synopsis - welcome to open access repository of indian...
TRANSCRIPT

Synopsis
1
The thesis entitled “Oxidative functionalization of alkenes by an intramolecular
sulfinyl nucleophile as the key step in the synthesis of (-)-tetrahydrolipstatin (THL),
analogues of mannostatin A and toward a synthesis of tetrahydroxy long chain base
(LCB)” consists of three chapters.
CHAPTER I
A brief account on synthesis of enantiomerically enriched sulfoxides and their
application to asymmetric synthesis of biologically active natural products
This sections deals with the preparation of homochiral sulfoxides and the
application of the sulfinyl chirality to asymmetric synthesis of bioactive target molecules as
reported by various research groups.
CHAPTER II
Stereoselective synthesis of (-)-tetrahydrolipstatin
In this chapter a brief account of the earlier synthesis of (-)-tetrahydrolipstatin
(THL) by various research groups and an elaborate account of the present work carried out
is described.
Tetrahydrolipstatin (THL) 2, a β-lactone antibiotic marketed as anti-obesity drug
under the trade name of Xenical®, functions as a potent and irreversible inhibitor of
pancreatic lipase (Figure 1). THL 2 is a crystalline and non-natural derivative of Lipstatin 1
with a β-lactone moiety derived from 5-substituted-3,5-dihydroxy-2-hexylpentanoic acid
having S,S,S, absolute configuration. In view of the interesting structure and important
biological activity in vivo, a number of groups have reported the total synthesis of THL by
a variety of synthetic strategies.
Figure 1
O
O
O
n-C6H13
NHCHO
O
O
n-C11H23
OO
n-C6H13
NHCHO
O
Lipstatin 1 Tetrahydrolipstatin 2

Synopsis
2
The disconnective analysis shown in Scheme 1 is based on utilizing the sulfinyl
group in 6 as an intramolecular nucleophile to prepare bromohydrin 5. Epoxide formation,
hydroxy directed opening of epoxide followed by Pummerer and ene reactions would yield
triol derivative 4. The primary hydroxy group in 4 can be oxidized to the carboxylic acid
and subsequent transformations following previously established protocol would afford
THL 2. The envisaged route differs from earlier approaches in that the sulfoxide chirality
was to be utilized to introduce the allylic stereogenic center and this in turn other
stereocenters by asymmetric induction.
Scheme 1
The synthesis commenced with the reaction of the lithium anion of (R)-methyl p-
tolyl sulfoxide 9 with unsaturated ester 8 to furnish β-keto sulfoxide 10 (Scheme 2). The
ester 8 was readily elaborated from 1,3-propane diol 7 by a sequence of three steps.
Diastereoselective reduction of β-keto sulfoxide 10 using DIBAL in the presence of
anhydrous zinc chloride yielded the allylic alcohol 6 (dr >95:<5), which on treatment with
freshly recrystallized N-bromosuccinimide (NBS) afforded bromohydrin 5 as a single
isomer regio- and stereoselectively.
Me Me
OO
OO
HN Me
Me
OHC
10 52
MeO
OP1 OH O
Me
Me10
43
p-TolS
OBnO OH Br
5
p-TolS
OBnOP OP
4
p-TolS
OBnO OH
6
6
OH
P, P1 = Protecting groups
1 3 5

Synopsis
3
Scheme 2
The transformation of 1,2-diol 5 to the 1,3-syn-diol derivative 12 was envisioned by
epoxide formation and hydroxy directed reduction of the epoxide (Scheme 3). Attempted
reduction using Red-Al afforded a complex mixture of products. The reduction was non-
chemoselective when attempted in a variety of solvents and at different temperatures. The
reduction using titanocene(III) chloride afforded the 1,3-diol 12 cleanly though the isolated
yield was poor.
Scheme 3
The diol 12 was protected as its acetonide 13 using an excess of 2,2-
dimethoxypropane in the presence of catalytic amount of CSA (Scheme 4). Subjecting
acetonide 13 to Pummerer reaction using trifluoroacetic anhydride cleanly afforded the
1. NaH, PhCH2Br, TBAI, THF, 0 oC to rt, 12 h, 58%2. Oxalyl chloride, DMSO then Et3N, CH2Cl2, - 78 to 0 oC, 3 h
3. Ph3PCHCO2Et, CH2Cl2, rt, 12 h, 70%7
p-TolS
OBn
Op-Tol
SMe
O
O
EtO
9
O
10
OH OH
8
p-TolS
O OH
DIBAL, ZnCl2, THF
-78 oC, 2 h, 73%
OBn p-TolS
O OH
OBn
OH
Br
5, 5-exo opening product
NBS, H2O, Toluene
rt, 15 min, 81%6
OBn
LDA, THF, -40 oC, 9, 30 minthen 8 at 0 oC, 1 h, 60%
p-TolS
OBn
O OH
12
OH
Cp2TiCl2, Zn-powder, ZnCl2, THF-23 to 0 oC, 6 h, 41%
p-TolS
O OH
OH
Br
p-TolS
O OH
O5 11
K2CO3, CH3CN
0 oC to rt, 8 h, 80%OBn OBn
Red-Al, THF-23 to 0 oC
Complex mixture

Synopsis
4
Pummerer intermediate I which without isolation was allowed to react with an excess of 1-
decene in the presence of stoichiometric amount of anhydrous SnCl4. A complex mixture of
products resulted from which the desired homoallylic sulfide 14 could be isolated in poor
yields only.
Scheme 4
Difficulties encountered in the reduction of epoxy alcohol 11 and Pummerer
followed by ene reaction on acetonide 13 prompted exploration of an alternate strategy
wherein the decyl side chain would be introduced at the initial stages of the synthesis. In
the revised retrosynthetic plan detailed in Scheme 5, introduction of the decyl chain was
envisaged by alkylation of the dianion derived from 17 and subjecting the aduct 16 to
further transformations. Methyl phenyl sulfoxide was chosen instead of methyl p-tolyl
sulfoxide to avoid the potential deprotonation of the aromatic methyl group by the second
equivalent of the base, instead of the methylene protons directly bonded to sulfur, after the
first equivalent of the base has abstracted the hydroxy proton. In addition to that, 4-
methoxybenzyl group was chosen in lieu of the benzyl group (compare with 6) to
differentiate the secondary hydroxy groups in 20 by intramolecular acetal formation.
Scheme 5
p-TolS
OBn
O O
OC(O)CF3
p-TolS
OBn
O O
14
6
TFAA, CH2Cl2 then1-decene, SnCl4
0 oC, 45 min, 17%
I
p-TolS
OBn
O OH
12
OH, Cat. CSA, CH2Cl2
p-TolS
OBn
O O
13
O
OMe
OMe
rt, 6 h, 89%
PhS
OPMB
O OH OH
PhS
OPMB
O OH
15 169
THL 2
9
PhS
OPMB
O OH
17

Synopsis
5
The β-hydroxy sulfoxide 17 was prepared in a fashion analogous to 6. Treatment of
17 with 3.5 equivalents of LDA and a slight excess of n-decyl iodide yielded a mixture of
products in addition to recovered unreacted starting material (Scheme 6). PMR spectrum of
the crude reside indicated the presence of O-alkylated product and the diene sulfoxide. The
result was no better using methyl lithium, other bases (LiHMDS), varying the temperature
and in the presence of additives (HMPA, TMEDA).
Scheme 6
Being unsuccessful in introducing the side chain, Wacker-type reaction was
envisaged to prepare β-hydroxy ketones from β-hydroxy-γ,δ-unsaturated sulfoxides in a
key step of THL 2 synthesis. Hydroxy directed reduction of the keto group and introduction
of the decyl side chain by Pummerer ene reaction were plan in the revised retrosynthetic
design detailed in Scheme 7.
Scheme 7
The β-hydroxy sulfoxide 17 was subjected to palladium catalyzed oxidative
functionalization to furnish β-hydroxy ketone 19 with complete regioslectivity (Scheme 8).
Stereoselective reduction of 19 using sodium borohydride in the presence of
diethylmethoxyborane afforded 1,3-diol 20 (dr >95:<5).
PhS
OPMB
O OH
19
PhS
OO O
18
6
THL 2
H PMP
O
17
O
MeLi, THF, 0 oC, 2 h then
n-C10H22I, -78 to 0 oC, 5 hPh
S
O OH
17Complex mixtureOPMB

Synopsis
6
Scheme 8
Treatment of diol 20 with DDQ in anhydrous dichloromethane yielded the 4-
methoxybenzylidene acetal 21 (Scheme 9). The hydroxy group in 21 was protected as its
MOM-ether 22 using MOM-Cl in the presence of Hunig’s base. Attempted one-pot
Pummerer ene reaction afforded once again a complex mixture of products. In an alternate
route diol 20 was converted to the diacetate 23 and subjected to one-pot Pummerer
followed by ene reaction but without any success. The failure can be rationalized by the
competitive activation of the oxygens in 1,3-acetonide 13, the acetal 22 and the 4-methoxy
benzyl group in 23 by the Lewis acid leading to many side reactions.
Scheme 9
The decyl side chain was introduced by a different strategy that warranted the
transformation of the toluenesulfinyl moiety into a hydroxy group. Toward this end β-
hydroxy sulfoxide 21 was subjected to Pummerer reaction to afford an intermediate, which
without isolation was transformed to alcohol 24 in an one-pot operation by treatment with
PhS
O OH OH
PhS
OOH O
H PMP
DDQ, CH2Cl20 oC, 30 min, 61%
OMOM-Cl, iPr2NEt,
TBAI, CH2Cl20 oC to rt, 6 h, 66%
Ac2O, Et3N,
Cat. DMAP, CH2Cl20 oC to rt, 2 h, 82%
PhS
O OAc OAc
PhS
OO O
H PMP
O
O
OPMB OPMB
Complex mixture
20 23
2221
TFAA, CH2Cl2
thenMe
7and SnCl4
TFAA, CH2Cl2
thenMe
7and SnCl4
Complex mixture
NaBH4, THF, -78 oC, 2 h, 73%Ph
S
O OH OHB OMe
Et
Et
, MeOH, -78 oC, 30 min
20
PhS
O OH PdCl2, CuCl,O2, DMF, H2O
50 oC, 4 h, 70%Ph
S
O OH O
17 19OPMB
OPMB
OPMB

Synopsis
7
saturated aq. NaHCO3 followed by sodium borohydride (Scheme 10). The diol 24 was
selectively monotosylated using p-toluenesulfonyl chloride in the presence of Et3N and
dibutyltin oxide to furnish tosylate (not indicated) which on treatment with anhydrous
potassium carbonate in acetonitrile yielded the epoxide 25. This on treatment with an
excess of decylmagnesium bromide in the presence of catalytic quantities of CuCN yielded
the long chain alcohol 26. The same was more conveniently prepared in an one-pot
operation following the Forsyth protocol. Thus diol 24 on treatment with tosyl imidazole
using sodium hydride as the base furnished epoxide 25, which without isolation was reacted
with an excess of decylmagnesium bromide as before to furnish 26.
Scheme 10
The alcohol 26 was protected as its benzyl ether 27 by the treatment with sodium
hydride and benzyl bromide (Scheme 11). 4-Methoxybezylidine acetal residue was
deprotected using PPTS in methanol to furnish the diol 28. Chemoselective oxidation of the
primary hydroxy group with the use of iodobenzene diacetate and catalytic TEMPO yielded
aldehyde 29. Further oxidation of 29 employing the Pinnick protocol cleanly afforded the
acid 30 which was transformed into the methyl ester 31 using ethereal diazomethane.
PhS
O OH
21
O O
PMP H
HO
OH O O
PMP HTFAA, Et3N, CH2Cl2, 0 oC 15 min,
then aq. NaHCO3, NaBH4, 20 min, 50%
24
N
N
Ts
NaH, THF
0 oC to rt, 40 min
Me (CH2)9MgBr, cat. CuCN
-10 to 0 oC, 2 h, 73% (for two steps)
O O O
PMP H
Me
OH
26
O O
PMP H
1025

Synopsis
8
Scheme 11
Frater-Seebach alkylation of 31 with hexyl iodide using LiHMDS as the base
proceeded cleanly to afford the mono alkylated product 3. Hydrolysis of the methyl ester
using aq. LiOH yielded the acid 32, which was transformed to the β-lactone 33 with the use
of BOP-Cl in the presence of Et3N (Scheme 12). Hydrogenolysis of the benzyl ether
afforded the alcohol 34 which was coupled to N-formyl leucine 35 using reported
conditions to furnish tetrahydrolipstatin 2. The physical data of 2 were in full agreement to
those reported in the literature.
Scheme 12
Me
OH O O
PMP H
10
Me
OBn
28
OH OH
10
NaH, BnBr, cat. TBAI, THF
0 oC to rt, 12 h, 72%
cat. PPTS, aq. MeOH
reflux, 4 h, 60%Me
OBn O O
PMP H
1026 27
Me
OBn
29
OH O
Me
OBn OH O
OH
PhI(OAc)2, cat. TEMPO, CH2Cl2
rt, 2 h, 65%
NaClO2, t-BuOH, NaH2PO4.H2Ocyclohexene, H2O
0 oC to rt, 3 h, 75%
CH2N2, Et2O
rt, 15 min, 90%Me
OBn OH O
OMe
H
30 31
10
Me
OBn OH O
OMe
MeOMe
OBn OH O
Me
10
4
LiHMDS, THF, -55 oC,1 h, then n-C6H13I, HMPA
-55 to -15 oC, 3 h, 70%1031 3
Me Me
OO
OBn
10 533
Me Me
OO
OO
HN Me
Me
OHC
10 52
Aq. 1N LiOH, THF
0 to 65 oC, 2 h, 70%
BOP-Cl, Et3N, CH2Cl2
rt, 1 h, 65%
34
H2, Pd(OH)2/C, MeOH
rt, 4 h, 80%
DCC, DMAP, CH2Cl2rt, 24 h, 72%
HN Me
Me
OHC
HO2C
35
MeOH
OBn OH O
Me
10
4
Me Me
OO
OH
10 5
32

Synopsis
9
CHAPTER III
Section A: Stereoselective synthesis of analogues of mannostatin A
This section deals with a brief account of the synthesis of mannostatin A, reported
by the various research groups and an elaborate account of the present work carried out.
Mannostatin A 36, a member of the aminocyclopentitol family of natural products,
was isolated by Aoyagi and coworkers and exhibits strong mannosidase inhibitory activity
(Figure 2). The densely functionalized structure and interesting biological properties has
stimulated several studies focusing on the synthesis of mannostatin and their stereisomeric
analogs.
Figure 2
Synthetic potential of a sulfinyl moiety as an intramolecular nuleophile toward
oxidative heterofunctionalization of an alkene was envisaged for designing a stereoselective
route to phenylthio analogue of mannostatin A 38. By a retrosynthetic analysis shown in
Scheme 13, 38 could be derived from the triol derivative 40 via the intermediacy of an
oxime ether 39. Retron 40 can be secured from bromohydrin 41 and that in turn from allylic
alcohol 42.
Scheme 13
SMe
OH
OHHO
H2N
S
OH
OHHO
H2N
S
OP
OPPO
N
S
OP
OHHO
PO
Ph
P
O Ph PhO
36 38 39 40
PhS OP
O OP Br
OH
3 5Ph
S OP
O OH
41 42 [P = Protecting groups]
1
OHHO
HO
NH2
SCH3
4512
3
OHHO
HO
NH2
SCH3
45
123
O
mannostatin A36
mannostatin B37

Synopsis
10
The synthesis commenced with the condensation of the lithium carbanion of (R)-
methyl phenyl sulfoxide 46 with the unsaturated ester 45 to yield the β-keto sulfoxide 47
(Scheme 14). Trans Olefine 45 was readily elaborated from cis but-2-en-1,4-diol 43 in a
three step sequence that includes, protection as di-PMB-ether 44 followed by ozonolysis
and subsequent Wittig olefination. Diastereoselective reduction of 47 using DIBAL in the
presence of anhydrous zinc chloride yielded allylic alcohol 42 (dr >95:<5). Oxidative
functionalization of 42 using freshly recrystallized NBS as the electrophile furnished
bromohydrin 41 via intramolecular participation of the sulfinyl group.
Scheme 14
The anti, anti-triol motif present in mannostatin A, required that the stereogenic at
C3 and C4 be inverted. This was accomplished by a three step sequence. Treatment of 41
with anhydrous potassium carbonate in acetonitrile furnished epoxide 48 (Scheme 15).
Protection of the hydroxy group as its silyl ether 49 followed by borontrifluoride etherate
(BF3.Et2O) mediated 5-exo opening of the epoxide by the sulfinyl group furnished diol 50
possessing three heteroatoms that are mutually anti disposed.
PhS
O
OPMB
O
PhS
Me
OO
EtO
OHHO OPMBPMBO
OPMB
43 44
45
46
47
NaH, n-Bu4NF, THF0 oC, rt, 30 min, then PMB-Br
70 oC, 12 h, 77%
1. O3, CH2Cl2, -78 oC, 30 minthen Me2S, - 78 oC to rt, 6 h
2. PPh3CHCO2Et, benzenert, 6 h, 81% (for two steps)
LDA, THF, -40 oC, 46, 30 minthen 45 at 0 oC, 1 h, 60%
PhS
O OH
42
DIBAL, ZnCl2, THF
-78 oC, 2 h, 89%
OPMBPh
S
OOH
OH
Br
41
NBS, H2O, toluene
rt, 15 min, 80%
OPMB3 4

Synopsis
11
Scheme 15
Intramolecular addition of the α–sulfinyl carbanion to an imine derived from
aldehyde 53 was envisaged to form the C1–C5 bond to furnish the carbocycle 38. Since the
aldehyde was readily available and expected to be more reactive it was used to study the
carbocyclization. Protection of the hydroxy groups of 50 by treatment with methoxymethyl
chloride (MOM-Cl) in the presence of Hunig’s base afforded the di–MOM derivative 51
(Scheme 16). The PMB group of 51 was deprotected with DDQ to afford the alcohol 52
that on oxidation employing Swern protocol yielded aldehyde 53.
Scheme 16
Aldehyde 53 was subjected to the treatment with lithium diisopropylamide (LDA)
in THF. While no change could be observed at –78 oC, warming to higher temperature (–23 oC) led to the undesired elimination resulting in the formation of unsaturated aldehyde 54
(Scheme 17). It is likely that the bulky LDA is unable to abstract the methylene proton
PhS
O OH
OH
Br
OPMBPh
S
O OH
O
41 48
K2CO3, MeCN
0 oC to rt, 8 h, 88%
OPMBTBS-Cl, Imidazole, CH2Cl2
rt, 1 h, 93%
PhS
O OTBS
O
49
OPMBPh
S
OOTBS
OH
OH
42
BF3.Et2O, Et2O/ CH2Cl2
-78 to 0 oC, 4 h, 73%
50
OPMB
PhS
OH
OOTBS
OMOM
MOMO
DDQ, aq.CH2Cl2
0 oC to rt, 30 min, 87%
(C(O)Cl)2, DMSO, Et3N, CH2Cl2
-78 to -10 oC, 45 min, 93% PhS
OOTBS
OMOM
MOMO O
52 53
PhS
OOTBS
OH
OH
2
MOM-Cl, iPr2NEt, DMAP, CH2Cl2
0 oC to rt, 6 h, 82%Ph
S
OOTBS
OMOM
OMOM
3 5 OPMBOPMB4
50 51

Synopsis
12
directly bonded to sulfur, it instead abstracts the proton α to the aldehyde at higher
temperature resulting in epimerization and β–elimination. The outcome was however not
different with the use of a less bulky lithium diethylamide as the base in the presence or
absence of added HMPA. Therefore studies directed toward addition of sulfinyl carbanion
to the imine were not explored.
Scheme 17
In an alternate strategy, intramolecular Pinacol reaction was explored to synthesize
mannostatin A 36. By the revised retrosynthetic design depicted in Scheme 18, mannostatin
A 36 was envisaged to be obtained from the acetonide 55 which can be secured from the
dialdehyde 56 and that inturn from the dihydroxy sulfoxide 50.
Scheme 18
Thus diol 50 was protected as its acetonide 57 (Scheme 19). DDQ mediated
deprotection of 4-methoxybenzyl residue furnished alcohol 58. Alcohol 58 when subjected
to Pummerer reaction, followed by hydrolysis of the intermediate afforded an aldehyde that
was reduced in the same pot to furnish diol 59. Oxidation employing Swern conditions
yielded dialdehyde 56 which was subjected to the intramolecular Pinacol coupling using
catalytic Cp2VCl2 in combination with TMSCl and Zn as stoichiometric reductant
exploiting Hirao’s protocol yielded a complex mixture of products. The outcome was no
better under a variety of reaction conditions.
PhS
O
OOTBS
OMOM
MOMO
PhS
O
OOTBS
OMOM
53 54
LDA, THF
-78 to -23 oC, 1 h, 57%
SMe
OHHO
HO
NH2
PhS OPMB
O OTBS OH
OH
OH
OO
TBSO
OH
O
OO
TBSO
O
505636 55

Synopsis
13
Scheme 19
As the intramolecular Pinacol coupling proved unsuccessful, α-chlorosulfide 63 was
envisaged as the precursor to carbon centered radical to add onto the oxime ether to form
the desired carbocycle. Toward this end, alcohol 58 was oxidized with the use of Swern
protocol to yield the aldehyde 60 (Scheme 20). Reaction with O-benzylhydroxylamine
hydrochloride in presence of aq. pyridine furnished the oxime ether 61. Reduction of the
sulfinyl moiety using sodium iodide and TFAA in acetone afforded sulfide 62. α–
Chlorosulfide 63 was prepared insitu by the treatment of sulfide 62 with N-
chlorosuccinimide (NCS) in benzene and without isolation was subjected to free radical
cyclization using n–Bu3SnH conditions. A complex mixture of products resulted both in the
presence and absence of added Et3N.
PhS
O OTBS
OH
OPMB
OH50
DDQ, aq.CH2Cl2
0 oC to rt, 30 min, 85%
PhS
O OTBS
O
OH
O
58
TFAA, Et3N, CH2Cl2, 0 oC 15 min,
then aq. NaHCO3, NaBH4, 20 min, 75%
, Cat. CSA, CH2Cl2
OMe
OMe
rt, 6 h, 87%
PhS
O OTBS
O
OPMB
O57
HO
OTBS
O
OH
O
59
(C(O)Cl)2, DMSO, Et3N, CH2Cl2
-78 to -10 oC, 45 min
O
OTBS
O
O
O
56
Cp2VCl2, Zn, Me3SiCl, THF, -20 oC, 16 hComplex mixture
Complex mixture
Complex mixture
Cp2TiCl2, Zn, Me3SiCl, THF, -20 oC, 24 h
SmI2, Mg, Me3SiCl, THF, -20 oC to rt, 2 h

Synopsis
14
Scheme 20
Addressing the issue of operational difficulty associated with highly sensitive α–
chlorosulfides, corresponding α–chlorosulfoxide 64 was envisioned to be a stable and
alternate radical precursor. Thus sulfoxide 61 was converted to α-chlorosulfoxide 64 and
was subjected to the free radical cyclization using n–Bu3SnH (Scheme 21). Unfortunately, a
complex mixture of products was obtained.
Scheme 21
Exploring an alternate route sulfoxide 51 was subjected to Pummerer rearrengment,
the resulting intermediate was hydrolyzed in the same pot to furnish aldehyde 65 which
without purification was used in the next step (Scheme 22). Dithioacetal formation using
PhS
OTBS
O
N
O
NCS, CCl4
rt, 2 h, 80% PhS
OTBS
O
N
OCl
Complex Mixture
n-Bu3SnH, AIBN, PhH
reflux, 3 h
61
O O
64
OBn OBn
61
HCl.NH2OBn, aq. Py, DCE
reflux, 8 h, 73% PhS
O OTBS
O
N
O
TFAA, NaI, Acetone
0 oC, 3 h, 77%Ph
S
OTBS
O
N
O
62
PhS
O OTBS
O
OH
O
(C(O)Cl)2, DMSO, Et3N, CH2Cl2
-78 to -10 oC, 1 h, 81%
58
PhS
O OTBS
O
O
O
60
OBn OBn
NCS, PhH
rt, 1.5 h PhS
OTBS
O
N
OCl
Complex Mixture
n-Bu3SnH, AIBN, PhH
reflux, 3 h
OBn
63

Synopsis
15
dimethyl disulfide and tributylphosphine proceeded well to afford compound 66. The 4-
methoxybenzyl group was deprotected to afford the alcohol 67. Oxidation using Parikh-
Doering conditions cleanly yielded the aldehyde 68 while other conditions screened such as
a) Swern, b) buffered Dess-Martin periodinane and c) BAIB/TEMPO resulted in a partial
cleavage of dithioacetal residue. Oxime ether formation proceeded without incident to yield
compound 69.
Scheme 22
Subjecting the oxime ether 69 to free radical cyclization employing the conditions
reported by Roberts and coworkers afforded an inseparable mixture of two
aminocyclopentitol derivatives 70 and 71 in 6:4 ratio (Scheme 23). The structures were
assigned to the cyclized products based on precedent, PMR and 2D-NOESY studies.
Scheme 23
PhS
OPMB
OOTBS
OMOM
MOMO
O
OPMB
TBSO OMOM
MOMO
65
TFAA, Et3N, CH2Cl2, 0 oC, 15 min
then aq. NaHCO3, 20 min, 77%
51
PMBO
MeS
OTBS
OMOM
OMOMMeS
66
MeSSMe, PBu3
rt, 14 h, 69%
DDQ, aq. CH2Cl2
0 oC, 30 min, 86%
O
MeS
OTBS
OMOM
OMOMMeS
68
SO3.Py, DMSO, Et3N, CH2Cl2
rt, 3 h, 86 %
HCl.NH2OBn, aq. Py, DCE
refllux, 8 h, 74%
N
MeS
OTBS
OMOM
OMOM
BnO
MeS
69
HO
MeS
OTBS
OMOM
OMOMMeS
67
+
N
MeS
OTBS
OMOM
OMOM
BnO
MeS
n-Bu3SnH, AIBN, Toluene
reflux, 7 h, 68%
69
SMe
OTBSMOMO
MOMO
NHOBn
70
SMe
OTBSMOMO
MOMO
NHOBn
71(6:4)

Synopsis
16
Section B: Toward a synthesis of tetrahydroxy long chain base (LCB)
This chapter deals with a brief account of the synthesis of tetrahydroxy long chain
base (LCB) reported by various research groups and a detailed account of the present work.
Amino–tetrahydroxyoctadecene 72, constitutes the long chain base (LCB) of the
cerebroside 73 isolated from the latex of Euphorbia characias L (Figure 3). The unique
structure of LCB 72 in particular, with four contiguous chiral centers disposed mutually
anti to each other, (Z)–double bond and the biological importance of cerebrosides in
general, make it an attractive target for synthesis.
Figure 3
A stereoselective route to LCB 72 was designed utilizing the sulfinyl moiety as an
intramolecular nucleophile for the vicinal heterofunctionalization of an alkene in the key
step of the reaction sequence.
By a retrosynthetic disconnection detailed in Scheme 24, the aminotetrol 74 was
arrived at as a key intermediate that was envisaged to be obtained from the enantiomer of
the triol derivative ent-51 via the intermediacy of an imine 75. Retron ent-51 can be traced
back to allylic alcohol ent-42 via bromoydrine ent-41.
Scheme 24
n-C11H23
OHOHOH
OH NH2
2 46
PhS
OP4O OP2 OP3
OP3 NHP1
2 4 6
PhS OP1
O OP2 Br
OH
2 4 PhS OP
O OH
72 74
III -ent-41
III -ent-42
PhS OP1
O OP2 OP3
OP3
III -ent-51 [P, P1, P2, P3, P4= Protecting groups]
PhS
O OP2 OP3
OP3 NP1
2 4
75
HO
n-C11H23OHOH
NH2 OH
Tetrahydroxy-LCB72
O
n-C11H23OHOH
NHCOR
OHOHO
HO
OH
OH
73

Synopsis
17
Enantiomer of the aldehyde ent-53 was prepared from (S)–methyl phenyl sulfoxide
ent–46 in the same manner as detailed in Scheme 16. Of the many methods available for
the stereoselective synthesis of 1,2–amino alcohols, the route employing nucleophilic
addition to sulfinylimines was attractive. Aldehyde 53 on reaction with (S)–tert-
butylsulfinamide 76 in the presence of excess titanium tetraisopropoxide furnished
sulfinylimine 75 (Scheme 25). The choice of (S)–tert-butylsulfinamide was arbitrary since
it was not possible to unambiguously predict the influence of the sulfinamide configuration
on the newly created C-N stereogenic center due to the likelihood of the reaction
proceeding via chelation of the organometallic reagent to N-sulfinyl oxygen/OMOM groups
of the substrate or otherwise. Attempted reaction of 75 with one carbon synthons,
benzyloxymethyllithium, derived from n–Bu3SnCH2OBn/n–BuLi and
benzyloxymethylmagnesium bromide, prepared from BOM-Cl/Mg turnings, returned only
unreacted starting material. Reaction with sterically less bulky vinylmagnesium bromide
however, proceeded cleanly to yield the aminotetrol derivative 74 as the sole product.
Scheme 25
Although the absolute configuration at C5 was not known until later (C4,C5-syn),
further transformations were attempted. It remained to unmask the sulfinyl group to reveal
an aldehyde carbonyl that could be subjected to cis–selective Wittig olefination to introduce
the alkenyl side chain. Toward this goal, sulfoxide 74 was subjected to Pummerer reaction
with the use of trifluoroacetic anhydride (TFAA) in the presence of triethylamine (Et3N)
PhS
O
OOTBS
OMOM
MOMO
H PhS
N
OOTBS
OMOM
MOMOS
O
H
S
O
NH2
75
Ti(OEt)4, CH2Cl2rt, 5 h, 86%
ent-53
S
O
Ph Me
ent-46
76
PhS
OOTBS
OMOM
MOMO HN
MgBr
THF
-78 oC, 3 h, 73%
SO
74
N
H
S
O
Nu
II
OMOMR
H
45

Synopsis
18
(Scheme 26). A less polar spot observed during TLC examination, that was assumed to be
the intermediate, was subjected to hydrolysis with aq. saturated sodium bicarbonate in the
same pot. After work up, none of the desired aldehyde, only unsaturated ketone 81 could be
isolated. The formation of 81 can be rationalized by an intramolecular attack of the
sulfinamide on sulfenium ion 77 to afford thioacetal 78, which suffers elimination of the
thiophenyl moiety to yield iminium ion 79. Compound 79 probably affords the more stable
iminium ion 80 by a [1,3]H shift which on hydrolysis yields 81.
Scheme 26
It was thus clear that the double bond of compound 74 had to be transformed into a
hydroxymethyl derivative prior to attempting the Pummerer reaction. The sulfinamide
moiety in 74 was deprotected with anhydrous 4 N HCl/dioxane, the resulting amine
hydrochloride without isolation was treated in the same pot with acetic anhydride in the
presence of an excess of triethylamine to furnish amide 82 (Scheme 27). Ozonolysis of the
terminal alkene and reduction of the resultant aldehyde afforded alcohol 83. The hydroxy
protecting groups were removed using CSA in methanol to yield tetrol 84. Reprotection
using an excess of acetic anhydride furnished pentaacetate 85. Treatment of 85 with TFAA
in the presence of Et3N afforded a less polar product (TLC), assumed to be the intermediate
III . Further hydrolysis of the intermediate only returned the unreacted starting material.
PhS
OOTBS
OMOM
MOMO HNS
O
74
TFAA, Et3N,CH2Cl20 oC, 15 min.
then aq NaHCO3N
PhS
OTBS
OMOM
OMOMSO
HN
PhS
OTBS
OMOM
OMOMSO
N
OTBS
OMOM
OMOMSO N
OTBS
OMOM
OMOMSO S
HN
OTBSOMOM
MOMO O
O
77 78
79 80 81
Aq NaHCO3
39%

Synopsis
19
Scheme 27
Efforts to obtain an aldehyde from sulfoxide 74 were thus thrawted and therefore it
was decided to attempt the Pummerer reaction before introducing the amino stereogenic
center. Thus enantiomer of the acetonide ent-57 was subjected to Pummerer reaction, the
intermediate IV , was hydrolyzed and the resulting aldehyde (not indicated) was reduced in
the same pot to furnish alcohol 87 (Scheme 28). The hydroxy group was protected as its
silyl ether 88. Deprotection of the PMB group in 88 with DDQ afforded alcohol 89 that was
oxidized without incident under Swern conditions to yield aldehyde 90. Sulfinylimine 91
was readily obtained from 90 and was reacted with vinyl magnesium bromide to furnish a
separable mixture of sulfinamides 92 & 93 (dr 85:15).
PhS
OOTBS
OMOM
MOMO HNS
O
74
PhS
OOTBS
OMOM
MOMO NHAc
82
1a. 4 N HCl in dioxane, MeOH0 oC, 3 h
1b. Ac2O, Et3N, CH2Cl2rt, 14 h, 88% (for two steps)
PhS
OH
OOTBS
OMOM
MOMO NHAc
1. O3, CH2Cl2, then Me2Srt, 30 min.
2. NaBH40 oC, 10 min., 78% (for two steps)
83
PhS
O OH OH
OHNHAc
OH
CSA, MeOH
rt. 30 h
Ac2O, Et3N, DMAP, CH2Cl2
rt, 2 h, 62% (Over two steps)Ph
S
O OAc OAc
OAcNHAc
OAc
8584
PhS
OAc OAc
OAcNHAcF3C(O)CO
O
OAc
OAc OAc
OAc NHAc
86, Not Obtained
OAcAq NaHCO3
TFAA, Et3N, CH2Cl2
0 oC, 30 min
III

Synopsis
20
Scheme 28
The structure was assigned to major isomer 92 by single crystal X-ray diffraction of
the acetate 94 prepared by a three step sequence (Scheme 29). The formation of 92 (C4,C5-
anti) as the major product lends support to the structural assignment to 74 (C4,C5-syn).
Scheme 29
PhS
O OTBS
O
OPMB
O
TFAA, Et3N, CH2Cl2
0 oC, 15 min.
ent-57
PhS
OTBS
O
OPMB
O
F3C(O)CO
HO
OTBS
O
OPMB
O
87
Aq NaHCO3 then NaBH4
0 oC, 20 min., 79% (for two steps)
IV
TBSO
OTBS
O
PMB
O
TBS-Cl, Imidazole, CH2Cl2
rt, 1 h, 91%
(C(O)Cl)2, DMSO, Et3N, CH2Cl2
-78 to -10 oC, 1 h, 73 %
DDQ, aq. CH2Cl2
0 oC to rt, 30 min. 84%
88
TBSO
OTBS
O
OH
O89
TBSO
OTBS
O
O
O90
S
O
NH2
Ti(OEt)4, CH2Cl2
rt, 5 h, 86%
TBSO OTBS
OO
NS
O
H
TBSOOTBS
O O
HNS
O TBSOOTBS
O O
HNS
O
91 9392
MgBr , THF
+-78 oC, 3 h, 72%
6 : 1
TBSOOTBS
O O
HNS
O
OAc
92 94
TBSOOTBS
O O
HNS
O 1. OsO4, NaIO4, 2,6-Lutidine, aq dioxane2. NaBH4, 0 oC, 30 min., 73% (for two steps)
3. Ac2O, Et3N, DMAP, CH2Cl2, rt, 2 h, 87 %
Synopsis
21
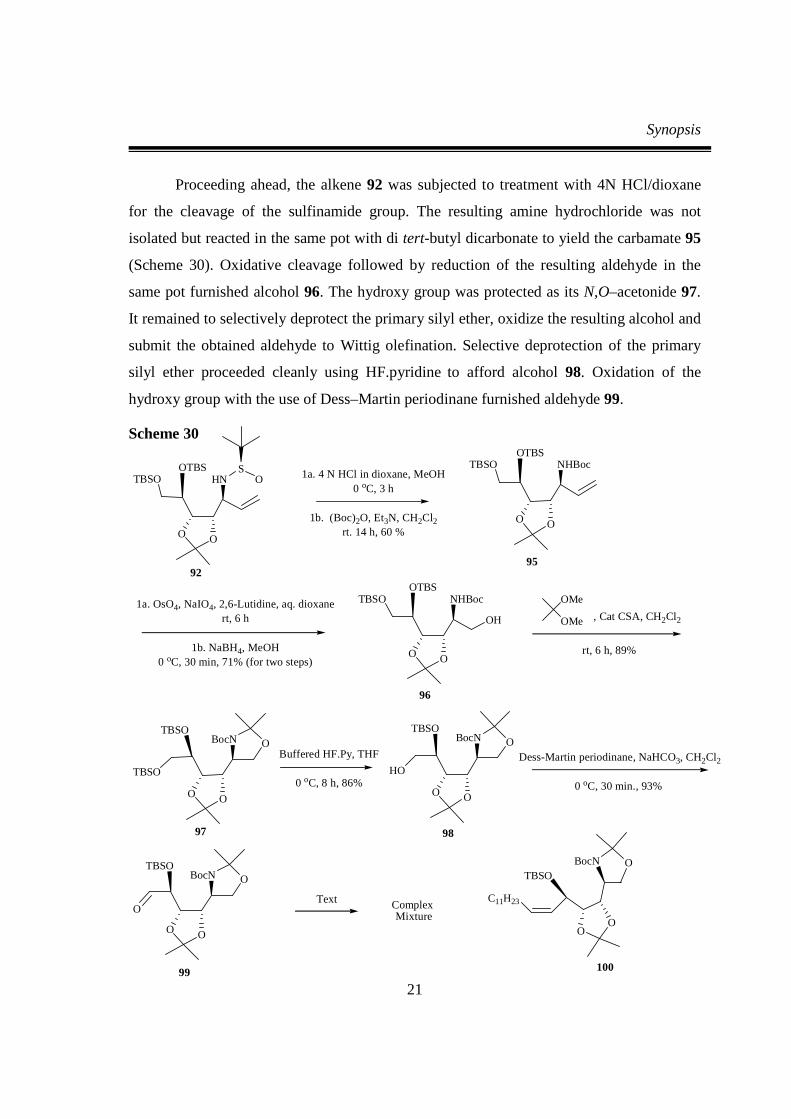
Proceeding ahead, the alkene 92 was subjected to treatment with 4N HCl/dioxane
for the cleavage of the sulfinamide group. The resulting amine hydrochloride was not
isolated but reacted in the same pot with di tert-butyl dicarbonate to yield the carbamate 95
(Scheme 30). Oxidative cleavage followed by reduction of the resulting aldehyde in the
same pot furnished alcohol 96. The hydroxy group was protected as its N,O–acetonide 97.
It remained to selectively deprotect the primary silyl ether, oxidize the resulting alcohol and
submit the obtained aldehyde to Wittig olefination. Selective deprotection of the primary
silyl ether proceeded cleanly using HF.pyridine to afford alcohol 98. Oxidation of the
hydroxy group with the use of Dess–Martin periodinane furnished aldehyde 99.
Scheme 30
TBSOOTBS
O O
HNS
O
92
TBSOOTBS
O O
NHBoc
95
1a. 4 N HCl in dioxane, MeOH0 oC, 3 h
1b. (Boc)2O, Et3N, CH2Cl2rt. 14 h, 60 %
TBSOOTBS
O O
NHBoc
OH
96
TBSO
TBSO
O O
BocN O
1a. OsO4, NaIO4, 2,6-Lutidine, aq. dioxanert, 6 h
1b. NaBH4, MeOH0 oC, 30 min, 71% (for two steps)
OMe
OMe , Cat CSA, CH2Cl2
97
rt, 6 h, 89%
HO
TBSO
O O
BocN O
O
TBSO
O O
BocN O
99
Buffered HF.Py, THF
0 oC, 8 h, 86%
Dess-Martin periodinane, NaHCO3, CH2Cl2
0 oC, 30 min., 93%
98
TBSO
OO
BocN O
Text Complex Mixture
C11H23
100

Synopsis
22
Wittig olefination using the ylid generated from n–dodecanyltriphenylphosphonium
bromide and n–BuLi in THF in the presence of HMPA afforded a complex mixture of
products. Wittig olefination using other bases like LiHMDS and KHMDS both in the
presence and absence of added HMPA afforded only a complex mixture of products. In the
meantime all the available sample of alcohol 98 was consumed. Thus it was unable to
explore alternate routes to prepare the alkene 100. Efforts are in progress to complete the
synthesis of LCB 72.