suitability of replacement markers for functional analysis studies insaccharomyces cerevisiae
TRANSCRIPT

. 13: 1563–1573 (1997)
Suitability of Replacement Markers for FunctionalAnalysis Studies in Saccharomyces cerevisiae
FRANK BAGANZ1, ANDREW HAYES1, DEREK MARREN2, DAVID C. J. GARDNER1 ANDSTEPHEN G. OLIVER1*1Department of Biomolecular Sciences, UMIST, PO Box 88, Manchester M60 1QD, U.K.2Pfizer Central Research, Sandwich, Kent CT13 9NJ, U.K.
Received 23 July 1997; accepted 4 October 1997
The complete yeast sequence contains a large proportion of genes whose biological function is completely unknown.One approach to elucidating the function of these novel genes is by quantitative methods that exploit the conceptsof metabolic control analysis. An important first step in such an analysis is to determine the effects of deletingindividual genes on the growth rate (or fitness) of Saccharomyces cerevisiae. Since the specific growth-rate effects ofmost genes are likely to be small, they are most readily determined by competition against a standard strain inchemostat cultures where the true steady state demanded by metabolic control analysis may be achieved. We haveconstructed two different standard strains in which the HO gene is replaced by either HIS3 or kanMX. Wedemonstrate that HO is a selectively neutral site for gene replacement. However, there is a significant marker effectassociated with HIS3 which, moreover, is dependent on the physiological conditions used for the competitionexperiments. In contrast, the kanMX marker exhibited only a small effect on specific growth rate (¦&4%). Thesedata suggest that nutritional markers should not be used to generate deletion mutants for the quantitative analysisof gene function in yeast but that kanMX replacements may be used, with confidence, for such studies.? 1997 John Wiley & Sons, Ltd.
Yeast 13: 1563–1573, 1997.
— Saccharomyces cerevisiae; functional analysis; gene replacement; competition experiments
which an individual member of the set makes to
*Correspondence to: S. G. Oliver, Department of BiomolecularSciences, UMIST, PO Box 88, Manchester M60 1QD, U.K.INTRODUCTION
The complete determination of the DNA sequenceof all 16 chromosomes which comprise the nucleargenome of the brewer’s and baker’s yeast, Sac-charomyces cerevisiae, has revealed the presence ofca. 6000 protein-encoding genes (Goffeau et al.,1996, 1997). Of these, some 38% encode a pre-dicted protein product whose function is com-pletely unknown (Mewes et al., 1997). This groupof nearly 2300 ‘orphan’ (Dujon, 1996) genesincludes 787 whose products show significantamino-acid sequence similarity to other gene
CCC 0749–503X/97/161563–11 $17.50? 1997 John Wiley & Sons, Ltd.
products which are also of unknown function. Itmay be that classical or ‘function-first’ (Oliver,1996a) molecular genetics has failed to identifythese genes because it employs an experimentalparadigm that prefers qualitative to quantita-tive assessments of phenotype. Another reason toconsider a quantitative approach to phenotypicanalysis is the high level of redundancy that isapparent in the yeast genome (Mewes et al., 1997).If a particular gene is a member of a paralogous setof identical (or nearly identical) genes then, pro-vided that they are all capable of being regulatedin a similar manner and their products are targetedto the same cellular location, the contribution

1564 . .
phenotype will, necessarily, be some fraction of thewhole.A conceptual and mathematical framework for
the quantitative analysis of gene function is pro-vided by metabolic control analysis (MCA; Kacserand Burns, 1973). The ‘top-down’ (Brand, 1996) ormodular (Rohwer et al., 1996) approach to such ananalysis has just the hierarchial features that arerequired for a systematic approach to the eluci-dation of gene function (Oliver, 1996a). Thisapproach aims to divide the metabolic map into anumber of large units or modules through, or in,which independent fluxes or metabolic concen-trations can be measured. Assignment of a novelgene as having its effect within a particular modulethen allows that module to be sub-divided into anumber of individual units that can be tested, byfurther flux or metabolic analyses, to determinein which unit the novel gene under examinationhas its effect. By moving down this hierarchy ofanalysis, a closer and closer approximation to thefunction of the novel gene is obtained.An important tool in MCA is the response
coefficient which is used to quantify the extent towhich a parameter affects a variable. The responsecoefficient is defined as the relative change in asteady-state variable (such as the flux through agiven metabolic pathway, or the concentration of aparticular metabolite) caused by a relative changein a parameter (such as the concentration of aspecific enzyme). The fact that relative changes,rather than absolute ones, are measured meansthat these response coefficients are dimensionless:they have no units (see Hofmeyr, 1995). Animportant feature of response coefficients is thatthey are measures of the impact of a given par-ameter on a system. At the highest level of atop-down MCA approach to yeast gene function,we would wish to define the system as being thewhole cell. This means that we can define the fluxcontrol coefficient of a particular gene productby manipulating its cellular concentration andmeasuring the impact of the change in growth rate,to obtain (in effect) a growth-rate control coeffi-cient. Metabolic control analysis tells us that fluxcontrol coefficients generally have values between0 and 1 (see Teusink et al., 1997 for a fullerexplanation), and experimental determinations ofsuch coefficients (e.g., see Niederberger et al.,1992) demonstrate that they usually have valuesmuch closer to 0 than to 1. This means that weneed a very sensitive method of measuring thechange in growth rate resulting from the deletion
? 1997 John Wiley & Sons, Ltd.
or disruption of an individual yeast gene ofunknown function. Competition experiments be-tween mutant and wild-type yeast have been shownto provide a very sensitive way of measuring smallgrowth rate differences (Danhash et al., 1991).A very attractive approach to competition
analysis has been employed by Smith and co-workers (Smith et al., 1995, 1996). They have usedpools of Ty-generated yeast mutants and employeda serial batch transfer approach to extend theperiod over which the competition takes place, Therelative proportions of the different mutant strainsin the population were monitored by carrying outPCR reactions using a common primer comp-lementary to the Ty sequence and a series ofprimers complementary to the flanking sequencesof the genes whose quantitative phenotype was tobe assessed. A sequencing machine equipped withGeneScan= software was used to perform theanalysis.There are a number of drawbacks to this system
if one is adopting an MCA approach to theelucidation of gene function. Each mutant linecarries mutations in genes additional to the onemonitored by the PCR reaction. It is assumed thatthese mutations occur at random and that theireffects are averaged out on a population basis.However, no individual mutant line can be carriedforward for further analyses since it will have itsown peculiar set of additional mutations. More-over, the averaging-out process does not allow theaccurate quantitation of the phenotypic impact ofinactivating a single gene that we would wish toexploit using MCA. For these reasons, we preferto use mutant strains in which specific ORFsof unknown function have been deleted byPCR-mediated gene replacement (Baudin et al.,1993; Wach et al., 1994). The EUROFAN project(Oliver, 1996b) will generate an extensive library ofdeletants for use in such an analysis. Moreover, alarge number of these mutants can be includedin any particular competition experiment, thusretaining the ‘multiplexing’ advantages of thegenetic footprinting approach. The proportion ofall of the mutants in a sample of the populationtaken at a given time in the competition exper-iment may be determined by carrying out a seriesof PCR reactions on DNA extracted from thesample. The fact that the PCR products are of adefined size that is determined by the experimenterprovides opportunities to increase the efficiency ofanalysis which are not available with the geneticfootprinting (Smith et al., 1995) technique. By
. 13: 1563–1573 (1997)

1565
choosing to generate different sized PCR pro-ducts, a number of mutants can be analysed on asingle gel track even when the analysis is done bysimple densitometry. Use of an ABI sequencingmachine equipped with GeneScan= softwarepermits further improvements in throughput byusing primers labelled with different fluoro-phors. The recent development of methods to givedeletion mutants specific oligonucleotide tags(so-called ‘molecular bar-codes’; Shoemaker et al.,1996) offers the best prospects for efficient analysisof the proportions of mutants in population byemploying hybridization-array technology (Schenaet al., 1996).The employment of serial transfer between
batch cultures also has the important limitationthat growth occurs for the most part in the pres-ence of excess nutrients. This situation might beagreed to be far from that obtaining in a naturalenvironment. Therefore, we prefer to use chemo-stat cultures since they allow competition to beperformed under defined nutrient limitations intrue steady states over an indefinite period.To determine the specific growth rate effects of
novel genes through the type of competition exper-iment described above, it is important to have astandard strain that is included in all the compe-titions and against which the mutant strains arecompared. This standard strain should be isogenicto the test strains and should have the ORF of acompletely dispensable gene deleted by replace-ment with the same marker as is used to generatethe test deletions. For the gene replacement in thestandard strain to be without significant pheno-typic effect, it is important that the replacementmarker should also not have any appreciableimpact on the organism’s growth rate under arange of physiological conditions. We have chosento generate a standard strain in which the codingsequence of the HO gene is deleted. HO has noknown role, apart from mating-type switching,and it has been used as the site of insertion ofheterologous genes in brewing yeasts without anyperceptible effect on the fermentation characteris-tics of the organism or the quality of the product(Hammond et al., 1994; Yocum, 1986). Wheneverpossible, a diploid strain, homozygous for theho deletion, is employed—both because HOis inactive in diploids and because the matingof independently derived haploid transformantsallows any transformation-induced genetic lesions(Danhash et al., 1991) to be nullified throughcomplementation.
? 1997 John Wiley & Sons, Ltd.
In this paper, we report on the evaluation of ourchoice of HO as a neutral site and the phenotypicimpact of two popular replacement markers(kanMX; Wach et al., 1994; and HIS3; Baudinet al., 1993) by competition between ho deletantsand their wild-type parents. These competitionexperiments have been performed in chemostatculture under some or all of the following physi-ological conditions: glucose-limited/anaerobic,glucose-limited/aerobic, ethanol-limited, N-limited,P-limited, S-limited. It was found that theho::kanMX deletion was without significant effecton growth rate whereas the ho::HIS3 replacementhad a profound effect which, moreover, was depen-dent on the physiological conditions employed. Thedata presented below confirm our choice ofHO as aneural site for replacement in standard strains, butalso indicate that resistance genes (such as kanMX)should be used in preference to nutritional markersin constructing such strains for use in the determi-nation of growth-rate control coefficients for novelgenes.
MATERIALS AND METHODS
Yeast strains and mediaThe S. cerevisiae strains used were BMA41-1B
(MATá ura 3-1 trp1-Ä2 leu2-3,112 his 3-11,15 ade2-1 can1-100; Baudin et al., 1993); FY23 (MATaura3-52 trp1-Ä63 leu2-Ä1) and FY73 (MATá his3-Ä200 ura3-52; Winston et al., 1995).S. cerevisiae was grown on 1% (w/v) yeast
extract, 2% (w/v) peptone, and 2% (w/v) glucose(YPD) or on complete synthetic media, contain-ing per litre: 1·7 g yeast nitrogen base (DifcoLaboratories), 5 g ammonium sulphate, and 20 gglucose (SC). The medium was supplemented asrequired with 20 mg uracil, adenine and the aminoacids -histidine, -tryptophan, and 100 mg-leucine. Solid media contained, in addition, 2%(w/v) agar (Difco Laboratories). G418-resistantstrains were grown on YPD plates containing200 mg/l of geneticin (Life Technologies).
Construction of HO deletion cassettes by PCRThe HIS3 disruption cassette was generated
using a three-stage PCR strategy based on themethod by Baudin et al. (1993). In the first stage ofPCR, a 1·1 kb HIS3 fragment was amplified byusing two primers (Table 1, oligonucleotides 1 and2) consisting of 40 nucleotides homologous to theHO flanking regions, followed by 21 nucleotides
. 13: 1563–1573 (1997)
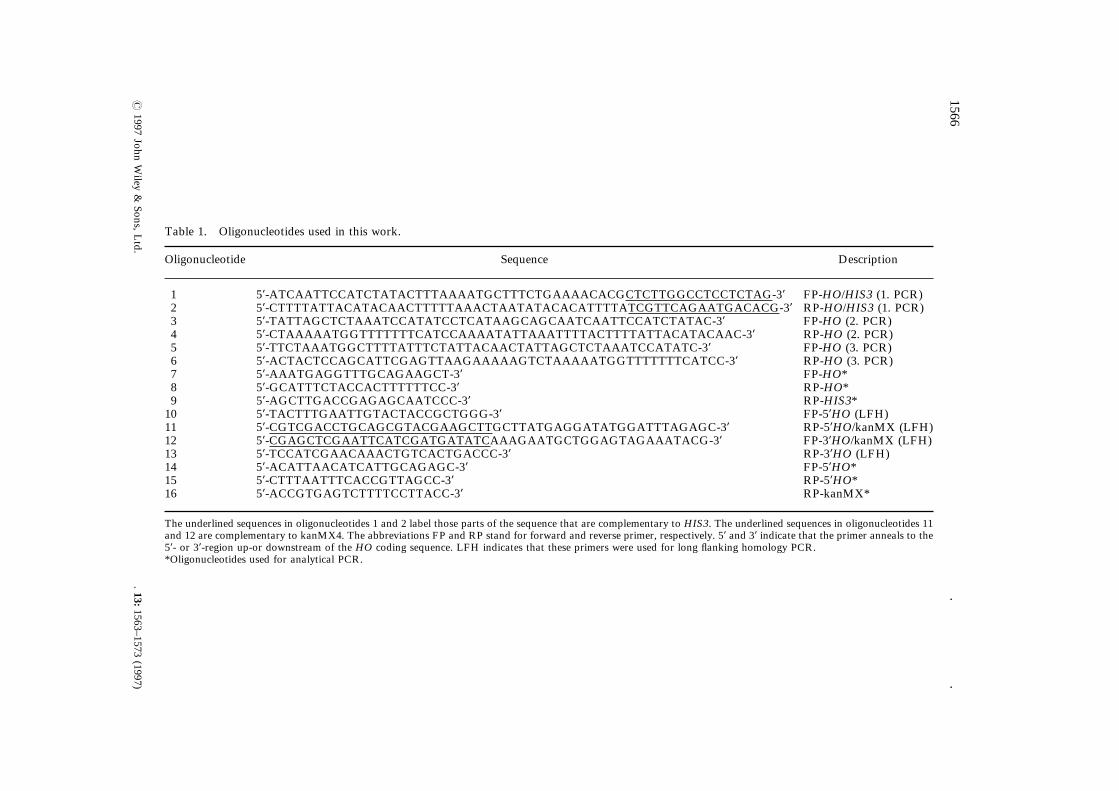
Table 1.
Oligonucl Description
1 CACGCTCTTGGCCTCCTCTAG-3* FP-HO/HIS3 (1. PCR)2 TTTTATCGTTCAGAATGACACG-3* RP-HO/HIS3 (1. PCR)3 CAATTCCATCTATAC-3* FP-HO (2. PCR)4 ACTTTTATTACATACAAC-3* RP-HO (2. PCR)5 CTAAATCCATATC-3* FP-HO (3. PCR)6 ATGGTTTTTTTCATCC-3* RP-HO (3. PCR)7 FP-HO*8 RP-HO*9 RP-HIS3*10 FP-5*HO (LFH)11 ATATGGATTTAGAGC-3* RP-5*HO/kanMX (LFH)12 GAGTAGAAATACG-3* FP-3*HO/kanMX (LFH)13 RP-3*HO (LFH)14 FP-5*HO*15 RP-5*HO*16 RP-kanMX*
The underl re complementary to HIS3. The underlined sequences in oligonucleotides 11and 12 are reverse primer, respectively. 5* and 3* indicate that the primer anneals to the5*- or 3*-reg imers were used for long flanking homology PCR.*Oligonucl
1566.
.
?1997
JohnWiley
&Sons,
Ltd.
.13:
1563–1573(1997)
Oligonucleotides used in this work.
eotide Sequence
5*-ATCAATTCCATCTATACTTTAAAATGCTTTCTGAAAA5*-CTTTTATTACATACAACTTTTTAAACTAATATACACA5*-TATTAGCTCTAAATCCATATCCTCATAAGCAGCAAT5*-CTAAAAATGGTTTTTTTCATCCAAAATATTAAATTTT5*-TTCTAAATGGCTTTTATTTCTATTACAACTATTAGCT5*-ACTACTCCAGCATTCGAGTTAAGAAAAAGTCTAAAA5*-AAATGAGGTTTGCAGAAGCT-3*5*-GCATTTCTACCACTTTTTTCC-3*5*-AGCTTGACCGAGAGCAATCCC-3*5*-TACTTTGAATTGTACTACCGCTGGG-3*5*-CGTCGACCTGCAGCGTACGAAGCTTGCTTATGAGG5*-CGAGCTCGAATTCATCGATGATATCAAAGAATGCTG5*-TCCATCGAACAAACTGTCACTGACCC-3*5*-ACATTAACATCATTGCAGAGC-3*5*-CTTTAATTTCACCGTTAGCC-3*5*-ACCGTGAGTCTTTTCCTTACC-3*
ined sequences in oligonucleotides 1 and 2 label those parts of the sequence that acomplementary to kanMX4. The abbreviations FP and RP stand for forward andion up-or downstream of the HO coding sequence. LFH indicates that these preotides used for analytical PCR.

1567
(underlined sequences in Table 1) which allowedamplification of the HIS3 cassette from the tem-plate, vector YDp-H (Berben et al., 1991). Thesecond and third pairs of primers (Table 1, oligo-nucleotides 3 and 4, and 5 and 6, respectively)extend the HO flanking region to ca. 90 nucle-otides on each side. After each stage of PCR,primers and unincorporated dNTPs were removedby Chromaspin-400 columns (Clontech) and 10 ìlof eluate was used as template in the next stage ofPCR. In the first and second stages, the reactionmix was submitted to two cycles of PCR (de-naturation for 1 min at 95)C, annealing for 1 minat 50)C and extension for 1 min at 72)C). In thethird stage of PCR, 30 cycles were performed,followed by an extension step for 5 min. Thereaction mix consisted of 2·5 units of Taq DNApolymerase, 10 m–Tris-HCl (pH 9·0), 50 m–KCl, 1·5 m–MgCl2, 0·2 m of each dNTP, 0·2 ìof each primer and ca. 10 ng of template DNA in afinal volume of 50 ìl.For the construction of the kanMX4 disruption
cassette, long flanking homology PCR (LFH-PCR; Wach, 1996) was used. In the first PCR, thefollowing fragments from the 5*- and 3* regionof the HO gene were generated in two separatereactions: a 300 bp 5*-fragment with primers 10and 11, and a 500 bp 3*-fragment with primers 12and 13 (see Table 1) using ca. 50 ng genomic DNAof S. cerevisiae as template. In the second PCR,100–300 ng of each product from the first PCRtogether with 1·0 ì of each of the outermost 5*-and 3*-primers (Table 1, oligonucleotides 10 and13) were used to synthesize the 2·1 kb kanMX4disruption cassette with long flanking HO homol-ogy regions from 100–500 ng template (pFA6-kanMX4, NotI digested; see Wach et al., 1994).Other PCR conditions and purification, concen-tration, and quantitation of PCR products were asdescribed by Wach (1996).
Yeast transformation and selection oftransformantsAliquots (0·5–1·0 ìg) of PCR products were
used to transform lithium acetate-treated cellsaccording to the protocol by Gietz et al. (1995).His+ transformants were selected on minimalmedium lacking histidine. For the selection ofG418-resistant colonies, transformed cells wereresuspended in 1 ml YPD and pre-incubated for2–3 h at 30)C with shaking. Subsequently, cellswere collected by centrifugation, resuspended in
? 1997 John Wiley & Sons, Ltd.
300 ìl of the supernatant and plated in 100 ìlaliquots on YPD-G418 (200 mg/l).
Verification of His+ and G418r transformants byPCRCorrect targeting of the disruption cassettes into
the HO locus was verified by analytical PCR withwhole yeast cells and three primers per reaction(Table 1, oligonucleotides 7, 8 and 9 for His+
transformants and oligonucleotides 14, 15 and 16for G418r transformants) as described by Wachet al. (1994).
Competition experiments in chemostat cultureCultivation of yeast strains was carried out at
30)C in Applikon fermenters with a 1 litre workingvolume and a stirring speed of 750 rpm. The pHwas automatically controlled at 4·5 by addition of2·5 –NaOH. Under aerobic conditions, the airflow rate was set at 1 l/min. For anaerobic cultiva-tion, the air flow rate was reduced to maintaina minimal dissolved oxygen tension of ca. 1%saturation, compared to ca. 100% under aerobicconditions. S. cerevisiae requires a small amount ofoxygen for lipid biosynthesis in the absence ofprecursors (Verduyn et al., 1990). The mineralmedium, supplemented with trace elements andvitamins, contained per litre: 2·0 g KH2PO4 (forP-limitation 54 mg KH2PO4 was added togetherwith 1·1 g KCl), 0·55 g MgSO4 . 7H2O (exceptthat 0·45 g MgCl2 . 2H2O was used forS-limitation), 0·1 g NaCl, 90 mg CaCl2 . 2H2O,70 ìg ZnSO4 . 7H2O, 50 ìg FeCl3 . 6H2O, 10 ìgCuSO4 . 5H2O, 10 ìg H3BO3, 10 ìg KI, 62 mginositol, 14 mg thiamine/HCl, 4 mg pyridoxine,4 mg Ca-pantothenate, and 0·3 mg biotin. Themedium was supplemented with 20 mg uracil,adenine and the amino acids -histidine,-tryptophan, and 100 mg -leucine as required.The carbon source (i.e. glucose or ethanol) wasseparately prepared and sterilized (in the case ofglucose) before adding it to the medium. Vitaminswere filter-sterilized and added after heat steriliz-ation of the medium. The glucose concentration inthe medium was 2·5 g/l for C-limitation and 21 g/lfor all other nutrient limitations, except ethanol.Under this condition, the final concentration ofethanol was 2·51 g/l. Ammonium sulphate wasused as nitrogen source, the concentration was3·13 g/l, except for N-limitation (0·46 g/l) andS-limitation (24 mg/l). Under the latter condition,NH Cl (2·5 g/l) was used as nitrogen source.
4. 13: 1563–1573 (1997)

1568 . .
A 1 : 1 mixture of two strains, grown to similarcell density, was used as inoculum. A batch com-petition was carried out before continuous cultureconditions were established. Once a steady statehad been obtained, samples were taken once aday for determination of cell density andreplica-plating.
Measurement of cell densityOptical density was measured at a wavelength
of 600 nm using a spectrophotometer (CecilInstruments, U.K.).
Determination of the proportion of ho deletants byreplica-platingSamples from competition experiments were
diluted to give a final concentration of ca. 1000cells/ml and 100 ìl aliquots of these dilutions werespread onto YPD plates. After 1 day of incubationthe colonies were replica-plated onto selectivemedia (i.e. SC without histidine for ho::HIS3deletants and YPD-G418 for ho::kanMX4deletants). The proportion of ho deletants wasdetermined as the number of colonies on selectiveplates divided by the total number of colonies onYPD from triplicate measurements.
Calculation of growth rate differentialThe specific growth rate differential (ó) was
calculated according to the following formula(Dykhuizen and Hartl, 1983): ln [x1(t)/x2(t)]=ln[x1(t0)/x2(t0)]+ó t, where x1(t) and x2(t) representthe relative proportion of the two competingstrains at time t, and ó is a measure of differentialgrowth rate also referred to as ‘selection per hour’.By plotting ln [x1(t)/x2(t)] against t, ó was obtainedas the slope of the ‘best fitted’ line from linearregression on the data.
RESULTS AND DISCUSSION
Standard strain constructionFor the construction of standard strains to be
used in competition experiments, we chose toreplace the HO locus with either of two selectablemarkers (HIS3 and kanMX4) in different geneticbackgrounds. The homologous HIS3 marker wasused for PCR-mediated gene replacement based onthe method by Baudin and colleagues (1993) togenerate a standard strain in the W303 back-ground. This strain and marker were chosen to
? 1997 John Wiley & Sons, Ltd.
permit the deletion mutants from the Europeanpilot project on the functional analysis of novelyeast genes (Baudin-Ballieu et al., 1997) to beincluded in our competition experiments. TheHIS3 gene replacement cassette, bearing 90 bpsequences homologous to HO at each end, wastransformed into the haploid strain BMA41-1Bwhich carries the double mutation his3-11,15. In5% of His+ transformants (3/60), analysed bycolony PCR, the HIS3 marker was correctlyintegrated. This frequency is in agreement withthe results of Rieger et al. (1997), who foundthat the frequency of homologous integration was1–4% using a HIS3 disruption cassette with 50 bpof homology to the target loci in the samebackground.The heterologous kanMX module (Wach et al.,
1994), which renders S. cerevisiae resistant to thedrug geneticin (G418), was used for strain con-struction in the EUROFAN diploid strainFY1679. This marker and strain were chosen toallow full exploitation of the deletion mutantsgenerated by the EUROFAN project (Oliver,1996b). The kanMX4 disruption cassette wasgenerated using LFH-PCR. This method (Wach,1996) was employed because of the much highertransformation efficiency obtained in comparisonto short flanking homology PCR. The LFH-PCRproduct, with long flanking homologous sequences5* and 3* to the HO gene (300 and 500 bp, respect-ively), was used for transformation of the haploidstrains FY23 and FY73. Correct integration wasfound in >80% of G418-resistant transformantsanalysed by colony PCR. In accordance with theresults of Wach et al. (1994), the level of illegiti-mate integration was very low, underlining theadvantages of using heterologous markers forgene replacement. A homozygous ho::kanMX4/ho::kanMX4 standard strain was generated bycrossing FY23 and FY73 deletion mutants.
Trial competition experimentsIn order to evaluate the phenotypic effect of
the two replacement markers chosen, competitionexperiments between the haploid ho deletantsand their wild-type parents were carried out incontinuous culture under glucose and ammoniumlimitation. The initial ratio of parent : mutant was1 : 1 and the dilution rate was set at D=0·1 h"1.An upshift in dilution rate to D=0·2 h"1 wasperformed in order to determine whether anyphenotypic effect of the replacement marker was
. 13: 1563–1573 (1997)

1569
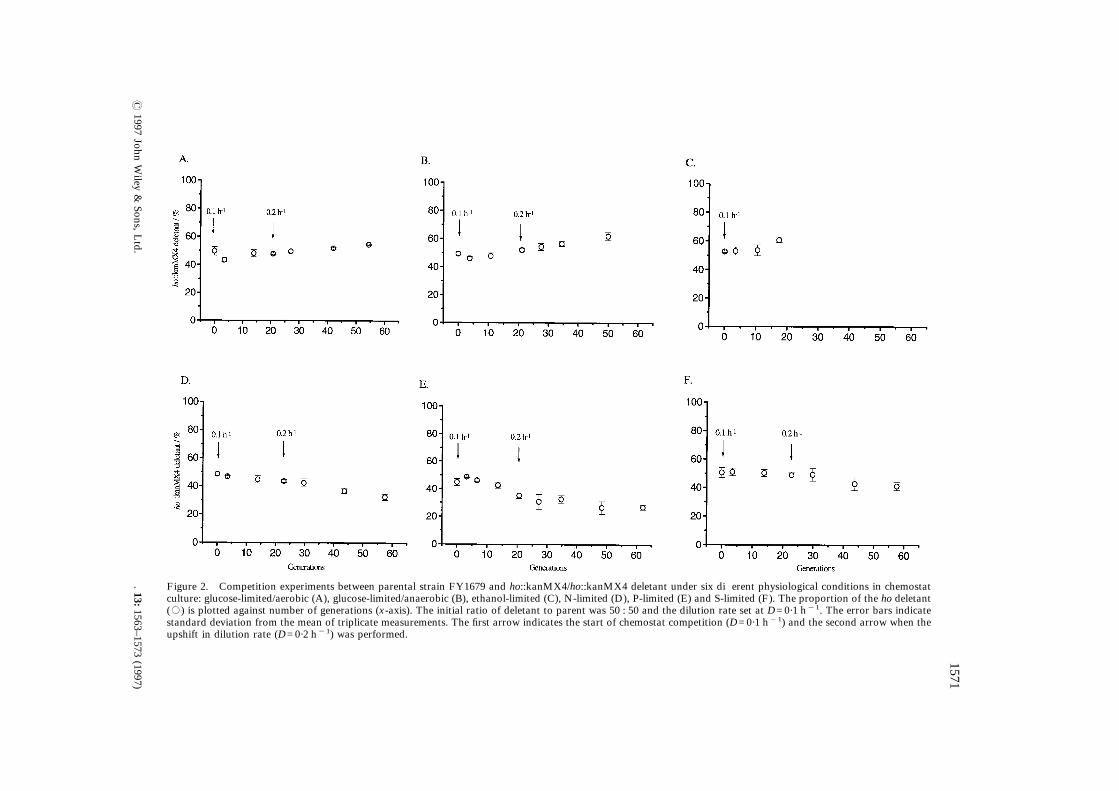
Figure 1. Competition experiments between haploid ho deletants and wild-type parental strains in chemostat culture:ho::kanMX4 deletant against FY23 under glucose and nitrogen limitation (A and B, respectively) and ho::HIS3 deletantagainst BMA41-1B under glucose and nitrogen limitation (C and D, respectively). The initial ratio of deletant to parent was50 : 50 and the dilution rate set at D=0·1 h"1. The error bars indicate standard deviation from the mean of triplicatemeasurements. The first arrow indicates the start of chemostat competition (D=0·1 h"1) and the second arrow when theupshift in dilution rate (D=0·2 h"1) was performed.
specific-growth-rate dependent. Replica platingwas used to measure the change in the relativeproportions of the two strains in the culture. Theresults of these competitions (Figure 1) show alinear increase in the proportion of theho::kanMX4 deletant of ca. 0·2% per generationover at least 60 generations of steady-state growthunder either glucose or ammonium limitation(Figure 1A, B). Two independent experimentsexhibited similar kinetics and showed no directcorrelation between the increase in the proportionof the ho deletant and the upshift in dilutionrate. This suggests that the observed effect isgrowth-rate independent.In order to determine if this increase was signifi-
cant, the growth rate differential was calculated.The results in Table 2 show that the ho::kanMX4deletant has only a small growth rate advantage of2–4% relative to the parent strain under either
? 1997 John Wiley & Sons, Ltd.
condition, indicating that the phenotypic impact ofthe kanMX4 marker is negligible. However, this isnot true for theHIS3marker. In three independentcompetition experiments carried out under glucoselimitation, the proportion of the ho::HIS3 deletantincreased, against its BMA41-1B parent, from50% to approximately 80% after less than 10generations in continuous culture (see Figure 1Cfor an example). In all three experiments, a furtherincrease to >90% was seen before the upshift indilution rate, with virtually complete take-over ofthe deletant after ca. 20 generations. From thesedata, an average growth rate advantage of 23%was calculated for the ho::HIS3 deletant (Table 2).In contrast, three independent experiments underammonium limitation showed the proportion ofthe ho deletant to decrease by ca. 30% over the first10 generations (see Figure 1D for an example). Inthis experiment, a further decrease to <5% was
. 13: 1563–1573 (1997)

1570 . .
Table 2. Specific growth rate differential (ó) forhaploid ho deletants relative to wild-type parental strainunder glucose and nitrogen limitation.
Strain Limitation ó*(%)
ho::kanMX4 Glucose/aerobic +2·2/+4·2Nitrogen +1·7/+3·0
ho::HIS3 Glucose/aerobic +23·5&1·8Nitrogen "25·3&4·9
ó is given as a percentage of D=0·1 h"1. The correlationcoefficient of the ‘best-fitted’ line from linear regression on thedata was >0·9 in all cases.*Values represented are averages followed by the standarderror of the mean of three independent experiments. The actualvalues are given, when only two experiments were performed.
observed before the dilution rate upshift, suggest-ing a complete take-over of the parental strainafter 30 generations. In the other two compe-titions, the proportion of the ho deletant remainedapproximately constant between 10–20% of thetotal population. The large decrease in the pro-portion of the ho deletant is equivalent to a dis-advantage in specific growth rate of some 25%(Table 2). The dilution-rate upshift had no effecton the proportion of the two strains in the cultureunder either nutrient limitation. However, at thetime of the shift, one cell type had virtually out-competed the other such that the experimentwould have had to have been extended to a verylarge number of generations in order to discernany growth-rate-specific effect.These large changes in selective fitness obtained
with the HIS3 marker may be explained in termsof differences in the kinetics and energetics ofamino acid uptake for the auxotrophic parentalstrain, as compared to those of intracellular aminoacid synthesis in the histidine-prototrophic hodeletant (see Pronk et al., 1996, for review).Although no direct comparison between these twosets of competition experiments is possible becauseof the different strains used, it is likely that theprofound phenotypic effects seen with theho::HIS3 deletion are due to the nutritionalmarker employed.It cannot be excluded that phenotypic effects
observed in haploid deletion mutants are the con-sequences of secondary mutations induced by thetransformation process. For this reason, we intendto use diploid strains in our competition exper-
iments, and so the homozygous ho::kanMX4/? 1997 John Wiley & Sons, Ltd.
ho::kanMX4 deletant was constructed andcompeted against the parental diploid FY1679.The competition experiments were carried out inchemostats under the following physiological con-ditions: glucose-limited/anaerobic, glucose-limited/aerobic, ethanol-limited, N-limited, P-limited,S-limited (Figure 2). Under glucose limitation(Figure 2A, B), the graphs show small increasesof ¦10% in the proportion of the ho deletant over50 generations. In agreement with the results of thehaploid competition with the kanMX marker, theincrease appeared to be linear and independentof the dilution rate upshift (and, hence, of thespecific growth rate). Under ethanol limitation(Figure 2C), the proportion of the ho deletantincreased by ca. 8% over 17 generations before theD-upshift. No data have been obtained at thehigher dilution rate, since the culture was washedout after the upshift, indicating that the maximumspecific growth rate of both strains was below0·2 h"1.Under all other nutrient limitations, a decrease
of 10–20% in the proportion of the ho deletant wasobserved over 50 generations of steady-stategrowth (Figure 2D, E, F). As with the experimentsunder the different carbon limitations, the decreaseappeared to be linear and independent of thedilution rate shift. However, the decrease in theproportion of the ho deletant in the populationgrown under ammonium limitation contrasts withthe results of the haploid competition experiment(Figure 1B) where an increase in the proportion ofthe ho deletant was found. These last results sug-gest that we are dealing with small variationswhich are not genetically determined.In order to determine whether the differences
in selective fitness were significant, the growthrate differential (ó) was calculated. The results(Table 3) reveal that the ho::kanMX4/ho::kanMX4deletion had a small, but measurable, effect ongrowth rate of ¦&4%. The estimated error in thecalculation of ó from triplicate measurements ofthe proportion of the ho deletants in one exper-iment was, for all conditions, ca.&10% of themean value. Thus, it can be concluded that thisstrain is suitable as a standard for our competitionexperiments since only a very small marker effectwas observed under the test conditions. Further-more, the results confirm our choice of HO as aneutral site, and indicate that resistance genes(such as kanMX) should be used in preferenceto nutritional markers for the construction ofstandard strains and deletion mutants for use in
. 13: 1563–1573 (1997)
nt under six different physiological conditions in chemostatited (E) and S-limited (F). The proportion of the ho deletantthe dilution rate set at D=0·1 h"1. The error bars indicateat competition (D=0·1 h"1) and the second arrow when the
1571
?1997
JohnWile
Figure 2. Competition experiments between parental strain FY1679 and ho::kanMX4/ho::kanMX4 deletaculture: glucose-limited/aerobic (A), glucose-limited/anaerobic (B), ethanol-limited (C), N-limited (D), P-lim(,) is plotted against number of generations (x-axis). The initial ratio of deletant to parent was 50 : 50 andstandard deviation from the mean of triplicate measurements. The first arrow indicates the start of chemostupshift in dilution rate (D=0·2 h"1) was performed.
y&Sons,
Ltd.
.13:
1563–1573(1997)

1572 . .
competition experiments. Moreover, the resultswith the kanMX4 marker indicate that competi-tion experiments in chemostat culture are a verysensitive way to measure changes in specificgrowth rate and that this method will be suitableas a first step for the quantitative analysis of genefunction based on the ‘top-down’ approach ofMCA.
ACKNOWLEDGEMENTS
We are grateful to Achim Wach and ChristophCullin for gifts of strains and plasmids. BasTeusink, Phil Butler and Paul Lane are thankedfor their critical reading of the manuscript and forstimulating discussions. Work on the functionalanalysis of the yeast genome, in our laboratory, issupported by the Chemicals and PharmaceuticalsDirectorate of the BBSRC, the EUROFANproject of the EC, the Wellcome Trust, PfizerCentral Research, Applied Biosystems, andAmersham International. F.B. would like to thankPfizer Central Research for their generous provi-sion of a Studentship and Ronnie Farquhar for hisstimulating and encouraging input as industrialsupervisor.
Table 3. Specific growth rate differential (ó) of thehomozygous ho::kanMX4/ho::kanMX4 diploid relativeto the HO/HO wild-type diploid.
Limitation ó (%)
Glucose/aerobic +1·8Glucose/anaerobic +2·9Ethanol +3·2Nitrogen "2·3Phosphorus "4·0Sulphur "1·6
ó is given as percentage of D=0·1 h"1. The correlationcoefficient of the ‘best-fitted’ line from linear regression on thedata was >0·9 in all cases.
REFERENCES
Baudin, A., Ozier-Kalogeropoulos, O., Denouel, A.,Lacroute, F. and Cullin, C. (1993). A simple andefficient method for direct gene deletion in Sac-charomyces cerevisiae. Nucl. Acids Res. 21, 3329–3330.
Baudin-Baillieu, A., Guillemet, E., Cullin, C andLacroute, F. (1997). Construction of a yeast straindeleted for the TRP1 promoter and coding region that
? 1997 John Wiley & Sons, Ltd.
enhanced the efficiency of the polymerase chainreaction-disruption method. Yeast 13, 353–356.
Berben, G., Dumont, J., Gillequet, V., Bolle, P. A. andHilger, F. (1991). The YDp plasmids: a uniform set ofvectors bearing versatile gene disruption cassettes forSaccharomyces cerevisiae. Yeast 7, 475–477.
Brand, G. C. (1996). Top-down metabolic controlanalysis. J. Theor. Biol. 182, 351–360.
Danhash, N., Gardner, D. C. J. and Oliver, S. G. (1991).Heritable damage to yeast caused by transformation.Bio/Technology 9, 179–182.
Dujon, B. (1996). The yeast genome project: What didwe learn? Trends in Genet. 12, 263–270.
Dykhuizen, D. E. and Hartl, D. L. (1983). Selection inchemostats. Microbial Rev. 47, 150–168.
Gietz, R. D., Schiestl, R. H., Willems, A. R. and Woods,R. A. (1995). Studies on the transformation of intactyeast cells by LiAc/SS-DNA/PEG procedure. Yeast11, 355–360.
Goffeau, A., Barrell, B. G., Bussey, H., et al. (1996). Lifewith 6000 genes. Science 274, 546–567.
Goffeau, A., et al. (1997). The yeast genome directory.Nature 387 (Suppl.), 5–105.
Hammond, J. R. M., Lancashire, W. D., Meaden, P. G.,Oliver, S. G., Smith, N. A. (1994). Stability ofgenetically modified yeast in relation to beer ofgood and consistent quality. MAFF (Ministry ofAgriculture, Fisheries and Food; UK) Report 07/63M.
Hofmeyr, J.-H. S. (1995). Metabolic regulation: a con-trol analytic perspective. J. Bioenerget. Biomembr. 27,479–490.
Kacser, H. and Burns, J. A. (1973). The control of flux.Symp. Soc. Exp. Biol. 27, 65–104.
Mewes, H. W., Albermann, K., Bahr, M., et al. (1997)Overview of the yeast genome. Nature 387 (Suppl.),7–65.
Niederberger, P., Prasad, R., Miozzari, G. and Kacser,H. (1992). A strategy for increasing an in vivo flux bygenetic manipulations—the tryptophan system ofyeast. Biochem. J. 287, 473–479.
Oliver, S. G. (1996a). From DNA sequence to biologicalfunction. Nature 379, 597–600.
Oliver, S. G. (1996b). A network approach to thesystematic analysis of yeast gene function. Trends inGenet. 12, 241–242.
Pronk, J. T., Steensma, Y. H. and Van Dijken, J. P.(1996). Pyruvate metabolism in Saccharomycescerevisiae. Yeast 12, 1607–1633.
Rieger, K.-J., Kaniak, A., Coppee, J.-Y., et al. (1997).Large-scale phenotypic analysis—the pilot project onyeast chromosome III. Yeast 13, 1547–1562.
Rohwer, J. M., Schuster, S. and Westerhoff, H. V.(1996). How to recognize monofunctional units in ametabolic system. J. Theor. Biol. 179, 213–228.
Schena, M., Shalon, D., Heller, R., Chai, A., Brown,P. O. and Davis, R. W. (1996). Parallel human
genome analysis—microarray-based expression. 13: 1563–1573 (1997)

1573
monitoring of 1000 genes. Proc. Natl. Acad. Sci. USA93, 10614–10619.
Shoemaker, D. D., Lashkari, D. A., Morris, D.,Mittmann, M. and Davis, R. W. (1996). Quantitativephenotypic analysis of yeast deletion mutants using ahighly parallel molecular bar-coding strategy. NatureGenetics 14, 450–456.
Smith, V., Botstein, D. and Brown, P. O. (1995). Geneticfootprinting—a genomic strategy for determining agene’s function given its sequence. Proc. Natl. Acad.Sci. USA 92, 6479–6483.
Smith, V., Chou, K. N., Lashkari, D., Botstein, D. andBrown, P. O. (1996). Functional analysis of the genesof yeast chromosome V by genetic footprinting.Science 274, 2069–2074.
Teusink, B., Baganz, F., Westerhoff, H. V., and Oliver,S. G. (1997). Metabolic control analysis as a tool inthe elucidation of the function of novel genes. InTuite, M. F. and Brown A. J. (Eds), Methods in
? 1997 John Wiley & Sons, Ltd.
Microbiology: Yeast Gene Analysis. Academic Press,London. (In press.)
Verduyn, C., Postma, E., Scheffers, W. A. and VanDijken, J. P. (1990). Physiology of Saccharomycescerevisiae in anaerobic glucose-limited chemostatcultures. J. Gen. Microbiol. 136, 395–403.
Wach, A., Brachat, A., Pohlmann, R. and Philippsen, P.(1994). New heterologous modules for classical orPCR-based gene disruptions in Saccharomycescerevisiae. Yeast 10, 1793–1808.
Wach, A. (1996). PCR-synthesis of marker cassetteswith long flanking homology regions for genedisruptions in S. cerevisiae. Yeast 12, 259–265.
Winston, F., Dollard, C. and Ricuperohovasse, S. L.(1995). Construction of a set of convenient Sac-charomyces cerevisiae strains that are isogenic toS288C. Yeast 11, 53–55.
Yocum, R. (1983). Genetic engineering of industrialyeasts. Proc. Bio. Expo. 86, 17.
. 13: 1563–1573 (1997)