supplementary material for...charged with 0.236 g (0.205 mmol) of sl-a109-2, 0.028 g (0.215 mmol)...
TRANSCRIPT

www.sciencemag.org/content/full/342/6162/1076/suppl/DC1
Supplementary Material for
Cobalt Precursors for High-Throughput Discovery of Base Metal Asymmetric Alkene Hydrogenation Catalysts.
Max R. Friedfeld, Michael Shevlin, Jordan M. Hoyt, Shane W. Krska, Matthew T. Tudge, Paul J. Chirik
Published 29 November 2013, Science 342, 1076 (2013) DOI: 10.1126/science.1243550
This PDF file includes:
Materials and Methods
Figs. S1 to S27
Tables S1 to S6
Full Reference List

2
Table of Contents
Experimental Section. S3 I. General Considerations. S3 II. Synthesis of Cobalt Precatalysts. S5 III. Representative Procedures for Catalytic Hydrogenation. S7 IV. Preparation and Purification of Alkene Substrates. S9 V. Characterization of Alkane Products. S10 VI. NMR Spectra of Alkane Products. S15
VII. Crystallographic Data. S19 VIII. Phosphine Evaluation with Methyl 2-acetamidoacrylate (MAC) S20 IX. Phosphine Evaluation with Acetomidostyrene. S29 X. Phosphine Evaluation with trans-(α)-Methylstilbene S37 XI. Evaluation of Cobalt Sources for the Hydrogenation of MAC. S52 XII. GC and SFC Chromatograms of Enantiopure Alkanes. S53 XIII. References. S56

3
I. General Considerations. All air- and moisture-sensitive manipulations were carried
out using standard high vacuum line, Schlenk or cannula techniques or in an M. Braun
intert atmosphere drybox containing an atmosphere of purified nitrogen. The M. Braun
drybox was equipped with a cold well designed for freezing samples in liquid nitrogen.
Solvents for air- and moisture-sensitive manipulations were dried and deoxygenated
using literature procedures.(42) Deuterated solvents for NMR spectroscopy were distilled
from sodium metal under an atmosphere of argon and stored over 4 Å molecular sieves.
Anhydrous cobalt dichloride and anhydrous cobalt(II) benzoate were purchased from
Acros Organics, Alfa Aesar or Aldrich and used as received. Bidentante chiral
phosphines were obtained from commercial sources including Aldrich, Strem, Solvias,
Takasago, and Chiral Quest.
1H NMR spectra were recorded on a Varian Inova 400 Spectrometer operating at
399.860 MHz or a Bruker AVANCE-500 equipped with a TCI cryoprobe optimized for 1H
detection operating at 500.62 MHz. All chemical shifts are reported relative to SiMe4
using 1H (residual) chemical shifts of the solvent as a secondary standard. 13C NMR
spectra were recorded on a Bruker 500 spectrometer equipped with a DCH cryoprobe
optimized for 13C detection operating at 125.71 MHz. 13C chemical shifts are reported
relative to SiMe4 using chemical shifts of the solvent as a secondary standard where
applicable. Elemental analyses were performed at Robertson Microlit Laboratories, Inc.,
in Ledgewood, NJ. Solid-state magnetic moments were determined using a Johnson
Matthey Magnetic Susceptibility Balance, collected at room temperature, unless
otherwise noted. Solution magnetic moments were determined by the method of Evans
at 22 ºC using a ferrocene standard unless otherwise noted.(43) High-resolution mass
spectra were measured using an Agilent 6220 Accurate-Mass TOF LC/MS. The mass
spectrometer was calibrated externally before each use with purine and the Agilent ES-

4
TOF tuning mix (part number = G1969-85000). These compounds were assigned a
(M+H)+ m/z ratio of 121.050873 and 922.009798 respectively. Optical rotations were
recorded on a Perkin Elmer Model 341 polarimeter (1-mL cell, 1 dm path length);
concentration (c) is in g / 100 mL and [α]D values are in degrees. The optical rotations for
methyl 2-acetamido-3-phenylacrylate and 1,2-diphenylpropane were compared to
literature data.(34, 44 ) Racemic alkanes were independently synthesized using a
heterogeneous Pd/C catalyst to obtain chiral analysis and for comparison to
enantioselective catalytic experiments. Representative NMR spectra are included.
Gas chromatography for the alkane products was performed on a Shimadzu GC-
2010 gas chromatograph. GC analyses were performed using a Restek 15 m x 0.25 mm
RTX-5 5% diphenyl/95% dimethyl polysiloxane column with a film thickness of 0.25 μm.
The following temperature program was used: 60 ºC, 1 min ; 15 ºC/min to 250 ºC; hold 1
min. Chiral gas chromatography for the alkane products was performed on a Shimadzu
GC-2010 gas chromatogram using a Supelco 30 m x 0.25 mm BETA DEX 120 capillary
column as noted for each product. Supercritical fluid chromatography (SFC) was
performed on a Berger Minigram equipped with a diode array UV detector (λ = 214-300
nm) using a chiral column (25 cm) and guard column (5 cm) as noted for each
compound.
Single crystals suitable for X-ray diffraction were coated with polyisobutylene oil
in a drybox, transferred to a nylon loop and then quickly transferred to the goniometer
head of a Bruker X8 APEX2 DUO diffractometer equipped with a molybdenum X-ray
tube (λ = 0.71073). Preliminary data revealed the crystal system. The data collection
strategy was optimized for completeness and redundancy using the Bruker COSMO
software suite. The space group was identified, and the data were processed using the
Bruker SAINT+ program and corrected for absorption using SADABS. The structures

5
were solved using direct methods (SHELXS) completed by subsequent Fourier synthesis
and refined by full-matrix least-squares procedures.
II. Synthesis of Cobalt Precatalysts.
Preparation of (SL-A109-2)CoCl2. A 20 mL scintillation vial was
charged with 0.236 g (0.205 mmol) of SL-A109-2, 0.028 g (0.215
mmol) anhydrous cobalt dichloride and 4 mL of THF. The reaction
mixture was stirred for 30 minutes at room temperature. Excess
cobalt dichloride was removed by filtration through Celite and the
filtrate solvent was removed in vacuo. The resulting green solid was washed with 1 mL
of pentane and dried in vacuo to yield 0.221 g (0.205 mmol, 81%) of (SL-A109-2)CoCl2.
Analysis for C74H104Cl2P2O6Co: Calc. C, 69.36; H, 8.18. Found C, 68.84; H, 8.45. Solid-
state magnetic susceptibility μeff (293 K) = 3.8 μB.
Preparation of (R,R-iPr-Duphos)CoCl2. A 20 mL scintillation vial was
charged with 0.103 g (0.246 mmol) of R,R-iPr-Duphos and 0.034 g
(0.258 mmol) of anhydrous cobalt dichloride and 4 mL of THF. The
reaction mixture was stirred for 30 minutes at room temperature. Excess cobalt
dichloride was removed by filtration through Celite and the filtrate solvent was removed
in vacuo. The resulting red solid was washed with 1 mL of pentane and dried in vacuo
yielded 0.131 g (0.239 mmol, 97%) of (R,R-iPr-Duphos)CoCl2. Cooling a concentrated
diethyl ether/tetrahydrofuran (1:4) solution to -35 ºC furnished red crystals suitable for X-
ray diffraction. Analysis for C26H44Cl2P2Co: Calc. C, 56.94; H, 8.09. Found C, 56.72; H,
7.81. Solid-state magnetic susceptibility μeff (293 K) = 2.2 μB.
P
PCo
iPr iPr
iPr iPr
Cl
Cl
OO
PP
OMet-But-Bu
OMet-Bu t-Bu
Ar
ArCo
Cl
Cl

6
Preparation of (R,R-iPr-Duphos)Co(CH2SiMe3)2. A 20 mL
scintillation vial was charged with 0.029 g (0.069 mmol) of R,R-iPr-
Duphos and 2 mL of toluene. To this solution, 346 μL of a 0.2 M
solution (0.069 mmol) of (py)2Co(CH2SiMe3)2 in toluene were added and rapid formation
of an orange solution was observed. After 15 minutes of stirring at room temperature, the
volume of the orange solution was reduced in vacuo. The resulting orange film was
extracted into 5 mL of pentane and filtered through Celite. The orange filtrate was
evaporated to dryness and yielded 0.043 g (0.066 mmol, 95%) of an orange solid
identified as (R,R-iPr-Duphos)Co(CH2SiMe3)2. Cooling a concentrated pentane solution
of (R,R-iPr-Duphos)Co(CH2SiMe3)2 to -35 ºC furnished crystals suitable for X-ray
diffraction. Analysis for C34H66P2Si2Co: Calc. C, 62.64; H, 10.20. Found C, 62.58; H,
10.25. 1H NMR (C6D6, 400 MHz): -44.90 (614 Hz), -34.19 (689 Hz), -29.85 (627 Hz), -
17.12 (85 Hz), -14.80 (166 Hz), -13.84 (104 Hz), -6.68 (214 Hz), 1.27 (54 Hz), 2.22 (173
Hz), 6.32 (68 Hz), 7.50 (12 Hz), 16.08 (30 Hz), 25.80 (114 Hz). μeff (293 K, Evans,
benzene-d6) = 2.0 μB.
Preparation of (R,R-iPr-Duphos)Co(OBz)2. A 20 mL scintillation
vial was charged with 0.046 g (0.110 mmol) of R,R-iPr-Duphos and
0.035 g (0.115 mmol) of anhydrous cobalt (II) benzoate. The solids
were suspended in 5 mL of toluene. The reaction mixture was stirred for one hour at
room temperature. Excess cobalt (II) benzoate was removed by filtration through Celite
and the filtrate solvent was removed in vacuo. The resulting red solid was washed with
pentane and dried in vacuo to furnish 0.068 g (0.094 mmol, 86% yield) of a red solid
identified as (R,R-iPr-Duphos)Co(OBz)2. Cooling a concentrated toluene solution of
(R,R-iPr-Duphos)Co(OBz)2 at -35 ºC furnished crystals suitable for X-ray diffraction.
P
PCo
iPr iPr
iPr iPr
OC(O)Ph
OC(O)Ph
P
PCo
iPr iPr
iPr iPrSiMe3
SiMe3

7
Analysis for C40H54O4P2Co: Calc. C, 66.75; H, 7.56. Found C, 67.01; H, 7.43. Solid-state
magnetic susceptibility μeff (293 K) = 1.9 μB.
III. Representative Procedures for Catalytic Hydrogenation.
High Throughput Experiments: trans-Methylstilbene Screen. Each well of a 1 mL x
96 well plate array containing 4.1 x 10-4 mmol each of pre-dispensed phosphine ligands
was charged with 100 μL of a 4 mM toluene solution of (py)2Co(CH2SiMe3)2. After mixing
for 20 minutes, the volatile components were removed in vacuo using a Genevac
apparatus. Each well was then charged with 100 μL of a 40 mM toluene solution of
trans-methylstilbene. The 96 well plate was then placed in a high-pressure block and
purged with nitrogen gas and then was connected to a 1 L gas buret, followed by purging
with hydrogen gas. The block and buret were both pressurized to 500 psi. The apparatus
was agitated with mechanical shaking at room temperature for 24 hours, at which point
the block was purged with nitrogen gas and the 96 well plate was removed. The samples
were then analyzed by SFC.
High Throughput Experiments: Methyl 2-Acetamidoacrylate (MAC) Screen. Each
well of a 1 mL x 96 well plate array containing 4.1 x 10-4 mmol each of pre-dispensed
phosphine ligands was added 50 μL of an 8 mM THF solution of CoCl2. After mixing for
20 minutes, 50 μL of a 16 mM THF solution of LiCH2SiMe3 was added to each reaction
well. After 20 minutes of stirring, the volatile components were removed in vacuo using a
Genevac apparatus. Each reaction well was then charged with 100 μL of a 40 mM THF
solution of methyl 2-acetamidoacrylate. The 96 well plate was then placed in a high-
pressure block and purged with nitrogen gas and then was connected to a 1 L gas buret,
followed by purging with hydrogen gas. The block and buret were both pressurized to

8
500 psi. The apparatus was agitated via mechanical shaking at room temperature for 24
hours, at which point the block was purged with nitrogen gas and the 96 well plate was
removed. The samples were then analyzed by SFC.
Pre-formed Cobalt Precursor, Method A: A 45 mL glass well was charged with 0.047
g (0.328 mmol) of methyl 2-acetamidoacrylate, 0.011 g (0.016 mmol) of (R,R)-iPr-
DuphosCo(CH2SiMe3)2 and 8 mL of THF. Formation of an orange solution was observed.
In the glovebox, the glass well was charged into 45 mL Parr Reactor which was then
sealed, pressurized to 1000 psi H2, vented, re-pressurized to 1000 psi, and vented to the
desired pressure (500 psi). The reaction solution was mechanically stirred under the H2
pressure for the appropriate time, at which point the vessel was vented and the reaction
solution was retrieved from the vessel. The solvent was evaporated and the product was
purified by flash chromatography.
Pre-formed Cobalt Precursor, Method B: A 45 mL glass well was charged with 300 μL
of a 0.372 M solution (0.016 mmol) of (R,R)-iPr-DuphosCoCl2 and 3.7 mL of THF. A blue
solution was observed. A solution of LiCH2SiMe3 (88 μL of a 0.372 M solution, 0.033
mmol, 2 eq. relative to [Co], 10% relative to substrate) was added and a blue solution
was observed. Methyl 2-acetamidoacrylate (47 mg, 0.328 mmol) and 4 mL of THF was
added to the reaction solution. The glass well was then transferred to the Parr reactor,
which was sealed, pressurized to 1000 psi H2, vented, re-pressurized to 1000 psi, and
vented to the desired pressure (500 psi). The reaction solution was mechanically stirred
under the H2 pressure for the appropriate time, at which point the vessel was vented and
the reaction solution was retrieved from the vessel. The solvent was evaporated and the
product was purified by flash chromatography.

9
Method C: A 20 mL scintillation vial was charged with 0.007 g (0.016 mmol) of (R,R)-iPr-
Duphos, 1 mL of toluene and 82 μL of a 0.2 M solution (0.016 mmol) of
(py)2Co(CH2SiMe3)2. The reaction mixture was stirred for 10 minutes until the reaction an
orange solution was observed and the solvent was then removed under reduced
pressure. The orange residue was reconstituted in 5 mL of pentane, filtered through
celite and evaporated to afford (R,R)-iPr-DuphosCo(CH2SiMe3)2. This material was
transferred to a 45 mL glass well with 4 mL of THF. Methyl 2-aceamidoacrylate (47 mg,
0.328 mmol) was then added with an additional 4 mL of THF. The glass well was then
transferred to the Parr reactor, which was sealed, pressurized to 1000 psi H2, vented, re-
pressurized to 1000 psi, and vented to the desired pressure (500 psi). The reaction
solution was mechanically stirred under the H2 pressure for the appropriate time, at
which point the vessel was vented and the reaction solution was retrieved from the
vessel. The solvent was evaporated and the product was purified by flash
chromatography.
IV. Preparation and Purification of Alkene Substrates. Trans-methylstilbene was
purchased from Aldrich. The solid was dried in vacuo over night and brought into a glove
box. A homogenous toluene solution was passed through a plug of activated alumina
and trans-methylstilbene was recrystallized at -35 ºC from a concentrated toluene
solution. Methyl 2-acetamidoacrylate was purchased from Aldrich. The solid was dried in
vacuo over night and brought into a glove box. A homogenous tetrahydrofuran solution
was passed through a plug of activated alumina and methyl 2-acetamidoacrylate was
recrystallized at -35 ºC from the concentrated tetrahydrofuran solution. Methyl 2-
acetamido-3-phenylacrylate was prepared according the literature method(45) and the

10
solid was dried in vacuo over night and brought into a glove box. A homogenous
tetrahydrofuran solution was passed through a plug of activated alumina and methyl 2-
acetamido-3-phenylacrylate was recrystallized at -35 ºC from the concentrated
tetrahydrofuran solution. N-(1-phenylvinyl)acetamide,( 46 ) (Z)-ethyl 3-acetamido-3-
phenylacrylate(47) were prepared according to literature procedures and purified using
techniques described above.
V. Characterization of Alkane Products.
(rac)-1,2-diphenylpropane. To a thick-walled glass vessel was added 0.057
g (0.293 mmol) of α-trans-methylstilbene, 0.020 g of 5% Pd/C and
approximately 10 mL ethanol. The reaction mixture was stirred under 4 atm H2 at room
temperature for 24 hours. The Pd/C was removed through Celite filtration and the filtrate
solvent was removed in vacuo, affording 0.048 g (0.245 mmol, 83%) of a pale yellow oil
identified as 1,2-diphenylpropane. (48 mg, 0.245 mmol, 83% yield) as a pale yellow oil.
1H NMR (CDCl3, 500.15 MHz): δ 1.24 (d, 3JHH = 6.78 Hz, 3H, PhCHCH3), 2.77 (dd, 2JHH =
13.07 Hz, 3JHH = 8.17 Hz, 1H, PhCH2, one set of diastereotopic resonances), 2.95 (dd,
2JHH = 13.07 Hz, 3JHH = 6.40 Hz, 1H, PhCH2, one set of diastereotopic resonances), 3.00
(m, 1H, PhCHCH3, overlapping signal), 7.09 (m, 2H, Ph-CH), 7.22 (m, 8H, Ph-CH,
overlapping resonances with residual solvent resonance). 13C{1H} NMR (CDCl3, 125.88
MHz): δ 21.17 (PhCHCH3), 41.90 (PhCHCH3), 45.06 (PhCH2), 125.86 (Ph-CH), 126.04
(Ph-CH), 127.07 (Ph-CH), 128.12 (Ph-CH), 128.33 (Ph-CH), 129.19, (Ph-CH), 140.85
(ipso Ph-C), 147.07 (ipso Ph-C). GC-MS: Calcd for C8H9, [M-PhCH2], m/z 105.07.
Found, m/z 105.0. Chiral SFC analysis of 1,2-diphenylpropane: (OJ-H, 10% MeOH/CO2,
3.0 mL/min, 300 nm) enantiomer retention times: 3.16 min (R)-(–)-1,2-diphenylpropane;
3.85 min (S)-(+)-1,2-diphenylpropane; alkene retention time: 6.94 min. Absolute
PhPh

11
configuration measurement: multiple catalytic samples containing 1,2-diphenylpropane in
~89%ee were combined to afford 0.33 g of alkane that was used for the optical rotation
measurements. [α]22D = -56.92º (c = 16.7, CH2Cl2), corresponding to (R)-(–)-1,2-
diphenylpropane. The optical rotation measurent for (R)-(–)-1-methoxy-4-(1-
phenylpropan-2-yl)benzene was compared as a reference.44
(rac)-Methyl 2-acetamido-3-phenylpropanoate. To a thick-walled glass
vessel was added 0.091 g (0.415 mmol) of (Z)-methyl 2-acetamido-3-
phenylacrylate, 0.020 g of 5% Pd/C and approximately 10 mL of ethanol. The
reaction mixture was stirred under 4 atm H2 at room temperature for 12 hours. The Pd/C
was removed through Celite filtration and the filtrate solvent was removed in vacuo
affording 0.083 g (0.375 mmol, 90%) of a while solid identified as methyl 2-acetamido-3-
phenylpropanoate. 1H NMR (CDCl3, 500.15 MHz): δ 1.41 (d, 3JHH = 7.21 Hz, 3H,
CHCH3), 2.03 (s, 3H, HNC(O)CH3), 3.75 (s, 3H, CO2CH3), 4.60 (dq, 3JHH = 7.28 Hz, 3JHH
= 7.28 Hz, 1H, HNCHCH3), 6.11 (br s, 1H, NHCHCH3). 13C{1H} NMR (CDCl3, 125.89
MHz): δ 18.73 (HNCHCH3), 23.32 (HNC(O)CH3), 48.19 (HNCHCH3), 52.69 (CO2CH3),
169.81 (HNC(O)CH3), 173.75 (CO2CH3). HR-MS (+EI): Calcd for C6H11NO3Na, [M+Na]+,
m/z 168.063657. Found, m/z 168.06358. The product was analyzed on a Supelco 30 m
(S)(S)
(+)-(S)-1,2-diphenylpropane
(R)(R)
(–)-(R)-1,2-diphenylpropane
HN
O
O
O

12
x 0.25 mm BETA DEX 120 capillary column using the following temperature program: 90
ºC for 75 min, 20 ºC/min until 200 ºC, hold 200 ºC for 6 min.. Enantiomer retention times:
58.03 min (S)-methyl 2-aceamidopropanoate, 59.56 min (R)-methyl 2-
acetamidopropanoate. Alkene retention time: 44.38 min. Enantiopure samples of alkane
were purchased and analyzed using this separation method to determine the absolute
configuration.
Methyl 2-acetamido-3-phenylpropanoate. (Z)-methyl 2-acetamido-3-
phenylacrylate (91 mg, 0.415 mmol), 5% Pd/C (20 mg) in approximately
10 mL of ethanol was stirred at room temperature under 4 atm H2. After
12 hours, the mixture was filtered through celite and the solvent was removed under
reduced pressure affording methyl 2-acetamido-3-phenylpropanoate (83 mg, 0.375
mmol, 90% yield) as a colorless oil (Rf = 0.28, 50% EtOAc/hexanes) 1H NMR (CDCl3,
500.15 MHz): δ 1.99 (s, 3H, C(O)CH3), 3.09 (dd, 2JHH = 13.85 Hz, 3JHH = 5.74 Hz, 1H,
PhCH2 diastereotopic CH2 resonances are overlapping), 3.15 (dd, 2JHH = 13.85 Hz, 3JHH
HNO
O
O
(S)(S)
HNO
O
O
(R)(R)
HNO
O
O

13
= 5.74 Hz, 1H, PhCH2) 3.73 (s, 3H, CO2CH3), 4.88 (dt, 2JHH = 7.76 Hz, 3JHH = 5.76 Hz,
1H, PhCH2CH), 5.97 (br d, 3JHH = 6.26 Hz, 1H, NH), 7.09 (m, 2H o-Ph-CH), 7.27 (m, 3H,
m-Ph-CH and p-Ph-CH). 13C{1H} NMR (CDCl3, 125.89 MHz): δ 23.25 (NC(O)CH3),
37.95 (PhCH2), 52.49 (CO2CH3), 53.25 (PhCH2CH), 127.29 (Ph-CH), 128.73 (Ph-CH),
129.37 (Ph-CH), 135.91 (ipso Ph-C), 169.87 (NC(O)CH3), 172.22 (CO2CH3). HR-MS
(+EI): Calcd for C12H15NO3Na, [M+Na]+, m/z 221.10519. Found, m/z 221.10498. The
product was analyzed on a Supelco 30 m x 0.25 mm BETA DEX 120 capillary column
using the following temperature program: 130 ºC for 140 min, 20 ºC/min until 200 ºC,
hold 200ºC for 6 min. Enantiomer retention times: 129.49 min (R)-(–), 129.66 min (S)-(+).
Alkene retention time: 125.55 min. Absolute configuration measurement: a catalytic
experiment resulting in >99% conversion and 92.7% ee was purified using column
chromatography and used for an optical rotation measurement. [α]22D = +13.3º (c = 1.5,
methanol), corresponding to (S)-(+) methyl 2-acetamido-3-phenylacrylate.
(rac)-N-(phenylethyl)-acetamide. To a thick-walled glass vessel was
added 0.100 g (0.620 mmol) of N-(1-phenylvinyl)-acetamide (100 mg,
0.62 mmol), 0.020 g of 5% Pd/C and approximately 10 mL ethanol. was stirred at room
temperature under 4 atm H2. The reaction mixture was stirred under 4 atm H2 at room
temperature for 12 hours. The Pd/C was removed through Celite filtration and the filtrate
solvent was removed in vacuo, affording 0.089 g (0.545 mmol, 88% yield) of a clear oil
NH
O
HNO
O
O

14
identified as N-(phenylethyl)-acetamide.1H NMR (CDCl3, 500.15 MHz): δ 1.50 (d, 3JHH =
6.89 Hz, 3H, CHCH3), 1.99 (s, 3H, C(O)CH3), 5.14 (m, JHH = 7.36 Hz, 1H, CHCH3), 5.67
(br s, 1H, NH), 7.31 (m, 5H, C6H5 overlapping signals). 13C{1H} NMR (CDCl3, 125.89
MHz): δ 21.79 (CHCH3), 23.56 (C(O)CH3), 48.95 (CHCH3), 126.33 (Ph-CH), 127.54 (Ph-
CH), 128.82 (Ph-CH), 143.20 (ipso Ph-C), 169.34 (C(O)CH3). HR-MS (+EI): Calcd for
C10H13NO, [M]+, m/z 163.09971. Found, m/z 163.09968. The product was analyzed on a
Supelco 30 m x 0.25 mm BETA DEX 120 capillary column using the following
temperature program: 130 ºC for 140 min, 20 ºC/min until 200 ºC, hold 200ºC for 6 min.
Enantiomer retention times: 59.41 min (S), 62.33 min (R). Alkene retention time: 125.94
min. An authentic sample of (S)-N-(phenylethyl)-acetamide was independently prepared
and analyzed with this chiral GC method.
NH
O
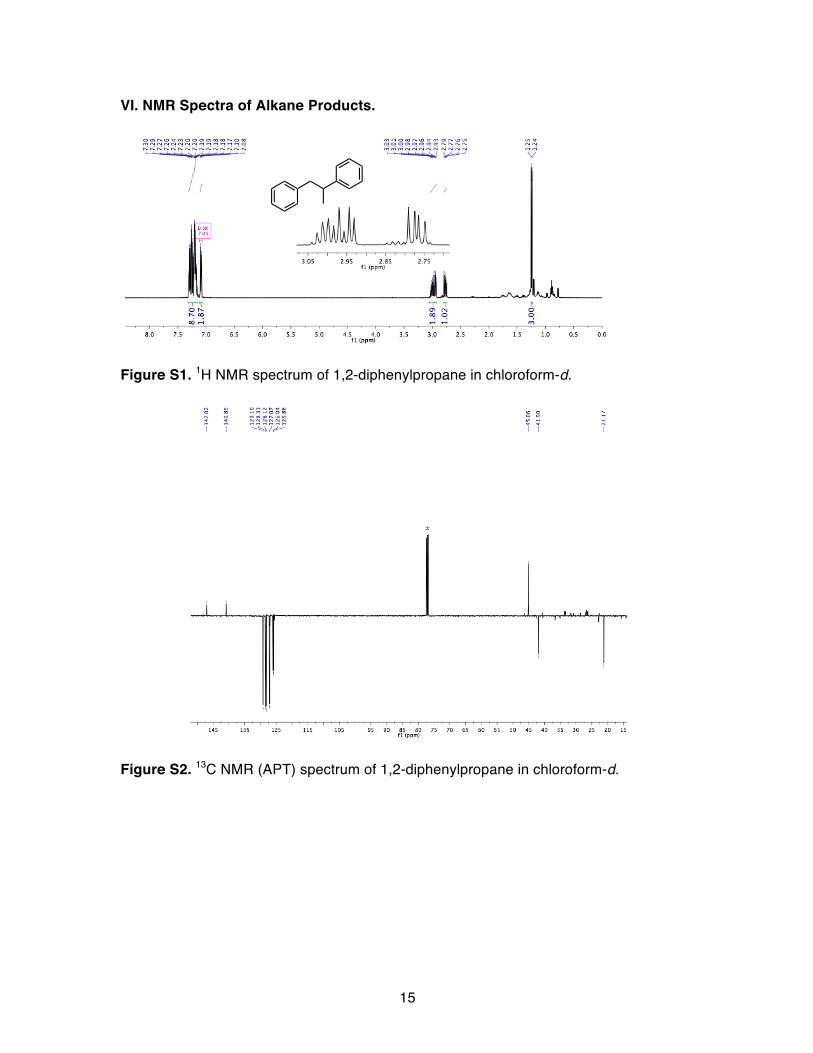
15
VI. NMR Spectra of Alkane Products.
Figure S1. 1H NMR spectrum of 1,2-diphenylpropane in chloroform-d.
Figure S2. 13C NMR (APT) spectrum of 1,2-diphenylpropane in chloroform-d.

16
Figure S3. 1H NMR spectrum of methyl 2-acetamidopropanote in chloroform-d.
Figure S4. 13C NMR (APT) spectrum of methyl 2-acetamidopropanoate in chloroform-d.
NH O
OO
NH O
OO

17
Figure S5. 1H NMR spectrum of methyl 2-acetamido-3-phenylpropanoate in chloroform-d.
Figure S6. 13C NMR spectrum of methyl 2-acetamido-3-phenylpropanoate in chloroform-d.
HNO
O
O
HNO
O
O

18
Figure S7. 1H NMR spectrum of N-(1-phenylethyl)acetamide in chloroform-d.
NH
O

19
VII. Crystallographic Data
Figure S8. Solid state structures of (R,R)-(iPrDuPhos)Co(CH2SiMe3)2 (left), (R,R)-(iPrDuPhos)CoCl2 (middle) and (R,R)-(iPrDuPhos)Co(OBz)2 (right) at 30% probability ellipsoids. Hydrogen atoms and additional molecules present in the unit cell omitted for clarity.
Figure S9. Additional representations of the solid state structure of (R, R)-(iPrDuphos)CoCl2 at 30% probability ellipsoids. Hydrogen atoms and additional molecules in the unit cell omitted for clarity.
! !
!
!!

20
VIII. Phosphine Evaluation Screen with Methyl 2-acetamidoacrylate (MAC).
Figure S10. Evaluation of various enantiopure phosphine ligands in combination with (py)2Co(CH2SiMe3)2 for the asymmetric hydrogenation of methyl 2-acetamidoacrylate.
N CoN
SiMe3
SiMe3
chiral phosphine +
in situ generated catalyst
0.041 M substrate, 10% [Co] + ligand, 500 psi H2, 0.1 mL THF, 20 hr
O
NH O
OO
NH O
O

21
Figure S11. Evaluation of various enantiopure phosphine ligands in combination with (py)2Co(CH2SiMe3)2 for the asymmetric hydrogenation methyl 2-acetamidoacrylate.

22
Figure S12. Bubble plot representing results of high throughput evaluation of chiral phosphines in combination with (py)2Co(CH2SiMe3)2 for the asymmetric hydrogenation of methyl 2-acetamidoacrylate (MAC).
(R,R)-iPr-Duphos!
(S,S)-Et-DuPhos! (S,S)-Me-DuPhos!
(S)-Binapine!
(S,S)-1,2-(Me,PhP)Ph!
-20!
0!
20!
40!
60!
80!
100!
120!
>99% conversion >50% conversion 10% conversion
DuPhos
P
P
R R
RR
(S,S)-1,2-(Me,PhP)Ph
P
P
H3C Ph
Ph CH3
(S)-Binapine
PtBu
PtBu
H
H
N CoN
SiMe3
SiMe3
chiral phosphine +
in situ generated catalyst
0.041 M substrate, 10% [Co] + ligand, 500 psi H2, 0.1 mL THF, 20 hr
O
NH O
OO
NH O
O

23
Table S1. Percent conversion and enantioselectivity of various chiral phosphines in combination with (py)2Co(CH2SiMe3)2 for the asymmetric hydrogenation of methyl 2-acetamidoacrylate (MAC).
Ligand %ee product
conversion (%)
(R,R)-iPr-Duphos
94.0% (S) 92.3%
(S,S)-Et-Duphos
25.9% (S) 3.7%
(S,S)-Me-Duphos
23.5% (S) 20.9%
(S)-Binapine
14.6% (R) 5.6%
(S,S)-1,2-(Me,PhP)2Ph
6.7 (R) >99%

24
Figure S13. Evaluation of various enantiopure phosphine ligands in combination with CoCl2 and Me3SiCH2Li for the asymmetric hydrogenation of methyl 2-acetamidoacrylate.
chiral phosphine +
in situ generated catalyst
0.041 M substrate, 10% CoCl2 + ligand, 20% Me3SiCH2Li, 500 psi H2, 0.1 mL THF, 20 hr
CoCl2 + Me3SiCH2LiO
NH O
OO
NH O
O
!"!#
$!"!#
%!"!#
&!"!#
'!"!#
(!"!#
)!"!#
*!"!#
+!"!#
,!"!#
$!!"!#
-./.0123456789#
-./.01:;1<8:#
=>;?@43A#BCC-.0#
-./.0148D1E38F6G#
-./.01<H5I89#
-@/@/./.01J>5K8F6G#
-./.01:;1E38F6G#
-@/@01<4G89#%L<M'#
@N18!(&1%#
-@/@01$/%1-B
H/8F808F#
-./.01BH1E38F6G#
-@01<45>O45H#
@N1P(!*1$#
-./.01BH1<8:#
@N1P%!&1%#
QHH#
Q#=65RHDG465#

25
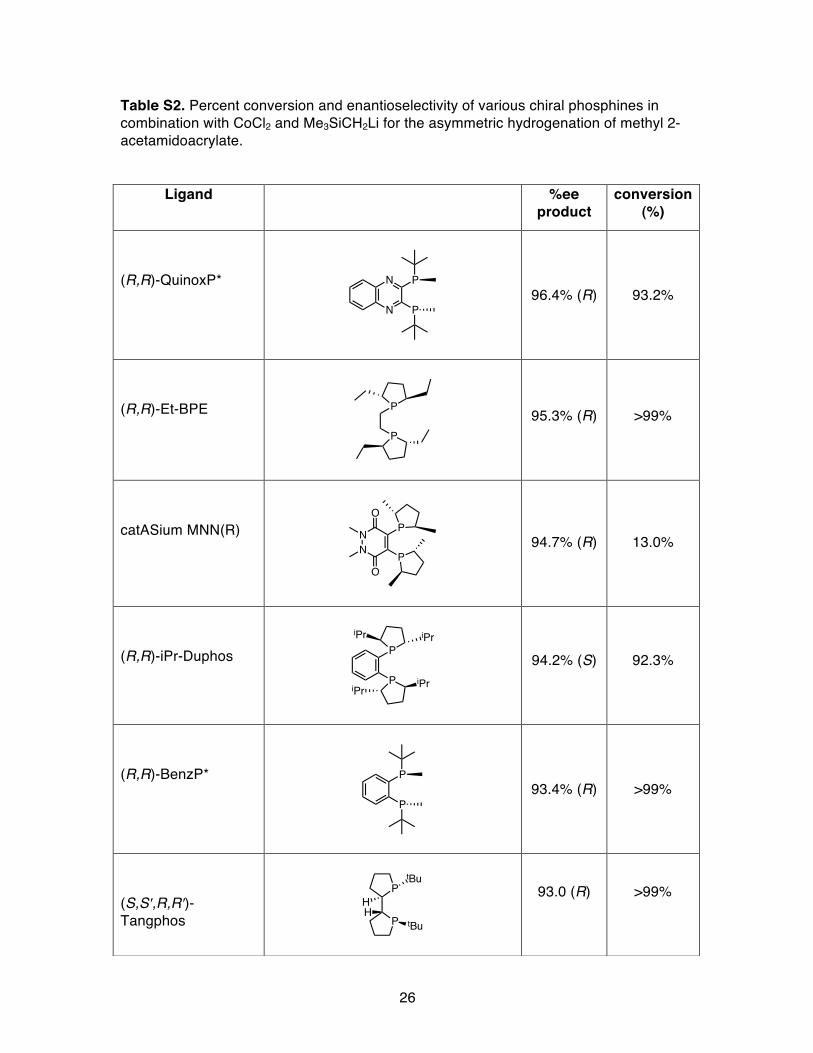
Figure S14. Evaluation of various enantiopure phosphine ligands in combination with CoCl2 and Me3SiCH2Li for the asymmetric hydrogenation of methyl 2-acetamidoacrylate (MAC).
chiral phosphine +
in situ generated catalyst
0.041 M substrate, 10% CoCl2 + ligand, 20% Me3SiCH2Li, 500 psi H2, 0.1 mL THF, 20 hr
CoCl2 + Me3SiCH2LiO
NH O
OO
NH O
O
BPE:
(R,R)-Me-BPE: >99% (52.9% ee, R)(R,R)-Et-BPE: >99% (95.3% ee, R)
P
P
R R
R RN
N P
P
(R,R)-QuinoxP*: 93.2% (96.4% ee, R)
Duphos:
(R,R)-Me-Duphos: 9.0% (72.5% ee, R)(R,R)-Et-Duphos: 80.1% (90.3%ee, S)(R,R)-iPr-Duhpos: 92.3% (94.2% ee, S)
P
P
R R
R R
NN
P
P
O
O
catASium MNN(R):13.0% (94.7% ee, R)
P
P
(R,R)-BenzP*: >99% (93.4% ee, R)
P
P
tBu
tBu
HH
(SS!,RR!)-Tangphos: >99% (93.0% ee, R)
P
P
tBu
tBu
(S,S)-BisP* 2HBF4: >99% (86.5% ee, R)(ligand treated with 2 eq. KOtBu prior to use)
2 HBF4
FeP PPh2
SL-P053-2 (S,S,R)-Me-Kephos: 2.7% (78.0% ee, S)
P
P
H3C Ph
Ph CH3
(S,S)-1,2-(MePPh)2Ph: >99% (77.0% ee, S)
PtBu
PtBu
H
H
(S)-Binapine: 22.5% (70.9% ee, R)
Josiphos:
SL-J203-2: 3.8% (23.1% ee, R)SL-J507-1: 2.4% (63.4% ee, S)
Fe PR22R12P
26
Table S2. Percent conversion and enantioselectivity of various chiral phosphines in combination with CoCl2 and Me3SiCH2Li for the asymmetric hydrogenation of methyl 2-acetamidoacrylate.
Ligand %ee product
conversion (%)
(R,R)-QuinoxP*
96.4% (R) 93.2%
(R,R)-Et-BPE
95.3% (R) >99%
catASium MNN(R)
94.7% (R) 13.0%
(R,R)-iPr-Duphos
94.2% (S) 92.3%
(R,R)-BenzP*
93.4% (R) >99%
(S,S′,R,R′)-Tangphos
93.0 (R) >99%
N
N P
P
P
P
NN
P
P
O
O
P
P
iPr iPr
iPriPr
P
P
P
P
tBu
tBu
HH

27
(R,R)-Et-Duphos
90.3% (S) 80.1%
(S,S)-BisP* 2HBF4
86.5% (R) >99%
SL-P053-2
78.0% (S) 2.7%
(S,S)-1,2-(MePPh)2Ph:
77.0% (S) >99%
(R,R)-Me-Duphos
72.5% (R) 9.0%
(S)-Binapine
70.9% (R) 22.5%
SL-J507-1
63.4% (R) 2.4%
P
P
P
P
tBu
tBu
2 HBF4
FeP PPh2
P
P
H3C Ph
Ph CH3
P
P
PtBu
PtBu
H
H
FePP

28
(R,R)-Me-BPE
52.9% (R) >99% P
P

29
IX. Phosphine Evaluation Screen with Acetomidostyrene.
Figure 15. Evaluation of various enantiopure phosphine ligands in combination with (py)2Co(CH2SiMe3)2 for the asymmetric hydrogenation of N-(1-phenylvinyl)acetamide.
N CoN
SiMe3
SiMe3
chiral phosphine +
in situ generated catalyst
0.041 M substrate, 10% [Co] + ligand, 500 psi H2, 0.1 mL THF, 20 hr
NH
O
NH
O
!"!#
$!"!#
%!"!#
&!"!#
'!"!#
(!"!#
)!"!#
*!"!#
+!"!#
,!"!#
$!!"!#
-./01234#56#5789:;<#
1=>?)++>$#
:1@1<>$@%>:5
7@ABA<%AB#
:;@;<>57>C3ABD8#
:1<>E2F.G2F7#
:;<>E2F.GB.F7#
:;@;<>2AH>C3ABD8#
:;@;<>I32FDJAK#
:;<>EL6M=>A>M2AH#
:1@1@;@;<>N.FOABD8#
:;@;<>E7FPAK#
Q77#
Q#-DFR7H82DF#

30
Figure S16. Evaluation of various enantiopure phosphine ligands in combination with (py)2Co(CH2SiMe3)2 for the asymmetric hydrogenation of N-(1-phenylvinyl)acetamide.
N CoN
SiMe3
SiMe3
chiral phosphine +
in situ generated catalyst
0.041 M substrate, 10% [Co] + ligand, 500 psi H2, 0.1 mL THF, 20 hr
NH
O
NH
O
P
P
catASium MN MesF(R):36.8% (83.1% ee, S)
N
O
OF3C
F3C FePH PO
SL-J688-1:6.2%% (81.2% ee, R)
P
P
H3C Ph
Ph CH3
(S,S)-1,2-(MePPh)2Ph: 17.8% (54.1% ee, R)
Duphos:
(R,R)-Me-Duphos: 99% (54.1% ee, R)(R,R)-iPr-Duhpos: 9.8% (8.7% ee, S)
P
P
R R
R R
PtBu
PtBu
H
H
(S)-Binapine: 4.8% (26.4% ee, R)
P
P
(R)-Binaphane: 4.1% (10.3% ee, S)N
N P
P
(R,R)-QuinoxP*: 4.2% (5.1% ee, R)
O
OP O
(R)-BINOL-P-OiPr:3.3% (3.8% ee, R)
P
P
tBu
tBu
HH
(SS!,RR!)-Tangphos: 27.9% (2.7% ee, R)
P
P
(R,R)-BenzP*: 14.4% (2.6% ee, R)

31
Table S3. Percent conversion and enantioselectivity of various chiral phosphines in combination with (py)2Co(CH2SiMe3)2 for the asymmetric hydrogenation of N-(1-phenylvinyl)acetamide.
Ligand %ee product
conversion (%)
catASium MN MesF(R)
83.1% (S) 36.8%
SL-J688-1
81.2% (R) 6.2%
(S,S)-1,2-(Me,PhP)2Ph
54.1% (R) 17.8%
(R,R)-Me-Duphos
31.1% (R) 99.0%
(S)-Binapine
26.4% (S) 4.8%
(R)-Binaphane
10.3% (R) 4.1%
P
PN
O
OF3C
F3C
FePH PO
P
P
H3C Ph
Ph CH3
P
P
PtBu
PtBu
H
H
PP

32
(R,R)-iPr-Duphos
8.7% (S) 9.8%
(R,R)-QuinoxP*
5.1% (R) 4.2%
(R)-BINOL-P-OiPr
3.8% (R) 3.3%
(S,S′,R,R′)-Tangphos
2.7% (R) 27.9%
(R,R)-BenzP*
2.6% (R) 14.4%
P
P
iPr iPr
iPr iPr
N
N P
P
O
OP O
P
P
tBu
tBu
HH
P
P
33
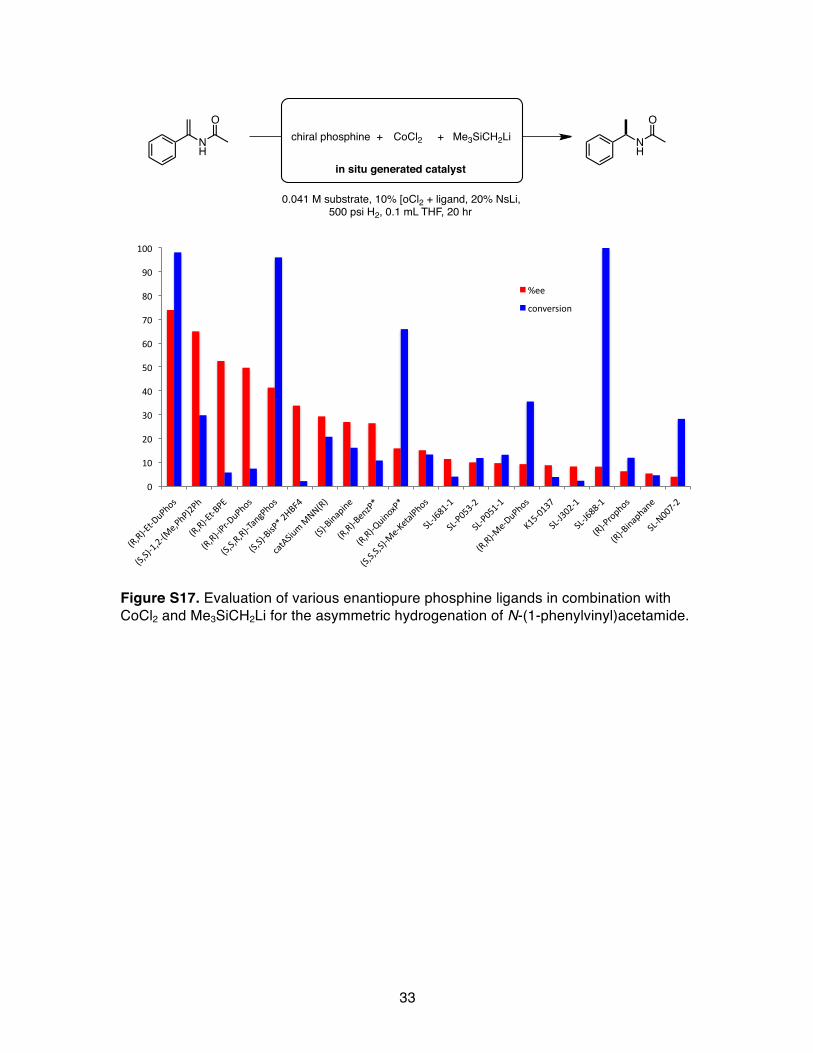
Figure S17. Evaluation of various enantiopure phosphine ligands in combination with CoCl2 and Me3SiCH2Li for the asymmetric hydrogenation of N-(1-phenylvinyl)acetamide.
chiral phosphine +
in situ generated catalyst
0.041 M substrate, 10% [oCl2 + ligand, 20% NsLi, 500 psi H2, 0.1 mL THF, 20 hr
NH
O
NH
OCoCl2 + Me3SiCH2Li
!"
#!"
$!"
%!"
&!"
'!"
(!"
)!"
*!"
+!"
#!!"
,-.-/0120345678"
,9.9/0#.$0,:
;.565/$56"
,-.-/0120<51"
,-.-/0=5>0345678"
,9.9.-.-/0?@AB5678"
,9.9/0<=85C"$D<E&"
F@2G9=4H":II,-/"
,9/0<=A@J=A;"
,-.-/0<;AK5C"
,-.-/0L4=A7M5C"
,9.9.9.9/0:;0N;2@O5678"
9P0Q(*#0#"
9P05!'%0$"
9P05!'#0#"
,-.-/0:;0345678"
N#'0!#%)"
9P0Q%!$0#"
9P0Q(**0#"
,-/05>7J678"
,-/0<=A@J6@A;"
9P0I!!)0$"
R;;"
F7AS;>8=7A"

34
Figure S18. Evaluation of various enantiopure phosphine ligands in combination with CoCl2 and Me3SiCH2Li for the asymmetric hydrogenation of N-(1-phenylvinyl)acetamide.
chiral phosphine +
in situ generated catalyst
0.041 M substrate, 10% [oCl2 + ligand, 20% NsLi, 500 psi H2, 0.1 mL THF, 20 hr
PtBu
PtBu
H
H
(S)-Binapine: 16.1% (26.9% ee, R)
NH
O
NH
OCoCl2 + Me3SiCH2Li
NN
P
P
Duphos:
(R,R)-Me-Duphos: 35.5% (9.2%, S)(R,R)-Et-Duphos: 98.1% (73.9% ee, R)(R,R)-iPr-Duphos: 7.3% (49.7% ee, S)
P
P
R R
R R
(R,R)-Et-BPE: 5.7% (52.5% ee, R)
P
P
H3C Ph
Ph CH3
(S,S)-1,2-(MePPh)2Ph: 64.9% (29.7% ee, R)
P
P
tBu
tBu
HH
(SS!,RR!)-Tangphos: 96.0% (41.4% ee, R)
P
P
tBu
tBu
(S,S)-BisP* 2HBF4: 2.1% (33.8% ee, S)(ligand treated with 2 eq. KOtBu prior to use)
2 HBF4
O
O
catASium MNN(R):20.7% (29.2% ee, S)
P
P
R R
R R
P
P
(R,R)-BenzP*: 10.7% (26.4% ee, S)
N
N P
P
(R,R)-QuinoxP*: 65.9% (15.8% ee, R)
PPO
OO
O
(S,S,S,S)-Me-ketalphos: 13.2% (15.0% ee, S)
FeHP PR2
R2
R1
O
JoSPOphos:
SL-J681-1: 4.0% (11.4% ee, R)SL-J688-1: >99% (8.2% ee, S)
FeP PPh2
SL-P051-1 (R,R,R)-Me-Kephos: 13.1% (-9.7% R)SL-P053-2 (S,S,R)-Me-Kephos: 11.8% (9.9% ee, S)
PPh2
NH2(1R,2R)-2-(diphenylphosphino)cyclohexanamine:3.8% (8.7% ee, S)
FeP P
SL-J302-1: 2.3% (8.2% ee, S)
PPh2
PPh2
(R)-Prophos: 11.9% (6.2% ee, R)
PP
(R)-Binaphane: 4.6% (5.3% ee, S)
Fe
PN
O
F3C
F3CSL-N007-2: 28.2% (3.9% ee, S)

35
Table S4. Percent conversion and enantioselectivity of various chiral phosphines in combination with CoCl2 and Me3SiCH2Li for the asymmetric hydrogenation of N-(1-phenylvinyl)acetamide.
Ligand %ee product
conversion (%)
(R,R)-Et-Duphos
73.9% (R) 98.1%
(S,S)-1,2-(Me,PhP)2Ph
64.9% (R) 29.7%
(R,R)-Et-BPE
52.5% (R) 5.7%
(R,R)-iPr-Duphos
49.7% (S) 7.3%
(S,S′,R,R′)-Tangphos
41.4% (R) 96.0%
(S,S)-BisP*-2HBF4 (treated with 2 eq. KOtBu prior to use)
33.8% (S) 2.1%
catASium MNN(R) 29.2% (S) 20.7%
P
P
P
H3C Ph
Ph CH3
P
P
P
iPr iPr
iPriPr
P
P
tBu
tBu
HH
P
P
tBu
tBu
2 HBF4

36
(S)-Binapine
26.9% (R) 16.1%
(R,R)-BenzP*
26.4% (S) 10.7%
(R,R)-QuinoxP*
15.8% (R) 65.9%
(S,S,S,S)-MeKetalPhos
15.0% (S) 13.2%
SL-J681-1
11.4% (R) 4.0%
SL-P053-2
9.9% (S) 11.8%
NN
P
P
O
O
PtBu
PtBu
H
H
P
P
N
N P
P
PPO
OO
O
FeHP PO
FeP PPh2

37
SL-P051-1
9.7% (R) 13.1%
(R,R)-MeDuphos
9.2% (S) 35.5%
(1R,2R)-2-(diphenylphosphino)cyclohexanamine
8.7% (S) 3.8%
SL-J302-1
8.2% (S) 2.3%
SL-J688-1
8.2% (S) >99%
(R)-Prophos
6.2% (R) 11.9%
(R)-Binaphane
5.3% (S) 4.6%
FeP PPh2
P
P
PPh2
NH2
FePP
FePH PO
PPh2
PPh2
PP

38
SL-N007-2
3.9% (S) 28.2%
Fe
PN
O
F3C
F3C
39
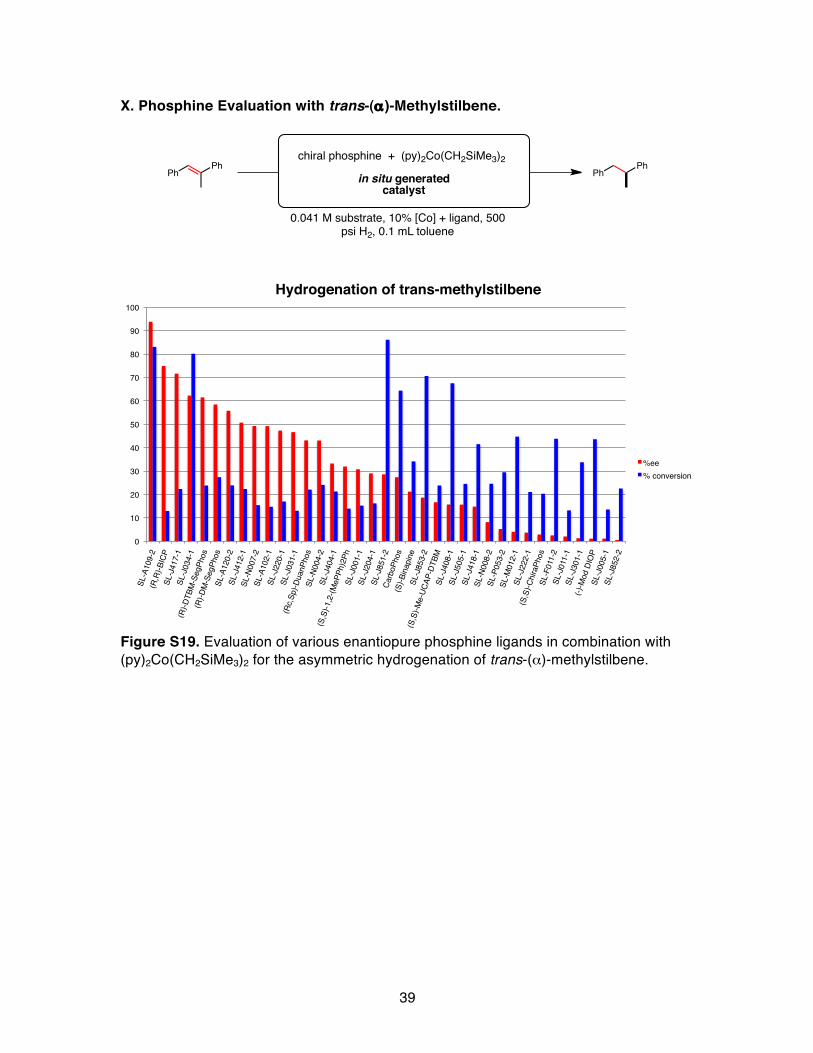
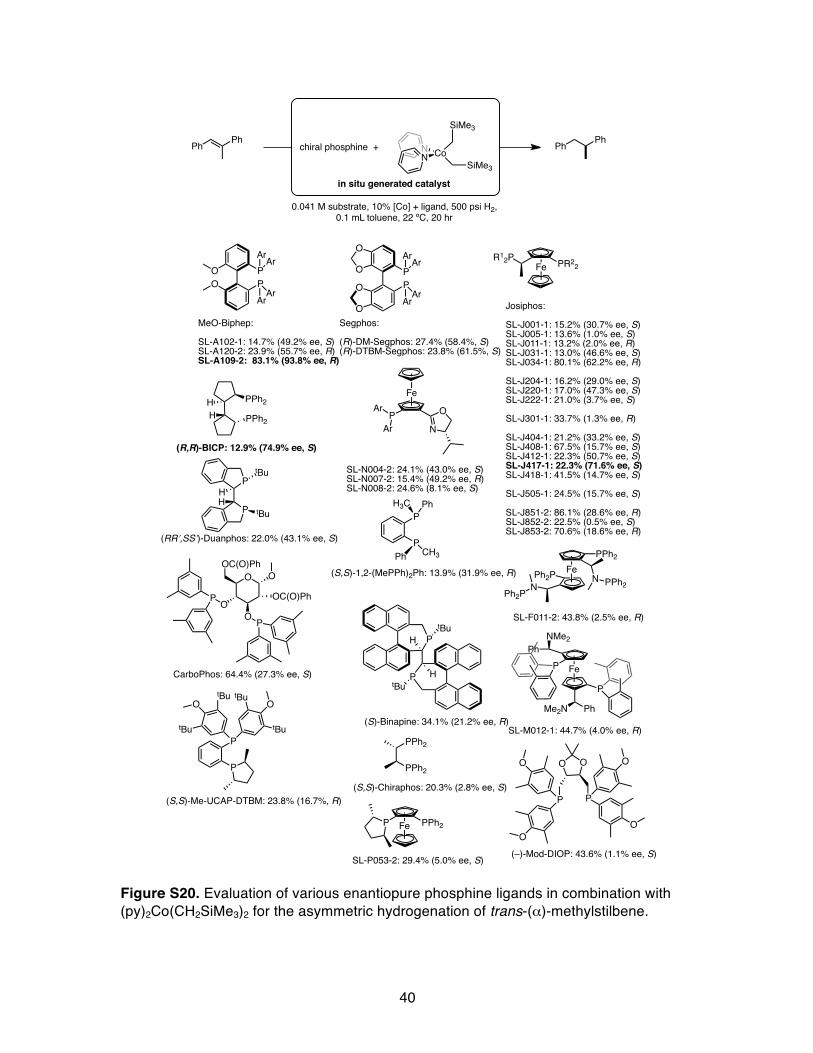
X. Phosphine Evaluation with trans-(α)-Methylstilbene.
Figure S19. Evaluation of various enantiopure phosphine ligands in combination with (py)2Co(CH2SiMe3)2 for the asymmetric hydrogenation of trans-(α)-methylstilbene.
chiral phosphine + (py)2Co(CH2SiMe3)2
0.041 M substrate, 10% [Co] + ligand, 500 psi H2, 0.1 mL toluene
in situ generated catalyst
PhPh
PhPh
0!
10!
20!
30!
40!
50!
60!
70!
80!
90!
100!
SL-A
109-
2!(R
,R)-B
ICP!
SL-J
417-
1!SL
-J03
4-1!
(R)-D
TBM
-Seg
Phos!
(R)-D
M-S
egPh
os!
SL-A
120-
2!SL
-J41
2-1!
SL-N
007-
2!SL
-A10
2-1!
SL-J
220-
1!SL
-J03
1-1!
(Rc,S
p)-D
uanP
hos!
SL-N
004-
2!SL
-J40
4-1!
(S,S
)-1,2
-(MeP
Ph)2
Ph!
SL-J
001-
1!SL
-J20
4-1!
SL-J
851-
2!Ca
rboP
hos!
(S)-B
inapin
e!SL
-J85
3-2!
(S,S
)-Me-
UCAP
-DTB
M!
SL-J
408-
1!SL
-J50
5-1!
SL-J
418-
1!SL
-N00
8-2!
SL-P
053-
2!SL
-M01
2-1!
SL-J
222-
1!(S
,S)-C
hiraP
hos!
SL-F
011-
2!SL
-J01
1-1!
SL-J
301-
1!(-)
-Mod
DIO
P!SL
-J00
5-1!
SL-J
852-
2!
Hydrogenation of trans-methylstilbene!
%ee!% conversion!
40
Figure S20. Evaluation of various enantiopure phosphine ligands in combination with (py)2Co(CH2SiMe3)2 for the asymmetric hydrogenation of trans-(α)-methylstilbene.
N CoN
SiMe3
SiMe3
chiral phosphine +
in situ generated catalyst
Ph Ph Ph Ph
0.041 M substrate, 10% [Co] + ligand, 500 psi H2, 0.1 mL toluene, 22 ºC, 20 hr
OO
PPAr
ArAr
Ar
PPAr
ArAr
ArO
O
O
O
MeO-Biphep:
SL-A102-1: 14.7% (49.2% ee, S) SL-A120-2: 23.9% (55.7% ee, R)SL-A109-2: 83.1% (93.8% ee, R)
Segphos:
(R)-DM-Segphos: 27.4% (58.4%, S)(R)-DTBM-Segphos: 23.8% (61.5%, S)
P
P
tBu
tBu
HH
(RR!,SS!)-Duanphos: 22.0% (43.1% ee, S)
Fe
PAr
Ar N
O
SL-N004-2: 24.1% (43.0% ee, S)SL-N007-2: 15.4% (49.2% ee, R)SL-N008-2: 24.6% (8.1% ee, S)
Fe PR22R12P
Josiphos:
SL-J001-1: 15.2% (30.7% ee, S)SL-J005-1: 13.6% (1.0% ee, S)SL-J011-1: 13.2% (2.0% ee, R)SL-J031-1: 13.0% (46.6% ee, S)SL-J034-1: 80.1% (62.2% ee, R)
SL-J204-1: 16.2% (29.0% ee, S)SL-J220-1: 17.0% (47.3% ee, S)SL-J222-1: 21.0% (3.7% ee, S)
SL-J301-1: 33.7% (1.3% ee, R)
SL-J404-1: 21.2% (33.2% ee, S)SL-J408-1: 67.5% (15.7% ee, S)SL-J412-1: 22.3% (50.7% ee, S)SL-J417-1: 22.3% (71.6% ee, S)SL-J418-1: 41.5% (14.7% ee, S)
SL-J505-1: 24.5% (15.7% ee, S)
SL-J851-2: 86.1% (28.6% ee, R)SL-J852-2: 22.5% (0.5% ee, S)SL-J853-2: 70.6% (18.6% ee, R)
PPh2HPPh2
H
(R,R)-BICP: 12.9% (74.9% ee, S)
P
P
H3C Ph
Ph CH3
(S,S)-1,2-(MePPh)2Ph: 13.9% (31.9% ee, R)O OOC(O)Ph
OC(O)Ph
O P
OP
CarboPhos: 64.4% (27.3% ee, S) PtBu
PtBu
H
H
(S)-Binapine: 34.1% (21.2% ee, R)
P
P
tBu
tBu tBu O
tBu
O
(S,S)-Me-UCAP-DTBM: 23.8% (16.7%, R)
FeP PPh2
SL-P053-2: 29.4% (5.0% ee, S)
FePPh
NMe2
P
Me2N Ph
SL-F011-2: 43.8% (2.5% ee, R)
PPh2
PPh2
(S,S)-Chiraphos: 20.3% (2.8% ee, S)
FePh2PNPh2P
PPh2
N PPh2
OO
PP
O
OO
O
(–)-Mod-DIOP: 43.6% (1.1% ee, S)
SL-M012-1: 44.7% (4.0% ee, R)

41
Table S5. Percent conversion and enantioselectivity of various chiral phosphines in combination with (py)2Co(CH2SiMe3)2 for the asymmetric hydrogenation of trans-(α)-methylstilbene.
Ligand %ee product conversion (%)
SL-A109-2
93.8 (R) 83.1
(R,R)-BICP
74.9 (S) 12.9
SL-J417-1
71.6 (S) 22.3
SL-J034-1
62.2 (R) 80.1
(R)-DTBM-SegPhos
61.5 (S) 23.8
OO
PP
OMetButBu
OMetBu tBu
Ar
Ar
PPh2HPPh2H
FePP
Fe PPh2P
Me3Si
PP
OMetButBu
OMetBu tBu
Ar
ArO
O
O
O

42
(R)-DM-SegPhos
58.4 (S) 27.4
SL-A120-2
55.7 (R) 23.9
SL-J412-1
50.7 (S) 22.3
SL-N007-2
49.2 (R) 15.4
SL-A102-1
49.2 (S) 14.7
PP
Ar
ArO
O
O
O
OO
PP
Ar
Ar
FePP
CF3
CF3
CF3F3C
Fe
PF3C
CF3
F3C CF3
N
O
OO
PP
Ar
Ar

43
SL-J220-1
47.3 (S) 17.0
SL-J031-1
46.6 (S) 13.0
(Rc,Sp)-DuanPhos
43.1 (S) 22.0
SL-N004-2
43.0 (S) 24.1
SL-J404-1
33.2 (S) 21.2
(S,S)-1,2-(MePPh)2Ph
31.9 (R) 13.9
FeP P
O
O
FePP
P
P
tBu
tBu
HH
Fe
PN
O
Fe PP
P
P
H3C Ph
Ph CH3

44
SL-J001-1
30.7 (S) 15.2
SL-J204-1
29.0 (S) 16.2
SL-J851-2
28.6 (R) 86.1
CarboPhos
27.3 (S) 64.4
(S)-Binapine
21.2 (R) 34.1
Fe PPh2P
FePP
F3C
F3C
FePh2P
PCy2
PPh2
PCy2
O OOC(O)Ph
OC(O)Ph
O P
OP
PtBu
PtBu
H
H

45
SL-J853-2
18.6 (R) 70.6
(S,S)-Me-UCAP-DTBM
16.7 (R) 23.8
SL-J408-1
15.7 (S) 67.5
SL-J505-1
15.7 (S) 24.5
SL-J418-1
14.7 (S) 41.5
FeP
PtBu2
P
PtBu2
O
O
O
O
P
P
tBu
tBu tBu O
tBu
O
FePP
FePPtBu
tBu
FeP P
O
O

46
SL-N008-2
8.1 (S) 24.6
SL-P053-2
5.2 (S) 29.4
SL-M012-1
4.0 (R) 44.7
SL-J222-1
3.7 (S) 21.0
(S,S)-ChiraPhos
2.8 (S) 20.3
SL-F011-2
2.5 (R) 43.8
SL-J011-1
2.0 (R) 13.2
Fe
PN
O
O
O
FeP PPh2
FeP
PhNMe2
P
Me2N Ph
FeP P
O
O
tBu
tBu
PPh2
PPh2
FePh2PN
Ph2P
PPh2
N PPh2
FePP
F3C
F3C
tBu
tBu

47
SL-J301-1
1.3 (R) 33.7
(-)-Mod DIOP
1.1 (S) 43.6
SL-J005-1
1.1 (S) 13.6
SL-J852-2
0.5 (S) 22.5
FePP tBu
tBu
O
O
P
P
O O
OO
FePP
Fe
PPh2
PPh2
P P

48
Figure S21. Evaluation of various enantiopure phosphine ligands in combination with CoCl2 and two equivalents of LiCH2SiMe3 for the asymmetric hydrogenation of trans-(α)-methylstilbene.
chiral phosphine +
in situ generated catalyst
0.041 M substrate, 10% CoCl2 + ligand, 20% LiCH2SiMe3, 500 psi H2, 0.1 mL THF, 24 hr
CoCl2 + LiCH2SiMe3
!"
#!"
$!"
%!"
&!"
'!"
(!"
)!"
*!"
+!"
#!!"
SL-A104-1!
SL-J222-1!
(R,R)-BenzP*!
SL-J605-1!
(S,S,R,R)-TangPhos!
SL-J004-1!
(R,R)-iPr-DuPhos!
(S)-DTBM-Garphos!
(R,R)-iPr-BPE!
SL-J001-1!
(S)-Binapine!
SL-J203-2!
(R,R)-Me-Ferrolane!
SL-J688-1!
SL-J408-1!
SL-J220-1!
(S)-C5-TunePhos!
SL-P051-1!
SL-J005-1!
SL-P053-2!
,--"
,"./01-234/0"

49
Figure S22. Evaluation of various enantiopure phosphine ligands in combination with CoCl2 and two equivalents of LiCH2SiMe3 for the asymmetric hydrogenation of trans-(α)-methylstilbene.
chiral phosphine +
in situ generated catalyst
0.041 M substrate, 10% CoCl2 + ligand, 20% LiCH2SiMe3, 500 psi H2, 0.1 mL THF, 24 hr
CoCl2 + LiCH2SiMe3
OO P
MeOOMe
OMeOMe
OMe
OMeP
MeOOMe
OMeOMe
OMe
OMe
SL-A104-1: 17.4% (-70.3% ee)
Josiphos:
SL-J001-1: 6.9% (32.5% ee)SL-J004-1: 7.1% (-49.7% ee)SL-J203-2: 6.2% (17.0% ee)SL-J220-1: 11.4% (-9.0% ee)SL-J222-1: 7.3% (69.8% ee)SL-J408-1: 7.0% (-10.4% ee)
Fe PR22R12P
P
P
(R,R)-BenzP*: 11.1% (-60.0% ee)
FeP PCy2
Ph
SL-J605-1: 38.4% (-57.8% ee)
P
P
tBu
tBu
HH
(SS!,RR!)-Tangphos: 6.2% (-51.8% ee)
P
P
iPr iPr
iPriPr
(R,R)-iPrDuphos:7.6% (37.1% ee)
OO P
t-BuOMe
t-But-Bu
OMe
t-BuP
t-Bu OMet-But-Bu
OMe
t-Bu
MeO
MeO
(S)-DTBM-Garphos:7.7% (34.6% ee)
P
P
iPr iPr
iPriPr
(R,R)-iPrBPE:7.5% (33.5% ee)
PtBu
PtBu
H
H
(S)-Binapine: 11.5% (18.1% ee)
FeP
P
(R,R)-Me-Ferrolane:8.6% (-14.7% ee )
FePH P
Ph
O
SL-J688-1: 13.8% (-13.2% ee)
OO P
P (S)-C5-TunePhos: 6.0% (7.4% ee)
FeP PPh2
SL-P051-1 (R,R,R)-Me-Kephos: 7.4% (4.2% ee)SL-P053-2 (S,S,R)-Me-Kephos: 7.0% (2.9% ee)

50
Table S6. Percent conversion and enantioselectivity of various chiral phosphines in combination with CoCl2 and two equivalents of LiCH2SiMe3 for the asymmetric hydrogenation of trans-(α)-methylstilbene.
Ligand %ee product conversion (%)
SL-A104-1
-70.3% 17.4%
SL-J222-1
69.8% 7.3%
(R,R)-BenzP*
-60.0% 11.1%
SL-J605-1
-57.8% 38.4%
(SS′,RR′)-TangPhos
-51.8% 6.2%
SL-J004-1
-49.7% 7.1%
OO P
MeOOMe
OMeOMe
OMe
OMeP
MeOOMe
OMeOMe
OMe
OMe
FeP P
MeO
MeO
P
P
FePPCy2
Ph
P
P
tBu
tBu
HH
FePPPh
Ph

51
(R,R)-iPrDuphos
37.1% 7.6%
(S)-DTBM-Garphos
34.6% 7.7%
(R,R)-iPrBPE
33.5% 7.5%
SL-J001-1
32.5% 6.9%
(S)-Binapine
18.1% 11.5%
SL-J204-1
17.0% 6.2%
P
P
iPr iPr
iPriPr
OO P
t-BuOMe
t-But-Bu
OMe
t-BuP
t-Bu OMet-But-Bu
OMe
t-Bu
MeO
MeO
P
P
iPr iPr
iPriPr
FePPPh
Ph
PtBu
PtBu
H
H
FePP
F3C
F3C

52
(R,R)-Me-Ferrolane
-14.7% 8.6%
SL-J688-1
-13.2% 13.8%
SL-J408-1
-10.4% 7.0%
SL-J220-1
-9.0% 11.4%
(S)-C5-TunePhos
7.1% 6.0%
SL-P051-1
4.2% 7.4%
FeP
P
FePH P
Ph
O
FePP
FeP P
MeO
MeO
OO P
P
FeP PPh2

53
SL-J005-1
4.0% 5.3%
SL-P053-2
2.9% 7.0%
FePP
FeP PPh2

54
XI. Evaluation of Cobalt Sources for the Hydrogenation of MAC.
Figure S23. Evaluation of various cobalt sources and additives for the asymmetric hydrogenation of MAC.
0.0 (--)!
0.0!(--)!
0.0!(--)!
>99 (38.4)!
0.0 (--)!
0.0!(--)!
0.0 (--)!
64.8 (85.9)!
0.0!(--)!
>99 (90.7)!
>99 (84.7)!
>99 (78.1)!
>99 (40.4)!
>99 (76.6)!
>99 (84.9)!
>99 (86.1)!
>99 (80.6)!
0.0!(--)!
>99 (80.2)!
96.5 (83.7)!
92.3 (67.8)!
46.8 (81.5)!
29.5 (70.0)!
>99 (87.9)!
>99 (83.6)!
>99 (84.9)!
0.0!(--)!
96.8 (75.8)!
49.4 (87.7)!
81.4 (86.4)!
>99 (70.3)!
33.0 (89.0)!
>99 (89.5)!
>99 (84.7)!
>99 (95.6)!
0.0 !(--)!
0.0 (--)!
0.0!(--)!
>99 (67.1)!
92.6 (81.7)!
>99 (77.4)!
0.0!(--)!
>99 (77.2)!
0.0!(--)!
>99 (-5.3)!
93.0 (93.5)!
>99 (43.5)!
95.2 (68.3)!
>99 (80.4)!
17.2 (88.0)!
>99 (51.5)!
98.3 (90.4)!
>99 (65.4)!
51.4 (6.4)!
97.0 (95.6)!
>99 (24.3)!
>99 (66.5)!
97.7 (68.5)!
>99 (83.3)!
>99 (61.0)!
>99 (93.0)!
>99 (41.6)!
0.0!(--)!
91.4 (57.5)!
92.4 (72.9)!
85.4 (39.4)!
94.9 (73.7)!
94.5 (52.8)!
93.3 (48.5)!
96.1 (86.2)!
>99 (96.4)!
94.4 (87.8)!
94.5 (86.8)!
>99 (47.2)!
>99 (46.8)!
>99 (67.6)!
54.5 (85.4)!
>99 (-28.6)!
>99 (84.0)!
>99 (55.3)!
>99 (88.4)!
>99 (85.5)!
0.0!(--)!
0.0!(--)!
>99 (81.3)!
0.0 (--)!
>99 (84.1)!
0.0 (--)!
>99 (91.6)!
>99 (91.3)!
>99 (38.4)!
>99 (90.0)!
>99 (82.8)!
>99 (23.9)!
>99 (77.4)!
>99 (72.5)!
>99 (83.6)!
>99 (79.4)!
>99 (88.2)!
>99 (91.9)!
>99 (92.4)!
>99 (93.5)!
>99 (89.0)!
>99 (87.3)!
>99 (92.1)!
>99 (89.4)!
>99 (91.4)!
>99 (83.6)!
Me 3SiCH 2L
i!Me 3S
iCH 2MgC
l!MeM
gCl!
Me 3Al!
PhMgB
r!
Et 2Zn!
i PrMgC
l!2-F
-BnZnC
l!PhZ
nCl!
No add
itive!
CoF2!
CoCl2!
CoBr2!
CoI2!
Co(acac)3!
Co(OAc)2!
Co(OBz)2!
(py)2Co(CH2SiMe3)2!
Co(OAc)2*4H2O!
Co(BF4)2*6H2O!
CoCl2*6H2O!
Co(ClO4)2*6H2O!
>90% ee!
80-90% ee!
70-80% ee!
<70% ee!

55
XII. GC and SFC Chromatograms of Enantiopure Alkanes.
Figure S24. Hydrogenation of MAC with (R,R)-QuinoxP*Co(CH2SiMe3)2.
Figure S25. Hydrogenation of (Z)-methyl 2-acetamido-3-phenylacrylate with (R,R)-iPrDuPhosCo(CH2SiMe3)2.
N
N P
PCo
CH2SiMe3
CH2SiMe3
4 atm H2
0.041 M substrate, 1% [Co], 8 mL THF, 18 hr, 25 ºC
NH
OO
ONH
OO
O>99% conversion, 99.0% ee
P
P
iPr iPr
iPriPr
CoCH2SiMe3
CH2SiMe3
500 psi H20.041 M substrate, 5% [Co], 8 mL THF, 12 hr, 25 ºC
HNO
O
O
HNO
O
O
>99% conversion, 92.7% ee

56
Figure S26. Hydrogenation of α-acetamidostyrene with (R,R)-EtDuPhosCo(CH2SiMe3)2.
P
P
Et Et
EtEt
CoCH2SiMe3
CH2SiMe3
500 psi H20.041 M substrate, 5% [Co], 8 mL THF, 6 hr, 25 ºC
NH
O
NH
O
>99% conversion, 82.0% ee

57
Figure S27. Hydrogenation of α-trans-methylstilbene with SL-A109-2 and (py)2Co(CH2SiMe3)2.
OO P
tBuOMe
tButBu
OMe
tBuP
tBuOMe
tButBu
OMe
tBu N CoN
SiMe3
SiMe3
0.041 M substrate, 5% [Co], 8 mL PhMe, 1 hr, 25 ºC
500 psi H2
>98% conversion, 89.4% ee

References and Notes 1. W. S. Knowles, Asymmetric hydrogenations (Nobel lecture) Angew. Chem. Int. Ed. 41, 1998
(2002). doi:10.1002/1521-3773(20020617)41:12<1998::AID-ANIE1998>3.0.CO;2-8
2. R. Noyori, Asymmetric catalysis: Science and opportunities (Nobel lecture) Angew. Chem. Int. Ed. 41, 2008 (2002). doi:10.1002/1521-3773(20020617)41:12<2008::AID-ANIE2008>3.0.CO;2-4
3. C. A. Busacca, D. R. Fandrick, J. J. Song, C. H. Senanayake, The growing impact of catalysis in the pharmaceutical industry. Adv. Synth. Catal. 353, 1825–1864 (2011). doi:10.1002/adsc.201100488
4. H.-U. Blaser, B. Pugin, F. Spindler, M. Thommen, From a chiral switch to a ligand portfolio for asymmetric catalysis. Acc. Chem. Res. 40, 1240–1250 (2007). doi:10.1021/ar7001057 Medline
5. W. S. Knowles, M. J. Sabacky, B. D. Vineyard, Chem. Commun. 1972, 10 (1972).
6. G. Hoge, Synthesis of both enantiomers of a P-chirogenic 1,2-bisphospholanoethane ligand via convergent routes and application to rhodium-catalyzed asymmetric hydrogenation of CI-1008 (pregabalin). J. Am. Chem. Soc. 125, 10219–10227 (2003). doi:10.1021/ja034715o Medline
7. Y. Hsiao, N. R. Rivera, T. Rosner, S. W. Krska, E. Njolito, F. Wang, Y. Sun, J. D. Armstrong, 3rd, E. J. Grabowski, R. D. Tillyer, F. Spindler, C. Malan, Highly efficient synthesis of beta-amino acid derivatives via asymmetric hydrogenation of unprotected enamines. J. Am. Chem. Soc. 126, 9918–9919 (2004). doi:10.1021/ja047901i Medline
8. H. U. Blaser, H.-J. Federsel, Eds., Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions (Wiley-VCH, Weinheim, ed. 2, 2010).
9. C. S. Shultz, S. W. Krska, Unlocking the potential of asymmetric hydrogenation at Merck. Acc. Chem. Res. 40, 1320–1326 (2007). doi:10.1021/ar700141v Medline
10. N. B. Johnson, I. C. Lennon, P. H. Moran, J. A. Ramsden, Industrial-scale synthesis and applications of asymmetric hydrogenation catalysts. Acc. Chem. Res. 40, 1291–1299 (2007). doi:10.1021/ar700114k Medline
11. A. M. Rouhi, Chem. Eng. News 82, 47 (2004). doi:10.1021/cen-v082n024.p047
12. J. A. DiMasi, C. Paquette, The economics of follow-on drug research and development: Trends in entry rates and the timing of development. Pharmacoeconomics 22, (Suppl 2), 1–14 (2004). doi:10.2165/00019053-200422002-00002 Medline
13. A. M. Thayer, Centering on chirality. Chem. Eng. News 85, 11 (2007). doi:10.1021/cen-v085n016.p011
14. W. S. Knowles, M. J. Sabacky, J. Chem. Soc. Chem. Commun. 1445 (1968).
15. L. Horner, H. Siegel, H. Büthe, Asymmetric catalytic hydrogenation with an optically active phosphinerhodium complex in homogeneous solution. Angew. Chem. Int. Ed. Engl. 7, 942 (1968). doi:10.1002/anie.196809422
16. R. R. Schrock, J. A. Osborn, J. Am. Chem. Soc. 93, 2397 (1971). doi:10.1021/ja00739a006

17. J. Halpern, Mechanism and stereoselectivity of asymmetric hydrogenation. Science 217, 401–407 (1982). doi:10.1126/science.217.4558.401
18. M. T. Ashby, J. Halpern, Kinetics and mechanism of catalysis of the asymmetric hydrogenation of. alpha.beta.-unsaturated carboxylic acids by bis(carboxylato) 2,2′-bis(diphenylphosphino)-1,1′-binaphthylruthenium(II), [RuII(BINAP) (O2CR)2]. J. Am. Chem. Soc. 113, 589–594 (1991). doi:10.1021/ja00002a029
19. S. Bell, B. Wüstenberg, S. Kaiser, F. Menges, T. Netscher, A. Pfaltz, Asymmetric hydrogenation of unfunctionalized, purely alkyl-substituted olefins. Science 311, 642–644 (2006). doi:10.1126/science.1121977
20. J. Mazuela, P. O. Norrby, P. G. Andersson, O. Pàmies, M. Diéguez, Pyranoside phosphite-oxazoline ligands for the highly versatile and enantioselective ir-catalyzed hydrogenation of minimally functionalized olefins. A combined theoretical and experimental study. J. Am. Chem. Soc. 133, 13634–13645 (2011). doi:10.1021/ja204948k Medline
21. M. C. Perry, X. Cui, M. T. Powell, D. R. Hou, J. H. Reibenspies, K. Burgess, Optically active iridium imidazol-2-ylidene-oxazoline complexes: Preparation and use in asymmetric hydrogenation of arylalkenes. J. Am. Chem. Soc. 125, 113–123 (2003). doi:10.1021/ja028142b Medline
22. B. Plietker, Ed., Iron Catalysis in Organic Chemistry: Reactions and Applications (Wiley-VCH, Weinheim, 2008).
23. U. Leutenegger, A. Madin, A. Pfaltz, Enantioselective reduction of α,β-unsaturated carboxylates with NaBH4 and catalytic amounts of chiral cobalt semicorrin complexes. Angew. Chem. Int. Ed. Engl. 28, 60–61 (1989). doi:10.1002/anie.198900601
24. L. O. Nindakova, F. M. Lebed, Z. Y. Zamazei, B. A. Shainyan, New C 2-symmetric optically active salen ligands and their cobalt(II) complexes. Hydridoborate reduction of prochiral C=O and C=C bonds. Russ. J. Org. Chem. 43, 1322–1329 (2007). doi:10.1134/S1070428007090102
25. L. O. Nindakova, B. A. Shainyan, Borohydride reduction of acetophenone and esters of dehydrocarboxylic acids in the presence of chiral cobalt(II) diamine complexes. Russ. Chem. Bull. Int. Ed. 54, 348–353 (2005). doi:10.1007/s11172-005-0258-8
26. Y. Ogho, S. Takeuchi, Y. Natori, J. Yoshimura, Bull. Chem. Soc. Jpn. 54, 2124 (1981). doi:10.1246/bcsj.54.2124
27. A. Corma, M. Iglesias, C. del Pino, F. Sánchez, Optically active complexes of transition metals (RhI, RuII, CoII and NiII) with 2-aminocarbonylpyrrolidine ligands. Selective catalysts for hydrogenation of prochiral olefins. J. Organomet. Chem. 431, 233–246 (1992). doi:10.1016/0022-328X(92)80121-D
28. Q. Knijnenburg, A. D. Horton, H. Heijden, T. M. Kooistra, D. G. H. Hetterscheid, J. M. M. Smits, B. Bruin, P. H. M. Budzelaar, A. W. Gal, Olefin hydrogenation using diimine pyridine complexes of Co and Rh. J. Mol. Cat. A. 232, 151–159 (2005). doi:10.1016/j.molcata.2004.12.039

29. G. Zhang, B. L. Scott, S. K. Hanson, Mild and homogeneous cobalt-catalyzed hydrogenation of C=C, C=O, and C=N bonds. Angew. Chem. Int. Ed. 51, 12102–12106 (2012).doi:10.1002/anie.201206051 Medline
30. G. Zhang, K. V. Vasudevan, B. L. Scott, S. K. Hanson, Understanding the mechanisms of cobalt-catalyzed hydrogenation and dehydrogenation reactions. J. Am. Chem. Soc. 135, 8668–8681 (2013). doi:10.1021/ja402679a Medline
31. S. Monfette, Z. R. Turner, S. P. Semproni, P. J. Chirik, Enantiopure C1-symmetric bis(imino)pyridine cobalt complexes for asymmetric alkene hydrogenation. J. Am. Chem. Soc. 134, 4561–4564 (2012). doi:10.1021/ja300503k Medline
32. D. Zhu, F. F. B. J. Janssen, P. H. M. Budzelaar, (Py)2 Co(CH2SiMe3)2 as an easily accessible source of “CoR2”. Organometallics 29, 1897–1908 (2010). doi:10.1021/om901045s
33. M. J. Burk, J. E. Feaster, W. A. Nugent, R. L. Harlow, Preparation and use of C2-symmetric bis(phospholanes): Production of α-amino acid derivatives via highly enantioselective hydrogenation reactions. J. Am. Chem. Soc. 115, 10125–10138 (1993). doi:10.1021/ja00075a031
34. S. J. Roseblade, A. Pfaltz, Iridium-catalyzed asymmetric hydrogenation of olefins. Acc. Chem. Res. 40, 1402–1411 (2007). doi:10.1021/ar700113g Medline
35. P. G. Cozzi, N. Zimmermann, R. Hilgraf, S. Schaffner, A. Pfaltz, Chiral phosphinopyrrolyl-oxazolines: A new class of easily prepared, modular P,N-ligands. Adv. Synth. Catal. 343, 450–454 (2001). doi:10.1002/1615-4169(200107)343:5<450::AID-ADSC450>3.0.CO;2-7
36. X. Cui, K. Burgess, Catalytic homogeneous asymmetric hydrogenations of largely unfunctionalized alkenes. Chem. Rev. 105, 3272–3296 (2005). doi:10.1021/cr0500131 Medline
37. T. L. Church, P. G. Andersson, Iridium catalysts for the asymmetric hydrogenation of olefins with nontraditional functional substituents. Coord. Chem. Rev. 252, 513–531 (2008). doi:10.1016/j.ccr.2007.09.015
38. The last number in the name indicates the enantiomer of the ligand. SL-A109-1 corresponds to the (R) enantiomer, whereas SL-A109-2 is the (S) antipode.
39. S. Gischig, T. M. Schmid, G.Consiglio, http://webcsd.ccdc.cam.ac.uk/display_csd_search_results.php?xml_temp_file=/temp/text_numeric_query_041631900137230271451cbad7a67a84.xml&identifier=NALPIA
40. Similarly, performing the hydrogenation of trans-methylstilbene with 5 mol % each of SL-A109-2 and (py)2Co(CH2SiMe3)2 without removal of the volatiles, and hence in the presence of two equivalents of pyridine, lowered the conversion and enantioselectivity to 50 and 51%, respectively, indicating that incomplete removal of the volatile byproducts in catalyst generation could also be deleterious to overall performance.
41. A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen, F. J. Timmers, Safe and convenient procedure for solvent purification. Organometallics 15, 1518–1520 (1996). doi:10.1021/om9503712
42. S. K. Sur, J. Magn. Reson. 169, 82 (1989).

43. A. Lightfoot, P. Schnider, A. Pfaltz, Enantioselective Hydrogenation of olefins with iridium-phosphanodihydrooxazole catalysts. Angew. Chem. Int. Ed. 37, 2897–2899 (1998). doi:10.1002/(SICI)1521-3773(19981102)37:20<2897::AID-ANIE2897>3.0.CO;2-8
44. Y. Zheng, X. Li, C. Ren, D. Zhang-Negrerie, Y. Du, K. Zhao, Synthesis of oxazoles from enamides via phenyliodine diacetate-mediated intramolecular oxidative cyclization. J. Org. Chem. 77, 10353–10361 (2012). doi:10.1021/jo302073e Medline
45. M. van den Berg, R. M. Haak, A. J. Minnaard, A. H. M. de Vries, J. G. de Vries, B. L. Feringa, Rhodium/monophos-catalysed asymmetric hydrogenation of enamides. Adv. Synth. Catal. 344, 1003–1007 (2002). doi:10.1002/1615-4169(200210)344:9<1003::AID-ADSC1003>3.0.CO;2-M
46. Y.-G. Zhou, W. Tang, W.-B. Wang, W. Li, X. Zhang, Highly effective chiral ortho-substituted BINAPO ligands (o-BINAPO): Applications in Ru-catalyzed asymmetric hydrogenations of beta-aryl-substituted beta-(acylamino)acrylates and beta-keto esters. J. Am. Chem. Soc. 124, 4952–4953 (2002). doi:10.1021/ja020121u Medline