summer students reports 2013
DESCRIPTION
Read the reports from students who undertook a Summer Vacation Studentship in 2013.TRANSCRIPT

Student:Aaron HoyleInvestigating the involvement of programmed cell death in seed viability loss duringartificial ageingSupervisor - Dr Louise Colville, Biochemist, Royal Botanic Gardens, Kew
Background: Anthropogenic climatechange and habitat degradation hasalready had profound impacts onnumerous plant species, leading todistribution shifts, population declinesand ultimately extinction (Thomas 2004).Kew’s Millennium Seed Bank’s (MSB) aimto conserve plant life is achieved byspecialisation in seed storage whichoccurs under specific conditions (-20°C,Relative Humidity (RH) 15%). Currently,the MSB has successfully banked nearly 2billion seeds representing over 30,000species from all continents. This collection
holds vast potential for agriculture, medicine and climate change mitigation. However, seed longevity inthe bank is not indefinite with seeds becoming inviable with time, consequently unusable for conservation.Studies into seed ageing have revealed that programmed cell death (PCD) may be important in the loss ofseed viability.
Aims: The objective of this study is to determine the role of PCD in the loss of seed viability in the modelplant species Pisum sativum.
Protocols:Treating and artificial ageing of seedsP. sativum seeds were initially treated with a range of inhibitors associated with caspase and proteasomeactivity. MG132 (50µM, 100µM) and Epoxomicin (1µM) both proteasome inhibitors, Ac-YVAD-CHO (50µM)a caspase inhibitor and DMSO (1%) a control. Post treatment seeds were artificial ageing at 60% RH and60°C for 0 days and 2 days. Seed viability was assessed via germination testing.
StainsPropidium iodide/Fluorescein diacetate (PI/FDA) - displays cell viability, staining the nuclei of inviablecells red and the cytoplasm of viable cells green (only visible under UV light), a solution containing
gml ¹ PI (dissolved and diluted in PBS) and 25 gml ¹ FDA (dissolved in acetone and diluted in PBS).Diaminobenzidine (DAB) - detects hydrogen peroxide by turning brown, a 5mM solution was used(dissolved and diluted in PBS).Nitroblue tetrazolium (NBT) - detects superoxides by turning dark purple, a 3mM solution was used(dissolved and diluted in PBS).
Staining ProceduresPrior to staining of the samples, trials were performed optimizing the procedure by experimenting withstain concentrations, PBS incubation time, stain incubation time and post staining PBS wash. Hand cutsections of P.sativum seeds were placed into 2ml eppendorf tubes containing 500 l PBS to preventdesiccation, before staining PBS was removed and 500 l of stain added. For each treatment sections weretaken from the same seed and all three stains applied. Photographs were taken at the tip of the axis, axisbase and at the edge of the cotyledon. At each point, 3 photographs were taken; brightfield (Exposure(E):1/3rdsec), triple filter (E: 1sec) and green filter (E: 4sec).

Student:Aaron Hoyle
Fig 1. Photograph taken with the triple filter at the edge ofthe cotyledon, showing green alive cells and red dead cells.
Protein Extraction and Caspase Activity Assay100 mg of ground seed was mixed with 1ml of extraction buffer and centrifuged. The pellet was discardedand the protein content was determined using the Bradford assay. This was then used in the caspase-3activity assay; 15µg protein was used in the assay alongside a fluorogenic caspase substrate.
DNA Extraction and Gel ElectrophoresisDNA was extracted from 100mg of ground seed using a standard protocol for DNA extraction. DNA wasthen quantified spectrophotometrically, the ratio of A260/A230 was used to check for polysaccharide andprotein contaminations and the ratio of A260/A280 to check for DNA purity. Gel electrophoresis was thenperformed using 50µg DNA. The gel was then imaged and analysed using GeneSnap software.
Results: After analysis, our findings opposed what wasexpected; the inhibitors had little effect on seed viabilityand vigour after artificial ageing. Therefore we canconclude that PCD was not having pronounced effectsunder the artificial ageing conditions. Consequently, theresults from the other protocols did not revealdifferences in accumulation of ROS (NBT and DABstaining), DNA laddering or caspase activity between thetreatments, indicating that cell death under theseconditions may involve necrosis rather than PCD.Interestingly, previous studies (Kranner et al., 2006, Huet al., 2012) have demonstrated that use of suchinhibitors can reduce seed viability loss. However, inthese studies the artificial ageing was carried out at a lower temperature (50°C) indicating that the type ofcell death occurring may be dependent on ageing conditions.
Future Studies: The experiment is going to be repeated with different artificial ageing conditions (60% RH,50°C), using the protocols refined during my studentship.
Value of the Studentship:Student: Prior to this two month studentship my laboratory experience consisted of 3-6 hours a week atuniversity. I have seen vast improvements in my general laboratory practice, protein assay technique,stoichiometry, microscopy skills and I have acquired new skills including; DNA extraction, quantificationand use of gel electrophoresis. My time at the MSB has been an invaluable experience and will shapedecisions I will make later in life including; further education and career choices. Therefore, I would like tothank the Biochemical Society for making such an opportunity available.
Lab: Aaron has made great progress with this project. He has developed and optimised protocols forviability and ROS staining of seed sections and DNA gel electrophoresis for visualising DNA laddering, andalso tested procedures for caspase activity assays. This has been a tremendous help and will form the basisof further studies in this area.
References:Hu, D., Ma, G., Wang, Q., Yao, J., Wang, Y., Pritchard, H.W. & Wang, X. (2011) Spatial and temporal nature of reactive oxygen speciesproduction and programmed cell death in elm (Ulmus pumila L.) seeds during controlled detioration. Plant, Cell and Environment, 11, 2045-2059.Kranner, I., Britic, S., Anderson, K.M., Pritchard, H.W. (2006) Glutathione half-cell reduction potential: a universal stress marker and modulatorof programmed cell death? Free Radical Biology and Medicine, 40, 2155-2165.Thomas, C.D., Cameron, A., Green, R.E., Bakkenes, M., Beaumont, L.J., Collingham, Y.C., Erasmus, B.F.N., Siqueira, M.F., Grainger, A., Hannah,L., Hughes, L., Huntley, B., Jaarsveld, A.S., Midgley, G.F., Miles, L., Ortega-Huerta, M.A., Peterson, A.T., Phillips, O.L. & Williams, S.E. (2004)Extinction risk from climate change. Nature, 427, 145-148.

1
Biochemical society summer studentship
Investigating the effect of Aβ on tau distribution in neuroblastomas.
Alex Foley, Youssra Al-Hilaly, Julian Thorpe, Louise Serpell
Affiliation: School of Life Sciences University of Sussex, Falmer, BN1 9QG
Email: [email protected]

2
INTRODUCTION Alzheimer’s disease (AD) is a crippling neurodegenerative disease that affected 26.6 million people worldwide in 2006, and is projected to affect 1 in 85 people by 2050 (Brookmeyer, 2007). Treatment on this scale is clearly unsustainable, making Alzheimer’s a critical disease for research. Alzheimer’s disease was first identified as a separate pathology from other forms of presenile dementia by Alois Alzheimer in 1907 (Alzheimer, 1907). Alzheimer described senile plaques and neurofibrillary tangles, protein aggregates now known to be hallmarks of the disease. Neurofibrillary were discovered to comprise paired helical filaments (Kidd, 1963) of the protein tau (Kosik et al., 1986) and the plaques were shown to be made up of the β-amyloid protein (Aβ) (Glenner and Wong, 1984b). Central to Alzheimer’s disease pathogenesis is the amyloid cascade hypothesis that states that Aβ aggregation is the initial insult to the cell in Alzheimer’s, and that the rest of the pathogenesis stems from this event (Hardy & Selkoe, 2002). It appears tau may also have a neuroprotective role before it contributes to neurodegeneration. This protection seems to depend on a nuclear translocation. Overexpressing tau in cells and, in turn, increasing the amount of hyperphsophorylated tau has been shown to antagonise the phosphorylation of β-catenin by GSK-3, allowing β-catenin to translocate to the nucleus, where it activates pro-survival factors (Li et al., 2007). Beyond this function as an indirect nuclear transcription regulator, tau has been shown to exist in a small amount in the nuclei of cells of both normal and AD brains (Brady et al., 1995), and that reversible accumulation of nuclear tau occurs in response to oxidative stress and heat shock and confers protection to DNA from heat stress-induced damage (Sultan et al., 2011). The current study aimed to determine whether Aβ treatment induces nuclear relocalisation of tau from a mainly cytoplasmic protein to a nuclear protein. Previous pilot work has shown that neuroblastoma cells treated with 10μM oligomeric Aβ are damaged and that tau is found to be enriched in the nucleus. Here we have examined the effect of a much lower concentration of Aβ (1μM) and have utilised immunogold labelling to follow the changes in the amounts of tau found within cells. Understanding tau’s role in locations other than the axon is vital in the quest to find a cure for the disease, as it may well be that a usually neuroprotective function is being high jacked in disease conditions, resulting in misfolded tau propagation and cell death. METHODS & MATERIALS Sample Preparation Aβ42 HFIP (1,1,1,3,3,3-hexafluoro-2-propanol), >97% purity, and undifferentiated human neuroblastoma SH-SY5Y cells, provided by Ms. M. Stewart, were acquired and prepared in accordance with the procedure detailed in Soura et al. (2012). The cells were then seeded in 12-well plates (density: 3x105 cells/well) and subsequently treated with either buffer only or Aβ42 for 24 or 48 hours. Treated cells were then pelleted and fixed in resin, and immunogold labelled using a polyclonal anti-tau antibody (1:10 dilution) using the methods described in Soura et al. (2012). TEM Imaging These samples were then examined in a Hitachi-7100 transmission electron microscope at 100kV. The images were captured digitally on ImageJ via an axially mounted (2000x2000 pixel) Gatan Ultrascan 1000 charge-coupled device camera (Gatan UK). ‘Mugshot’ images of the cells (chosen at random) were taken at 1k x magnification, and then random sections of the cell nucleus and the cytoplasm were photographed at 20k x magnification. RESULTS Electron micrographs were inspected and the numbers of gold particles were counted to examine the localisation of tau. 20 images were examined for each condition. Figure 1 shows the mean number of gold particles per mm2 for each of the untreated versus those cells treated with 1 µM Aβ(1-42) found in the nucleus or cytoplasm The results revealed a significant difference between the 48 hour untreated and 48 hour 1μM Aβ nuclear samples (p<.05). Figure 1 shows that nuclear tau was greater in the untreated cells than in the treated cells at the 24 hour incubation, but that this was reversed at the 48 hour mark. Cytoplasmic tau levels also increased from 24 hours of treatment to 48 hours, but this difference was not significant.

3
DISCUSSION The results of the current study echo the findings of previous pilot work in the Serpell lab, which showed that treatment of neuroblastomas with 10 μM oligomeric Aβ resulted in an increase in nuclear tau (unpublished data, Grant Butcher & Nathan Hill). However, this study was performed with significantly lower concentrations of Aβ, and the increased nuclear localisation of tau occurred over a longer time period (48 hours). Accordingly, it was only in the 48 hour samples that a significant increase in nuclear tau levels was observed in the current study, whilst the data obtained with 10 μM oligomeric Aβ treated cells showed significant increases after 24 hour incubation. This suggests Aβ exerts its effect on tau in a dose dependent manner. The 24 hour untreated result is believed to be extraneous, and it is thought that with a larger sample, the result would fit the trend better. That cytoplasmic tau also increases after 48 hours as well as nuclear tau suggests increased transcription of tau, but further research will be conducted to verify this. References Alzheimer, A. (1907).. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-Gerichtisch Medizin, 64, 146-148. Brookmeyer, et al. (2007). Alzheimer’s & Dimentia, 3(3), 186-191. Glenner, G.G., & Wong, C.W. (1984). BBRC, 120, 885-890 Hardy & Selkoe (2002).. Science, 297, 353-356. Kidd, M. (1963). Nature, 197, 192-193. Kosik, K.S., et al. (1986). PNAS USA, 83, 4044-4048. Li, et al., (2007).. PNAS USA, 107(9), 3591-1596. Soura, et al.,. (2012). Biochemical Journal, 441, 579-590. Sultan, et al. (2011).. The Journal of Biological Chemistry, 286(6), 4566-4575.

Biochemical Society Summer Internship 2013 Report
Heparan sulfate disaccharide analysis using fluorescence labellingand High Performance Liquid Chromatography
Student: André Lavin Supervisor: Dr Andrew Powell
Background and Aims
Heparan sulphate (HS) is a linear sulfated polysaccharide thathas been shown to interact with more than 400 humanproteins (Ori, et al 2011). Its structure is highly heterogeneousand dynamic (Lindahl, Li, 2009), which makes it a difficultmolecule to accurately quantify and analyse. The structure ofHS is made of repeating diverse disaccharides (Table 1). Theabundance and sequence of these disaccharides have beenshown to influence temporal, spatial and pathologicallydynamic interactions of HS (Shriver et al, 2012).
High performance liquid chromatography (HPLC) is adopted toenable disaccharide compositional analysis of HS (Deakin,Lyon, 2008; Skidmore et al, 2010). The aim of this study is todevelop a Reverse Phase (RP)-HPLC protocol for HSdisaccharide analysis using disaccharide labelling with thefluorophore Borodipyromethene (BODIPY) to increasesensitivity (Skidmore et al, 2010). RP-HPLC would enable in-line mass spectrometry analysis (Galeotti & Volpi, 2011) andremove maintenance and efficiency issues surrounding stronganionic exchange (SAX)-HPLC which requires elution with agradient of sodium chloride.
Results and Discussion
SAX-HPLC analysis of BODIPY labelled HS disaccharides.
The resulting study found that the SAX-HPLC protocol(Skidmore et al, 2010) was successful and reproduciblewith clear peaks that could be readily assigned (Figure 1).
Thin layer chromatography (TLC) sample purification
A method of TLC was adapted to remove uncongugatedBODIPY from the labelled HS disaccharide standards. Thisreduced the amount of free BODIPY (Figure 2), henceenabling the wash phase to be shortened before elution ofHS disaccharides in the gradient phase.
Table1. Unit formula and degree of sulfation ofHS disaccharide reference standards.
Figure1. Comparative analysis of SAX-HPLC runs of BODIPYlabelled HS disaccharide standards 1-8 .
Figure2. Comparative analysis of pre and post TLC protocolfor free BODIPY reduction, with pre TLC denoted in RED andpost TLC denoted in BLUE.

Biochemical Society Summer Internship 2013 Report
RP-HPLC analysis of BODIPY labelled HS disaccharides
BODIPY labelled HS disaccharide reference standards wereanalysed by RP-HPLC using several different solventgradients (Figure 3). The approach was successful inseparating peaks (Figure 3A) based upon the HSdisaccharide level of sulfation (Table1). Lengthening thegradient improved resolution of structural isomers(disaccharides 4, 5, & 8 and 2, 3 and 7: Figure 3A) but didnot achieve complete resolution (Figure 3B).
RP-HPLC analysis of AMAC labelled HS disaccharides
To determine if this was a BODIPY or a column issue thefluorophore 2-aminoacridone (AMAC) was adopted as thishas been shown to be successful by Deakin and Lyon (2008).However labelled HS disaccharides and unconjugatedAMAC were found to elute together and peaks could not bedistinguished (data not shown). As unconjugated BODIPYdid not elute with labelled HS reference disaccharides thissuggests that method development should be continued.
Future Directions
This study provided credence that SAX-HPLC in conjunctionfluorescent label BODIPY is still the most consistentapproach to disaccharide analysis and in the short termshould be applied to biological samples which will help toconfirm its robustness and generate novel disaccharidecomposition data linked to biological function. However,development of RP-HPLC was promising and should becontinued via testing
Even shallower elution gradientsIon-pairing RP-HPLCColumn specifications - Column length, capacity,particle size, pore size and hydrophobicityAlternate solvents. e.g Acetonitrile
Value of Studentship
Student
The studentship experience enhanced my laboratory, problem solving and method development skills andenabled me to become an expert on the operation and maintenance of an HPLC and analysis ofchromatograms using HPLC software and Excel. During the studentship I worked alongside a graduatedeveloping methodology for purification of heparan sulfate from urine and conditioned cell culture media. Iattended a conference at the University of Liverpool with speakers from around the UK and abroad where Imet PhD’s, Post Doc’s and principal investigators from the University of Liverpool and Keele who are world-leading in HS chromatography and analysis and had a tour of the University of Liverpool laboratories andequipment.
Laboratory
My time spent in Dr Powell’s Laboratory helped to further embed BODIPY labelled disaccharide analysis withSAX-HPLC. I also generated positive preliminary data on a novel methodology for BODIPY labelled disaccharideanalysis using RP-HPLC for a manuscript, exploitation of the departmental autosampler-driven HPLC suite andincorporation with laser induced fluorescence equipment development at Keele University.
Figure3. (A) RP-HPLC analysis of BODIPY labelled HS disaccharidereference standards 1-8 with varying elution gradients.(B) Comparative zoomed in analysis of (A) with labelled HSstandards 2, 3 & 7 overlaid.

Biochemical Society Summer Internship 2013 Report
References
Galeotti F & Volpi N. (2011). Online reverse phase high performance liquid chromatography fluorescence detectionelectrospray ionisation mass spectrometry separation and characterisation of heparan sulfate, heparin and lowmolecular weight heparin disaccharides derivatized with 2-aminoacridone. Analytical chemistry. 83 (1), 6770-6777.
Lindahl U & Li JP. (2009). Interactions between heparan sulfate and proteins-design and functional implications.International review of cell and molecular biology. 276 (1), 105-159.
Ori A, Wilkinson MC & Fernig D. (2011). A systems Biology approach for the investigation of the Heparin/Heparansulfate interactome . J BIOL CHEM. 286 (22), 19892-19904.
Shriver ZN, Capila I, Ventrataraman G & Sasisekharan R. (2012). Heparin and Heparan Sulfate: Analyzing Structure andMicroheterogeneity. Handbook of Experimental Pharmacology. 207 (1), 159-176.
Skidmore MA, Guimond S, Dumax-Vorzet AF, Yates EA & Turnbull JE. (2010). Disaccharide compositional analysis ofheparan sulfate and heparin polysaccharides using UV or high sensitivity fluorescence (BODIPY) detection. Natureprotocols. 5 (12), 1983-1991.

Biochemical Society summer vacation studentship 2013
_______________________________________________________
Enzymes of Mycobacterial Glucan Metabolism
Andrii Gorelik, Biological Chemistry Department, John Innes Centre, Norwich, UK
Project leader: Dr. Stephen Bornemann
Background Until recently, it was thought that bacteria biosynthesise the glucose polymer glycogen (a type of glucan) as a carbon storage molecule using a well-established metabolic pathway involving the enzymes GlgC and GlgA. More recently it became apparent that the human pathogen Mycobacterium tuberculosis also makes a glycogen-like molecule that coats the outside of the bacterial cell to form a capsule and contributes to immune evasion. Furthermore, a third methylglucose lipopolysaccharide glucan is generated in mycobacteria. While the genes encoding the enzymes associated with this third pathway were identified a few years ago, it was only very recently that those likely to be associated with capsular glucan were identified. It turned out that not only was the key enzyme of this new pathway, GlgE, a potential drug target, but also that the emergence of resistance could be minimised by simultaneously blocking the pathway to methylglucose lipopolysaccharides. There remain many unanswered questions about the other enzymes of mycobacterial glucan metabolism and this project was about initiating studies to address some of these.
OtsA is a trehalose-6-phosphate synthase and catalyzes the reaction: NDP-α-D-glucose + α-D-glucose 6-phosphate <=> NDP + trehalose 6-phosphate
GlgC is an ADP-glucose pyrophosphorylase and catalyzes the reaction: α-D-glucose 1-phosphate + ATP <=> ADP-α-D-glucose + pyrophosphate
Rv3032 is predicted to be a glucosyltransferase that catalyzes the reaction: NDP-glucose + glucann <=> ADP + glucann+1
Aims The aim of this project was to investigate whether OtsA, GlgC or Rv3032 are inhibited by maltose 1-phosphate, a metabolic intermediate that builds up to toxic levels in glgE mutants. In addition, an understanding of the substrate specificity of Rv3032 has implications for its role in methylglucose lipopolysaccharide biosynthesis.
Description of work Genes encoding GlgC, OtsA and Rv3032 enzymes were sub-cloned from pUC57 into pET21a. Escherichia coli BL21(DE3)* cells were transformed using the resulting plasmids. Plasmid toxicity tests with glgC in pET21a were carried out by plating transformed cells onto LB agar containing either carbenicillin, carbenicillin with IPTG, IPTG or no additions. Cell cultures were put into expression trials on a 5 ml scale in 24-well racks containing IPTG, IPTG
with 1 or 2% ethanol, IPTG with 0.2 or 0.4% DMSO or no additions on three media; L, LB and AIM (auto induction medium). The trials were carried out at 37 and 18 °C in
incubators with shaking at 200 rpm. Soluble and insoluble protein fractions were analyzed using SDS-PAGE. The best conditions were scaled up to 4 × 1 L. Proteins were expressed, extracted and purified using Ni-affinity and, with OtsA, gel filtration chromatographies (Fig. 1). The glgC gene was amplified using Touchdown PCR and sub-cloned into the pBAD43 vector, expressed in Top10 E. coli cells, and protein was extracted and purified using the abovementioned
methods (Fig. 2). For enzyme assays, 50 mM Tris-HCl, at either pH 7 or 8, containing either 4 or 50 mM MgCl2, and 50 mM MES, pH 6, containing 50 mM MgCl2, were prepared. The activity and substrate specificity of the enzymes were
Fig. 1. OtsA enzymeat ~75 kDa after gelfiltration chromatography
Fig. 2. GlgC in pBAD43vector at ~57 kDapurified using Ni-affinitychromatography

measured using TLC, NMR spectroscopy and MALDI mass spectrometry. TLC was carried out with silica plates and a mobile phase of 85:20:50:50 acetonitrile:ethyl acetate:isopropanol:water. NMR assays contained 250 µl of buffer, 100 µl of enzyme, 10 µl of the first substrate, 10 µl of the second substrate, 130 µl of H2O and were analysed after 1, 24 or 72 h after the addition of 50 µl of D2O. MALDI mass spectrometry assays contained 25 µl of buffer, 10 µl of enzyme, 1 of the µl first substrate, 1 of the µl second substrate and 13 µl of H2O, and were incubated for 1 or 24 h.
Results OtsA was successfully produced (Fig. 1) using the pET21a vector according to mass spectrometry of the protein. According to TLC and NMR spectroscopy (Fig. 3), OtsA was most active at pH 8. It was specific for glucose 6-phophate and either ADP-glucose or UDP-glucose, and was not inhibited by maltose 1-phosphate. OtsA generated an unknown side-product according to the appearance of an unexpected doublet of doublets in NMR spectra.
A pET21a vector containing the glgC gene was toxic to E. coli but the equivalent pBAD43-based vector was not due to tighter control of gene expression, allowing the successful production of GlgC (Fig. 2) according to mass spectrometry of the protein and TLC of assays (Fig. 4). GlgC was most active at pH 6 and was specific for glucose 1-phosphate and ATP, and was not inhibited by maltose 1-phosphate according to NMR spectroscopy. Whether GlgC could use other acceptors such as glucose or glucose-6-phosphate was tested but these proved negative as expected.
Expression of active Rv3032 appeared not to be successful with either vector because extracts did not contain the protein according to mass spectrometry. In addition, extracts did not exhibit any activity with maltooligosaccharides with a degree of polymerization of 2, 3, 4, 6 or 7 with either UDP-glucose or ADP-glucose according to mass spectrometry.
Departures from the original proposal The effect of other metabolites on enzyme activity was not tested because we heard through a personal communication that these experiments had recently been done in another laboratory. TLC proved to be a useful method for monitoring OtsA and GlgC assays. The enzymes were not entered into crystallization trials because conditions for generating large quantities of stable enzyme were not yet identified.
Future directions The Rv3032 enzyme needs to be analyzed using different approaches such as sub-cloning into different vectors and/or optimizing the conditions as well as checking other potential substrates. If the unknown product is confirmed to be a product of OtsA, it would be interesting to understand what it is and how it is produced.
Value of the studentship It has been a wonderful opportunity to test my skills, which I acquired the year before, and my university training in genetics and biochemistry. Not only have I become more confident doing more familiar work, but I also learnt a lot about protein expression trials, basic molecular biology methods and NMR spectroscopy. This is certainly a step forward for me, because a PhD has become my ultimate goal after graduation. I am very happy to have contributed to such a broad research project and feel that my work in the lab will be helpful for future studies on the glucan pathways.
Figure 3. NMR spectra of OtsA enzyme reaction.
Figure 4. GlgC activity determination using TLC

Biochemical Society Vacation Studentship Report
Determining the role of the SLK and LOK kinases in melanoma cell migrationand invasion using quantitative 3D live-cell imaging
Student: Andy Russell, University of OxfordSupervisor: Dr. Chris Bakal, Institute of Cancer Research, London.
Introduction
Lymphocyte orientated kinase (LOK) and Ste-20 like kinase(SLK) are two closely related members of the STE-20 family.LOK and SLK have been implicated as Polo-like kinasekinases,i,ii mediators of apoptosisiii,iv and ERM(Ezrin/radixin/moesin) kinases.v,vi ERM proteins exist in aninactive conformation where the C-terminus binds to the FERMdomain. Phosphorylation near the C terminus converts anERM protein into an active form where the C terminus can bindfilamentous actin and the N terminus binds to the plasmamembrane through phospholipid and transmembrane proteininteractionsvii (figure 1). A recent cell-based RNAi screenperformed in the Bakal laboratory found that gene depletion ofLOK and SLK increase the percentage of elongated cells inmouse and human cell populations, suggesting these kinasesmake important contributions to the shape of metastatic cancercells.viii Due to the fact ERM proteins promote cell roundingand migrationix,x LOK and SLK could be important drivers ofcancer metastasis.
Fig. 1. Shows the role of ERM proteins and their kinases.
Aims
The aims of this project were the quantification of the effect ofsiRNA knockdown of LOK and SLK on cell shape andphospho-ezrin localisation and to identify downstream targetsof LOK by phosphoproteomics.
Materials and Methods
Cell Lines and Culture Conditions
Human cervical carcinoma (HeLa) cells and human metastaticmelanoma WM-266-4 cells were cultured in ‘complete media’at 37 °C in 5% CO2. Cells were passaged at 80% confluency.Complete media consists of Dulbecco’s modified Eagle’smedium (DMEM, GIBCO) with 10% v/v SIGMA heat
inactivated fetal bovine serum (FBS) and 5% v/vpenicillin/streptomycin.
HeLa cells for phosphoproteomics experiments were grown inheavy or light SILAC media under the same conditions. SILACmedia consists of DMEM-F12 SILAC media (Caisson labs),10% v/v dialyzed FBS (GIBCO) with the addition of heavy orlight arginine and lysine.
Transfection and immunostaining
2000 HeLa or WM-266-4 cells were plated per well in Optimemreduced serum medium (GIBCO) on a 384-well plate andtransfected with LOK/SLK siRNA at 74nM using LipofectamineRNAiMax (Invitrogen) according to the manufacturers protocol.Two days post transfection cells were fixed in 4%formaldehyde / PBS (Thermo) before permeabilization with0.2% Triton x-100. Cells were blocked for 1 hour in 2% BovineAlbumin Serum (BSA). Primary anti-bodies -tubulin (mouse1:1000 in BSA) and phospho-ezrin (rabbit 1:400 in BSA) fromInvitrogen were incubated for 1 hour at room temperature.Secondary antibodies goat anti-mouse 488 and goat anti-rabbit647 were used at 1:1000 in BSA and phalloidin 568 1:400 inBSA, incubated for 2 hours at RT. Finally, Hoechst was usedat 1:5000 in PBS to stain nuclei for 15 minutes. Triplicatewashes in PBS were performed before each stage of staining.
The plate was imaged using a 20X magnification air lens on anOPERA UHTS spinning disc confocal microscope. 12 Z-stackswere taken 1um apart for 32 fields per treatment. Images wereuploaded to the Columbus Image Data Storage and AnalysisSystem or transferred to Volocity (Perkin Elmer). Imagesegmentation was used to define different cellular regionsbefore quantitative analysis of fluorescence intensity andgeometrical measurements of cell shape features. Figure 2shows how the ‘membrane region’ was defined during imageanalysis.
Fig. 2. Segmentation of cells in Columbus and the definition of ‘membraneregion’. 2A. HeLa cells. 2B. WM-266-4 cells. Phospho-ezrin (red), -tubulin(green) and nuceli (blue).
Mitosis Experiments
Cortical actin
Plasmamembrane
CERM Kinase
N
C
Biochemical Society Vacation Studentship Report
HeLa cells were cultured in ‘complete media’ and plated on a384-well plate with 2mM thymidine to synchronize cells in Sphase. 24 hours later this block was washed off, 4 hours laterthe cells were transfected with siRNAs as described above 4hours later a second 2 mM thymidine block was added. After afurther 16 hours the cells were released from the doublethymidine block. After 9 hours the mitotic population of thecells was increased and the cells were fixed and stained asdetailed above.
Phosphoproteomics (SILAC)
Cells were grown for 10 passages in SILAC media to ensurethey were fully labelled. Heavy and light labelled cells grown in15cm dishes were either mock transfected or transfected usingLOK siRNA using Lipofectamine RNAi max. 48 hours post-transfection cells were lysed using 2% sodium dodecylsulphate (SDS) lysis buffer, protein was extracted and itsconcentration was measured using a bicinchoninic acid (BCA)assay. The treatments were mixed in a 1:1 ratio to give 2samples: light LOK siRNA transfected vs. heavy mocktransfected & light mock transfected vs. heavy LOK siRNAtransfected. Samples were purified using a filter aided samplepreparation (FASP) protocol and then phosphoenriched usinga TiO2 Titansphere Phos-TiO kit. Finally, samples were run ona LC-MS mass spectrometer.
Results and Discussion
Slik, the Drosophila homolog of LOK and SLK is an importantregulator of mitosis,xi and so I investigated the localisation ofSLK and LOK in mitotic HeLa cells (Figures 2 and 3).
Fig. 3. 3.A. Phospho-ezrin localisation throughout mitosis (representative imagesat each stage). 3.B. Image from Perkin Elmer Volocity 3D Image AnalysisSoftware displaying Z-stacks to show phospho-ezrin localisation in 3D.
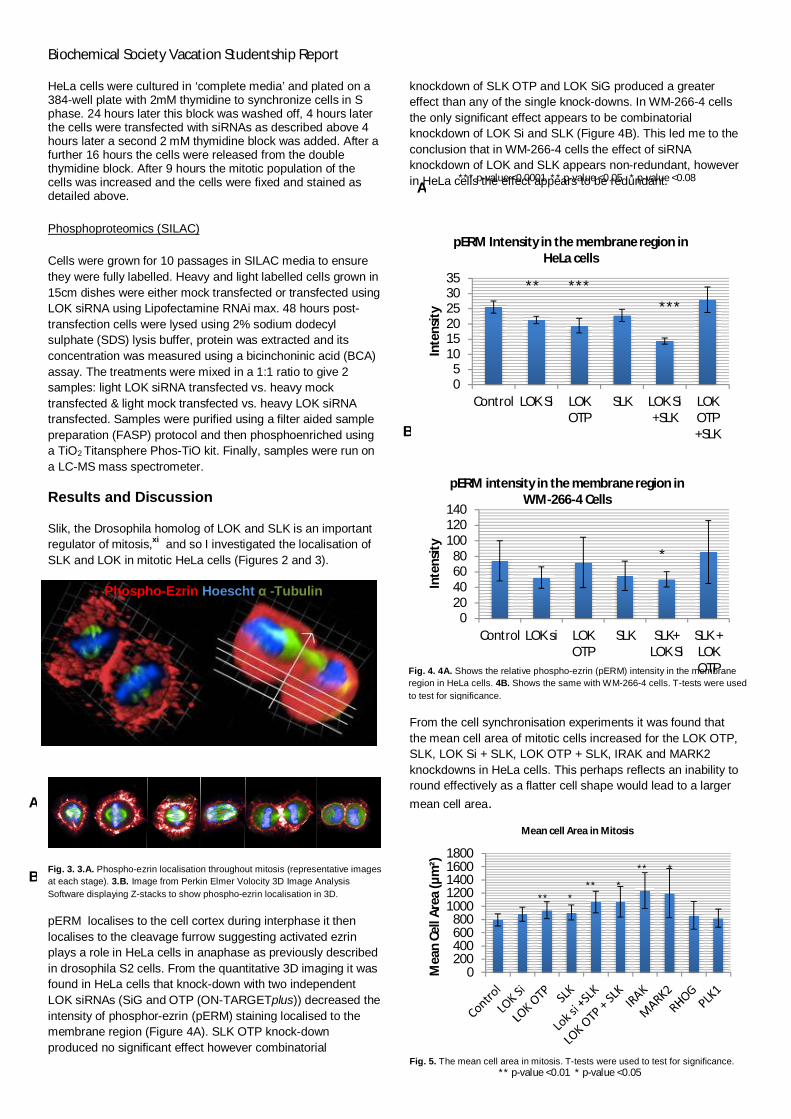
pERM localises to the cell cortex during interphase it thenlocalises to the cleavage furrow suggesting activated ezrinplays a role in HeLa cells in anaphase as previously describedin drosophila S2 cells. From the quantitative 3D imaging it wasfound in HeLa cells that knock-down with two independentLOK siRNAs (SiG and OTP (ON-TARGETplus)) decreased theintensity of phosphor-ezrin (pERM) staining localised to themembrane region (Figure 4A). SLK OTP knock-downproduced no significant effect however combinatorial
knockdown of SLK OTP and LOK SiG produced a greatereffect than any of the single knock-downs. In WM-266-4 cellsthe only significant effect appears to be combinatorialknockdown of LOK Si and SLK (Figure 4B). This led me to theconclusion that in WM-266-4 cells the effect of siRNAknockdown of LOK and SLK appears non-redundant, howeverin HeLa cells the effect appears to be redundant.
From the cell synchronisation experiments it was found thatthe mean cell area of mitotic cells increased for the LOK OTP,SLK, LOK Si + SLK, LOK OTP + SLK, IRAK and MARK2knockdowns in HeLa cells. This perhaps reflects an inability toround effectively as a flatter cell shape would lead to a larger
mean cell area.
Fig. 5. The mean cell area in mitosis. T-tests were used to test for significance.
05
101520253035
Control LOK Si LOKOTP
SLK LOK Si+SLK
LOKOTP+SLK
Inte
nsity
pERM Intensity in the membrane region inHeLa cells
020406080
100120140
Control LOK si LOKOTP
SLK SLK+LOK Si
SLK +LOKOTP
Inte
nsity
pERM intensity in the membrane region inWM-266-4 Cells
0200400600800
10001200140016001800
Mea
nCe
llAr
ea(µ
m²)
Mean cell Area in Mitosis
**
*
****
**
A
B
** ******
*
Fig. 4. 4A. Shows the relative phospho-ezrin (pERM) intensity in the membraneregion in HeLa cells. 4B. Shows the same with WM-266-4 cells. T-tests were usedto test for significance.
Phospho-Ezrin Hoescht -Tubulin
*** p-value <0.0001 ** p-value <0.05 * p-value <0.08
** p-value <0.01 * p-value <0.05
A
B

Biochemical Society Vacation Studentship Report
The SILAC experiment results were analysed by Maxquantsoftware that identified ~300 peptides, of which 200 appearedin
both experiments. In total, 95 proteins were identified in theSILAC experiment. 13 of the proteins with functions associatedwith the cytoskeleton are shown as coloured nodes in Figure 5
linked by known physical interactions between proteins.
Fig. 5. A STRING (Search Tool for the Retrieval of Interacting Genes/Proteins)network for 11 proteins (nodes in colour) identified in the phosphoproteomicsexperiments that are involved in the regulation of the cytoskeleton. Arrowsrepresent if phosphorylation is decreasing with siRNA treatment or increasing.Nodes without arrows indicate inconclusive results due or multiple sites.
Future Directions
The phosphoproteomic experiments will be repeated with theaim of capturing more peptides; this will make the outlieranalysis more reliable and provides the opportunity to identifyother targets of LOK that may have been lost in the initialexperiment. The same experiment will also be performed onSLK. Further experiments will be performed to determine ifknock-down of LOK, SLK and LOK+SLK contribute to thedelocalisation of pERM from the membrane. The earlyinvestigations of pERM’s localisation throughout the cell cycleappear promising and further experiments should beperformed. It could be possible the ERM and PLKK functionsof LOK and SLK are temporally regulated with respect to thecell cycle. A GFP construct of LOK could be used to determineif the localisation of LOK and SLK themselves changethroughout the cell cycle. Experiments could be repeated in a
soft matrix such as collagen where contractile forces arereduced and effects on cell shape seem more pronounced.
Collagen is also more realistic as an in vivo substrate thanplastic.
Departures from Original Proposal
My original plan was to use 3D live-cell imaging however cellswere first fixed and then imaged as this allowed me to visualisepERM.
Value of the Studentship
The studentship allowed me to explore what it is like to work ina research laboratory. It afforded me the opportunity to learn aplethora of new skills and gave me a platform to apply theskills that I had already learned during my first year atuniversity. I believe this studentship has only served to cementmy desire to pursue a career in research science andundertake a PhD at the end of my 4-year degree course. Iwould like to thank Dr. Chris Bakal for accepting me into hislab and providing me with shrewd insight and guidancewhenever I required it. I would additionally like to thank the restof the Bakal lab for always assisting me in my project,irrespective of what they were doing or when. I wouldespecially like to thank Vicky Bousgouni who showed me a lotof the techniques that I have learnt and spent a lot of her timeoverseeing my work during my studentship.
References
i Walter S.A. et al., Stk10, a new member of the polo-like kinase kinase familyhighly expressed in hematopoietic tissue. J Biol Chem, 2003. 278(20): p.18221-8.

Biochemical Society Vacation Studentship Report
ii Ellinger-Ziegelbauer H. et al., Ste20-like kinase (SLK), a regulatory kinase forpolo-like kinase (Plk) during the G2/M transition in somatic cells. Genes Cells,2000. 5(6): p.491-8.iii Sabourin L.A., Rudnicki M. A., Induction of apoptosis by SLK, a Ste20-relatedkinase. Oncogene, 1999. 18(52): p.7566-75.iv Fukumura K. et al., STK10 missense mutations associated with anti-apoptoticfunction. Oncol Rep, 2013. 30(4): p.1542-8.v Belkina N.V., LOK is a major ERM kinase in resting lymphocytes and regulatescytoskeletal rearrangement through ERM phosphorylation. Proc Natl Acad Sci US A, 2009. 106(12):4707-12.vi Viswanatha R, Ohouo PY, Smolka MB, Bretscher A., Local phosphocyclingmediated by LOK/SLK restricts ezrin function to the apical aspect of epithelialcells. J Cell Biol, 2012. 199(6): p. 969-84.vii Bretscher A, Edwards K, Fehon RG., ERM proteins and merlin: integrators atthe cell cortex. Nat Rev Mol Cell Biol. 2002 Aug; 3(8): p.586-99.viii Bakal, C. et al., A screen for morphological complexity identifies regulators ofswitch-like transitions between discrete cell shapes. Nat Cell Biol, 2013. 15(7): p.860–871.ix Lee J.H,et. al., Roles of p-ERM and Rho-ROCK signalling in lymphocytepolarity and uropod formation. J Cell Biol, 2004. 167: p.327–337.x Brown MJ, et al. Chemokine stimulation of human peripheral blood T lympho-cytes induces rapid dephosphorylation of ERM proteins, which facilitates loss ofmicrovilli and polarization. Blood. 2003. 102: p.3890–3899
xi Carreno S, Kouranti I, Glusman ES, Fuller MT, Echard A, Payre F., Moesin andits activating kinase Slik are required for cortical stability and microtubuleorganization in mitotic cells. J Cell Biol. 2008 Feb 25;180(4): p.739-46

Anna MilesSupervisor: Professor Stephen PerkinsDaily Supervision: Elizabeth Rodriguez, Jayesh Gor and Gary Fung
Role of the molecular interactions between complement C3d and Factor H in regulating the complement cascade of innateimmunity
Background
The complement system comprises a large number of plasma proteins which have the ability to opsonize pathogens and induce aninflammatory response. Complement is activated by three activation pathways; the classical pathway, lectin pathway andalternative pathway which all converge at C3. Initiation of the alternative pathway proceeds following the spontaneous hydrolysis ofC3. This particular pathway generates a distinct convertase; C3bBb which cleaves C3 into active C3b. It is critical to havemechanisms in place to ensure that the complement response occurs in a targeted manner towards pathogenic surfaces and toprevent damage to host cells.
The regulatory protein being examined is Factor H which rapidly dissociates C3bBb to limit amplification of the complementresponse whilst competing with factor B for C3b binding. In addition, Factor H acts as a cofactor to Factor I, degrading C3b to iC3band finally C3d. C3d is the final degradation product of C3 and a proteolytic fragment of C3b. C3d binds to host cell membranesthrough its thioester group providing additional binding sites for Factor H when further regulation is required. Ternary complexformation has been observed between Factor H, C3b/C3d and glycosaminoglycans. More specifically, Factor H SCR 19 and 20 areshown to be critical for discriminating self from non self.
Two conflicting crystal structures have been presented revealing a 1:1 complex of C3d and Factor H SCR 19/20 (FH 19/20) [1] aswell as a 2:1 complex of C3d and FH 19/20 [2]. Determining the correct stoichiometry for Factor H interacting with its ligands isessential for fully understanding the regulation of complement.
Aims
To investigate which crystal structure is most representative of the complex formed between FH 19/20 and C3d under physiologicalconditions using isothermal titration calorimetry, fluorescence detection analytical ultracentrifugation and surface plasmonresonance studies. Furthermore, to establish a KD for both binding sites; SCR 19 with C3d and SCR 20 with C3d.
Description of work
Expression and Purification of recombinant C3d
Recombinant C3d containing a glutathione S transferase tag was expressed using an Escherichia coli expression system. Cellculture preparations took place in Luria broth before transfer to 2xYT medium. Optical density measurements monitored the growthof E. coli prior to induction by Isopropyl -D-1-thiogalactopyranoside (IPTG), an inducer of the Lac operon. Following overnightinduction, the cells were harvested by centrifugation and re-suspended in Tris buffer for sonication. Vital protease inhibitors wereadded including leupeptin, pepstatin A and Pefabloc to prevent degradation of C3d. Further centrifugation placed recombinant C3din the supernatant. The lysate was passed through a GSTrap FF column for affinity chromatography using an AKTA purificationsystem. Glutathione and GST have relatively slow binding kinetics and so the flow rate was set to 0.5 ml/min to increase bindingcapacity. C3d now bound to glutathione in the GSTrap FF column was eluted using thrombin which cleaves the GST-tag. Finally,size exclusion chromatography using gel filtration further purified our protein by separation according to size.
Expression and Purification of FH 19/20
Expression of FH 19/20 was carried out in a Pichia pastoris expression system. P. pastoris cells transformed with a pPICZ Avector containing a zeocin resistance gene were streaked onto YPD agar plates. Incubation for 48 hours ensured cell growth hadbeen achieved by observing colony growth. Inoculation of buffered complex glycerol media then allowed for the large scale growthof P. pastoris before transfer to buffered complex methanol media. pPICZ A contains an alcohol oxidase gene with a stronglyinducible promoter, and so cells were fed with methanol to induce protein expression. Following 3 days’ incubation, centrifugation ofthe media resulted in FH 19/20 being secreted into the supernatant. FH 19/20 purification was achieved using Heparin affinitypurification in an AKTA purification system using Tris buffer with a NaCl gradient. Finally gel filtration removed other unwanted celland media debris.
Fluorescence detection analytical ultracentrifugation
Analytical ultracentrifugation (AUC) is a non-destructive technique which monitors the sedimentation of a sample in real time as it iscentrifuged at high speeds. Previous studies using conventional AUC experiments revealed that unbound C3d masks the 1:1complex as these share a similar sedimentation coefficient and so complex formation could not be monitored. To overcome this,Factor H was labelled using fluorescein isothiocyanate (FITC). Labelled FH 19/20 and C3d were mixed at varying ratios and spunat 40k rpm in a fluorescence detection system, a novel technique in this laboratory. Results were analysed using SEDFIT.
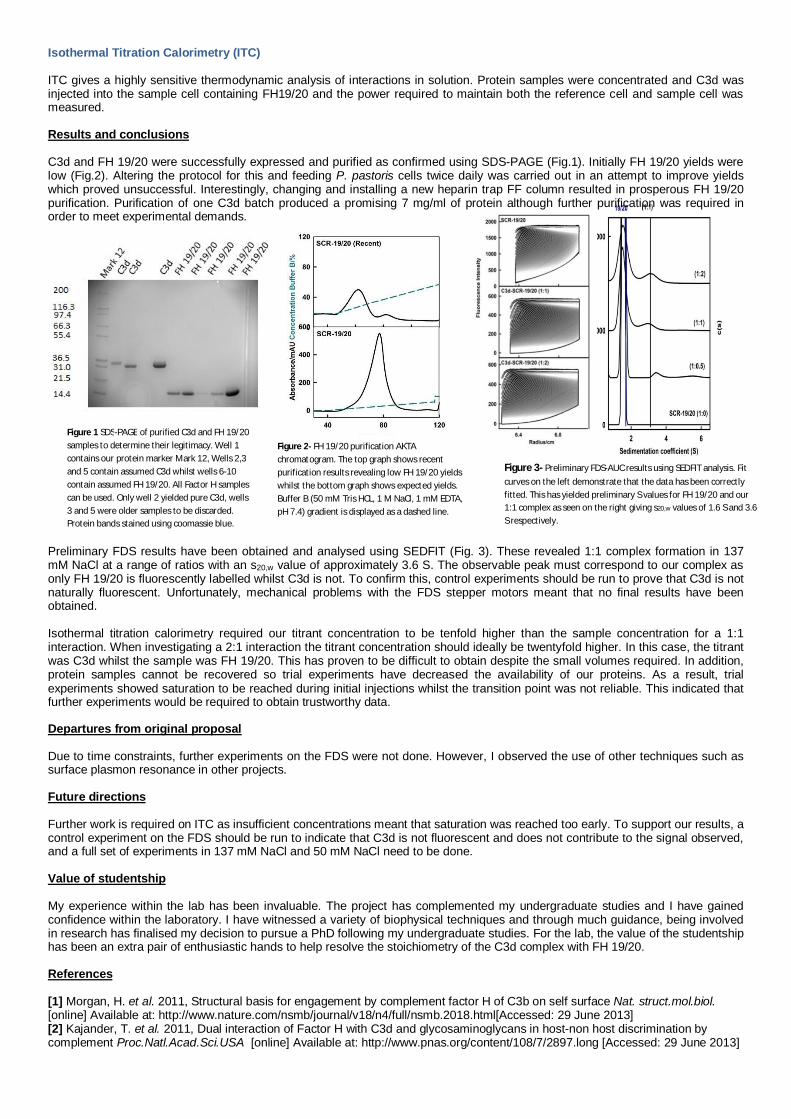
Figure 3- Preliminary FDS-AUC results using SEDFIT analysis. Fitcurves on the left demonstrate that the data has been correctlyfitted. This has yielded preliminary S values for FH 19/20 and our1:1 complex as seen on the right giving s20,w values of 1.6 S and 3.6S respectively.
Isothermal Titration Calorimetry (ITC)
ITC gives a highly sensitive thermodynamic analysis of interactions in solution. Protein samples were concentrated and C3d wasinjected into the sample cell containing FH19/20 and the power required to maintain both the reference cell and sample cell wasmeasured.
Results and conclusions
C3d and FH 19/20 were successfully expressed and purified as confirmed using SDS-PAGE (Fig.1). Initially FH 19/20 yields werelow (Fig.2). Altering the protocol for this and feeding P. pastoris cells twice daily was carried out in an attempt to improve yieldswhich proved unsuccessful. Interestingly, changing and installing a new heparin trap FF column resulted in prosperous FH 19/20purification. Purification of one C3d batch produced a promising 7 mg/ml of protein although further purification was required inorder to meet experimental demands.
Preliminary FDS results have been obtained and analysed using SEDFIT (Fig. 3). These revealed 1:1 complex formation in 137mM NaCl at a range of ratios with an s20,w value of approximately 3.6 S. The observable peak must correspond to our complex asonly FH 19/20 is fluorescently labelled whilst C3d is not. To confirm this, control experiments should be run to prove that C3d is notnaturally fluorescent. Unfortunately, mechanical problems with the FDS stepper motors meant that no final results have beenobtained.
Isothermal titration calorimetry required our titrant concentration to be tenfold higher than the sample concentration for a 1:1interaction. When investigating a 2:1 interaction the titrant concentration should ideally be twentyfold higher. In this case, the titrantwas C3d whilst the sample was FH 19/20. This has proven to be difficult to obtain despite the small volumes required. In addition,protein samples cannot be recovered so trial experiments have decreased the availability of our proteins. As a result, trialexperiments showed saturation to be reached during initial injections whilst the transition point was not reliable. This indicated thatfurther experiments would be required to obtain trustworthy data.
Departures from original proposal
Due to time constraints, further experiments on the FDS were not done. However, I observed the use of other techniques such assurface plasmon resonance in other projects.
Future directions
Further work is required on ITC as insufficient concentrations meant that saturation was reached too early. To support our results, acontrol experiment on the FDS should be run to indicate that C3d is not fluorescent and does not contribute to the signal observed,and a full set of experiments in 137 mM NaCl and 50 mM NaCl need to be done.
Value of studentship
My experience within the lab has been invaluable. The project has complemented my undergraduate studies and I have gainedconfidence within the laboratory. I have witnessed a variety of biophysical techniques and through much guidance, being involvedin research has finalised my decision to pursue a PhD following my undergraduate studies. For the lab, the value of the studentshiphas been an extra pair of enthusiastic hands to help resolve the stoichiometry of the C3d complex with FH 19/20.
References
[1] Morgan, H. et al. 2011, Structural basis for engagement by complement factor H of C3b on self surface Nat. struct.mol.biol.[online] Available at: http://www.nature.com/nsmb/journal/v18/n4/full/nsmb.2018.html[Accessed: 29 June 2013][2] Kajander, T. et al. 2011, Dual interaction of Factor H with C3d and glycosaminoglycans in host-non host discrimination bycomplement Proc.Natl.Acad.Sci.USA [online] Available at: http://www.pnas.org/content/108/7/2897.long [Accessed: 29 June 2013]
Figure 1 SDS-PAGE of purified C3d and FH 19/20samples to determine their legitimacy. Well 1contains our protein marker Mark 12, Wells 2,3and 5 contain assumed C3d whilst wells 6-10contain assumed FH 19/20. All Factor H samplescan be used. Only well 2 yielded pure C3d, wells3 and 5 were older samples to be discarded.Protein bands stained using coomassie blue.
Figure 2- FH 19/20 purification AKTAchromatogram. The top graph shows recentpurification results revealing low FH 19/20 yieldswhilst the bottom graph shows expected yields.Buffer B (50 mM Tris HCL, 1 M NaCl, 1 mM EDTA,pH 7.4) gradient is displayed as a dashed line.

Biochemical Society Studentship Report 2013Athina Rigalou
Supervisor:Dr Maria Tuohy Lab mentor: Dr Martina Wernecke
‘Investigating the biochemical basis for the prebiotic effects of a novel seaweed extract onbeneficial bacteria’
Background and AimsFood and health are inextricably linked areas and due to increasing demand, there is abundantongoing research on supplements with health promoting effects which can be part of the dailydiet. Bacteria belonging to the Lactobacillus family are some of the most commonly usedprobiotics, found in a range of food stuffs and beverages. Although there has been extensiveresearch on probiotics, we know very little about the mechanism of action of prebiotics (Quigley,2010). Prebiotics are defined as "selectively fermentable ingredients that allow specific changes,both in the composition and/or activity in the gastrointestinal microbiota that confers benefitsupon host wellbeing and health"(Gibson et al., 2004). Recent group research has demonstratedthat an algal polysaccharide-rich extract promotes the growth of probiotic organisms in vitrowhile down-regulating the growth of gut pathogens in vitro. Our working hypothesis is that thebacteria metabolize the extract to produce oligosaccharides which act like known prebiotics. (egGOS and FOS)Description of work carried outDose experimentLactobacillus acidophilus (La) was grown on different concentrations of the algal polysaccharide-rich extract; 0.15% extract, 0.3% extract and 0.75%(final conc. Respectively,0.05%,0.1% and0.25%) as well as GOS, glucose and water (i.e. no supplement) over 68 hours. The optimalconcentration of extract for use in future experiments was determined.Time Series experiment and Live/Dead analysisOur organism was grown in M9+0.3% extract, M9+glucose and M9+no supplement over 39hours. Samples were taken at t0,tmid-log (15 hours) and tstat (39 hours) and bacterial cell growth wasmonitored using nephelometry at 595 nm. Live/Dead analysis was also performed on tmid and tstatsamples for bacteriostatic effects.Dubois assayThe overall sugar concentration of our Time Series samples was determined using a Duboisassay; La T0 extract, La Tmid extract, La Tstat extract, La T0 glucose, La Tmid glucose, La Tstatglucose, La T0 no suppl., La Tmid no suppl. and La Tstat no suppl.HPLCUtilization of the carbohydrates by L. acidophilus was monitored by HPLC analysis.Glucanase AssayCultures harvested at specific time points were then assayed for polysaccharide-modifyingenzyme activity.Results
Fig.1 Fig.2
The 0.3% extract was chosen forsubsequent experiments, based on apanel of probiotic and potentiallypathogenic microorganisms.
The extract was shown to stimulate andenhance the growth of L. acidophilus. (SeeFig.2) The bacteria are metabolizing theextract and supplying the nutrients they need.

Biochemical Society Studentship Report 2013Athina Rigalou
Supervisor:Dr Maria Tuohy Lab mentor: Dr Martina Wernecke
Fig.3The Live/Dead assay showed that 51.03% of bacteria grown on the extract are alive in mid-log phase(significantly higher than our controls).As expected, by stationary phase, the nutrients have run out andbacterial numbers have begun to decline (8% live).
0
0.1
0.2
0.3
La t0 La tmid La tstat
Enzymeactivity(IU/ml)
Glucanase Activity
Fig.5Fig.4
Departures from original proposalUnfortunately, time did not permit us to run transcriptomics or bioinformatics.Future directionsFull proteomics, transcriptomics and bioinformatics analysis will be conducted.Value of studentship to the studentThe studentship has been quite an enjoyable and rewarding experience. I have acquired a range ofnew skills and techniques in the lab which I feel will be of huge advantage in my future studies. Ifeel that I have had a valuable insight into a research career and I now hope to seek a PhD orresearch position after my undergraduate studies.
L.acidophilus HPLC
µg/ml T0 Tmid Tstat
LAM2 1.442 n.a. n.a.
LAM3 6.084 1.231 1.845
LAM4 1.575 1.175 2.121
LAM5 n.a. 1.005 8.333
LAM6 6.137 7.215 9.478
References1) Quigley, EMM.(2010) ‘Prebiotics and probiotics; modifying and mining the microbiota’, Pharmalogical Research,61,213-2182) Gibson, G.R., Probert H.M., Van Loo J., Rastall R.A. and Roberfroid, M.B. (2004) ‘Dietary modulation of the human colonic
microbiota; updating the concept of prebiotics’, Nutrition Research Reviews ,17(02), 259-275
HPLC analysis determined the quantity ofoligosaccharides in each of our samples.As expected, we see production ofdifferent types of laminaranoligosaccharides.
T0 activity is presumed to be due to MRS mediumcarryover.Great difference in enzyme activity between T0 andmid log phase. As the polymer is metabolized, a lot ofreducing sugars are being produced; hence the greatincrease in enzyme activity.The reduction in enzyme activity in stationary phaseshows utilization of sugars and corresponds to theincrease in bacterial cell density (See Fig,1)

Biochemical Society Studentship Report 2013
Expression, isolation and characterisation of Arf family small G-proteins with novel post-translational modifications
Bethan Wolfenden, supervised by Professor Geraint ThomasDepartment of Cellular and Developmental Biology, University College London
Background and aimsADP-ribosylation factors (Arfs) play an important role in diseases related tomembrane traffic and organelle structure. Arf6 in particular is critical in thecoordination of membrane trafficking; affecting immune system signalling, cell growthand human insulin sensitivity. A GTP-binding protein of the Ras superfamily, Arf6 isinvolved in cargo loading, membrane recycling and regulation of lipid modifyingsignalling enzymes such as phospholipase D (PLD). Arf6 is typically regulated byGTPase activating proteins (GAPs), but research indicates that it may be a target forkinases, and of particular interest to this project, the proto-oncogene Src kinase thattransmits signals by supressing Arf6 activity. In order to better understand the linkbetween Arf6 and Src signaling, the aim of this project was to purify phosphorylatedArf6 for crystal analysis by co-expressing the Src kinase domains Lck and Arf6 insideE.coli and resolving the phosphorylated from non-phosphorylated forms of theprotein by ion exchange chromatography.
MethodsBacterial transformation
An engineered ARF6+Lck-KD-pETDuet-1 plasmid (Novagen) was constructed byProf. Thomas’ lab. This construct was transformed into BL21 (DE3) pLysS E. colicells (Invitrogen). Plasmid uptake was confirmed by mini-prep (Qiagen) isolation ofplasmids, digestion with Cla1 and visualization by gel electrophoresis.
Large-scale expression trial
A 1L culture of the transformed BL21 (DE3) pLysS E.coli strain was grown at 37°.When an optical density of 0.4 was reached, protein expression was induced byIPTG of final concentration 1mM. After centrifugation, pellets were resuspended inlysis buffer, before the addition of lysozyme and a second lysis buffer. The cell lysatewas centrifuged, and the supernatant was purified by ion exchange chromatography.
Ion exchange chromatography
The recombinant pETDuet plasmid contained a His6 tag and an S tag. A Ni-NTAcolumn was used to bind the polyhistidine-tag, and the Arf6 protein eluted with animidazole buffer overnight, resulting in twenty-two 10mL fractions. An SDS-Page gelwas run with the fractions 9 to 17, and probed by Western blotting with anti-ARF6and anti-phosphotyorsine (PY20) antibodies (Fig. 1). Fractions were further purifiedwith a mono-S column, which was used to bind the S tag of the recombinant protein.The subsequent twenty-six 10mL fractions were similarly probed with anti-ARF6 andanti-phosphotyorsine (PY20) antibodies by Western blotting (Fig. 2).
ResultsArf6 was successfully expressed and purified. Ponceau S staining and Coomassieblue staining of the fraction samples in Fig. 1 (a) and (d) show high levels of proteinexpression REFINED. Anti-phosphotyorsine antibody probing of the Western blotindicates a strong presence of phosphorylated Arf6 in fractions 6 to 9. Subsequentanalysis of Western blot data allowed comparison of anti-phosphotyorsine and anti-Arf6 staining, and the generation of graph showing the ratio of anti-phosphotyorsineto anti-Arf6 staining over the range of fractions (Fig. 2). This indicated the greatestconcentration of phosphorylated Arf6 is present in fraction 8.

Biochemical Society Studentship Report 2013
Fig. 1: Western blot of Mono-S fractions. (a) Ponceau S (b) Coomassie Blue (c) Anti-PY20 (d) Anti-Arf6
Future directionsIn future, the phosphorylated and non-phosphorylated forms could be resolved by ionexchange chromatography to produce a sampleof purified phosphorylated Arf6 for crystalanalysis. Mass spectrometry of the proteinsample would be another continuation as thiswas not possible during the project due to timerestrictions.
Departures from the original proposalIn addition to the work presented, the constructwas ligated into three additional plasmids andtransformed into XL10 Gold cells. Theseplasmids – pet47b[+], pmmb66EH and pQR445 – covered a range of high and lowcopy numbers to determine the best expression conditions to obtain phosphorylatedArf6.
Contribution to career aspirationsDuring my studentship, I was exposed to an interdisciplinary research group thatcombined with wet lab work with systems and synthetic biology. I had the opportunityto attend two computational biology meetings with my lab group, both in the UK andDenmark. My summer experience has not only contributed to my motivation inpursuing graduate study, but also highlighted the benefit and need forinterdisciplinary and international collaboration. As a result, I hope to pursue aninterdisciplinary PhD at a European institute.
AcknowledgmentsI would like to thank Prof. Geraint Thomas and Helina Marshall for their guidance andtime, and the Biochemical Society for their support.
Lane 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Fraction no. 6 7 8 9 10 11 12 13 14 15 16 17
0123456789
6 7 8 9 10 11 12 13 14 15 16 17
pY/Arf6
Fig. 2: Ratio of anti-phosphotyorsine to anti-Arf6 stainingover fractions 6 to 17

Student: Brendan RogersSupervisor: Jennifer Potts
Engineering a domain-domain interface in a novel rod-like protein report
The Staphylococcus aureus surface protein G (SasG) is a rod-like protein involved in protein mediated biofilm formation (Corrigan et al,2007). SasG has a repetitive structure of nine G5 domains interspersed by E regions (Gruszka et al. 2012)(Fig 1A) and is stabilised byinterdomain interfaces. The G5² (78 amino acids) region on its own forms a stable structure however the E region (50 amino acids)doesn’t. In an E1- G5² construct there is a stabilising interface and a larger folded structure is formed (Fig 1B). However, in a G5²-E2 (B2repeat) construct, the G5²region forms a stable fold while the E segment does not.
Figure 1A- The SasG protein. The B repeat region is shown with nine G5 domains interspersed with E regions. 1B shows the E-G5 interfaceand the stable structures of the two regions. Also shown is G5-E with no stable interface and the E region unfolded.
Protein denaturation by increasing urea concentration experiments have found that E1-G5² with a tyrosine to tryptophan mutation at theinterface (Y547W) on E1 was more stable than the wild type E1-G5². The E1-G5² Y625W construct was found to be destabilised comparedto the wild-type. The aim study for the summer studentship is to see if the homologous mutation (Y625W) on the G5² will stabilise theG5²-E2 interface so that E2 stably folds. The interfaces of E1-G5² and G5²-E2 are very similar and so if the Y547W mutation stabilised theE1- G5², the Y625W mutation should stabilise the G5²-E2 interface. A purified 15N labelled B2 Y625W will be used in a 1H-15N HSQC to see ifthe E2 segment stably folds.
Method
The work during the studentship involved the preparation of a 15N labelled B2 Y625W sample for1H-15N HSQC. All protein constructs wereinserted into pET 28b (pSKB2) vectors. The main features of the plasmid included an N terminal 6XHis tag with thrombin cleavage site,kanamycin resistance and the lac operator region to regulate expression of the B2Y625W construct. pSKB2-B2WT underwent sitedirected mutagenesis by PCR to insert the Y625W mutation. BL21 competent cells were transformed with pSKB2-B2Y625W.The BL21competent cells were grown in 15N M9 media at 37oC until the OD600 reached 0.6. IPTG was added to the media (final concentration of0.1mM) to induce the expression of the B2Y625W. The cells were lysed in a French press at 25 kpsi. The lysate produced was then clarifiedby centrifugation at 48,000 xg.
The first purification step involved nickel affinity purification where the lysate was loaded onto a 5ml HisTrap column. Forty 2ml fractionswere collected as elution buffer was loaded onto the column. SDS PAGE electrophoresis was used to assess that the protein of interestwas present in the fractions. Fractions thought to contain the B2Y625W were pooled. After the initial purification stage 6XHis taggedthrombin was added to the sample overnight to cleave the 6XHis tag off the B2Y625W. Nickel affinity purification was also used toremove the 6XHis tagged thrombin from the sample. The sample was loaded onto the HisTrap column and the flow-through collectedcontained untagged B2Y625W. The B2Y625W sample was then concentrated to 600 µM and the buffer was exchanged ready for1H-15NHSQC NMR.
Results
1H-15N HSQC is a common method used to see if a protein construct has a stable fold. The 1H-15N HSQC spectra of the B2Y625W producedin this studentship (Fig 2) shows that there are peaks which are widely distributed along the 1H dimension and are consistent withpreviously obtained spectra of G5². The wide distribution of peaks suggests a stably folded protein is present. There are also a collection ofpeaks close together along the 1H dimension. This suggests that there is an unfolded structure present within the construct. The clustersof peaks are likely to correspond to the E2 region. This suggests the E segment has not folded and there is no interface between G5²-E2.The Y625W mutation has not caused a stabilisation effect at the G5²-E2 interface as Y547W for E1-G5², even though the E1-G5² and G5²-E2interfaces are very similar.

Figure 2- The 1H-15N HSQC spectra of the B2 Y625W construct. The peaks clustered together along the 1H dimension are likely tocorrespond to the E2 region. The peaks that are widely distributed are consistent with previous HSQC spectra of G5².
To further access if the undistributed peaks along the 1H dimension do correspond to the E2 region, a comparison was made with apreviously-acquired 1H-15N HSQC spectrum for the wild- type B2 repeat under similar conditions (Fig. 3). While there are some smalldifferences between the two spectra, overall the chemical shifts of both well-dispersed and weak (G5) and poorly-dispersed and intensepeaks is very similar, suggesting that the mutation has had little effect on the stability of E2.
Figure 3- The 1H-15N HSQC spectra of the B2 Y625W construct (black) compared with previous HSQC spectra of G5²-E (red).
The studentship has given me the opportunity to see for myself what it is really like working in a research environment. I have been ableto gain key laboratory skills such as PCR and affinity chromatography, which will be crucial for any research position I undertake in thefuture. Whilst at the placement I have also had the opportunity to participate in laboratory meetings and present my work to the group.The placement has given me a sense of what will be expected of me in a lab post degree from day to day. Overall the studentship has beena fantastic experience which has given be the skills and confidence in the lab I will need in a future research position.This project is part of ongoing work in the lab to understand the structure and function of this very unusual bacterial surface protein. Weare now testing other mutations in a similar way to identify the key factors accounting for the difference in the stability of the E region inthe E-G5 and G5- context.
Corrigan RM, Rigby D, Handley P, Foster TJ. The role of Staphylococcus aureus surface protein SasG in adherence andbiofilm formation. 2007. Microbiology 153, 2435–2446.Gruszka, D.T., Wojdyla, J. Bingham, R.J., Turkenburg, J.P., Manfield, I., Steward A., Leech, A.P., Geoghegan, J.A., Foster, T.J.,Clarke, J. and Potts, J.R.A staphylococcal biofilm-forming protein has a contiguous rod-like structure.2012. Proc. Natl. Acad.Sci. (USA) 109, 1011-1018

Biochemical Society Studentship Report 2013
Study of Human Papillomavirus E2 Protein Interactions with Human TopBP1
Student: Calum Robertson
Supervisor: Dr. Brian Smith
Introduction
Human papillomavirus (HPV) is one of the major causative agents of cervical cancer
in the world, a disease which is responsible for approximately 2% of death of
adolescent and middle-aged women [1]. While a vaccine already exists, and is
widely distributed, this only affects the most common serotypes (16 and 18), so a
better drug which works against all, including the most virulent, serotypes, along with
new diagnostic techniques, could be a dramatic shift in the field of HPV infection
prevention.
This project aimed to study the interaction between the protein E2 from HPV, and
TopBP1 from human cells. This interaction has been previously shown to be vital for
initiating replication of the viral genome in host cells.[2] These two interact via BRCA
C-terminal (BRCT) domains, which exist on the n-terminus of TopBP1 [3]. There are
three reported interacting points which cover BRCT 012, BRCT 5 and BRCT 6. This
was achieved by using surface plasmon resonance chips, which use the degree to
which light bends when reflected by a very thin gold surface changes as protein
complexes are formed on the other side of the layer to detect protein-protein
interactions. This project studied both BRCT012 and BRCT6, hoping to measure the
strength of their interaction with E2.
Project Aims
In 8 weeks, we aimed to study the interaction between BRCT012 and BRCT6fragments of TopBP1 with HPV E2.

Results
Initial expression tests showed strong levels of BRCT012 expression in BL21 E.Coli.(Fig.1)
Initially, most of the expressed BRCT012 was insoluble, and therefore not useable infurther tests. This issue was solved by lowering the expression temperature to 15oc,and leaving the liquid cultures overnight.
27kDa
66.4kDa
34.6kDaBand ofInterest
Figure 1: Test Expression of BRCT012 in BL21 E.Coli. Conducted with 1mM IPTG at37oc.
Post IPTGExpression
SolublePelletSample
InsolublePelletSample
Pre IPTGExpression
Post IPTGExpression

BRCT012 then needed to be purified. Being His-tagged, nickel column purificationwas used, resulting in a pure protein sample (Fig.2 and 3)
As shown, BRCT012 was bound to the nickel resin very well, so a sample of highpurity was easily obtained
Figure 2: Ni2+ Resin in GravityColumn
Figure 3: SDS PAGE of Ni2+ Column Fractions.
27kDa
66.4kDa
Cell Lysate Binding Buffer Wash Buffer Elution Buffer 1M Imidazole
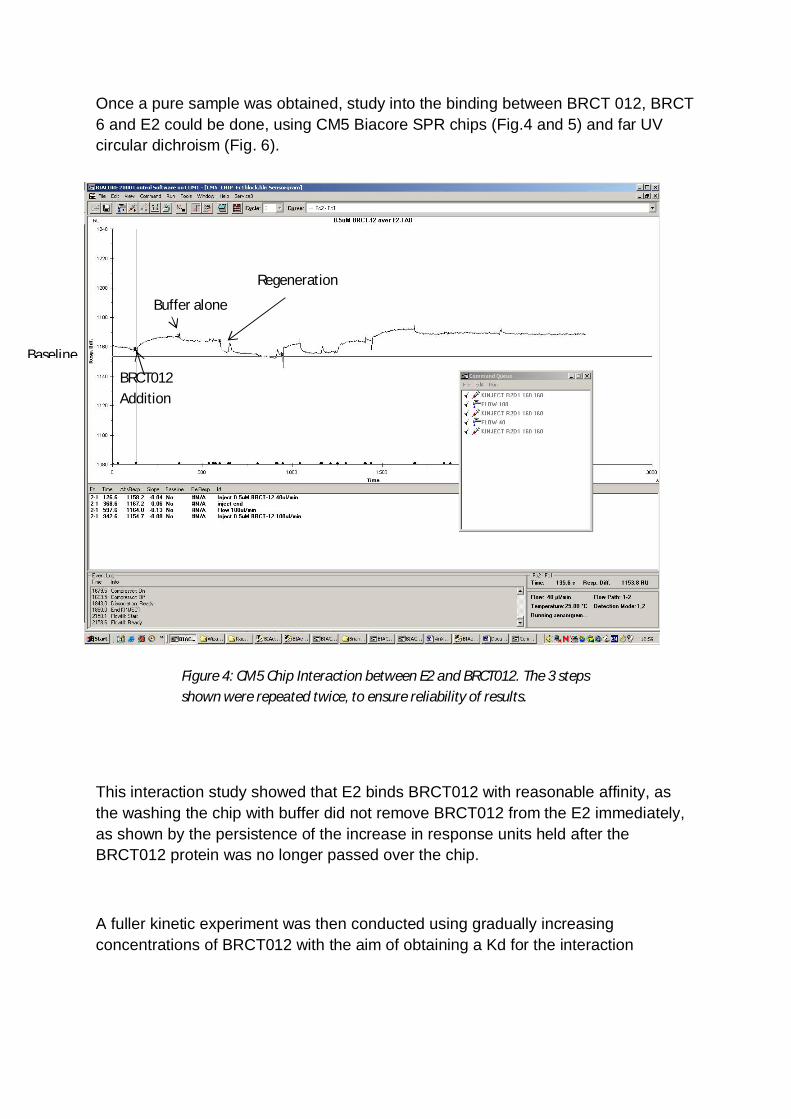
Once a pure sample was obtained, study into the binding between BRCT 012, BRCT6 and E2 could be done, using CM5 Biacore SPR chips (Fig.4 and 5) and far UVcircular dichroism (Fig. 6).
This interaction study showed that E2 binds BRCT012 with reasonable affinity, asthe washing the chip with buffer did not remove BRCT012 from the E2 immediately,as shown by the persistence of the increase in response units held after theBRCT012 protein was no longer passed over the chip.
A fuller kinetic experiment was then conducted using gradually increasingconcentrations of BRCT012 with the aim of obtaining a Kd for the interaction
Figure 4: CM5 Chip Interaction between E2 and BRCT012. The 3 stepsshown were repeated twice, to ensure reliability of results.
BaselineBRCT012Addition
Buffer alone
Regeneration

The Biacore software interpreted these data to give a Kd of approximately 30uM.These results are still considered preliminary as the binding phases did not reachsteady state, the titration did not show clear saturation as the concentration ofBRCT012 was increased, and the fits for the on- and off-rates were not well fitted. Inorder to be more sure, this experiment would need to be conducted again.
Figure 5: Kinetic Study of E2 and BRCT012. The concentrations used were 1,2,3,7.5and 10 M BRCT012

Along with these studies, far UV circular dichroism was also conducted.
It is clear from the difference in the spectra that there is a conformational change inducedby mixing of E2 with BRCT 012. It is not possible to discern from CD whether the spectralchange is due to conformational changes in E2 or BRCT012 or possibly both.
Figure 6: Far UV CD spectra of: summmed spectra of E2 and BRCT-12 (red) recordedseparately; and, spectrum of mixed solution of E2 and BRCT012 (blue).

Changes to Initial Protocol
Initially, both Ion affinity and gel filtration purification methods were planned as additionalpurification steps for BRCT012. However, the protein did not bind to the ion exchange resinunder the conditions tested. Gel filtration also could not be carried out since it requiressmall volumes of concentrated protein which were not achieved due to problems duringconcentration process after the nickel affinity purifcation. Similarly, BRCT012 could not beobtained at a sufficient concentration to perform analytical gel filtration analysis of bindingto E2.
Value of Studentship: Student
Personally, I found the studentship to be a very fulfilling and interesting experience. Itallowed me to develop lab techniques and etiquette, which I will carry on to my futurestudy. Knowledge of techniques like bacterial transformations, growth and expression ofproteins in transformed bacteria, nickel affinity chromatography, SDS-PAGE and severalother common lab tests. This knowledge will be invaluable for my upcoming final yearproject/dissertation, along with any other future research I may pursue. Dr Smith and histeam were incredibly accommodating, and a pleasure to work with.
Value of Studentship: Supervisor
Calum was able to pursue an area of active research in my lab and obtain encouragingpreliminary data that makes a real contribution to our research into the interaction betweenTopBP1 and proteins from Human Papilloma Virus.

References
1. CDC Information (found at http://www.cdc.gov/std/hpv/stdfact-hpv.htm)
2. TopBP1 Regulates Human Papillomavirus Type 16 E2 Interaction with Chromatin. MaryM. Donaldson, Winifred Boner, and Iain M. Morgan. J. Virol. April 2007 vol. 81 no. 8. 4338-4342
3. An Interaction between Human Papillomavirus 16 E2 and TopBP1 Is Required forOptimum Viral DNA Replication and Episomal Genome Establishment. Mary M. Donaldson,Lorna J. Mackintosh, Jason M. Bodilyb, Edward S. Dornan, Laimonis A. Laimins and Iain M.Morgan. J. Virol. December 2012 vol. 86 no. 23, 12806-12815

Investigating the expression of BACE2 in mouse models of Downsyndrome
Carlos Siganporia
Supervised by Frances K. Wiseman, Institute of Neurology, University College London
_________________________________________________________________________________
Introduction
People who have Down syndrome (DS), causedby trisomy of chromosome 21 (Hsa21), havegreatly increased risk of developing Alzheimerdisease (AD). By the age of 40 people who haveDown syndrome will have developed amyloidplaques and neurofibrillary tangles, and by theage of 60 around 60 will have dementia. APP isencoded on Hsa21 and trisomy of this gene ishighly likely to significantly contribute to the
development of AD, in people who have DS. Indeed, duplication of the APP locus in the absenceof DS is sufficient to cause early on-set Alzheimer disease. However, we have recently shownusing transgenic mouse models, that Hsa21 gene or genes, other than APP are crucial to theincreased risk of AD associated with DS. We used our unique Tc1 mouse model of Downsyndrome to demonstrate that trisomy of gene or genes on Hsa21 significantly exacerbatesAPP/Abeta pathology in the mouse brain.
Aims
There were two principal aims to the project. Firstly, previous Abeta accumulation findingsshowed significant increase in area of Abeta deposition in Tc1/J20 crosses compared to J20mice (J20 mice express an increased number of copies of human APP). This was observed inboth the cortex and hippocampus. Therefore, the first aim was to examine whetherrelationship is observed by counting the number of plaques. The second aim of this studentshipwas to examine whether Beta-site APP cleaving enzyme (BACE2), candidate gene located onHsa21, was expressed differently when exposed to Abeta or APP and where this difference inexpression occurs and therefore may provide the reasoning as to why trisomy exacerbatesAbeta pathology.
Methods
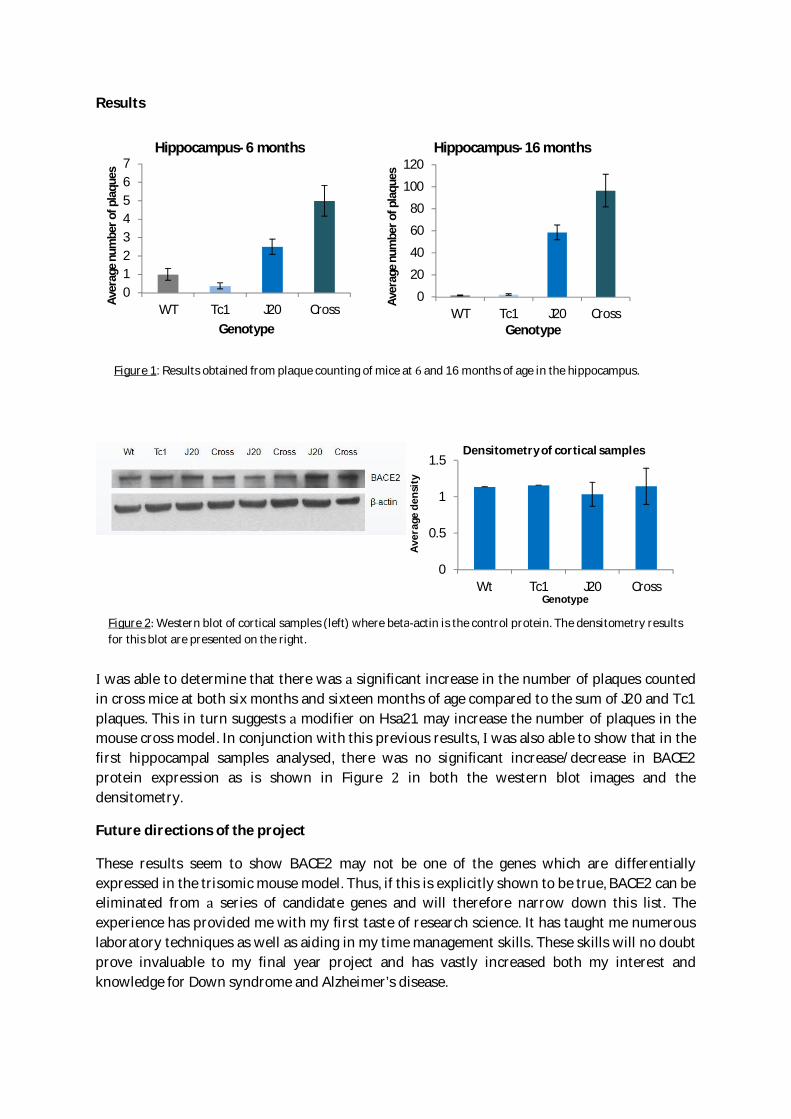
For the initial part of the project, manual counting of the amyloid plaques was carried outamong the four genotypes. Once this was completed, the averages amongst the genotypes weretested for statistical differences. graph presenting the results could then be produced with theerrors calculated for each genotype (Figure 1). Western blotting was carried out in order toobserve whether BACE2 was expressed differently amongst the genotypes. This required plentyof optimisation in terms of both the dilutions of antibodies used and the type of buffer used.Once optimised, Image J, protein densitometry program, was used to determine significantdeviation in BACE2 protein levels.
Results
was able to determine that there was significant increase in the number of plaques countedin cross mice at both six months and sixteen months of age compared to the sum of J20 and Tc1plaques. This in turn suggests modifier on Hsa21 may increase the number of plaques in themouse cross model. In conjunction with this previous results, was also able to show that in thefirst hippocampal samples analysed, there was no significant increase/decrease in BACE2protein expression as is shown in Figure in both the western blot images and thedensitometry.
Future directions of the project
These results seem to show BACE2 may not be one of the genes which are differentiallyexpressed in the trisomic mouse model. Thus, if this is explicitly shown to be true, BACE2 can beeliminated from series of candidate genes and will therefore narrow down this list. Theexperience has provided me with my first taste of research science. It has taught me numerouslaboratory techniques as well as aiding in my time management skills. These skills will no doubtprove invaluable to my final year project and has vastly increased both my interest andknowledge for Down syndrome and Alzheimer’s disease.
0
0.5
1
1.5
Wt Tc1 J20 Cross
Aver
age
dens
ity
Genotype
Densitometry of cortical samples
Figure 1: Results obtained from plaque counting of mice at and 16 months of age in the hippocampus.
Figure 2 Western blot of cortical samples (left) where beta-actin is the control protein. The densitometry resultsfor this blot are presented on the right.
01234567
WT Tc1 J20 Cross
Aver
age
num
bero
fpla
ques
Genotype
Hippocampus- 6 months
0
20
40
60
80
100
120
WT Tc1 J20 Cross
Aver
age
num
bero
fpla
ques
Genotype
Hippocampus- 16 months

Value of studentship to student and to supervisor
Carlos not only contributed useful data to our project to understand Alzheimer’s disease inpeople who have Down syndrome but also his enthusiasm and well thought-out questionsadded greatly to our research group this summer. We would welcome Carlos back again towork with us and he so much impressed my mentor Professor Fisher that she has asked Carlosto consider undertaking PhD with us. Carlos gained valuable experience of bench-work,experimental planning and optimisation, data-analysis, group and University research meetingsand data presentation. believe he now has rounded understanding of the different options for
research career; including how to combine research with further study of medicine.

Undergraduate Summer Studentship 2013 – Claire Cattermole
(Supervisor: Dr Michael Ladomery at the University of the West of England)
Aims of the project.
The project was centred around the question of determining the effects of hypoxia on the
alternative splicing particularly of cancer associated genes.
Description of the work.
The framework of the project was as follows: expose prostate cancer cell lines to 1%
hypoxia for 48 hrs, and assess the consequences on alternative splicing of cancer
associated genes. The first few weeks were spent watching my supervisor and other
laboratory members to learn the different techniques I would need in order to work
independently on my project. I learned two methods of RNA isolation; Qiagen miniprep spin
columns and the widely used Trizol method. I was trained in tissue culture, learning to grow
three independent prostate cancer cell lines (PC3, DU145 and VCaP). I also learned how to
make cDNA, perform PCR and analyse amplicons on agarose gels. Towards the end of the
project I attempted a full experiment (i.e. expose cells to hypoxia, extract RNA and perform
RT-PCR). I performed PCR on the following genes: carbonic anhydrase IX (CA9) and VEGF,
two key markers of hypoxia; and in terms of looking at consequences on alternative splicing
I focused on the oncogene RON. RON is a tyrosine kinase member of the MET family of
receptors. Exon 11 of RON is often skipped in cancer as it produces a constitutively active
tyrosine kinase. Towards the end of the project I also treated cells with DMOG, a chemical
inducer of hypoxia, which can be used to determine whether or not any downstream
changes are due to the HIF pathway (hypoxia inducible factor).
Assessment of results and outcomes of studentship.
The eight weeks spent in the laboratory allowed me to master a wide range of techniques,
gaining confidence in basic tissue culture techniques, cell counts, cell viability assays, RNA
extraction, cDNA synthesis, PCR and agarose gel electrophoresis. I also gained further
understanding of how a research group operates as a team, and also in health and safety
procedures and guidelines.
By measuring and upregulation in CAIX and VEGF I was able to confirm that the prostate
cancer cells were adapting to hypoxia as expected. I then looked at RON alternative splicing
focusing on the inclusion of its exon 11. Figure 1 shows a sample of the data obtained. I saw
indications that in hypoxia there might be a greater proportion of exon 11 skipping.
LNCa
PH
(3)
RT-

Figure 1. Effect of hypoxia on the alternative splicing of RON pre-mRNA. The prostate cancer PC3 cell line
was exposed to 1% oxygen hypoxia for 48 hrs, RNA extracted and RT-PCR performed. The proportion of RON
exon 11 skipping appeared to increase in hypoxic PC3 cells.
Future directions in which the project could be taken.
The increased proportion of exon 11 skipping in RON pre-mRNA is potentially very
significant because tumours often encounter hypoxia. The next steps will be to confirm and
quantify the measurements with statistical repeats. Then it will be interesting to see what
splice factors might be involved in altering RON splicing. The oncogenic splice factor SRSF1
is already known to be involved in exon 11 skipping so its role could be looked at – might
SRSF1 activity be changed in response to hypoxia? Or perhaps additional splice factors?
Departures from original proposal.
- No significant alterations.
Value of studentship to the student and to the lab.
As a student on this Summer project I was able to learn a several important techniques and
how they can be applied to a range of experiments. I am now also confident in the use of
equipment and operating procedures. I am now starting my independent final year project
where I can already make use of all that I have learned. The experience has also shown me
that a PhD in this area of research is very much appealing to me now, and I intend to pursue
a career in science as a result.

Student: Csilla SzundiSupervisor: Eva Szegezdi
Background: Cytokines of the Tumour Necrosis Factor (TNF) family, especially the so-called death ligands (TNF, TRAIL, FasL) have been considered for the treatment of cancersince their discovery. Their utility is however hampered because of either toxic side effects(for TNF and FasL) or low potency due to the NF- B-mediated inflammatory response thatthey can also induce (TRAIL, TNF). Activation of NF- B drives the expression of anti-apoptotic genes, cytokines and chemokines that promote tumour drug resistance andinvasiveness. The actual biological response (cell death or inflammation) induced by TRAILand TNF is determined by the complement of the adaptor proteins recruited to the activateddeath receptor.The supervisor’s laboratory, as well as others, has found that inhibition of NF- B or thedeficiency of the adaptor protein, TRAF2, involved in TRAIL-mediated NF- B activation,can sensitise tumour cells to TRAIL-induced apoptosis. Based on these results the laboratoryis developing inhibitors that can disrupt the recruitment of TRAF2 to the signal transducingprotein complex (DISC) of the TRAIL receptors to block TRAIL-mediated inflammatorysignalling. Recent discoveries on the mechanism of signal transduction by the foundingmember of the TNF ligand family, TNF itself and the development of new formulations ofrecombinant TNF have reopened the opportunity to use TNF for cancer therapy.New discoveries in the signal transduction pathways induced by TNF receptor 1 (TNFR1)indicate that similar to the TRAIL receptors, disruption of the TRADD-TRAF2 interactionmay promote TNF-induced cell death over TNF-induced NF- B activation and thus theinhibitors being developed in the host laboratory for the TRAIL receptor complex may havebroader therapeutic applications (Gyrd-Hansen and Meier 2010, Nat Rev Cancer). Equallyimportantly, a large number of tumours secrete TNF and maintain an autocrine TNFsignalling loop (Wu, Boyer et al. 1993, Cancer Res; Stifter, Heiss et al. 2005, Eur JHaematol). This loop drives constitutive NF- B and AP-1 activities, promoting tumourinvasiveness, drug resistance as well as proliferation. Breaking this cycle by targeting thepro-inflammatory/proliferative signalling may offer a means to sensitise tumour cells to TNF-induced cell death allowing the use of lower, non-toxic, doses of TNF that still produce thedifferent anti-tumorigenic effects of TNF both on the tumour cells themselves as well as onthe tumour vasculature.
Aim: The aim of the current project was to determine whether blocking the recruitment ofTRAF2 to the TNFR-DISC can prevent TNF-mediated inflammatory and proliferativesignalling and promote TNF-mediated cell death. For the studies, cell lines engineered toexpress a TRADD-binding deficient TRAF2 will be utilised. These studies aimed todetermine whether the TRADD-TRAF2 interaction inhibitors have a broad therapeuticpotential and to provide important data to asses the feasibility of the development of theinhibitors into lead molecules.
Methods HCT116 and Colo205 cells were used for the study. Both of these colon carcinomacell lines respond to TNF with prolonged NF-kB activation. We have previously establishedTRAF2 stable knockdown strains of these cell lines using an shRNA-expressing lentiviralconstruct.NF- B-mediated pro-inflammatory signalling has been monitored by detecting NF- Btransactivating activity using an NF- B reporter construct (from SystemBio, lentiviral

transduction) after 4-24 h of TNF treatment, I B phosphorylation and degradation withWestern blotting after 5 -120 min of TNF treatment.TNF-induced cell death signalling has been quantified and the mechanism determined bymeasuring loss of mitochondrial transmembrane potential or Annexin V and propidiumiodide staining.To detect the interaction between TRADD and TRAF2 a novel protein-protein interactionassay has been developed. We generated and purified recombinant TRADD and TRAF2proteins with penta-histidine and Strep-II tags respectively. The interaction between the twoproteins was monitored using AlphaScreen technology.TRADD was linked to a Nickel-coated acceptor bead while TRAF2 was connected to an anti-TRAF2 antibody coated Donor bead. Upon excitation of the donor bead, the singlet oxygenreleased by the donor bead has reached the acceptor bead and generated luminesce indicatingthat the two proteins have bound to each other.We could use this system to assess the ability of novel small molecule inhibitors of theTRADD-TRAF2 interaction designed by the host laboratory.The cell line assays showed that TNF induced prolonged NF-kB activity in cells. In cellsdeficient for TRAF2 TNF induced high levels of cell death while TRAF2 proficient cellswere fully protected.Overall the results of the summer project highlighted the TRADD-TRAF2 interaction as apotential target for drug design to interfere with death ligand-induced pro0inflammarotysignalling.
Recent relevant publications by the supervisor:Szegezdi E, van der Sloot AM, Mahalingam D, O'Leary L, Cool RH, Muñoz IG, Montoya G, Quax WJ, de JongS, Samali A and Serrano L. (2012) Kinetics in Signal Transduction Pathways Involving PromiscuousOligomerizing Receptors Can be Determined by Receptor Specificity: Apoptosis Induction by TRAIL. Mol CellProteomics. 1: M111.013730.
van der Sloot AM, Tur V, Szegezdi E, Mullally MM, Samali A, Serrano L and Quax W. (2006) Computationaldesign of a DR5 receptor selective TRAIL variant. Proc. Natl. Acad. Sci. USA, 103: 8634–8639.
Mahalingam D, Natoni A, Keane M, Samali A and Szegezdi E. (2010) Early growth response-1 is a regulator ofDR5-induced apoptosis in colon cancer cells. Br J Cancer. 102:754-64.
Mahalingam D, Keane M, Pirianov G, Mehmet H, Samali A and Szegezdi E. Differential activation of JNK1isoforms by TRAIL receptors modulate apoptosis of colon cancer cell lines. Br J Cancer. 2009, 100(9):1415-24.

Danielle de Bourcier Biochemical Society Summer Studentship Report 2013
Dynamics of the coiled-coil Fos-W homodimer
Danielle de BourcierUniversity of East Anglia, Norwich, UKSupervisor: Tharin Blumenschein
Background and aims of the project
This project focused on the Jun-Fos Activator Protein-1 (AP-1) which is a heterodimeric transcription factorcomposed of the proteins, c-Fos and c-Jun. AP-1 has been found to be responsible for the regulation of anumber of key genes and has subsequently led to the discovery of AP-1’s involvement in various diseasesincluding cancer. Because of this, there is interest in finding peptides capable of sequestering key componentsof AP-1. This would make it possible to prevent its function and therefore is a foundation for drug design withthe potential to combat diseases. Fos-W is a new binding partner which was developed to bind with a higheraffinity to the corresponding coiled coil regions of c-Jun than the cellular protein c-Fos. This prevents c-Junfrom binding c-Fos and therefore from interacting with DNA.i
The aim of my project was to generate double labelled Fos-W peptide using bacterial transformation,expression, purification and digestion. The Fos-W peptide could then be used to acquire triple-resonance NMRexperiments for backbone assignment in the presence and absence of c-Jun. ULP1 was also produced as it wasrequired for the digestion of the fusion protein to His-tagged SUMO (small ubiquitin-like modifier) to give thepeptide we desire. This data can then be processed using computer software to fully assign Fos-W. NMRrelaxation experiments can also be performed.
Work carried out
Media for bacterial growth (minimal and LB) was prepared along with solutions and buffers fordialysis and other general lab work.Transformation of bacteria.Expression in E. coli as N15 labelled protein and purified using Ni-affinity chromatography.Preparation of samples for NMR experiments.Acquiring 1D, 2D and 3D nuclearmagnetic resonance spectra using 500and 800 MHz Bruker NMRspectrometer.Processing, analysis and assignment oftriple-resonance NMR spectra andNMR relaxation measurements.
Results
I acquired and processed the 2D [1H, 15N]-HSQCand 3D CBCA(CO)NH and HNCACB spectra ofFos-W at 25 C (Figure 1), which led to theassignment of the Fos-W backbone.
T1, T2 and NOE relaxation experiments were performed at500 MHz (red) and 800 MHz (blue) (Figure 2). Generallyuniform dynamics are apparent throughout the majority ofthe sequence with the C terminal showing greater flexibility than the N terminal end of the peptide.
Figure 1 - Assigned 2D [1H, 15N]-HSQC spectrumof FosW homodimer.

Danielle de Bourcier Biochemical Society Summer Studentship Report 2013
There was insufficient time to perform experiments on Fos-W in the presence of c-Jun. However relaxationdata for the peptide was acquired which wasn’t in the original proposal.
Figure 2 – A. NOE Relaxation data of FosW at 800 (blue)and 500 (red) MHz B. T1 Relaxation data of FosW at 800(blue) and 500 (red) MHz. C. T2 Relaxation data of FosWat 800 (blue) and 500 (red) MHz
Future directions in which the project can be taken
To repeat the NMR experiments on Fos-W in the presence of c-Jun to compare against the results acquired forthe homodimer and determine how it is affected.
Value of studentship
I thoroughly enjoyed taking part in this summer placement and I feel that my practical laboratory skills andconfidence have improved considerably. Many of the techniques I learnt were completely new to me and willhelp me with final year research project. I have found it extremely interesting to delve into more biologicalaspects of chemistry and have enjoyed broadening my knowledge around the subject of NMR. Gaining first-hand experience of research in a laboratory, alongside PhD students has influenced me to aspire to complete aPhD once I finish my Undergraduate Master’s degree. I was also given the opportunity to put together a posterof my research for the CCPN conference in Leicester. The experience I have gained over the summer has beeninvaluable and I would highly recommend the scheme to anyone thinking of pursuing a career in research.
Value to the Laboratory
Danielle has acquired and analysed essential experiments in a project that is mainly being performed byundergraduate students, either during their final year projects, or as summer students. Once the analysis ofthe cJun-FosW pair is completed, it will result in a publication, which will also be used as preliminary results forgrant applications. In short, Danielle directly contributed to advance the research in my group.
i Jonathan A. R. Worrall, Jody M. Mason, Thermodynamic analysis of Jun-Fos coiled coil peptideantagonists, FWBS Journal 278 (2011) 663-672
A
C
B

Danielle de Bourcier Biochemical Society Summer Studentship Report 2013

Can modulation of amine oxidase derived H2O2 play a role inabrogating oral squamous cell carcinoma progression?
Student: David Naughton
Supervisor: Dr Jeff O’Sullivan, School of Dental Science, Trinity College Dublin
Background: Oral cancer is the sixth mostcommon form of cancer with an incident rateequivalent to that of cervical cancer.Dysregulation of apoptotic pathways play amajor role in the progression of tumours andcan involve hydrogen peroxide (H2O2), whichcan modulate the regulation of thesepathways. A source of H2O2 particularlypertinent to cancer is produced via the amineoxidase group of enzymes which oxidiseamine substrates with a stoichiometricrelease of H2O2. Many oral cancers presentwith a p53 mutation and as H2O2 has theability to drive p53-mediated apoptosis it isimportant to try to understand thismechanism. Should amine oxidase activityprove to be an apoptotic inducer in these oralcancer cell lines, it may lead to personalisedtreatments in patients who present with anon-mutated p53 phenotype.
Aims: The aims of this project were toestablish if primary amine oxidase (PrAO)activity can induce apoptosis in oral squamouscell carcinoma (OSCC) cells through theoxidation of exogenous and endogenousamine substrates. Characterisation of PrAOwas explored utilising a range of substrates.Further examination of a role for aminederived H2O2 in oral cancer cell lines where ap53 mutation exists was also explored.
Description of Work Carried Out:
PrAO assay: Spectrophotometric analysis ofPrAO activity was determined by following theassay described by Holt et al. Cells (2 X104
cells per well) were seeded in a 96-well plateovernight. Cells were treated with a range of
concentrations of primary amine oxidase(PrAO) and amines in a solution of vanillic acid(1mM), 4-aminoantipyrine (500µM) andhorse-radish peroxidase (4 U/ml) in apotassium phosphate buffer (0.2M, pH7.6).Reactions were followed in a microplatereader at 37 C at 490nm. Absorbance wasread every 10 s for 30 min.
Cell Culture: Ca9-22 and Tr146 cells wereused as models of OSSC for this study. Ca9-22cells were maintained in minimum essentialmedium supplemented with 10% heatinactivated fetal bovine serum (FBS), 1%penicillin and streptomycin and 2mM L-glutamine. Tr146 cells were maintained inDulbecco’s modified eagle’s medium withGlutamax supplemented with 1% penicillinand streptomycin and 10% heat inactivatedFBS. Both cell lines were cultured at 37 C in anincubator maintained at 5%CO2.
Cell viability: Cells were seeded in a 96-wellplate (2 X104 cells per well) and maintainedfor 24h. Prior to the prescribed treatmentsthe cells were serum starved in mediacontaining 1%FBS for 6 hours. After exposureto the prescribed treatment for 20h in fullmedia, Alamar Blue was added to a finalconcetration of 10% (Invitrogen, Carlsbad, CA,USA) to determine viability by measuringrelative absorbance of the cells.
Cell cycle analysis: Cells were seeded in 6-wellplates (3 X 105 cells per well) and serumstarved overnight with 1%FBS. They weretreated as described for 24h, in full media.The cells were harvested and suspended inice-cold ethanol overnight at 4 C. They were
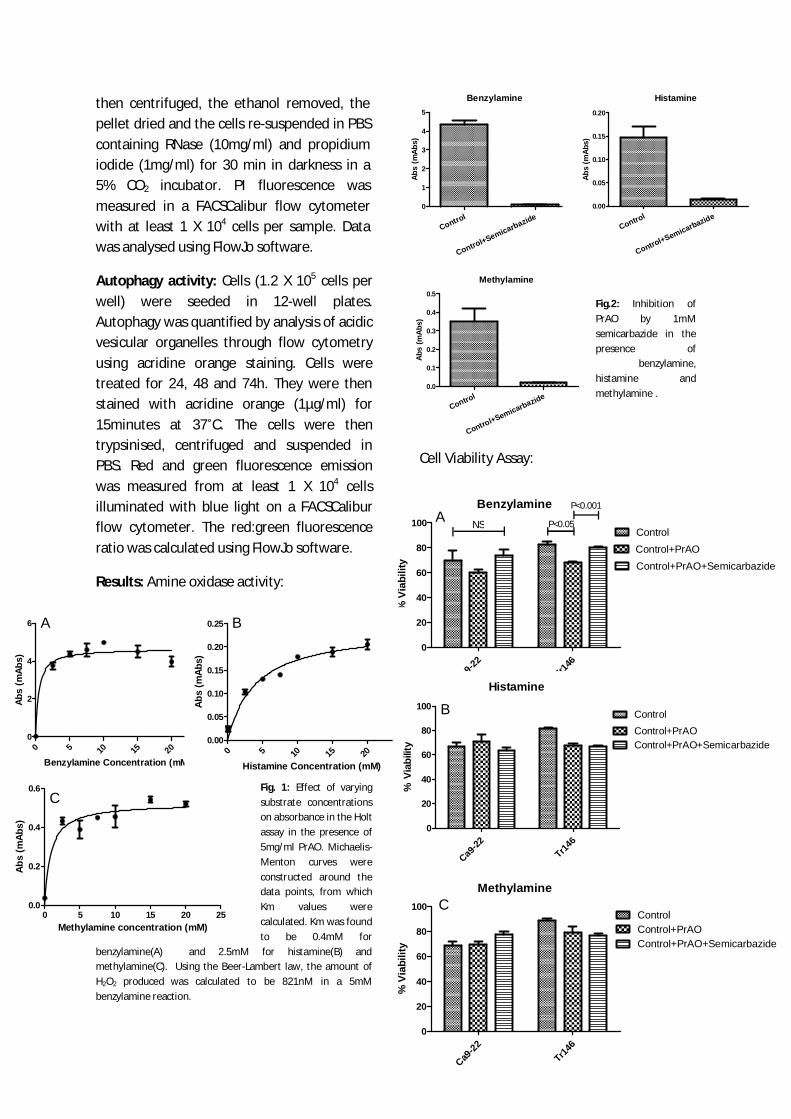
Benzylamine
Ca9-2
2
Tr146
0
20
40
60
80
100
Control+PrAO
Control+PrAO+Semicarbazide
Control
%V
iab
ility
0 5 10 15 20 250.0
0.2
0.4
0.6
Methylamine concentration (mM)
Ab
s(m
Ab
s)
NS P<0.05
P<0.001
then centrifuged, the ethanol removed, thepellet dried and the cells re-suspended in PBScontaining RNase (10mg/ml) and propidiumiodide (1mg/ml) for 30 min in darkness in a5% CO2 incubator. PI fluorescence wasmeasured in a FACSCalibur flow cytometerwith at least 1 X 104 cells per sample. Datawas analysed using FlowJo software.
Autophagy activity: Cells (1.2 X 105 cells perwell) were seeded in 12-well plates.Autophagy was quantified by analysis of acidicvesicular organelles through flow cytometryusing acridine orange staining. Cells weretreated for 24, 48 and 74h. They were thenstained with acridine orange (1µg/ml) for15minutes at 37 C. The cells were thentrypsinised, centrifuged and suspended inPBS. Red and green fluorescence emissionwas measured from at least 1 X 104 cellsilluminated with blue light on a FACSCaliburflow cytometer. The red:green fluorescenceratio was calculated using FlowJo software.
Results: Amine oxidase activity:
Fig. 1: Effect of varyingsubstrate concentrationson absorbance in the Holtassay in the presence of5mg/ml PrAO. Michaelis-Menton curves wereconstructed around thedata points, from whichKm values werecalculated. Km was foundto be 0.4mM for
benzylamine(A) and 2.5mM for histamine(B) andmethylamine(C). Using the Beer-Lambert law, the amount ofH2O2 produced was calculated to be 821nM in a 5mMbenzylamine reaction.
Fig.2: Inhibition ofPrAO by 1mMsemicarbazide in thepresence of
benzylamine,histamine andmethylamine .
Cell Viability Assay:
0 5 10 15 20 250
2
4
6
Benzylamine Concentration (mM)
Ab
s(m
Ab
s)
0 5 10 15 20 250.00
0.05
0.10
0.15
0.20
0.25
Histamine Concentration (mM)
Ab
s(m
Ab
s)
Benzylamine
Control
Control+Semicarbazide
0
1
2
3
4
5
Ab
s(m
Ab
s)
Histamine
Control
Control+Semicarbazide
0.00
0.05
0.10
0.15
0.20
Ab
s(m
Ab
s)
Methylamine
Control
Control+Semicarbazide
0.0
0.1
0.2
0.3
0.4
0.5
Ab
s(m
Ab
s)
Histamine
Ca9-2
2
Tr146
0
20
40
60
80
100Control
Control+PrAOControl+PrAO+Semicarbazide
%V
iab
ility
Methylamine
Ca9-2
2
Tr146
0
20
40
60
80
100ControlControl+PrAOControl+PrAO+Semicarbazide
%V
iab
ility
A B
C
A
B
C

Fig 3: Cell viability for both cell lines with 2mMbenzylamine (A), 10mM histamine (B) and 10mMmethylamine (C). For each amine substrate, the Tr146cells show more resistance to cell death than the Ca9-22cells. There is also a significant drop in viability observedin the Tr146 cell line when exposed to benzylamine andPrAO.
Cell Cycle Analysis:
Ca9-22 cells showed an increase in the Sub-G1phase, representing apoptosis however this isnot significant. Tr146 cells also showed anincrease in the Sub-G1 phase along with acorresponding increase in the G2/M phasefurther signifying apoptosis (Fig 4).
Induction of autophagy: Both cell linesshowed no significant autophagy activity inthe presence of benzylamine (Fig 5).
Discussion:
Benzylamine derived H2O2 was the only amineproduct to elicit a response, to a significantlevel, of apoptosis by the OSSC cell lines.Histamine showed a significant drop in cellviability but this could not be solely attributedto the presence of H2O2. Tr146 cells, whichhave no known p53 mutation showed asignificant increase in apoptosis which wasnot observed in the Ca9-22 cells, which have awell documented p53 mutation. Anobserverable arrest of the G2/M phasecoupled to an increase in the sub-G1 phaseserves as confirmation that apoptoticmechanisms are activated. This observationmay be a result of the functioning p53 andconfirms a role for H2O2 in the apoptoticpathway while suggesting that p53 mediatedapopotosis does not function in patientspresenting with p53 mutation. It also opens
Fig. 4: Cell cycle analysis of Ca9-22 and TR146 cells aftertreatment with 2mM benzylamine.
Fig. 5: Flow cytometric analysis of Ca9-22 and Tr146 cells after treatmentwith 2mM benzylamine for 24, 48 and 72h followed by staining withacridine orange.
P<0.05 P<0.05

up a potential therapeutic avenue utilising apersonal treatment plan dependent on thep53 status of the patient and makes possiblethe opportunity to develop an amine basedtherapy.
How the grant has shaped my future careerprogression/what I got from the grant.
As well as being a thoroughly enjoyable 8weeks, I also felt my time in the lab was ofgreat benefit. I was fortunate to be able towork alongside many PhD students withwhom I could discuss results and from whom Icould get a good insight into the type of workwhich was going on around me. Furthermore,I found it interesting to see the differentoutlooks I had, having a scientific but alsoclinical background in dentistry, compared tothose who were studying and researching oralcancers from a more laboratory basedstandpoint.
In the long term I hope to integrate my clinicalpractice of dentistry and research. Thetechniques I have learned and practicedduring this studentship will hopefully be animportant building block in a career centredaround patient care, research and continuing-education.
Selected References:
Holt A, Sharman DF, Baker GB, Palcic MM; AContinuous Spectrophotometric Assay forMonoamine Oxidase and Related Enzymes inTissue Homogenates; Analytical Biochemistry,1997 Jan; 244(2): 384-392
O'Sullivan J, Unzeta M, Healy J, O'Sullivan MI,Davey G, Tipton KF; Semicarbazide-sensitiveamine oxidases: enzymes with quite a lot todo; Neurotoxicology, 2004 Jan; 25(1-2):303-15.
Greene LM, Nolan DP, Regan-Komito D,Campiani G, Williams DC, Zisterer DM;
Inhibition of late-stage autophagysynergistically enhances pyrrolo-1,5-benzoxazepine-6-induced apoptotic cell deathin human colon cancer cells. InternationalJournal of Oncology, 2013 Sep; 43(3):927-35.

Biochemical Society Summer Studentship Report 2013Modulating the cellular phenotype of collagen IV mutations by targetingprotein degradation pathways.
Student: Emma Campbell,Molecular and Cellular Biology,University of GlasgowSupervisor: Dr Tom Van Agtmael,University of Glasgow
1. The aims of your project
Collagen IV is the major structural component of the basement membrane and COL4A1/COL4A2mutations (collagen IV alpha chains 1 and 2) cause cerebrovascular diseases such as haemorrhagicstroke. Mutations lead to intracellular accumulation of collagen IV which causes ER stress andapoptosis in cells. The aim of this project was to investigate and modulate mutant proteindegradation within primary dermal fibroblast cells containing a COL4A2 mutation, in comparisonwith control cells. It was hypothesised that increased protein degradation in COL4A2 mutant patientcells may reduce the cellular phenotype asociated with COL4A1/COL4A2 mutations and thus posepotential therepeutic benefits.
2. A description of the work carried out
The aim of the project was to investigate if promotion of proteasomal activity would decrease theintracellular collagen IV accumulation. However, no FDA approved drugs are available that promoteproteasome activity without leading to apoptosis. Experiments in the lab were performed before Ijoined which tested a compound, betulinic acid, that was reported to promote proteasomal acivity.These experiments failed to show any activity of this compound. Thus this project focused on thrstimulation of autophagy.Patient and control cells were cultured in media containing ascorbic acidsto promote collagen expression. Cells were then individually treated with several autophagyinhibitors and one FDA approved compound, carbamazepine, which promotes autophagy. Autphagywas inhibited using LY294002 and Wortmannin which both acts of phagosome formation whiletreatment with E64D and Pepstatin A was used to inhibit protein degradation within the lysosome.Protein samples were collected of treated and untreated cells in RIPA buffer and analysed bywestern blot analysis. The expression of the ER stress marker BiP will be investigated, alongsidecollagen (using the M26 antibody) for assessing the intracellular accumulation. The levels ofautophagy were tested using using antibody against LC3.
3. An assessment of your results and the outcomes of the studentship
The dermal fibroblasts expressing mutant COL4A2 showed a defect in cell proliferation andincreased levels of apoptosis. This severely delayed the collection of cells and the preparation ofprotein samples. The initial collection of protein samples was performed on cells that did not have asufficient level of confluence. Having to repeat the cell collection and treatment severely impactedon the progress during my project. New protein samples were collected and western blot analysiswas performed for the autophagy marker LC-3. Preliminary data showed that treatment withcarbamazepine had little effect on autophagy levels in control cells. However increased levels of LC3were observed in patient cells. This may indicate that in WT cells LC3 is not elevated as there is no

need for this to occur as there in co accumualtion of Col4a2. However in patients cells the increaseautophagy may indicate and attempt by the cell to degrade intracellular Col4a2. There was notenough time to investigate the levels of Col4a2 and Bip to confirm whether this treatmenteffectively reduced Col4a2 accumulation and ER stress.
I also assisted in the analysis of the kidney disease caused by Col4a1 mutations. Work wasperformed to investigate which sodium channels in the kidney were responsible for the increasedsodium re-absorption in 4 month old Col4a1 mutant mice. Increased levels of the NHE3 sodiumchannel revealed that increased sodium re-absorption is achieved in the proximal tubule of thekidney.
4. Future directions in which the project can be taken
If time had permitted, the described experiments would have been repeated in order to gain anappropriate number of results to draw more accurate conclusions. Protein collection from patientcells would be optimised, as difficulty was exhibited in cell density due to their nature of growth. Theobtained results suggest that the described drug treatments may provide a therepeutic role indegradation of protein accumulant in patients exhibiting a COL4A2 mutation.
5. Any departures from the original proposal
Progress was delayed due to the reduced cell proliferation of the mutant cells and the collection ofprotein samples from cells that were not confluent enough. The period during which cells weregrowing, I assisted with other project in the lab which included the immortalisation of patient cellsby continuous passaging. Work was also performed on the analysis of the kidney disease caused byCol4a1 mutations.
6. Benefit of Studentship
The Biochemical Summer Studentship has awarded me the opportunity to work within cutting-edgeand novel scientific research, while undertaking an individually focused project. Prior to commencingthe project I was certain that a PhD was the next logical step in my career. My research project hasreinforced this, and has allowed me first hand experience which will be of the utmost importancewhen selecting a PhD. The reality of scientific research has been enforced; the time frames, day-to-day work, and the importance of troubleshooting and motivation skills when things are not going asplanned.
Within research it is of benefit to have a large repertoire of skills, and my summer placement hasallowed me to vastly increase this e.g. the use of human cell culture, something which I have not hadthe opportunity to do within my undergraduate degree. My daily planning, excecution,interpretation and troubleshooting of experiments has been developed, and through this a new wayof thinking has evolved which differs vastly from lecture based learning.
I have been involved in bi-weekly interdepartmental meetings and presentations, which haveallowed me to gain insight from many different academic perspectives and utilise their expertise. Asa result I have come to appreciate the benefits of cross disciplinary research.
I would highly recommend the Biochemical Summer Studentship for undergraduates interested inpersuing a career in research. The personal and professional benefits are countless, and provide asolid grounding for University final year projects.

Student: Evie Rejnowicz Supervisor: Dr Helen O’Hare University of Leicester
My summer studentship project focused on investigating the conserved patch on the surface of the RNApolymerase binding protein A (RbpA) in M. tuberculosis. It was postulated by Bortoluzzi, A., et al., 2013that this patch could be involved in the binding of subunit of RNA polymerase. The project’s aim wasto analyze the importance of the amino acids within the patch in binding RNA polymerase by utilizingsite – directed mutagenesis technique and subsequently assessing the binding between RbpA and RNApolymerase.
The first stage of the studentship was to select amino acids to be mutated and design the primers thatwould enable the insertion of the mutations into RbpA. Overall, six amino acids were selected formutation (A48M, D47K E49K, W54F, R27E, A45Q, and W54I). Rv2050 (gene encoding RbpA in M.tuberculosis) with a hexahistidine tag had been cloned into pLEICS 01 vector that would serve as atemplate for PCR. Three mutations were chosen to be inserted into the gene via long PCR of the wholeplasmid (R27E, A45Q and W54I) whereas the other three were inserted by using university-based facility(Protex) utilizing 2-step PCR and ligase-independent cloning. Due to unsuccessfulness of the first methodeventually all the mutations were inserted within the gene exploiting Protex technique.Having received the complete set of plasmids, they were sequenced and transformed into E. coli BL21strain. All of the mutated proteins had their solubility and level of expression tested which served as themajor criterion for selecting mutants for large scale expression. Four proteins (D47K E49K, R27E, W54Iand W54F) were designated for this step. Purification of the proteins was performed by using Ni – NTAagarose. Furthermore, to confirm the proteins purified are of the right nature we performed secondarystructure studies using circular dichroism technique on two of the mutants: D47K E49K and R27E andcompared them with the previously established secondary structure of wild type RbpA.
The last stage of the project investigated the role the mutations in RbpA and their impact on RNApolymerase interaction. This assessment was done by using pull down assay, successfully performed byDey, et al., 2010. The last two weeks were focused on the optimization of the protocol found in thepaper. The project concluded at the level of optimization of the pull down assay using wild – type RbpAand partially purified RNA polymerase. Additionally, to confirm the presence of subunit in the pulldown assay, Western blot was performed using mouse antibodies raised against subunit of E. coli RNApolymerase.
It has been established that, surprisingly, all of the mutated proteins were sufficiently soluble to beexpressed on a large scale. Mutations do not seem to have an effect on the structure of RbpA, evenwhen the most conserved amino acid – tryptophan is altered. Circular dichroism indicates that at leasttwo of the mutants purified (R27E, D27K E49K) are suitable for testing with RNA polymerase. Fourmutants expressed were purified in quite high yields: 4.2 mg, 15.2 mg, 16.0 mg, 10.6 mg from 1 litre forR27E, D47K E49K, W54F and W54I, respectively. It is worth noting that D47K E49K mutant wasexpressed at very high levels in E.coli when grown at 30°C for 4 hours.
Unfortunately we were unable to reproduce the pull-down method described in Dey et al., 2009. Certainsteps were taken to optimize the pull down assay which is necessary for further investigation of RbpAmutants and their impact on RNA polymerase binding. These steps included use of M. smegmatis lysateas a negative control, use of lysate rather than partially purified RNA polymerase to investigate the

Student: Evie Rejnowicz Supervisor: Dr Helen O’Hare University of Leicester
interaction; varying the NaCl and imidazole concentration in the pull down buffer. However, Westernblotting using an antibody against RNA polymerase subunit showed that this subunit did not bindRbpA under the assay conditions.
Based on the findings the project could be expanded in term of short – term and long – term directions.The priority lies within the optimization of the pull down assay. As this is an integral part testing theimportance of the conserved patch in binding of RbpA to RNA polymerase it is reasonable to firstoptimize the pull down assay using wild type RbpA and then repeat the process on the purified proteinmutants.Long term directions regarding the project could involve utilizing new methods to confirm the outcomeof the pull down assay. These methods could include yeast two – hybrid system, transcription assay orpromoter binding assay. The project utilizes M. smegmatis RNA polymerase and RbpA coming from M.tuberculosis. Future work may include expressing mutated RbpA in M. tuberculosis cells and testing theviability of the microorganism in these conditions and its virulence.
The studentship was a great opportunity to broaden my knowledge in both Biochemistry as well asMicrobiology. As second year practical classes mainly comprised of physiology techniques this projectwas a chance to learn a different skill set related to recombinant protein production. I also learntexperimental techniques associated with genetics (gel electrophoresis of DNA, plasmid extractions) andBiochemistry (SDS PAGE, Western blot, column chromatography). To familiarize myself with theresearch and the project I had to read scientific articles. It was invaluable to be able to get used to thelanguage used in these publications, as well as critically analyze published resources. All of these aspectsprepared me for the challenge of the third year project that I will be undertaking next academic year. Asa person who wishes to pursue PhD as a future career, the studentship gave me an insight intochallenging world of the scientific research and enabled me to choose my potential area of research as amore aware candidate. Additionally, the studentship improved my CV by the relative work experienceand made me a more competitive applicant for research – based jobs.
Besides contributing to my career aspiration, the studentship has also contributed to the ongoingproject on RbpA within the lab. The expression plasmids for the six variants of RbpA, and the pureprotein samples produced will be used to complete the investigation of the role of the conserved patchon the surface of RbpA.
References:
1. Dey, A., Verma, A, K., Chatterji, D., 2010. Role of an RNA polymerase interacting protein,MsRbpA, from Mycobacterium smegmatis in phenotypic tolerance to rifampicin. Microbiology, 156, 873– 883.
2. Bortoluzzi, A., Muskett, F. W., Waters, L. C., Addis, P. W., Rieck, B., Munder, T., Schleier, S.,Forti, F., Ghisotti, D., Carr, M. D., O’Hare, H. M., 2013. Mycobacterium tuberculosis RNA Polymerase-binding Protein A (RbpA) and Its Interactions with Sigma Factors. The Journal of Biological Chemistry,288, 14438 – 14450.

Biochemical Society Studentship Report 2013Ferrochelatase Activity in Human Lymphocytes: Influence onPhotodynamic Therapy (PDT).Student: Fatima Ulhuq, Supervisor: Dr Julie Woods, Photobiology Unit, University of Dundee
Background and Aims
PDT is based on the activation of photosensitizers (PS) that are localized in target tissues. It involves administration of anon-toxic photosensitizing drug, which accumulates in tumour cells, and subsequently these PS can be activated with theappropriate wavelength of light corresponding to the PS absorption wavelength. This then generates active molecularspecies, such as free radicals and singlet oxygen that are toxic to cells and tissues. (1)
The addition of precursors such as 5-aminolevulinic acid (ALA) increases the selectivity of PDT. ALA is a precursor for thenatural photosensitizer protoporphyrin IX. Illumination of the tumor site with red light activates PpIX, around 630nm is thepreferred wavelength due to its tissue penetration, triggers the oxidative damage and induces cytotoxicity. Fluorescencediagnosis (FD) of non-melanoma skin cancers can be used diagnostically by exciting the PpIX with blue light (400nm). Whichthen causes release of energy in the form of red light at 635nm and so a red fluorescence is observed. (2)
However, there can be significant individual variation in the amount of PpIX formed from the prodrug, which may effect theeffectiveness of PDT, the FD of the lesion, and the amount of pain (in topical PDT) experienced during treatment. Thisvariation might be partly due to the rate-limiting enzyme ferrochelatase (FECH), which inserts a ferrous (Fe2+) ion into PpIXto produce haem. If FECH is inhibited, haem is not synthesized and PpIX begins to accumulate. The primary aim of this studywas to isolate lymphocytes from individuals and treat them with the prodrug ex-vivo. The amount of PpIX synthesized bythe cells and their sensitivity to PDT was determined. This was to help us understand if there is a clear link between FECHactivity inside cells, PpIX fluorescence (amount) and sensitivity to PDT treatment. (3)
Experimental Procedures
Cell Lines and Maintenance: U937 cells from human histocytic lymphoma were maintained in RPMI containing 10% (v/v)FCS. Mononuclear cells (human lymphocytes) were isolated by density gradient separation on Ficoll. Blood was obtainedfrom healthy volunteers from the department. The isolated lymphocytes were then split up and used for % viability, FD andthe FECH activity assay. U937 cells were used initially for method development.
% Viability: The effect of the irradiation and subsequent damage to the cells by PpIX is measured by calculating thepercentage viability of the cells, compared to sham (dark) samples. Working solutions of ALA were made in EBSS, andcontrol solution by adding DMSO to EBSS. 100uL of cells were aliquoted into the wells of a 96 well plate. Two duplicateplates were prepared so one was irradiated and the second was the sham (dark) sample. The samples were covered intinfoil to protect them from the light and placed in the incubator for 4 hours to allow PpIX to form. Then the samples wereirradiated with light using the PDT 1200 light source, used for patients during PDT treatment, initially at two differentintensities of 10J/cm2 and 25J/cm2. After the experiments using U937 cells it was found that the required dose to treat theisolated lymphocytes would be 4mM ALA, irradiated at a dose of 25J/cm2 with a cell density of 2 x 106cells/ml.
FECH Assay: FECH catalyzes the final step in the heme biosynthetic pathway by inserting ferrous ion into the PpIX ring toform heme. Zinc can be used as an alternative to ferrous ion. Mesoporphyrin was used as the alternative substrate to FECHas it is more chemically stable than PpIX. The cell extract was prepared by sonicating in an extraction buffer. The reactionbuffer contained 50uM mesoporphyrin in Tris/Palmitate/Tween buffer. One sample contained 20uL of cell homogenate inreaction buffer. Another sample without cell homogenate was the non-enzymatic control samples. Another sample wasprepared, cell homogenate, reaction buffer, however no zinc acetate was added. After the incubation period adding 500uLof stop solution terminated the reaction. The samples were analysed using the spectrofluorimeter with fluorescenceemission at an excitation wavelength of 410nm, and an emission range between 500-750nm. (4)
Data Analysis: The fluorescence detected in the FECH activity assay in the presence of lymphocytes was quantified to nMper mg protein using BCA assay and zinc-mesoporphyrin standards.

Results
By using the U937 cells first important factors were determined before using human lymphocytes. As the concentration ofALA was increased from 0-2mM, there was a continuous decrease in % viability. However, as the concentration was thenfurther increased the response came to a plateau. This allowed us to determine the concentration of ALA to be used withthe lymphocytes would be between the plateau phase of 2.5 to 5mM. As the light dose was increased from 10J/cm2 to25J/cm2 there was a further decrease in the percentage of viable cells. The viability of the cells kept in the dark remainedfairly constant around the 100%, so PpIX was not toxic in the dark. These results show that there is an effect between thosecells exposed to light + PpIX and those in the dark + PpIX. This suggests that PpIX is being formed in the cells. To supportthis, the fluorescence of the samples was taken. The fluorescence intensity of the samples kept in the dark was high, thiswas expected as there was no light and so the PpIX was not photo-bleached. The FECH assay parameters were alsodetermined in the U937 cells. FECH activity showed a linear increase in the amount of Zinc-Mesoporphyrin produced inincreasing U937 cell numbers; 1e6 cells produced 10nM Zinc-Mesoporphyrin, 2e6 produced 18nM Zinc-Mesoporphyrin and5e6 cells produced 38nM Zinc-Mesoporphyrin. (5)
The human lymphocytes showed:1. There was high inter-individual variation in fluorescence intensity despite adding the same amount of ALA to the same
number of cells, which confirms what is seen in skin.2. There was a correlation between fluorescence intensity and % viability.3. The link between fluorescence intensity and FECH activity was not clear.
Figure 1: Results from analysis of human lymphocytes
Future directionsFurther work can be done to improve these results by repeating the experiments again in more subjects and collecting moredata from the lymphocytes.
Departures from the proposalNot all the objectives could be met because of lack of time, including the skin fluorescence in the patients however otherworkers in the lab are continuing this project.
Value of the studentshipMy experience while working within the lab has been very enjoyable, while learning and carrying out new techniques neverexplored in the University laboratory. I understood the true nature of scientific research and this has definitely enhanced mypassion to continue in the field of research and pursue a PhD after my studies. The skills, which I have gained, will beextremely important during my 4th year Honours project. I would like to thank the Biochemical Society for giving me thisexcellent opportunity.
LaboratoryFatima’s project is part of a clinical study being undertaken in the Photobiology Unit. Fatima established all of the proceduresto be used in the study using the U937 cell line, and also made a start on analyzing the clinical samples. Her work is a veryimportant contribution to the clinical study, which will be submitted for publication once all the analysis, is complete. Wefound the experience to be very beneficial and enriching to the lab, and were delighted to be able to host Fatima.

References
(1) Yoon I, Zhu Li J, Shim Y, ‘Advance in Photosensitizers and Light Delivery for Photodynamic Therapy’ Clin Endosc. 2013 January; 46(1): 7–23.Published online2013 January 31. doi: 10.5946/ce.2013.46.1.7
(2) Hasan T, Moor ACE, Ortel B. Photodynamic Therapy of Cancer. In: Bast RC Jr, Kufe DW, Pollock RE, et al., editors. Holland-Frei Cancer Medicine. 5thedition. Hamilton (ON): BC Decker; 2000.
(3) Wachowska M, Muchowicz A, Firczuk M, Gabrysiak M.’ Aminolevulinic Acid (ALA) as a Prodrug in Photodynamic Therapy of Cancer’ Molecules 2011, 16,4140-4164; doi:10.3390/molecules16054140
(4) Taketani, S. ‘Measurement of Ferrochelatase Activity’, Current protocols in Toxicology.(5) Valentine RM, Ibbotson SH, Brown CT, Wood K, Moseley H: A quantitative comparison of 5-aminolaevulinic acid- and methyl aminolevulinate-induced
fluorescence, photobleaching and pain during photodynamic therapy. Photochem Photobiol 2011, 87(1):242-249.

Biochemical Society Summer Studentship Report 2013
Involvement of the transcription factor Nrf2 in the regulation ofmitochondrial uncoupling by oxidative stress
Student: Guillermo Serrano Nájera
Supervisor: Dr Susana Cadenas, Department of Molecular Biology, Universidad Autónoma de Madrid
Background and project aims
Uncoupling proteins (UCPs) are transporter proteins present in the inner mitochondrialmembrane. These proteins are able to reduce the electrochemical gradient of themitochondrial inner membrane. The function of UCP1 is adaptive thermogenesis in brownadipose tissue in mammals. However, the role of UCP2 and UCP3 is controversial. Severalreports support a role for these proteins in the control of reactive oxygen species (ROS)production and against oxidative damage.
Our laboratory has recently reported the increase in UCP3 expression in cells exposed to theoxidant hydrogen peroxide (H2O2) through the activation of the transcription factor Nrf2(nuclear factor erythroid 2-related factor 2). In this project we wanted to test whether or notother oxidant, 4-hydroxynonenal (HNE), as well as a reduced oxygen concentration (hypoxia),also induce UCP3 expression via Nrf2.
In addition, UCP3 could have therapeutic potential. This protein is expressed in brown adiposetissue, skeletal muscle and the heart. UCP3 could be important in attenuating ischemia-reperfusion damage. Therefore, other research lines in this project are the study of samples ofischemic hearts of patients, and of mice heart subjected to ischemia-reperfusion using aLangendorff perfusion system. These lines are aimed at studying the cardioprotective role ofUCP3.
Description of the work carried out
To reach our goals we have used the following techniques:
Cell culture: mouse heart muscle cell line (HL1) and mouse skeletal muscle cell line(C2C12).Separation of cytosolic, mitochondrial and nuclear protein fractions.Immunoblotting of cellular fractions to detect HNE-protein adducts, UCP3 or Nrf2.Cell treatments with 4-HNE or antioxidant compounds (MitoQ or NAC), and hypoxia(1% O2).Gene silencing of Nrf2 using RNA interference in HL1 cell culture.RNA extraction from the silenced cells and from tissues.Functional studies of oxygen consumption using the XF24 Seahorse Analyzer.Protein extraction from tissue.Processing of samples of heart of patients for immunoblotting.Setting up a Langendorff perfusion system.

Results
The lipid peroxidation product 4-HNE is able to bind histidine residues in proteins giving HNE-protein adducts. We used specific antibodies against HNE-protein adducts to detect oxidativestress after treatment of the cells with 4-HNE (20 µM) or hypoxia (1% O2). Our preliminaryresults show that these treatments increase UCP3 and HNE-protein adducts. However, theantioxidant NAC prevented the increase in UCP3 and HNE-protein adducts, which suggests thatROS are involved. We studied the involvement of the antioxidant transcription factor Nrf2 inUCP3 induction. Our results in siNrf2 cells suggest that Nrf2 plays a role in 4-HNE-mediatedinduction of UCP3. Functional studies of oxygen consumption with an XF24 Seahorse Analyzerare currently under way in the laboratory.
We also tested whether HNE-protein adducts were augmented in cardiac tissue of patientsduring cardioplegic arrest (a temporary interruption of blood flow to the heart due to aorticcross-clamping during cardiac surgery). We confirmed an increase in HNE-protein adductsduring cardioplegia.
Finally, I have also participated in the satisfactory setting up of a Langendorff perfusion systemand learnt how to apply protocols of ischemia-repefusion and to determine heart damage.
Future directions
The planned future experiments include ex vivo cardiac ischemia-reperfusion using theLangendorff perfusion system, with the aim of studying the protective role of UCP3 against IRinjury. Reperfusion after a ischemic period increases ROS generation. These studies will becarried out in hearts of UCP3 knockout mice and Nrf2 knockout mice, so we can alsodetermine the importance of the Nrf2/UCP3 pathway in the cardioprotective effect of ischemicpreconditioning.
Departures from the original proposal
Apart from the planned HL1 cell cultures, I have also used C2C12 cells from mice muscle andanalyzed samples obtained from the human heart.
Value of the studentship to the student
This studentship has given to me the opportunity to apply the knowledge acquired in myBiochemistry Degree into a research project, to gain experience and also, to learn a lot ofimportant things such as different laboratory techniques or how to organize my work.Moreover, I have learnt how to argue the results in the meetings with people of my group andother laboratories. I have learnt a lot of new things about the electron transport chain,mitochondria, oxidative stress, and, especially, about the role of uncoupling proteins and thephysiology of the heart in relation to interesting topics as ischemia-reperfusion damage orischemic preconditioning. Finally, I have felt very welcomed in the research group and thisstudentship has been a great personal experience.

Value of the studentship to the lab
Guillermo Serrano has worked with dedication and enthusiasm during the time he spent in thelab. He has learnt several techniques in a very short time and managed to complete somespecific goals. The data he obtained confirm and complement previous results from the lab. Hehas integrated perfectly in the team and is highly motivated and involved in the project,participating in the lab meetings in which he has presented and discussed his results.Guillermo now continues working in the lab part-time carrying out his final-year Degreeproject.

Biochemical Society Studentship Report 2013
BIOCHEMICAL SOCIETY SUMMER VACATION STUDENTSHIP REPORT 2013
ROLE OF HYPOXIA INDUCIBLE TRANSCRIPTION FACTORS HIF-1 AND HIF-2 INMACROPHAGE DIFFERENTIATION TOWARDS PRO-INFLAMMATORY M1 OR PRO-RESOLUTION M2 PHENOTYPES
Student: MR HARMONY UWADIAESupervisor: Dr Debra Higgins, UCD Conway Institute, University College Dublin, Belfield,Dublin 4, Ireland
Introduction:
Hypoxia occurs when oxygen supply is insufficient for cellular demand. Hypoxia inducibletranscription factors (HIF-1 and HIF-2 ) play a primary role in mediating cellularadaptation to hypoxia and inflammation through regulation of a number of genes.
When adequate oxygen is present, HIF is hydroxylated by prolyl hydroxylase (PHD)enzymes affording interaction with pVHL, the substrate recognition component of a ubiquitinligase complex. However, in hypoxia, the oxygen co-factor of PHD is limiting, inhibitingPHD function and resulting in stabilisation and nuclear translocation of HIF where it bindsHIF1 forming functional HIF-1 or HIF-2, depending on the HIF -moiety of the complex.Recent studies have shown evidence of differing roles between HIF-1 and HIF-2 in thehypoxic and inflammatory process.
The aim of this study was to determine whether macrophage differentiation towards eitherpro-inflammatory M1 or pro-resolution M2 is influenced by HIF-1 and HIF-2, which mayimpact progression or regression of inflammation in renal injury.
Work carried out:
Bacterial cell cultures were grown and transformed with DNAplasmids containing coding sequences for HIF1 , HIF2 ,empty vector, epo-HRE luciferase or TK Renilla luciferase.Three strategies were employed to optimise transfection-efficiency of THP-1 monocytes and THP-1 derivedmacrophages (Figure 1) using either Fugene HD (Promega),Lipofectamine 2000 (Invitrogen) or HiPerfect (Qiagen) andinvolved transfecting HIF-1 or HIF-2 expression plasmidsinto THP-1 monocytes which were either (1) maintained asmonocytes (left column), (2) differentiated into M with PMA(centre column) or (3) transfected after differentiation into M(right column). Transfection-efficiency was monitored using theepo-HRE reporter luciferase assay for HIF-transcriptionalactivity and normalised to renilla luciferase using DualLuciferase assay (Promega).
Effect of over-expression of HIF-1 and HIF-2 was determined by monitoring changes inexpression of M1 marker; MCP-1, and the M2 markers; TGF and IL-10, using ELISA andquantitative real time PCR analysis.
Figure 1: Schematic depicting the threetransfection strategies employed with eitherFugene HD, Lipofectamine 2000 or HiPerfect.
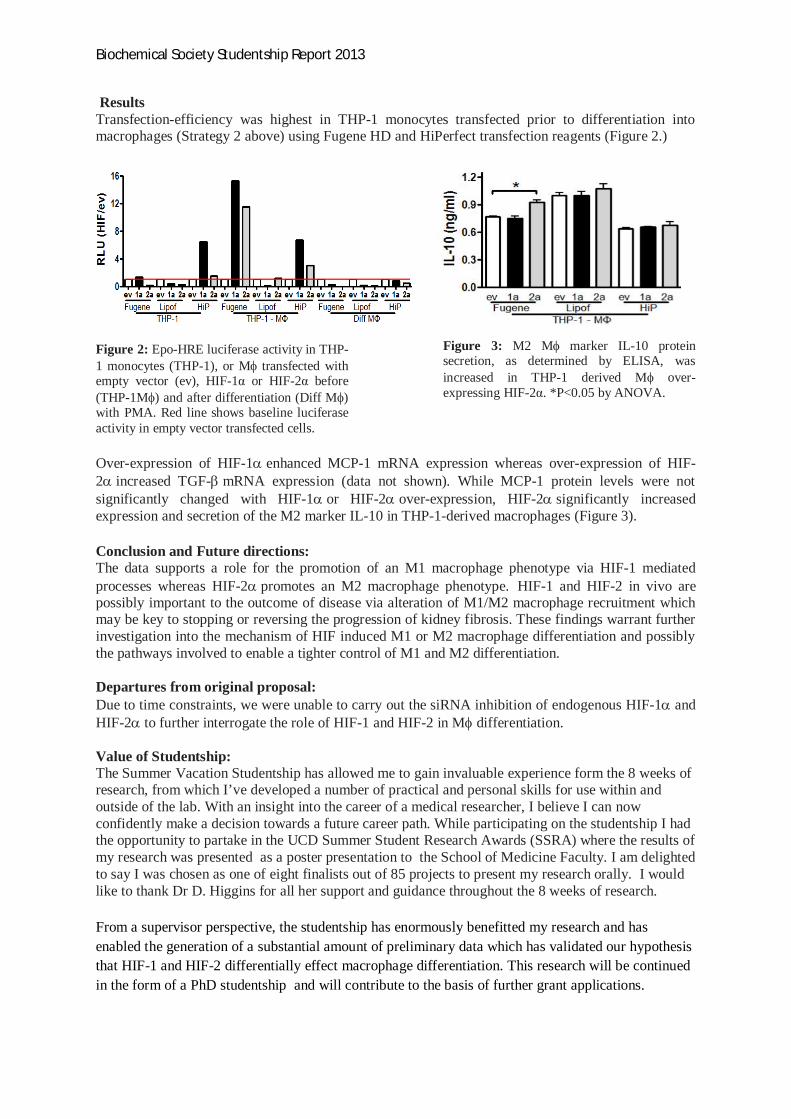
Biochemical Society Studentship Report 2013
ResultsTransfection-efficiency was highest in THP-1 monocytes transfected prior to differentiation intomacrophages (Strategy 2 above) using Fugene HD and HiPerfect transfection reagents (Figure 2.)
Figure 2: Epo-HRE luciferase activity in THP-1 monocytes (THP-1), or M transfected withempty vector (ev), HIF-1 or HIF-2 before(THP-1M ) and after differentiation (Diff M )with PMA. Red line shows baseline luciferaseactivity in empty vector transfected cells.
Figure 3: M2 M marker IL-10 proteinsecretion, as determined by ELISA, wasincreased in THP-1 derived M over-expressing HIF-2 . *P<0.05 by ANOVA.
Over-expression of HIF-1 enhanced MCP-1 mRNA expression whereas over-expression of HIF-2 increased TGF- mRNA expression (data not shown). While MCP-1 protein levels were notsignificantly changed with HIF-1 or HIF-2 over-expression, HIF-2 significantly increasedexpression and secretion of the M2 marker IL-10 in THP-1-derived macrophages (Figure 3).
Conclusion and Future directions:The data supports a role for the promotion of an M1 macrophage phenotype via HIF-1 mediatedprocesses whereas HIF-2 promotes an M2 macrophage phenotype. HIF-1 and HIF-2 in vivo arepossibly important to the outcome of disease via alteration of M1/M2 macrophage recruitment whichmay be key to stopping or reversing the progression of kidney fibrosis. These findings warrant furtherinvestigation into the mechanism of HIF induced M1 or M2 macrophage differentiation and possiblythe pathways involved to enable a tighter control of M1 and M2 differentiation.
Departures from original proposal:Due to time constraints, we were unable to carry out the siRNA inhibition of endogenous HIF-1 andHIF-2 to further interrogate the role of HIF-1 and HIF-2 in M differentiation.
Value of Studentship:The Summer Vacation Studentship has allowed me to gain invaluable experience form the 8 weeks ofresearch, from which I’ve developed a number of practical and personal skills for use within andoutside of the lab. With an insight into the career of a medical researcher, I believe I can nowconfidently make a decision towards a future career path. While participating on the studentship I hadthe opportunity to partake in the UCD Summer Student Research Awards (SSRA) where the results ofmy research was presented as a poster presentation to the School of Medicine Faculty. I am delightedto say I was chosen as one of eight finalists out of 85 projects to present my research orally. I wouldlike to thank Dr D. Higgins for all her support and guidance throughout the 8 weeks of research.
From a supervisor perspective, the studentship has enormously benefitted my research and hasenabled the generation of a substantial amount of preliminary data which has validated our hypothesisthat HIF-1 and HIF-2 differentially effect macrophage differentiation. This research will be continuedin the form of a PhD studentship and will contribute to the basis of further grant applications.

Biochemical Society Summer Studentship 2013 Hayley Anne MouldingCharacterisation of novel protein-protein interactions within the nonhomologous end-joining machinery
Student: Hayley Anne Moulding (University of Bristol)Supervisors: Gabrielle Grundy and Stuart Rulten (Genome Damage and Stability Centre, University ofSussex)
INTRODUCTION
DNA double-stranded breaks (DSB), induced byionising radiation or V(D)J recombination, are dependenton the Ku heterodimer for repair by the classical nonhomologous end-joining (c-NHEJ) machinery (1). TheNHEJ pathway requires multiple different proteins andprotein interactions to ensure genetic integrity includingKu/DNA-PKcs (DNA-PK), XRCC4/LigIV, XLF/Cernnuos,Artemis and DNA polymerases (2). The Ku heterodimer,comprised of 70kDa (Ku70) and 80kDa (Ku80) subunits,directly interacts with DNA-dependent protein kinasecatalytic subunit (DNA-PKcs) generating the pivotalcomplex for DNA DSB repair, DNA-dependent proteinkinase (DNA-PK). Ku binds to DNA double-strand breaks(3), recruiting the other c-NHEJ proteins to enabledamage repair. Ku exhibits many interactions, notjust with DNA-PKcs.
Fig.1 Ku heterodimer (Ku70 (blue) and Ku80 (beige). Ku80-APLF binding site (pink) (4). Figured generated by using Pymol.
Both Ku70 and 80 possess von Willebranddomains, which are positioned at the N-terminus.Protein-protein interactions have been mapped to theKu80 vWA domain; however the vWA domain of Ku70has not been explored. Ku80 vWA recruits Aprataxin-and-PNK-Like Factor (APLF) incorporating it into Ku-DNAcomplex interactions (5). APLF acts as a scaffold proteinenabling ligation of the DSB by recruitment ofXRCC4/LigIV and XLF. By mapping protein interactionswith the Ku70 vWA domain, it will help to elucidate itsrole in assisting the NHEJ pathway and presumablyprotein recruitment to Ku-DNA complexes.
Fig.2 Ku heterodimer binding DNA (green). This is the accesspoint for proteins interacting with the DNA end. Ku70-vWAdomain (light blue) and Ku80-vWA domain (beige). Ku70-hydrophobic pocket (red) (4, 6).
AIMS
The objective of this investigation is toidentify novel protein-protein interactions with thevWA domain of Ku70 by undertaking yeast-2-hybridscreens. The vWA domain of Ku80 was used to probea human brain cDNA library for novel proteinsinteracting with the Ku70 vWA domain. The Ku70-vWA domain construct was ligated into the pGBKT7plasmid, acting as the ‘bait’ protein with the GAL4DNA-binding domain upstream of it. The human braincDNA library was ligated into the pACT plasmid, actingas the ‘prey’ protein which possesses the DNA-activation domain upstream. The concept of yeast-2-hybrids depends on the interaction of the DNA-binding domain with the activation domain, and as aresult the ‘bait’ attracting the ‘prey’ resulting intranscriptional processing. Successful interactionsbetween the Ku70-vWA and prey are detected via twomarkers: histidine synthetase (His3) gene expressionand -galactosidase expression that are stablyincorporated within the Y190 strain. DH5 E.coli canbe transformed with the recovered plasmid and theselected pACT plasmid can be sequenced to identifythe particular protein interaction. This will revealnovel protein-protein interactions with the Ku70-vWAdomain. Interactions will ultimately be tested too ifthey are mediate by an equivalent hydrophobicpocket to the APLF binding on Ku80.

Biochemical Society Summer Studentship 2013 Hayley Anne Moulding
1(www.clontech.com/xxclt_ibcGetAttachment.jsp?cItemId=17639)2http://www.qiagen.com/products/ Catalog/Sample-Technologies/DNA-Sample-Technologies/Plasmid-DNA/QIAprep-Spin-Miniprep-Kit#resources
MATERIALS AND METHODS
Site directed mutagenesis
Oligonucleotide primers possessing the site-specific mutations were used to amplify pGBKT7-Ku70-vWA.v2 and pGBKT7-Ku80-vWA plasmids bypolymerase chain reaction (PCR) using KODpolymerase. Parental wild-type plasmid was removedby digestion with DpnI. Running the PCR digestalongside the non-PCR product on a 0.8% agarose gelillustrated if the mutagenesis was effective.
Fig.3 pGBKT7 ‘bait’ protein plasmid. Ku70-vWA.v2 andKu80-vWA constructs were ligated into the multiple-cloningsite (MCS) prior to site-directed mutagenesis1.
Bacterial transformation
1.5ml eppendorfs were placed on ice prior tothe transformation. DH5 cells were obtained fromthe -80°C freezer and immediately put on ice. Thealiquots of DH5 cells thawed on ice and 50µl werethen added to the pre-chilled eppendorf. Care had tobe taken to avoid disturbing the cells. 5µl of the PCR-digest product was added to the DH5 cells in theeppendorf. Tapping the tube mixed them gently. Thetransformation was left on ice for 20mins then heatshocked for 45secs at 42°C, before being returned toice for 2mins. 500 µl of Luria-broth (LB) was added toeach of the eppendorfs, before incubating the tubesfor 1h at 37°C in the shaker. The transformations werespread evenly on LB-agar plates containing 50 g/mlkanamycin. The plates dried and were incubated at37°C overnight to allow for colony growth. A steriletechnique was maintained throughout.
Culture and bacterial mini-prep
The emerged colonies were picked andinoculated 5ml of LB-Kan in universal tubes. These
were then incubated at 37°C in the shaker. Thecolonies grew overnight before the DNA was purified.This was done following the QIAGEN QIAprepprotocol2. The eluted DNA concentration wasmeasured and a 10µl, 100ng/µl sample was producedfor sequencing.
Yeast 2-hybridStreak, culture and transformation of library DNAinto Y190-Ku70vWA cells
Y190 cells, already transfected with Ku70-vWA construct, were streaked onto Minimal media10cm agar plates and incubated for 3-4 days at 30°C.Once the colonies have grown, patches were pickedusing a sterile loop into 2 x 10ml yeast minimalmedium (YMM: His+, Ade+, Leu+) in 50ml falcon tubes.The tubes were incubated in the 30°C shaker (180 rpm)during the day before being transferred to 100ml ofYMM and cultured overnight in the 30°C shaker. Theoptical density at 600nm (OD600) was read todetermine how many cells were to inoculate 2x 200mlof yeast-extract (YE) media, starting at 0.05 OD. Thecells were grown for up to 6 hours, measuring the ODthroughout to ensure it didn’t exceed 0.4 OD.
Once the cells reached between 0.2-0.4 OD,they were harvested into falcon tubes and spun at3000rpm for 5mins at 4°C and after the media wasremoved with an aspirator. The cells were washedwith 25ml 1x TE (10mM Tris and 1mM EDTA) and spun,then aspirated similarly. The cells were re-suspendedin 125µl of TE/Lithium acetate which was preparedprior to transfection. The tubes were placed on ice.1µg of pACT-cDNA library DNA was added to aneppendorf and was mixed with 100µg of denaturedsonicated salmon sperm DNA. 100µl of competentcells were added to the tubes and 600µl of pre-prepared 40% polyethylene glycol (PEG)/100 mMLithium acetate was added to the tubes and mixedwell. The tubes were incubated at 30°C for 30mins.
70µl of DMSO was added and mixed byinverting before the cells were heat shocked in 42°Cwater bath for 15mins. The tubes were returned to icefor 2mins before being spun at 3000rpm for 5mins atroom temperature. All supernatant was removed andthe cells were re-suspended carefully in 500µl 1x TE.Transformation efficiency was tested by diluting atransfection in 1x TE 1:10, 1:100 and 1:1000 whichwere plated onto pre-warmed 10cm His+ plates. 250µlof transfected cells were added to a quarter of pre-warmed 25cm His- plate and distributed evenly. Allplates were incubated for 3 days at 30°C. Whencolonies had grown these were picked from the His-

Biochemical Society Summer Studentship 2013 Hayley Anne Mouldingplates and added to 100µl 1x TE. 10µl of the cells werethen re-streaked onto pre-warmed 10cm His+ and His-plates using a sterile loop. These colonies wereincubated again for 3 days at 30°C. Re-streaks ofemerging colonies were done in the following weeks.
Fig.4 pACT ‘prey’ protein plasmid. The c-DNA librarycontaining multiple different proteins which potentiallyinteract with Ku70-vWA are inserted downstream of theDNA-activating domain.
-Gal colony lifts and test
Once the re-streaked colonies had grown, theHis+ plates were used for colony lifts to test for -Galexpression, whilst colonies from the His- were picked,added to 5ml YMM (Ade+, His+) and cultured at 30°Cfor 16-24h for plasmid DNA extraction to identify theinteracting plasmid. The colony lifts required 2x filterpapers cut to fit 10cm plates. These filters weredivided into quarters and labelled with the clonenumber. The labelled filter was placed on top of thecolonies on the His+ plate applying pressure to ensurethe colonies were transferred to the filter. The filterwas added to liquid nitrogen for 5secs using longtweezers, and then thawed. This was repeated twicemore.
10ml of Z buffer and, in the fume hood, 27µlof -mercaptoethanol and 83.5µl of X-gal was made-up. 3ml of the mix was added to an empty 10cm platecontaining the blank filter. The filter was allowed tosoak in the solution before the colony lift was added,colonies-upward, to the soaked filter. The plate wasincubated for at least 5h at 30°C. Interactions withKu70-vWA turned blue and the filter was removedand placed in the fume hood overnight to dry.
Recovery of plasmid DNA from Y190, andtransformation of DH5 cells
To recover the plasmid for sequencing, theovernight cultures were harvested at 3000rpm for5mins, with the media removed by an aspirator. Thecells were transferred using 1ml dH2O to a 1.5ml
eppendorf, spun at 3000rpm for 5mins. The cells werere-suspended in 640µl zymolase buffer (0.9Msorbitol/0.1M EDTA) and 6.4µl -mercaptoethanolwas added along with 4µl 20T zymolase (-20°C stock).The tubes were incubated for 1h in 37°C water bath.
The tubes were spun at 3000rpm for 5minsand supernatant removed before being re-suspendedfor mini-prep (same as bacterial mini-prep bar 200µlof P1 and P2 were added, and 300µl of N3 addedinstead, with 30µl of EB buffer being pre-warmed at60°C to elute the plasmid DNA.10µl of the plasmidDNA transformed 50µl of DH5 cells (followingbacterial transformation protocol) and were added toLB-Ampicillin (Amp) plates, selecting for pACT plasmid.These were incubated overnight at 37°C.
Bacterial mini-prep and digest, and sequencing
Colonies were picked from the LB-Amp platesinto 5ml LB-Amp cultures and incubated overnight at37°C. The DNA was eluted from the bacterial culturesusing mini-prep. 2µl of eluted DNA was added to 10µlBgIII digest mix (Fermentas enzyme in 1x orangebuffer) for 1h at 37°C. The digests were run on a 1%agarose/TAE gel to identify distinct clones. Cloneswere prepared for sequencing using the nanodrop(Thermo Scientific) to measure DNA concentration ofthe overall recovered DNA. 10µl of 100ng/µl plasmidwas sent for sequencing to Beckman CoulterGenomics.
Ku70-vWA interactions verification
Verification follows a similar transformationprotocol that was undertaken into Y190 cells (withoutKu70.vWA.) Co-transformations of the purified preyplasmid with pGBKT7 empty vector or pGBKT7-Ku70were made, excluding the DMSO step.Transformations were plated onto His+ plates andincubated for 3 days. 4 colonies were picked into 100
l TE and 10 l was streaked onto His+ and His- plates.After a 2 day incubation, growth on His- plates wasassessed and -gal colony lifts were used to verifywhether there is an interaction between the Ku70-vWA and the clones.
Human tissue culture and laser-tracking
Laser tracking was also undertaken exploringthe interactions of Ku and GFP-clone9. This is anadditional experiment to the primary focus, but isrelevant in the characterization of a novel Ku80interactor. It also provided an opportunity to developanother technique.

Biochemical Society Summer Studentship 2013 Hayley Anne MouldingU2OS (human osteosarcoma cells expressing WT p53and retinoblastoma protein, but not p16) weretransfected. 100 l of optiMEM plus 6 l of Genejuiceper 2 g DNA were added to a 1.5 ml tube. The tubeswere incubated for 5 mins at RT. DNA was added (2 g)and the tubes labelled 1-6 were assembled dependenton their contents. After the DNA was added, the tubeswere incubated for 20 mins at RT. After this, all of thesamples were added to their respected U2OS cells, totransfect them. The dishes had to be rocked to ensurethe transfected cells were evenly distributed over thecircular cover slip in the dishes, enabling the lasertracking. The cells were incubated at 37 °C for 24 h.
Laser tracking was accomplished using anOlympus microscope fitted with a spinning discconfocal microscopy system and 405, 488 and 565 nmlasers (Intelligent Imaging Solutions.) Light wasfocussed through a 60x oil immersion objective.Fluorescence is based on the concept of absorbinghigh excitation energy (short wavelength) of light,which is then re-emitted at lower emission energy(longer wavelength). Fluorescence recovery afterphotobleaching (FRAP) is used to track thefluorescently-tagged proteins and DNA. DNA islabelled with Hoechst dye and UV light (405 nm) isused to excite this dye, inducing DNA breaks, creatinga linear track across the cells. The clone 9 protein isfluorescently tagged with GFP so that its recruitmentto the laser track can be measured. This techniqueshows how some proteins co-migrate with others tothe break, hinder migration or sequester otherproteins to enable faster migration themselves. Ithelped to elucidate the role which Ku and WRN playat the DSB and how they interact.
RESULTS and DISCUSSION
Yeast 2-hybrid
Interactions from the pACT c-DNA librarywere found with the Ku70-vWA. BLAST analysisconfirmed this. 16 individual clones were successfullyre-streaked onto His- plates. Only three of the clones,clone 3, 10 and 78 provided a positive result in the colony lift, turning blue. However, plasmids were onlyrecovered from 11 clones of which 4 of the clones didnot show successful His- growth. Overall, 10 of therecovered plasmids were successfully sequenced, with3 of the plasmids providing in-frame sequences:
clones 65, 78 and 100. Clones 65 and 78 successfullygrew on the His- plates, however clone 100 did not. Inspite of this, plasmid was recovered. Other sequenceswere either reverse or not in-frame. All of the cloneshowever did not provide a successful verificationstage.
Different sequences were obtained in-framewith pACT. Clone 65 and 100 contained the ribosomalprotein L7 sequence whereas clone 78 contained thein-frame sequence for attractin isoform 4.Ribosomal protein L7 is a factor of the 60S largesubunit of ribosomes. L7 belongs to the L30P familycontaining an N-terminal basic region-leucine zipper-like domain. This motif enables stable DNA and RNAbinding. DNA binding could suggest implications inrepair. Also, L7 has been implicated as an autoantigen,similar to Ku80, in autoimmune disease. L7 and Ku70were both obtained from an Ago-2 interaction study,supporting the possibility that they may be part of acommon complex (7). Verification however, didn’tconfirm the interaction.
Fig.5 His+ and His- plates showing library clones 1, 2, 3, 4, 9,10, 11 and 12. B-gal colony lifts show a positive protein-protein interaction between the library clone and Ku70-vWA by the blue stain. Library clones 3 and 10 were shownto have positive interactions.
More clones are currently undergoing investigationsuggesting Ku70-vWA interactions.
As a result of positive His- growth and -gal colonyassays, other clones potentially interact with Ku70-vWa. The lab will endeavour to verify these clones.Interactions from the pACT c-DNA library were foundwith the Ku70-vWA.

Biochemical Society Summer Studentship 2013 Hayley Anne Moulding
Fig.6 1% agarose gel shows the uncut and cut versions ofclones 3, 78, 81, 99 and 100. Faint bands can be seentowards the bottom of the gel. Uncut clones are in lane 2, 4,6, 8, 10 and 12 and cut in 3, 5, 7, 9, 11 and 13. Clone 10,known to contain plasmid DNA is in lane 14 as a control.
Verification -gal colony lifts of clone-9 confirminteraction with Ku80.
pGBKT7-Ku80 mutants and pACT-clone9 (froma previous Ku80 screen) were used to transform Y190cells. Colonies from the transformation were re-streaked on His+ and His- plates and -gal colony liftswere done to verify interactions between clone-9 andeither Ku80 or Ku80 mutants. There was a positive liftfor the clone9-Ku80 WT and Ku80 F127R interaction,and negative lifts for the mutants L68R, Y74R andI112R, the conserved binding site for APLF to Ku80-vWA. This indicated that the specific APLF binding siteon Ku80 is also involved in binding to the clone9protein. This confirmation was supported by the lasertracking results (below).
Laser Tracking
The intensity of GFP-clone9 tracks is increased in thepresence of Ku.
Laser tracking shows 1) U2-OS cellstransfected with GFP-clone9 have a clear, strong trackafter 1 min. 2) transfected Ku80-/- MEFs with GFP-clone9 and RFP-Ku at 1 min have stronger tracks,suggesting Ku contributes to clone9 recruitment tothe DSB (Fig.7). The addition of human RFP-Ku to theMEFs shows a distinct track across the nucleus. Fig.8shows that there is an increase in fluorescence in thepresence of Ku compared to without Ku or mutant Ku(L68R). Fig.9 confirms this theory showing a strong,positive correlation between the percentage of clone9and Ku at the break. This backs-up the theory ofincreased clone9 binding in the presence of Ku.Comparing the tracks of the MEFs with and withoutKu, it is evident that the intensity of the track
increases with Ku. There are still very subtle trackswithout the Ku suggesting an alternate pathway whenKu is not available. This will be investigated moreclosely and further by the Caldecott lab.
FUTURE WORK
The lab will continue the re-streaking ofemerging colonies from my initial transformation todetermine whether there are other novel proteininteractions that can be identified. Verification of thealready recovered plasmid DNA suggesting novelprotein interactions will be done, as will sequencingfor these. Work will continue with regards to the lasertracking to confirm the interaction between WRN andKu and its implication in DNA DSB repair.
COMMENTS
This studentship has provided me with theopportunity to broaden my practical and researchskills. It has confirmed solidly that I endeavour tocomplete a PhD. This experience has assured me thatbiochemical research is the direction I wish to pursue.
SUPERVISORS COMMENTS
Hayley has undertaken an Y2H library screenusing a Ku70 domain known to be important inprotein-protein interactions in non-homologous endjoining, but as yet no specific interactions have beenmapped. Thus, any findings are of great importance toour lab and the wider DNA repair community. She hasalready obtained several potential candidates whichwere identified by sequencing and are currently beingverified using appropriate controls. She has a clearunderstanding of the complete process and hasmanaged the labour-intensive project extremelyefficiently in 7 weeks. Further to gaining skills in yeastmicrobiology, Hayley has also produced 10 site-specific Ku70 and Ku80 mutants which will she hasused to map the binding site of a novel interactor withKu80. She also has undertaken live-cell imagingexperiments to demonstrate recruitment of some Kuinteractors to sites of laser-induced DNA damage inmammalian cells. In addition to a broad range oflaboratory skills, Hayley has also been of great help inpreparing figures and sourcing references for a reviewarticle of the Ku heterodimer, which will be in press in2014. Hayley will be rewarded for her contributionswith authorship on our review and possibly futureresearch papers arising from these experiments,which will further her promising career in science.

Biochemical Society Summer Studentship 2013 Hayley Anne MouldingACKNOWLEDGEMENTS
Special thanks go directly to the BiochemicalSociety for awarding me with the opportunity toundertake research at the Genome Damage andStability Centre at the University of Sussex. Particularthanks go to Professor Keith Caldecott for hosting meat the Caldecott lab and providing me with aphenomenal opportunity to further my knowledgeand research skills. To my supervisors Dr GabrielleGrundy and Dr Stuart Rulten, who were incredibleguides and advisors. Thank you for dedicating yourtime and expertise to ensure that I obtained the mostout of my studentship. It was an honour to haveworked in your company.
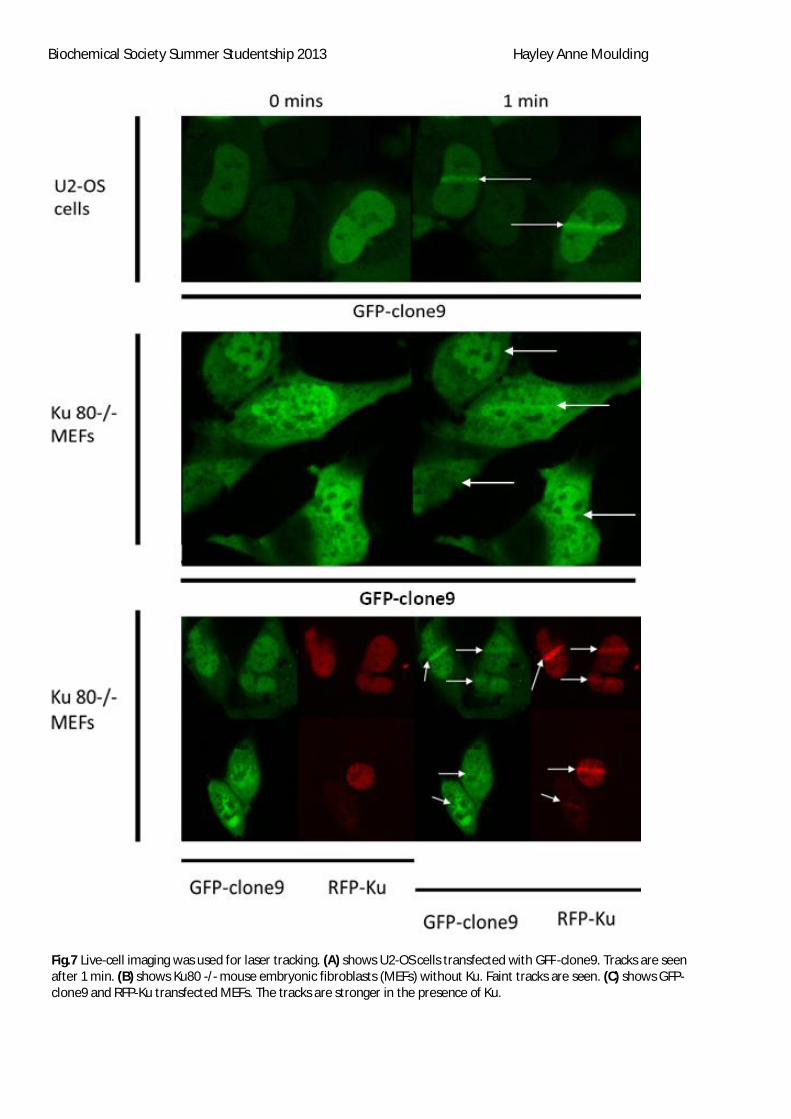
Biochemical Society Summer Studentship 2013 Hayley Anne Moulding
Fig.7 Live-cell imaging was used for laser tracking. (A) shows U2-OS cells transfected with GFP-clone9. Tracks are seenafter 1 min. (B) shows Ku80 -/- mouse embryonic fibroblasts (MEFs) without Ku. Faint tracks are seen. (C) shows GFP-clone9 and RFP-Ku transfected MEFs. The tracks are stronger in the presence of Ku.

Biochemical Society Summer Studentship 2013 Hayley Anne Moulding
Fig.8 Graph showing the percentage change in fluorescence after the addition of WT and L68R Ku. This shows thatfluorescence increases at the track with addition of Ku, suggesting Ku aids clone9 migration.
Fig.9 Graph showing the correlation between the amount of Ku and clone9 recruited to laser-induced damage. There is arelatively strong, positive correlation between the two.

Biochemical Society Summer Studentship 2013 Hayley Anne MouldingREFERENCES
1. Gu, Y., Jin, S., Gao, Y., Weaver, D. T., and Alt, F. W. (1997) Ku70-deficient embryonic stem cells haveincreased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)Jrecombination, Proc Natl Acad Sci U S A 94, 8076-8081.
2. Lieber, M. R., Gu, J., Lu, H., Shimazaki, N., and Tsai, A. G. (2010) Nonhomologous DNA end joining (NHEJ) andchromosomal translocations in humans, Subcell Biochem 50, 279-296.
3. Hammel, M., Yu, Y., Mahaney, B. L., Cai, B., Ye, R., Phipps, B. M., Rambo, R. P., Hura, G. L., Pelikan, M., So, S.,Abolfath, R. M., Chen, D. J., Lees-Miller, S. P., and Tainer, J. A. (2010) Ku and DNA-dependent protein kinasedynamic conformations and assembly regulate DNA binding and the initial non-homologous end joiningcomplex, J Biol Chem 285, 1414-1423.
4. Walker, J. R., Corpina, R. A., and Goldberg, J. (2001) Structure of the Ku heterodimer bound to DNA and itsimplications for double-strand break repair, Nature 412, 607-614.
5. Grundy, G. J., Rulten, S. L., Zeng, Z., Arribas-Bosacoma, R., Iles, N., Manley, K., Oliver, A., and Caldecott, K. W.(2013) APLF promotes the assembly and activity of non-homologous end joining protein complexes, Embo J32, 112-125.
6. Fell, V. L., and Schild-Poulter, C. (2012) Ku regulates signaling to DNA damage response pathways throughthe Ku70 von Willebrand A domain, Mol Cell Biol 32, 76-87.
7. http://www.genecards.org/cgi-bin/carddisp.pl?gene=RPL7&search=ribosomal+protein+L78. Höck J, Weinmann L, Ender C, Rüdel S, Kremmer E, Raabe M, Urlaub H, Meister G. (2007) Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells, Embo J Rep 11, 1052- 60.
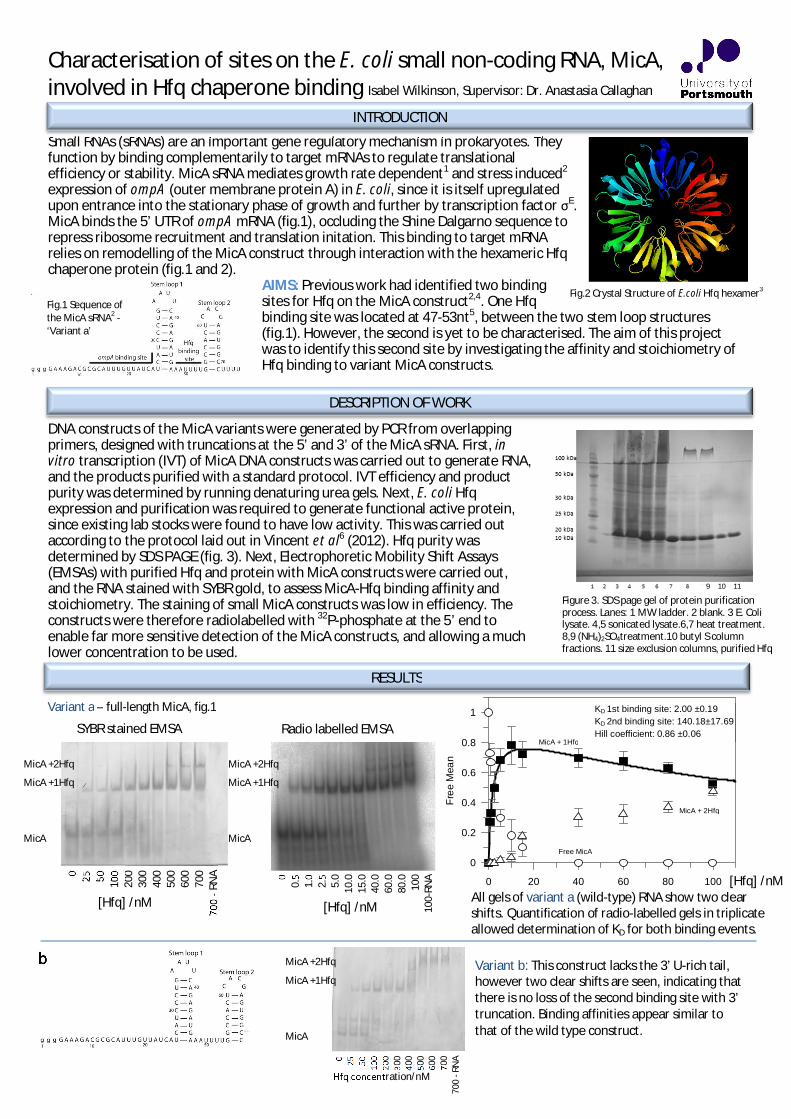
Fig.2 Crystal Structure of E.coli Hfq hexamer3
Characterisation of sites on the E. coli small non-coding RNA, MicA,involved in Hfq chaperone binding Isabel Wilkinson, Supervisor: Dr. Anastasia Callaghan
Small RNAs (sRNAs) are an important gene regulatory mechanism in prokaryotes. Theyfunction by binding complementarily to target mRNAs to regulate translationalefficiency or stability. MicA sRNA mediates growth rate dependent1 and stress induced2
expression of ompA (outer membrane protein A) in E. coli, since it is itself upregulatedupon entrance into the stationary phase of growth and further by transcription factor E.MicA binds the 5’ UTR of ompA mRNA (fig.1), occluding the Shine Dalgarno sequence torepress ribosome recruitment and translation initation. This binding to target mRNArelies on remodelling of the MicA construct through interaction with the hexameric Hfqchaperone protein (fig.1 and 2).
AIMS: Previous work had identified two bindingsites for Hfq on the MicA construct2,4. One Hfqbinding site was located at 47-53nt5, between the two stem loop structures(fig.1). However, the second is yet to be characterised. The aim of this projectwas to identify this second site by investigating the affinity and stoichiometry ofHfq binding to variant MicA constructs.
DNA constructs of the MicA variants were generated by PCR from overlappingprimers, designed with truncations at the 5’ and 3’ of the MicA sRNA. First, invitro transcription (IVT) of MicA DNA constructs was carried out to generate RNA,and the products purified with a standard protocol. IVT efficiency and productpurity was determined by running denaturing urea gels. Next, E. coli Hfqexpression and purification was required to generate functional active protein,since existing lab stocks were found to have low activity. This was carried outaccording to the protocol laid out in Vincent et al6 (2012). Hfq purity wasdetermined by SDS PAGE (fig. 3). Next, Electrophoretic Mobility Shift Assays(EMSAs) with purified Hfq and protein with MicA constructs were carried out,and the RNA stained with SYBR gold, to assess MicA-Hfq binding affinity andstoichiometry. The staining of small MicA constructs was low in efficiency. Theconstructs were therefore radiolabelled with 32P-phosphate at the 5’ end toenable far more sensitive detection of the MicA constructs, and allowing a muchlower concentration to be used.
INTRODUCTION
DESCRIPTION OF WORK
Figure 3. SDS page gel of protein purificationprocess. Lanes: 1 MW ladder. 2 blank. 3 E. Colilysate. 4,5 sonicated lysate.6,7 heat treatment.8,9 (NH4)2SO4treatment.10 butyl S columnfractions. 11 size exclusion columns, purified Hfq
Fig.1 Sequence ofthe MicA sRNA2 -‘Variant a’
RESULTS
0 25 50 100
200
300
400
500
600
700
700
-RN
A
[Hfq] /nM
MicA +2Hfq
MicA +1Hfq
MicA
00.
51.
02.
55.
010
.015
.040
.060
.080
.010
010
0-RN
A
[Hfq] /nM
MicA +2Hfq
MicA +1Hfq
MicA
Variant b: This construct lacks the 3’ U-rich tail,however two clear shifts are seen, indicating thatthere is no loss of the second binding site with 3’truncation. Binding affinities appear similar tothat of the wild type construct.
0 25 50 100
200
300
400
500
600
700
700
-RN
A
Hfq concentration/nM
MicA +2Hfq
MicA +1Hfq
MicA
Variant a – full-length MicA, fig.1
SYBR stained EMSA Radio labelled EMSA
All gels of variant a (wild-type) RNA show two clearshifts. Quantification of radio-labelled gels in triplicateallowed determination of KD for both binding events.
Hfq [nM]0 20 40 60 80 100
Fre
eM
ean
0
0.2
0.4
0.6
0.8
1 KD 1st binding site: 2.00 ±0.19KD 2nd binding site: 140.18±17.69Hill coefficient: 0.86 ±0.06
MicA + 1Hfq
MicA + 2Hfq
Free MicA
[Hfq] /nM
9 10 11

The generated data suggests that the second Hfq binding site is not located at either of the extreme ends of MicA,but perhaps instead at the first stem loop, such as the junction with the 5’ sequence. This is supported by similarfindings with other Hfq binding sRNAs, including DsrA and OxyS7,8,9 which suggest that Hfq typically binds to A/U richregions adjacent to one or more hairpin structures5.Future directions for this work include continuing to radiolabel constructs for accurate quantification, anddetermination of specific location of RNA:protein interaction by hydroxyl radical footprinting.Departures from the original proposal included the requirement for a new protein preparation of Hfq. This meant Igained experience in working with E. coli cultures, chromatography and making and running SDS PAGE gels. However,although SYBR labelled EMSA gels were produced in triplicate for all six constructs, there was insufficient time forradiolabelling to be carried out for each as well.Value of Studentship Student: This summer studentship has been an extremelyvaluable experience of life in academic research. I have really enjoyed my time in theCallaghan lab, learning many new skills and techniques and working alongside someof the most friendly PhD students and postdocs. I’m very grateful for the supportprovided by the Biochemical society.Lab: It has been an absolute pleasure to host Isabel in my group for a vacationstudentship. She is an extremely bright and motivated individual who has made afantastic contribution to the project. Her results have allowed her to draw importantconclusions and these key findings will contribute towards a manuscript as well asprovide preliminary data for a grant proposal. Isabel’s enthusiasm and commitmenthave been evident throughout the placement and she will be greatly missed by thewhole research team. Isabel has the makings of an excellent research scientist and iswished all the very best for her future careerREFERENCES 1 Udekwu KI, Darfeuille F, Vogel J, Reimegård J, Holmqvist E, Wagner EG (2005) Hfq-dependent regulation of OmpAsynthesis is mediated by an antisense RNA. Genes Dev. 19: 2355-2366. 2 Henderson C, Vincent H, Stone C, Phillips J, Cary P, Gowers D, and Callaghan A (2013) Characterization of MicAinteractions suggests a potential novel means of gene regulation by small non-coding RNAs. Nucleic Acids Research, 41 (5). pp. 3386-3397 3 Sauter, C., Basquin, J., Suck, D. (2003) Sm-like proteins in Eubacteria: the crystal structure of the Hfq protein from Escherichia coli. Nucleic Acids Res. 31: 4091 4 Andrade JM, Pobre V, Arraiano CM (2013) Small RNA ModulesConfer Different Stabilities and Interact Differently with Multiple Targets. PLoS ONE 8(1): e52866 5 Rasmussen, A. A., Eriksen, M., Gilany, K., Udesen, C., Franch, T., Petersen, C. andValentin-Hansen, P. (2005), Regulation of ompA mRNA stability: the role of a small regulatory RNA in growth phase-dependent control. Molecular Microbiology, 58: 1421–14296Vincent H,Henderson C, Ragan T, Garcia A, Cary P, Gowers D, Malfois M, Driscoll P, Sobott F, Callaghan A. Characterization of Vibrio cholerae Hfq Provides Novel Insights into the Roleof the Hfq C-Terminal Region. J. Mol. Biol. (2012) 420, 56–69 7Møller, T., Franch, T., Højrup, P., Keene, D.R., Bächinger, H.P., Brennan, R.G., Valentin-Hansen, P. (2002a) Hfq: a bacterialSm-like protein that mediates RNA–RNA interaction. Mol Cell 9: 23–30. 8Zhang, A.X., Wassarman, K.M., Ortega, J., Steven, A.C., Storz, G. (2002) The Sm-like Hfq protein increases OxySRNA interaction with target mRNAs. Mol Cell 9: 11–22. 9Brescia, C.C., Mikulecky, P.J., Feig, A.L., Sledjeski, D.D. (2003) Identification of the Hfq-binding site on DsrA RNA: Hfq bindswithout altering DsrA secondary structure. RNA 9: 33–43.
CONCLUSIONS AND FUTURE DIRECTIONS
Variant c: Here, despite the loss of the first 25nts of the MicA sRNA, two discrete bindingevents are observed. Binding affinity appearsweaker, with KD ~150nM for first shift.Therefore, it appears the second binding site isnot found exclusively in the 5’, but perhaps inthe stem loop regions.
0 25 50 100
200
300
400
500
600
700
700
-RNA
[Hfq]/nM
MicA +2Hfq
MicA +1Hfq
MicA
0 25 50 100
200
300
400
500
600
700
700
-RN
A
[Hfq]/nM
Aggregates
MicA +1Hfq
MicA
0 25 50 100
200
300
400
500
600
700
700
-RNA
MicA +1Hfq
MicA
[Hfq]/nM
Variant d: The 3’ truncation removes the knownHfq binding site, giving a single binding event. Thusthe second binding site must be found in these first44 nts. Combined with the result from c, it suggeststhe second binding site is located in the 1st stemloop.
0 25 50 100
200
300
400
500
600
700
700-
RNA
[Hfq]/nM
Variant e: This construct is very similar to variantd, and again one clear shift is seen. However, atthe higher concentrations of Hfq, the stainingforms smears indicative of protein aggregation inunknown stoichiometry.
Variant f: The exchange of the two stem loopsappears to reduce KD for the first binding eventrelative to wild type. However, again at higher Hfqconcentrations, aggregation is seen. Radiolabellingthis construct would enable detection at lower Hfqconcentration.
Aggregates
MicA +1Hfq
MicA

Structural investigation of the interaction between SGTAand the androgen receptor
Biochemical Society Summer Vacation Studentship 2013
Student: Jaspreet Khaira Supervisor: Dr Rivka Isaacson, King’s College, London.
Aims and BackgroundSmall, glutamine-rich tetratricopeptide repeat protein alpha (SGTA) is a human co-chaperone with roles in membrane protein targeting, prostate cancer, HIV infection andpolycystic ovary syndrome. Dr Isaacson’s lab had already substantially characterised theinteraction between SGTA and two other proteins Bag6 and Ubl4a, which facilitates the firststep in a post-translational membrane targeting pathway for tail-anchored proteins. Theaim of the studentship was to extend this study to investigate suspected interactionbetween SGTA and the androgen receptor, which could shed some light on its role inprostate cancer. The project involved expressing the ligand-binding domain of the humanandrogen receptor and characterising its interaction with SGTA using biophysicalmethodology.
Description of WorkThe ligand binding domain (LBD) of the androgen receptor had already been cloned.1. Transformation of androgen receptor LBD on pET-46 (plasmid) into Rosetta cells andexpression in E.Coli.Cells were spread over agar plate and colonies found the next day from cells containing theantibiotic-resistance plasmid and hence the androgen receptor LBD.One colony was used to inoculate 5ml of starter culture and this was subsequently used in 1lof YT medium. This was allowed to grow until the cells produced an optical density of 0.6-0.8. Protein expression was induced using IPTG.2. Protein purification.Cells were lysed by sonication and then centrifuged to separate soluble (supernatant) andinsoluble (pellet) fractions. The protein was then purified by Cobalt-Affinity Chromatographyand fractions of the supernatant, flow-through, wash, elution, pellet and resin were taken.The testosterone analogue, androstan, was also obtained and added to act as a ligand forthe androgen receptor and encourage it to remain in its folded conformation.3. Gel Electrophoresis for Expression Tests.Stacking and separating gels were initially made according to protocol (12% acrylamide) and30 l samples of the above fractions were loaded, along with 10 l loading dye. The gels wererun to ascertain the location of the protein within the fractions.4. Protein concentration.The protein was concentrated and buffer exchanged using dialysis.5. NMR analysis and titration.The protein was examined by 1D NMR spectroscopy to look for evidence of a foldedmonomeric domain. 2D NMR was used to examine the interaction of SGTA with theandrogen receptor. Since the backbone amides of the N-terminal dimerization domain ofSGTA had already been assigned, unlabelled androgen receptor was titrated in and theinteraction studied on a residue-by-residue basis.

ResultsOver the course of the project, we demonstratedthat there was indeed an interaction betweenSGTA and AR. Although protein expression of ARproved difficult and it often aggregated orremained in inclusion bodies, we eventuallyobtained a sample for analysis by 2D NMR. The gel,right, shows the presence of AR in the elution andresin. Therefore we lysed the pellet again to extractas much AR as possible. The sample from theelution was concentrated and analysed by 2D NMRwith labelled SGTA. The spectrum, right, wasproduced and confirmed the hypothesisthat SGTA and the androgen receptorinteract with one another. Many of theresidues of the labelled SGTA show a clearshift after addition of unlabelledandrogen receptor. As the backbone ofSGTA was already known, this informationwas used to analyse which residues hadshifted to increase our understanding ofthe interaction. The binding surface ismore extensive than that of the twopreviously characterised binding partnerswhich raises interesting biologicalquestions to address in the future.
Future DirectionsIf the expression of AR can be improved further, this will be a highly promising area of study.Further experiments could include crystallisation trials, atomic force microscopy andisothermal titration calorimetry to fully characterise the interaction and solve the complexstructure.
Departures from Original ProposalDue to poor protein expression and air-conditioning failures in the lab, we had to repeatmany of the experiments and subsequently did not have sufficient time to performcrystallisation trials or isothermal titration calorimetry.
Value of StudentshipStudent: This studentship has been an extremely valuable taster of scientific research andhas encouraged me to pursue this further within my medical career. I have very muchenjoyed the experience and learnt much, not only about the theory and practical aspectsconcerning this area of biochemistry, but also about the nature of research in general,including the creativity and patience required. I have had the opportunity to learn manynew techniques and practice these with the support of the lab.Lab: Jaspreet was a wonderful addition to our lab over the summer. Her medicalbackground and general curiosity helped us view our research from new angles. Her resultswill now be used as preliminary data for a grant application.

1
00.20.40.60.8
11.21.4
0 5 10 15 20
LS498
LS473
Time (hours)
Abso
rban
ceat
600
nm
Expression and Purification of Hydrogenase from Shewanella oneidensisStudent: Jayne Watson; Supervisor: Prof Julea Butt
Background: Hydrogen has great potential as a renewable energy source. It is considered a fuel of lowenvironmental impact as its combustion product is water, which is non-polluting. Industrial methods ofproducing hydrogen are costly and the search is on for a cheaper way of producing hydrogen. Shewanellaoneidensis MR-1 (MR-1) has the unusual capacity to conduct electrons across its outer membrane by aporin:cytochrome complex MtrCAB. Here, we wished to begin studies to allow investigations of electrontransfer from MtrCAB to the periplasmic [NiFe]-hydrogenase of MR-1 within a proteoliposome model.Aim: The initial aim of the project was to purify the [NiFe]-hydrogenase enzyme from a strain of Shewanellaoneidensis which had been transformed with a plasmid containing the hyaAB genes. If this was successful itwas hoped to incorporate the hydrogenase into proteo-liposomes to establish if it could receive electronsfrom the membrane spanning porin:cytochrome conduit MtrCAB.Experiments: To purify the Ni-Fe hydrogenase of MR-1 we made use of the LS498 strain previouslyreported to produce periplasmic [NiFe]-hydrogenase. The MR-1 strains LS498 and LS473 were kindlyprovided by Dr Liang Shi (Pacific Northwest National Laboratory, USA) (Shi et al, 2011). The LS473 strain hadthe hyaB- hyaA genotype, so it could not express the [NiFe]- or [Fe-Fe]-hydrogenases, and no kanamycinresistance. The LS498 strain was LS473 containing the phyaAB plasmid. This plasmid carried genes forkanamycin resistance and arabinose induction of the genes for [Ni-Fe]-hydrogenase consisting of a largesubunit of 62 kDa, which contains the [NiFe] centre and a His-tag on its N-terminus, and a small subunit of41 kDa which contains 3 Fe-S clusters.
The initial experiments to purify [Ni-Fe]-hydrogenase from a large scale grow up of LS498 wereunsuccessful. As a consequence a series of expression trials were conducted allowing a protocol to bedeveloped for purification of [Ni-Fe]-hydrogenase from smaller scale grow-ups. The protocol for successfulpurification of hydrogenase is outlined below. LB (50 ml) was inoculated with overnight cultures of eitherLS473 or LS498. The LS498 strain was grown with 50 g/ml kanamycin; both cultures were left shakingovernight at 30°C and then used to inoculate 100 ml of M72 media. This media contained 15 g/L of caseindigest peptone, 5 g/L of papaic digest of soybean meal and 5 g/L, pH 7.8; after autoclaving the followingreagents were added to give the stated concentrations in the media, 1 mM NiCl + 20 mM lactate + 20 mMHepes, pH 7.9. The cultures were grown at 30°C with shaking at 180 rpm, until the OD reached 0.6. Two100 ml cultures were combined, protein production was induced with 1 mM L-arabinose and the bacteriaallowed to grow anaerobically overnight without shaking.
The cells were harvested by centrifugation. The pellet was suspended in 5 ml of buffer (containing20 mM Hepes, 150 mM NaCl, pH 7.8) and sonicated to break the cells open. Ultracentrifugation was usedto pellet the membrane, 40,000 rpm for 1 hour at 4°C. An aliquot of 1000 l of the supernatant was appliedto 100 l of magnetic beads carrying Ni-NTA, these were left to shaking at 4°C for 30 minutes so that theprotein could bind to the beads. The supernatant wasremoved. The beads were washed 3 times in 20 mMimadzole (150 mM NaCl, 20 mM Hepes). Proteins wereeluted in 50 l buffer with 500 mM imidazole. Fractionswere analysed by SDS-PAGE. In attempt to expresshydrogenase with the MtrCAB protein the strains weregrown with 4.9 g/L iron (II) citrate in M72 media. Othersteps were the same.Results: As can be seen from the growth curves, Fig.1,LS498 and LS473 grew at similar rates in 100 ml
Fig1. Compares the growth curves of LS498 and LS473.
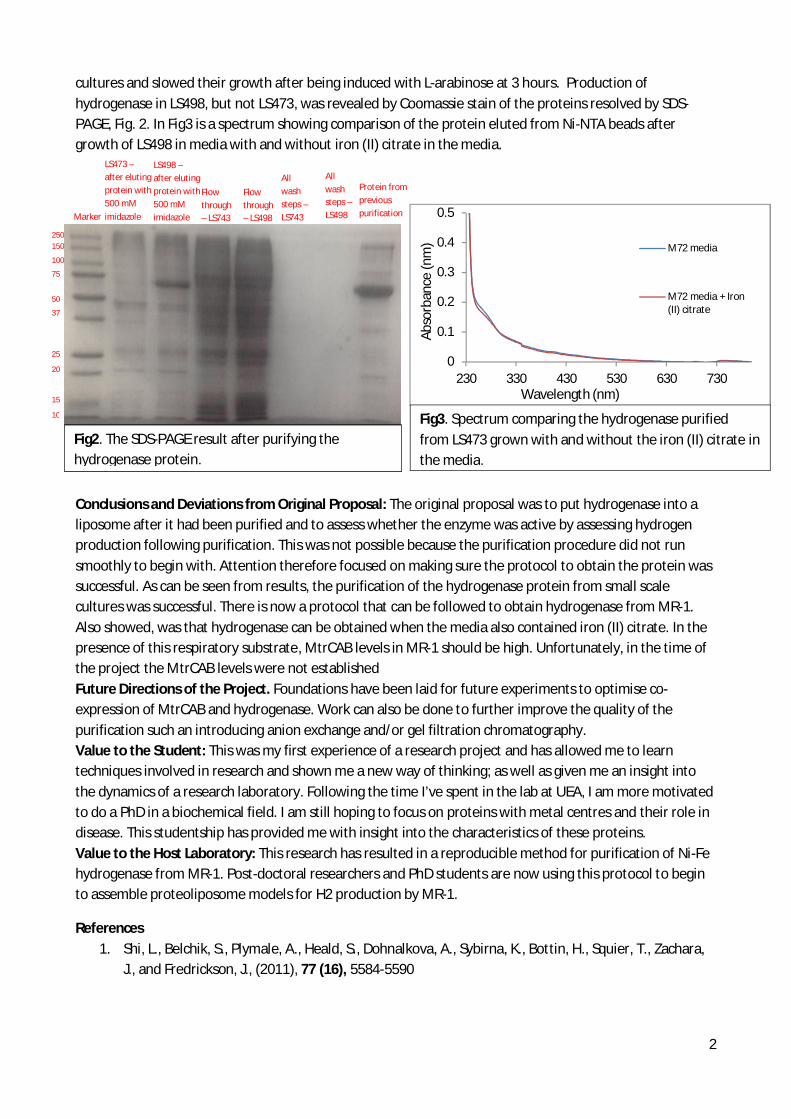
2
0
0.1
0.2
0.3
0.4
0.5
230 330 430 530 630 730
M72 media
M72 media + Iron(II) citrate
Wavelength (nm)
Abso
rban
ce(n
m)
cultures and slowed their growth after being induced with L-arabinose at 3 hours. Production ofhydrogenase in LS498, but not LS473, was revealed by Coomassie stain of the proteins resolved by SDS-PAGE, Fig. 2. In Fig3 is a spectrum showing comparison of the protein eluted from Ni-NTA beads aftergrowth of LS498 in media with and without iron (II) citrate in the media.
Conclusions and Deviations from Original Proposal: The original proposal was to put hydrogenase into aliposome after it had been purified and to assess whether the enzyme was active by assessing hydrogenproduction following purification. This was not possible because the purification procedure did not runsmoothly to begin with. Attention therefore focused on making sure the protocol to obtain the protein wassuccessful. As can be seen from results, the purification of the hydrogenase protein from small scalecultures was successful. There is now a protocol that can be followed to obtain hydrogenase from MR-1.Also showed, was that hydrogenase can be obtained when the media also contained iron (II) citrate. In thepresence of this respiratory substrate, MtrCAB levels in MR-1 should be high. Unfortunately, in the time ofthe project the MtrCAB levels were not establishedFuture Directions of the Project. Foundations have been laid for future experiments to optimise co-expression of MtrCAB and hydrogenase. Work can also be done to further improve the quality of thepurification such an introducing anion exchange and/or gel filtration chromatography.Value to the Student: This was my first experience of a research project and has allowed me to learntechniques involved in research and shown me a new way of thinking; as well as given me an insight intothe dynamics of a research laboratory. Following the time I’ve spent in the lab at UEA, I am more motivatedto do a PhD in a biochemical field. I am still hoping to focus on proteins with metal centres and their role indisease. This studentship has provided me with insight into the characteristics of these proteins.Value to the Host Laboratory: This research has resulted in a reproducible method for purification of Ni-Fehydrogenase from MR-1. Post-doctoral researchers and PhD students are now using this protocol to beginto assemble proteoliposome models for H2 production by MR-1.
References1. Shi, L., Belchik, S., Plymale, A., Heald, S., Dohnalkova, A., Sybirna, K., Bottin, H., Squier, T., Zachara,
J., and Fredrickson, J., (2011), 77 (16), 5584-5590
Fig2. The SDS-PAGE result after purifying thehydrogenase protein.
LS473 –after elutingprotein with500 mMimidazole
LS498 –after elutingprotein with500 mMimidazole
Fig3. Spectrum comparing the hydrogenase purifiedfrom LS473 grown with and without the iron (II) citrate inthe media.
Flowthrough– LS743
Flowthrough– LS498
Allwashsteps –LS743
Allwashsteps –LS498
Protein frompreviouspurification
250150
100
75
50
37
25
20
15
10
Marker

Characterization of zebrafish plakoglobin TALEN mutants as a model forarrhythmogenic right ventricular cardiomyopathy
Jenny Feehan
Supervisor: Dr. Maura Grealy
Lab: Grealy Lab, biochemical wing. National University of Ireland, Galway.
IntroductionArrythmogenic Right Ventricular Cardiomyopathy is a genetic condition caused by mutations in desmosomalproteins. One such protein is plakoglobin, or gamma-catenin (Martin et al., 2009). In this project the newlydeveloped transcription activator-like effector nucleases (TALENs) were used to target and cause an alteration inthe plakoglobin gene in the model organism zebrafish.AimsTo cross F0 TALEN founder mutant zebrafish with wild types and:
Collect and compare the embryos in terms of morphological defects and heart rates.Use qRT-PCR to compare expression of the following genes; Wnt1, Wnt5b, Bone morphogenic protein4 (Bpm4) and Plakoglobin in embryos at 72 hours post fertilisation (hpf).To raise the F1 fish to adulthood, in order to back-cross and produce F2 stable line.
Materials and methodsTALEN F0 mutant zebrafish were crossed with wild types and their embryos (TALEN embryos) were collectedimmediately. Wild types were crossed with wild types to provide control embryos (Wild Type embryos). Theembryos were then cleaned and stored in petri dishes (50 per dish) at 28°C. At 48hpf embryos still in the chorionwere dechorionated. At 48- and 72-hpf embryos were scored, under the dissecting microscope, for morphologicaldefects in the heart, head, brain, tail, and yolk sac using a predetermined scoring sheet. Heart rates were counted.At 72 hpf embryos (50 per tube) were stored in Trizol -80°C, for RNA extraction. cDNA was synthesized bystandard protocol, diluted to 100ng/ l, and amplified by qRT-PCR (Sybr green detection). The cT (Pfaffelmethod) was used to quantify relative expression of Wnt1, Wnt5b, Bmp4 and Plakoglobin. Remaining embryoswere grown to 5 dpf, when locomotor activity was assessed using Daniovison. Each week one set of embryoswere raised to obtain adult F1 fish.ResultsMorphology and heart rateEmbryos were scored and classed as: normal, mild, intermediate and severely affected (Fig. 1) at 48 hpf (n=45)and 72 hpf ((n=33). All wild-type embryos were normal, whereas a few TALEN embryos had severe cardiacoedema, heart-string, yolk sac and tail defects (Fig. 1). However, there was no significant difference between theTALEN and wild type embryos, at 48- or 72-hpf (Fig. 2). Heart rates were similar in TALEN and wild-typeembryos at both 48 hpf (Wild type; 119.3±1.5. TALEN; 118.8±1.6) and 72 hpf (Wild Type; 121.27±1.5. TALEN;117.0±3.2).
Normal Mild Intermediate SevereFig 1: Morphological classifications of zebrafish embryos.
A B
Fig 2: Percentage of embryos in each classification at 48 hpf (A) and 72 hpf (B).
Locomotor ActivityAt 5 dpf DanioVision was used to assess locomotor activity. There were no significant differences seen betweengroups (Fig. 3)
Cardiac oedemaHeart string defect
Yolk sac defect
Tail defect

Fig. 3: The total distance moved of 5 dpf TALEN and Wild Type embryos per 1 minute intervals in a 50 minutelight/dark cycle using Daniovison.
qRT-PCRqRT-PCR was used to assess the relative quantities of Wnt1, Wnt 5b, Bmp4 and Plakoglobin in 72 hpf TALENembryos compared to 72 hpf Wild Type (Fig. 4). From the graph we can see that compared to the control Bmp4appears to be decreased in the mutant embryos, however this would have to be repeated to obtain significantresults.
Relative gene expression in Talen PG mutants
Control
Wnt
1
Wnt 5
b
BMP4
PG
0.0
0.5
1.0
1.5
Gen
eex
pre
ssio
nfo
ldch
ang
e(m
ean
sd)
Fig. 4: The relative quantities of Wnt1, Wnt 5b, Bmp4 and Plakoglobin in 72 hpf TALEN embryos compared to 72hpf Wild Type as a control.
Raising F1 fishFrom each batch of TALEN embryos collected a fraction were kept to be raised to adulthood. Out of theapproximately 200 embryos we attempted to raise, 3 successfully reached adulthood. These F1 fish will be used inthe lab to hopefully produce F2 fish.
DiscussionThe TALEN embryos appeared to show no differences, in terms of morphological defects, heart rate and mobility at the timepoints tested. A possible reason for this would be that the defects caused by the plakoglobin mutated gene did not show untilafter 5 dpf. The fact that only 3 out of the 200 embyros kept to be raised to adulthood survived suggests that defects appeared at alater stage than we could test at.
The value of the studentship to myself, the studentThis grant has allowed me to gain valuable experience in a working laboratory and a true insight into howscientific research is carried out. By working on my own project and by observing others in the lab I feel I nowhave a very good idea of what the role of a biomedical researcher entails. This has allowed be to decide that Iwould like to pursue a career in research, however I still have yet to decide on whether I would prefer to work inmolecular/developmental biology or in the field of neuropharmacology but I hope to have a better idea by the endof my undergraduate degree.
The value of the studentship to the labThe studentship allowed Jenny to gain valuable laboratory experience in a variety of techniques including embryomanipulation and observation, raising zebrafish embryos and larvae, qRTPCR, and behavioural tests in zebrafishlarvae. She also gained an insight into the research process, from writing the proposal to planning theexperiments, to doing the work each day and finally writing up the report. Jenny’s work confirmed preliminaryfindings by a PhD student Rebecca Ryan wrt gene expression in the F1 TALEN embryos. We have had greatdifficulty in raising the F1 embryos to adulthood and had only successfully raised three F1s. Jenny managed toraise another three F1s to adulthood, thus doubling our stock. This will have a big impact on our ability to produceF2 fish and thus a stable line for future work on an ARVC model
ReferencesMARTIN, E. D., MORIARTY, M. A., BYRNES, L. GREALY, M. 2009. Plakoglobin has both structural and
signalling roles in zebrafish development. Dev Biol, 327, 83-96.
Locomotor activity of TALEN and Wild Type 5 dpf Embryos
0
100
200
300
400
500
10 20 30 40 50Dark Light Dark Light Dark
To
talD
ista
nce
Mo
ve
d(m
m)
TALEN
Time (min)
Wild Type

Biochemical Society Summer Studentship Report 2013
Fascin and its Protein Binding Partners
Student: Jennifer Gurnett Supervisor: Professor Josephine Adams
Background and aims of the projectFascin is an actin bundling protein that is found in cells. Fascin is present in filopodia, which are cellsurface protrusions that extend beyond the leading edge of the cell and enable a cell to migrate.Filopodia structure and hence function are determined by the actin filaments which are bundledtogether by fascin. Fascin has a molecular weight of 55 kDa, it is absent in most normal adultepithelia but is present in many cancerous cells because it promotes cell migration.
The aim of my project was to use immunoprecipitation and Western blotting to see what bound tofascin by analysing PVDF membranes and probing with different proteins to see whether a complexhad been formed with the fascin. We know that actin binds to fascin but we want to find out moreabout what binds to fascin to form a complex. For example we wanted to find out whether LIMK andPKC could both bind to fascin at the same time.
Description of the workI carried out 4 experiments all of which followed the same methodology steps described below butwere varied by using different plasmid DNA for transfection and then by probing with differentproteins in the western blot.Cell cultureThe cells I worked with were cos-7 cells, these are monkey kidney cells and grow very quickly. I grewup my cells in a T25 flask feeding them 3 times a week with fresh medium of Dulbecco’s ModifiedEagle Medium with 10% Fetal Bovine Serum added.Plating out and TransfectionOnce a good number of healthy cos-7 cells had grown I detached the cells from the bottom of theflask by trypsinising them. I plated out my cells onto two p90 dishes, one which would containtransfected cells and one which wouldn’t. I carried out several transfections and transfected the cellswith either wild type GFP-fascin, S39D mutant GFP-Fascin or PKC.mRFP plasmid DNA.ImmunoprecipitationOnce lots of protein had been expressed as a result of the insertion of plasmid DNA into the cos-7cells this protein was then collected through immunoprecipitation. Immunoprecipitation involvedlysing the cos-7 cells to release the protein from within the cells so that it could then bind to aspecific antibody such as an anti-GFP or RFP antibody. This enabled the protein to be precipitatedout and any unspecific proteins would not bind.SDS-PAGEThe proteins that were collected through immunoprecipitation were then run on an SDS-PAGE gel.This allowed the protein if it was present to separate out into distinct bands on the gel. The proteinswere then transferred onto a PVDF membrane using a semi-dry membrane transfer.Western BlottingThese membranes were then blocked and incubated with specific primary and secondary antibodiessuch as fascin, actin and LIMK to analyse what proteins bound to the protein on the membrane.Films were produced in the dark room showing what bound to fascin and PKC
Results and outcomesExperiment 1-GFP-Fascin (WT) -probed with fascin, reprobed with actin.I got a GFP-Fascin band when I probed with fascin but no bands seen on reprobe with actin.(Experimental error)Experiment 2 -S39D mutant –

Biochemical Society Summer Studentship Report 2013
SET A - probed with fascin, reprobed with actin.I got no GFP-bands and no bands for actin. Error?SET B (S39D mutant) – probed with LIMK, re-probed with actin.Got LIMK bands at 70kDa, therefore fascin binds to LIMK. Got a band on reprobe with actin whereactin binds to fascin.Experiment 3 - PKC.mRFP – probed with LIMK, reprobed with fascin.Saw bands in tubes 3 and 4 which both had RFP beads in them. Difficult to tell whether there is aband for LIMK at 70kDa as there are very strong heavy and light chain antibody bands. On reprobewith fascin no bands were seen.Experiment 4 – PKC.mRFP – probed with fascin - No bands were seen.
Figure 1: Exp1(lanes 1-7), Exp 2(lanes 11-15) – fascin Figure 2: Experiment 2 (SET B) – LIMK
Figure 3: Experiment 2 (SETB) - actin Figure 4: Experiment 3 – LIMK
Future DirectionsFuture experiments based on my work could involve carrying out similar experiments using moreproteins and repeating the experiments to get better results where my experiments failed. There is alot more that can be found out and my project was definitely inconclusive in places, with more time Iwould have carried out more repeats of experiments to make sure my results were valid.
Contributions to my Career AspirationsThis studentship has been very worthwhile and has shown me that I am definitely interested infollowing a career path in science and research. I found the experiments and hands-on work veryrewarding and loved the day to day life as a researcher. Following the success of this studentship Ihave decided that I would like to continue my career in research and apply for a PhD.
Value of the StudentshipThe studentship has helped me to see that I very much enjoy research and that I should now pushand motivate myself harder to succeed in carrying out research in a similar field. My presence in thelab hopefully helped the others in the lab as my work was closely related to what other PhD studentswere doing in the lab, therefore it would have been relevant to their projects too. This furtherinformation could have provided openings in the research and allowed for more research options.

Student: Jocelyn MacDonaldSupervisor: Robin Plevin
Testing the potential of PAR-2 as a therapeutic target in arthritis
Abstract – PAR-2 is a G-protein coupled receptor that is associated with inflammatory diseases, in particularRheumatoid Arthritis and Osteoarthritis. Activation of PAR-2 is initiated by serine proteinase-mediated N-terminal cleavage. Once PAR-2 is activated it is then desensitized and internalised through phosphorylationand -arrestin binding. DM/8/36 and MW016 are both potential inhibitors of PAR-2 and these wereinvestigated using a variety of techniques to determine if they can block PAR-2 signalling events. It wasfound that MW016 can prevent activation of PAR-2 whereas DM/8/36 was expressing agonist properties andactivating PAR-2.
Background –A novel subgroup of the seven transmembrane G-protein coupled receptor family is theprotease activated receptors (PARs) of which there are four members i.e. PAR1-4 (1). These receptors have aunique method of activation in which serine proteases cleave the N-terminus of the receptor to expose atethered ligand which binds to the 2nd extracellular loop of the receptor initiating activation of downstreamsignalling events (2). The main serine protease is Trypsin, and in the process of inflammation mast cellsrelease Tryptase which leads to the activation of PAR-2 (3). Trypsin was used to determine the physiologicalroles of PAR-2 and it has been found that PAR-2 activation mediates several downstream signalling eventsincluding ERK, JNK and p38 phosphorylation and activation of the NFkB transcription pathway which isstrongly associated with inflammation (1). A recent study has shown the orally available antagonist GB88 isa relatively potent drug which reversibly inhibits PAR-2 activation thus, there is potential to determineexperimentally the effect of PAR-2 blockade in inflammation (2). Due to this new discovery regarding GB88the lab have generated their own novel PAR-2 inhibitors DM/8/36 and MW016 based on the GB88 scaffoldin order to determine if these have the ability to inhibit PAR-2 therefore prevent activation of the downstreamsignalling cascades leading to inflammation. Different aspects of the pathway were tested to ensure thepotential inhibitors weren’t specific to one part of the pathway and not another.Aims – During this studentship I have:1) Characterized PAR-2 activation and signalling in a keratinocyte cell line stably over expressing the PAR-2receptor, using serine proteases and synthetic ligands.2) Analysed the properties of two PAR-2 compounds on their ability to inhibit events associated with PAR-2activation and signalling, such as receptor internalisation, increases in InsP3, phosphorylation of ERK andactivation of NFkB transcription.
Results – A luciferase assay was conducted to determine the effect of DM/8/36 on PAR-2 activation byTrypsin. The drug DM/8/36 had a fold increase which was larger than Trypsin which was unexpected as theinhibitor is activating PAR-2 more than the agonist. We predicted that as the inhibitor concentration wasincreased PAR-2 activation would be deceased as there would be more inhibitor to block the receptor bindingsites preventing activation but the results have contradicted this and PAR-2 activation actually increased.Hence, DM/8/36 is acting like an agonist. The effect of the inhibitor MW016 on PAR-2 activation by Trypsinwas also investigated. MW016 had a lower fold value than Trypsin and the control indicating that it wasinhibiting PAR-2. This is further seen when Trypsin is in the presence of varying inhibitor concentrations asthe highest MW016 concentration had the greatest inhibition although the fold values were slightly varied anddid not show a general trend but they showed that MW016 is by and large inhibiting PAR-2.
Western Blotting was conducted to determine if DM/8/36 was activating the PAR-2 receptor initiating thestimulation of a signalling pathway leading to the phosphorylation of ERK (4). The blot displayed that p-ERK had activity between 5 and 15 minutes with the maximum activity being seen at 5mins. Owing to thepresence of p-ERK this has confirmed that DM/8/36 has activated PAR-2 leading to the phosphorylation ofERK, therefore it is acting similar to an agonist.
The inositol phosphate assay measures the level of inositol phosphate within cells as a result of PAR-2triggering a down-stream signalling pathway. DM/8/36 was investigated to discover if it behaves like theagonist Trypsin, in that it activates PAR-2 leading to the formation of inositol phosphate. A Trypsin

concentration course was conducted which showed there was little deviation from the control until 30nMwhere there was a rapid increase in fold followed by the maximum fold value at 50nM. As expected as theconcentration of Trypsin is increased the inositol phosphate level also increases due to an elevated activationof PAR-2. In addition as the concentration of DM/8/36 was increased the fold value also increased in a linearmanner until it reached the maximum fold value at 30µM. Therefore, increasing the concentration increasesthe level of inositol phosphate as a result of an elevated PAR-2 activation.
Trypsin cleaves PAR-2 initiating the binding of -arrestins which desensitize and internalise the receptor andsort it to lysosomes for degradation (5). Uncleaved naïve PAR-2 from intracellular stores then migrates to thecell surface to replenish the PAR-2 loss (6). Cells were transfected with PAR2-YFP thus the PAR-2 receptorswere tagged yellow. When these cells were stimulated with Trypsin for 5mins this initiated PAR-2 activation,desensitization and internalization as seen by the yellow PAR-2 receptors redistributing from the cellmembrane to the cell cytosol. It was found that DM/8/36 was behaving similarly to Trypsin as PAR-2 wasdesensitized and internalised at 5mins indicating that DM/8/36 was acting more like an agonist rather than aninhibitor of the PAR-2 receptor.
By collating the results obtained from each of the methods described previously, it can be seen that:1. The novel drug MW016 is a true antagonist which inhibits PAR-2 activation therefore prevents the
signalling cascades leading to inflammation. This may be due to MW016 being a competitiveinhibitor with a high affinity for the receptor binding sites therefore it will block these sites preventingagonists from activating PAR-2. It may also be a result of MW016 binding to the receptor andinactivating it therefore the receptor can no longer carry out its desired effects.
2. The novel drug DM/8/36 is expressing unusual characteristics as it is activating PAR-2 more thanwhat the agonists are therefore it is amplifying the signalling cascades leading to inflammation. Thismay be caused by DM/8/36 having a combination effect with Trypsin: thus they are working inunison to activate the receptor and increase its activity.
Future directions regarding this project could be to do more tests on DM/8/36 to determine why it isactivating PAR-2 and to also test all the labs synthetic PAR-2 compounds based on the GB88 scaffold. Thefuture goal of this project is to uncover an effective drug to inhibit the effects of the PAR-2 receptor and tomake this orally available to test on arthritic mice to see if it halts the process of Arthritis.
I feel I have gained valuable laboratory experience due to my active involvement with my supervisor and labcolleagues. Due to working in a committed and lively research laboratory this has increased my interest inresearch and has pushed me in the direction of completing a PhD after my Honours Degree as I havethoroughly enjoyed being part of a team and making new discoveries.
References-
1. 1. Barry, G.D. et al (2010) Novel Agonists and Antagonists for Human Protease Activated Receptor2. Journal of Medicinal Chemistry. 28; 53 (20) 7428-7440
2. Suen, J.Y. et al (2011) Modulating human proteinase activated receptor 2 with a novel antagonist(GB88) and agonist (GB110). British Journal of Pharmacology. 165, 1413-1423
3. MacFarlane, S.R. et al (2001) Proteinase-Activated Receptors. Pharmacological Reviews. 53: 245-282
4. Melo,M. (2006) Phosphorylated Extracellular Signal-Regulated Kinases are significantly Increased inMalignant Mesothelioma. J Histochem Cytochem. 54 (8) 855-861
5. Soh, U.J.K. (2010) Signal transduction by protease-activated receptors. British Journal ofPharmacology. 160, 191-203
6. Arora, P. et al (2007) Protease Activated Receptor Signalling, Endocytic Sorting and Dysregulation inCancer. Journal of Cell Science. 120, 921-928

Investigating the chromatin association of the SMC5/6 complexthroughout the cell cycle and in response to replicative stress
Student-Jonathan Goult Supervisor- Dr Elaine Taylor, University of Lancaster
Introduction and Aims
The SMC5/6 complex is important in several aspects of chromosomal maintenance including the repair of DNAdamage, tolerance of replication stress and accurate chromosome segregation during mitosis. This complex isable to post translationally modify other proteins through subunits that possess ubiquitin and SUMOconjugating activities, with these functions being essential in the preservation of genome integrity andtherefore considered a key role of the SMC5/6 complex within the cell.
The aim of this project is to characterise the SMC5/6 complex and analyse the modification of its constituentsubunits in Xenopus egg extract; an excellent model system as it can undergo cell cycle events in vitro. It isknown that an analogous SMC5/6 complex exists in Xenopus egg extract which will be confirmed thoughWestern blotting and immunoprecipitation of the complexes components. The pattern of chromatinassociation of each of the SMC5/6 components will also be examined throughout the cell cycle and in responseto DNA damaging agents that generate replication stress.
Description of the Work carried out
Immunoprecipitation of the SMC5/6 complex in Xenopus
Antibodies specific for some of the SMC5/6 complexes components (Nse1, Nse3 and Nse4) were crosslinked toProtein A Dynabeads and added to Xenopus egg extract (SXt), allowing them to bind to their specific antigen.The unbound fraction was then discarded and any proteins bound to the beads were then eluted using a pH2.0glycine solution. An acrylamide gel was then run using samples from various stages of theimmunoprecipitation; the eluted protein fraction and the flow through fraction. These were compared againsta negative control; immunoprecipitation fractions from Dynabeads with none specific IgG’s crosslinked tothem (M1 and M2 in Figure 1). This was then blotted onto a nitrocellulose membrane and probed usingantibodies specific for the complex components to verify their presence on the blot.
Cell cycle analysis of the SMC5/6 complexes chromatin association
Xenopus egg extract was mixed with sperm nuclei and after gentle resuspension to ensure equal nucleidistribution, pipetted into labelled chromatin aliquots. These were placed in a 21°c water bath and removed ata defined time point during replication. After stopping replication and releasing the chromatin, the sampleswere spun in an aliquot containing a sucrose cushion. This pelleted the chromatin bellow the cushion, allowingus to obtain samples of chromatin and any proteins associated with it. This protocol was modified to allowinvestigation into the effect of replication stress on the cell cycle. Addition of inhibitors such as aphidicolin,roscovitine and geminin that affect replication at different points before initiation or DNA damaging agentssuch as restriction enzymes, MMS and UV radiation generate chromosomal damage and allow us to explorewhether replicative stress alters the association of the chromosomal maintenance proteins such as SMC5/6complex with chromatin.
These experiments were supported by replication assays that allowed us to measure the incorporation of aradioisotope P32-dCTP into the DNA of replicating extract, thus indicating the amount of replication the extractis undergoing at each specific time point. The replication assay was also a good negative control for replicationinhibition; addition of replication inhibitors should prevent DNA replication and therefore decrease theamount of radiolabelled isotope that is incorporated into the DNA.

Figure 2- Western Blot showing proteins associated with Xenopus egg extract chromatin with differentreplication inhibitors (untreated, geminin, roscovitine, aphidicolin and aphidicolin+caffeine) and DNAdamaging agents (untreated, restriction enzyme double strand breaks (D1), restriction enzyme doublestrand breaks after 15 minutes of replication (D2), MMS and UV). Each DNA damaging agent sample hascorresponding one containing both the damaging agent and caffeine (indicated by the subscript c)
Conclusions
From these results we can conclude thatthere is an analogous SMC5/6 complex inXenopus egg extract. Immunoprecipitation ofthe extract (Figure 1) shows that depletion ofone of the subunits also depletes the others,indicating that all six subunits IP as a complexand thus are associated with one another inextract
From the cell cycle analysis, we can concludethat the SMC5/6 complex does interact withchromatin over the course of replication. Whenthe extract is placed under conditions ofreplication stress due to DNA damaging elements(Figure 2), we can see the levels of the SMC5/6complex componentsare down whencompared to theuntreated (NT) control.The levels of chromatinassociation of thecomplex components issomewhat restored bythe addition of caffeine,a molecule that isknown to uncoverdormant replicationorigins, allowing anincrease in the amountof replication.
Future Directions
Some of the results seem tosuggest that SMC5/6 complex components bind to chromatin under conditions of replicative stress in differentconcentrations. However the changes detected are marginal, and thus further investigation and repeats of theexperiment will need to be undertaken in order to elucidate whether SMC5/6 subunit binding is affected bychromosomal damage. Another direction for further study would be the post translational modificationactivity of some of the SMC5/6 subunits. Due to time constraints, we were unable to focus much attention onthis aspect of the SMC5/6 complex; a complex that is known to have subunits with ubiquitin and SUMOconjugating activity. However proteins could be tagged with labelled (eg His-tag) ubiquitin and SUMOmodifications, allowing them to be isolated through pull down assays.
Value of the studentship
Through undertaking this studentship I feel I have definitely grown in confidence with my practical skills.Initially I didn’t feel comfortable working without supervision, as many of the lab techniques I was performingwere new to me. However I was able to become more independent over the duration of the project andperform many of the experiments my supervisor and I planned on my own. I also feel that the studentship has
Figure 1- Western Blot showing the proteins from Xenopus egg extract treated withantibody beads targeting different subunits of the SMC5/6 complex. Lanes 3-7 areantibody flow through (depleted) samples and lanes 8-12 are proteinsimmunoprecipitated from the beads

helped me improve my basic laboratory skills as I have been introduced to many standard biochemicaltechniques that I may have to use in the future. Before I began the studentship, I was unsure on whetherundertaking a career in scientific research was right for me. However the last eight weeks in a lab have beenthoroughly enjoyable and rewarding; I am certain that I would now like to pursue a research or PhD positionwhen I complete my undergraduate’s degree.

An Octanoylated Protein Substrate for Lipoyl Synthase
Student: Josh Prince Supervisor: Prof. Peter Roach
Background and Aims:
LipA is part of the radical SAM enzyme family, which used radical chemistry to activateinert carbons. LipA inserts a sulphur atom into positions C6 and C8 of it octanoyl substrates.Substrates for this enzyme include E2 and H proteins. The aims of my project were to clonesmall fragments of E2 and H proteins containing the octanoyl chain in the pET16b plasmid.Then to transform these plasmids into BL21 (DE3) cells and to grow 5 L cultures of each toobtain the protein fragments. Also to complete a large scale growth of cells containing thelplA gene to obtain a stock of the LplA protein.
Work Carried Out:
Firstly Stocks of the plasmids pEX::SsE2, SsE2l, TeE2, TeH and pET16b in XL-10gold strain of E. coli were grown and isolated using mini-prep techniques. Nano-dropreadings were then taken to measure the concentration of the DNA.
Secondly the desired DNA fragments were isolated. This was done originally usinganalytical digests to test the restriction enzymes cut the plasmid in the desired place.Then prep digests were completed of the same reaction mixture but on a larger scale.Then using gel extraction techniques the DNA fragments were isolated.
Then Next step was to ligate the isolated DNA fragments into the pET16b backbonethis was done using DNA ligase, DNA buffer and adding an additional amount ofATP. Ligations were done using an insert to backbone ratio of 3:1, 6:1 and 9:1. Theligation mixtures were spread onto agar plates containing ampicillin resistance.
The colonies that grew on the plates were tested to confirm the presence of the desiredplasmid using analytical digests. Successful colonies were transformed intoBL21(DE3) cells.
Small scale growths of cells containing the plasmids were completed using 100 mLsamples. The cell pellets were then used for batch purifications.
After successful results from the batch purifications, large scale growths (5 L) werecompleted of the plasmids pET16b::SsE2, SsE2l, TeH, TeE2. The cell pellets werethen re-suspended in Buffer A (HEPES 0.025 M, Imidazole 0.05 M, NaCl 0.5 M,glycerol 10 %). After the cell suspension was sonicated for 10 minutes, it was loadedonto a Ni column so that the protein fragment can be isolated.
Lastly 1 µL of pCA24N::lplA was transformed into 50 µL of JM109 and Bl21(DE3)cells and grown on agar plates. Three colonies from each transformation were grownin 10 mL 2YT (10 µL Chloramphenicol 34 mg/mL) and incubated at 37 °C overnight.The JM109 cultures were used to obtain the plasmid via Mini-prep techniques. TheBL21(DE3) cells were used for a large scale growth (5 L). The cell pellet was thenused for a Ni column so that the LplA protein can be isolated.

Results:
Plasmids pET16b::SsE2, SsE2l, TeH, TeE2 were successfully created by ligating the pET16bbackbone with the insert (figure 1). Furthermore SsE2, SsE2l and TeE2 protein fragmentswere successfully isolated using a Ni column.
Figure 1: Shows analytical digests (using Nco1 and Xho1) of the pET16b::SsE2, SsE2l, TeH and TeE2plasmids.
The batch purification of the TeH cell pellet showed no isolated protein in the elution.However in the cell pellet samples, broad bands were seen at the same size of the interestedprotein, which could suggest that the protein fragment couldn’t fold in the correct way, andso was stored in vesicles.
Future Directions:
The successful isolation of these protein fragments containing the octanoyl group willhopefully lead to more successful attempts at producing a crystal image of LipA interactingwith its substrate. The extended protein structure should be more stable at higherconcentrations than previous fragments used, which should hopefully increase the chances ofacquiring the image.
Value of Studentship:
Working in a research environment has been an excellent opportunity to learn new techniqueswhich I haven’t previously seen, learn to work more independently to solve problems as theyarise and to learn more about a PhD which I am now very interested in studying after mycourse. During my studentship I made protein fragments of SsE2, SsE2l, TeH, TeE2 whichthe lab could use to interact with the LipA enzyme and hopefully obtain a crystal image.
100008000600050004000300025002000
1500
1000
750
500
250
100008000600050004000300025002000
1500 1000 750500
250

Biochemical Society 2013 ReportKatharine LockeInvestigating Host Cell Interactions of the Foot-and-Mouth Disease Virus 3A Protein.Supervisor: Prof. Martin RyanSchool of Biomolecular Science, St Andrews University
Background and Aims:Foot-and-Mouth Disease Virus (FMDV) is one of the most important animal pathogens known to man. The positive, single strandedRNA genome of FMDV comprises one long open reading frame. The genome codes for the structural proteins 1A, 1B, 1C and 1D andnon-structural proteins such as 3A, the role of which is not fully understood1 (Figure 1). However, 3A is thought to play a role indetermining the host range of the virus2. In this 2 month studentship, it was aimed to discover cellular-interacting partners of FMDV3A or 3AB. This was achieved byamplifying sequences encoding the FMDV-3A or 3AB proteins, and creating N- or C-terminal GFP-fusion proteins. Subsequently,plasmids containing these proteins weretransfected and imaged using EVOS andIncuCyte ZOOM fluorescent microscopes tomonitor GFP expression, which wouldindicate successful translation of associatedFMDV 3A or 3AB. Following this total cellprotein was extracted and purified using allama GFP antibody conjugated to agarosebeads. Lysates containing GFP-3A/3ABwere then separated by SDS-PAGE andstained to look for potential interactions.
Description of Work:Construction of 3A/B GFP Expressing PlasmidsA PCR reaction was set up to amplify sequences containingFMDV 3A and FMDV 3AB, using a cDNA encoding theFMDV genome i.e. the 'FMDV Replicon'. Primers usedduring PCR were designed to contain restriction enzymesites (Table 1). To generate the GFP fusion proteins,plasmids JN1 or JC3 (N or C-terminal, respectively) andPCR products were digested with the appropriate restrictionenzymes (Table 1). These fragments were separated byagarose gel electrophoresis, purified and then ligatedtogether to generate the plasmids (Figure 2). The aim ofthis was to determine whether having GFP fused to eitherthe N or C-terminal of the 3A/AB sequence affected thefunction of the 3A/AB protein in the cell.
Transfection into BHK cellsGFP expression was monitored by transfecting plasmid DNA into cells. BHK cells were seeded 24hr prior to transfection into a 12-well plate. Plasmid DNA was diluted with Opti-MEM reduced-serum media. This mixture was added to lipofectamine 2000 andincubated at room temperature for 5 min. Total solution was added drop-wise to BHK monolayers, which were 70-80% confluent.Cells were imaged every 2hr for 24hr using an IncuCyte ZOOM (see Figure 3). For the final IP procedure, cells were seeded into a9cm plate and volumes “up-scaled”. Cells were harvested 24hr post-transfection using trypsin-EDTA and ice-cold PBS.
Immunopreciptation ProtocolCells were harvested, as described above, and pelleted by centrifugation. IP buffer (50 mM Tris-HCl, 150 mM NaCl, 5mM NEM, 1%NP-40, 10% glycerol, 10mM EDTA, complete protease inhibitors EDTA-free and PhosStop) was used to re-suspend the cell pelletand the lysate was sonicated Lysates were incubated with GFP-trap agarose beads (kind gift of Professor Angus Lamond, Universityof Dundee) and RIPA. The beads were then washed three times in IP buffer before eventual elution in LDS buffer with 25mM TCEP.All steps were carried out at 4°C. The eluted samples were run on an SDS-PAGE 4-20 % gradient gel and then stained with SYPRO-Ruby Rapid Protein Gel Stain. The gel was then re-stained using coomassie protein stain and imaged on an Odyssey imaging system(LI-COR) (Figure 4).
Results:Both the FMDV 3A and 3AB genes were cloned and inserted into vectors to generate GFP fusion proteins (C-term or N-term). Theseconstructs were successfully transfected into BHK cells and fluorescent images showed GFP expression, indicating the plasmids werefunctional (Figure 3). Fluorescent data also showed GFP expression peaked at approximately 24hr, allowing optimal harvesting duringIP experiments. An initial IP experiment was carried out on a smaller scale (6-well plate, reagents scaled down) to test constructs,reagents, technique etc. These samples were run on a 12% SDS-PAGE gel and stained using coomassie. This was then 'scaled-up' touse 9cm dishes. Eluates from the larger pull-down experiment were run on a pre-cast gradient gel and stained with SYPRO-Rubyprotein stain, which is more sensitive than coomassie. This gave higher resolution and increased the extent of protein visualisation.Large protein bands could be seen at approximately 15kDa (Figure 4) from samples containing the 3A C-term plasmid. This suggests

an interaction was present, possibly a complexof proteins due to the presence of more than asingle band. To increase the resolution further,the gel was re-stained with coomassie andimaged using an Odyssey imaging system (LI-COR).
Value of Studentship:On commencing my studentship I felt nervousabout completing more complex experimentsunsupervised and the use of specialisedequipment was daunting. However, by theend of the 2 months I was often workingon procedures independently and evenworked on a separate project for 1 week,almost entirely on my own. This helped tofurther increase my confidence within thelab. In addition to this, as a chemistrystudent, I feel this experience has givenme an invaluable understanding of theoverlap between the biology andchemistry boundary and that I aminterested in pursuing a career in researchwithin a multidisciplinary area.
Future Work: Fig 2: Plasmid maps of the JN1 3A/B ligation (N-terminal fusion) and JC3 3A/B ligation.
It is planned to ‘scale up’ the experiment furtherby using 2x 9cm dishes for each IP. Theexperiment needs to be repeated to check thatthe bands we attained in our SDS-PAGE gel didnot arise by experimental flaw, and are actuallya true result. If the bands are found to besignificant they will be cut out and sent to ourcollaborators at The University of Dundee formass spectrometry analysis. This couldpotentially lead to more experiments toinvestigate any possible interactions present.
Departures from the original proposal:Due to an on-going collaboration, it wasdecided that a GFP fusion protein tag would beused in place of a TAP-TAG method. Thebenefit of this was that GFP expression could beseen using the IncuCyte ZOOM, which allowsfor the plasmids to also be used duringfluorescence microscopy analysis.
Acknowledgements:I would like to thank Prof. Ryan's team: Fiona,Claire, Garry and John for supporting and encouraging me throughout theduration of my time in the lab and also Prof. Ryan himself for allowingme this opportunity.
References:1 M. J. Grubman and B. Baxt, Clinical Microbiology Reviews, 2004, 17(2), 465-4932 N. J. Knowles et al., Journal of Virology, 2001, 75 (3), 1551-1556

Validation of microRNA biomarkers for motor neurone disease and investigation of their expression inneurones
Biochemical Society Summer Studentship report 2013Student: Laura Francis
Supervisor: Dr. Emily Goodall, Sheffield Institute for Translational Neuroscience
Background and aims:Motor neurone disease (MND) is a progressive neurodegenerative disorder. Patients become paralysed, as musclesweaken and waste due to selective loss of motor neurones. Patients have a life expectancy of 2-5 years from symptomonset and MND kills on average five people per day in the UK. Currently there is no diagnostic test for MND anddiagnosis relies upon excluding mimic diseases that also cause muscle weakness, such as myopathy. The causes ofMND are unknown, it is believed that there are multiple pathways that contribute.MicroRNA (miRNA) disregulation is one pathway thought to be involved in MND pathogenesis. MiRNAs are small non-coding RNAs 18-22 nucleotides long. Mature miRNA are recruited to a silencing complex to direct post-transcriptionalgene regulation via sequence directed binding to mRNA targets.The aim of my project was to validate miRNAs as biomarkers for MND that could distinguish MND from myopathy usingquantitative PCR (QPCR). The project also investigated the miRNA expression in motor neurones, which has not beenpreviously explored in the published literature. We hypothesised that miRNAs are dysregulated in MND and will act asinformative biomarkers. Findings may help to develop a blood test for MND, to diagnose patients earlier and give themthe care they need.
Methods:
Validation by QPCRHuman serum samples were used from control individuals, MND and myopathy patients. MiRNA was extracted using thetotal RNA purification micro kit (Qiagen). MiRNA from 6 samples was then pooled for the myopathy group, 18 for theMND group and 12 for the control group, to allow a quick screen of candidate miRNAs of interest. Previous experimentsfrom this laboratory involving expression profiling of miRNAs informed the choice of miRNAs to investigate as potentialbiomarkers. These miRNAs were then tested in the pooled samples using QPCR which measured TaqMan probesignals. Relative concentrations were calculated using the 2 CT method.
Laser Capture Microdissection (LCM)Post mortem spinal cord tissue from the lumbar region were sectioned onto slides and stained with toluidine blue tohighlight cell bodies. Using laser capture microdissection, motor neurones were isolated from the spinal cord tissue.MiRNA was then extracted using the RNA purification micro kit (Norgen). TaqMan QPCR assays were used toinvestigate expression of target miRNAs within the motor neurones. Both control individuals and MND patients spinalcord tissue were used.
Results:
Fig 1: QPCR results of serum samplesfor miR-196b and 143 in Control, MNDand myopathy patients.
Fig 2: QPCR results of serum samplesfor miR-133a, 133b and 206 in Control,MND and myopathy patients.

The QPCR results for miRNA extracted from serum samples of control, MND and myopathy patients showed that miR-196b and miR-133a are highly expressed in MND compared to the myopathy and control group. Whereas miR-143 andmiR-133b have similar expression between all three groups (Fig 1 & 2). However miR-206 is highly expressed in bothMND and myopathy groups compared to the control (Fig 2). It can be concluded that miR-196b and 133a may be goodbiomarkers to distinguish MND from myopathy. MiR-206 may not be a good biomarker to distinguish between MND andmyopathy as it is highly expressed in both, however it may be useful to discern between MND and other mimic diseases.
Future directions:The results gained from this project will be used as a screening process to decide which miRNAs to pursue in futurework. These findings will lead to miRNA 206, 196b and 133a being studied in individual samples, to validate them asbiomarkers. In future work the sample size needs to be increased to confirm results and also be expanded to includeother mimics, for example inflammatory neuropathy. This would hopefully lead to a panel of miRNAs being developed toproduce a diagnostic blood test for MND. Data produced during this project will form part of a scientific publication andmay also be used as pilot data for further grant funding.
Departures from original proposal:The original proposal stated that 10 MND cases and 10 myopathy patients serum samples would be used to validatepotential miRNA biomarkers, however a different number of samples for each group were pooled. This project would beused as a screening process to decide which miRNAs to pursue in further work and to then investigate in individualsamples. Due to the pooling of samples, it was not possible to perform statistical analysis. This would be conductedonce results from individual samples have been obtained. The potential miRNA biomarkers had been looked at inindividual samples. As LCM is a time consuming technique, only one of the target miRNAs was tested via QPCR in theLCM material. However, results were inconclusive due to variability in controls (data not shown). LCM material will beused in a more comprehensive study to investigate miRNA dysregulation in motor neurones from MND patients.
Value of studentship:The studentship has been an invaluable experience that I have thoroughly enjoyed. I have learnt how to use lasercapture technology and QPCR which will help in future experiments. My laboratory skills have improved and I now feelconfident in conducting experiments whilst working independently. The research project has reinforced my interest inresearch and has confirmed to me that I would love to pursue a scientific career. Before the placement, my experienceof working in a lab during my degree was completely different to how it is in a real research career and I now know Idefinitely want to complete a PhD in the future. I have found working in a laboratory, towards a goal that will hopefully bebeneficial to the medical field, extremely rewarding.

Biochemical Society Summer Vacation Studentship Report 2013
Biochemical Society Studentship Report: Lenka Stejskal
Department of Chemical and Biological Sciences, University of Huddersfield, Queensgate,Huddersfield,
Supervisor: Dr. Richard Bingham
Project Aims
Lyme Disease is an infectious diseasetransmitted to humans by the Borreliaburgdorferi sensu lato-infested ticks. The threemost common strains found to infect humansare B. burgdorferi, B. afzelii and B. garinii. Thesebacteria are able to evade the host immuneresponse and disseminate throughout the bodyto different organs and tissues, resulting in anextremely wide variety of symptoms. A numberof Borrelial surface proteins are known to bindthe human protein Factor-H, a regulator of thecomplement immune response. The aim of thisproject was to gain further understanding of aputative Factor-H binding protein from B. afzelii.We aim to determine a low-resolution structureby Small Angle X-ray Scattering, and to test theFactor-H binding using pure recombinantprotein.
Material and Methods
Protein Expression
A pET47b+ construct containing the putative outermembrane protein from Borrelia afzelii (BaOmp)fused to an N-terminal 6x-His tag and HRV-3Crecognition sequence was provided. The full lengthBorrelia protein has an N-terminal signal sequencethat would normally result in translocation acrossthe inner membrane. Replacing the signal sequencewith the 6x-His tag and HRV-3C cleavage siteprevents the translocation to the membrane andresults in the formation of inclusion bodiescontaining the protein of interest. Expression wasinduced by the addition of IPTG. SDS-PAGE ofsoluble and insoluble extracts showed significantoverexpression and large band in the insolublefraction (data not shown).
Protein Purification
Cells were disrupted by sonication in a Lysis bufferconsisting of 0.3M NaCl, 20mM sodium phosphatepH 8 (10 ml per gram wet weight of cells) andcentrifuged to separate the soluble and insolublefractions. The pellet was then washed by a series offour buffers, each wash step required the pellet tobe resuspended by brief sonication followed bycentrifugation. Two washes with Lysis buffersupplemented with 10mM DTT, 1mM EDTA and 5%Triton X-100. One wash in Lysis buffersupplemented with 5% Triton X-100. Finally, onewash with Lysis buffer. The pellet was thenresuspended in 8 M urea 20 mM NaCl, 20 mM tris,0.01% LDAO, pH 8 and loaded onto the Q-sepharose ion exchange column.
Refolding on the ion exchange column
Protein was refolded while bound to the Q-sepharose ion exchange column. Protein wasloaded in Equilibration Buffer (8 M urea, 20 mMNaCl, 20 mM tris, 0.01% LDAO, pH 8), and refoldedby running a gradient over 10 column volumes intoRefold buffer (1 M urea, 20 mM NaCl, 20 mM tris,0.1% LDAO, pH 8) followed by gradient elution overanother 10 column volumes into Elution buffer (1 MNaCl, 20 mM tris, 0.1% LDAO, pH 8) all at 0.5ml/min. SDS-PAGE analysis showed pure protein inall fractions (Figure 1).
Optimisation of Factor-H Binding Assay
An affinity binding immunoblot assay was used todetermine if the Borrelial protein had Factor-Hbinding activity. The Borrelia protein was run onnon-reducing SDS-PAGE alongside human factor H(20 µg) and bovine serum albumin (20 µg) aspositive and negative controls. The gel wasimmunoblotted, trimmed and blocked in 5% milkpowder made up in TBS. Following three, 5 minutewashes with TBS-Tween (0.05%) the membranewas incubated with human factor H at aconcentration of 88 µg/ml in TBS, for two hours withgentle agitation. The membrane was again washedand incubated with primary antibody (1:1000 ofmouse monoclonal anti-human factor H – Abcam)for one hour. The membrane was washed with thesecondary antibody for one hour (1:5000 of goatanti-mouse polyclonal alexafluor 680 – Abcam).The membrane was then visualised on a LI-COROdyssey infrared imaging device at 680nm
Initial results were poor with very faints bandsindicating either a weak interaction with Factor-H orsome non-specific binding. We optimized theprocedure, increasing the concentration of Factor-Hduring the washes and decreasing the amount ofFactor-H in the positive control (Figure 2B).
Figure 1. Fractions eluted from Q-sepharoseion exchange chromatography.

Biochemical Society Summer Vacation Studentship Report 2013
Small Angle X-ray Scattering
High concentrations of detergent are known tointerfere with SAXS results. In order to decrease theconcentration of LDAO in fractions collected after
Ni-NTA column purification (0.1% LDAO) they weretransferred into Amicon Ultra-15 Centrifugal FilterUnits tubes, topped up to 12 ml with SAXS Buffer(0.3 M NaCl, 50 mM Tris and 0.04% LDAO) andcentrifuged at 5000G for 10 minutes. This wasrepeated twice and the protein concentration was4.4 mg/ml (determined by Abs280nm). A sampleof 250 ul was transferred to a capillary and X-rayscattering was collected for 12 hours in triplicate ona Bruker Nanostar (36 hours total datacollection). X-ray scattering was also measured inthe same way from a buffer blank. The scatteringfrom the buffer blank was subtracted from thesample data and the triplicates were averaged. Aconstant value was subtracted to correct for thermalbackground scattering.
The results (Figure 3) showed a significant increasein scattering at low q values, indicative of sampleaggregation. This is problematic because themolecular envelope calculations are only valid if thesystem is monodisperse. We truncated the data toremove this portion of the curve and attempted themolecular envelope calculations using the ATSASpackage. The results consistently produced aspherical molecule (data not shown), which isprobably not realistic as proteins (includingmembrane proteins) are generally asymmetric. Thiswill need to be repeated using different conditions.
Departures from the original proposal
Because the protein was produced as a fusion tothe 6xHis tag there are numerous non-native aminoacids at the N-terminus. One departure from theoriginal aims was to remove this tag using the HRV-3C protease. The protein was incubated with HRV-3C over a weekend at room temperature. Thisenzymatically removed the tag resulting in a nativeprotein. The 6xHis tag and the contaminating HRV-3C were then removed by passing the materialthrough the Ni-NTA column. The enzyme and thetag remained bound, and the protein of interestpassed through.
Attempts were made to crystallize the protein for X-ray crystallography. Some hanging-drop vapourdiffusion trays were set up and small crystalsproduced.
Future directions
The small crystals produced from the sparse matrixscreens will be optimized for high-resolutionstructural studies by X-ray crystallography.
The aggregation observed during the small angle X-ray scattering prevented high quality data collection;this will be repeated using more dilute samples, andperhaps higher detergent concentrations.
The Factor-H binding assay will be repeating usinghuman serum. This may be more successfulbecause the concentration of Factor-H isapproximately 3x higher than used in our assay.
Value of the studentship
Student: The studentship allowed me to becomefamiliar with a research laboratory environment andgain confidence in my lab skills. I have gainedexperience of X-ray data collection, ALBI assay andPCR; techniques I would not normally use duringmy undergraduate degree. I met and haddiscussions with several Lyme disease patients andmedical practitioners at the Lyme Disease ActionAnnual Conference 2013 in Guildford, Surrey. Ienjoyed working with a protein that may beimportant for virulence of this curious pathogen.
Supervisor: I have been impressed by Lenka’scommitment and enthusiasm throughout this project.Lenka has exceeded my expectations and made alasting contribution to this project. Thank you Lenka.
Acknowledgments
We would like to thank Gemma Brown for her skillsand guidance in the lab. Thanks go to Dr Peter Laityand Dr Adam Dyer for assistance in collecting andprocessing SAXS data.
Thanks to the Biochemical Society and theDepartment of Chemical and Biological Sciences atthe University of Huddersfield for Funding.
Figure 3.Small angle X-ray scattering profile ofBaOmp. Some aggregation is seen at low qvalues.
Factor -H
BaOmp
Figure 2. The ALBI assay indicates theBorrelia protein (BaOmp) binds Factor-H.
A B

Summer Project 2013 – Purification and analysis of HEAT repeat domains of DNA-dependent protein kinase A, aDNA repair protein
Student: Lorna Jackson, Supervisor: Dr Laura Spagnolo
Background
DNA dependent protein kinase (DNA-PK), which comprisesthe catalytic subunit (DNA-PKcs) and the Ku70/Ku80heterodimer, is an enzyme involved in DNA double strandbreak repair by non-homologous end joining (1). It istherefore important for maintenance of genomic integrity.The crystal structure of DNA-PKcs has been solved to aresolution of 6.6A° (2)(figure 1), revealing a 66 HEAT repeat
loop, which was the topic of study for this project. DNA binding activity has previously beendemonstrated in DNA-PKcs (3), and a possible DNA binding domain has been recognised in theseHEAT repeats (figure1) which, if confirmed, could give insight into the molecular mechanism ofDNA-PKcs in NHEJ.
Figure 1: 6.6A crystal structure of DNA-PKcs. HEAT repeat loop is shown in green. Putative DNA binding domain is shown in cyan. (Sibanda, B., Chirgadze, D. AndBlundell, T., 2010. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats . Nature, 463; pp118-122)
Aims:-To overexpress and purify protein constructs of the HEAT repeat loop of DNA-PKcs.-To possibly achieve a high enough purity for subsequent structural analysis.-To use these constructs in DNA binding assays for investigation of regional DNA binding.
Work carried out and results:The focus of the project was mainly on protein purification. Various plasmids containing sections of DNA-PKcs HEAT repeatswere already available in the lab. All these constructs have a poly-histidine tag, allowing them to be purified using nickel affinitychromatography. Initially construct 127, which covers a section of the HEAT repeats, and construct 226, which covers theputative DNA binding domain described earlier, were expressed and their purification attempted.
Protein expression:Arabinose inducible BL21 cells were transformed with plasmids containing 127 and 226 constructs using heat shock. Cells werecultured, and protein production induced using IPTG and arabinose. The cells were harvested by centrifugation and the pelletslysed using BugBuster® and nuclease. Protease inhibitors were added to reduce degradation of the target proteins.
Overexpression of 127 and 226 was successful (Fig2), and 127 was present in thesupernatant after lysis and centrifugation, indicating that it is soluble. However 226appears to precipitate in the conditions used, and indeed subsequent purificationfailed. For this reason purification and further analysis of 226 was not possible.
The same methods were used for overexpression and extraction of two differentconstructs; 213 and 235, which also cover the putative DNA binding domain in DNA-PKcs, in the hope that these would be soluble and therefore suitable for furtherexperimentation, however the same result was observed as for 226. Due to thisinsolubility and time constraints I focussed on construct 127 for the remainder of theproject.Protein purification
Large scale cultures of BL21 transformed with 127 were performed for high protein yield. The cells were lysed, centrifuged, andthe protein extracted from the supernatant using an AKTA® machine and three different purification steps:
-Nickel affinity chromatography, eluted in a gradient of 20 to 500mM imidazole-Ion exchange chromatography using Q anion exchange column, eluted in a gradient of 50 to 1000mM NaCl-After Ion exchange the protein was concentrated-Gel filtration chromatography
The results of this purification are shown in Figure3.
Figure2: Overexpression of 127 and 226 constructs inBL21 1h, 2h and ca.18h after induction, anduninduced calls (NI). Band positions of 127 and 226are indicated

Figure 3: SDS-PAGE of fractions from protein purification of 127 using a) nickel affinity chromatography, b) ion exchange chromatography and c) gel filtrationchromatography, successively.
The majority of contamination was removed through these steps. The bands at different sizes around the molecular weight of127 indicate degradation, possibly at a vulnerable site such as a linker.
DNA binding assay – Electrophoretic Mobility Shift Assay
DNA was synthesized using PCR of PST39 as template with primers T7 and STO768. The purified 127 protein from gel filtrationwas concentrated, and aliquots mixed with increasing concentrations of DNA in the presence of MgCl2. This was left to incubatethen run on a native-PAGE gel. The results of this assay were inconclusive. It appears that the protein is still present in thestacking gel, indicating that a DNA-127 complex may have formed but may have been too large to enter the native gel (resultsnot shown), however this is not sufficient evidence and further study is needed.
Future directionsIn an attempt to obtain highly purified 127, and avoid degradation, the above processes were repeated, using protein inhibitorsat every step. A sample of this newly purified 127 has been sent for mass spectrometry analysis to confirm that the correctprotein has been purified (awaiting results); if so this purified protein will be used for further analysis of DNA binding activity.The DNA binding assay gave promising but inconclusive results, so future efforts will be aimed at investigating this further. Asthe previously described method produced inconclusive results this could be done by using immobilised DNA for a pull downexperiment.Further attempts could be made to purify one of the constructs spanning the putative DNA binding region (226, 213 and 235) totest it for DNA binding. Using different conditions may allow the constructs to stay stable in solution. If these constructs, as wellas 127, can be purified to a high enough level they could be sent for x-ray crystallography to try to resolve their structure to abetter resolution than has been achieved in the DNAPKcs molecule so far.
Departures from the original proposalThe scaling up of the culture and protein production was more complex than we anticipated, so it took us some time to performlarge scale purification of 127.
After completing this summer project I feel that a career in scientific research is definitely right for me. I really enjoyed my timehere, and it has persuaded me to apply for PHDs this year, with a view to starting straight from graduation next year. Theexperience I have gained has been invaluable, and I now feel ready to start the next stage of my career.
References1) Ma, Y., Pannicke, U., Schwarz, K. and Lieber, M. R., 2002. Hairpin opening and overhang processing by an Artemis/DNA-dependent
protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell, 108; pp781–7942) Sibanda, B., Chirgadze, D. And Blundell, T., 2010. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT
repeats. Nature, 463; pp118-1223) Yaneva, M., Kowalewski, T. and Lieber, M. R., 1997. Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical
and atomic force microscopy studies. EMBO Journal, 16; pp5098–5112
a b c

Characterising the DNA binding specificity of a novel bacterial RNA polymerase ECF sigma factor
Project undertaken by Lucy CLARK, in the School of Biological Sciences, University of East Anglia (UEA),Norwich, UK
Supervisors: Dr Richard BOWATER & Dr Matt HUTCHINGS
Background to projectAntimycins are a family of molecules discovered over 60 years ago and are produced by Streptomyces bacteria. Theyare currently exploited for their antifungal properties due to their ability to inhibit the electron transport chain [1].Antimycins are also potent inhibitors of the mitochondrial Bcl-2/Bcl-xL related anti apoptotic proteins, suggestingmodified antimycins that no longer inhibit the electron transport chain could be effective anti-cancer drugs [2].Although antimycins were discovered in the 1950’s their biosynthesis was poorly understood until several geneclusters were recently identified in S. albus S4 [3]. The gene clusters contain four operons: antAB, antCDE, antFG andantHIJKLMNO. The antA gene encodes a unique ECF RNA polymerase sigma factor, referred to as AntA, which has thesole function of regulating antimycin synthesis by activating transcription of the antFG and antHIJKLMNO genes. Allknown antimycin gene clusters encode AntA and have conserved binding regions within their promoter sequences [4].
Aims of projectThe aim of the summer project was to begin characterisation of the regulatory mechanisms of the novel ECF RNApolymerase sigma factor, with an ultimate objective to aid further work into engineering synthetic pathways to allowfor overproduction of antimycins.
Material and methodsProtein Production in Esherichia coli BL21 Rosetta:The plasmid pET28AntA was previously generated in the laboratory of Dr Matt Hutchings at UEA, and contains thegene that encodes for AntA under the control of the promoter for T7 RNA polymerase. The gene for AntA is cloneddownstream of a His-tag, which aids affinity purification of the recombinant protein. Plasmid pET28AntA wastransformed into E. coli BL21 Rosetta cells and grown on LB Agar plates containing kanamycin to select for theplasmid. Colonies were inoculated into 10 ml LB with kanamycin and grown at 37 °C. Half the cultures were theninduced with IPTG at a concentration of 0.4 mM to over express the sigma factor.Protein Purification and AnalysisThe cells were harvested, lysed using sonication and separated into insoluble and soluble fractions. The solublefractions from the protein expression were purified using a His-Mag solution to select the His-tagged- AntA. Both theunpurified and purified soluble samples were analysed by 10% or 15% SDS-PAGE and stained with instant blue,which identifies proteins present in the samples.Cloning and Analysis of DNAA second plasmid was cloned to contain the gene encoding neomycin resistance under control of the antG promotersequence. DNA samples were purified using the Wizard Mini-prep kit, as specified by the manufacturer, and analysedby electrophoresis using a 1% agarose gel. This plasmid along with pET28AntA was transformed into E. coli BL21cells to allow further characterisation of the sigma factor.Cloning and Analysis of DNA:
ResultsProtein Production:Colonies were visible on the plates containing E. coli BL21 Rosetta cells that had been transformed with plasmidpET28AntA.Protein AnalysisThe 10% SDS-PAGE showed that the soluble fractions induced with IPTG contained a protein at the expected size forthe sigma factor, Fig 1.Protein Purification:The 15% SDS-PAGE showed the sigma factor had been purified at a relatively low yield, Fig 2.
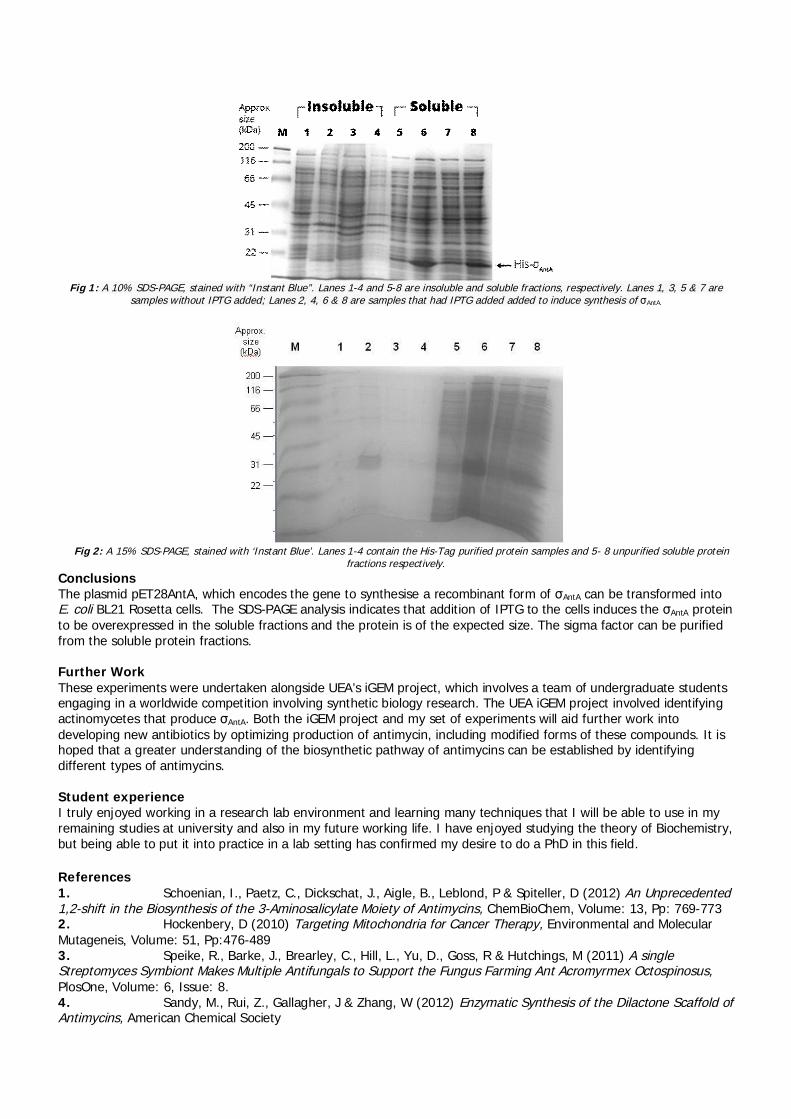
Fig 1: A 10% SDS-PAGE, stained with “Instant Blue”. Lanes 1-4 and 5-8 are insoluble and soluble fractions, respectively. Lanes 1, 3, 5 & 7 aresamples without IPTG added; Lanes 2, 4, 6 & 8 are samples that had IPTG added added to induce synthesis of AntA.
Fig 2: A 15% SDS-PAGE, stained with ‘Instant Blue’. Lanes 1-4 contain the His-Tag purified protein samples and 5- 8 unpurified soluble proteinfractions respectively.
ConclusionsThe plasmid pET28AntA, which encodes the gene to synthesise a recombinant form of AntA can be transformed intoE. coli BL21 Rosetta cells. The SDS-PAGE analysis indicates that addition of IPTG to the cells induces the AntA proteinto be overexpressed in the soluble fractions and the protein is of the expected size. The sigma factor can be purifiedfrom the soluble protein fractions.
Further WorkThese experiments were undertaken alongside UEA’s iGEM project, which involves a team of undergraduate studentsengaging in a worldwide competition involving synthetic biology research. The UEA iGEM project involved identifyingactinomycetes that produce AntA. Both the iGEM project and my set of experiments will aid further work intodeveloping new antibiotics by optimizing production of antimycin, including modified forms of these compounds. It ishoped that a greater understanding of the biosynthetic pathway of antimycins can be established by identifyingdifferent types of antimycins.
Student experienceI truly enjoyed working in a research lab environment and learning many techniques that I will be able to use in myremaining studies at university and also in my future working life. I have enjoyed studying the theory of Biochemistry,but being able to put it into practice in a lab setting has confirmed my desire to do a PhD in this field.
References1. Schoenian, I., Paetz, C., Dickschat, J., Aigle, B., Leblond, P & Spiteller, D (2012) An Unprecedented1,2-shift in the Biosynthesis of the 3-Aminosalicylate Moiety of Antimycins, ChemBioChem, Volume: 13, Pp: 769-7732. Hockenbery, D (2010) Targeting Mitochondria for Cancer Therapy, Environmental and MolecularMutageneis, Volume: 51, Pp:476-4893. Speike, R., Barke, J., Brearley, C., Hill, L., Yu, D., Goss, R & Hutchings, M (2011) A singleStreptomyces Symbiont Makes Multiple Antifungals to Support the Fungus Farming Ant Acromyrmex Octospinosus,PlosOne, Volume: 6, Issue: 8.4. Sandy, M., Rui, Z., Gallagher, J & Zhang, W (2012) Enzymatic Synthesis of the Dilactone Scaffold ofAntimycins, American Chemical Society

Photograph: Lucy Clark, with supervisor Dr Richard Bowater, in the CMSB Laboratory in the School of Biological Sciences, UEA.

Biochemical Society Summer Studentship Report 2013 - Luke Daniel Hutchinson
Investigating the cell cycle regulation of the deubiquitylase UCHL5Supervisor: Dr. Judy Coulson (Cellular & Molecular Physiology, University of Liverpool)
Introduction: Ubiquitylation is a reversible post-translational modification involved in multiplecellular processes including protein degradation, signal transduction cascades and regulation oftranscription. Ubiquitin, a 76-amino acid polypeptide, can be covalently conjugated to lysineresidues of protein substrates via the sequential action of E1, E2 and E3 ligases. Ubiquitin moietiescan be subsequently removed from protein substrates via deubiquitylases (DUBs), a familycomprised of approximately 90 enzymes(1). Ubiquitin carboxyl-terminal hydrolase L5 (UCHL5) is adeubiquitylase enzyme associated with the 19S regulatory particle of the proteasome and thehuman INO80 chromatin-remodelling complex in the nucleus(2, 3). UCHL5 is a cysteine protease thatspecifically cleaves Lys-48-linked polyubiquitin chains. Previous research in Dr. Coulson’slaboratory had identified several DUBs that exhibit differential affinity towards ubiquitin-active-siteprobes in mitotic or interphase cell extracts. Based on molecular weight and cellular abundance,UCHL5 was predicted as one DUB that may exhibit cell-cycle dependent catalytic activity.
Aims: The aim of this project was to investigate the differential regulation of UCHL5 throughout thecell cycle, by profiling the transcript level, protein expression, phosphorylation status and ubiquitin-active-site probe affinity for UCHL5 at specific cell cycle phases.
Description of Work: A549 human lung carcinoma cells were cultured in DMEM supplementedwith 10% FBS at 37°C with 5% CO2. For synchronisation, cells were seeded at different densities:1.0x106 cells per 10cm dish for asynchronous, G1/S and late S cells, or 1.5x106 cells for G2/M andearly G1 cells, and arrested at different stages of the cell cycle using standard thymidine/nocodazoleprotocols. Various extracts were prepared for different analyses.
For immunoblotting, cells were lysed in Laemmli buffer and protein concentrations determined byBCA assay (Pierce). Equal amounts of protein were separated by gel electrophoresis on 12%polyacrylamide gels (BioRad) and transferred to nitrocellulose membranes. The membranes wereincubated with primary antibodies (anti-UCHL5, ab133508; anti-beta Actin, ab6276; anti-Cyclin B1,05-373) and IRDye secondary antibodies for imaging on a LI-COR Odyssey 2.1. Protein extractswere also run on a 10% polyacrylamide PhosTag gel, and immunoblotted for UCHL5 and Actin.
For quantitative RT-PCR, total RNA was purified on RNeasy columns (Qiagen); yields and puritywere determined on a NanoDrop 1000. From the RNA extracts that were obtained, cDNA wassynthesised and amplified with PCR primers specific for UCHL5 or Actin using SYBR green.
For ubiquitin activity probe analysis, detergent-free protein extracts were prepared by mechanicaldisruption and quantified by BCA assay. Extracts (asynchronous, G2/M, G1) were incubated withHA-Ubiquitin-VME (vinyl methyl ester), and immunoblotted on 4-12% gradient gel (NuPAGE).
Results and Outcomes: Preliminary data had suggested a potential decrease in UCHL5 activity atG2/M relative to that in interphase cells. We set out to validate this observation, explore thepotential causes and extrapolate analysis to additional cell cycle phases. One reason for adecrease in UCHL5 activity may be a decrease in its abundance, however quantitativeimmunoblotting indicated increased UCHL5 protein expression at G2/M relative to asynchronousextracts (Figure 1A). On quantitative PCR analysis, no corresponding increase in UCHL5 mRNAexpression was observed at G2/M (Figure 1b), suggesting this is may be the result of post-transcriptional regulation. In contrast, we found that UCHL5 protein expression decreased at G1/Srelative to asynchronous cells, again without a corresponding change in the UCHL5 mRNA level(Figure 1A, 1B). As a measure of UCHL5 activity, we incubated extracts with the ubiquitin-active-site probe HA-Ub-VME. Immunodetection of UCHL5 indicated that the proportion of ‘active’ (i.e.probe-bound) and ‘inactive’ (i.e. free) UCHL5 remained relatively consistent between theasynchronous and G2/M cell extracts. Thus we did not validate the original observation.Interestingly however, there was a substantial decrease in the proportion of catalytically activeUCHL5 in the early G1 cell extract (Figure 1C). As phosphorylation of proteins may alter theiractivity, we separated protein extracts on a PhosTag gel to enhance the mobility shift forphosphorylated proteins. Immunodetection of UCHL5 indicated that it is phosphorylated andprovided preliminary evidence that some forms of phospho-UCHL5 vary during the cell cycle,
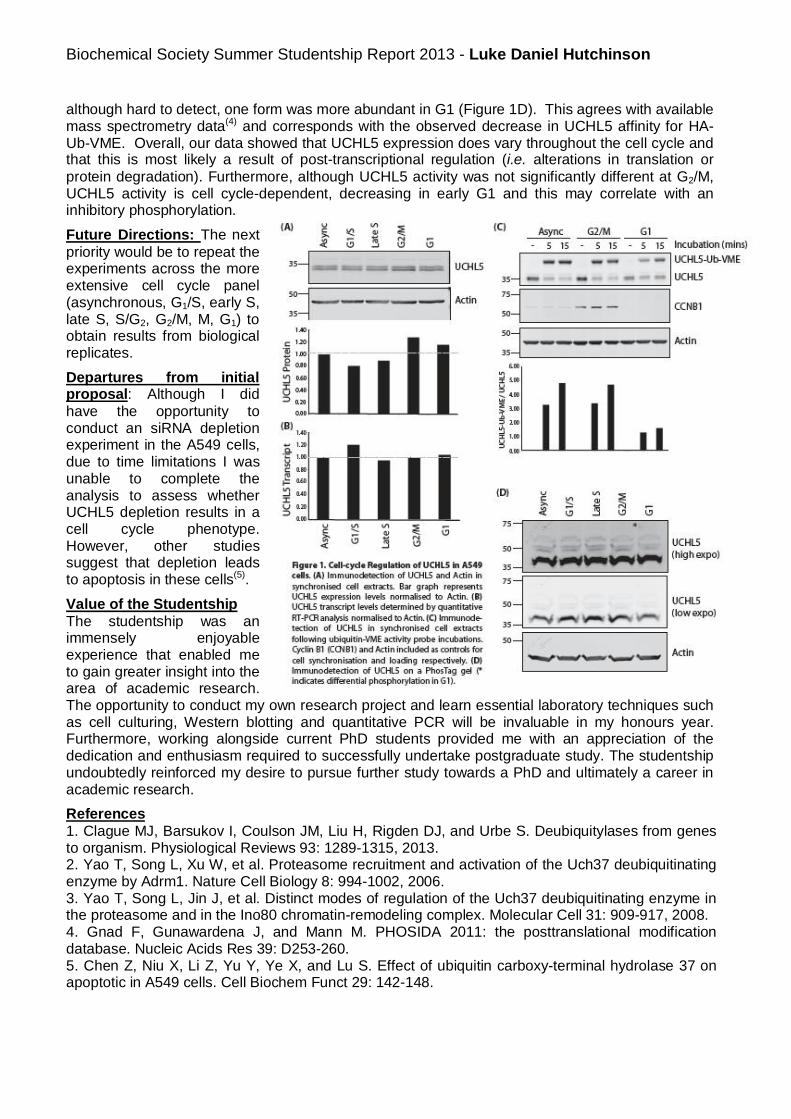
Biochemical Society Summer Studentship Report 2013 - Luke Daniel Hutchinson
although hard to detect, one form was more abundant in G1 (Figure 1D). This agrees with availablemass spectrometry data(4) and corresponds with the observed decrease in UCHL5 affinity for HA-Ub-VME. Overall, our data showed that UCHL5 expression does vary throughout the cell cycle andthat this is most likely a result of post-transcriptional regulation (i.e. alterations in translation orprotein degradation). Furthermore, although UCHL5 activity was not significantly different at G2/M,UCHL5 activity is cell cycle-dependent, decreasing in early G1 and this may correlate with aninhibitory phosphorylation.
Future Directions: The nextpriority would be to repeat theexperiments across the moreextensive cell cycle panel(asynchronous, G1/S, early S,late S, S/G2, G2/M, M, G1) toobtain results from biologicalreplicates.
Departures from initialproposal: Although I didhave the opportunity toconduct an siRNA depletionexperiment in the A549 cells,due to time limitations I wasunable to complete theanalysis to assess whetherUCHL5 depletion results in acell cycle phenotype.However, other studiessuggest that depletion leadsto apoptosis in these cells(5).
Value of the StudentshipThe studentship was animmensely enjoyableexperience that enabled meto gain greater insight into thearea of academic research.The opportunity to conduct my own research project and learn essential laboratory techniques suchas cell culturing, Western blotting and quantitative PCR will be invaluable in my honours year.Furthermore, working alongside current PhD students provided me with an appreciation of thededication and enthusiasm required to successfully undertake postgraduate study. The studentshipundoubtedly reinforced my desire to pursue further study towards a PhD and ultimately a career inacademic research.
References1. Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, and Urbe S. Deubiquitylases from genesto organism. Physiological Reviews 93: 1289-1315, 2013.2. Yao T, Song L, Xu W, et al. Proteasome recruitment and activation of the Uch37 deubiquitinatingenzyme by Adrm1. Nature Cell Biology 8: 994-1002, 2006.3. Yao T, Song L, Jin J, et al. Distinct modes of regulation of the Uch37 deubiquitinating enzyme inthe proteasome and in the Ino80 chromatin-remodeling complex. Molecular Cell 31: 909-917, 2008.4. Gnad F, Gunawardena J, and Mann M. PHOSIDA 2011: the posttranslational modificationdatabase. Nucleic Acids Res 39: D253-260.5. Chen Z, Niu X, Li Z, Yu Y, Ye X, and Lu S. Effect of ubiquitin carboxy-terminal hydrolase 37 onapoptotic in A549 cells. Cell Biochem Funct 29: 142-148.

0%
20%
40%
60%
80%
100%
120%
Control 2Hrs 4Hrs
%C
ells
Time after Ciliobrevin addition
ERGICp53 localisation to the Golgiapparatus
Highlocalisation
Mediumlocalisation
lowlocalisation
0%
20%
40%
60%
80%
100%
120%
Control 2Hrs 4Hrs
%C
ells
Time after Ciliobrevin addition
Lysosome DistributionPeripheral
EvenlyDistributed andPeripheralEvenlyDistributed
Central andEvenlyDistributedCentralLocalisation
Biochemical Studentship Report 2013
Background
Dynein is a motor protein that carries intracellular cargo towards the minus end of microtubules, hencemoving cargo towards the centre of the cell. It has also been shown to play a role in ciliary movement andspindle formation (Carter et al, 2011). In 2012 Firestone et al. identified a group of compounds calledciliobrevins, which disrupted spindle formation and the transport of components within cilia. Because of thisFirestone et al hypothesized that ciliobrevins act as an inhibitor of the dynein motor protein. Ciliobrevins havesince been used as dynein inhibitors in other studies (Clippinger & Alwine, 2012; Wen et al. 2013).
Aims
The aims of this project were to identify any effects of ciliobrevin on the organisation of certain organelles inthe cell, and to test the theory that ciliobrevin has an inhibitory effect on dynein.
Project description
This was originally going to be achieved by using phase contrast and fluorescence live cell imaging toobserve changes in organelle movement before and after ciliobrevin addition. This method was not used inthe final experiments as the MRC5 and U2OS cells used in preliminary tests appeared to react badly to themedia used for imaging and also showed that the addition of ciliobrevin had no clear effects on the cell.
A timed experiment was used to identify the effects of ciliobrevin on VERO cells. The cells were plated ontocoverslips, and treated with or without 50µM of ciliobrevin for two or four hours. Cells were then fixed andlabelled with anibodies to a range of cellular organelles, and obseved by fluorescence microscopy.
Results
Ciliobrevin had very little effect on the morphology of the Golgi apparatus, early endosomes, transferrinreceptors, lysosomes and microtubules. The only exception was ERGICp53, a marker of the ER-Golgiintermediate compartment (ERGIC) that recycles between the ER, the Golgi apparatus and the ERGIC(Fujiwara et al., 1998). ERGICp53 showed a decrease in staining at the Golgi apparatus after the addition ofciliobrevin. This change in localisation was quantified by scoring 100 cells as having low, medium or highlocalisation of ERGICp53 to the Golgi apparatus (see Fig. 1: 300 cells scored in 3 experiments). This dataclearly showed that the number of cells with low Golgi apparatus localisation of ERGICp53 increase withtime after ciliobrevin addition, whereas the number of cells with high ERGICp53 localisation to the Golgiapparatus decrease with time after ciliobrevin addition. Chi squared analysis with 2 degrees of freedom gavethe changes in low and high localisation in both time intervals an x2 value greater than the 5.991 criticalvalue. This shows that they are significantly different to the control results, suggesting that the regulardynamic recycling of ERGIC53 between the ER and the Golgi apparatus is being disrupted.
Fig1: The amount of ERGICp53 localized to the Golgiapparatus with time after the addition of ciliobrevin.Although there is no significant change in the amount ofmedium localization, the number of low localisation cellsincreases significantly, whilst the number of highlocalization cells decreases over time.
Fig 2: Lysosome distribution in the cell after treatment withciliobrevin. 100 cells from each repeat experiment weregraded. Whilst there is a slight increase in cells withevenly distributed lysosomes, the number of cells withcentrally localised lysosomes decreases. None of thesechanges are significant however, so they are probably justcaused by natural variation.
Student: Madeleine StanleySupervisor: Victoria Allan, University of Manchester

The localisation of lysosomes (labelled with antibodies to LAMP1) was also examined because lysosomesmove using dynein. An increase in peripheral lysosomes after ciliobrevin addition would show that it has aninhibitory effect on the dynein motor protein. The position of lysosomes was scored before or after ciliobrevintreatment for 2 or 4 hours (Fig. 2: 300 cells scored in 3 experiments). The results show that ciliobrevin leadsto a slight reduction in centrally-located lysosome, and a concomitant increase in evenly-distributedlysosomes. A chi squared test with 4 degrees of freedom has shown that none of the changes observed inlysosome distribution after ciliobrevin addition have a x2 statistic that exceeds the critical value for the 0.05significance level. This means the changes are not significant, which suggests that ciliobrevin does not havean inhibitory effect on the dynein motor protein.
Departures from the original proposal
The original aim of this project was to study the motion of endosomes within the cell using live cell imaging.However, a paper on this topic was published just before the start of the project (Zajac et al. 2013), makingthat goal redundant: the aim of the project was therefore changed.
Future directions for the project
The results of this project clearly show that ciliobrevin is causing ERGICp53 to delocalise from the Golgiapparatus, however there is no indication as to how or why this is occurring. Identifying the mechanism bywhich these results have come about is one way in which this project can be continued. Another directionthis project could take is to study the effects ciliobrevin has on other cell lines. A preliminary experiment withU2OS cells failed to show the change in ERGICp53 localisation seen in VERO cells. This could suggest thatciliobrevin has different effects in different cell types.
Value of the studentship
This studentship has been a very enjoyable experience. Working in a cell biology lab has given me a betterunderstanding of what working in a lab is like, as well as in which direction I personally want my career to go.I’ve learnt the importance of planning experiments ahead of time as there is always the possibility of thingsgoing wrong. Most of all working as part of a lab has shown me the importance of communicating with othersand has increased my confidence greatly.
Value to the lab
Preliminary experiments in the Allan laboratory had already suggested that ciliobrevin was not a particularlygood inhibitor of dynein, although it seems to be accepted by the community as a useful tool. It was thereforevery useful to have Madeleine perform some simple experiments to test ciliobrevin’s effects further. Shegenerated some surprising results which show that the drug is indeed having an effect on cells, but not thoseyou would expect from a dynein inhibitor. She has generated some excellent data which will be combinedwith our other results to generate a paper that questions ciliobrevin’s specificity and usefulness. Such apaper will be important for the research community to prevent others from paying large sums for a compoundthat simply does not do what it is supposed to.
References
Carter A.P., Cho C., Jin L. & Vale R.D. (2011) Crystal Structure of the Dynein Motor Domain. Science. New Series. 331.pp 1159-1165
Clippinger A.J. & Alwine J.C. (2012). Dynein mediates the localisation and activation of mTOR in normal and humancytomegalovirus-infected cells. Genes and Development. 26:2015-2026
Firestone A. J., Weinger J. S., Maldonado M., Barlan. K., Langston L. D., O’Donnell M., Gelfrand V. I., Kapoor T. M. &Chen T. M. (2012) Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature. 484:125-129
Fujiwara T., Misumi Y. & Ikehara Y. (1998) Dynamic Recycling of ERGIC53 between the Endoplasmic Reticulum and theGolgi Complex Is Disrupted by Nordihydroguaiaretic Acid. Biochem. Biophys. Res. Commun. 253:869-876
Lu W., Fox P., Lakonishok M., Davidson M.W. & Gelfand V.I. (2013) Initial Neurite Outgrowth in Drosophila Neurons isDriven by Kinesin-Powered Microtubule Sliding. Curr. Biol. 23:1018-1023
Tan S.C., Scherer J. & Vallee R.B. (2011) Recruitment of dynein to late endosomes and lysosomes through lightintermediate chains. Molecular Biology of the Cell. 22:467-477
Zajac A.L., Goldman Y.E., Holzbaur E.L.F. & Ostap E.M. (2013) Local Cytoskeletal and Organelle Interactions ImpactMolecular-Motor-Driven Early Endosomal Trafficking. Curr. Biol. 23:1173-1180

Investigation of Short Chain Fatty Acid (SCFA) Receptor GPR-43 Expression and Function inthe Islets of Langerhans
Student- Ming Yang, BSc. Physiology, King’s College LondonSupervisor- Professor Shanta Persaud, Division of Diabetes and Nutritional Sciences, King’s College London
Introduction and AimsA diverse number of endocrine, paracrine and nutrient signals activate islet beta cell G-protein coupledreceptors (GPCRs) to regulate insulin secretion. Short chain fatty aAcids (SCFAs) are a product of fermentationof dietary fibres from intestinal microbiota, which are released into the blood where they are known to act onfree fatty acid receptors (FFARs) such as GPR-41 and GPR-43. They play a role in the regulation of variousphysiological processes including gastrointestinal motility, leukocyte recruitment and, in particular, nutrientsensing and energy homeostasis. Therefore, FFARs may provide potential pharmacological targets for thetreatment of type 2 diabetes, which is characterized by dysfunctional metabolic homeostasis and impairedinsulin secretion. The aims of this 6 week Biochemical Society-funded summer studentship project were toinvestigate the localisation of GPR-43 in the pancreas and, since GPR-43 is known to couple to the Gq signaltransduction pathway, the effects of SCFA agonists on beta cell calcium levels were also investigated.
MethodsDouble Fluorescence ImmunolabellingParaffin embedded mouse pancreatic samples were longitudinally sectioned to 7µm. Antigen retrieval wasperformed via microwave treatment of the sections with boiling sodium citrate (pH 6) for 10min. The sectionswere then immunostained with guinea pig anti-insulin monoclonal antibody (1:50 dilution, DAKO) for 1 hour atroom temperature and with rabbit anti-GPR-43 polyclonal antibody (1:10 dilution, Santa-Cruz) overnight at 4°C.Fluorescent detection of GPR-43 and insulin immunoreactivity was accomplished by treatment of sections withsecondary antibodies to GPR-43 (1:250 dilution, Alexa-Fluor) and insulin (1:250 dilution, Alexa-Fluor) for 1 hourrespectively. The sections were viewed using a Nikon TE2000-U fluorescent microscope.
Tissue CultureMIN6 beta cells were incubated in T75 flasks in Dulbecco’s Modified Eagle Medium (DMEM) containing 25mMglucose and supplemented with 10% fetal bovine serum (FBS), 1% L-glutamate, 2% penicillin/streptomycin at37°C, 5% CO2 in a humid atmosphere. The medium was changed every 3-4 days. When the cells had reached80-90% confluency they were trypsinized to detach the monolayer cells from tissue culture plastic and eitherused for experiments or passaged for further growth.
Ca2+ MicrofluorimetryMIN6 beta cells were seeded onto circular borosilicate glass coverslips at a density of 50,000 cells per slip andallowed to adhere in serum-free DMEM overnight in standard tissue culture conditions. The coverslips wereincubated for 40min at 37°C in 5 M Fura-2/AM then cells were perifused with a Na+ rich physiological saltsolution containing 2mM glucose, 2mM CaCl2, 10mM HEPES and various agents of interest at a flow rate of0.5mL/min. In order to record real time changes in intracellular calcium, cells were illuminated at 340nm and380nm alternately, emitted light was filtered at 510nm and data were collected every 3s using a CCD camera.
ResultsGPR-43 is localised to islet beta cellsThe double labelling immunohistochemistry experiments shown in Figure 1 indicated that GPR-43 receptorsare highly expressed in the endocrine pancreas, with much lower levels in the exocrine cells. Furthermore, theyellow staining (right panel) obtained by merging of the images of insulin expression (red, left panel) with GPR-
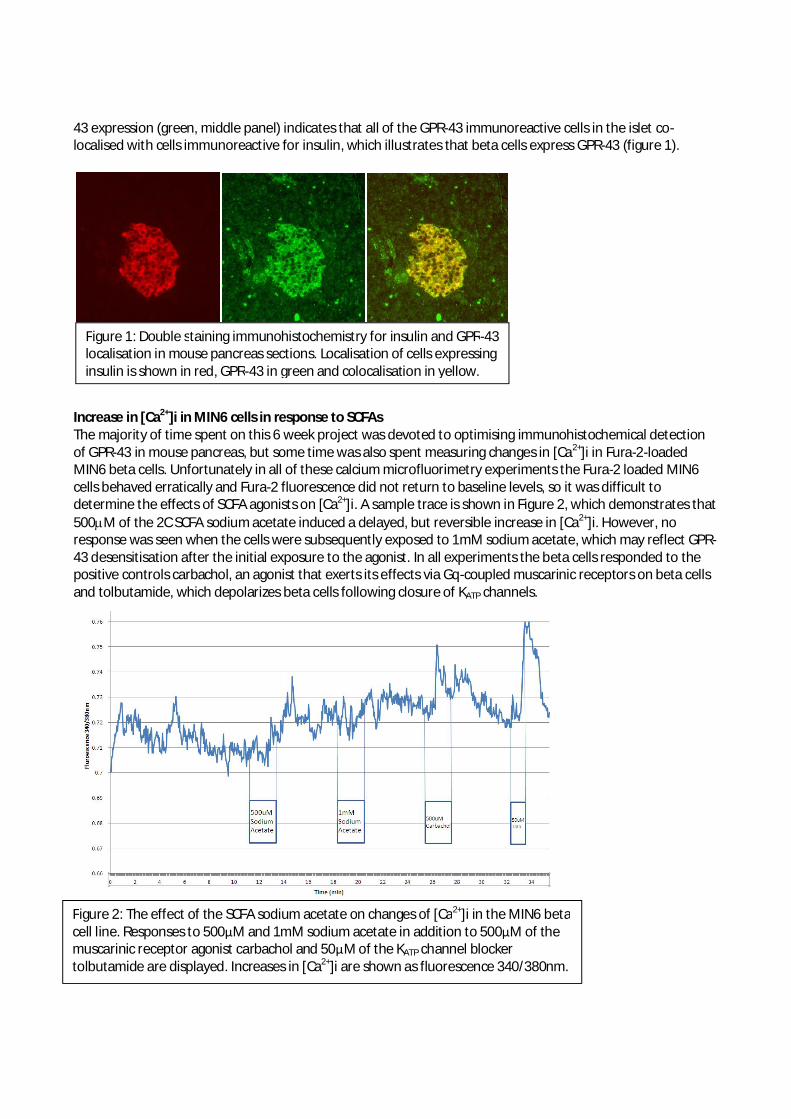
43 expression (green, middle panel) indicates that all of the GPR-43 immunoreactive cells in the islet co-localised with cells immunoreactive for insulin, which illustrates that beta cells express GPR-43 (figure 1).
Increase in [Ca2+]i in MIN6 cells in response to SCFAsThe majority of time spent on this 6 week project was devoted to optimising immunohistochemical detectionof GPR-43 in mouse pancreas, but some time was also spent measuring changes in [Ca2+]i in Fura-2-loadedMIN6 beta cells. Unfortunately in all of these calcium microfluorimetry experiments the Fura-2 loaded MIN6cells behaved erratically and Fura-2 fluorescence did not return to baseline levels, so it was difficult todetermine the effects of SCFA agonists on [Ca2+]i. A sample trace is shown in Figure 2, which demonstrates that500 M of the 2C SCFA sodium acetate induced a delayed, but reversible increase in [Ca2+]i. However, noresponse was seen when the cells were subsequently exposed to 1mM sodium acetate, which may reflect GPR-43 desensitisation after the initial exposure to the agonist. In all experiments the beta cells responded to thepositive controls carbachol, an agonist that exerts its effects via Gq-coupled muscarinic receptors on beta cellsand tolbutamide, which depolarizes beta cells following closure of KATP channels.
Figure 1: Double staining immunohistochemistry for insulin and GPR-43localisation in mouse pancreas sections. Localisation of cells expressinginsulin is shown in red, GPR-43 in green and colocalisation in yellow.
Figure 2: The effect of the SCFA sodium acetate on changes of [Ca2+]i in the MIN6 betacell line. Responses to 500 M and 1mM sodium acetate in addition to 500 M of themuscarinic receptor agonist carbachol and 50 M of the KATP channel blockertolbutamide are displayed. Increases in [Ca2+]i are shown as fluorescence 340/380nm.ratio.

Departures from Original Research ProposalThere were no major departures from the original research proposal.
Future DirectionsFuture experiments should investigate whether islet alpha cells and delta cells express GPR-43. In addition,further microfluorimetry experiments should be conducted to clearly indicate the effect of the SCFAs on betacell [Ca2+]i. The effect of GPR-43 on [cAMP]i levels should also be determined via cAMP immunoassay,fluorimetry or live cell imaging, since GPR-43 is also known to transduce via the Gi pathway to inactivateadenylate cyclase and lower [cAMP]i. Furthermore, the effects of SCFAs on insulin secretion should also beinvestigated via static incubation and dynamic perifusion experiments.
Value of the StudentshipThis six week studentship has given me an insight into many of the molecular and cellular techniques that areused in diabetes research, as well as an understanding of the molecular physiology behind these techniques. Incontrast to my practical classes during my university studies, in which all of the experiments have beenprepared for us, working in the lab on a defined project has allowed me to prepare and plan my ownexperiments, in addition to managing my time. Not only has this experience given me confidence in working inthe lab, the PhD students and the postdoctorate researchers have encouraged me to solve problems whenexperiments went wrong, in addition to scientific reasoning. Certainly, this experience would be invaluable formy third year research project (which is on a diabetes-related project with another member of the same lab)and it has inspired me further to pursue a career in research: I will be applying for PhD positions in diabetesresearch when I complete my BSc.
Supervisor’s CommentsMing picked up the necessary technical skills with relative ease and always asked when he was unsure aboutanything in the protocols. He optimised immunohistochemical detection of GPR43 expression in mousepancreas and obtained some convincing data to demonstrate its expression in islet -cells. He has a keen andenquiring mind and shows aptitude for lab-based research – I think that he gained a lot from the BiochemicalSociety summer studentship and he has solid future prospects as a research scientist.
AcknowledgementsI would like to thank Professor Shanta Persaud for accepting me into the lab and providing detailed structureand instructions for my summer studentship project. I would also like to thank the PhD students MustafaDogan and Zoheb Hassan for providing technical expertise and teaching me the techniques that enabled me tocomplete my project.
ReferencesBindels LB, Dewulf EM, Delzenne NM. 2013. GPR43/FFA2: physiopathological relevance and therapeuticprospects. Trends Pharmacol Sci. doi: 10.1016/j.tips.2013.02.002.
Layden BT, Angueira AR, Brodsky M, Durai V, Lowe WL Jr. 2013. Short chain fatty acids and their receptors: newmetabolic targets Transl Res. doi: 10.1016/j.trsl.2012.10.007. Epub 2012 Nov 9. Review.
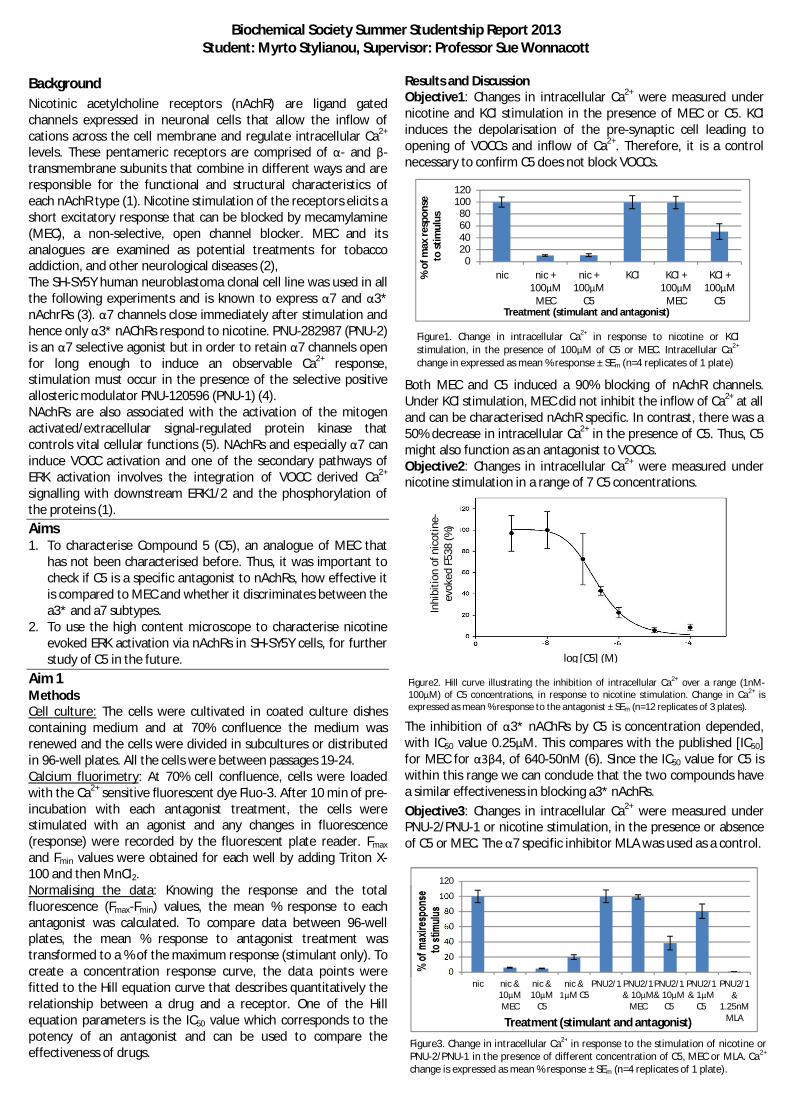
Biochemical Society Summer Studentship Report 2013Student: Myrto Stylianou, Supervisor: Professor Sue Wonnacott
Figure2. Hill curve illustrating the inhibition of intracellular Ca2+ over a range (1nM-100 M) of C5 concentrations, in response to nicotine stimulation. Change in Ca2+ isexpressed as mean % response to the antagonist ± SEm (n=12 replicates of 3 plates).
Figure3. Change in intracellular Ca2+ in response to the stimulation of nicotine orPNU-2/PNU-1 in the presence of different concentration of C5, MEC or MLA. Ca2+
change is expressed as mean % response ± SEm (n=4 replicates of 1 plate).
020406080
100120
nic nic +100 M
MEC
nic +100 M
C5
KCl KCl +100 M
MEC
KCl +100 M
C5
%of
max
resp
onse
tost
imul
us
Treatment (stimulant and antagonist)
0
20
40
60
80
100
120
nic nic &10 MMEC
nic &10 M
C5
nic &M C5
PNU2/1 PNU2/1& 10 M
MEC
PNU2/1& 10 M
C5
PNU2/1& 1 M
C5
PNU2/1&
1.25nMMLATreatment (stimulant and antagonist)
Inhi
bitio
nof
nico
tine-
evok
edF5
38(%
)
log [C5] (M)
BackgroundNicotinic acetylcholine receptors (nAchR) are ligand gatedchannels expressed in neuronal cells that allow the inflow ofcations across the cell membrane and regulate intracellular Ca2+
levels. These pentameric receptors are comprised of - and -transmembrane subunits that combine in different ways and areresponsible for the functional and structural characteristics ofeach nAchR type (1). Nicotine stimulation of the receptors elicits ashort excitatory response that can be blocked by mecamylamine(MEC), a non-selective, open channel blocker. MEC and itsanalogues are examined as potential treatments for tobaccoaddiction, and other neurological diseases (2),The SH-SY5Y human neuroblastoma clonal cell line was used in allthe following experiments and is known to express 7 and 3*nAchrRs (3). 7 channels close immediately after stimulation andhence only 3* nAChRs respond to nicotine. PNU-282987 (PNU-2)is an 7 selective agonist but in order to retain 7 channels openfor long enough to induce an observable Ca2+ response,stimulation must occur in the presence of the selective positiveallosteric modulator PNU-120596 (PNU-1) (4).NAchRs are also associated with the activation of the mitogenactivated/extracellular signal-regulated protein kinase thatcontrols vital cellular functions (5). NAchRs and especially 7 caninduce VOCC activation and one of the secondary pathways ofERK activation involves the integration of VOCC derived Ca2+
signalling with downstream ERK1/2 and the phosphorylation ofthe proteins (1).Aims1. To characterise Compound 5 (C5), an analogue of MEC that
has not been characterised before. Thus, it was important tocheck if C5 is a specific antagonist to nAchRs, how effective itis compared to MEC and whether it discriminates between thea3* and a7 subtypes.
2. To use the high content microscope to characterise nicotineevoked ERK activation via nAchRs in SH-SY5Y cells, for furtherstudy of C5 in the future.
Aim 1MethodsCell culture: The cells were cultivated in coated culture dishescontaining medium and at 70% confluence the medium wasrenewed and the cells were divided in subcultures or distributedin 96-well plates. All the cells were between passages 19-24.Calcium fluorimetry: At 70% cell confluence, cells were loadedwith the Ca2+ sensitive fluorescent dye Fluo-3. After 10 min of pre-incubation with each antagonist treatment, the cells werestimulated with an agonist and any changes in fluorescence(response) were recorded by the fluorescent plate reader. Fmax
and Fmin values were obtained for each well by adding Triton X-100 and then MnCl2.Normalising the data: Knowing the response and the totalfluorescence (Fmax-Fmin) values, the mean % response to eachantagonist was calculated. To compare data between 96-wellplates, the mean % response to antagonist treatment wastransformed to a % of the maximum response (stimulant only). Tocreate a concentration response curve, the data points werefitted to the Hill equation curve that describes quantitatively therelationship between a drug and a receptor. One of the Hillequation parameters is the IC50 value which corresponds to thepotency of an antagonist and can be used to compare theeffectiveness of drugs.
Results and DiscussionObjective1: Changes in intracellular Ca2+ were measured undernicotine and KCl stimulation in the presence of MEC or C5. KClinduces the depolarisation of the pre-synaptic cell leading toopening of VOCCs and inflow of Ca2+. Therefore, it is a controlnecessary to confirm C5 does not block VOCCs.
Both MEC and C5 induced a 90% blocking of nAchR channels.Under KCl stimulation, MEC did not inhibit the inflow of Ca2+ at alland can be characterised nAchR specific. In contrast, there was a50% decrease in intracellular Ca2+ in the presence of C5. Thus, C5might also function as an antagonist to VOCCs.Objective2: Changes in intracellular Ca2+ were measured undernicotine stimulation in a range of 7 C5 concentrations.
The inhibition of 3* nAChRs by C5 is concentration depended,with IC50 value 0.25 M. This compares with the published [IC50]for MEC for 4, of 640-50nM (6). Since the IC50 value for C5 iswithin this range we can conclude that the two compounds havea similar effectiveness in blocking a3* nAchRs.Objective3: Changes in intracellular Ca2+ were measured underPNU-2/PNU-1 or nicotine stimulation, in the presence or absenceof C5 or MEC. The 7 specific inhibitor MLA was used as a control.
Figure1. Change in intracellular Ca2+ in response to nicotine or KClstimulation, in the presence of 100 M of C5 or MEC. Intracellular Ca2+
change in expressed as mean % response ± SEm (n=4 replicates of 1 plate)
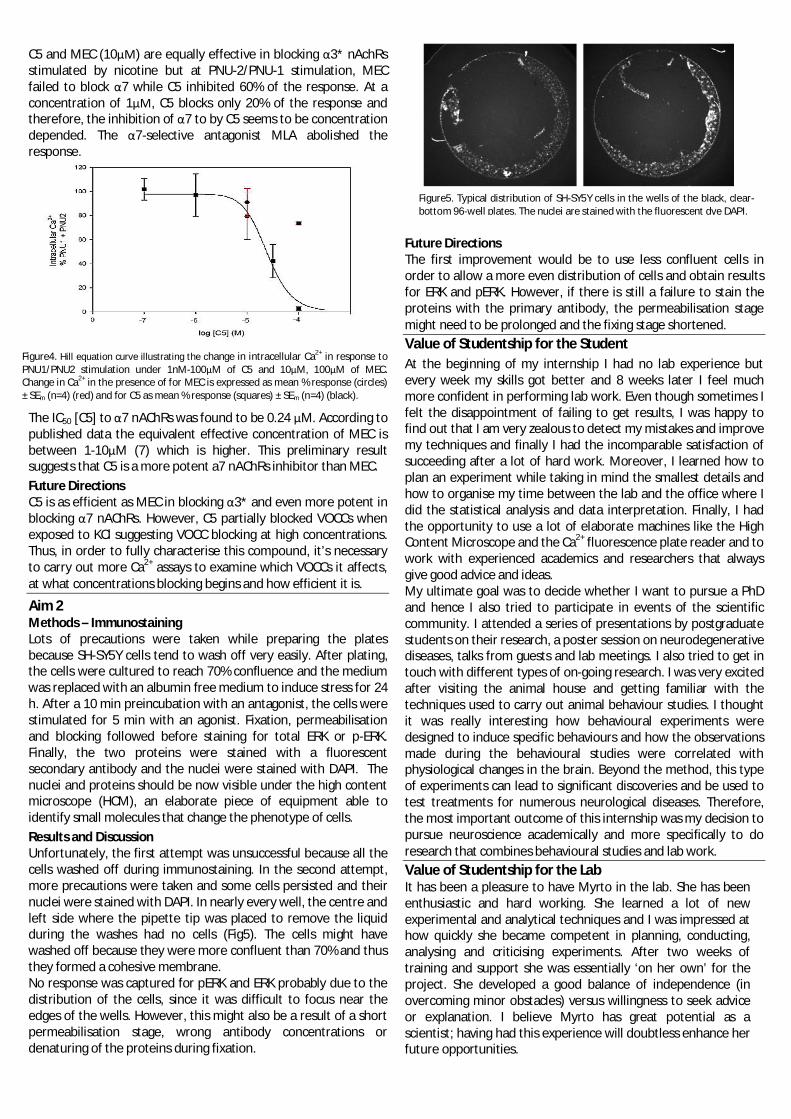
Figure4. Hill equation curve illustrating the change in intracellular Ca2+ in response toPNU1/PNU2 stimulation under 1nM-100 M of C5 and 10 M, 100 M of MEC.Change in Ca2+ in the presence of for MEC is expressed as mean % response (circles)± SEm (n=4) (red) and for C5 as mean % response (squares) ± SEm (n=4) (black).
Figure5. Typical distribution of SH-SY5Y cells in the wells of the black, clear-bottom 96-well plates. The nuclei are stained with the fluorescent dye DAPI.
C5 and MEC (10 ) are equally effective in blocking 3* nAchRsstimulated by nicotine but at PNU-2/PNU-1 stimulation, MECfailed to block 7 while C5 inhibited 60% of the response. At aconcentration of 1 , C5 blocks only 20% of the response andtherefore, the inhibition of 7 to by C5 seems to be concentrationdepended. The 7-selective antagonist MLA abolished theresponse.
The IC50 [C5] to 7 nAChRs was found to be 0.24 M. According topublished data the equivalent effective concentration of MEC isbetween 1-10 M (7) which is higher. This preliminary resultsuggests that C5 is a more potent a7 nAChRs inhibitor than MEC.Future DirectionsC5 is as efficient as MEC in blocking 3* and even more potent inblocking 7 nAChRs. However, C5 partially blocked VOCCs whenexposed to KCl suggesting VOCC blocking at high concentrations.Thus, in order to fully characterise this compound, it’s necessaryto carry out more Ca2+ assays to examine which VOCCs it affects,at what concentrations blocking begins and how efficient it is.
Aim 2Methods – ImmunostainingLots of precautions were taken while preparing the platesbecause SH-SY5Y cells tend to wash off very easily. After plating,the cells were cultured to reach 70% confluence and the mediumwas replaced with an albumin free medium to induce stress for 24h. After a 10 min preincubation with an antagonist, the cells werestimulated for 5 min with an agonist. Fixation, permeabilisationand blocking followed before staining for total ERK or p-ERK.Finally, the two proteins were stained with a fluorescentsecondary antibody and the nuclei were stained with DAPI. Thenuclei and proteins should be now visible under the high contentmicroscope (HCM), an elaborate piece of equipment able toidentify small molecules that change the phenotype of cells.Results and DiscussionUnfortunately, the first attempt was unsuccessful because all thecells washed off during immunostaining. In the second attempt,more precautions were taken and some cells persisted and theirnuclei were stained with DAPI. In nearly every well, the centre andleft side where the pipette tip was placed to remove the liquidduring the washes had no cells (Fig5). The cells might havewashed off because they were more confluent than 70% and thusthey formed a cohesive membrane.No response was captured for pERK and ERK probably due to thedistribution of the cells, since it was difficult to focus near theedges of the wells. However, this might also be a result of a shortpermeabilisation stage, wrong antibody concentrations ordenaturing of the proteins during fixation.
Future DirectionsThe first improvement would be to use less confluent cells inorder to allow a more even distribution of cells and obtain resultsfor ERK and pERK. However, if there is still a failure to stain theproteins with the primary antibody, the permeabilisation stagemight need to be prolonged and the fixing stage shortened.Value of Studentship for the StudentAt the beginning of my internship I had no lab experience butevery week my skills got better and 8 weeks later I feel muchmore confident in performing lab work. Even though sometimes Ifelt the disappointment of failing to get results, I was happy tofind out that I am very zealous to detect my mistakes and improvemy techniques and finally I had the incomparable satisfaction ofsucceeding after a lot of hard work. Moreover, I learned how toplan an experiment while taking in mind the smallest details andhow to organise my time between the lab and the office where Idid the statistical analysis and data interpretation. Finally, I hadthe opportunity to use a lot of elaborate machines like the HighContent Microscope and the Ca2+ fluorescence plate reader and towork with experienced academics and researchers that alwaysgive good advice and ideas.My ultimate goal was to decide whether I want to pursue a PhDand hence I also tried to participate in events of the scientificcommunity. I attended a series of presentations by postgraduatestudents on their research, a poster session on neurodegenerativediseases, talks from guests and lab meetings. I also tried to get intouch with different types of on-going research. I was very excitedafter visiting the animal house and getting familiar with thetechniques used to carry out animal behaviour studies. I thoughtit was really interesting how behavioural experiments weredesigned to induce specific behaviours and how the observationsmade during the behavioural studies were correlated withphysiological changes in the brain. Beyond the method, this typeof experiments can lead to significant discoveries and be used totest treatments for numerous neurological diseases. Therefore,the most important outcome of this internship was my decision topursue neuroscience academically and more specifically to doresearch that combines behavioural studies and lab work.Value of Studentship for the LabIt has been a pleasure to have Myrto in the lab. She has beenenthusiastic and hard working. She learned a lot of newexperimental and analytical techniques and I was impressed athow quickly she became competent in planning, conducting,analysing and criticising experiments. After two weeks oftraining and support she was essentially ‘on her own’ for theproject. She developed a good balance of independence (inovercoming minor obstacles) versus willingness to seek adviceor explanation. I believe Myrto has great potential as ascientist; having had this experience will doubtless enhance herfuture opportunities.

Final year PhD student Jack Brown, Myrto Stylianou and ProfessorSue Wonnacott
References1. Dajas-Bailador F.A, Mogg A.J, Wonnacott S., 2002, Intracellular Ca2+ signals evoked by stimulation of nicotinic
acetylcholine receptors in SH-SY5Y cells: contribution of voltage-operated Ca2+ channels and Ca2+ stores, Journal ofNeurochemistry, Vol. 81, Pp 606-614
2. Batcher I. et al, Wu B., Shytle D.R., George T.P., 2009, Mecamylamine – a nicotinic acetylcholine receptor antagonist withpotential for the treatment of neuropsychiatric disorders, Informa healthcare, Vol. 10, No. 16 , Pp 2709-2721
3. Lukas R.J., Norman S.A., Lucero L., 1993, Characterization of Nicotinic Acetylcholine Receptors Expressed by Cells of theSH-SY5Y Human Neuroblastoma Clonal Line, Molecular and Cellular Neurosciences, Pp. 1-12
4. Livingstone P.D., Dickinson J.A., Srinivasan J., Kew J.N. C., Wonnacott S., 2010, Glutamate–Dopamine Crosstalk in theRat Prefrontal Cortex is Modulated by Alpha7 Nicotinic Receptors and Potentiated by PNU-120596, Journal of MolecularNeuroscience, Vol. 40, Issue 1-2, Pp 172-176
5. Caunt C.J., Armstrong S.P., McArdle C.A., 2010, Using high-content microscopy to study gonadotrophin-releasing hormoneregulation of ERK, Methods Mol Biol., Vol 661, 2010, Pp 507-524
6. Parket. L., Sanberg P. R., and Shytle R. D., 2001, Analysis of Mecamylamine Stereoisomers on Human Nicotinic ReceptorSubtypes, THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 297, No. 2, JPET 297:646–656,.
7. Wonnacott S., Barik J., 2007, Nicotinic Ach Receptors, Tocris bioscience, No.28

Natalie BarberBen Berks Lab (Oxford)
The twin-arginine translocation (Tat) pathway is a protein export system found in most bacteria, inwhich proteins are transported in their fully folded state. Proteins are targeted to this pathway via anamino terminal signal peptide, which comprises3 domains: the basic amino region, which containsthe consensus twin-arginine S-R-R-x-F-L-K motif , a hydrophobic central domain (h-region) and apolar carboxy-terminal domain (c-region).
TatA assembly is an important step in the Tat translocation cycle, and the TatA oligomeric state canact as a good indicator of activity of the Tat export pathway. When the Tat pathway is not active, theTatA exists as protomers distributed evenly throughout the membrane creating a “halo” image. Whena protein is targeted to the Tat pathway however, the TatA protomers assemble together and soappear as discrete spots on the membrane. These were the two phenotypes that needed to bedistinguished (Figure 1).
The aim of my project was to determine whether features of the signal peptide outside the twin-arginine motif are necessary to trigger TatA assembly.
Firstly, working with the signal peptide of the Streptomyces coelicolor Rieske iron-sulphur protein,the effects of truncating the signal peptide on the targeting of this protein to the Tat pathway wereinvestigated. Site-directed mutagenesis was performed to introduce premature stop codons in thesignal peptide to produce three truncations of varying lengths (Table 1). Once the mutants had beenproduced, the plasmids were transformed into ELV16 Ary which synthesised TatA-YFP so thatTatA assembly could be observed through fluorescence microscopy. The expression of the constructswas monitored by immune blotting and assembly was assessed by fluorescence imaging. The resultwas that the full length signal peptide control induced spots (as expected); that truncation to 11 wassufficient to abolish TatA oligomerization (Figure 1C) but that the effects of a less severe truncationto 15 could not be assessed because the protein was not expressed.
Secondly, using the Tat substrate CueO, I performed site-directed mutagenesis to incrementallyincrease the polarity of the signal peptide core h-region by substituting sections with one or moreGGG or GGGS sequences (Table 2).
Once the mutant had been produced, cell fractionation was used to see if proteins with these mutatedsignal peptides could still result in the successful export of CueO. For this assay, the pQE-CueOplasmids were transformed into MC4100 competent cells, induced and grown. Successful CueOexport would show that the mutant was still targeted and transported via the Tat system.
Construct name SequenceFull length pQE80-TM123Cys ..GRRKLIRNTMLGALTLVPLSGVVLLRCpQE80 Rsk 7 STOP ..GRRKLIRNTMpQE80 Rsk 11 STOP ..GRRKLIRNTMLGALpQE80 Rsk 15 STOP ..GRRKLIRNTMLGALTLVP
Table 1. Truncations of the signal sequence produced by site-directed mutagenesis
Figure 1. Fluorescence microscopy images of ELV16 Ary cells showing A) Examples of spotty cells in whichTatA polymerisation has occurred. B) Examples of halo cells in which TatA polymerisation has not occurred.C) ELV16 Ary cells transformed with Rsk 11 STOP construct, showing TatA polymerisation does not occur.

Natalie BarberBen Berks Lab (Oxford)
Fluorescence microscopy was also used in the same way as described above to determine whetherthe different constructs caused TatA assembly. The results are shown in Table 2.
The major outcome of this project was discovering, thatmy most minimal change in the h-region of the signalpeptide (GS3(1)) abolishes both export and TatAassembly. This is important since previously it wasthought that the h-region had no significant role intargeting of a protein to the Tat pathway. From Figure 2it is clear that GS3(1) is not transported as there is noprotein in the periplasmic (PGS3(1)) fraction, unlike theWT in which there is a band representing themature transported protein in the periplasm(PWT).
Further directions:Further fluorescence microscopy data, as well as more in depth quantitative analysis is needed inorder to come to more definitive conclusions regarding TatA oligomerisation. Also, looking at theCueO export results, it would be interesting to research how few of the GS3(4) changes are needed tostop export and assembly. In addition, if there were not time constraints I would ideally performanother experiment to see whether the variants are blocked in binding to TatBC. In this way I couldconclude whether the changes introduced here act through blocking the signal peptide binding orwhether they allow signal peptide binding but block downstream effects like TatA oligomerisation.
Having completed this studentship and experiencing what academic research is actually like, I amnow seriously considering embarking upon a PhD after my degree. I thoroughly enjoyed the wholeexperience as it gave me an understanding of what independent research entails which is so differentfrom the practical laboratory work I have undertaken on my degree course.
(Written by Ben Berks). This was a challenging and important project on an aspect of Tat transportthat we had not previously explored and with which Natalie made outstanding progress. Earlier workhad shown that the hydrophobic core of Tat signal peptides (the h-region) is not essential forrecognition by the Tat system. It is, therefore, remarkable, that Natalie’s data from two experimentalsystems implies that the h-region is required to mediate TatA assembly – which is the keydownstream effects of signal peptide binding to the Tat system. Natalie’s data raises the veryexciting possibility that the binding and downstream effector functions of Tat signal peptides areseparable and are associated with different parts of the signal peptide. Natalie’s project has openedup an entirely new research direction for the laboratory. With further development her results shouldform the basis of a publication in a major journal.
Construct name G/S substitutions CueO export? TatAassembly
Wild type pQE80 CueOh MQRRDFLKYSVALGVASALPLWSRAVFA Yes YespQE80 CueO GS3(1) MQRRDFLKYSGGGGVASALPLWSRAVFA No NopQE80 CueO GS3(2) MQRRDFLKYSGGGGSGGALPLWSRAVFA No NopQE80 CueO GS3(3) MQRRDFLKYSGGGGSGGGGSLWSRAVFA Probably not NopQE80 CueO GS3(4) MQRRDFLKYSGGGGSGGGGSGGGGAVFA Probably not -pQE80 CueO GS3(5) MQRRDFLKYSVALGVASALPLGGGAVFA Yes -
Table 2. G/S substitutions that were introduced into the signal peptide core, and the interpretation of the results of cellfractionation (column 3) and fluorescence microscopy assays (column 4) for each construct.
Figure 2. CueO export gel, comparing WT with GS3(1)T=total fraction, P=periplasmic fraction andS=spheroplastic fraction

Student: Nikolay Martyushenko, [email protected]: Jonathon Pines
Summer Project Report:
The aims of my project1) To investigate how the localization and activity of cyclinB1-cdk1 affect the latest time point
(“point of no return”) at which a cell can successfully activate the Spindle Assembly Checkpoint.2) To perform an assessment of the turnover rates of Mad2, Cdc20 and CyclinB1 between
kinetochores and the cytosol in living cells under different conditions, using FluorescenceRecovery After Photobleaching (FRAP) in “knock-in” cell lines.
3) To prepare a stable transfection of RPE cells with a fluorescently tagged inducible phosphomimeticmutant of Mps1to investigate the relative importance of cyclinB1-cdk1 and Mps1 in the formationof the mitotic checkpoint complex (MCC).
Results and discussionI began by making 4 new RPE cell lines with 4 different fluorescent (tagged with the Cyan FluorescentProtein, Cerulean) cyclinB1-cdk1 fusions. These fusions cannot be degraded in mitosis because they have anon-degradable mutant of cyclin B1, and have an altered Cdk1 such that they can be specifically inhibited. Imade the new cell lines by stable transfection and Flp-FRT recombination into an existing cell line thatalready had an endogenously tagged cyclinB1 allele, tagged with the Yellow Fluorescent Protein, Venus.Three of these fusions also had additional domains to localize them at different places inside the cell. Thesedomains were the CaaX box for plasma membrane localization, histone H3.1 for chromosome/chromatinlocalization, and Mis12 for kinetochore localization during mitosis. All these constructs were under thecontrol of the tetracyclin repressor, which prevents their expression (and possible toxic effects) untiltetracyclin is added.
We had to change my initial plan to transiently transfect cells that already expressed endogenously taggedMad2 with the above constructs. The reason was that the transfection was never very efficient and theproliferation of the RPE cells was dramatically reduced. We also observed the appearance of stress granulesthat were fluorescent in the spectra of all of our fluorescent proteins, making a quantitative assessment oftheir brightness virtually impossible. Therefore, stable transfections were made as outlined above. We alsodiscovered that 1NM-PP1 did not inhibit the activity of our constructs, probably due to our constructsinteracting with the endogenous cdk1 pool.
Using live cell microscopy we discovered that cells expressing our cyclinB1-cdk1 constructs were arrestedin metaphase, while all the endogenous cyclinB1 was degraded over time. We also found the untetheredversion of our construct localised to the same places as endogenous cyclin B1 except for thechromosomes/kinetochores. The new cell lines did not have a fluorescently tagged Mad2, whoserecruitment to kinetochores we initially planned to use to assess whether or not the SAC is activated.Therefore we decided to use immunofluorescence (IF) to detect Mad2 recruitment. I performed the IFanalysis on tetracyclin-induced (and therefore metaphase – arrested) cells. I created unattachedkinetochores adding nocodazole to the cell medium for a set amount of time before fixing the cells. Wefound that cells expressing the CaaX tagged and the untethered versions of our constructs could not recruitMad2 to kinetochores in the absence of endogenous CyclinB1. The cells expressing the Mis12 – tetheredconstructs (which localised to kinetochores) did, however, recruit detectable amounts of Mad2 to thekinetochores, although less than normal. Unfortunately we did not have time to perform an investigation ofthe “point of no return” of our new cell lines.

Student: Nikolay Martyushenko, [email protected]: Jonathon Pines
Though we did not have enough time to perform an assessment of the Mad2 recruiting capabilities of ourH3.1 tagged constructs, our current working model is that active cyclinB1-cdk1 complexes need to belocalized to the kinetochores and or chromosomes in order for the kinetochores to initiate a SAC response.If this is confirmed by future experiments, it will mean that cyclinB1-cdk1 is required for the SAC byphosphorylating or binding some non-diffusible kinetochore localized molecule. Therefore futureexperiments could be performed to try to identify this target of Cyclin B1-Cdk1. One candidate is Mps1 butunfortunately we did not have time to perform any experiments on our phosphomimetic mutant.
To perform FRAP analysis of Mad2, Cdc20 and Cyclin B1, RPE cells with endogenously Venus-taggedalleles of these proteins were used. I developed a macro for the ImageJ image processing program thatautomatically quantified the recovery of these proteins, correcting for bleaching due to filming and incidentlight intensity variations. The macro also automatically normalized, plotted and made an exponential fit tothe data, and wrote the resulting parameters automatically into spreadsheet files. The half-life of theproteins and their recovery fractions were the parameters derived from the fits. I performed FRAP analysison cells treated with nocodazole, or a CenpE inhibitor (GSK923295), or a combination of nocodazole withvarious concentrations of the Mps1 inhibitor AZ 3146. Each experiment was repeated on at least 15 cells,and 30 at most. Most effort was concentrated on the Mad2 cell line.
We found the average half-life of Mad2 on kinetochores in untreated RPE cells to be 1.3 seconds. This issignificantly different from previous estimations by Howell et al. (19 seconds) and Vink et al. (4 seconds)who measured Mad2 turnover in PtK2 cells and in vitro respectively. We found out that the number ofunattached kinetochores inversely affects the turnover rate and the recovery of Mad2 on kinetochores.Using the value of the presumed Kd for Mad2 dissociation from Mad1-Mad2 complexes (10 M) measuredby Vink et al. coupled with the Mad2 recovery data, we calculated, using the simple ligand-receptorbinding model (Mad2 + Mad2Mad1 Mad2-Mad2Mad1) proposed by Vink et al., that there should beabout 4*107 cytosolic Mad2 molecules per RPE cell during full kinetochore Mad2 recruitment. In our lab,however we have determined by repeated experiment that the total number of Mad2 molecules in one RPEcell to be about 5*105. This implies that either the Kd or the model proposed by Vink et al. is not valid.Therefore we propose a new, enzymatic model of Mad2 kinetochore recruitment:
Mad2O + Mad2Mad1 Mad2-Mad2Mad1 Mad2C + Mad2Mad1
Here Mad2O and Mad2C refer to the open and closed conformations of Mad2 respectively. The freeenergy difference corresponding to the Kd measured by Vink et al. (-33.9 kJ/mol) may possibly refer to thefree energy difference between the closed and open forms of Mad2. If true, it fits well with the modelproposed by Westhorpe et al., where p31comet promotes conversion of Mad2C back into Mad2O, possiblywith the help of an ATPase. The free energy difference of 33.9 kJ/mol of such a conversion fits well withthe free energy released during ATP hydrolysis (~50kJ/mol). To further test and possibly improve ordiscard our new model, we will need to perform in vitro experiments similar to those done by Vink et al.
How has the grant contributed to my career aspirations and skills?The grant gave me the opportunity to work in a high profile laboratory, and find out for myself how it is towork independently in a scientific research environment. The positive research experience I got from doingthis summer project contributed amongst other things to strengthen my determination to work as a scientistin the future.The studentship has also been of great value to me technically. I learned how to culture cells and develop

Student: Nikolay Martyushenko, [email protected]: Jonathon Pines
new cell lines. I also learned how to do FRAP, how to prepare cells for IF microscopy, and I greatlyimproved my skills of western blotting. As for the lab, I developed a FRAP workflow from elucidatingoptimal microscope settings to developing a macro that speeded up the processing of data by an order ofmagnitude compared to conventional manual processing. Now other people in the lab also have begundoing FRAP using this workflow.
References1. Howell B.J., Moree B., Farrar E.M., Stewart S., Fang G., SalmonE.D. Spindle Checkpoint Protein
Dynamics at Kinetochores in Living Cells. Curr. Biol. 14, 953–64, (2004).2. Vink M, Simonetta M, Transidico P, Ferrari K.,Mapelli M, De Antoni A., Massimiliano L.,
Ciliberto A., Faretta M., Salmon E. D., Musacchio A. In Vitro FRAP Identifies the MinimalRequirements for Mad2 Kinetochore Dynamics. Curr. Biol. 16, 755-66, (2006)
3. Westhorpe F. G. , Tighe A., Lara-Gonzalez P., Taylor S. S. p31comet-mediated extraction of Mad2from the MCC promotes efficient mitotic exit. J Cell Sci. 124(22): 3905–16, (2011)

Student: Oliver HydeSupervisor: Mark Shepherd
Biochemical Society Summer Vacation Studentship Report
Elucidating the molecular switch for nitric oxide sensing inCampylobacter jejuni
Introduction:Campylobacter jejuni is a bacterial pathogen found in the gut, where it is exposed to nitrate, nitricoxide (NO) and reactive nitrogen species during colonisation. Nitric oxide can penetrate the cell wallsof bacterial cells and cause internal damage to proteins. Nitric oxide nitrosylates proteins that haveimportant functions such as respiratory proteins, therefore inhibiting cellular growth and causing celldeath. There are several mechanisms by which the bacterial cell is exposed to nitric oxide: it isproduced in the gut by commensal microorganisms and can also be produced by cells such asmacrophages that utilise a nitric oxide synthase mechanism to defend the host from invadingbacteria. Defence against nitric oxide is crucial to bacterial survival during infection. To achieve this,C. jejuni expresses a single-domain globin (Cgb) and a truncated globin (Ctb) which detoxify NO. Theexpression of these globins is controlled by the Nitrosative stress sensing Regulator (NssR) protein.The exact mechanism by which NssR sense NO is unknown, so the focus of this study was toinvestigate potential mechanisms via which NssR protein may interact with NO.
Aims:Expression and purification of NssRInvestigate the association of NssR with haem and potential ligandsMeasure the impact of intermediates of nitrogen metabolism upon NssR activity in vivo.Measure direct binding of apo-NssR and cofactor-bound derivatives to the cgb promoter
Description of work carried out:
1) Purification of an NssR:haem complexNssR was purified by affinity chromatography as previously described (Smith, H. K., et al. 2011 NitricOxide 25, 234-41), and purity was confirmed using SDS-PAGE. Purified NssR was incubated withhaemin at a ratio of 1:2, with the intention of saturating NssR with a haem cofactor. The sample waspassed through a DEAE column, which was predicted to retain the free haemin while allowing theprotein-cofactor complex to pass through and be collected. The experiment was a success withNssR:Haem complex being produced, and confirmed by absorption spectroscopy. However theconcentration of NssR:Haem was low (3.16µM), and the binding of this complex to the cgb promoterusing gel shifts could not be demonstrated.
2) Gel-shift analysis of NssR:haem binding to the cgb promoterThis approach involved saturation of NssR with haemin just prior to performing gel shift analysis ofcgb promoter binding. In order to do this, we required a method to verify that haem was indeedbinding to the NssR complex. This was achieved by exploiting the ‘CO difference spectrum’ of theNssR:haem complex to monitor cofactor binding. The NssR was incubated with Haemin at variousratios for 1 hour, and then several spectra were then recorded including raw, reduced (with sodiumdithionite), and CO-reduced (reduced and pumped with carbon monoxide for 5 minutes). CO issimilar in structure to NO, so it was predicted to bind to the NssR:haem complex. The CO differencespectra showed that the interaction of haemin with NssR occurred with high affinity, with clearresults at an NssR:Haemin ratio of 0.5:1. When this ratio was increased, the shoulder peak at 407nmrepresenting free haemin diminished further in favour of the 424nm peak, representing furtherformation a an NssR:haem complex.

Student: Oliver HydeSupervisor: Mark Shepherd
Once the saturating concentration of haemin had been elucidated, several gel shift assays wereperformed to determine the effect of the Haem cofactor and the presence of NO had on the bindingof NssR to the promoter region of the cgb gene. Each gel shift reaction contained 10nM of cgbpromoter DNA. Those with haemin present were incubated with 3 M haemin for 30 minutes, andthose with NO added had a NOC-12 (an NO donor) concentration of 100µM. Various concentrationsof the protein were added to each gel, all used a 1kb Promega DNA ladder as a marker.Successful binding to the promoter region was represented by the DNA band disappearing from thebottom of the gel and appearing closer to the top. This represents the protein binding to the DNAregion causing the DNA to move slower through the gel due to the additional size. The gel was “Non-Denaturing” to maintain the functionality of the protein, and the ethidium bromide staining wasused to visualise the position of the cgb promoter DNA. The data demonstrated that an NssR:haemcomplex binds more readily to the cgb promoter than NssR alone, although the presence of NO hasno influence upon promoter:NssR interactions.
Assessment of results and outcomes:In conclusion, the results demonstrate that the NssR protein readily binds the haem cofactor.Furthermore, the binding of the protein:cofactor complex is rapid and saturation occurs at low ratiosof protein:cofactor. The gel shift experiments indicate that addition of haem to NssR increases thebinding affinity for the cgb promoter. However, the addition of an NO-donating compound (such asNOC-12) does not increase the binding to DNA with haem present. NssR positively regulates cgbexpression in response to NO, and our results suggest that this occurs via an alternative mechanismthan direct binding of NO to the haem of the NssR:Haem complex.
Future directions for the project:Future work for this project may include investigating the influence of derivatives of NO (e.g. nitrite,peroxynitrite) upon the promoter binding capabilities of the NssR:haem complex.
Departures from the original proposal:There was a small deviation from the original project proposal, in which we intended to investigateshifting with a number of nitrogen pathway intermediates. However due to verification of haem asan NssR cofactor, NO binding became the primary focus.
Influence upon career aspirations:This summer studentship has greatly reinforced my desire to pursue a PhD placement as I have beenable to experience firsthand the joys of the academic career pathway. Although I am still undecidedas to whether I will continue down the academic route after PhD, it has been a valuable experiencethat sparked my interests and passions. Finally I wish to thank the society for allowing me to havethis amazing opportunity and to everyone in the Shepherd Lab for their assistance and kindness, alsoto Mark Shepherd himself for his diligent teachings and encouraging persona.
Value of the studentship:This project has provided robust evidence that NssR binds a haem cofactor, and has developed aspectroscopic tool to monitor this process. This will contribute to the elucidation of the molecularmechanism of NssR-mediated transcriptional regulation, and has advanced our knowledge of NO-sensing in Campylobacter.
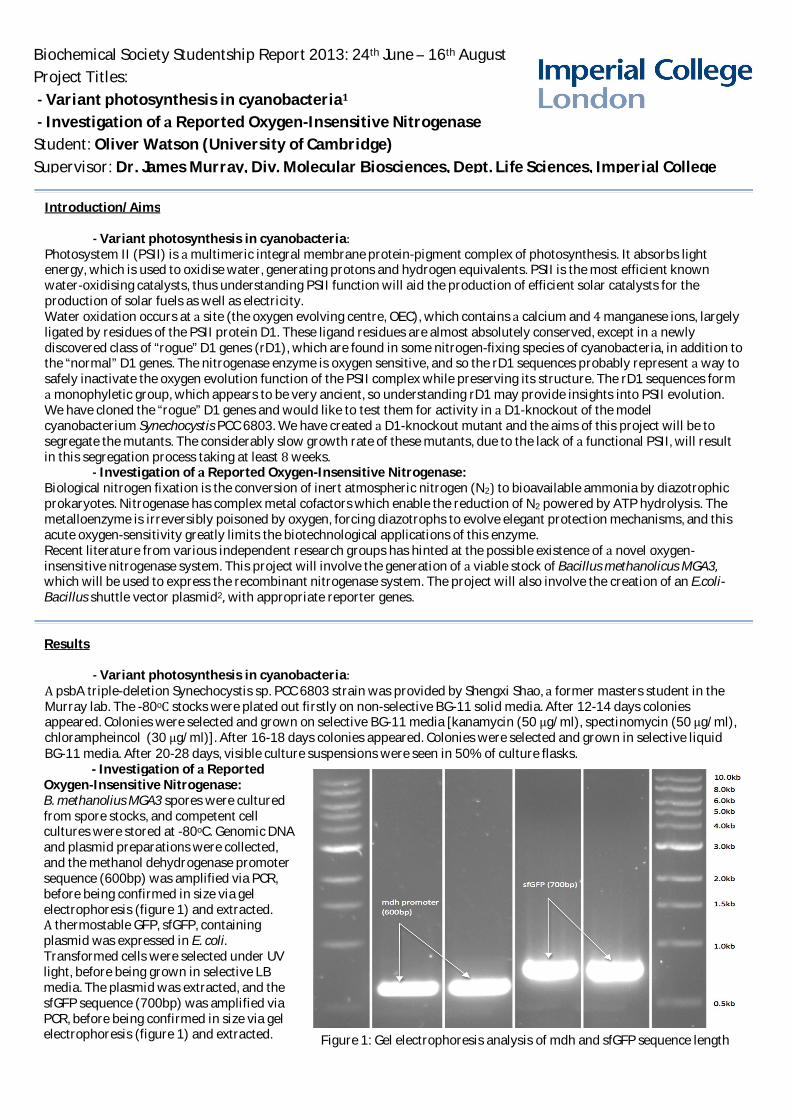
Biochemical Society Studentship Report 2013: 24th June – 16th AugustProject Titles:
Variant photosynthesis in cyanobacteriaInvestigation of Reported Oxygen-Insensitive Nitrogenase
Student: Oliver Watson (University of Cambridge)Supervisor: Dr. James Murray, Div. Molecular Biosciences, Dept. Life Sciences, Imperial College
Introduction/Aims
Variant photosynthesis in cyanobacteriaPhotosystem II (PSII) is multimeric integral membrane protein-pigment complex of photosynthesis. It absorbs lightenergy, which is used to oxidise water, generating protons and hydrogen equivalents. PSII is the most efficient knownwater-oxidising catalysts, thus understanding PSII function will aid the production of efficient solar catalysts for theproduction of solar fuels as well as electricity.Water oxidation occurs at site (the oxygen evolving centre, OEC), which contains calcium and manganese ions, largelyligated by residues of the PSII protein D1. These ligand residues are almost absolutely conserved, except in newlydiscovered class of “rogue” D1 genes (rD1), which are found in some nitrogen-fixing species of cyanobacteria, in addition tothe “normal” D1 genes. The nitrogenase enzyme is oxygen sensitive, and so the rD1 sequences probably represent way tosafely inactivate the oxygen evolution function of the PSII complex while preserving its structure. The rD1 sequences form
monophyletic group, which appears to be very ancient, so understanding rD1 may provide insights into PSII evolution.We have cloned the “rogue” D1 genes and would like to test them for activity in D1-knockout of the modelcyanobacterium Synechocystis PCC 6803. We have created D1-knockout mutant and the aims of this project will be tosegregate the mutants. The considerably slow growth rate of these mutants, due to the lack of functional PSII, will resultin this segregation process taking at least weeks.
Investigation of Reported Oxygen-Insensitive Nitrogenase:Biological nitrogen fixation is the conversion of inert atmospheric nitrogen (N2 to bioavailable ammonia by diazotrophicprokaryotes. Nitrogenase has complex metal cofactors which enable the reduction of N2 powered by ATP hydrolysis. Themetalloenzyme is irreversibly poisoned by oxygen, forcing diazotrophs to evolve elegant protection mechanisms, and thisacute oxygen-sensitivity greatly limits the biotechnological applications of this enzyme.Recent literature from various independent research groups has hinted at the possible existence of novel oxygen-insensitive nitrogenase system. This project will involve the generation of viable stock of Bacillus methanolicus MGA3which will be used to express the recombinant nitrogenase system. The project will also involve the creation of an E.coli-Bacillus shuttle vector plasmid2 with appropriate reporter genes.
Results
Variant photosynthesis in cyanobacteriapsbA triple-deletion Synechocystis sp. PCC 6803 strain was provided by Shengxi Shao, former masters student in the
Murray lab. The -80o stocks were plated out firstly on non-selective BG-11 solid media. After 12-14 days coloniesappeared. Colonies were selected and grown on selective BG-11 media [kanamycin (50 g/ml), spectinomycin (50 g/ml),chlorampheincol (30 g/ml)]. After 16-18 days colonies appeared. Colonies were selected and grown in selective liquidBG-11 media. After 20-28 days, visible culture suspensions were seen in 50% of culture flasks.
- Investigation of ReportedOxygen-Insensitive Nitrogenase:B. methanolius MGA3 spores were culturedfrom spore stocks, and competent cellcultures were stored at -80oC. Genomic DNAand plasmid preparations were collected,and the methanol dehydrogenase promotersequence (600bp) was amplified via PCR,before being confirmed in size via gelelectrophoresis (figure 1) and extracted.
thermostable GFP, sfGFP, containingplasmid was expressed in E. coliTransformed cells were selected under UVlight, before being grown in selective LBmedia. The plasmid was extracted, and thesfGFP sequence (700bp) was amplified viaPCR, before being confirmed in size via gelelectrophoresis (figure 1) and extracted. Figure 1: Gel electrophoresis analysis of mdh and sfGFP sequence length

Figure 2: pNW33N E.coli-Bacillus shuttleplasmid with incorporated mdh promoter andsfGFP sequence
The isolated sfGFP and mdh sequences were assembled via Gibson3 assembly. The Gibson product was amplified by PCRbefore being restriction ligated into the E.coli-Bacillus shuttle vector plasmid, pNW33N. The resultant plasmid (figure 2)was amplified in E.coli competent cells. The created plasmid was initially analysed for correct incorporation of the Gibsonproduct by restriction digest, which yielded promising results (not shown here). The created plasmid was then sent forsequencing, before it is to be used in the future for the expression of the recombinant nitrogenase system.
Future Work
Variant photosynthesis in cyanobacteriaThe triple deletion psbA mutant will continue to be grown to produce sufficiently large culture size ready fortransformation with the isolated “rD1” gene. The “normal” D1 genes will be used as positive control for activity in thenew PSII background. We would also wish to show that the rD1 genes are expressed in the native hosts, and will useWestern blots with specific rD1 antibody to show this. In this way the functional role of the “rD1” protein can be greaterelucidated.
Investigation of Reported Oxygen-Insensitive Nitrogenase:The created plasmid will have to first be checked for sequence accuracy from the results of the purchased sequence.The associated genes for the novel nitrogenase system need to then be sequenced and incorporated into the createdplasmid before being used to transform the created competent B. methanolicus MGA3 cells.
Value of the Studentship to the Student
Most important to mention is how my preconceptions about what life in research would be like were hugely incorrect.University does not prepare you very well for career in research. This is not the fault of the university, but simply that in
three year degree there is simply not enough time. Having the opportunity to spend weeks working on these projectsgave me an appreciation of many aspects of research that had been lost in the short practical classes at University; theimportance for methodical, well documented and careful work. It is this time consuming, repetitive style of work thatoriginally thought would not fascinate me, but fortunately was proven wrong. think it was the ability to follow myhunches when trying to understand why certain tests weren’t working, why certain gels were blank, and so on, that madethe project more enjoyable than initially thought it would be. It was this opportunity to have more freedom, rather thanfollowing laid out protocols at university, which made these weeks more enjoyable. This enjoyment was also greatlyenhanced by having an incredibly supportive and friendly supervisor in Dr. James Murray, who always made sure thathad plenty to work on by giving me multiple projects to work on. He was excellent at helping to clear up any confusionhad, and helped me explore an area of biochemistry that have never studied (as don’t in fact study biochemistry). amgreatly indebted to him, and the other members of his lab group.
References
[1] J. W. Murray, Photosynthesis Research (2012) 110:177-184. PMID: 22187288; “Sequence variation at the oxygen-evolving centre of photosystem II: new class of 'rogue' cyanobacterial D1 proteins.”
[2] D. Nilasari et al., American Institute of Chemical Engineers Biotechnology (2012) Prog., 28: 662–668, 2012; “Expressionof Recombinant Green Fluorescent Protein in Bacillus methanolicus.
[3] D. Gibson et al., Nature Methods (2009), Vol.6 No.5; “Enzymatic assembly of DNA molecules up to several hundredkilobases.”

Structure, mechanism and application of Oxidised Polyvinyl Alcohol Hydrolase (OPVAH)
Student Rebecca Hodgson, Biochemist at University of BirminghamSupervisor Gideon Grogan, York Structural Biology Laboratory, Department of Chemistry, University of
York
BackgroundEnzymes involved in the formation and cleavage of carbon-carbon bonds have had their fair share ofscientific research over the years. This is due to the significance of creating and destroying carbon-carbonbonds as they are present in so many biochemical structures; for example they are found in every aminoacid and therefore in the backbone of every single protein. Bidirectional enzymes which form and cleaveC-C bonds mainly rely on cofactor such as biotin and thDPi These enzymes have been studied with theintention to be used as catalysts when forming C-C bonds in synthetic chemistry and in essence removethe harsh conditions of purely chemical methodsii Less well researched enzymes are analogous toheteroatom hydrolyses, such as lipases and proteases. These enzymes seem to be dependent on serineresidue in order to cleave the C-C bonds. The serine creates charge relay system by way of catalytictriad in order to cleave the bondiii Due to their activity being primarily concerned with the cleavage andnot the formation of the bonds little research on them exists. However, one such enzyme, oxidisedpolyvinyl alcohol hydrolase (OPVAH) from Pseudomonas strain VM15C, has been proposed to beadvantageous for the breakdown of PVA used in textile and paper industriesiv (scheme 1).
Aims:As can be seen in the schematic, the enzyme OPVAH is -diketone hydrolase which cleaves the C-C bondto produce carboxylic acid, such as butyric acid 2 and methyl ketone, such as pentan-2-one 3However, there is very little known about the mechanism of action for this enzyme. The aim of thisproject is to investigate the kinetics of the enzyme and its structure through the use of UV-visspectrophotometry and GC along with crystallography. The overall goal for this project is to try to reversethe direction of the enzyme, making it create C-C bonds instead of destroying them.
Description of Work and resultsTo begin with the main bulk of the work was purifying the enzyme from cells of E.coli so that furtherexperiments could take place. was given gene of Gst-1 fusion of OPVAH which had only just beencreated as stable, reproducible, soluble form of the enzyme. The first step was to transform the cells ofE.coli with the gene and allow the colonies to grow. We could then pick suitable colony and inoculateovernight cultures containing the necessary substances to allow for growth. The following day LB wasinoculated with the overnight cultures and left to allow the cells to grow to sufficient density. Oncereached, induction took place using IPTG to force the E.coli into transcribing our desired protein. Thefollowing steps involved sonication and centrifugation in order tobreak open the cells and release our protein. The following weekwould involve purifying the protein using the FPLC and variouscolumns, including Ni column and gel filtration column. Once wehad finally made our pure protein we then were able to carry outnumber of different experiments.With sample of the pure protein we ran an SDS-Page gel todetermine its purity. As can be seen from the gel in Fig. the pureprotein in lane contains fewer impurities than the precleavedprotein in lane 2. However, upon closer observation it can be notedthat the band for the protein is around 60kDa in both the cleaved andprecleaved lanes. If cleavage was taking place the final pure protein
Fig. – SDS-Page gelshowing themarker in lane1, thePrecleavedprotein (PC) inlane and thecleaved ‘pure’(C) protein inlane

should only have weight of about 30kDa as the Gst fusion would be removed. This led us to believe thatthe cleavage step was not working correctly and raised the question of whether it needed to be done at allif it was just going to mean losing more protein along the way.
We set up crystal screens with some of the protein in order to see if crystal growth occurred. Once weobserved crystals growing in some of the wells we were able to use more protein in order to optimise thescreens and grow larger crystals in the hope of growing some that could defract. Some of the crystalswhich grew can be seen in fig.2. The discovery of the cleavage step failing meant that were started towonder what was responsible for the crystal formation, whether it was the protein or simply the Gstwhich was forming crystals. Unfortunately when we came to shoot the crystals no defraction patterncould be seen so we were unable to decipher the origin of the crystals.
We tried to use some of the protein for UV-Vis Spectrophotometry assays but investigation of theabsorbance of the substrate, nonane,4-6,dione, revealed it to be around the 270-280nm region. Everybiochemist knows this isn’t the region you want your substrate to absorb at! We then used some of theprotein for GC assay to try to determine the kinetic this way. This involved running four experimentsside by side for 24 hours taking samples at 0, 0.5, 1, 2, 4, 6, 8, 24 hours. The reaction vessels contained:
1. Enzyme (OPVAH) and substrate (nonane,4-6,dione)2. No enzyme, just substrate3. Biphasic Enzyme and substrate4. Biphasic No enzyme, just substrate
This was very successful in that we found product peak in some of the assays. However the peak wassmall and conclusions could not easily be drawn from it. When the results of the biphasic reaction werealso studied closely it could be seen that the area under the peak was decreasing, but no product peakwas formed. This led us to believe that the product could be being formed in the other constituent of thebiphasic reaction; luckily when this fraction was put through the GC an obvious (but still small!) productpeak could be seen.
Departure from original proposal:During my placement we came across some pitfalls in the original plan mainly due to the crystals notdefracting. This meant we were unable to determine structure for the enzyme. We also had difficultywith the enzyme kinetics as the substrate absorbed in the same region as the protein. Essentially thesedrawbacks meant we weren’t able to get very far with trying to reverse the action of the enzyme.
Future work:In terms of getting somewhere with determining the kinetics of the enzyme we were fairly successfulwith the GC as we were able to see product peak forming. This was close to the end of the weekproject however and was only an initial trial run so the results were difficult to draw conclusions from.Hopefully if this project were to be continued this experiment could be tweaked and refined in order toproduce valuable data. It may also be valuable to do further work on the purification of the enzyme to seewhether the cleavage step is really making valuable difference to the whole procedure.
Value of Studentship:The weeks spent in the lab at YSBL allowed me to improve many of my basic lab techniques. now feelcompetent at recombinant gene expression, protein purification using FPLC and making and running SDS-PAGE gels. also was able to use many of the larger machines such as the UV-vis Spectrophotometer, theGC, machines for crystallisation (such as the Mosquito) as well as observing data collection on thecrystals.
Fig. – This figure shows examples ofthe crystals which formed of OPVAH. Theone on the left shows the crystals in theinitial screen, these are much too small forshooting. However the ones on the rightshow crystals which formed in theoptimisation trays, these are much largerand plate like and were used for trying todetermine the structure.

The lab at York is friendly and dynamic working environment and one in which felt was able to carryout my lab project confidently knowing that if needed advice or supervision someone was willing to helpme.From an employability aspect the lab techniques which have developed will help stand me in good steadwhen looking for job in the future however think more importantly the attitude you develop towardsscience whilst being in the lab is the most valuable as you learn to question why things are happening.feel like am now much more confident at asking why something does what it is doing and finding ananswer to it than felt like was at the beginning of the placement. feel that everything that did at Yorkwas valuable learning experience from which have gained great deal.
would like to acknowledge the help and support given by all the members of the Grogan Group at the YSBL.
i Resch et al. (2011) Curr. Opin. Biotechnol 22 793-799iiWolf-Dieter. (1998) Curr. Opin. In Chem Bio 2 85-97iiiShimao et al. (2000) Microbiology 146, 649-657iv Siirola et al. (2013) Adv. Synth. Catal. 355, 1677-1691

1
Optimising the expression in E. coli of the PGRL1 protein involved in photosynthetic cyclic electronflow
Student: Rebecca Tregent
Supervisor: Prof Peter J Nixon (Imperial College London)
Aim:To establish an expression system for the production of the PGRL-1 protein in E. coli for structuralstudies
Introduction:Cyclic electron flow around the photosystem I complex in plants, cyanobacteria and algae has longbeen recognised as an important route for the conversion of solar energy to ATP (reviewed byArnon, 1971). However the proteins involved in this process and the mechanism of electrontransport still remain elusive. Very recently, the membrane bound PGRL-1 protein was identified byHertle and colleagues (Hertle et al. 2013) as the elusive antimycin-sensitiveferredoxin:plastoquinone reductase (or FQR protein) originally described by Tagawa et al. in 1963.PGRL1 is thought to bind Fe but its structure is currently not known. Three PGRL1 sequences werechosen for expression in E. coli: PGRL-1A and PGRL-1B from the model plant Arabidopsis thalianaand a PGRL1 homologue from the thermophilic red alga Cyanidioschyzon merolae.
Work carried out:
Codon-optimised synthetic DNA sequences encoding the precursor forms of each gene product werecloned into the expression vector pRSET-A (Invitrogen) with the following features suitable forexpression of foreign proteins in E. coli: a strong T7 RNA promoter and upstream sequencesencoding a cleavable N-terminal His6-tag for immobilised metal affinity purification, an Xpressepitope tag suitable for immunodetection and an enterokinase cleavage site for liberating the targetprotein. The three different expression vectors were transformed into two different types of E. colicells capable of inducible expression of the T7 RNA polymerase: KRX and LEMO cells. Total proteinfrom induced and uninduced E. coli extracts was separated by SDS-PAGE. No significant expressionof PGRL1 was detected, either by Coomassie Blue staining or by immunoblotting using an Anti-Xpress-HRP antibody (Invitrogen). Altering the concentration of rhamnose used to induce expressthe T7 RNA polymerase (0.2 and 1 mM) in the KRX cells did not improve expression of PGRL1.
In the second stage of the project, sequences encoding the predicted mature forms of PGRL1 wereamplified by PCR and cloned into the pRSET-A expression vector using the ligation independent In-Fusion technique. Positive clones were confirmed by restriction digestion and DNA sequencesconfirmed by sequencing.
Future directions
The groundwork has now been laid to assess the expression of the mature forms of each of thePGRL1 proteins in E. coli. Ultimately, the purified proteins will be subjected to crystallisation trials inorder to produce crystals of sufficient quality to generate a crystal structure. Such a structure willprovide important insights into the function of this very important plant protein.

2
Departures from the original proposal:
Originally the plan was to optimise the expression of the Ycf39 PSII assembly factor in E. coli.However, this work had already been done by the Nixon group at the outset of the project so it wasdecided to shift the focus on to PGRL1.
Importance of the studentship
This internship gave me a once in a lifetime opportunity to work with leading individuals in the fieldof photosynthesis and biochemistry. Throughout my time at imperial, I learnt a variety of skills andtechniques that cannot be gained through my current degree course. I now have the ability to use allof the things that I learnt to not only benefit my final year project, but to also vastly benefit mycareer, giving me a head start in an industry that is difficult to get into as a young graduate. I feelthat working with Professor Nixon and his team has given me a greater knowledge andunderstanding of the level of high standard needed in a practicallaboratory, also equipping me with the confidence to work on my ownwhen needed. I am very thankful for the opportunities that ProfessorNixon has given me and for the skills that I have learnt whilst workingwith him.
References
Arnon, D. I. (1971) Proc. Natl Acad. Sci. USA 68:2883-2892
Hertle, A. P., Blunder, T., Wunder, T., Pesaresi, P., Pribil, M., Armbruster, U. and Leister, D. (2013)Mol Cell 49: 511-523
Tagawa, K., Tsujimoto, H. Y. and Arnon, D. I. (1963) Proc. Natl Acad. Sci. USA 49:567-572

Biochemical Society Summer Vacation Studentship Report 2013
Sadhbh Tennant: University College Dublin. Supervisor: Dr D Higgins
1
Elucidating the molecular mechanism underlying BMP-7 protection against renal injury
Background: Bone morphogenetic proteins, BMPs, are glycosylated ECM-associated molecules
belonging to TGF- super family and are crucial for normal developmental processes such as bone
growth. Loss of BMP-7 signalling is associated with the pathophysiology of renal disease. Numerous
studies had revealed that exogenous BMP-7 administration protects against development of renal
fibrosis in vivo however the precise molecular mechanisms underlying this protective role were not
clearly defined.
The research team I worked with had recently completed an in vivo renal injury model, UUO-induced
fibrosis, with and without BMP-7 treatment and found a significant reduction in fibrotic marker
expression in kidneys treated with BMP-7. Interestingly, they found specific effects of BMP-7 on
certain aspects of TGF- mediated signalling pathways. My project involved investigating effects of
BMP-7 on TGF- signalling in two in vitro models of renal injury. The aims of the project were to 1)
Examine effects of BMP-7 on TGF- -induced signalling in human renal tubular epithelial HK-2 cells.
2) Examine effects of BMP-7 on hypoxia-induced signalling in HK-2 cells and (3) to determine if
BMP-7 can inhibit TGF- - or hypoxia- induced pro-fibrotic gene expression by qPCR in HK-2 cells.
The significance of this study is that it may offer incredible insight into the mechanisms underlying
the renoprotection offered through activation of BMP-7 signalling.
Departures from Original Proposal: The original proposal included analysis of BMP-7 effects on
TGF- mediated signalling in tubular epithelial HK-2 cells and collecting duct mIMCD cells,
however the mIMCD work had been completed by the time the studentship started.
Work carried out: Tubular epithelial HK-2 cell responses to either TGF- (5ng/ml) or hypoxia (1%
O2) stimulation were initially monitored over time (0-120 minutes) by analysing activation of a
number of signalling pathways including PI3 kinase (Akt and GSK3 phosphorylation), MAP kinase
(ERK and p38 phosphorylation) and SMAD 2 phosphorylation using western blot analysis. Overall,
the various pathways were activated at different times ranging between 0 to 30 minutes, however the
majority remained activated at 60 minutes, and thus this time was chosen for subsequent experiments
with BMP-7.
To investigate the effect of BMP-7 on each signalling pathway above, HK-2 cells were pre-treated
with BMP-7 (0-100ng/ml) for 30 minutes prior to stimulation with TGF or hypoxia as described
above for 60 minutes. Phosphorylated and total isoforms of Akt, GSK3 , ERK, p38 and SMAD2
were analysed by western blot. GAPDH was analysed and used as a loading control for densitometric
analysis which was performed using NIH Image J software, freely available for download from the
NIH website (http://rsbweb.nih.gov/ij/).
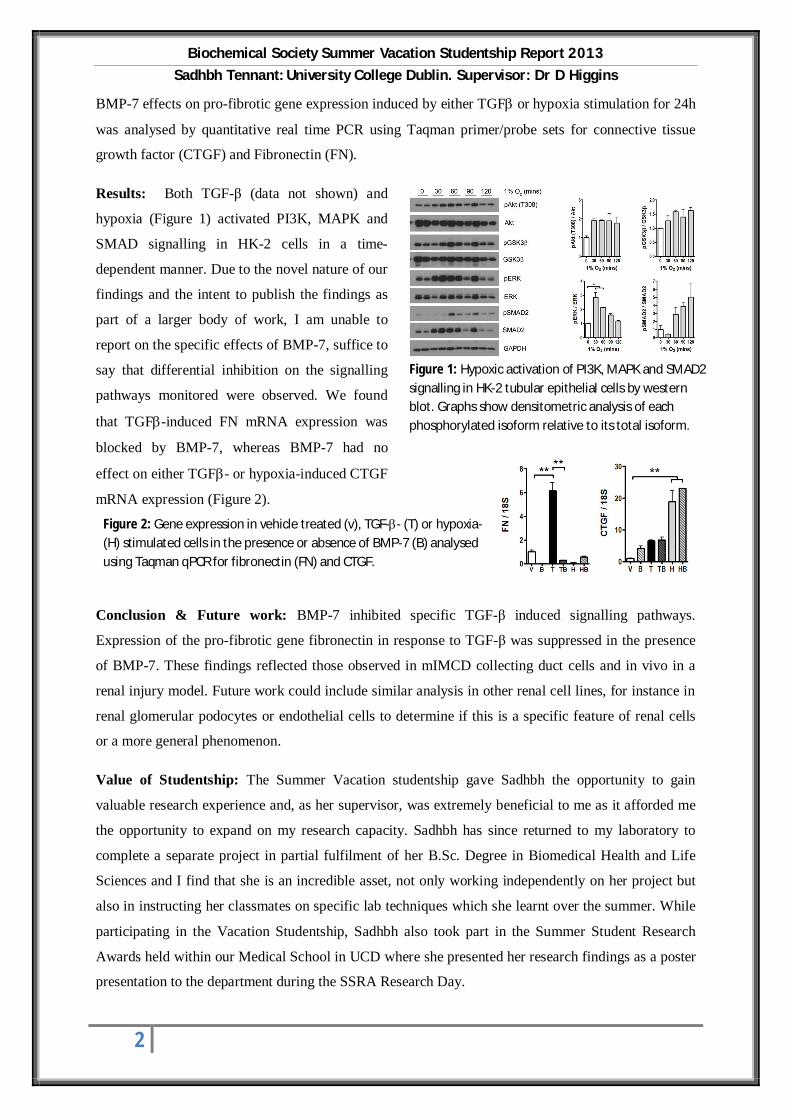
Biochemical Society Summer Vacation Studentship Report 2013
Sadhbh Tennant: University College Dublin. Supervisor: Dr D Higgins
2
BMP-7 effects on pro-fibrotic gene expression induced by either TGF or hypoxia stimulation for 24h
was analysed by quantitative real time PCR using Taqman primer/probe sets for connective tissue
growth factor (CTGF) and Fibronectin (FN).
Results: Both TGF- (data not shown) and
hypoxia (Figure 1) activated PI3K, MAPK and
SMAD signalling in HK-2 cells in a time-
dependent manner. Due to the novel nature of our
findings and the intent to publish the findings as
part of a larger body of work, I am unable to
report on the specific effects of BMP-7, suffice to
say that differential inhibition on the signalling
pathways monitored were observed. We found
that TGF -induced FN mRNA expression was
blocked by BMP-7, whereas BMP-7 had no
effect on either TGF - or hypoxia-induced CTGF
mRNA expression (Figure 2).
Conclusion & Future work: BMP-7 inhibited specific TGF- induced signalling pathways.
Expression of the pro-fibrotic gene fibronectin in response to TGF- was suppressed in the presence
of BMP-7. These findings reflected those observed in mIMCD collecting duct cells and in vivo in a
renal injury model. Future work could include similar analysis in other renal cell lines, for instance in
renal glomerular podocytes or endothelial cells to determine if this is a specific feature of renal cells
or a more general phenomenon.
Value of Studentship: The Summer Vacation studentship gave Sadhbh the opportunity to gain
valuable research experience and, as her supervisor, was extremely beneficial to me as it afforded me
the opportunity to expand on my research capacity. Sadhbh has since returned to my laboratory to
complete a separate project in partial fulfilment of her B.Sc. Degree in Biomedical Health and Life
Sciences and I find that she is an incredible asset, not only working independently on her project but
also in instructing her classmates on specific lab techniques which she learnt over the summer. While
participating in the Vacation Studentship, Sadhbh also took part in the Summer Student Research
Awards held within our Medical School in UCD where she presented her research findings as a poster
presentation to the department during the SSRA Research Day.
Figure 1: Hypoxic activation of PI3K, MAPK and SMAD2signalling in HK-2 tubular epithelial cells by westernblot. Graphs show densitometric analysis of eachphosphorylated isoform relative to its total isoform.
Figure 2: Gene expression in vehicle treated (v), TGF- - (T) or hypoxia-(H) stimulated cells in the presence or absence of BMP-7 (B) analysedusing Taqman qPCR for fibronectin (FN) and CTGF.

Biochem Soc Summer Vacation Studentship Report 2013
Student: Ms. Sandy Hartmann
Principal Supervisor: Dr. Tim Palmer (Institute of Cardiovascular and Medical Sciences,University of Glasgow).
Day-to-Day Supervisor: Ms. Claire Speirs
Project: Modulation of the JAK-STAT pathway by AMP-activated protein kinase (AMPK)
Background AMP-activated protein kinase (AMPK) is a serine/threonine protein kinase proposed tobe a therapeutic target for the treatment of Type 2 diabetes due to its role in regulating energybalance. AMPK-activating drugs such as metformin, which is widely prescribed to people with Type2 diabetes, are known to have beneficial anti-inflammatory effects but the mechanisms responsibleare still unclear. The basis for my project in Dr Palmer’s lab was trying to find out how AMPK caninhibit signalling through the JAK-STAT pathway, which is an important driver of inflammation indiseases such as atherosclerosis and rheumatoid arthritis.
Departures from the original proposal One of the JAK family of proteins downstream of the type Iinterferon receptor complex had emerged as a potential mediator of AMPK’s effects so thestudentship focused on that aspect.
Aims The aim of my project was to characterise the cytokine responses and test the potential useof fibrosarcoma cells with specific alterations in JAK1 expression as a model system to examineAMPK-mediated inhibition of pro-inflammatory signalling.
Methods
Cell culture Human umbilical vein endothelial cells were grown in a humidified 5% (v/v) CO2
atmosphere in endothelial growth medium-2 (EGM-2) supplemented with foetal bovine serum,hydrocortisone, ascorbate and recombinant growth factors as recommended by the supplier(Lonza). Human fibrosarcoma cell lines 2C4 (wild type), U4C (JAK1-null) and U4C.JAK1 (rescuedby stable expression of mouse JAK1) were kindly provided by Dr. Ana Costa-Pereira (ImperialCollege, London) and were grown in DMEM supplemented with 10% (v/v) foetal bovine serum and1 mM L-glutamine and appropriate selection antibiotics.
Preparation of soluble cell lysates Confluent cells in six-well plates were treated as described inthe figures prior to washing in ice-cold phosphate-buffered saline and solubilisation by scrapinginto 0.2 ml/well detergent lysis buffer containing protease and phosphatase inhibitors. Insolublematerial was removed by microcentrifugation and the supernatant assayed for protein contentusing a bicinchonic acid assay.
SDS-PAGE and immunoblotting Samples equalised for protein content were run on SDS-PAGEon 8% (w/v) acrylamide resolving gels. Following transfer to nitrocellulose, membranes wereblocked for one hour at room temperature in blocking buffer (5% (w/v) skimmed milk in Tris-HCl(pH 7.5) containing 0.1% (v/v) Tween-20) and incubated overnight at 4oC with primary antibodydiluted in fresh blocking buffer. Following three washes, membranes were incubated for one hourat room temperature with appropriate horseradish peroxidase-conjugated secondary antibody at a1 in 1000 dilution. After further washes, immunoreactive proteins were visualised and quantitated.
Results
Initial experiments looking at the inhibitory effects of AMPK activation on IL-6 signalling werecarried out in HUVEC cells: this was done to familiarise myself with the techniques I would beusing (cell culture, drug treatments, preparation of cell lysates, immunoblotting) and to make sure Icould reproduce findings already established in the lab. Once this had been achieved, I thendetermined several aspects of the fibrosarcoma cell lines that were to be the main model for mywork. Initially, I characterised the expression of JAK isoforms in these cells. I was able to confirmthat while JAK1 was detectable in 2C4 and expressed at a high level in U4C.JAK1 cells, it wasundetectable in absent in U4C cells (Fig. 1). Importantly, I was also able to show that the loss of

JAK1 expression in 2C4 cells did not alter the levels of related protein JAK2 but did result in a two-fold increase in the expression of Tyk2 (Fig. 2).
Fig. 1: JAK1 expression in 2C4, U4C and U4C.JAK1 cells
Fig. 2: Expression of JAK1, JAK2 and Tyk2 in 2C4 and U4C fibrosarcoma cells
Based on these data, I then tested whether activation of AMPK by the selective activator A769662could inhibit IL-6-stimulated phosphorylation of STAT3 on Tyr705, which is a surrogate marker ofSTAT3 activation. I found that while an IL-6 trans-signalling complex (sIL-6R /IL-6) increasedSTAT3 phosphorylation, this effect was dramatically reduced (but not abolished) in JAK1-null cellsand was enhanced upon high level expression of mouse JAK1. Overexpression of JAK1 alsoresulted in detectable basal STAT3 phosphorylation. Importantly, while A769662 did not have anyeffect on its own, it was able to inhibit IL-6-trans-signalling complex stimulation of STAT3phosphorylation in each cell line. I was able to show that A769662 could activate AMPK to thesame level in each cell line by blotting with an antibody that recognises phosphorylated acetyl coAcarboxylase (phospho-ACC), a validated AMPK substrate.
Figure 3: Effect of AMPK activator A769662 on STAT3 phosphorylation in 2C4, U4C andU4C.JAK1 cells

Conclusions My experiments suggest that JAK1 is the main kinase responsible for IL-6-stimulatedphosphorylation of STAT3 in the fibrosarcoma cell line used. Along with other data from the lab, mywork has also shown that U4C cells could be a useful system to examine exactly how AMPKinhibits JAK-STAT signalling by allowing re-introduction of JAK1 proteins mutated at specific sites.
Value of the experience This 8 week project allowed me to get an insight into the actual researchcarried out in a lab on a day to day basis. I had to think and design my own experiments inadvance, which improved my planning skills, and I was fully responsible for their correct execution.I enjoyed learning many new techniques that are applicable to a future career in science and dueto the limited project period developed ways to work efficiently and accurately. Working in the labenvironment also gave me a chance to talk to many other scientists about their research. Ithoroughly enjoyed the chance to contribute to current research and would love to continue doingso.
Value of the studentship to the lab Sandy proved to be a hard working and enthusiastic student.Having had limited practical experience before starting, she proved herself very accomplished bythe time the placement finished. She also produced some invaluable data for us by characterisingthe 2C4-derived cell lines, and establishing them as a very useful cell system for taking the workforward. We anticipate that some of her data will form part of a publication and if so we will includeSandy as an author and acknowledge the Biochem Soc Summer Vacation Studentship scheme forsupport.

Biochemical Society Studentship Report
Shahana Sengupta 1
Student: Shahana Sengupta, University of YorkSupervisor: Dr John Pascall, Dr. Geoff ButcherBabraham Institute, Cambridge
Background
GIMAP6 is a member of the GTPase immunity-associated protein family. It is thought to relocatefrom the cytosol to the autophagosomal membrane during autophagy and shown to have aninteraction with a member of the Atg8 family. It has also been shown to be involved with thepathogenesis and immune response to non-small cell lung cancer (Shiao et al, 2008). GABArapl2 isanother autophagosomal protein seen to have an interaction with GIMAP6, although it is unknownthe exact specifics or function of this interaction. With autophagy being such a key process in cellsurvival, it is important to understand these intricate mechanisms and interactions.
Aims
To investigate the effect of knocking down endogenous GABArapl2 and GIMAP6 on the process ofautophagy in Jurkat cells.
Description of Work
I used a combination of SDS-PAGE, Western Blotting and Gbox imaging using GeneSys software toinvestigate the effects of different shRNA knockdowns in Jurkat cells on the process of autophagy. Ialso helped to try to establish an inducible C1498 myeloid leukaemia cell line for further studies inthis field.
Results
LC3 is a commonly used marker forautophagy that is present in the membraneof the autophagosome. It is actually thecleavage of LC3(1) to LC3(2) that is key tomonitoring the effect on the autophagicprocess. Actin is such an abundant protein inthe cell that it serves as an accurate internalcontrol and is important to ascertaining theLC3: actin ratio.As shown by Figure 1, the preliminaryknockdowns proved to be quite successful.This is particularly visible with the GABArapl2knockdown as there is a significant reductionin the amount of GABArapl2 in lanes 5-7,(these lanes contain GABArapl2 KO Clone 4lysed cells.)The GIMAP6 knockdown appears to have had some effect. However, from Figure 1, it is not possibleto ascertain for certain whether the knockdown is entirely successful – although the 10% SDS-PAGE
Figure 1SDS-PAGE gels of lysed cells from the 23rd August starvationexperiments. The figure shows the effect that GIMAP6 and GABAknockdown has on LC3 cleavage. The GIMAP6 KO seems to havegreater effect.

Biochemical Society Studentship Report
Shahana Sengupta 2
gel shows some reduction in the amount of GIMAP6 protein in lanes 8-11. The decline of LC3(1)cleavage in these lanes cannot therefore be accurately attributed to the GIMAP6 knockdown.The gels produced from the second starvation experiments were not as effective, as visible in Figure2. The gels are not as clean and the controls (lanes 1-4) do not exhibit the expected results – clearLC3 cleavage and uniform expression of both endogenous GABArapl2 and GIMAP6. Hence, anychanges to LC3 abundance or cleavage cannot be attributed to the knockdowns of GIMAP6 andGABArapl2.
Departure from Original Proposal
Due to time constraints I did not have timeto do much confocal microscopy. Therunning and probing of SDS-PAGE gels tookup the majority of time.
Future Work
Repeats of each of the knockdowns andstarvation experiments need to be carriedout to improve reliability if the dataproduced. In addition, it would be useful tofind a method of minimising the leakage ofexpression that occurs with the inducible (Doxycycline) Jurkat Cell line. At the moment, this must betaken into account when analysing the data and could reduce the accuracy of the results.
Value of the Studentship
I think that this studentship has been the perfect opportunity to explore a career in academicresearch further. It has allowed me to become more confident with experimental design and basiclaboratory techniques. Through the placement, I have also become familiar with the pace of labwork and this has really emphasized how science is not so fast-paced as is the commonmisconception.
Value to the lab
“There are definitely benefits to hosting a studentship and assisting undergraduate students ingaining research experience. Proactive students usually bring their contagious desire to learn andthey ask many questions that help us to re-familiarize with the fundamentals of experimentalprotocols. The lab encourages students to talk freely and lose their inhibitions and allow theinstructor to improve communication skills.Communication is an important part of being a scientist and hosting an undergraduate studentstrengthens the instructor’s ability to teach scientific concepts and reach out to educate non-scientists.”
BibliographyShiao YM, Chang YH, Liu YM, Li JC, Su JS, Liu KJ, Liu YF, Lin MW, Tsai SF. (2008). Dysregulation of GIMAP genesin non-small cell lung cancer. Lung Cancer. 62 (3), 287-94
Figure 2SDS-PAGE gels of lysed cells from the 30th August starvationexperiments. As shown above, this is a less successful experimentas compared with that of Figure 1.

Stephanie Ward- 2013 Summer PlacementSupervisor: Claire Friel
Is Loop 8 of the kinesin motor domain critical in microtubule endrecognition by the microtubule depolymerising kinesin 13s?
Backgrounds and aimsKinesin 13s are a family of motor proteins which are characterised by their unusualinteractions with microtubules. Unlike conventional kinesins, kinesin 13s do nottranslocate along microtubules but migrate to the end and cause depolymerisation.Kinesin 13 plays a crucial role in mitosis as it enables the separation of the sisterchromatids during anaphase and is also able to correct any inappropriate kinteophore-microtubule attachments during the segregation of the chromosomes. These kinesinsshow a similar structure to conventional kinesins, however kinesin 13s have the ability torecognise the end of the microtubule whilst regular kinesins don’t. This project focuseson a kinesin 13 called MCAK. It is currently proposed that part of MCAKs motor domainknown as loop 8, is responsible for the recognition of the microtubule end.
The aim of my project was to investigate the importance of the interaction between loop8 and the microtubules using two mutants, K426A and K418A. The mutants’ ability todepolymerise microtubules was compared to wild type MCAK using a turbidity assay. Ouraim for the end of the placement was to determine whether or not these mutationsaffected MCAKs ability to depolymerise microtubules.
Description of workProtein Purification: The MCAK was H6-tagged allowing it to be purified in one stepusing Ni-NTA beads. The purified protein was collected in fractions and the concentrationdetermined using the Bradford assay. The purity of the protein fractions was assessedusing SDS-PAGE. The protein samples were run on a 12% acrylamide gel forapproximately 90 minutes at 125V.
ATP Turnover Assay: The ability of the mutants to utilise ATP was compared to wildtype MCAK. This was achieved by measuring the amount of inorganic phosphate produceper second. The MCAK was exposed to 2 mM of ATP and samples were taken every 15minutes for 2 hours. The phosphate produced was measured using Biomol phosphategreen and compared to a phosphate standard curve.
Buffer exchange: The buffer which MCAK was purified in contained high amounts ofNaCl which would affect its ability to interact with microtubules. Therefore it wasnecessary to change the buffer that MCAK was in before using it in the turbidity assay.This was achieved using buffer exchange columns.
Turbidity Assay: The assay was carried out using a fluorimeter to measure theturbidity. 5 nM of MCAK was added to 1 uM of double cycled microtubules after 2.5minutes. The microtubules were fully depolymerised by the addition of 5 mM CaCl after12.5 minutes.
ResultsThe ATP turnover assay (figure 1) showed that the K426A mutation and the K418Amutation had no significant effect on its ability to hydrolyse ATP. The data was comparedto wild type MCAK and a two tailed t-test was used to determine that there was nosignificant difference in the ATP turnover rate of the mutants. These were the expectedresults as the mutations were not in the domain responsible for the MCAK’s ATPaseactivity. This indicated that any changes seen in the ability of MCAK to depolymerisemicrotubules was not due to the fact that its ability to hydrolyse ATP had been reduced.

Stephanie Ward- 2013 Summer PlacementSupervisor: Claire Friel
The turbidity assay (figure 2) showed some interesting results. The K426A mutant wasseen to depolymerise the microtubules at much slower rate, showing a similar trend tothe blank which contained no MCAK. Interestingly the K418A mutant depolymerisedmicrotubules at a much faster rate than the wild type MCAK. These results suggest thatthe loop 8 is important for the interactions between microtubules and MCAK.
Future DirectionsThe mutants both displayed interesting activity and will be investigated further by theFriel lab using techniques such as Stopped-flow and Fluorescence microscopy to try anddetermine exactly how the mutations in the loop 8 have affected MCAK’s ability todepolymerise microtubules.
Value of the studentshipStudent: The 8 weeks in the lab proved to be a very valuable and enjoyable experience.I gained important insight into the reality of a working lab, and it has confirmed for methat a PhD is what I want to do once I have graduated. I was fortunate to be involved inthe designing and development of an experiment which I would not have had theopportunity to experience otherwise. I was able to build on skills I had learnt in teachinglabs and apply them to the experiments I was working on, as well has increasing myrepertoire of lab techniques. I am very grateful for the Biochemical society and the FrielLab for providing me with this opportunity.
Lab: It was great having Steph in the lab over the summer. I think everyone in thegroup enjoyed having her around. She also worked hard and produced some valuabledata, which we will certainly follow up on. She showed an aptitude for lab work, whichwould stand her in good stead if she wishes to pursue lab-based research in the future.
0
50
100
150
0 50 100 150Phos
phat
eco
ncen
trat
ion
µm
Time seconds
Phosphate concentration over time
Wild Type
K426A
K418A
Figure 1: Graph showing rate of inorganicphosphate production per second for wildtype, K426A and K418A. Average rate of ATPturnover was 0.0009 s-1 +/- 0.0001 (mean ±Std n=2) 0.001 +/- 0.0002 (mean ± Std n=3)and 0.0015 +/- 0.0003 (mean ± Std n=3)respectively.
Figure 2: Graph showing depolymerisationagainst time. Addition of CaCl is indictedwith a .

Biochemical Society Studentship Report 2013
Investigating the pro-tumour effects of the mucosal-associated epithelialchemokine CCL28 on oesophageal adenocarcinoma cells
Student: Stephen TaylorSupervisor: Dr Stephen Maher
University of Hull, UK
Background
Oesophageal cancer is the UK’s ninth most common cancer, and the sixth most lethal (CRUK,2013). Males are three times more likely to develop oesophageal cancer, and cases have risen by68% since 1975 (CRUK). Prognosis is often poor due to the late presentation of the primarysymptom, dysphagia, with only one in ten patients surviving ten years post-diagnosis (CRUK).
CCL28 is a mucosal-associated epithelial chemokine expressed by columnar epithelial cells invarious mucosal tissues, including the colon, salivary glands and lungs. It facilitates the chemotacticmovement of cells that express the CCL28 receptors, CCR3 and CCR10, and also serves as a potentanti-microbial agent. It has been shown that an upregulation of CCL28 within tumours enables hostimmune evasion (Facciabene et al, 2011). Past research has shown CCL28 is upregulated inmalignant ovarian cells and that it co-localises with HIF-1 to promote migration of T-regulatory cellsinto the tumour to stimulate proliferation (Facciabene et al, 2012). It is hypothesised that cancercells exploit CCL28 and its effects to promote their own survival by indirectly stimulatingangiogenesis, proliferation, and chemoresistance.
Aims of Project
It has been established by Dr Stephen Maher that CCL28 overexpression in oesophagealadenocarcinoma is associated with resistance to chemoradiotherapy. The aims of this project are toinvestigate the effect of CCL28 on pro-tumour pathways. The effects of CCL28 on CCR3/CCR10receptor expression, proliferation, apoptosis, and regulation of survival pathways will beinvestigated.
Description of Work
Cell Culture
Two separate oesophageal adenocarcinoma cell lines to maintain: OE19 and OE33.Cells grown in RPMI-1640 medium supplemented with 10% foetal bovine serum, astreptomycin-penicillin cocktail, and GlutaMax. Cells were maintained at 37°C in a humidified5% CO2 incubator.Regular maintenance performed: media changes, cell passaging, cell straining, daily confluencychecks.
SDS-PAGE / Western Blotting

Blots were performed using 10% hand-cast polyacrylamide gels. SDS-PAGE experiments wererun at 120 V for one hour. Western blot transfers were run at 100 V for one hour.Levels of phosphorylated AKT (p-AKT) were compared in CCL28-treated cells and un-treatedcells. CCL28 was applied for fifteen minutes to the relevant cells before a protein extraction wasperformed. Beta-actin was used as a loading control.
Flow Cytometry
Cytometry was performed on a FACSCalibur cytometer and analysed using CellQuest softwareversion 6.0. Both CCL28-treated and untreated cells were scraped from T75 flasks to ensure minimalmembrane protein damage, and each aliquoted into five separate falcon tubes: cells only, FL-2 IgG1isotype antibody, CCR3 FL-1 antibody, CCR10 FL-2 antibody, and both CCR3 and CCR10 antibodies.The relevant antibody (20 µL) was applied from a stock containing 25 µG antibody in 1 mL saline, andleft in dark at 4°C for one hour. Cytometry was performed to measure the intensity of thefluorophores and subsequent receptor expression on both OE33 and OE19 cell lines. Analysis wasperformed, which compared the levels of receptor expression in both treated and untreated cells.
Clonogenics
A 5x103 OE19 cell suspension was created and seeded at varying densities into six-well plates andleft to incubate for 24 hours. Varying levels of cisplatin, from 10 M to .01 M, were applied to thewells in the presence/absence of 500 ng/mL CCL28 and incubated for seven days. Analysis ofcolonies was conducted by fixing and staining the cells with methanol and Crystal Violet beforecounting the colonies using a Gelcount instrument (Oxford Optronix).
General lab skills
MTS assays, BCA protein assays, annexin-V and propidium iodide apoptosis assays, introductionto ELISA, introduction to liposomal-based transfection.
Results
Relative to unstimulated controls of both OE19 and OE33 cells with CCL28 (500ng/mL), increased cellsurface CCR10 receptor expression was determined via flow cytometry. This effect was not observedon either cell line for the CCR3 receptor. The upregulation of the CCR10 receptor in response toCCL28 stimulation was more pronounced in OE33 compared with OE19 cells (Mean ChannelFluorescence (MCF) 3.7 versus 6.1 in OE33, compared with MCF 3.3 versus 4.7 in OE19. See figure 1.

Fig. 1 The left panel demonstrates the increase in CCR10 receptor expression in OE33 cells treated with CCL28 for 24 h (green histogram)compared with unstimulated control (purple histogram). The right panel demonstrates the increase in CCR10 receptor expression in OE19cells treated with CCL28 for 24 h (green histogram) compared with unstimulated control (purple histogram). An isotype control was usedto detect and correct for non-specific binding.
CCL28 induces AKT phosphorylation in oesophageal adenocarcinoma cells (OE33).
The signalling cascade activated following CCL28-mediated CCR10 ligation is unknown. Western blotanalysis revealed that AKT is phosphorylated in the presence of CCL28 (500 ng/mL), indicating thatCCL28 has a potential role in cell survival. Further testing, such as CCR10 neutralisation, will berequired to demonstrate that CCL28 is stimulating AKT activation via the CCR10 receptor directly.
CCL28 inhibits OE33 cell proliferation.
MTS analysis demonstrated that proliferation was reduced in response to CCL28 exposure in a dose-dependent manner, which opposed the original hypothesis that it may stimulate cell proliferation.However, the reduction in proliferation is likely a key survival strategy, allowing cells to resistchemotherapeutics, which primarily work on dividing cells. Clonogenic analysis examining sensitivityto cisplatin in the presence/absence of CCL28 was inconclusive due to time constraints.
Departures from Original Proposal
Due to time constraints, it was not possible to treat cells with deoxycolic acid, a component ofgastric refluxate that has been demonstrated to stimulate CCL28 production from OE19 and OE33cells.
Future Directions
Treatment of cells with deoxycolic acid to simulate exposure of the oesophagus to gastric bileacids; potentially measuring levels of p-AKT as well as looking at rates of proliferation.Using p-AKT inhibitors alongside treatment with CCL28 to test apoptosis in presence ofchemotherapeutics.Clonogenics performed with chemotherapeutic drugs (such as cisplatin and 5-FU, the othermajor chemotherapeutic used in the treatment of oesophageal cancer).
Personal Reflection
This studentship has been more valuable to me than I can possibly articulate here. It hasfirmly cemented that cancer research is the field I would like to develop my career in. I have gonefrom wobbly uneasiness at the start, where almost all lab techniques were entirely new to me, tofeeling confident about undertaking any experiment given to me. Whilst I have come to realise thatyour results are not always what you expect and your experiments do not always go according toplan, I have learnt so much during my time here, and for that I am grateful. My supervisor, DrStephen Maher, has ensured that I not only understand how to conduct experiments, but that I alsocomprehend them. He has encouraged me to think of how and why we perform certain techniques,and to think of future directions for each experiment. He has also taken some time to ensure that Iam up to speed with calculations – something I was particularly worried about – and I can see theimprovement in this area myself. I am feeling very confident about my ability to conduct a researchproject in my third year, and it is entirely down to this studentship. My theoretical background ofcancer has been broadened substantially and I have been introduced to new ways of thinking aboutcancer that I doubt I would have gained without the help of this project. I haven’t just learnt awealth of knowledge here; I’ve also very much enjoyed myself. It has been fantastic to work in such

a supportive and friendly environment. I am indebted to both Dr Stephen Maher and theBiochemical Society for giving me the opportunity to conduct this research.
Value to the lab
Stephen has been a refreshing addition to the lab this summer. Commendably he hasworked in the lab for the remainder of the summer in order to continue the project and gain asmuch from the experience as possible. As a supervisor it has been fulfilling to see passion for thesubject in the student, and his development in the research lab from a place of uncertainty to one ofdecisiveness and confidence. Stephen has worked hard on the project, which forms part of a muchlarger programme of research on CCL28. Indeed, some of the work he has done for the summerstudentship is being included as part of a research poster at the 2013 National Cancer ResearchInstitute (NCRI) annual meeting, on which Stephen is listed as a co-author. This has been a beneficialexperience for both student and supervisor, and the Biochemical Society should be applauded forproviding these opportunities in support of the next wave of researchers.
References
Cancer Research UK, 2013. Oesophageal Cancer Incidence Statistics. [Online] Available at:http://www.cancerresearchuk.org/cancer-info/cancerstats/types/oesophagus/incidence/[Accessed 29th September 2013].Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, LalP, Zhang L, Coukos G. 2011. Tumour hypoxia promotes tolerance and angiogenesis via CCL28and Treg cells. Nature 475:7355, pp. 226-30.Facciabene A, Santoro S, Coukos G. 2012. Know Thy Enemy: Why are tumour-infiltratingregulatory T-cells so deleterious? Oncoimmunology 1:4, pp. 575–577.

1
What controls redox stabilisation in Student: Vlad Antonthermophilic Photosystem II? Supervisor: A. William Rutherford
Introduction
Photosystem II (PS II), the water-oxidizing enzyme of plants and cyanobacteria, is able to split water intomolecular oxygen, protons and electrons thanks to its manganese cluster that can accumulate positive charges(oxidising power). With every successfully absorbed photon a charge separation occurs in P680, a specialchlorophyll at the heart of PS II. Then, the electron is transferred all the way to the final electron acceptor QB(plastoquinone) while the electron hole (positive charge) eventually ends up on the manganese cluster ([Mn]4).A temperature-dependent back-reaction is possible where the charges recombine and light is emitted. Inthermoluminescence, PS II is heated at a constant rate and, when a sufficiently high temperature is reached, thecharges recombine and give rise to luminescence. The stability of the charge separation (i.e. temperature atwhich recombination occurs) is higher in thermophiles relative to mesophiles but it can be diminished or lost,depending on the purification of PS II.
This project started based on an older observation that in PS II purified by Rögner et al in (1) the chargeseparation has a lower stability than in the PS II purified by Nickel affinity chromatography (2). The two maindifferences between the purifications are that, in Rögner’s, PS II is extracted from the thylakoid memtranes witha 1.2% (m/m) DDM (n-Dodecyl- -D-maltopyranoside) buffer (instead of 1%) and then passed through ahydrophobic interaction chromatographic column instead of the relatively inert Nickel column.
Aims
Since this aspect had been left unpursued, the aim of my project was to reproduce the Rögner purification and,using thermoluminescence (TL) as a redox stability assay, to identify what treatment affects the thermostabilityand consequently what renders the thermophilic PS II more stable.
Materials and Methods
PS II Purification – CP47 His-tagged PS II was purified from 15 l of Thermosynechococcus elongatus culturegrown at 45 C to an OD730 of 1.2. The purification was performed as described in (2): PS II was extracted fromthe thylakoid membranes using buffer (40 mM MES pH 6.5, 20 mM MgCl2, 20 mM CaCl2 and 20% glycerol)containing 1% (m/m) DDM and then passed through a Nickel column. Eluted PS II was washed andconcentrated using Amicon 100 kDa cut-off filters to a final concentration of 2.37 mg Chl a/ml with an oxygenevolution of 1710 mol O2/mg Chl a · h.
Rögner Purification Reproduction. Hydrophobic Interaction Chromatography – Purified PS II was passedthrough a 1 ml GE Healthcare HiTrap Butyl FF hydrophobic interaction chromatography (HIC) column using anFPLC machine and an ammonium sulphate gradient from 3 M to 0 M. PS II eluted very slowly and for a longtime after the ammonium sulphate concentration reached zero.Higher DDM concentration treatment – Purified PS II was once again stuck to a 1 ml Nickel column andwashed with 25 ml of buffer containing 1.2 % (m/m) DDM before elution and concentration. For thethermoluminescence measurements, part of the resulting PS II was further incubated in 1.2 % DDM for another2 h.
Thermoluminescence – Single flash thermoluminescence experiments (3) were performed on PS II samples at150 g Chl a/ml using a ‘home-made’ thermoluminescence instrument built by Jean-Marc Ducruet. Sampleswere dark-adapted for several hours before the measurements. A heating rate of 0.35 C/s was used throughout.The temperature gradients started at 0 C for S2 – QB
- recombination measurements and at -10 C for S2 – QA-
recombination measurements (when 10 M DCMU (3-(3,4-dichlorophenyl)-1,1-dimethylurea) was added).
Results and Discussion
Performing the Rögner purification steps with already purified PS II allowed for a reliable control for allexperiments. There were no differences in terms of peak position between the control and the post-HIC PS II.

2
Washing the PS II with more concentrated detergent, on the other hand, did destabilize the charge pair. Inconclusion, in all likelihood, the stability difference seen between the two purifications is caused by theexposure of PS II to different DDM concentrations when extracted from the thylakoid membranes. Figure 1shows the TL peaks of control PS II (54.4 C), PS II washed with 1.2% DDM (50.1 C) and PS II washed andfurther incubated with 1.2% DDM (47.5 C). DCMU experiments also showed a loss of stability of the S2 – QA
-
pair, from 17.5 C to 16.7 and then 15.8. Since the differences between the DCMU peak positions are smaller,the measurements will have to be repeated before any conclusions can be drawn. In any case, it is clear that notonly DDM concentration but also duration of exposure to DDM affect the thermostability of the S2 – QB
- pair,which is reduced gradually. The intensity of the DDM-incubated peak is lower than the other two, possiblybecause of the presence of DDM during the measurement or because a longer exposure to detergent has adamaging effect on PS II, reducing the number of functional centres in the sample.
Figure 1. Single flashthermoluminescence measurements ofpurified PS II (control), DDM-washedPS II and DDM-washed and incubatedPS II, all at 150 g Chl a/ml and at a0.35 C/s heating rate.
The stability loss may be due to thedetergent stripping off or replacingsome special lipids with redox tuningroles in the PS II complex. The fact thatthe change is gradual might simply bedue to the fact that the peak position isan average and, with longer exposuresto detergent, the average shifts towardsthe less stable state.
Future Directions
Washing PS II with a wider range of DDM concentrations and incubating it for different periods of time will benecessary to get a better overview of this process and see what the lowest achievable stability is. Then, toidentify the stabilizing component (presumably a lipid), the washate could be separated from the concentrateddetergent via thin layer chromatography, for example, and the fractions subjected to mass spectrometry analysis.
References
1. Kuhl H., Rögner M. et al, 2000, Towards Structural Determination of the Water-splitting Enzyme, J.Biol. Chem. 2000, 275:20652-20659.
2. Sugiura M, Inoue Y., 1999, Highly Purified Thermo-Stable Oxygen-Evolving Photosystem II CoreComplex from the Thermophilic Cyanobacterium Synechococcus elongatus Having His-Tagged CP43Plant Cell Physiol. 40(12): 1219-1231
3. Rutherford A. W., Crofts, A.R. and Inoue, Y. (1982) Biochim. Biophys. Acta 682, 457-465.Thermoluminescence as a probe of Photosystem II photochemistry: the origin of the flash inducedglow peaks.
Value of the Studentship
This grant has enabled me to have my first real lab experience and I loved it. Before undertaking thisproject, I already knew I wanted to become a researcher and professor, and working in this beautifulenvironment has only reinforced my choice. Most importantly, with this occasion, I learned that I canenjoy research regardless of the field of study. I found myself very happy and content over theduration of the project, feeling that the things I did were bringing me closer to a good result. Then,when the good result arrived (two days before the end of the project), I was truly ecstatic. I plan oncontinuing with these experiments this year, alongside my course, and hope to sum them up in what isgoing to be my first research paper.