synthesis and density functional theoretical study of steroidal spiro-triazolidinone
TRANSCRIPT

Journal of Steroid Biochemistry & Molecular Biology 110 (2008) 278–283
Contents lists available at ScienceDirect
Journal of Steroid Biochemistry and Molecular Biology
journa l homepage: www.e lsev ier .com/ locate / j sbmb
Synthesis and density functional theoretical studyof steroidal spiro-triazolidinone
Kamlesh Sharma ∗, Bhajan Lal ∗∗
5�-ciro-1the bdenschanctiontrostw ofr proinedtal fr
Department of Chemistry, University of Pretoria,Pretoria 0002, South Africa
a r t i c l e i n f o
Article history:Received 12 July 2007Accepted 14 November 2007
Keywords:Stereoselective synthesisSteroidal triazolidinoneA free radical reaction mechanismDensity functional theory
a b s t r a c t
The reaction of 3�-chloro-chloro-5�-cholestan-6-spproduct was confirmed onwere performed by usinga free radical reaction meThe mechanism of the readensity map, encoded elecfrontier orbitals and the flomechanism. The moleculastructure, were also explatheir respective fundamen
1. Introduction
The introduction of a heteroatom in the steroidal moleculeaffects the chemical properties of that particular steroid and oftenresults in alterations of its biological activities, which sometimesmay be useful. The syntheses of triazolidinone [1–7], a heteroatomcompound, have attracted considerable interest particularly inpharmaceutical and agricultural field. Many herbicidal activitieshave been shown by triazolidinone compounds [8–10]. Literaturesurvey revealed that most emphasis has been given on synthesesof triazolidinone, but there is the lack of information regarding itscomputational molecular modeling study, which may be useful inenhancing the yield of biologically active compounds at molecularlevel.
Computational chemistry is a set of methodologies employed tostudy chemical problems with the aid of a computer. The informa-tion on geometries and energies of reactants, intermediates and
∗ Corresponding author. Present address: Department of Chemistry, University ofWisconsin-Madison, 1101 University Avenue, WI 53706-1396, USA.Tel.: +1 608 265 8285.∗∗ Corresponding author. Present address: Computational Chemistry, University of
Cape Town, Cape Town 7701, South Africa. Tel.: +27 21 650 2530;fax: +27 21 686 4333.
E-mail addresses: [email protected] (K. Sharma),[email protected], [email protected] (B. Lal).
0960-0760/$ – see front matter © 2008 Elsevier Ltd. All rights reserved.doi:10.1016/j.jsbmb.2007.11.005
holestan-6-one semicarbazone 1 with hydrogen peroxide at 0 ◦C gives 3�-′,2′,4′-triazolidine-3′-one 2 as a product. The structural assignment of theasis of its elemental, analytical and spectral data. The ab initio calculations
ity functional theory (DFT) at B3LYP/6-31G* basis set in order to describeism. The reaction proceeds through two radical intermediates formation.was explained by using frontier molecular orbital (FMO), spin electronic
atic potential map and atomic charges. It was found that the localization ofatomic charges of all the calculated structures support the present reactionperties like total energy, dipole moment and hardness of each optimized. Stability of all the optimized structures in this study was supported byequencies and energy minima.
© 2008 Elsevier Ltd. All rights reserved.
products provide the microscopic perspective of chemical reac-tions; all this information can be obtained by using quantummechanics method. In recent years, density functional theory hasbeen a shooting star in molecular quantum mechanics. The devel-opment of better and better exchange-correlation functionals made
it possible to calculate many molecular properties with compa-rable accuracies to traditional correlated ab initio methods, withmore favorable computational costs [11]. Literature survey revealedthat the DFT has a great accuracy in reproducing the experimentalvalues of quadrupole hyperfine coupling constants [12], molec-ular structural properties [13], Infrared (IR) frequencies and IRintensities [14,15] etc. During previous theoretical studies [16–18]toward steroid derivatives amenable for biological chemistry appli-cations, we utilized Hartree-Fock/6-31G* method. However, thereis no theoretical investigation has yet been reported where morereliable method, B3LYP functional [19] is used to explain thereaction mechanism of steroidal triazolidinone. These significantfacts prompted us to undertake the present study of the titlecompound.In this article, we report synthesis and a free radical reactionmechanism of formation of 3�-chloro-5�-cholestan-6-spiro-1′,2′,4′-triazolidine-3′-one from respective ketone semicarbazonemoiety. To gain insight into the factors influencing reactivity, feasi-bility and stability of the reaction molecules, we focus on evaluationof the electronic effects by considering the results of B3LYP/6-31G*density functional models.

mistry
closure from one side of the ring at C6 carbon atom, which mightbe explained on the basis that –NH–CO– group is more bulky interms of molecular weight than –NH–NH– group. Therefore, theheterocyclic ring closes at C6 carbon atom by the attack of –CO–HN•
(being a radical) preferably in the way that –NH–CO– group becameequatorial to the C6 position to avoid 1,3-diaxial interaction due tothe C10�–CH3 group. As a result, only one isomer of this reaction,in which –NH–CO– group is cis with respect to the C5�–H, wasselectively obtained.
3.4. Theoretical investigation
The geometry of all the four steroid molecules; ketone
K. Sharma, B. Lal / Journal of Steroid Bioche
2. Experimental
2.1. General
1H NMR spectrum (CDCl3) was recorded on a Bruker BZH-200instrument. Tetramethylsilane (TMS) served as an internal stan-dard (ı = 0) for 1H NMR. Multiplicities of proton resonance weredesignated as singlet (s) and multiplet (m). The Infrared (KBr) spec-trum recorded on a PerkinElmer 782 IR spectrophotometer. Meltingpoint was recorded on a Kofler apparatus and is uncorrected. All thechemicals were of analytical grade and were used without furtherpurification. Reaction was monitored by thin layer chromatography(TLC) using 0.25 mm E. Merk precoated silica gel 60 (particle size0.040–0.063 mm).
2.2. 3ˇ-Chloro-5˛-cholestan-6-spiro-1′,2′,4′-triazolidine-3′-one2
To synthesize compound 2, the solution of 3�-chloro-5�-cholestan-6-one semicarbazone [20] (1.00 g, 2.10 mmol) inchloroform (25 mL) was treated with excess of hydrogen perox-ide (30%, 4 mL) and stirred for 3 h at 0 ◦C. During this period thesolution turned light yellow. The organic layer was separated,dried over anhydrous sodium sulphate and evaporated to drynessunder reduced pressure. The crude product obtained was puri-fied over silica gel column (light petroleum ether–ethyl acetate;4:1). Recrystallisation from methanol gave a white crystalline prod-uct 2 in high yield (85%), mp 162–163 ◦C. IR �max (KBr): 3438(N–H), 1442–1407 (N–H), 1699 (C O), and 715 cm−1 (C–Cl). 1H NMR(200 MHz, CDCl3): ı 7.84 (s, 1H, CONH), 10.58 (s, 2H, –NH–NH–)and 4.52 (m, 1H, w½ 18 Hz, axial, C3�–H). Analysis. Calculated forC28H48N3OCl: C, 70.33; H, 10.12; N, 8.79%. Found: C, 70.34; H, 10.16;N, 8.83%.
2.3. Computational methods
The geometry of 3�-chloro-5�-cholestan-6-one semicarbazone1, two radical intermediates (A and B) and 3�-chloro-5�-cholestan-6-spiro-1′,2′,4′-triazolidine-3′-one 2, were optimized forthe description of reaction mechanism of formation of steroidalspiro-1′,2′,4′-triazolidine-3′-one.
The optimized geometries of all the molecules (1, A, B and 2)
were calculated by using quantum chemical calculations includingsemi-empirical and ab initio method. The computational calcula-tions of all the molecules started with semi-empirical PM3 method,followed by the minimal STO-3G basis set Hartree-Fock (HF) model.The resulting energy, Hessian matrix, wave function and optimizedgeometry of the calculated molecule were used to perform the nextlevel of calculation with split-valence basis set 3-21G(*). The pro-cedure was applied further for a calculation at the HF/6-31G* levelof theory. Finally, the density functional calculation using B3LYPmethod [19] with 6-31G* basis set [21] was performed. Analyti-cal vibrational frequency calculations were performed for all themolecules. Exact zero imaginary vibrations characterize stationarypoints. The asterisk means that “d” polarized functions were addedfor C, O and N atoms.To obtain the real picture of the reaction mechanism, the fol-lowing molecular and electronic properties of all the structureswere calculated: total energy, atomic charges, frontier molecularorbital (FMO), hardness (�), electrostatic potential map, spin elec-tronic density map and dipole moment. Spartan’04 Windows [22],quantum mechanics software, was used for computation. All thecalculations were carried out on a Pentium 4 (4.2 GHz) based com-puter.
& Molecular Biology 110 (2008) 278–283 279
3. Results and discussion
3.1. Synthesis
The synthesis of requisite steroidal 3�-chloro-5�-cholestan-6-spiro-1′,2′,4′-triazolidine-3′-one 2 from the reaction of 3�-chloro-5�-cholestan-6-one semicarbazone [20] 1 with hydrogen peroxideat 0 ◦C was accomplished as summarized in Scheme 1. Overall yieldwas excellent (85%).
3.2. Spectroscopic study
The selected diagnostic bands of IR spectrum of compound 2provided useful information for determining its structure. The IRspectrum exhibited characteristic absorption bands at 3438 forN–H stretching of amide, 1442–1407 for N–H bending of –NH–NH–group and 715 cm−1 for C–Cl. The presence of amide carbonylstretching frequency at 1699 cm−1, which is the evidence of 5 mem-ber cyclic amide ring, also supports the structure 2. The 1H NMRspectrum showed a one-proton singlet of –CONH– at ı 7.84, a two-proton singlet of –NH–NH– at ı 10.58 and one-proton multiplet at ı4.52 of C3�–H. All the obtained spectral data are in good agreementwith the desired structure steroidal spiro-1′,2′,4′-triazolidine-3′-one 2.
3.3. Stereochemistry
The steroidal ketone semicarbazone 1 on reaction with H2O2provides respective steroidal spiro-1′,2′,4′-triazolidine-3′-one 2.There is a considerable amount of steric hindrance to the ring-
semicarbazone 1, intermediates (A and B) and the spiro-1′,2′,4′-triazolidine-3′-one 2, was fully optimized using DFT atB3LYP/6-31G* level of theory, to obtain the detain picture of thereaction mechanism of the formation of compound 2.
The optimized geometry from Hartree-Fock/6-31G* basis setwas used as the starting point for the DFT optimization. As is wellknown, DFT and HF structures are not identical, but we will notdiscuss this point in this article. We focus our attention on the
Scheme 1.
mistry
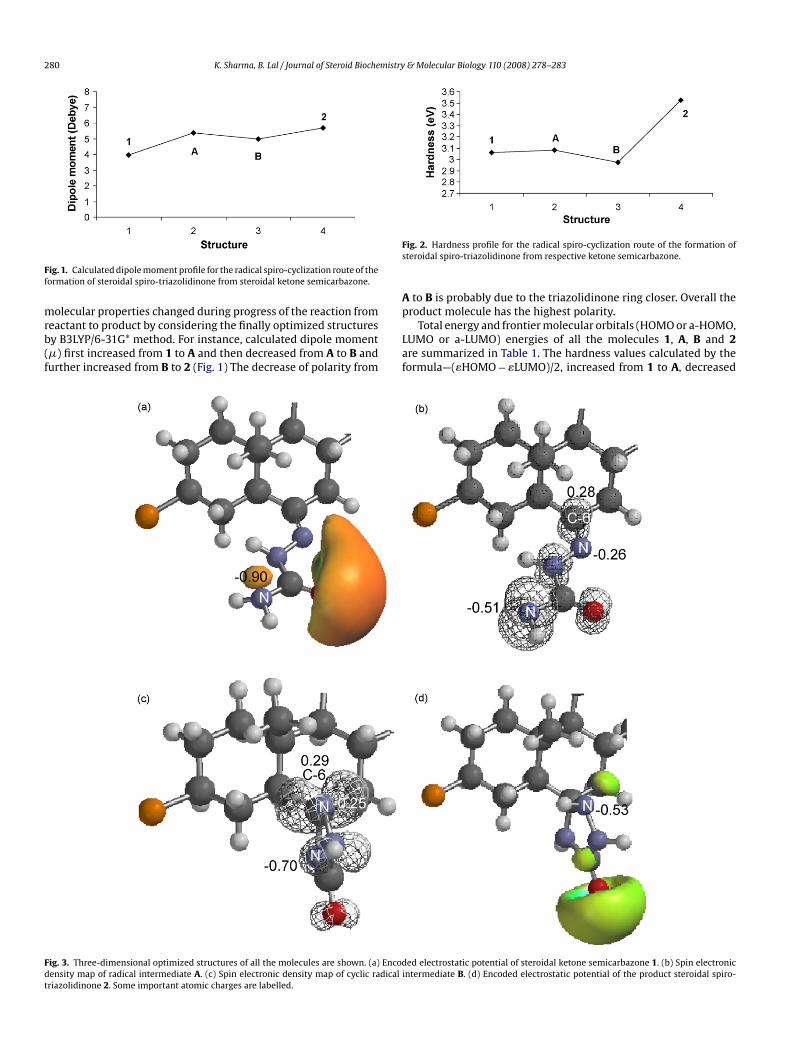
280 K. Sharma, B. Lal / Journal of Steroid BiocheFig. 1. Calculated dipole moment profile for the radical spiro-cyclization route of theformation of steroidal spiro-triazolidinone from steroidal ketone semicarbazone.
molecular properties changed during progress of the reaction fromreactant to product by considering the finally optimized structuresby B3LYP/6-31G* method. For instance, calculated dipole moment(�) first increased from 1 to A and then decreased from A to B andfurther increased from B to 2 (Fig. 1) The decrease of polarity from
Fig. 3. Three-dimensional optimized structures of all the molecules are shown. (a) Encodensity map of radical intermediate A. (c) Spin electronic density map of cyclic radicaltriazolidinone 2. Some important atomic charges are labelled.
& Molecular Biology 110 (2008) 278–283
Fig. 2. Hardness profile for the radical spiro-cyclization route of the formation ofsteroidal spiro-triazolidinone from respective ketone semicarbazone.
A to B is probably due to the triazolidinone ring closer. Overall theproduct molecule has the highest polarity.
Total energy and frontier molecular orbitals (HOMO or a-HOMO,LUMO or a-LUMO) energies of all the molecules 1, A, B and 2are summarized in Table 1. The hardness values calculated by theformula—(�HOMO − �LUMO)/2, increased from 1 to A, decreased
ded electrostatic potential of steroidal ketone semicarbazone 1. (b) Spin electronicintermediate B. (d) Encoded electrostatic potential of the product steroidal spiro-
K. Sharma, B. Lal / Journal of Steroid Biochemistry
Table 1Molecular properties of all the structures involved in the synthesis of steroidal spiro-1′ ,2′ ,4′-triazolidine-3′-one
Structure Energya HOMO/a-HOMOb LUMO/a-LUMOb
1 −1126726.8 −6.15 −0.03A −1126306.3 −6.52 −0.36B −1126340.8 −5.26 0.692 −1126731.6 −6.51 0.54
a Total energy is in kcal/mol.b Frontier molecular orbital energies are in electron volts (eV).
from A to B and further increased from B to 2 (Fig. 2). Hardnessprofile indicates the softness of the reactant than product. This isin agreement with Pearson [23] as the soft molecules are morereactive than hard molecules.
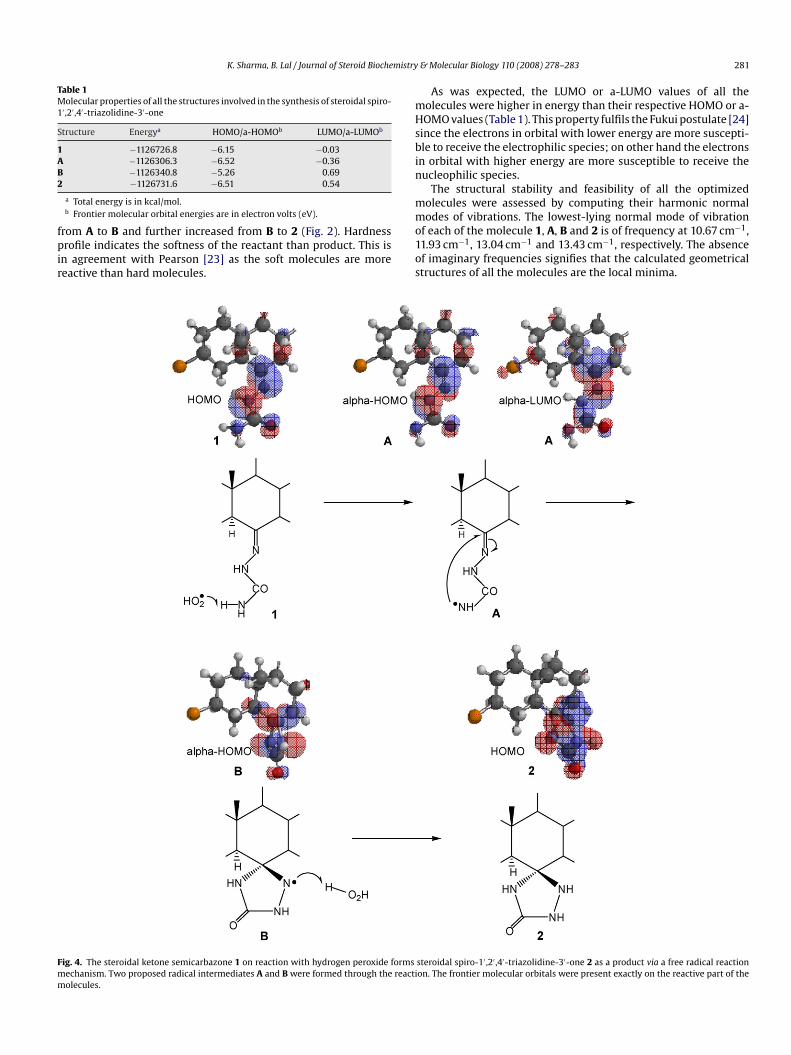
Fig. 4. The steroidal ketone semicarbazone 1 on reaction with hydrogen peroxide formsmechanism. Two proposed radical intermediates A and B were formed through the reactimolecules.
& Molecular Biology 110 (2008) 278–283 281
As was expected, the LUMO or a-LUMO values of all themolecules were higher in energy than their respective HOMO or a-HOMO values (Table 1). This property fulfils the Fukui postulate [24]since the electrons in orbital with lower energy are more suscepti-ble to receive the electrophilic species; on other hand the electronsin orbital with higher energy are more susceptible to receive thenucleophilic species.
The structural stability and feasibility of all the optimizedmolecules were assessed by computing their harmonic normalmodes of vibrations. The lowest-lying normal mode of vibrationof each of the molecule 1, A, B and 2 is of frequency at 10.67 cm−1,11.93 cm−1, 13.04 cm−1 and 13.43 cm−1, respectively. The absenceof imaginary frequencies signifies that the calculated geometricalstructures of all the molecules are the local minima.
steroidal spiro-1′ ,2′ ,4′-triazolidine-3′-one 2 as a product via a free radical reactionon. The frontier molecular orbitals were present exactly on the reactive part of the

282 K. Sharma, B. Lal / Journal of Steroid Biochemistry & Molecular Biology 110 (2008) 278–283
eme
Sch3.4.1. Formation of radical intermediate ASome interesting characteristics of intermediate A formation
can be classified into two groups: structural and electronic fea-tures. The molecule 1 must have a reactive semicarbazone group(–NNHCONH2) that could release H• radical, to be abstracted bythe peroxyl radical (•O2H). In terms of electronic features, the semi-carbazone moiety 1 must have electrostatic potential (Fig. 3) andHOMO (Fig. 4) at amino group (–NH2) to release H• radical. The–NH2 group must have spin electronic density after releasing H•,
due to the formation of unpaired electron (HN•–), it was observedexactly on the reactive part as shown by the theoretical model A inFig. 3.An important feature of the radical formation from structure 1 tostructure A is the change in total energy. The molecule 1 forms inter-mediate A due to the abstraction of its hydrogen by •O2H free radicalas shown in Scheme 2. As was expected the total energy increasedfrom structure 1 to structure A. This is in good agreement withthe Hartree-Fock energies for the atoms, where it was observedthat energy diminishes when the number of atoms increases [25].The change in atomic charge on nitrogen atom from structure 1 tostructure A is also supportive to carry out this step of the reaction(Fig. 3).
3.4.2. Formation of cyclic radical intermediate BThe spiro-cyclization of semicarbazone moiety at C6 carbon
atom of intermediate A, results to the formation of triazolidinonering at C6 and thus generate intermediate B. For this step of thereaction to be feasible, it is essential to have a-HOMO as well asspin electronic density on HN•– and the presence of a virtual a-LUMO orbital at C6 carbon atom of intermediate A. The localization
2.
of frontier molecular orbitals in Fig. 4 shows that the molecule Ahas molecular orbitals (MOs) just on its reactive part and work inthe same way as the frontier molecular orbital analysis based onperturbation theory (PT) works. The said theory has been widelyused for explaining the relative reactivity patters in the differenttype of reactions [26–28]. Optimized structure of intermediate Ashowed that HN•– radical group possesses nitrogen with higheratomic charge (−0.51) that could share charge with C6, because ofthe lack of atomic charge (0.28) at C6. Accordingly, nitrogen atom
of HN•– radical group forms bond with C6, which further reducesthe C N �-bond to form C–N• radical at heterocyclic ring moietyand thus generate intermediate B. The changes in atomic chargesconfirm the polarization of �-bond from A to B and thus supportthe order of the present reaction mechanism.3.4.3. Formation of steroidal spiro-1′,2′,4′-triazolidine-3′-one 2A common feature of the product formation from intermediate
B to 2 is the change in total energy. The –N•– radical of cyclic inter-mediate B interacts with H• radical from the environment of thereaction system and thus converts into spiro-1′,2′,4′-triazolidine-3′-one. Due to the fact mentioned above, it could be consider thatthis step of the reaction occurs only if a-HOMO and spin electronicdensity is located on –N•– radical of the intermediate B. These prop-erties were observed exactly on the reactive part of B as shownby the spin electronic density map (Fig. 3) and frontier molecularorbital analysis (Fig. 4).
Based upon all the above results; a free radical mechanism isproposed for the preparation of steroidal spiro-triazolidinone fromrespective ketone semicarbazone using H2O2 (Scheme 2). The HO2
•
induced radical cyclization of semicarbazone moiety at C6 atom of

mistry
[
[
[
[
[
[
[catalytic mechanism of the �5-3-ketosteroid isomerase reaction, Steroids 71
K. Sharma, B. Lal / Journal of Steroid Bioche
steroid. The abstraction of H• as a radical from molecule 1 by peroxylradical (•O2H), initiates the reaction causes homolytic cleavage ofN–H bond. In the final step, the abstraction of H as a radical bytriazolidinone ring of radical intermediate B causes the formation ofN–H bond and thus completes the reaction. The energy released byfragmentation of N–H bond in first step must be counter balancedby the energy gained by the formation of N–H bond in the last step;this is found in good agreement with the energy values obtainedby the computational calculations in this article.
The calculated physicochemical parameters: total energy,atomic charges, electrostatic potential map, spin electronic densitymap, dipole moment, frontier MOs like HOMO, a-HOMO, LUMO,a-LUMO and hardness were highly supportive to provide a logicalexplanation of the reaction mechanism of the formation of steroidalspiro-1′,2′,4′-triazolidine-3′-one. All these calculated propertiescould be useful to design, optimize and synthesize further newbiologically active compounds.
4. Conclusions
We have synthesized 3�-chloro-5�-cholestan-6-spiro-1′,2′,4′-triazolidine-3′-one selectively from respective semicarbazonemoiety by a simple method. Heterocyclic ring closes at C6 carbonatom of steroid. The only one isomer as a product of this reaction, inwhich –NH–CO– group is cis with respect to C5�–H, was selectively
obtained. Present reaction proceeds via a free radical mechanism.By using calculated density functional theoretical models, it is con-cluded that the present reaction undergoes through the formationof two reaction intermediary states.Acknowledgments
Authors are grateful to the University of Pretoria for finan-cial support. We thank the help of Dr. J.R. Pasqualini, Institut dePuericulture, 75014 Paris, France, who reviewed this manuscript.
References
[1] M.S. Novikov, A.F. Khlebnikov, A.A. Stepanov, R.R. Kostikov, N-Tert-butyl-N-(2,2-dichlorovinyl) carbamoyl chloride: a novel building block for the synthesis ofnitrogen heterocycles, Synthesis 1994 (1994) 782–784.
[2] J.G. Schantl, H. Gstach, Geminale azo- und heteroelement-funktionen,I: Einwirkung von Grignard-reagentien auf 1-(4-chlorphenylazo)-1-methylethylisocyanat, Monatsh. Chem. 116 (1985) 1051–1064.
[3] S.M. Ramsh, Y.G. Basova, A.I. Ginak, Production of alkali salts of 2-phenylimino-4-triazolidinone, Chem. Heterocyclic Compds. 20 (1984) 734–735.
[4] J. Schantl, P. Hebeisen, 2-Aryl-5,5-dimethyl-1,2,4-triazolidin-3-onederivatives[2-aryl-5,5-dimethyl-1,2,4-triazolidin-3-on-erivate], Sci. Pharm. 51(1983) 379–390.
[
[
[
[
[
[
[
[
[
[
& Molecular Biology 110 (2008) 278–283 283
[5] M. Komatsu, N. Nishikaze, M. Sakamoto, Y. Ohshiro, T. Agawa, Reaction ofdiaziridines with diphenylketene and isocyanates, J. Org. Chem. 39 (1974)3198–3205.
[6] K. Koyano, C.R. McArthur, Formation and reactions of (1-chloroalkyl) carbamoylchlorides. Synthesis of 1,2,4-triazolidine-5-ones, Can. J. Chem/Rev. Can. Chim.51 (1973) 333–337.
[7] M. Komatsu, Y. Ohshiro, T. Agawa, Chemistry of cumulated double-bond com-pounds. XI. Reaction of nitrones with diphenylcarbodiimide, J. Org. Chem. 37(1972) 3192–3194.
[8] J. Krenzer, 2-Alkyl-4-aryl-1,2,4-triazolidin-3-ones. U.S. Pat. 3,922,162, Chem.Abstr. 84 (1976) 121834.
[9] J. Krenzer, Increasing the yield of plants having storage organs by treatmentwith triazolidinones, U.S. Pat. 3,925,054, Chem. Abstr. 84 (1976) 55326.
10] J. Krenzer, 1,2,3-Triazolidin-3-ones. Ger. Offen. Pat. 2,510,573, Chem. Abstr. 84(1976) 31095.
[11] F.D. Proft, P. Geerlings, Conceptual and computational DFT in the study of aro-maticity, Chem. Rev. 101 (2001) 1451–1464.
12] W.C. Bailey, DFT and HF–DFT calculations of 14N quadrupole coupling constantsin molecules, Chem. Phys. 252 (2000) 57–66.
13] H.L. Skriver, Crystal structure from one electron theory, Phys. Rev. B31 (1985)1909–1923.
14] R. Kolos, A.L. Sobolewski, The infrared spectroscopy of HNCCC: matrix isolationand density functional theory study, Chem. Phys. Lett. 344 (2001) 625–630.
15] S.R. Polo, M.K. Wilson, Infrared intensities in liquid and gas phases, J. Chem.Phys. 23 (1955) 2376–2377.
16] K. Sharma, B. Lal, An experimental and theoretical approach to 5�-cholestan-6-spiro-1′ ,2′ ,4′-triazolidine-3′-one, J. Steroid Biochem. Mol. Biol. 107 (2007)270–276.
[17] K. Sharma, C. Kubli-Garfias, Theoretical mechanism of the formation ofcholesteryl chloride from cholesterol and thionyl chloride, J. Mol. Model. 11(2005) 135–140.
18] K. Sharma, R. Vazquez-Ramırez, C. Kubli-Garfias, A theoretical model of the
(2006) 549–557.19] (a) A.D. Becke, Density-functional thermochemistry. III. The role of exact
exchange, J. Chem. Phys. 98 (1993) 5648–5652;(b) C. Lee, W. Yang, R.G. Parr, Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B 37 (1988)785–789;(c) A.D. Becke, A new mixing of Hartree-Fock and local density-functional the-ories, J. Chem. Phys. 98 (1993) 1372–1377.
20] Shafiullah, S.A. Ansari, Reaction of lead (IV) acetate with steroidal semicar-bazones, J. Indian Chem. Soc. 67 (1990) 431–432.
21] T.J. Dwyer, P.G. Jasien, Electronic effects in a prototype push–pull ethlylene: astudy of the rotational barriers in C4H4N4 isomers, J. Mol. Struct. (THEOCHEM)363 (1996) 139–150.
22] Spartan’04 Version 1.0.3, Wavefunction, Inc. 18401 Von Karman Ave., Suite 370,Irvine, CA 92612, USA.
23] R.G. Pearson, Recent advances in the concept of hard and soft acids and bases,J. Chem. Educ. 64 (1987) 561–567.
24] K. Fukui, Role of frontier orbital in chemical reactions, Science 218 (1982)747–754.
25] S.R. Gadre, S.B. Sears, S.J. Chakravorty, R.D. Bengale, Some novel characteristicsof atomic information entropies, Phys. Rev. 32A (1985) 2602–2606.
26] K. Fukui, Recognition of stererochemical path by orbital interactions, Acc. Chem.Res. 4 (1971) 57–64.
27] K.N. Houk, Frontier molecular orbital theory of cycloaddition reactions, Acc.Chem. Res. 8 (1975) 361–369.
28] I. Fleming, Frontier Orbitals and Organic Chemical Reactions, Wiley, New York,1976.