supplementary methods - media.nature.com · steady-state kinetics for the single-nucleotide...
TRANSCRIPT

Supplementary Methods
I. General methods and materials for synthesis
II. Synthesis of nucleotide derivatives of Ds and Pa
III. Synthesis of Bio-PaTP
IV. Steady-state kinetics for the single-nucleotide insertion experiments with KF
exo–
V. Sequences of DNA fragments for the PCR, sequencing, and transcription
experiments
VI. Dye terminator sequencing of the PCR products from DNA
VII. T7 transcription for 17-mer RNA fragments
VIII. Selectivity of the Ds-Pa pair in transcription
1

I. General methods and materials for synthesis
Reagents and solvents were purchased from standard suppliers and used without further
purification. Reactions were monitored by thin-layer chromatography (TLC) using 0.25
mm silica gel 60 plates impregnated with 254 nm fluorescent indicator (Merck). 1H NMR, 13C NMR, and 31P NMR spectra were recorded on JEOL EX270 and BRUKER (300-AVM
and AVANCE-600) magnetic resonance spectrometers. Nucleoside purification was
performed on a Gilson HPLC system with a preparative C18 column (Waters Microbond
Sphere, 150 × 19 mm). The triphosphate derivatives were purified with a DEAE-Sephadex
A-25 column (300 × 15 mm) and a C18 column (Synchropak RPP, 250 × 4.6 mm, Eichrom
Technologies). High resolution mass spectra (HRMS) and electrospray ionization mass
spectra (ESI-MS) were recorded on a JEOL HX-110 or JM 700 mass spectrometer and a
Waters micromass ZMD 4000 equipped with a Waters 2690 LC system, respectively. The
compounds for the deoxynucleoside derivatives of pyrrole-2-carbaldehyde1 and
4-propynylpyrrole-2-carbaldehyde2 were synthesized according to the previously reported
literature.
References
1. Mitsui, T., Kitamura, A., Kimoto, M., To, T., Sato, A., Hirao, I. & Yokoyama, S. An unnatural hydrophobic base pair with shape complementarity between pyrrole-2-carbaldehyde and 9-methylimidazo[(4,5)-b]pyridine. J. Am. Chem. Soc. 125, 5298-5307 (2003).
2. Mitsui, T., Kimoto, M., Sato, A., Yokoyama, S. & Hirao, I. An unnatural hydrophobic base, 4-propynylpyrrole-2-carbaldehyde, as an efficient pairing partner of 9-methylimidazo[(4,5)-b]pyridine. Bioorg. Med. Chem. Lett. 13, 4515-4518 (2003).
2
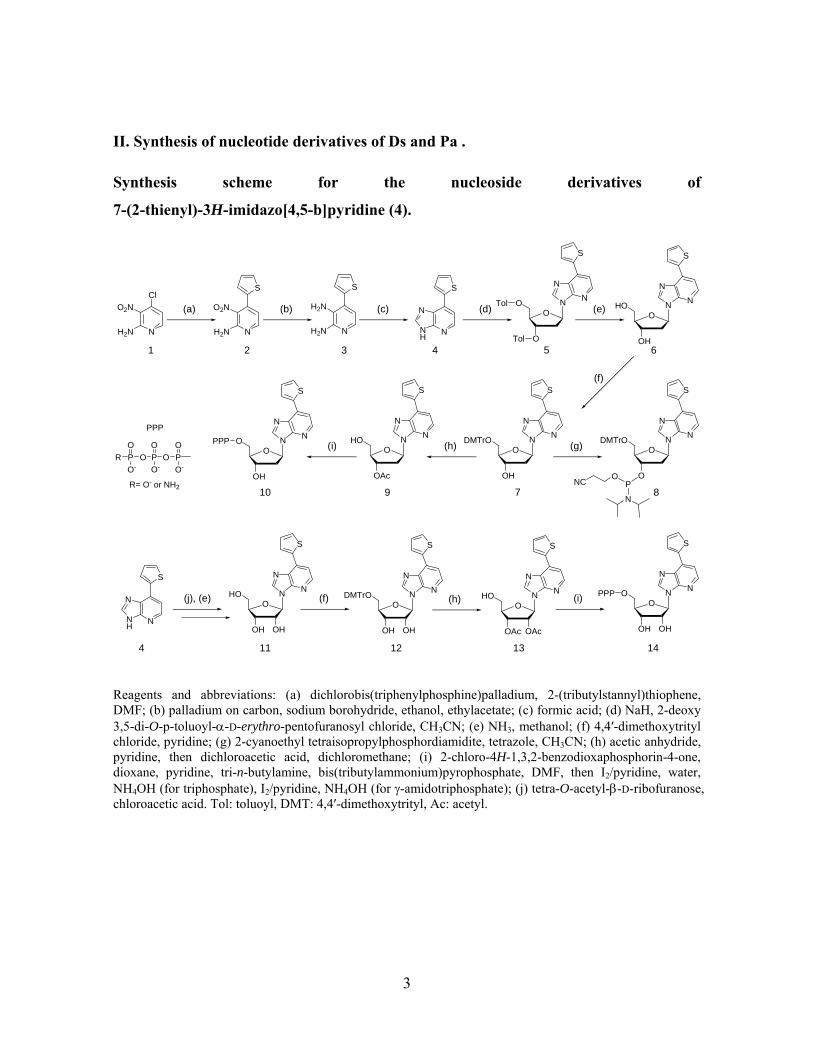
II. Synthesis of nucleotide derivatives of Ds and Pa .
Synthesis scheme for the nucleoside derivatives of
7-(2-thienyl)-3H-imidazo[4,5-b]pyridine (4).
N
O2N
H2N
Cl
N
O2N
H2N
S
N
H2N
H2N
S
O
OH
HO N
N
N
S
N
S
N
NH
O
O
O N
N
N
S
Tol
Tol
O
OH
DMTrO N
N
N
S
O
O
DMTrO N
N
N
S
P
N
ONC
O
OAc
HO N
N
N
S
O
OH
O N
N
N
S
PPP
P OOO
O-PPR
O
O- O-
O
PPP
R= O- or NH2
N
S
N
NH
O
OH
HO N
N
N
S
OH
O
OH
DMTrO N
N
N
S
OH
O
OAc
HO N
N
N
S
OAc
O
OH
O N
N
N
S
PPP
OH
(a) (b) (c) (d) (e)
(i) (h) (g)
(f)
(j), (e) (f) (h) (i)
1 2 3 4 5 6
7 8910
4 11 12 13 14
Reagents and abbreviations: (a) dichlorobis(triphenylphosphine)palladium, 2-(tributylstannyl)thiophene, DMF; (b) palladium on carbon, sodium borohydride, ethanol, ethylacetate; (c) formic acid; (d) NaH, 2-deoxy 3,5-di-O-p-toluoyl-α-D-erythro-pentofuranosyl chloride, CH3CN; (e) NH3, methanol; (f) 4,4′-dimethoxytrityl chloride, pyridine; (g) 2-cyanoethyl tetraisopropylphosphordiamidite, tetrazole, CH3CN; (h) acetic anhydride, pyridine, then dichloroacetic acid, dichloromethane; (i) 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one, dioxane, pyridine, tri-n-butylamine, bis(tributylammonium)pyrophosphate, DMF, then I2/pyridine, water, NH4OH (for triphosphate), I2/pyridine, NH4OH (for γ-amidotriphosphate); (j) tetra-O-acetyl-β-D-ribofuranose, chloroacetic acid. Tol: toluoyl, DMT: 4,4′-dimethoxytrityl, Ac: acetyl.
3
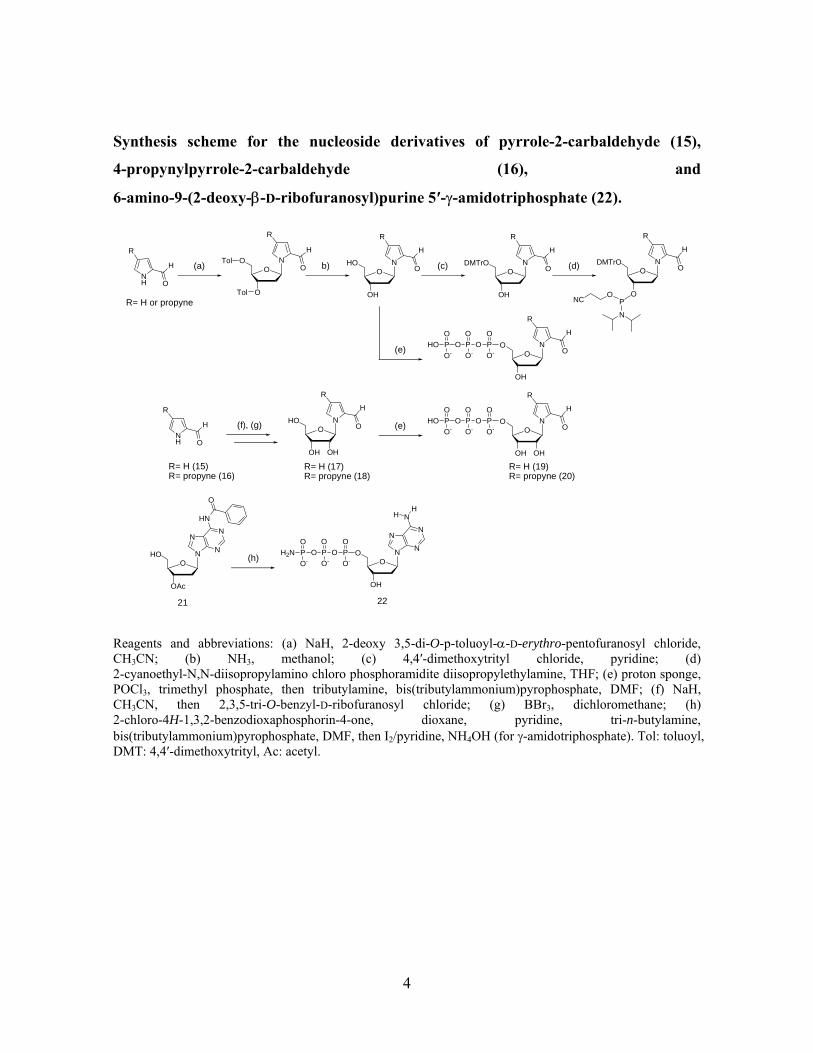
Synthesis scheme for the nucleoside derivatives of pyrrole-2-carbaldehyde (15),
4-propynylpyrrole-2-carbaldehyde (16), and
6-amino-9-(2-deoxy-β-D-ribofuranosyl)purine 5′-γ-amidotriphosphate (22).
O
OH
HO NO
O
O NTol
Tol
O
OAc
HO N
N
N
N
HN
b)
(h)
21
(a)
H
O
H
O
R R
O
OH
DMTrO N(c)
H
O
R
(d) O
O
DMTrO N
P
N
ONC
O
H
R
NH
H
O
R
R= H or propyne
(e) O
OH
O NP OOO
O-PPHO
O
O- O-
O H
O
R
O
OH
HO N
OH
R= H (17)R= propyne (18)
H
O
R
(f), (g)NH
H
O
R
R= H (15)R= propyne (16)
(e) O
OH
O NP OOO
O-PPHO
O
O- O-
O H
O
R
R= H (19)R= propyne (20)
O
O
OH
O N
N
N
NN
22
P OOO
O-PPH2N
O
O- O-
O
OH
HH
Reagents and abbreviations: (a) NaH, 2-deoxy 3,5-di-O-p-toluoyl-α-D-erythro-pentofuranosyl chloride, CH3CN; (b) NH3, methanol; (c) 4,4′-dimethoxytrityl chloride, pyridine; (d) 2-cyanoethyl-N,N-diisopropylamino chloro phosphoramidite diisopropylethylamine, THF; (e) proton sponge, POCl3, trimethyl phosphate, then tributylamine, bis(tributylammonium)pyrophosphate, DMF; (f) NaH, CH3CN, then 2,3,5-tri-O-benzyl-D-ribofuranosyl chloride; (g) BBr3, dichloromethane; (h) 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one, dioxane, pyridine, tri-n-butylamine, bis(tributylammonium)pyrophosphate, DMF, then I2/pyridine, NH4OH (for γ-amidotriphosphate). Tol: toluoyl, DMT: 4,4′-dimethoxytrityl, Ac: acetyl.
4

The synthesis of the nucleoside derivatives of 7-(2-thienyl)-3H-imidazo[4,5-b]pyridine (4)
was accomplished by the reactions described in the above scheme. The
7-(2-thienyl)-3H-imidazo[4,5-b]pyridine (4) was synthesized from
2-amino-3-nitro-4-chloro-pyridine (1)3 in 3 steps (72%). Deoxyribonucleoside 6 was
obtained by the coupling reaction of
1-chloro-2-deoxy-3,5-di-O-toluoyl-α-D-erythro-pentofranose4 with the sodium salt of 4,
followed by deprotection of the toluoyl groups of 5 with methanolic ammonia as a single
product in a 61% yield from 4. The ribonucleoside 11 was synthesized by a fusion coupling
reaction of tetra-O-acetyl-β-D-ribofuranose and 4 at 200°C with a catalytic amount of
chloroacetic acid, and was obtained as a single product in a 29% yield after deprotection by
methanolic ammonia and purification by RP-HPLC.
The structures of nucleosides 6 and 11 were confirmed by NMR and high resolution
mass spectroscopy. The aromatic proton peaks of compounds 6 and 11 showed the same
chemical shifts. The HMBC and HSQC spectra of compounds 6 and 11 revealed the
presence of an N-glycoside bond between the sugar C1′ and N-3 positions of the
imidazo[4,5-b]pyridine base moiety. Furthermore, we found that the thienyl moiety of
compounds 6 and 11 was attached at position 7 in the imidazo[4,5-b]pyridine ring,
according to the 2D NOESY and 2D HMBC spectra. The anomeric configurations of
compounds 6 and 11 were confirmed by the 2D NOESY and 1D NOE spectra
(Supplementary Data). The key results of the 1D NOE experiments were that the
irradiation of the H1′ proton gives 2% and 3% enhancement of the H4′ signal and 8% and
9% enhancement of the H2 signal of compounds 6 and 11, respectively. When the H2
proton was irradiated, enhancements of 9% and 10% of the H1′ signal and 3-5% of the H2′
and H3′ signals were obtained in the differential NOE experiments. Thus, the anomeric
configurations of compounds 6 and 11 were assigned as β, on the basis of NOE
experiments. Compound 6 was converted to the amidite by the conventional method, and
the trimer of d(Tp6pT) [d(TpDspT)] was confirmed by electrospray ionization mass
spectrometry (ESI-MS: see Supplementary Data). Nucleoside 5′-triphosphates 10 and 14,
5

as substrates for the enzymatic reaction, were synthesized by the previously reported
procedure5.
The syntheses of the nucleoside derivatives of pyrrole-2-carbaldehyde (15) and
4-propynylpyrrole-2-carbaldehyde (16) were accomplished by the reactions described in
the above scheme. The syntheses of the deoxynucleoside derivatives of 15 and 16 were
reported1,2. The ribonucleosides of 15 and 16 were synthesized by the coupling reaction of
2,3,5-tri-O-benzyl-D-ribofuranosyl chloride6 and the sodium salt of respective 15 and 16,
followed by a treatment with BBr3 to deprotect the benzyl groups to give the
ribonucleosides of 17 (15%) and 18 (7%) in 2 steps, respectively. The structures of 17 and
18 were confirmed by NMR and high resolution mass spectroscopy. The HMBC and
HSQC spectra of compounds 17 and 18 showed that the N-glycoside bond had formed
between the sugar and the pyrrole base moiety at the C1′ carbon (see Supplementary
Data). The anomeric configurations of 17 and 18 were confirmed by the NOE experiments
(differential NOE and NOESY spectra, Supplementary Data). The key results of the
differential NOE experiments are as follows. The irradiation of the H1′ proton gives a 3-4%
enhancement of the H4′ signal. When the H2′ (and/or H3′) proton was irradiated, 9-10%
enhancement of the H5 signal was obtained. The NOESY spectra of compounds 17 and 18
showed cross peaks between H1′ and H4′, between H1′ and the CHO proton, and between
H5 and H2′ (and/or H3′). Thus, the anomeric configurations of 17 and 18 were assigned as
β, on the basis of the NOE NMR studies. The ribonucleosides of 17 and 18 were converted
to the ribonucleside 5′-triphosphates by the conventional method7.
The nucleoside 5′-triphosphates were synthesized according to the literature5,7. The
synthesis of 5′-γ-amidotriphosphate was accomplished by a slight modification of the
conventional method5. Protected nucleosides 9 and 218 were phosphitylated by
2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one. The protected nucleoside phosphites were
converted to the P2,P3-dioxo-P1-5′-nucleosidylcyclotriphosphites by a treatment with
pyrophosphate. After oxidation with iodine/water, the resulting 5′-trimetaphosphate was
treated with concentrated ammonia to give the nucleoside of 5′-γ-amidotriphosphate. The
6

purification of the 5′-γ-amidotriphosphates was carried out by anion exchange DEAE
Sephadex column chromatography and RP-HPLC. The DEAE column elution pattern and
the electrospray ionization mass spectrum of deoxyadenosine 5′-γ-amidotriphosphate (22)
are shown in Supplementary Data. In the synthesis of the 5′-γ-amidotriphosphate, the
normal 5′-triphosphate of deoxyadenosine was also formed, and the ratio of
5′-γ-amidotriphosphate:5′-triphosphate was 4.8:1, from the calculation of the fraction
absorbances. Compound 22 eluted quickly, as compared to the 5′-triphosphate of
deoxyadenosine (dATP), and was separated by the DEAE column purifacation. We
confirmed the moleculer weight of the 5′-γ-amidotriphosphate by the ESI-MS spectrum (the
difference between 22 and dATP was only 1 m/z, see Supplementary Data). After the
final purification by HPLC, the nucleoside 5′-γ-amidotriphosphates were obtained as
triethylammonium salts and characterized by NMR spectroscopy (1H and 31P NMR). The
γ-phosphate signals of the 5′-γ-amidotriphosphates of deoxyadenosine and 6 were shifted
downfield (at –0.50 and –0.52 p.p.m.), as compared to those of the 5′-triphosphates of 6, 11,
17, and 18 (Supplementary Data). This phenomenon was also observed with the
5′-γ-amidotriphosphate of guanosine9,10.
2-Amino-3-nitro-4-(2-thienyl)pyridine (2). To a solution of
2-amino-3-nitro-4-chloropyridine (1) (1.74 g, 10 mmol)3 and
dichlorobis(triphenylphosphine)palladium (II) (350 mg, 0.50 mmol) in DMF (50 ml) was
added 2-(tributylstannyl)thiophene (3.82 ml, 12 mmol) under an argon atmosphere. The
resulting mixture was stirred for 4 h at 100°C. The mixture was poured into water (250 ml)
and extracted with ethyl acetate (250 ml × 3). After drying over Na2SO4, the solvent was
evaporated in vacuo. The residue was subjected to flash silica gel chromatography using
methylene chloride : ethyl acetate (100:0 to 49:1, v/v) as an eluent, to afford 2.07 g of 2 (Rf
0.30 on methylene chloride : ethyl acetate = 19:1, v/v) in 93% yield. 1H NMR (270 MHz,
CDCl3) δ 8.17 (d, 1H, J = 5.1 Hz), 7.45 (dd, 1H, J = 5.0 and 1.1 Hz), 7.12 (dd, 1H, J = 3.6
and 1.1 Hz), 7.07 (dd, 1H, J = 5.0 and 3.6 Hz), 6.77 (d, 1H, J = 5.1 Hz), 5.66 (bs, 2H).
HRMS (FAB, 3-NBA matrix) for C9H8N3O2S (M+1): calcd, 222.0337; found, 222.0337.
7

2,3-Diamino-4-(2-thienyl)pyridine (3). To a mixture of 2 (2.06 g, 9.3 mmol) and 466 mg
palladium 10% wt. on carbon, in 130 ml of ethanol and 65 ml of ethyl acetate, 28 ml of 1 M
aqueous sodium borohydride was added at 0°C. The resulting mixture was stirred for 1 h at
0°C. To the mixture was added 43 ml of 5% aqueous ammonium chloride. The mixture was
filtered with celite. To the filtrate was added 500 ml water. After the ethanol and ethyl
acetate were evaporated, the mixture was extracted with ethyl acetate (250 ml × 3). After
drying over Na2SO4, the solvent was evaporated. The residue was subjected to flash silica
gel chromatography using methylene chloride : ethanol (19:1 to 93:7, v/v) as an eluent, to
afford 1.46 g of 3 (Rf 0.24 on methylene chloride : ethanol = 9:1, v/v) in 82% yield. 1H
NMR (270 MHz, CDCl3) δ 7.64 (d, 1H, J = 5.1 Hz), 7.40 (dd, 1H, J = 5.1 and 1.1 Hz), 7.23
(dd, 1H, J = 3.5 and 1.1 Hz), 7.14 (dd, 1H, J = 5.1 and 3.5 Hz), 6.74 (d, 1H, J = 5.1 Hz),
4.26 (bs, 2H), 3.72 (bs, 2H). HRMS (FAB, 3-NBA matrix) for C9H10N3S (M+1): calcd,
192.0595; found, 192.0588.
7-(2-Thienyl)-3H-imidazo[4,5-b]pyridine (4). A solution of 3 (956 mg, 5.0 mmol) in
formic acid (15 ml) was refluxed for 12 h. To the reaction mixture was added 24 ml of 28%
NH4OH on an ice cold bath. The resulting precipitate was filtered, washed with H2O and
ethyl eter, and dried at 60°C for 12 h to give 7-(2-thienyl)-3H-imidazo[4,5-b]pyridine (970
mg, 96%). 1H NMR (300 MHz, DMSO-d6) δ 13.20 (s, 1H), 8.48 (s, 1H), 8.30 (m, 2H),
7.78 (dd, 1H, J = 1.0 and 5.1 Hz), 7.54 (d, 1H, J = 5.1 Hz), 7.25 (dd, 1H, J = 3.8 and 4.9
Hz). 13C NMR (75 MHz, DMSO-d6) δ 148.66, 144.09, 143.44, 137.60, 131.16, 130.00,
129.24, 128.71, 128.00, 113.12. HRMS (FAB, 3-NBA matrix) for C10H8N3S (M+1): calcd,
202.0439; found, 202.0444.
7-(2-Thienyl)-3-[2-deoxy-3,5-di-O-(toluoyl)-β-D-ribofuranosyl]imidazo[4,5-b]pyridine
(5). To a mixture of 4 (403 mg, 2.0 mmol) and 32 ml of CH3CN was added NaH (96 mg,
2.4 mmol, 60% dispersion in mineral oil). The resulting mixture was stirred for 1 h at room
temperature. To the mixture was added
2-deoxy-3,5-di-O-p-toluoyl-α-D-erythro-pentofuranosyl chloride (933 mg, 2.4 mmol)4.
After stirring for 2.5 h at room temperature, the reaction mixture was separated by ethyl
8

acetate and water. The organic phase was washed three times with saturated NaCl, dried
with Na2SO4, and evaporated in vacuo. The product was purified by silica gel column
chromatography (0.5% methanol in CH2Cl2) to give 5 (714 mg, 65%). 1H NMR (270 MHz,
CDCl3) δ 8.32 (d, 1H, J = 5.3 Hz), 8.26 (s, 1H), 8.16 (dd, 1H, J = 3.8 and 1.2 Hz), 7.93 (m,
4H), 7.50 (dd, 1H, J = 5.1 and 1.2 Hz), 7.47 (d, 1H, J = 5.3 Hz), 7.22 (m, 5H), 6.68 (dd, 1H,
J = 8.6 and 5.8 Hz), 5.82 (m, 1H), 4.69 (m, 3H), 3.18 (ddd, 1H, J = 14.2, 8.6 and 6.4 Hz),
2.86 (ddd, 1H, J = 14.2, 5.8 and 2.0 Hz), 2.43 (s, 3H), 2.37 (s, 3H). HRMS (FAB, 3-NBA
matrix) for C31H28N3O5S (M+1): calcd, 554.1750; found, 554.1748.
7-(2-Thienyl)-3-(2-deoxy-β-D-ribofuranosyl)imidazo[4,5-b]pyridine (6). To 1.33 g (2.40
mmol) of 5 was added 120 ml methanolic ammonia that was saturated at 0°C. The solution
was stirred for 2 days at room temperature. The solvent was evaporated. The residue was
subjected to flash silica gel chromatography, using methylene chloride : ethanol (97:3 to
93:7, v/v) as an eluent, to afford 717 mg of 6 in 94% yield. 1H NMR (300 MHz, DMSO-d6)
δ 8.75 (s, 1H), 8.35 (d, 1H, J = 5.1 Hz), 8.30 (d, 1H, J = 3.7 Hz), 7.83 (d, 1H, J = 5.1 Hz),
7.65 (d, 1H, J = 5.1 Hz), 7.28 (t, 1H, J = 4.2 Hz), 6.54 (t, 1H, J = 6.9 Hz), 5.34 (d, 1H, J =
4.1 Hz), 5.11 (t, 1H, J = 5.7 Hz), 4.46 (m, 1H), 3.91 (m, 1H), 3.60 (m, 2H), 2.80 (m, 1H),
2.37 (m, 1H). 13C NMR (75 MHz, DMSO-d6) δ 147.10, 144.04, 143.62, 137.06, 132.00,
131.00, 129.78, 129.06, 128.07, 113.93, 87.88, 83.72, 70.80, 61.74, 39.40. HRMS (FAB,
3-NBA matrix) for C15H16N3O3S (M+1): calcd, 318.0912; found, 318.0905. UV λmax; 311
nm; ε = 2.04 × 104 in 25 mM sodium phosphate buffer (pH 6.8).
7-(2-Thienyl)-3-[2-deoxy-5-O-(4,4′-dimethoxytrityl)-β-D-ribofuranosyl]imidazo[4,5-b]
pyridine (7). A 317 mg (1.0 mmol) portion of 6 was co-evaporated with dry pyridine three
times. To the residue in 5.0 ml of dry pyridine was added 356 mg (1.1 mmol) of
4,4′-dimethoxytrityl chloride. The resulting mixture was stirred overnight at room
temperature. The mixture was poured into water (50 ml) and extracted with methylene
chloride (50 ml × 3). After drying over Na2SO4, the solvent was evaporated in vacuo. The
residue was subjected to flash silica gel chromatography, using methylene chloride : ethyl
acetate (9:1 to 13:7, v/v) as an eluent, to afford 550 mg of 7 in 89% yield. 1H NMR (270
9

MHz, CDCl3) δ 8.29 (d, 1H, J = 5.1 Hz), 8.22 (s, 1H), 8.17 (dd, 1H, J = 3.8 and 1.1 Hz),
7.49 (dd, 1H, J = 5.1 and 1.1 Hz), 7.44 (d, 1H, J = 5.1 Hz), 7.37 (m, 2H), 7.27 (m, 5H), 7.20
(m, 3H), 6.78 (m, 4H), 6.57 (dd, 1H, J = 6.5 and 6.2 Hz), 4.66 (m, 1H), 4.12 (m, 1H), 3.75
(s, 6H), 3.42 (dd, 1H, J = 10.1 and 4.6 Hz), 3.38 (dd, 1H, J = 10.1 and 5.4 Hz), 2.86 (m,
1H), 2.56 (ddd, 1H, J = 13.8, 6.5 and 4.6 Hz), 2.06 (d, 1H, J = 3.5 Hz). HRMS (FAB,
3-NBA matrix) for C36H34N3O5S (M+1): calcd, 620.2219; found, 620.2230.
7-(2-Thienyl)-3-[2-deoxy-5-O-(4,4′-dimethoxytrityl)-β-D-ribofuranosyl]imidazo[4,5-b]
pyridine 2-cyanoethyl-N,N-diisopropylphosphoramidite (8). A 425 mg (0.69 mmol)
portion of 7 was co-evaporated with dry pyridine three times and then with dry acetonitrile
three times. To the residue in 4.6 ml of dry acetonitrile was added 262 µl (0.82 mmol) of
2-cyanoethyl tetraisopropylphosphorodiamidite and 1.68 ml of 0.45 M tetrazole in
acetonitrile. The resulting mixture was stirred for 1 h at room temperature. To the mixture
was added 90 µl of dry methanol. The mixture was poured into water (50 ml) and extracted
with methylene chloride containing 1% triethylamine (50 ml × 3). After drying over
Na2SO4, the solvent was evaporated in vacuo. The residue was subjected to flash silica gel
chromatography using hexane : ethyl acetate (4:1 to 3:2, v/v) containing 2% triethylamine
as an eluent, to afford 490 mg of 8 in 87% yield. 1H NMR (270 MHz, DMSO-d6) δ 8.65 (m,
1H), 8.27 (m, 1H), 8.23 (m, 1H), 7.81 (m, 1H), 7.62 (m, 1H), 7.26 (m, 3H), 7.18 (m, 7H),
6.75 (m 4H), 6.54 (m, 1H), 4.81 (m, 1H), 4.13 (m, 1H), 3.84-3.45 (m, 10H), 3.21 (m, 3H),
2.82-2.48 (m, 3H) 1.13 (m, 12H). 31P NMR (109 MHz, DMSO-d6) δ 148.76 ppm and
148.14 ppm. HRMS (FAB, 3-NBA matrix) for C45H51N5O6SP (M+1): calcd, 820.3298;
found, 820.3325.
7-(2-Thienyl)-3-(2-deoxy-3-O-acetyl-β-D-ribofuranosyl)imidazo[4,5-b]pyridine (9). A
124 mg (0.20 mmol) portion of 7 was co-evaporated with dry pyridine three times. To the
residue in 2.0 ml of dry pyridine was added 38 µl (0.40 mmol) of acetic anhydride. The
resulting mixture was stirred for 2 days at room temperature. The mixture was poured into
water (50 ml) and extracted with methylene chloride (50 ml × 3). After drying over Na2SO4,
the solvent was evaporated in vacuo. To the residue in 20 ml of dry methylene chloride was
10

added 200 µl of dichloroacetic acid at 0°C. The resulting mixture was stirred for 15 min at
0°C. The mixture was poured into 8 ml of sat. aqueous sodium hydrogen carbonate and 42
ml of water and extracted with methylene chloride (50 ml × 3). After drying over Na2SO4,
the solvent was evaporated in vacuo. The residue was subjected to flash silica gel
chromatography using methylene chloride : ethyl acetate (9:1 to 3:2, v/v) as an eluent to
afford 65 mg of 9 in 88% yield. 1H NMR (270 MHz, CDCl3) δ 8.29 (d, 1H, J = 5.4 Hz),
8.18 (dd, 1H, J = 3.8 and 1.1 Hz), 8.13 (s, 1H), 7.53 (dd, 1H, J = 5.0 and 1.1 Hz), 7.50 (d,
1H, J = 5.4 Hz), 7.20 (dd, 1H, J = 5.0 and 3.8 Hz), 6.69 (m, 1H), 6.37 (dd, 1H, J = 10.1 and
5.4 Hz), 5.58 (m, 1H), 4.28 (m, 1H), 3.96 (m, 2H), 3.32 (ddd, 1H, J = 15.9, 10.1, and 5.9
Hz), 2.41 (m, 1H), 2.12 (s, 3H). HRMS (FAB, 3-NBA matrix) for C17H18N3O4S (M+1):
calcd, 360.1018; found, 360.0993.
7-(2-Thienyl)-3-(β-D-ribofuranosyl)imidazo[4,5-b]pyridine (11). A mixture of 4 (80 mg,
0.4 mmol), tetra-O-acetyl-β-D-ribofuranose (130 mg, 0.4 mmol), and chloroacetic acid (2
mg) was fused at 200°C for 5 min. The resulting dark syrup was treated with methanolic
ammonia (40 ml). The reaction mixture was stirred at room temperature for 18 h. The
volatile fraction was evaporated in vacuo. To the residue was added 10 ml of 30% CH3CN
in H2O, and the product was purified by reversed phase HPLC to give 11 (39 mg, 29%, 2
steps). 1H NMR (300 MHz, DMSO-d6) δ 8.78 (s, 1H), 8.36 (d, 1H, J = 5.2 Hz), 8.31 (dd,
1H, J = 1.0 and 3.7 Hz), 7.84 (dd, 1H, J = 0.9 and 5.1 Hz), 7.66 (d, 1H, J = 5.2 Hz), 7.28
(dd, 1H, J = 3.7 and 5.0 Hz), 6.08 (d, 1H, J = 5.8 Hz), 5.49 (d, 1H, J = 6.0 Hz), 5.26 (t, 1H,
J = 6.5 Hz), 5.20 (d, 1H, J = 4.9 Hz), 4.67 (q, 1H, J = 5.8 Hz), 4.20 (q, 1H, J = 4.9 Hz), 4.00
(m, 1H), 3.71 (m, 1H), 3.59 (m, 1H). 13C NMR (75 MHz, DMSO-d6) δ 147.29, 144.07,
143.91, 137.00, 132.13, 131.08, 129.86, 129.14, 128.10, 114.02, 87.76, 85.57, 73.48, 70.43,
61.43. HRMS (FAB, 3-NBA matrix) for C15H16N3O4S (M+1): calcd, 334.0862; found,
334.0871.
7-(2-Thienyl)-3-[5-O-(4,4′-dimethoxytrityl)-β-D-ribofuranosyl]imidazo[4,5-b]pyridine
(12). Compound 11 (99 mg, 0.29 mmol) was co-evaporated with dry pyridine three times
and was dissolved in pyridine (3.0 ml). To the solution was added 4,4'-dimethoxytrityl
11

chloride (106 mg, 0.31 mmol), and the mixture was stirred at room temperature for 2 h. The
reaction mixture was poured into 5% NaHCO3 in H2O and extracted with ethyl acetate. The
organic phase was washed with saturated NaCl three times, dried with Na2SO4, and
evaporated in vacuo. The product was purified by silica gel column chromatography (1%
methanol in CH2Cl2) to give 12 (131 mg, 71%). 1H NMR (600 MHz, CDCl3) δ 8.39 (s, 1H),
8.28 (d, 1H, J = 5.2 Hz), 8.25 (d, 1H, J = 3.4 Hz), 7.52 (t, 2H, J = 5.7 Hz), 7.22 (m, 2H),
7.14 (m, 7H), 6.69 (m, 5H), 6.04 (d, 1H, J = 6.2 Hz), 4.77 (m, 1H), 4.48 (m, 1H), 4.34 (d,
1H, J = 4.6 Hz), 3.70 (s, 6H), 3.46 (dd, 1H, J = 3.4 and 10.5 Hz), 3.23 (dd, 1H, J = 2.9 and
10.5 Hz). HRMS (FAB, 3-NBA matrix) for C36H34N3O6S (M+1): calcd, 636.2168; found,
636.2173.
7-(2-Thienyl)-3-(2,3-di-O-acetyl-β-D-ribofuranosyl)imidazo[4,5-b]pyridine (13). A 120
mg (0.19 mmol) portion of 12 was co-evaporated three times with dry pyridine. To the
residue in 1.9 ml of dry pyridine was added 72 µl (0.76 mmol) of acetic anhydride. The
resulting mixture was stirred for 7 h at room temperature. The mixture was poured into 5%
NaHCO3 (50 ml) and ethyl acetate (50 ml). The organic phase was washed once with
saturated NaCl. After the organic phase was dried over Na2SO4, the solvent was evaporated
in vacuo. The residue was co-evaporated twice with toluene. To the residue, in 19 ml of
CH2Cl2, was added 190 µl of dichloroacetic acid at 0°C. The resulting reaction mixture was
stirred for 15 min at 0°C. The mixture was poured into 5% NaHCO3 and extracted with
CH2Cl2. The organic phase was washed with saturated NaCl, and after the organic phase
was dried over Na2SO4, the solvent was evaporated in vacuo. The product was purified by
silica gel column chromatography, using 2% methanol in CH2Cl2 as an eluent, to afford 77
mg of 13 in 93% yield. 1H NMR (300 MHz, DMSO-d6) δ 8.79 (s, 1H), 8.37 (d, 1H, J = 5.2
Hz), 8.29 (dd, 1H, J = 1.2 and 3.7 Hz), 7.84 (dd, 1H, J = 1.1 and 5.1 Hz), 7.68 (d, 1H, J =
5.2 Hz), 7.27 (dd, 1H, J = 3.7 and 5.1 Hz), 6.38 (d, 1H, J = 6.8 Hz), 6.02 (dd, 1H, J = 5.6
and 6.6 Hz), 5.54 (m, 2H), 4.26 (m, 1H), 3.71 (m, 2H), 2.14 (s, 3H), 1.98 (s, 3H). 13C NMR
(75 MHz, DMSO-d6) δ 169.55, 169.22, 146.98, 144.39, 143.85, 136.74, 132.50, 131.02,
12

130.11, 129.32, 128.14, 114.36, 85.20, 83.63, 72.34, 71.23, 61.05, 20.44, 20.13. HRMS
(FAB, 3-NBA matrix) for C19H20N3O6S (M+1): calcd, 418.1073; found, 418.1049.
1-(β-D-Ribofuranosyl)pyrrole-2-carbaldehyde (17). To a solution of
pyrrole-2-carboxaldehyde (330 mg, 3.5 mmol) in CH3CN (18 mL) was added NaH (60%
oil dispersion, 152 mg, 3.8 mmol). The reaction mixture was stirred at room temperature
for 45 min. A solution of 2,3,5-tri-O-benzyl-D-ribofuranosyl chloride6 (3.1 mmol) in
CH3CN (13 ml), prepared from 2,3,5-tri-O-benzyl-1-O-p-nitrobenzoyl-D-ribofuranose (1.8
g 3.1 mmol), was then added. The reaction mixture was stirred at room temperature for 4 h.
The product was separated by ethyl acetate and water, and the organic phase was washed
with saturated NaCl three times, dried with Na2SO4, and evaporated in vacuo. The residue
was purified by silica gel column chromatography (eluted by 20% ethyl acetate in hexane)
to give crude 1-(2,3,5-tri-O-benzyl-D-ribofuranosyl)pyrrole-2-carbaldehyde (506 mg).
After the crude 1-(2,3,5-tri-O-benzyl-D-ribofuranosyl)pyrrole-2-carbaldehyde was
co-evaporated with toluene three times, CH2Cl2 (17 ml) was added to the residue. To the
solution was added BBr3 (1 M solution, 3.0 ml) at –78°C. The reaction mixture was stirred
for 2.5 h, and then 50% methanol in CH2Cl2 (25 ml) was added. After the solution was
stirred for 10 min at –78°C, 28% NH4OH (0.5 ml) was added and the reaction mixture was
stirred until it reached room temperature. The product was separated by CH2Cl2 and H2O,
and the water phase was washed with CH2Cl2 three times, and evaporated in vacuo. The
product was purified by reversed phase C18 HPLC to give
1-(β-D-ribofuranosyl)pyrrole-2-carbaldehyde (108 mg, 15%, 2 steps). 1H NMR (270 MHz,
DMSO-d6) δ 9.54 (s, 1H), 7.74 (s, 1H), 7.06 (dd, 1H, J = 1.6 and 4.0 Hz), 6.39 (d, 1H, J =
4.3 Hz), 6.30 (dd, 1H, J = 3.0 and 4.0 Hz), 5.27 (d, 1H, J = 5.6 Hz), 5.05 (d, 1H, J = 4.9 Hz),
5.00 (t, 1H, J = 5.3 Hz), 4.02 (m, 2H), 3.85 (m, 1H), 3.52 (m, 2H). 13C NMR (75 MHz,
DMSO-d6) δ 179.41, 131.68, 128.24, 124.94, 110.23, 89.34, 84.33, 75.64, 69.50, 60.82.
HRMS (FAB, 3-NBA matrix) for C10H14NO5 (M+1): calcd, 228.0872; found, 228.0863.
4-Propynyl-1-(β-D-ribofuranosyl)pyrrole-2-carbaldehyde (18).
4-Propynyl-1-(β-D-ribofuranosyl)pyrrole-2-carbaldehyde was synthesized by using
13

4-propynylpyrrole-2-carbaldehyde (16)2 (266 mg, 2.0 mmol) according to the synthesis of
17, and yielded 18 (39 mg, 7%, 2 steps) after purification by RP-HPLC. 1H NMR (300
MHz, DMSO-d6) δ 9.50 (s, 1H), 7.91 (s, 1H), 7.09 (d, 1H, J = 1.8 Hz), 6.32 (d, 1H, J = 3.6
Hz), 5.32 (d, 1H, J = 5.5 Hz), 5.07 (t, 1H, J = 5.2 Hz), 5.05 (d, 1H, J = 4.2 Hz), 4.01 (m,
1H), 3.86 (m, 1H), 3.66 (ddd, 1H, J = 3.4, 5.3, and 11.9 Hz), 3.55 (ddd, 1H, J = 3.6, 4.9,
and 12.1 Hz), 1.97 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 179.60, 131.15, 130.43,
126.13, 106.22, 89.62, 85.26, 84.49, 75.86, 73.17, 69.30, 60.53, 3.79. HRMS (FAB, 3-NBA
matrix) for C13H16NO5 (M+1): calcd, 266.1028; found, 266.1023.
Synthesis of nucleoside 5′-triphosphates 10 and 14. The protected nucleoside (0.1 mmol,
9 or 13) was dissolved in pyridine and evaporated to dryness in vacuo. The residue was
dissolved in pyridine (100 µl) and dioxane (300 µl). A 1 M solution of
2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one in dioxane (110 µl, 0.11 mmol) was added.
After 10 min, tri-n-butylamine (100 µl) and 0.5 M bis(tributylammonium)pyrophosphate in
DMF (300 µl, 0.15 mmol) were quickly added to the reaction mixture. The reaction mixture
was stirred at room temperature for 10 min. A solution of 1% iodine in pyridine/water (98/2,
v/v) (2.0 ml) was then added. After 15 min, 150 µl of a 5% aqueous solution of NaHSO3,
followed by 5.0 ml of water, was added to the reaction mixture. The solution was stirred at
room temperature for 30 min, and then 20 ml of concentrated ammonia was added.
Ammonolysis was carried out at room temperature for 2 h. After the reaction solution was
concentrated in vacuo, the product was purified by DEAE Sephadex (A-25) column
chromatography (eluted by a linear gradient of 50 mM to 1 M TEAB), and then by
C18-HPLC (eluted by a gradient of 0% to 30% CH3CN in 100 mM triethylammonium
acetate) to give the nucleoside 5′-triphosphate.
7-(2-Thienyl)-3-(2-deoxy-β-D-ribofuranosyl)imidazo[4,5-b]pyridine 5′-triphosphate
(10). 1H NMR (270 MHz, D2O) δ 8.51 (s, 1H), 8.09 (d, 1H, J = 5.3 Hz), 7.78 (d, 1H, J = 3.6
Hz), 7.56 (d, 1H, J = 4.9 Hz), 7.36 (d, 1H, J = 5.3 Hz), 7.12 (t, 1H, J = 4.9 Hz), 6.41 (t, 1H,
J = 7.3 Hz), 4.16 (m, 1H), 4.04 (m, 2H), 3.01 (q, 18H, J = 7.3 Hz), 2.72 (m, 1H), 2.46 (m,
1H), 1.09 (t, 27H, J = 7.3 Hz). 31P NMR (109 MHz, D2O) δ –9.94 (d, 1P, J = 20.1 Hz),
14

–10.72 (d, 1P, J = 20.1 Hz), –22.58 (t, 1P, J = 20.1 Hz). Electrospray ionization-mass
spectroscopy (ESI-MS) for C15H18N3O12P3S; calcd, 555.97 (M-H)–; found, 555.69 (M-H)–.
7-(2-Thienyl)-3-(β-D-ribofuranosyl)imidazo[4,5-b]pyridine 5′-triphosphate (14). 1H
NMR (300 MHz, D2O) δ 8.74 (s, 1H), 8.32 (d, 1H, J = 5.4 Hz), 8.01 (d, 1H, J = 3.5 Hz),
7.68 (dd, 1H, J = 1.1 and 5.1 Hz), 7.64 (d, 1H, J = 5.1 Hz), 7.25 (dd, 1H, J = 3.5 and 5.1
Hz), 6.25 (d, 1H, J = 6.0 Hz), 4.82 (m, 1H), 4.57 (m, 1H), 4.36 (m,1H), 4.20 (m, 2H), 3.11
(q, 18H, J = 7.3 Hz), 1.19 (t, 27H, J = 7.3 Hz). 31P NMR (109 MHz, D2O) δ –9.80 (d, 1P, J
= 20.1 Hz), –11.03 (d, 1P, J = 18.9 Hz), –22.78 (t, 1P, J = 20.1 and 18.9 Hz). ESI-MS for
C15H18N3O13P3S; calcd, 571.97 (M-H)–; found, 571.74 (M-H)–.
Synthesis of nucleoside 5′-γ-amidotriphosphates 10 and 22.5,9,10 The protected
nucleoside (0.1 mmol, 9 or 218) was dissolved in pyridine and evaporated to dryness in
vacuo. The residue was dissolved in pyridine (100 µl) and dioxane (300 µl). A 1 M solution
of 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one in dioxane (110 µl, 0.11 mmol) was
added. After 10 min, tri-n-butylamine (100 µl) and 0.5 M
bis(tributylammonium)pyrophosphate in DMF (300 µl, 0.15 mmol) were quickly added to
the reaction mixture. The reaction mixture was stirred at room temperature for 10 min. A
solution of 1% iodine in pyridine/water (98/2, v/v) (2.0 ml) was then added. After 15 min,
150 µl of a 5% aqueous solution of NaHSO3 was added. After the solvent was evaporated,
20 ml of 28% aqueous ammonia was added to the residue. Ammonolysis was carried out at
60°C for 5 h (for the γ-amidotriphosphate of deoxyadenosine) or at room temperature 2 h
(for the γ-amidotriphosphate of 6). After the reaction solution was concentrated in vacuo,
the product was purified by DEAE Sephadex (A-25) column chromatography (eluted by a
linear gradient of 50 mM to 1 M TEAB), and then by C18-HPLC (eluted by a gradient of
0% to 30% CH3CN in 100 mM triethylammonium acetate) to give the nucleoside
5′-γ-amidotriphosphate.
6-Amino-9-(2-deoxy-β-D-ribofuranosyl)purine 5′-γ-amidotriphosphate (22). 1H NMR
(270 MHz, D2O) δ 8.35 (s, 1H), 8.11 (s, 1H), 6.37 (t, 1H, J = 6.9 Hz), 4.16 (m, 1H), 4.03
15

(m, 2H), 3.05 (q, 18H, J = 7.3 Hz), 2.70 (m, 1H), 2.48 (m, 1H), 1.13 (t, 27H, J = 7.3 Hz). 31P NMR (109 MHz, D2O) δ –0.50 (d, 1P, J = 19.5 Hz), –10.77 (d, 1P, J = 19.5 Hz), –22.14
(t, 1P, 20.1 Hz). ESI-MS for C10H17N6O11P3; calcd, 489.01 (M-H)–; found, 488.98 (M-H)–.
7-(2-Thienyl)-3-(2-deoxy-β-D-ribofuranosyl)imidazo[4,5-b]pyridine
5′-γ-amidotriphosphate (5′-γ-amidotriphosphate of 10). 1H NMR (270 MHz, D2O) δ
8.57 (s, 1H), 8.16 (d, 1H, J = 5.3 Hz), 7.85 (d, 1H, J = 3.6 Hz), 7.58 (d, 1H, J = 4.9 Hz),
7.45 (d, 1H, J = 5.3 Hz), 7.15 (t, 1H, J = 4.6 Hz), 6.48 (t, 1H, J = 6.9 Hz), 4.18 (m, 1H),
4.05 (m, 2H), 3.03 (q, 18H, J = 7.3 Hz), 2.75 (m, 1H), 2.50 (m, 1H), 1.11 (t, 27H, J = 7.3
Hz). 31P NMR (109 MHz, D2O) δ –0.52 (d, 1P, J = 20.1 Hz), -10.75 (d, 1P, J = 19.5 Hz),
-22.14 (t, 1P, J=20.8 Hz). ESI-MS for C15H19N4O11P3S; calcd, 554.99 (M-H)–; found,
555.01 (M-H)–.
Synthesis of nucleoside 5′-triphosphates 19 and 20.7 To a solution of
1-(β-D-ribofuranosyl)pyrrole-2-carbaldehyde (17) or
4-propynyl-1-(β-D-ribofuranosyl)pyrrole-2-carbaldehyde (18) (0.1 mmol) and a proton
sponge (33 mg, 0.15 mmol) in trimethyl phosphate (500 µl) was added POCl3 (12 µl, 0.13
mmol) at 0°C. The reaction mixture was stirred at 0°C for 2 h. Tri-n-butylamine (120 µl,
0.5 mmol) was added to the reaction mixture, followed by 0.5 M
bis(tributylammonium)pyrophosphate in a DMF solution (1.0 ml, 0.5 mmol). After 5 min,
the reaction was quenched by the addition of 0.5 M triethylammonium bicarbonate (TEAB,
500 µl). The resulting crude product was purified by DEAE Sephadex A-25 column
chromatography (eluted by a linear gradient of 50 mM to 1 M TEAB), and then by
C18-HPLC (Synchropak RPP, Eichrom Technologies, eluted by a gradient of 0% to 30%
CH3CN in 100 mM triethylammonium acetate).
1-(β-D-Ribofuranosyl)pyrrole-2-carbaldehyde 5′-triphosphate (19). 1H NMR (270 MHz,
D2O) δ 9.28 (s, 1H), 7.64 (s, 1H), 7.08 (d, 1H, J = 3.9 Hz), 6.45 (d, 1H, J = 4.1 Hz), 6.32 (m,
1H), 4.32 (m, 2H), 4.10 (m, 3H), 3.03 (q, 18H, J = 7.3 Hz), 1.11 (t, 27H, J = 7.3 Hz). 31P
16

NMR (109 MHz, D2O) δ –10.51 (d, 1P, J = 19.5 Hz), –11.3 (d, 1P, J = 20.1 Hz), –22.91 (t,
1H, J = 20.1 Hz). ESI-MS for C10H16NO14P3: calcd, 465.97 (M-H)–; found, 465.85 (M-H)–.
4-Propynyl-1-(β-D-ribofuranosyl)pyrrole-2-carbaldehyde 5′-triphosphate (20). 1H
NMR (270 MHz, D2O) δ 9.28 (s, 1H), 7.70 (s, 1H), 7.08 (s, 1H), 6.40 (d, 1H, J = 4.0 Hz),
4.30 (m, 2H), 4.13 (m, 3H), 3.06 (q, 18H, J = 7.3 Hz), 1.86 (s, 3H), 1.14 (t, 27H, J = 7.3
Hz). 31P NMR (109 MHz, D2O) δ –10.10 (d, 1P, J = 19.5 Hz), –11.02 (d, 1P, J = 19.5 Hz),
–22.82 (t, 1P, J = 20.1 Hz). ESI-MS for C13H18NO14P3: calcd, 503.99 (M-H)–; found,
503.94 (M-H)–.
References
3. De Roos, K. B. & Salemink, C. A. Deazapurine derivatives. V, A new synthesis of 1- and 3-deaza-adenine and related compound. Recueil. 88, 1263-1274 (1963).
4. Rolland, V., Kotera, M. & Lhomme, J. Convenient preparation of 2-deoxy 3,5-di-O-p-toluoyl-α-D-erythro-pentofuranosyl chloride. Synthetic commun. 27, 3505-3511 (1997).
5. Ludwig, J. & Eckstein, F. Rapid and efficient synthesis of 5′-O-(1-thiotriphosphates), 5′-O-triphosphates and 2′,3′-cyclophosphorothioates using 2-chloro-4H-1,3,2-benzosioxaphosphorin-4-one. J. Org. Chem. 54, 631-635 (1989).
6. Stevens, J. D., Ness, R. K. & Fletcher. Jr, H. G. Syntheses with partially benzylated sugars. XI. Studies on the synthesis of the anomeric 5,6-dimethyl-1-D-ribofuranosylbenzimidazole (Ribazoles). Comparison of the condensation of 2,3,5-tri-O-benzoyl-D-ribofuranosyl bromide and 2,3,5-tri-O-benzoyl-D-ribofuranosyl chloride with 5,6-dimethylbenzimidazole. J. Org. Chem. 33, 1806-1810 (1968).
7. Kovacs, T. & Otvos, L. Simple synthesis of 5-vinyl- and 5-ethynyl-2′-deoxyuridine-5′-triphosphates. Tetrahedron Lett. 29, 4525-4528 (1988).
8. Ti, G. S., Gaffney, B. L. & and Jones, R. A. Transient protection: Efficient One-Flask Syntheses of Protected Deoxynucleosides. J. Am. Chem. Soc. 104, 1316-1319 (1982).
9. Stumber, M., Herrmann, C., Wohlgemuth, S., Kalbitzer, H. R., Jahn, W. & Geyer, M. Synthesis, characterization and application of two nucleoside triphosphate analogues, GTPγNH2 and GTPγF. Eur. J. Biochem. 269, 3270-3278 (2002).
10. Knorre, D. G., Kurbatov, V. A. & Samukov, V. V. General method for the synthesis of ATP gamma-derivatives. FEBS Lett. 70, 105-108 (1976).
17

III. Synthesis of Bio-PaTP
Synthesis scheme for 1-(β-D-ribofuranosyl)-4-[(3-biotinamido-1-propynyl)]
pyrrole-2-carbaldehyde 5′-triphosphate (28).
(a), (b), (c)
NH
H
O
I
23
O
OH
HO N
OH
24
H
O
NH
Cl2HCO
(d) O
OH
DMTrO N
OH
25
H
O
NH
Cl2HCO
(e) O
OAc
HO N
OAc
26
H
O
NH
Cl2HCO
O
OH
O N
OH
27
H
O
H2N
(f)
P OOO
O-PPHO
O
O- O-
O(g)
O
OH
O N
OH
28
H
O
N
P OOO
O-PPHO
O
O- O-
O
H
OS
NN
O
H
H
Reagents and abbreviations: (a) NaH, CH3CN, then 2,3,5-tri-O-benzyl-D-ribofuranosyl chloride; (b) BBr3, dichloromethane; (c) 3-(dichloroacetamido)-1-propyne, tetrakis(triphenylphosphine)palladium, CuI, triethylamine, DMF; (d) 4,4′-dimethoxytrityl chloride, pyridine; (e) acetic anhydride, pyridine, then dichloroacetic acid, dichloromethane; (f) 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one, dioxane, pyridine, tri-n-butylamine, bis(tributylammonium)pyrophosphate, DMF, then I2/pyridine, water, NH4OH; (g) biotin-N-hydroxysuccinimide, DMF, NaHCO3 and Na2CO3 buffer (pH 8.6) then NH4OH. DMTr: 4,4′-dimethoxytrityl, Ac: acetyl.
18

1-(β-D-Ribofuranosyl)-4-[(3-dichloroacetamido)-1-propynyl]pyrrole-2-carbaldehyde (24)
was synthesized by coupling 4-iodopyrrole-2-carbaldehyde (23) with
2,3,5-tri-O-benzyl-D-ribofuranosyl chloride, which was prepared by chlorination11 of
2,3,5-tri-O-benzyl-D-ribofuranose, using CCl4 and tris(dimethylamino)phosphane, followed
by a treatment with BBr3. The ribonucleoside of 4-iodopyrrole-2-carbaldehyde was
converted to the ribonucleoside of
4-[(dichloroacetamide)-1-propynyl]pyrrole-2-carbaldehyde (24) by palladium-mediated
cross-coupling according to the literature12. The structure of 24 was confirmed by NMR
and high resolution mass spectroscopy. The HMQC and HMBC spectra of 24 revealed that
the N-glycoside bond is formed between the sugar and the pyrrole base moiety at the C1′
carbon. From the NOE experiment (NOESY spectrum in Supplementary Data), the
anomeric configuration of 24 was assigned as β; the cross peaks of the NOESY spectrum of
24 were similar to those of 18, and showed a cross peak between the H1′ and H4′ protons.
Compound 24 was converted to the 2′,3′-O-acetylated ribonucleoside 26 in two steps (87%
from 24), and then the nucleoside 5′-triphosphate (27) was prepared by a conventional
method5. Finally, compound 27 was converted to Bio-PaTP (28) in a 14% yield from 26.
The structure of 28 was confirmed by 1H NMR, 31P NMR and mass spectrometry. The
proton signals of the biotin moiety in 28 were identical to those of the previously
synthesized biotinylated-yTP12, and the 31P NMR spectrum in D2O showed a typical
phosphorus signal corresponding to nucleoside 5′-triphosphates.
1-(β-D-Ribofuranosyl)-4-[(3-dichloroacetamido)-1-propynyl]pyrrole-2-carbaldehyde
(24). To a THF solution (4.6 ml) containing 2,3,5-tri-O-benzyl-D-ribofuranose (1.0 g, 2.3
mmol) and CCl4 (344 µl, 3.6 mmol), hexamethylphosphorous triamide (562 µl, 3.0 mmol)
was added at –78°C. The solution was stirred for 2 h at –78°C and for 30 min at room
temperature (solution A). To a solution of 4-iodopyrrole-2-carboxaldehyde (830 mg, 3.7
mmol) in CH3CN (25 mL) was added NaH (60% oil dispersion, 150 mg, 3.7 mmol). The
reaction mixture was stirred at room temperature for 30 min. A solution of
2,3,5-tri-O-benzyl-D-ribofuranosyl chloride in THF (solution A) was then added. The
19

reaction mixture was stirred at room temperature for 12 h. The product was separated by
ethyl acetate and water. The organic layer was washed with saturated NH4Cl three times,
dried with Na2SO4, and evaporated in vacuo. The product was purified by silica gel column
chromatography (eluted by 1% methanol in dichloromethane) to give
1-(2,3,5-tri-O-benzyl-D-ribofuranosyl)-4-iodopyrrole-2-carbaldehyde. To a solution of
1-(2,3,5-tri-O-benzyl-D-ribofuranosyl)-4-iodopyrrole-2-carbaldehyde in dichloromethane
(15 ml) was added BBr3 (1 M solution, 8.5 ml) at –78°C. The reaction mixture was stirred
for 2 h, and then 50% methanol in CH2Cl2 (30 ml) was added. After stirring the solution at
–78°C for 10 min, 28% NH4OH (4 ml) was added, and the reaction mixture was stirred
until it reached room temperature. The solution was added to CH2Cl2 and H2O. The water
layer was isolated and washed with CH2Cl2 three times, and the residue was evaporated in
vacuo. The product was purified by reversed phase C18 HPLC to give
1-(β-D-ribofuranosyl)-4-iodopyrrole-2-carbaldehyde (330 mg).
1-(β-D-Ribofuranosyl)-4-iodopyrrole-2-carbaldehyde (176 mg, 0.5 mmol, containing the α
anomer) was co-evaporated with pyridine and toluene. To a solution of
1-(β-D-ribofuranosyl)-4-iodopyrrole-2-carbaldehyde (176 mg),
tetrakis(triphenylphosphine)palladium (29 mg, 0.025 mmol), CuI (15 mg, 0.08 mmol), and
triethylamine (105 µl, 0.75 mmol) in DMF (1.8 ml), a 1 M solution (750 µl) of
3-(dichloroacetamido)-1-propyne (0.75 mmol) in DMF was added. The reaction was stirred
at room temperature for 12 h. The product was separated with EtOAc/H2O, and the organic
layer was dried with Na2SO4, and evaporated in vacuo. The product was purified by silica
gel column chromatography (10% methanol in dichloromethane) and RP-HPLC to give
compound 24 (123 mg, 26%, 3 steps total yield) as the β isomer. Compound 24: 1H NMR
(300 MHz, DMSO-d6) δ 9.54 (d, 1H, J = 0.8 Hz), 9.10 (t, 1H, J = 5.2 Hz), 8.01 (s, 1H),
7.18 (d, 1H, J = 1.8 Hz), 6.49 (s, 1H), 6.34 (d, 1H, J = 3.5 Hz), 5.35 (d, 1H, J = 5.6 Hz),
5.10 (m, 2H), 4.17 (d, 2H, J = 5.4 Hz), 4.02 (m, 2H), 3.88 (m, 1H), 3.68 (ddd, 1H, J = 3.4,
5.3, and 12.1 Hz), 3.57 (ddd, 1H, J = 3.5, 5.0, and 12.1 Hz). 13C NMR (75 MHz,
DMSO-d6) δ 180.28, 163.78, 131.88, 131.49, 126.85, 105.23, 90.23, 85.26, 85.04, 76.83,
20

76.41, 69.75, 67.01, 60.96, 30.20. HRMS (FAB, 3-NBA matrix) for C15H17N2O6Cl2 (M+1):
calcd, 391.0464; found, 391.0462.
1-(2,3-Di-O-acetyl-β-D-ribofuranosyl)-4-[(3-dichloroacetamido)-1-propynyl]pyrrole-2-
carbaldehyde (26). Compound 24 (118 mg, 0.3 mmol) was co-evaporated with pyridine
three times. To the residue in pyridine (3.0 ml) was added 4,4′-dimethoxytrityl chloride
(113 mg, 0.33 mmol). The mixture was stirred for 1 h at room temperature, and was added
to EtOAc and 5% NaHCO3. The organic layer was washed with saturated NaCl, dried over
Na2SO4, and evaporated in vacuo. The product was purified by silica gel chromatography
(1% methanol in CH2Cl2) to give 197 mg of 25 in a 95% yield. Compound 25 (188 mg,
0.27 mmol) was co-evaporated with pyridine three times. To the residue in pyridine (2.7
ml) was added acetic anhydride (103 µl, 1.1 mmol). The mixture was stirred for 12 h at
room temperature, and added to EtOAc and 5% NaHCO3. The organic layer was washed
with saturated NaCl, dried over Na2SO4, and evaporated in vacuo. To the residue in
dichloromethane (27 ml) was added dichloroacetic acid (270 µl) at 0°C. The mixture was
stirred for 15 min at 0°C, poured into 5% aqueous sodium hydrogen carbonate, and
extracted with dichloromethane. After it was dried over Na2SO4, the solution was
evaporated in vacuo. The product was purified by silica gel chromatography (1% methanol
in CH2Cl2) to give 118 mg of 26 in a 92% yield. Compound 25: 1H NMR (500 MHz,
DMSO-d6) δ 9.56 (s, 1H), 9.05 (t, 1H, J = 5.0 Hz), 7.72 (s, 1H), 7.38-7.20 (m, 10H), 6.87
(d, 4H, J = 7.1 Hz), 6.47 (s, 1H), 6.34 (d, 1H, J = 3.3 Hz), 5.47 (d, 1H, J = 5.3 Hz), 5.13 (d,
1H, J = 5.7 Hz), 4.12-4.00 (m, 5H), 3.73 (s, 6H), 3.22 (m, 2H). HRMS (FAB, 3-NBA
matrix) for C36H35N2O8Cl2 (M+1): calcd, 693.1770; found, 693.1721. Compound 26: 1H
NMR (500 MHz, DMSO-d6) δ 9.50 (s, 1H), 9.10 (bs, 1H), 8.06 (s, 1H), 7.24 (d, 1H, J = 1.3
Hz), 6.64 (d, 1H, J = 5.0 Hz), 6.48 (s, 1H), 5.43 (t, 1H, J = 5.0 Hz), 5.35 (t, 1H, J = 5.2 Hz),
5.32 (t, 1H, J = 4.8 Hz), 4.17 (m, 3H), 3.73 (m, 1H), 3.62 (m, 1H), 2.07 (s, 3H), 2.01 (s,
3H). HRMS (FAB, 3-NBA matrix) for C19H21N2O8Cl2 (M+1): calcd, 475.0675; found,
475.0687.
21

1-(β-D-Ribofuranosyl)-4-[(3-biotinamido-1-propynyl)]pyrrole-2-carbaldehyde
5′-triphosphate (28). The protected nucleoside 26 (47 mg, 0.1 mmol) was dissolved in
pyridine and evaporated in vacuo. The residue was dissolved in pyridine (100 µl) and
dioxane (300 µl). A 1 M solution of 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one in
dioxane (110 µl, 0.11 mmol) was added. After 10 min, aliquots of tri-n-butylamine (100 µl)
and 0.5 M bis(tributylammonium)pyrophosphate in DMF (300 µl, 0.15mmol) were quickly
added to the reaction mixture. The mixture was stirred at room temperature for 10 min. A
solution of 1% iodine in pyridine/water (98/2, v/v) (2.0 ml) was then added. After 15 min, a
5% aqueous solution (150 µl) of NaHSO3 was added. The volatile components were
removed by evaporation, and then water (5.0 ml) was added to the residue. The solution
was stirred at room temperature for 30 min, and then was treated with concentrated
ammonia (20 ml) at room temperature for 12 h. The solution was evaporated in vacuo, and
the product was purified by DEAE Sephadex (A-25) column chromatography (eluted by a
linear gradient of 50 mM to 1 M TEAB), and then by C18-HPLC (eluted by a gradient of
0% to 30% CH3CN in 100 mM triethylammonium acetate) to give the nucleoside
5′-triphosphate 27. After lyophilization, compound 27, in 0.1 M NaHCO3–Na2CO3 buffer
(5 ml, pH 8.6), was reacted with biotin-N-hydroxysuccinimide (900 µl, 0.14 M in a DMF
solution) at room temperature for 3 h. The mixture was treated with 28% NH4OH (2 ml) for
1 h. The product was purified by DEAE Sephadex (A-25) column chromatography (eluted
by a linear gradient of 50 mM to 1 M TEAB), and then by C18-HPLC (eluted by a gradient
of 0% to 30% CH3CN in 100 mM triethylammonium acetate) to give the nucleoside
5′-triphosphate 28 in a 14% yield from 26. Compound 27: ESI-MS for C13H19O14N2P3:
calcd, 519.00 (M-H)-; found, 518.98 (M-H)-. Compound 28: 1H NMR (300 MHz, D2O)
δ 9.36 (d, 1H, J = 0.9 Hz), 7.86 (s, 1H), 7.20 (d, 1H, J = 1.7 Hz), 6.44 (d, 1H, J = 4.1 Hz),
4.39-4.33 (m, 3H), 4.23-4.15 (m, 4H), 4.06 (d, 2H, J = 3.7 Hz), 3.18 (m, 1H), 3.12 (q, 24H,
J = 7.3 Hz), 2.82 (dd, 1H, J = 4.9 and 13.1 Hz), 2.60 (d, 1H, J = 13.0 Hz), 2.23 (t, 2H, J =
7.0 Hz), 1.60 (m, 4H), 1.32 (m, 2H), 1.20 (t, 36H, J = 7.3 Hz). 31P NMR (107 MHz,
D2O) δ –8.96 (d, 1P, J = 16.5 Hz), –10.67 (d, 1H, J = 20.1 Hz), –22.36 (t, 1P, J = 20.1 Hz).
ESI-MS for C23H33O16N4P3S: calcd, 745.07 (M-H)-; found, 745.07 (M-H)-. UV-vis
22

spectrum (in 10 mM sodium phosphate buffer, pH 7.0), λmax = 258 nm (ε = 1.1 × 104),
308 nm (ε = 9.5 × 103).
Referemces
11. Rosemeyer, H., & Seela, F. 171. Stereoselective synthesis of pyrrolo[2,3-d]pyrimidine α- and β-D-ribonucleosides from anomerically pure D-ribofuranosyl chlorides: solid-liquid phase-transfer glycosylation and 15N-NMR spectra. Helv. Chim. Acta 71, 1573-1585 (1988).
12. Moriyama, K., Kimoto, M., Mitsui, T., Yokoyama, S. & Hirao, I. Site-specific biotinylation of RNA molecules by transcription using unnatural base pairs. Nucleic Acids Res. 33, e129 (2005).
23

IV. Steady-state kinetics for the single-nucleotide insertion experiments with KF exo–.
A primer (20-mer) labeled with 6-carboxyfluorescein at the 5'-end was annealed with a
template (35-mer), in 100 mM Tris-HCl (pH 7.5) buffer containing 20 mM MgCl2, 2 mM
DTT, and 0.1 mg/ml bovine serum albumin. The primer-template duplex solution (10 µM,
5 µl) was mixed with 2 µl of an enzyme solution containing the exonuclease-deficient
Klenow fragment, KF exo– (Amersham USB). The mixture was incubated for more than 2
min, and then the reactions were initiated by adding each dNTP solution (3 µl) to the
duplex-enzyme mixture at 37°C. The amount of enzyme used (5–50 nM), the reaction time
(1–35 min), and the gradient concentration of dNTP (0.3–1500 µM) were adjusted to give
reaction extents of 25% or less. The reactions were quenched with 10 µl of a stop solution
(95% formamide and 20 mM EDTA), and the mixtures were immediately heated at 75°C
for 3 min. The diluted products were analyzed on an automated ABI 377 DNA sequencer
equipped with the GeneScan software (version 3.0). Relative velocities (v0) were calculated
as the extents of the reaction divided by the reaction time, and were normalized to the
enzyme concentration (20 nM) for the various enzyme concentrations used. The kinetic
parameters (KM and Vmax) were obtained from Hanes-Woolf plots of [dNTP]/v0 against
[dNTP]. The entire list of kinetic parameters can be found as Supplementary Table 1a and
1b.
24

V. Sequences of DNA fragments for the PCR, sequencing, and transcription experiments.
(The sequences of F1-F14 are presented on the next page.)
25

Sequences of chemically synthesized DNA fragments F1(91-mer) 5'-GAAATTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACCATGGGCTCCAAGAAGCCGGTCCCCATCATN1N2N3N4N5GCAACCGC-3' F2 (81-mer) 5'-TTTCACACAGGAAACAGCTATGACCCGGGTTATTACATGCGCTGGCACTTGCCCGTACGGCGGTTGCN6N7N8N9N10ATGATGGGG-3' F3 (66-mer) 5'-GAAATTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACCATGGGCTCCAAGAAGCC-3' F4 (47-mer) 5'-pGGTCCCCATCATN1N2N3N4N5GCAACCGCCGTACGGGCAAGACGCAGCGCG-3' F5 (61-mer) 5'-pGGGGTTCTCATCATCATCATCATCATTAATAACCCGGGTCATAGCTGTTTCCTGTGTGAAA-3' F6 (53-mer) 5'-pCCATGGTATATCTCCTTCTTAAAGTTAACCCTATAGTGAGTCGTATTAATTTC-3' F7 (50-mer) 5'-pCTTGCCCGTACGGCGGTTGCN6N7N8N9N10ATGATGGGGACCGGCTTCTTGGAGC-3' F8 (71-mer) 5'-TTTCACACAGGAAACAGCTATGACCCGGGTTATTAATGATGATGATGATGATGAGAACCCCCGCGCTGCGT-3' F9 (66-mer) 5'-GAAATTAATACGACTCACTATAGGGCCAACCAGAAGAAGGAGACAGACCAAGGGCACCAAGAAGCC-3' F10 (47-mer) 5'-pGGACCCCAACAAN1N2N3N4N5GCAACCGCCGAACGGGCAAGACGCAGCGCG-3' F11 (61-mer) 5'-pGGGGAACACAACAGCACCAACAGCACCAACAACCCGGGACAAAGCAGCAACCAGGGAGAAA-3' F12 (53-mer) 5'-pCCTTGGTCTGTCTCCTTCTTCTGGTTGGCCCTATAGTGAGTCGTATTAATTTC-3' F13 (50-mer) 5'-pCTTGCCCGTTCGGCGGTTGCN6N7N8N9N10TTGTTGGGGTCCGGCTTCTTGGTGC-3'
26

F14 (71-mer) 5'-TTTCTCCCTGGTTGCTGCTTTGTCCCGGGTTGTTGGTGCTGTTGGTGCTGTTGTGTTCCCCCGCGCTGCGT-3' Primers for sequencing and PCR Primer1 (25-mer, 5'-primer) 5'-GAAATTAATACGACTCACTATAGGG-3' Primer2 (24-mer, 3'-primer) 5'-TTTCACACAGGAAACAGCTATGAC-3'
27

VI. Dye terminator sequencing of the PCR products from DNA.
The cycle sequencing reactions (20 µl) were performed on a PTC-100 Programmable
Thermal Controller (MJ Research) with Cycle Sequencing Mix (8 µl) of BigDye
Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) containing 0.3 pmol template
and 4 pmol Primer1 or Primer2, in the presence or absence of 1 nmol dPa'TP. After 25
cycles of PCR (96°C, 10 sec; 50°C, 5 sec; 60°C, 4 min), the residual dye terminators were
removed from the reaction with CENTRI-SEP columns (Princeton Seperations), and the
solutions were dried. The residues were resuspended in a formamide solution (4 µl) and
fractionated on the ABI 377 DNA sequencer, using a 6% polyacrylamide-6 M urea gel. The
sequence data were analyzed with the Applied Biosystems PRISM sequencing analysis
v3.2 software.
PCR was performed in 20 mM Tris-HCl buffer (pH 8.8), with 10 mM KCl, 10
mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100, 0.3 mM each dNTP (N = Pa, G, C, and
T) and dNTPN (N = Ds and A), 1 µM each Primer1 and Primer2, 2.3-2.4 nM
double-stranded DNA fragment (DNA1-9), and 0.04 unit/µl Vent DNA polymerase (NEB)
on the PTC-100 Controller. The PCR cycle was as follows: 94°C, 0.5 min; 45°C, 0.5 min;
65°C, 4 min. For sequencing analysis, the PCR products were purified by gel
electrophoresis (7% polyacrylamide-7 M urea gel) or filtration using Microcon YM-30
(Millipore) and Micropure-EZ (Millipore). After sequencing reactions with or without
dPa'TP, the sequences were analyzed on the ABI 377 DNA sequencer, using a 6%
polyacrylamide-6 M urea gel (see Supplementary Figs. 1 and 2).
Some DNA templates (DNA2, 4, 5, 7-9) with different sequence contexts around
the unnatural bases did not yield a clear gap on the sequencing pattern obtained in the
presence of dPa'TP. For example, in the sequencing of DNA templates (DNA2) containing
a 5'-TDsG-3'/3'-APaC-5' sequence, the resulting fragment yielded by the incorporation of
the dye-terminator A, at a position following the Pa' incorporation, showed unusual
mobility on the sequencing gel, and the peak corresponding to the A was shifted forward
(Supplementary Fig. 2a). Thus, the positions of the unnatural bases in DNA fragments
28

should be confirmed by comparing both of the peak patterns obtained from sequencing in
the presence and absence of dPa'TP.
In addition, sequencing of the original DNA fragments, DNA8 and DNA9,
containing a 5'-GDsT-3'/3'-CPaA-5' sequence, revealed unusual patterns. In the sequencing
in the presence of dPa'TP, strong peaks of the dye-terminators of dideoxy-A and -C
appeared around the unnatural base position. Similarly, in the sequencing in the absence of
dPa'TP, the peaks following the unnatural base position did not disappear, although this
phenomenon was improved by heating the sample in the PCR buffer (pH 8.8)
(Supplementary Fig. 2g) or a concentrated NH4OH solution. In spite of this abnormality,
the amplification specificity and efficiency of DNA8 and DNA9 were as high as those of
the other DNA fragments (Supplementary Fig. 2g).
29

VII. T7 transcription for 17-mer RNA fragments.
Transcription (20 µl) was performed in 40 mM Tris-HCl buffer (pH 8.0) containing 24 mM
MgCl2, 2 mM spermidine, 5 mM DTT, 0.01% Triton X-100, 1 mM each natural NTP, 0–3
mM PaTP, 0–3 mM Pa'TP, 0–3 mM DsTP, 10 mM GMP, 2 µM template, and 2.5 units/µl
T7 RNAP (Takara). For the efficiency analysis, the reactions were carried out in the
presence of 2 µCi [γ-32P]GTP (Perkin Elmer, instead of GMP). After an incubation at 37°C
for 3 h, the reactions were quenched by the addition of the dye solution. The mixtures were
heated at 75°C for 3 min, and were loaded onto a 15–20% polyacrylamide–7 M urea gel.
The products on the gels were analyzed with a bio-imaging analyzer (BAS2500, Fuji Film).
For the nucleotide-composition analysis, transcripts were internally labeled with
0.1 µCi/µl [α-32P]UTP or [α-32P]ATP (Amersham). The transcripts were digested by 0.75
unit of RNase T2 at 37°C for 2 h, in 10 µl of 15 mM sodium acetate buffer (pH 4.5) with
0.05 A260 unit of E. coli tRNA (Sigma). The digestion products were analyzed by 2D-TLC
(HPTLC plate, 100 x 100 mm, Merck) with the following developing solvents: isobutyric
acid-ammonia-water (66:1:33 v/v/v) for the first dimension, and isopropyl
alcohol-HCl-water (70:15:15 v/v/v or 75:15:10 v/v/v for Pa'-transcripts) for the second
dimension. The spots on the TLC plates were analyzed with a bio-imaging analyzer,
BAS2500 (Fuji Film) (see Supplementary Fig. 3 and Table 2).
30

VIII. Selectivity of the Ds–Pa pair in transcription.
In the nucleotide-composition analysis of the 17-mer transcripts, 1% of the total
misincorporation rate in the transcripts can be detected, and thus, the misincorporation of
Pa or Ds opposite the natural bases corresponded to less than 0.25% per position on average,
as determined by the formula: (the Pa or Ds composition) / [(total numbers of nucleotides at
5' neighbor of A or U) = 4].
By using transcripts with a long chain length, small amounts of misincorporation
can be detected. Thus, the analysis of the biotinylated 152-mer transcripts is more useful for
detecting the subtle misincorporation of the unnatural substrate, Bio-PaTP, opposite the
natural bases. The average misincorporation rates per position were determined by using
the formula: y = [1-(1-0.01x)152]×100, where y is the yield (%) of the complex of the
biotinylated transcripts with streptavidin (nearly equal to the percent of the total Bio-Pa
misincorporation) and x is the misincorporation rate (%) per position. For example, when
the misincorporation (x) is 0.25%, the mobility-shifted band yield (y) corresponds to 31%.
31