summary of safety and effectiveness …€¢ tubing components and container ports of the intercept...
TRANSCRIPT

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 1
SUMMARY OF SAFETY AND EFFECTIVENESS DATA (SSED) I. GENERAL INFORMATION
Device Generic Name: Pathogen Reduction System for Platelets
Device Trade Name: INTERCEPT Blood System for Platelets
Device Procode: PJF
Applicant’s Name and Address: Cerus Corporation 2550 Stanwell Drive Concord, CA 94520
Date(s) of Panel Recommendation: None
Premarket Approval Application (PMA) Number: BP140143
Office’s Signatory Authority: Jay S. Epstein, MD
Director, OBRR/CBER □ I concur with the summary review. □ I concur with the summary review and include a separate review to add
further analysis. □ I do not concur with the summary review and include a separate review. Date of FDA Notice of Approval: December 18, 2014

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 2
II. INTENDED USE
The INTERCEPT Blood System for Platelets is intended to be used for ex vivo preparation of pathogen-reduced apheresis platelet components in order to reduce the risk of transfusion-transmitted infection (TTI) including sepsis, and to potentially reduce the risk of transfusion-associated graft versus host disease (TA-GVHD).
III. CONTRAINDICATIONS
• Contraindicated for preparation of platelets intended for patients with a history of hypersensitivity reaction to amotosalen or other psoralens.
• Contraindicated for preparation of platelets intended for neonatal patients treated with phototherapy devices that emit wavelengths less than 425 nm due to the potential for erythema resulting from interaction between ultraviolet light and amotosalen. Note: Information about these contraindications needs to be included in the labeling provided with transfusible platelets prepared using the INTERCEPT Blood System for Platelets.
IV. WARNINGS AND PRECAUTIONS
• Only INTERCEPT Processing Sets for platelets are approved for use with the INTERCEPT Blood System. Use only the INTERCEPT INT100 Illuminator for UVA illumination of amotosalen-treated platelet components. No other source of UVA light may be used. Please refer to the Operator’s Manual for the INT100 Illuminator. Discard any platelet components not exposed to the complete INT100 illumination process.
• Tubing components and container ports of the INTERCEPT Blood System contain polyvinyl chloride (PVC). Di(2-ethylhexyl)phthalate (DEHP) is known to be released from PVC medical devices, and increased leaching can occur with extended storage or increased surface area contact. Blood components will be in contact with PVC for a brief period of time (approximately 15 minutes) during processing. The risks associated with DEHP released to into the blood components must be weighed against the benefits of therapeutic transfusion.
• Pulmonary events: Acute Respiratory Distress Syndrome
An increased incidence of Acute Respiratory Distress Syndrome (ARDS) was reported in a randomized trial for recipients of IBS processed platelets, 5/318 (1.6%), compared to recipients of conventional platelet components (0/327). Monitor patients for signs and symptoms of ARDS. Note: Information about these Warnings and Precautions needs to be included in the labeling provided with transfusible platelets prepared using the INTERCEPT Blood System for Platelets.

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 3
V. DEVICE DESCRIPTION The INTERCEPT Blood System for Platelets, LV Processing Set, contains a sterile, non-pyrogenic, single-use, integrated, fluid path platelet processing set (INT22) comprised of four key components (See Table below) and an ultraviolet (UVA) illumination device (INT100) for the ex vivo preparation and storage of pathogen reduced apheresis platelet components. The INT100 is a microprocessor controlled device designed to deliver a controlled amount of UVA light, wavelength 320 to 400 nm, to up to two illumination containers simultaneously. The device is programmed to be able to control, deliver, record and store intensity and duration of light dose for each cycle.
Components of the INTERCEPT Platelet LV Processing Set (INT22)
Component Description Amotosalen (S-59, psoralen derivative) solution container
17.5 mL, 3 mM amotosalen in 0.924% NaCl packaged in a 20 mL, flexible, heat-sealed plastic container, with an integral light-protective overwrap and sleeve
Illumination container Heat-sealed, plastic bag Compound Adsorption Device (CAD)
Immobilized beads (wafer) in mesh pouch
Platelet storage containers
One platelet storage bag – 1300 mL storage capacity
The operating principle for the INTERCEPT Blood System for Platelets is illustrated below. Platelets collected by apheresis in a container are sterile connected to the Platelet LV Processing Set [INT22]. The platelets flow through the amotosalen container into the illumination container. The illumination container is placed into the INT100 illumination device for UVA treatment while being mixed with horizontal agitation. Inactivation of potential pathogen or leukocyte contaminants in platelet components is achieved through a photochemical treatment process. Amotosalen (S-59, psoralen derivative), a chemical capable of binding to nucleic acids is added to platelets. UVA illumination (320 – 400 nm wavelengths) of amotosalen-treated platelet components induces covalent cross-linking of any nucleic acids to which amotosalen is bound; thereby, preventing replication of infectious agents. Treated platelets are then transferred to the CAD container for 4 to 16 hours depending on the disposable set used (Small Volume, Large Volume, or Dual Storage) to reduce the levels of residual amotosalen and its free photoproducts. Finally, the platelet components are transferred through the in-line filter to the storage container for use or storage at 20-24 °C with continuous agitation for up to 5 days from the time of collection.

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 4
INTERCEPT Blood System for Platelets and Treatment Process
VI. ALTERNATIVE PRACTICES AND PROCEDURES
The current strategy for prevention of transfusion-transmitted infectious diseases utilizes pre-donation screening questionnaires as well as serologic and nucleic acid based testing of the donors for infection with a limited number of known pathogens. In the U.S., blood donor screening tests include tests for syphilis, HBV, HCV, HIV, HTLV WNVand T. cruzi. Currently, under AABB standards, accredited institutions perform bacterial culture testing of platelets. Currently, there are no approved methods for pathogen reduction of platelets in the U.S.
VII. MARKETING HISTORY The IBS for platelets was CE marked in 2002 and is commercially available in many countries in Europe, Asia, and the Middle East.
VIII. SUMMARY OF PRECLINICAL STUDIES
A. INTERCEPT Blood System processed platelets: In vitro Characterization
In vitro platelet function characteristics were evaluated in a prospective, randomized, paired, controlled, in vitro study in healthy subjects of IBS processed platelets
compared to unprocessed (control) platelets. Each subject completed two donations. From each donor either a single or a double unit was collected at each donation. From each donor one donation was randomized to be processed as a control and the other as Test platelets. In vitro platelet function of the platelet components was

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 5
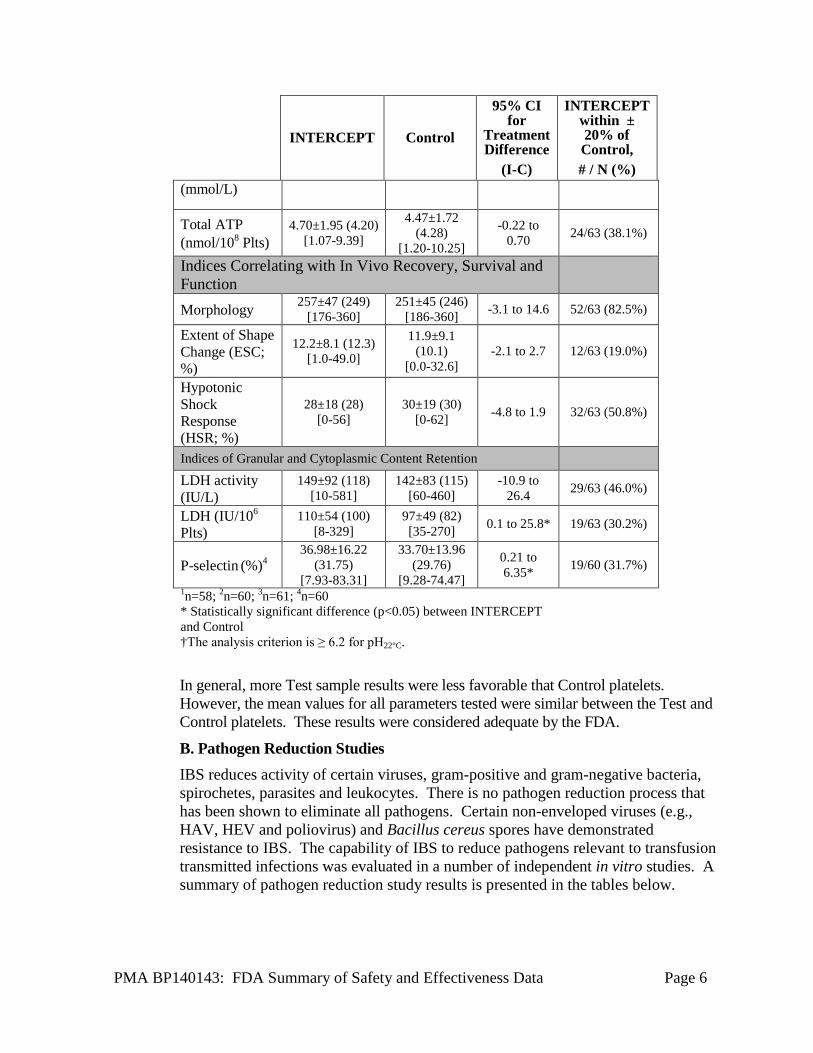
evaluated on day 5 after apheresis donation. The physical and metabolic characteristics for evaluation included: component volume, platelet count, platelet volume (MPV), pH22◦C, pO2, pCO2, HCO3, morphology, supernatant LDH activity, P-selectin, total ATP, extent of shape change, hypotonic shock response, supernatant glucose, and supernatant lactate. Single-dose and double-dose platelet collections containing 2.9 – 8.0×1011 platelets treated with the IBS processed platelets and stored for 5 days retained a presumptively therapeutically effective mean platelet doses (the lower bound of the 95% CI for the proportion of IBS processed platelet components retaining >3.0×1011 platelets/component was 78%) maintaining an average dose after INTERCEPT treatment of 91%, and with retention of adequate in vitro metabolic and functional properties. This study, along, with other in vitro studies, was used to establish the specifications for the IBS processed platelets.
In Vitro Platelet Function Characteristics of INTERCEPT (I) and Control (C) Platelets After 5 Days of Storage (Mean +/- SD (median) [range]; n=63)
INTERCEPT Control
95% CI for
Treatment Difference
(I-C)
INTERCEPT within ± 20% of Control, # / N (%)
Platelet Component Characteristics
Component volume (mL)
279±74 (327) [162-382]
288±74 (317) [152-400]
-14.4 to -3.7* 60/63 (95.2%)
Platelet count (×103/µl)
1383±341(1368) [717-2002]
1482±375 (1408)
[730-2250]
-148.1to -51.0* 53/63 (84.1%)
Platelet dose (×1011 cells/unit)
3.7±0.8 (3.5) [2.5-6.1]
4.0±0.6 (3.9) [2.3-5.5] -0.5 to -0.2* 51/63 (81.0%)
MPV (fL)1 8.1±0.8 (8.1) [6.7-10.6]
8.1±0.9 (8.0) [6.7-10.4] -0.1 to 0.2 58/58 (100%)
Indices of Platelet Metabolism
pH at 22°C 7.02±0.11(7.04) [6.78-7.20]
7.03±0.12 (7.03)
[6.73-7.25]
-0.04 to 0.02 63/63 (100%)†
pO2 (mm Hg) 130±21(134) [66-168]
124±20 (128) [70-154] -1.0 to 13.2 39/63 (61.9%)
pCO2 (mm Hg) 20±6 (20) [10-32]
24±6 (24) [13-35] -4.4 to -2.4* 36/63 (57.1%)
HCO3−
(mmol/L)2 3.2±1.1 (3.1)
[1.0-5.3] 3.8±1.3 (4.0)
[2.0-7.0] -0.9 to -0.3* 23/60 (38.3%)
Supernatant glucose (mg/dL)3
21.5±24.0 (18.5) [0.0-127.8]
15.5±23.3 (2.0)
[0.0-120.6] 1.2 to 10.9* 29/61 (47.5%)
Supernatant lactate
11±2 (11) [7-15]
12±3 (13) [7-19]
-1.99 to -0.82* 43/63 (68.3%)
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 6
INTERCEPT Control
95% CI for
Treatment Difference
(I-C)
INTERCEPT within ± 20% of Control, # / N (%)
(mmol/L)
Total ATP (nmol/108 Plts)
4.70±1.95 (4.20) [1.07-9.39]
4.47±1.72 (4.28)
[1.20-10.25]
-0.22 to 0.70 24/63 (38.1%)
Indices Correlating with In Vivo Recovery, Survival and Function
Morphology 257±47 (249) [176-360]
251±45 (246) [186-360] -3.1 to 14.6 52/63 (82.5%)
Extent of Shape Change (ESC; %)
12.2±8.1 (12.3) [1.0-49.0]
11.9±9.1 (10.1)
[0.0-32.6] -2.1 to 2.7 12/63 (19.0%)
Hypotonic Shock Response (HSR; %)
28±18 (28) [0-56]
30±19 (30) [0-62] -4.8 to 1.9 32/63 (50.8%)
Indices of Granular and Cytoplasmic Content Retention
LDH activity (IU/L)
149±92 (118) [10-581]
142±83 (115) [60-460]
-10.9 to 26.4 29/63 (46.0%)
LDH (IU/106
Plts) 110±54 (100)
[8-329] 97±49 (82) [35-270] 0.1 to 25.8* 19/63 (30.2%)
P-selectin (%)4 36.98±16.22
(31.75) [7.93-83.31]
33.70±13.96 (29.76)
[9.28-74.47]
0.21 to 6.35* 19/60 (31.7%)
1n=58; 2n=60; 3n=61; 4n=60 * Statistically significant difference (p<0.05) between INTERCEPT and Control †The analysis criterion is ≥ 6.2 for pH22°C.
In general, more Test sample results were less favorable that Control platelets. However, the mean values for all parameters tested were similar between the Test and Control platelets. These results were considered adequate by the FDA.
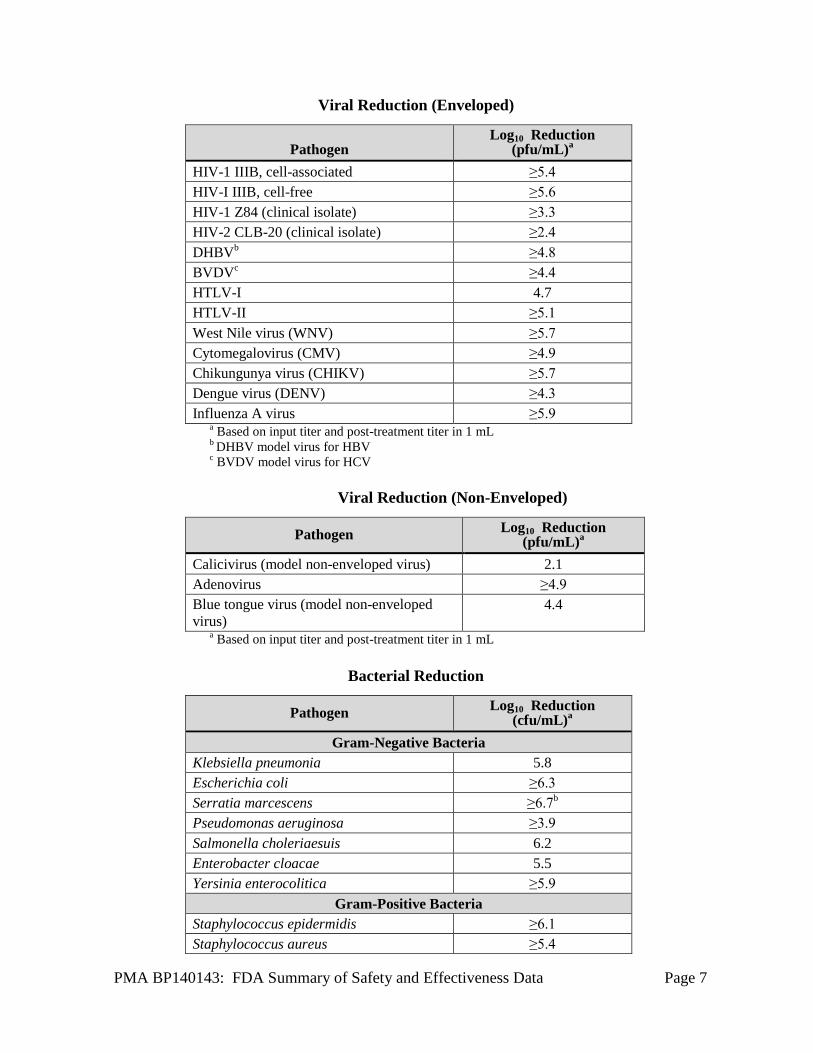
B. Pathogen Reduction Studies IBS reduces activity of certain viruses, gram-positive and gram-negative bacteria, spirochetes, parasites and leukocytes. There is no pathogen reduction process that has been shown to eliminate all pathogens. Certain non-enveloped viruses (e.g., HAV, HEV and poliovirus) and Bacillus cereus spores have demonstrated resistance to IBS. The capability of IBS to reduce pathogens relevant to transfusion transmitted infections was evaluated in a number of independent in vitro studies. A summary of pathogen reduction study results is presented in the tables below.
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 7
Viral Reduction (Enveloped)
Pathogen Log10 Reduction
(pfu/mL)a HIV-1 IIIB, cell-associated ≥5.4 HIV-I IIIB, cell-free ≥5.6 HIV-1 Z84 (clinical isolate) ≥3.3 HIV-2 CLB-20 (clinical isolate) ≥2.4 DHBVb ≥4.8 BVDVc ≥4.4 HTLV-I 4.7 HTLV-II ≥5.1 West Nile virus (WNV) ≥5.7 Cytomegalovirus (CMV) ≥4.9 Chikungunya virus (CHIKV) ≥5.7 Dengue virus (DENV) ≥4.3 Influenza A virus ≥5.9
a Based on input titer and post-treatment titer in 1 b DHBV model virus for HBV
mL
c BVDV model virus for HCV
Viral Reduction (Non-Enveloped)
Pathogen Log10 Reduction (pfu/mL)a
Calicivirus (model non-enveloped virus) 2.1 Adenovirus ≥4.9 Blue tongue virus (model non-enveloped 4.4 virus)
a Based on input titer and post-treatment titer in 1 mL
Bacterial Reduction
Pathogen Log10 Reduction (cfu/mL)a
Gram-Negative Bacteria Klebsiella pneumonia 5.8 Escherichia coli ≥6.3 Serratia marcescens ≥6.7b Pseudomonas aeruginosa ≥3.9 Salmonella choleriaesuis 6.2 Enterobacter cloacae 5.5 Yersinia enterocolitica ≥5.9
Gram-Positive Bacteria Staphylococcus epidermidis ≥6.1 Staphylococcus aureus ≥5.4
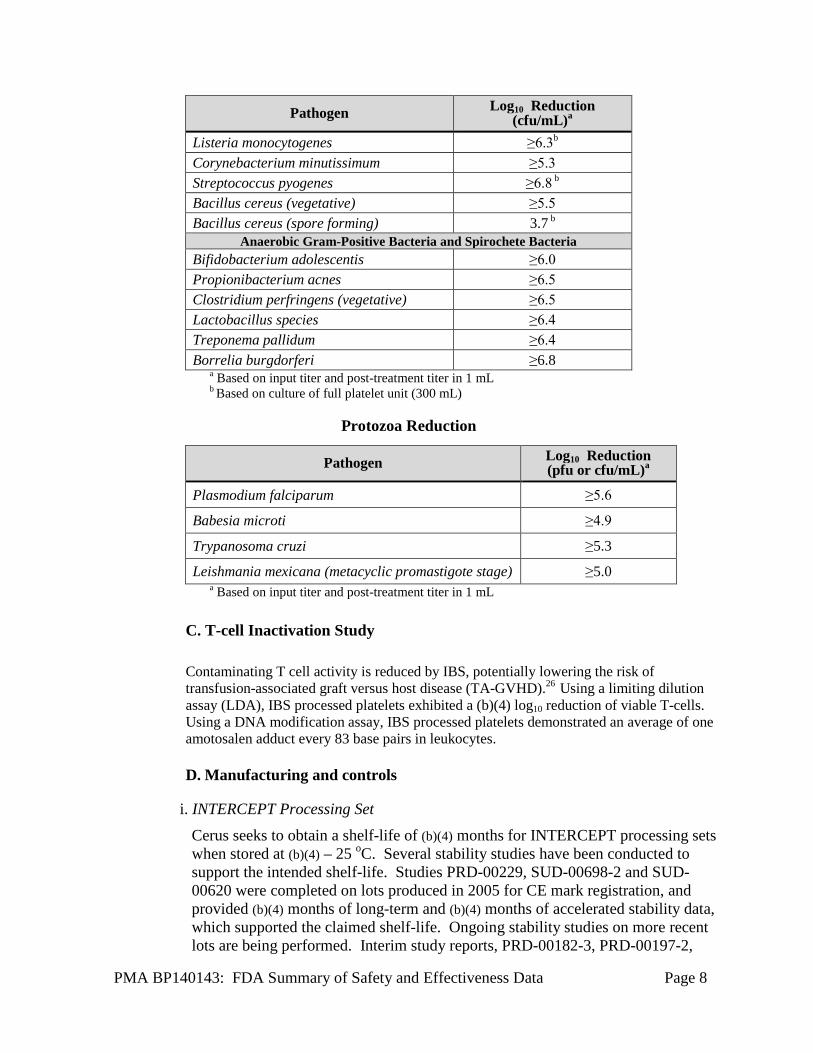
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 8
Pathogen Log10 Reduction (cfu/mL)a
Listeria monocytogenes ≥6.3b Corynebacterium minutissimum ≥5.3
Streptococcus pyogenes ≥6.8 b Bacillus cereus (vegetative) ≥5.5
Bacillus cereus (spore forming) 3.7 b Anaerobic Gram-Positive Bacteria and Spirochete Bacteria
Bifidobacterium adolescentis ≥6.0
Propionibacterium acnes ≥6.5 Clostridium perfringens (vegetative) ≥6.5
Lactobacillus species ≥6.4
Treponema pallidum ≥6.4 Borrelia burgdorferi ≥6.8
a Based on input titer and post-treatment titer in 1 mL b Based on culture of full platelet unit (300 mL)
Protozoa Reduction
Pathogen Log10 Reduction (pfu or cfu/mL)a
Plasmodium falciparum ≥5.6
Babesia microti ≥4.9
Trypanosoma cruzi ≥5.3
Leishmania mexicana (metacyclic promastigote stage) ≥5.0
a Based on input titer and post-treatment titer in 1 mL
C. T-cell Inactivation Study Contaminating T cell activity is reduced by IBS, potentially lowering the risk of transfusion-associated graft versus host disease (TA-GVHD).26 Using a limiting dilution assay (LDA), IBS processed platelets exhibited a (b)(4) log10 reduction of viable T-cells. Using a DNA modification assay, IBS processed platelets demonstrated an average of one amotosalen adduct every 83 base pairs in leukocytes.
D. Manufacturing and controls
i. INTERCEPT Processing Set Cerus seeks to obtain a shelf-life of (b)(4) months for INTERCEPT processing sets when stored at (b)(4) – 25 oC. Several stability studies have been conducted to support the intended shelf-life. Studies PRD-00229, SUD-00698-2 and SUD-00620 were completed on lots produced in 2005 for CE mark registration, and provided (b)(4) months of long-term and (b)(4) months of accelerated stability data, which supported the claimed shelf-life. Ongoing stability studies on more recent lots are being performed. Interim study reports, PRD-00182-3, PRD-00197-2,

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 9
PRD-00223-1 provided data from amotosalen solution stability monitoring, CAD functional testing and plasma bag UV transmission measurements. Results from monitoring amotosalen dose and degradation products as a function of storage temperature and time were recorded. Interim stability data revealed trends in amotosalen dose that reflected -----------------(b)(4)----------------. Interim stability data indicated acceptable stability for up to (b)(4) months, at temperatures up to 25 oC. Temperatures above 25 oC, however, negatively impacted amotosalen stability; therefore, the product will be labeled, “Do not store above 25 oC.”
Cerus and --(b)(4)-- have established an --(b)(4)-- stability monitoring program to monitor ongoing processing set stability and to qualify manufacturing changes. The stability specification for the amotosalen solution container includes tests for amotosalen dose, amotosalen assay, (b)(4), impurities and microbial safety. Photostability studies supported storage and handling conditions for the amotosalen solution in its container closure, --(b)(4)-- plastic container with integral -------------------(b)(4)------------------------------------ sleeve with label. Cerus has committed to conducting prospective stability studies to confirm stability of the intended U.S. commercial configuration of assembled processing sets. Study PRD-00235 will monitor amotosalen solution stability and study PRD-00218 will monitor the processing set dry side. Cerus committed to completing each (b)(4) month study.
ii. INTERCEPT Illuminator
The INT100 Illuminators were tested and certified according to EN ISO 61000-3-2, EN ISO 61000-3-3, EN ISO 61010-1, and EN ISO 61326-1 electrical and EMC safety standards.
Reliability testing was conducted which includes a variety of stress, aging, mechanism and life tests conducted on major subassemblies of the INTERCEPT Illuminator. At a 95% confidence level, the calculated mean time between failures (MTBF) is >1 year. The INTERCEPT Illuminator is calibrated and preventive maintenance is performed every 6 months after successful INTERCEPT Illuminator installation by a trained service personnel.
iii. Facility Inspection
There are two main components of the INTERCEPT Blood System for Platelets which are manufactured by contract manufacturers for Cerus Corporation. Specifically, the Processing Set for Platelets is manufactured by -----(b)(4)----- ---- and the INTERCEPT Illuminator is manufactured by ------(b)(4)--------. Inspections of both contract manufacturers were performed under a separate PMA from Cerus Corporation, namely, the INTERCERPT Blood System for Plasma. Details regarding the facilities that were inspected under the alternate PMA are listed in the table below.
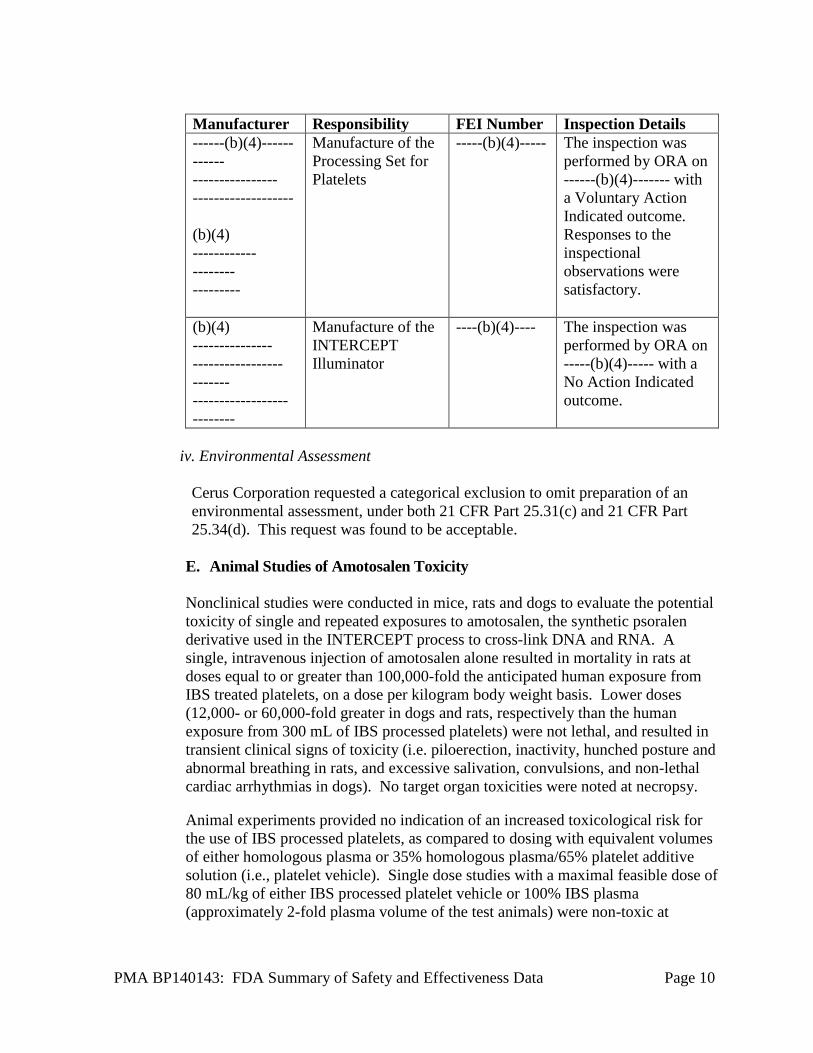
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 10
Manufacturer Responsibility FEI Number Inspection Details ------(b)(4)------ ------ ---------------- ------------------- (b)(4) ------------ -------- ---------
Manufacture of the Processing Set for Platelets
-----(b)(4)----- The inspection was performed by ORA on ------(b)(4)------- with a Voluntary Action Indicated outcome. Responses to the inspectional observations were satisfactory.
(b)(4) --------------- ----------------- ------- ------------------ --------
Manufacture of the INTERCEPT Illuminator
----(b)(4)---- The inspection was performed by ORA on -----(b)(4)----- with a No Action Indicated outcome.
iv. Environmental Assessment
Cerus Corporation requested a categorical exclusion to omit preparation of an environmental assessment, under both 21 CFR Part 25.31(c) and 21 CFR Part 25.34(d). This request was found to be acceptable.
E. Animal Studies of Amotosalen Toxicity Nonclinical studies were conducted in mice, rats and dogs to evaluate the potential toxicity of single and repeated exposures to amotosalen, the synthetic psoralen derivative used in the INTERCEPT process to cross-link DNA and RNA. A single, intravenous injection of amotosalen alone resulted in mortality in rats at doses equal to or greater than 100,000-fold the anticipated human exposure from IBS treated platelets, on a dose per kilogram body weight basis. Lower doses (12,000- or 60,000-fold greater in dogs and rats, respectively than the human exposure from 300 mL of IBS processed platelets) were not lethal, and resulted in transient clinical signs of toxicity (i.e. piloerection, inactivity, hunched posture and abnormal breathing in rats, and excessive salivation, convulsions, and non-lethal cardiac arrhythmias in dogs). No target organ toxicities were noted at necropsy.
Animal experiments provided no indication of an increased toxicological risk for the use of IBS processed platelets, as compared to dosing with equivalent volumes of either homologous plasma or 35% homologous plasma/65% platelet additive solution (i.e., platelet vehicle). Single dose studies with a maximal feasible dose of 80 mL/kg of either IBS processed platelet vehicle or 100% IBS plasma (approximately 2-fold plasma volume of the test animals) were non-toxic at

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 11
amotosalen doses of approximately 18,000-fold greater than the expected clinical amotosalen exposure. Repeated, daily dosing of rats and dogs with IBS processed platelet vehicle or amotosalen alone for 28 days, or 3 times weekly dosing for up to 13 weeks resulted in transient hematologic changes at doses approximately 15,000-fold greater than the expected human exposure to amotosalen in a 300 mL transfusion of IBS processed platelets. Minimal toxicities, including transient, minor changes in hematology profiles with no correlating histopathology findings or major organ toxicities were reported in dogs dosed 3 times weekly for 13 weeks with autologous platelets processed with the IBS, at a cumulative amotosalen exposure of approximately 115-fold the anticipated clinical exposure following a single transfusion of 300 mL of IBS processed platelets. Electrocardiogram, hemodynamic, renal and CNS safety pharmacology findings were unremarkable in dogs or ----(b)(4)---- monkeys dosed with a single intravenous infusion of IBS-processed platelet vehicle, 100% IBS treated plasma, or 25 mg/kg amotosalen alone, compared animals dosed with the control platelet vehicle with no IBS or amotosalen treatment. There were no cardiac, central nervous system or kidney safety pharmacology findings in rats dosed with a single intravenous infusion of IBS-processed platelet vehicle or 100% homologous IBS plasma. IBS processed platelets were not tested in rodent safety pharmacology studies; however, there were no treatment-related cardiac safety signals following infusion of IBS processed human platelets in ----(b)(4)---- monkeys, or after 13 weeks of repeated, 3 times weekly dosing of dogs with autologous platelets or platelet vehicle that were processed with the IBS for platelets. Although no specific pulmonary or gastrointestinal safety pharmacology studies were conducted, no adverse macroscopic or histopathologic findings were observed in the single-dose or repeat-dose toxicology studies, at doses of up to 15000-fold the anticipated clinical amotosalen exposure. Amotosalen was rapidly eliminated in mice and rats with an initial plasma t1/2 of less than 1 hour. There was no evidence of amotosalen accumulation after repeated exposures over periods as long as 13 weeks. The primary route of excretion of amotosalen and its photoproducts was fecal.
No effects on fertility parameters were noted in male or female rats repeatedly dosed with amotosalen. In studies evaluating the effects of amotosalen dosing of pregnant rats or rabbits on embryo-fetal or peri-postnatal development, and in one study of neonatal rats dosed with amotosalen, IBS processed platelet vehicle or homologous IBS processed plasma, there was no evidence of teratogenicity, or other reproductive or developmental toxicities. No evidence of genotoxicity or mutagenicity was observed in the in vitro or in vivo mutagenicity studies of amotosalen. In transgenic mice heterozygous for the p53 tumor suppressor gene, there was no evidence of carcinogenicity after repeated, three times weekly dosing for 6 months with amotosalen in IBS-processed platelet vehicle, at cumulative

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 12
weekly doses approximately 450 times the human exposure from a single infusion of IBS platelets.
F. Additional Studies
All nonclinical studies conducted with the extractable and leachable components of the IBS met the criteria for passing their specific tests, as outlined in the ISO 10993 -----(b)(4)----- standards. There were no adverse findings in in vitro cytotoxicity and biocompatibility testing of either the plastics themselves or of aqueous or aliphatic extracts of the storage containers, labels, or the CAD wafer in its container closure system in cytotoxicity studies with (b)(4) cells, hemolysis of -(b)(4)- whole blood and Ames assay for mutagenicity testing, or in in vivo local tolerability in rabbits, dermal sensitization in guinea pigs, and single and repeat-dose intravenous toxicity studies in mice and rats, respectively. Additionally, the same testing performed on --------------------(b)(4)-------------------- extracts of the IBS did not result in detectable, treatment-related toxicities in any of the test systems assayed. A comprehensive risk assessment based on the identity, quantity and toxicity of the individual extractable chemicals from the plastic storage containers and the CAD did not reveal significant risk of toxicity, mutagenicity, carcinogenicity or teratogenicity at the exposure levels present in a predicted human daily dose of 1,000 mL IBS processed plasma per day, with safety margins between 125-fold and 130,000-fold between the anticipated human daily exposures and the non-toxic, intravenous dose levels of the individual chemicals. Cerus adheres to a number of national and international voluntary standards that identify industry recommendations for medical device quality systems, development and manufacturing processes applicable to the Intercept Blood System for Platelets.
IX. SUMMARY OF PRIMARY CLINICAL STUDIES
Description of the Clinical Development Program
The U.S. clinical development program under BB-IDE 6200 was initiated in 1995 and was concluded in 2001. Six U.S. clinical trials conducted in Phases 1, 2 and 3 were included in the PMA.
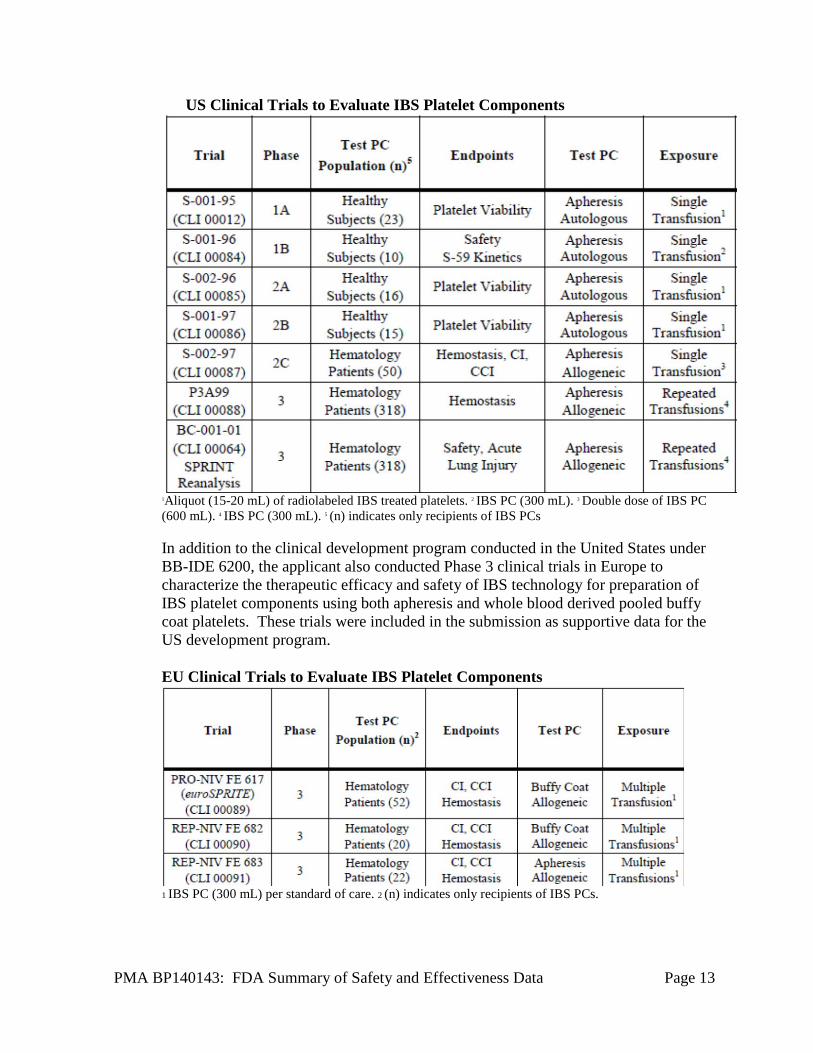
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 13
US Clinical Trials to Evaluate IBS Platelet Components
1Aliquot (15-20 mL) of radiolabeled IBS treated platelets. 2 IBS PC (300 mL). 3 Double dose of IBS PC (600 mL). 4 IBS PC (300 mL). 5 (n) indicates only recipients of IBS PCs In addition to the clinical development program conducted in the United States under BB-IDE 6200, the applicant also conducted Phase 3 clinical trials in Europe to characterize the therapeutic efficacy and safety of IBS technology for preparation of IBS platelet components using both apheresis and whole blood derived pooled buffy coat platelets. These trials were included in the submission as supportive data for the US development program. EU Clinical Trials to Evaluate IBS Platelet Components
1 IBS PC (300 mL) per standard of care. 2 (n) indicates only recipients of IBS PCs.
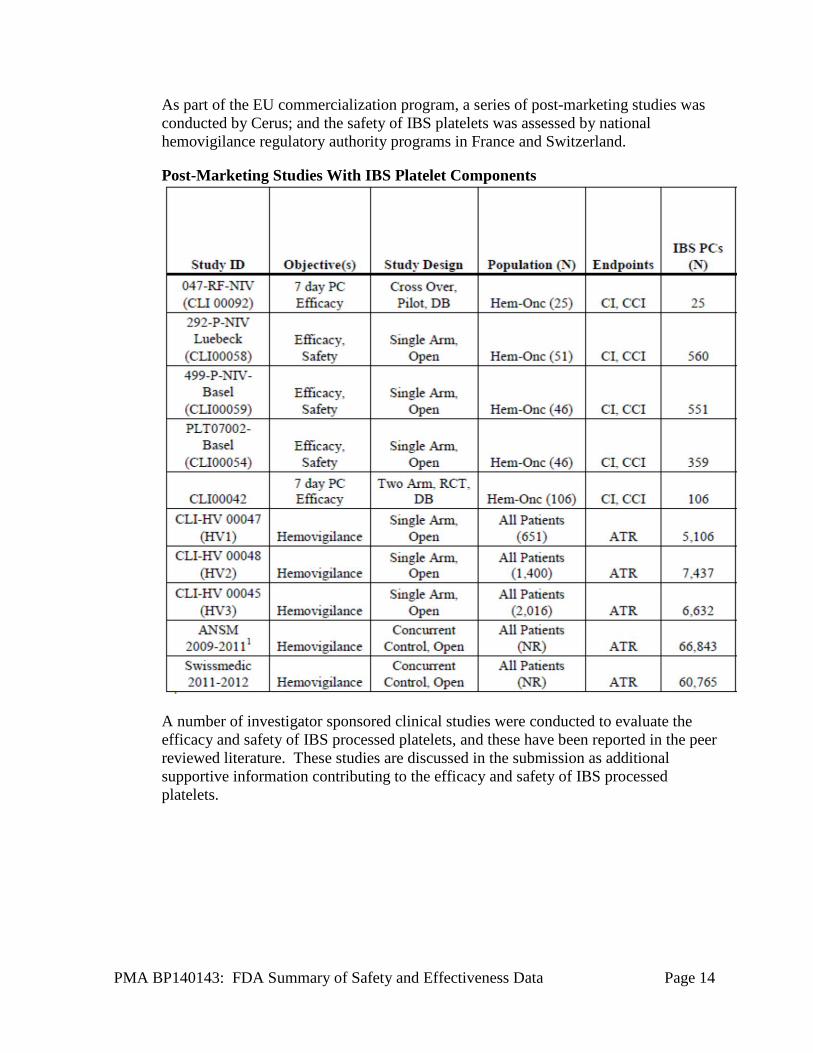
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 14
As part of the EU commercialization program, a series of post-marketing studies was conducted by Cerus; and the safety of IBS platelets was assessed by national hemovigilance regulatory authority programs in France and Switzerland.
Post-Marketing Studies With IBS Platelet Components
A number of investigator sponsored clinical studies were conducted to evaluate the efficacy and safety of IBS processed platelets, and these have been reported in the peer reviewed literature. These studies are discussed in the submission as additional supportive information contributing to the efficacy and safety of IBS processed platelets.
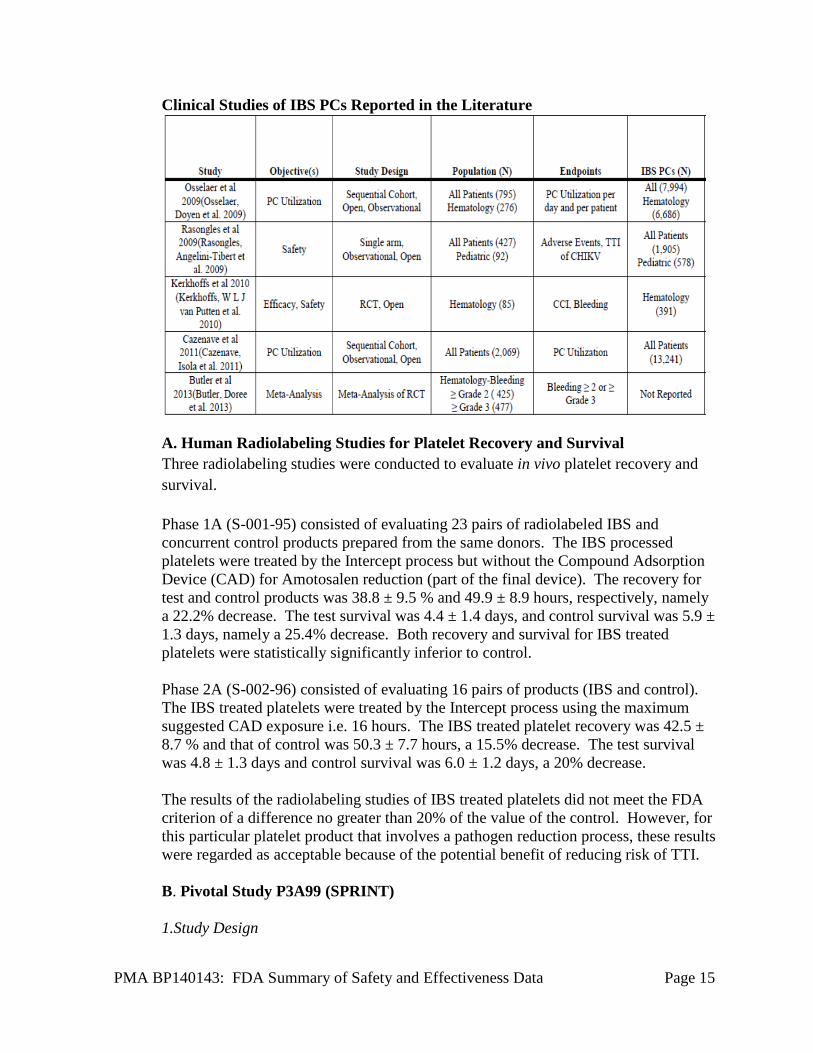
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 15
Clinical Studies of IBS PCs Reported in the Literature
A. Human Radiolabeling Studies for Platelet Recovery and Survival Three radiolabeling studies were conducted to evaluate in vivo platelet recovery and survival.
Phase 1A (S-001-95) consisted of evaluating 23 pairs of radiolabeled IBS and concurrent control products prepared from the same donors. The IBS processed platelets were treated by the Intercept process but without the Compound Adsorption Device (CAD) for Amotosalen reduction (part of the final device). The recovery for test and control products was 38.8 ± 9.5 % and 49.9 ± 8.9 hours, respectively, namely a 22.2% decrease. The test survival was 4.4 ± 1.4 days, and control survival was 5.9 ± 1.3 days, namely a 25.4% decrease. Both recovery and survival for IBS treated platelets were statistically significantly inferior to control.
Phase 2A (S-002-96) consisted of evaluating 16 pairs of products (IBS and control). The IBS treated platelets were treated by the Intercept process using the maximum suggested CAD exposure i.e. 16 hours. The IBS treated platelet recovery was 42.5 ± 8.7 % and that of control was 50.3 ± 7.7 hours, a 15.5% decrease. The test survival was 4.8 ± 1.3 days and control survival was 6.0 ± 1.2 days, a 20% decrease.
The results of the radiolabeling studies of IBS treated platelets did not meet the FDA criterion of a difference no greater than 20% of the value of the control. However, for this particular platelet product that involves a pathogen reduction process, these results were regarded as acceptable because of the potential benefit of reducing risk of TTI.
B. Pivotal Study P3A99 (SPRINT) 1.Study Design

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 16
The pivotal trial to evaluate the safety and efficacy of IBS processed platelets was designed as a prospective, randomized, controlled, double blind, non-inferiority phase 3 trial (SPRINT, 2001). The primary objective was to compare the efficacy and safety of IBS processed apheresis platelets (Test group) to conventional apheresis platelets (Reference group) in thrombocytopenic patients. The primary efficacy endpoint was the proportion of subjects with Grade 2 bleeding, as defined by the World Health Organization (WHO) bleeding scale. Secondary endpoints included incidence of WHO Grade 2 or higher bleeding, proportion with Grade 3 or 4 bleeding, mean number of days with Grade 2 bleeding, mean days between study platelet transfusions, mean number of platelet and red cell transfusions, rate of refractoriness to platelet transfusions, and the mean 1- and 24-hour post-transfusion count increments (CI) and corrected platelet count increment (CCI). Safety endpoints included Amotosalen levels after multiple transfusions, incidence of bacteremia attributable to platelet transfusion, acute platelet transfusion reactions, and antibody directed against potential treatment -induced plasma protein and platelet neoantigens. Differences between treatment groups for the primary endpoint (the proportion of subjects with Grade 2 bleeding) and one secondary endpoint (the proportion of subjects with Grade 3 or 4 bleeding) were analyzed using one-sided tests of non-inferiority with pre-specified non-inferiority margins of 12.5% and 7%, respectively.
2. Efficacy Results
2.1 Demographics and baseline characteristics of the study groups The mean age was not significantly different between treatment groups, with 46.9 years in the Test group and 45.7 years in the Reference group. Seven of 318 (2.2%) Test subjects and 16/327 (4.9%) Reference subjects were in the pediatric age category (≤16 years) and 28/318 (8.8%) Test subjects and 31/327 (9.5%) Reference subjects were in the geriatric age category (≥65 years). Distribution by gender was similar in both treatment groups, with 172/318 (54.1%) males in the Test group and 168/327 (51.4%) males in the Reference group. Distribution of subjects by ethnic origin was similar in both treatment groups; the vast majority of the subjects were Caucasian with 290/318 (91.2%) in the Test group and 299/327 (91.4%) in the Reference group. Demographics were similar among the 12 study sites where the study was conducted. However, only 3 sites (sites 802 [N=20], 807 [N=1], and 808 [N=2]) enrolled subjects in the pediatric age category due to difficulties in obtaining IRB approvals at the pediatric hospitals and logistical difficulties in assessing this population of subjects. The relatively low number of geriatric subjects in both groups may be attributed to the fact that intensive chemotherapy and stem cell transplantation with resultant need for platelet transfusion support is not frequently undertaken in subjects in this age category.

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 17
Baseline clinical characteristics were similar in both treatment groups. The two most common diagnoses were acute leukemia (28%) and lymphoma (27%). 78% of the enrolled subjects were undergoing stem cell transplantation, 19% chemotherapy in the absence of stem cell support, and 3% had thrombocytopenia as a result of their underlying disease. Approximately 70% of the stem cell transplants were from peripheral blood, 27% from bone marrow, and 3% were from cord blood. The donor source of stem cells in patients who received stem cell transplants was similar in the 2 treatment groups. Approximately two-thirds of the stem cells were autologous and approximately one-third were allogeneic. The majority of platelet transfusions were administered for prophylaxis of bleeding based on the transfusion threshold (Test = 93.5%, Reference = 90.1%). The pre-transfusion platelet count was similar between the two groups (15.1 × 109/L). The study population was broadly representative of patients who receive multiple platelet transfusions.
2.2 Analyses of Primary efficacy endpoint
The study met the primary endpoint of the proportion of subjects with Grade 2 bleeding: 58.5% in the Test group compared to 57.5% in the Reference group (upper bound of the one-sided 95% confidence interval of difference 7.3%, which was within the prespecified non-inferiority margin of 12.5%). 2.3 Analyses of Secondary Endpoints The outcome of the following secondary endpoints were all statistically significant and against the Test platelets. Secondary Endpoints with Statistically Significant Differences between Treatment Groups
Secondary Endpoints Test (n=318)
Reference (n=327)
p value
Mean Days of Grade 2 bleeding
3.2 2.5 0.023
Mean Days between transfusion
1.9 2.4 < 0.001
Mean number of platelet transfusion per subject
8.4 6.2 < 0.001
Mean CI at 1 hour (× 109/L)
21.4 34.1 < 0.001
Mean CCI at 1 hour (× 103/L)
11.1 16.0 < 0.001
Mean CI at 24 hour (× 109/L)
13.2 21.5 < 0.001
Mean CCI at 24 hours 6.7 10.1 < 0.001 Clinical platelet refractoriness (%)
21.4 7.0 < 0.001
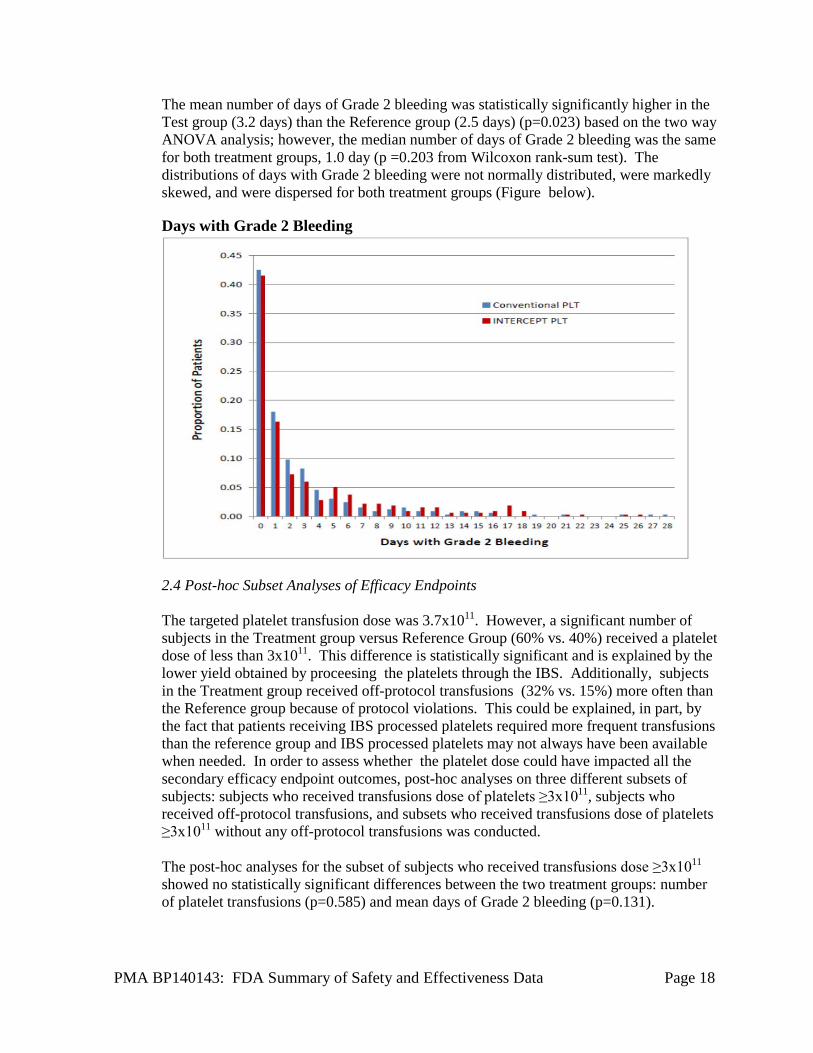
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 18
The mean number of days of Grade 2 bleeding was statistically significantly higher in the Test group (3.2 days) than the Reference group (2.5 days) (p=0.023) based on the two way ANOVA analysis; however, the median number of days of Grade 2 bleeding was the same for both treatment groups, 1.0 day (p =0.203 from Wilcoxon rank-sum test). The distributions of days with Grade 2 bleeding were not normally distributed, were markedly skewed, and were dispersed for both treatment groups (Figure below). Days with Grade 2 Bleeding
2.4 Post-hoc Subset Analyses of Efficacy Endpoints The targeted platelet transfusion dose was 3.7x1011. However, a significant number of subjects in the Treatment group versus Reference Group (60% vs. 40%) received a platelet dose of less than 3x1011. This difference is statistically significant and is explained by the lower yield obtained by proceesing the platelets through the IBS. Additionally, subjects in the Treatment group received off-protocol transfusions (32% vs. 15%) more often than the Reference group because of protocol violations. This could be explained, in part, by the fact that patients receiving IBS processed platelets required more frequent transfusions than the reference group and IBS processed platelets may not always have been available when needed. In order to assess whether the platelet dose could have impacted all the secondary efficacy endpoint outcomes, post-hoc analyses on three different subsets of subjects: subjects who received transfusions dose of platelets ≥3x1011, subjects who received off-protocol transfusions, and subsets who received transfusions dose of platelets ≥3x1011 without any off-protocol transfusions was conducted. The post-hoc analyses for the subset of subjects who received transfusions dose ≥3x1011 showed no statistically significant differences between the two treatment groups: number of platelet transfusions (p=0.585) and mean days of Grade 2 bleeding (p=0.131).

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 19
The post-hoc analyses for the subset of subjects who did not receive off-protocol transfusions showed no statistically significant differences between the two treatment groups in the number of platelet transfusions (p=0.647) and the number of days of Grade 2 bleeding (p=0.516). The post-hoc analyses for the subset of subjects who received transfusions dose ≥3x1011 without any off-protocol transfusions showed no statistically significant differences between the two treatment groups in number of platelet transfusions (p=0.833), the number of days of Grade 2 bleeding (p=0.582) and refractoriness (p=0.095). In summary, the posthoc analyses for all three subject subsets showed no statistically significant difference in number of platelet transfusions and number of days of Grade 2 bleeding whereas results of the other secondary endpoints remained the same as the original analyses. These results are taken with caution. While potentially informative, the power of these posthoc studies was reduced compared with the original analysis due to smaller numbers . Additionally, posthoc subset selection may have introduced undefined bias. 2.5 Efficacy Subpopulation Analyses Age and Sex The proportion of subjects with Grade 2 bleeding across all organ systems by age group and sex was studied. Grade 2 bleeding was similarly distributed between treatment groups in all age categories. However, Grade 2 bleeding was higher in the pediatric and geriatric categories compared to the adult category (67.1% for pediatric and geriatric combined versus 56.7% for adult). The proportion of subjects with Grade 2 bleeding was higher in the female category (62.3% female versus 54.1% male), but similarly distributed between treatment groups in both gender categories. These differences do not impact the overall conclusion of the primary endpoint.
3. Safety Results
3.1 Overall results No confirmed platelet transfusion-attributed bacteremia was reported in any subject. No antibodies to amotosalen (S-59) related neoantigens were detected. There was no evidence of significant amotosalen accumulation in the plasma of repeatedly exposed subjects. The most common AEs (>25% ) were petechiae (39%), diarrhea (38%), hematuria (37%), epistaxis (36%), hypokalemia (36%), rigors (35%), epistaxis (33%), fecal occult blood positive (33%), decreased magnesium (28%), anemia NOS(28%), vomiting (27%), and pyrexia (26%).
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 20
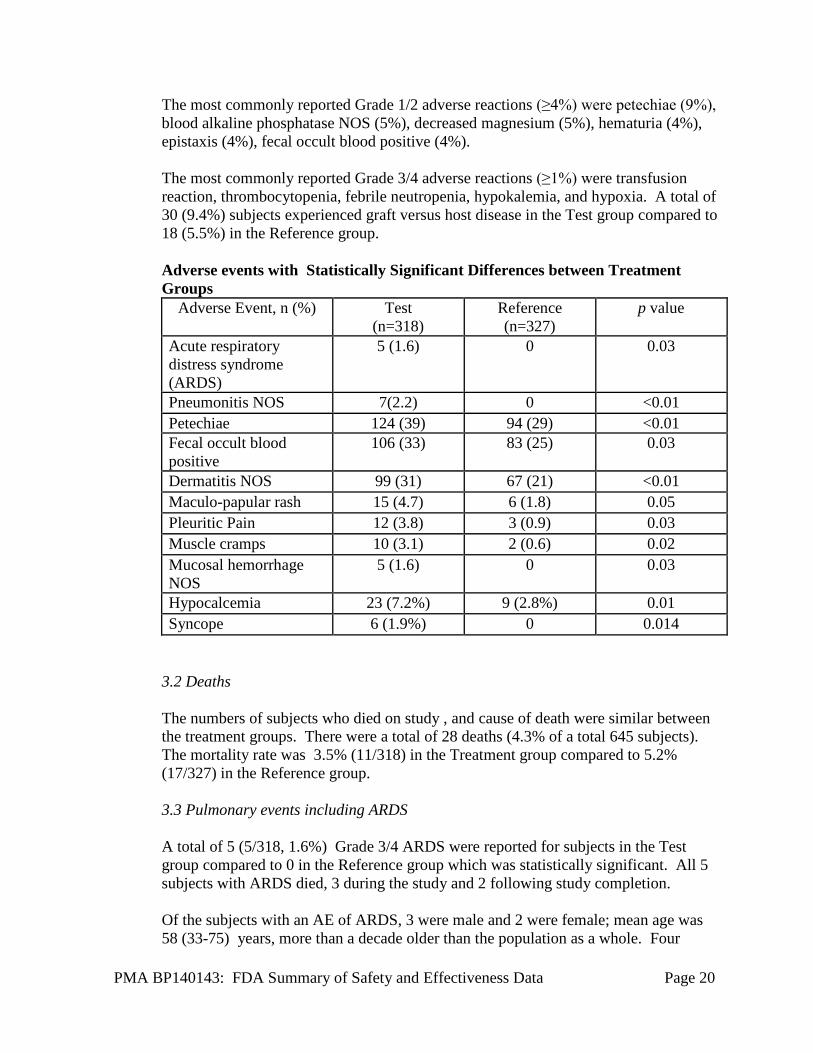
The most commonly reported Grade 1/2 adverse reactions (≥4%) were petechiae (9%), blood alkaline phosphatase NOS (5%), decreased magnesium (5%), hematuria (4%), epistaxis (4%), fecal occult blood positive (4%). The most commonly reported Grade 3/4 adverse reactions (≥1%) were transfusion reaction, thrombocytopenia, febrile neutropenia, hypokalemia, and hypoxia. A total of 30 (9.4%) subjects experienced graft versus host disease in the Test group compared to 18 (5.5%) in the Reference group. Adverse events with Statistically Significant Differences between Treatment Groups
Adverse Event, n (%) Test (n=318)
Reference (n=327)
p value
Acute respiratory distress syndrome (ARDS)
5 (1.6) 0 0.03
Pneumonitis NOS 7(2.2) 0 <0.01 Petechiae 124 (39) 94 (29) <0.01 Fecal occult blood positive
106 (33) 83 (25) 0.03
Dermatitis NOS 99 (31) 67 (21) <0.01 Maculo-papular rash 15 (4.7) 6 (1.8) 0.05 Pleuritic Pain 12 (3.8) 3 (0.9) 0.03 Muscle cramps 10 (3.1) 2 (0.6) 0.02 Mucosal hemorrhage NOS
5 (1.6) 0 0.03
Hypocalcemia 23 (7.2%) 9 (2.8%) 0.01 Syncope 6 (1.9%) 0 0.014
3.2 Deaths The numbers of subjects who died on study , and cause of death were similar between the treatment groups. There were a total of 28 deaths (4.3% of a total 645 subjects). The mortality rate was 3.5% (11/318) in the Treatment group compared to 5.2% (17/327) in the Reference group.
3.3 Pulmonary events including ARDS A total of 5 (5/318, 1.6%) Grade 3/4 ARDS were reported for subjects in the Test group compared to 0 in the Reference group which was statistically significant. All 5 subjects with ARDS died, 3 during the study and 2 following study completion. Of the subjects with an AE of ARDS, 3 were male and 2 were female; mean age was 58 (33-75) years, more than a decade older than the population as a whole. Four
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 21
subjects had leukemia (2 acute and 2 chronic) and 1 subject had multiple myeloma. All 5 subjects received intensive myeloablative chemotherapy; 3 also received total body irradiation and underwent allogeneic stem cell transplantation (2 PBSCT and 1 bone marrow). The average number of study platelet transfusions given to these 5 subjects was 22, considerably higher than the average number given to the Test group as a whole (8.4), and higher than the average number given to subjects who died on study (13 in the Test group and 6 in the Reference group).
3.3.1 Post Hoc safety analyses: BC-001-01 (SPRINT reanalysis) ARDS was not a prespecified adverse event for safety assessment and therefore the study protocol did not have a prespecified definition to qualify this event. These were captured by the investigators in the overall safety assessment. Cerus conducted a reanalysis of all clinically serious pulmonary events from the SPRINT trial. The purpose of the reanalysis was to address concerns regarding grade 3/4 pulmonary adverse events reported in the trial and to address FDA’s request to perform a subject-oriented safety review and provide a comparison between cohorts focused on serious respiratory adverse events. Three analyses were conducted to address this:
• Original P3A99 clinical study analyses as defined prospectively in the Statistical Analysis Plan
• Subset reanalysis performed by an expert engaged by Cerus of those subjects [N=28] who died during the P3A99 clinical study
• Study BC-001-01, the blinded re-analysis of safety data performed by an independent Expert Physicians Panel [EPP] of subjects [N=148] with potentially clinically serious pulmonary events.
Outcomes of original safety analyses This analysis is summarized above. Outcomes of Subset re-analysis Medical records data for the 28 subjects who died during the study (n=28; 11 test and 17 reference) was collected to assess the true incidence of ARDS using the objective American-European ARDS Consensus Conference (AEACC) criteria, which utilized assessment of bilateral pulmonary infiltrates, PaO2/FiO2 ratios and exclusion of cardiogenic pulmonary edema for the diagnosis of ALI and the subset of ARDS: AEACC Criteria for Acute Lung Injury

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 22
The rationale for selection of this subset of subjects for further review was based on the observation that ARDS in the setting of HSCT is associated with a high mortality rate. The findings of this review was that the incidence of ARDS in this subset of subjects was comparable between treatment groups (8/11 [73%] Test vs. 12/17 [71%] Reference, p=1.00). Outcomes of the Study BC-001-01 The primary objective of the study BC-001-01 was to obtain an independent and objective assessment by an external Expert Pulmonary Panel (EPP) blinded to treatment assignment, to determine the occurrence of clinically serious pulmonary AEs in Test and Reference group subjects of the SPRINT trial. For the purposes of this reanalysis primary medical records were reviewed by the EPP. Adverse pulmonary events reported for all subjects were reviewed and the EPP identified all subjects experiencing a clinically serious pulmonary AE for review of primary medical records. These events were further categorized by the EPP as to the category of pulmonary event by respiratory anatomical and disease categories, including acute lung injury (ALI) and ARDS, and the incidence rates were compared between Test and Reference groups. In the re-analysis study, AEs were coded using MedDRA 6.0, whereas version 3.3 was used in the original trial. The EPP determinations of ALI and ARDS were based on the AEACC criteria. Differences in the SPRINT Trial (P3A99) and BC-001-01 Study

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 23
The primary analysis of the BC-001-01 study, based upon EPP determinations, found no differences between treatment groups for clinically serious pulmonary AEs, either overall, or by respiratory anatomical or disease category. Specifically, no statistically significant differences were observed for pneumonitis or acute lung injury, including ARDS; however, a trend was observed for a slightly higher incidence of ARDS in the test group 12 (n=78) vs. 5 (n=70) in a subset subjects with pulmonary AEs (n = 148). When precise PaO2/FiO2 ratio measurements were not available, the EPP used a conservative approach and assigned the diagnosis of ALI. In addition, ARDS diagnoses were only given to ventilated subjects even if they met the AEACC criteria. The true incidence of ARDS may have been underestimated beacuse of the lower power of the reanalysis. Among subjects included in the reanalysis, there were no differences in exposure to chemotherapy associated with pulmonary toxicity (e.g. Carmustine, Bleomycin, Methotrexate, Busulfan)
4. Summary and Conclusion on the Efficacy and Safety of IBS Processed Platelets in the SPRINT Trial
The SPRINT trial met its primary efficacy endpoint of proportion of subjects with Grade 2 bleeding using WHO criteria. However, statistically significant differences were found in the following secondary endpoints between the two treatment groups: mean number of days with Grade 2 bleeding, mean days between study platelet transfusions, mean number of platelet and red cell transfusions, rate of refractoriness to platelet transfusions, the 1- and 24-hour post-transfusion count increments (CI), and corrected platelet count increment (CCI). The outcomes of these secondary efficacy endpoints all favored the Reference group. Posthoc analyses showed that when corrected for the platelet dose, therewas no statistically significant difference in number of platelet transfusions and mean number of days with Grade 2 bleeding. Therefore, adjusting the platelet dose per unit to ≥ 3x1011would be expected to overcome the differences in the secondary efficacy outcomes observed in this study.
The numbers of subjects who died on study and cause of death were similar between the two treatment groups. There appeared to be no significant differences between treatment groups for severity of AEs or the incidence of AEs considered to be related to study platelet transfusion. The incidence of SAEs was similar between the two treatment groups. However, the incidence of 10 AEs were statistically significantly different between the two treatment groups and the results all favored the Reference group. Particularly, there were five cases of ARDS in the Test group and zero in the Reference group. This difference was statistically significant. ARDS is a syndrome and acute lung injury may have been misclassified as ARDS as there was no prespecified protocol definition. This may have introduced confounding into the reanalysis conducted by the expert and the EPP. This is exemplified by the fact that, based upon interpretation of the AEACC criteria, some subjects were assessed by Cerus expert as having ARDS while the EPP assessed them as having ALI, and vice

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 24
versa. In addition, a selection bias may have been introduced by provided the EPP with preselected subject medical records. It is important to note that although there were no significant differences noted by the EPP, there still remained a trend of increased ARDS in the Test group. The casuality of these events remain uncertain.
In summary, the U.S. pivotal study (SPRINT) met its primary efficacy endpoint. Although statistically significant differences were found in some secondary endpoints in favor of the Reference group, the specific differences in the mean number of days with Grade 2 bleeding, and the difference in the days between transfusions, were not considered clinically significant. The number of deaths and incidence of SAEs between the two treatment groups were similar. Based on the SPRINT study an increased risk of ARDS with the IBS processed platelets cannot be ruled out.
C. Overview of Supportive Studies in Comparison with SPRINT Three additional phase 3 trials and five post-marketing studies were conducted in Europe. These studies provided additional evidence of decreased platelet viability for IBS platelets that is independent of the dose of platelets administered. There was a trend towards increased hemorrhagic AEs in the phase 3 European trial (euroSPRITE). Unlike SPRINT, increased pulmonary events were not seen in these other studies. However, only two of the European phase 3 trials (REP-NIV FE 683 and euroSPRITE) and one European post-marketing study (TESSI) were randomized controlled trials (RCT) and all were small studies and likely would not have been able to detect the differences in ARDS that were observed in SPRINT. One case of ARDS was reported in the Reference arm of euroSPRITE. The remaining post-marketing studies were observational trials to evaluate IBS processed platelets during routine clinical use, and therefore were not adequately designed to evaluate the potential increased risk of ARDS in IBS processed platelets. A single case of ARDS was reported in each of the following observational trials: CLI 00058 (1/51 [2%] subjects; 1/560 (0.17%) transfusions); CLI00059 (1/46 [2%] subjects; 1/551 (0.18%) transfusions). No additional safety concerns were identified in the hemovigilance (HV) studies/reports submitted in the PMA. National surveillance programs conducted by France and Switzerland indicate that IBS platelets reduce transfusion-related sepsis, which supports the findings of the clinical trials. Hemovigilance reports from the French and Swiss agencies reported frequencies of acute transfusion reactions that were similar for IBS and conventional platelets and consistent with what was observed in the clinical trials. However, the adverse event reporting systems for both countries is non-exhaustive, and underreporting is likely. D. Limitations Clinical Endpoints in Regard to TTI Because of the relatively low frequency of contamination of platelets by infectious pathogens it was not feasible to conduct clinical trials to evaluate the efficacy of the IBS technology for prevention of TTI. Thus, the focus of the clinical development program was primarily directed to characterize the therapeutic efficacy and safety of

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 25
IBS processed platelets in comparison to conventional platelets. The primary clinical indication for platelet transfusion is prevention or treatment of bleeding associated with thrombocytopenia. Therefore the clinical development program for the IBS processed platelets was focused on the evaluation of the efficacy of treated platelets to prevent bleeding in transfusion-dependent patients. The evaluation of efficacy was based upon in vivo viability after maximal storage duration and the assessment of hemostatic efficacy in profoundly thrombocytopenic patients. For assessment of hemostatic efficacy, multiple surrogate clinical endpoints, were utilized in studies of thrombocytopenic patients including platelet count increments, transfusion requirements (platelets and RBCs), and the interval between platelet transfusions. E. Study Populations
The clinical development program included study designs to characterize the therapeutic efficacy of IBS processed platelets in comparison to conventional platelets. The primary clinical indication for platelet transfusion is prevention or treatment of bleeding associated with thrombocytopenia. Therefore, the clinical development program for the IBS was focused on the evaluation of the efficacy of treated platelets to prevent bleeding in transfusion-dependent patients in relevant clinical settings.
In certain clinical settings such as hematopoetic stem cell transplant (HSCT) patients without active bleeding at time of transfusion require repeated exposures to platelet for prophylaxis against bleeding during multiple therapeutic cycles to treat malignant disorders, and these patients received the highest exposures to IBS processed platelets in all the conducted clinical trials. The hematology-oncology patients accounted for 95% of the total patients enrolled in all RCT studies which accounted for 60% of platelets transfused. In the SPRINT trial, 85% of the subjects were stem cell transplant cases. The observational studies of IBS platelets in routine use and investigator studies included a higher proportion of patients with non-hematology-oncology disorders.
XI. FINANCIAL DISCLOSURE
The pivotal clinical study included 12 investigators. None of the clinical investigators had disclosable financial interests/arrangements as defined in sections 54.2(a), (b), (c), and (f). The information provided does not raise any questions about the reliability of the data.
XII. SUMMARY OF SUPPLEMENTAL CLINICAL INFORMATION
Safety outcomes of the submitted postmarketing data provided significant supplemental clinical information. The post-market studies submitted included three single-arm observational studies (funded but not conducted by the sponsor), three observational studies described as “active hemovigilance studies” (conducted by the sponsor), National Hemovigilance data from two European countries (France 2009-2012 and Switzerland 2011-2012), andone small RCT conducted in the US (TESSI trial)(n=211). Additional annual reports from the National Hemovigilance systems of
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 26
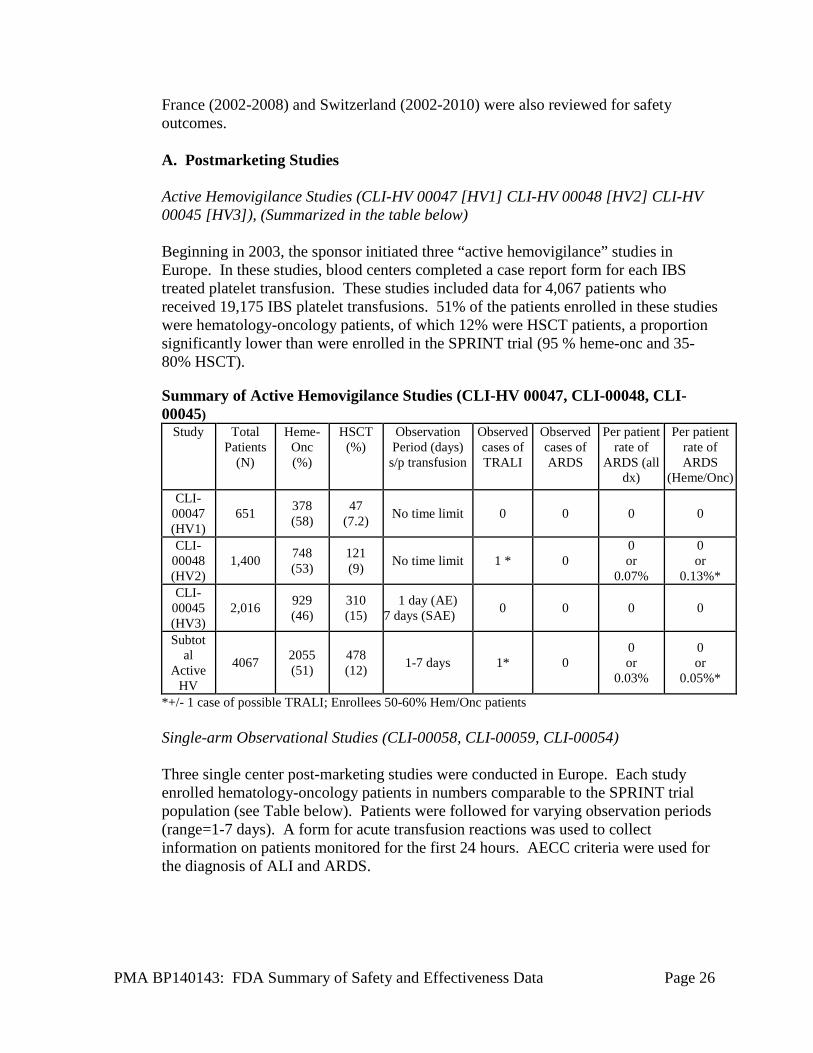
France (2002-2008) and Switzerland (2002-2010) were also reviewed for safety outcomes. A. Postmarketing Studies Active Hemovigilance Studies (CLI-HV 00047 [HV1] CLI-HV 00048 [HV2] CLI-HV 00045 [HV3]), (Summarized in the table below) Beginning in 2003, the sponsor initiated three “active hemovigilance” studies in Europe. In these studies, blood centers completed a case report form for each IBS treated platelet transfusion. These studies included data for 4,067 patients who received 19,175 IBS platelet transfusions. 51% of the patients enrolled in these studies were hematology-oncology patients, of which 12% were HSCT patients, a proportion significantly lower than were enrolled in the SPRINT trial (95 % heme-onc and 35-80% HSCT).
Summary of Active Hemovigilance Studies (CLI-HV 00047, CLI-00048, CLI-00045)
Study Total Patients
(N)
Heme-Onc (%)
HSCT (%)
Observation Period (days)
s/p transfusion
Observed cases of TRALI
Observed cases of ARDS
Per patient rate of
ARDS (all dx)
Per patient rate of ARDS
(Heme/Onc) CLI-
00047 (HV1)
651 378 (58)
47 (7.2) No time limit 0 0 0 0
CLI-00048 (HV2)
1,400 748 (53)
121 (9) No time limit 1 * 0
0 or
0.07%
0 or
0.13%* CLI-
00045 (HV3)
2,016 929 (46)
310 (15)
1 day (AE) 7 days (SAE) 0 0 0 0
Subtotal
Active HV
4067 2055 (51)
478 (12) 1-7 days 1* 0
0 or
0.03%
0 or
0.05%*
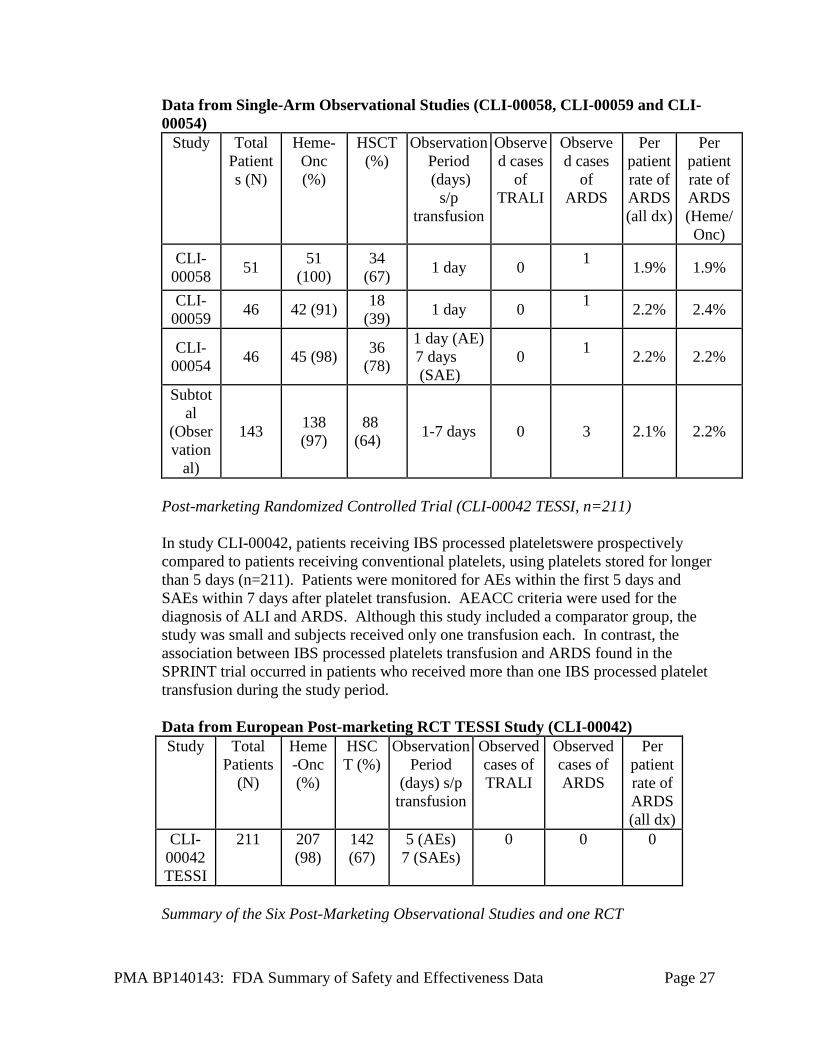
*+/- 1 case of possible TRALI; Enrollees 50-60% Hem/Onc patients Single-arm Observational Studies (CLI-00058, CLI-00059, CLI-00054) Three single center post-marketing studies were conducted in Europe. Each study enrolled hematology-oncology patients in numbers comparable to the SPRINT trial population (see Table below). Patients were followed for varying observation periods (range=1-7 days). A form for acute transfusion reactions was used to collect information on patients monitored for the first 24 hours. AECC criteria were used for the diagnosis of ALI and ARDS.
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 27
Data from Single-Arm Observational Studies (CLI-00058, CLI-00059 and CLI-00054)
Study Total Patients (N)
Heme-Onc (%)
HSCT (%)
Observation Period (days)
s/p transfusion
Observed cases
of TRALI
Observed cases
of ARDS
Per patient rate of ARDS (all dx)
Per patient rate of ARDS (Heme/ Onc)
CLI-00058 51 51
(100) 34
(67) 1 day 0 1 1.9% 1.9%
CLI-00059 46 42 (91) 18
(39) 1 day 0 1 2.2% 2.4%
CLI-00054 46 45 (98) 36
(78)
1 day (AE) 7 days (SAE)
0 1 2.2% 2.2%
Subtotal
(Observation
al)
143 138 (97)
88 (64) 1-7 days 0 3 2.1% 2.2%
Post-marketing Randomized Controlled Trial (CLI-00042 TESSI, n=211) In study CLI-00042, patients receiving IBS processed plateletswere prospectively compared to patients receiving conventional platelets, using platelets stored for longer than 5 days (n=211). Patients were monitored for AEs within the first 5 days and SAEs within 7 days after platelet transfusion. AEACC criteria were used for the diagnosis of ALI and ARDS. Although this study included a comparator group, the study was small and subjects received only one transfusion each. In contrast, the association between IBS processed platelets transfusion and ARDS found in the SPRINT trial occurred in patients who received more than one IBS processed platelet transfusion during the study period. Data from European Post-marketing RCT TESSI Study (CLI-00042) Study Total
Patients (N)
Heme-Onc (%)
HSCT (%)
Observation Period
(days) s/p transfusion
Observed cases of TRALI
Observed cases of ARDS
Per patient rate of ARDS (all dx)
CLI-00042 TESSI
211 207 (98)
142 (67)
5 (AEs) 7 (SAEs)
0 0 0
Summary of the Six Post-Marketing Observational Studies and one RCT

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 28
The 6 post-marketing observational trials collected data on 20,754 IBS platelet transfusions. A total of three ARDS cases were reported. No case of ARDS was observed in either arm of the TESSI RCT. Rate of ARDS (per 10K transfusions) in Seven Post-marketing Studies IBS Platelet Transfusions (n)
Cases of ARDS
Cases of Platelet Related Bacteremia
Rate of ARDS (per 10k Platelet Transfusions)
20,754 3 0 1.4 4* 0 1.9
*+Adding patient B01-201 (Reported case of TACO, possible acute lung injury) The definitive ARDS cases came from 3 separate observational studies with proportions of Hem/Onc and HSCT patients similar to the SPRINT trial population (90-100% Heme/Onc and 40-75% HSCT). No cases were observed in the remaining 3 studies that contained a lower proportion of Hem/Onc patients, than in the SPRINT study population. No cases of ARDS occurred in the TESSI RCT in a population similar to the SPRINT trial. However, conclusions regarding ARDS are limited by the small size of this trial. The results from the six observational, uncontrolled studies as well as the TESSI RCT are not able to confirm or refute the signal observed in the randomized, controlled SPRINT study. While the seven studies included over 4,000 patients, it is difficult to draw conclusions from the pooled study results given the differences in study sites, study year and proportion of subjects with Hem/Onc diagnoses and the inherent limitations of observational and uncontrolled studies. B. National Hemovigilance Systems Data (France and Switzerland)
Structure and Attributes of National Hemovigilance Systems, postmarketing surveillance systems in France and Switzerland The National Hemovigilance (HV) reporting systems in France and Switzerland are designed to collect information on known, prespecified transfusion associated (TA) AEs (e.g., TRALI, TACO, TAD, etc.). AE reporting is mandated for Transfusion Associated (TA) AEs for all blood products, regardless of the causality assessment (possible, probable, certain), observed within 6-24 hours of transfusion. Outcomes that clinicians typically recognize as TA AEs are the well-known, pre-specified outcomes of Transfusion Related Acute Lung Injury (TRALI), Transfusion Associated Cardiac Output (TACO), and Transfusion Associated Dyspnea (TAD). TRALI and other forms of ALI (e.g., ARDS) occur on a continuum and have a similar clinical presentation. If IBS platelets are associated with a non-immune mediated form of ALI, these events could potentially be reported by clinicians as possible TRALI. Examining the National HV system data for an increase in the frequency of TRALI (as a surrogate for ALI/ARDS) associated with IBS platelet use is somewhat relevant. A

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 29
major limitation of the HV system lies in the fact that pulmonary adverse events not considered to be TA AEs may not be reported to the HV system. Patients requiring platelet transfusions often have underlying co-morbidities or ALI risk factors that may preclude clinicians from associating subsequent pulmonary injury to a recent platelet transfusion. Adverse event (AE) reporting data for IBS platelets is available from the National Hemovigilance reporting systems in France (ANSM) and Switzerland (SwissMedic). Widespread post-marketing use of IBS platelets has been implemented in parts of France and in all of Switzerland. Since 2006, four regions in France have implemented IBS platelets: this represents approximately 7% of platelet use in France (2006-2012). Annual data on the number of platelet transfusions (conventional vs IBS platelets) and TRALI rates is available from the French HV system for the years 2006-2012 (Table 8). Switzerland transitioned from use of conventional platelets to IBS platelets during 2011, thus, in 2012 and 2013, all platelet transfusions were IBS platelets. Annual data on the number of platelet transfusions (conventional vs IBS platelets) and TRALI rates is available from the Swiss HV system for the years 2002-2013 (Table 9). Case Ascertainment in the French National HV system (Afssaps and ANSM) Beginning in 2002, the French National HV system (ANSM) used a specific definition for the diagnosis of TRALI: “new onset ALI during or within 6 hours of transfusion which is not related to a competing etiology for ALI…there should be no temporal relation to an existing ALI risk factor to definitively conclude that the new onset ALI is transfusion-related.” Additionally, the sponsor notes that the system does recognize the occurrence of other causes of ALI in platelet transfusion recipients and captures those diagnoses by providing a definition of “possible TRALI” defined as “ALI in which the evidence is indeterminate for relation to the transfusion or other causes for ALI.”
Case Ascertainment in the Swiss HV System (SwissMedic) -------------------------------------------(b)(3)------------------------------------------------ --------------------------------------------------------------------------------------------- ------------------------------------------------------------------------------------------------------ ---------------------------------------------------------------------------------------------------- ------------------------------------------------------------------------------------------------ ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- ---------------------------------------------------------------------------------------------------- ------------------------------------------------------------------------------------------------------- ----------------------------------------------------------------------------------------------------------------------------------------------------------------------- --------------------------------------------(b)(3)----------------------------------------------------------------------------------------------------------------------------------------------------- -------------------------------------------------------------------------------------------

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 30
----------------------------------------------(b)(3)------------------------------------------------ ----------------------------------------------------------------------------------------------- ---------------------------------------------------------------------------- -------------------------------------------(b)(3)------------------------------------------- --------------------------------------------------------------------------------------------- ----------------------------------------------------------------------------------------------------------------------------------------------------------------- Rates of TRALI from French and Swiss HV Systems (Note: TRALI cases include “possible, probable or certain” imputabilities) Tables below show a comparison of the number of platelet transfusions (conventional vs. IBS processed platelets) by year, and the rates of TRALI reported to the French HV system, and Swiss HV systems
Comparison of the rate of TRALI after Conventional vs. IBS processed platelets reported to the National HV, France, 2002-2012
Year
Conventional Platelet
Transfusions (n)
IBS Platelet Transfusions
(n)
Rate of TRALI s/p Conventional Platelets (per 10k plt transfusions)
Rate of TRALI s/p IBS Platelets (per 10k plt transfusions)
2002 197,750 0 0.15 IBS not in use 2003 209,594 0 0.43 IBS not in use
2004 203,680 0
TRALI cases not
distinguished by product
transfused IBS not in use 2005 188,327 0 0.85 IBS not in use 2006 231853 6,420 0.38* 2007 232708 15,393 0.28* 2008 239379 15,544 0.31* 2009 241,634 21,767 0.50 0.46 2010 256,200 21,897 0.43 0.46 2011 269,467 23,179 0.15 0 2012 275,834 24,849 0.33 0
*For 2006-2008, no distinction was made between conventional and IBS platelets
PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 31
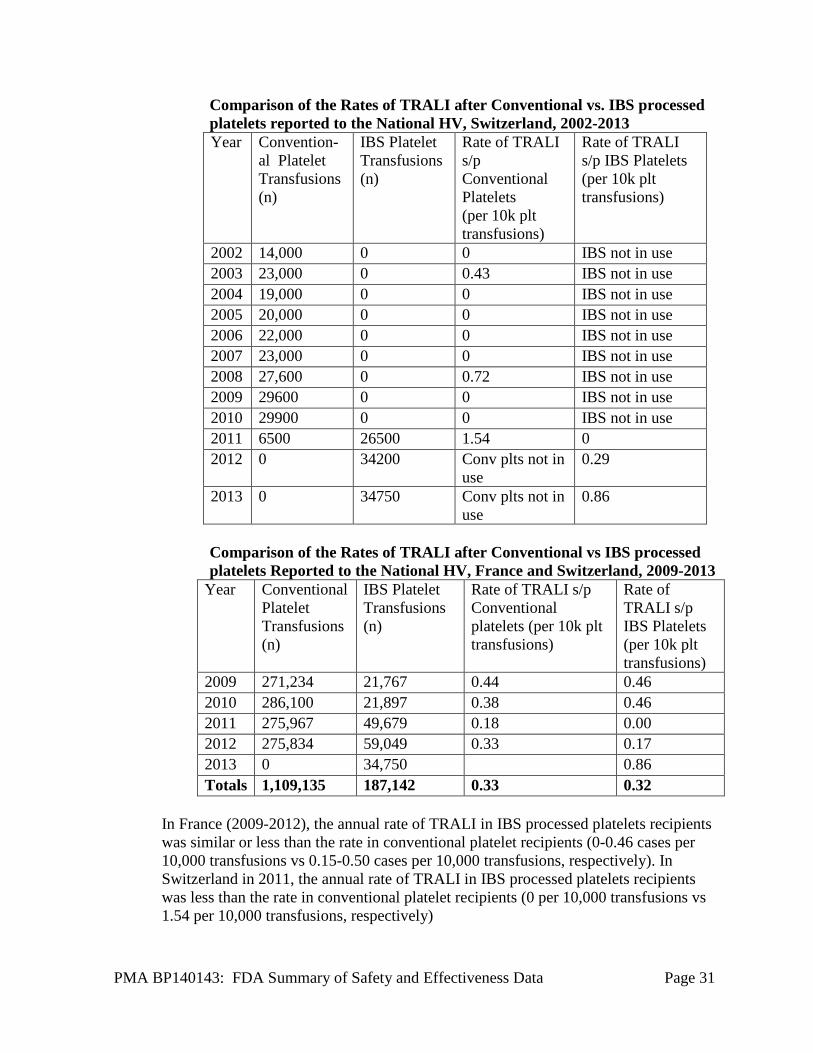
Comparison of the Rates of TRALI after Conventional vs. IBS processed platelets reported to the National HV, Switzerland, 2002-2013 Year Convention-
al Platelet Transfusions (n)
IBS Platelet Transfusions (n)
Rate of TRALI s/p Conventional Platelets (per 10k plt transfusions)
Rate of TRALI s/p IBS Platelets (per 10k plt transfusions)
2002 14,000 0 0 IBS not in use 2003 23,000 0 0.43 IBS not in use 2004 19,000 0 0 IBS not in use 2005 20,000 0 0 IBS not in use 2006 22,000 0 0 IBS not in use 2007 23,000 0 0 IBS not in use 2008 27,600 0 0.72 IBS not in use 2009 29600 0 0 IBS not in use 2010 29900 0 0 IBS not in use 2011 6500 26500 1.54 0 2012 0 34200 Conv plts not in
use 0.29
2013 0 34750 Conv plts not in use
0.86
Comparison of the Rates of TRALI after Conventional vs IBS processed platelets Reported to the National HV, France and Switzerland, 2009-2013
Year Conventional Platelet Transfusions (n)
IBS Platelet Transfusions (n)
Rate of TRALI s/p Conventional platelets (per 10k plt transfusions)
Rate of TRALI s/p IBS Platelets (per 10k plt transfusions)
2009 271,234 21,767 0.44 0.46 2010 286,100 21,897 0.38 0.46 2011 275,967 49,679 0.18 0.00 2012 275,834 59,049 0.33 0.17 2013 0 34,750 0.86 Totals 1,109,135 187,142 0.33 0.32
In France (2009-2012), the annual rate of TRALI in IBS processed platelets recipients was similar or less than the rate in conventional platelet recipients (0-0.46 cases per 10,000 transfusions vs 0.15-0.50 cases per 10,000 transfusions, respectively). In Switzerland in 2011, the annual rate of TRALI in IBS processed platelets recipients was less than the rate in conventional platelet recipients (0 per 10,000 transfusions vs 1.54 per 10,000 transfusions, respectively)

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 32
A total of six cases of TRALI were reported after use of IBS processed platelets (four in Switzerland, two in France). The four TRALI cases (three in 2013 and one in 2012) reported by Swissmedic were all graded as “probable” cases. Three of the four cases were classified as immune-mediated (including two of the three cases in 2013 and the single 2012 case); the symptoms in all four cases started during or within 30 minutes of transfusion. Data on the immune-mediated status of thetwo French TRALI cases (one in 2009 and one in 2010) are not available. Combining data from both systems, the annual rate of TRALI for the years 2009-2013 was similar in both the IBS processed platelet and conventional platelet recipient groups. There were no cases of ARDS reported to the French or Swiss HV database. The limitations of the French and Swiss National HV systems are those inherent to passive surveillance systems including underreporting, lack of a comparator group, incomplete data, and reporting bias. The number of TRALI reports received by the HV systems is small. ALI cases not recognized by clinicians as platelet-associated events might not be reported to these HV systems. Review of the data for the years for which the data is available reveals the TRALI reporting rates are similar between both the IBS platelet and conventional platelet recipient groups. The slight increase in the Swiss TRALI rate in 2013 does not appear to represent a safety signal for ARDS since this rate represents three TRALI cases, two of which were found to be immune-mediated cases of TRALI. The French and Swiss HV systems provided useful information from an estimated 250,000 platelet transfusions. Conclusions regarding the ARDS risk in this population are limited by: 1) factors inherent to passive surveillance reporting systems (underreporting, lack of comparator group), and 2) the HV systems are designed to collect data on TA AEs (including TRALI, an ALI diagnosis of exclusion), rather than ARDS. During the periods reviewed, no reports of ARDS were received by the HV system, thus, the true rate of ARDS in this population is not accounted for, and most likely indicates failure of the HV system to capture this event. Summary: National HV Database Results The National HV database requires reporting of TA AEs after receipt of blood components including IBS and conventional platelets. The data from this review did not find increased rates of TRALI in IBS platelet recipients. The number of TRALI reported to the HV systems during the years 2009-2013 is small, and the TRALI rates were similar in both groups. There were 6/187,142 TRALI cases per IBS platelet transfusions, for a TRALI rate of 0.33 per 10,000 platelet transfusion, compared to 37/1,109,135 TRALI cases per conventional platelet transfusions, for a rate of 0.32 per 10,000 platelet transfusions. Limitations of the HV system include: 1) data collection is limited to TA AEs (TRALI, TACO, TAD, etc.), 2) there is a high incidence of co-morbidity in patients receiving IBS platelets, thus ALI may be attributed to a patient’s underlying condition rather than the transfusion and may not be reported, 3) some reports of TRALI

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 33
captured data through 24 hours (“possible TRALI”) may in fact be cases of ALI/ARDS, however, we are unable to define which were true ALI/ARDS cases, or quantify the extent of the potential misclassification. Summary of all Post marketing data for ARDS and TRALI The populations studied in the three CLI observational trials appear the most similar to patients enrolled in the SPRINT trial (90% Hem/Onc patients). These observational studies (n=143) found a per patient ARDS rate of 1.9-2.4%, which is consistent with the results from the SPRINT trial (ARDS rate of 1.6%). The post-marketing National HV experience in France and Switzerland (IBS processed platelets in use eight years in France; 3 years in Switzerland) of ~200,000 IBS processed platelets transfusions did not reveal a trend towards an increase in TRALI (which could include some cases of ALI/ARDS); however, this study assessed the rate of TRALI and other TA AEs, not ARDS. Of note, the National HV system did not receive any reports of ARDS during the study period, suggesting that ARDS cases are not well captured by the system. The studies noted above have the following limitations including: 1) they are uncontrolled studies without a comparator group, 2) the populations reporting to the National HV programs (France, Switzerland) were more heterogeneous with potentially lower rates of ALI, as opposed to the predominantly HSCT population evaluated in the SPRINT trials 3) reports submitted to the HV programs are primarily TA AEs and as such, may not include all cases of ALI (i.e., HSCT patients are medically complex with multiple co-morbidities and risk factors for ALI that were not considered transfusion-associated may not be reported to the HV system). In summary, the six observational studies, and the National HV data from France and Switzerland are uncontrolled studies without a comparator group, the results of which are not able to confirm or refute the signal observed in the SPRINT RCT, which found a statistically significant association with ARDS in patients who received Cerus platelets; re-analysis found no statistically significant difference between groups, though a trend of increased incidence remained in the IBS group. Currently, theer are two ongoing RCT trials, in France and Italy to primarily evaluate efficacy of IBS processed platelets. Safety data will also be captured.
XIII. HEMOVIGILANCE PLAN
The clinical safety database reviewed by FDA to evaluate the potential safety signal of increased frequency of ALI/ARDS included the pivotal phase 3 trial (SPRINT), seven post-market studies (1 randomized controlled trial, 3 observational and three active hemovigilance studies) and national hemovigilance data from France and Switzerland 2009-2012. Following approval routine hemovigilance activities will be performed such as,spontaneous adverse reaction reporting, the ongoing European post-market active hemovigilance study (CLI-HV 00079) and regular monitoring of the literature and national hemovigilance reports. In addition to routine hemovigilance, a post-market active hemovigilance study, CLI-HV 00112 will also be conducted to evaluate the incidence of acute lung injury, with

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 34
an emphasis on ARDS to obtain additional information on this event The following information was used to determine the background rate of ALI in hematology oncology patients in the US and was used to design and calculate the sample size for the phase 4 study (CLI-HV 00112).
Background rates for ALI and TRALI
The sponsor used 5 data sources to estimate the background incidence of ALI: 1) data from the ---------------------------(b)(4)---------------------------- database in predominantly HSCT patients using assisted ventilation as a surrogate marker for ALI, 2) an assessment of ALI in HSCT patients who received platelets from 39 large US transplant centers using assisted ventilation as a surrogate, 3) a prospective study of TRALI at 2 U.S. medical centers (Toy et al. 2011), 4) a review study conducted by a working group of the French National HV (Ozier et al 2011), and 5) cumulative data from multiple years of ANSM annual reports as discussed above. These data are summarized in the table below.
Background Rates for ALI and TRALI Population Rate Assessment: ALI in HSCT patients who received platelets from the -------------------(b)(4)---------------------- Population: Primarily allogeneic transplant patients Endpoint: Assisted ventilation (intubation, tight fitting face mask with continuous positive pressure)
5.6%
Assessment: ALI in HSCT patients who received platelets from 39 large U.S. transplant Centers (Combined data) Population: Autologous and allogeneic transplant patients in National Donor Matching Program Endpoint: Assisted ventilation (intubation, tight fitting face mask with continuous positive pressure)
3.7%
TRALI at 2 tertiary care centers (Toy, et al) Population: Heterogenous population Endpoint: TRALI
0.81 -2.57 per 10k blood components transfused *
TRALI from French HV, 2 years of data, apheresis platelets (Ozier, et al)
0.77 per 10k transfusions of apheresis platelets only 0.17 per 10k transfusions of all blood components
*Rate declined from 2.57 in 2006 to 0.81 in 2009, which was attributed to use of male donor plasma.

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 35
The -------------(b)(4)--------------- population typically receives more allogeneic transplants than other centers, resulting in a higher morbidity and corresponding increased rate of ALI. The range of background rates provided by the sponsor is extremely wide and varies by patient population, type of blood components transfused, and the observation method (study vs. HV reporting system). The rate of TRALI in non-IBS platelet exposed populations was consistent with or higher than the rates observed in the National HV reporting and the 7 post-marketing observational studies submitted by the sponsor.
Platelet exposure and concurrent risk factors for non-platelet associated ALI is expected to be higher at tertiary care centers and at transplant centers. The background rate of TRALI in the Toy study and assisted ventilation in -(b)(4)- is higher than or similar to the observed rate of ARDS in IBS treated patients from the SPRINT trial (1.6% initial and 3.8% EPP reanalysis). All -(b)(4)- patients meeting the AECC ALI criteria required assisted ventilation, but not all patients receiving assisted ventilation had ALI. The use of assisted ventilation as a surrogate for ALI while inclusive, is likely an over representation of lung injury associated with platelet transfusion, and most likely represents an upper bound estimate. The degree of variation from the true rate is unknown.
Design of the Phase 4 study
This study is a prospective, open-label, multi-center, non-randomized, non-inferiority Phase IV surveillance study following transfusion of INTERCEPT processed platelets. The primary outcome measure is the proportion of patients requiring treatment-emergent assisted mechanical ventilation during the study observation period. Assisted mechanical ventilation is selected as the primary outcome as it provides an objective measure of severe, clinically significant pulmonary injury. Additionally, the study will characterize the frequency of transfusion associated AEs and SAEs [including acute respiratory distress syndrome (ARDS) and clinically serious pulmonary AEs (CSPAEs)] and transfusion reactions (TR) in patients receiving at least one study platelet component (PC) transfusion. ARDS is defined by the Berlin criteria; and will be classified as related to platelet transfusion or as related to concurrent clinical conditions associated with ARDS in patients receiving platelet transfusion support. The patient population will be hematology-oncology patients, including those undergoing hematopoietic stem cell transplant (HSCT), expected to require one or more PC transfusions. Patients will be assessed for the primary outcome measure for up to 21 days of platelet transfusion support.
The study will be powered on the primary endpoint of treatment-emergent assisted ventilation [administered by intubation or tight fitting mask with positive end-expiratory pressure (PEEP) or continuous positive airway pressure (CPAP) ≥ 5 cm H2O]. The study is designed to use a cohort-sequential design with a comparative

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 36
Control group supported with conventional PCs prior to implementation of INTERCEPT processed platelets in centers likely to adopt the INTERCEPT Blood System for Platelets technology. Patients will be stratified by type of primary disease therapy (autologous HSCT, allogeneic HSCT, cord blood HSCT, and chemotherapy). The study will be conducted in two phases: 1) the control phase during which study patients will receive only conventional platelet components; and 2) the INTERCEPT phase during which patients will receive only INTERCEPT processed platelet components. For both phases, patients will be supported with study PC transfusions for a maximum of 21 days or until they become platelet independent. Patients are considered to have achieved platelet independence if more than 5 days elapse after exposure to a study PC transfusion. When a patient achieves platelet independence, the patient will have completed the study PC transfusion period (even if they subsequently resume another cycle of platelet support). Patients who have achieved at least 14 days of platelet independence after his/her first Control phase may be enrolled into the INTERCEPT phase.
A minimum of 1466 patients per cohort period will be enrolled, which will provide approximately 90% power at one-sided alpha level of 0.025 to reject the null hypothesis of inferiority for the primary endpoint. For the primary endpoint, the following statistical hypotheses will be used.
H0 (null hypothesis): p_Test - p_Control ≥ 0.023 vs.
H1 (alternative hypothesis): p_Test - p_Control < 0.023,
where p_Test and p_Control are the proportion of patients requiring treatment-emergent assisted ventilation during the study observation period for the Test and Control groups, respectively. The non-inferiority test will be assessed by comparing the upper bound of the two-sided 95% exact confidence interval for the treatment difference (Test - Control) in the proportions with the non-inferiority margin of 0.023.
The proportions of patients with any ARDS by the Berlin criteria, any CSPAE, any AE, any TR, and any SAE will also be compared between treatment groups using the two-sided Fisher’s exact test with a significance level of 0.05.
The median time to assisted ventilation from the first study PC transfusion within each study period will be estimated using the Kaplan-Meier method, and the treatment difference will be explored by the log-rank test.
Baseline data on primary disease (e.g., types of HSCT received) will be considered as covariates for association analysis.
The proportion of INTERCEPT PC containing platelet doses ≥ 3.0 ×1011 platelets will be summarized. Patient demographics, exposure to study PCs, and primary indication for transfusion will be summarized.
A progress report will be submitted to the FDA every six months until the completion of the study.

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 37
XIII. PANEL MEETING RECOMMENDATION AND FDA’S POST-PANEL ACTION
A panel meeting was not convened based on an assessment that FDA did not require additional external expert advice on review of the submitted data in the absence of identification of new issues since the BPAC discussion in 2009.
XIV. BENEFIT-RISK CONCLUSIONS
Emerging infectious diseases can have a major impact on the safety of platelet transfusions, especially when there are no FDA-approved screening tests for the disease. For example, in 2002 “[t]wenty-three patients were confirmed to have acquired WNV through transfused leukoreduced and non-leukoreduced red cells, platelets, or fresh-frozen plasma.” Eight of those cases involved platelet transfusions. Even after mini-pool nucleic acid testing of blood donations for WNV was introduced in 2003, 12 transfusion-transmitted cases have been reported. (MMWR, 2003) This includes a fatal case of WNV that was linked to a platelet transfusion in 2012. The use of IBS processed platelets will likely reduce the risk of platelet related transfusion-transmitted infections that are not detected with screening tests.
Dengue Fever, Chikungunya, and other possible emerging infectious diseases (EID) could become endemic in the continental United States and already are endemic in Puerto Rico. While blood screening tests for these EIDs may eventually be available to minimize the potential for transfusion-transmitted EIDs, the availability of IBS processed platelets and other developing technologies would provide an alternative or additive safeguard to donor testing. The difficulty in setting a quantitative benefit for the ability to mitigate many DNA/RNA-based EIDs is that the future number of cases cannot be estimated with much certainty. While the number of future cases avoided by use of IBS processed platelets is unknown, the availability of the technology is a decided benefit. There is the added, but small, advantage that IBS processed platelets also could mitigate window period contamination of platelets by pathogens for which donors are tested.
ARDS and sepsis are both serious adverse events that often cause death. There have been improvements in care of both ALI and ARDS since the SPRINT study was conducted. The rate of sepsis related to contaminated platelets has gone down as testing procedures identify contaminated doses more effectively. The rate of hospital acquired ARDS in critically ill patients has apparently dropped in half from 0.08% to 0.04% since the late 90’s when the SPRINT trial was conducted. The improvement in ARDS rates is likely due to advances in the diagnosis and treatment. The mortality of ARDS has remained constant over time approximately 40%, despite the reduction of hospital acquired ARDS in critically ill patients.
There is uncertainty about the validity of both the ARDS safety signal from the clinical trial and the more recent results from the observational studies. Some of the cases may have been misclassified, as suggested by the post-hoc re-analysis of the data.

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 38
The coverage properties for the various methods of estimating confidence intervals when the probability of the event is very small and the sample size is small (approx. 350) are poor and a risk difference of 1.6% could be observed by chance. A simulation was conducted where two binomial variables were set to an identical probability of 0.8% (which approximates the rate of ALI in the early 2000’s generated 10 million iterations, and calculated the risk difference. The risk difference was greater than 1.6% in 2.1% of the simulations. The risk difference was greater than 1.26% (4/318) in over 5.5% of the simulations. This is consistent with the reported confidence interval.
The French and Swiss national hemovigilance data have their strengths and limitations. While there is uncertainty, the national hemovigilance data are not consistent with a risk difference of 1.6% per patient and 0.19% per transfusion. All of the ARDS cases in the SPRINT trial occurred within 24 hours of transfusion, and an increased risk of 1.6% per patient could have generated 420 cases (242 + 179) of ARDS potentially within 24 hours of transfusion in the Swiss and French hemovigilance data. Assuming a 40% mortality rate for ARDS, this would have caused more than 160 excess deaths among IBS processed platelet recipients. This compares to six reported cases of TRALI among the 187,142 IBS processed platelet transfusions in France and Switzerland since 2009. While we expect some underreporting of ARDS, this would be a relatively large signal to miss. The national hemovigilance data cannot rule out a small increased risk for ARDS given the difficulties in linking platelet transfusions with cases of ARDS.
The patients in the SPRINT trial were all hematology-oncology patients, so there is some uncertainty about generalizing from the SPRINT results to the general population of platelet transfusion recipients. In particular, if the increased risk of ARDS seen in the SPRINT trial is restricted to hematology-oncology patients and to those who received large number of platelets units, the ARDS risk in the general population would be smaller and hemovigilance results would be less likely to detect the smaller increase in risk.
There is less uncertainty about the benefit of reduced bacterial infections and sepsis. Currently there are approximately 200 cases of sepsis caused by platelet transfusions reported every year, and some of those cases cause death. In vitro testing with pathogen-spiked platelets has shown that the IBS platelets have significantly reduced pathogen counts and limited clinical trials and hemovigilance data are consistent with the IBS platelets claimed effect of reducing transfusion transmitted bacterial infections. The French and Swiss observational data show no cases of bacterial infection in recipients receiving the IBS platelets compared to a combined rate of 0.30 per 10,000 recipients who received conventional platelets.
It is not known whether the next EID will spread to the U.S. or how many platelet transfusion-transmitted cases could be prevented if the IBS platelets are on the market. Several EIDs, including Dengue and Chikungunya are currently epidemic in

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 39
areas geographically close to the continental U.S. and could become endemic in the U.S. WNV is a recent example that caused several platelet transfusion- transmitted cases.
There is evidence that IBS reduces T cell numbers and thereby lowers the risk of TA-GVHD. The risk of TA-GVHD is very low since the use of gamma irradiation. Whether IBS can effectively replace gamma irradiation is not known.
The recovery and survival of IBS platelets are lower than conventional platelets. This probably explains the finding of increased platelet requirements in subjects receiving IBS compared to control platelets, though there was also underdosing with IBS processed platelets due to platelet losses in processing. However, it is likely that this deficiency can be overcome by increasing the platelet dosage to at least 3 x 1011.
In conclusion, the known benefits, mainly of reducing bacterial infection including sepsis, outweigh the risks, which are somewhat uncertain, particularly the pulmonary adverse events.
XV. CONCLUSIONS DRAWN FROM PRECLINICAL AND CLINICAL STUDIES
A. Effectiveness Conclusions
The efficacy of IBS processed platelets was evaluated using clinical parameters such as CI and/or CCI and hemostatic efficacy in thrombocytopenic patients. The mean 1- and 24-hr CI and CCI following administration of IBS processed platelets were lower compared to conventional platelets, likely implying reduced quality of the IBS platelets. Nevertheless, the US pivotal study (SPRINT) met its efficacy primary endpoint that assessed hemostatic efficacy (proportion of patients with grade 2 bleeding). Although statistically significant differences were found in some secondary hemostatic endpoints in favor the control group, the differences were not considered clinically significant.
B. Safety Conclusions
The significance of the main safety concern of ARDS seen in the SPRINT study remains uncertain. The postmarketing studies and hemovigilance data are not able to confirm or refute the signal observed in the SPRINT RCT, though they suggest that the true rate of ARDS with IBS prepared platelets may be lower than was seen in the SPRINT study. The apparent risk of ARDS will be noted in labeling and this or other potential safety concerns will be evaluated in a postmarketing period and site controlled observational study.
XVI. OVERALL CONCLUSIONS
The data in this application support the reasonable assurance of safety and effectiveness of this device when used in accordance with the indications for use.

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 40
XVII. CBER DECISION
CBER issued an approval order on December18, 2014. The final conditions of approval cited in the approval order are described below.
The applicant’s manufacturing facility(ies) has/have been inspected and found to be in compliance with the device Quality System (QS) regulation (21 CFR 820).
XVIII. APPROVAL SPECIFICATIONS
Directions for use: See device labeling.
Hazards to Health from Use of the Device: See Indications, Contraindications, Warnings, Precautions, and Adverse Events in the device labeling.
Post-approval Requirements and Restrictions: See approval order.
XIX. REFERENCES 1. Schmidt M et al. Efficiency of the Pathogen Inactivation System INTERCEPT under
Experimental Conditions. ISBT 2011 Lisbon, Portugal. 2. Nussbaumer W et al. Transfusion 2007;47:1125-1133. 3. (AABB 2011). Standards for Blood Banks and Transfusion Services. Process Control.
Bethesda, MD, AABB. 5.1.5.1.). 4. Wollowitz, S. Fundamentals of the psoralen-based Helinx technology for inactivation of
infectious pathogens and leukocytes in platelets and plasma. Semin Hematol 2001 Oct;38(4 Suppl 11):4-11.
5. Lin L, Hanson CV, Alter HJ, Jauvin V, Bernard KA, Murthy KK, Metzel P, Corash L. Inactivation of viruses in platelet concentrates by photochemical treatment with amotosalen and long-wavelength ultraviolet light. Transfusion 2005;45:580-590.
6. Dupuis K, Arnold DJ, Sawyer L. 2012. Transfusion 52(3S):225A 7. Tsetsarkin KA, Sampson-Johannes A, Sawyer L, Kinsey J, Higgs S, Vanlandingham DL.
Photochemical inactivation of chikungunya virus in human apheresis platelet components by amotosalen and UVA light. Am J Trop Med Hyg. 2013;88(6):1163-9.
8. Sawyer L, Dupuis K, et al. Inactivation of influenza A H5N1 and Lymphocytic Choriomenigitis virus (LCMV) by the INTERCEPT Blood System (IBS). Transfusion 2008;48(2S): 88A.
9. “>” refers to inactivation below the limit of detection of the assay; “≥” refers to inactivation equal to or below the limit of detection of the assay.
10. Lin L, Dikeman R, Molini B, Lukehart SA, Lane R, Dupuis K, Metzel P, Corash L. Photochemical treatment of platelet concentrates with amotosalen and long-wavelength ultraviolet light inactivates a broad spectrum of pathogenic bacteria. Transfusion 2004;44:1496-1504.
11. Grellier P, Benach J, Labaied M, Charneau S, Gil H, Monsalve G, Alfonso R, Sawyer L, Lin L, Steiert M, Dupuis K. Photochemical inactivation with amotosalen and long-

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 41
wavelength ultraviolet light of Plasmodium and Babesia in platelet and plasma components. Transfusion. 2008;48:1676-84.
12. Van Voorhis WC, Barrett LK, Eastman RT, Alfonso R, Dupuis K. Trypanosoma cruzi Inactivation in Human Platelet Concentrates and Plasma by a Psoralen (Amotosalen HCl) and Long-Wavelength UV. Antimicrobial Agents and Chemotherapy 2003;47:475–479.
13. Eastman RT, Barrett LK, et al. Leishmania inactivation in human pheresis platelets by a psoralen (amotosalen HCl) and long-wavelength ultraviolet irradiation. Transfusion 2005;45(9): 1459-1463.
14. Corash L, Lin L. Novel processes for inactivation of leukocytes to prevent transfusion-associated graft-versus-host disease. Bone Marrow Transplant 2004;33: 1-7.
15. Lin L, Corash L, Osselaer J. Protection against TA-GVHD due to platelet transfusion by using pathogen inactivation with the INTERCEPT blood system gamma irradiation is not the only answer. Haematologica 2010;95, Extra 1: 230-7.
16. Hei DJ, Grass J, Lin L, Corash L, Cimino G. Elimination of cytokine production in stored platelet concentrate aliquots by photochemical treatment with psoralen plus ultraviolet A light. Transfusion 1999;39: 239-48.
17. Grass JA, Hei DJ, Metchette K, Cimino GD, Wiesehahn GP, Corash L, Lin L. Inactivation of leukocytes in platelet concentrates by photochemical treatment with psoralen plus UVA. Blood 1998;91: 2180-8.
18. Osselaer JC, Cazenave JP, Lambermont M, Garraud O, Hidajat M, Barbolla L, Tardivel R, Defoin L, Waller C, Mendel I, Raidot JP, Kandel G, De Meuter R, Fabrigli P, Dehenau D, Arroyo JL, Padrón F, Gouezec H, Corral M, Jacquet M, Sundin D, Lin L, Corash L. An active haemovigilance programme characterizing the safety profile of 7437 platelet transfusions prepared with amotosalen photochemical treatment. Vox Sang 2008;94(4):315-323.
19. Osselaer JC, Messe N, Hervig T, Bueno J, Castro E, Espinosa A, Accorsi P, Junge K, Jacquet M, Flament J, Corash L.. A prospective observational cohort safety study of 5106 platelet transfusions with components prepared with photochemical pathogen inactivation treatment. Transfusion 2008;48(6): 1061-1071.
20. Snyder E, McCullough J, et al. Clinical safety of platelets photochemically treated with amotosalen HCl and ultraviolet A light for pathogen inactivation: the SPRINT trial. TRANSFUSION 2005;45:1864-1875.
21. Agence Francaise de Securite Sanitaire des Produits de Sante, 2009. 22. Jutzi M and Amsler L (2014). Swissmedic Haemovigilance Annual Report 2013. Lucerne,
Switzerland, Swissmedic. 23. van Rhenen D, Gulliksson H, Cazenave JP, Pamphilon D, Ljungman P, Klüter H, Vermeij
H, Kappers-Klunne M, de Greef G, Laforet M, Lioure B, Davis K, Marblie S, Mayaudon V, Flament J, Conlan M, Lin L, Metzel P, Buchholz D, Corash L. Transfusion of pooled buffy coat platelet components prepared with photochemical pathogen inactivation treatment: the euroSPRITE trial. Blood 2003;101:2426-2433.
24. McCullough J1, Vesole DH, Benjamin RJ, Slichter SJ, Pineda A, Snyder E, Stadtmauer EA, Lopez-Plaza I, Coutre S, Strauss RG, Goodnough LT, Fridey JL, Raife T, Cable R, Murphy S, Howard F 4th, Davis K, Lin JS, Metzel P, Corash L, Koutsoukos A, Lin L, Buchholz DH, Conlan MG.. Therapeutic efficacy and safety of platelets treated with a photochemical process for pathogen inactivation: the SPRINT Trial. Blood 2004;104(5):1534-1541.

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 42
25. Janetzko K, Cazenave JP, Klüter H, Kientz D, Michel M, Beris P, Lioure B, Hastka J, Marblie S, Mayaudon V, Lin L, Lin JS, Conlan MG, Flament J.. Therapeutic efficacy and safety of photochemically treated apheresis platelets processed with an optimized integrated set. Transfusion 2005;45(9):1443-1452.
26. Ciaravino V, McCullough T, et al. The role of toxicology assessment in transfusion medicine. Transfusion 2003;43(10): 1481-1492.
27. Ciaravino V, McCullough T, Dayan AD. Pharmacokinetic and toxicology assessment of INTERCEPT (S-59 and UVA treated) platelets. Human and Experimental Toxicology 2001;20:533-550.
28. Ciaravino V, Hanover J, Lin L, Sullivan T, Corash L. Assessment of safety in neonates for transfusion of platelets and plasma prepared with amotosalen photochemical pathogen inactivation treatment by a 1-month intravenous toxicity study in neonatal rats. Transfusion 2009;49:985-994.
29. Tice RR, Gatehouse D et al. The pathogen reduction treatment of platelets with S-59 HCl (Amotosalen) plus ultraviolet A light: Genotoxicity profile and hazard assessment. Mutation Research Mutation Research 2007: 630; 50–68.
30. Snyder E, Raife T, Lin L, Cimino G, Metzel P, Rheinschmidt M, Baril L, Davis K, Buchholz DH, Corash L, Conlan MG.. Recovery and Lifespan of 111 Indium radiolabeled platelets treated with pathogen inactivation using amotosalen HCl (S-59) and UVA light. Transfusion 2004; 44:1732-1440.
31. Corash, L,Paton V, et al. S-59 clearance and kinetics after transfusion of platelets treated with Helinx™ Technology. Transfusion 2000;40(S10):137.
32. Slichter SJ, Raife TJ, Davis K, Rheinschmidt M, Buchholz DH, Corash L, Conlan MG. Platelets photochemically treated with amotoslaen HCl and ultraviolet A light correct prolonged bleeding times in thrombocytopenic patients. Transfusion 2006;46:731-740.
33. Slichter SJ, Kaufman RM, Assmann SF, McCullough J, Triulzi DJ, Strauss RG, Gernsheimer TB, Ness PM, Brecher ME, Josephson CD, Konkle BA, Woodson RD, Ortel TL, Hillyer CD, Skerrett DL, McCrae KR, Sloan SR, Uhl L, George JN, Aquino VM, Manno CS, McFarland JG, Hess JR, Leissinger C, Granger S. Dose of prophylactic platelet transfusions and prevention of hemorrhage. N Engl J Med 2010;362:600-613.
34. Schlenke P, Hagenah W, Irsch J, Sundin D, Corash L, Lin L, Kirchner H, Wagner T.. Safety and clinical efficacy of platelet components prepared with pathogen inactivation in routine use for thrombocytopenic patients. Ann Hematol 2011;90(12):1457-1465.
35. Infanti L, Stebler C, Job S, Ruesch M, Gratwohl A, Irsch J, Lin L, Buser A.. Pathogen-inactivation of platelet components with the INTERCEPT Blood System : a cohort study. Transfus Apher Sci 2011;45(2):175-181.
36. Sigle JP, Infanti L, Studt JD, Martinez M, Stern M, Gratwohl A, Passweg J, Tichelli A, Buser AS.. Comparison of transfusion efficacy of amotosalen-based pathogen-reduced platelet components and gamma-irradiated platelet components. Transfusion 2013;53(8):1788-1797.
37. Sweeney J, Lozano M. Platelet Transfusion Therapy. Bethesda: AABB Press, 2013. 38. French National Agency for Medicine and Health Product Safety/ANSM, Hemovigilance
Activity Report, 2009. 39. French National Agency for Medicine and Health Product Safety/ANSM, Hemovigilance
Activity Report, 2010.

PMA BP140143: FDA Summary of Safety and Effectiveness Data Page 43
40. French National Agency for Medicine and Health Product Safety/ANSM, Hemovigilance Activity Report, 2011.
41. Cazenave JP, Isola H, et al. (2013). Pathogen Inactivation of Platelets. Platelet Transfusion Therapy. J. D. Sweeney and M. Lozano. Bethesda, MD, AABB Press: 119-176.
42. SwissMedic Haemovigilance Annual Report, 2010. 43. SwissMedic Haemovigilance Annual Report, 2011. 44. SwissMedic Haemovigilance Annual Report, 2012. 45. Osselaer JC, Doyen C, Defoin L, Debry C, Goffaux M, Messe N, Van Hooydonk M,
Bosly A, Lin JS, Lin L, Corash L. Universal adoption of pathogen inactivation of platelet components: impact on platelet and red blood cell component use. Transfusion 2009;49(7):1412-1422.
46. Cazenave JP, Isola H, Waller C, Mendel I, Kientz D, Laforêt M, Raidot JP, Kandel G, Wiesel ML, Corash L. Use of additive solutions and pathogen inactivation treatment of platelet components in a regional blood center: impact on patient outcomes and component utilization during a 3-year period. Transfusion 2011;51(3):622-629.
47. Pelszynski MM, et al. Blood. 1994;83:1683-9. 48. Luban NL, et al. Transfusion 2000;40:348-352. 49. Lin L, et al. Haematologica 2010;95,Extra 1:230-237. 50. Roback JD et al. Transfusion 2007a;47(S3):23A. 51. Roback JD et al. 2007b;Blood,110(11):849A. 52. Jordan CT et al. Transfusion 2004;44:1159-1165. 53. Lin L., Seminars in Hematology 2001;38:27-33.] 54. aabb (2014). Standards for Blood Banks and Transfusion Services 29th Edition. Process
Control. Bethesda, MD, AABB. 5.1.5.1. 55. Corash, L., J. S. Lin, et al. (2011). "Determination of acute lung injury after repeated
platelet transfusions." Blood 117(3): 1014-1020. 56. Janetzko, K., J. P. Cazenave, et al. (2005). "Therapeutic efficacy and safety of
photochemically treated apheresis platelets processed with an optimized integrated set." Transfusion 45(9): 1443-1452.
57. Snyder, E., J. McCullough, et al. (2005). "Clinical safety of platelets photochemically treated with amotosalen HCl and ultraviolet A light for pathogen inactivation: the SPRINT trial." Transfusion 45(12): 1864-1875.
58. Snyder, E., T. Raife, et al. (2004). "Recovery and Lifespan of 111 Indium radiolabeled platelets treated with pathogen inactivation using amotosalen HCl (S-59) and UVA light." Transfusion 44: 1732-1440.