sulfur isotope geochemistry and fractionation between coexisting
TRANSCRIPT

Mineral. Soc. Amer. Spec. Pap. 3, 133-144 (1970).
SULFUR ISOTOPE GEOCHEMISTRY AND FRACTIONATIONBETWEEN COEXISTING SULFIDE MINERALS
H. G. THODE
Department of Chemistry, McMaster University, Hamilton, Ontario, Canada
ABSTRACT
The study of sulfur isotope abundances in nature is rewarding because of the ubiquity of the element and the varietyof chemical forms in which it is found. Theoretical calculations and laboratory investigations of sulfur isotope fractionationin chemical, microbiological and mineralogical systems lead to an understanding of the isotope distribution patterns innature and give information regarding the modes of formation and subsequent histories of many kinds of sulfur bearingmaterials.
A broad distinction may be made between biogenic and primary sulfur. Biogenic sulfur is characterized by a widerange of isotope ratio values as a result of the variable fractionation introduced during the reduction of sulfate to sulfideby anaerobic bacteria, while primary sulfur tends to have a comparatively narrow range of isotope ratio values. The simplenature of these patterns may be altered either by homogenization of isotope ratios by metamorphic heating events or bycontamination with sulfur from other sources.
There is considerable current interest in the comparison of the sulfur isotope ratios in coexisting sulfide minerals. Thedifference in isotope ratios between mineral types may represent equilibrium sulfur isotope effects or may be related tokinetic isotope effects during the emplacement of the minerals with later modification by metamorphic heating events. Ineither case such differences can be used as a geothermometer by utilizing the predictable and measurable temperaturedependences of sulfur isotope exchange equilibria. The types of observational, theoretical and experimental studies per-formed to date are mentioned, as are some of the problems which remain to be solved in order to develop this method ofgeothermometry.
INTRODUCTION
Isotope geochemistry has as its basis the small differ-ences in chemical behavior shown by the isotopes of anelement. The concept of an isotope was introduced bySoddy (1910) and Fajans (1911) as a consequence of radio-activity in heavy elements and isotopes were consideredto be element pairs which could not be separated chemi-cally. For twenty years all attempts at chemical separa-tion failed. It was the discovery of the isotopes of the lightelements around 1930, particularly that of deuterium byUrey et al. (1932) that lead to success in this endeavour.Since then differences in the chemical properties of theisotopes of hydrogen, carbon, nitrogen, oxygen, sulfur,and other elements have been both calculated using themethods of statistical mechanics and measured experi-mentally. These differences can lead to significant isotopefractionation in chemical reactions, both in nature and inthe laboratory, and are the cause of changes in the rela-tive isotope abundances of these elements from sampleto sample.
Since the war extensive studies have been made ofnatural isotope abundance variations and, together withtheoretical and laboratory work, such studies have beenused to help in the solution of a wide variety of geochemi-cal problems. Sulfur isotope geochemistry has proved tobe a particularly rewarding field because of the reasonablylarge percentage mass difference between the two principalisotopes, the variety of the chemical forms of sulfur andtheir widespread occurrence in such diverse materials assea water, sedimentary rocks, petroleum, primary igneousrocks, volcanic gases and mineral deposits. Studies withsulfur isotopes have been concerned with such problemsas the isotope fractionation in the biological sulfur cycleand in the sulfur bearing gases of volcanoes, the isotopic
composition of present day and ancient oceans, the possibleorigins of oil and the modes of formation and post deposi-tional histories of sulfide mineral deposits.
Sulfur has four stable isotopes (32. 33. 34. 36S) whose per-centage abundances are approximately 95.0, 0.75, 4.20 and0.017 respectively. Isotope abundance variations are gen-erally considered in terms of the abundance ratio, 34S/32S,of the two principal isotopes, although for special purposes,such as the measurement of cosmic ray effects in meteor-ites, use may be made of variations of both 33S/32Sand 36S/32Sratios (Hulston and Thode, 1965). The range of varia-tion of 34S/32S in nature is approximately 10% and whenreferring to differences of isotope abundance ratios be-tween samples it is common practice to use the "del"notation where
((3'Sj32S)sampJe. ),,3'S = _ 1
(3'S/32S)standard
(534S)X 1000, therefore, gives the permil difference in iso-tope ratio between a sample and a standard.
The generally accepted standard is the 34S/32Sratio oftroilite from the Canyon Diablo meteorite. There areseveral reasons for expressing sulfur isotope ratios in thismanner. First, the mass spectrometric techniques usedfor sulfur isotope abundance measurements make the de-termination of differences of 34S/32S between samplesmuch more precise than the determination of the absolutevalue of 34S/32Sfor either sample. Using a few mg of sulfuras sulfur dioxide or sulfur hexafluoride it is possible tocompare samples with a precision of the order of 0.1 0/00.
Second, the use of troilite from Canyon Diablo as a stan-dard makes the interlaboratory comparison of resultsmuch easier than if a number of personal standards areused. Finally, the ratio 34S/32S is remarkably uniform inmeteorites (Macnamara and Thode, 1950; Vinogradov,
133

134 H. G. THODE
1958; Thode et al., 1961; Kaplan and Hulston, 1965) andthere is a considerable body of evidence to suggest thatthe primordial, or initial, terrestrial value of 3'S/32S is thesame as that of Canyon Diablo troilite. Thus this valuerepresents a logical base level from which to measurefractionation effects in nature.
Isotope geochemistry then involves observations ofsulfur isotope distribution patterns in nature, theoreticalcalculations of equilibrium and kinetic isotope effects andlaboratory measurements of isotope effects in chemical,biological, and mineralogical processes. All these studiesare important in the interpretation of natural isotopedistribution patterns and in the solution of geochemicalproblems. This paper deals briefly with these various as-pects of sulfur isotope geochemistry and discusses specif-ically recent work with coexisting sulfide minerals.
THE THEORY OF ISOTOPE EFFECTS
Predictions of the differences in behaviour of isotopicsubstances are made in two ways. For systems in equilib-rium the calculations involve the properties of the equili-brating substances but do not depend on the particularpathways or mechanisms involved in the achievement ofequilibrium. For systems undergoing change, as in uni-directional chemical reactions, the situation is somewhatmore complicated and attention must be paid to the reac-tion mechanism and possible intermediates involved inthe formation of the final product material.
Equilibrium isotope effects. Equilibrium constants forisotope exchange reactions may be calculated in terms ofthe characteristic vibrational frequencies of the variousisotopic species. A typical isotope exchange reaction maybe written
Al+B2~A2+Bl
where the subscripts 1 and 2 indicate that the molecules Aand B with one element as a common constituent containonly the light and heavy isotope respectively of that ele-ment.
Using statistical mechanics the isotope equilibrium con-stant may be expressed in terms of the partition functions,Q, of the equilibrating species:
K = Q(A2) /Q(B~.Q(Al) Q(B1)
The equilibrium constant then is simply the product orquotient of two partition function ratios, one for the twoisotopic species of A, the other for B. The partition func-tion of a molecule is given by:
Q = L gi exp (- E;/kT).
Here the summation is over all the allowed energy levelsof the molecules, and gi is the statistical weight of the ithlevel Ei. It has been shown (Bigeleisen and Mayer, 1947;Urey, 1947) that for the purpose of calculating partitionfunction ratios of isotopic molecules, other than H2 for
which rotational energies must be taken into account, it issufficient to consider a modified partition function, Q',such that
Qf = IIV; L exp( - E y;b/kT)
where Vi is the ith zero order vibrational frequency andEvib is a vibrational energy state of the molecule.
If the harmonic oscillator approximation is used formolecular vibrations then:
Qf = IIU; exp(- u;/2)/(1 - exp(- u;))
where
u; = hv;/kT.
For many molecules only the observed fundamentalfrequencies are known. If, however, zero order frequenciesand anharmonicity coefficients are available then moreprecise estimates of Q' may be made. The harmonic oscil-lator approximation is most seriously in error at high tem-peratures where vibrational energy levels other than theground state have appreciable populations.
The calculation of a partition function ratio for a pair ofisotopic molecules thus requires a knowledge of the vibra-tional frequencies of each of them. When fundamentalvibrational frequencies (and possibly zero order frequenciesand anharmonicity coefficients) are obtained from infraredand Raman spectroscopy for the most abundant isotopicspecies it is possible to set up a force constant model of themolecule and to write equations relating the vibrationalfrequencies, force constants, atomic masses and bondangles. Force constants are invarient under isotopic sub-stitution so that once they are determined it is possible toevaluate the vibrational frequencies for the rare isotopicspecies by simply substituting the appropriate masses inthe force constant equations. Then with the vibrationalfrequencies of the two species, the one measured, the othercalculated, the partition function ratio for the two iso-topic species is determined.
Using these methods the equilibrium constant K for theexchange reaction
H23'S + a2S 0, ~ H23'S + 3'S O2
has been calculated to be 1.008 at 500°C indicating thatunder equilibrium conditions there is 8%
0 more 3'S inthe SO, than in the H,S (Cragg et al., in prep.). Thus the de-parture of this equilibrium constant from unity is ameasure of the difference in the equilibrium properties ofthe two isotopes in this process.
When solid materials are considered the evaluation ofpartition function ratios becomes more complicated. It isno longer sufficient to consider the independent vibra-tions of each molecule, instead the lattice vibrationsmust be taken into account and the phonon spectrum ofthe lattice determined. This topic will be considered inmore detail in a later section.
Kinetic isotope effects. The isotope fractionation in-troduced in the course of a unidirectional reaction may be

SULFUR ISOTOPES IN COEXISTING SULFIDES
considered in terms of the ratio of rate constants for theisotopic substances. According to the transition state the-ory of reaction kinetics (Eyring, 1935; Evans and Po-lanyi, 1935), reactant molecules are in equilibrium withmolecules in a transition state through which reaction pro-ceeds from reactants to products. Using the framework ofthis theory the ratio of rate constants, kl/k" for two com-peting isotopic reactions:
and
may be written (Bigeleisen, 1949; Bigeleisen and Wolfs-berg, 1958):
The ratio of rate constants for the reaction of light andheavy isotopic species is therefore expressed, as in thecase of equilibrium constants, simply in terms of two par-tition function ratios, one for the two isotopic reactantsspecies A, and one for the two isotopic species of acti-vated complex or transition state At. The factor Vl/V2 inthe expression is a mass term ratio for the two isotopicspecies. v may be related to a vibrational mode in the acti-vated complex which becomes imaginary as the reactioncoordinate is traversed and a bond broken. The determina-tion of the ratio of rate constants is therefore formallythe same as the determination of an equilibrium constant,although the calculations are not so precise because of theneed for detailed knowledge of the transition state. How-ever, by making certain assumptions concerning the natureof the transition state estimates for the ratio of rate con-stants for two competing isotopic reactions can be made.
Fractionation factors. For the purpose of understandingisotope abundance variations it is important to distinguishbetween the simple process fractionation factors given byequilibrium constants, or by ratios of rate constants inunidirectional reactions, and fractionation factors ob-served for systems where the simple process factor may havebeen multiplied many times. Commercial processes for theseparation of isotopes depend on this kind of multiplica-tion. Where such systems occur in nature isotope fractiona-tion factors will be observed that are significantly largerthan those calculated or measured for the simple processes.
A batch process or batch distillation is one such systemthat can give rise to large fractionation factors. For exam-ple, in the chemical reduction of sulfate to hydrogen sul-fide the isotopic cases may be written as:
kl32S042- .......H232S
and
135
The ratio of rate constants, kl/k2, is about 1.024 at roomtemperature (Harrison and Thode, 1957). Since the 3'SOrspecies reacts 24 0/00 faster than 34S04~ the H,S producedat any instant is enriched in 32S, or depleted in 34Sby thisamount (240
/00) relative to the remaining SO,~.The isotope effects resulting from this reaction may be
estimated using the mathematical treatment of the Ray-leigh distillation process. For small fractions of reaction thesulfide formed will be at - 240
/00 with respect to thestarting material, but as the reaction proceeds to com-pletion this difference will decrease until at the time ofcomplete reaction the sulfide produced must of coursehave the same isotopic composition as the initial sulfate.Similarly for small fractions of reaction the residual sul-fate isotopic composition will be virtually unaltered, butas the reaction proceeds, because of the continuing prefer-ential reduction of 32S0,~, the remaining sulfate will be-come more and more enriched in 34S0,~. Thus the residualmaterial will attain higher and higher 5"S values relativeto the starting material. After 75 percent of the sulfate hasreacted, it may be shown that the residual material hasa del value of +350
/00 so that a ratio of rate constantsof 1.024 has here produced a fractionation factor of 1.035.
Clearly batch processes of this type can occur in natureand give rise to a variety of observable fractionation fac-tors which will depend upon the degree of completenessof reaction. This must be taken into account in interpreta-tions of the natural isotope distribution pattern.
THE SULFUR ISOTOPE DISTRIBUTION IN NATURE
Figure 1 shows the ranges of 53'S values found in naturefor a number of different forms of sulfur: It may be seenthat the overall range is about 1000
/00• It is this total
8545 'Yoo+!lO +40 +30 +20 +10 -/0 -20 -30 -40 -50
METEORITES
•• BAS I C SILLS
IGNEOUS ROCKS- - - ..' ...---- -S' 0' VOLCANIC ORIGIN
'"SEA WATER
EVAPORITES.. RAIN ond SNOW
SEDIMENTARY SUL.FHIOES-- - - ...PETROLEUM
COAL
FIG. 1. The sulfur isotope distribution in nature.

136 H. G. THODE
spread and its fine details which must be explained interms of isotope fractionation effects.
The two most common forms of sulfur in the earth'scrust are sulfates and sulfides. The isotope exchange be-tween sulfate and sulfide may be written
and the theoretical value of the exchange constant is1.075 at 25°C (Tudge and Thode, 1950). Therefore if thisexchange takes place or is partially established in natureit should lead to sulfides being in general depleted in 34Srelative to sulfates by amounts up to 750/00. Although nosimple mechanism for the establishment of this exchangeis known spreads in isotope ratios of this order of magnitudeare seen to occur. Evaporites represent the largest concen-tration of sulfates and they have 534Svalues ranging from+350/00 to +50/00 while sedimentary sulfides mave 534Svalues ranging from +400/00 to - 50%0. This gives anoverall spread of 85%
0 between sulfates and sulfides.Since complete oxidation of a sulfide or the complete reduc-tion of a sulfate will not result in any change in isotopiccomposition it is possible in special situations to have sul-fate depleted, or sulfide enriched, in 34S. A case in point isthe pyrite of the Onwatin Slate in the Sudbury basin whichhas 534S values of up to around +30%0 (Thode et al.,1962) .
The types of material which exhibit the widest spread of534Svalues are secondary in nature, that is to say they havebeen involved in the sedimentary cycle. Other forms ofsulfur bearing materials such as basic sills, primary igneousrocks and volcanic gases show distinctly narrower rangesof 5~4S values which tend to be disposed symmetricallyabout zero (see Fig. 1). This distinction provides a basicdiagnostic tool in sulfur isotope geochemistry. When anigneous rock is found which displays a wide range of 534Svalues or a narrower range well removed from zero it ispossible to state with some certainty that the sulfur is notsolely of primary origin but contains at least a componentof reworked sedimentary sulfur.
The wide spread in 534S values for cyclic sulfur is theresult of biological activity. The major biological processinvolving sulfur isotope fractionation is the reduction ofsulfate to sulfide by the bacterium Desulphovibrio desul-phuricans in the muds or developing sediments on the seabottom (Thode et al., 1951). Many laboratory experimentshave been performed to elucidate the fractionation pro-duced by these bacteria.
Experiments on the chemical reduction of sulfate tosulfide (Harrison and Thode, 1957) have shown that thefractionation in this case may be adequately explained if itis assumed that the step in the reaction which controls theisotope fractionation produced is one involving the cleav-age of a sulfur-oxygen bond in the sulfate ion.
The situation for the bacterial reduction of sulfate ismuch more complex. In the course of experiments wheresuch parameters as temperature, hydrogen donor type andconcentration, sulfate concentration and bacterial popula-
tion density were varied (Jones and Starkey, 1957; Harri-son and Thode, 1958; Nakai and Jensen, 1964; Kaplan andRittenberg, 1964) it was found that the measured fraction-ation factor introduced during the conversion of a smallfraction of the available sulfate to sulfide could vary from0.996 to 1.050. In a recent paper, Kemp and Thode (1968)list the possible steps that are involved in the reductionprocess as: (a) the uptake of sulfate, (b) the organic com-plexing of sulfate, (c) the reduction to organically boundsulfite and (d) the production of hydrogen sulfide. Underdifferent conditions the net isotope fractionation producedwill depend on the isotope effects in each of these steps, therelative speeds of the steps and the extent to which thesulfate reservoir is depleted. When the bacteria are ex-tremely active, and the speeds of steps (b), (c), and (d)do not limit the rate of sulfide formation, it is the isotopiccompetition in step (a) which regulates the net isotopefractionation produced. Under these circumstances theisotope fractionation is small and fractionation factorsbetween sulfate and sulfide of the order of unity are likelyto be found.
The other extreme occurs when the bacteria producesulfide under conditions of very low metabolic rate andwhen the uptake of sulfate is no longer the slow step inthe overall reduction process. Under such circumstances itseems likely that the net isotope fractionation producedis the superimposition of an equilibrium isotope effect be-tween bound sulfate and bound sulfite and the kinetic iso-tope effect involved in step (d). In this case overall frac-tionation factors produced can approach the upper limitof 1.050. However, in case of intermediate metabolic rates,a condition prevailing most often in laboratory experi-ments and in nature, intermediate fractionation factorsare obtained (1.010 to 1.025).
Sulfate is reduced to sulfide in nature in two distinctsituations, open and closed systems. In the open system,the sulfate reducing bacteria are in good contact with seawater and the sulfate which they reduce is constantly re-plenished. In such circumstances the product sulfide willhave a 534Svalue removed from that of the ocean by anamount corresponding to the ratio of the rates of productionof H232Sand H234S.In a closed system the bacteria are notin contact with the ocean sulfate reservoir. This may becaused by the isolation of a small volume of sea waterfrom the well mixed ocean or by the overlaying of stillactive bacteria by muds holding interstitial sulfate. Insuch cases the 534Svalues of the sulfide produced and ofthe residual sulfate depend on the extent of reaction of theavailable sulfate as in the batch processes mentionedpreviously.
534Svalues are remarkably constant for all present-dayoceans and seas and it is reasonable to assume that theyhave been geographically uniform at any particular timein the past. Thus the 534S value of the ocean provides aconvenient base level from which fractionation effects inthe formation of sedimentary sulfides can be reckoned.The study of marine evaporites which were associated
SULFUR ISOTOPES IN COEXISTING SULFIDES 137
0
+5
+ 10
8345"00
j +20
+25
+30
lib~O-O-r\o IA~ It- 0 0 I I I \ I', I
1:----\ \/k~O,j~/~~;~_!__ \. 0
\--j\0
---- ----- - --- ----- ---
" 0-
~o-en p
o"o~:::> 6>- 0 z CDa: W . <I . if ~<I U en Z a: > . <I._; j::: ~ ~ . z . o CDz en ~ :::> o ~ o~ ffi ~ § a:ffi W !!? _, WW (i) a: <I a:o I- u ..,I- Cl. Cl. ~ 0 o u Cl.
<,
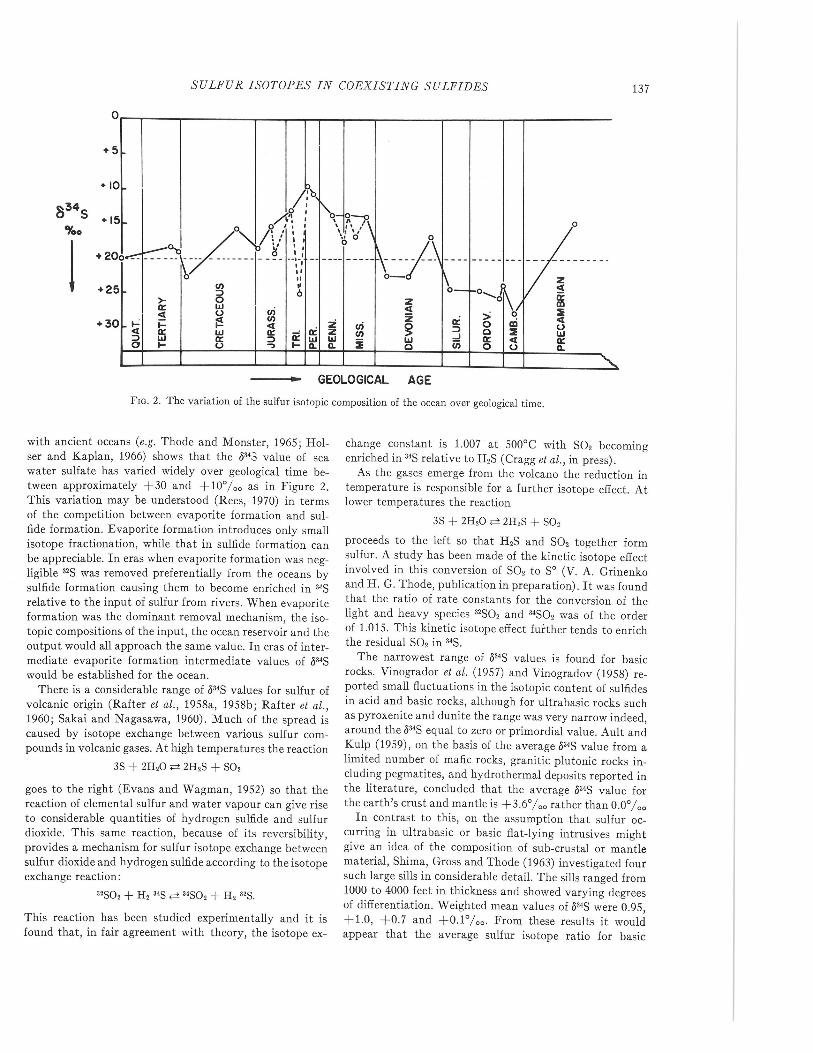
FIG. 2. The variation of the sulfur isotopic composition of the ocean over geological time.
GEOLOGICAL AGE
with ancient oceans (e.g. Thode and Monster, 1965; Hol-ser and Kaplan, 1966) shows that the 5343 value of seawater sulfate has varied widely over geological time be-tween approximately +30 and +10%0 as in Figure 2.This variation may be understood (Rees, 1970) in termsof the competition between evaporite formation and sul-fide formation. Evaporite formation introduces only smallisotope fractionation, while that in sulfide formation canbe appreciable. In eras when evaporite formation was neg-ligible 32Swas removed preferentially from the oceans bysulfide formation causing them to become enriched in 3'Srelative to the input of sulfur from rivers. When evaporiteformation was the dominant removal mechanism, the iso-topic compositions of the input, the ocean reservoir and theoutput would all approach the same value. In eras of inter-mediate evaporite formation intermediate values of 534Swould be established for the ocean.
There is a considerable range of 534Svalues for sulfur ofvolcanic origin (Rafter et al., 1958a, 1958b; Rafter et al.,1960; Sakai and Nagasawa, 1960). Much of the spread iscaused by isotope exchange between various sulfur com-pounds in volcanic gases. At high temperatures the reaction
3S + 2H20 <=± 2H,S + SO,
goes to the right (Evans and Wagman, 1952) so that thereaction of elemental sulfur and water vapour can give riseto considerable quantities of hydrogen sulfide and sulfurdioxide. This same reaction, because of its reversibility,provides a mechanism for sulfur isotope exchange betweensulfur dioxide and hydrogen sulfide according to the isotopeexchange reaction:
3'SO, + H, 3'S <=± 3'SO, + H, 3'S.
This reaction has been studied experimentally and it isfound that, in fair agreement with theory, the isotope ex-
change constant is 1.007 at 500°C with S02 becomingenriched in 34Srelative to H2S (Cragg et al., in press).
As the gases emerge from the volcano the reduction intemperature is responsible for a further isotope effect. Atlower temperatures the reaction
3S + 2H20 <=± 2H2S + SO,
proceeds to the left so that H2S and S02 together formsulfur. A study has been made of the kinetic isotope effectinvolved in this conversion of SO, to So (V. A. Grinenkoand H. G. Thode, publication in preparation). It was foundthat the ratio of rate constants for the conversion of thelight and heavy species 32S02 and 34S02 was of the orderof 1.015. This kinetic isotope effect further tends to enrichthe residual S02 in 34S.
The narrowest range of 534S values is found for basicrocks. Vinogrador et al. (1957) and Vinogradov (1958) re-ported small fluctuations in the isotopic content of sulfidesin acid and basic rocks, although for ultrabasic rocks suchas pyroxenite and dunite the range was very narrow indeed,around the 534Sequal to zero or primordial value. Ault andKulp (1959), on the basis of the average 534Svalue from alimited number of mafic rocks, granitic plutonic rocks in-cluding pegmatites, and hydrothermal deposits reported inthe literature, concluded that the average 53'S value forthe earth's crust and mantle is +3.6%
0 rather than 0.0%0In contrast to this, on the assumption that sulfur oc-
curring in ultrabasic or basic flat-lying intrusives mightgive an idea of the composition of sub-crustal or mantlematerial, Shima, Gross and Thode (1963) investigated foursuch large sills in considerable detail. The sills ranged from1000 to 4000 feet in thickness and showed varying degreesof differentiation. Weighted mean values of 534Swere 0.95,+1.0, +0.7 and +0.1%0. From these results it wouldappear that the average sulfur isotope ratio for basic

138 H. G. THODE
magmas is very close to that for meteorites, i.e. 534Sequalto zero.
The isotope ratios found in granitic intrusives and sul-fide ore bodies have to be interpreted in terms of the frac-tionation patterns mentioned so far. This is not alwayssimple, although detailed sulfur isotope studies have per-mitted the classification of sulfur in specific instances asprimary or secondary or mixed, and has given evidencefor subdividing many hydrothermal deposits into mag-matic hydrothermal, metamorphic hydrothermal andground water hydrothermal (Jensen, 1957, 1959).
Because of the complexities involved it is difficult to laydown strict rules for determining the origin of mineral de-posits. Each particular case must be treated separatelywith due regard being given to the local geological condi-tions.
As the techniques of mass spectrometry have improvedso has the range of problems which isotope geochemistsmay usefully study. In sulfur isotope geochemistry it wasthe large fractionation effects, such as in the bacterial re-duction of sulfate, that were first investigated but as theprecision of measurement of 534Svalues improved it becamepossible to investigate systems such as ultrabasic rocksand the meteorites where only small isotope fractionationeffects are found.
The latter part of this paper is devoted to the isotopefractionation between coexisting sulfide minerals. Theisotope effects involved are rarely greater than 50
/00 sothat measurement of these important effects requires care-ful sample preparation and precise mass spectrometry.
FRACTIONATION BETWEEN SULFIDE MINERALS
Observation of fractionation effects. In 1957 in the courseof a broad survey of sulfur isotopes in nature Sakai (1957)suggested that isotopic fractionation between differentmetallic sulfides would bring about a slight variation oftheir isotope ratios during their deposition. The observa-tional data he presented seemed to support this view, foramong nine sulfide pairs he examined, in three cases out ofthree marcasite was enriched in 34Srelative to chalcopyrite,and in three cases out of three pyrite was enriched in 34Srelative to sphalerite. In one case sphalerite was enrichedrelative to galena and the remaining two cases were en-richment and depletion of sphalerite relative to chalcopy-rite. However, Sakai was obliged to state that no firmconclusions could be drawn from these data since the magni-tudes of the effects noted were at the limits of the precisionof the measurements. Since 1957 many cases of sulfurisotope fractionation bewteen coexisting sulfide mineralshave been noted.
Gavelin, Parwel and Ryhage (1960) in the course of acritical study of the isotope fractionation involved in sul-fide mineralization noted that for sulfides in a single handspecimen there was a sequence pyrite-sphalerite-galenafrom higher values of 534Sto lower values.
In two studies of the HeathSteele Ore deposits of New-castle, New Brunswick, Dechow (1960) and Tupper (1960)
noted possible isotope fractionation between sulfides.Dechow suggested that there was a tendency for pyrite tohave a higher 1)3'Svalue than chalcopyrite while remarkingthat this trend was not statistically significant. Tuppernoted that the group means of 534S values for pyrite andchalcopyrite were very similar and that both were higherthan those for sphalerite and galena.
Buschendorf et al. (1963) in a study designed to de-termine the origins of sulfide ore bodies at Meggen,Germany, again noted a high to low 534S trend for pairsamong the three minerals, pyrite, sphalerite and galena.
Smitheringale and Jensen (1963) in a study of sulfidesfrom igneous rocks and genetically associated mineral de-posits of the Triassic Newark group of the Eastern UnitedStates noted that with coexisting pyrite and chalcopyritethere was a clear tendency for the pyrite 53'S value to behigh.
Friedrich, Schachner and Nielsen (1964) in their studyof ore deposits in the Sierra de Cartagena, Spain, notedthat the 53·S values of the different coexisting sulfides differonly slightly from one another but that a high to lowsequence could be written FeS, FeS2, ZnS, PbS.
The work reported by Tatsumi (1965) seems to be thefirst study specifically concerned with sulfur isotope frac-tionation between sulfides. Taking his data from about adozen suites of coexisting sulfide minerals from Japanesemetallic deposits, he was able to say that 53·S values forpyrite tend to be high, those for galena low, and those forchalcopyrite, sphalerite, bornite and tetrahedrite inter-mediate. The fractionation factors that he noted betweenpyrite-sulfide and sulfide-galena pairs ranged from 1.001to 1.003, with deposits genetically connected with Tertiaryvolcanism giving higher factors than those of contactmetasomatic or regionally metamorphic origin. The degreeof fractionation is higher in the ore deposits formed atlower temperatures, which is in accord with the theory ofisotope exchange equilibrium. This suggested that equilib-rium had been established and that these effects might beused as a geothermometer. The principle of such a ther-mometer is based on the temperature dependence of theisotope equilibrium constant for reactions such as:
Fe3'S + Pb3·S <=' Fe3'S + Pb3'S
Measurement of the fractionation factor, which at equilib-rium would be equal to the isotope equilibrium constant,would give a measure of the temperature at which equi-librium of the two sulfides took place. Such a geotherrnom-eter would be of particular interest and utility, since,because of the nature of isotope effects, it would be pressureindependent.
Speelman and Schwarcz (1966) have examined the sulfurisotope composition of coexisting pyrite-pyrrhotite pairsin metamorphic rocks in the Hailburton-Madoc area,Ontario. In 90 percent of their samples pyrite has a higher534Svalue than pyrrhotite by between 0 and 1.40
/00•
In a study of the origins of the Quemont ore body inNorthwestern Quebec, Ryznar, Campbell and Krouse

SULFUR ISOTOPES IN COEXISTING SULFIDES 139
(1967) noted two isotope effects connected with sulfides.Firstly, there is a tendency for the group mean 53·S valueof pyrite plus pyrrhotite to be higher than that for chalco-pyrite plus sphalerite and that, secondly, the differencebetween the group means increases slightly with depth inthe ore body. These effects are explained by the authorsin terms of isotope fractionation during the course ofmineralization from a sulfide melt. That is to say theisotope abundance patterns involved are the result ofkinetic isotope effects associated with mineralization andnot directly related to the equilibrium isotope effectsbetween minerals.
The possibility of depositional effects is also mentionedby Stanton and Rafter (1967). For samples of coexistingsphalerite and galena from Broken Hill, New South Wales,they note that in general sphalerite has a higher 534S valuethan galena and that the difference between pairs is relatedto the degree of metamorphism which the minerals haveexperienced. They point out that this might representpurely metamorphic partitioning (an equilibrium isotopeeffect) or a substantial depositional partitioning (a kineticisotope effect) that has been lessened by aging and meta-morphism.
Lusk and Crocket (1969) in a new study of sulfur iso-topes from the Heath Steele B-1 orebody of New Bruns-wick and from five other stratiform deposits in the areahave made careful observations of the difference in 534Svalues between pairs of sulfides. They note that the frac-tionation factors between given pairs of sulfide mineralsare relatively constant throughout all six deposits and thatfor pyrite-sphalerite, sphalerite-galena and pyrite-galenahave mean values of 1.001, 1.002 and 1.003 respectively.From these observations they conclude that sulfur isotopeequilibrium was closely approached at a fairly uniformtemperature and that the deposits studied have undergonelow temperature regional metamorphism which has gener-ated the observed fractionations and caused localizedisotopic homogenization with respect to given minerals.
Sasaki and Krouse (1969) in their study of the PinePoint lead-zinc mineralization again raise the suggestionthat the isotopic composition of sulfides may be deter-mined more by the circumstances of their emplacementthan by post-emplacement equilibration. They point outthat the previously reported studies on coexisting sulfideminerals seem to include many examples of successivemineralization as opposed to coprecipitation but that evenso the observed isotopic trend is in accord with the min-erals in question being in sulfur isotopic equilibrium. Theysuggest that this might be an indication that the sulfurisotope composition of the ore forming fluid in these de-posits may have been comparatively uniform during thecourse of mineralization and that in such circumstances theisotope ratio variations among different sulfide specieswould have the same trend as if the minerals had formedat the same time and under conditions of isotopic equi-librium.
Further examples of isotope fractionation between co-
existing sulfides have been reported following studies onore bodies in Tasmania (Solomon, Rafter, and Jensen,1969; Both et al., 1969). These studies reinforce the pre-viously observed ordering of 534S values with pyrite,sphalerite, chalcopyrite and galena forming a high to lowsequence. The authors point out again that this trend isconsistent with either depositional or later metamorphiceffects.
The existence of systematic differences in 534S betweencoexisting sulfides in nature is now well established as isthe idea that these differences reflect in some way thethermal histories of the sulfides in question. The theoreticaland experimental work that has been done and is now inprogress is largely concerned with the determination ofthe variations with temperature of sulfur isotope exchangeconstants for pairs of sulfides and the application of suchdata to geothermometry.
Theory. The first theoretical study of sulfur isotope frac-tionation between metallic sulfides was made by Sakai(1968). In addition, Bachinski (1969) has reported a studyof the bond strengths of sulfide minerals and their rela-tionship to isotope fractionation.
It is possible to make qualitative estimates of the frac-tionation to be expected between coexisting sulfides byusing the simple idea that the process of equilibration isone where the total free energy of a system is minimized.If the rearrangement of isotopes between two phases isconsidered, it may be shown that in order to bring aboutthis free energy minimization the heavier isotopes will tendto accumulate at sites with higher bond energies. Thus inorder to predict the way in which sulfur isotopes will par-tition themselves in a multiple mineral assembly, it isnecessary to have some knowledge of the force constantsof the mineral lattices. These atomic scale parameters arereflected in many ways in the macroscopic properties of acrystal, for example volume compressibility, heat capacityat constant volume, free energy and heat of formation,lattice energy, solid state reaction rate and activationenergy of diffusion.
Sakai and Bachinski have between them considered allthe above macroscopic properties for a number of metallicsulfides with a view to determining the expected order ofincreasing 534S values in equilibrated sulfide mixtures.Bachinski has pointed out the use of such indicators is mostsuccessful for minerals with identical structures, but usefulinformation may still be obtained for minerals of differentstructures in cases where the differences in the chosenmacroscopic parameters are so large as to outweigh effectsdue to structural distortions.
Using these techniques it may be shown that in anequilibrium mixture the sequence in order of decreasing534S value for the more important sulfide minerals shouldbe:
pyrite> sphalerite> chalcopyrite> galena
This is in agreement with the trends noted for naturallyoccurring samples.

140 H. G. THODE
TABLE1. VALUESOF (K -1) X 1000 FORSULFIDE-SULFIDEPAIRS.FROMSAKAI(1968)
Temp. OK 300 400 500 600 700 SOO
Pyrite Galena 14.S S.9 5.7 3.9 3.0 2.2Sphalerite Galena 10.7 6.0 3.9 2.7 2.0 1.5Pyrite Sphalerite 4.1 2.9 1.S 1.2 1.0 0.7
In order to employ the isotope fractionation betweensulfides as a geothermometer it is of course necessary todetermine quantitatively the isotope equilibrium constantsfor pairs of sulfides and the variations of these equilibriumconstants with temperature. As was mentioned earlier, inorder to do this, it is necessary to know the phonon spectraof the lattices involved.
Sakai has gone part of the way to determining thenecessary parameters by adopting a simplified model forlattice vibrations. He assumes firstly that the phononspectrum of a sulfide mineral is made up of Einstein andDebye frequencies, corresponding to atomic and latticevibrations. By further assuming the Debye frequencyspectrum to be replaced by a single frequency he thendevelops simplified expressions for partition functionratios which may be related to measurable parameters suchas the Debye temperatures of the crystals. The values ofequilibrium isotope effects (K-l) he determines in this way,which are shown in Table 1, are only likely to be semi-quantitative because of the simplifying assumptions madebut at least give some indication of the magnitudes andtemperature dependences which may be expected.
According to a recent paper by Bottinga (1969), in orderto determine explicitly the phonon spectrum of a lattice itis necessary to choose a physically plausible potential whichdescribes the atomic interactions in the unit cell as a func-tion of interatomic distances and angles. Then the equa-tions of motion of the particles in the unit cell may bewritten and solved. As a check on the correctness of theassumed interparticle potential, the specific heat at con-stant volume must be calculated as a function of tempera-ture and compared with the measured values. If agreementis not satisfactory, then the constants in the assumed inter-particle potential must be adjusted and the calculationsrepeated. Bottinga has followed these procedures forgraphite and diamond with respect to 12C and 13C isotopeeffects. Where experimental results are available, hiscalculations appear to be reasonably accurate. It seemslikely that if precise calculations are to be applied to sulfurisotope fractionation between sulfide minerals, then it willbe necessary to use this sort of technique.
At the present time then the equilibrium isotope effectsbetween coexisting sulfide minerals can be understood ina qualitative way and the calculations performed so fargive at least an indication of the magnitudes of the effectsand of their temperature coefficients. More work is re-quired, however, before theoretical estimates of the isotope
equilibrium constants can be used with any degree of con-fidence.
Experimental work. Because of the difficulties and un-certainties involved in the calculation of partition functionratios for sulfides it seems that the best method of deter-mining isotope exchange constants and thus calibratingthe geothermometer is by laboratory experiments. A num-ber of such studies are currently under way.
Ideally, binary mixtures of minerals in intimate contact(direct method) should be used for such determinations;but in order to obtain accurate results, it is essential toeffect complete separation of the two components afterequilibration. Anything less than 100 percent separationwill lead to spuriously low values of estimated equilibriumconstants. In order to overcome this problem, two essen-tially similar approaches have been used. The first, or in-direct approach, is to have both sulfides present in theequilibration vessel but to keep them physically separatedand to effect isotope exchange between them via transportof sulfur vapour. This may be written as:
M1St=±St=±M,S.
The second, or separate approach, is to measure the isotopeexchange constants of individual minerals with elementalsulfur separately. If two such exchanges are written:
KlMl3'S + 3'St=±MI3'S + 32S
K2M,3'S + 34St=±M23'S + 32S
then the sulfide-sulfide exchange may be written as:
If K, and K2 are isotope exchange constants for the twoseparate sulfide-sulfur exchanges, then the isotope exchangeconstant for the two coexisting metallic sulfides will besimply:
Before discussing briefly the results of such experimentsit is worthwhile to mention a study of a similar nature inpreparation by H. Puchelt and G. Kullerud in which theisotope effect in the formation of PbS from lead and sulfurwas studied. The results of the series of experiments areshown in Figure 3.
Without going into the details of the mechanisms in-volved, it may be seen qualitatively that there is a kineticisotope effect involved in the formation of lead sulfide andthat 32Sreacts faster than 34Sso that the 53'S value of theproduct sulfide is less then that of the starting material at5=o. Also, because of the preferential reaction of 32S, theremaining unreacted sulfur becomes enriched in 34S, thisenrichment reaching a maximum as the last of the leadreacts. After this the excess free sulfur and the lead sulfidestart to equilibrate and with time the difference betweentheir 53'S values decreases to finally reach a value corre-
141SULFUR ISOTOPES IN COEXISTING SULFIDES
+8
+7 '" '""~+6
+5 .. ~0~ +4
/ ~
0
(J).,.+~
. ._..._'"zo
+2
TIME (MINUTES)
10'10' 10' _.-10' ._-- .....10
....--»>: ---2
~ ~'" ,--~'-,,...--. LEAD SULFHIDE
-4
FIG. 3. Sulfur isotope fractionation during the reactionPb+S<=,PbS.
sponding to the isotope ecxhange constant of the reaction:
Pb3'S + 3'S <=' Pb3'S + 32S.
The important point to note concerning these experi-ments is that the reaction in nature of elemental sulfur withlead could under special circumstances produce appreciableisotope effects.
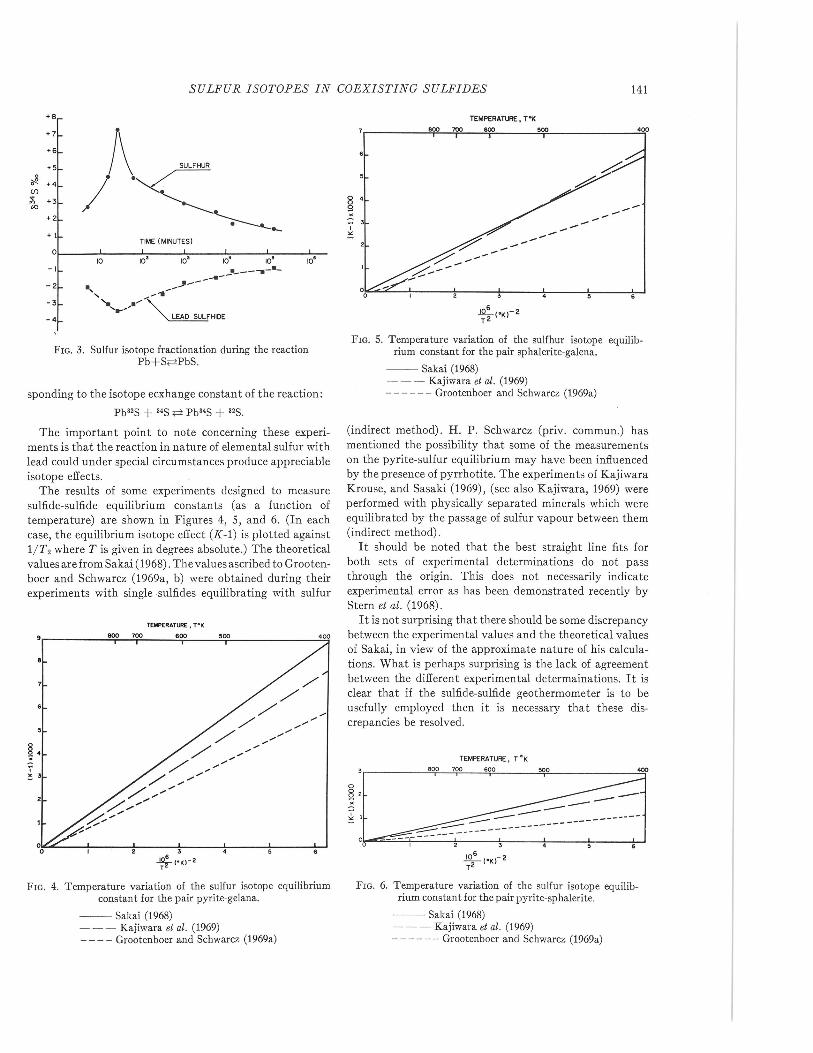
The results of some experiments designed to measuresulfide-sulfide equilibrium constants (as a function oftemperature) are shown in Figures 4, 5, and 6. (In eachcase, the equilibrium isotope effect (K-1) is plotted against1/T2 where T is given in degrees absolute.) The theoreticalvalues are from Sakai (1968). The values ascribed to Grooten-boer and Schwarcz (1969a, b) were obtained during theirexperiments with single ·sulfides equilibrating with sulfur
TEMPERATURE. PI(
800 700 600 !SOO
FIG. 4. Temperature variation of the sulfur isotope equilibriumconstant for the pair pyrite-gelana,
-- Sakai (1968)- - - Kajiwara et al. (1969)____ Grootenboer and Schwarcz (1969a)
TEMPERATURE, T"I<
I
'"
BOO 700 400600
10'
8 4g ---::: 3' _--_-__
_ -
FIG. 5. Temperature variation of the sulfhur isotope equilib-rium constant for the pair sphalerite-galena.
-- Sakai (1968)- - - Kajiwara et al. (1969)- - - - - - Grootenboer and Schwarcz (1969a)
(indirect method). H. P. Schwarcz (priv. commun.) hasmentioned the possibility that some of the measurementson the pyrite-sulfur equilibrium may have been influencedby the presence of pyrrhotite. The experiments of KajiwaraKrouse, and Sasaki (1969), (see also Kajiwara, 1969) wereperformed with physically separated minerals which wereequilibrated by the passage of sulfur vapour between them(indirect method).
It should be noted that the best straight line fits forboth sets of experimental determinations do not passthrough the origin. This does not necessarily indicateexperimental error as has been demonstrated recently byStern et al. (1968).
It is not surprising that there should be some discrepancybetween the experimental values and the theoretical valuesof Sakai, in view of the approximate nature of his calcula-tions. What is perhaps surprising is the lack of agreementbetween the different experimental determainations. It isclear that if the sulfide-sulfide geothermometer is to beusefully employed then it is necessary that these dis-crepancies be resolved.
TEMPERATURE, T OK
FIG. 6. Temperature variation of the sulfur isotope equilib-rium constant for the pair pyrite-sphalerite.
-- Sakai (1968)- - - Kajiwara et al. (1969)- - - - - - Grootenboer and Schwarcz (1969a)
142 H. G. THODE
--------- = A
TIME
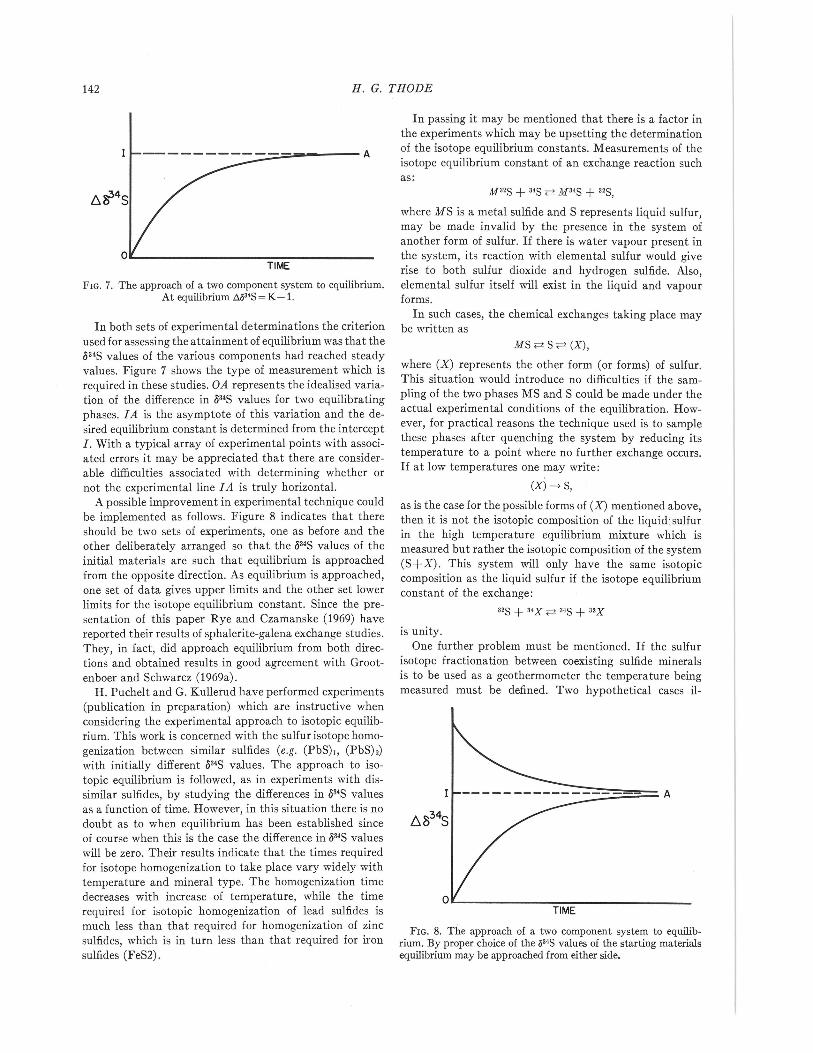
FIG. 7. The approach of a two component system to equilibrium.At equilibrium t,03'S = K-1.
In both sets of experimental determinations the criterionused for assessing the attainment of equilibrium was that the534S values of the various components had reached steadyvalues. Figure 7 shows the type of measurement which isrequired in these studies. OA represents the idealised varia-tion of the difference in 534S values for two equilibratingphases.IA is the asymptote of this variation and the de-sired equilibrium constant is determined from the interceptI. With a typical array of experimental points with associ-ated errors it may be appreciated that there are consider-able difficulties associated with determining whether ornot the experimen tal line I A is truly horizontal.
A possible improvement in experimental technique couldbe implemented as follows. Figure 8 indicates that thereshould be two sets of experiments, one as before and theother deliberately arranged so that the 534S values of theinitial materials are such that equilibrium is approachedfrom the opposite direction. As equilibrium is approached,one set of data gives upper limits and the other set lowerlimits for the isotope equilibrium constant. Since the pre-sentation of this paper Rye and Czamanske (1969) havereported their results of sphalerite-galena exchange studies.They, in fact, did approach equilibrium from both direc-tions and obtained results in good agreement with Groot-enboer and Schwarcz (1969a).
H. Puchelt and G. Kullerud have performed experiments(publication in preparation) which are instructive whenconsidering the experimental approach to isotopic equilib-rium. This work is concerned with the sulfur isotope homo-genization between similar sulfides (e.g. (PbSh , (PbS)2)with initially different 534S values. The approach to iso-topic equilibrium is followed, as in experiments with dis-similar sulfides, by studying the differences in 534S valuesas a function of time. However, in this situation there is nodoubt as to when equilibrium has been established sinceof course when this is the case the difference in 534S valueswill be zero. Their results indicate that the times requiredfor isotope homogenization to take place vary widely withtemperature and mineral type. The homogenization timedecreases with increase of temperature, while the timerequired for isotopic homogenization of lead sulfides ismuch less than that required for homogenization of zincsulfides, which is in turn less than that required for ironsulfides (FeS2).
In passing it may be mentioned that there is a factor inthe experiments which may be upsetting the determinationof the isotope equilibrium constants. Measurements of theisotope equilibrium constant of an exchange reaction suchas:
M32S + a.s <=± M3'S + 3'S,
where MS is a metal sulfide and S represents liquid sulfur,may be made invalid by the presence in the system ofanother form of sulfur. If there is water vapour present inthe system, its reaction with elemental sulfur would giverise to both sulfur dioxide and hydrogen sulfide. Also,elemental sulfur itself will exist in the liquid and vapourforms.
In such cases, the chemical exchanges taking place maybe written as
MS <=± S <=± (X),
where (X) represents the other form (or forms) of sulfur.This situation would introduce no difficulties if the sam-pling of the two phases MS and S could be made under theactual experimental conditions of the equilibration. How-ever, for practical reasons the technique used is to samplethese phases after quenching the system by reducing itstemperature to a point where no further exchange occurs.If at low temperatures one may write:
(X) .......S,
as is the case for the possible forms of (X) mentioned above,then it is not the isotopic composition of the liquid sulfurin the high temperature equilibrium mixture which ismeasured but rather the isotopic composition of the system(S+X). This system will only have the same isotopiccomposition as the liquid sulfur if the isotope equilibriumconstant of the exchange:
3'S + 3.X <=± 3'S + 32X
is unity.One further problem must be mentioned. If the sulfur
isotope fractionation between coexisting sulfide mineralsis to be used as a geothermometer the temperature beingmeasured must be defined. Two hypothetical cases il-
FIG. 8. The approach of a two component system to equilib-rium. By proper choice of the ,,3·S values of the starting materialsequilibrium may be approached from either side.

SULFUR ISOTOPES IN COEXISTING SULFIDES
lustrating this point are shown in Figure 9. In case A,the temperature stays at some value T long enough forisotope equilibrium to be established among coexistingsulfides. At time t the temperature falls sharply to a valuewhere no further exchange takes place. This case is straightforward and clearly the 'temperature' measured by thegeothermometer is T.
In case B, the temperature again stays at temperature Tlong enough for equilibrium to be established, but thenstarts to decrease very slowly. The decrease is so slow thatisotopic equilibrium continues to be maintained, until atemperature T, is reached at which there is no longer amechanism or pathway available to permit exchange tocontinue and the exchange system becomes 'frozen'.
Other more complicated cases may be envisaged. Forexample, it may be that a pyrite-galena geothermometerand a sphalerite-galena geothermometer might under cer-tain circumstances give different answers for the very goodreason that because of their different rates of exchange theybecome 'frozen' at different temperatures.
CONCLUSIONS
The fractionation of the sulfur isotopes between coexist-ing sulfide minerals has been discussed from the point ofview of the observation of such effects in nature and thetheoretical and experimental work done so far on the de-termination of isotope equilibrium constants for such sys-tems.
Work in this area is largely directed towards the de-velopment of a geothermometer for sulfide ore bodies.Before such a geothermometer can be successfully used anumber of problems must be solved.
The differences in isotopic composition of coexistingsulfides may result either from simple equilibrium isotopeeffects or from depositional isotope effects followed bypartial or complete equilibration. It is commonly supposedthat concordance between two or more sulfide geother-mometers is a necessary and sufficient condition for therecognition of isotope equilibrium, but it seems likelythat the assumptions underlying this criterion will needcritical examination in the future.
Theoretical values of isotope equilibrium constants for
AULT, W. U., AND J. L. KuLP (1959) Isotopic geochemistry ofsulphur. Geochim. Cosmochim. Acta 16, 201-235.
BACHINSKI, D. J. (1969) Bond strength and sulfur isotopic frac-tionation in coexisting sulfides. Econ. Geol. 64, 56-65.
BIGELEISEN, J. (1949) The relative reaction velocities of isotopicmolecules. J. Chem. Phys. 17,675-678.
---, AND M. G. MAYER (1947) Calculation of equilibriumconstants for isotopic exchange reactions. J. Chem. Phys. 15,261-267.
---, ANDM. WOLFSBERG (1958) Theoretical and experimentalaspects of isotope effects in chemcial kinetics. Advan. Chem.Phy. 1, 15-76.
BOTH, R. A., T. A. RAFTER, M. SOLOMON, AND M. L. JENSEN(1969) Sulphur isotopes and zoning of the Zeehan mineral field,Tasmania. Econ. Ceol. 64, 618-628.
BOTTINGA, Y. (1969) Carbon isotope fractionation betweengraphite, diamond and carbon dioxide. Earth Planet. Sci. Lett.5,301-307.
143
TEMPERATURE TEMPERATURE
Tt----,
CASE A
TIME TIME
t
FIG. 9. Hypothetical temperature variations with time forsulfide mineral deposits.
coexisting sulfides have so far been evaluated only in aqualitative or semiquantitative fashion. Much remains tobe done in this type of approach.
The experimental determination of equilibrium con-stants performed so far do not agree well with each other.Before such determinations can be used as satisfactorycalibrations of geothermometers it is clearly necessary thatthe discrepancies be removed.
Because of the foregoing considerations, no attempt hasbeen made here to list the instances where individualworkers have used such techniques to determine sulfidetemperatures for specific ore bodies. Clearly until there isagreement on the calibrations of the temperature scalesthere will be no agreement on the temperature determined.It is felt that such applications, except when made in aqualitative manner, are somewhat premature.
However, with the wealth of observational materialavailable, and the wide interest being shown in the tech-nique by many isotope geochemists it seems likely thatwith time the problems outlined will be overcome and thetechnique developed into a satisfactory and useful methodof pressure independent geothermometry.
ACKNOWLEDGEMENTS
I would like to acknowledge the financial support of the NationalResearch Council of Canada, and the assistance of C. E. Rees inthe preparation of the manuscript. I would also like to thank thefollowing for pre-publication information: Both, et al., Grooten-boer and Schwarcz, Kajiwara et al., Puchelt and Kullerud, Rees,Sasaki and Krouse.
REFERENCESBUSCHENDORF, FR., H. NIELSEN, H. PUCHELT, AND W. RICKE
(1963) Schwefel-Isotopen-Untersuchungen am Pyrit-Sphalerit-Baryt-Lager Meggeri/Lenne (Deutschland) und an verschie-denen Devon-Evaporiten. Geochim. Cosmochim. Acta 27, 501-523.
DECHOW, E. (1960) Geology, sulfur isotopes and the origin of theHeath Steele Ore Deposits, Newcastle, N. B., Canada. Econ.Ceol. 55, 539-556.
EVANS, M. G., AND M. POLANYI (1935) Some applications of thetransition state method to the calculation of reaction velocities,especially in solution. Trans. Farad. Soc. 31, 875-894.
EVANS, W. H., AND D. P. WAGMAN (1952) Thermodynamics ofsimple sulphur-containing molecules. J. Res. Nat. Bur. Stand.[U.S.] 49, 141-148.
EYRING, H. (1935) The activated complex in chemical reactions.J. Chem, Phys. 3,107-115.
FA]ANS, K. (1911) The complex nature of radium C. Physik Z.12, 369-378.

144 H. G. THODE
FRIEDRICH, G., D. SCHACHNER, AND H. NIELSEN (1964) Sch-wefelisotopen-Untersuchungen an Sulfiden aus den Erzvorkom-men del Sierra de Cartagena in Spanien. Geochim. Cosmochim.Acta 28, 683-698.
GAVELIN, S., A. PARWEL, AND R. RYHAGE (1960) Sulfur isotopefractionation in sulfide mineralisation. Econ. Ceol. 55, 510-530.
GROOTENBOER, J., AND H. P. SCHWARCZ(1969a) Experimentallydetermined sulfur isotope fractionations between sulfide min-
---, AND--- (1969b) Temperature dependent sulfur iso-tope fractionations between sulfide minerals. Ceol. Soc. A mer.Abstr. Atlantic City Meet., 1969, p. 85.
HARRISON, A. G., AND H. G. THODE (1957) The kinetic isotopeeffect in the chemical reduction of sulphate. Trans. Faraday Soc.53, 1-4.
---, AND--- (1958) Mechanism of the bacterial reductionof sulphate from isotope fractionation studies. Trans. FaradaySoc. 54,84-92.
HOLSER, W. T., ANDI. R. KAPLAN (1966) Isotope geochemistry ofsedimentary sulfates. Chem, Ceol. 1,93-135.
HULSTON, l R., AND H. G. THODE (1965) Variations in the S33,S3' and S3Gcon ten ts of meteorites and their relation to chemicaland nuclear effects. J. Geophys. Res. 70,3475-3484.
JENSEN, M. L. (1957) Sulfur isotopes and mineral paragenesis.Econ. Ceol. 52, 269-281.
--- (1959) Sulfur isotopes and hydrothermal mineral deposits.Econ. Ceol. 54, 374-393.
JONES, G. E., AND R. L. STARKEY (1957) Fractionation of stableisotopes of sulphur by micro-organisms and their role in nativedeposition of sulphur. J. Appl. Microbiol. 5,111-115.
KAJIWARA, y. (1969) Experimental study of sulfur isotope frac-tionation between coexistent sulfide minerals. (abstr.) Prog.Abstr. Ceol. Soc. Amer. 1, 118.
---, H. R. KROUSE, ANDA. SASAKI (1969) Experimental studyof sulfur isotope fractionation between coexistent sulfide min-erals. Earth Planet. Sci. Lett. 7, 271-277.
KAPLAN, I. R., ANDJ. R. HULSTON (1965) The isotopic abundanceand content of sulfur in meteorites. Geochim. Cosmochim. Acta30, 479-496.
---, AND S. C. RITTENBERG (1964) Microbiological fractiona-tion of sulphur isotopes. J. Cen. Microbiol. 34, 195-212.
KEMP, A. L. W., ANDH. G. THODE (1968) The mechanism of thebacterial reduction of sulphate and of sulphite from isotopefractionation studies. Ceochim. Cosmochim. Acta 32,71-91.
LUSK, J., AND J. H. CROCKET (1969) Sulfur isotope fractionationin coexisting sulfides from the Heath Steel B-1 Orebody, NewBrunswick, Canada. Econ. Ceol. 64, 147-155.
MACNAMARA,l, ANDH. G. THODE (1950) Comparison of isotopicconstitution of terrestrial and meteoritic sulphur. Phys. Rev.78, 307-308.
NAKAI, N., ANDM. L. JENSEN (1964) The kinetic isotope effect inthe bacterial reduction and oxidation of sulphur. Ceochim.Cosmochim. Acta 28,1893-1912.
RAFTER, T. A., I. R. KAPLAN, ANDJ. R. HULSTON (1960) Sulphurisotopic variations in nature-7. Sulphur isotopic measurementson sulphur and sulphates in New Zealand geothermal and vol-canic areas. N. Z. J. Sci. 3,209-218.
---, S. H. WILSON, AND B. W. SHILTON (1958a) Sulfur iso-topic variations in nature-S, Sulfur isotopic variations inNew Zealand geothermal bore waters. N. Z. J. Sci. 1, 103-126.
---, ---, AND--- (1958b) Sulphur isotopic variations innature-6. Sulphur isotopic measurements on the discharge fromfumaroles on White Island. N. Z. J. Sci. 1, 154-171.
REES, C. E. (1970) The sulphur isotope balance of the ocean: animproved model. Earth Planet. Sci. Lett. 7, 366-370.
RYE, R. 0., AND G. K. CZAMANSKE(1969) Experimental deter-mination of sphalerite-galena sulfur isotope fractionation andapplication to the ores at Providencia, Mexico. (abstr.) Prog.Abstr. Ceol. Soc. A mer. 1, 85.
RYZNAR, G., F. A. CAMPBELL, AND H. R. KROUSE (1967) Sulfurisotopes and the origin of the Quemont ore body. Econ. Ceol. 62,664-678.
SAKAI, H. (1967) Fractionation of sulphur isotopes in nature.Ceochim. Cosmochim. Acta 12,150-169.
--- (1968) Isotopic properties of sulfur compounds in hydro-thermal processes. Geochem. J. 2, 29-49.
---, AND H. NAGASAWA (1958) Fractionation of sulfur iso-topes in volcanic gases. Geochim, Cosmochim. Acta 15, 32-39.
SASAKI, A., AND H. R. KROUSE (1969) Sulfur isotopes and thePine Point lead-zinc mineralization. Econ. Ceol. 64, 718-730.
SHIMA, M., W. H. GROSS, AND H. G. THODE (1963) Sulphur iso-tope abundances in basic sills, differentiated granites andmeteorites. J. Geophys. Res. 68, 2835-2848.
SMITHERINGALE,W. G., AND M. L. JENSEN (1963) Sulfur isotopiccomposition of the Triassic igneous locks of Eastern UnitedStates. Ceochim. Cosmochim. Acta 27,1183-1207.
SODDY, F. (1910) The relation between uranium and radium.Phil. Mag. 18, 846-858.
SOLOMON,M., T. A. RAFTER, AND M. L. JENSEN (1969) Isotopicstudies on the Rosebery, Mount Farrell and Mount Lyell ores,Tasmania. Mineral. Deposita 4, 172-200.
SPEELYANN, E. L., ANDH. P. SCHWARCZ(1966) Metamorphic sul-'fur isotope studies in the Haliburton-Madoc Area, GrenvilleSubprovince, Canada. Geol, Soc. Amer. Spec. Pcp., 101, 209.
STANTON,R. L., ANDT. A. RAFTER (1967) Sulfur isotope ratios inco-existing galena and sphalerite from Broken Hill, New SouthWales. Econ. Ceol. 62, 1088-1091.
STERN, M. J., W. SPINDEL, ANDE. U. MONSE (1968) Temperaturedependences of isotope effects. J. Chem. Phys. 48,2908-2919.
TATSUMI, T. (1965) Sulfur isotopic fractionation between co-existing sulfide minerals from some Japanese ore deposits.Econ. Ceol. 60, 1645-1659.
THODE, H. G., G. B. DUNFORD, AND M. SIHMA (1962) Sulfur iso-tope abundances in rocks of the Sudbury District and theirgeological significance. Econ. Ceol. 57, 565-578.
---, H. KLEEREKOPER, AND D. McELCHEREN (1951) Isotopefractionation in the bacterial reduction of sulphate. Research(London) 4, 581.
---, AND l MONSTER (1965) Sulfur isotope geochemistry ofpetroleum, evaporties and ancient seas. Amer. Assoc. Petrol.Ceol., Mem. 4, 367-377.
---, ---, AND H. B. DUNFORD (1961) Sulphur isotopegeochemistry. Geochim. Cosmochim. Acta 25,150-174.
TUDGE, A. P., ANDH. G. THODE (1950) Thermodynamic proper-ties of isotopic compounds of sulphur. Can. J. Res. B, 28, 567-578.
TUPPER, W. M. (1960) Sulfur isotopes and the origin of the sulfidedeposits of the Bathurst-Newcastle Area of Northern NewBrunswick. Econ. Ceol. 55,1676-1707.
UREY, H. C. (1947) The thermodynamic properties of isotopicsubstances. J. Chem. Soc. 562-581.
---, AND F. BRICKWEDDE (1932) A hydrogen isotope of mass2. Phys. Rev. 39,164-165.
VINOGRADOV, A. P. (1958) Isotopic composition of sulphur inmeteorites and in the earth. In R. C. Extermann (ed.) Radio-isotopes in Scientific Research, II, Pergamon Press, New York.p.581-591.
VINOGRADOV, A. P., M. S. CHUPAKHIN, AND V. A. GRINENKO(1957) Some data on the isotopic composition of the sulphur ofsulphides. Ceokhimiya 3, 183-186.