is likely to be stabilized by hydrogen bond formation with Ser165. As mentioned above, Ser165 is located in the highly variable loop F and as such differential stabilization of the rotameric states of the gatekeeper tyrosine residue may play a role in the observed nAChR subtype-selectivity of lobeline. A thermodynamic and structural study that includes site-directed mutagenesis and fragment-growing into the ligand-induced subpocket, to investigate the role of loop F in stabilization of the tyrosine-flip is described in more detail in Chapter 4. Conclusion This introductory chapter exemplifies that AChBP has proven its value in gaining new structural insights into the homologous nicotinic receptor LBDs. However, it must be remembered that the sequence homology remains moderate and caution should be taken when using AChBP as a structural template for the nAChR extracellular domains. As such, translation of AChBP-derived hypotheses to the nicotinoid receptors should always be validated by for example, site-directed mutagenesis experiments or by using carefully designed molecular probes. It is noteworthy to mention that AChBP can also be considered an excellent tool to study ligand-protein interactions in general. Due to its water-soluble nature, AChBP provides relative facile access to X-ray co-crystal structures. In addition, compared to membrane-bound proteins, AChBP is easier to incorporate in biological assays and better suited for biophysical analyses such as surface plasmon resonance (SPR) biosensor analysis and isothermal titration calorimetry (ITC). References 1. Irons-Georges, T. Magill's Medical Guide Revised Edition, (Salem Press,
Pasadena, (1998). 2. Drachman, D.A. Do we have brain to spare? Neurology 64, 2004-5 (2005).

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
29
3. Alonso-Nanclares, L., Gonzalez-Soriano, J., Rodriguez, J.R. & DeFelipe, J. Gender differences in human cortical synaptic density. Proc Natl Acad Sci U S A 105, 14615-9 (2008).
4. Bennett, M.V. & Zukin, R.S. Electrical coupling and neuronal synchronization in the Mammalian brain. Neuron 41, 495-511 (2004).
5. Lukas, R.J. et al. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 51, 397-401 (1999).
6. Jensen, A.A., Frolund, B., Liljefors, T. & Krogsgaard-Larsen, P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem 48, 4705-45 (2005).
7. http://en.wikipedia.org/wiki/Chemical_synapse. 8. Gotti, C. et al. Structural and functional diversity of native brain neuronal
nicotinic receptors. Biochem Pharmacol 78, 703-11 (2009). 9. Millar, N.S. & Gotti, C. Diversity of vertebrate nicotinic acetylcholine
receptors. Neuropharmacology 56, 237-46 (2009). 10. Ramirez-Latorre, J. et al. Functional contributions of alpha5 subunit to
neuronal acetylcholine receptor channels. Nature 380, 347-51 (1996). 11. Grinevich, V.P. et al. Heterologous expression of human
{alpha}6{beta}4{beta}3{alpha}5 nicotinic acetylcholine receptors: binding properties consistent with their natural expression require quaternary subunit assembly including the {alpha}5 subunit. J Pharmacol Exp Ther 312, 619-26 (2005).
12. Albuquerque, E.X., Pereira, E.F., Alkondon, M. & Rogers, S.W. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89, 73-120 (2009).
13. Azam, L., Winzer-Serhan, U. & Leslie, F.M. Co-expression of alpha7 and beta2 nicotinic acetylcholine receptor subunit mRNAs within rat brain cholinergic neurons. Neuroscience 119, 965-77 (2003).
14. Khiroug, S.S. et al. Rat nicotinic ACh receptor alpha7 and beta2 subunits co-assemble to form functional heteromeric nicotinic receptor channels. J Physiol 540, 425-34 (2002).
15. Palma, E., Maggi, L., Barabino, B., Eusebi, F. & Ballivet, M. Nicotinic acetylcholine receptors assembled from the alpha7 and beta3 subunits. J Biol Chem 274, 18335-40 (1999).
16. Yu, C.R. & Role, L.W. Functional contribution of the alpha5 subunit to neuronal nicotinic channels expressed by chick sympathetic ganglion neurones. J Physiol 509 ( Pt 3), 667-81 (1998).
17. Elgoyhen, A.B., Johnson, D.S., Boulter, J., Vetter, D.E. & Heinemann, S. Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79, 705-15 (1994).
18. Elgoyhen, A.B. et al. alpha10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A 98, 3501-6 (2001).
19. Moroni, M., Zwart, R., Sher, E., Cassels, B.K. & Bermudez, I. alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity:

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
30
pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol 70, 755-68 (2006).
20. Nelson, M.E., Kuryatov, A., Choi, C.H., Zhou, Y. & Lindstrom, J. Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol 63, 332-41 (2003).
21. Alkondon, M. & Albuquerque, E.X. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther 265, 1455-73 (1993).
22. Castro, N.G. & Albuquerque, E.X. alpha-Bungarotoxin-sensitive hippocampal nicotinic receptor channel has a high calcium permeability. Biophys J 68, 516-24 (1995).
23. Broide, R.S. & Leslie, F.M. The alpha7 nicotinic acetylcholine receptor in neuronal plasticity. Mol Neurobiol 20, 1-16 (1999).
24. Suzuki, T. et al. Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res 83, 1461-70 (2006).
25. Sher, E. et al. Physiological roles of neuronal nicotinic receptor subtypes: new insights on the nicotinic modulation of neurotransmitter release, synaptic transmission and plasticity. Curr Top Med Chem 4, 283-97 (2004).
26. Romanelli, M.N. et al. Central nicotinic receptors: structure, function, ligands, and therapeutic potential. ChemMedChem 2, 746-67 (2007).
27. Conti-Fine, B.M., Navaneetham, D., Lei, S. & Maus, A.D. Neuronal nicotinic receptors in non-neuronal cells: new mediators of tobacco toxicity? Eur J Pharmacol 393, 279-94 (2000).
28. Sharma, G. & Vijayaraghavan, S. Nicotinic receptor signaling in nonexcitable cells. J Neurobiol 53, 524-34 (2002).
29. Gahring, L.C. & Rogers, S.W. Neuronal nicotinic acetylcholine receptor expression and function on nonneuronal cells. AAPS J 7, E885-94 (2005).
30. Chavez-Noriega, L.E. et al. Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors h alpha 2 beta 2, h alpha 2 beta 4, h alpha 3 beta 2, h alpha 3 beta 4, h alpha 4 beta 2, h alpha 4 beta 4 and h alpha 7 expressed in Xenopus oocytes. J Pharmacol Exp Ther 280, 346-56 (1997).
31. Vijayaraghavan, S., Pugh, P.C., Zhang, Z.W., Rathouz, M.M. & Berg, D.K. Nicotinic receptors that bind alpha-bungarotoxin on neurons raise intracellular free Ca2+. Neuron 8, 353-62 (1992).
32. Gotti, C. et al. Heterogeneity and complexity of native brain nicotinic receptors. Biochem Pharmacol 74, 1102-11 (2007).
33. Newhouse, P., Singh, A. & Potter, A. Nicotine and nicotinic receptor involvement in neuropsychiatric disorders. Curr Top Med Chem 4, 267-82 (2004).
34. Ripoll, N., Bronnec, M. & Bourin, M. Nicotinic receptors and schizophrenia. Curr Med Res Opin 20, 1057-74 (2004).
35. Shytle, R.D. et al. Nicotinic acetylcholine receptors as targets for antidepressants. Mol Psychiatry 7, 525-35 (2002).
36. D'Hoedt, D. & Bertrand, D. Nicotinic acetylcholine receptors: an overview on drug discovery. Expert Opin Ther Targets 13, 395-411 (2009).

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
31
37. Marrero, M.B., Bencherif, M., Lippiello, P.M. & Lucas, R. Application of Alpha7 Nicotinic Acetylcholine Receptor Agonists in Inflammatory Diseases: An Overview. Pharm Res.
38. Wang, H. et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421, 384-8 (2003).
39. Pavlov, V.A. et al. Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med 35, 1139-44 (2007).
40. Changeux, J.P. Allosteric Interactions Interpreted in Terms of Quaternary Structure. Brookhaven Symp Biol 17, 232-49 (1964).
41. van Nierop, P. The nicotinic acetylcholine receptor subunit family expressed in the central nervous system of the freshwater snail Lymnaea stagnalis: Molecular and functional diversity of the neuronal nicotinic acetylcholine receptors in a molluscan species, Doctoral Thesis, http://hdl.handle.net/1871/10839. 13-15 (2007).
42. Monod, J., Wyman, J. & Changeux, J.P. On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol 12, 88-118 (1965).
43. Taly, A., Corringer, P.J., Guedin, D., Lestage, P. & Changeux, J.P. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov 8, 733-50 (2009).
44. Bertrand, D. et al. Unconventional pharmacology of a neuronal nicotinic receptor mutated in the channel domain. Proc Natl Acad Sci U S A 89, 1261-5 (1992).
45. Brejc, K. et al. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411, 269-76 (2001).
46. Bourne, Y., Talley, T.T., Hansen, S.B., Taylor, P. & Marchot, P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake alpha-neurotoxins and nicotinic receptors. EMBO J 24, 1512-22 (2005).
47. Miyazawa, A., Fujiyoshi, Y. & Unwin, N. Structure and gating mechanism of the acetylcholine receptor pore. Nature 423, 949-55 (2003).
48. Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol 346, 967-89 (2005).
49. Smit, A.B. et al. A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature 411, 261-8 (2001).
50. Celie, P.H. et al. Crystal structure of acetylcholine-binding protein from Bulinus truncatus reveals the conserved structural scaffold and sites of variation in nicotinic acetylcholine receptors. J Biol Chem 280, 26457-66 (2005).
51. Hansen, S.B. et al. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J 24, 3635-46 (2005).
52. Bouzat, C. et al. Coupling of agonist binding to channel gating in an ACh-binding protein linked to an ion channel. Nature 430, 896-900 (2004).
53. Dellisanti, C.D., Yao, Y., Stroud, J.C., Wang, Z.Z. & Chen, L. Crystal structure of the extracellular domain of nAChR alpha1 bound to alpha-bungarotoxin at 1.94 A resolution. Nat Neurosci 10, 953-62 (2007).

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
32
54. Edink, E. et al. Fragment Growing Induces Conformational Changes in Acetylcholine-Binding Protein: A Structural and Thermodynamic Analysis. J Am Chem Soc 133, 5363-5371 (2011).
55. unpublished results. 56. Hansen, S.B. et al. Tryptophan fluorescence reveals conformational
changes in the acetylcholine binding protein. J Biol Chem 277, 41299-302 (2002).
57. Peng, J.H. et al. High-affinity epibatidine binding of functional, human alpha7-nicotinic acetylcholine receptors stably and heterologously expressed de novo in human SH-EP1 cells. J Pharmacol Exp Ther 313, 24-35 (2005).
58. Brams, M. et al. A structural and mutagenic blueprint for molecular recognition of strychnine and d-tubocurarine by different cys-loop receptors. PLoS Biol 9, e1001034 (2011).
59. Servent, D. et al. Molecular characterization of the specificity of interactions of various neurotoxins on two distinct nicotinic acetylcholine receptors. Eur J Pharmacol 393, 197-204 (2000).
60. Gotti, C. et al. Alpha7 and alpha8 nicotinic receptor subtypes immunopurified from chick retina have different immunological, pharmacological and functional properties. Eur J Neurosci 9, 1201-11 (1997).
61. Hilf, R.J. & Dutzler, R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375-9 (2008).
62. Hilf, R.J. & Dutzler, R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 457, 115-8 (2009).
63. Bocquet, N. et al. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111-4 (2009).
64. Celie, P.H. et al. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41, 907-14 (2004).
65. Ihara, M. et al. Crystal structures of Lymnaea stagnalis AChBP in complex with neonicotinoid insecticides imidacloprid and clothianidin. Invert Neurosci 8, 71-81 (2008).
66. Celie, P.H. et al. Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an alpha-conotoxin PnIA variant. Nat Struct Mol Biol 12, 582-8 (2005).
67. Ulens, C. et al. Structural determinants of selective alpha-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc Natl Acad Sci U S A 103, 3615-20 (2006).
68. Hansen, S.B. & Taylor, P. Galanthamine and non-competitive inhibitor binding to ACh-binding protein: evidence for a binding site on non-alpha-subunit interfaces of heteromeric neuronal nicotinic receptors. J Mol Biol 369, 895-901 (2007).
69. Dutertre, S. et al. AChBP-targeted alpha-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J 26, 3858-67 (2007).

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
33
70. Talley, T.T. et al. Atomic interactions of neonicotinoid agonists with AChBP: molecular recognition of the distinctive electronegative pharmacophore. Proc Natl Acad Sci U S A 105, 7606-11 (2008).
71. Ulens, C. et al. Use of acetylcholine binding protein in the search for novel alpha7 nicotinic receptor ligands. In silico docking, pharmacological screening, and X-ray analysis. J Med Chem 52, 2372-83 (2009).
72. Hansen, S.B., Wang, H.L., Taylor, P. & Sine, S.M. An ion selectivity filter in the extracellular domain of Cys-loop receptors reveals determinants for ion conductance. J Biol Chem 283, 36066-70 (2008).
73. Hibbs, R.E. et al. Structural determinants for interaction of partial agonists with acetylcholine binding protein and neuronal alpha7 nicotinic acetylcholine receptor. EMBO J 28, 3040-51 (2009).
74. Brams, M. et al. Crystal structures of a cysteine-modified mutant in loop D of acetylcholine binding protein. J Biol Chem 286, 4420-8 (2011).
75. Bourne, Y. et al. Structural determinants in phycotoxins and AChBP conferring high affinity binding and nicotinic AChR antagonism. Proc Natl Acad Sci U S A 107, 6076-81 (2010).
76. Dougherty, D.A. Cys-loop neuroreceptors: structure to the rescue? Chem Rev 108, 1642-53 (2008).
77. Dougherty, D.A. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science 271, 163-8 (1996).
78. Mecozzi, S., West, A.P., Jr. & Dougherty, D.A. Cation-pi interactions in aromatics of biological and medicinal interest: electrostatic potential surfaces as a useful qualitative guide. Proc Natl Acad Sci U S A 93, 10566-71 (1996).
79. Xiu, X., Puskar, N.L., Shanata, J.A., Lester, H.A. & Dougherty, D.A. Nicotine binding to brain receptors requires a strong cation-pi interaction. Nature 458, 534-7 (2009).
80. www.uniprot.org, the displayed residue numbers are corrected for the amount of signalling peptide residues.
81. Blum, A.P., Lester, H.A. & Dougherty, D.A. Nicotinic pharmacophore: the pyridine N of nicotine and carbonyl of acetylcholine hydrogen bond across a subunit interface to a backbone NH. Proc Natl Acad Sci U S A 107, 13206-11 (2011).
82. Wu, J. et al. Roles of nicotinic acetylcholine receptor beta subunits in function of human alpha4-containing nicotinic receptors. J Physiol 576, 103-18 (2006).
83. Miller, D.K., Crooks, P.A. & Dwoskin, L.P. Lobeline inhibits nicotine-evoked [(3)H]dopamine overflow from rat striatal slices and nicotine-evoked (86)Rb(+) efflux from thalamic synaptosomes. Neuropharmacology 39, 2654-62 (2000).
84. Dwoskin, L.P. & Crooks, P.A. A novel mechanism of action and potential use for lobeline as a treatment for psychostimulant abuse. Biochem Pharmacol 63, 89-98 (2002).
85. Hibbs, R.E., Talley, T.T. & Taylor, P. Acrylodan-conjugated cysteine side chains reveal conformational state and ligand site locations of the acetylcholine-binding protein. J Biol Chem 279, 28483-91 (2004).

Chapter 1 An introduction to nicotinic acetylcholine receptors and acetylcholine-binding protein
34
86. Shi, J., Koeppe, J.R., Komives, E.A. & Taylor, P. Ligand-induced conformational changes in the acetylcholine-binding protein analyzed by hydrogen-deuterium exchange mass spectrometry. J Biol Chem 281, 12170-7 (2006).
87. Gao, F. et al. Solution NMR of acetylcholine binding protein reveals agonist-mediated conformational change of the C-loop. Mol Pharmacol 70, 1230-5 (2006).
88. Geitmann, M. et al. Interaction kinetic and structural dynamic analysis of ligand binding to acetylcholine-binding protein. Biochemistry 49, 8143-54.
89. Akdemir, A. et al. Acetylcholine binding protein (AChBP) as template for hierarchical in silico screening procedures to identify structurally novel ligands for the nicotinic receptors. Bioorganic & Medicinal Chemistry In Press, Accepted Manuscript.
90. Utsintong, M., Talley, T.T., Taylor, P.W., Olson, A.J. & Vajragupta, O. Virtual screening against alpha-cobratoxin. J Biomol Screen 14, 1109-18 (2009).
91. Babakhani, A., Talley, T.T., Taylor, P. & McCammon, J.A. A virtual screening study of the acetylcholine binding protein using a relaxed-complex approach. Comput Biol Chem 33, 160-70 (2009).
92. Jones, G., Willett, P., Glen, R.C., Leach, A.R. & Taylor, R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267, 727-48 (1997).
93. Sander, T., Bruun, A.T. & Balle, T. Docking to flexible nicotinic acetylcholine receptors: a validation study using the acetylcholine binding protein. J Mol Graph Model 29, 415-24 (2010).
94. Geitmann, M. et al. Interaction kinetic and structural dynamic analysis of ligand binding to acetylcholine-binding protein. Biochemistry 49, 8143-54 (2010).
95. Grutter, T. et al. An H-bond between two residues from different loops of the acetylcholine binding site contributes to the activation mechanism of nicotinic receptors. EMBO J 22, 1990-2003 (2003).
96. Stokes, C. et al. The structural basis for GTS-21 selectivity between human and rat nicotinic alpha7 receptors. Mol Pharmacol 66, 14-24 (2004).
97. Miller, D.K. et al. Lobeline analogs with enhanced affinity and selectivity for plasmalemma and vesicular monoamine transporters. J Pharmacol Exp Ther 310, 1035-45 (2004).

35
Chapter 2
Aim and scope of the thesis

Chapter 2 Aim and scope of the thesis
36
Fragment-based Drug Discovery In comparison with traditional high-throughput screening (HTS), fragment-based drug discovery (FBDD) is characterized by the screening of smaller libraries of compounds (typically containing 1000-5000 fragments) with lower molecular weight (i.e., smaller than 300 Da).
1 The essence of FBDD is that small molecular
fragments are ideally suited for probing protein binding sites for key binding interactions but small enough to minimize the chances of unfavorable interactions that prevent them from binding.
2 Because the size of chemical space increases
exponentially with molecular size, fragment-based screening results in substantially better coverage of chemical space and typically results in higher hit rates than HTS.
3-5 Furthermore, fragment hits often provide better starting points in terms of
physicochemical properties.6 Due to its attractiveness in terms of a highly efficient
drug discovery process, FBDD has transformed in less than a decade, from a niche area of research that was only applied by small biotech companies to a serious alternative for HTS that is currently being applied (often in parallel with HTS) by every major drug discovery firm. Illustrative of the impact that FBDD is making on the drug development process, is the increasing number of FBDD-derived compounds that are in phase I and II clinical trials.
5,7 Very recently, the first FBDD-
derived drug, Zelboraf® (vemurafenib) has been approved by the FDA and may be considered a case study on how FDBB can enable a more efficient drug discovery as it took only 6 years from the start of the program to FDA approval whereas it has recently been estimated that this process takes 13 years on average.
8-10
A crucial aspect of FBDD is the efficient optimization of the binding affinity of fragment hits towards high affinity clinical candidates. Structural biology has been shown to be extremely helpful to guide the optimization of fragment hits towards novel and potent lead compounds.
7,11-14 As AChBP provides relative easy access
to X-ray co-crystal structures, we embarked on FBDD approaches to develop new ligands that bind to AChBP. Next to finding novel ligands with affinity for AChBP and to improve our understanding of ligand-protein interactions, the studies were also aimed at increasing our understanding of FBDD. Thermodynamic Analysis Although structure-activity relationship studies have almost exclusively been using affinity and activity data, there is also growing awareness that this experimental data is an indirect measure of measuring the complementarity between ligand and protein. A more direct way of describing intermolecular interactions is by considering the thermodynamic properties of these interactions. Thermodynamic analysis provides access to the constituents of the Gibbs energy of binding (ΔG°), enthalpy (ΔH°) and entropy (ΔS°). The dissection of binding energy into separate enthalpic and entropic contributions adds an additional dimension to binding affinity and can provide crucial insights into the physical forces that drive ligand-protein interactions. For example, favorable changes in enthalpy may result from direct binding forces such as formation of hydrogen bonds, van der Waals contacts and
- interactions. On the other hand, favorable changes in entropy often result from changes in the ligand or protein‟s conformational freedom and the displacement of water molecules as apolar regions of the binding site and the ligand are brought together.

Chapter 2 Aim and scope of the thesis
37
Determination of thermodynamic binding profiles may be especially useful in a FBDD context. Typically, fragment hits bind with low affinity to their respective protein partners, and as a consequence, extensive increases in binding affinity need to be realized to obtain low nM clinical candidates. As will be discussed in a survey of scientific literature (Chapter 3), it is easier to improve binding affinity by optimizing entropy, with the addition of hydrophobic groups, than it is through enthalpy, which requires polar interactions to be optimized. Therefore, choosing a fragment hit in which binding is enthalpically driven as a starting point and adding hydrophobic groups during optimization, may provide an efficient route to high affinity compounds with both favorable changes in enthalpy and entropy and a reduced risk of attrition. In addition, thermodynamic analysis may be very useful in guiding and monitoring (structure-based) fragment optimization. In combination with structural data, the stepwise fragment growing process provides an ideal dataset with which to improve our understanding of the thermodynamics of binding. In Chapter 3, the techniques that enable thermodynamic analysis of fragment-protein complexes are discussed, the currently available thermodynamic data on fragment-protein complexes are summarized and several key studies that highlight the role of thermodynamics in FBDD are discussed in more detail. Research Aims AChBP can be considered an excellent research tool to study FBDD approaches. Due to its water soluble nature, this nAChR ligand binding domain homolog provides relative facile access to X-ray co-crystal structures enabling structure-based optimization. Moreover, compared to membrane-bound proteins, water-soluble proteins are easier to incorporate in biological assays and better applicable in biophysical techniques such as SPR biosensor analysis and isothermal titration calorimetry (ITC). For these reasons, we have embarked on a fundamental study in which AChBP is used to increase our knowledge on how to efficiently optimize fragment hits. In addition, we have studied which techniques are suitable to monitor the fragment-optimization process in terms of efficiency and information content. In the process, we kept an eye on translating our findings with AChBP to the homologous but therapeutically relevant nicotinic receptors. Thus, at the beginning of this project we set ourselves with the following research aims:
Increase our knowledge on how to efficiently optimize fragments towards high affinity binders using AChBP as a model protein.
Investigate if thermodynamic analysis can contribute to a more efficient fragment-optimization process using AChBP as a model protein.
Investigate if our fragment-optimization studies on AChBP can contribute to our understanding on how to design subtype-selective ligands for the therapeutically relevant human nAChRs.
Outline of the thesis In Chapter 3, the technologies that enable thermodynamic analysis of fragment-protein complexes are discussed. In addition, the available thermodynamic data of fragment-protein complexes is summarized and several key studies that highlight the role of thermodynamics in FBDD are discussed in more detail. Chapter 4 describes the successful optimization of a fragment hit by growing into AChBP‟s

Chapter 2 Aim and scope of the thesis
38
lobeline-pocket. The fragment optimization was monitored with X-ray co-crystal structures and thermodynamic analysis using SPR biosensor analysis and ITC. Chapter 5, focuses on the structure-based design, synthesis and structure-activity relationships of dibenzosuberyl- and benzoate substituted tropines as ligands for
acetylcholine binding protein. These studies resulted in the identification of an 7 nAChR selective fragment hit. The optimization of this fragment towards a potential dual action anti-inflammatory agent is described in chapter 6. The final chapter 7 summarizes the most important conclusions of this thesis and evaluates the outcome of our research aims. References 1. Congreve, M., Carr, R., Murray, C. & Jhoti, H. A 'rule of three' for fragment-
based lead discovery? Drug Discov Today 8, 876-7 (2003). 2. Hann, M.M., Leach, A.R. & Harper, G. Molecular complexity and its impact
on the probability of finding leads for drug discovery. J Chem Inf Comput Sci 41, 856-64 (2001).
3. Bohacek, R.S., McMartin, C. & Guida, W.C. The art and practice of structure-based drug design: a molecular modeling perspective. Med Res Rev 16, 3-50 (1996).
4. Fink, T., Bruggesser, H. & Reymond, J.L. Virtual exploration of the small-molecule chemical universe below 160 Daltons. Angew Chem Int Ed Engl 44, 1504-8 (2005).
5. Chessari, G. & Woodhead, A.J. From fragment to clinical candidate--a historical perspective. Drug Discov Today 14, 668-75 (2009).
6. Keseru, G.M. & Makara, G.M. The influence of lead discovery strategies on the properties of drug candidates. Nat Rev Drug Discov 8, 203-12 (2009).
7. de Kloe, G.E., Bailey, D., Leurs, R. & de Esch, I.J. Transforming fragments into candidates: small becomes big in medicinal chemistry. Drug Discov Today 14, 630-46 (2009).
8. Bollag, G. et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596-9 (2010).
9. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ ucm268241.htm.
10. Bunnage, M.E. Getting pharmaceutical R&D back on target. Nat Chem Biol 7, 335-9 (2011).
11. Hajduk, P.J. & Greer, J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov 6, 211-9 (2007).
12. Sledz, P. et al. Optimization of the interligand overhauser effect for fragment linking: application to inhibitor discovery against Mycobacterium tuberculosis pantothenate synthetase. J Am Chem Soc 132, 4544-5 (2010).
13. Wang, Y.S. et al. Application of fragment-based NMR screening, X-ray crystallography, structure-based design, and focused chemical library design to identify novel microM leads for the development of nM BACE-1 (beta-site APP cleaving enzyme 1) inhibitors. J Med Chem 53, 942-50 (2010).
14. Zhu, Z. et al. Discovery of cyclic acylguanidines as highly potent and selective beta-site amyloid cleaving enzyme (BACE) inhibitors: Part I--inhibitor design and validation. J Med Chem 53, 951-65 (2010).

39
Chapter 3
Thermodynamic analysis in fragment-based drug discovery
Ewald Edink#, Chimed Jansen
#, Rob Leurs and Iwan J.P. de Esch
# These authors contributed equally.
Abstract Thermodynamic analysis provides access to the determinants of binding affinity, enthalpy and entropy. In fragment-based drug discovery (FBDD), thermodynamic analysis provides a powerful tool to discriminate fragments based on their potential for successful optimization. The thermodynamic data generated by FBDD studies can in turn be used to better understand the forces that drive biomolecular interactions. In this review, the technologies that enable thermodynamic analysis of fragment-protein complexes are discussed. In addition, the available thermodynamic data on fragment-protein complexes is summarized and several key studies which highlight the role of thermodynamics in FBDD are discussed in more detail. Although, thermodynamic analysis is not yet applied widely within the FBDD field, the first success stories are starting to appear, exemplifying its value in the development of a more efficient fragment optimization process and a better understanding of ligand-protein interactions.

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
40
Introduction Fragment-based drug discovery (FBDD) is making an impact as an efficient and effective drug discovery method.
1 With small libraries of a few thousand structures
and hit rates reaching 10%, FBDD provides a valuable alternative to HTS.2 A
crucial aspect of FBDD is the efficient optimization of the binding affinity of fragment hits towards high affinity clinical candidates. Ultimately, binding affinity is governed by the changes in enthalpy and entropy (see Glossary) that occur upon formation of a ligand-protein complex. The small size of fragments simplifies the process of relating the thermodynamic profile of fragments to the interactions made during binding. When combined with structural data, the stepwise fragment growing process provides an ideal dataset with which to improve our understanding of the thermodynamics of binding. In this manner both FBDD, and our understanding of the thermodynamics of binding, have much to gain from the application of thermodynamic analysis in FBDD. Thermodynamics in drug discovery Thermodynamic analysis provides access to the constituents of the Gibbs energy of binding (ΔG°), enthalpy (ΔH°) and entropy (ΔS°) (see Glossary). Enthalpy, or heat energy, is associated with direct binding forces such as hydrogen bonding, van der Waals forces and π-π interactions. Entropy, a term determined by the number of accessible states of a system, is associated with conformational freedom and the hydrophobic effect (see Glossary). The thermodynamic changes that occur when a ligand binds to its respective protein binding site are schematically depicted in Figure 1. Improving the enthalpic contribution to binding generally involves optimizing the polar interactions made by a ligand. This may be achieved by strengthening already existing interactions within the binding site or by forming new interactions by adding additional polar groups to a ligand. However, a poorly optimized polar interaction may not simply provide a lower favorable contribution to the enthalpy change on binding, it may actually result in an unfavorable contribution to enthalpy. Desolvation of polar functionalities comes at a cost, which can only be overcome when the positioning of interacting groups obeys strict angle and distance requirements.
3 As a consequence, enthalpic optimization is a difficult route by
which to improve affinity. However, due to the specificity of polar interactions, enthalpy driven ligand binding may provide a major selectivity advantage.
4
Interestingly, retrospective analysis of the thermodynamics of HIV-protease inhibitors and cholesterol lowering statins, shows that the later best in class compounds, which outperformed earlier first in class compounds, had been enthalpically optimized.
5
While enthalpic optimization can provide highly selective high affinity drugs, medicinal chemists tend to optimize entropy, as shown in a recent study comparing synthetic and natural drugs.
6 Unfortunately, too much focus on entropic
optimization by constraining ligands in their bioactive conformation, and by the addition of hydrophobic groups, may in the end result in poorly soluble compounds, with reduced selectivity and higher chances of attrition.
7,8 A further complication to
thermodynamic optimization results from enthalpy-entropy compensation. As a polar interaction becomes tighter (enthalpically favorable), the binding atoms

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
41
become locked into a tighter conformational geometry (entropically unfavorable).9,10
This means that optimizing enthalpy necessarily adversely affects entropy and vice versa. Since we are looking at interactions occurring in water, desolvation and the ordering or disordering of water surrounding the binding partners may play an important role in enthalpy-entropy compensation.
10 As a result, large favorable
changes in enthalpy or entropy often result in only minor gains in binding affinity.
Figure 1: In this simplified schematic diagram, four stages of ligand binding are shown. The colors indicate the degree of conformational freedom of the ligand, protein and water as indicated on the key. 1) Prior to complex formation, the ligand and the protein binding site are solvated by water. 2) and 3) As the ligand approaches the binding pocket, the ligand and the protein’s conformational freedom become increasingly restricted, resulting in an unfavorable entropic contribution. In addition, the polar moieties become partially desolvated, resulting in an unfavorable change in enthalpy. At the same time, the formation of enthalpic van der Waals interactions, as well as the hydrophobic effect (see Glossary), ensure that this process is favorable. 4) Eventually, the ligand reaches a binding conformation and a significant favorable contribution to enthalpy is made as the ligand and protein become locked together, resulting in an unfavorable entropy contribution. At this crucial stage, the net effect of the polar interactions, desolvation, conformational constraint and the hydrophobic effect, will determine the final thermodynamic profile of the ligand.

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
42
For an extensive overview on the use of thermodynamics in drug discovery, the reader is referred to the book on drug-receptor thermodynamics edited by Raffa.
11
An overview of the studies in which isothermal titration calorimetry (ITC) (see below) has been applied to attain thermodynamic data is published annually, and represents a good starting point for those interested in an overview on more recent studies.
12-18 Finally, several online databases that contain data on the
thermodynamics of ligand-protein interactions exist, providing access to relevant studies.
6,19,20
Glossary Gibbs energy change (ΔG°): The energy change on binding can be divided into enthalpy (ΔH°) and entropy (ΔS°): ΔG° = ΔH° –TΔS°. Or looked at as a function of
the binding affinity (e.g., KD,): D
o KRTG ln . In which R is the gas constant
and T is the absolute temperature. Van ‘t Hoff Equation: Combining the two Gibbs equations results in the van „t Hoff
equation: R
S
RT
HK
oo
D
ln .This equation can be used to derive enthalpy
and entropy from affinity measurements at a range of temperatures. Enthalpy change (ΔH°): A measure for the heat that is released or absorbed upon binding, often associated with polar interactions. Entropy change (ΔS°): A measure of the change in order/disorder or in the number of configurations available to the system upon binding, associated with conformational freedom and the hydrophobic effect. Heat capacity change (ΔCp°): A measure for the change in the ability of a system to absorb heat. The van „t Hoff equation can only be used when ΔH° does not vary with temperature (ΔCp° = 0). When ΔH° has temperature dependence (ΔCp° ≠ 0), the integrated form of the van „t Hoff equation that includes a ΔCp° term can be
applied to derive ΔH°, ΔS° and ΔCp°(where and are constants):
)ln()(
11ln TCTC
TRK o
p
o
pD
Values of ΔH° and ΔS° can subsequently be obtained using the following equations: ΔH°(T) = ΔCp°·T + α
ΔS°(T) = ΔCp°·lnT + β

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
43
Determination of ΔCp° can provide additional information about biomolecular interactions, for example a negative ΔCp° in combination with favorable entropy changes is indicative of hydrophobic interactions. The hydrophobic effect: A favorable change in entropy during ligand binding resulting from the displacement of water molecules as apolar regions of the binding site are brought together.
61
Ligand Efficiency (LE): A metric that normalizes the binding affinity of molecules by their size. LE is calculated by dividing the Gibbs energy change (ΔG°) by the number of heavy atoms (HA): LE = ΔG°/HA.
62
Enthalpic Efficiency (EE): A molecular size corrected measure of the enthalpic contribution to binding, calculated by dividing the enthalpy (ΔH°) by the number of heavy atoms (HA): EE = ΔH°/HA.
21
Application of thermodynamics in FBDD Determination of thermodynamic binding profiles may be especially useful in a FBDD context. In general, the identified fragment hits exhibit mM to μM affinity towards their respective protein targets, and as a consequence extensive increases in binding affinity are necessary to obtain low nM binders that can enter the clinic. As mentioned in the previous section, it is easier to improve binding affinity by optimizing entropy, with the addition of hydrophobic groups, than it is through enthalpy, which requires polar interactions to be optimized. Therefore, choosing a fragment in which binding is enthalpically driven as a starting point and adding hydrophobic groups during optimization, may provide the easiest route to a high affinity compound with favorable changes in both enthalpy and entropy and a reduced risk of attrition. The measure, enthalpic efficiency (EE, see Glossary), has recently been postulated as a tool for chemists and may prove useful as a hit selection criterion in FBDD, to supplement established criteria such as ligand efficiency (LE, see Glossary), logP, polar atom count, and synthetic accessibility to rank identified fragment hits.
21
Measuring the thermodynamics of binding requires a suitable system. Here we describe two methods, used to acquire thermodynamic data, calorimetry and van „t Hoff, and provide examples of their application through the technologies, Isothermal Titration Calorimetry (ITC) and Surface Plasmon Resonance (SPR) biosensor analysis, respectively. Isothermal Titration Calorimetry (ITC) The most direct method of acquiring thermodynamic data is via calorimetry. The technology most commonly used for this purpose in drug discovery is ITC (systems available from e.g., microcal, which is now part of GE Healthcare, www.microcal.com, or TA Instruments, www.tainstruments.com). During an ITC experiment, a solution containing one of the binding partners is titrated into a sample cell containing the second binding partner. During the ligand binding event heat will either be released (exothermic) or absorbed (endothermic). This is measured through changes in the amount of energy required to retain a constant temperature in the sample cell. ITC does not require labeling or immobilization of

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
44
the protein or ligand and allows determination of the enthalpy and entropy change, binding constant and binding stoichiometry in a single experiment. Performing experiments at multiple temperatures allows determination of the heat capacity change (ΔCp°) (see Glossary) , which may provide further insight into the mode of binding, e.g., manifestation of the hydrophobic effect. A problem encountered in using ITC within FBDD is the low binding affinity of the small fragments, which may preclude direct measurement of all the binding parameters. This problem can be circumvented by performing competition experiments.
22 However, two major disadvantages of ITC remain; the amount of
protein required and the time taken per experiment. As a consequence, ITC is not suitable for primary fragment screening, but may be considered as a secondary screening tool, able to validate identified fragment hits and obtain the thermodynamic parameters of binding at the same time. Nevertheless, recent progress in downsizing the ITC sample cell and automation of sample handling enables a throughput of 75 samples per day with as little as of 10 µg of protein per sample.
23 However, It should be noted that the amount of protein required for
analysis is dependant on the affinity and thermodynamic binding profile of the specific interaction that is under investigation. Furthermore, reducing the amount of protein per sample or the time per experiment comes at a cost to accuracy.
24
Van ’t Hoff method using SPR biosensor analysis Any method which is able to provide affinity data over a range of temperatures can be deployed to retrieve thermodynamic data using the van ‟t Hoff equation (see Glossary). In practice this has mainly been done using radioligand binding assays, however, spectroscopic techniques such as NMR, mass spectroscopy and chromatographic methods have all been employed to obtain data for the van „t Hoff method.
11 In this review we will focus on a relatively new technology used to obtain
affinity data, surface plasmon resonance (SPR) biosensor analysis, to highlight some of the differences between using calorimetry and the van ‟t Hoff method (systems available from e.g., Biacore which is now (also) part of GE Healthcare, www.biacore.com, Reichert, www.reichertspr.com, Ibis, www.ibis-spr.nl, Metrohm, www.metrohm.com, and Xantec, www.xantec.com). SPR biosensor analysis measures mass changes resulting from binding of an immobilized binding partner to a binding partner in solution. This is done by measuring the change in the incidence angle, of light reflected off the surface on which binding occurs, required to achieve resonance with surface plasmon.
25
Modern SPR biosensor systems provide integrated van ‟t Hoff analysis software to retrieve thermodynamic data. When a limited amount of the target protein is available, SPR biosensor analysis is particularly attractive as a technique for thermodynamic analysis, since it requires small amounts of protein. Furthermore, besides determination of the thermodynamics behind a binding event, SPR biosensor analysis also enables retrieval of the kinetics (kon and koff) of binding, although kinetic analysis may not be possible for low affinity fragments because the kinetic rates are too fast.
26 If kinetic and thermodynamic data can be obtained,
investigation of transition state thermodynamics, e.g., enthalpy of association, becomes a possibility and may provide additional insights into binding mechanisms.

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
45
Comparison between ITC and SPR biosensor analysis Much has been written on differences in results obtained by ITC and van ‟t Hoff experiments.
27-34 Several studies were performed to test for consistent differences
between calorimetric and van ‟t Hoff determinations of binding thermodynamics using ITC and SPR at different laboratories. In total 61 SPR experiments were compared with 26 ITC experiments. Results from ITC and SPR experiments were found to be highly consistent, deviation between the technologies averaged just 4%.
35-37 In principle this proves that there is no difference in accuracy between
thermodynamic experiments performed by ITC and those performed by SPR. However, only two protein targets were used in the experiments, so other targets might show greater variability. The studies found significant differences across locations and experimental results from some labs were not included because of the presence of artifacts, which were presumed to have resulted from a lack of maintenance and/or thorough calibration of the equipment. Indeed, in thermodynamic studies which compare experimental conditions it has been found that, salt levels,
38 protein sources,
39,40 metal oxidation states,
41 cofactor presence,
42
species source,43
pH,44
and temperature45
can all have significant influence on the measured thermodynamic profile. This presents a warning for those comparing thermodynamic data from different sources and illustrates the need for standardized protocols to be used in the measurement of thermodynamic data. ITC is a near universally applicable technology for thermodynamic analysis, requiring no special sample preparation steps and having a broad affinity range. Since ITC is a direct method for determination of enthalpy changes, it is considered to be more accurate than the indirect van „t Hoff method.
46 However, ITC requires
large amounts of sample, making its use in studies looking at hard to obtain proteins untenable. On the other hand, SPR biosensor analysis is efficient in sample consumption and analysis times. It should be realized though, that switching between measurement temperatures, which is required for thermodynamic analysis, significantly lengthens the measurement times. Furthermore, the required immobilization of one of the binding partners represents an additional initial effort, which may not always be successful and requires subsequent validation. In addition, retrieval of thermodynamic binding parameters using SPR biosensor analysis may be complicated by the manifestation of non-linear van „t Hoff plots which have to be fit over a large temperature range with a substantial amount of data-points in order to include heat capacity (ΔCp°) in the model. However, curvature in the van „t Hoff plots may be masked by experimental noise, compromising the accuracy of the determined thermodynamic binding parameters.
33 A comparative overview of both technologies is shown in Table 1.
Examples of the application of thermodynamics in FBDD Despite its potential, the use of thermodynamic analysis in a FBDD context, is currently far from common and there are only a few examples in literature in which both technologies have been combined (see below). However, since FBDD is a relatively young discipline, thermodynamic data on fragment-like molecules may be available that has not been labeled as FBDD. Inspection of literature and the databases mentioned earlier, revealed that indeed already a substantial amount of thermodynamic data on fragments is available. In order to increase our understanding of the thermodynamic aspects of fragment-protein binding, we

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
46
constructed a database containing 162 unique ligand-protein complexes in which the molecular weight of the ligand was smaller than 300 Da.
47 The thermodynamic
data that was collected using ITC is summarized in Figure 2a and data that was obtained using van „t Hoff analysis is summarized in Figure 2b. Several trends can be observed from the enthalpy-entropy plots depicted in Figure 2. First of all, van „t Hoff data is characterized by a greater amount of entropy driven binding. This is not a trend resulting from the techniques, but rather a trend resulting from the different targets being tested. The van ‟t Hoff experiments involving fragments were almost exclusively performed on membrane-bound receptors (see Figure 2b), while the ITC experiments tended to be performed on water-soluble enzymes (see Figure 2a). When applying van „t Hoff analysis on membrane-bound proteins, temperature effects on the membrane itself (e.g., membrane fluidity) may influence the determined thermodynamic binding parameters. As such, these effects may therefore play a role in the observed differences between Figures 2a and 2b. Nevertheless, the trend of wider distribution of thermodynamic binding signatures in the case of membrane-bound receptors is also likely to originate from the conformational changes that are required for receptors to switch between active and inactive states. This explanation is in line with an additional trend that is visible in Figure 2b; the thermodynamic discrimination between agonists and antagonists in many of the tested receptors. This phenomenon, in which agonist binding to a receptor is entropy-driven and the binding of its antagonist is enthalpy-driven, or vice versa, has been observed for G-protein-coupled receptors (GPCRs), such as β-adrenoceptors, adenosine A1 and A2A, as well as for ligand-gated ion channels (LGICs), such as glycine, GABAA, 5-HT3 and nicotinic acetylcholine receptors.
48,49
Although the exact origin of thermodynamic discrimination remains unclear, for adenosine A1 receptors it has been suggested that binding of an antagonist results in the displacement of a water network from the binding site, whereas the binding of agonists does not, providing an explanation for the distinct thermodynamic profiles of binding.
50 In the case of LGICs, the conformational change that takes
place upon receptor-activation to induce channel opening, has been linked to the discrimination between agonists and antagonists.
51 Although fragments that exhibit
enthalpy driven complex formation may be the preferred candidates for further optimization, Figure 2 illustrates that this may depend on the target protein being an enzyme, or a receptor, and on what kind of functional profile (antagonist or (partial) agonist) is required to obtain the anticipated therapeutic effect. It is important to note that the van „t Hoff analysis on receptors was not performed using SPR biosensor systems, but rather by traditional affinity assays (radioligand displacement) performed at a range of temperatures. A third trend that can be observed is that the distribution in Gibbs energy of binding (ΔΔG° = 11 kcal·mol
-1) is significantly smaller than the distribution in enthalpy and
entropy (ΔΔH° = TΔΔS° = 63 kcal·mol-1
). These differences reflect enthalpy-entropy compensation; large favorable changes in enthalpy are compensated by unfavorable changes in entropy, and vice versa.
9,10

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
47

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
48
Figure 2: Enthalpy vs. entropy plot of ligand-protein complexes that were obtained using ITC (a) and van ‘t Hoff analysis (b). The two diagonal lines show Gibbs energy changes of -4.1 kcal·mol
-1 and -12.4 kcal·mol
-1 equivalent to affinities of 1
mM and 1 nM, respectively. Several series of structurally related fragments were identified upon searching the literature and databases. This data may provide an opportunity to increase our understanding of the thermodynamic aspects of fragment optimization. By taking the smallest common scaffold as a starting point in a hypothetical fragment optimization process, the enthalpic and entropic contributions to any subsequent fragment growing or linking step could be calculated. Here, we provide a selection of examples from our analysis. Adenosine A1 receptor Borea and co-workers have studied the binding thermodynamics of xanthine derivatives binding to the G-protein coupled adenosine A1 receptor.
50,52 These
compounds which include theophylline and caffeine act as non-selective antagonists on the A1 and A2A adenosine receptors.
53 Measuring binding affinity
constants at six different temperatures in the range from 0-35 °C, enabled van „t Hoff analysis of the thermodynamic binding parameters of 16 xanthine derivatives. In the example shown in Figure 3, the fragment theophylline (MW = 180) was chosen as a starting point. Theophylline exhibits a good LE and EE (see Glossary) for the adenosine A1 receptor and its binding is completely enthalpy driven (ΔH° = -7.2 and -TΔS°
= +0.5 kcal·mol
-1). Binding affinity can be increased by substituting
the 8-position with hydrophobic moieties such as phenyl (8-PT) and cyclopentyl (CPT) moieties, resulting in 125- and 900-fold increases in affinity, respectively. While addition of a phenyl moiety increases binding affinity solely due to a favorable increase in entropy (-TΔΔS° = -4.8 kcal·mol
-1), growing the fragment from
the 8-position with a cyclopentyl moiety results in favorable changes in enthalpy, as well as in entropy (ΔΔH° = -1.6 and -TΔΔS
0 = -2.5 kcal·mol
-1). An additional
increase in the binding affinity of the cyclopentyl substituted fragment can be achieved by extending both methyl substituents with two carbons each (DPCPX). This modification affords a 6-fold increase in binding affinity due to a favorable increase in entropy (-TΔΔS°
= -2.1 kcal·mol
-1). Figure 3 shows that introducing or
increasing hydrophobic substituents can result in increases in binding affinity due to favorable increases in entropy, illustrative of the hydrophobic effect. More interesting is the 900-fold increase in affinity upon addition of a cyclopentyl moiety to theophylline‟s 8-position. This extensive increase in affinity results from favorable changes in entropy as well as in enthalpy. As optimization of both thermodynamic binding parameters at the same time is the most efficient way of increasing binding affinity, this specific example may warrant further investigation. For example the generation of X-ray co-crystal structures of the starting fragment and its 8-cyclopentyl substituted derivative may provide insights into this unusual simultaneous enthalpy and entropy optimization.

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
49
Figure 3: Changes in Gibbs energy (ΔΔG0), enthalpy (ΔΔH
0) and entropy (-TΔΔS
0)
in kcal·mol-1
upon growing the fragment theophylline towards high affinity adenosine A1 antagonists. For each compound, the LE and EE have been calculated in kcal·mol
-1 per heavy atom.
Matrix Metalloproteinases Bertini and coworkers have performed a thorough study on matrix metalloproteinase 12 (MMP12) inhibitors in which they obtained co-crystal structures, as well as thermodynamic parameters of binding of a large number of inhibitors using ITC.
54 The common scaffold of all the inhibitors analyzed in this
study is N-hydroxy-2-(phenylsulfonamido)acetamide (1). This fragment (MW = 230) binds with a Kd of 61 nM (ΔG° = -9.8 kcal·mol
-1) and exhibits a high LE for MMP12
(0.65 kcal·mol-1
per heavy atom). In a later study, the p-methoxy substituted analog of fragment 1 (2) was further deconstructed to acetohydroxamic acid (AHA) and p-methoxybenzene-sulfonamide (PMS), see Figure 4.
55 This approach allows

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
50
determination of the thermodynamic contributions to binding affinity when two separate fragments are linked. Thermodynamic analysis shows that the sum of the binding enthalpies of the separate fragments is quite similar to the binding enthalpy of 2. The significant gain in binding affinity upon linking the fragments seems to be completely due to a gain in entropy (-TΔΔS = -4.4 kcal·mol
-1). This favorable
increase in entropy upon linking fragments arises because, upon binding to the protein binding site, ligands lose translational and rotational degrees of freedom. When the linked fragments bind, the entropy cost of restricting ligand rotation and translation only needs to be paid once. As a consequence, the linking of fragments that occupy different spots within the protein binding site can yield significant increases in binding affinity. Unfortunately, in practice the expected increases in affinity are often not obtained due to perturbation of the binding modes of the separate fragments upon linking, or because of strain in the linker that is used to connect the fragments.
56,57
From a thermodynamic point of view, the common scaffold 1 can be considered as an ideal starting point for further optimization, as complex formation is almost exclusively enthalpy driven (ΔH° = -9.1 kcal·mol
-1). X-ray analysis shows that the
hydroxamic group interacts with the catalytic zinc ion. Furthermore, the hydroxamic and sulfonamide moieties are involved in additional enthalpic interactions with the binding site via direct and water-mediated hydrogen bonds. Extending the 4-position of the phenyl moiety of fragment (1) with a methoxy (2) or phenyl moiety (4) results in additional hydrophobic interactions in the S1‟ pocket (see Figure 5) affording 3- and 25-fold increases in binding affinity, respectively. These increases in binding affinity by addition of hydrophobic moieties are in both cases due to more favorable entropy (4-MeO: -TΔΔS° = -1.3 kcal·mol
-1 and 4-Phenyl: -TΔΔS° = -
2.2 kcal·mol-1
). Analysis of the X-ray structures shows that extending the 4-position with a methoxy or phenyl moiety, worsens the interactions of the hydroxamic group with the zinc ion and the hydrogen bonds formed by the sulfonyl moiety. Bertini and co-workers suggest that this weakening of enthalpic interactions results in a more favorable entropy as the ligand and protein gain more freedom of movement. It is also likely that growing into the hydrophobic S1‟ pocket results in the displacement of water molecules affording additional favorable increases in entropy.
Capping the sulfonamide nitrogen of 2 with an isobutyl group (3) results in a 5-fold increase in binding affinity resulting entirely from a favorable increase in entropy (TΔΔS° = -1.2 kcal·mol
-1). In contrast, a 3-fold increase in affinity by decoration of
the sulfonamide nitrogen with a hydroxyethyl moiety (5), is established by a favorable decrease in enthalpy (ΔΔH° = -1.2 kcal·mol
-1). X-ray analysis shows that
the favorable decrease in enthalpy may originate from van der Waals contacts of the ethyl spacer with Pro238 and hydrogen bond formation with a network of water molecules that is interacting with residues that align the entrance of the binding site (see Figure 5). In terms of binding affinity, compounds 3 and 4 can be considered the best compounds for further optimization. Nevertheless, the compound most optimized in enthalpy is the N-hydroxyethyl substituted derivative 5, which may be the best choice for further optimization when EE is considered.

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
51

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
52
Figure 4: Changes in Gibbs energy (ΔΔG0), enthalpy (ΔΔH
0) and entropy (-TΔΔS
0)
in kcal·mol-1
upon linking and growing fragments towards high affinity MMP12 inhibitors. For each compound, the LE and EE have been calculated in kcal·mol
-1
per heavy atom.
Figure 5: The binding mode of compound 5 (black ball and sticks) in complex with MMP-12 as determined by X-ray crystallography (PDB: 3NX7). The S1’ pocket is indicated by the grey arrow whereas the water network which interacts with the hydroxyethyl moiety is indicated by the white arrow. Major mouse urinary protein The mouse major urinary proteins (MUPs) are pheromone-binding proteins, a complex of protein isoforms encountered in mice which appear to serve as signal-modulating agents for pheromones. MUPs have been used in several studies as a model protein to investigate the thermodynamics of ligand-protein interactions.
45,58,59 X-ray crystallography and NMR studies have shown that the
interior of MUPs contain a hydrophobic cavity, which forms the ligand binding site.
45 Interestingly, increasing the size of hydrophobic substituents of two identified
ligands for MUP-I, 2-methyl-4,5-dihydrothiazole (MT) and 2-isopropyl-3-methoxypyrazine (IPMP), results in favorable decreases in enthalpy (ΔΔH° = -1.5 and – 1.2 kcal·mol
-1, resp.), see Figure 6. These findings are in contrast with the
classical view on the hydrophobic effect, in which increased burial of hydrophobic substituents in a hydrophobic cavity results in more favorable entropy. In an initial study by Sharrow et al. three possible explanations were provided for the favorable change in enthalpy that was observed upon increasing the size of the hydrophobic substituents on the 2-position of 4,5-dihydrothiazoles: (1) formation of a buried water-mediated hydrogen bond network between the ligand and protein
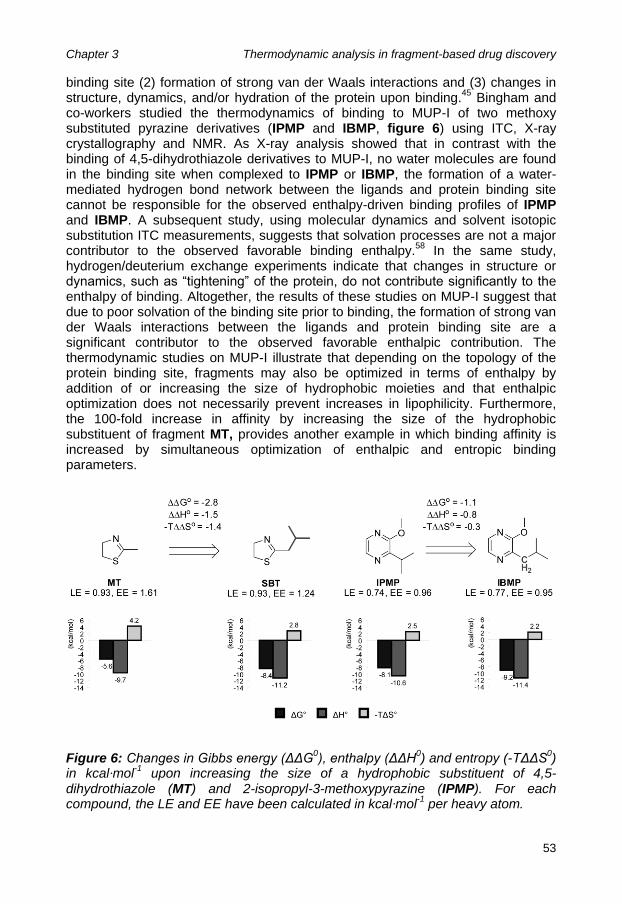
Chapter 3 Thermodynamic analysis in fragment-based drug discovery
53
binding site (2) formation of strong van der Waals interactions and (3) changes in structure, dynamics, and/or hydration of the protein upon binding.
45 Bingham and
co-workers studied the thermodynamics of binding to MUP-I of two methoxy substituted pyrazine derivatives (IPMP and IBMP, figure 6) using ITC, X-ray crystallography and NMR. As X-ray analysis showed that in contrast with the binding of 4,5-dihydrothiazole derivatives to MUP-I, no water molecules are found in the binding site when complexed to IPMP or IBMP, the formation of a water-mediated hydrogen bond network between the ligands and protein binding site cannot be responsible for the observed enthalpy-driven binding profiles of IPMP and IBMP. A subsequent study, using molecular dynamics and solvent isotopic substitution ITC measurements, suggests that solvation processes are not a major contributor to the observed favorable binding enthalpy.
58 In the same study,
hydrogen/deuterium exchange experiments indicate that changes in structure or dynamics, such as “tightening” of the protein, do not contribute significantly to the enthalpy of binding. Altogether, the results of these studies on MUP-I suggest that due to poor solvation of the binding site prior to binding, the formation of strong van der Waals interactions between the ligands and protein binding site are a significant contributor to the observed favorable enthalpic contribution. The thermodynamic studies on MUP-I illustrate that depending on the topology of the protein binding site, fragments may also be optimized in terms of enthalpy by addition of or increasing the size of hydrophobic moieties and that enthalpic optimization does not necessarily prevent increases in lipophilicity. Furthermore, the 100-fold increase in affinity by increasing the size of the hydrophobic substituent of fragment MT, provides another example in which binding affinity is increased by simultaneous optimization of enthalpic and entropic binding parameters.
Figure 6: Changes in Gibbs energy (ΔΔG0), enthalpy (ΔΔH
0) and entropy (-TΔΔS
0)
in kcal·mol-1
upon increasing the size of a hydrophobic substituent of 4,5-dihydrothiazole (MT) and 2-isopropyl-3-methoxypyrazine (IPMP). For each compound, the LE and EE have been calculated in kcal·mol
-1 per heavy atom.

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
54
v-Src homology (SH2) domain The studies described above did not deliberately apply thermodynamic analysis to guide and/or monitor the fragment optimization process. However, recently two studies have been published in which thermodynamic analysis played a significant role in deciding which fragment to pursue or, monitoring the fragment optimization. The group of Ladbury has applied thermodynamic analysis on two hits that were identified using an NMR-based fragment screening on the Src SH2 domain.
60
Dissection of binding affinity into its separate thermodynamic parameters revealed that although both fragments exhibit similar affinity, one of the compounds binds with a significantly more favorable enthalpy (ΔΔH° = -3.3 kcal·mol
-1) and is
therefore considered to be a better fragment hit to optimize. Unfortunately, results on further optimization of the identified fragment-hits have so far not been disclosed. Carbonic anhydrase The value of using thermodynamic analysis in FBDD is well illustrated by a study performed by Scott and co-workers in which the thermodynamic parameters of 20 benzene sulfonamide derivatives binding to human carbonic anhydrase were determined using ITC.
4 Substitution of benzene sulfonamide (6) with a fluorine
atom at the 2- or 3-position affords a 3- and 7-fold increase in affinity, respectively (see Figure 7). Based on LE, the 3-substituted fragment 8 can be considered to be more appropriate for further optimization. However, thermodynamic analysis reveals that the increase in affinity by fluorine substitution at the 2-position (7) is caused by a favorable change in enthalpy (ΔΔH° = -1.9 kcal·mol
-1, -TΔΔS° = + 1.3
kcal·mol-1
), whereas the increase in affinity for 3-fluorobenzene sulfonamide results solely from a favorable change in entropy (ΔΔH° = 0 kcal·mol
-1, -TΔΔS° = -1.1
kcal·mol-1
). X-ray analysis of co-crystal structures shows that the fluorine atom in the 2-substituted fragment is engaged in a specific interaction with the backbone N-H of Thr200, whereas the fluorine atom of the 3-substituted benzene sulfonamide is pointing towards a hydrophobic part of the binding site. Growing both fragments with a 4-benzylamide substituent affords a 55-fold increase in affinity for the 2-fluorobenzene sulfonamide (9), but only a modest 6-fold increase in affinity for its 3-substituted isomer (11). In addition, the binding of 2-fluorobenzene sulfonamide remains entirely enthalpically driven. In this specific study, the most enthalpy efficient fragment became the most potent ligand upon optimization and retained its favorable thermodynamic signature. Conclusion Recent findings suggest that in drug development, compounds that have been enthalpically optimized, outperform compounds in which binding is based on favorable entropy changes. However, it is easier to improve binding affinity by optimizing entropy than it is by optimizing enthalpy. It therefore seems a good strategy to determine the thermodynamic profiles of fragment hits and include EE as one of the hit selection criteria. Choosing a fragment in which binding is enthalpically driven as a starting point and adding hydrophobic groups during optimization, may provide the easiest route to a selective high affinity compound. In addition, determination of thermodynamic profiles may be very useful in guiding and monitoring of (structure-based) fragment optimization. When combined with structural data, the stepwise fragment growing process provides an ideal dataset

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
55
Figure 7: Changes in Gibbs energy (ΔΔG
0), enthalpy (ΔΔH
0) and entropy (-TΔΔS
0)
in kcal·mol-1
upon growing benzene sulfonamide (1) towards more potent carbonic anhydrase inhibitors. For each compound, the LE and EE have been calculated in kcal·mol
-1 per heavy atom.

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
56
with which to improve our understanding of the thermodynamics of binding. This is exemplified by the case on MUP that shows that extending a fragment in a poorly solvated binding pocket can afford considerable increases in binding affinity by favorable changes in enthalpy as well as entropy. Another example of the unusual simultaneous of enthalpy and entropy by fragment growing has been identified for the Adenosine A1 receptor and may provide important lessons on how to optimize fragments in a most efficient manner. These examples emphasize that both FBDD, and our understanding of the thermodynamics of binding, have much to gain from the application of thermodynamic analysis in FBDD. The two main techniques that are used to determine thermodynamic binding parameters are ITC and van „t Hoff analysis. The main advantage of ITC is its easy set-up, as calorimetric measurements do not require immobilization or labeling. Disadvantages are the large amounts of protein required and the relatively long time required to perform an experiment. It should be noted that progress has recently been made in resolving these issues. Measuring binding affinity at a range of different temperatures (0 – 37 °C) enables van „t Hoff analysis, providing access to the thermodynamic parameters. Although discrepancies between calorimetry and van „t Hoff analysis have been reported, more recent comparison studies show that application of both techniques result in similar values for the thermodynamic binding parameters. Nevertheless, retrieval of thermodynamic binding parameters using van „t Hoff analysis may be complicated when the heat capacity change is not equal to zero (ΔCp° ≠ 0). A convenient technique enabling van „t Hoff analysis is SPR biosensor analysis, since it allows accurate temperature control, requires much smaller amounts of target-protein than ITC and is a label-free technique. In addition to thermodynamic data, kinetic parameters of binding may be obtained as well, providing an additional dimension to binding affinity. Both ITC- and SPR biosensor-based thermodynamic analysis are hampered by low throughput and are therefore not suitable for primary fragment screening. Instead, these techniques are more appropriate for secondary screening, to confirm and characterize the thermodynamics of fragment hits. If target-protein production is not limiting, one may consider ITC, since measurements can be performed without the need for immobilization and all binding parameters (n, KB, ΔH
o and ΔS
o) are
determined in a single experiment. If the target protein is more difficult to obtain, SPR biosensor analysis may be a more sensible technique to use, requiring only small amounts of protein. Although, the use of thermodynamic analysis within FBDD is still far from common, the first cases of successful application are becoming apparent, exemplifying its value in the development of a more efficient fragment optimization process. Acknowledgements This work was supported by grants D2-103 and T4-302, from the Top Institute Pharma. We would like to thank Chris Oostenbrink for reading the manuscript and providing us with useful suggestions.

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
57
References and notes 1. de Kloe, G.E., Bailey, D., Leurs, R. & de Esch, I.J. Transforming fragments
into candidates: small becomes big in medicinal chemistry. Drug Discov Today 14, 630-46 (2009).
2. Schulz, M.N. & Hubbard, R.E. Recent progress in fragment-based lead discovery. Current Opinion in Pharmacology 9, 615-621 (2009).
3. Bissantz, C., Kuhn, B. & Stahl, M. A medicinal chemist's guide to molecular interactions. J Med Chem 53, 5061-84 (2010).
4. Scott, A.D. et al. Thermodynamic optimisation in drug discovery: a case study using carbonic anhydrase inhibitors. ChemMedChem 4, 1985-9 (2009).
5. Freire, E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov Today 13, 869-74 (2008).
6. Olsson, T.S., Williams, M.A., Pitt, W.R. & Ladbury, J.E. The thermodynamics of protein-ligand interaction and solvation: insights for ligand design. J Mol Biol 384, 1002-17 (2008).
7. Leeson, P.D. & Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov 6, 881-90 (2007).
8. Wenlock, M.C., Austin, R.P., Barton, P., Davis, A.M. & Leeson, P.D. A comparison of physiochemical property profiles of development and marketed oral drugs. J Med Chem 46, 1250-6 (2003).
9. Dunitz, J.D. Win some, lose some: enthalpy-entropy compensation in weak intermolecular interactions. Chem Biol 2, 709-12 (1995).
10. Whitesides, G.M. & Krishnamurthy, V.M. Designing ligands to bind proteins. Q Rev Biophys 38, 385-95 (2005).
11. Raffa, R.B., ed. Drug-Receptor Thermodynamics: Introduction and Applications, New York: John Wiley and Sons. (2001).
12. Ababou, A. & Ladbury, J.E. Survey of the year 2004: literature on applications of isothermal titration calorimetry. J Mol Recognit 19, 79-89 (2006).
13. Ababou, A. & Ladbury, J.E. Survey of the year 2005: literature on applications of isothermal titration calorimetry. J Mol Recognit 20, 4-14 (2007).
14. Bjelic, S. & Jelesarov, I. A survey of the year 2007 literature on applications of isothermal titration calorimetry. J Mol Recognit 21, 289-312 (2008).
15. Cliff, M.J., Gutierrez, A. & Ladbury, J.E. A survey of the year 2003 literature on applications of isothermal titration calorimetry. J Mol Recognit 17, 513-23 (2004).
16. Cliff, M.J. & Ladbury, J.E. A survey of the year 2002 literature on applications of isothermal titration calorimetry. J Mol Recognit 16, 383-91 (2003).
17. Falconer, R.J., Penkova, A., Jelesarov, I. & Collins, B.M. Survey of the year 2008: applications of isothermal titration calorimetry. J Mol Recognit 23, 395-413 (2010).

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
58
18. Okhrimenko, O. & Jelesarov, I. A survey of the year 2006 literature on applications of isothermal titration calorimetry. J Mol Recognit 21, 1-19 (2008).
19. Li, L., Dantzer, J.J., Nowacki, J., O'Callaghan, B.J. & Meroueh, S.O. PDBcal: a comprehensive dataset for receptor-ligand interactions with three-dimensional structures and binding thermodynamics from isothermal titration calorimetry. Chem Biol Drug Des 71, 529-32 (2008).
20. Liu, T., Lin, Y., Wen, X., Jorissen, R.N. & Gilson, M.K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res 35, D198-201 (2007).
21. Ladbury, J.E., Klebe, G. & Freire, E. Adding calorimetric data to decision making in lead discovery: a hot tip. Nat Rev Drug Discov 9, 23-7 (2010).
22. Turnbull, W.B. & Daranas, A.H. On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc 125, 14859-66 (2003).
23. www.microcal.com/products/itc/itc200.asp. 24. Holdgate, G.A. Thermodynamics of binding interactions in the rational drug
design process. Expert Opinion on Drug Discovery 2, 1103-1114 (2007). 25. Huber, W. & Mueller, F. Biomolecular interaction analysis in drug discovery
using surface plasmon resonance technology. Curr Pharm Des 12, 3999-4021 (2006).
26. Danielson, U.H. Fragment library screening and lead characterization using SPR biosensors. Curr Top Med Chem 9, 1725-35 (2009).
27. Chaires, J.B. Possible origin of differences between van't Hoff and calorimetric enthalpy estimates. Biophys Chem 64, 15-23 (1997).
28. Horn, J.R., Brandts, J.F. & Murphy, K.P. van't Hoff and calorimetric enthalpies II: effects of linked equilibria. Biochemistry 41, 7501-7 (2002).
29. Horn, J.R., Russell, D., Lewis, E.A. & Murphy, K.P. Van't Hoff and calorimetric enthalpies from isothermal titration calorimetry: are there significant discrepancies? Biochemistry 40, 1774-8 (2001).
30. Liu, Y. & Sturtevant, J.M. Significant discrepancies between van't Hoff and calorimetric enthalpies. II. Protein Sci 4, 2559-61 (1995).
31. Liu, Y. & Sturtevant, J.M. Significant discrepancies between van't Hoff and calorimetric enthalpies. III. Biophys Chem 64, 121-6 (1997).
32. Naghibi, H., Tamura, A. & Sturtevant, J.M. Significant discrepancies between van't Hoff and calorimetric enthalpies. Proc Natl Acad Sci U S A 92, 5597-9 (1995).
33. Tellinghuisen, J. Van't Hoff analysis of K degrees (T): how good...or bad? Biophys Chem 120, 114-20 (2006).
34. Tellinghuisen, J. Calibration in isothermal titration calorimetry: heat and cell volume from heat of dilution of NaCl(aq). Anal Biochem 360, 47-55 (2007).
35. Day, Y.S., Baird, C.L., Rich, R.L. & Myszka, D.G. Direct comparison of binding equilibrium, thermodynamic, and rate constants determined by surface- and solution-based biophysical methods. Protein Sci 11, 1017-25 (2002).
36. Myszka, D.G. et al. The ABRF-MIRG'02 study: assembly state, thermodynamic, and kinetic analysis of an enzyme/inhibitor interaction. J Biomol Tech 14, 247-69 (2003).

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
59
37. Navratilova, I. et al. Thermodynamic benchmark study using Biacore technology. Anal Biochem 364, 67-77 (2007).
38. Harper, E.A. & Black, J.W. Histamine H3-receptor agonists and imidazole-based H3-receptor antagonists can be thermodynamically discriminated. Br J Pharmacol 151, 504-17 (2007).
39. Borea, P.A., Dalpiaz, A., Gessi, S. & Gilli, G. Thermodynamics of 5-HT3 receptor binding discriminates agonistic from antagonistic behaviour. Eur J Pharmacol 298, 329-34 (1996).
40. Maksay, G. Distinct thermodynamic parameters of serotonin 5-HT3 agonists and antagonists to displace [3H]granisetron binding. J Neurochem 67, 407-12 (1996).
41. Pham, C., Jankun, J., Skrzypczak-Jankun, E., Flowers, R.A., 2nd & Funk, M.O., Jr. Structural and thermochemical characterization of lipoxygenase-catechol complexes. Biochemistry 37, 17952-7 (1998).
42. Ciulli, A., Chirgadze, D.Y., Smith, A.G., Blundell, T.L. & Abell, C. Crystal structure of Escherichia coli ketopantoate reductase in a ternary complex with NADP+ and pantoate bound: substrate recognition, conformational change, and cooperativity. J Biol Chem 282, 8487-97 (2007).
43. Wittmann, H.-J., Seifert, R. & Strasser, A. Contribution of Binding Enthalpy and Entropy to Affinity of Antagonist and Agonist Binding at Human and Guinea Pig Histamine H1-Receptor. Molecular Pharmacology 76, 25-37 (2009).
44. Ciulli, A. et al. pH-tuneable binding of 2'-phospho-ADP-ribose to ketopantoate reductase: a structural and calorimetric study. Acta Crystallogr D Biol Crystallogr 63, 171-8 (2007).
45. Sharrow, S.D., Novotny, M.V. & Stone, M.J. Thermodynamic analysis of binding between mouse major urinary protein-I and the pheromone 2-sec-butyl-4,5-dihydrothiazole. Biochemistry 42, 6302-9 (2003).
46. Zhukov, A. & Karlsson, R. Statistical aspects of van't Hoff analysis: a simulation study. J Mol Recognit 20, 379-85 (2007).
47. Only unique fragment-protein complexes were incorporated. In cases where complexes were measured under different conditions (concentration of salts, t., presence of co-factor etc.), only the data from one set of conditions were included.
48. Borea, P.A., Dalpiaz, A., Varani, K., Gilli, P. & Gilli, G. Can thermodynamic measurements of receptor binding yield information on drug affinity and efficacy? Biochem Pharmacol 60, 1549-56 (2000).
49. Maksay, G. Activation of ionotropic receptors and thermodynamics of binding. Neurochem Int 46, 281-91 (2005).
50. Borea, P.A., Dalpiaz, A., Varani, K., Gessi, S. & Gilli, G. Binding thermodynamics at A1 and A2A adenosine receptors. Life Sci 59, 1373-88 (1996).
51. Borea, P.A., Varani, K., Gessi, S., Gilli, P. & Gilli, G. Binding thermodynamics at the human neuronal nicotine receptor. Biochem Pharmacol 55, 1189-97 (1998).
52. Borea, P.A., Varani, K., Guerra, L., Gilli, P. & Gilli, G. Binding thermodynamics of A1 adenosine receptor ligands Mol Neuropharmacol 2, 273-281 (1992).

Chapter 3 Thermodynamic analysis in fragment-based drug discovery
60
53. Ferre, S. An update on the mechanisms of the psychostimulant effects of caffeine. J Neurochem 105, 1067-79 (2008).
54. Bertini, I. et al. Exploring the subtleties of drug-receptor interactions: the case of matrix metalloproteinases. J Am Chem Soc 129, 2466-75 (2007).
55. Borsi, V., Calderone, V., Fragai, M., Luchinat, C. & Sarti, N. Entropic contribution to the linking coefficient in fragment based drug design: a case study. J Med Chem 53, 4285-9 (2010).
56. Chung, S., Parker, J.B., Bianchet, M., Amzel, L.M. & Stivers, J.T. Impact of linker strain and flexibility in the design of a fragment-based inhibitor. Nat Chem Biol 5, 407-13 (2009).
57. Howard, N. et al. Application of fragment screening and fragment linking to the discovery of novel thrombin inhibitors. J Med Chem 49, 1346-55 (2006).
58. Barratt, E. et al. Van der Waals interactions dominate ligand-protein association in a protein binding site occluded from solvent water. J Am Chem Soc 127, 11827-34 (2005).
59. Bingham, R.J. et al. Thermodynamics of binding of 2-methoxy-3-isopropylpyrazine and 2-methoxy-3-isobutylpyrazine to the major urinary protein. J Am Chem Soc 126, 1675-81 (2004).
60. Taylor, J.D., Gilbert, P.J., Williams, M.A., Pitt, W.R. & Ladbury, J.E. Identification of novel fragment compounds targeted against the pY pocket of v-Src SH2 by computational and NMR screening and thermodynamic evaluation. Proteins 67, 981-90 (2007).
61. Williams, D.H. & Bardsley, B. Estimating binding constants - The hydrophobic effect and cooperativity. Perspectives in Drug Discovery and Design 17, 43-59 (1999).
62. Hopkins, A.L., Groom, C.R. & Alex, A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today 9, 430-1 (2004).

61
Chapter 4
Fragment growing induces conformational changes in
acetylcholine-binding protein:
A structural and thermodynamic analysis
Ewald Edink#, Prakash Rucktooa
#, Kim Retra, Atilla Akdemir, Tariq Nahar,
Obbe Zuiderveld, René van Elk , Elwin Janssen, Pim van Nierop, Jacqueline van Muijlwijk-Koezen, August B. Smit, Titia K. Sixma,
Rob Leurs and Iwan J.P. de Esch
1*
# These authors contributed equally.
Abstract Optimization of fragment hits towards high affinity lead compounds is a crucial aspect of fragment-based drug discovery (FBDD). In the current study, we have successfully optimized a fragment by growing into a ligand-inducible subpocket of the binding site of acetylcholine-binding protein (AChBP). This protein is a soluble homolog of the ligand binding domain (LBD) of Cys-loop receptors. The fragment optimization was monitored with X-ray structures of ligand complexes and systematic thermodynamic analyses using surface plasmon resonance (SPR) biosensor analysis and isothermal titration calorimetry (ITC). Using site-directed mutagenesis and AChBP from different species, we find that specific changes in thermodynamic binding profiles, are indicative of interactions with the ligand-inducible subpocket of AChBP. This study illustrates that thermodynamic analysis provides valuable information on ligand binding modes and is complementary to affinity data when guiding rational structure- and fragment-based discovery approaches.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
62
Introduction In the past decade, fragment-based drug discovery (FBDD) has become a well-established method in the field of drug discovery.
1-3 Compared to traditional high-
throughput screening (HTS), FBDD is based on screening of smaller libraries of compounds (typically containing 1000-5000 fragments) with lower molecular weights (i.e., smaller than 300 Da).
4 Fragment-based screening results in a better
coverage of chemical space and usually higher hit rates than HTS.3,5
Identified fragment hits are either optimized by linking or growing. In fragment linking, two simultaneously binding fragments are connected via a chemical linker. Unfortunately, the expected increase in affinity is often compromised by perturbation of the binding modes of the separate fragments or strain in the linker used to connect the fragments.
6,7 The preferred hit-optimization strategy in FBDD
has become fragment growing.1,8
In iterative cycles, additional features are added to the hit fragment, leading to more potent compounds. Structural biology has been shown to be crucial to guide the optimization of fragment hits towards novel and potent drugs.
1,3,9-11 In the current study, we employ FBDD approaches to develop
new ligands that bind to acetylcholine-binding protein (AChBP). This water-soluble pentameric protein is widely recognized as a structural homolog of the ligand binding domain (LBD) of Cys-loop receptors.
12,13 Currently, AChBPs from different
species such as Lymnaea stagnalis (Ls-AChBP)14-16
, Aplysia californica (Ac-AChBP)
17,18 and Bulinus truncatus (Bt-AChBP)
19 have been identified and
crystallized, providing high-resolution structural models of the extracellular domains of the pentameric ligand-gated ion channels (LGICs) of the Cys-loop family, such as nicotinic acetylcholine receptors (nAChRs), GABAA-, serotonin 5-HT3-, and glycine receptors.
13
In a recent study, tropine derivatives were identified as AChBP ligands using an in silico screening protocol.
18 A structural analog, the benzoate substituted nortropine
fragment 1, exhibits good ligand efficiency (LE)20
of 0.43 kcalmol-1
per heavy atom
for Ac-AChBP and is therefore considered to be a good starting point for further optimization (Figure 1). In the current study, a co-crystal complex of fragment 1 and Ac-AChBP was generated thereby enabling structure-based optimization. Comparison with previously obtained co-crystal complexes reveals conformational changes of the target protein upon ligand binding, mainly with respect to the loop C which closes the binding site upon agonist binding.
15,17-19,21 A unique
conformational change is observed for AChBP while binding to lobeline, leading to the opening of a subpocket that enables the binding of the α-hydroxyphenetyl moiety of the ligand.
17 More specifically, the subpocket that we will refer to as the
lobeline pocket, becomes accessible after a change in the rotameric state of Tyr91 (g- to t conformation
22, hereafter referred to as the tyrosine-flip). Considering the
partially overlapping binding modes of hit fragment 1 and lobeline (2), we designed a fragment growing optimization study to induce the tyrosine-flip and grow the fragment into the lobeline pocket (Figure 1). The ligand-induced opening of binding site subpockets represents an interesting challenge and a potentially rewarding drug discovery opportunity.
23 Using X-ray analysis of co-crystal structures,
molecular modelling, site-directed mutagenesis, AChBP species differences, surface plasmon resonance (SPR) biosensor analysis and isothermal titration calorimetry (ITC), we show that the thermodynamic signature of ligand binding

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
63

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
64
Figure 1. Fragment optimization strategy. Surface representations of the crystal structures of fragment 1-bound Ac-AChBP (a) and of lobeline-bound Ac-AChBP (PDB 2BYS)
16 (b). (a) In the fragment 1-Ac-AChBP complex, Tyr91 (orange) is
stabilized in the g- conformation through a hydrogen bond with Ser144, rendering the lobeline pocket inaccessible. (b) However Tyr91 adopts a t conformation in the lobeline-Ac-AChBP complex interacting with Tyr53 and Ser165 through hydrogen bonds, thus leading to the opening of the lobeline pocket. (c) The superposition of the fragment 1 and lobeline (2) molecules indicates that the fragment may be grown into the lobeline pocket by extending its tropine nitrogen with the α-hydroxyphenetyl moiety of lobeline generating compound (3).
changes drastically when the lobeline pocket is addressed. The obtained results illustrate that thermodynamic analysis of ligand binding provides important information about the binding mode and reveal the value of monitoring thermodynamic aspects during fragment growing. Results and Discussion
Chemistry The designed compounds were prepared according to the route depicted in Scheme 1. Acylation of tropine with benzoyl chloride afforded tropine benzoate (7) in an excellent yield.
24 Subsequent demethylation using α-chloroethyl
chloroformate gave the corresponding nortropinyl ester (1).25
The α-hydroxylphenetyl extended fragment was synthesized by heating 1 with the (R)-enantiomer of phenyloxirane in the microwave.
26 The enantiomeric excess (ee) of
compound 3 was determined at 98% by chiral HPLC. Reductive amination of phenylacetaldehyde with nortropine benzoate 1 using sodium triacetoxyborohydride as the reducing agent gave compound 4.
27 Finally, treatment
of 3 with iodomethane followed by recrystallization from chloroform resulted in isolation of the endo-α-(R)-hydroxyphenetyl substituted quaternary ammonium derivative (6). The endo-configuration of the α-(R)-hydroxyphenetyl substituent was confirmed by 2D NMR.
Structure-Based Design of a Novel ligand that Interacts with the Lobeline Pocket Fragment 1, which is a structural analog of hits that have been identified in an earlier study,
18 exhibits good LE (0.43 kcal·mol
-1 per heavy atom) and was co-
crystallized with Ac-AChBP. A 3.65 Å resolution crystal structure was obtained, with good density for the ligand (Figure 1a, 2a and 3a) despite the low resolution. The ability to use five-fold restraints in crystallographic refinement allowed confident building of the compound into electron density, thus enabling structure-based optimization. The structure displays one Ac-AChBP pentamer per asymmetric unit where the C-loops adopt a closed conformation over all five binding sites. Fragment 1 is present at all five protomer-protomer interfaces, with similar orientations. The ligand is stabilized through hydrophobic interactions with Cys188, Cys189, Gln55, Ile116 and Tyr91, and through a hydrogen bond between the protonated amine atom of 1 and the backbone carbonyl from Trp145 (Figure

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
65
1a, 2a and 3a). When compared with lobeline-bound Ac-AChBP (Figure 1b, PDB: 2BYS), the fragment‟s benzoate group is tilted ~50˚ with respect to lobeline‟s 1-phenylethanone moiety and stacked against the vicinal cysteines at the tip of loop C, placing the loop in a more open conformation (2.7 Å when comparing Cys188 Cα positions). The structure further reveals that gatekeeper residue Tyr91 is in the g- conformation, making the lobeline pocket inaccessible. From the complex structures we predicted that further optimization of the fragment could be achieved by opening of and growing into the lobeline pocket (Figure 1). To address this subpocket, the fragment was merged with the α-hydroxyphenetyl group of lobeline (2), resulting in compound 3 (Figure 1). Molecular docking using GOLD (version 4.0)
28 suggests that the extended fragment 3 can adopt a binding mode in which
the lobeline pocket is being addressed by the α-hydroxyphenetyl moiety (Figure 4). Because these in silico results seemed promising, the designed compound was synthesized and screened for AChBP affinity.
Scheme 1: Synthetic route towards target molecules

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
66
X-Ray Structures Confirm Insertion into the Lobeline Pocket Extension of fragment 1 with an α-(R)-hydroxyphenetyl moiety yields compound 3 and resulted in a ~50-fold increase in affinity (pKi = 7.0 ± 0.1), although the LE drops slightly from 0.43 to 0.37 kcal·mol
-1 per heavy atom (Figure 6a). In order to
confirm successful insertion into the lobeline pocket, a crystal structure of the optimized fragment (3) bound to Ac-AChBP was generated. The 3.59 Å resolution crystal structure shows that fragment 1 was successfully grown into the lobeline pocket (Figure 2b and 3b). The structure displays one pentamer in the asymmetric unit with each of the protomer-protomer interfaces binding one ligand molecule. The benzoate substituted nortropine moiety adopts an orientation similar to 1, although displaced ~1Å deeper into the binding site, hence bringing loop C into a conformation intermediate between those observed for fragment 1 and lobeline Ac-AChBP complexes. Interestingly, an ion, most likely a chloride, could be refined in the position occupied by the lobeline 1-phenylethanone moiety from the lobeline-Ac-AChBP complex. Extension of fragment 1 with an α-(R)-hydroxyphenetyl moiety (resulting in 3), induces a change in rotameric state of Tyr91 (g- to t conformation) that opens the lobeline pocket. This tyrosine-flip is stabilized by hydrogen bond formation between the phenolic oxygen of Tyr91 and the side chain hydroxyl groups of Tyr53 and Ser165. The α-(R)-hydroxyphenetyl moiety responsible for the insertion into the lobeline pocket displays an orientation similar to that observed in the Ac-AChBP-lobeline complex and is involved in extensive hydrophobic interactions with Asp195, Tyr91, Lys141, Gly143 and Thr89 (Figure 2b and 3b). As a result, the experimentally determined binding mode corresponds very well to the predicted binding mode that was obtained by molecular docking (rmsd of 1.1 Å). Nevertheless, the experimentally determined binding mode differs from the predicted binding mode in that the hydroxyl group of 3 is not engaged in hydrogen bonding to the backbone carbonyl oxygens of Ser144 or Trp145 but instead is involved in van der Waals interactions with Tyr193 (Figure 4). Since, the hydroxyl group of the optimized fragment 3 is not involved in formation of hydrogen bonds with the binding site, it was expected that its removal would lower the desolvation penalty and thereby result in an increase in binding affinity. The norhydroxyl derivative (4) was therefore synthesized and screened for AChBP affinity. Indeed, 4 exhibited higher AChBP affinity (pKi = 7.5 ± 0) than 3 and when compared with the starting fragment 1, a 150-fold increase in binding affinity is observed (Figure 6). To determine if 4 induces the tyrosine-flip and interacts with the lobeline pocket, an additional co-crystal structure was generated. The compound 4-Ac-AChBP structure was solved with a resolution of 3.30 Å and shows an almost identical binding mode to 3 (rmsd of 0.6 Å), exemplifying that a hydroxyl functionality is not required for opening of and insertion into the lobeline pocket (Figure 2c and 3c).

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
67
Figure 2. Growing fragment 1 into or away from the lobeline pocket. Cartoon representations of the Ac-AChBP complexes with fragment 1 (a), optimized fragments 3 (b) and 4 (c) and quaternary ammonium derivative 6 (d). The Sigma A weighted 2Fo-Fc electron density map at a 1σ level, carved 2Å around the ligand is depicted as a mesh. The small sized fragment (1) does not interact with or induce an opening of the lobeline pocket and maintains the gatekeeper Tyr91 into a g- state stabilized by a hydrogen bond with the Ser144 carbonyl (a). Growing into the lobeline pocket with compound 3 (b) or 4 (c) induces a change in rotameric state of Tyr91 (g- to t conformation) which opens the lobeline pocket. This tyrosine-flip is stabilized by hydrogen bond formation with the side chains of Tyr53 and Ser165 (b, c). The small sphere near the carbonyl oxygen of the ligand in (b) corresponds to a chloride ion. Quaternization of the tropine nitrogen of 3 with a methyl group, producing 6 locks the α-(R)-hydroxyphenetyl moiety in an endo configuration, unable to induce the opening of the lobeline pocket (d).

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
68
Figure 3. Binding site with different orientation. A 60 degree rotation of structures of Ac-AChBP in complex with (a) fragment 1, (b) compound 3, (c) compound 4 and (d) compound 6, with respect to the respective representations in Figure 2. Interesting loop C residues are now displayed as sticks.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
69
Figure 4. Predicted binding mode (light grey colored sticks) versus the experimentally determined binding mode (dark grey colored sticks) of optimized fragment 3 (rmsd = 1.1 Ǻ). The orientation of the α-hydroxyl moiety is different than was predicted by molecular docking. Instead of hydrogen bond formation with the carbonyl backbone of Ser144 (predicted hydrogen bonds are shown in black dashed lines), it is pointing towards a lipophilic part of the binding site; Tyr193.
Site-Directed Mutagenesis Study on Stabilization of the Tyrosine-Flip Visual inspection of the Ac-AChBP co-crystal structures of the optimized fragments 3 and 4 and the structure of the Ac-AChBP-lobeline complex by Hansen and co-workers
17 shows that the side chains of two residues (Tyr53 and Ser165) are likely
to be involved in stabilization of the flipped state of gatekeeper residue Tyr91 by hydrogen bond formation with the phenolic oxygen of Tyr91 (Figure 1b, 2b, 2c, 3b and 3c). Superposition of X-ray structures of Ac-AChBP (PDB: 2BYS)
17 with its
species variant Ls-AChBP (PDB: 1UW6)16
shows that the tyrosine-flip stabilizing residues, Tyr53 and Ser165, correspond to a tryptophan and a tyrosine (Trp53 and Tyr164) respectively in Ls-AChBP (Figure 5). The side chains of Trp53 and Tyr164 in Ls-AChBP are positioned in such a way that these residues are not able to stabilize the tyrosine-flip and hamper opening of the lobeline pocket in Ls-AChBP (Figure 3). To study the importance of Tyr53 and Ser165 in stabilizing the tyrosine-flip in Ac-AChBP, site-directed mutagenesis experiments were performed

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
70
in which these residues were substituted for their Ls-AChBP counterparts, a tryptophan and a tyrosine, respectively. Affinity measurements of Ac-AChBP Y53W and S165Y mutants as well as wild-type Ac- and Ls-AChBPs were performed with nicotine and acetylcholine, which cannot interact with the lobeline pocket because of their small size, and with optimized fragment 3 and lobeline, which have been shown to interact with the lobeline pocket in Ac-AChBP from co-crystal structures. The results summarized in Table 1, indicate that Ser165 is essential for stabilizing the tyrosine-flip. Substitution of Tyr53 with a non-stabilizing tryptophan decreases the affinity of compound 3, 5-fold and generates a small increase in affinity for the three other ligands. The switch from Ser165 to a non-stabilizing tyrosine has a more dramatic effect. The affinity for lobeline and compound 3 decrease by >400- and 25-fold respectively, whereas the affinities of acetylcholine and nicotine which do not interact with the lobeline pocket are hardly affected. Strikingly, this single point mutation in Ac-AChBP (S165Y) renders the affinity of 3 and lobeline very similar to wild type Ls-AChBP. These results provide strong evidence for a less accessible lobeline pocket in Ls-AChBP compared to Ac-AChBP. Thermodynamic Analysis of Fragment Optimization using SPR and ITC The thermodynamic aspects of growing a fragment into the ligand-induced lobeline pocket of Ac-AChBP were studied by surface plasmon resonance (SPR) biosensor analyses, using an assay developed by Geitmann et al.,
29,30 at five different
temperatures (15, 20, 25, 30 and 35 °C) and isothermal titration calorimetry (ITC). SPR at different temperatures allows dissection of binding affinity into the separate enthalpic and entropic contributions using van „t Hoff analysis
31-33 whereas ITC
analysis is directly related to enthalpic contribution and free energy and entropy are derived through titration curves
34. Our results on a large set of ligands show that
these fundamentally different techniques result in very comparable profiles (Figure 6c and 6d, Table 2). A major advantage of using SPR biosensor analysis is that this method requires substantially lower amounts of target protein compared to ITC. The van „t Hoff plots that were derived from the SPR biosensor analysis are depicted in Figure 7 whereas the ITC titration curves are depicted in Figure 8.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
71
Figure 5. AChBP species differences in stabilization of the tyrosine-flip. The superposition of crystal structures of Ac-AChBP (in grey) and Ls-AChBP (in blue) suggests that Trp53 and Tyr164 in Ls-AChBP (represented as blue sticks) cannot stabilize the tyrosine-flip contrary to Tyr53 and Ser165 in Ac-AChBP (represented as grey sticks).
Table 1. Investigation of AChBP species differences using site-directed mutagenesis
Ac-wt pKi ± SEM
a Ac-Y53W
pKi ± SEMa
Ac-S165Y pKi ± SEM
a Ls-wt
pKi ± SEMa
α-lobeline (2) 8.5 ± 0.1 8.7 ± 0.1 5.9 ± 0.1 6.2 ± 0.1 3 7.0 ± 0.1 6.3 ± 0.1 5.6 ± 0.1 6.2 ± 0.1
Acetylcholine 4.0 ± 0.1 4.5 ± 0.1 4.3 ± 0.1 5.2 ± 0.1 nicotine (5) 5.6 ± 0.1 6.3 ± 0.1 5.5 ± 0.1 6.4 ± 0.1

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
72
Figure 6. (a) Chemical structures and binding affinities for Ac-AChBP and Ls-AChBP as determined by [
3H]-epibatidine displacement of the compounds that
were evaluated using SPR biosensor analysis and ITC. Ligand efficiency (LE) of each compound is depicted between brackets in kcal·mol
-1 per heavy atom. (b)
Representative sensorgrams of the compounds binding to Ac-AChBP at different concentrations at 25˚C. Thermodynamic profiles for ligand binding to Ac-AChBP (c) and Ls-AChBP (d) were obtained using SPR biosensor analysis (dark bars ± SEM) and ITC (light bars ± fitting errors) and are represented in bar charts. Shown are the changes that occur upon ligand binding in Gibbs energy (∆G˚) (SPR: dark blue; ITC: light blue), enthalpy (∆H˚) (SPR: dark green; ITC: light green) and entropic contributions (-T∆S˚) (SPR: dark red; ITC: light red). All thermodynamic parameters shown are in kcal·mol
-1 (e) Representative sensorgrams of the four compounds
binding to Ls-AChBP at different concentrations at 25˚C.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
73

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
74

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
75
Figure 7. SPR biosensor analysis at different temperatures allows dissection of binding affinity into the separate thermodynamic contributions (ΔH° and -TΔS°). (a) Chemical structures of the compounds that were evaluated using SPR biosensor analysis. (b) Representative sensorgrams of compounds binding to Ac-AChBP at different concentrations at 25˚C. The inserts show the determination of the KD by steady-state analysis. (c) Representative sensorgrams of compounds binding to Ls-AChBP at different concentrations at 25˚C. The inserts show the determination of the KD by steady-state analysis (d) Representative van „t Hoff plots of compounds for Ls- and Ac-AChBP. (e) Thermodynamic profiles of compounds for binding to Ac- (left) and Ls-AChBP (right). Shown are ∆G° (kcal·mol
-1, left bar), ∆H°
(kcal·mol-1
, bar in the middle) and -T∆S° (kcal·mol-1
, right bar).
Figure 8. ITC titration curves for the binding of ligands to Ac- and Ls-AChBP respectively : (a) and (b) fragment 1, (c) and (d) compound 3, (e) and (f) compound 4, (g) and (h) compound 6, (i) and (j) (S)-nicotine (5), (k) and (l) α-lobeline (2).

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
76
Extending fragment 1 with an α-(R)-hydroxyl substituted phenetyl moiety (leading to 3) results in a significant shift in thermodynamic binding signature where a large increase in favourable enthalpic contribution to the binding is observed for the optimized fragment 3 (Figure 6c and Table 2). An even more enhanced increase in favourable enthalpic contribution is observed when extending the fragment with a phenetyl moiety (leading to 4). These favourable enthalpic contributions are compensated to some extent by an unfavourable entropic contribution to the binding for both optimized ligands. A rationale for the difference in the enthalpic contributions to the binding between the two optimized ligands can be derived from crystal structures of the complexes of the two ligands to Ac-AChBP. These show that the hydroxyl group of 3 displays an unsatisfied hydrogen bond whereas the buried surface area is similar in both cases (~460Ų calculated using the PISA webserver
35), possibly causing the small difference in enthalpy.
Combining the thermodynamic analysis with the identified species differences, it was anticipated that complex formation to Ls-AChBP (non-stabilized tyrosine-flip) of lobeline, and optimized fragments 3 and 4 would be driven by less favourable enthalpy compared to Ac-AChBP (stabilized tyrosine-flip). Thermodynamic analysis using SPR biosensor analysis and ITC, confirmed our hypothesis. Binding of ligands that interact with the lobeline pocket in Ac-AChBP (3, 4 and lobeline) is characterized by more favourable enthalpy when compared to Ls-AChBP (Figure 6c, 6d and Table 2). This is in contrast with ligands that do not interact with the lobeline pocket. Their binding to Ls-AChBP is driven by similar (nicotine) or more (fragment 1) favourable enthalpy compared with binding to Ac-AChBP. In addition, these results show that growing fragment 1 into the ligand-induced lobeline-pocket in Ac-AChBP renders the fragment selective for Ac-AChBP over Ls-AChBP. A plausible explanation for these observations that is in line with the site-directed mutagenesis results, is a differential stabilization of the t-conformation of gatekeeper residue between the AChBP species variants. As such, the lobeline pocket is better accessible in Ac-AChBP compared to Ls-AChBP and can be targeted as a species-selectivity subpocket.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
77
Table 2. Thermodynamic parameters of binding at 25 °Ca
Compound Method pKD ΔG°
(kcalmol-1
)
ΔH°
(kcalmol-
1)
-TΔS°
(kcalmol-
1)
Ac
-AC
hB
P
1 SPR 4.7 ± 0.1 -6.5 ± 0.1 -2.1 ± 0.1 -4.5 ± 0.1
ITC 5.9 ± 0.2 -8.0 ± 0.2 -4.5 ± 0.4 -3.5 ± 0.7
3 SPR 6.5 ± 0.1 -8.8 ± 0.2 -14.2 ± 0.3 5.4 ± 0.5
ITC 7.4 ± 0.1 -10.1 ± 0.1 -12.3 ± 0.1 2.3 ± 0.1
4 SPR 6.9 ± 0.1 -9.3 ± 0.1 -15.0 ± 0.5 5.6 ± 0.6
ITC 7.3 ± 0.2 -9.9 ± 0.2 -16.0 ± 0.3 6.1 ± 0.5
α-lobeline (2)
SPR 7.5 ± 0.1 -10.2 ± 0.1 -8.4 ± 0.8 -1.9 ± 0.8
ITC 8.3 ± 0.3 -11.3 ± 1.0 -13.7 ± 0.3 2.3 ± 1.4
(S)-nicotine (5)
SPR 5.3 ± 0.1 -7.3 ± 0.1 -11.1 ± 1.0 3.8 ± 1.1
ITC 6.1 ± 0.1 -8.3 ± 0.1 -12.5 ± 0.3 4.2 ± 0.2
6 SPR 5.5 ± 0.1 -7.5 ± 0.1 -4.8 ± 1.1 -2.7 ± 1.2
ITC 6.0 ± 0.4 -8.2 ± 0.4 -3.8 ± 0.5 -4 ± 1
1
SPR 6.1 ± 0.1 -8.4 ± 0.1 -7.5 ± 0.5 -0.9 ± 0.4
Ls
-AC
hB
P
ITC 6.7 ± 0.2 -9.1 ± 0.2 -12.3 ± 0.3 3.2 ± 0.5
3 SPR 6.0 ± 0.1 -8.2 ± 0.1 -5.5 ± 1.0 -2.7 ± 1.0
ITC 6.6 ± 0.2 -9.0 ± 0.2 -7.0 ± 0.2 -2.0 ± 0.3
4 SPR 6.8 ± 0.1 -9.3 ± 0.1 -11.1 ± 0.6 1.8 ± 0.5
ITC 7.2 ± 0.2 -9.8 ± 0.3 -10.9 ± 0.3 1.1 ± 0.5
α-lobeline (2)
SPR 6.1 ± 0.1 -8.4 ± 0.1 -5.4 ± 0.5 -3.0 ± 0.5
ITC 6.6 ± 0.1 -9.0 ± 0.2 -7.4 ± 0.2 -1.6 ± 0.5
(S)-nicotine (5)
SPR 6.5 ± 0.1 -8.9 ± 0.1 -11.0 ± 1.2 1.3 ± 1.3
ITC 7.3 ± 0.2 -10.0 ± 0.2 -14.5 ± 0.3 4.5 ± 0.5
6 SPR 7.0 ± 0.1 -9.6 ± 0.1 -8.6 ± 1.1 -1.0 ± 1.1
ITC 7.2 ± 0.3 -9.8 ± 0.4 -11.0 ± 0.3 1.2 ± 0.6
a SPR values are ± SEM over multiple experiments (n = 3-6). ITC values are ± fitting errors.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
78
Ligand-based chemical validation Next to binding mode characterization by considering site-directed mutagenesis and species differences of the protein target, a ligand-based chemical validation of the conclusions was pursued. The X-ray co-crystal structure of the optimized fragment 3 shows that in order to interact with the lobeline pocket the α-(R)-hydroxyphenetyl moiety takes an exo-configuration with respect to the tropine moiety, i.e., the substituent that is inserted into the lobeline pocket is pointing towards the tropine ethylene bridge (Figure 2b). Quaternization of the tropine nitrogen atom of 3 by introduction of an additional methyl substituent, prevents pyramidal inversion and locks the α-(R)-hydroxyphenetyl moiety in the opposite endo-configuration, which is anticipated to prevent this derivative from interacting with the lobeline pocket. Interestingly, pharmacological screening using [
3H]-
epibatine displacement shows that upon quaternization of the basic amine of 3 affording compound 6, the affinity for Ac-AChBP is lowered 10-fold whereas affinity for Ls-AChBP increases 6-fold (Figure 6a). This minor modification, i.e., addition of a single methyl substituent renders compound 3 from 8-fold Aplysia-selective to 8-fold selective for the Lymnaea-AChBP species. The co-crystal structure of the quaternary methylammonium derivative 6 with Ac-AChBP was solved to a resolution of 3.25Å and provides an explanation for the observed change in AChBP species selectivity. Quaternary ammonium derivative 6 was present in the five protomer-protomer interfaces with similar orientations, showing that the quaternized ligand does not interact with the lobeline pocket. In this complex, Tyr91 adopts a g- conformation with a χ2 value of -30°, and is stabilized through a hydrogen bond between the phenolic hydroxyl group and the carbonyl group from the Ser144 backbone (Figure 2d and 3d). Such an orientation of the Tyr91 side chain is indicative of an inaccessible lobeline pocket. Furthermore, in this complex Tyr91 adopts a different rotamer state from the one observed in the Ac-AChBP- complex with fragment 1 (χ2 = -85°) likely to accommodate the methyl group tethered to the amine of VUF11438. The quaternized nitrogen of ligand 6 can make cation-π interactions with Trp145, Tyr91, Tyr186, Tyr193 and Tyr53 whereas a hydrogen bond is made between the hydroxyl group of the ligand and of Tyr53. The benzoate moiety of 6 is oriented differently in the binding site in an intermediate position relative to the benzoate of optimized fragments 3 or 4 and the lobeline 1-phenylethanone moiety, where it interacts via hydrophobic contacts with Ile116, Met114 and the vicinal Cys188 and Cys 189 at the tip of loop C.
In line with these findings is the thermodynamic data that reveals that the favourable enthalpy of binding of compound 3 to Ac-AChBP is dramatically reduced upon quaternization, leading to ligand 6. On the other hand, the entropy of binding becomes more favourable limiting the loss in affinity upon quaternization of 3 to 10-fold when binding to Ac-AChBP (Figure 6a and 6c). In the case of Ls-AChBP and in contrast with Ac-AChBP, quaternization of compound 3 affords a favourable change in enthalpy. As a result, the quaternary ammonium derivative 6 binds with more favourable enthalpy to Ls-AChBP compared to Ac-AChBP. Similar to what is observed with ligands not interacting with the lobeline pocket such as fragment 1, but not with ligands addressing the lobeline pocket such as lobeline (2), 3 and 4. The more favourable enthalpic contribution possibly arises from the differences in the complementary binding sites of Ls- and Ac-AChBP, such as the

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
79
replacement of Tyr53 by a tryptophan or of Ile116 by a methionine. Such variations are likely to significantly alter the interaction interface with the binding site and may be of influence to the binding mode.
Integrating the X-ray data on the Ac-AChBP co-crystal complexes of the optimized fragments 3 and 4 with the thermodynamic analysis and site-directed mutagenesis results, provides evidence that the change in thermodynamic binding signature upon fragment optimization is indicative of interactions with the ligand-induced AChBP subpocket. Ligand 4 binds with similar affinity to Ac-AChBP and Ls-AChBP. The affinity data therefore does not provide any indication for a difference in binding modes. However, the changes in binding enthalpy upon extending fragment 1 with a phenetyl moiety (resulting in 4) are substantially different for Ac-AChBP (ΔΔH° = -12.9 kcal·mol
-1 (SPR), -11.5 kcal·mol
-1 (ITC)) compared to Ls-
AChBP (ΔΔH° -3.6 kcal·mol-1
(SPR), +1.4 kcal·mol-1
(ITC)). The significant favourable change in enthalpy for Ac-AChBP, upon extending fragment VUF10663 with a phenetyl moiety is likely to result from interactions with the ligand-induced AChBP subpocket. The site-directed mutagenesis results strongly indicate that the lobeline pocket is less accessible in Ls-AChBP compared to Ac-AChBP. Therefore, we propose, that due to a distinct binding mode in Ls-AChBP, in which the lobeline pocket is not addressed, no significant favourable change in enthalpy, upon extension of fragment 1 by a phenetyl moiety is observed. In line with this proposal, are the thermodynamic changes upon growing fragment 1 into the quaternary ammonium derivative 4. This chemical modification prevents the ligand from interacting with the lobeline pocket in Ac-AChBP, and yields similar changes in enthalpy for Ac-AChBP (ΔΔH° -2.7 kcal·mol
-1 (SPR), -1.1 kcal·mol
-1 (ITC)) and Ls-
AChBP (ΔΔH° -1.1 kcal·mol-1
(SPR), +1.3 kcal·mol-1
(ITC)) (Figure 9). As such, these results illustrate that more than affinity data alone, thermodynamic analysis and focus on enthalpic and entropic contributions during fragment optimization can provide valuable information on the binding mode of a ligand, thereby better guiding the rational design process. In the current study we have grown a hit fragment into a protein binding pocket that can be induced by triggering conformational changes of the protein. X-ray analysis confirmed the successful design strategy. Thermodynamic binding analysis showed that insertion into the lobeline pocket by extending fragment 1 with a hydrophobic phenetyl moiety resulting in compound 4 affords a considerable favourable change in enthalpy of ~-12 kcal·mol
-1, that is partly compensated by an
entropic penalty . This is in contrast with the classical view on the hydrophobic
effect, in which increased burial of hydrophobic moieties in a hydrophobic pocket results in favourable changes in entropy. Studies on major mouse urinary protein suggest that enthalpy driven hydrophobic association results from poor solvation of the binding site, prior to complex formation.
36-38 The apo-Ac-AChBP X-ray structure
(pdb: 2W8E)18
shows that residue Tyr91 is in the g-conformation and functions as a gatekeeper making the lobeline pocket inaccessible. It is therefore likely that before complex formation, the lobeline pocket is poorly solvated. A significant part of the extensive favourable change in enthalpy may therefore result from strong van der Waals interactions between the phenetyl moiety of compound 4 and the lobeline pocket that are not compensated by the solvent.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
80

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
81
Figure 9. Thermodynamic analysis provides indication of successful insertion into the lobeline pocket. Chemical structures and binding modes as determined by X-ray analysis of Ac-AChBP co-crystal structures and the thermodynamic binding signatures for Ac-AChBP and Ls-AChBP of optimized fragment 4 (a) fragment 1 (b) and quaternary ammonium derivative 6 (c) as determined by SPR biosensor analysis (dark bars ± SEM) and ITC (light bars ± fitting errors). Shown are the changes that occur upon ligand binding in Gibbs energy (∆G˚) (SPR: dark blue; ITC: light blue), enthalpy (∆H˚) (SPR: dark green; ITC: light green) and entropic contributions (-T∆S˚) (SPR: dark red; ITC: light red). All thermodynamic parameters shown are in kcal·mol
-1.
Besides providing insight into the thermodynamic aspects of fragment-growing, our investigations also reveal that growing the fragment 1 into the lobeline pocket renders the fragment selective for Ac-AChBP whereas introduction of an additional methyl substituent preventing interactions with the lobeline pocket reverses the AChBP species selectivity back to Lymnaea. Thus, selectivity for Ac-AChBP over Ls-AChBP can be achieved by addressing the lobeline pocket. The subtle differences in protein conformational changes that induce the lobeline pocket may be of interest in the design of subtype-selective ligands for human nicotinic receptors as well. The gatekeeper tyrosine residue is conserved amongst the human nAChR subtypes whereas the residue (Ser165 in Ac-AChBP) that stabilizes the open lobeline pocket conformation is located in a highly variable region. As can be seen from Figure 6a, lobeline exerts a 250-fold selectivity for Ac-AChBP (accessible lobeline pocket) over Ls-AChBP (inaccessible lobeline pocket) and ~1000-fold selectivity for α4β2 over α7 nAChRs.
39,40 Differential stabilization of the
rotameric states of the gatekeeper tyrosine residue may provide an explanation for the observed nAChR subtype selectivity of lobeline and the lobeline pocket may be targeted as a subtype-selectivity pocket. Conclusion The work described here illustrates that fragment growing can trigger ligand-induced conformational changes of the target protein. The obtained results strongly indicate that the distinct changes in thermodynamic binding signatures upon fragment optimization between Ac-AChBP and Ls-AChBP, result from a difference in binding modes of the optimized fragments. We conclude that more than affinity data alone, dissection of binding affinity into the separate enthalpic and entropic contributions provides valuable information with regards to the binding mode of a ligand. Furthermore, our studies show that thermodynamic analysis enabled by state-of-the-art technologies such as ITC and SPR biosensor analysis, in our hands give comparable results. Altogether, this study illustrates that in combination with detailed structural information (X-ray of co-crystals), thermodynamic data provides crucial insights that enable efficient fragment optimization. All of the crystal structures with fragments described in this paper have been deposited in the Protein Data Bank (PDB codes 2Y54, 2Y56, 2Y57 and 2Y58).

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
82
Acknowledgement. This work was supported by a grant of the Top Institute Pharma D2-103 (to ABS), and received funding from the European Union Seventh Framework Programme under grant agreement n° HEALTH-F2-2007-202088 ("NeuroCypres" project). PR was supported by a long term fellowship from the European Molecular Biology Organization. We thank Eric Karssen for chemical synthesis of compound 3, Frans J.J. de Kanter for performing 2D NMR measurements and Chris Oostenbrink and Chimed Janssen for useful discussions on thermodynamic aspects of ligand-protein interactions. We thank staff from SLS PX1 and ESRF ID23-2 for their assistance during data collection, Robbie Joosten for useful discussion during structure validation steps and Patrick Celie for help with ITC measurements.
Materials and Methods
Chemistry General Chemicals and reagents were obtained from commercial suppliers and were used without further purification. DCM, THF and toluene were distilled under an atmosphere of nitrogen from CaH2 prior to use. Yields given are isolated yields. Microwave reactions were performed with Biotage Initiator microwave system. All melting points are uncorrected and were measured on an Optimelt automated melting point system from Stanford research systems.
1H NMR and
13C NMR
spectra were measured on a Bruker 200, 250 and 500. Analytical HPLC-MS analyses were conducted using Shimadzu LC-20AD liquid chromatography pump system with a Shimadzu SPD-M20A Diode Array detector with the MS detection performed with a Shimadzu LCMS-2010 liquid chromatography mass spectrometer. The buffer is a 0.4% (w/v) NH4CO3 solution in water, adjusted to pH 8.0 with NH4OH. The analyses were performed using the following conditions: an Xbridge (C18) 5 μm column (100 mm × 4.6 mm) with solvent A (90% acetonitrile-10% buffer) and solvent B (90% water-10% buffer), flow rate of 1.0 mL/min, start 5% A, linear gradient to 90% A in 8 min, then linear gradient to 5% A in 0.5 min, then 6.5 min at 5% A, total run time of 15 min. Compound purities under both conditions were calculated as the percentage peak area of the analyzed compound by UV detection at 228 nm.
8-methyl-8-azabicyclo[3.2.1]octan-3α-yl benzoate (7) Benzoyl chloride (19.1 mL, 165 mmol) was added in a dropwise manner to a boiling solution of α-tropine (21,2 g, 150 mmol) and TEA (22.9 mL, 165 mmol) in freshly distilled toluene (200 mL). The mixture was heated at reflux temperature for 4 h. The mixture was washed with a 2 times diluted NaHCO3(sat) solution (3 x 75 mL), with brine (75 mL) and dried with MgSO4. Upon drying, a white precipitate starts to form and the mixture was directly filtered over paper under suction. The residue was extensively washed with DCM. The filtrate was concentrated in vacuo to afford 33.2 g (90%) of a white solid. For biological characterization purposes, a small amount was recrystallized from toluene/acetonitrile to obtain pure material. Mp: 265.6-267.0°C.
1H NMR (500
MHz, MeOD) δ (ppm) 8.06 (d, J = 8.2 Hz, 2H), 7.68 (t, J = 6.9 Hz, 1H), 7.63-7.51 (m, 2H), 5.31 (t, J = 5.0 Hz,1H), 4.01-3.94 (m, 2H), 2.86 (s, 3H), 2.61-2.20 (m, 8H); 13
C NMR (75 MHz, MeOD) δ (ppm) 166.87, 134.65, 121.26, 130.48, 129.88, 66.25,

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
83
63.62, 25.23; HRMS (m/z): [M+H]
+ calcd. for C15H20NO2, 246.1489; found,
246.1496.
8-azabicyclo[3.2.1]octan-3α-yl benzoate hydrochloride (1) 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl benzoate (7) (3.80 g, 16 mmol) was suspended in 40 mL DCE. Upon cooling in an ice/water bath and under an atmosphere of nitrogen, 1-chloroethyl chloroformate (3.5 mL, 32 mmol) was added in a dropwise manner. The mixture was heated to reflux temperature and stirred for 5 h. The mixture was concentrated in vacuo and dissolved in MeOH (40 mL). Stirring at room temperature for 1 h results in formation of a white precipitate which was isolated by filtration over glass under suction. The obtained hydrochloride salt was washed with MeOH and dried in a vacuum stove to afford 2.77 g (67%) of fine white solid. Further concentration of the mother liquor afforded 0.42 g (10%) of a white solid. Total isolated yield 77%. Decomposes at > 280
oC;
1H NMR (500 MHz, MeOD) δ
(ppm) 8.03 (d, J = 8.5 Hz, 2H), 7.65 (t, J = 7.5 Hz, 1H), 7.55-7.50 (m, 2H), 5.33 (t, J = 4.9 Hz, 1H), 4.16-4.01 (m, 2H), 2.56-2.36 (m, 4H), 2.36-2.12 (m, 4H);
13C NMR
(50 MHz, D2O) δ (ppm) 169.03, 135.45, 130.85, 130.77, 130.30, 68.01, 55.31, 34.52, 27.01; HRMS (m/z): [M+H]
+ calcd. for C15H18NO2, 232.1332; found,
232.1343.
8-((R)-2-hydroxy-2-phenylethyl)-8-azabicyclo[3.2.1]octan-3α-yl benzoate (3) Under an atmosphere nitrogen, a solution of (R)-phenyloxirane (230 μL, 2.0 mmol) in NMP (2 mL) was added dropwise to a stirring solution of 8-azabicyclo[3.2.1]octan-3α-yl benzoate (1, free base) (463 mg, 2.0 mmol) in NMP (2 mL). Subsequently, the mixture was heated for 1 h at 150 °C in the microwave. The clear mixture was mixed with water (50 mL) and stirred for 30 min at room temperature. Subsequently, the white suspension that has formed is set aside in the fridge for 3 h. A white precipitate is isolated by filtration over glass, washed with water and dried in the vacuum stove overnight. 548 mg of a white solid was obtained. Recrystallization from water/EtOH afforded 482 mg (69%) of fine white needle-shaped crystals. Mp: 128.0 – 128.7 °C; [α]D = - 48 (c = 1, CHCl3);
1H NMR
(500 MHz, MeOD) δ (ppm) 8.02 (d, J = 8.0 Hz 2H), 7.61 (t, J = 7.5 Hz, 1H), 7.53-7.47 (m, 2H), 7.41-7.37 (m, 2H), 7.36-7.30 (m, 2H), 7.28-7.22 (m, 1H), 5.23 (t, J = 5.2 Hz, 1H), 4.76-4.72 (m, 1H), 3.39-3.27 (m, 2H) 2.69-2.62 (m, 1H), 2.60-2.53 (m, 1H), 2.35-2.19 (m, 2H), 2.18 – 1.97 (m, 4H), 1.88 – 1.75 (m, 2H).
13C NMR (125
MHz, CDCl3) δ (ppm) 167.35, 144.68, 134.23, 131.99, 130.36, 129.69, 129.30, 128.46, 127.18, 73.55, 69.91, 60.98, 59.58, 37.48, 37.39, 27.51, 26.87; HRMS (m/z): [M+H]
+ calcd. for C22H26NO3, 352.1907; found, 352.1916. Chiral HPLC: ee =
98% (Chiralcel OD-H column (250x4.6 mm), eluent: n-hexane (+0.05% diethylamine) : 2-propanol; 95 : 5, 40
oC, 0.7 mlmin
-1).
8-phenethyl-8-azabicyclo[3.2.1]octan-3α-yl benzoate hydrochloride (4) Freshly grinded NaBH(OAc)3 (400 mg, 1.9 mmol) was added to a round bottom flask containing the free base of 8-azabicyclo[3.2.1]octan-3α-yl benzoate (1) (290 mg, 1.3 mmol) and phenylacetaldehyde (170 μL, 90%, 1.3 mmol) dissolved in DCE (5 mL). The mixture was stirred overnight at room temperature under an atmosphere of nitrogen. Saturated NaHCO3 solution (5 mL) was added and the mixture was extracted with DCM (2 x 5 mL). The combined organics were washed with

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
84
NaHCO3(sat) (5 mL) and dried with MgSO4. Concentration in vacuo afforded 331 mg of colorless oil. The crude product was dissolved in Et2O (5 ml) and 5 ml of a 2M HCl solution in Et2O was added dropwise. A white precipitate formed that was isolated by decantation. The residue was washed with Et2O (2 x 10 ml) and recrystallized from abs. EtOH to yield 232 mg (50%) of a white cubic shaped crystals. Decomposes at > 280
oC;
1H NMR (250 MHz, MeOD) δ (ppm) 8.06 (d, J
= 8.0 Hz, 2H), 7.67 (t, J = 7.1 Hz, 1H), 7.60-7.49 (m, 2H), 7.44 – 7.25 (m, 5H), 5.42-5.31 (m, 1H), 4.21-4.04 (m, 2H), 3.43-3.25 (m, 2H), 3.23–3.07 (m, 2H), 2.68–2.17 (m, 8H);
13C NMR (50 MHz, MeOD) δ (ppm) 166.83, 137.71, 134.65, 131.23,
130.48, 130.02, 129.87, 128.34, 66.64, 32.21, 25.44; HRMS (m/z): [M+H]+ calcd.
for C22H26NO2, 336.1958; found, 336.1955.
3α-(benzoyloxy)-8β-((R)-2-hydroxy-2-phenylethyl)-8α-methyl-8-azoniabicyclo-[3.2.1]octane methiodide (6) Under an atmosphere of nitrogen, iodomethane (450 μL, 7.2 mmol) was added dropwise to a solution of 8-((R)-2-hydroxy-2-phenylethyl)-8-azabicyclo[3.2.1]octan-3α-yl benzoate (3) (228 mg, 0.7 mmol) in DCM (2 ml). The mixture was protected from light by wrapping the vial in aluminium foil and set aside at room temperature, overnight. TLC analysis indicated ~70% conversion (DMC : MeOH : TEA = 100 : 10 : 1). A supplement of iodomethane was added (100 μL, 1.6 mmol) and the mixture was heated for 15 min at 100 °C in the microwave. Subsequently, the mixture was concentrated in vacuo to afford 371 mg of a slight orange colored solid. Recrystallization from chloroform results in formation of 91 mg (28%) of fine white needle-shaped crystals. Mp: 215.2 – 216.1 °C; [α]D = - 72 (c = 1, MeOH);
1H NMR (500 MHz, MeOD) δ (ppm) 8.04 (d, J = 8.5
Hz, 2H), 7.65 (t, J = 7.5 Hz, 1H), 7.61 – 7.49 (m, 4H), 7.46-7.39 (m, 2H), 7.36 (t, J = 7.4 Hz, 1H), 5.44 - 5.31 (m, 2H), 4.54-4.46 (m, 1H), 4.06-43.97 (s, 1H), 3.93 – 3.75 (m, 1H), 3.54 -3.45 (m, 1H), 3.37 (s, 3H), 3.16 - 3.07 (m, 1H), 2.80 - 2.71 (m, 1H), 2.63 – 2.45 (m, 4H), 2.29 – 2.05 (m, 2H).
13C NMR (126 MHz, MeOD) δ
166.78, 142.39, 134.61, 131.25, 130.46, 129.84, 129.83, 129.59, 127.43, 71.22, 69.91, 68.85, 64.84, 61.95, 47.06, 33.71, 32.87, 26.44, 25.20; HRMS (m/z): [M]
+
calcd. for C23H28NO3, 366.2064; found, 366.2064.
The epimeric configuration was confirmed by performing 2D NMR. The multiplet at 2.61-2.48 ppm can be ascribed to the tropine ethylene bridge due to the presence of COSY coupling with H(1) and H(5) and absence of COSY coupling to H(3). The identification of the ethylene bridge
1H NMR signals allows determination of the
configuration of the quaternary nitrogen. NOESY coupling of the methyl substituent with the tropine ethylene bridge hydrogens and NOESY coupling of CH2(9) with the axial hydrogens of C(2) and C(4) shows that the α-hydroxyphenetyl substituent is in an endo- whereas the methyl substituent is in an exo-configuration (Scheme 2).

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
85
Scheme 2. NOESY couplings that were observed using 2D 1H NMR measurements on compound 6.
Table 3. Purity, detected masses and retention times of the synthesized compounds that were biologically assessed, determined by analytical HPLC-MS.
No. Retention time
(min) Purity [M+H]
+
1 4.14 98% 231.8
3 6.77 98% 352.0
4 9.82 >99% 336.1
6 5.09 >99% 366.0 [M+]

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
86
Protein expression and purification of wild type andmutant AChBPs Ac-AChBP and Ls-AChBP genes were cloned in the pFastbac vector (Invitrogen, San Diego, CA, USA). Constructs used for SPR and radioligand binding assays coded for C-terminally His-tagged protein whereas constructs used for ITC and crystallization coded for untagged protein. The different constructs were used to generate baculoviruses, following the manufacturer‟s recommendations. These baculoviruses were used to infect SF9 or SF21 cells for protein expression. Ac-AChBP S165Y was constructed using a QuickChange approach (Stratagene, La Jolla, CA, USA) and verified by DNA sequencing. Secreted his-tagged protein was purified from the medium on an ÄKTA purifier, using a HisTrap HP Cartridge (GE Healthcare, Uppsala, Sweden). Protein purity was assayed using SDS-PAGE and protein concentration was determined by Bradford analysis. Untagged protein secreted by the cells was purified from the medium using anion exchange chromatography and size exclusion chromatography. This protein was concentrated to 5 mg/ml in a buffer containing 10mM TRIS pH 8 and 50mM NaCl. Protein aliquots were stored at -80˚C.
Radioligand binding assays Competition binding assays were performed with His-tagged Ls-AChBP, Ac-AChBP, Ac-Y53W or Ac-S165Y-AChBP diluted in PBS-Tris binding buffer (final concentration per well; 1.4 mM KH2PO4, 4.3 mM Na2HPO4, 137.0 mM NaCl, 2.7 mM KCl, 20 mM Trizma-base, 4% DMSO, 0.05% Tween 20. pH 7.4 at 25°C). A constant concentration of [
3H]epibatidine (specific activity ~ 56.3 Ci/mmol Perkin-
Elmer Life Scince, Inc., USA.) was used for Ls-AChBP and Ac-AChBP. The amount of protein and radioligand was chosen to obtain a clear window for the target protein. For Ls-AChBP approximately 1.3 ng protein / well and 1nM [3H]epibatidine (Kd = 0.875). For Ac-AChBP 4.5 ng protein / well and 4 nM
[3H]epibatidine (Kd = 12.43). The AChBPs were incubated with 1.10
-4 to 1.10
-11M of
the ligands (stock concentrations; 10mM in DMSO and further diluted in a 20% DMSO H2O solution) together with 0.2mg PVT Copper His-tag SPA beads (GE Healthcare) per well. Final well volume was 100 µL and incubated for 1h at room temperature under continuous shaking followed by 3 hours storage in the absence of light before counting. The label-bead complex was counted in the Microbeta Trilux (PerkinElmer, USA). All radioligand binding data were evaluated by a non-
linear, least squares curve fitting procedure using Graphpad Prism (version 4.01, GraphPad Software, Inc., San Diego, CA). All data are represented as mean ± SEM from at least three independent experiments.
Crystallization Untagged Ac-AChBP was incubated with 1 mM ligand on ice for 1 hour before crystallization trials were setup. Co-crystals grew in 0.6-1.2 M ammonium sulfate, 0.1 M MMT buffer at pH ranging from 6 to 8. The crystals were cryoprotected by soaking in a mother liquor supplemented by 25% glycerol and 1 mM ligand, and then flash frozen in liquid nitrogen.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
87
Structure and solution refinement Data collection was carried out on beamline PX1 at the SLS synchrotron (Switzerland) or on beamline ID23-2 at the ESRF (France). Wavelengths used for datacollection are reported in the table below. The different co-crystals were from space group I23 and diffracted to a resolutions ranging from 3.25 to 3.65 Å. Data was processed using XDS and scaled using XSCALE
42. The structures of the
different complexes were solved with Phaser in molecular replacement trials using 2BR7
15 as the search model. All the different Ac-AChBP-ligand complex structures
displayed one pentamer in the asymmetric unit, with all five protomer-protomer interfaces occupied by a ligand molecule. Initial electron density maps were already of very good quality despite the low resolution of the data recorded. Non-crystallographic symmetry restraints were maintained on most of the structure during refinement except for deviating loop regions. Iterative cycles of structure refinement were performed using either REFMAC from the CCP4 suite
43 or
Phenix44
. Refinement cycles were interspersed with manual building using COOT
45. Final refinement cycles were performed with BUSTER
46, using Local
Structure Similarity (LSSR) Restraints. Ligands were introduced only at the end of refinement and were clearly defined in the electron density. The refined structures were validated using the Molprobity server
47. The data collection, refinement and
Ramachandran statistics are reported in the tables below.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
88
Table 4. Data collection and refinement statistics
AcAChBP- fragment 1
AcAChBP- compound 3
AcAChBP- compound 4
AcAChBP- compound 6
Data collection Space group I23 I23 I23 I23 Cell dimensions a, b, c (Å) 216.42 218.93 210.74 213.33
α, β, () 90, 90, 90 90, 90, 90 90, 90, 90 90, 90, 90
Resolution (Å) 16.33-3.65 (3.75-3.65)
16.05-3.59 (3.68-3.59)
14.74-3.30 (3.38-3.30)
14.53-3.25 (3.33-3.25)
Rmerge (%) 17.6(65.5) 13.7(59.5) 16.9(84.8) 17.9(74.3)
I / I 10.19(2.78) 12.31(2.87) 11.60(1.81) 10.81(2.05)
Completeness (%) 100(100) 99.8(99.7) 99.4(95.3) 99.6( 97.1) Redundancy 5.0(4.9) 5.4(5.0) 5.1(5.0) 4.8(4.4) Refinement Resolution (Å) 48.39-3.65 48.95-3.59 49.67-3.30 47.70-3.25 No. reflections 18858 20507 23584 25505 Rwork / Rfree 0.20/0.22 0.17/0.20 0.17/0.20 0.18/0.20 No. atoms Protein 8180 8180 8180 8180 Ligand/ion 95 292 217 151 Water 0 0 0 0 B-factors Protein 97 98 95 71.114 Ligand/ion 84.022 106 74 76.146 Water - - - - R.m.s. deviations Bond lengths (Å) 0.008 0.009 0.009 0.008
Bond angles () 0.88 0.99 1.06 1.03
*Data was recorded from a single crystal for each structure. *Values in parentheses are for highest-resolution shell.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
89
Table 5. Ramachandran statistics
1 3 4 6
Wavelength (Å) 1.282 1.282 0.9184 0.8726
Ramachandran
Favoured (%) 98.5 98.3 97.3 99.6
SPR biosensor assay SPR interaction experiments were performed at various temperatures on a Biacore T100 (GE Healthcare, Uppsala, Sweden). Ls- and Ac-AChBP were diluted to 0.03-0.1 mg/ml in 50 mM NaAc (pH 5.5) and immobilized to a CM5 sensor chip (research grade, GE Healthcare, Uppsala, Sweden) by amine coupling according to the manufacturer recommendations as described before.
30 A closed dextran
surface was used as a reference surface. A phosphate buffer (10 mM phosphate, pH 7.4, 137 mM NaCl, 3 mM KCl, with addition of 5% DMSO and 0.005% v/v surfactant P20 (GE Healthcare, Uppsala, Sweden) was used as a running buffer at a flow rate of 90 μL/min. All compounds were prepared as 10 mM stock solutions in pure DMSO and diluted in the running buffer. Typically, samples were injected in concentration series for 60 s and the dissociation was recorded for 300 s. Signals were corrected for nonspecific binding to the surface by subtracting signals of the reference surface from those of the AChBP surfaces (reference subtraction). In addition, corrections for minor differences between AChBP and reference surface interactions with DMSO were performed by using a series of solvents standards (solvent correction). Moreover, signals were corrected for background by subtracting signals from a blank injection from those of compound injections (blank subtraction). The affinity was determined by fitting a 1:1 model to steady state binding signals at different concentrations.
Van ‘t Hoff analysis Equilibrium dissociation constants determined at 15, 20, 25, 30 and 35 °C were used to obtain thermodynamic parameters by plotting ln(KD) versus 1/T using Biacore T100 evaluation software version 2.0.1. Due to a lack of curvature all van „t Hoff plots were plotted linear. ∆H°, ∆S° and ∆G° values were obtained using the nonintegrated form of the van „t Hoff equation ln(KD)=∆H˚/RT - ∆S˚/R. Linear plots were based on dissociation constants of at least 4 temperatures and were rejected

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
90
when the linear regression had a value of R
2 < 0.85. All thermodynamic parameters
of binding are represented as mean ± SEM from at least three independent van „t Hoff plots.
Isothermal titration calorimetry Isothermal titration calorimetry experiments were performed on a VP-ITC Microcalorimeter (Microcal) at 25°C. Untagged Ls- and Ac- AChBPs used in these experiments were dialyzed against a buffer containing 20 mM Tris-Cl pH 8, 150 mM NaCl, 1% DMSO. The different ligands investigated were dissolved in the same buffer. For a typical experiment, 100 µM of ligand was titrated into 10 µM of AChBP monomer. Titration experiments of the ligands into buffer alone were also performed to determine the change in enthalpy caused by the dilution of the ligands. This background was subtracted from the AChBP-ligand binding experiments. Corrected data were analyzed using the software supplied by the manufacturer and were fitted using the non-linear least squares method, with a single binding site model. To control whether differences could arise from the use of differently tagged AChBPs in the ITC and SPR experiments, ITC data was also collected for the binding of compound 3 to His-tagged Ac-AChBP. This data was consistent with results obtained using untagged Ac-AChBP in a similar assay.
References
1. de Kloe, G.E., Bailey, D., Leurs, R. & de Esch, I.J. Transforming fragments into candidates: small becomes big in medicinal chemistry. Drug Discov Today 14, 630-46 (2009).
2. Erlanson, D.A. Fragment-based lead discovery: a chemical update. Curr Opin Biotechnol 17, 643-52 (2006).
3. Hajduk, P.J. & Greer, J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov 6, 211-9 (2007).
4. Congreve, M., Carr, R., Murray, C. & Jhoti, H. A 'rule of three' for fragment-based lead discovery? Drug Discov Today 8, 876-7 (2003).
5. Schuffenhauer, A. et al. Library design for fragment based screening. Curr Top Med Chem 5, 751-62 (2005).
6. Chung, S., Parker, J.B., Bianchet, M., Amzel, L.M. & Stivers, J.T. Impact of linker strain and flexibility in the design of a fragment-based inhibitor. Nat Chem Biol 5, 407-13 (2009).
7. Howard, N. et al. Application of fragment screening and fragment linking to the discovery of novel thrombin inhibitors. J Med Chem 49, 1346-55 (2006).
8. Congreve, M., Chessari, G., Tisi, D. & Woodhead, A.J. Recent developments in fragment-based drug discovery. J Med Chem 51, 3661-80 (2008).
9. Sledz, P. et al. Optimization of the interligand overhauser effect for fragment linking: application to inhibitor discovery against Mycobacterium tuberculosis pantothenate synthetase. J Am Chem Soc 132, 4544-5 (2010).
10. Wang, Y.S. et al. Application of fragment-based NMR screening, X-ray crystallography, structure-based design, and focused chemical library

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
91
design to identify novel microM leads for the development of nM BACE-1 (beta-site APP cleaving enzyme 1) inhibitors. J Med Chem 53, 942-50 (2010).
11. Zhu, Z. et al. Discovery of cyclic acylguanidines as highly potent and selective beta-site amyloid cleaving enzyme (BACE) inhibitors: Part I--inhibitor design and validation. J Med Chem 53, 951-65 (2010).
12. Rucktooa, P., Smit, A.B. & Sixma, T.K. Insight in nAChR subtype selectivity from AChBP crystal structures. Biochem Pharmacol 78, 777-87 (2009).
13. Sixma, T.K. & Smit, A.B. Acetylcholine binding protein (AChBP): a secreted glial protein that provides a high-resolution model for the extracellular domain of pentameric ligand-gated ion channels. Annu Rev Biophys Biomol Struct 32, 311-34 (2003).
14. Brejc, K. et al. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411, 269-76 (2001).
15. Celie, P.H. et al. Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an alpha-conotoxin PnIA variant. Nat Struct Mol Biol 12, 582-8 (2005).
16. Celie, P.H. et al. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41, 907-14 (2004).
17. Hansen, S.B. et al. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. Embo J 24, 3635-46 (2005).
18. Ulens, C. et al. Use of acetylcholine binding protein in the search for novel alpha7 nicotinic receptor ligands. In silico docking, pharmacological screening, and X-ray analysis. J Med Chem 52, 2372-83 (2009).
19. Celie, P.H. et al. Crystal structure of acetylcholine-binding protein from Bulinus truncatus reveals the conserved structural scaffold and sites of variation in nicotinic acetylcholine receptors. J Biol Chem 280, 26457-66 (2005).
20. Hopkins, A.L., Groom, C.R. & Alex, A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today 9, 430-1 (2004).
21. Dutertre, S. et al. AChBP-targeted alpha-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. Embo J 26, 3858-67 (2007).
22. Lovell, S.C., Word, J.M., Richardson, J.S. & Richardson, D.C. The penultimate rotamer library. Proteins 40, 389-408 (2000).
23. Kallblad, P. & Dean, P.M. Efficient conformational sampling of local side-chain flexibility. J Mol Biol 326, 1651-65 (2003).
24. Maksay, G., Nemes, P. & Biro, T. Synthesis of tropeines and allosteric modulation of ionotropic glycine receptors. J Med Chem 47, 6384-91 (2004).
25. Howarth, N.M., Malpass, J.R. & Smith, C.R. Manipulation of substituents at nitrogen in tropanes, homotropanes, and dehydro- derivatives. Tetrahedron 54, 10899-10914 (1998).

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
92
26. Couturier, M. & Tucker, J.L. Efficient synthesis of the k-opioid rceptor
agonist CJ-15,161: four stereospecific inversions at a single aziridinium stereogenic center Tetrahedron Asymmetry 14, 3517-3523 (2003).
27. Abdel-Magid, A.F., Carson, K.G., Harris, B.D., Maryanoff, C.A. & Shah, R.D. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures(1). J Org Chem 61, 3849-3862 (1996).
28. Verdonk, M.L., Cole, J.C., Hartshorn, M.J., Murray, C.W. & Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 52, 609-23 (2003).
29. Geitmann, M. et al. Interaction kinetic and structural dynamic analysis of ligand binding to acetylcholine-binding protein. Biochemistry 49, 8143-54.
30. Retra, K. et al. Development of surface plasmon resonance biosensor assays for primary and secondary screening of acetylcholine binding protein ligands. Anal Biochem 407, 58-64.
31. Roos, H., Karlsson, R., Nilshans, H. & Persson, A. Thermodynamic analysis of protein interactions with biosensor technology. J Mol Recognit 11, 204-10 (1998).
32. Shuman, C.F., Hamalainen, M.D. & Danielson, U.H. Kinetic and thermodynamic characterization of HIV-1 protease inhibitors. J Mol Recognit 17, 106-19 (2004).
33. Zhukov, A. & Karlsson, R. Statistical aspects of van't Hoff analysis: a simulation study. J Mol Recognit 20, 379-85 (2007).
34. O‟Brien, R., Ladbury, J.E. & B.Z., C. Isothermal titration calorimetry of biomolecules. Protein-Ligand Interactions, Oxford University Press, 263-284 (2000).
35. Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774-97 (2007).
36. Barratt, E. et al. Van der Waals interactions dominate ligand-protein association in a protein binding site occluded from solvent water. J Am Chem Soc 127, 11827-34 (2005).
37. Bingham, R.J. et al. Thermodynamics of binding of 2-methoxy-3-isopropylpyrazine and 2-methoxy-3-isobutylpyrazine to the major urinary protein. J Am Chem Soc 126, 1675-81 (2004).
38. Sharrow, S.D., Novotny, M.V. & Stone, M.J. Thermodynamic analysis of binding between mouse major urinary protein-I and the pheromone 2-sec-butyl-4,5-dihydrothiazole. Biochemistry 42, 6302-9 (2003).
39. Miller, D.K. et al. Lobeline analogs with enhanced affinity and selectivity for plasmalemma and vesicular monoamine transporters. J Pharmacol Exp Ther 310, 1035-45 (2004).
40. Zheng, G., Dwoskin, L.P., Deaciuc, A.G., Norrholm, S.D. & Crooks, P.A. Defunctionalized lobeline analogs: structure-activity of novel ligands for the vesicular monoamine transporter. J Med Chem 48, 5551-60 (2005).
41. Wallace, A.C., Laskowski, R.A. & Thornton, J.M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 8, 127-34 (1995).
42. Kabsch, W. Xds. Acta Crystallogr D Biol Crystallogr 66, 125-32.

Chapter 4 Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis
93
43. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D
Biol Crystallogr 50, 760-3 (1994). 44. Adams, P.D. et al. PHENIX: a comprehensive Python-based system for
macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213-21.
45. Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126-32 (2004).
46. Blanc, E. et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr 60, 2210-21 (2004).
47. Davis, I.W., Murray, L.W., Richardson, J.S. & Richardson, D.C. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 32, W615-9 (2004).

94

95
Chapter 5
Structure-based design, synthesis and structure-activity
relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
Ewald Edink#, Atilla Akdemir
#, Chimed Jansen, René van Elk,
Obbe Zuiderveld, Frans J.J. de Kanter, Jacqueline E. van Muijlwijk-Koezen, August B. Smit, Rob Leurs and Iwan J.P. de Esch
# These authors contributed equally.
Abstract Using structure-based optimization procedures on previously identified in silico hits, two novel classes of compounds, i.e., dibenzosuberyl- and benzoate substituted tropines were designed and synthesized as ligands for acetylcholine-binding protein (AChBP). This protein is a homolog to the ligand binding domain of the nicotinic acetylcholine receptor (nAChR). The hit optimization was monitored and guided by ligand and group efficiency (LE and GE) considerations as well as structural information. Distinct SAR of the novel compound series is observed between two AChBP species variants and between the α7 and α4β2 nAChR subtype. The AChBP species differences are indicative of a difference in accessibility of a ligand-inducible subpocket between AChBPs from different species. Hereby, we have identified a region within the ligand binding site that can be scrutinized to achieve selectivity for nicotinic receptor subtypes.

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
96
Introduction The neuronal nicotinic acetylcholine receptors (nAChRs) belong to the Cys-loop receptor family of the ligand-gated ion channels (LGICs). The Cys-loop receptors are characterized by a pentameric assembly of subunits. Until now, 12 human
neuronal nAChR subunits (2-10 and 2-4) have been identified. These
subunits combine to form either homopentamers (e.g., 7) or heteropentamers
(e.g., 42), resulting in different pharmacological characteristics.1-3
The most
abundant nAChR subtypes in the central nervous system (CNS) are the 42 and
the 7 receptors.2 The 42 receptor is characterized by high affinity for nicotine,
high permeability for Na+, K
+, and low permeability for Ca
2+ ions. In contrast, the 7
receptor has lower affinity for nicotine, but high affinity for the snake toxin -
bungarotoxin and the alkaloid methyllycaconitine (MLA).2 In addition, the 7
receptor is permeable for Ca2+
and Na+ ions.
1, 2
Several important physiological and mental processes are regulated by nicotinic receptors and they are therefore potential therapeutic targets for a wide variety of
neurodegenerative and psychiatric disorders. The human 42 and 7 receptors play a role in the pathophysiology of Alzheimer‟s disease, Parkinson‟s disease,
schizophrenia, epilepsy and anxiety.1-3
In addition, the human 42 nAChR is
involved in nicotine addiction and pain.2 Furthermore, the human 7 may also be of
value as a pharmacological target in inflammation.4, 5
Several nicotinic receptor ligands are being investigated for clinical use. The first clinical breakthrough was
reported in 2006, when Varenicline, a partial agonist on the 42 nAChR, was approved as a drug for smoking cessation.
6, 7
Due to high sequence identity of the binding pockets of the different subtypes and lack of detailed structural information on nAChRs, the development of selective ligands remains a challenge. Furthermore, the lack of high resolution crystal structures of eukaryotic LGICs hampers a structure-based design approach to tackle this challenge. Fortunately, the water-soluble acetylcholine binding protein (AChBP) from the fresh water snail Lymnaea stagnalis (Ls-AChBP) has been characterized and crystallized. This protein is a widely accepted structural homolog of the extracellular domain (ECD) of nicotinic receptors
8, 9 The binding pocket of
this homopentameric protein is situated at the interface of two adjacent subunits and is formed by the principal and complementary side, (Figure 1A-D).
8 The
principal side is formed by mostly aromatic residues of loops A (Tyr89, Ls-AChBP amino acid numbering), B (Trp143) and C (Tyr185 and Tyr192). In addition, the flexible loop C has two vicinal cysteines (Cys187-188) that form a disulfide bond. The complementary side is formed by loops D, E and F and only loop D donates an aromatic residue (Trp53) (Figure 1C-E). Co-crystal structures of Ls-AChBP in complex with nicotine and carbamylcholine have revealed important interactions between these nicotinic receptor ligands and Ls-AChBP.
10 This includes the cation-
interactions between a cationic center in nicotinic receptor ligands and Trp143. In addition, nicotine forms a hydrogen bond between its pyrolidine nitrogen atom and the carbonyl backbone of Trp143 (Figure 1F).

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
97
Figure 1: Cartoon and surface representation of 2.2 Å X-ray structure of AChBP from Lymnaea stagnalis in complex with nicotine (PDB: 1UW6) shown in side view (A) and top view (B). The binding site is located at the interface of two subunits (indicated by a black circle) and consists of a principal (C) and a complementary side (D). The AChBP homopentamer has five orthosteric and identical ligand binding sites. (E) A schematic representation showing the loops A-F that shape the orthosteric binding site. (F) Close up on the binding pose of nicotine bound to Ls-AChBP.

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
98
After the identification of Ls-AChBP, similar binding proteins have been identified in
other molluscan species, e.g., Aplysia californica (Ac-AChBP) and Bulinus truncatus AChBP (Bt-AChBP) and these proteins have been co-crystallized in the presence of nicotinic receptor ligands or buffer molecules.
11, 12 The crystal
structures of the three different AChBPs show a conserved architectural fold that has been recognized as a template to understand the ligand binding domains of nicotinic receptors and other mammalian Cys-loop receptors. Crucial information on ligand-receptor interactions has been obtained from agonist-bound structures of
AChBP, i.e., carbamylcholine, nicotine and epibatidine.10, 12
Similar cation- interactions between a conserved tryptophan (loop B) and cationic centers of several nonpeptidic ligands are observed in these co-crystal structures. Experimental evidence has been obtained that identical interactions are present in neuronal nicotinic receptors, illustrating the use of AChBP in nAChR research.
13
Recently, we have described a study in which a benzoate substituted tropine-containing fragment (18, Scheme 3) and several derivatives that interact with a ligand-inducible subpocket of the binding site of AChBP were thoroughly characterized using thermodynamic and structural analysis.
14 In another recent
study, we have reported an in silico screening protocol that resulted in the identification of ligands with affinity for AChBP and the α7 but no affinity for the α4β2 nAChR.
15 Two of the identified hits contained a dibenzosuberyl-substituted
tropine scaffold (1 and 2, Table 1) and were co-crystallized with Ac-AChBP. In the current study, we describe the structure-based design and synthesis of dibenzosuberyl- and benzoate substituted tropine derivatives and their SAR on Ac- and Ls-AChBP and on α7 and α4β2 nicotinic receptors. The hit optimization was monitored and guided by ligand efficiency and group efficiency considerations (LE and GE) as well as structural information.
Results and Discussion
Design
The previously obtained co-crystal structures of Ac-AChBP in complex with hit compounds 1 and 2 (PDB: 2W8F and 2W8G, respectively) show that the ligands
have a comparable binding pose with their cationic nitrogen atoms forming cation- interactions with Trp145 (Ac-AChBP amino acid numbering, Trp143 in Ls-AChBP).
15 Structural comparison with previously obtained co-crystal complexes
exemplifies the conformational flexibility of the AChBP binding site, in particular with respect to Loop C. The bulky dibenzosuberyl group of the in silico hits pushes loop C away from the binding pocket into an intermediate open conformation. Comparing the structures of Ac-AChBP in complex with 1 ( Figure 2A) and lobeline (3, PDB: 2BYS, Figure 2B)
12, reveals the opening of the lobeline pocket by the
rotameric change of Tyr91 (Tyr93 in 2BYS corresponds to Tyr91 in 2W8F), see Figure 2C. In addition, the superposition of both X-ray structures shows that the cationic nitrogen atoms of compound 1 and lobeline are at a similar position and
engaged in cation- interactions with Trp145 (Figure 2B). The structural overlay

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
99
inspired us to pursue a fragment merging approach
16 with the aim of increasing
the binding affinity of in silico hit 1 (Figures 3A-C). It was anticipated that the
merging of lobeline‟s -hydroxyphenetyl moiety with the dibenzosuberyl substituted tropine part of 1 would afford a hybrid ligand (e.g., 5) with improved binding affinity due to additional interactions with the lobeline pocket. We note that the diastereomeric configuration of lobeline, which has been fitted in the electron density of the obtained co-crystal structure by Hansen and co-workers, is exactly opposite to the naturally occurring enantiomer (-)-lobeline.
12 Since lobeline was
obtained from Tocris Biosciences (supplying the (-)-enantiomer) it is questionable whether this proposed binding pose is correct. Our docking studies with (-)-lobeline show that a similar binding pose as the published pose of (+)-lobeline, and retaining the key ligand protein interactions, could be obtained (rmsd: 0.9 Å, over all ligand atoms). Therefore, we assumed that this discrepancy did not influence our structure-based hit-optimization procedures.
Figure 2. A) Binding mode of compound 1 (shown in grey balls and sticks) in Ac-AChBP. B) Superposition of Ac-AChBP in complex with 1 (shown in grey balls and sticks) and lobeline (3, black balls and sticks) shows that fragment merging may afford a novel chemotype capable of addressing the lobeline pocket. C) Lobeline addresses the lobeline pocket after a change in rotameric state of Tyr91 (Y-flip, bent arrow).
To further optimize the interactions of the -hydroxyphenetyl moiety of hybrid compound 5 with the lobeline pocket, we designed analogs of 5 (4-7, Figure 3A, Table 1). These analogs were subsequently docked into the crystal structure of 1 (PDB: 2W8F) after opening of the lobeline pocket by manually changing the rotameric state of Tyr91 from the g- to t conformation)
17. The obtained binding
poses indicate that the lobeline pocket can indeed be addressed by the -hydroxyphenetyl moiety (Figure 3B). Further investigation of the docking results suggests that additional interactions with the binding site can be established by incorporation of hydroxyl moieties at the meta positions (one or both) of the phenyl ring. By doing so, it was anticipated that hydrogen bonds could be formed with the backbone carbonyl oxygen of Thr89 and the side chain of Asp195 (Figure 3C). Our in silico screening study has provided an indication that compounds 1 and 2 bind to both the orthosteric site and the ion pore of nAChRs.
15 The interaction with the ion

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
100

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
101
Figure 3. A) Adding the -hydroxyphenetyl moiety of -lobeline to the tropine nitrogen atom of the previously obtained hit compound 1 is suggested to result in a new class of hybrid compounds (4-6, Table 1) that can address the lobeline pocket. B) Docking studies suggest that compound 4 (ball and sticks) can form hydrogen bonds with the backbone carbonyls of Trp147 and Ser146, and can insert its phenyl moiety into the lobeline pocket. C) Close-up of the lobeline pocket shows possibilities for hydrogen bonding (dashed lines) with the backbone carbonyl of Thr89 and the side chain of Asp195 by introduction of hydroxyl moieties at the 3 and 5 position of the phenyl moiety.
pore may arise from the structural resemblance to tricyclic antidepressants that have been shown to block Na
+ channels.
18 In order to abolish the putative channel
blockade of the tricyclic ligands, we performed an additional fragment-merging exercise affording new ligands, in which the dibenzosuberyl moiety was replaced by a smaller benzoate group (Table 2). Thus, a set of benzoate substituted analogs was synthesized in order to determine the influence on the binding affinity of 1) the configuration of the epimeric C(3) position of the tropine spacer, 2) the quaternization of the tropine nitrogen atom, and 3) the introduction of hydroxyl substituted phenetyl moieties.
Chemistry
The dibenzosuberyl substituted tropinyl ethers were synthesized according to Scheme 1. Treatment of tropine with 5-chlorodibenzosuberane followed by oxidative demethylation with KMnO4 afforded dibenzosuberyl ether 9.
19, 20 The
corresponding phenetylamine 4 was obtained by reductive amination of 9 with phenylacetaldehyde.
21 Methylation using iodomethane afforded the quaternary
ammonium salt 10 with the phenetyl moiety in an endo-configuration as determined by 2D NMR spectroscopy at 393 K. The mono and di-hydroxyl substituted phenetylamines 5 and 6 were obtained via TBTU-mediated coupling with the corresponding racemic mandelic acids, followed by LAH reduction. β-tropine (13) was prepared via Meerwein-Ponndorf-Verley reduction of tropinone using Al(i-OPr)3.
22 The benzoate esters of α- and β-tropine (7, 8) were obtained by acylation
with benzoyl chloride.23
Subsequent demethylation using α-chloroethylchloro-formate afforded the corresponding nortropinyl esters (16, 17).
24 Similar as in the
tricyclic series, phenetylamines 20 and 21 were obtained by reductive amination with phenylacetaldehyde. Treatment of endo-epimer 20 with iodomethane resulted in formation of the endo-phenetyl substituted quaternary ammonium derivative 22. The epimeric configuration was confirmed by performing 2D NMR (supplementary data). The mono and di-hydroxyl substituted phenetylamines 24-26 were synthesized via TBTU-mediated coupling with the corresponding phenylacetic
acids, followed by BH3 reduction. Reaction of -hydroxyl substituted phenetylamine 24 with iodomethane afforded similar as before the endo-phenetyl substituted quaternary ammonium derivative 23 as confirmed by 2D NMR (supplementary data). The tri-hydroxyl substituted endo- and exo-benzoate substituted N-phenetyltropines (29, 30) were obtained through alkylation of nortropinyl esters 18 and 19 with 2-bromo-1-(3,5-dihydroxyphenyl)ethanone (14) followed by BH3 reduction of the obtained ketone to the corresponding alcohol. 2-Bromo-1-(3,5-

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
102
dihydroxyphenyl)ethanone (14) was prepared from the corresponding acetophenone using CuBr.
25
Scheme 1. Reagents and conditions: (a) toluene, rt, 16h, reflux, 1h; (b) KMnO4, KOH, pyridine/water, rt, 3h; (c) NaBH(OAc)3, DCE, rt, 16h; (d) MeI, toluene, rt, 24h; (e) TBTU, Et3N, DMF, rt, 1h; (f) LiAlH4, THF, rt, 2h.
Scheme 2. Reagents and conditions: a) Al(i-OPr)3, 2-propanol, reflux, 24h; b) CuBr2, EtOAc/DCM, reflux, 6.5h.

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
103
Scheme 3. Reagents and conditions: a) TEA, toluene, reflux, 4-6 h; b) 1-chloroethyl chloroformate, DCE, reflux, 5-19 h; c) MeOH, reflux/rt, 1-4 h d) NaBH(OAc)3, DCE, rt, 16-48 h; e) MeI, acetonitrile, rt, 16h; f) TBTU, Et3N, DMF, rt, 3-16 h; g) BH3.THF, THF, rt, 16 h; h) DIPEA, acetonitrile, rt, 2-16h; i) BH3.THF, THF, 0 °C, 0.75-1.5 h; j) MeI, toluene, rt, 2 weeks; k) NMP, microwave 150
oC, 50
min.
Pharmacology
The binding affinity data of the dibenzosuberyl-substituted compounds shows that removing methyl groups from the quaternary tropine nitrogen atoms reduces binding affinity for Ls-AChBP, see Table 1 (compare 1, 8 and 9). Extension of the methylene spacer of benzyl derivative 2 with one methylene unit affording the phenetyl substituted compound 4 resulted in a 0.4 log unit increase in affinity. Subsequent quaternization of the tropine nitrogen atom of 4 gave rise to an additional 0.4 log unit increase in binding affinity. In total, the affinity of the in silico identified hit 1 was increased 6-fold (10, pKi = 7.4). Introduction of hydroxyl groups
at the - and meta positions of the phenetyl group of compound 4 diminished the
affinity. It should be noted that both -hydroxyl substituted derivatives (5 and 6) were tested as racemic mixtures. Taken together, these results illustrate that for Ls-AChBP affinity, a quaternary tropine nitrogen atom is preferred and a lipophilic

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
104
Table 1. The binding affinity (pKi) dibenzosuberyl substituted tropines.
Cpd R1 R2
Ls-AChBP
pKi ± SEMa
Ac-AChBP
pKi ± SEMa
α7
pKi ±
SEMb
α4β2
pKi ± SEMa
Nicotine 6.5 ± 0.1 5.6 ± 0.1 6.0 ± 0.1 7.9 ± 0.1
α-lobeline 6.2 ± 0.1 8.6 ± 0.1 5.1 ± 0.1 8.3 ± 0.1
9 H H 6.0 ± 0.1 5.0 ± 0.1 n.d. n.d.
8 Me H 5.7 ± 0.1 5.2 ± 0.1 n.d. n.d.
1 Me Me 6.5 ± 0.1 6.0 ± 0.1 6.0 ± 0.1 < 4.5
2
H 6.6 ± 0.1 5.0 ± 0.1 n.d. n.d.
4
H 7.0 ± 0.1 5.0 ± 0.1 4.9 ± 0.1 < 4.5
10
Me 7.4 ± 0.1 6.2 ± 0.1 5.8 ± 0.1 < 4.5
5
H 5.7 ± 0.1 4.9 ± 0.1 4.7 ± 0.1 < 4.5
6
H 5.7 ± 0.1 4.5 ± 0.1 4.4 ± 0.1 < 4.5
a) [3H]epibatidine displacement studies, pH 7.4; b) [
3H]MLA displacement studies, pH 7.4; n.d. = not
determined; Cpd = compound

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
105
Table 2. The binding affinity (pKi) of benzoate substituted tropines
Cpd R1 R2 Ls-AChBP
pKi ± SEMa
Ac-AChBP
pKi ± SEMa
α7
pKi ± SEMb
α4β2
pKi ± SEMa
1814
endo H H 6.1 ± 0.1 5.3 ± 0.1 5.5 ± 0.1 < 4.5
19 exo H H 5.1 ± 0.1 4.8 ± 0.1 n.d. n.d.
16 endo Me H 5.7 ± 0.1 5.4 ± 0.1 4.8 ± 0.1 < 4.5
17 exo Me H 5.2 ± 0.1 4.8 ± 0.1 n.d. n.d.
2014
endo
H 7.1 ± 0.1 7.5 ± 0.1 5.1 ± 0.1 < 4.5
21 exo
H 6.4 ± 0.1 6.0 ± 0.1 5.3 ± 0.1 < 4.5
22 endo
Me 7.5 ± 0.1 6.8 ± 0.1 5.2 ± 0.1 < 4.5
24 endo
H 6.2 ± 0.1 6.8 ± 0.1 4.9 ± 0.1 < 4.5
31 endo
H 5.9 ± 0.1 6.1 ± 0.1 n.d. n.d.
3214
endo
H 6.1 ± 0.1 7.0 ± 0.1 n.d. n.d.
23 endo
Me 6.9 ± 0.1 5.7 ± 0.1 5.0 ± 0.1 < 4.5
3314
endo
Me 6.9 ± 0.1 6.1 ± 0.1 n.d. n.d.

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
106
Table 2, continued. The binding affinity (pKi) of benzoate substituted tropines
Cpd R1 R2 Ls-AChBP
pKi ± SEMa
Ac-AChBP
pKi ± SEMa
α7
pKi ± SEMb
α4β2
pKi ± SEMa
26 endo
H 6.1 ± 0.1 6.2 ± 0.1 4.6 ± 0.1 < 4.5
29 endo
H 6.4 ± 0.1 5.1 ± 0.1 4.8 ± 0.1 < 4.5
30 exo
H 5.7 ± 0.1 4.7 ± 0.1 4.9 ± 0.1 < 4.5
25 endo
H 7.0 ± 0.1 6.7 ± 0.1 4.9 ± 0.1 < 4.5
a) [3H]epibatidine displacement studies, pH 7.4; b) [
3H]MLA displacement studies, pH 7.4; n.d. = not
determined; Cpd = compound
substituent on the nitrogen atom such as a phenetyl moiety is beneficial for affinity. Introduction of hydroxyl functionalities on the phenetyl moiety decreases affinity significantly. For the benzoate-substituted series, we observed that the endo-epimers exhibit higher affinity than the exo-epimers for Ls-AChBP. This preference is observed for 4 different sets of epimers (16-21, 25, 26 and 29, 30). Interestingly, for Ls-AChBP, the SAR of the endo-benzoate tropine esters coincides with the SAR of the dibenzosuberyl-substituted tropinyl ethers. Similar binding affinities are observed but due to a decrease in molecular weight, the benzoate esters exhibit better ligand efficiencies
26 (LE) than the dibenzosuberyl ethers (Figure 4).
Particularly, fragment 18 with an LE > 0.4 kcal·mol-1
for both AChBP species variants, serves as an ideal starting point for further optimization. Similar to the tricyclic series, quaternization of the tropine nitrogen atom of benzoate substituted tropines results in increased affinity for Ls-AChBP (21 vs. 22 and 24 vs. 23). Extending fragment 18 with a phenetyl moiety on the tropine nitrogen atom affords compound 20 with a 10-fold increase in affinity. As seen with the dibenzosuberyl
substituted derivatives, introduction of an -hydroxyl group to the phenetyl moiety results in a significant loss in affinity. This effect is observed for an unsubstituted phenetyl moiety (20 and 24) and for the meta hydroxyl-substituted phenetyl moiety
O H O H

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
107
as well (compounds 25 and 26). Additional introduction of hydroxyl groups at the
meta positions to -hydroxyphenetyl-substituted endo-epimers 24 resulted in minor or no increases in affinity (24 vs. 26 vs. 29). As such, the affinity of the tri-hydroxyl-substituted phenetylamine with an endo-configuration (29 pKi = 6.4) is lower than the affinity of the corresponding unsubstituted phenetylamine (compound 20, pKi = 7.1). The highest affinity (pKi = 7.5) was observed for the quaternary ammonium derivative 22.
Distinct SAR is observed for Ac-AChBP. One of the best dibenzosuberyl-substituted compounds in the Ls-AChBP assay (1) binds with 100-fold lower affinity to Ac-AChBP, suggesting that in contrast to Ls-AChBP, large hydrophobic substituents on a tertiary nitrogen atom are not allowed. Similar to Ls-AChBP, quaternization of the tropine nitrogen atom of N-phenetyl derivative 8 (affording 10) is beneficial, although the increase in affinity upon quaternization is more pronounced for Ac-AChBP, i.e., 1.2 log units compared to 0.4 log units, respectively. Similar to Ls-AChBP, introduction of hydroxyl functionalities on the N-
phenetyl group‟s and meta position (5 and 6) does not result in increases in binding affinity. For Ac-AChBP binding affinity, the endo-epimer of benzoate-substituted tropines is also preferred. However, in contrast to Ls-AChBP, quaternization of the tropine nitrogen atom results in significant decreases of affinity for Ac-AChBP (20 vs. 22 and 24 vs. 23). Introduction of a hydroxyl group at
the or two meta positions of the phenetyl moiety is not tolerated and the highest Ac-AChBP affinity is obtained for the unsubstituted phenetylamine 20 (pKi of 7.5). An interesting observation is that in contrast to all the other ligands, benzoate esters 20 and 24 exhibit a preference for Ac- over Ls-AChBP. Since 24 was tested as a racemic mixture, both enantiomers ((S)-31 and (R)-32) were synthesized revealing that only the (R)-enantiomer shows a preference for Ac-AChBP.
14
Whereas for Ls-AChBP, the SAR of the tropine substituents is almost identical for the dibenzosuberyl and benzoate series, in the case of Ac-AChBP clear SAR differences were observed when focusing on the tropine substituent. To exemplify these SAR differences, we have calculated both the ligand efficiency (LE)
26 and the
group efficiency (GE)27
of several of our newly designed ligands for both AChBP species variants (Figure 4). For Ls-AChBP, we observed an identical GE value of 0.17 kcal·mol
-1 per heavy atom for the addition of a phenetyl moiety to
dibenzosuberyl nortropinyl ether 9 (affording 4) and to nortropinyl benzoate 18 (affording 20). Adding an additional methyl group to the tropine nitrogen atom of N-phenetyl derivatives (to obtain compounds 10 and 22, respectively) results again in identical GE values (0.55 kcal·mol
-1 per heavy atom, (Figure 4). Remarkably, for
Ac-AChBP, we observed significant differences in the trends for the dibenzosuberyl-substituted tropines and benzoate-substituted tropines (Figure 4). Addition of a phenetyl group to ether 9 does not result in an increase in Ac-AChBP binding affinity (GE = 0 kcal·mol
-1 per heavy atom) whereas the same modification
for benzoate ester 18 yields a substantial GE of 0.38 kcal·mol-1
per heavy atom. Even more pronounced differences are observed for the subsequent methylation of

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
108
Figure 4. Calculation of LE and GE allows easy comparison of the average affinity contributions per heavy atom of compounds and functional groups of different sizes. In this example, comparison of LE’s of 18 and 9 points out that 18 is a much better starting point for further optimization than 9. In addition, comparison of GE’s between Ls- and Ac-AChBP underlines that there are clear SAR differences between both proteins.
the tropine nitrogen atom of ether 4 and ester 20. In the case of ether 1, this minor modification affords a significant increase in Ac-AChBP binding affinity (GE = 1.65 kcal·mol
-1 per heavy atom), whereas quaternization of ester 20 results in a large
drop in binding affinity (GE = -0.96 kcal·mol-1
per heavy atom). These SAR differences between dibenzosuberyl tropinyl ethers 9, 4 and 10 and the benzoate tropine esters 18, 20 and 22 are indicative of different binding modes between the two compound series. Since we have provided structural evidence that benzoate ester 20 ((R)-enantiomer) is interacting with the lobeline pocket in Ac-AChBP (2Y57.pdb),
14 a likely explanation for the observed SAR differences between the
compound series is that dibenzosuberyl ether 4 is not interacting with the lobeline pocket. In the same study, we have shown that the loss of Ac-AChBP affinity upon quaternization of the α-hydroxyl- substituted analog of 24 (23, (R)-enantiomer) is due to loss of interactions with the lobeline pocket. As such, the beneficial effect of quaternization of the dibenzosuberyl ether 4, is another indication that 4 is not interacting with the lobeline pocket in Ac-AChBP. In addition, we have provided

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
109
strong evidence that due to the lack of stabilization of the tyrosine-flip of Tyr91, the lobeline pocket in Ls-AChBP is less accessible compared to Ac-AChBP.
14 The
current results indicate that in Ls-AChBP, the N-phenetyl substituents of ligands 4-6 are likely to be accommodated by a (hydrophobic) part of the binding site different than the lobeline pocket.
Figure 5. Overlay of lobeline (PDB: 2BYS, black ball and sticks) and compound 21 ((R)-enantiomer, PDB: 2Y56, light grey ball and sticks) bound to Ac-AChBP
showing that similar to lobeline the -hydroxyphenetyl group of 21 is interacting
with the lobeline pocket. However, in contrast to lobeline the -hydroxyl group is not engaged in the formation of hydrogen bonds with the carbonyl backbones of Ser144 or Trp145 but is pointing in an opposite direction and involved in van der Waals interactions with Tyr193.
Our docking efforts suggested that incorporation of hydroxyl substituents at the α- or meta positions of the N-phenetyl substituents of the dibenzosuberyl as well as the benzoate series could increase binding affinity by the formation of additional hydrogen bonds with the lobeline pocket. However, for both series of compounds, introduction of hydroxyl moieties at the α- and meta- positions of the N-phenetyl

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
110
moiety diminishes binding affinity for Ls-AChBP as well as Ac-AChBP. For Ls-AChBP and the dibenzosuberyl-substituted tropines, this can be explained by a different binding mode than predicted, i.e., no interaction of the N-phenetyl moiety with the lobeline pocket. It is noted that the N-phenetyl substituted tropine benzoates 20 and 24 have been shown by X-ray co-crystal structures to interact with the lobeline pocket in Ac-AChBP. Nevertheless, the co-crystal complex of AChBP with 24, shows that the α-hydroxyl group of 24 ((R)-enantiomer) is not engaged in a hydrogen bond with the backbone carbonyl oxygen atoms of Ser144 or Trp145 but instead, is involved in van der Waals interactions with Tyr193, providing an explanation for the observed detrimental effect on binding affinity of
the -hydroxyl group (Figure 5). Due to desolvation penalties, the positioning of hydrogen-bonding groups needs to be near optimal in order to be beneficial in terms of binding affinity.
28, 29 Apparently, upon introduction of one or two meta-
hydroxyl groups to benzoate ester 12, the desolvation penalty dominates, indicating that the positioning of the α-hydroxyphenetyl moiety in the lobeline pocket does not allow for strong hydrogen bond formation with the carbonyl backbone of Thr89 and/or the side chain of Asp195. Interestingly, ionic and hydrogen bond interactions with Asp195 have been observed for α-conotoxins in complex with Ac-AChBP with Tyr91 in a g- conformation (closed lobeline pocket, PDB: 2UZ6 and 2BYP), showing that it is possible for ligands to interact with this residue.
12, 30
These ligands were also tested on the 7 and 42 nicotinic receptors. None of
these compounds showed any affinity for the 42 receptor. However, the binding
affinities of the dibenzosuberyl-substituted tropines for the 7 receptor were similar to the affinities determined for Ac-AChBP. As seen with the AChBPs, quaternization of the tropine nitrogen atom increases the binding affinity. None of
the novel derivatives had higher affinity for the 7 nAChR than the initial in silico hit 1. No large changes in affinity between the benzoate esters 16, 18, 20-26, 29 and 30 for the α7 nAChR were observed. All binding affinities for the N-phenetyl substituted benzoate esters are within 0.9 log units (pKi = 4.6 – 5.5). Interestingly,
the compound in this series with the highest affinity for the 7 nAChR is fragment 5 (pKi = 5.5). Apparently, all the introduced substituents on the tropine nitrogen atom even as small as methyl decrease affinity for this nAChR subtype, indicating that the lobeline pocket in the α7 nAChR subtype is not being addressed. It is noted that the optimization of the initial in silico hits that has been achieved for both AChBPs does not translate to increased affinities for the α7 and α4β2 receptors. Even though it has been shown that AChBP can be used to identify new ligands for nicotinic receptors, the current study indicates its limitations in directly using AChBP X-ray structures for structure-based optimization of nicotinic receptor ligands. Nevertheless, the current and our previous studies have provided strong evidence that interactions with the lobeline pocket can render ligands selective for Ac-AChBP. These findings may be of interest in the design of subtype-selective ligands for human nicotinic receptors. The gatekeeper tyrosine residue is conserved among the human nAChR subtypes, whereas the residue (Ser165 in Ac-AChBP) that stabilizes the open lobeline pocket conformation is located in a

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
111
highly variable region. As a consequence, due to differential stabilization of rotameric states of the gatekeeper tyrosine residue, there may exist pronounced differences in the accessibility of the lobeline pocket between nAChR subtypes.
Conclusion In summary, novel dibenzosuberyl- and benzoate substituted tropines were designed using a structure-based fragment-merging hit-optimization approach. Distinct SAR of the novel compound series was observed between two AChBP species variants and between the α7 and α4β2 nAChR subtype. The AChBP species differences were indicative of a difference in accessibility of a ligand-inducible subpocket between AChBPs from different organisms. Hereby, we have identified a region within the pocket that can be scrutinized to unravel its importance for obtaining selectivity amongst the fast number of nicotinic receptor subtypes. In addition, our studies underline that structure-based fragment merging can be an efficient method for increasing binding affinity. The novel compounds described in this study and the obtained structural understanding can be used to focus on the subtle differences between the AChBPs and nicotinic receptors, possibly the lobeline pocket accessibility, and binding pose differences, that cause the observed selectivity profiles and SAR differences.
Acknowledgments
This work was supported by a grant of the Top Institute Pharma D2-103 (to ABS), and received funding from the European Union Seventh Framework Programme under grant agreement n° HEALTH-F2-2007-202088 ("NeuroCypres" project). AA was supported by a Mozaik research grant from the Netherlands Organization for Scientific Research (NWO, project number 017.001.132). We thank Eric Karssen for chemical synthesis of compound 31. Materials and Methods Molecular docking Ligand preparation Three-dimensional structures of the ligands were generated using MOE (v2008.10, Chemical Computing Group, Montreal, Canada). Subsequently, the ligands were protonated according to physiological pH and stereoisomers were generated when necessary. Partial atomic charges were calculated and the molecules were energy minimized in vacuo using the MMFF94x force field in MOE. Ligands were stored as mol2 files.
Template preparation Co-crystal structures of Ac-AChBP in complex with lobeline (2BYS, chains A and E) and compound 31 (2W8F, chains A and B) were used for our docking studies. The ligands and all water molecules except for water molecules number 92 and 96 (2BYS, chain V) were removed. These water molecules are involved in a hydrogen bond network that interacts with the carbonyl oxygen atom of lobeline and were expected to be able to interact with the carbonyl oxygen atoms of the benzoate

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
112
substituted series in a similar fashion. Hydrogen atoms were added, partial atomic charges were calculated and a steepest-descent energy minimization was performed using the AMBER99 force field in MOE. In the 2W8F structure, we manually changed the rotamer of Y91 (numbering in 2W8F; corresponds to Y93 in 2BYS) in such a way that it was similar to Y93 of the lobeline-bound structure (2BYS), i.e., from the g- to t conformation. To this end, Y93 was assigned another rotamer that allowed for a hydrogen bond between Y93 and S167 of the complementary side.
Docking procedure Docking studies were performed using the GOLD docking program (version 4.0, CCDC, Cambridge, UK)
25 and the ChemScore scoring function and default
settings. The binding pocket was defined within a radius of 16 Å from W147 (atom NE1).
Expression and purification of AChBPs Hexahis-Lymnaea stagnalis-acetylcholine binding protein (Ls-AChBP) and hexahis-Aplysia californica-acetylcholine binding protein (Ac-AChBP) were expressed using the Bac-to-Bac Baculovirus Expression System (Invitrogen, Carlsbad, CA, USA) following the manufacturer‟s recommendations. Secreted protein was trapped on an HIS-Select Cartridge (Sigma, St. Louis, MO, USA). The cartridge was washed with 24 ml 10 mM imidazole, left overnight at 4ºC in 250 mM imidazole and thereafter eluted with 5 ml of 250 mM imidazole. A Slide-A-Lyzer Dialysis Cassette (Pierce, Rockford, IL, USA) was used according to manufactures recommendations to strongly decrease the concentration of imidazole. The purity of the protein was checked on a SDS gel and the protein concentration was determined by a Bradford analysis. Protein aliquots were stored at -80˚C until use.
Expression and purification of nicotinic receptors
Human neuroblastoma cells (SH-SY5Y) expressing human 7 receptor were obtained from Christian Fuhrer (Department of Neurochemistry, Brain Research
Institute, University of Zurich).26
Human 42 receptors were obtained using a transient transfection of HEK293t cells. To this end, HEK293t cells were maintained in Dulbecco‟s Modified Eagle Medium (DMEM) supplemented with 10%
Fetal Calf Serum (FCS), 50 IU/mL penicillin, and 50 g/mL streptomycin in 5% CO2 humidified atmosphere at 37 °C. Approximately 2 million cells were seeded in a 10 cm dish and cultured overnight before transfection. For transfection of each dish of
cells, the transfection mixture was prepared in 0.6 mL PBS and contained 0.3 g of
human 4 subunit plasmid, 2.7 g of human 2 subunit plasmid, 3.0 g of ric3 and
24 L of 1 mg/mL 25 kDa linear polyethyleneimine (Polyscience, Inc., USA). The mixture was incubated for 10–15 minutes at room temperature before it was added into the monolayer cell culture loaded with 6 mL of fresh and pre-warmed cell culture medium. Two days after transfection, the cells were washed with PBS, collected as pellet by centrifuging, and stored at –80 °C until use.

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
113
Radioligand binding assay on AChBPs Competition binding assays were performed with His-tagged Ls-AChBP or Ac-AChBP in buffer (PBS, 20 mM Tris, 1 mg/mL BSA, pH 7.4) in a final assay volume
of 120 L. A constant concentration of [3H]epibatidine (GE Healthcare, specific
activity ~49.1 Ci/mmol) was used for Ls-AChBP and Ac-AChBP, respectively. The concentrations of radioligand were set close to the Kd value for the target protein, i.e., 1 nM for Ls-AChBP and 8 nM for Ac-AChBP. The amount of protein and [3H]epibatidine were chosen as such that we obtained a clear window in the
displacement curve, sufficient amount of counts in our scintillation counting and a radioligand depletion of less than 10%. Ligands were added together with PVT Copper His-Tag SPA beads (GE Healthcare), which were diluted (1:1) with PBS, and incubated for 1.5 hours at room temperature under continuous shaking. Afterwards, the SPA beads were allowed to precipitate during 4 hours and the radioactivity was measured in a Wallac 1450 MicroBeta liquid scintillation counter.
Radioligand binding assay on nicotinic receptors
Binding assays for human 42 and 7 receptors were performed in a similar way as described for Ls-AChBP, but with a classical filtration assay. The cells were
homogenized immediately before use. In the 42 assay, [3H]epibatidine was used
at a final concentration of 2 nM and [3H]MLA (American Radiolabeled Chemicals,
Inc, specific activity ~100 Ci/mmol) was used at a final concentration of 2 nM for
the 7 assay. Bound radioligand was collected on 0.3% polyethyleneimine-pretreated Unifilter-96 GF/C filters (Perkin Elmer) using ice-cold 50 mM Tris buffer at pH 7.4. After drying the filters, scintillation fluid (MicroScint, Perkin Elmer) was added and the radioactivity was measured in a Wallac 1450 MicroBeta liquid scintillation counter. Radioligand saturation experiments were performed with nicotine to determine non-specific binding. The concentration of nicotine was 100
M for the 42 receptor and 1 mM for the 7 receptor.
Data analysis All radioligand binding data were evaluated by a non-linear, least squares curve
fitting procedure using Graphpad Prism (version 4.01, GraphPad Software, Inc., San Diego, CA). All data are represented as mean ± SEM from at least three independent experiments. Chemistry General Chemicals and reagents were obtained from commercial suppliers and were used without further purification, unless mentioned otherwise. DCM, THF and toluene were distilled under an atmosphere of nitrogen from CaH2 prior to use. Yields given are isolated yields. Flash column chromatography was typically carried out on a Flashmaster or Biotage flash chromatography system, using prepacked columns with the UV detector operating at 254 nm. All melting points are uncorrected and were measured on an Optimelt automated melting point system from Stanford research systems. All
1H NMR and
13C NMR spectra were measured on a Bruker
250 or Bruker 400 at T = 300 K or on a Bruker 500 at T = 296 K unless mentioned otherwise. Analytical HPLC-MS analyses were conducted using two different

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
114
methods. In both methods an Xbridge (C18) 5 μm column (100 mm x 4.6 mm) was used and the buffer used was a 0.4% (w/v) NH4CO3 solution in water, adjusted to pH 8.0 with NH4OH. Solution A consists of 90% acetonitrile and 10% buffer. Solution B consists of 90% water and 10% buffer. Method I) Shimadzu LC-8A preparative liquid chromatography pump system with a Shimadzu SPD-10AV UV-vis detector with the MS detection performed with a Shimadzu LCMS-2010 liquid chromatography mass spectrometer. Flow rate of 2.0 mL/min, start 5% A, linear gradient to 90% A in 10 minutes, then 10 minutes at 90% A, then 10 minutes at 5% A, total run time of 30 minutes. Compound purities were calculated as the percentage peak area of the analyzed compound by UV detection at 254 nm. Method II) Shimadzu LC-20AD prominence liquid chromatography pump system with a Shimadzu M20A prominence diode array detector with the MS detection performed with a Shimadzu LCMS-2010 liquid chromatography mass spectrometer. Flow rate of 1.0 mL/min, start 5% A, linear gradient to 90% A in 8 minutes, then linear gradient to 5%A in 0.5 minutes, then 6.5 minutes at 5% A, total run time of 15 minutes. Compound purities were calculated as the percentage peak area of the analyzed compound by UV detection at 230 nm.
3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-methyl-8-aza-bicyclo[3.2.1]octane maleate (8). 5-Chlorodibenzosuberane (11.9 g, 96%, 50.0 mmol) was added to a round bottom flask containing tropine (14.4 g, 98%, 100 mmol) dissolved in freshly distilled toluene (50 mL). The mixture was stirred overnight at room temperature. The next day, a very thick white suspension had formed. Toluene (50 mL) was added and the mixture was heated at reflux for 1 hours. When cooled down to room temperature, the mixture was filtered over glass under suction. Evaporation of the filtrate afforded 18.2 g of a slight yellow colored oil. A solution of maleic acid (5.64 g, 48.6 mmol) and crude product in EtOAc (750 mL) was concentrated in vacuo to ~400 mL and set aside. Overnight, white needle-shaped crystals formed that were isolated by filtration over glass. The obtained maleate salt was washed with EtOAc and dried in a vacuum stove to afford 16.4 g (73%) of fine white needled-shaped crystals. Further concentration of the mother liquor afforded 4.04 g (18%) of fine white needle-shaped crystals. Total isolated yield 91%. Mp: 79.0-80.2
oC;
1H NMR (250 MHz, MeOD) δ (ppm) 7.38-7.30 (m,
2H), 7.27-7.07 (m, 6H), 6.25 (s, 2H), 5.38 (bs, 1H), 3.89-3.48 (m, 5H), 3.09-3.02 (m, 2H), 2.76 (s, 3H), 2.50-2.04 (m, 8H);
13C NMR (62.5 MHz, MeOD) δ (ppm)
173.00, 136.75, 131.56, 129.48, 127.06, 67.62, 64.33, 39.47, 33.35, 25.00; HRMS (m/z): [M+H]
+ calcd. for C23H28NO, 334.2165; found, 334.2164.
3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-azabicyclo[3.2.1]-octane maleate (9). KMnO4 (6.53 g, 41.3 mmol) was added to a round bottom flask containing the free base of 3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-methyl-8-azabicyclo[3.2.1]octane (8) (6.66 g, 20.0 mmol) dissolved in pyridine (400 mL). The mixture was cooled in an ice/water bath and a solution of KOH (4 g, 71.3 mmol) in water (400 mL) was added at such a rate that the internal temperature did not exceed 25
oC. The resulting mixture was stirred for 5 hours at
room temperature. Formed MnO2 was removed by filtration over paper under suction. The slightly yellow colored filtrate was concentrated in vacuo to ~50 mL.

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
115
Water (50 mL) was added and the mixture was extracted with DCM (3 x 100 mL), followed by extraction with EtOAc (2 x 100 mL). The combined organics were dried with Na2SO4(s) and concentrated in vacuo to afford 5.82 g of a yellow colored oil. Crude product was dissolved in EtOAc (~150 mL) and a solution of maleic acid (1.78 g, 15.3 mmol) in EtOAc (~125 mL) was added. After a few seconds, a white precipitate starts to form. Further precipitation was established by storing the mixture in the freezer overnight. The crude product was isolated by filtration over glass under suction, washed with EtOAC and dried in a vacuum stove to afford 4.14 g (46%) of a white solid. The obtained maleate salt was used without further purification in the experiments described below. However, for biological characterization purposes, a small amount of the maleate salt was recrystallized from acetone to obtain pure material. Mp: 188.0-188.9
oC;
1H NMR (250 MHz,
MeOD) δ (ppm) 7.38-7.29 (m, 2H), 7.28 – 7.07 (m, 6H), 6.25 (s, 2H), 5.43 (bs, 1H), 4.05-3.39 (m, 5H), 3.13-2.75 (m, 2H), 2.52 – 1.70 (m, 8H);
13C NMR (62.5 MHz,
CDCl3) δ (ppm) 171.07, 136.80, 131.60, 129.53, 127.13, 68.53, 55.63, 34.38, 33.80, 27.15; HRMS (m/z): [M+H]
+ calcd. for C22H26NO, 320.2009; found,
320.2009.
3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8α-phenetyl-8-aza-bicyclo[3.2.1]octane maleate (4). NaBH(OAc)3 (318 mg, 1.50 mmol) was added to a round bottom flask containing the free base of 3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-azabicyclo[3.2.1]octane (9) (431 mg, 1.35 mmol) and phenylacetaldehyde (170 μL, 90%, 1.35 mmol) dissolved in DCE (5 mL). The mixture was stirred overnight at room temperature under an atmosphere of nitrogen. Saturated NaHCO3 (10 mL) was added, phases were separated and aqueous phase was extracted with DCM (2 x 10 mL). The combined organics were dried with Na2SO4(s) and concentration in vacuo afforded 513 mg (90%) of a slight yellow colored oil. For biological characterization purposes, 200 mg of the crude product was converted into its maleate salt by addition of a saturated ethereal solution of maleic acid to an ethereal solution of the crude product. Recrystallization of the formed precipitate from abs. EtOH afforded white needle-shaped crystals. Decomposes at > 190
oC;
1H NMR (200 MHz, CDCl3) δ (ppm)
7.55-6.98 (m, 13H), 6.28 (s, 2H), 5.19 (bs, 1H), 4.15-2.81 (m, 11H), 2.72 – 1.87 (m, 8H);
13C NMR (50 MHz, CDCl3) δ (ppm) 169.31, 135.89, 135.57, 130.38, 128.91,
128.49, 127.22, 125.96, 60.7, 52.94, 34.24, 32.00, 30.95, 24.54; HRMS (m/z): [M+H]
+ calcd. for C30H34NO, 424.2635; found, 424.2636.
3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8α-phenetyl-8-aza-bicyclo[3.2.1]octane methiodide (10). The free base of 3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8α-phenetyl-8-azabicyclo[3.2.1]octane (4) (200 mg, 0.71 mmol) was dissolved in freshly distilled toluene (1.2 mL). Iodomethane (500 μL, 8 mmol) was added and the mixture was set aside at room temperature over the weekend. Concentration of the mixture in vacuo afforded 481 mg of a yellow colored oil. Isolation of the endo-phenetyl isomer was achieved using reversed phase preparative chromatography (C18 derivatized (17%), endcapped silicycle column with gradient elution from 10 to 60% acetonitrile in water). Concentration of the relevant fractions afforded 77 mg (19%) of a slight orange

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
116
colored solid. Decomposes at > 110
oC;
1H NMR (400 MHz, T = 393 K, DMSO) δ
(ppm) 7.42 – 7.05 (m, 13H), 5.57 (s, 1H), 4.38-4.19 (m, 2H), 3.79 (t, J = 5.4 Hz, 1H), 3.65 – 3.53 (m, 2H), 3.45 (m, 2H), 3.18 – 2.94 (m, 7H), 2.53 – 2.39 (m, 2H), 2.39 – 2.19 (m, 4H), 2.14-2.03 (m, 2H);
13C NMR (100 MHz, CDCl3) δ (ppm)
135.30, 130.57, 129.09, 128.99, 127.51, 126.13, 67.00, 56.19 , 47.42, 32.21, 30.15, 25.17; HRMS (m/z): [M]
+ calcd. for C31H36NO, 438.2791; found, 438.2791.
1-(3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-azabicyclo-[3.2.1]octan-8-yl)-2-hydroxy-2-phenylethanone 11). D,L-mandelic acid (84 mg, 0.55 mmol) and TBTU (190 mg, 0.58 mmol) were dissolved in DMF (5 mL) containing 3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-azabicyclo-[3.2.1]octane maleate (9) (150 mg, 0.47 mmol) and TEA (80 μL, 0.57 mmol). The mixture was stirred for 1.5 hours at room temperature. Subsequently, 3 drops of NH4OH (25%) and water (10 mL) were added. The resulting suspension was extracted with Et2O (3 x 5 mL). The combined organics were washed with brine (5 mL) and dried with MgSO4(s). Concentration in vacuo afforded 161 mg of a yellowish oil that was purified over SiO2 (20 g, n-hexane/EtOAc = 2/1 with 5% TEA) to yield 100 mg (47%) of a colourless oil.
1H NMR (400 MHz, 363K, DMSO) δ
(ppm) 7.44 – 7.22 (m, 7H), 7.21 – 7.06 (m, 6H), 5.52 (bs, 1H), 5.30-5.23 (m, 2H), 4.59-4.35 (m, 2H, incl. OH), 3.70-3.28 (m, 3H), 3.12-2.91 (m, 2H) 2.09-1.42 (m, 8H); MS (ESI) m/z 454 (M+H)
+.
1-(3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-azabicyclo-[3.2.1]octan-8-yl)-2-hydroxy-2-(3-hydroxyphenyl)ethanone (12). Racemic 2-hydroxy-2-(3-hydroxyphenyl)acetic acid (93 mg, 0.55 mmol) and TBTU (190 mg, 0.58 mmol) were dissolved in DMF (5 mL) containing 3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-azabicyclo[3.2.1]octane maleate (9) (150 mg, 0.47 mmol) and TEA (80 μL, 0.57 mmol). The mixture was stirred for 1.5 hours at room temperature. Subsequently, 3 drops of NH4OH (25%) and water (10 mL) were added. The resulting suspension was extracted with Et2O (3 x 5 mL). The combined organics were washed with brine (5 mL) and dried with MgSO4(s). Concentration in vacuo afforded 164 mg of a yellowish oil that was purified over SiO2 (20 g, n-hexane/EtOAc = 1/3 with 5% TEA) to yield 68 mg (31%) of a colourless oil.
1H NMR (400 MHz, 363K, DMSO) δ (ppm) 9.06 (bs, 1H), 7.33 (d, J =
7.2 Hz, 2H) 7.21-7.08 (m, 7H), 6.81-6.76 (m, 2H), 6.68-6.62 (m, 1H), 5.53 (s, 1H), 5.21-5.11 (m, 2H), 4.71-4.27 (m, 2H, incl. OH), 3.70-3.29 (m, 3H), 3.13-2.94 (m, 2H) 2.05-1.42 (m, 8H).
2-(3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-azabicyclo-[3.2.1]octan-8-yl)-1-phenylethanol (5). LiAlH4 (10 mg, 0.26 mmol) was added to a round bottom flask containing 1-(3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-azabicyclo[3.2.1]octan-8-yl)-2-hydroxy-2-phenylethanone (11) (100 mg, 0.22 mmol) dissolved in freshly distilled THF (5 mL). The mixture was stirred for 2 hours at room temperature under an atmosphere of nitrogen. Subsequently, a 1:1 mixture of THF and water (1 mL), 3 drops of NH4OH and Et2O (3 mL) were added. After stirring for a few minutes, the suspension was filtered over paper under suction. Concentration of the filtrate in vacuo afforded 79 mg of a white solid that

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
117
was purified over SiO2 (10 g, n-hexane/EtOAc = 5/2 with 5% TEA) to yield 75 mg (76%) of a colourless oil that crystallized over time. Mp: 111.4-112.6
oC;
1H NMR
(400 MHz, T = 363 K, DMSO) δ (ppm) 7.39 – 7.24 (m, 6H), 7.23 – 7.06 (m, 7H), 5.56 (bs, 1H), 4.78 – 4.52 (m, 2H, incl. OH), 3.62-3.54 (m, 1H), 3.48-3.30 (m, 2H), 3.19-2.97 (m, 4H) 2.55-2.48 (m, 1H), 2.47-2.35(m, 1H), 2.06 – 1.59 (m, 8H);
13C
NMR (100 MHz, T = 360 K, DMSO) δ (ppm) 144.08, 139.39, 138.43, 129.84, 127.63, 127.37, 126.55, 125.83, 125.49, 71.29, 69.42, 60.53, 58.90, 57.93, 35.55, 35.43, 31.30, 26.34, 25.79; HRMS (m/z): [M+H]
+ calcd. for C30H34NO2, 440.2584;
found, 440.2583.
3-(2-(3α-((10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)oxy)-8-azabicyclo-[3.2.1]octan-8-yl)-1-hydroxyethyl)phenol (6). LiAlH4 (15 mg, 0.40 mmol) was added to a round bottom flask containing 1-(3α-((10,11-dihydro-5H-dibenzo-[a,d]cyclohepten-5-yl)oxy)-8-azabicyclo[3.2.1]octan-8-yl)-2-hydroxy-2-(3-hydroxy-phenyl)ethanone (12) (170 mg, 0.36 mmol) dissolved in freshly distilled THF (5 mL). The mixture was stirred for 2 hours at room temperature under an atmosphere of nitrogen. Subsequently, a 1:1 mixture of THF and water (1 mL), 3 drops of NH4OH and Et2O (3 mL) were added. After stirring for a few minutes, the suspension was filtered over paper under suction. Concentration of the filtrate in vacuo afforded 115 mg of a white solid that was purified over SiO2 (10 g, n-hexane/EtOAc = 1/3 with 5% TEA) to yield 92 mg (56%) of a white solid. Mp: 88.7-90.3
oC;
1H NMR (400 MHz, 363 K, DMSO) δ (ppm) 7.39-7.33 (m, 2H), 7.20-7.10
(m, 6H), 7.10-7.03 (m, 1H), 6.84 – 6.70 (m, 2H), 6.65-6.58 (m, 1H), 5.56 (bs, 1H), 4.53-4.44 (m, 1H), 3.62-3.55 (m, 1H), 3.47-3.33 (m, 2H), 3.16-2.93 (m, 4H), 2.53-2.45 (m, 1H), 2.41-2.31 (m, 1H), 2.03 – 1.68 (m, 8H);
13C NMR (100 MHz, T = 360
K, DMSO) δ (ppm) 156.66, 145.44, 139.47, 138.18, 139.47, 138.18, 129.53, 128.15, 127.02, 125.18, 116.28, 113.32, 112.68, 70.92, 69.20, 60.37, 58.66, 57.63, 35.38, 35.25, 31.05, 26.12, 25.54; HRMS (m/z): [M+H]
+ calcd. for C30H34NO3,
456.2533; found, 456.2538.
3β-hydroxy-8-methyl-8-azabicyclo[3.2.1]octane (13). Aluminium isopropoxide (61.03 g, 300 mmol) was placed in three neck round bottom flask. To this 2-propanol (200 mL) was added and the mixture was brought to reflux with stirring. Tropinone (13.95 g, 100 mmol) was dissolved in 2-propanol (100 mL) and added dropwise to the refluxing solution over a period of 45 minutes. Refluxing continued 24 hours and the reaction was allowed to continue boiling 45 minutes without a condenser to remove acetone produced during the reaction. Water (18 mL) was added dropwise to the boiling solution, resulting in foam formation and formation of a white precipitate. The mixture was filtered and the residue was washed with hot 2-propanol and EtOH. The filtrate was filtered once more and then evaporated under reduced pressure to give a red oil which crystallised over time. This residue was recrystallized from hexane (170 mL) and EtOAc (7 mL) affording 8.82 g (62%) of white needle-shaped crystals.
1H NMR (250 MHz, CDCl3) δ (ppm) 3.88 (m, 1H),
3.21-3.12 (m, 2H), 2.29 (s, 3H) 2.01-1.49 (m, 8H).
3α-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane (16). Benzoyl chloride (19.1 mL, 165 mmol) was added in a dropwise manner to a boiling solution of α-tropine

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
118
(15) (21,2 g, 150 mmol) and TEA (22.9 mL, 165 mmol) in freshly distilled toluene (200 mL). The mixture was heated at reflux temperature for 4 hours. The mixture was washed with a 2 times diluted saturated NaHCO3 solution (3 x 75 mL), with brine (75 mL) and dried with MgSO4(s). Upon drying, a white precipitate starts to form and the mixture was immediately filtered over paper under suction. The residue was extensively washed with DCM. The filtrate was concentrated in vacuo to afford 33.2 g (90%) of a white solid. For biological characterization purposes a small amount was recrystallized from toluene/acetonitrile to obtain pure material. Mp: 265.6-267.0°C.
1H NMR (500 MHz, MeOD) δ (ppm) 8.06 (d, J = 8.2 Hz, 2H),
7.68 (t, J = 6.9 Hz, 1H), 7.63-7.51 (m, 2H), 5.31 (t, J = 5 Hz,1H), 4.01-3.94 (m, 2H), 2.86 (s, 3H), 2.61-2.20 (m, 8H);
13C NMR (75 MHz, MeOD) δ (ppm) 166.87,
134.65, 121.26, 130.48, 129.88, 66.25, 63.62, 25.23; HRMS (m/z): [M+H]+ calcd.
for C15H20NO2, 246.1489; found, 246.1496.
3β-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane (17). Benzoyl chloride (7.7 mL, 66 mmol) was added in a dropwise manner to a boiling solution of β-tropine (13) (8.46 g, 60 mmol) and TEA (9.2 mL, 66 mmol) in freshly distilled toluene (175 mL). The mixture was heated at reflux temperature for 4 hours and stirred at room temperature overnight. The reaction mixture was washed with a saturated NaHCO3 solution (3x 60 mL) and subsequently dried with Na2SO4(s) . Upon drying, a white precipitate starts to form and the mixture was directly filtered over paper under suction. The residue was extensively washed with DCM. The filtrate was concentrated in vacuo to afford a white solid. This was partially dissolved in refluxing 1:1 hexane and ethyl acetate (400 mL) and filtered while hot to give 2.27 g (15%) of free base 8. The filtrate was evaporated to a solid under reduced pressure. This was dissolved in Et2O (40 mL) and a 2M solution of HCl in Et2O (40 mL) was added. A white precipitate formed that was isolated by filtration over paper affording 10.51 g (62%) of a white solid. Mp: 274.1-275.3 °C.
1H NMR (400MHz,
CDCl3) δ (ppm) 8.03 (d, J = 7.4 Hz, 2H), 7.67-7.46 (m, 1H), 7.45-7.41 (m, 2H), 5.34-5.29 (m, 1H), 3.95-3.80 (m, 2H), 2.82 (s, 3H), 2.62-2.09 (m, 8H).
13C NMR (50
MHz, MeOD) δ (ppm) 167.5, 134.6, 130.9, 130.6, 129.7, 65.8, 64.5, 36.5, 35.9, 25.3; HRMS (m/z): [M+H]
+ calcd. for C15H20NO2 246.1489; found, 246.1491.
3α-benzoyloxy-8-azabicyclo[3.2.1]octane hydrochloride (18). 3α-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane (16) (3.80 g, 15.5 mmol) was suspended in 40 mL DCE. Upon cooling in an ice/water bath and under an atmosphere of nitrogen, 1-chloroethyl chloroformate (3.5 mL, 32 mmol) was added in a dropwise manner. The mixture was heated to reflux temperature and stirred for 5 hours. The mixture was concentrated in vacuo and dissolved in MeOH (40 mL). Stirring at room temperature for 1hour results in formation of a white precipitate that was isolated by filtration over glass under suction. The obtained hydrochloride salt was washed with MeOH and dried in a vacuum stove to afford 2.77 g (67%) of fine white solid. Further concentration of the mother liquor afforded 0.42 g (10%) of a white solid. Total isolated yield 77%. Decomposes at > 280
oC;
1H NMR (500 MHz, MeOD) δ
(ppm) 8.03 (d, J = 8.5 Hz, 2H), 7.65 (t, J = 7.5 Hz, 1H), 7.55-7.50 (m, 2H), 5.33 (t, J = 4.9 Hz, 1H), 4.16-4.01 (m, 2H), 2.56-2.36 (m, 4H), 2.36-2.12 (m, 4H);
13C NMR
(50 MHz, D2O) δ (ppm) 169.03, 135.45, 130.85, 130.77, 130.30, 68.01, 55.31,

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
119
34.52, 27.01; HRMS (m/z): [M+H]
+ calcd. for C15H18NO2, 232.1332; found,
232.1343.
3β-benzoyloxy-8-azabicyclo[3.2.1]octane hydrochloride (19). 3β-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane (17) (9.02 g, 35.5 mmol) was suspended in 50 mL DCE. Upon cooling in an ice/water bath and under an atmosphere of nitrogen, 1-chloroethyl chloroformate (7.8 mL, 72 mmol) was added in a dropwise manner. The mixture was heated to reflux temperature and stirred for 19 hours. The mixture was concentrated in vacuo and dissolved in MeOH (50 mL) and left refluxing for 4 and stirring at room temperature for 18 hours. The mixture was then concentrated in vacuo leaving yellow crystals. These were recrystallized from 2-propanol. The resulting crystals were then dissolved in DCM (150 mL), mixed with a solution of 10% NaHCO3 in water (150 mL) and separated. The aqueous phase was extracted with DCM (2 x 75 mL). The combined organic phases were dried with Na2SO4(s), filtered and concentrated in vacuo to give crude free base in a yield of 76%. A portion of the crude product (1.00 g, 4.32 mmol) was purified over SiO2 using flash chromatography (gradient from 1% MeOH to 10% MeOH in EtOAc containing 1% TEA) resulting in an isolated yield of 65%. Mp: 63.1-64.8 °C.
1H NMR (400MHz,
MeOD) δ (ppm) 7.95 (dd, J = 7.6, 0.8 Hz, 2H), 7.57 (t, 1H), 7.47-7.41 (m, 2H), 5.33-5.21 (m, 1H), 3.64-3.52 (m, 2H), 2.12-2.01 (m, 2H), 1.92-1.66 (m, 6H).
13C
NMR (50 MHz, MeOD) δ (ppm) 167.4, 134.2, 131.6, 130.4, 129.6, 69.3, 55.3, 38.8, 29.6. HRMS (m/z): [M+H]
+ calcd. for C15H18NO2, 232.1332; found, 232.1333.
3α-benzoyloxy-8-phenetyl-8-azabicyclo[3.2.1]octane hydrochloride (20). Freshly grinded NaBH(OAc)3 (400 mg, 1.9 mmol) was added to a round bottom flask containing the free base of 3α-benzoyloxy-8-azabicyclo[3.2.1]octane (18) (290 mg, 1.3 mmol) and phenylacetaldehyde (170 μL, 90%, 1.3 mmol) dissolved in DCE (5 mL). The mixture was stirred overnight at room temperature under an atmosphere of nitrogen. Saturated NaHCO3 solution (5 mL) was added and the mixture was extracted with DCM (2 x 5 mL). The combined organics were washed with saturated NaHCO3 (5 mL) and dried with MgSO4(s). Concentration in vacuo afforded 331 mg of colourless oil. The crude product was dissolved in Et2O (5 ml) and 5 ml of a 2M HCl solution in Et2O was added dropwise. A white precipitate formed that was isolated by decantation. The residue was washed with Et2O (2 x 10 ml) and recrystallized from abs. EtOH to yield 232 mg (50%) of white cubic shaped crystals. Decomposes at > 280
oC;
1H NMR (250 MHz, MeOD) δ (ppm)
8.06 (d, J = 8.0 Hz, 2H), 7.67 (t, J = 7.1 Hz, 1H), 7.60-7.49 (m, 2H), 7.44 – 7.25 (m, 5H), 5.42-5.31 (m, 1H), 4.21-4.04 (m, 2H), 3.43-3.25 (m, 2H), 3.23–3.07 (m, 2H), 2.68–2.17 (m, 8H);
13C NMR (50 MHz, MeOD) δ (ppm) 166.83, 137.71, 134.65,
131.23, 130.48, 130.02, 129.87, 128.34, 66.64, 32.21, 25.44; HRMS (m/z): [M+H]+
calcd. for C22H26NO2, 336.1958; found, 336.1955.
3β-benzoyloxy-8-phenetyl-8-azabicyclo[3.2.1]octane hydrochloride (21). 3β-benzoyloxy-8-azabicyclo[3.2.1]octane (19) (231 mg, 1 mmol) was dissolved in DCE (5 mL) with stirring under an atmosphere of nitrogen. Phenylacetaldehyde (0.17 mL, 1.5 mmol) was added, followed by NaBH(OAc)3 (383 mg, 1.8 mmol). The reaction mixture was left stirring at room temperature under nitrogen atmosphere

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
120
for 48 hours before being evaporated to a solid under reduced pressure. The crude product was suspended in Et2O (25 mL) and the resulting suspension was washed with a solution of 10% NaHCO3 (3 x 20 mL, pH 10). The combined aqueous phases were extracted with Et2O (2 x 50 mL). The combined organics were dried with Na2SO4(s) , filtered and evaporated under reduced pressure to give a clear gel. This was dissolved in Et2O (5.5 mL) and mixed with a 2M solution of HCl in Et2O (5.5 mL). The resulting precipitate was collected by filtration and washed with Et2O before drying in vacuo. This gave 10 (232mg, 0.624 mmol) as very fine hair-like crystals with a yield of 62%. Decomposes at > 285°C.
1H NMR (400MHz,
MeOD) δ (ppm) 8.03-8.00 (m, 2H), 7.66-7.58 (m, 1H), 7.53-7.44 (m , 2H), 7.39-7.24 (m, 5H), 5.49-5.36 (m, 1H), 4.22-4.11 (m, 2H), 3.37-3.21 (m, 2H), 3.16-3.06 (m, 2H), 2.49-2.28 (m, 4H), 2.29-2.10 (m, 4H).
13C NMR (100 MHz, MeOD) δ
(ppm) 167.3, 137.9, 134.7, 131.1, 130.7, 130.1, 130.0, 129.8, 128.4, 66.4, 63.0, 32.7, 25.7; HRMS (m/z): [M+H]
+ calcd. for C22H26NO2, 336.1958; found, 336.1955
3α-benzoyloxy-8α-phenetyl-8-azabicyclo[3.2.1]octane methiodide (22). In a round bottom flask which was shielded from light, the free base of 3α-benzoyloxy-8-phenetyl-8-azabicyclo[3.2.1]octane (20) (285 mg, 0.85 mmol) was dissolved in acetonitrile (1.2 mL). Iodomethane (500 μL, 8.0 mmol) was added and the mixture was set aside at room temperature overnight. Upon shaking of the flask, a white precipitate formed. The precipitate was isolated by filtration over glass under suction, washed with Et2O and dried in a vacuum stove to afford 231 mg (57%) of a white crystalline solid. Mp: 211.8-213.7
oC;
1H NMR (250 MHz, CDCl3) δ (ppm)
8.01-7.89 (m, 2H), 7.70-7.16 (m, 8H), 5.40 (t, J = 5.6 Hz, 1H), 4.42-4.48 (m, 2H), 4.00-3.87 (m, 2H), 3.43 (s, 3H), 3.33-3.19 (m, 2H), 2.85-2.42 (m, 8H).;
13C NMR
(62.5 MHz, CDCl3) δ (ppm) 165.1, 135.3, 133.6, 129.3, 129.1, 129.0, 128.7, 127.6, 66.6, 62.5, 56.5, 47.5, 32.2, 29.9, 25.1; HRMS (m/z): [M]
+ calcd. for C23H28NO2,
350.2115; found, 350.2123.
3α-benzoyloxy-8-(2-hydroxy-2-phenylethyl)-8-azabicyclo[3.2.1]octane (24). D,L-mandelic acid (1.52 g, 10 mmol), 3α-benzoyloxy-8-azabicyclo[3.2.1]octane hydrochloride (18) (2.67 g, 10 mmol) and TBTU (3.21 g, 10 mmol) were dissolved in DMF (100 mL) containing TEA (2.8 mL, 20 mmol). The mixture was stirred for 3 hours at room temperature. Water (100 mL) was added and the resulting suspension was extracted with Et2O (3 x 75 mL). The combined organics were washed with brine (75 mL) and dried with MgSO4(s). Concentration in vacuo afforded 3.20 g (88%) of a white solid that was dissolved in freshly distilled THF (90 mL). Upon cooling in an ice/water bath and under an atmosphere of nitrogen, 1M BH3
.THF in THF (27 mL, 27 mmol) was added in a dropwise manner. The mixture
was heated to room temperature and stirred overnight. MeOH (20 mL) was added with caution. The mixture was concentrated in vacuo and dissolved in MeOH (~10 mL) and re-concentrated in vacuo. This procedure was repeated two more times in order to remove borane as its methyl ester. 2.62 g of slight opaque oil was obtained which crystallized over time. Recrystallization from EtOH/water afforded 1.31 g (36%) of a white crystalline solid. Mp: 110.7-112.0
oC;
1H NMR (400 MHz,
CDCl3) δ (ppm) 8.06-8.00 (m, 2H), 7.60-7.54 (m, 1H), 7.50-7.43 (m, 2H), 7.42 – 7.31 (m, 4H), 7.31 – 7.26 (m, 1H), 5.31 (t, J = 5.4 Hz, 1H), 4.70-4.58 (m, 1H), 3.45-

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
121
3.37 (m, 1H), 3.23-3.15 (m, 1H), 2.77-2.67 (m, 1H), 2.37-1.84 (m, 10H);
13C NMR
(125 MHz, CDCl3) δ (ppm) 165.97, 142.31, 133.05, 130.85, 128.81, 128.62, 128.49, 127.63, 126.02, 70.43, 68.32, 61.83, 60.46, 57.46, 37.41, 37.23, 27.38, 25.82; HRMS (m/z): [M+H]
+ calcd. for C22H26NO3, 352.1907; found, 352.1907.
3α-benzoyloxy-8-(2-(S)-hydroxy-2-phenylethyl)-8-azabicyclo[3.2.1]octane (31). Under an atmosphere nitrogen, a solution of (S)-phenyloxirane (76 μL, 0.67 mmol) in NMP (0.5 mL) was added dropwise to a stirring solution of 8-azabicyclo[3.2.1]octan-3α-yl benzoate (1, free base) (154 mg, 0.67 mmol) in NMP (0.5 mL). Subsequently, the mixture was heated for 20 minutes at 150 °C in the microwave. A supplement of (S)-phenyloxirane (76 μL, 0.67 mmol) was added and the reaction mixture was heated for an additional 30 minutes in the microwave. The clear mixture was mixed with a 1M Na2CO3 solution (5 ml) and extracted with EtOAc (3 x 5 ml). The combined organics were dried with Na2SO4(s) and concentrated in vacuo. The residue was mixed with water (20 mL) and stirred for 3 hours at room temperature. The formed white precipitate is isolated by filtration over glass, washed with water and dried in the vacuum stove overnight. 165 mg of a white solid (71%) was obtained. Mp: 122.1–124.0 °C; [α]D = + 38 (c = 1, CHCl3); 1H NMR (250 MHz, CDCl3) δ (ppm) 7.96 (d, J = 7.1 Hz, 2H), 7.55 – 7.15 (m, 8H),
5.24 (t, J = 5.3 Hz, 1H), 4.55-4.33 (m, 2H), 3.42-3.26 (m, 1H), 3.21-3.05 (m, 1H), 2.70-2.55 (m, 1H), 2.40 – 1.67 (m, 9H).
13C NMR (63 MHz, CDCl3) δ (ppm) 165.99,
142.39, 133. 05, 130.92, 129.58, 128.63, 128.50, 127.64, 126.04, 70.48, 68.37, 61.84, 60.45, 57.52, 37.44, 37.24, 27.41, 25.87. HRMS (m/z): [M+H]+ calcd. for C22H26NO3, 352.1907; found, 352.1905. Chiral HPLC: ee = 85% (Chiralcel OD-H column (250x4.6 mm), eluent: n-hexane (+0.05% diethylamine) : 2-propanol; 95 : 5, 40
oC, 0.7 ml/min
-1).
3α-(benzoyloxy)-8α-(2-hydroxy-2-phenylethyl-azabicyclo[3.2.1]octane methiodide (23). 3α-benzoyloxy-8-(2-hydroxy-2-phenylethyl)-8-azabicyclo[3.2.1]-octane (18) (110 mg, 0.3 mmol) was dissolved in freshly distilled toluene (1 mL). Iodomethane (30 μL, 0.5 mmol) was added and the mixture was set aside at room temperature over the weekend. A white precipitate (~30 mg, 20%) formed and was isolated by decantation. A supplement of iodomethane (100 μL, 1.6 mmol) was added to the remaining supernatant and the mixture was set aside for 2 weeks. Again a white precipitate (~90 mg) had formed that was isolated by decantation and combined with the first crop. The crude product was washed with toluene and recrystallized from 2-propanol/methanol affording 70 mg (47%). Concentration of the mixture in vacuo afforded 481 mg of a yellow colored oil. Mp: 234.9-236.0
oC;
1H NMR (250 MHz, MeOD) δ (ppm) 8.12 – 7.95 (m, 2H), 7.74 – 7.25 (m, 8H), 5.47
– 5.27 (m, 2H), 4.53-4.46 (m, 1H), 4.09-4.00 (m, 1H), 3.93 – 3.77 (m, 1H), 3.56 – 3.44 (m, 1H), 3.36 (s, 3H), 3.21 – 3.00 (m, 1H), 2.93 – 2.65 (m, 1H), 2.61-2.48 (m, 4H), 2.28-2.06 (m, 2H);
13C NMR (62.5 MHz, MeOD) δ (ppm) 165.36, 140.95,
133.17, 129.82, 129.03, 128.40, 128.16, 126.00, 125.36, 69.78, 68.47, 67.45, 63.40, 60.55, 40.67, 32.29, 31.47, 29.23, 25.01, 23.73; HRMS (m/z): [M]
+ calcd. for
C22H28NO3, 366.2064; found, 366.2062.

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
122
3α-benzoyloxy-8-(3-hydroxyphenethyl)-8-azabicyclo[3.2.1]octane (25). 3-hydroxyphenylacetic acid (1.52 g, 10 mmol), 3α-benzoyloxy-8-azabicyclo-[3.2.1]octane hydrochloride (18) (2.67 g, 10 mmol) and TBTU (3.21 g, 10 mmol) were dissolved in DMF (100 mL) containing TEA (2.8 mL, 20 mmol). The mixture was stirred for 3 hours at room temperature. Water (100 mL) was added and the resulting suspension was extracted with Et2O (3 x 75 mL). The aqueous phase was acidified to pH 5-6 and extracted once more with Et2O (3 x 75 mL). The combined organics were washed with brine (75 mL) and dried with MgSO4(s). Concentration in vacuo afforded 2.35 g (64%) of a white solid that was dissolved in freshly distilled THF (15 mL). Upon cooling in an ice/water bath and under an atmosphere of nitrogen, 1M BH3
.THF in THF (4.2 mL, 4.2 mmol) was added in a dropwise
manner. The mixture was heated to room temperature and stirred overnight. MeOH (20 mL) was added with caution. The mixture was concentrated in vacuo and dissolved in MeOH (~5 mL) and re-concentrated in vacuo. This procedure was repeated two more times in order to remove borane as its methyl ester. 576 mg of a white solid was obtained. The crude product was purified using reversed phase preparative chromatography (C18 derivatized (17%), endcapped silicycle column with gradient elution from 10 to 90% acetonitrile in 0.04% NH4OH, pH 8.0). Concentration of the relevant fractions afforded 410 mg (36%) of a white solid. Mp: 148.2-148.9
oC;
1H NMR (200 MHz, CDCl3) δ (ppm) 8.01 (d, J = 6.9 Hz, 2H), 7.59-
7.40 (m, 3H), 7.20-7.08 (m, 1H), 6.78-6.62 (m, 3H), 5.28 (s, J = 5.1 Hz 1H), 3.52-3.28 (m, 1H), 2.98-2.63 (m, 4H), 2.36–1.66 (m, 8H).;
13C NMR (50 MHz, CDCl3) δ
(ppm) 165.72, 156.45, 141.38, 132.79, 130.51, 129.58, 129.27, 129.15, 128.34, 128.20, 120.11, 115, 63, 113.60, 68.05, 57.68, 53.62, 35.37, 34.72, 26.02; HRMS (m/z): [M+H]
+ calcd. for C22H26NO3, 352.1907; found, 352.1907.
3α-benzoyloxy-8-(2-hydroxy-2-(3-hydroxyphenyl)ethyl)-8-azabicyclo[3.2.1]-octane maleate (26). Racemic 2-hydroxy-2-(3-hydroxyphenyl)acetic acid (336 mg, 2.0 mmol), 3α-benzoyloxy-8-azabicyclo[3.2.1]octane hydrochloride (18) (336 mg, 2 mmol) and TBTU (706 mg, 2.2 mmol) were dissolved in DMF (20 mL) containing TEA (1.4 mL, 10 mmol). The mixture was stirred overnight at room temperature. 10% NH4Cl (20 mL) was added and the aqueous phase was extracted with Et2O (3 x 45 mL). TLC analysis indicated incomplete extraction and the aqueous phase was saturated with NH4Cl and extracted with EtOAc (3 x 25 mL). The combined organics were washed with a saturated solution of NH4Cl (75 mL) and dried with Na2SO4(s). Concentration in vacuo afforded 840 mg (110%) of a yellowish solid that was dissolved in freshly distilled THF (10 mL). Upon cooling in an ice/water bath and under an atmosphere of nitrogen, 1M BH3
.THF in THF (3.5 mL, 3.5 mmol)
was added in a dropwise manner. The mixture was heated to room temperature and stirred overnight. MeOH (2 mL) was added with caution. The mixture was concentrated in vacuo and dissolved in MeOH (~10 mL) and re-concentrated in vacuo. This procedure was repeated two more times in order to remove borane as its methyl ester. 296 mg of a white solid was obtained that was purified over SiO2 using flash chromatography (gradient from 2% MeOH to 20% MeOH in DCM) to yield 87 mg (20%) of colourless oil. The purified product and maleic acid (55 mg, 0.47 mmol) were dissolved in EtOAc (~5 mL) and heated to reflux. Such an amount of n-hexane was added that the maleate salt nearly precipitated. Slowly cooling

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
123
down to room temperature afforded 98 mg (17%) of a white crystalline solid. Mp: 195.5-196.6
oC;
1H NMR (250 MHz, MeOD) δ (ppm)
1H NMR (250 MHz, MeOD) δ
(ppm) 8.01 (d, J = 8.0 Hz, 2H), 7.63 (t, J = 7.4 Hz, 1H), 7.57-7.43 (m, 2H), 7.22- 7.11 (m, 1H), 6.90-6.72 (m, 2H), 6.74-6.68 (m, 1H), 6.23 (s, 2H), 5.32 (s, 1H), 5.10-4.98 (m, 1H), 4.35-4.12 (m, 2H), 3.28-3.09 (m, 2H), 2.72 – 2.12 (m, 8H).;
13C NMR
(63 MHz, MeOD) δ (ppm) 170.58, 166.93, 159.03, 143.93, 136.76, 134.65, 131.29, 130.87, 130.48, 129.88, 118.13, 116.33, 114.00, 70.02, 66.68, 62.00, 25.53; HRMS (m/z): [M+H]
+ calcd. for C22H26NO4, 368.1856; found, 368.1859.
2-bromo-1-(3,5-dihdroxyphenyl)ethanone (14). 3,5-dihydroxyacetophenone (1.57 g, 97%, 10 mmol) in 1:1 EtOAC/DCM (20 mL) was added to a refluxing suspension of CuBr2 (4,91 g, 22 mmol) in a mixture of EtOAC (10 mL) and CHCl3 (5 mL). The mixture was stirred for 2 hours at reflux temperature. A supplement of CuBr2 (2,23 g, 10 mmol) was added and the mixture was stirred at reflux temperature for another 3 hours. Another supplement of CuBr2 (1,12 g, 5 mmol) was added and the mixture was refluxed for an additional 1.5 hours. After a reaction time of 6.5 hours in total, two small spoons of active carbon were added and the suspension was stirred for a few minutes. While still hot, the mixture was filtered over celite. Concentration of the filtrate in vacuo afforded 3.35 g of a dark brown colored oil that was purified over SiO2 using flash chromatography (gradient from 10% EtOAc to 100% EtOAc in n-hexane) to yield 1.53 g (67%) of a yellow solid.
1H NMR (400 MHz, DMSO) δ (ppm) 9.66 (s, 2H), 6.81 (d, J = 2.1 Hz, 2H),
6.48 (t, J = 2.1 Hz, 1H), 4.77 (s, 2H).
3α-benzoyloxy-8-(2-(3,5-dihydroxyphenyl)-2-oxoethyl)-8-azabicyclo[3.2.1]-octane (27). A solution of 2-bromo-1-(3,5-dihdroxyphenyl)ethanone (14) (393 mg, 1.7 mmol) in dry acetonitrile (1.5 mL) was added to a round bottom flask (shielded from light) containing the free base of 3α-benzoyloxy-8-azabicyclo[3.2.1]octane (18) (347 mg, 1.5 mmol) and DIPEA (300 μL, 1.7 mmol) dissolved in dry acetonitrile (1.5 mL). The mixture was stirred for 2 hours at room temperature under an atmosphere of nitrogen. Concentration in vacuo afforded 550 mg of a slight yellow colored oil that was purified over SiO2 using flash chromatography (gradient from 2% MeOH to 20% MeOH in DCM) to yield 340 mg (50%) of a slight yellow colored solid. Decomposes at > 105
oC;
1H NMR (250MHz, MeOD) δ (ppm)
8.09-8.00 (m, 2H), 7.71-7.42 (m, 3H), 6.94 (d, J = 2.2 Hz, 2H), 6.53 (t, J = 2.2 Hz, 1H), 5.36-5.27 (m, 1H), 3.35 (s, 2H) 3.33-3.27 (m, 2H), 2.45-1.85 (m, 8H). MS (ESI) m/z 382 (M+H)
+.
3β-benzoyloxy-8-(2-(3,5-dihydroxyphenyl)-2-oxoethyl)-8-azabicyclo[3.2.1]-octane (28). The free base of 3β-benzoyloxy-8-azabicyclo[3.2.1]octane (19) (231 mg, 1.0 mmol) was added to a round bottom flask containing a solution of 2-bromo-1-(3,5-dihdroxyphenyl)ethanone (14) (231 mg, 1.0 mmol) and DIPEA (175 μL, 1.0 mmol) in dry acetonitrile (3 mL). The mixture was stirred overnight at room temperature. Concentration in vacuo afforded 861 mg of a brown colored oil that was purified over SiO2 using flash chromatography (gradient from 2% MeOH to 20% MeOH in DCM) to yield 318 mg (83%) of a yellow crystalline solid.
1H NMR
(250 MHz, DMSO) δ (ppm) 9.54 (bs, 2H), 7.91 (d, J = 7.8 Hz, 2H), 7.72-7.42 (m,

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
124
3H), 6.82 (d, J = 2.2 Hz, 2H), 6.41 (t, J = 2.1 Hz, 1H), 5.29-5.03 (m, 1H), 3.80 (s, 2H), 2.01 – 1.52 (m, 8H). 3α-benzoyloxy-8-(2-hydroxy-2-(3,5-dihydroxyphenyl)ethyl)-8-azabicyclo-[3.2.1]octane (29). 3α-benzoyloxy-8-(2-(3,5-dihydroxyphenyl)-2-oxoethyl)-8-azabicyclo[3.2.1]octane (27) (190 mg, 0.5 mmol) was suspended in freshly distilled THF (4 mL). Upon cooling in an ice/water bath and under an atmosphere of nitrogen, 1M BH3
.THF in THF (1.0 mL, 1.0 mmol) was added in a dropwise
manner. The mixture was stirred for 1.5 hours under cooling in the ice/water bath. MeOH (5 mL) was added with caution. The mixture was concentrated in vacuo and dissolved in MeOH (~10 mL) and re-concentrated in vacuo. This procedure was repeated two more times in order to remove borane as its methyl ester. A slight orange colored solid was obtained that was purified over SiO2 using flash chromatography (gradient from 5% MeOH to 20% MeOH in DCM) to yield 146 mg (76%) of an off-white solid. Decomposes at > 150
oC;
1H NMR (400 MHz, MeOD) δ
(ppm) 8.02 (d, J = 7.1 Hz, 2H), 7.62 (t, J = 7.2 Hz, 1H), 7.55-7.47 (m, 2H), 6.37 (s, 2H), 6.19 (s, 1H), 5.29-5.21 (m, 1H), 4.78-4.66 (m, 1H), 3.72-3.52 (m, 2H), 2.92-2.72 (m, 2H), 2.50-1.88 (m, 8H);
13C NMR (63 MHz, MeOD) δ (ppm) 167.27,
159.63, 146.34, 134.35, 131.82, 130.40, 129.75, 102.82, 72.54, 69.06, 61.52, 61.10, 60.24, 36.85, 36.71, 26.99, 26.57; HRMS (m/z): [M+H]
+ calcd. for
C22H26NO5, 384.1804; found, 384.1805.
3β-benzoyloxy-8-(2-hydroxy-2-(3,5-dihydroxyphenyl)ethyl)-8-azabicyclo-[3.2.1]octane maleate (30). 3β-benzoyloxy-8-(2-(3,5-dihydroxyphenyl)-2-oxoethyl)--8-azabicyclo[3.2.1] octane (28) (141 mg, 0.4 mmol) was suspended in freshly distilled THF (2.5 mL). Upon cooling in an ice/water bath and under an atmosphere of nitrogen, 1M BH3
.THF in THF (1.0 mL, 1.0 mmol) was added in a
dropwise manner. The mixture was stirred for 45 minutes under cooling in the ice/water bath. MeOH (1 mL) was added with caution. The mixture was concentrated in vacuo and dissolved in MeOH (~5 mL) and re-concentrated in vacuo. This procedure was repeated two more times in order to remove borane as its methyl ester. 157 mg of colourless oil was obtained that was purified over SiO2 using flash chromatography (gradient from 2% MeOH to 20% MeOH in DCM) to yield 55 mg (39%) of a colourless oil. For biological characterization purposes (easier handling), the oily freebase was converted into its maleate salt by dissolving the freebase in Et2O followed by addition of an ethereal solution of maleic acid. Mp: 195.5-196.6
oC
;
1H NMR (400 MHz, MeOD) δ (ppm) 8.01 (d, J =
8.0 Hz, 2H), 7.63 (t, J = 7.4 Hz, 1H), 7.51-7.42 (m, 2H), 6.41 d, J = 2.3 Hz, 2H), 6.24 (t, J = 2.3 Hz, 1H), 6.23 (s, 2H), 5.50-5.38 (m, 1H), 5.02-4.92 (m, 1H), 4.45-4.32 (m, 1H) 4.31-4.21 (m, 1H) 4.18, 3.36-3.10 (m, 2H), 2.49 – 2.09 (m, 8H).
13C
NMR (63 MHz, MeOD) δ (ppm) 170.82, 167.12, 160.05, 144.64, 136.66, 134.59, 131.01, 130.57, 129.67, 105.43, 103.45, 70.09, 66.89, 66.00, 63.14, 25.52 HRMS (m/z): [M+H]
+ calcd. for C22H26NO5, 384.1804; found, 384.1804.

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
125
Supplementary Scheme 1. NOESY couplings that were observed using 2D 1H
NMR measurements on 2, 11 and 13.
Configuration of quaternary ammonium salts 10, 22 and 23. Methylation using iodomethane afforded the quaternary ammonium salt 10 with the phenetyl moiety in an endo-configuration as determined by 2D NMR spectroscopy at 393 K. The
1H
NMR multiplet from 2.39-2.19 ppm can be ascribed to the tropine ethylene bridge due to the absence of COSY and NOESY coupling to H(3). In contrast, COSY coupling of H(3) with hydrogens of C(2) and C(4) is observed. The identification of the ethylene bridge allows determination of the configuration of the quaternary nitrogen. NOESY coupling of the methyl substituent hydrogens with the tropine ethylene bridge (C(6) and C(7)) and NOESY coupling of CH2(9) with hydrogens of C(2) and C(4) shows that the phenetyl substituent has an endo- and that the methyl substituent must have an exo-configuration. Treatment of endo-epimer 20 with iodomethane resulted in formation of the endo-phenetyl substituted quaternary ammonium derivative 22 (Supplementary Scheme 4). The epimeric configuration was confirmed by performing 2D NMR. The singlet at 2.56 ppm can be ascribed to the tropine ethylene bridge due to the absence of NOESY coupling to H(3). Similar to compound 2, NOESY shows coupling of H(3) with the axial and equatorial hydrogens of C(2) and C(4). The identification of the ethylene bridge
1H NMR
signals allows determination of the configuration of the quaternary nitrogen. NOESY coupling of the methyl substituent with the tropine ethylene bridge hydrogens and NOESY coupling of CH2(9) with the axial hydrogens of C(2) and C(4) shows that the phenetyl substituent must have an endo- while the methyl substituent is in an exo-configuration. Reaction of alpha-hydroxyl substituted phenetylamine 21 with iodomethane afforded similar as before the endo-phenetyl substituted quaternary ammonium derivative 23 as confirmed by 2D NMR. As with 10 and 22, the
1H NMR signal of the tropine ethylene bridge can be identified due
to the absence of NOESY coupling of to H(3). This signal (multiplet at 2.61-2.48 ppm) shows clear NOESY coupling with the methyl substituent indicative of an exo-configuration. The lack of NOESY coupling of CH2(9) with the ethylene bridge hydrogens is indicative of an endo-configuration of the α-hydroxyphenetylmoiety. See Supplementary Scheme 1.

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
126
Table 1: Purity, detected masses and retention times of the synthesized compounds determined by analytical HPLC-MS. Rt: Retention time (minutes).
Method Rt Purity [M+H] Method Rt Purity [M+H]
1 II 7.40 91% 319.9 17 I 15.44 >99% 336.2
2 I 13.92 >99% 348.1 [M+] 18 I 15.72 >99% 336.2
8 I 17.64 >99% 424.4 19 II 7.58 >99% 350.0 [M+]
10 II 8.46 >99% 438.0 [M+] 21 I 15.10 97% 352.1
5 I 17.42 99% 440.3 20 I 12.96 96% 366.1 {M+]
6 I 15.78 >99% 456.3 23 I 13.28 >99% 368.1
15 I 10.58 97% 232.1 26 I 12.07 99% 385.1
16 II 6.58 >99% 231.9 27 I 12.21 91% 384.1
13 I 11.32 96% 246.1 20 I 13.59 97% 352.1
14 II 6.25 >99% 246.1 31 II 6.76 95% 351.9
References 1. Gotti, C. & Clementi, F. Neuronal nicotinic receptors: from structure to
pathology. Prog Neurobiol 74, 363-96 (2004). 2. Jensen, A.A., Frolund, B., Liljefors, T. & Krogsgaard-Larsen, P. Neuronal
nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem 48, 4705-45 (2005).
3. Kalamida, D. et al. Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J 274, 3799-845 (2007).
4. Bencherif, M., Lippiello, P.M., Lucas, R. & Marrero, M.B. Alpha7 nicotinic receptors as novel therapeutic targets for inflammation-based diseases. Cell Mol Life Sci 68, 931-49 (2011).
5. de Jonge, W.J. & Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 151, 915-29 (2007).
6. Coe, J.W. et al. Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem 48, 3474-7 (2005).
7. Potts, L.A. & Garwood, C.L. Varenicline: the newest agent for smoking cessation. Am J Health Syst Pharm 64, 1381-4 (2007).
8. Brejc, K. et al. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411, 269-76 (2001).
9. Smit, A.B. et al. A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature 411, 261-8 (2001).
10. Celie, P.H. et al. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41, 907-14 (2004).
11. Celie, P.H. et al. Crystal structure of acetylcholine-binding protein from Bulinus truncatus reveals the conserved structural scaffold and sites of

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
127
variation in nicotinic acetylcholine receptors. J Biol Chem 280, 26457-66 (2005).
12. Hansen, S.B. et al. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. Embo J 24, 3635-46 (2005).
13. Xiu, X., Puskar, N.L., Shanata, J.A., Lester, H.A. & Dougherty, D.A. Nicotine binding to brain receptors requires a strong cation-pi interaction. Nature 458, 534-7 (2009).
14. Edink, E. et al. Fragment Growing Induces Conformational Changes in Acetylcholine-Binding Protein: A Structural and Thermodynamic Analysis. J Am Chem Soc 133, 5363-5371 (2011).
15. Ulens, C. et al. Use of Acetylcholine Binding Protein in the Search for Novel alpha7 Nicotinic Receptor Ligands. In Silico Docking, Pharmacological Screening, and X-ray Analysis (dagger). J Med Chem (2009).
16. Whittaker, M., Law, R.J., Ichihara, O., Hesterkamp, T. & Hallett, D. Fragments: past, present and future. Drug Discovery Today: Technologies 7, e163-e171 (2010).
17. Lovell, S.C., Word, J.M., Richardson, J.S. & Richardson, D.C. The penultimate rotamer library. Proteins 40, 389-408 (2000).
18. Pancrazio, J.J., Kamatchi, G.L., Roscoe, A.K. & Lynch, C., 3rd. Inhibition of neuronal Na+ channels by antidepressant drugs. J Pharmacol Exp Ther 284, 208-14 (1998).
19. van der Stelt, C., Harms, A.F. & Nauta, W.T. The effect of alkylsubstitution in drugs. V. Synthesis and chemical properties of some dibenzo [a,d]1,4-cycloheptadienyl ethers. J Med Pharm Chem 4, 335-49 (1961).
20. van der Stelt, C.F., A.B.,; Tersteege, H.M.; Nauta, W.T. The effect of alkyl substitution in drugs. XVI. Basic ethers of 10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-ol and some related compounds. Arzneimittel Forschung 16, 1342-1345 (1966).
21. Abdel-Magid, A.F., Carson, K.G., Harris, B.D., Maryanoff, C.A. & Shah, R.D. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures(1). J Org Chem 61, 3849-3862 (1996).
22. Kovacs, E., Schneider, J. & Ures, F. Mechanism of the isomerization of tropine into pseudotropine. Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 320-326 (1964).
23. Maksay, G., Nemes, P. & Biro, T. Synthesis of tropeines and allosteric modulation of ionotropic glycine receptors. J Med Chem 47, 6384-91 (2004).
24. Howarth, N.M., Malpass, J.R. & Smith, C.R. Manipulation of substituents at nitrogen in tropanes, homotropanes, and dehydro-derivatives. Tetrahedron 54, 10899-10914 (1998).
25. King, L.C. & Ostrum, G.K. Selective Bromination with Copper(2) Bromide. Journal of Organic Chemistry 29, 3459-& (1964).

Chapter 5 Structure-based design, synthesis, and structure-activity relationships of dibenzosuberyl- and benzoate substituted
tropines as ligands for acetylcholine-binding protein
128
26. Hopkins, A.L., Groom, C.R. & Alex, A. Ligand efficiency: a useful metric for
lead selection. Drug Discov Today 9, 430-1 (2004). 27. Verdonk, M.L. & Rees, D.C. Group efficiency: a guideline for hits-to-leads
chemistry. ChemMedChem 3, 1179-80 (2008). 28. Edink, E., Jansen, C., Leurs, R. & de Esch, I.J.P. The heat is on:
thermodynamic analysis in fragment-based drug discovery Drug Discov Today: Technologies 7, e189-e201 (2010).
29. Bissantz, C., Kuhn, B. & Stahl, M. A medicinal chemist's guide to molecular interactions. J Med Chem 53, 5061-84 (2010).
30. Dutertre, S. et al. AChBP-targeted alpha-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J 26, 3858-67 (2007).

129
Chapter 6
Structure-based design of novel NSAID ester prodrugs:
Dual targeting of cyclooxygenase-2 (COX-2)
and α7 nicotinic receptors
Ewald Edink, Obbe Zuiderveld, Ingrid J. de Vries-van Leeuwen, Albertus H. de Boer, Jacqueline E. van Muijlwijk-Koezen, Aletta D. Kraneveld,
August B. Smit, Rob Leurs and Iwan J.P. de Esch
Abstract Non-steroidal anti-inflammatory drugs (NSAIDs) are a common treatment for chronic inflammatory disorders but suffer from several unwanted side effects, particularly in the gastrointestinal (GI) tract. The GI-related side-effects can be reduced by masking of the carboxylate moiety of the NSAID, e.g., by chemical
conversion into esters. Recently, it has been shown that activation of 7 nicotinic acetylcholine receptors (nAChRs) on macrophages and other immune cells, inhibits the release of proinflammatory mediators, thereby suppressing inflammatory processes. In the current work, we have used crystal structure data of acetylcholine-binding protein (AChBP), a ligand-binding domain (LBD) homolog to the α-7 nAChR, to design NSAID ester prodrugs that are also capable of
activating 7 nicotinic acetylcholine receptors (nAChRs). Here, we describe the structure-based design, chemical synthesis and the pharmacological evaluation of these novel dual action anti-inflammatory NSAID prodrugs.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
130
Introduction Inflammation is the complex biological response of tissues to harmful stimuli, such as pathogens, damaged cells or irritants and is part of the body‟s natural defense system against injury and disease. However, in some cases the defensive response may be inappropriately deployed against tissues of the body itself. In this case, the inflammatory response can produce damage and may be part of the disease process itself, like for example in asthma, rheumatoid arthritis and atherosclerosis.
160 The most common treatment for these disorders are non-
steroidal anti-inflammatory drugs (NSAIDs). In general, NSAIDs exert their effects through inhibition of arachidonate cyclooxygenase type 2 (COX-2) enzymes, attenuating the production of proinflammatory mediators such as prostaglandins and thromboxanes.
161 However, the chronic use of NSAIDs may lead to side
effects, particularly in the gastrointestinal (GI) tract but also in the liver, kidney, spleen, blood and bone marrow.
162 It has become clear that the GI-related side
effects of NSAIDs are associated with inhibition of COX-1 and not COX-2.163
As a consequence, several selective COX-2 inhibitors such as valdecoxib, celecoxib, etoricoxib and rofecoxib were approved and introduced to the market in the first few years of the new millennium. Unfortunately, shortly after their introduction, reports of cardiovascular side effects of the “coxibs” began to emerge and ultimately rofecoxib (Vioxx®) and valdecoxib (Bextra®) were withdrawn from the market in 2004 and 2005, respectively.
164,165 In an alternative approach to reduce
or abolish GI toxicity, considerable efforts have been focused on prodrugs in which the carboxylic group of non-selective NSAIDs is temporarily masked by converting their carboxylate functionalities into esters. This strategy aims at chemical and/or enzymatic transformation of the prodrug, resulting in release of the active NSAID after passage of the GI tract. A large amount of studies exemplify the success of this approach as reduced toxicity and similar or improved anti-inflammatory activity have been reported. These studies have recently been reviewed by Halen and co-workers.
166 So far, these efforts have not translated into clinical utility and the ideal
NSAID prodrug with a superior therapeutic advantage remains to be identified.166
The current practice in drug discovery focuses mainly on a “one target, one disease” approach. However, the redundancy that exists in biological networks may mean that targeting single proteins will not be sufficient to tackle complex diseases.
167 As such, approaches in which compounds are being developed that
modulate multiple targets are gaining interest.167-171
These approaches are known as “polypharmacology”, “designed multiple ligands (DMLs)”, “multi-target drugs (MTDs)” or “hybrid drugs” and have been applied in inflammation-related disorders as well. Examples include, dual COX-2 and 5-lipoxygenase (5-LOX) inhibitors
172,173, dual inhibitors of 5-LOX and TxA2 synthase
174 and nitric oxide (NO)
donating NSAIDS175
. Recently, studies have emerged that show that the nervous system controls peripheral inflammatory responses via efferent vagal nerves.
176-178 Vagal nerve
stimulation has been shown to result in the release of peripheral acetylcholine. The
released acetylcholine subsequently activates 7 nAChRs that are expressed on macrophages and other immune cells.
37,179 Activation of this “cholinergic anti-
inflammatory pathway” inhibits the production of TNF- and other pro-inflammatory

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
131
cytokines and as a consequence inflammatory processes are being
suppressed.37,145
The important role of the 7 nAChR in inflammatory processes is underlined by studies that show that the anti-inflammatory effects of nicotine on
macrophages can be abolished by selective 7 antagonists such as -bungarotoxin.
145,179 In contrast to wildtype mice, vagus nerve stimulation does not
result in attenuation of TNF- release in 7-knockout mice.37
Notably, treatment
with 7 (partial) agonists such as nicotine and GTS-21 (DMXBA), has been shown to be effective in several animal models of inflammation.
180-182 These findings
prompted us to examine if we could combine the advantages of NSAID ester pro-drugs with α7 nAChR active compounds. Our strategy aims on derivatization of commercially available NSAIDs with moieties that would render the resulting
prodrug capable of activating the 7 nAChR. In this manner, we expect to obtain dual action anti-inflammatory compounds with a reduced GI toxicity profile, see figure 1.
Figure 1: Dual action approach to treat inflammatory disorders. I. Activation of α7 nAChRs on macrophages (or other immune cells) by the NSAID ester prodrug will suppress the release of proinflammatory cytokines. II. Subsequent hydrolysis releases the parent NSAID that will exert an additional anti-inflammatory effect by acting on COX-2, lowering the formation of proinflammatory prostaglandins.
Nizri and coworkers have recently shown that dual targeting of 7 nAChRs and COX-2 may indeed increase therapy effectiveness as a dual action compound
consisting of the 7 nicotinic agonist cytisine and the traditional NSAID ibuprofen was significantly more effective in a central nervous system (CNS) inflammatory model than when both compounds were dosed separately, or both unconjugated.
183 Their approach, however, differed significantly from the approach
that is described in this work, as an octyl spacer was used to connect the NSAID and the nicotinic agonist. Very recently, we have disclosed
184 a fragment (VUF10663, Chapter 4) that
exhibits good ligand efficiency118
(LE = 0.43) for AChBP, as well as for the 7

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
132
nicotinic receptor (LE = 0.44, Chapter 5). This fragment is an ester of benzoic acid
and nortropinyl. Since the fragment VUF10663 displays affinity for the 7 receptor and the benzoic acid part exhibits high structural resemblance to certain NSAIDs or structural fragments of NSAIDs, we investigated if esterification of NSAIDs with
tropine-like moieties would afford NSAID prodrugs with intrinsic 7 nAChR activity, see Figure 2. Figure 2: Fragment VUF10663 and the 5 NSAID derivatives that were selected in
order to obtain NSAID prodrugs with an additional 7 nAChR mediated anti-inflammatory effect. Upon analyzing the available literature on NSAID prodrugs, two publications by Yadav and co-workers were considered very interesting as they report on the anti-inflammatory action of tropine esters of several NSAIDs including ibuprofen and
ketoprofen. Their aim, however, was not to target 7 nAChRs but to construct ester prodrugs that display reduced GI toxicity by targeting muscarinic acetylcholine receptors in the gut
185 or to achieve selective localization of the ester prodrug in
inflamed joints caused by rheumatoid arthritis.186
As anticipated, the NSAID tropine esters exhibited reduced GI toxicity, as well as selective localization in inflamed joints when derivatized with a quaternary nitrogen atom and comparable anti-inflammatory activity to their parent NSAIDs in a chronic arthritis model. To select which NSAIDs are most suitable for modification towards α7 nAChR activation we have used molecular docking in X-ray structures of acetylcholine-binding protein (AChBP) in complex with (partial) agonists for the α7 nAChRs.
63 AChBP is widely
recognized as a water soluble structural homolog of the ligand binding domain (LBD) of nAChRs, and in particular of the α7 nAChR subtype.
116,117,187 The
obtained structural information on AChBP, therefore enables structure-based design of the potential dual action anti-inflammatory agents. Based on the in silico results, a selection of NSAIDs was chemically modified with tropine moieties to obtain ester prodrugs with potential α7 nAChR activity. Here we describe the structure-based design, chemical synthesis and the pharmacological evaluation of these potential dual action anti-inflammatory NSAID prodrugs.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
133
Results Structure-based Design Over 20 currently marketed NSAIDs that contain a carboxylate functionality (Table 1) were derivatized in silico with tropine moieties containing an unsubstituted (NH), methyl substituted (NMe) and dimethyl substituted nitrogen atom (N
+(Me)2).
Subsequently, the NSAID tropine esters were docked using Gold 4.0125
in the X-ray structures of Lymnaea stagnalis AChBP (Ls-AChBP) in complex with nicotine (PDB: 1UW6) and in the structure of fragment VUF10663 in complex with AChBP from Aplysia californica (Ac-AChBP).
53,63
The obtained binding modes were visually inspected, and evaluated on complementarity towards the AChBP binding site and a conserved water molecule (that is involved in hydrogen bonds between the AChBP binding site and the
pyridine nitrogen atom of nicotine)63
in terms of polar, cation- and hydrophobic interactions while maintaining low energy conformations. In addition, our previously obtained co-crystal complex of VUF10663 with Ac-AChBP was particularly useful to evaluate the NSAIDs of the salicylate (e.g., salicylic acid) and fenamate class (e.g., flufenamic acid), since their respective tropine esters contain VUF10663 as a structural fragment (Figure 2). Assuming that these NSAID tropine esters would take similar binding modes as VUF10663, the VUF10663-Ac-AChBP co-crystal structure revealed that a hydroxyl moiety at the 2-position of VUF10663‟s phenyl moiety, as would be the case for the salicylates, is in close proximity to Ac-AChBP‟s Met114. Sequence alignments between AchBPs and human nAChR subunits (Chapter 1, Table 2) suggest that in the human α7 nAChR, a glutamine (Gln117) is located at this specific position. In silico mutation of Met114 for a glutamine, followed by an exploration of putative rotamers revealed that a glutamine is capable of forming hydrogen bonds with the 2-hydroxyl functionality of salicylate tropine esters (Figure 4). This finding was considered interesting as the formation of additional hydrogen-bonds with the α7 nAChR binding site is likely to be beneficial in terms of affinity and selectivity. Furthermore, inspection of the VUF10663-AChBP complex showed that between the tip of loop C (Cys188-Cys189) and Gln55 on the complementary side, space is available that can accommodate larger NSAIDs such as flufenamic acid, ketoprofen and diflunisal. Moreover, the positioning of the ketone functionality of the ketoprofen tropinyl ester also indicated putative hydrogen bond formation with Gln117 in the α7 nAChR binding site. Based on these molecular modeling and docking results, and their commercial availability, 5 NSAIDs were selected for chemical derivatization, see Figure 2,3 and 4.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
134
Table 1: Chemical structures of the NSAIDs that were evaluated for chemical derivatization to obtain α7 nAChR active NSAID ester prodrugs
NSAID Chemical structure NSAID Chemical structure
Salicylic acid
Aspirin
Diflunisal
Flurbiprofen
Naproxen
Fenoprofen
Ketoprofen
Ibuprofen
Carprofen
Ketorolac
Flunoxaprofen
Fenbufen

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
135
Table 1 continued. Chemical structures of the NSAIDs that were evaluated for chemical derivatization to obtain α7 nAChR active NSAID ester prodrugs
NSAID Chemical structure NSAID Chemical structure
Indomethacin:
Sulindac:
Alclofenac:
Tolmetin:
Diclofenac:
Bromfenac:
Meclofenamic
acid:
Flufenamic
acid:
Tolfenamic
acid:
Mefenamic
acid:
Etodolac

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
136
Figure 3: Predicted binding modes in Ls-AChBP (PDB: 1UW6) of quaternary methylammonium tropine derivatives of three NSAIDs; ibuprofen, flufenamic acid and ketoprofen, respectively.
Figure 4: Analysis of the VUF10663-AC-AChBP crystal structure reveals that the 2-hydroxyl moiety of salicylate tropinyl esters may be positioned in the vicinity of Met114 (shown in sticks). The equivalent residue in the α7 nAChR is Gln117 and exploration of putative rotamers indicates that the glutamine side chain is likely to be involved in hydrogen bond formation with the 2-hydroxyl group of salicylate tropinyl esters.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
137
Chemical Synthesis The designed NSAID dual action anti-inflammatory agents were synthesized according to the synthetic routes that are depicted in Scheme 1,2 and 3.
Scheme 1: Synthesis of tropinyl esters of ibuprofen (a), ketoprofen (b) and flufenamic acid (c). The synthesis of the tropine esters of ibuprofen, ketoprofen and flufenamic acid from their parent NSAIDs was initiated by their conversion into the corresponding acyl chlorides with thionyl chloride (Scheme 1). Subsequent reaction with an excess of tropine (2-5 equivalents) afforded the NSAID tropine esters 1a-c in moderate yields. The corresponding secondary amine derivatives of 2a-c were obtained by demethylation of the tropine nitrogen atom using α-chloroethylchloroformate.
188 Since the purification of ibuprofen derivative 2a by
solid phase extraction with propylsulfonic acid derivatized silica resulted in a very low yield of 18%, the ketoprofen and flufenamic acid derivatives 2b and 2c were isolated by basic extraction followed by column chromatography, resulting in significant better yields. The quaternary ammonium derivatives 3a-c were obtained from 2a-c by the use of iodomethane in reasonable to excellent yields. The salicylic acid tropine esters were synthesized from commercially available 2-methyoxybenzoyl chloride and tropine (Scheme 2). The acylation (82% yield of 4), was followed by a demethylation of the methoxy substituent using BBr3, affording target molecule 1d in 64% yield. Using similar procedures as described above, the tertairy amine 1d was converted into its corresponding secondary amine (2d) and quaternary ammonium (3d) derivatives in reasonable to excellent yields. A similar approach as in the synthesis of the salicylic acid tropine esters was used to synthesize the target diflunisal derivatives (1e, 2e and 3e, Scheme 3). Methyl protected diflunisal (6) was obtained from diflunisal by a double methylation step

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
138
using iodomethane and subsequent hydrolysis of the methyl ester using NaOH in water/methanol. As described above, the acid moiety was converted into the corresponding acid chloride using thionyl chloride. The next step in the synthesis of the target diflunisal derivatives was reacting the acid chloride with an excess of tropine. However, in this case the reaction suffered from incomplete conversion and 0.1 eq. of DMAP was added followed by refluxing the mixture for an additional 3 hours. Nevertheless, conversion did not seem to improve. Despite the presence of triethylamine in the eluent (5 %) and similar to the purification of other tropine esters, the flash chromatography purification of 8 resulted in a low recovery of wanted product (31% isolated yield). The yield (21%) in the subsequent O-demethylation using BBr3 was compromised by hydrolysis of the tropine ester bond and again low recovery in the flash chromatography purification. From the diflunisal tropine ester 1e, the secondary amine and quaternary ammonium derivative were synthesized using similar procedures as described for 2a and 3b. Purification of the secondary amine derivative 2e was cumbersome and two columns and one solid phase extraction were required in order to obtain pure material. NMR experiments have shown that diflunisal tropine esters are not stable in deuterated methanol and the use of methanol in the flash chromatography purification may therefore provide an explanation for the low yields that were observed. In D2O and deuterated dmso, the diflunisal esters were more stable and no signs of breakdown products were observed after 2 days
Scheme 2: Synthesis of tropinyl esters of salicylic acid.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
139
Scheme 3: Synthesis of tropinyl esters of diflunisal.
. Biological evaluation A [
3H]-methyllycaconitine (MLA) displacement assay with human α7 nAChRs
expressed in human neuroblastoma cells was performed, to determine if the NSAID tropine ester prodrugs display the anticipated affinity for the α7 nAChR. As shown in Table 3 and in line with our molecular docking results, all NSAID tropinyl
esters have affinity for the 7 nAChR (pKi > 4.8). Table 3 shows that quaternization of the tropine nitrogen atom with an additional methyl substituent
(compounds 3a-e) is beneficial for 7 nAChR affinity, and all the quaternary ammonium ligands exhibit nicotine-like affinity (pKi = 6.0 ± 0.1, data not shown). Tertairy (1a-e) and secondary amine derivatives (2a-e) bind in general with 10-fold lower affinities. An exception is the secondary amine salicylic acid derivative 2d,
which displays similar affinity (pKi = 6.1) towards the 7 nicotinic receptor compared to its quaternary ammonium analog 3d.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
140
Table 3. A selection of NSAIDs (top row) and 3 tropine alcohols (left column) have been combined and the resulting esters were screened for affinity for the human
7 nAChR, pKi ± SEMa.
6.1 ± 0.1 5.2 ± 0.1 5.1 ± 0.1 4.8 ± 0.1 5.0 ± 0.1
5.2 ± 0.1 5.0 ± 0.1 5.1 ± 0.1 5.0 ± 0.1 5.3 ± 0.1
6.2 ± 0.1 5.9 ± 0.1 6.0 ± 0.1 5.8 ± 0.1 5.8 ± 0.1
a [
3H]MLA displacement studies, pH 7.4; n = 3.
Table 3 shows that several NSAID tropine esters were identified that bind with
nicotine-like affinity to the 7 nAChR. Nevertheless, in order to inhibit the release of proinflammatory mediators such as TNF-α from macrophages and other immune cells, the compounds should not only bind to the α7 nAChR but also activate the receptor, i.e., induce opening of the α7 ion channel. To determine if the highest affinity compounds (2d, 3a-e) posses the required (partial) agonistic activity,
electrophysiological recordings from Xenopus leavis oocytes expressing human 7 nAChRs were determined. The results in Table 4 show that most NSAID tropine esters do not activate the human α7 nicotinic receptors. However, the salicylic acid derivatives 2d and 3d activate the α7 nAChR with low µM EC50‟s , albeit with limited efficacy, i.e., they induce maximal currents of 7-10% compared to the maximal current that is induced by the endogenous ligand acetylcholine.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
141
Table 4: Human α7 nAChR concentration-response parameters for acetylcholine, tropisetron and NSAID tropine esters.
Compound EC50 ± s.e.m.
(µM)
Maximal
Response (%) n
Acetylcholine
(control) 31 ± 8 100 5
Tropisetron
(control) 3.3 ± 1.1 80 6
2d 4.0 ± 2.1 7 3
3a n.a. 0 4
3b n.a. 0 3
3c >100 4@500µM 3
3d 5.7 ± 1.4 10 3
3e n.a. 0 3
Concentration-response parameters were determined by fitting to a one site saturation equation. Data are shown as mean ± s.e.m. for EC50 and maximal response releative to acetylcholine (500 µM). n.a. means not applicable. The values for n are the number of determinations.
The pharmacological results show that two salicylate tropine esters (2d, 3d) have been obtained that bind with nicotine-like affinity and act as partial agonists at the
7 nAChRS. As such, these derivatives may produce a dual anti-inflammatory
action by activating 7 nAChRs on immune cells and subsequently lower COX-2 mediated production of proinflammatory prostaglandins. However, in order to exert a dual action anti-inflammatory effect, the NSAID ester prodrug should be hydrolyzed in vivo, preferably at its site of action. In order to determine if the required hydrolysis is likely to occur, we have measured hydrolysis rates of salicylate ester 3d in 49% human serum and pure PBS pH 7.4 buffer. The NSAID ester prodrug 3d is hydrolyzed slightly faster in the presence of serum compared to pure buffer, with half times of 36 ± 3 and 43 ± 1 hours, respectively.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
142
Discussion Using AChBP co-crystal structures, 15 potential dual action NSAID prodrugs were designed and subsequently synthesized. Radioligand displacement studies on the human α7 nAChR showed that all 15 tropine esters displayed affinity for this nicotinic receptor subtype, validating our molecular modeling efforts. Interestingly, the 0.6 log unit increase in affinity of salicylate ester 2d, compared to fragment VUF10663 is in line with its predicted binding pose. Compound 2d has an additional hydroxyl functionality that is anticipated to form a hydrogen bond with Gln117 in the α7 nAChR binding site. As such, the observed increase in α7 nAChR affinity indicates that 2d is indeed interacting with Gln117. Nevertheless, it cannot be excluded that other factors, e.g., a switch in binding mode or stabilization of the bioactive conformation through intramolecular hydrogen bond formation, are involved in the observed increase in binding affinity. From the compounds that displayed nicotine-like affinity (2d, 3a-e), electrophysiological recordings from Xenopus leavis oocytes expressing the human α7 nAChR were determined. These experiments showed that most compounds blocked the α7 nAChR instead of activating the receptor. However, it was found that the salicylate tropine esters 2d and 3d act as partial agonists and induce maximal currents of 7-10% compared to the maximal current that is induced by the endogenous ligand acetylcholine. It is not known how much receptor activation is required for an α7 nicotinic partial agonist to reduce the release of proinflammatory mediators from macrophages in vivo. GTS-21 (DMXBA), an α7 nAChR partial agonist with reported efficicacies between 9 and 32% (depending on which species is studied)
95,189 inhibits the release of proinflammatory cytokines in vitro and in vivo
and improved survival in a mouse inflammatory model.38
The minimal efficacy of α7 nAChR partial agonists that is required to inhibit the release of proinflammatory mediators is likely to be dependent on the frequency of acetylcholine release near macrophages by the “cholinergic anti-inflammatory pathway”. When this frequency is relatively high, a low efficacious agonist that is bound to the receptor, prevents acetylcholine from activating the α7 receptor and may not lower but instead increase the release of proinflammatory mediators from immune cells. On the other hand, if the frequency of “cholinergic anti-inflammatory pathway” stimulation is low, which is likely to be the case in chronic inflammatory diseases, a low efficacious agonist will still be able to lower the release of proinflammatory cytokines from macrophages and other immune cells. As mentioned in the introduction, Yadav and co-workers have prepared quaternary ammonium tropinyl esters of NSAIDs, aiming on selective localization of the ester prodrug in inflamed tissue for the treatment of rheumatoid arthritis (RA)
186. Our
current study shows that the quaternary ammonium tropine ester of ibuprofen (3a) and ketoprofen (3b) antagonize human α7 nAChRs, and as a result may display a proinflammatory component by preventing endogenous acetylcholine from activating α7 receptors on macrophages and other immune cells. As such, next to slow hydrolysis of the ester linkage, antagonism on α7 nAChRs may play a role in the observed limited anti-inflammatory activity in the initial phase of the in vivo study that has been performed by Yadav et al.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
143
Having identified esters of salicylic acid that are capable of activating the α7 nAChR, we subsequently investigated if the ester linkage is likely to be hydrolyzed in vivo. We found that 50% of compound 3d is hydrolyzed in ~36 hours when human serum is present (49%) and in ~43 hours in pure PBS buffer pH 7.4. These results indicate that the salicylate ester prodrug is likely to be present long enough to exert its effect on the α7 nicotinic receptor but eventually will be hydrolyzed to release salicylic acid producing an additional anti-inflammatory effect by affecting COX-2. The difference in observed hydrolysis rates between 49% serum and pure buffer indicates that the ester hydrolysis is catalyzed by certain serum constituents, e.g., esterases. It is noted that unlike most NSAIDs, salicylic acid does not exert its anti-inflammatory effects through direct COX-2 inhibition but instead inhibits the transcription of the COX-2 gene in a dose-dependent manner.
190
191,192 However,
the kinetic aspects in an in vivo model of these potential dual action compounds remain to be studied. Conclusion and outlook Using structure-based design, two NSAID tropinyl esters have been obtained that
are capable of activating 7 nicotinic receptors. As such, these salicylate ester prodrugs may exert an anti-inflammatory effect by modulation of macrophages and other immune cells that are expressing α7 nicotinic receptors. In addition, we have shown that the salicylate ester prodrugs are hydrolyzed in human serum, resulting in the release of salicylic acid. The liberated salicylic acid will exert an additional anti-inflammatory effect by inhibition of the transcription of the COX-2, attenuating the production of proinflammatory mediators such as prostaglandins and thromboxanes. Therefore, the NSAID ester prodrugs 2d and 3d are likely to exhibit a dual anti-inflammatory action combined with an improved GI toxicity profile compared to the parent NSAID. Furthermore, one of the NSAID ester prodrugs (3d) contains a quaternary ammonium nitrogen atom which is likely to result in selective localization of the compound in inflamed joints which is of interest for treating rheumatoid arthritis. At this moment, we are performing in vitro
experiments to determine if the dual targeting of 7 nAChRS and COX-2 translates into an improved anti-inflammatory action compared to the parent NSAID. Ultimately, the kinetic properties of the compounds including hydrolysis of the ester bond, key for the COX-2 activity, needs to be evaluated in vivo. It is noted that the salicylic acid derivatives 2d and 3d may serve as ideal starting points for further optimization. Considering their small size, these ligands exhibit
very good binding affinities for the 7 nAChR. This provides room to further
improve the activity against COX-2 and the 7 receptor without ending up with too large molecules that are likely to exhibit unfavorable ADMET properties.
193
To conclude, using structure-based design, a novel class of NSAID ester prodrugs was obtained in which the prodrug can exert an additional anti-inflammatory effect by activating α7 nicotinic receptors on macrophages and other immune cells. Acknowledgements This work was supported by a grant of the Top Institute Pharma D2-103 (to ABS) and we thank Giano Leeuwe for chemical synthesis.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
144
Materials and methods Molecular docking Ligand preparation Three-dimensional structures of the ligands were generated using MOE (v2008.10). Subsequently, the ligands were protonated according to physiological pH and stereoisomers were generated when necessary. Partial atomic charges were calculated and the molecules were energy minimized in vacuo using the MMFF94x force field in MOE. Ligands were stored as mol2-files. Protein preparation Co-crystal structure of Ls-AChBP in complex with nicotine (PDB: 1UW6, chain A and B) was used for our docking studies. The ligands and all water molecules were removed, except for the water molecule that is involved in hydrogen bonds between the pyridine nitrogen atom of nicotine and the backbone carbonyl oxygen atom of Leu102 and backbone nitrogen atom of Met114. Hydrogen atoms were added and a steepest-descent energy minimization was performed using the AMBER99 force field in MOE. Docking procedure Docking studies were performed using the GOLD docking program (version 4.0)
125
and the ChemScore scoring function and default settings. The binding pocket was defined within a radius of 15Å from W147 (atom NE1). Docking was performed in the presence as well as absence of the water molecule mentioned above. Chemistry General Chemicals and reagents were obtained from commercial suppliers. α-Tropine was dried by azeotropic removal of water with toluene. DCM, THF and toluene were freshly distilled under nitrogen from CaH2. Acetonitrile was dried over 4 Å molecular sieves. All other reagents were used without further purification. Yields given are isolated yields. Flash column chromatography was typically carried out on a Biotage flash chromatography system, using prepacked columns with the UV detector operating at 254 nm. All melting points are uncorrected and were measured on an Optimelt automated melting point system from Stanford research systems. All
1H NMR and
13C NMR spectra were measured on a Bruker 250, 400
at T = 300 K or Bruker 500 at T = 296 K. Analytical HPLC-MS analyses were conducted using Shimadzu LC-20AD liquid chromatography pump system with a Shimadzu SPD-M20A Diode Array detector with the MS detection performed with a Shimadzu LCMS-2010 liquid chromatography mass spectrometer. The analyses were performed using an Xbridge (C18) 5 μm column (100 mm × 4.6 mm), flow rate of 1.0 mL/min and two different methods with solvent A (acetonitrile containing 0.1% (v/v) formic acid) and solvent B (water containing 0.1% (v/v) formic acid). Method I) start 5% A, linear gradient to 90% A in 4.5 min, then 1.5 min at 90% A, followed by linear gradient to 5% A in 0.5 min, then 1.5 min at 5% A, total run time of 8 min. Method II) start 5% A, linear gradient to 90% A in 4.5 min, then 4.5 min at 90% A, followed by linear gradient to 5% A in 0.5 min, then 2.5 min at 5% A, total

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
145
run time of 12 min. Compound purities under both conditions were calculated as the percentage peak area of the analyzed compound by UV detection at 230 nm. 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(4-isobutylphenyl)propanoate (1a) Under an atmosphere of nitrogen and stirring, 2-(4-isobutylphenyl)propanoic acid (ibuprofen, 1.03 g, 5 mmol) was added to SOCl2 (3 mL) and heated at reflux for 5 min. After cooling down to room temperature, the mixture was concentrated under reduced pressure. To the residue, DCM (~ 25 mL) was added and removed in vacuo. This procedure was repeated two more times in order to completely remove SOCl2, yielding 1.10 g 2-(4-isobutylphenyl)propanoyl chloride as an opaque oil which was used without further purification. Under an atmosphere of nitrogen and stirring, dry α-tropine (1.41 g, 10 mmol) was added to a solution of the racemic 2-(4-isobutylphenyl)propanoyl chloride in dry toluene (20 ml). The mixture was stirred for 2 h at rt. After cooling down to rt, toluene (20 ml) was added. The mixture was washed with 0.5M Na2CO3 (3 x 20 ml) and NaHCO3(sat), dried with Na2SO4. Concentration of the filtrate under reduced pressure afforded an opaque oil. Purification of the crude using flash chromatography (gradient 1% 10% MeOH in DCM, both solvents contain 1% TEA) afforded 681 mg (37%) of an opaque oil. A small amount was converted into a hydrochloride salt by dissolving the oil in dioxane and the dropwise addition of 4M HCl in dioxane. Concentration under reduced pressure afforded a white crystalline solid. Mp: 160.5-162.0
oC;
1H NMR
(250 MHz, DMSO) δ (ppm) 10.72 (bs, 1H), 7.20 (d, J = 8.1 Hz, 2H), 7.12 (d, J = 8.1 Hz, 2H), 4.87 (t, J = 4.4 Hz, 1H), 3.88 – 3.59 (m, 3H), 2.74 – 2.32 (m, 5H), 2.12 – 1.00 (m, 12H), 0.86 (d, J = 6.60 Hz, 6H);
13C NMR (63 MHz, DMSO) δ 173.15,
140.37, 138.09, 129.64, 127.61, 64.89, 61.33, 44.81, 44.64, 38.50, 34.29, 30.10, 23.84, 23.53, 22.57, 18.12; [M+H]
+ calcd. for, C21H32NO2, 330.2428; found
330.2411. 8-azabicyclo[3.2.1]octan-3α-yl 2-(4-isobutylphenyl)propanoate (2a) 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(4-isobutylphenyl)propanoate (1a, 247 mg, 0.75 mmol) was dissolved in DCE (2 ml). Under an atmosphere of nitrogen and stirring, DIPEA (260 μL, 1.5 mmol) followed by 1-chloroethyl chloroformate (160 μL, 1.5 mmol) were added in a dropwise manner. The mixture was heated at 150 °C for 30 min in the microwave. The mixture was concentrated under reduced pressure and the residue dissolved in MeOH (2 ml). This solution was heated at 150 °C for 30 min in the microwave. Before opening, the septum that closes the microwave vial is punctured with a needle, resulting in the release of CO2(g). Propylsulfonic derivatized silica (3.0 g, 0.67 mmol/g) was added and stirred for 30 min. The suspension was filtered over an empty flash chromatography cartridge and washed with MeOH (100 ml), followed by elution with a 10x diluted saturated NH3(g) solution in methanol (100 ml). The eluate was concentrated under reduced pressure affording 42 mg (18%) of a colorless oil.
1H NMR (500 MHz, CDCl3) δ (ppm) 7.18
(d, J = 8.1 Hz, 2H), 7.08 (d, J = 8.1 Hz, 2H), 4.99 (t, J = 5.1 Hz, 1H), 3.64 (q, J = 7.2 Hz, 1H), 3.46 – 3.36 (m, 1H), 3.36 – 3.28 (m, 1H), 2.44 (d, J = 7.2 Hz, 2H), 2.06 – 1.34 (m, 14H), 0.87 (d, J = 6.6 Hz, 6H).
13C NMR (126 MHz, CDCl3) δ 173.84,
140.62, 137.80, 129.45, 127.29, 68.32, 53.27, 53.22, 45.67, 45.10, 37.47, 37.23, 30.33, 29.09, 28.87, 22.42, 22.41, 17.95; HRMS (m/z): [M+H]
+ calcd. for
C20H30NO2, 316.2271; found 316.2270.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
146
8,8-dimethyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(4-isobutylphenyl)propanoate iodide (3a) 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(4-isobutylphenyl)-propanoate (1a, 82 mg, 0.25 mmol) was dissolved in chloroform (2 ml). Under an atmosphere of nitrogen and stirring, iodomethane (310 μL, 5.0 mmol) was added slowly. The reaction vial was wrapped in aluminum foil and set aside overnight at rt. MTBE (10 ml) was added in a dropwise manner, which resulted in the formation of a white precipitate. After 1 h, the precipitate was isolated by filtration over glass, followed by 3 consecutive washings with MTBE. Finally, remaining MTBE was co-evaporated by dissolving the residue in MeOH, followed by concentration under reduced pressure, affording 67 mg (57%) of a white solid. Mp: 257.3-259.2
oC.
1H
NMR (250 MHz, CDCl3) δ (ppm) 7.21 – 6.98 (m, 4H), 5.12 (t, J = 5.5 Hz, 1H), 4.26 (m, 1H), 4.10 (m, 1H), 3.65 (q, J = 7.1 Hz, 1H), 3.49 (s, 3H), 3.31 (s, 3H), 2.83 – 2.49 (m, 2H), 2.43 (d, J = 7.2 Hz, 2H), 2.31 – 1.60 (m, 6H), 1.43 (m, 4H), 0.87 (d, J = 6.6 Hz, 6H).
13C NMR (63 MHz, CDCl3) δ 172.99, 141.22, 137.13, 129.79,
127.10, 67.41, 67.37, 62.38, 51.29, 45.72, 45.34, 45.03, 32.47, 32.40, 30.27, 24.94, 24.50, 22.47, 22.43, 17.67. [M]
+ calcd. for C22H34NO2, 344.2584; found
344.2573. 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(3-benzoylphenyl)propanoate hydrochloride (1b) Under an atmosphere of nitrogen and stirring, 2-(3-benzoylphenyl)propanoic acid (ketoprofen, 1.27 g, 5 mmol) was added to SOCl2 (3 mL) and heated at reflux for 15 min. After cooling down to room temperature, the mixture was concentrated under reduced pressure. To the residue, DCM (~ 25 mL) was added and removed in vacuo. This procedure was repeated two more times in order to completely remove SOCl2, yielding 1.31 g of 2-(3-benzoylphenyl)propanoyl chloride as an opaque oil which was used without further purification. Under an atmosphere of nitrogen and stirring, dry α-tropine (1.41 g, 10 mmol) was added to a solution of the 2-(3-benzoylphenyl)propanoyl chloride in dry toluene (20 ml). The mixture was stirred for 2 h at rt. After cooling down to rt, toluene (30 ml) was added. The mixture was washed with 0.5M Na2CO3 (3 x 20 ml) and NaHCO3(sat), dried with Na2SO4 and filtered over paper. Concentration of the filtrate under reduced pressure afforded opaque oil. Purification of the crude using flash chromatography (gradient 1% 10% MeOH in DCM, both solvents contain 1% TEA) afforded 1.18 g (63%) of a colorless oil. A small amount was converted into a hydrochloride salt by dissolving the oil in dioxane and the dropwise addition of 4M HCl in dioxane. Concentration under reduced pressure afforded a colorless oil that upon stirring in Et2O became a white solid. Mp: 161.1 - 161.8
oC;
1H NMR (250
MHz, DMSO) δ (ppm) 10.41(bs, 1H), 7.79-7.48 (m, 9H), 4.92 (t, J = 4.9 Hz 1H), 4.00 (q, J = 7.2 Hz, 1H), 3.84-3.56 (m, 2H), 2.69-2.31 (m, 5H), 2.14 – 1.29 (m, 9H). 13
C NMR (63 MHz, DMSO) δ (ppm) 195.58, 172,24, 140.86, 137.34, 136.92, 132.78, 131.73, 129.49, 128.92, 128.65, 128.61, 128.48, 64.72, 60.87, 44.43, 23.54, 17.51; [M+H]
+ calcd. for C24H28NO3; found 378.2052.
8-azabicyclo[3.2.1]octan-3α-yl 2-(3-benzoylphenyl)propanoate (2b) 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(3-benzoylphenyl)propanoate (1b, 380 mg, 1.0 mmol) was dissolved in DCE (2 ml). Under an atmosphere of nitrogen and stirring, 1-chloroethyl chloroformate (250 μL, 2.3 mmol) was added in a dropwise manner. The mixture was heated at 150 °C for 30 min in the microwave. DIPEA (175 μL, 1

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
147
mmol) and 1-chloroethyl chloroformate (200 μL, 1.9 mmol) were added in a dropwise manner. The mixture was heated at 150 °C for 30 min in the microwave. To the reaction mixture, 1 M Na2CO3 (2 ml) was added. After vigorous mixing, the phases were separated and aqueous phase was extracted with DCM (2 x 2 ml). Combined organic extracts were concentrated under reduced pressure and the residue dissolved in MeOH (10 ml). The mixture was heated to reflux for 3 h. Concentration of the filtrate under reduced pressure afforded 506 mg of a brown oil. Purification of the crude using flash chromatography (gradient 10% 40% MeOH in DCM, both solvents contain 1% TEA) afforded 160 mg (60%) of a colorless oil.
1H NMR (250 MHz, CDCl3) δ (ppm) 7.82 – 7.70 (m, 3H), 7.70 – 7.37
(m, 6H), 5.02 (t, J = 5.1 Hz, 1H), 3.75 (q, J = 7.2 Hz, 1H), 3.49 – 3.29 (m, 2H), 2.09 – 1.35 (m, 11H);
13C NMR (126 MHz, CDCl3) δ (ppm) 196.26, 172.65, 140.56,
138.11, 137.32, 132.64, 131.32, 129.94, 129.23, 129.03, 128.69, 128.38, 66.77, 53.24, 53.17, 45.69, 34.64, 34.49, 26.83, 26.59, 17.78. [M+H]
+ calcd. for
C23H26NO3; 364.1907 found 364.1889. 8,8-dimethyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(3-benzoylphenyl)propanoate iodide (3b) 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(3-benzoylphenyl)-propanoate (1b, 94 mg, 0.25 mmol) was dissolved in dry acetonitrile (2 ml). Under an atmosphere of nitrogen and stirring, iodomethane (310 μL, 5.0 mmol) was added slowly. The reaction vial was wrapped in aluminum foil and set aside for 40h. MTBE (15 ml) was added in a dropwise manner, which resulted in the formation of a white precipitate. After 1 h, the precipitate was isolated by filtration over glass, followed by 3 consecutive washings with MTBE. The residue was dried in the vacuum stove, overnight affording 91 mg (70%) of a white solid. Mp: 210.4-212.2
oC;
1H NMR (250 MHz, CDCl3) δ (ppm) 7.81 – 7.69 (m, 3H), 7.67 – 7.56 (m,
2H), 7.56 – 7.40 (m, 4H), 5.18 (t, J = 5.5 Hz, 1H), 4.34 - 4.07 (m, 2H), 3.81 (q, J = 7.1 Hz, 1H), 3.49 (s, 3H), 3.34 (s, 3H), 2.84 – 2.57 (m, 2H), 2.39 – 1.42 (m, 9H); 13
C NMR (126 MHz, CDCl3) δ (ppm) 196.35, 172.39, 140.42, 138.43, 137.23, 133.00, 131.34, 130.13, 129.62, 129.08, 128.96, 128.65, 67.46, 67.42, 62.70, 51.33, 45.65, 45.32, 32.60, 27.11, 25.11, 24.78, 17.71; [M]
+ calcd. for C25H30NO3;
392.2220 found 392.2220. 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(3-(trifluoromethyl)phenylamino)-benzoate hydrochloride (1c) Under an atmosphere of nitrogen and stirring, 2-(3-(trifluoromethyl)phenylamino)benzoic acid (flufenamic acid, 1.13 g, 4 mmol) was
added to SOCl2 (5 mL) and heated to 60C for 3 h. After cooling down to room temperature, the mixture was concentrated under reduced pressure. To the residue, CHCl3 (~ 10 mL) was added and removed in vacuo. This procedure was repeated two more times in order to completely remove SOCl2. This yielded 1.10 g of 2-(3-(trifluoromethyl)phenylamino)benzoyl chloride as a light brown solid which was used without further purification. Under an atmosphere of nitrogen and stirring, a solution of the 2-(3-(trifluoromethyl)phenylamino)benzoyl chloride in dry toluene (4 ml) was added in a dropwise manner to a solution of dry α-tropine (1.24 g, 8.8 mmol) in dry toluene (20 mL). The mixture was stirred for 16 h at rt. Propylsulfonic derivatized silica (15 g, 0.67 mmol/g) was added to the organic phase and stirred for 30 min. The suspension was filtered over an empty flash chromatography cartridge and washed with MeOH (100 ml), followed by elution with a 10x diluted

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
148
saturated NH3(g) solution in methanol (100 ml). The eluate was concentrated under reduced pressure. Purification of the crude using flash chromatography (gradient 1% 34% MeOH in EtOAC, both solvents contain 5% TEA) afforded 687 mg (42%) of a slight yellow colored oil. A hydrochloride salt was obtained by dissolving the freebase in Et2O (25 ml), followed by the addition of 1M HCl in Et2O (2.5 ml). The resulting white suspension was heated to reflux and absolute ethanol was added until a clear solution was obtained. Cooling down to room temperature resulted in the formation of a white crystalline solid that was isolated by filtration over glass. After washing with Et2O and drying in a vacuum stove, 515 mg (29%) of white needle-shaped crystals were obtained. Mp: 198.3-199.3
oC;
1H NMR (400
MHz, MeOD) δ (ppm) 7.97 (dd, J = 8.0, 1.5 Hz, 1H), 7.55 – 7.27 (m, 6H), 6.97-6.87 (m, 1H), 5.33 (t, J = 4.8 Hz, 1H), 4.01-3.98 (m, 2H), 2.85 (s, 3H), 2.60 – 2.24 (m, 8H);
13C NMR (126 MHz, CDCl3) δ 168.50, 148.09, 143.21, 135.80, 132.94 (q, J =
32.2 Hz) 132.36, 131.47, 125.53, 120.50, 120.01, 118.40, 116.03, 114.33, 66.07, 63.89, 39.58, 36.12, 32.42, 25.00; [M+H]
+ calcd. for C22H24F3N2O3, 405.1784; found
405.1766. 8-azabicyclo[3.2.1]octan-3-yl 2-(3-(trifluoromethyl)phenylamino)benzoate (2c) 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(3-(trifluoromethyl)phenylamino)-benzoate (1c, 239 mg, 0.6 mmol) was suspended in 2 mL DCE. Under an atmosphere of nitrogen and stirring, DIPEA (210 μL, 1.2 mmol) was added, followed by the dropwise addition of 1-chloroethyl chloroformate (130 μL, 1.2 mmol). The mixture was heated at 150 °C for 30 min in the microwave. The mixture was concentrated under reduced pressure and the residue dissolved in MeOH (2 ml). This solution was heated at 150 °C for 30 min in the microwave. Before opening, the septum that closes the microwave vial is punctured with a needle, resulting in the release of CO2(g). The mixture was concentrated under reduced pressure. EtOAc (10 ml) and 1M Na2CO3 (10 ml) were added to the residue. When the residue was completely dissolved upon swirling, the phases were separated and the aqueous phase was extracted with EtOAc (2 x 10 ml). The combined organics were dried with Na2SO4, filtered over paper, and concentrated under reduced pressure. Purification of the crude using flash chromatography (gradient 1% 36% MeOH in EtOAC, both solvents contain 5% TEA) afforded 155 mg (66%) of a colorless oil.
1H NMR (500 MHz, CDCl3) δ (ppm) 9.72 (s, 1H), 7.95 (dd,
J = 8.0, 1.5 Hz, 1H), 7.48 (s, 1H), 7.46 – 7.34 (m, 3H), 7.32-7.27 (m, 2H), 6.87-6.80 (m, 1H), 5.30 (t, J = 5.0 Hz, 1H), 3.63 (s, 2H), 2.99 (s, 1H), 2.26 – 2.12 (m, 4H), 2.00 – 1.84 (m, 4H).
13C NMR (126 MHz, CDCl3) δ 167.92, 147.10, 141.66, 134.33,
131.92 (q, J = 32.2 Hz), 131.42, 130.01, 124.66, 124.06 (q, J = 272.5 Hz), 119.68 ( J = 3.8 Hz), 118.46, 118.12 (J = 3.8 Hz), 114.41, 113.32, 68.59, 53.46, 37.30, 29.34; [M+H]
+ calcd. for C21H22F2N2O2, 391.1628; found 391.1626.
8,8-dimethyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(3-(trifluoromethyl)phenyl-amino)benzoate iodide (3c) 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-(3-(trifluoromethyl)phenylamino)benzoate (1c, 101 mg, 0.25 mmol) was dissolved in acetonitrile (2 ml). Under an atmosphere of nitrogen and stirring, iodomethane (310 μL, 5.0 mmol) was added slowly. The mixture was stirred for 20 h at rt. MTBE (10 ml) was added in a dropwise manner which resulted in the formation of a white precipitate. After 1 h, the precipitate was isolated by filtration over glass, followed

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
149
by 3 consecutive washings with MTBE. Finally, remaining MTBE was co-evaporated by dissolving the residue in MeOH, followed by concentration under reduced pressure, affording 127 mg (93%) of a white solid. Mp: 253.3-254.1
oC;
1H
NMR (250 MHz, CDCl3) δ (ppm) 9.49 (s, 1H), 7.79 (dd, J = 8.0, 1.3 Hz, 1H), 7.55 – 7.19 (m, 6H), 6.88-6.74 (m, 1H), 5.49 (t, J = 5.5 Hz, 1H), 4-51-4.40 (m, 2H), 3.68 (s, 3H), 3.54 (s, 3H), 2.98-2.79 (m, 2H), 2.76 – 2.31 (m, 4H), 2.26-2.12 (m, 2H);
13C
NMR (63 MHz, CDCl3) δ 167.08, 147.67, 141.25, 135.11, 132,32 (q, J = 32.3 Hz) 130.93, 130.17, 125.29, 120.29 (q, J = 3.8 Hz), 118.71 (q, J = 3.7 Hz), 118.61, 114.62, 111.88, 67.73, 62.69, 51.75, 45.95, 32.93, 25.61; [M]
+ calcd. for
C23H26F3N2O2, 419.1941; found 419.1934. 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-methoxybenzoate (4) Under an atmosphere of nitrogen and stirring, 2-methoxybenzoyl chloride (1.35 mL, 10 mmol) was added in a dropwise manner to a solution of dry α-tropine (3.11 g, 22 mmol) in dry toluene (40 mL). The mixture was heated to reflux temperature. After 2 h, the mixture was cooled down to rt and mixed with EtOAc (40 ml). The mixture was washed with 1M Na2CO3 (30 ml) and dried with Na2SO4. After filtration over paper, the filtrate was concentrated under reduced pressure. Purification of the crude using flash chromatography (gradient 4% 36% MeOH in EtOAC, both solvents contain 5% TEA) afforded 2.25 g (82%) of a white solid. Mp: 45.9 – 47.0 °C;
1H NMR (400 MHz, CDCl3) δ (ppm) 7.80 (dd, J = 7.6, 1.4 Hz, 1H), 7.50 – 7.40
(m, 1H), 7.04-6.95 (m, 2H), 5.23 (t, J = 5.4 Hz, 1H), 3.89 (s, 3H), 3.12 (bs, 2H), 2.30 (s, 3H), 2.26 – 2.15 (m, 2H), 2.15 – 1.98 (m, 4H), 1.92-1.81 (m, 2H);
13C NMR
(63 MHz, CDCl3) δ 165.72, 159.31, 133.43, 131.68, 120.61, 120.16, 111.99, 67.81, 59.86, 55.69, 40.45, 36.68, 25.64; HRMS (m/z): [M+H]
+ calcd. for C16H22NO3
276.1594; found, 276.1593. 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-hydroxybenzoate (1d) 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-methoxybenzoate (4, 1,38 mg, 5.0 mmol) was dissolved in DCM (50 ml). Under an atmosphere of nitrogen, the mixture was cooled in an acetonitrile/liquid nitrogen bath (~ -40°C). Under stirring, 1M BBr3 (15 ml, 15 mmol) was added in a dropwise manner. The mixture was slowly warmed to rt and stirred for an additional hour. Subsequently, the mixture was quenched in ice-cold 1M Na2CO3 (100 ml). After warming up to rt, the phases were separated and the aqueous phase was extracted twice with DCM (2 x 50 ml). The combined organics were dried with Na2SO4 and filtered over paper. Concentration under reduced pressure afforded a brown colored oil which was purified by flash chromatography (gradient 4% 36% MeOH in EtOAC, both solvents contain 5% TEA) affording 880 mg (64%) of a white crystalline solid. Mp: decomposes >215 °C;
1H NMR (400 MHz, CDCl3) δ (ppm) 10.92 (s, 1H), 7.80 (dd, J = 8.0, 1.7 Hz,
1H), 7.53 – 7.39 (m, 1H), 7.04 – 6.83 (m, 2H), 5.30 (t, J = 5.3 Hz, 1H), 3.17 (s, 2H), 2.32 (s, 3H), 2.29 – 1.97 (m, 6H), 1.89-1.81 (m, 2H);
13C NMR (63 MHz, CDCl3) δ
169.59, 161.88, 135.53, 129.46, 119.20, 117.67, 112.93, 68.92, 59.72, 40.49, 36.66, 25.82. HRMS (m/z): [M+H]
+ calcd. for C15H21NO3, 262.1438; found
262.1432.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
150
8-azabicyclo[3.2.1]octan-3α-yl 2-hydroxybenzoate (2d) 8-methyl-8-azabicyclo- [3.2.1]octan-3α-yl 2-hydroxybenzoate (1d, 261 mg, 1.0 mmol) and DIPEA (350 μL, 1.0 mmol) were dissolved in DCE (2 ml) in a microwave vial. Under an atmosphere of nitrogen and stirring, 1-chloroethylchloroformate (220 μL, 2.0 mmol) was added in dropwise manner. The reaction mixture was heated for 30 min at 150 °C in the microwave and subsequently concentrated under reduced pressure. The residue was dissolved in MeOH (10 ml) and heated for 15 min at 150 °C in the microwave. The microwave vial‟s septum was punctured with a needle to liberate formed CO2. The mixture was concentrated under reduced pressure and the residue was suspended in EtOAc (10 ml), 1M Na2CO3 (10 ml) was added and phases were separated. Aqueous phase was extracted with EtOAc (2 x 10 ml) and the combined organics are dried with Na2SO4. Filtration over paper followed by concentration of the filtrate under reduced pressure affords an off-white solid that was purified by flash chromatography (gradient 4% 36% MeOH in EtOAc, both solvents contain 5% TEA) yielding 140 mg (57%) of an off-white solid.
Mp: 92.9 -93.4 °C;
1H NMR
(250 MHz, CDCl3) δ (ppm) 7.72 (dd, J = 8.0, 1.5 Hz, 1H), 7.45-7.32 (m, 1H), 6.98 – 6.75 (m, 2H), 5.29 (t, J = 5.0 Hz, 1H), 3.60-3.42 (m, 2H), 2.29 – 1.97 (m, 4H), 1.92 – 1.73 (m, 4H);
13C NMR (63 MHz, CDCl3) δ 169.54, 161.82, 135.50, 129.42,
119.16, 117.62, 112.86, 69.52, 53.23, 37.33, 29.41; HRMS (m/z): [M+H]+ calcd. for
C14H18NO3, 248.1281; found, 248.1286.
8,8-dimethyl-8-azabicyclo[3.2.1]octan-3α-yl 2-hydroxybenzoate iodide (3d) Under an atmosphere of nitrogen and stirring at rt, iodomethane (310 μL, 5.0 mmol) was added slowly to a solution of 8-methyl-8-azabicyclo[3.2.1]octan-3α-yl 2-hydroxybenzoate (1d, 131 mg, 0.5 mmol) in DCM (2 ml). After stirring for 3 h at rt, MTBE (2 ml) was added in dropwise manner which resulted in the formation of a white precipitate. Filtration over glass was followed by 3 consecutive washings with MTBE. Drying in the vacuum stove afforded 187 mg (93%) of a white solid. Mp: decomposes > 251 °C;
1H NMR (400 MHz, DMSO) δ (ppm) 10.52 (s, 1H), 7.77 (dd,
J = 7.8, 1.7 Hz, 1H), 7.59 – 7.49 (m, 1H), 6.99 (m, 2H), 5.30 (t, J = 5.6 Hz, 1H), 3.98-3.86 (m, 2H), 3.17 (s, 3H), 3.07 (s, 3H), 2.71-2.61 (m, 2H), 2.46 – 2.31 (m, 4H), 2.10 (t, J = 17.0 Hz, 2H);
13C NMR (63 MHz, DMSO) δ 167.75, 160.09, 135.61,
130.03, 119.50, 117.40, 113.53, 66.55, 63.87, 50.29, 43.74, 31.52, 24.51. HRMS (m/z): [M]
+ calcd. for C16H22NO3, 276.1594; found 276.1595.
methyl 2',4'-difluoro-4-methoxybiphenyl-3-carboxylate (5) Under an atmosphere of nitrogen and stirring, iodomethane (5.0 ml, 80 mmol) was added portion wise to a solution of diflunisal (5.00 g, 20 mmol) and K2CO3 (6.91 g, 50 mmol) in dry acetone (50 ml). The mixture was heated at reflux overnight. Filtration over celite and concentration under reduced pressure afforded 4.39 g of a white solid. Recrystallization of the crude from MeOH/H2O afforded 4.44 g (80%) of a white crystalline solid.
1H NMR (250 MHz, CDCl3) δ (ppm) 7.96 - 7.90 (m, 1H), 7.68
– 7.56 (m, 1H), 7.47 – 7.30 (m, 1H), 7.05 (d, J = 8.7 Hz, 1H), 7.00 – 6.81 (m, 2H), 3.95 (s, 3H), 3.91 (s, 3H).

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
151
2',4'-difluoro-4-methoxybiphenyl-3-carboxylic acid (6) Under stirring, 6M NaOH (13 ml) was added slowly to a solution of methyl 2',4'-difluoro-4-methoxybiphenyl-3-carboxylate (5, 4.17 g, 15 mmol) in MeOH (20 ml). The mixture was heated at reflux for 2h. Concentration under reduced pressure afforded a white solid that was mixed with EtOAc (50 ml) and water (50 ml). Under vigorous stirring, the pH was adjusted to 2-3 by slowly adding conc. HCl (~ 10 ml). Phases were separated and aqueous phase was extracted with EtOAc (2 x 20 ml). Combined organics were dried with Na2SO4, filtered over paper, and concentrated under reduced pressure. The residue was recrystallized form n-heptane affording 3.69 g (93%) of a white crystalline solid.
1H NMR (250 MHz, CDCl3) δ (ppm) 8.40 – 8.21 (m, 1H), 7.74 (dt, J
= 8.7, 2.2 Hz, 1H), 7.47 - 7.34 (m, 1H), 7.14 (d, J = 8.7 Hz, 1H), 7.01 – 6.83 (m, 2H), 4.13 (s, 3H). 8-methyl-8-azabicyclo[3.2.1]octan-(3α-yl)-2',4'-difluoro-4-methoxybiphenyl-3-carboxylate (7) 2',4'-difluoro-4-methoxybiphenyl-3-carboxylic acid (6, 1.03 g, 4
mmol) was added to SOCl2 (5 mL) and heated to 60C for 3 h, under a N2 atmosphere. After cooling down to room temperature, the mixture was concentrated under reduced pressure. To the residue, CHCl3 (~ 10 mL) was added and removed in vacuo. This procedure was repeated two more times in order to completely remove SOCl2, yielding 1.06 g of 2',4'-difluoro-4-methoxybiphenyl-3-carbonyl chloride as a light brown solid which was used without further purification. Under an atmosphere of nitrogen and stirring, a solution of the 2',4'-difluoro-4-methoxybiphenyl-3-carbonyl chloride in a 5 : 1 mixture of dry toluene and dry acetonitrile (6 ml) was added in a dropwise manner to a solution of dry α-tropine (1.24 g, 8.8 mmol) in dry toluene (20 mL). The mixture was heated at reflux temperature for 15 h. DMAP (73 mg, 0.6 mmol) was added and the mixture was heated at reflux for an additional 3 h. After cooling down to rt, EtOAc (20 ml) was added. The mixture was washed with 1M Na2CO3 (10 ml), dried with Na2SO4 and filtered over paper. Concentration of the filtrate under reduced pressure afforded a yellow oil. Purification of the crude using flash chromatography (gradient 1% 34% MeOH in EtOAC, both solvents contain 5% TEA) afforded 481 mg (32%) of a slight yellow colored oil.
1H NMR (250 MHz, CDCl3) δ (ppm) 7.96 (dd, J = 2.3, 1.4
Hz, 1H), 7.83 – 7.52 (m, 1H), 7.52 – 7.16 (m, 1H), 7.04 (d, J = 8.7 Hz, 1H), 7.00 – 6.82 (m, 2H), 5.27 (t, J = 5.3 Hz, 1H), 3.93 (s, 3H), 3.35-3.18 (m, 2H), 2.43 – 1.80 (m, 11H).
13C NMR (63 MHz, CDCl3) δ 165.47, 162.52 (dd, J = 249.0, 11.8 Hz),
159.80 (dd, J = 250.2, 11.9 Hz), 158.88, 133.87 (d, J = 3.2 Hz), 132.26 (d, J = 2.9 Hz), 131.09 (dd, J = 9.4, 4.8 Hz), 127.03 (d, J =1.2 Hz), 124.05 (dd, J = 13.4, 3.8 Hz), 120.60, 112.27, 111.79 (dd, J = 21.1, 3.8 Hz), 104.56 (dd, J = 26.6, 23.7 Hz), 67.65, 60.23, 55.93, 40.18, 36.21, 25.45; HRMS (m/z): [M+H]
+ calcd. for
C22H24F2NO3, 388.1719; found, 388.1719.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
152
Scheme 4: Assigned
13C NMR shifts of compound 1e
8-methyl-8-azabicyclo[3.2.1]octan-(3α-yl)-2',4'-difluoro-4-hydroxybiphenyl-3-carboxylate (1e) 8-methyl-8-azabicyclo[3.2.1]octan-(3α-yl)-2',4'-difluoro-4-methoxybiphenyl-3-carboxylate (8, 852 mg, 2.2 mmol) was dissolved in DCM (20 ml). Under an atmosphere of nitrogen, the mixture was cooled in an acetone/liquid nitrogen bath (~ -78°C). Under stirring, 1M BBr3 (6.6 ml, 6.6 mmol) was added in a dropwise manner. The mixture was slowly warmed to rt and stirred for an additional hour. Subsequently, the mixture was quenched in ice-cold 1M Na2CO3 (20 ml). After warming up to rt, the phases were separated and the aqueous phase was extracted with DCM (2 x 50 ml). The combined organics were dried with Na2SO4 and filtered over paper. Concentration under reduced pressure afforded a brown colored oil which was purified by flash chromatography (1
st column: gradient 1%
34% MeOH in EtOAC, both solvents contain 5% TEA; 2nd column: step 1, 10 column volumes EtOAc : MeOH 9 :1, step 2, gradient 10% 34% MeOH in EtOAc, both solvents contain 5% TEA). The product containing fractions were concentrated under reduced pressure affording 250 mg of a colorless oil. The residue was dissolved in MeOH (10 ml) and propylsulfonic derivatized silica (1.5 g, 0.67 mmol/g) was added and stirred for 30 min. The suspension was filtered over an empty flash chromatography cartridge and washed with MeOH (50 ml), followed by elution with a 10x diluted saturated NH3(g) solution in methanol (50 ml). The eluate was concentrated under reduced pressure affording 172 mg (21%) of a colorless oil.
1H NMR (500 MHz, CDCl3) δ (ppm) 10.93 (s, 1H), 7.96 (s, 1H), 7.56
(d, J = 8.7 Hz, 1H), 7.37-7.29 (m, 1H), 7.01 (d, J = 8.7 Hz, 1H), 6.96 – 6.83 (m, 2H), 5.28 (t, J = 5.0 Hz, 1H), 3.16-3.07 (m, 2H), 2.29 (s, J, 3H), 2.25 – 1.93 (m, 6H), 1.83 (d, J = 15.1 Hz, 2H).
13C NMR (126 MHz, CDCl3) δ 169.33, 162.10 (dd, J
= 249.0, 11.8 Hz), 161.35, 159.64 (dd, J = 250.2, 11.8 Hz), 135.76, 130.69 (dd, J = 9.4, 4.8 Hz), 129.79 (d, J = 4.0 Hz), 125.86, 123.99 (dd, J = 13.3, 3.9 Hz), 117.96, 112.90, 111.69 (dd, J = 21.1, 3.8 Hz), 104.48 (dd, J = 26.46, 25.54 Hz), 66.19,

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
153
59.64, 40.45, 36.59, 25.66; HRMS (m/z): [M+H]
+ calcd. for C21H22F2NO3, 374.1562;
found, 374.1565. 8-azabicyclo[3.2.1]octan-3α-yl 2',4'-difluoro-4-hydroxybiphenyl-3-carboxylate (2e) 8-methyl-8-azabicyclo[3.2.1]octan-(3α-yl)-2',4'-difluoro-4-hydroxybiphenyl-3-carboxylate (1e, 90 mg, 0.25 mmol) was dissolved in DCE (0.5 ml). Under an atmosphere of nitrogen and stirring, DIPEA (100 μL, 0.57 mmol) followed by 1-chloroethyl chloroformate (60 μL, 0.56 mmol) were added in a dropwise manner. The mixture was heated at 150 °C for 30 min in the microwave. The mixture was concentrated under reduced pressure and the residue dissolved in MeOH (2 ml). This solution was heated at 150 °C for 30 min in the microwave. Before opening, the septum that closes the microwave vial is punctured with a needle. Propylsulfonic derivatized silica (1.0 g, 0.67 mmol/g) was added and stirred for 30 min. The suspension was filtered over an empty flash chromatography cartridge and washed with MeOH (50 ml), followed by elution with a 10x diluted saturated NH3(g) solution in methanol (50 ml). The eluate was concentrated under reduced pressure. Purification of the crude using flash chromatography (gradient 1% 36% MeOH in EtOAC, both solvents contain 5% TEA) afforded 21 mg (23%) of a colorless oil.
1H NMR (500 MHz, CDCl3) δ (ppm) 7.97-7.92 (m, 1H), 7.66 – 7.58 (m,
1H), 7.44-7.31 (m, 1H), 7.08 (d, J = 8.67 Hz, 1H), 7.00 – 6.88 (m, 2H), 5.41 (t, J = 4.68 Hz, 1H), 3.89-3.81 (m, 2H), 2.52-2.42 (m, 2H), 2.29-2.08 (m, 4H), 2.03 (d, J = 15.42 Hz, 2H).
13C NMR (126 MHz, CDCl3) δ 167.31, 162.47 (dd, J = 249.4, 11.9
Hz), 161.61 159.83 (dd, J = 250.0, 11.8 Hz), 136.27 (d, J = 1.8 Hz), 130.85 (dd, J = 9.4, 4.8 Hz), 129.79 (d, J = 4.4 Hz), 126.20 (d, J = 1.1 Hz), 124.01 (dd, J = 13.3, 3.9 Hz), 118.39, 112.65, 111.99 (dd, J = 21.1, 3.9 Hz), 104.76 (d, J = 1.2 Hz), 104.76 (dd, 26.7, 25.4 Hz), 68.57, 53.44, 36.63, 28.02; [M+H]
+ calcd. for
C20H20F2NO3, 360.1406; found 360.1396. 8,8-methyl-8-azabicyclo[3.2.1]octan-(3α-yl)-2',4'-difluoro-4-hydroxybiphenyl-3-carboxylate iodide (3e) 8-methyl-8-azabicyclo[3.2.1]octan-(3α-yl)-2',4'-difluoro-4-hydroxybiphenyl-3-carboxylate (1c, 74 mg, 0.20 mmol) was dissolved in dry DCM (2 ml). Under an atmosphere of nitrogen and stirring, iodomethane (200 μL, 3.2 mmol) was added slowly. The reaction vial was wrapped in aluminum foil and set aside overnight at rt. A white precipitate had formed, Et2O (2 ml) was added in a dropwise manner. After 1 h, the precipitate was isolated by filtration over glass, followed by 3 consecutive washings with Et2O. Drying in the vacuum stove afforded 88 mg (85%) of a white solid. Mp: 253.3 -253.9
oC;
1H NMR (500 MHz, DMSO) δ
(ppm) 10.65 (s, 1H), 7.92 (s, 1H), 7.70 (d, J = 9.4 Hz, 1H), 7.63-7.55 (m, 1H), 7.41-7.34 (m, 1H), 7.24-7.17 (m, 1H), 7.12 (d, J = 8.6 Hz, 1H), 5.30 (t, J = 5.5 Hz, 1H), 3.94-3.85 (m, 2H), 3.17 (s, 3H), 3.06 (s, 3H), 2.70-2.60 (d, 2H), 2.37 (s, 4H), 2.18-2.07 (m, 2H).
13C NMR (126 MHz, CDCl3) δ 165.09, 161.55 (dd, J = 246.9, 12.2
Hz), 159.48, 159.05 (dd, J = 247.6, 12.2 Hz), 135.56, 131.43 (dd, J = 9.7, 4.5 Hz), 130.34 (d, J = 4.2 Hz), 125.17, 123.61 (dd, J = 12.9, 3.9 Hz), 118.04, 114.18, 112.20 (dd, J = 21.1, 3.5 Hz), 104.63 (app. t, 26.5 Hz), 66.59, 63.99, 50.27, 43.70, 31.48, 24.42; [M]
+ calcd. for C22H24F2NO3, 388.1719; found 388.1700.

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
154
Table 4: Compound purities and molecular weights as determined by HPLC-MS
Compound Method retention time Purity [M+H]+/[M]
+
1a II 3.50 94% 331.10
1b II 3.89 >99% 378.05
1c II 4.57 97% 405.05
2a II 4.63 97% 317.10
2b II 3.8 99% 364.00
2c II 4.04 >99% 391.05
3a II 4.08 98% 344.15
3b II 3.86 >99% 392.05
3c II 4.76 95% 419.15
5 I 2.95 >99% 262.10
6 I 2.85 97% 248.05
7 I 2.96 >99% 276.10
11 I 4.66 98% 374.05
12 II 4.07 98% 360.00
13 II 4.05 >99% 388.00
Radioligand binding assay on α7 nicotinic receptors Human neuroblastoma cells (SH-SY5Y) expressing human α7 receptor were obtained from Christian Fuhrer (Department of Neurochemistry, Brain Research Institute, University of Zurich).
45 The cells were homogenized immediately before
use. Competition binding assays were performed in buffer (PBS, 20 mM Tris, 1
mg/mL BSA, pH 7.4) in a final assay volume of 120 L and [3H]MLA (American
Radiolabeled Chemicals, Inc, specific activity ~100 Ci/mmol) was used at a final concentration of 2 nM. Bound radioligand was collected on 0.3% polyethyleneimine-pretreated Unifilter-96 GF/C filters (Perkin Elmer) using ice-cold 50 mM Tris buffer at pH 7.4. After drying the filters, scintillation fluid (MicroScint, Perkin Elmer) was added and the radioactivity was measured in a Wallac 1450 MicroBeta liquid scintillation counter. Radioligand saturation experiments were performed with nicotine (1 mM) to determine non-specific binding. All radioligand binding data were evaluated by a non-linear, least squares curve fitting procedure using GAll data are represented as mean ± SEM from at least three independent experiments.
Electrophysiological assay on α7 nicotinic receptors Human α7 nACh receptor cloned in the pMXT expression vector was kindly provided by Dr Jon Lindstrom of the University of Pennsylvania. The pMXT expression vector was linearized by XbaI to create template cDNA. Capped cRNA was synthesized in vitro from the cDNA using the SP6 RNA polymerase kit (SP6

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
155
mMESSAGE mMACHINE KIT™, Ambion, Austin, TX) following the manufacturer‟s recommended protocol. Oocytes were obtained from Ecocyte Biosciences (Germany), and injected with 20 ng cRNA using a Nanoject-II injector (Drummond Scientific, Broomall, PA). Injected oocytes were incubated at 16-18°C in ND96 solution (96 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM HEPES-TRIS, pH 7.4) supplemented with 2.5 mM Na-pyruvate and 50 μg/ml gentamycin for 2–4 days, until the start of the electrophysiological experiments. Electrophysiological recordings were made using the two-electrode voltage-clamp technique using the Axoclamp 2A (Axon Instruments). Oocytes were impaled with 0.5-2 MΩ electrodes filled with 3 M KCl and voltage-clamped at -70 mV. Extracellular ND96 solution was continuously perfused at 3-5 ml/min. Various concentration of the compounds were dissolved in DMSO and diluted in ND96 till the desired concentration, containing a minimum of 0.1% DMSO. All compounds were perfused for a few seconds, and applications were separated by intervals of at least one minute to eliminate receptor desensitization. All electrophysiological experiments were performed at room temperature. Peak amplitudes of the elicited current were measured and related to the 500 μM acetylcholine-induced current on the same oocyte. Data were then fitted with an one site saturation ligand binding curve. Rate of hydrolysis of 3α-(2-hydroxybenzoyloxy)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane iodide (3d) 1.0 mg 3α-(2-hydroxybenzoyloxy)-8,8-dimethyl-8-azoniabicyclo[3.2.1]octane iodide (3d) was dissolved in a mixture of 490 µL human serum, 490 microL PBS buffer pH 7.4 ([NaCl] = 137 mM, [KCl] =2.7 mM, [NaHPO4] = 8.1 mM, [KH2PO4] = 1.8 mM, pH 7.4) and 20 µL of a solution of (S)-naproxen in dmso (2.5 mg/ml) (internal standard). Mixture was incubated at 35 ˚C. Hydrolysis of the ester to salicylic acid was determined using HPLC-MS method I with UV detection at 239 nM by injection of 1µl of the incubation mixture, every hour. The resulting chromatogram was background corrected with a chromatogram resulting from 1 µl injections of a similar incubation mixture lacking compound 3d and naproxen. The conversion of compound 3d into salicylic acid was calculated by dividing the peak area of 3d by the area of 3d at t = 0 whereas both peak areas were corrected by the peak area of the internal standard naproxern. The rate of hydrolysis in pure PBS pH 7.4 buffer was determined using an identical method in which the serum was replaced by 490
L PBS buffer pH 7.4. References 1. Rang, H.P., Dale, M.M., Ritter, J.M. & Moore, P.K. Pharmacology, 5th ed.
Elsevier Science Ltd., 217 (2003). 2. Rang, H.P., Dale, M.M., Ritter, J.M. & Moore, P.K. Pharmacology, 5th ed.
Elsevier Science Ltd., 244 (2003). 3. Rang, H.P., Dale, M.M., Ritter, J.M. & Moore, P.K. Pharmacology, 5th ed.
Elsevier Science Ltd., 248 (2003). 4. Warner, T.D. et al. Nonsteroid drug selectivities for cyclo-oxygenase-1
rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci U S A 96, 7563-8 (1999).

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
156
5. Amer, M., Bead, V.R., Bathon, J., Blumenthal, R.S. & Edwards, D.N. Use
of Nonsteroidal Anti-Inflammatory Drugs in Patients With Cardiovascular Disease A Cautionary Tale. Cardiology in Review 18, 204-212 (2010).
6. Fitzgerald, G.A. Coxibs and cardiovascular disease. N Engl J Med 351, 1709-11 (2004).
7. Halen, P.K., Murumkar, P.R., Giridhar, R. & Yadav, M.R. Prodrug designing of NSAIDs. Mini Rev Med Chem 9, 124-39 (2009).
8. Morphy, R. & Rankovic, Z. Fragments, network biology and designing multiple ligands. Drug Discov Today 12, 156-60 (2007).
9. Morphy, R. & Rankovic, Z. Designing multiple ligands - medicinal chemistry strategies and challenges. Curr Pharm Des 15, 587-600 (2009).
10. Morphy, R. & Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem 48, 6523-43 (2005).
11. Metz, J.T. & Hajduk, P.J. Rational approaches to targeted polypharmacology: creating and navigating protein-ligand interaction networks. Curr Opin Chem Biol 14, 498-504.
12. Hopkins, A.L. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 4, 682-90 (2008).
13. Charlier, C. & Michaux, C. Dual inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) as a new strategy to provide safer non-steroidal anti-inflammatory drugs. Eur J Med Chem 38, 645-59 (2003).
14. Akhter, M. et al. Synthesis and biological evaluation of 2,5-disubstituted 1,3,4-oxadiazole derivatives with both COX and LOX inhibitory activity. J Enzyme Inhib Med Chem.
15. Hibi, S. et al. Novel dual inhibitors of 5-lipoxygenase and thromboxane A2 synthetase: synthesis and structure-activity relationships of 3-pyridylmethyl-substituted 2-amino-6-hydroxybenzothiazole derivatives. J Med Chem 37, 3062-70 (1994).
16. Wallace, J.L., Reuter, B.K. & Cirino, G. Nitric oxide-releasing non-steroidal anti-inflammatory drugs: a novel approach for reducing gastrointestinal toxicity. J Gastroenterol Hepatol 9 Suppl 1, S40-4 (1994).
17. Borovikova, L.V. et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458-62 (2000).
18. Bernik, T.R. et al. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med 195, 781-8 (2002).
19. Tracey, K.J. The inflammatory reflex. Nature 420, 853-9 (2002). 20. Wang, H. et al. Nicotinic acetylcholine receptor alpha7 subunit is an
essential regulator of inflammation. Nature 421, 384-8 (2003). 21. Bencherif, M., Lippiello, P.M., Lucas, R. & Marrero, M.B. Alpha7 nicotinic
receptors as novel therapeutic targets for inflammation-based diseases. Cell Mol Life Sci 68, 931-49 (2010).
22. de Jonge, W.J. & Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 151, 915-29 (2007).
23. van Westerloo, D.J. et al. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 130, 1822-30 (2006).

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
157
24. van Westerloo, D.J. et al. The cholinergic anti-inflammatory pathway
regulates the host response during septic peritonitis. J Infect Dis 191, 2138-48 (2005).
25. de Jonge, W.J. et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 6, 844-51 (2005).
26. Nizri, E. et al. IBU-octyl-cytisine, a novel bifunctional compound eliciting anti-inflammatory and cholinergic activity, ameliorates CNS inflammation by inhibition of T-cell activity. Int Immunopharmacol 7, 1129-39 (2007).
27. Edink, E. et al. Fragment Growing Induces Conformational Changes in Acetylcholine-Binding Protein: A Structural and Thermodynamic Analysis. J Am Chem Soc 133, 5363-5371 (2011).
28. Hopkins, A.L., Groom, C.R. & Alex, A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today 9, 430-1 (2004).
29. Halen, P.K., Chagti, K.K., Giridhar, R. & Yadav, M.R. Combining anticholinergic and anti-inflammatory activities into a single moiety: a novel approach to reduce gastrointestinal toxicity of ibuprofen and ketoprofen. Chem Biol Drug Des 70, 450-5 (2007).
30. Yadav, M.R. et al. Site specific chemical delivery of NSAIDs to inflamed joints: synthesis, biological activity and gamma-imaging studies of quaternary ammonium salts of tropinol esters of some NSAIDs or their active metabolites. Bioorg Med Chem 16, 9443-9 (2008).
31. Celie, P.H. et al. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41, 907-14 (2004).
32. Rucktooa, P., Smit, A.B. & Sixma, T.K. Insight in nAChR subtype selectivity from AChBP crystal structures. Biochem Pharmacol 78, 777-87 (2009).
33. Smit, A.B. et al. Acetylcholine-binding proteins: functional and structural homologs of nicotinic acetylcholine receptors. J Mol Neurosci 30, 9-10 (2006).
34. Sixma, T.K. & Smit, A.B. Acetylcholine binding protein (AChBP): a secreted glial protein that provides a high-resolution model for the extracellular domain of pentameric ligand-gated ion channels. Annu Rev Biophys Biomol Struct 32, 311-34 (2003).
35. Verdonk, M.L., Cole, J.C., Hartshorn, M.J., Murray, C.W. & Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 52, 609-23 (2003).
36. Edink, E. et al. Fragment Growing Induces Conformational Changes in Acetylcholine-Binding Protein: A Structural and Thermodynamic Analysis. J Am Chem Soc 133, 5363-5371 (2011).
37. Olofson, R.A. & Martz, J.T. A New Reagent for the Selective, High-Yield N-Dealkylation of Tertiary Amines: Improved Syntheses of Naltrexone and Nalbuphine. J. Org. Chem. 49, 2081-2082 (1984).
38. de Fiebre, C.M. et al. Characterization of a series of anabaseine-derived compounds reveals that the 3-(4)-dimethylaminocinnamylidine derivative is a selective agonist at neuronal nicotinic alpha 7/125I-alpha-bungarotoxin receptor subtypes. Mol Pharmacol 47, 164-71 (1995).

Chapter 6 Structure-based design of novel NSAID ester prodrugs: Dual targeting of cyclooxygenase-2 (COX-2) and α7 nicotinic receptors
158
39. Stokes, C. et al. The structural basis for GTS-21 selectivity between
human and rat nicotinic alpha7 receptors. Mol Pharmacol 66, 14-24 (2004). 40. Pavlov, V.A. et al. Selective alpha7-nicotinic acetylcholine receptor agonist
GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med 35, 1139-44 (2007).
41. Mitchell, J.A., Akarasereenont, P., Thiemermann, C., Flower, R.J. & Vane, J.R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci U S A 90, 11693-7 (1993).
42. Wu, K.K., Sanduja, R., Tsai, A.L., Ferhanoglu, B. & Loose-Mitchell, D.S. Aspirin inhibits interleukin 1-induced prostaglandin H synthase expression in cultured endothelial cells. Proc Natl Acad Sci U S A 88, 2384-7 (1991).
43. Xu, X.M. et al. Suppression of inducible cyclooxygenase 2 gene transcription by aspirin and sodium salicylate. Proc Natl Acad Sci U S A 96, 5292-7 (1999).
44. Lipinski, C.A., Lombardo, F., Dominy, B.W. & Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46, 3-26 (2001).
45. Charpantier, E. et al. Alpha7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J Neurosci 25, 9836-49 (2005).

159
Chapter 7
Summary and conclusions
Ewald Edink, Rob Leurs and Iwan J.P. de Esch

Chapter 7 Summary and conclusions
160
1. Research aims The work described in this thesis focuses on structure- and fragment-based approaches on acetylcholine-binding protein (AChBP). This water-soluble pentameric protein is widely recognized and used as a structural homolog of the ligand-binding domain (LBD) of the nicotinic acetylcholine receptor (nAChR).
1,2 Due
to its water-soluble nature, AChBP provides relative easy access to X-ray co-crystal structures enabling structure-based optimization. Moreover, compared to its structural analog, the membrane bound nAChR, AChBP is easier to incorporate in biological assays and better suited for studying ligand-protein interactions with biophysical techniques such as surface plasmon resonance (SPR) biosensor analysis and isothermal titration calorimetry (ITC). For these reasons, we have embarked on a fundamental study in which AChBP is used to increase our knowledge on how to efficiently optimize fragment- and virtual screening hits and in the process gain more understanding on ligand-protein interactions. In addition, we have investigated which techniques are most suitable for monitoring the hit optimization process. While generating new compounds to increase our molecular insights in ligand-AChBP molecular interactions, we also considered translational aspects by comparing our AChBP findings with the therapeutically relevant nicotinic acetylcholine receptors. At the beginning of this project, we set ourselves with the following research aims:
Increase our knowledge on how to efficiently optimize fragments towards high affinity binders using AChBP as a model protein.
Investigate if thermodynamic analysis can contribute to a more efficient fragment-optimization process using AChBP as a model protein.
Investigate if our fragment-optimization studies on AChBP can contribute to our understanding on how to design subtype-selective ligands for the therapeutically relevant human nAChRs.
In this final chapter, the most important findings in this thesis will be summarized and the outcome of the research aims will be evaluated.
2 Efficient fragment hit optimization The water-soluble nature of AChBP facilitates studying ligand-protein interactions and having x-ray co-crystal structures at hand, we decided to embark on a structure-based optimization project. The aim of this project was to increase our understanding on how to efficiently optimize fragment- and virtual screening hits. Several aspects are known to complicate fragment hit optimization. For example, maintaining the original binding mode of the fragment hit throughout the optimization process is often difficult as the ligand adjusts the orientation of the different substructures to find the pose that is the energetically most favorable pose for the complete molecule.
3,4 At the same time, the protein target can adjust its
conformation, leading to induced-fit phenomena.5,6
The conformational changes of the protein are difficult to predict and most computational tools consider the protein as rigid entities.
7 Acurate first principle (QM) calculations are possible for bigger
systems, such as AChBP-fragment complexes but these studies remain computationally too expensive to guide chemistry programs.

Chapter 7 Summary and conclusions
161
Another factor that complicates fragment optimization is that establishing additional interactions with the protein binding site not always translates into higher binding affinities. This is because binding affinity does not correlate directly to the physical forces that drive complex formation (enthalpy) but rather quantifies an average value that also originates from conformational changes of the ligand and its binding partner and changes in their solvation shells (entropy) (Chapter 3). Interestingly, biophysical approaches, such as the well established isothermal titration calorimetry (ITC) and the newer surface plasmon resonance (SPR) biosensor analysis, are able to determine the thermodynamical aspects of interactions, and allows dissection of binding affinity into the separate enthalpic and entropic contributions. Biophysical analysis of molecular recognition events will therefore increase our understanding of ligand-protein interactions and may ultimately lead to a more efficient fragment optimization process. Clearly, structure- and fragment-based approaches can reach a next level of efficiency if the dynamic and thermodynamic aspects of ligand-receptor binding can be addressed. 2.1 Conformational changes induced by fragment growing By comparison of previously obtained AChBP co-crystal structures with an X-ray structure of lobeline bound to Ac-AChBP, an interesting conformational change of the binding site was observed that leads to the opening of a subpocket, enabling the binding of the α-hydroxyphenetyl moiety of the ligand. This subpocket, referred to as the lobeline-pocket, becomes accessible after a change in rotameric state of Tyr91 (g- to t conformation
8, hereafter referred to as the tyrosine-flip). At the time
the various crystal structures became available, lobeline was the only ligand known to be able to induce this conformational change in the protein and address this sub-pocket. Considering the partially overlapping binding modes of fragment 1 and lobeline (2), we designed a fragment growing optimization study to induce the tyrosine-flip and grow the fragment into the lobeline-pocket (Figure 1). This structure-based fragment optimization study that is described in Chapter 4 was found to be an efficient method to increase binding affinity for Ac-AChBP. The compound designed in the first iteration displayed a 50-fold improvement in binding affinity (pKi = 7.0, Table 1) compared to the starting fragment. The successful insertion into the ligand-inducible lobeline-pocket was confirmed by an X-ray co-crystal structure and the experimentally determined binding mode corresponded reasonably well to the predicted binding mode that was obtained by molecular docking (rmsd of 1.1 Å). The most pronounced deviation was that in the experimentally determined binding mode the hydroxyl group of compound (3) was not engaged in hydrogen bonding to the backbone carbonyl oxygen atom of Ser144 or Trp145, but instead was facing the hydrophobic ring of Tyr193 (Figure 2). Since the hydroxyl group of the optimized fragment 3 is not involved in formation of hydrogen bonds with the binding site, it was expected that its removal would lower the desolvation penalty and thereby result in an increase in binding affinity. Indeed, nor-hydroxyl derivative 4 exhibited higher Ac-AChBP affinity (pKi = 7.5, Table 1) than 3 and when compared with the starting fragment 1, a 150-fold increase in binding affinity was obtained (Figure 4). To determine if optimized fragment 4 induces the tyrosine-flip and interacts with the lobeline-pocket, an additional co-crystal structure was generated that shows an almost identical binding mode to 3

Chapter 7 Summary and conclusions
162
Figure 1. Fragment optimization strategy. Surface representations of the crystal structures of fragment 1-bound Ac-AChBP (a) and of lobeline-bound Ac-AChBP (PDB 2BYS)
16 (b). (a) In the fragment 1-Ac-AChBP complex, Tyr91 is stabilized in
the g- conformation through a hydrogen bond with Ser144, rendering the lobeline-pocket inaccessible. (b) However, Tyr91 adopts a t conformation in the lobeline-Ac-AChBP complex interacting with Tyr53 and Ser165 through hydrogen bonds, thus leading to the opening of the lobeline-pocket. (c) The superposition of the fragment 1 and lobeline (2) molecules indicates that the fragment may be grown into the lobeline-pocket by extending its tropine nitrogen atom with the α-hydroxyphenetyl moiety of lobeline generating compound (3).A version of this figure in full color is shown on page 63.

Chapter 7 Summary and conclusions
163
Figure 2. Predicted binding mode (light grey-colored sticks) versus the experimentally determined binding mode (dark grey-colored sticks) of optimized fragment 3 (rmsd = 1.1 Ǻ). The orientation of the α-hydroxyl moiety is different than was predicted by molecular docking. Instead of hydrogen bond formation with the carbonyl backbone of Ser144 (predicted hydrogen bonds are shown in black dashed lines), it facing a lipophilic part of the binding site; Tyr193. (rmsd of 0.6 Å), exemplifying that a hydroxyl functionality is not required for opening of and insertion into the lobeline-pocket. In the performed hit optimization procedures, we found that that binding affinity for AChBP could be readily increased by the introduction of lipophilic phenetyl moieties. It was also found that optimization by the formation of additional hydrogen bonds with the binding site residues was substantially more difficult. Molecular docking studies had suggested that incorporation of hydroxyl substituents at the α- or meta positions of the N-phenetyl substituents of the dibenzosuberyl as well as the benzoate series could enable the formation of additional hydrogen bonds with the lobeline-pocket (Chapter 5). However, for both series of compounds, introduction of hydroxyl moieties at the α- or meta-positions of the N-phenetyl moiety diminished binding affinity for Ls-AChBP as well as Ac-AChBP (Table 1 and 2). For Ls-AChBP and the dibenzosuberyl substituted tropines, this can be explained by a different binding mode than predicted, i.e., no interaction of the N-phenetyl moiety with the lobeline-pocket (see section 2.2 in this chapter). However, for substituted tropine benzoates 3 and 4, it has been shown by X-ray co-crystal structures that their N-phenetyl moieties were interacting with

Chapter 7 Summary and conclusions
164
Table 1. The binding affinity (pKi) of benzoate substituted tropines
Cpd R1 R2 Ls-AChBP
pKi ± SEMa
Ac-AChBP
pKi ± SEMa
α7
pKi ± SEMb
α4β2
pKi ± SEMa
Nicotine 6.5 ± 0.1 5.6 ± 0.1 6.0 ± 0.1 7.9 ± 0.1
α-lobeline 6.2 ± 0.1 8.6 ± 0.1 5.1 ± 0.1 8.3 ± 0.1
1 H H 6.1 ± 0.1 5.3 ± 0.1 5.5 ± 0.1 < 4.5
3
H 6.1 ± 0.1 7.0 ± 0.1 4.9 ± 0.1c
< 4.5c
4
H 7.1 ± 0.1 7.5 ± 0.1 5.1 ± 0.1 < 4.5
5
Me 6.9 ± 0.1 6.1 ± 0.1 5.0 ± 0.1c
< 4.5c
a) [3H]epibatidine displacement studies, pH 7.4; b) [
3H]MLA displacement studies, pH 7.4; c)
tested as a racemic mixture n.d. = not determined; Cpd = compound
the lobeline-pocket in Ac-AChBP (Chapter 4). As discussed in Chapter 3, it can be anticipated that due to desolvation penalties, the positioning of hydrogen-bonding groups needs to be near-optimal in order to be beneficial in terms of binding affinity.
9,10 Apparently, upon introduction of one or two meta-hydroxyl
groups to benzoate ester 3, the desolvation penalty dominates. This indicates that the positioning of the α-hydroxyphenetyl moiety in the lobeline-pocket does not allow for strong hydrogen bond formation with the carbonyl backbone of Thr89 and/or the side chain of Asp195. Interestingly, ionic and hydrogen bond interactions with Asp195 have been observed for α-conotoxins in complex with Ac-AChBP with Tyr91 in a g- conformation (closed lobeline-pocket, PDB: 2UZ6 and 2BYP), showing that it is possible for ligands to interact with this residue.
11,12
In these studies that explore structure-activity relationships between different series of compounds, we found the use of Group Efficiency (GE) measurements very useful. For Ls-AChBP, the GE‟s of the tropine substituents are almost identical for the dibenzosuberyl and benzoate series (Figure 3). These similar GE‟s that were observed for modifications on the tropine nitrogen atom of the dibenzosuberyl- and benzoate derivatives, are indicative of similar binding modes between the two compound series in Ls-AChBP. In contrast, for Ac-AChBP clear GE differences are

Chapter 7 Summary and conclusions
165
Table 2. The binding affinity (pKi) of dibenzosuberyl substituted tropines
Cpd R1 R2
Ls-AChBP
pKi ± SEMa
Ac-AChBP
pKi ± SEMa
α7
pKi ± SEMb
α4β2
pKi ± SEMa
6
H 7.0 ± 0.1 5.0 ± 0.1 4.9 ± 0.1 < 4.5
7
H 5.7 ± 0.1 4.9 ± 0.1 4.7 ± 0.1 < 4.5
8
H 5.7 ± 0.1 4.5 ± 0.1 4.4 ± 0.1 < 4.5
a) [3H]epibatidine displacement studies, pH 7.4; b) [3H]MLA displacement studies, pH 7.4; n.d. = not determined; Cpd = compound
observed when focusing on the tropine substituent, indicative of different binding modes between the two compound series. Since we have provided structural evidence that benzoate ester 4 is interacting with the lobeline-pocket in Ac-AChBP (2Y57.pdb, Chapter 4),
13 a likely explanation for the observed SAR differences
between the compound series is that dibenzosuberyl ether 6 is not interacting with the lobeline-pocket. In Ls-AChBP, the N-phenetyl substituents of the dibenzosuberyl- and benzoate- substituted ligands are likely to be accommodated by a (hydrophobic) part of the binding site different than the lobeline pocket.

Chapter 7 Summary and conclusions
166
Figure 3. Calculation of ligand efficiency (LE) and group efficiency (GE) allows easy comparison of and the average affinity contributions per heavy atom of compounds and functional groups of different sizes. Comparison of GE’s underlines that there are clear SAR differences between both proteins and the two compound series, indicative of binding mode differences. For each compound, the LE and EE have been calculated in kcal·mol-
1 per heavy atom. ΔG
0 = Gibbs
energy change, and HA = number of heavy atoms. 2.2 Thermodynamic analysis in Fragment-based drug discovery In Chapter 3, we analyzed a significant number of datasets and studied the information that can be gained from thermodynamic analysis in a fragment-based drug discovery (FBDD) context. The small size of fragments simplifies the process of relating the thermodynamic profile of fragments to the interactions made during binding. When combined with structural data, the stepwise fragment growing process provides an ideal dataset with which to improve our understanding of the thermodynamics of binding. In this manner both FBDD, and our understanding of the thermodynamics of binding, have much to gain from the application of thermodynamic analysis in FBDD. As exemplified by our studies on AChBP (Chapter 4 and 5), enthalpic optimization by the formation of additional polar interactions with the binding site is a difficult route to improve binding affinity. A poorly optimized polar interaction may not simply provide a lower favorable contribution to the enthalpy change on binding, it may actually decrease binding affinity. Desolvation of polar functionalities comes at a cost, which can only be overcome when the positioning of interacting groups obeys strict angle and distance requirements.
10,14 Nevertheless, due to the

Chapter 7 Summary and conclusions
167
specificity of polar interactions, enthalpy driven ligand binding may provide a major selectivity advantage. The examples of HIV-protease inhibitors and cholesterol lowering statins illustrate that the best in class compounds had been better optimized with respect to enthalpy compared to the first in class compounds (Chapter 3).
15 While enthalpic optimization can provide highly selective high affinity
drugs, medicinal chemists tend to optimize entropy, as shown in a recent study comparing synthetic and natural drugs.
16 Unfortunately, too much focus on entropic
optimization by constraining ligands in their bioactive conformation, and by the addition of hydrophobic groups, may in the end result in poorly soluble compounds, with reduced selectivity and higher chances of attrition.
17,18 Very appropriately,
Michael Hann has recently introduced the term “Molecular Obesity” to describe our tendency to build potency into molecules by the inappropriate use of lipophilicity, leading to the premature demise of drug candidates.
19
Since enthalpic optimization is difficult to achieve, choosing a fragment in which binding is enthalpically driven as a starting point and adding hydrophobic groups during optimization, may provide the easiest route to a high affinity compound with favorable changes in both enthalpy and entropy and a reduced risk of attrition. In this respect, nicotine can be considered a fragment that is better suited for further optimization than the fragment that we optimized in Chapter 4, both in terms of ligand efficiency (LE) and enthalpic efficiency (EE). Since we had not determined the thermodynamic profiles before initiating our fragment optimization study, the thermodynamic profiles of binding were not taken into account upon selecting which fragment to pursue. At that time, fragment 1 was selected as it represented a novel chemotype with affinity for AChBP. Nevertheless, the future synthesis of a hybrid molecule of nicotine and lobeline would be a nice opportunity to challenge the above consideration. In order to increase our understanding of the thermodynamic aspects of fragment-protein binding, we have constructed a database containing 162 unique ligand-protein complexes in which the molecular weight of the ligand was smaller than 300 Da (Chapter 3). The thermodynamic data that was collected using ITC is summarized in Figure 4a and data that was obtained using van „t Hoff analysis is summarized in Figure 4b. Several trends were observed from the enthalpy-entropy plots depicted in Figure 4. First of all, data obtained using van „t Hoff analysis is characterized by a greater amount of entropy driven binding. This is not a trend resulting from the techniques, but rather a trend resulting from the different targets being tested. The van ‟t Hoff experiments were almost exclusively performed on membrane-bound receptors, while the ITC experiments tend to be performed on water-soluble enzymes. The trend of wider distribution of thermodynamic binding signatures in the case of membrane-bound receptors is likely to originate from the conformational changes that are required for receptors to switch between active and inactive states. This explanation is in line with an additional trend that is visible in Figure 4b; the thermodynamic discrimination between agonists and antagonists in many of the tested receptors. This phenomenon, in which agonist binding to a receptor is entropy-driven and the binding of its antagonist is enthalpy-driven, or vice versa, has been observed for different receptor families such as G-protein-coupled

Chapter 7 Summary and conclusions
168
receptors (GPCRs), and ligand-gated ion channels (LGICs).20-27
The phenomenon of thermodynamic discrimination illustrates that fragments that exhibit enthalpy driven complex formation are not always the preferred candidates for further optimization. The preferred thermodynamic profile of the fragment may depend on the target protein being an enzyme, or a receptor, and on what kind of functional profile (antagonist or (partial) agonist) is required to obtain the anticipated therapeutic effect. A third trend that can be observed is that the distribution in Gibbs energy of binding (ΔΔG° = 11 kcal·mol
-1) is significantly smaller than the
distribution in enthalpy and entropy (ΔΔH° = TΔΔS° = 63 kcal·mol-1
). These differences reflect enthalpy-entropy compensation; large favorable changes in enthalpy are compensated by unfavorable changes in entropy, and vice versa. The analysis described in Chapter 3 also revealed that sometimes fragment growing can result in simultaneous enthalpic and entropic optimization. An example on the adenosine A1 receptor illustrates that enormous increases in binding affinity (900-fold) can be obtained by the introduction of relative small substituents (cyclopentyl) (Figure 5).
21 The unusual simultaneous optimization of enthalpy and
entropy by extending the fragment with a cyclopentyl moiety may result from additional flexibility in the ligand-protein complex (favorable ΔS°) from which the loss in enthalpy by enthalpy-entropy compensation is overcompensated by the formation of strong enthalpic van der Waals interactions. An alternative explanation may be that the introduction of the cyclopentyl substituent, displaces water molecules from a hydrophobic subpocket that are not engaged in hydrogen bonds prior to complex formation. As optimization of both thermodynamic binding parameters at the same time is the most efficient way of increasing binding affinity, this specific example warrants further investigation. For example, the generation of X-ray co-crystal structures of the starting fragment and its 8-cyclopentyl substituted derivative may provide insights in how to successfully escape from enthalpy-entropy compensation and thereby optimize compounds in the most efficient manner.

Chapter 7 Summary and conclusions
169
Figure 4: Enthalpy vs. entropy plot of ligand-protein complexes that were obtained using ITC (a) and van ‘t Hoff analysis (b). The two diagonal lines show Gibbs energy changes of -4.1 kcal·mol
-1 and -12.4 kcal·mol
-1 equivalent to affinities of 1
mM and 1 nM, respectively. A version of this figure in full color is shown on page 47.

Chapter 7 Summary and conclusions
170
Figure 5: Changes in Gibbs energy (ΔΔG
0), enthalpy (ΔΔH
0) and entropy (-TΔΔS
0)
in kcal·mol-1
upon growing the fragment theophylline towards the high affinity adenosine A1 antagonist CPT. For each compound, the ligand efficiency (LE), enthalpic efficiency (EE) and group efficiency (GE) have been calculated in kcal·mol
-1 per heavy atom.
Polar contacts are generally associated with enthalpic rather than entropic interactions, and as a consequence, enthalpic binders are likely to be more hydrophilic than entropic binders. One may therefore argue that keeping control of lipophilicity, for example by taking LE as well as logP into account, is likely to be as useful and less complicated than the determination of thermodynamic binding parameters.
28 However, as exemplified in Chapter 3 and 4 this is not always true.
In our work that is described in Chapter 4 we have shown that enthalpic optimization can also be established by the incorporation of lipophilic substituents. Moreover, in Chapter 3, a thermodynamic study on carbonic anhydrase is analyzed, that shows that selecting the more enthalpy efficient (12) instead of the most ligand efficient fragment (14) for further optimization from two fragments with similar logP values, may ultimately result in a more potent compound with a better thermodynamic profile (compare 15 and 17, Figure 6). These examples show that, compared to binding affinity data alone, thermodynamic data can provide additional information and can be utilized in a more efficient hit optimization process.

Chapter 7 Summary and conclusions
171
Figure 6: Changes in Gibbs energy (ΔΔG
0), enthalpy (ΔΔH
0) and entropy (-TΔΔS
0)
in kcal·mol-1
upon growing benzene sulfonamide (6) towards more potent carbonic anhydrase inhibitors. For each compound, the LE and EE have been calculated in kcal·mol
-1 per heavy atom.

Chapter 7 Summary and conclusions
172
2.3 Thermodynamic analysis provides novel insights in ligand recognition Our site-directed mutagenesis work on AChBP suggest differences in accessibility of the lobeline-pocket and therefore indicates that the benzoate esters 3 and 4 are likely to exhibit different binding modes between Ac- and Ls-AChBP. Nevertheless, ligand 4 binds with similar affinity to Ac-AChBP and Ls-AChBP. The affinity data therefore do not provide any indication for a difference in binding modes. However, the thermodynamic analysis that is described in Chapter 4, reveals significant differences that are indicative of different binding modes between both protein species (Figure 7). We therefore conclude that more than affinity data alone, dissection of binding affinity into the separate enthalpic and entropic contributions provides valuable information with regards to the binding mode of a ligand and has therefore a high added value in structure- and fragment-based hit optimization. Another interesting observation from the thermodynamic analysis experiments in Chapter 4 is that the favorable change in enthalpy upon growing fragment 1 into the lobeline-pocket is partially compensated by an entropic penalty, illustrative of the phenomenon known as enthalpy-entropy compensation. In our case, it is likely that the favorable enthalpic interactions that are made upon growing into the lobeline-pocket, restrict the ligand as well as the protein binding site in their conformational flexibility, resulting in the observed entropic penalty. Furthermore, comparing the thermodynamic profiles of binding to Ac-AChBP of optimized fragments 3 and 4 shows that binding of 4 is driven by more enthalpic contributions than its hydroxyl substituted analog 3 (Chapter 4). This difference in thermodynamic binding profiles is likely to originate from a desolvation penalty for 3 from which its hydroxyl group is not engaged in hydrogen bonds with the AChBP binding site (Figure 2). Inspection of the co-crystal structures of lobeline and optimized fragment 4 with Ac-AChBP reveals that lobeline is involved in four hydrogen bonds with the Ac-AChBP binding site whereas 4 only forms one hydrogen bond. Therefore, one would expect that the binding of lobeline that is driven by extensive polar interactions would be characterized by more favorable changes in enthalpy than 4. Surprisingly, this is not the case (Chapter 4). This moderate enthalpy driven binding of lobeline to Ac-AChBP may be explained by a more flexible lobeline-Ac-AChBP complex compared to the compound 3-Ac-AChBP complex, reducing enthalpic contributions but also preventing a significant entropic penalty. Accordingly, lobeline can be considered a more flexible molecule than 4 as it contains more rotable bonds and the center of the molecule consists of a flexible piperidine ring instead of a rigid bicyclic tropane moiety. As a consequence, lobeline may be better able to adopt to the dynamic movements of the Ac-AChBP binding site (low entropic penalty) whereas the more rigid compound 4 locks the protein in a minimal ensemble of conformations (high entropic penalty). As lobeline binds to Ac-AChBP with higher affinity than 4, these results are in line with the hypothesis postulated by Williams and Whitesides that a so-called „sloppy‟ fit, one that is sufficiently complementary in shape to allow some favorable enthalpy, but loose enough not to be entropically too unfavorable may result in the strongest associations.
29,30

Chapter 7 Summary and conclusions
173
Figure 7. Thermodynamic analysis provides indication of successful insertion into the lobeline-pocket. Chemical structures and binding modes as determined by X-ray analysis of Ac-AChBP co-crystal structures and the thermodynamic binding signatures for Ac-AChBP and Ls-AChBP of optimized fragment 4 (a) fragment 1 (b) and quaternary ammonium derivative 5 (c) as determined by SPR biosensor analysis (dark bars ± SEM) and ITC (light bars ± fitting errors). Shown are the changes that occur upon ligand binding in Gibbs energy (∆G˚) (SPR: dark blue; ITC: light blue), enthalpy (∆H˚) (SPR: dark green; ITC: light green) and entropic contributions (-T∆S˚) (SPR: dark red; ITC: light red). All thermodynamic parameters shown are in kcal·mol
-1. A version of this figure in full color is shown on page 72.

Chapter 7 Summary and conclusions
174
The obtained thermodynamic results also show that the fundamentally different biophysical techniques van „t Hoff analysis (by SPR) and ITC result in very comparable thermodynamic binding profiles (Figure 8). Previously, discrepancies between calorimetry and van „t Hoff analysis have been reported,
31-35 but our
current work and other recent comparison studies36-39
show that application of both techniques result in similar values for the thermodynamic binding parameters (Chapter 3 and 4), at least when using these protein targets and sets of compounds. When comparing both techniques, the main advantages of ITC experiments are that set-up is straight forward, as calorimetric measurements do not require immobilization or labeling. Furthermore, all binding parameters (n, KB, ΔH
o and ΔS
o) are determined in a single experiment. A major disadvantage is the
large amounts of protein needed for a single experiment. If the target protein is more difficult to obtain, SPR biosensor analysis is a more sensible technique to use, as the protein consumption is very low. SPR biosensor analysis has an additional advantage of being able to provide information about the kinetic parameters of binding (kon and koff). This enables combining kinetic and thermodynamic data providing insights into transition state thermodynamics, e.g., enthalpy of association. Unfortunately, both ITC- and SPR biosensor-based thermodynamic analysis are hampered by low throughput and are therefore not suitable for primary screening purposes. Instead, these techniques are more appropriate for secondary screening and are excellent ways for hit validation and to obtain more information about the process of binding by characterization of the thermodynamic binding profile.
3 AChBP: A close mimic or far homolog to the nAChR LBD?
The first chapter provides an introduction to the nAChRs and their water-soluble LBD structural homolog, AChBP. In Chapter 1, various studies are summarized that exemplify how AChBP X-ray structures have significantly improved our understanding on the overall molecular structure of the nAChR LBD and its molecular interactions with nicotinic ligands. Particularly, the validation of AChBP-derived hypotheses by site-directed mutagenesis with unnatural amino acids in nAChRs is a powerful method to increase our understanding on the nAChR LBD.
40-
43 Examples of insights in the interactions between ligands and the nicotinic
receptors that have been obtained using this strategy include; the formation of cation-π interactions of cationic ligands with specific aromatic residues within the binding pocket,
40,41 the formation of charged hydrogen bonds with the carbonyl
backbone oxygen atom of Trp145 (Ls-AChBP numbering),41
and the formation of a hydrogen bond between the pyridine nitrogen atom of nicotine and a conserved water molecule (Figure 8).
42
An important issue that has been raised in this thesis, is the role of amino acid residues located outside the first shell of binding site residues in molecular recognition processes. It is tempting to focus solely on residues directly aligning the protein binding site but site-directed mutagenesis work on AChBP (see also Chapter 4) as well as nAChRs illustrate that in certain cases amino acid residues in the second or third shell should be considered as well. Structural comparisons between AChBPs originating from different species revealed differences in stabilization of the ligand-induced tyrosine-flip. Site-directed mutagenesis

Chapter 7 Summary and conclusions
175
experiments were performed in which the tyrosine-flip stabilizing residues in Ac-AChBP were substituted for their Ls-AChBP counterparts (Figure 9). The results strongly indicate that the side chain of a serine residue (Ser165) is essential for stabilizing the tyrosine-flip and that the lobeline-pocket is less accessible in Ls-AChBP compared to Ac-AChBP. It is worth mentioning that the residues that putatively stabilize the tyrosine-flip are not directly aligning the binding site (i.e., are not in contact with ligands) and the obtained results therefore underline the important role that residues outside the first shell of binding site residues may play in ligand recognition. In line with the site-directed mutagenesis results is the affinity data that is depicted in Table 1. Ligands that are known to interact with the lobeline-pocket in Ac-AChBP (2, 3 and 4) have higher affinity for Ac-AChBP whereas ligands that do not interact with the lobeline-pocket (nicotine, 1 and 5) have higher affinity for Ls-AChBP. Thus, our investigations reveal that growing the fragment 1 into the lobeline-pocket shifts the species preference towards Ac-AChBP whereas introduction of an additional methyl substituent preventing interactions with the lobeline-pocket reverses the preference back to Ls-AChBP. These subtle differences in protein conformational changes that induce the lobeline-pocket may be of interest in the design of subtype-selective ligands for human nicotinic receptors as well. The gatekeeper tyrosine residue is conserved amongst the human nAChR subtypes whereas the residue (Ser165 in Ac-AChBP) that stabilizes the open lobeline-pocket conformation is located in a highly variable region (Chapter 1). As can be seen in Table 1, lobeline exerts a 250-fold selectivity for Ac-AChBP (accessible lobeline-pocket) over Ls-AChBP (inaccessible lobeline-pocket) and ~1000-fold selectivity for α4β2 over α7 nAChRs. Differential stabilization of the rotameric states of the gatekeeper tyrosine residue may provide an explanation for the observed nAChR subtype selectivity of lobeline and the lobeline-pocket may therefore be targeted as a nAChR subtype-selectivity pocket, as well. Alas, our initial site-directed mutagenesis studies that were aimed to validate this hypothesis give ambiguous results (unpublished results), indicating that other differences in the ligand binding pockets may play a complementary role. Additional experiments using site-directed mutagenesis and/or molecular probes are therefore required to determine if the lobeline-pocket may be of use in addressing nAChR subtype selectivity. Another example of the important role that amino acid outside the first shell of amino acid residues aligning the binding site may play in nAChR subtype selectivity was already discussed in Chapter 1. A backbone mediated hydrogen bond between two residues in the second shell is likely to be responsible for shaping the α4β2 nAChR binding site in such a way that nicotine establishes a very strong cation-π interaction with a tryptophan residue (Figure 8). These findings are in line with the selectivity of nicotine for the α4β2 over the α7 and muscle nAChR subtypes.

Chapter 7 Summary and conclusions
176
Figure 8: X-ray structure of Ls-AChBP in complex with nicotine (PDB: 1UW6) shows that the formation of a cation-π interaction between the charged pyrolidine nitrogen atom and the aromatic side chain of Trp143 is a major contributor to complex formation. In addition, two hydrogen bonds (black dashed lines) with the AChBP binding site are made upon binding. A charged hydrogen bond is established between the positively charged pyrolidine nitrogen atom of nicotine and the backbone carbonyl oxygen atom of Trp143 and the pyridine nitrogen atom is involved in hydrogen bond formation with a conserved water molecule. The residues Ser147 and Ala191 are engaged in a backbone mediated hydrogen bond (black dashed line indicated by the white arrow) between Ser147 and Ala191. Combining the results of molecular dynamics and site-directed mutagenesis experiments indicates that compared to the muscle-type (Ser147 = Gly175) and α7 nAChR (Ser147 = Gly148), this hydrogen bond is much stronger in the α4β2 nAChR (Ser147 = Lys150), shaping the binding pocket in such a way that nicotine establishes a very strong cation-π interaction with Trp143.

Chapter 7 Summary and conclusions
177
Figure 9. AChBP species differences in stabilization of the tyrosine-flip. The superposition of crystal structures of Ac-AChBP (in light grey) and Ls-AChBP (in dark grey) suggests that Trp53 and Tyr164 in Ls-AChBP (represented as blue sticks) cannot stabilize the tyrosine-flip contrary to Tyr53 and Ser165 in Ac-AChBP. A colored version of this figure is shown on page 71. It should be noted that due to its moderate overall sequence identity (24% with the human α7 nAChR),
44 AChBP cannot be considered an exact water-soluble mimic
of the nicotinic receptor LBD. Some of the work that is discussed in this thesis, exemplifies the limitations of AChBP in providing a structural template for nAChRs. Previously, using a virtual screening exercise based on AChBP X-ray structures, we have found that only a limited amount of AChBP derived in silico hits displays affinity for the α7 nAChR and none of the hits bind to the less homologous α4β2 nAChR LBD.
45 In our current work, we have found that ligands that have been
optimized for AChBP do not display affinity for the α4β2 nAChR subtype and that the SAR that was identified for AChBP often does not correlate with SAR for the α7 nAChRs (Chapter 5 and Table 1 in this chapter). Nevertheless, until structures of nAChR LBDs become available, AChBP can be considered a valuable tool to elucidate general features of the molecular structure of nicotinic receptors. However, when using AChBP as a structural template for nAChRs, one should be aware of its limitations and validate AChBP-derived hypotheses in the membrane bound nicotinic receptors. Obviously, an alternative to using AChBP directly for nAChRs research is to use the structural AChBP data for homology modeling. However, homology models often have small inaccuracies that compromise the success of the computational approaches. More importantly for the studies described in this thesis is that we would have lost the ability to validate and guide the fragment- and structure-based approaches by using the water soluble AChBP that allows structural and

Chapter 7 Summary and conclusions
178
biophysical evaluation next to determining binding affinities of the reference and the novel ligands. In an effort to combine the best of both worlds (i.e., nAChR ligand binding characteristics and water soluble AChBP properties), mutant AChBP proteins are being constructed and tested in our labs that are designed to better mimic the human nAChRs, but until now the results have been ambiguous when evaluating panels of reference nAChRs ligands (unpublished results). Until better nAChRs mimics are available, the direct use of AChBP as bait for nAChRs remains problematic. Ultimately, it can be anticipated that LBD or full length receptors of the nAChR subtypes themselves can be crystallized. A similar revolution is now taking place for the other membrane-bound receptors that are often targeted in drug development programs as in recent months several structures for G-protein coupled receptors have been elucidated.
46-50 At that time when more detailed
structural information for human nAChRs becomes available, the computational protocols and approaches described in this thesis can be revisited by replacing the AChBP structural information with the nAChRs data. 4 Fragment optimization towards dual-action NSAID ester prodrugs In the final chapter of this thesis, we have used crystal structure data of acetylcholine-binding protein (AChBP), to design NSAID ester prodrugs that are
capable of activating 7 nicotinic acetylcholine receptors (nAChRs) (Chapter 6). The α7 nAChR is expressed on macrophages and other immune cells and has recently been identified as a target for treating inflammation-related disorders.
51,52
Thus, by incorporating α7 nAChR activity, we hope to obtain dual-action anti-
inflammatory NSAID prodrugs. After suppressing inflammatory disorders via the 7 nicotinic receptor, hydrolysis of the prodrug will release the parent NSAID that will exert an additional anti-inflammatory effect by acting on COX-2. In chapter 4 and 5, we have described a fragment (1, Chapter 4) that exhibits good ligand efficiency
53
(LE = 0.43) for AChBP, as well as for the 7 nicotinic receptor (LE = 0.44, Chapter 5). This fragment is an ester of benzoic acid and nortropinol. Since the fragment
displays affinity for the 7 receptor and the benzoic acid part exhibits high structural resemblance to certain NSAIDs or structural fragments of NSAIDs, we investigated if esterification of NSAIDs with tropine-like moieties would afford
NSAID prodrugs with intrinsic 7 nAChR activity, see Figure 10. To select which NSAIDs are most suitable for modification towards α7 nAChR activation we have used molecular docking and X-ray structures of acetylcholine-binding protein (AChBP) in complex with (partial) agonists for the α7 nAChRs.
54
Although our later work has given indications that AChBP is compromised for use in structure-based optimization of ligands for the human nAChRs, at that time it was anticipated that AChBP can still be used to discriminate binders from non-binders of the α7 nicotinic receptors and therefore useful in designing the NSAID ester prodrugs. Based on the in silico results, a selection of NSAIDs was chemically modified with tropine moieties to obtain ester prodrugs with potential α7 nAChR activity.

Chapter 7 Summary and conclusions
179
Figure 10: Fragment VUF10663 and the 5 NSAID derivatives that were selected
in order to obtain NSAID prodrugs with an additional 7 nAChR mediated anti-inflammatory effect. Radioligand displacement studies showed that all designed NSAID tropinyl esters
exhibit affinity for the 7 nAChR (pKi > 4.8). Using electrophysiological recordings from Xenopus leavis oocytes expressing the human α7 nAChR, two salicylate
tropine esters were identified that acted as 7 partial agonists and induce maximal currents of 7-10% compared to the maximal current that is induced by the endogenous ligand acetylcholine. It is not known how much receptor activation is required for an α7 nicotinic partial agonist to reduce the release of proinflammatory mediators from macrophages in vivo. This minimal efficacy is likely to be dependent on the frequency of acetylcholine release near macrophages by the “cholinergic anti-inflammatory pathway”. When this frequency is relatively high, a low efficacious agonist that is bound to the receptor, prevents acetylcholine from activating the α7 receptor and may not lower but instead increase the release of proinflammatory mediators from immune cells. On the other hand, if the frequency of “cholinergic anti-inflammatory pathway” stimulation is low, which is likely to be the case in chronic inflammatory diseases, a low efficacious agonist will still be able to lower the release of proinflammatory cytokines from macrophages and other immune cells. Having identified esters of salicylic acid that are capable of activating the α7 nAChR, we subsequently investigated if the ester linkage is likely to be hydrolyzed in vivo. We found that the salicylate ester prodrug is likely to be present long enough to exert its effect on the α7 nicotinic receptor but eventually will be hydrolyzed to release salicylic acid producing an additional anti-inflammatory effect by affecting COX-2. Thus, the salicylate ester prodrugs are likely to exhibit a dual anti-inflammatory action combined with an improved GI toxicity profile. At the moment of finalizing this thesis, we are performing in vitro experiments to
determine if the dual targeting of 7 nAChRS and COX-2 translates into an improved anti-inflammatory action compared to the parent NSAID. Ultimately, the pharmacokinetic properties of the compounds including hydrolysis of the ester bond, key for the COX-2 activity, needs to be evaluated in vivo.

Chapter 7 Summary and conclusions
180
5 Conclusion The work described in this thesis exemplifies that AChBP is a very useful tool to study fragment- and structure-based optimization procedures. One of our research aims was to increase our knowledge on how to efficiently optimize fragments. Using an X-ray co-crystal structure of a fragment in complex with AChBP, an efficient fragment hit optimization was established. The obtained results show that fragment merging can be a very efficient method for increasing binding affinity. Furthermore, the work described in this thesis demonstrates that anticipating on ligand-induced conformational changes of the protein binding site can be of great value when optimizing fragment hits. The second research aim was to determine if thermodynamic analysis can contribute to a more efficient fragment-optimization process. Our findings exhibit that the monitoring of the optimization process by thermodynamic analysis provided novel insights into molecular recognition principles and in combination with detailed structural information (X-ray of co-crystals), thermodynamic data provides crucial insights that can enable efficient fragment optimization. In addition, our work underlines the importance in ligand recognition of amino acid residues that are outside of the first shell aligning the binding site. Our work also suggests that targeting ligand-inducible subpockets may be a fruitful strategy to obtain selectivity between highly homologous proteins. These findings may be of relevance to the human nicotinic receptors, as well, as sequence alignments indicate that the lobeline-pocket can be addressed to obtain selectivity between the human nAChR subtypes. As such, the third research aim that we set ourselves with at the beginning of this research has also been met. Although, not finalized yet, our AChBP-related findings concerning the lobeline pocket may be translated to the therapeutically relevant human nAChRs. Furthermore, the work that is described in this thesis has led to the development of novel potential dual-action anti-inflammatory compounds that are capable of activating α7 nicotinic receptors. These dual action NSAID ester prodrugs may have clinical benefit over traditional NSAIDs in treating chronic inflammatory disorders such as rheumatoid arthritis. 6 References 1. Rucktooa, P., Smit, A.B. & Sixma, T.K. Insight in nAChR subtype
selectivity from AChBP crystal structures. Biochem Pharmacol 78, 777-87 (2009).
2. Sixma, T.K. & Smit, A.B. Acetylcholine binding protein (AChBP): a secreted glial protein that provides a high-resolution model for the extracellular domain of pentameric ligand-gated ion channels. Annu Rev Biophys Biomol Struct 32, 311-34 (2003).
3. Chung, S., Parker, J.B., Bianchet, M., Amzel, L.M. & Stivers, J.T. Impact of linker strain and flexibility in the design of a fragment-based inhibitor. Nat Chem Biol 5, 407-13 (2009).
4. Howard, N. et al. Application of fragment screening and fragment linking to the discovery of novel thrombin inhibitors. J Med Chem 49, 1346-55 (2006).
5. Englert, L. et al. Fragment-based lead discovery: screening and optimizing fragments for thermolysin inhibition. ChemMedChem 5, 930-40 (2010).

Chapter 7 Summary and conclusions
181
6. Hartshorn, M.J. et al. Fragment-based lead discovery using X-ray crystallography. J Med Chem 48, 403-13 (2005).
7. Sotriffer, C.A. Accounting for induced-fit effects in docking: what is possible and what is not? Curr Top Med Chem 11, 179-91 (2011).
8. Lovell, S.C., Word, J.M., Richardson, J.S. & Richardson, D.C. The penultimate rotamer library. Proteins 40, 389-408 (2000).
9. Edink, E., Jansen, C., Leurs, R. & de Esch, I.J.P. The heat is on: thermodynamic analysis in fragment-based drug discovery Drug Discov Today: Technologies 7, e189-e201 (2010).
10. Bissantz, C., Kuhn, B. & Stahl, M. A medicinal chemist's guide to molecular interactions. J Med Chem 53, 5061-84 (2010).
11. Hansen, S.B. et al. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J 24, 3635-46 (2005).
12. Dutertre, S. et al. AChBP-targeted alpha-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J 26, 3858-67 (2007).
13. Edink, E. et al. Fragment Growing Induces Conformational Changes in Acetylcholine-Binding Protein: A Structural and Thermodynamic Analysis. J Am Chem Soc 133, 5363-5371 (2011).
14. Scott, A.D. et al. Thermodynamic optimisation in drug discovery: a case study using carbonic anhydrase inhibitors. ChemMedChem 4, 1985-9 (2009).
15. Freire, E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov Today 13, 869-74 (2008).
16. Olsson, T.S., Williams, M.A., Pitt, W.R. & Ladbury, J.E. The thermodynamics of protein-ligand interaction and solvation: insights for ligand design. J Mol Biol 384, 1002-17 (2008).
17. Leeson, P.D. & Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov 6, 881-90 (2007).
18. Wenlock, M.C., Austin, R.P., Barton, P., Davis, A.M. & Leeson, P.D. A comparison of physiochemical property profiles of development and marketed oral drugs. J Med Chem 46, 1250-6 (2003).
19. Hann, M.M. Molecular obesity, potency and other addictions in drug discovery. MedChemComm 2, 349-355 (2011).
20. Borea, P.A., Dalpiaz, A., Gessi, S. & Gilli, G. Thermodynamics of 5-HT3 receptor binding discriminates agonistic from antagonistic behaviour. Eur J Pharmacol 298, 329-34 (1996).
21. Borea, P.A., Dalpiaz, A., Varani, K., Gessi, S. & Gilli, G. Binding thermodynamics at A1 and A2A adenosine receptors. Life Sci 59, 1373-88 (1996).
22. Borea, P.A., Dalpiaz, A., Varani, K., Gilli, P. & Gilli, G. Can thermodynamic measurements of receptor binding yield information on drug affinity and efficacy? Biochem Pharmacol 60, 1549-56 (2000).
23. Borea, P.A., Varani, K., Gessi, S., Gilli, P. & Gilli, G. Binding thermodynamics at the human neuronal nicotine receptor. Biochem Pharmacol 55, 1189-97 (1998).

Chapter 7 Summary and conclusions
182
24. Borea, P.A., Varani, K., Guerra, L., Gilli, P. & Gilli, G. Binding thermodynamics of A1 adenosine receptor ligands Mol Neuropharmacol 2, 273-281 (1992).
25. Maksay, G. Thermodynamics of gamma-aminobutyric acid type A receptor binding differentiate agonists from antagonists. Mol Pharmacol 46, 386-90 (1994).
26. Maksay, G. Distinct thermodynamic parameters of serotonin 5-HT3 agonists and antagonists to displace [3H]granisetron binding. J Neurochem 67, 407-12 (1996).
27. Maksay, G. Activation of ionotropic receptors and thermodynamics of binding. Neurochem Int 46, 281-91 (2005).
28. http://practicalfragments.blogspot.com/2011/04/sixth-annual-fragment-based-drug.html.
29. Whitesides, G.M. & Krishnamurthy, V.M. Designing ligands to bind proteins. Q Rev Biophys 38, 385-95 (2005).
30. Williams, D.H., Stephens, E., O'Brien, D.P. & Zhou, M. Understanding noncovalent interactions: ligand binding energy and catalytic efficiency from ligand-induced reductions in motion within receptors and enzymes. Angew Chem Int Ed Engl 43, 6596-616 (2004).
31. Chaires, J.B. Possible origin of differences between van't Hoff and calorimetric enthalpy estimates. Biophys Chem 64, 15-23 (1997).
32. Horn, J.R., Russell, D., Lewis, E.A. & Murphy, K.P. Van't Hoff and calorimetric enthalpies from isothermal titration calorimetry: are there significant discrepancies? Biochemistry 40, 1774-8 (2001).
33. Liu, Y. & Sturtevant, J.M. Significant discrepancies between van't Hoff and calorimetric enthalpies. II. Protein Sci 4, 2559-61 (1995).
34. Liu, Y. & Sturtevant, J.M. Significant discrepancies between van't Hoff and calorimetric enthalpies. III. Biophys Chem 64, 121-6 (1997).
35. Naghibi, H., Tamura, A. & Sturtevant, J.M. Significant discrepancies between van't Hoff and calorimetric enthalpies. Proc Natl Acad Sci U S A 92, 5597-9 (1995).
36. Day, Y.S., Baird, C.L., Rich, R.L. & Myszka, D.G. Direct comparison of binding equilibrium, thermodynamic, and rate constants determined by surface- and solution-based biophysical methods. Protein Sci 11, 1017-25 (2002).
37. Myszka, D.G. et al. The ABRF-MIRG'02 study: assembly state, thermodynamic, and kinetic analysis of an enzyme/inhibitor interaction. J Biomol Tech 14, 247-69 (2003).
38. Wear, M.A. & Walkinshaw, M.D. Thermodynamics of the cyclophilin-A/cyclosporin-A interaction: a direct comparison of parameters determined by surface plasmon resonance using Biacore T100 and isothermal titration calorimetry. Anal Biochem 359, 285-7 (2006).
39. Navratilova, I. et al. Thermodynamic benchmark study using Biacore technology. Anal Biochem 364, 67-77 (2007).
40. Beene, D.L. et al. Cation-pi interactions in ligand recognition by serotonergic (5-HT3A) and nicotinic acetylcholine receptors: the anomalous binding properties of nicotine. Biochemistry 41, 10262-9 (2002).

Chapter 7 Summary and conclusions
183
41. Xiu, X., Puskar, N.L., Shanata, J.A., Lester, H.A. & Dougherty, D.A. Nicotine binding to brain receptors requires a strong cation-pi interaction. Nature 458, 534-7 (2009).
42. Blum, A.P., Lester, H.A. & Dougherty, D.A. Nicotinic pharmacophore: the pyridine N of nicotine and carbonyl of acetylcholine hydrogen bond across a subunit interface to a backbone NH. Proc Natl Acad Sci U S A 107, 13206-11 (2010).
43. Puskar, N.L., Xiu, X., Lester, H.A. & Dougherty, D.A. Two neuronal nicotinic acetylcholine receptors, alpha4beta4 and alpha7, show differential agonist binding modes. J Biol Chem 286, 14618-27 (2011).
44. Bourne, Y., Talley, T.T., Hansen, S.B., Taylor, P. & Marchot, P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake alpha-neurotoxins and nicotinic receptors. EMBO J 24, 1512-22 (2005).
45. Ulens, C. et al. Use of acetylcholine binding protein in the search for novel alpha7 nicotinic receptor ligands. In silico docking, pharmacological screening, and X-ray analysis. J Med Chem 52, 2372-83 (2009).
46. Lebon, G. et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521-5 (2011).
47. Rasmussen, S.G. et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175-80 (2011).
48. Shimamura, T. et al. Structure of the human histamine H1 receptor complex with doxepin. Nature 475, 65-70 (2011).
49. Warne, T. et al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 469, 241-4 (2011).
50. Xu, F. et al. Structure of an agonist-bound human A2A adenosine receptor. Science 332, 322-7 (2011).
51. de Jonge, W.J. & Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 151, 915-29 (2007).
52. Wang, H. et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421, 384-8 (2003).
53. Hopkins, A.L., Groom, C.R. & Alex, A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today 9, 430-1 (2004).
54. Celie, P.H. et al. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41, 907-14 (2004).

184

Nederlandse samenvatting
185
Structuur-gebaseerd ontwerp van AChBP liganden, nieuwe inzichten en toepassingen Uitgangspunt voor het ontwikkelen van een medicijn Het ontdekken van nieuwe geneesmiddelen begint vaak met het vaststellen van een doel-eiwit. Dit kan een receptor of een enzym zijn waarvan verwacht wordt dat het een belangrijke rol speelt in een specifiek ziekteproces. De volgende stap om tot een bruikbaar medicijn te komen is meestal het vinden van chemische uitgangspunten; vaak kleine moleculen die enige affiniteit (bindingssterkte) voor het desbetreffende eiwit bezitten. Deze kleine moleculen, liganden, kunnen dan vervolgens geoptimaliseerd worden door middel van chemische modificaties. Het doel van deze modificaties is om de bindingsaffiniteit te vergroten en tegelijkertijd de chemisch-fysische eigenschappen van het molecuul zodanig aan te passen dat er uiteindelijk een stof verkregen wordt dat geschikt is voor toediening als medicijn. Dat wil zeggen dat toediening van deze stof aan een patiënt zal resulteren in genezing danwel verlichting van symptomen zonder daarbij al te veel ongewenste bijwerkingen te bewerkstelligen. Er zijn verscheidene mogelijkheden om deze moleculaire uitgangspunten te vinden. Een van de mogelijkheden is om grote bibliotheken van chemische stoffen te testen op het desbetreffende eiwit. Dit gebeurt binnen de farmaceutische industrie vaak door middel van een volledig geautomatiseerd proces dat high-throughput screening (HTS) genoemd wordt. Dit is een kostbaar proces omdat deze methode een uitgebreide infrastructuur van robots en een grote collectie van chemische stoffen vereist. Mede daarom wordt er veel onderzoek gedaan naar alternatieve methoden om liganden voor therapeutisch relevante eiwitten te ontdekken. Een van de alternatieven is om eerst met behulp van geavanceerde software een voorspelling te doen welke chemische stoffen in staat zullen zijn om in de bindingsholte van het desbetreffende eiwit te passen. Deze benadering, die bekend staat als virtual screening, heeft als doel het aantal chemische verbindingen dat daadwerkelijk getest wordt aanzienlijk te verkleinen, maar een vergelijkbaar aantal actieve moleculen (hits) als bij HTS geeft. Een andere methode die recent in zwang is geraakt, richt zich op het testen van relatief kleine moleculen, fragmenten, en staat bekend als fragment-based drug discovery (FBDD). Het grote voordeel van deze methode is dat de kans op het vinden van hits groter is dan bij traditionele HTS. Er kan daardoor met relatief kleine stoffenbibliotheken gewerkt worden. Dit komt doordat het aantal mogelijke moleculen (chemical space) aanzienlijk kleiner is wanneer men zich beperkt tot kleine fragmenten. Zo zijn er met 10 atomen nu eenmaal veel minder mogelijkheden om moleculen te construeren dan met 50 atomen. Simpel gezegd, fragment chemical space is aanzienlijk kleiner dan HTS-liganden chemical space. Bovendien bestaat de mogelijkheid dat een relatief groot molecuul niet aan een eiwit bindt doordat slechts een klein gedeelte van het molecuul niet complementair aan de bindingsholte is. Wanneer in dit geval het molecuul eerst opgedeeld zou

Nederlandse samenvatting
186
worden in kleinere fragmenten en vervolgens de afzonderlijke fragmenten opnieuw getest zouden worden zou men wel een bruikbaar chemisch uitgangspunt voor het ontwikkelen van een medicijn kunnen ontdekken. Optimaliseren van bindingsaffiniteit Welke methode er ook gebruikt wordt om liganden te vinden, HTS, virtual screening of FBDD, in bijna alle gevallen dient de bindingsterkte van de gevonden moleculen geoptimaliseerd te worden. Het onderzoek dat beschreven is in dit proefschrift heeft zich gericht op het optimaliseren van de bindingsaffiniteit van fragment- en virtual screening-hits. Het optimaliseren van liganden wordt aanzienlijk vereenvoudigd wanneer men de beschikking heeft tot gedetailleerde structurele informatie van het gedeelte van het eiwit waar het ligand aan bindt; de bindingsholte. Deze informatie geeft toegang tot structuur-gebaseerde geneesmiddelenontwikkeling (structure-based drug design; SBDD). De in SBDD benodigde structurele informatie kan bijvoorbeeld verkregen worden door middel van Röntgen diffractie van een kristal van een eiwit-ligand complex. In vergelijking met de membraan-gebonden eiwitten zijn de voor SBDD benodigde eiwit-kristallen in het algemeen veel makkelijker te verkrijgen van volledig water-oplosbare eiwitten. Het eiwit dat in dit proefschrift centraal staat is het water-oplosbare acetylcholine-bindend eiwit (acetylcholine-binding protein; AChBP). AChBP is een algemeen geaccepteerd model-eiwit voor het ligand-bindingsdomein van de membraan-gebonden nicotinerge acetylcholine receptoren (nAChRs). Van deze receptoren is aangetoond dat zij een belangrijke rol spelen in de signaaltransductie in het centrale en perifere zenuwstelsel en er zijn sterke aanwijzingen dat zij als doel-eiwit kunnen fungeren om geneesmiddelen te ontwikkelen voor ziektes als Alzheimer, schizophrenie en reuma. In tegenstelling tot de nAChRs is AChBP een stabiel en water-oplosbaar eiwit en heeft daarmee ideale eigenschappen voor toepassing in SBDD. Hoofdstuk 1 bestaat uit een inleiding tot de nAChRs en bevat tevens een overzicht van hoe AChBP kristalstructuren hebben bijgedragen aan een beter inzicht in de structuur van nAChRs. Het overzicht richt zich met name op hoe AChBP inzicht heeft verschaft op de interacties die nAChRs met verscheidene liganden maken. Men heeft bijvoorbeeld mede door AChBP-kristalstructuren een zeer gedetailleerd beeld verkregen hoe nicotine, de verslavende stof in tabak aan het ligand-bindingsdomein van de nAChRs bindt. Zelfs wanneer men over gedetailleerde structurele informatie van de eiwit-bindingsholte bezit, kan het soms lastig zijn om de bindingsaffiniteit van fragmenten en virtual screening hits te vergroten. Chemische modificaties van hit moleculen resulteren soms in onverwachte veranderingen in oriëntatie van het molecuul binnen de eiwit-bindingsholte. Bovendien kan de vorm van een eiwit onder invloed van een gemodificeerd ligand onverwachts veranderen. Daarnaast resulteert het maken van additionele interacties met de bindingholte niet altijd in een toename van bindingsaffiniteit. Dit komt doordat bindingsaffiniteit niet rechtsreeks correleert met de fysieke interacties die een ligand met de bindingsholte maakt. Bindingsaffiniteit moet echter gezien worden als een gemiddelde waarde die ook afhankelijk is van veranderingen in de conformaties van het ligand en eiwit en veranderingen in de positie van aanwezige watermoleculen. Door middel van thermodynamische analyse is het mogelijk om bindingsaffiniteit onder te verdelen in een enthalpische en een entropische

Nederlandse samenvatting
187
bijdrage. De enthalpische bijdrage correspondeert met het aangaan en verbreken van fysieke interacties tussen ligand, eiwit en watermoleculen tijdens het bindingsproces. Terwijl entropische bijdragen voortkomen uit veranderingen in de conformatie van het ligand, het eiwit en veranderingen in de positionering van watermoleculen. Het moge duidelijk zijn dat het bepalen van de afzonderlijke thermodynamische parameters, enthalpie en entropie, meer inzichten verschaft in het bindingsproces tussen eiwitten en liganden dan het bepalen van bindingsaffiniteit alleen. In hoofdstuk 3 wordt beschreven hoe thermodynamische bindingsparameters van fragment-eiwit complexen bepaald kunnen worden en hoe de verkregen informatie kan bijdragen aan een meer efficiënt optimaliseringproces van fragmenten. Groeien van een fragment In hoofdstuk 4 wordt een studie beschreven waar we specifiek kijken naar de veranderingen in thermodynamische bindingsprofielen bij het optimaliseren van een fragment. Door het vergelijken van een kristalstructuur van een complex van AChBP met een fragment met de kristalstructuur van de natuurstof lobeline gebonden aan AChBP, werd een interessante conformationele verandering binnen de AChBP bindingsholte zichtbaar. Lobeline induceert namelijk een beweging van een specifiek tyrosine residue (tyrosine-flip) waardoor een nieuwe kleine bindingsholte (lobeline-holte) wordt geopend. Een gedeelte van het lobeline molecuul maakt vervolgens interacties met deze kleine bindingsholte. Door het vergelijken van beide kristal structuren kregen wij het idee dat we de bindingsaffiniteit van het fragment zouden kunnen vergroten door deze in de lobeline-holte te groeien. Deze op structuren gebaseerde fragment optimalisering bleek een efficiënte manier om de bindingsaffiniteit van het fragment te vergroten en er werd in korte tijd een 150-maal toename in bindingsterkte bewerkstelligt. Verkregen kristalstructuren lieten zien dat de ontwerpstrategie succesvol is geweest, aangezien het geoptimaliseerde ligand inderdaad de tyrosine-flip had geïnduceerd en interacties met de lobeline-holte werden waargenomen. Een thermodynamische analyse gaf aan dat de toename in bindingsaffiniteit toe te schrijven is aan additionele enthalpische interacties met de lobeline-holte, welke echter gedeeltelijk gecompenseerd werden door ongunstige veranderingen in entropie. Deze veranderingen in thermodynamisch bindingsprofiel kunnen verklaard worden met het gegeven dat de additionele interacties met de lobeline-holte enhalpisch gunstig zijn (extra fysieke interacties tussen ligand en eiwit), maar entropisch ongunstig doordat hierdoor ligand en eiwit meer beperkt zijn geraakt in hun bewegingsvrijheid. Daarmee komen we op de belangrijkste bevinding van deze studie en dat is dat de geobserveerde verandering in thermodynamisch bindingsprofiel bij het groeien van het fragment indicatief is voor interacties met de lobeline-holte. De behaalde resultaten laten zien dat thermodynamische analyse informatie kan geven over hoe een ligand bindt aan een eiwit-bindingsholte. Dit is belangrijke informatie die vervolgens gebruikt zou kunnen worden om de bindingsaffiniteit verder te vergroten. Bovendien liet de in hoofdstuk 4 beschreven studie zien dat er een significant verschil in toegankelijkheid van de lobeline-holte bestaat tussen AChBP afkomstig van de californische zeehaas (Aplysia californica) en AChBP afkomstig van de poelslak (Lymneae stagnalis). Door middel van mutagenese experimenten hebben

Nederlandse samenvatting
188
wij bewijs aangevoerd dat stabilisatie van de tyrosine-flip door een specifiek aminozuur residue in de bindingsholte van AChBP afkomstig van Aplysia californica (Ac-AChBP) in een toegankelijke lobeline-holte resulteert. Deze stabilisatie is afwezig in AChBP afkomstig van Lymneae stagnalis (Ls-AChBP). Onze bevindingen laten zien dat het hierdoor mogelijk is om de bindingsaffiniteit selectief voor Ac-AChBP te vergroten ten opzichte van Ls-AChBP. Een interessant gegeven is dat het tyrosine-flip stabiliserende aminozuur residue zich niet in de directe nabijheid van de bindingsplaats van het ligand bevindt en dus op afstand van invloed is op het bindingsproces. Er zijn aanwijzingen dat we deze resultaten kunnen vertalen naar de therapeutisch relevante humane nAChRs en dat het misschien mogelijk is om de lobeline-holte te gebruiken om meer selectieve nAChR liganden te ontwikkelen en daarmee bijwerkingen van toekomstige medicijnen die ingrijpen op de nAChRs te verminderen. In hoofdstuk 5 wordt een studie beschreven waarin een virtual screening hit geoptimaliseerd wordt met betrekking tot bindingsaffiniteit voor AChBP. Hiertoe wordt wederom gebruikt gemaakt van gedetailleerde structurele informatie die in een eerdere studie is verkregen door AChBP te kristalliseren in aanwezigheid van het virtual screening hit molecuul. Er werden twee series chemische verbindingen gesynthetiseerd. De bindingsaffiniteit experimenten lieten verschillende structuur-activiteits relaties (SAR) tussen de twee series moleculen zien, wat duidt op een verschil in bindingsoriëntatie. Eveneens werd er een verschil in SAR tussen Ac- en Ls-AChBP waargenomen. Deze SAR verschillen zijn indicatief voor een verschil in toegankelijkheid van de lobeline-holte tussen Ac- en Ls-AChBP. Bindingsaffiniteit studies op de humane nAChRS lieten zien dat de gerealiseerde toename in bindingsaffiniteit voor AChBP door chemische modificaties van de virtual screening hit, zich niet hebben vertaald naar de humane nAChRS. Deze resultaten laten zien dat AChBP limitaties heeft als een model-eiwit voor de nAChRs. Structurele informatie van AChBP kan gebruikt worden om door middel van virtual screening nieuwe liganden voor de nAChRs te vinden, maar heeft zijn beperkingen in het gebruik om de bindingsaffiniteit voor de nicotine receptoren te vergroten. Dubbele werking tegen ontstekingen In hoofdstuk 6, hebben we gebruik gemaakt van structurele informatie van AChBP, om bestaande niet-steroïde ontstekingsremmers (non-steroidal anti-inflammatory drugs; NSAIDS) zodanig te modificeren dat deze in staat zouden zijn om de humane α7 nAChRs te activeren. Van deze nicotinerge receptor is namelijk recent aangetoond dat het een rol speelt in ontstekings-gerelateerde ziektes zoals bijvoorbeeld reumatoïde artritis. Via deze weg, proberen wij nieuwe ontstekingsremmers te ontwikkelen die via twee verschillende eiwitten een ontstekingsremmend effect uitoefenen. Onze strategie is er op gericht dat de gemodificeerde NSAID in het lichaam van de patiënt eerst de α7 nAChR activeert om vervolgens uiteen te vallen tot de oorspronkelijke NSAID. De NSAID zal dan via een ander eiwit, cyclooxygenase-2, een tweede ontstekingsremmend effect uitoefenen. Op deze manier hopen we nieuwe ontstekingsremmers te verkrijgen die een sterker effect laten zien vergeleken met de oorspronkelijke NSAID. Om te bepalen welke NSAIDs het meest geschikt zijn om het gewenste α7 nAChR effect in te bouwen door middel van chemische modificaties, hebben we eerst

Nederlandse samenvatting
189
gebruik gemaakt van geavanceerde computersoftware om te voorspellen welke gemodificeerde NSAIDs aan het modeleiwit voor de α7 nAChR, AChBP, zouden kunnen binden. Op basis van deze resultaten zijn er uiteindelijk vijf NSAIDs geselecteerd en chemisch gemodificeerd. Deze aanpak bleek erg succesvol aangezien alle vijf gemodificeerde NSAIDs een vergelijkbare bindingsaffiniteit voor de α7 nAChR als nicotine vertoonden. Van nicotine is in eerder onderzoek al aangetoond dat het via de α7 nAChR een ontstekingsremmend effect uitoefent. Er is echter nog een tweede voorwaarde voor een molecuul om via de α7 nAChR een ontstekingsremmend effect uit te oefenen en dat is dat naast binding aan de receptor deze ook geactiveerd dient te worden. Door middel van additionele biochemische experimenten hebben we kunnen aantonen dat één van de gemodificeerde NSAIDs inderdaad in staat is om de α7 nAChR te activeren. Bovendien hebben we experimenten uitgevoerd die laten zien dat de gemodificeerde NSAID in menselijk serum uiteindelijk uiteen zal vallen tot de oorspronkelijke NSAID. Deze initiële resultaten laten zien dat onze ontwerpstrategie juist was en dat we een molecuul hebben ontwikkeld dat ontstekingen via twee verschillende eiwitten kan remmen. Op dit moment worden additionele studies uitgevoerd in celsystemen of deze dubbele werking werkelijk bijdraagt aan een betere remming van ontstekingen. Conclusies In het afsluitende hoofdstuk 7, worden de belangrijkste resultaten samengevat en geëvalueerd of de onderzoeksdoelstellingen uiteengezet in hoofdstuk 2 gehaald zijn. Op de eerste plaats wilden wij met het onderzoek beschreven in dit proefschrift onze kennis vergroten over hoe de bindingsaffiniteit van fragmenten efficiënt geoptimaliseerd kan worden. Uit onze studies blijkt dat het combineren van structuurelementen afkomstig van verschillende moleculen (fragment merging) een zeer efficiënte manier is om de bindingsaffiniteit te vergoten. Bovendien is gebleken dat anticiperen op mogelijke conformationele veranderingen van de bindingsholte eveneens een effectieve methode kan zijn om bindingsaffiniteiten te vergroten. Daarnaast waren we benieuwd of thermodynamische analyse kan bijdragen aan een meer efficiënt fragment optimaliserings proces. Aan de hand van de resultaten van hoofdstuk 4 kunnen wij concluderen dat dit inderdaad het geval is en dat thermodynamische analyse meer inzichten verschaft dan het meten van bindingsaffiniteit alleen. Bovendien hebben we aangetoond dat aminozuur residuen die zich niet direct in de bindingsholte bevinden, een belangrijke rol kunnen spelen in ligand-eiwit interacties. Uiteraard was één van onze doelstellingen om bevindingen op AChBP te transleren naar de therapeutisch relevante humane nAChRs. Dit is in twee opzichten gelukt. Ten eerste kan de ontdekking dat interacties met de lobeline-holte liganden selectief maken voor Ac-AChBP ten opzichte van Ls-AChBP, mogelijkerwijs gebruikt worden om selectiviteit voor specifieke subtypes van de nAChRs te bewerkstelligen. Ten tweede is in hoofdstuk 6 een fragment geoptimaliseerd tot een nieuw type onstekingsremmer dat een dubbele onstekingsremmende werking via zowel de α7 nAChR als COX-2 uitoefent. Er kan dus geconcludeerd worden dat aan alle onderzoeksdoelstellingen die in hoofdstuk 2 zijn uiteengezet is voldaan.

Nederlandse samenvatting
190

Dankwoord
191
Dankwoord
Na vijf jaren van noeste arbeid kan ik in ieder geval concluderen dat promoveren een hele klus is. Gelukkig is dat is dat niet de enige conclusie die ik aan al mijn inspanningen kan verbinden. Uiteraard heb ik het promotietraject niet alleen doorlopen en ik ben dan ook veel personen dank verschuldigd. Zonder deze mensen was ik er nooit aan begonnen, was het niet gelukt of in ieder geval veel minder leuk geweest. Op de eerste plaats wil ik mijn promotor bedanken. Rob, hartelijk dank voor het vertrouwen in mijn persoon en het geven van een kans om in jouw groep te mogen promoveren. Ik heb onze wetenschappelijke discussies altijd zeer waardevol gevonden en heb veel respect voor de manier waarop jij richting en leiding aan de vakgroep weet te geven. Iwan, je was mijn co-promotor en hebt een zeer grote bijdrage aan de totstandkoming van dit proefschrift geleverd. Allereerst gaf je mij het vertrouwen om er überhaupt aan te beginnen. Daarnaast hebben jouw visionaire inzichten wat betreft FBDD, SPR en thermodynamica een grote rol gespeeld in de uiteindelijke inhoud van mijn proefschrift. Ik heb erg veel van je geleerd, op vele vlakken. Bovendien ben ik jou en Rob dankbaar voor de ruimte die jullie mij gaven om mijn (wilde) ideeën na te jagen, zoals de nAChR subtype selectivity pocket en de NSAID dual action ester prodrugs. Maikel, van jou heb ik op het wetenschappelijk vlak enorm veel geleerd. Je leerde mij om op de juiste manier experimenten op te zetten, met de juiste controles. Bovendien kon ik altijd bij je terecht met synthetische vraagstukken. Onze buitenlandse trips heb ik als zeer gezellig ervaren, ook al zat ik de laatste keer voornamelijk ‟s avonds op mijn hotelkamer (oftewel suite) aan dit proefschrift te werken. Eveneens erg veel dank voor alle C&EN‟s! Jacqueline, jij hebt ook een groot aandeel gehad in de totstandkoming van dit proefschrift. Tijdens de nicotine meetings vond ik je altijd erg scherp en je commentaar uiterst waardevol. Chris, jij bent ongeveer halverwege mijn promotie naar de VU teruggekeerd en ik heb je input tijdens werkbesprekingen en het begeleiden van studenten als zeer nuttig ervaren. Merci beaucoup! Grote dank gaat eveneens uit naar mijn computationale leermeester, Atilla. Op de eerste plaats wil ik je bedanken voor het leren van de fijne MOE- en GOLD kneepjes. Ten tweede heb jij als ontdekker van de “lobeline pocket” een grote rol gespeeld in twee hoofdstukken van mijn proefschrift. Ik heb onze samenwerking als zeer prettig ervaren en ik wens je het allerbeste toe in Istanbul! Daarmee kom ik automatisch bij de andere ”nicotine-freaks”. Gerdien en Mark. Ik vond onze nicotine-meetings altijd erg nuttig. Daarnaast zijn jullie zeer fijne collega‟s en misschien moeten we daarom in de toekomst daad bij woord voegen en een slakkenverdelgingsmiddelenbedrijf (goed woord voor wordfeud, Mark?) gaan oprichten. Oscar, wij kennen elkaar nog uit de tijd dat we samen aan de master begonnen dat inmiddels wel een vorig leven lijkt te zijn. Ik wil je bedanken voor de fijne tijd tijdens de studie en het a.i.o.-schap. Kim, jij hebt een zeer grote rol gespeeld in de totstandkoming van Hoofdstuk 4. Behoorlijk zwanger heb jij alle SPR metingen bij een reeks van verschillende

Dankwoord
192
temperaturen voor elkaar gekregen en daarmee de thermodynamische analyse die essentieel is geweest voor ons artikel. Bovendien heb ik onze samenwerking als bijzonder gezellig ervaren! Obbe, jij hebt ook zeker een belangrijke bijdrage aan dit proefschrift geleverd. Betrouwbare farmacologische resultaten zijn onontbeerlijk voor het bedrijven van medicinal chemistry en in dat opzicht kon ik volledig op je rekenen. Bedankt voor het meten van de olifantjes en het altijd opbeurende commentaar (“Had ze graag wat potenter gezien, maar ik heb de shit, sorry, compounds niet gesynthetiseerd”). Hans, Andrea en Laura, jullie zijn fantastische collega‟s en houden onze afdeling draaiende. Bedankt hiervoor! Frans, veel dank voor de NMR metingen bij hoge temperaturen en de vele hulp bij het oplossen van complexe NMR-spectra. Uiteraard wil ik ook al mijn andere synthetic, computational, pharmacological en toxicological (ex-)collega‟s bedanken voor de goede werksfeer en de leuke tijd die ik met jullie heb beleefd. Er zijn ook een flink aantal studenten die een bijdrage aan dit proefschrift hebben geleverd. In willekeurige volgorde: Umit, Nok, Phoung, Giano, Eric en Thomas, bedankt voor jullie inspanningen en bijdragen. Chimed wil ik nog in het bijzonder bedanken aangezien ik naast de hoofdvakstage je ook heb mogen begeleiden bij jouw afstudeerscriptie. Het was altijd prettig om met je samen te werken en ik vond het dan ook erg leuk dat we in een later stadium toen jij inmiddels als a.i.o. bij ons werkzaam was, we samen een artikel hebben kunnen publiceren. Binnen het AChBP project zijn er ook een aantal mensen die ik graag wil bedanken voor de prettige samenwerking. Guus, René, Tariq en Pim, hartelijk dank voor de wetenschappelijke discussies, de voorraad AChBP op peil houden, het maken van de mutanten en het bepalen van bindingsaffiniteiten. Titia, Chris, en Patrick wil ik eveneens bedanken voor de wetenschappelijke input en de prettige samenwerking. Prakash, thanks a lot for the very nice cooperation that has resulted in Chapter 4. Verder wil ik Hein en Sara bedanken voor het commentaar op ons onderzoek vanuit een industrieel perspectief. Mijn vrienden en (schoon-)familie wil ik bedanken voor de feestjes, spelletjesavonden en etentjes die voor de broodnodige ontspanning hebben gezorgd. Mijn ouders wil ik bedanken voor de steun en het vertrouwen in mij. Gelukkig heb ik mij de afgelopen vijf jaren niet alleen maar met onderzoek bezig gehouden. Ik ben eveneens gepromoveerd tot een liefhebbende echtgenoot en vader van twee wonderschone dochters. Juliette, Ella en Dana, jullie zijn voor mij het allerbelangrijkst en al het andere is uiteindelijk maar bijzaak! Juliette, jou wil ik in het bijzonder bedanken voor je onvoorwaardelijke steun en liefde.
Ewald

Appendices
193
List of abbreviations 5-HT3 serotonin α-Bgt alpha-bungarotoxin Ac Aplysia californica AChBP acetylcholine-binding protein Ala alanine Asp aspartate Bt Bulinus truncatus CDCl3 deuterated chloroform CNS central nervous system COSY correlation spectroscopy Cys cysteine DA dopamine Da Dalton DCE dichloroethane DCM dichloromethane DIPEA N,N-diisopropylethylamine DMAP 4-dimethylaminopyridine DMF N,N-dimethyformamide DMSO dimethyl sulfoxide ECD extracellular domain EE enthalpic efficiency Et2O diethyl ether EtOAc ethyl acetate EtOH ethanol FBDD fragment-based drug discovery
GABA -aminobutyric acid GE group efficiency Gln glutamine Glu glutamate Gly glycine GI gastrointestinal GPCRs G protein-coupled receptors His histidine HPLC high performance liquid chromatography HR-MS high resolution mass spectroscopy HTS high-throughput screening ITC isothermal titration calorimetry LBD ligand-binding domain LE ligand efficiency LGIC ligand-gated ion channels Ls Lymnaea stagnalis Lys lysine MeOD deuterated methanol MeOH methanol MLA methyllycaconitine MMP-12 matrix metallopeptidase 12

Appendices
194
mp melting point MS mass spectroscopy MUP major urinary protein MW molecular weight nAChR nicotinic acetylcholine receptor NE norepinephrine NMP N-methylpyrrolidon NMR nuclear magnetic resonance spectroscopy NOESY nucleur Overhauser effect spectroscopy NSAID non-steroidal anti-inflammatory drug PBS phosphate-buffered saline PDB protein databank PNS peripheral nervous system RA rheumatoid arthritis SEM standard error of the mean Ser serine SPA scintillation proximity assay SPR surface plasmon resonance TEA N,N,N- triethylamine Thr threonine TLC thin layer chromatography THF tetrahydrofuran TNF-α tumour necrosis factor α TM transmembrane Trp tryptophan Tyr tyrosine

Appendices
195
List of publications
Ewald Edink, Atilla Akdemir, Chimed Jansen, René van Elk, Obbe Zuiderveld, Frans J.J. de Kanter, Jacqueline E. van Muijlwijk-Koezen, August B. Smit, Rob Leurs and Iwan J.P. de Escha, Structure-based design, synthesis and structure-activity relationships of dibenzosuberyl- and benzoate-substituted tropines as ligands for acetylcholine binding- protein, submitted to Bioorg. Med. Chem. Lett.
Jeroen Kool, Ferry Heus, Gerdien de Kloe, Henk Lingeman, August B. Smit, Rob Leurs, Ewald Edink, Iwan J.P. de Esch, Hubertus Irth and Wilfried M.A. Niessen, High-resolution bioactivity profiling of mixtures towards the acetylcholine binding protein using a nanofractionation spotter technology, J. Biomol. Screen. 2011, 917-924.
Ewald Edink, Prakash Rucktooa, Kim Retra, Atilla Akdemir, Tariq Nahar, Obbe Zuiderveld, René van Elk, Elwin Janssen, Pim van Nierop, Jacqueline van Muijlwijk-Koezen, August B. Smit, Titia K. Sixma, Rob Leurs, and Iwan J. P. de Esch, Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis, J. Am. Chem. Soc. 2011, 133(14), 5363-71.
Ewald Edink, Chimed Jansen, Rob Leurs and Iwan J.P. de Esch, The heat is on: Thermodynamic analysis in fragment-based drug discovery, Drug Discovery Today: Technologies 2010 7(3), e189-e201.
Alexander E. Ivanov, Ewald Edink, Ashok Kumar, Igor Yu. Galaev, Alexander F. Arendsen, Alle Bruggink and Bo Mattiasson, Conjugation of penicillin acylase with the reactive copolymer of N-isopropylacrylamide: A step toward a thermo-sensitive industrial biocatalyst, Biotechnol Prog. 2003 19(4): 1167-75.

Appendices
196
Curriculum Vitae Ewald Edink was born on May 4
th 1977 in Delft. In 1995 he graduated from Hugo
Grotius college in Delft. In 2000, Ewald obtained a bachelor degree in organic chemistry at the higher laboratory education in Rotterdam. After working at the technical university in Delft, DSM Anti-Infectives and DSM Food Specialties as a research technician, he started in 2003 with the master education Pharmacochemistry at the VU University in Amsterdam. He did his major in the division of medicinal chemistry under supervision of Dr. Iwan J.P. de Esch and ing. Andrea van de Stolpe. The research involved the synthesis of novel imidazole based antinociceptives. His major traineeship was extended with an external traineeship at the medicinal chemistry department of the R&D center Drug Discovery for Oncology of Boehringer Ingelheim in Vienna, Austria. In 2006 he recieved his MSc degree and initiated his PhD project in the same department of the Vrije Universiteit under supervision of Prof. Dr. Rob Leurs and Dr. Iwan J.P. de Esch. This project focused on the structure-based design of ligands for acetylcholine-binding protein. In 2010, Ewald continued being a researcher in the medicinal chemistry department of the VU University in Amsterdam where he currently works on the design and synthesis of small molecule inhibitors that interfere with key protein-protein interactions at the influenza ribonucleoprotein complex