synthesis of 2-boron substituted-1,3-dienes and …dienes are less stable than the tetra-coordinated...
TRANSCRIPT

SYNTHESIS OF 2-BORON SUBSTITUTED-1,3-DIENES AND THEIR
DIELS-ALDER/SUZUKI CROSS COUPLING REACTIONS
BY
LIQIONG WANG
A Dissertation Submitted to the Graduate Faculty of
WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES
in Partial Fulfillment of the Requirements
for the Degree of
DOCTOR OF PHILOSOPHY
Chemistry
August 2012
Winston Salem, North Carolina
Approved By:
Mark E. Welker, Ph.D., Advisor
Anne Glenn, Ph.D., Chair
Uli Bierbach, Ph.D.
Christa L. Colyer. Ph.D.
Paul B. Jones, Ph.D.

I
ACKNOWLEDGEMENTS
Firstly, I would like to thank the almighty God for all the love and guidance in the
process of pursuing my Ph.D. degree. All the love that HE has given is through these
generous people whom I will respect and love for the rest of my life.
I would like to extend my deepest and warmest appreciation to my advisor, Dr. Mark E.
Welker, who has instructed and led me in the research and study with great patience and
support. Dr. Welker always encouraged me to try new ideas and think about science in a
precise and concrete way. The encouragement has led me to break through the messy
results and make progress in the project. The benefit from your professional training and
personal fascination will eventually have great influence on my career and life.
Also, Dr. Marcus Wright has offered me much help in NMR, chromatography and mass
spectrometry. No matter what time and situation you were facing, you were always trying
your best to help me to resolve the problems. I would like to thank you for your great
patience and generosity.
The crystal analysis in my dissertation was from Dr. Cynthia Day. You also tried to
help me to get a good crystal when I could not figure out if it is a crystal. I would like to
thank you for all the help.
For the molecular modeling of the compound in the dissertation, I would like to thank
Dr. Fred Salsbury from the Physics Department. His work was a great help in the
progress of my research.
Thank you to my committee members, Dr.Ulrich Bierbach, Dr. Paul B. Jones, Dr. Anne
Glenn and Dr. Rebecca Alexander, for taking time out of your busy schedule to read and

II
correct my dissertation and give me professional instructions to finish my proposals. Also,
I thank Dr. Bierbach for the kind help and suggestions when I looked for a job.
Also, I would like to thank Dr. Christa L. Colyer for your great support in the
development of my professional career. You have given me a lot of wise opinions on the
job and career. Thank you for always being nice and patient.
To Dr. Al Rives, Dr. S. Bruce King, Dr. Lindsay R. Comstock, Dr. Amanda Jones, Dr.
Susan Tobey and Dr. Akbar Salam, I would like to thank you for the kind help in my
study and research.
Many thanks to Mike Thompson, Tommy Murphy, Linda Tuttle, Melissa Doub and
Nancy for your generous help in the past few years.
Also thank you, my former and current lab mates, Dr. Subhasis De, Dr. Ramakrishna R.
Pidaparthi, Maben Ying, Dr. Tanya Pinder, Dr. Sarmad Hindo, Dr. Christopher S. Junker,
Partha Choudhury, Kimberly M. Tillman, Dr. Ken Crook and undergraduates. Each of
you has been a source of advice and/or friendship and I have enjoyed working with you
all.
I would like to thank all the past and current Chinese students and postdocs in the
department: Xiuli, Weibin, Zhaoli, Zhidong, Huajun, Yue, Jiyan, Changkun, Yuhao, Lu,
Lei, Ye, Zhengrui, Zhong, Song, Yuyang, Xin, Lin and Mu, for the friendship and help in
these years.
Also, I could not have reached this point without the instruction and encouragement
from Dr. Jin Nie, my former advisor from Huazhong University of Science and
Technology. Thank you for helping me overcome all the frustrations and become strong.

III
Lastly, thank you to my big family: my great father and mother, brothers, sisters, nieces,
nephew and my lovely son. Thank you for the amazing love, support and sacrifice for me.
Without you, I could not go through all of this. The last kiss is to my son, Weiwei; my
sweetheart, you are always the power source for your mom. You never know how much
you mean to mom.

IV
TABLE OF CONTENTS
ACKNOWLEDGEMENTS ................................................................................................. I
TABLE OF CONTENTS .................................................................................................. IV
LIST OF FIGURES ......................................................................................................... VII
LIST OF TABLES ......................................................................................................... VIII
LIST OF ABBREVIATIONS ........................................................................................... IX
ABSTRACT ................................................................................................................... XIII
CHAPTER 1 Introduction ........................................................................................... 1
1.1 Diels-Alder Reactions .......................................................................................... 1
1.1.1 Mechanism of Diels-Alder Reactions ........................................................... 1
1.1.2 Regioselectivity of Diels-Alder Reactions.................................................... 4
1.1.3 Stereoselectivity of Diels-Alder Reactions ................................................... 5
1.2 Brief Review of Recent Boronate Substituted Dienes ....................................... 13
1.2.1 Preparation and Reactions of 1-Boron Substituted-1,3-Dienes .................. 17
1.2.2 Preparation and Reactions of 2-Boron Substituted-1,3-Dienes .................. 22
1.3 Suzuki-Miyaura Cross Coupling Reactions ....................................................... 26
1.4 Rhodium Reaction .............................................................................................. 30
1.4.1 Advantages of rhodium catalyzed 1,4-addition of enones .......................... 30

V
1.4.2 Catalytic Cycle and Mechanism ................................................................. 32
1.4.3 Reaction Properties ..................................................................................... 33
1.5 Aims of Current Project ..................................................................................... 43
CHAPTER 2 Preparation of Boron Substituted Dienes ............................................ 45
2.1 Results and Discussion ....................................................................................... 45
2.1.1 Preparation of 2- Diethanolamine Borate Diene 2.4 And 2.6 ..................... 45
2.1.2 Resolution of Dimerization Problems and Preparation of Tri-Coordinated
and Tetra-Coordinated Boron-1,3-Dienes ................................................................ 47
2.1.3 Conformation Analysis of 2-Boron Substituted-1,3-Dienes ....................... 51
2.2 Attempts to Synthesize Other Boron Substituted Dienes ................................... 52
2.2.1 Attempts to Synthesize Sodium 1-(but-1,3-dien-2yl)-5-methyl-2,8,9-trioxa-
1-borabicyclo[3,3,1]nonan-1-uide) ........................................................................... 53
2.2.2 Attempts to Synthesize 2-(Buta-1,3-Dien-2-yl)-6-Methyl-1,3,6,2 -
Dioxazaborocane-4.8-Dione ..................................................................................... 53
2.2.3 Attempts To Synthesize 1-Phenyl-2-Pinacol Boronate-1, 3-Diene ............ 54
2.2.4 Attempts To Synthesize 1-Substituted-2-Boron-1,3-Dienes By Cross-
Metathesis ................................................................................................................. 57
2.3 Conclusion .......................................................................................................... 60
2.4 Experimental Procedures and Characterization Data ......................................... 61
CHAPTER 3 Diels-Alder Reactions of 2-Boron Substituted-1,3-Dienes ................. 70
3.1 Results and Discussion ....................................................................................... 70

VI
3.1.1 Diels-Alder Reactions ................................................................................. 70
3.1.2 Dimerization of Boron Substituted Dienes ................................................. 77
3.2 Conclusion .......................................................................................................... 79
3.3 Experimental Procedures and Characterization Data ......................................... 79
CHAPTER 4 Suzuki Cross Coupling Reactions ....................................................... 87
4.1 Results and Discussion ....................................................................................... 87
4.1.1 Suzuki Cross Coupling Reactions ............................................................... 87
4.1.2 Tandem Diels-Alder/Suzuki Cross Coupling Reactions............................. 90
4.2 Conclusions ........................................................................................................ 99
4.3 Experimental and Characterization Data ............................................................ 99
CHAPTER 5 Unexpected Proton Deboronation Reactions ..................................... 116
5.1 Rhodium Catalyzed Chemistry With 1-Alkyl-3-Boronate-1,3-Dienes ............ 116
5.2 Conclusion ........................................................................................................ 123
5.3 Experimental and Characterization Data .......................................................... 124
CHAPTER 6 CONCLUSIONS ............................................................................... 128
APPENDIX A Crystal Structure Data of Diene 2.4 ....................................................... 147
APPENDIX B NOESY and COSY of Boron Substituted Dienes ................................. 169
CURRICULUM VITAE ................................................................................................. 179

VII
LIST OF FIGURES
Figure 1.1 Diels-Alder Reactions ....................................................................................... 1
Figure 1.2 Diels-Alder Molecular Orbital Diagram ........................................................... 2
Figure 1.3 Normal, Neutral and Inverse Electron Demand Diels-Alder Reactions ............ 3
Figure 1.4 “Zwitterionic” Model ........................................................................................ 5
Figure 1.5 General Catalytic Cycle for Suzuki-Miyaura, Heck And Stille Cross-Coupling
Reactions ........................................................................................................................... 27
Figure 1.6 General Catalytic Cycle for Suzuki-Miyaura Couplings................................. 29
Figure 1.7 Reductive Elimination of Suzuki-Miyaura Couplings .................................... 29
Figure 1.8 Applications of New BINAP Ligands ............................................................. 34
Figure 2.1 Mechanism of Ruthenium Catalyzed Cross Metathesis Reactions ................ 58
Figure 3.1 Crystal Structures of BF3 Substituted Diene and Diethanolamineborate-1,3-
Butadiene 2.4 .................................................................................................................... 74
Figure 3.2 NOESY of Diethanolaminoborate-1,3-Butadiene 2.4 ..................................... 75
Figure 3.3 Thermodynamic Diagram of Dimerization of 2-Boron Substituted Dienes ... 79
Figure 4.1 Comparison of NMR Chemical Shifts of Boron Substituted Diene and
Palladium Substituted Diene in CD3CN ........................................................................... 96
Figure 5.1 Catalytic Cycle of Rhodium Catalysis .......................................................... 116
Figure 5.2 Catalytic Cycle of Palladium Catalysis ......................................................... 124

VIII
LIST OF TABLES
Table I Attempts for Olefin Cross Metathesis Reactions ................................................. 59
Table II Attempts for Enyne Cross Metathesis ................................................................. 60
Table III Diels-Alder Reactions of 2-Boron Substituted-1,3-Dienes ............................... 71
Table IV Dimerization Experiments of 2-Boron Substituted-1,3-Dienes ........................ 78
Table V Optimization of Suzuki Cross Coupling Reaction ............................................. 88
Table VI Suzuki Cross Coupling Reactions of 2-Boron Substituted-1,3-Dienes ............. 88
Table VII Tandem Diels-Alder/Suzuki cross coupling reactions ..................................... 91
Table VIII Palladium Catalysts and 2-Palladium Substituted 1,3-Diene ......................... 97
Table IX Rhodium Catalyzed Reactions ......................................................................... 117

IX
LIST OF ABBREVIATIONS
Å Angstrom(s)
°C Degree(s) Celsius
Ac Acetyl
allyl 2-Propenyl
Ar Aryl
atm Atmosphere(s)
BINAP 2,2'-bis(diphenylphosphino)-1,1'-
binaphthalene
Bn Benzyl
bs Broad Singlet
Bz Benzoyl
C=O Carbonyl
Calcd Calculated
CD3CN Deuterated Acetonitrile
CDCl3 Deuterated Chloroform
cm-1
Wave Numbers
COSY Correlation Spectroscopy
Cp Cyclopentadienyl

X
Cy Cyclohexane
d Doublets
dba Dibenzyldeneacetone
D-A Diels-Alder
DCE 1,2-dichloroethane
DCM Dichloromethane
dd Double Doublets
ddd Double Double Doublets
ddt Double Double Triplet
DFT Density Functional Theory
DIPHOS 1,2-bis(diphenylphosphino)ethane
DME Dimethoxyethane
DMF N,N-Dimethylformamide
DMSO Dimethylsulfoxide
EDG Electron Donating Group
EWG Electron Withdrawing Group
Equiv Equivalents
Et Ethyl
Et2O Diethyl Ether

XI
g Grams
h Hours
HMBC Heteronuclear Multiple Bond
Coherence
HOMO Highest Occupied Molecular Orbital
HRMS High Resolution Mass Spectrometry
Hz Hertz(s)
iPr Isopropyl
J Coupling Constant
kcal Kilocalorie
LUMO Lowest Unoccupied Molecular Orbital
m Multiplet
m Meta
m.p. Melting Point
m/z Mass to Charge Ratio
Me Methyl
MHz Megahertz(s)
min Minutes
mol Moles

XII
N/A Not Available
nBu Normal Butyl
NMR Nuclear Magnetic Resonance
NOE Nuclear Overhauser Effect
NOESY Nuclear Overhauser Effect
Spectroscopy
o Ortho
OTf Trifluoromethanesulfonate
p Para
Ph Phenyl
Q Quartet
Rf Retention Factor
S Singlet
t Triplet
t1/2 Half Life
TLC Thin Layer Chromatography
TMS Trimethylsilyl
X Halide
δ Chemical Shift

XIII
ABSTRACT
Dissertation under the direction of Mark E. Welker, Ph.D., Professor of Department of
Chemistry, Wake Forest University
Clerodane diterpenoids represent a large group of secondary metabolites which are
distributed widely in nature. Over a thousand members have been isolated to date, many
of which possess interesting biological activities: antifeedant, antitumor, antifungal,
antibiotic, anti-peptic ulcer and so on. Although synthetic approaches to the trans-
clerodanes abound, cis-clerodane syntheses are rare. The long term objective of this
research is to make highly reactive dienes which would undergo Diels-Alder (D-A)
reactions with sterically crowded dienophiles to construct cis-clerodane compounds.
In previous work, 2-boron-1,3-dienes had shown a high propensity toward self D-A
dimerization. Different strategies were tried to prepare dienes without dimerization and
seven boron substituted dienes were prepared in good yields. The tri-coordinated boron
dienes are less stable than the tetra-coordinated dienes because the boron atom is
stabilized by donating electron pairs or groups in the latter type. The reactivity of these
boron substituted dienes (2-diethanolaminoborate-1,3-butadiene) in D-A and Suzuki
cross coupling reactions were tested and evaluated.
Testing concluded that in D-A reactions high yields (> 90%) were found in the
reactions of boron substituted 1,3-dienes and N-phenyl maleimide at room temperature or
lower. The fastest diene (2-diethanolaminoborate-1,3-butadiene) reacted with a t1/2 less
than 4 minutes at -10 °C. The 2-diethanolaminoborate-1,3-butadiene has a high HOMO
energy compared to its BF3 diene counterpart which accelerates its D-A reactions. High

XIV
regioselectivities (16:1) were found in the D-A reactions of unsymmetrical dienophiles
and 2-diethanolaminoborate-1,3-butadiene. Both high yields and regioselectivities were
achieved in Suzuki cross coupling reactions of the resulting boron substituted D-A
cycloadducts. Using this methodology, 2-diethanolaminoborate-1,3-butadiene is a
promising precursor in providing a potential tool to asymmetric synthesis.
Three boron substituted dienes reacted with n-phenylmaleimide very quickly, from
seconds to 2 hours. All three dienes are stable at 0℃ for more than 2 years. The research
also revealed that 4 dienes can be used in tandem D-A/Suzuki cross coupling reactions.
High yields and good selectivities were obtained. The possible mechanism of tandem
reactions showed that Pd(II) acts as a Lewis acid catalyst to catalyze the D-A reaction
between boron substituted dienes and dienophiles followed by Suzuki cross coupling
reactions. A possible unstable novel intermediate, a palladium substituted diene, was
observed in the reaction by NMR.

1
CHAPTER 1 Introduction
1.1 Diels-Alder Reactions
As the most thoroughly explored cycloaddition reactions since 1928, Diels-Alder
reactions have been essential methods to realize the simultaneous construction of
substituted cyclohexenes with a high degree of regioselectivity, diastereoselectivity and
enantioselectivity. Various ramifications of Diels-Alder reactions have been developed
with thousands of papers being published every year.1-3
Obviously, this concerted
(4+2) pericyclic reaction is already an indispensable way to form a carbon-carbon
bond in modern organic synthetic chemistry.
Although several reports on asymmetric Diels-Alder reactions have been published,4-6
they are still in the minority compared with many asymmetric oxidative and reductive
reactions. We are dedicated to improve the enantioselectivity and diastereoselectivity by
employing the main group metal substituted dienes in Diels-Alder reactions.
1.1.1 Mechanism of Diels-Alder Reactions
Diels-Alder reactions have been reported to have two mechanisms: concerted and two
step mechanisms. Although some cases occur by a two step mechanism that involve a
biradical intermediate, most thermal cycloadditions can be described by a symmetry-
allowed one step mechanism (Figure 1.1).7, 8
Figure 1.1 Diels-Alder Reactions

2
FMO ( Frontier Molecular Orbital ) theory, a powerful practical model, was developed
by Kenichi Fukui in the1950's and has been used successfully to explain reactivity and
selectivity phenomena in cycloaddition reactions.9, 10
The fundmental principle of FMO is
the focus on the highest occupied and lowest unoccupied molecular orbitals (HOMO and
LUMO). The smaller the HOMO-LUMO energy gap, the easier the Diels-Alder reactions
are because the lower HOMO-LUMO energy makes a greater contribution to the
transition state. For acrolein with an electron withdrawing carbonyl group, which
efficiently lowers the LUMO energy of the dienophile, the energy difference between
acrylaldehyde and diene are greatly reduced compared with ethylene. So, acrylaldehyde
is a better dienophile than ethylene in this case.
Figure 1.2 Diels-Alder Molecular Orbital Diagram
3
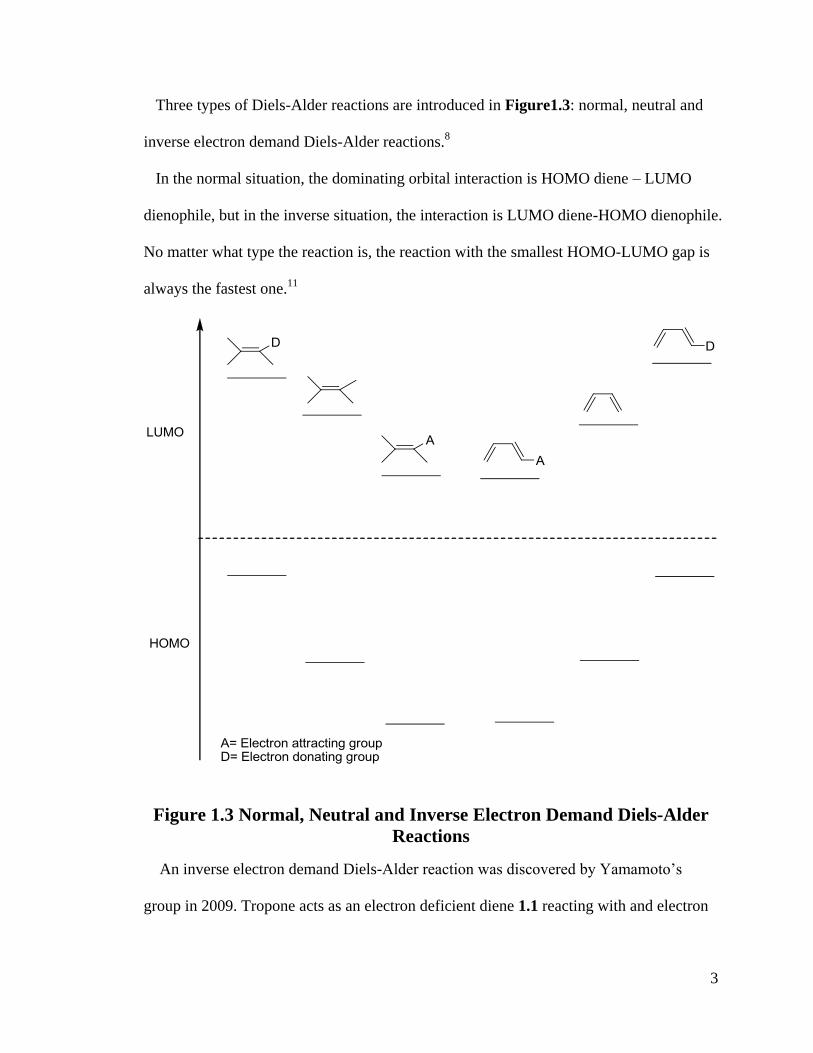
Three types of Diels-Alder reactions are introduced in Figure1.3: normal, neutral and
inverse electron demand Diels-Alder reactions.8
In the normal situation, the dominating orbital interaction is HOMO diene – LUMO
dienophile, but in the inverse situation, the interaction is LUMO diene-HOMO dienophile.
No matter what type the reaction is, the reaction with the smallest HOMO-LUMO gap is
always the fastest one.11
Figure 1.3 Normal, Neutral and Inverse Electron Demand Diels-Alder
Reactions
An inverse electron demand Diels-Alder reaction was discovered by Yamamoto’s
group in 2009. Tropone acts as an electron deficient diene 1.1 reacting with and electron
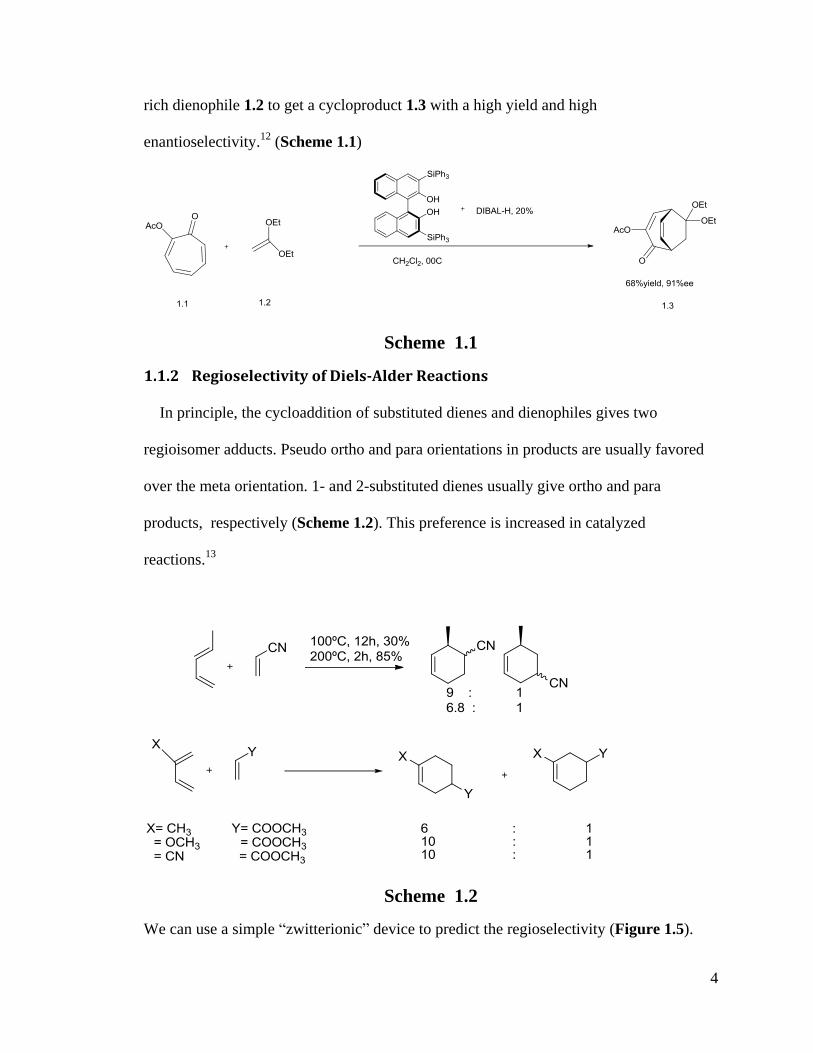
4
rich dienophile 1.2 to get a cycloproduct 1.3 with a high yield and high
enantioselectivity.12
(Scheme 1.1)
Scheme 1.1
1.1.2 Regioselectivity of Diels-Alder Reactions
In principle, the cycloaddition of substituted dienes and dienophiles gives two
regioisomer adducts. Pseudo ortho and para orientations in products are usually favored
over the meta orientation. 1- and 2-substituted dienes usually give ortho and para
products, respectively (Scheme 1.2). This preference is increased in catalyzed
reactions.13
Scheme 1.2
We can use a simple “zwitterionic” device to predict the regioselectivity (Figure 1.5).

5
Figure 1.4 “Zwitterionic” Model
For disubstituted dienes, relative amounts of each depend on electron donating strength
of substituents.
1.1.3 Stereoselectivity of Diels-Alder Reactions
The cis principle was found by Alder and Stein in 1937, after the observation that the
relative configuration of reactants was preserved in the final products.14
Cis principle: The stereochemistry of substituents in the starting material is retained in
the product.15
Since a trans double bond in a six-memebered ring is geometrically
impossible, the Diels-Alder cycloaddition can occur only when the diene possesses a
cisoid conformation. So, a number of cyclic transoid cyclic dienes have been shown to be
unavailable to dienophiles.
Endo principle: The two reactants approach each other in parallel planes which may
interact in two different orientations affording endo- and exo- adducts (Scheme 1.3 ).
The most stable transition state is the one with maximum orbital overlap.16
Endo is the
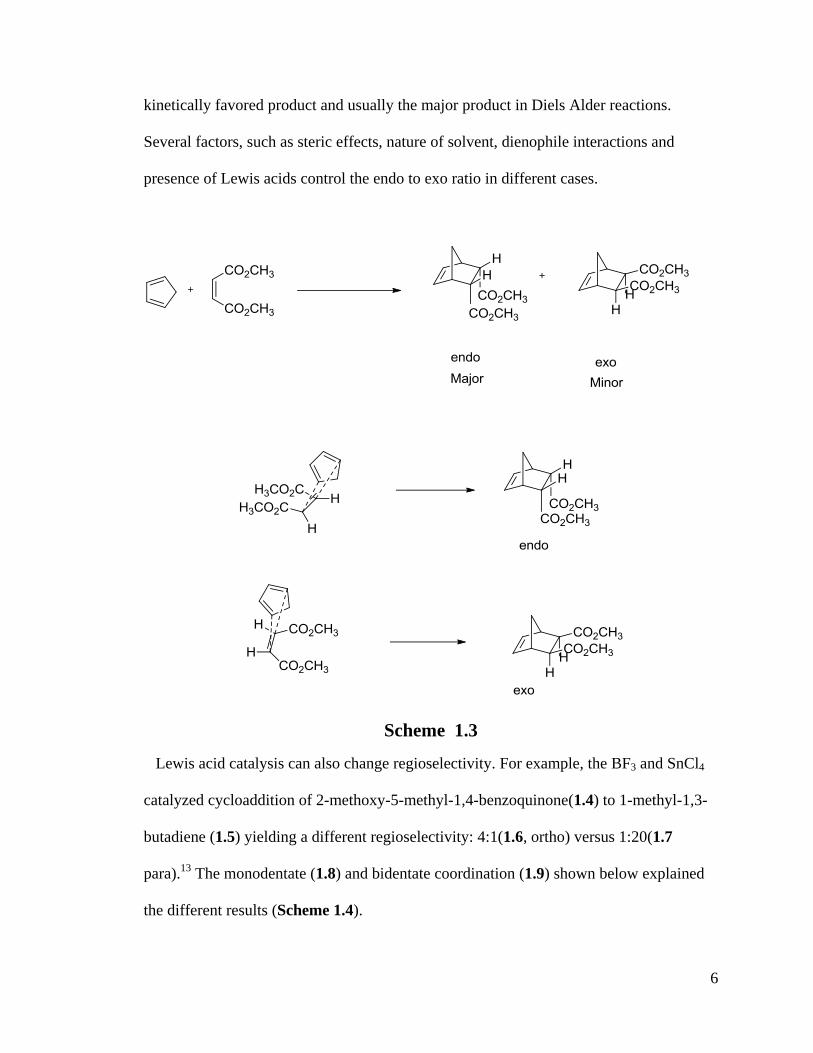
6
kinetically favored product and usually the major product in Diels Alder reactions.
Several factors, such as steric effects, nature of solvent, dienophile interactions and
presence of Lewis acids control the endo to exo ratio in different cases.
Scheme 1.3
Lewis acid catalysis can also change regioselectivity. For example, the BF3 and SnCl4
catalyzed cycloaddition of 2-methoxy-5-methyl-1,4-benzoquinone(1.4) to 1-methyl-1,3-
butadiene (1.5) yielding a different regioselectivity: 4:1(1.6, ortho) versus 1:20(1.7
para).13
The monodentate (1.8) and bidentate coordination (1.9) shown below explained
the different results (Scheme 1.4).
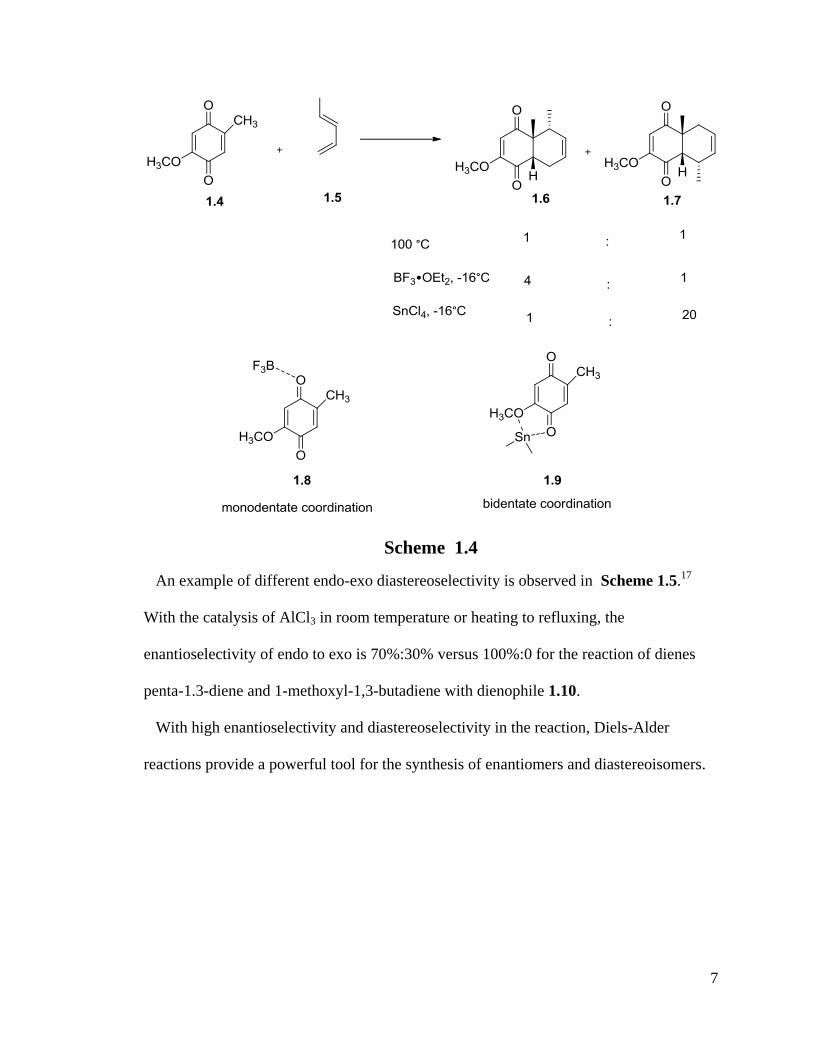
7
Scheme 1.4
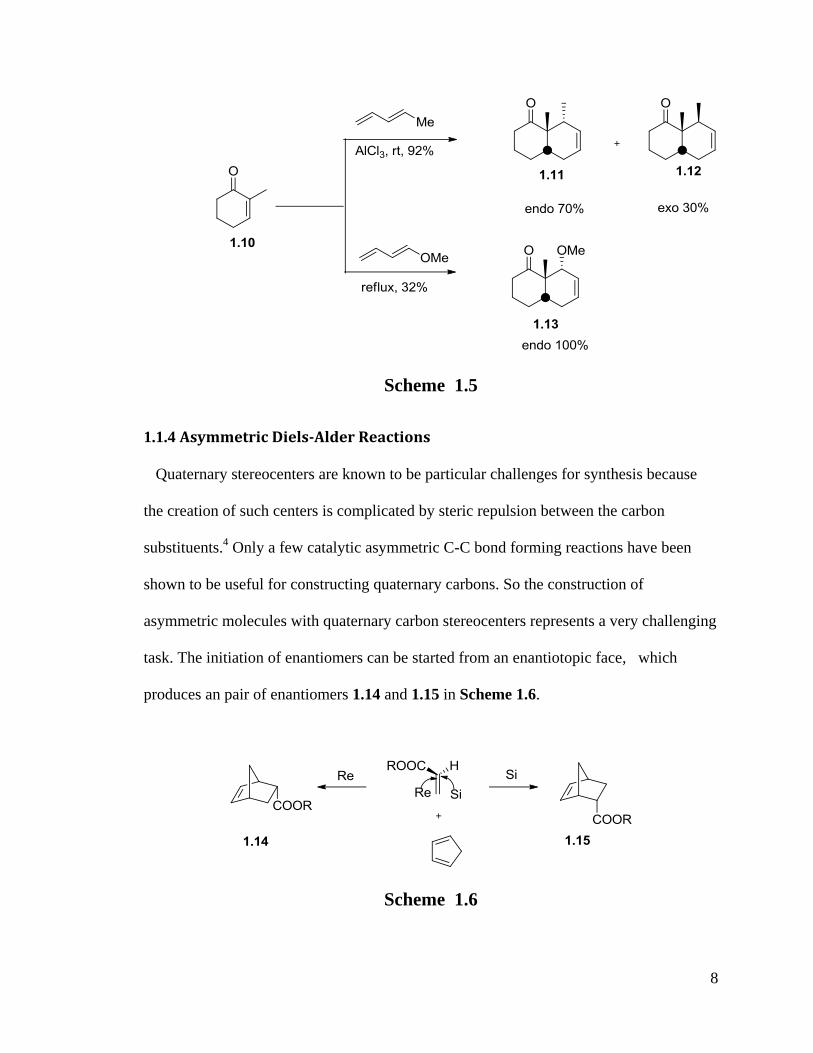
An example of different endo-exo diastereoselectivity is observed in Scheme 1.5.17
With the catalysis of AlCl3 in room temperature or heating to refluxing, the
enantioselectivity of endo to exo is 70%:30% versus 100%:0 for the reaction of dienes
penta-1.3-diene and 1-methoxyl-1,3-butadiene with dienophile 1.10.
With high enantioselectivity and diastereoselectivity in the reaction, Diels-Alder
reactions provide a powerful tool for the synthesis of enantiomers and diastereoisomers.
8
Scheme 1.5
1.1.4 Asymmetric Diels-Alder Reactions
Quaternary stereocenters are known to be particular challenges for synthesis because
the creation of such centers is complicated by steric repulsion between the carbon
substituents.4 Only a few catalytic asymmetric C-C bond forming reactions have been
shown to be useful for constructing quaternary carbons. So the construction of
asymmetric molecules with quaternary carbon stereocenters represents a very challenging
task. The initiation of enantiomers can be started from an enantiotopic face, which
produces an pair of enantiomers 1.14 and 1.15 in Scheme 1.6.
Scheme 1.6

9
Asymmetric induction of D-A reactions may be achieved by using chiral dienophiles,
chiral dienes or chiral catalysts. The use of a diene and/or a dienophile with a chiral
auxiliary is a common technique to make chiral dienes and dienophiles. In the reaction of
cis-2-neopentyloxyisobornyl acrylate 1.17 with cyclopentadiene 1.16,18
virtually
quantitative asymmetric induction can be explained by the staggered conformation of the
neopentyloxy chain, in which the re-face is shielded by the t-butyl group (Scheme 1.7).
Scheme 1.7
The chiral dienes have been investigated less extensively because of the difficulty of
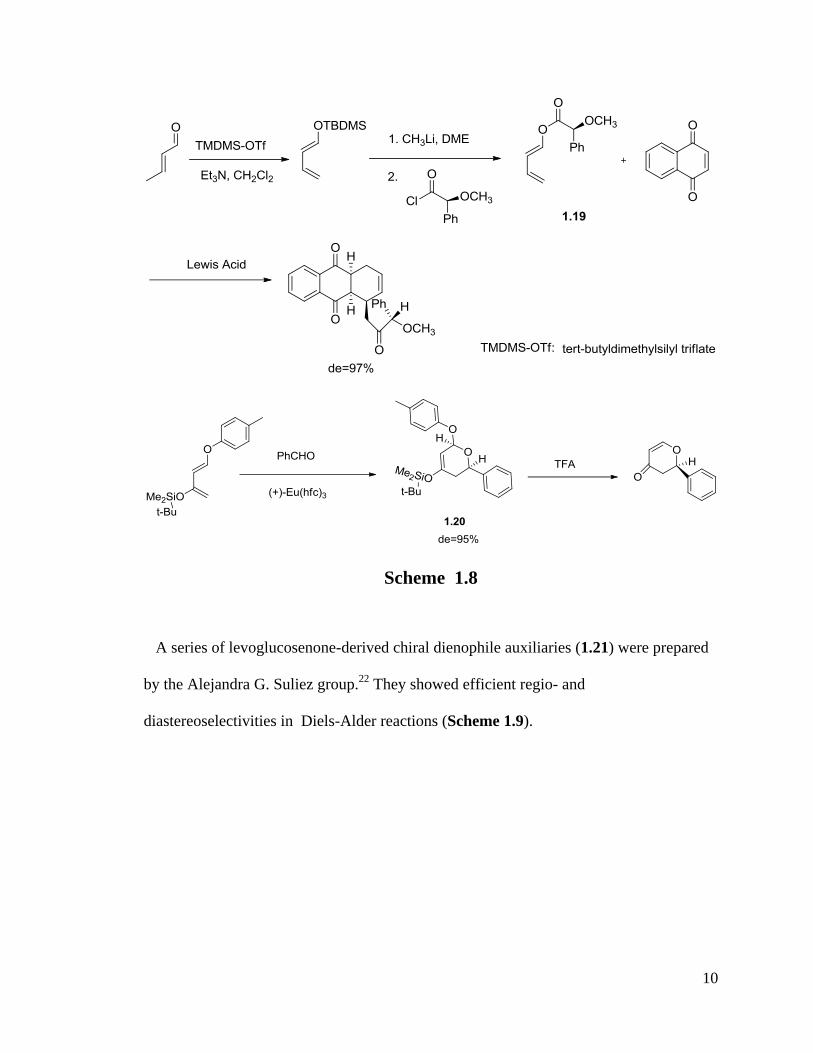
connecting a chiral group to the diene. A useful example has been developed by Trost et
al. by using an (s)-O-methylmandeloxy group as a chiral inducing agent to get a chiral
diene (Trost diene) 1.19,19, 20
which was described to be the first synthetically useful
chiral diene (Scheme 1.8).
An interesting result was reported by Danishefsky’s group about asymmetric Diels-
Alder reactions. They observed the dramatic increase of de value from 50% to 95%
(1.20), which was obtained by the combination of chiral diene and catalyst (+)-Eu(hfc)3.21
(Scheme 1.8)
10
Scheme 1.8
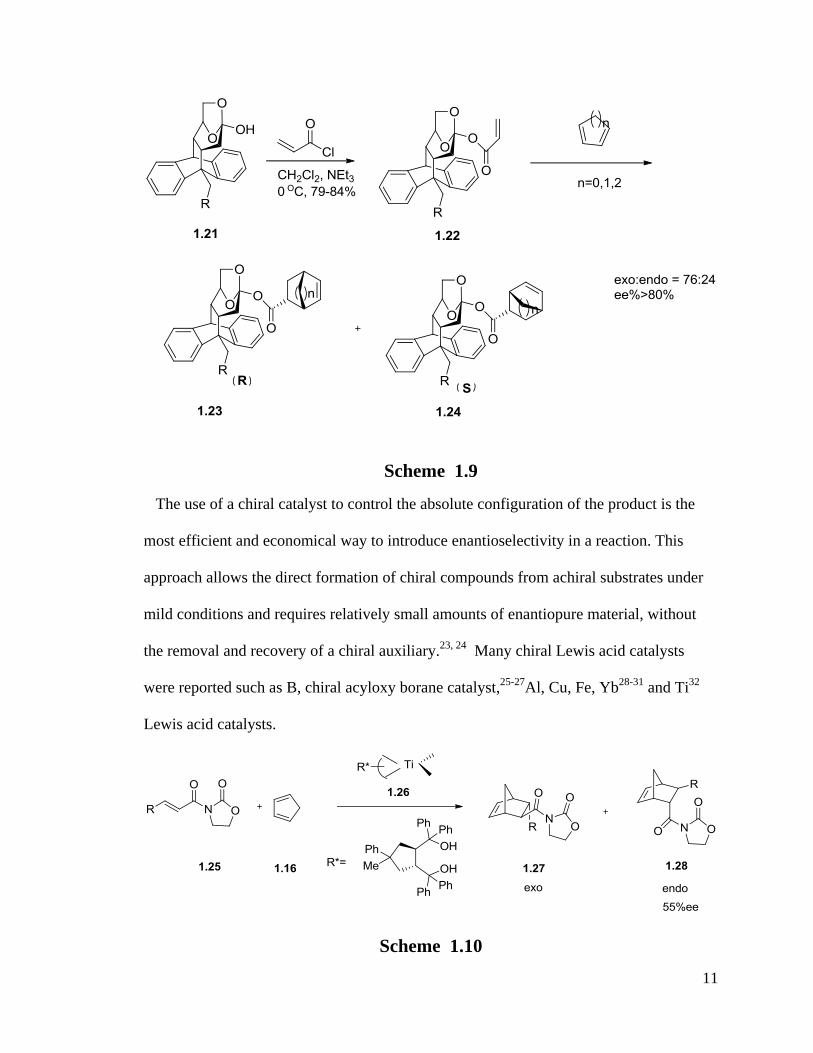
A series of levoglucosenone-derived chiral dienophile auxiliaries (1.21) were prepared
by the Alejandra G. Suliez group.22
They showed efficient regio- and
diastereoselectivities in Diels-Alder reactions (Scheme 1.9).
11
Scheme 1.9
The use of a chiral catalyst to control the absolute configuration of the product is the
most efficient and economical way to introduce enantioselectivity in a reaction. This
approach allows the direct formation of chiral compounds from achiral substrates under
mild conditions and requires relatively small amounts of enantiopure material, without
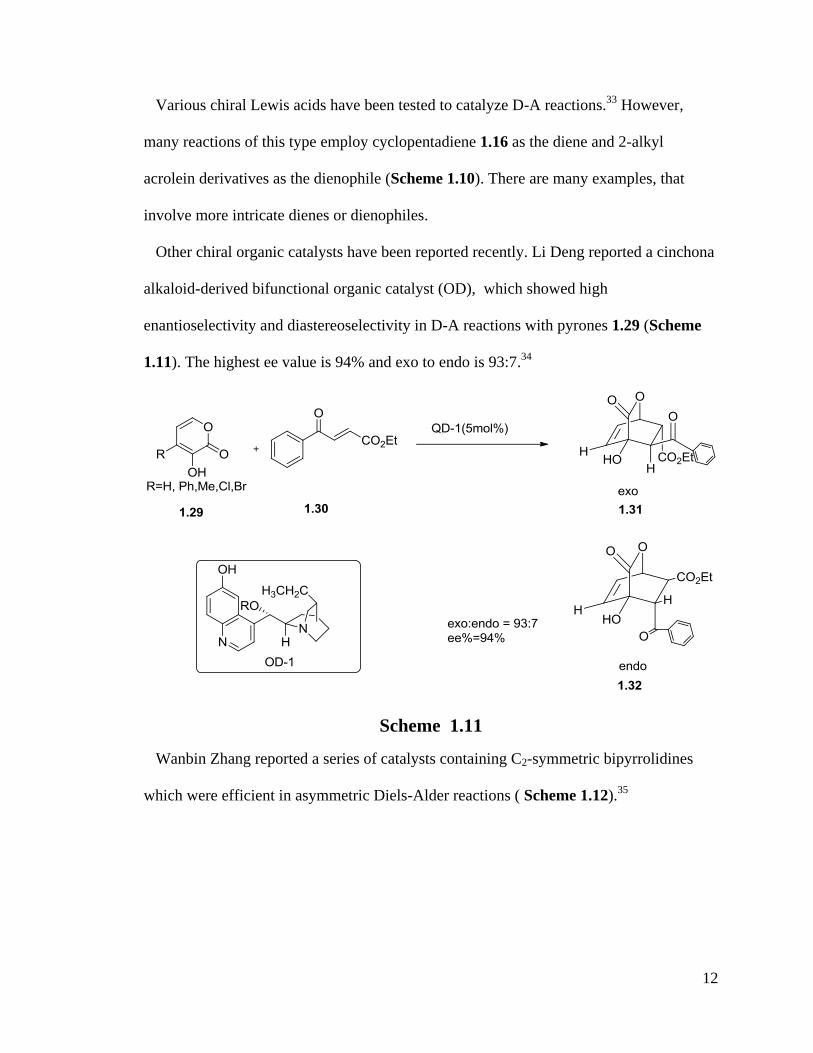
the removal and recovery of a chiral auxiliary.23, 24
Many chiral Lewis acid catalysts
were reported such as B, chiral acyloxy borane catalyst,25-27
Al, Cu, Fe, Yb28-31
and Ti32
Lewis acid catalysts.
Scheme 1.10
12
Various chiral Lewis acids have been tested to catalyze D-A reactions.33
However,
many reactions of this type employ cyclopentadiene 1.16 as the diene and 2-alkyl
acrolein derivatives as the dienophile (Scheme 1.10). There are many examples, that
involve more intricate dienes or dienophiles.
Other chiral organic catalysts have been reported recently. Li Deng reported a cinchona
alkaloid-derived bifunctional organic catalyst (OD), which showed high
enantioselectivity and diastereoselectivity in D-A reactions with pyrones 1.29 (Scheme
1.11). The highest ee value is 94% and exo to endo is 93:7.34
Scheme 1.11
Wanbin Zhang reported a series of catalysts containing C2-symmetric bipyrrolidines
which were efficient in asymmetric Diels-Alder reactions ( Scheme 1.12).35
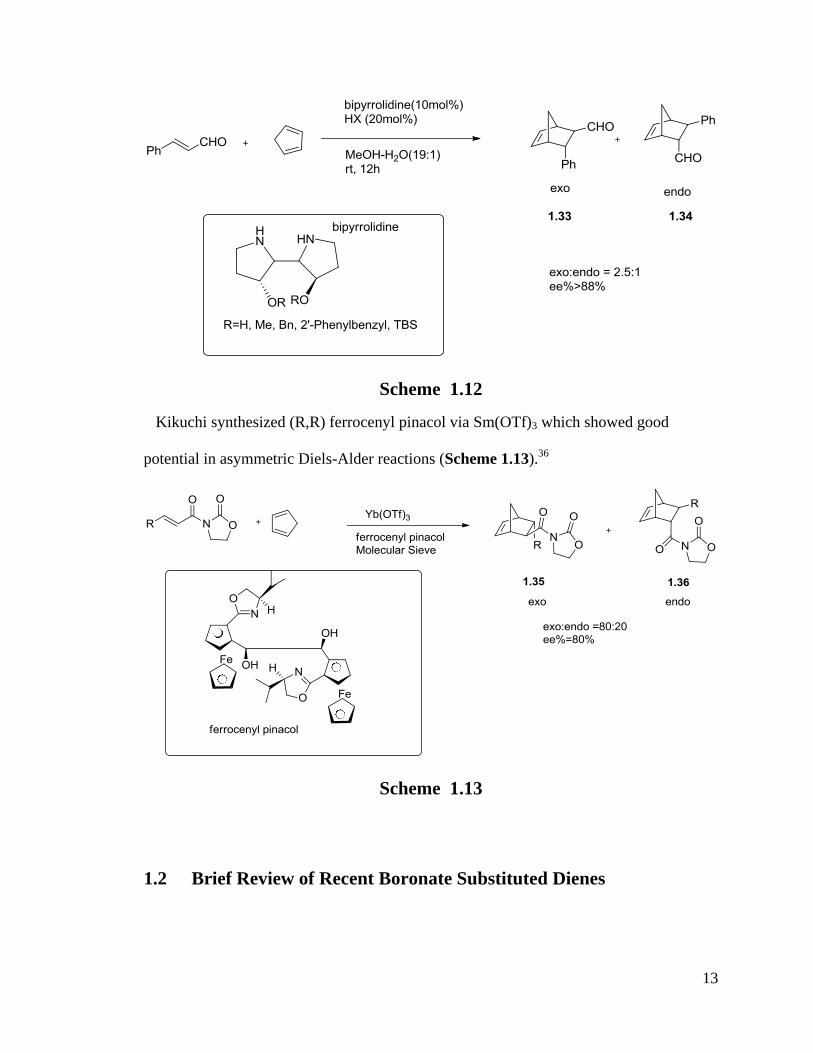
13
Scheme 1.12
Kikuchi synthesized (R,R) ferrocenyl pinacol via Sm(OTf)3 which showed good
potential in asymmetric Diels-Alder reactions (Scheme 1.13).36
Scheme 1.13
1.2 Brief Review of Recent Boronate Substituted Dienes

14
Organoboron compounds were not attractive until the extraordinary works of two
Nobel Prize winners: H. C. Brown and W. N. Lipscomb. They started the promising work
of the organoboron area by the discovery of hydroboration of alkenes and alkynes,
which leads to terminal boronate compounds.37, 38
Organoboron compounds are excellent precursors in organic synthesis. As the bridge to
construct different organic compounds by Suzuki coupling, rhodium catalysis and various
reactions, boron compounds have lots of advantages: readily available, easily handled,
some are stable in air and at room temperature, and the apparatus for handling these
compounds is simple. Also, boron compounds usually have no extreme toxicity hazards
although we need to treat them with care and avoid ingestion or contact.39
The traditional synthesis of aryl- and alkenylborates are from Grignard reagents or
lithium reagents and trialkyl borates.40
The general synthetic methods for preparing boron
compounds are transmetallation, hydroboration and haloboration.40-42
They are still the
basic methods for preparations of organoboron compounds nowadays(Scheme 1.14).
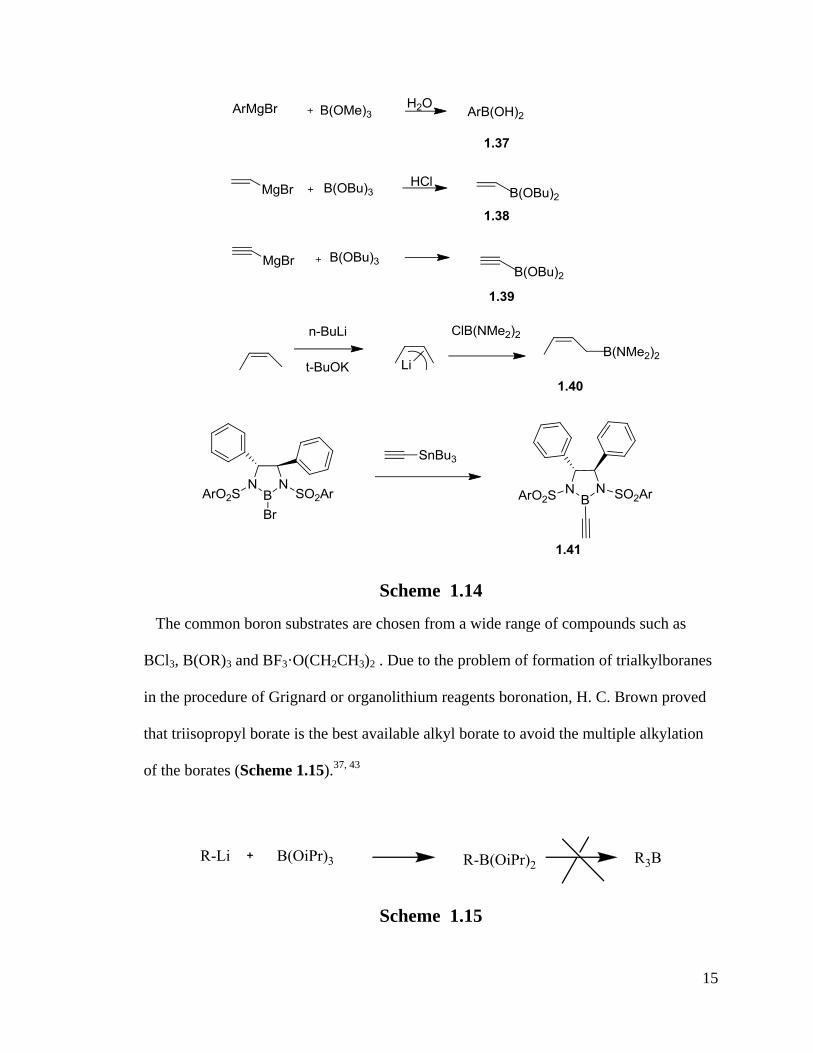
15
Scheme 1.14
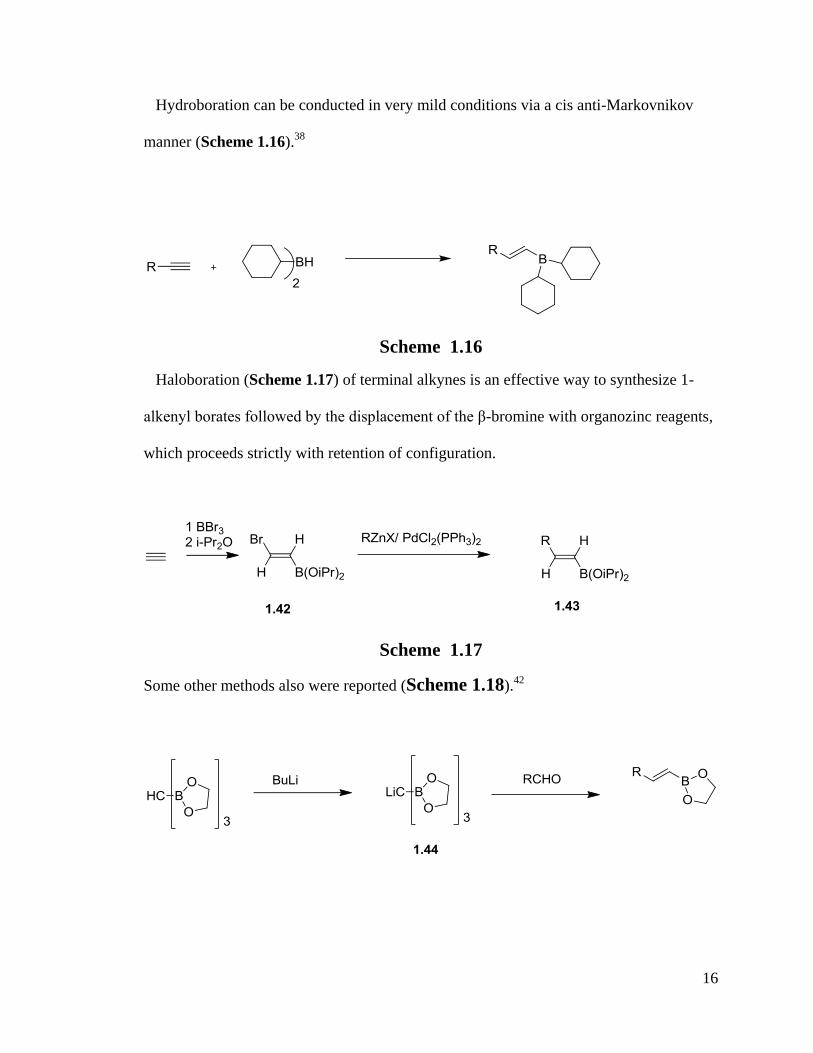
The common boron substrates are chosen from a wide range of compounds such as
BCl3, B(OR)3 and BF3·O(CH2CH3)2 . Due to the problem of formation of trialkylboranes
in the procedure of Grignard or organolithium reagents boronation, H. C. Brown proved
that triisopropyl borate is the best available alkyl borate to avoid the multiple alkylation
of the borates (Scheme 1.15).37, 43
Scheme 1.15
16
Hydroboration can be conducted in very mild conditions via a cis anti-Markovnikov
manner (Scheme 1.16).38
Scheme 1.16
Haloboration (Scheme 1.17) of terminal alkynes is an effective way to synthesize 1-
alkenyl borates followed by the displacement of the β-bromine with organozinc reagents,
which proceeds strictly with retention of configuration.
Scheme 1.17
Some other methods also were reported (Scheme 1.18).42

17
Scheme 1.18
Methods for preparation of organoboron dienes have been explored for a long time.
This dissertation briefly introduces some of the recent developments in this area.
High regioselectivity and stereoselectivity are always the goal scientists want to achieve
in D-A reactions. Heteroatom substituents can be placed on either the dienophile or diene
partner to get higher regioselectivity and stereoselectivity compared with unsubstituted
or alkyl substituted ones. That is also the goal of many substituted dienes such as
cobaloxime dienyl complexes,44-46
molybdenum and tungsten substituted dienes47, 48
and
silane and boron substituted dienes.49-52
1.2.1 Preparation and Reactions of 1-Boron Substituted-1,3-Dienes
Most reported boron substituted dienes are 1-boron substituted-1,3-dienes, especially
many 1-(dialkoxyboryl)-1,3-butadienes, sometimes termed 1,3-dienyl-1-boronates, which
can be easily prepared by hydroboration of enynes.53-56
Numerous reports of their Diels-
Alder/allylation reactions have been published and this sequence is usually called the
Vaultier tandem sequence. (Scheme 1.19)
Scheme 1.19
The sequence is very useful in the synthesis of clerodanes (Scheme 1.20).57
However,
this 1,3-dienylboronate has to react with activated dienophiles, which limits the wide
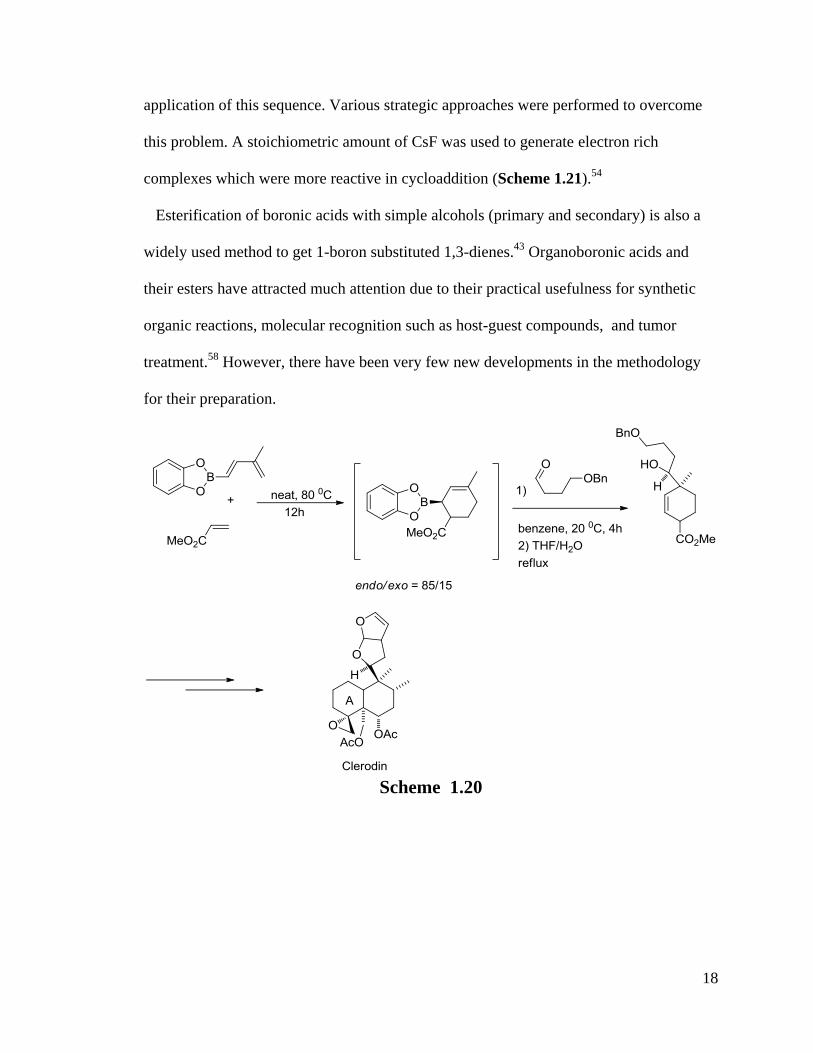
18
application of this sequence. Various strategic approaches were performed to overcome
this problem. A stoichiometric amount of CsF was used to generate electron rich
complexes which were more reactive in cycloaddition (Scheme 1.21).54
Esterification of boronic acids with simple alcohols (primary and secondary) is also a
widely used method to get 1-boron substituted 1,3-dienes.43
Organoboronic acids and
their esters have attracted much attention due to their practical usefulness for synthetic
organic reactions, molecular recognition such as host-guest compounds, and tumor
treatment.58
However, there have been very few new developments in the methodology
for their preparation.
Scheme 1.20
19
Scheme 1.21
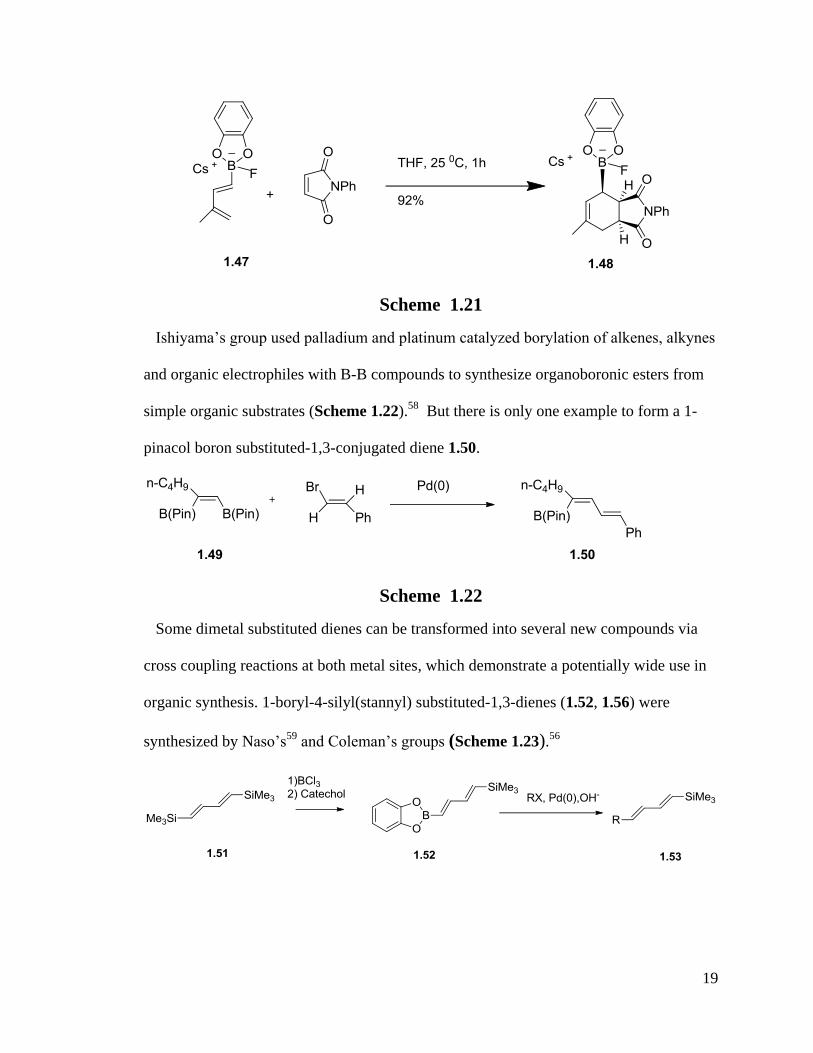
Ishiyama’s group used palladium and platinum catalyzed borylation of alkenes, alkynes
and organic electrophiles with B-B compounds to synthesize organoboronic esters from
simple organic substrates (Scheme 1.22).58
But there is only one example to form a 1-
pinacol boron substituted-1,3-conjugated diene 1.50.
Scheme 1.22
Some dimetal substituted dienes can be transformed into several new compounds via
cross coupling reactions at both metal sites, which demonstrate a potentially wide use in
organic synthesis. 1-boryl-4-silyl(stannyl) substituted-1,3-dienes (1.52, 1.56) were
synthesized by Naso’s59
and Coleman’s groups (Scheme 1.23).56

20
Scheme 1.23
1-Boron-substituted-1,3-dienes are usually easily prepared by the hydroboration of an
enyne.60
In 1999, Barrett and co-workers used this method to get a 1,3-dienylboronate,
which was hydrolyzed to dienyl boronic acid 1.58 and used in a cross coupling reaction
(Scheme 1.24).61
In 2002, Prandi and Venturello reported another way to prepare the
dienyl boronic ester1.60 from ,-unsaturated acetals 1.59 (Scheme 1.25).62
These
derivatives can be readily reacted with aryl substrates in very mild conditions to get
aromatic ketones.
Scheme 1.24
Scheme 1.25
21
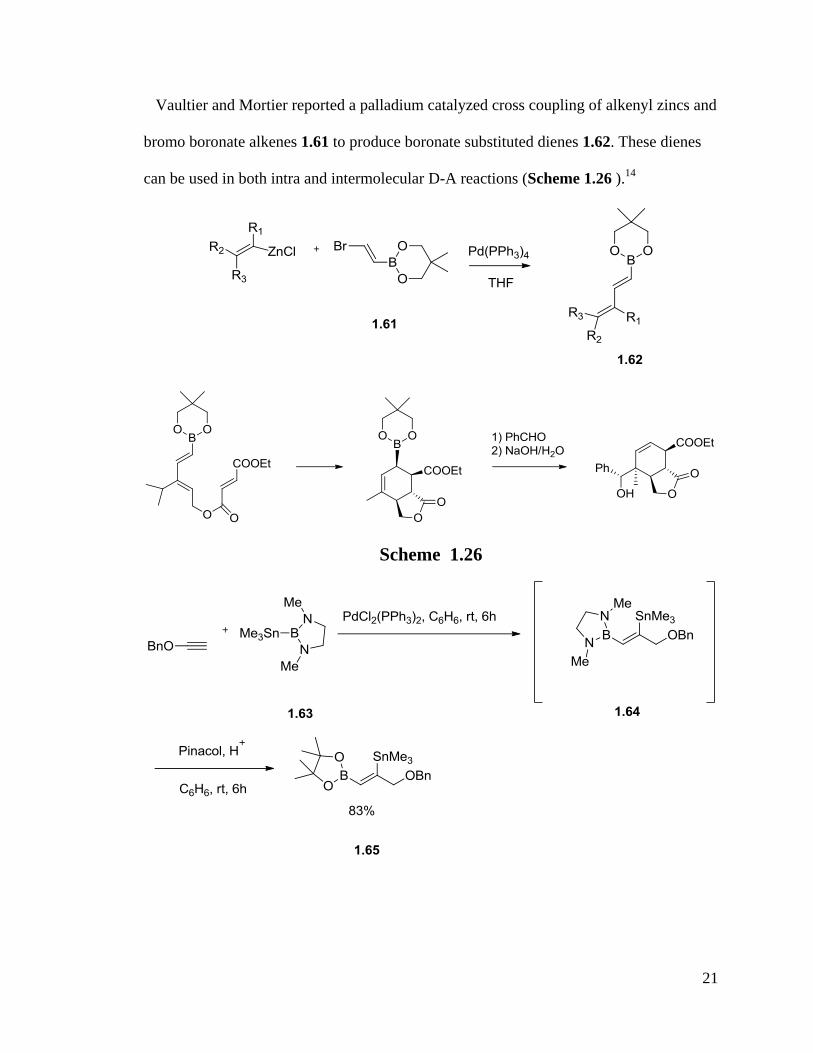
Vaultier and Mortier reported a palladium catalyzed cross coupling of alkenyl zincs and
bromo boronate alkenes 1.61 to produce boronate substituted dienes 1.62. These dienes
can be used in both intra and intermolecular D-A reactions (Scheme 1.26 ).14
Scheme 1.26

22
Scheme 1.27
RajanBabu found an efficient way to synthesize 1,4-disubstituted borylstannyl dienes
1.66 by borostannylation of an alkyne (Scheme 1.27).63
These bimetalated alkenes and
dienes were used in the Suzuki-Miyaura cross coupling reactions to prepare the polyene
side chains of some natural products.
1.2.2 Preparation and Reactions of 2-Boron Substituted-1,3-Dienes
Few reports were found on the preparation of 2-boron substituted-1,3-dienes in contrast
to the 1-boron substituted-1,3-dienes prior to 1998. Limited use of early members of this
class of compounds is presumably due to their high affinity towards dimerization even at
room temperature (Scheme 1.28).14,64
Scheme 1.28
Wrackmeyer and Vollrath reported 2-boron substituted 1,3-dienyl systems in 1998 by
the reaction of 1-propyl-1-boroindane 1.69 and dimethyl-di(1-propynyl)silane
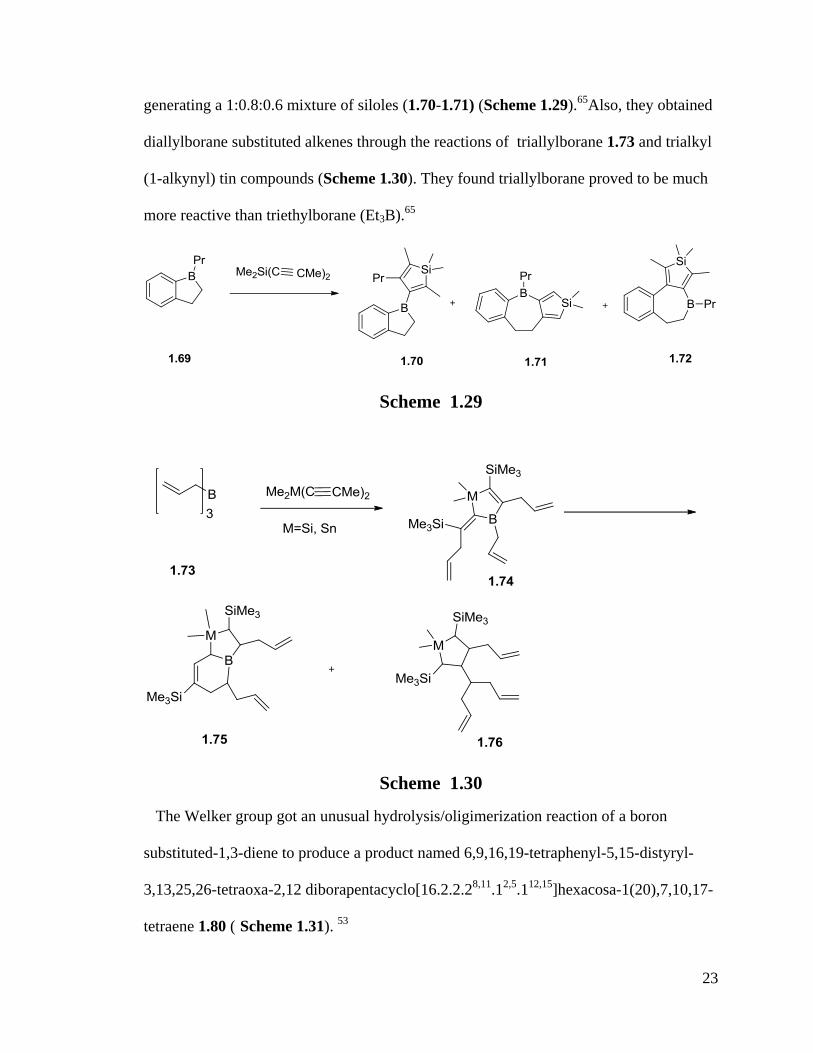
23
generating a 1:0.8:0.6 mixture of siloles (1.70-1.71) (Scheme 1.29).65
Also, they obtained
diallylborane substituted alkenes through the reactions of triallylborane 1.73 and trialkyl
(1-alkynyl) tin compounds (Scheme 1.30). They found triallylborane proved to be much
more reactive than triethylborane (Et3B).65
Scheme 1.29
Scheme 1.30
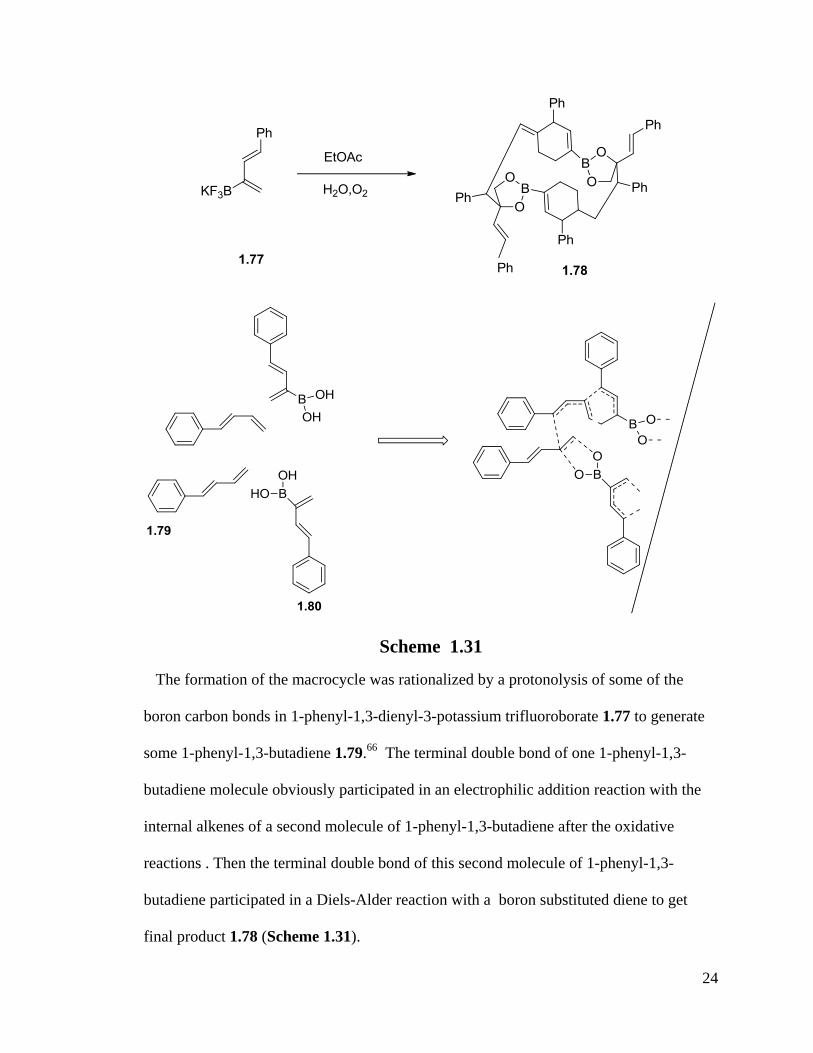
The Welker group got an unusual hydrolysis/oligimerization reaction of a boron
substituted-1,3-diene to produce a product named 6,9,16,19-tetraphenyl-5,15-distyryl-
3,13,25,26-tetraoxa-2,12 diborapentacyclo[16.2.2.28,11
.12,5
.112,15
]hexacosa-1(20),7,10,17-
tetraene 1.80 ( Scheme 1.31). 53
24
Scheme 1.31
The formation of the macrocycle was rationalized by a protonolysis of some of the
boron carbon bonds in 1-phenyl-1,3-dienyl-3-potassium trifluoroborate 1.77 to generate
some 1-phenyl-1,3-butadiene 1.79.66
The terminal double bond of one 1-phenyl-1,3-
butadiene molecule obviously participated in an electrophilic addition reaction with the
internal alkenes of a second molecule of 1-phenyl-1,3-butadiene after the oxidative
reactions . Then the terminal double bond of this second molecule of 1-phenyl-1,3-
butadiene participated in a Diels-Alder reaction with a boron substituted diene to get
final product 1.78 (Scheme 1.31).
25
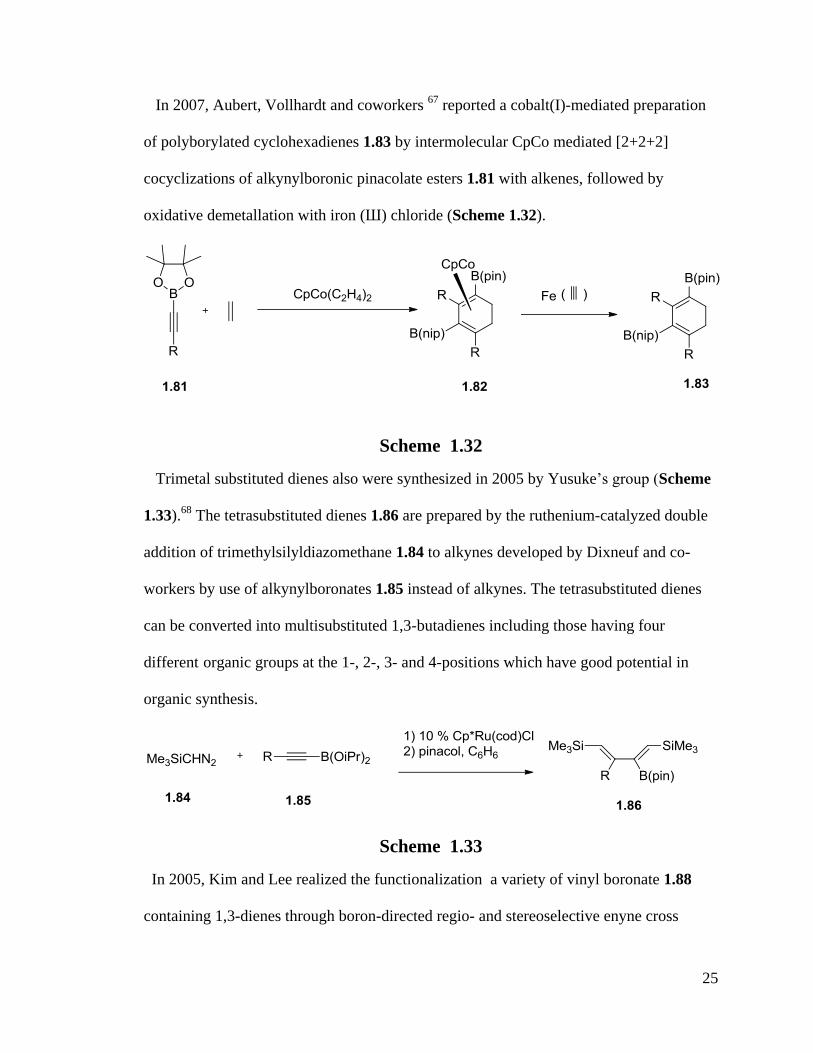
In 2007, Aubert, Vollhardt and coworkers 67
reported a cobalt(I)-mediated preparation
of polyborylated cyclohexadienes 1.83 by intermolecular CpCo mediated [2+2+2]
cocyclizations of alkynylboronic pinacolate esters 1.81 with alkenes, followed by
oxidative demetallation with iron (Ш) chloride (Scheme 1.32).
Scheme 1.32
Trimetal substituted dienes also were synthesized in 2005 by Yusuke’s group (Scheme
1.33).68
The tetrasubstituted dienes 1.86 are prepared by the ruthenium-catalyzed double
addition of trimethylsilyldiazomethane 1.84 to alkynes developed by Dixneuf and co-
workers by use of alkynylboronates 1.85 instead of alkynes. The tetrasubstituted dienes
can be converted into multisubstituted 1,3-butadienes including those having four
different organic groups at the 1-, 2-, 3- and 4-positions which have good potential in
organic synthesis.
Scheme 1.33
In 2005, Kim and Lee realized the functionalization a variety of vinyl boronate 1.88
containing 1,3-dienes through boron-directed regio- and stereoselective enyne cross
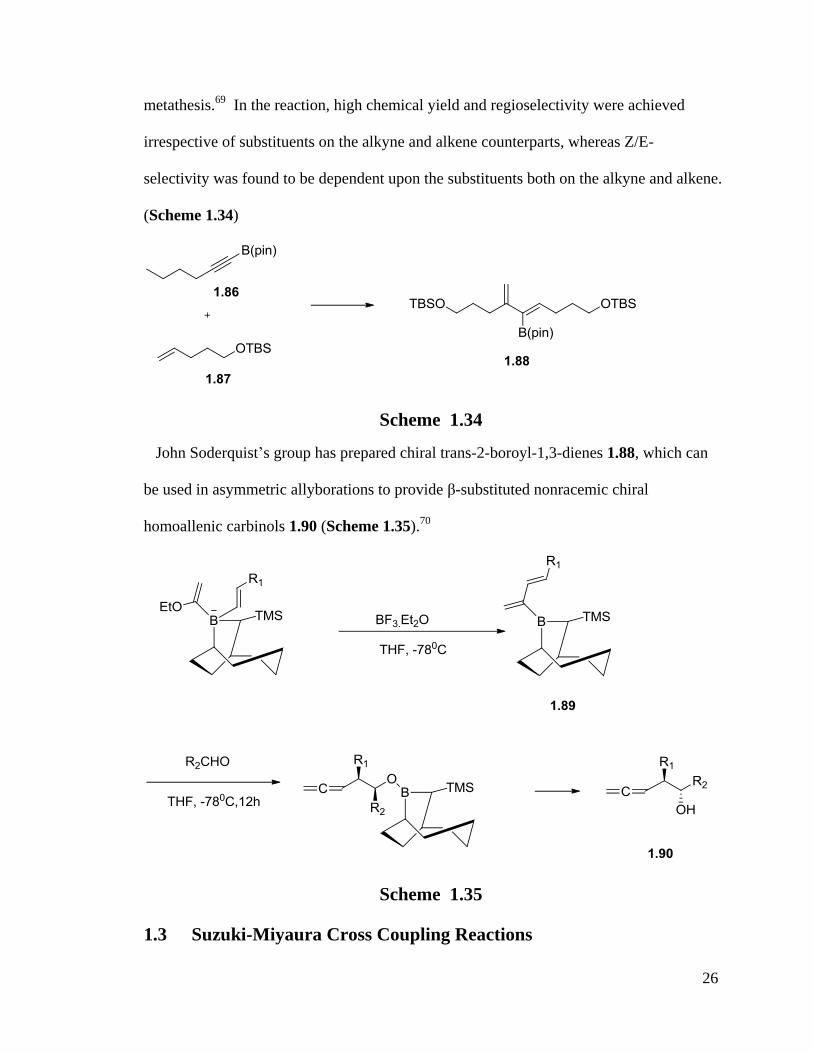
26
metathesis.69
In the reaction, high chemical yield and regioselectivity were achieved
irrespective of substituents on the alkyne and alkene counterparts, whereas Z/E-
selectivity was found to be dependent upon the substituents both on the alkyne and alkene.
(Scheme 1.34)
Scheme 1.34
John Soderquist’s group has prepared chiral trans-2-boroyl-1,3-dienes 1.88, which can
be used in asymmetric allyborations to provide β-substituted nonracemic chiral
homoallenic carbinols 1.90 (Scheme 1.35).70
Scheme 1.35
1.3 Suzuki-Miyaura Cross Coupling Reactions

27
The most utilized C-C bond forming cross coupling reactions are probably the Heck,
Stille and Suzuki-Miyaura reactions.71
In these reactions, palladium is a tremendously
effective catalyst which also plays an important role in a general catalytic cycle (Figure
1.5 ).71
This mechanism explains most cross coupling reactions including oxidative
addition of halide, transmetallation and reductive elimination. The Suzuki-Miyaura
reaction is preeminent due to its compatibility with a diverse range of functional groups.
It also has good tolerance to the presence of water and proceeds generally with good
regio- and stereoselectively with a non-toxic inorganic by-product which can be easily
removed from the reaction mixture. Therefore, the Suzuki coupling has been used not
only in the lab but also in industrial processes.72, 73
Figure 1.5 General Catalytic Cycle for Suzuki-Miyaura, Heck And
Stille Cross-Coupling Reactions

28
In Suzuki-Miyaura reactions, oxidative addition is usually the rate determining step in a
catalytic cycle that affords a stable trans-σ-palladium (B) complex. However, Gebbink
reported recently that transmetalation is the rate determining step when they used
Dendriphos ligands in Suzuki cross-coupling reactions.74
The relative reactivity of
oxidative addition decreases in the sequence of I > OTf > Br >> Cl for the halide
compounds. The structure of alkenyl halides can be preserved in the final products but
the inversion effects for allylic and benzylic halides were observed.42
Bulky and electron
donating ligands, which generate an electron-rich metal complex, were found to make
palladium complexes more reactive in oxidative addition reactions.75
Transmetallation is the hardest step to control in practical experiments since its
mechanism still remains obscure and highly dependent on organometallics or reaction
conditions used for the couplings.72
In the presence of a negatively charged base, the
transmetallation between organopalladium halides and organoboron compounds
apparently is accelerated. The reason is that the quaternization of boron with a negatively
charged base enhances the nucleophilicity of other organic groups on boron. Thus, the
role of the base is to facilitate the slow transmetalation of the boron compounds by
forming a more reactive boronate species that can interact with the Pd center and finish
transmetalation.76, 77
There is another explanation for the effects of base that the base
replaces the halide in the coordination sphere of the palladium complex and facilitates an
intramolecular transmetalation (path B).78
The bases often used are K3PO4, K2CO3, KOH
and KF. There is no general rule for the selection of bases except experience.
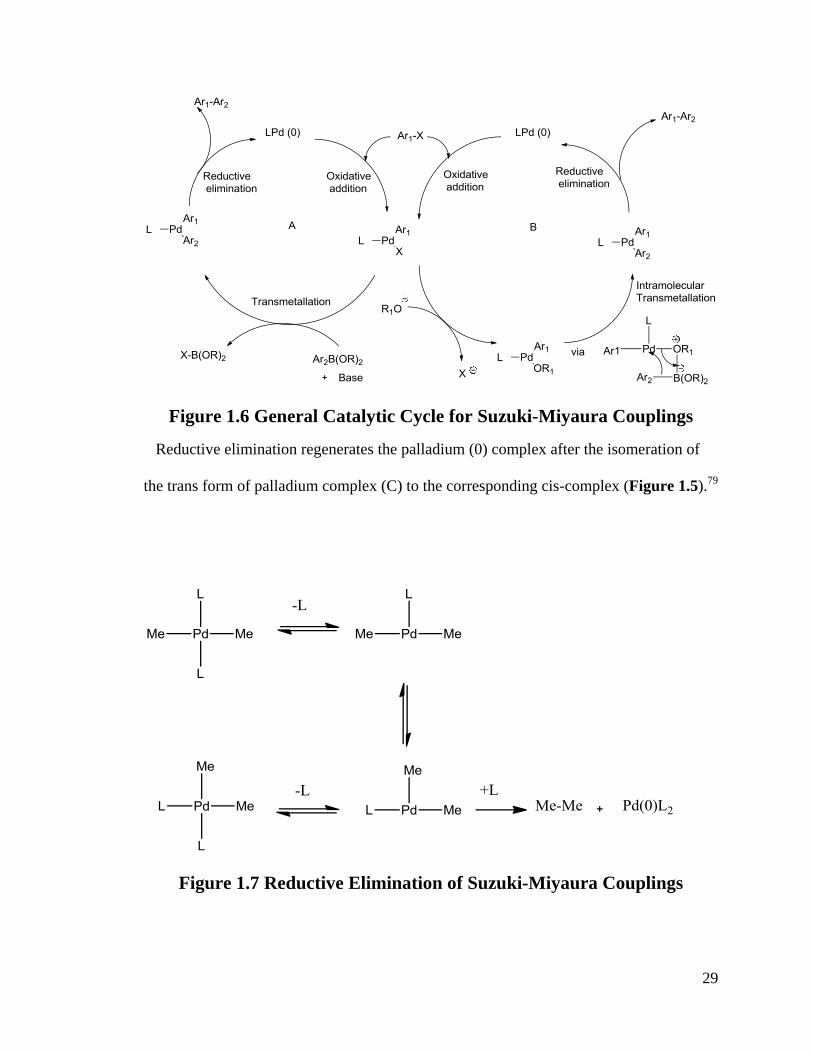
29
Figure 1.6 General Catalytic Cycle for Suzuki-Miyaura Couplings
Reductive elimination regenerates the palladium (0) complex after the isomeration of
the trans form of palladium complex (C) to the corresponding cis-complex (Figure 1.5).79
Figure 1.7 Reductive Elimination of Suzuki-Miyaura Couplings

30
It is generally accepted that this step is faster when palladium is coordinated to
electron-withdrawing and sterically bulky ligands. It has also been shown that for very
bulky ligands, the steric properties dominate over the electronic properties.78
1.4 Rhodium Reaction
The newly developed reaction of rhodium catalyzed 1,4-addition of substituted boronic
acids to enones has progressed rapidly in the last 10 years. The reaction was discovered
in 1997 by Miyaura and other chemists.80, 81
After that, many subsequent reports have
appeared.82
Asymmetric carbon-carbon bond forming is very important in asymmetric
synthesis but in most cases it is difficult to achieve high yields and excellent
enantioselectivities. However, the 1,4-addition of enones by boronic acids provides a
good way to this goal. It is already a powerful method to achieve stereoselective C-C
bonds and quaternary stereocenters.
1.4.1 Advantages of rhodium catalyzed 1,4-addition of enones
The reaction (Scheme 1.36) has several advantages over other 1,4-additions.
83
In this reaction, the organoboronic acids are not so sensitive to oxygen and moisture
compared with some other organometallic reagents, such as substituted boron esters.
Therefore, one can conduct reactions in protic media or aqueous solution, which is a good
choice for the requirements of green chemistry. The organoboronic acids are much less
reactive toward enones without a rhodium catalyst. Thus, we prevent the 1,2-addition to
enones by adding the rhodium catalyst.

31
Groups connecting to boron are generally added to the position with high
enantioselectivity. Both chiral ligands and chiral substituted boronic acids can be used as
a chiral resource to generate a new chiral center with high stereoselectivity. The reaction
has a large scope for substrates not only for ,-unsaturated ketones, but also for ,-
unsaturated aldehydes, esters, amides, imides, 84
and even some metal connecting
unsaturated ketones.
Scheme 1.36
Different rhodium catalysts, various ligands and the bases are used in this reaction.
Most rhodium catalysts contain BINAP groups83, 85
or BINAP derivatives, which are
essential for the generation of chiral centers e.g. [Rh(OH)(S)-binap]2. 83
Because the
temperature influences the hydrolysis of boronic acids, usually excess boronic acids are
used. A detailed process of the reaction is also shown in Scheme 1.37.86
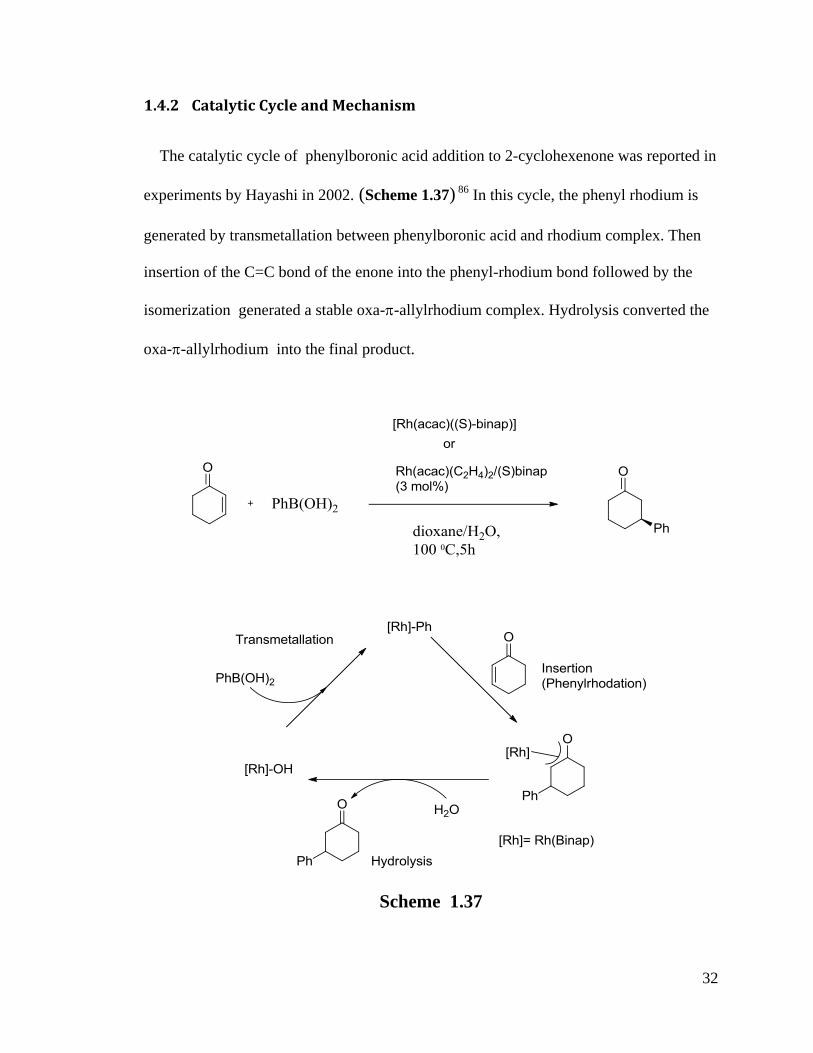
32
1.4.2 Catalytic Cycle and Mechanism
The catalytic cycle of phenylboronic acid addition to 2-cyclohexenone was reported in
experiments by Hayashi in 2002. (Scheme 1.37) 86 In this cycle, the phenyl rhodium is
generated by transmetallation between phenylboronic acid and rhodium complex. Then
insertion of the C=C bond of the enone into the phenyl-rhodium bond followed by the
isomerization generated a stable oxa--allylrhodium complex. Hydrolysis converted the
oxa--allylrhodium into the final product.
Scheme 1.37

33
1.4.3 Reaction Properties
Ligand Effects
In the first stage, Miyaura’s work was focused on the non-asymmetric 1,4-addition of
aryl- and alkenyl-boronic acids to ,-unsaturated ketones.81
Research followed by other
chemists which was rapidly transfered to asymmetric synthesis. Among several factors
that can influence the stereoselectivities, the chiral ligand has crucial effects on the
enantiomeric excess (ee) value and yields. This dissertation only discusses the two most
used ligand groups : BINAP and [2.2.2] bicyclooctadiene and their derivatives.
(a) 1,1’-Binaphtyl ligands and its derivatives : (R)or (S)-BINAP(2,2'-bis
(diphenylphosphino) -1,1'-binaphthyl) and some phosphorus ligands.
BINAP is a chiral bisphosphine ligand, which consists of two naphthyl groups linked
by a single bond with diphenylphosphino groups at the end of each naphthyl group
(Figure 1.9). Although BINAP has no stereogenic center, it is a chiral molecule. Rotation
about the single bond binding the two naphthyl groups is restricted because of the rigidity
of their π systems. Almost all 1,4-addition reactions using the BINAP groups produced
good yields and over 90% enantioselectivity.87, 88
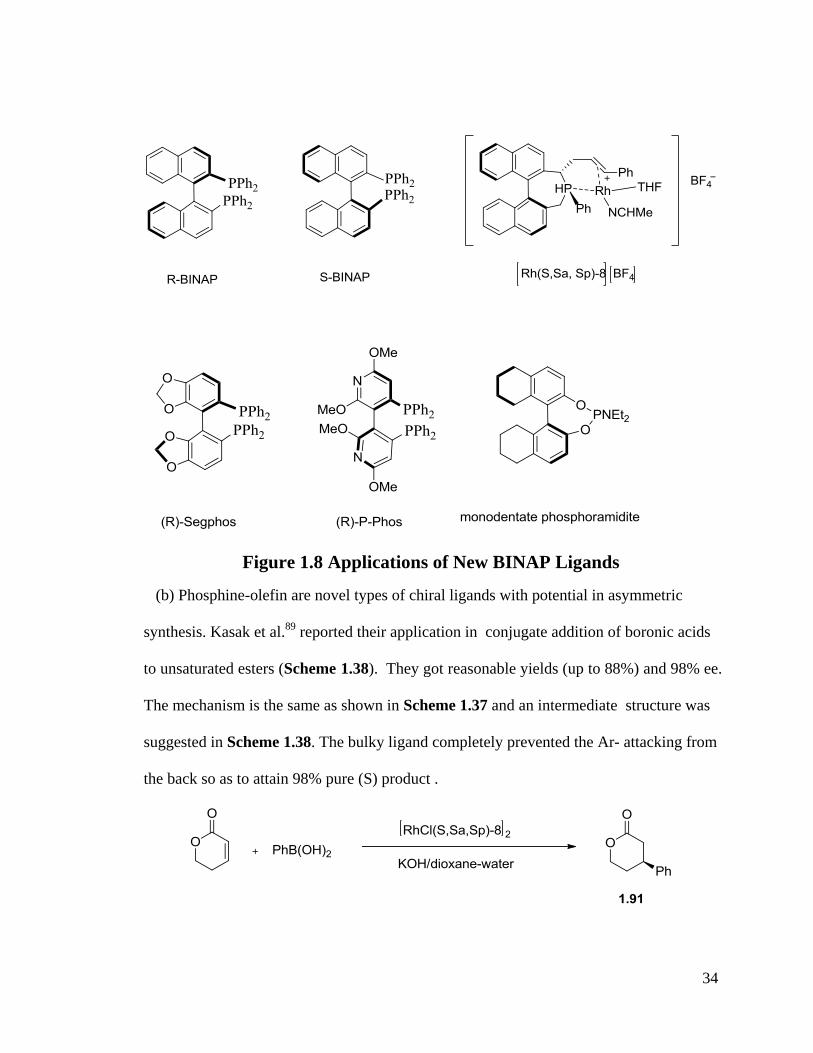
Some new applications of BINAP ligands employed since 2003 are also demonstrated
in Figure 1.8.
34
Figure 1.8 Applications of New BINAP Ligands
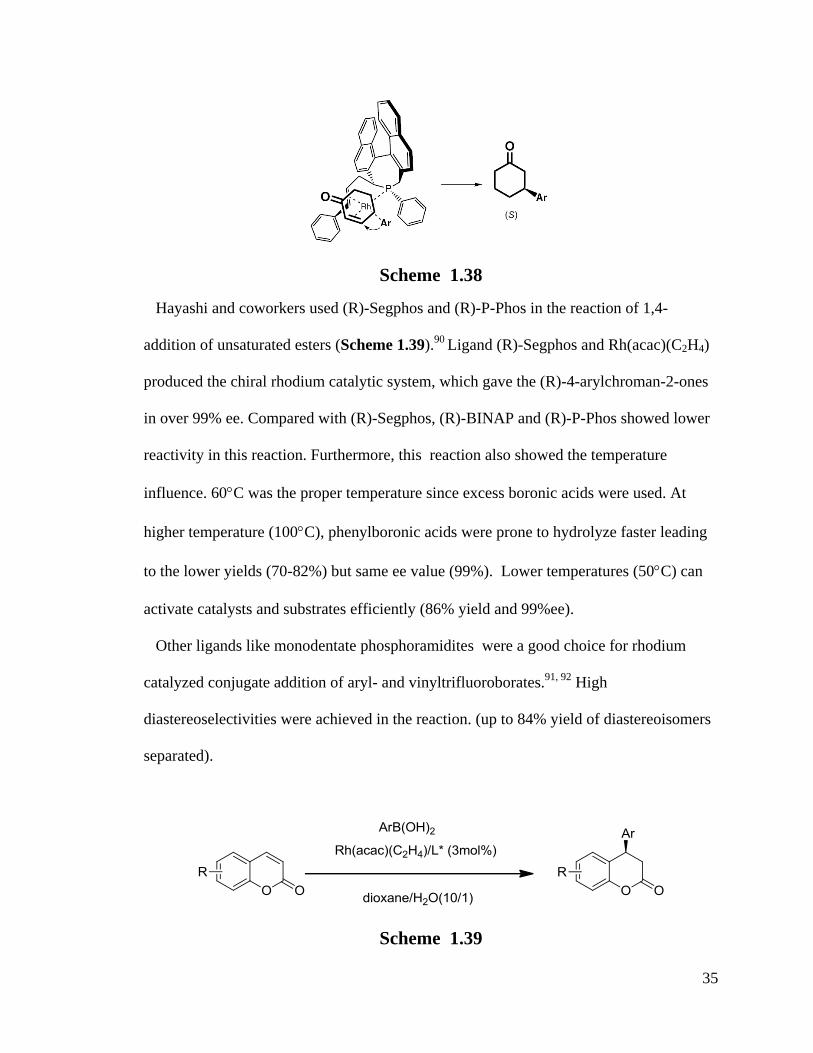
(b) Phosphine-olefin are novel types of chiral ligands with potential in asymmetric
synthesis. Kasak et al.89
reported their application in conjugate addition of boronic acids
to unsaturated esters (Scheme 1.38). They got reasonable yields (up to 88%) and 98% ee.
The mechanism is the same as shown in Scheme 1.37 and an intermediate structure was
suggested in Scheme 1.38. The bulky ligand completely prevented the Ar- attacking from
the back so as to attain 98% pure (S) product .
35
Scheme 1.38
Hayashi and coworkers used (R)-Segphos and (R)-P-Phos in the reaction of 1,4-
addition of unsaturated esters (Scheme 1.39).90
Ligand (R)-Segphos and Rh(acac)(C2H4)
produced the chiral rhodium catalytic system, which gave the (R)-4-arylchroman-2-ones
in over 99% ee. Compared with (R)-Segphos, (R)-BINAP and (R)-P-Phos showed lower
reactivity in this reaction. Furthermore, this reaction also showed the temperature
influence. 60C was the proper temperature since excess boronic acids were used. At
higher temperature (100C), phenylboronic acids were prone to hydrolyze faster leading
to the lower yields (70-82%) but same ee value (99%). Lower temperatures (50C) can
activate catalysts and substrates efficiently (86% yield and 99%ee).
Other ligands like monodentate phosphoramidites were a good choice for rhodium
catalyzed conjugate addition of aryl- and vinyltrifluoroborates.91, 92
High
diastereoselectivities were achieved in the reaction. (up to 84% yield of diastereoisomers
separated).
Scheme 1.39
36
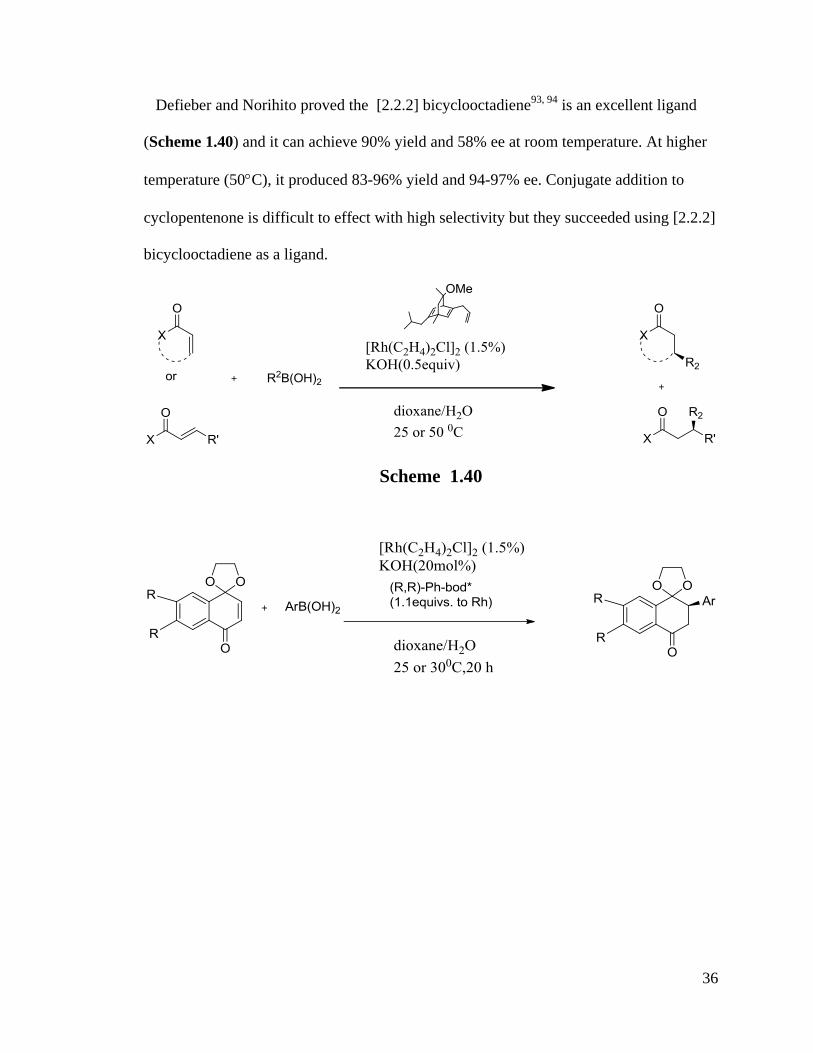
Defieber and Norihito proved the [2.2.2] bicyclooctadiene93, 94
is an excellent ligand
(Scheme 1.40) and it can achieve 90% yield and 58% ee at room temperature. At higher
temperature (50C), it produced 83-96% yield and 94-97% ee. Conjugate addition to
cyclopentenone is difficult to effect with high selectivity but they succeeded using [2.2.2]
bicyclooctadiene as a ligand.
Scheme 1.40
37
Scheme 1.41
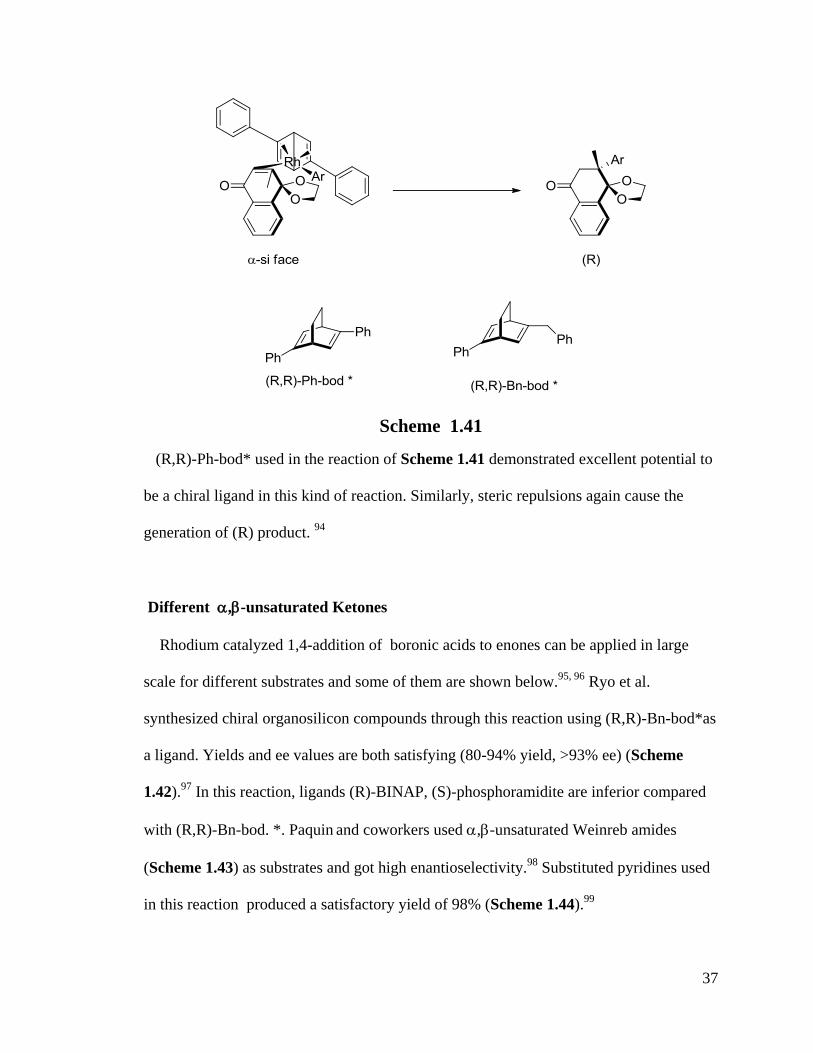
(R,R)-Ph-bod* used in the reaction of Scheme 1.41 demonstrated excellent potential to
be a chiral ligand in this kind of reaction. Similarly, steric repulsions again cause the
generation of (R) product. 94
Different ,-unsaturated Ketones
Rhodium catalyzed 1,4-addition of boronic acids to enones can be applied in large
scale for different substrates and some of them are shown below.95, 96
Ryo et al.
synthesized chiral organosilicon compounds through this reaction using (R,R)-Bn-bod*as
a ligand. Yields and ee values are both satisfying (80-94% yield, >93% ee) (Scheme
1.42).97
In this reaction, ligands (R)-BINAP, (S)-phosphoramidite are inferior compared
with (R,R)-Bn-bod. *. Paquin and coworkers used ,-unsaturated Weinreb amides
(Scheme 1.43) as substrates and got high enantioselectivity.98
Substituted pyridines used
in this reaction produced a satisfactory yield of 98% (Scheme 1.44).99
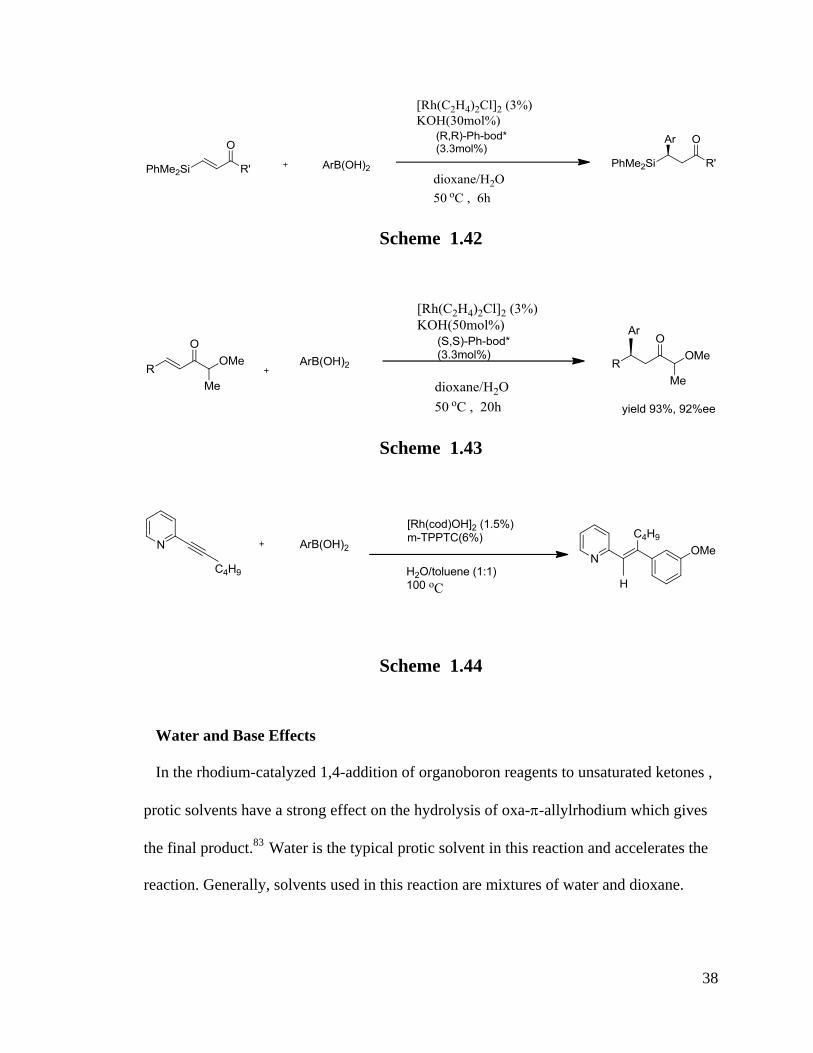
38
Scheme 1.42
Scheme 1.43
Scheme 1.44
Water and Base Effects
In the rhodium-catalyzed 1,4-addition of organoboron reagents to unsaturated ketones ,
protic solvents have a strong effect on the hydrolysis of oxa--allylrhodium which gives
the final product.83
Water is the typical protic solvent in this reaction and accelerates the
reaction. Generally, solvents used in this reaction are mixtures of water and dioxane.

39
Yasuhiro et al used heterogeneous catalysts ( rhodium complexes immobilized on the
polymer)100
in water catalyzing 1,4-addition of ArB(OH)2. Up to 93% yield was attained
and the catalysts can be recovered and reused for 3 times.
The reaction is strongly inhibited by acid.101
Both protic acids and Lewis acids act as
strong inhibitors. Introduction of base can remarkably enhance the reaction speed. A
proposed active intermediate is L2RhOH generated in-situ and accelerates
transmetallation (Scheme 1.45).
Scheme 1.45
The Rh(I) catalyzed conjugate addition of substituted boronic acids offers a general
solution for the synthesis of chiral compounds as mentioned before. However, in certain
conditions, 1,2-addition occurs competing with 1,4-addition in unsaturated aldehydes
(Scheme 1.45).102
Different ligands lead to different reaction directions. Generally speaking, the
polarization of the C-Rh bond plays a key role in the competition. Donating ligands
increase the nucleophilicity of the organic group on the rhodium metal. t-Bu3P does not
catalyze the conjugate 1,4-addition seletively but produces 1,2-addition product. A
reasonable catalytic cycle is shown in Scheme 1.46.102
Transmetallation is the key step
in the cycle. Because the dppf complex is more Lewis acidic than the tert-butylphosphine
complex then coordination of dppf complex and the nitro group retards the addition to
nitrobenzaldehydes.
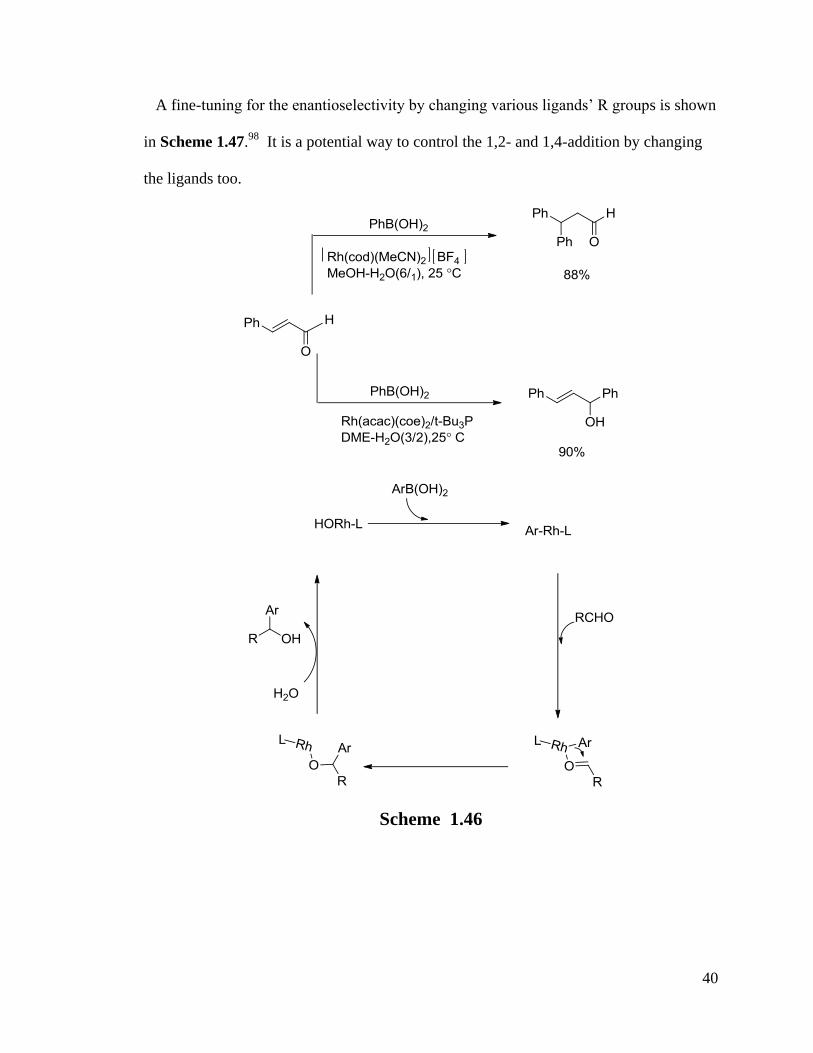
40
A fine-tuning for the enantioselectivity by changing various ligands’ R groups is shown
in Scheme 1.47.98
It is a potential way to control the 1,2- and 1,4-addition by changing
the ligands too.
Scheme 1.46
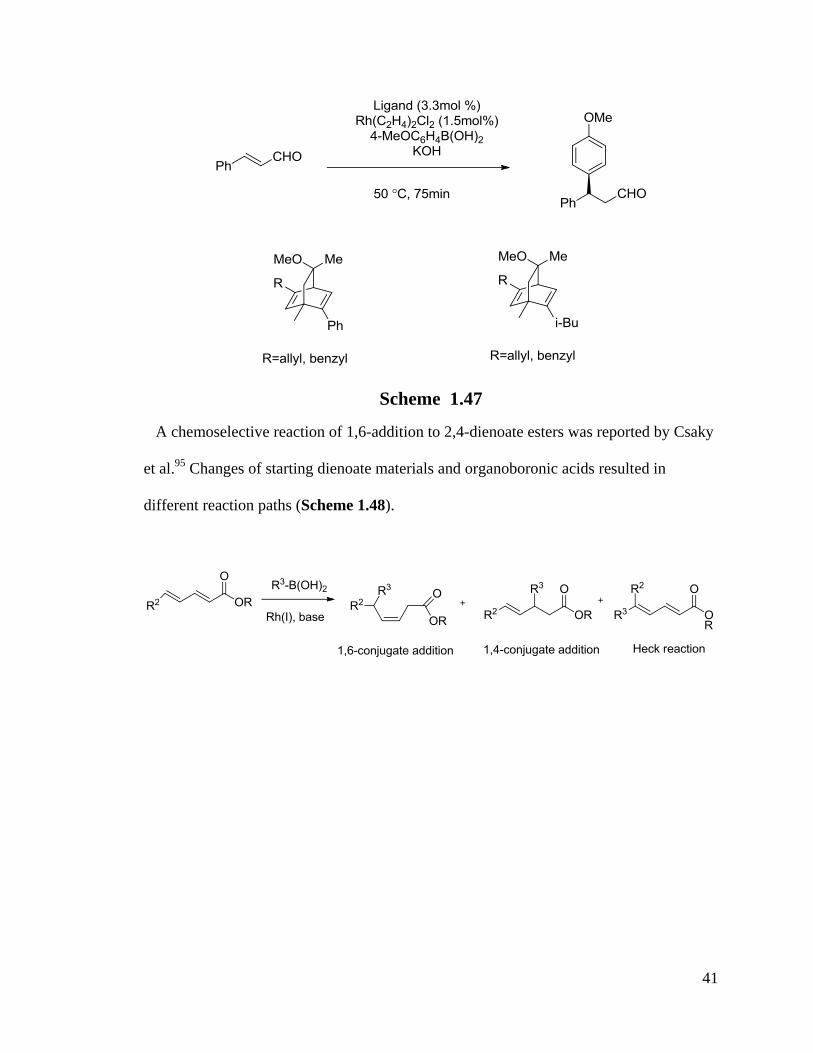
41
Scheme 1.47
A chemoselective reaction of 1,6-addition to 2,4-dienoate esters was reported by Csaky
et al.95
Changes of starting dienoate materials and organoboronic acids resulted in
different reaction paths (Scheme 1.48).
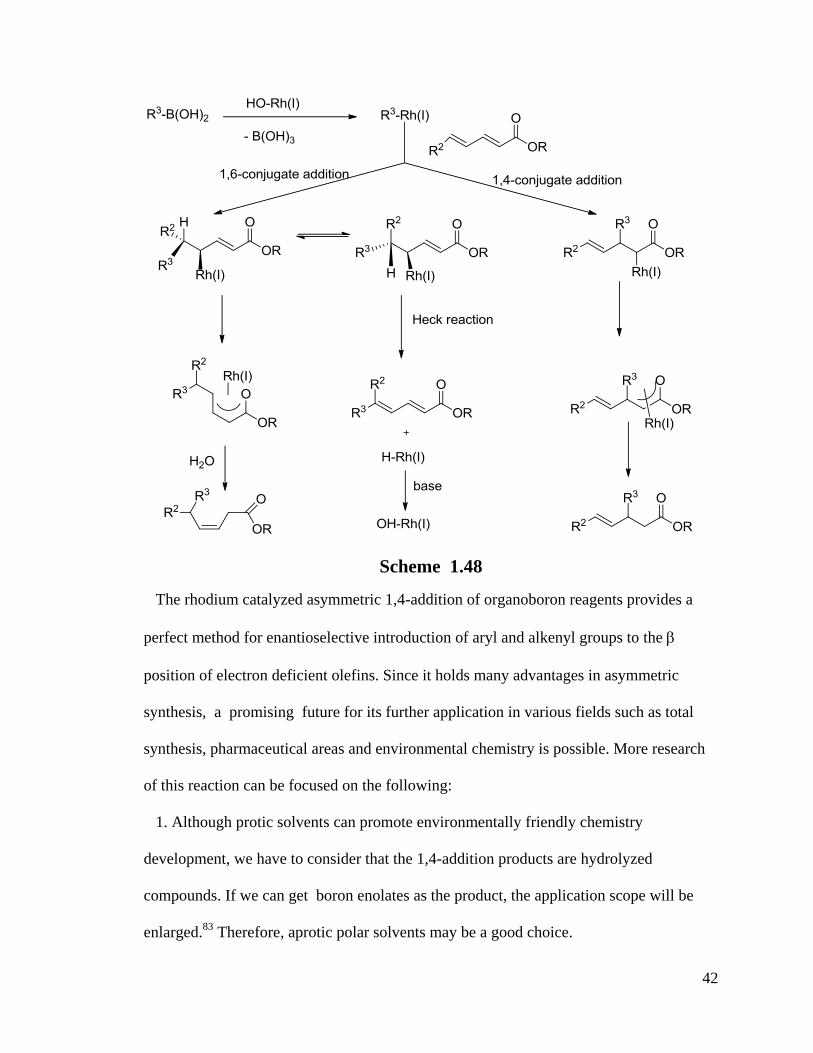
42
Scheme 1.48
The rhodium catalyzed asymmetric 1,4-addition of organoboron reagents provides a
perfect method for enantioselective introduction of aryl and alkenyl groups to the
position of electron deficient olefins. Since it holds many advantages in asymmetric
synthesis, a promising future for its further application in various fields such as total
synthesis, pharmaceutical areas and environmental chemistry is possible. More research
of this reaction can be focused on the following:
1. Although protic solvents can promote environmentally friendly chemistry
development, we have to consider that the 1,4-addition products are hydrolyzed
compounds. If we can get boron enolates as the product, the application scope will be
enlarged.83
Therefore, aprotic polar solvents may be a good choice.

43
2. Different ligands have different enantioselectivities in various reactions. The
research in this area is still a hotspot.
3. Water as solvent and heterogeneous catalysis in water are also attractions for green
chemistry workers. Especially asymmetric synthesis of heterogeneous catalysts in water
is a challenge. There are few reports about organometallic substrates in this reaction.
Research in this area is also promising. 1,2-and 1,4- addition competition is a good
method for chemists to design and control reaction products by tuning the ligands,
solvents and other factors.103
1.5 Aims of Current Project
Previous research by our group showed the superiority of 2-metal substituted 1,3-dienes
to 1-metal substituted 1,3-dienes104
both in terms of rate enhancement and
stereoselectivity in D-A reactions. As 2-boron-substituted dienes have wide applications
in D-A and cross coupling reactions, which will lead to decalin core structures synthesis,
our project will focus on their synthesis and cross coupling reactions. The other reasons
that we selected boron substituted dienes are the wide varieties of protocols in their
preparations, air and moisture stability, and easy handling and storage relative to other
main group metal substituted dienes.
However, the synthesis of 2-boron substituted dienes had proved difficult according to
the research of other groups since they all obtained a dimerization product of diene by
self D-A reactions.105,64
The current project begins with the synthesis and purification of
2-boron-substituted dienes. Although many different types of organoboron species are
known in the literature, dienyl borates are rarely reported because they are easily

44
polymerized in the preparation process and difficult to separate by the usual silica gel
column chromatography.
The long term goal of this project is to apply the methodology to access substituted cis-
fused decalin core structures that are otherwise difficult to make. Many natural products
have the decalin core, such as clerodane diterpenoids.106
Although reports in the trans-
clerodanes are abundant, cis-clerodanes syntheses are rare. Since we have already shown
the exo selective cobalt mediated D-A reactions could be used to synthesize cis-fused
bicyclic compounds related to known diterpenes, the same trends are expected to be
applicable to our main group substituted dienes.

45
CHAPTER 2 Preparation of Boron Substituted Dienes
2.1 Results and Discussion
2.1.1 Preparation of 2- Diethanolamine Borate Diene 2.4 And 2.6
Previous Welker group member Subhasis De prepared a monomeric 2-BF3-1,3-butyl
diene in high yield and studied its D-A/cross coupling reactions under mild conditions.49
This dienyltrifluoroborate also showed a high activity in rhodium catalysis. Therefore,
dienyltrifluoroborate can serve as a potential building block for the construction of other
complex molecules.
However, the purification of boron compounds is still a most difficult task. The
dienyltrifluoroborate is anionic with low organic solvent solubility. We began to search
for other neutral boron compounds. Diethanolamine dienylboronates (2.4) seemed
reasonable because they have a nitrogen lone electron pair donating to the boron atom
generating an electron rich diene.
In the first attempt at the preparation of diethanolamine dienylboronates we used the
dienyl trimethoxy boronate to react with diethanolamine in THF. We did get the product
(2.4) (NMR) but we could not separate it from the hard solid reaction mixture (Scheme
2.1). After that, efforts were made to prepare 2.4 via a direct esterification route from
dienyl boronic acid (2.5) and diethanolamine.
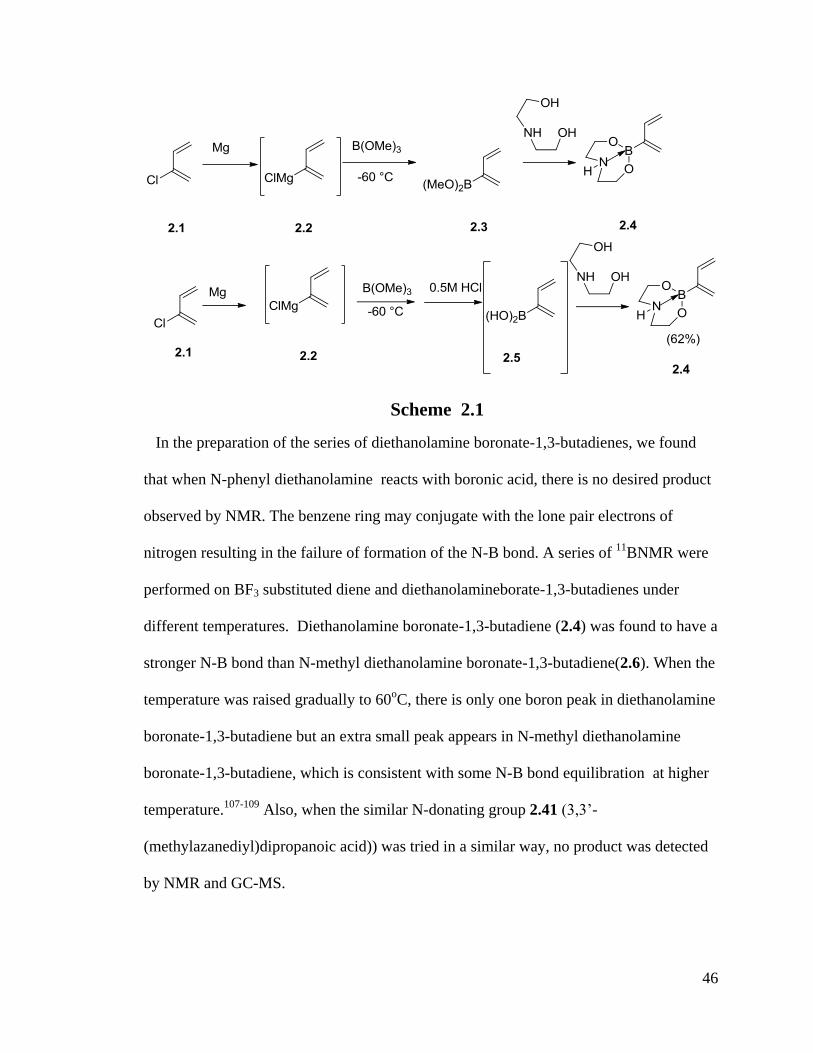
46
Scheme 2.1
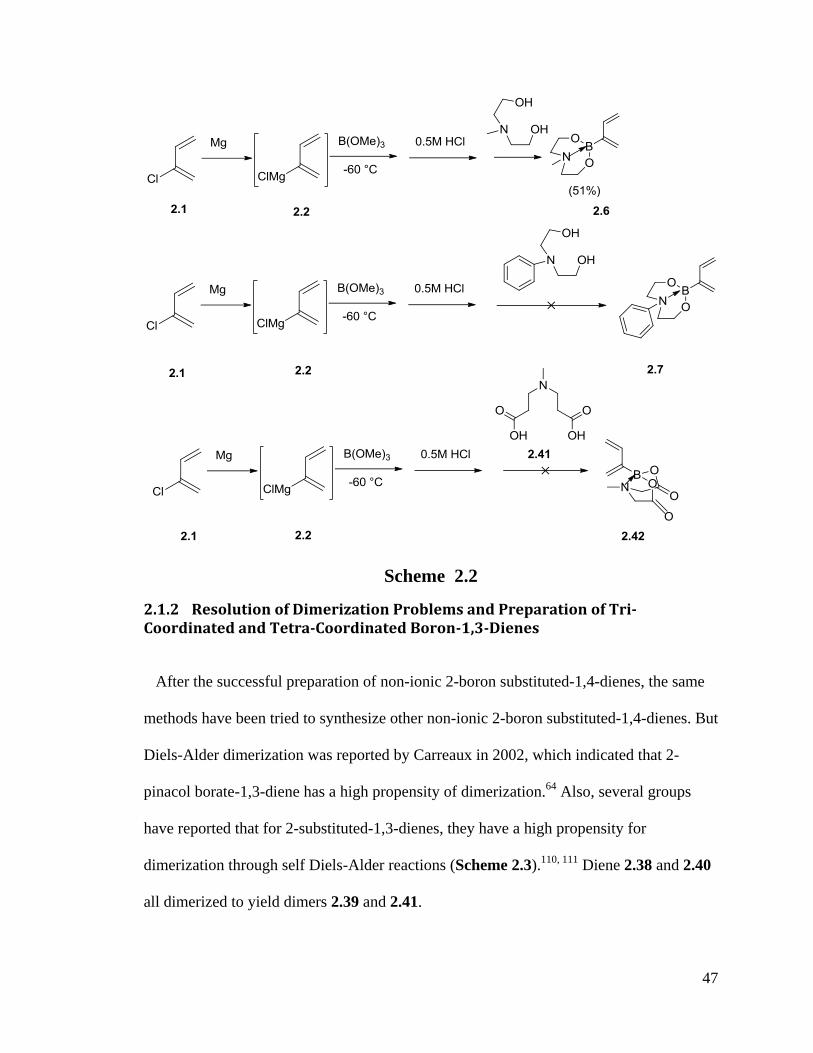
In the preparation of the series of diethanolamine boronate-1,3-butadienes, we found
that when N-phenyl diethanolamine reacts with boronic acid, there is no desired product
observed by NMR. The benzene ring may conjugate with the lone pair electrons of
nitrogen resulting in the failure of formation of the N-B bond. A series of 11
BNMR were
performed on BF3 substituted diene and diethanolamineborate-1,3-butadienes under
different temperatures. Diethanolamine boronate-1,3-butadiene (2.4) was found to have a
stronger N-B bond than N-methyl diethanolamine boronate-1,3-butadiene(2.6). When the
temperature was raised gradually to 60oC, there is only one boron peak in diethanolamine
boronate-1,3-butadiene but an extra small peak appears in N-methyl diethanolamine
boronate-1,3-butadiene, which is consistent with some N-B bond equilibration at higher
temperature.107-109
Also, when the similar N-donating group 2.41 (3,3’-
(methylazanediyl)dipropanoic acid)) was tried in a similar way, no product was detected
by NMR and GC-MS.
47
Scheme 2.2
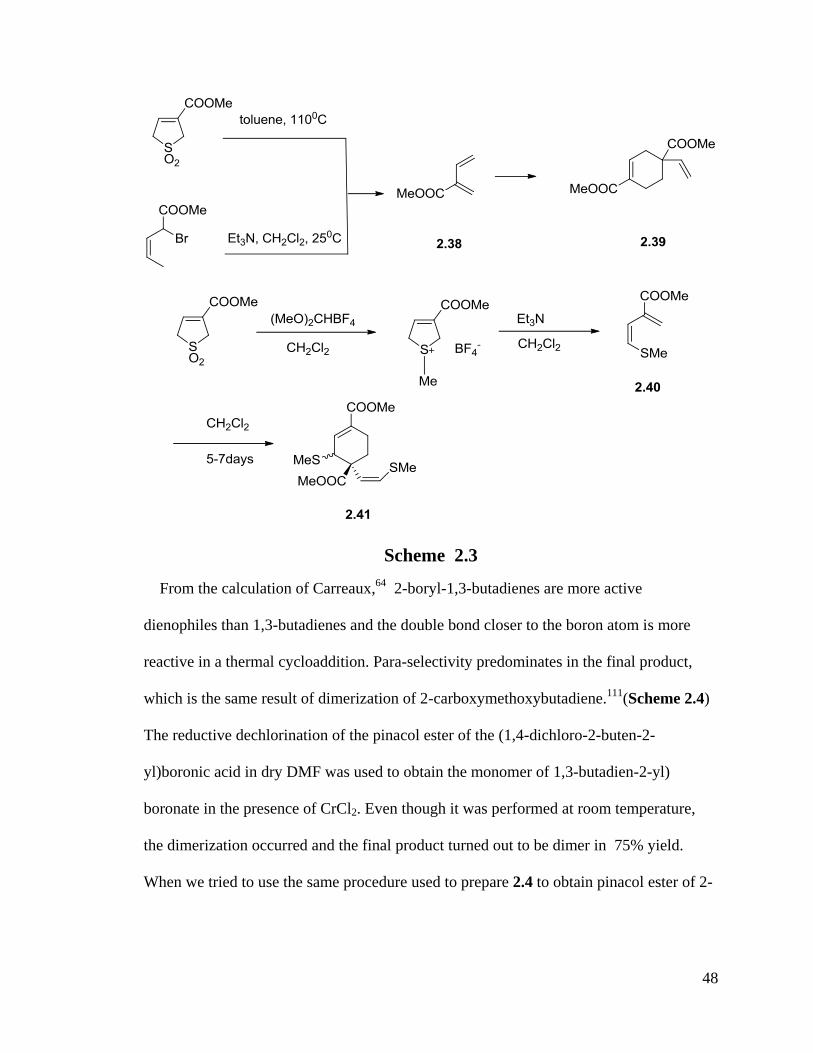
2.1.2 Resolution of Dimerization Problems and Preparation of Tri-Coordinated and Tetra-Coordinated Boron-1,3-Dienes
After the successful preparation of non-ionic 2-boron substituted-1,4-dienes, the same
methods have been tried to synthesize other non-ionic 2-boron substituted-1,4-dienes. But
Diels-Alder dimerization was reported by Carreaux in 2002, which indicated that 2-
pinacol borate-1,3-diene has a high propensity of dimerization.64
Also, several groups
have reported that for 2-substituted-1,3-dienes, they have a high propensity for
dimerization through self Diels-Alder reactions (Scheme 2.3).110, 111
Diene 2.38 and 2.40
all dimerized to yield dimers 2.39 and 2.41.
48
Scheme 2.3
From the calculation of Carreaux,64
2-boryl-1,3-butadienes are more active
dienophiles than 1,3-butadienes and the double bond closer to the boron atom is more
reactive in a thermal cycloaddition. Para-selectivity predominates in the final product,
which is the same result of dimerization of 2-carboxymethoxybutadiene.111
(Scheme 2.4)
The reductive dechlorination of the pinacol ester of the (1,4-dichloro-2-buten-2-
yl)boronic acid in dry DMF was used to obtain the monomer of 1,3-butadien-2-yl)
boronate in the presence of CrCl2. Even though it was performed at room temperature,
the dimerization occurred and the final product turned out to be dimer in 75% yield.
When we tried to use the same procedure used to prepare 2.4 to obtain pinacol ester of 2-
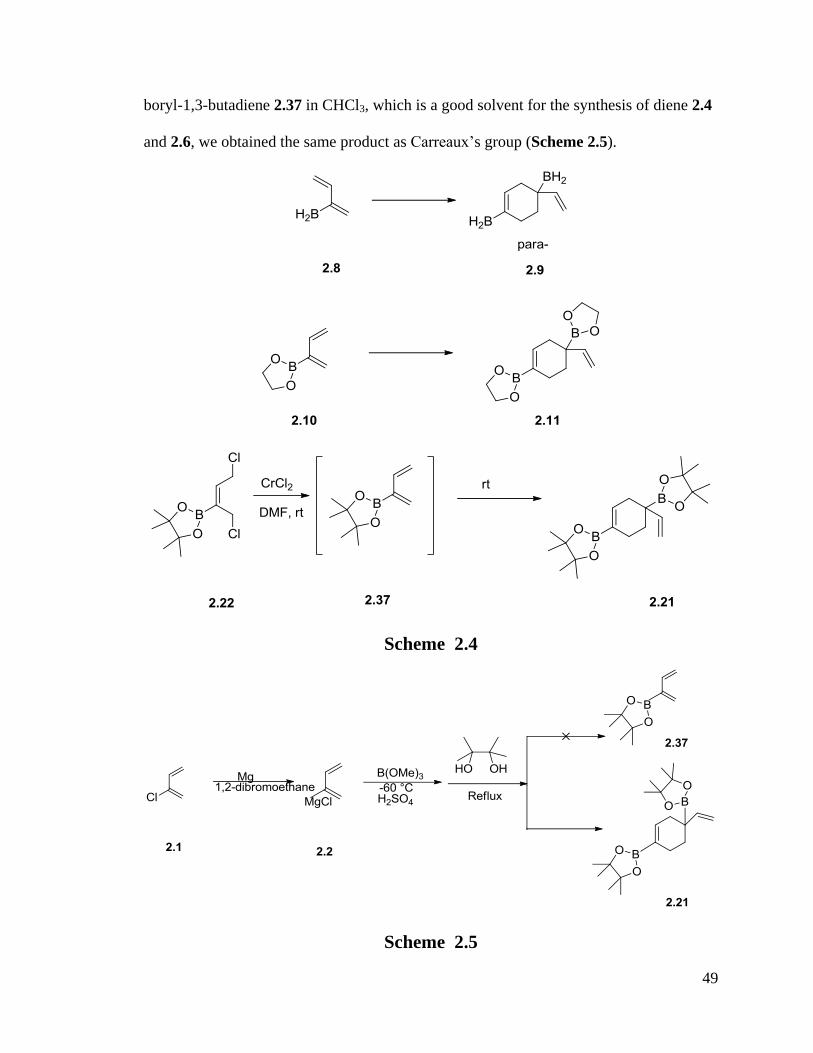
49
boryl-1,3-butadiene 2.37 in CHCl3, which is a good solvent for the synthesis of diene 2.4
and 2.6, we obtained the same product as Carreaux’s group (Scheme 2.5).
Scheme 2.4
Scheme 2.5
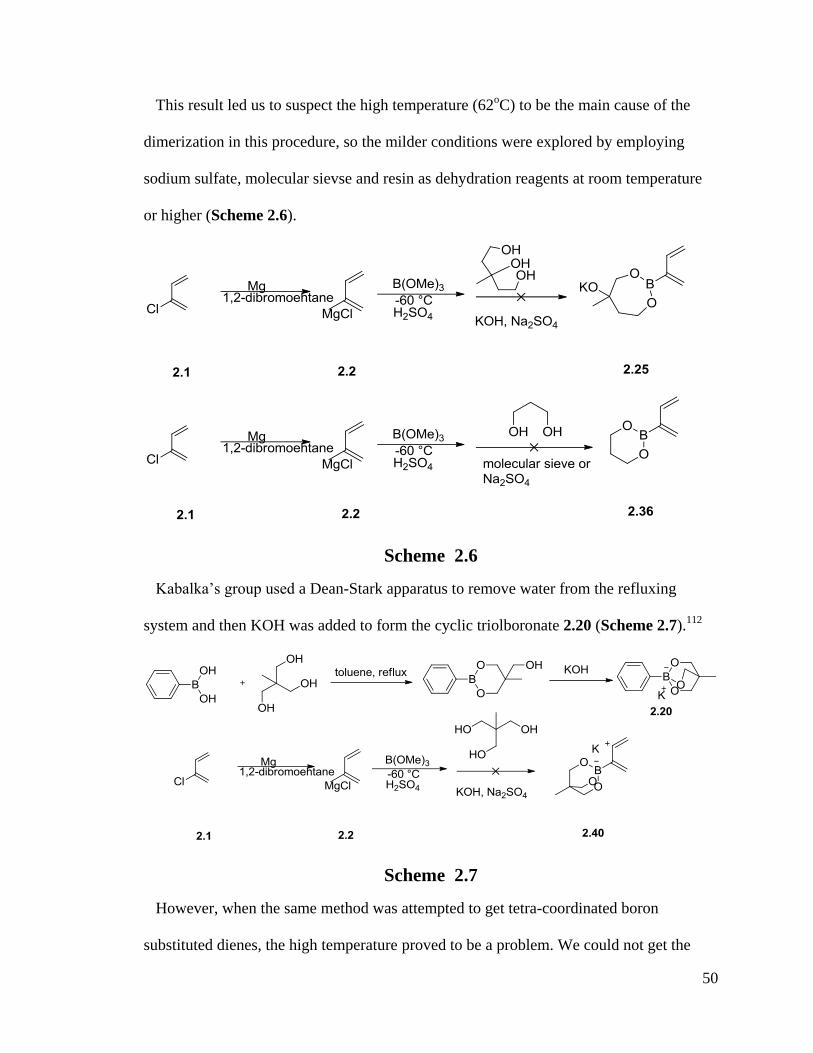
50
This result led us to suspect the high temperature (62oC) to be the main cause of the
dimerization in this procedure, so the milder conditions were explored by employing
sodium sulfate, molecular sievse and resin as dehydration reagents at room temperature
or higher (Scheme 2.6).
Scheme 2.6
Kabalka’s group used a Dean-Stark apparatus to remove water from the refluxing
system and then KOH was added to form the cyclic triolboronate 2.20 (Scheme 2.7).112
Scheme 2.7
However, when the same method was attempted to get tetra-coordinated boron
substituted dienes, the high temperature proved to be a problem. We could not get the
51
desired product 2.40; only decomposed compounds were obtained at 50 oC and no
monomer of 2-boron substituted dienes was found at low temperatures (Scheme 2.7).
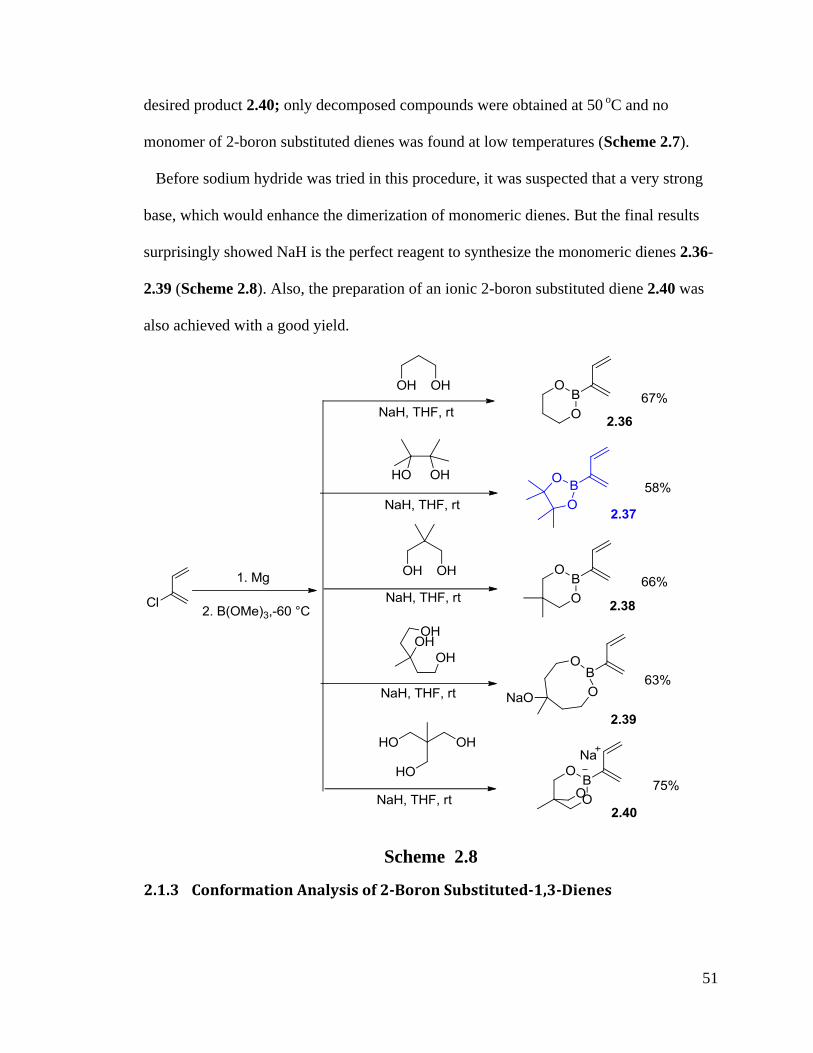
Before sodium hydride was tried in this procedure, it was suspected that a very strong
base, which would enhance the dimerization of monomeric dienes. But the final results
surprisingly showed NaH is the perfect reagent to synthesize the monomeric dienes 2.36-
2.39 (Scheme 2.8). Also, the preparation of an ionic 2-boron substituted diene 2.40 was
also achieved with a good yield.
Scheme 2.8
2.1.3 Conformation Analysis of 2-Boron Substituted-1,3-Dienes
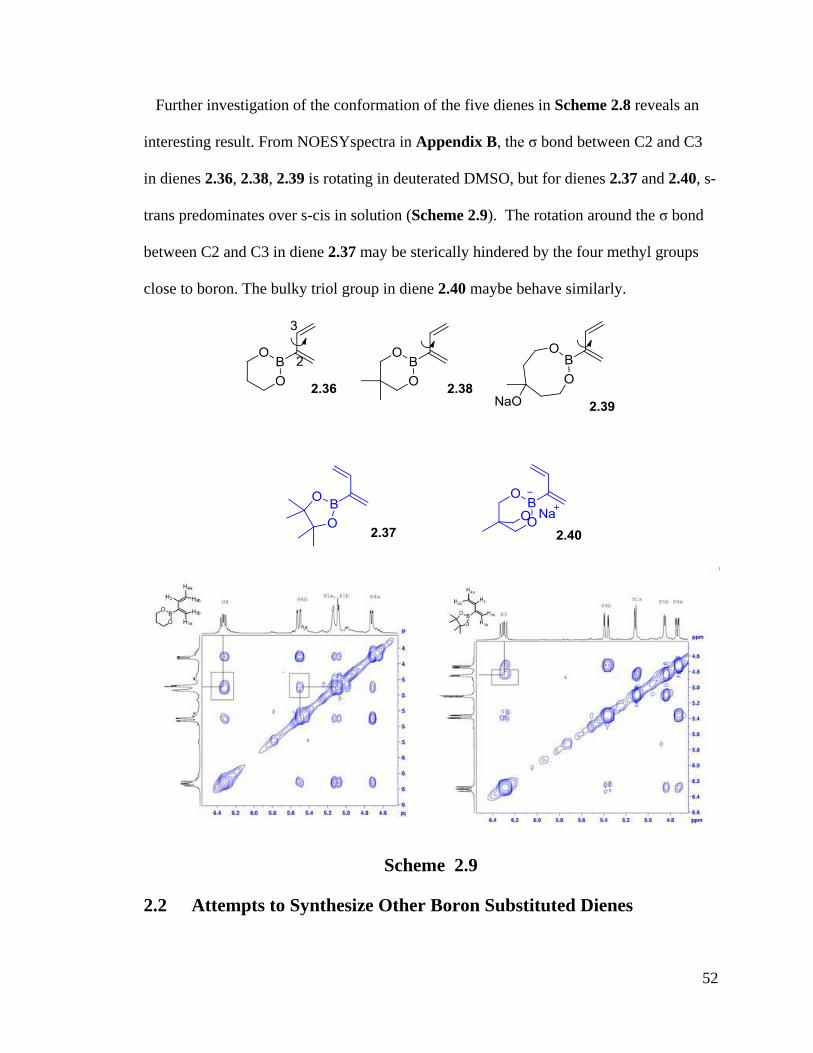
52
Further investigation of the conformation of the five dienes in Scheme 2.8 reveals an
interesting result. From NOESYspectra in Appendix B, the σ bond between C2 and C3
in dienes 2.36, 2.38, 2.39 is rotating in deuterated DMSO, but for dienes 2.37 and 2.40, s-
trans predominates over s-cis in solution (Scheme 2.9). The rotation around the σ bond
between C2 and C3 in diene 2.37 may be sterically hindered by the four methyl groups
close to boron. The bulky triol group in diene 2.40 maybe behave similarly.
Scheme 2.9
2.2 Attempts to Synthesize Other Boron Substituted Dienes

53
2.2.1 Attempts to Synthesize Sodium 1-(but-1,3-dien-2yl)-5-methyl-2,8,9-trioxa-1-borabicyclo[3,3,1]nonan-1-uide) The nucleophilicity of a boron atom is enhanced significantly by quaternization with
an anionic ligand.113
The cyclic triolborates are exceptionally stable in air and water.
Moreover, they are more soluble in organic solvents than potassium trifluoroborates. So,
we attempted to prepare tetracoordinated organoboron complexe 2.26 expecting the
tetra-coordinated boron atom to increase the electron density of diene by the following
sequence:
Scheme 2.10
However, we only obtained an intermediate product 2-(buta-1,3-dien-2-yl)-1,3,6,2-
dioxazaborocane (2.25) and failed in the last step in which we suspect the high steric
strain of the ring destabilizes the whole structure.
2.2.2 Attempts to Synthesize 2-(Buta-1,3-Dien-2-yl)-6-Methyl-1,3,6,2 -Dioxazaborocane-4.8-Dione
Scheme 2.11

54
In this reaction, esterification of the boronic acid failed at low temperature (rt) and high
temperature (refluxing ) caused the decomposition of diene. The reason is the same as
discussed above: the N-electron withdrawing group destabilized the structure. We also
tried the di-sodium salt of N-methyliminodiaacetic acid 2.43 to react with boronic acid in
refluxing CHCl3 (Scheme 2.12). However, we did not get 2.27 but rather decomposed
diene.
Scheme 2.12
2.2.3 Attempts To Synthesize 1-Phenyl-2-Pinacol Boronate-1, 3-Diene
Billingsley et al. used iodobenzene via Suzuki coupling to prepare a boronate
substituted benzene.114
They used high temperature (refluxing) and stable iodo
compounds to get product 2.28. We attempted to prepare a similar product, diene 2.29,
employing the same procedure but failed (Scheme 2.13). The reason might be the
decomposition of unstable iodo diene 2.42 at high temperature. We tried refluxing and
appeared to get polymerization. Finally, chloro diene was also tried but no positive
results were obtained.
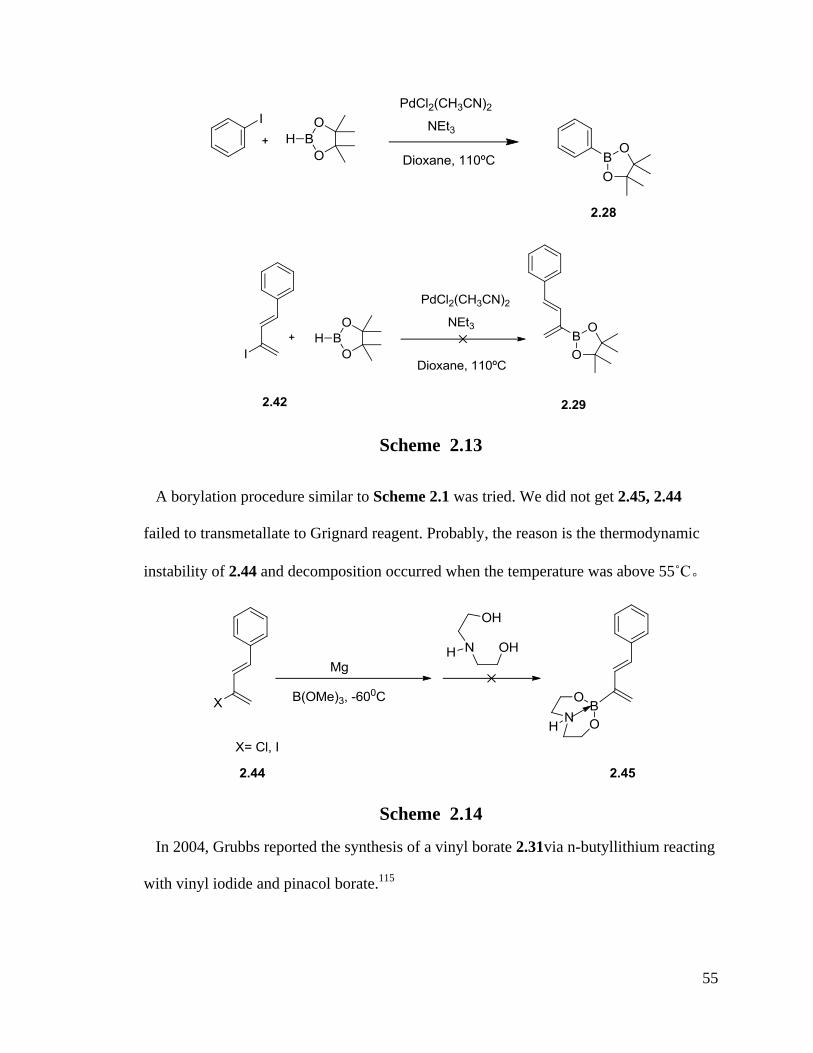
55
Scheme 2.13
A borylation procedure similar to Scheme 2.1 was tried. We did not get 2.45, 2.44
failed to transmetallate to Grignard reagent. Probably, the reason is the thermodynamic
instability of 2.44 and decomposition occurred when the temperature was above 55˚C。
Scheme 2.14
In 2004, Grubbs reported the synthesis of a vinyl borate 2.31via n-butyllithium reacting
with vinyl iodide and pinacol borate.115
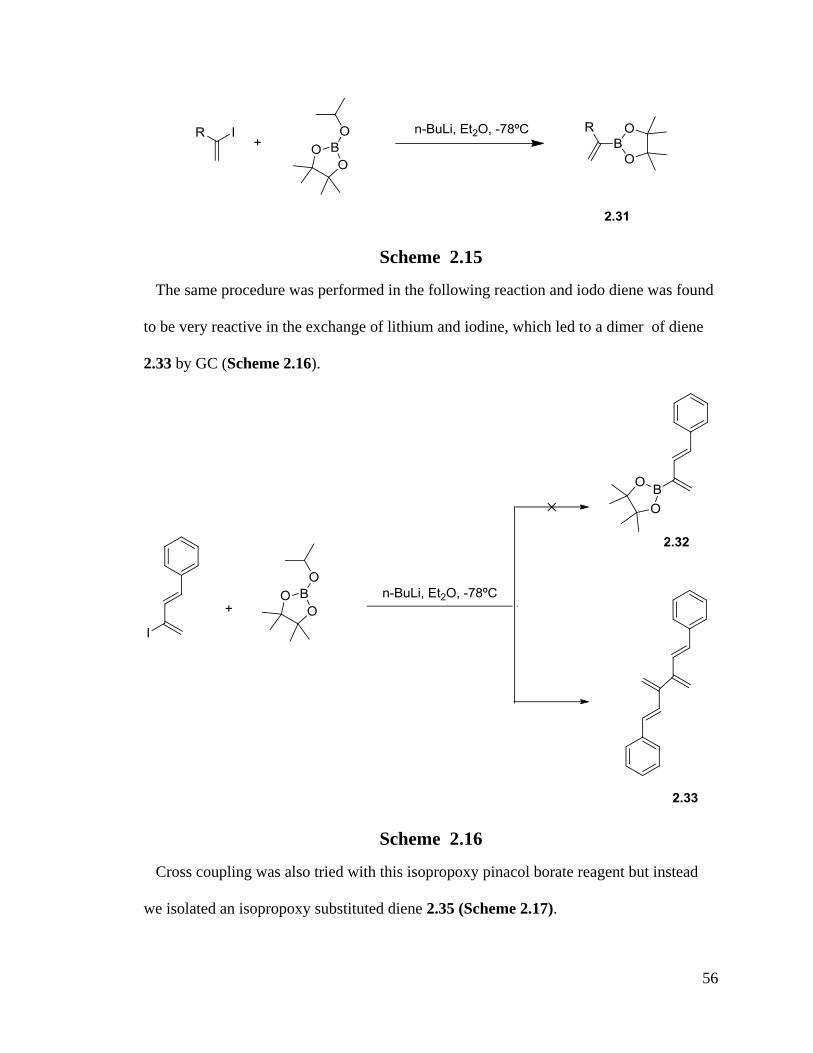
56
Scheme 2.15
The same procedure was performed in the following reaction and iodo diene was found
to be very reactive in the exchange of lithium and iodine, which led to a dimer of diene
2.33 by GC (Scheme 2.16).
Scheme 2.16
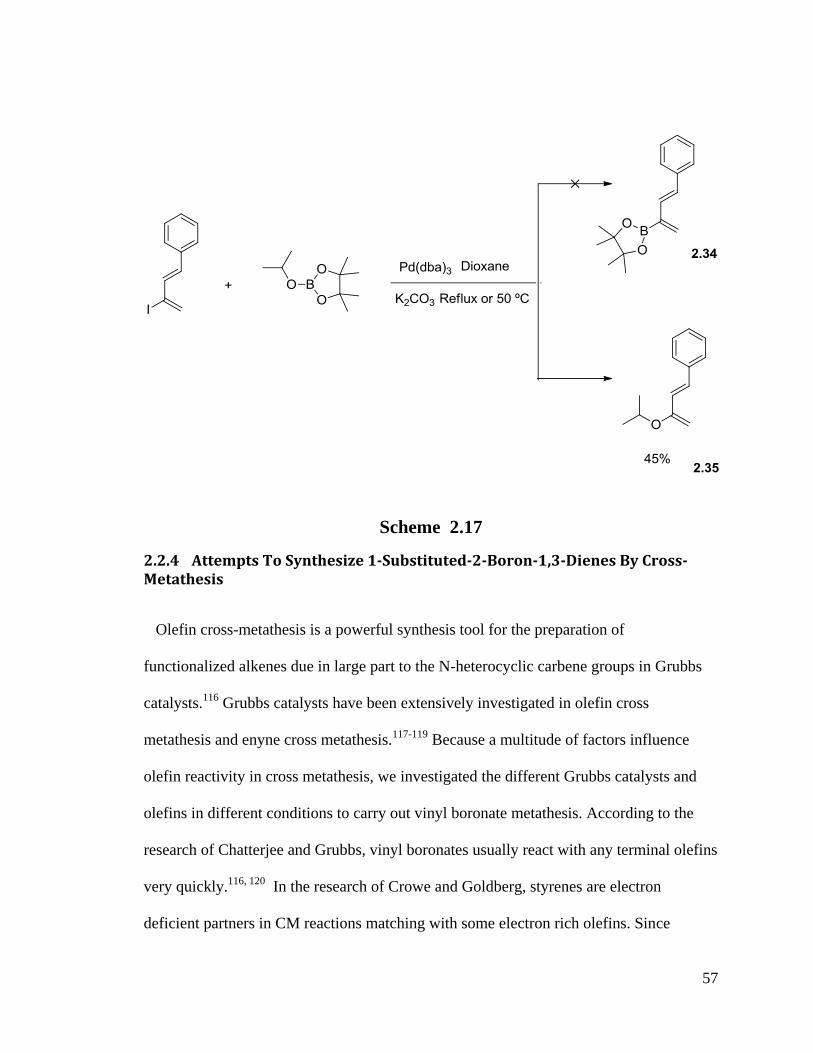
Cross coupling was also tried with this isopropoxy pinacol borate reagent but instead
we isolated an isopropoxy substituted diene 2.35 (Scheme 2.17).
57
Scheme 2.17
2.2.4 Attempts To Synthesize 1-Substituted-2-Boron-1,3-Dienes By Cross-Metathesis
Olefin cross-metathesis is a powerful synthesis tool for the preparation of
functionalized alkenes due in large part to the N-heterocyclic carbene groups in Grubbs
catalysts.116
Grubbs catalysts have been extensively investigated in olefin cross
metathesis and enyne cross metathesis.117-119
Because a multitude of factors influence
olefin reactivity in cross metathesis, we investigated the different Grubbs catalysts and
olefins in different conditions to carry out vinyl boronate metathesis. According to the
research of Chatterjee and Grubbs, vinyl boronates usually react with any terminal olefins
very quickly.116, 120
In the research of Crowe and Goldberg, styrenes are electron
deficient partners in CM reactions matching with some electron rich olefins. Since
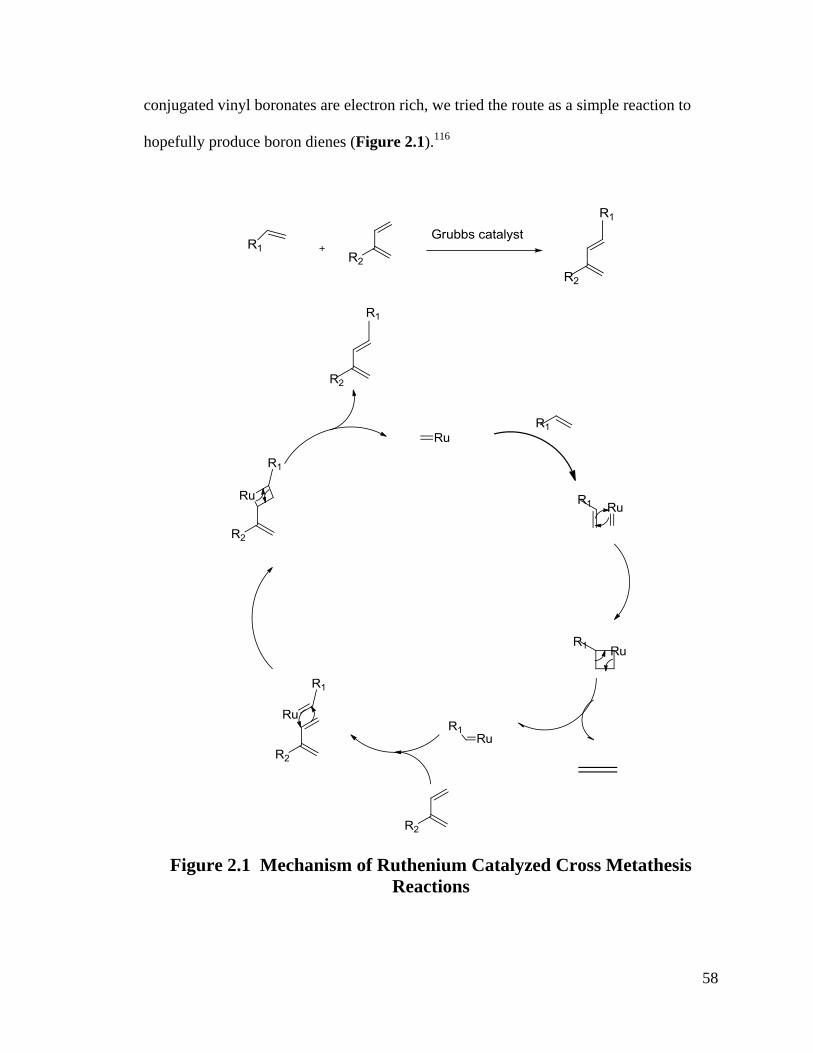
58
conjugated vinyl boronates are electron rich, we tried the route as a simple reaction to
hopefully produce boron dienes (Figure 2.1).116
Figure 2.1 Mechanism of Ruthenium Catalyzed Cross Metathesis
Reactions
59
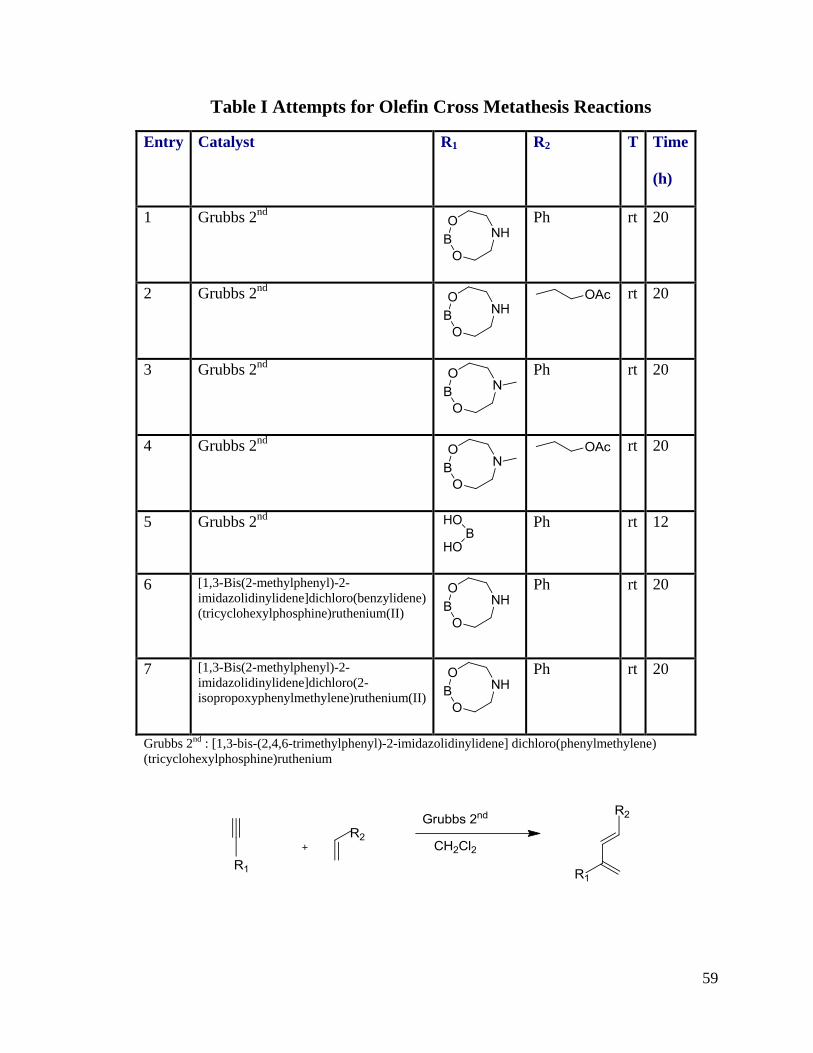
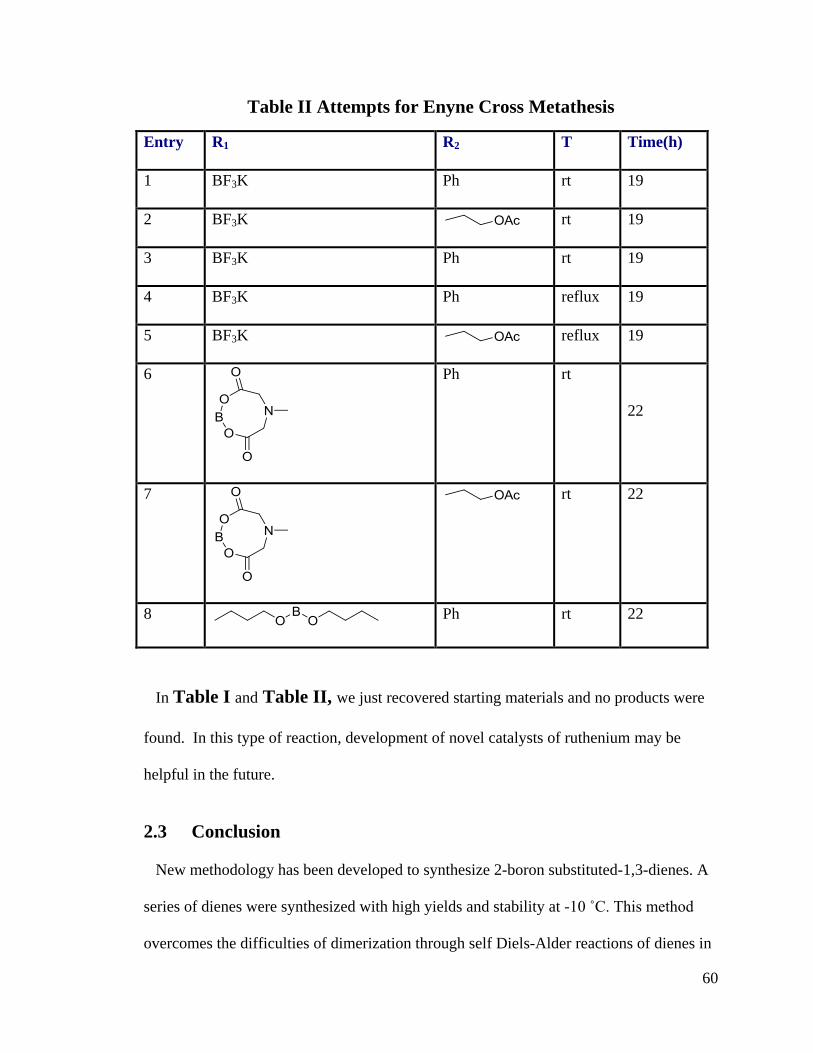
Table I Attempts for Olefin Cross Metathesis Reactions
Entry Catalyst R1 R2 T Time
(h)
1 Grubbs 2nd
Ph rt 20
2 Grubbs 2nd
rt 20
3 Grubbs 2nd
Ph rt 20
4 Grubbs 2nd
rt 20
5 Grubbs 2nd
Ph rt 12
6 [1,3-Bis(2-methylphenyl)-2-
imidazolidinylidene]dichloro(benzylidene)
(tricyclohexylphosphine)ruthenium(II)
Ph rt 20
7 [1,3-Bis(2-methylphenyl)-2-
imidazolidinylidene]dichloro(2-
isopropoxyphenylmethylene)ruthenium(II)
Ph rt 20
Grubbs 2nd : [1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene] dichloro(phenylmethylene)
(tricyclohexylphosphine)ruthenium
60
Table II Attempts for Enyne Cross Metathesis
Entry R1 R2 T Time(h)
1 BF3K Ph rt 19
2 BF3K rt 19
3 BF3K Ph rt 19
4 BF3K Ph reflux 19
5 BF3K reflux 19
6
Ph rt
22
7
rt 22
8
Ph rt 22
In Table I and Table II, we just recovered starting materials and no products were
found. In this type of reaction, development of novel catalysts of ruthenium may be
helpful in the future.
2.3 Conclusion
New methodology has been developed to synthesize 2-boron substituted-1,3-dienes. A
series of dienes were synthesized with high yields and stability at -10 ˚C. This method
overcomes the difficulties of dimerization through self Diels-Alder reactions of dienes in

61
previous methods. Synthesis of 1-substituted-3-boron-1,3-dienes through cross
metathesis requires new catalysts and methodology to realize the goal.
2.4 Experimental Procedures and Characterization Data
General: The 1H NMR were recorded on a Brucker Avance 500MHz spectrometer and
Brucker Avance 300MHz spectrometer operating at 500.13MHz and 300.13MHz
respectively. 13
C NMR were recorded on a Bruker Avance 300MHz spectrometer and
Bruker Avance 500MHz spectrometer operating at 75.48MHz and 125.77MHz
respectively. Chemical shifts were reported in parts per million () relative to
tetramethylsilane (TMS), or the residual proton resonances in the deuterated solvents:
chloroform (CDCl3) and dimethyl sulfoxide (DMSO). Coupling constants (J values) were
reported in hertz (Hz).
All elemental analyses were performed by Atlantic Microlabs Inc., GA. High
resolution mass spectrometric (HRMS) analyses were performed at Caudill Laboratories
of University of North Carolina at Chapel Hill and Mass Spectrometry Lab, School of
Chemical Sciences, University of Illinois at Urbana-Champaign.
All reactions were carried out under an inert atmosphere unless otherwise noted. Flash
chromatography was performed using thick walled glass chromatography columns and
ultrapure silica gel. Ether and pentane were distilled over Na. Absolute ethanol and
methanol were used without further purification. THF was purchased from Fischer
Scientific in the form of solvent kegs and purified using the centrally located solvent
dispensing system developed by J.C. Meyer. Deuterated solvents were purchased from
Cambridge Isotope Laboratories and dried over molecular sieves. Magnesium sulfate,
magnesium small turnings, iodobenzene, 4-iodobenzotrifluoride, 2-iodoanisole, 4-

62
iodoanisole, ethyl acrylate, N-phenylmaleimide and N-phenyl methyl maleimide were
purchased from Aldrich Chemical Company and used as received. 2-Chloro-1,3-diene, 50%
in xylene (Chloroprene) was purchased from Pfaltz & Bauer, INC and used as received.
General procedure 1 for preparing 2.4 and 2.6
A mixture of magnesium (1.0 g, 41.1 mmol), 1, 2-dibromoethane (0.5 mL,5.3 mmol), and
THF (10 mL) was refluxed under nitrogen for 15 min to activate the magnesium. To the
mixture anhydrous zinc chloride (0.6g, 4.1mmol) in THF (60 mL) was added and reflux
was continued for another 15 min. 2-Chloro-1,3-butadiene (4.9 mL, 25 mmol) (density
0.915 g/mL, 50 % in xylene) and 1,2-dibromoethane (0.95 g, 5 mmol) in THF (30 mL)
were added dropwise over a period of 30 min. This addition was controlled so as to bring
the mixture into a gentle reflux. The color of the contents changed gradually from grayish
white to greenish black. The mixture was heated to reflux for an additional 30 min after
completion of the addition. The Grignard reagent thus obtained was immediately added
dropwise to a solution of trimethoxyborane (4.25 mL, 38.5 mmol) in THF (25 mL) using
a double-ended needle. The addition was controlled in such a way that the internal
temperature of the mixture was maintained below –60 °C all the time. After completion
of the addition, the solution was allowed to warm to room temperature quickly. The
cloudy grey colored reaction mixture was stirred for 1 h . To the resulting mixture at
room temperature, 0.5 M HCl solution (100 mL) was added. The reaction mixture was
extracted with Et2O (2 × 75 mL). The combined colorless clear organic layers were dried
over MgSO4, and the volatiles were removed by rotovap (30 °C, 20 Torr) to yield diene
boronic acid.

63
Preparation of 2.4
General procedure 1 was followed to get a diene boronic acid, then the boronic acid was
added at once to a solution of diethanolamine (0.8 equiv, 22.5 mmol, 8.411g) dissolved in
THF (100 mL). Sodium sulfate (8 g) was added and refluxed for 6 hours. At the end of
the reaction, the flask was cooled to room temperature. Solid Na2SO4 was separated from
the solution by filtration. The solution 250 mL was reduced by 200 mL using a rotary
evaporator.
A cold bath was used to induce crystallization. After 4 h, the solid was filtered and
washed with cold chloroform. The product 2 was obtained as white needles following
drying under vacuum (2.40 g, 14.4 mmol, 62.4%).
1H NMR (300 MHz, CDCl3) 6.51 (dd, J =17.9, 10.9 Hz, 1H-H3), 5.46–5.40 (m, 3H),
4.98 (dd, J=17.9, 1.9 Hz, 1H-H4), 5.18 (s, 1H-H7), 4.05 (m, 2H-H5,9), 3.89 (m, 2H-
H5,9), 3.31 (m, 2H-H6,8), 2.76 (m, 2H- H6,8). 13
C NMR (300, MHz, CDCl3) 143.6-C3,
124.3-C4, 114.6- C1, 63.4-C5,9, 52.1-C6,8, the signal of carbon (C2) next to a tetravalent
boron is generally not observed due to quadrupolar broadening. 121
Elemental anal. calcd
for C8H14BNO2: C, 57.53; H, 8.45. Found: 57.06, 8.44.
Preparation of 1,3-Butadiene-2-diethanolamine borate 2.6

64
General procedure 1 was followed to get the boronic acid, then the boronic acid was
added at once to a solution of N-methyl diethanolamine (0.8 equiv, 22.5mmol, 4.545g)
dissolved in THF (100 mL). Sodium sulfate (8 g) was added and refluxed for 6 hours. At
the end of the reaction, the flask was cooled to room temperature. Solid Na2SO4 was
separated from the solution by filtration. The solution 200 mL was reduced by 150 mL
using a rotary evaporator. Pentane (10 mL) was added to precipitate the product and
product 2.6 was obtained as a white solid following filtration and washing with cold
pentane under vacuum. (2.7 g, 15 mmol, 60%).
1H NMR (300 MHz, CDCl3) 6.58 (dd, J =17.6, 10.7Hz, 1H), 5.58 (m, 1H), 5.52-5.55
(m, 2H), 4.95 (dd, J=10.9, 2.6 Hz, 1H), 4.09 (m, 2H), 3.99 (m, 2H), 3.09 (m, 2H), 2.96
(m, 2H), 2.62 (s, 3H) 13
C NMR (300, MHz, CDCl3) 143.6, 124.3, 114.6, 63.4, 52.1
13C NMR (300, MHz, DMSO) 143.5, 124.0, 113.6, 61.5, 59.8, 45.7
HRMS calcd for C9H16BNO2: 181.1274+Na=204.1172, found M+Na=204.1172
General procedure 2 for preparing 2.36-2.40
A mixture of mixture of magnesium (1.0 g, 41.1 mmol), 1, 2-dibromoethane (0.5 mL,
5.3 mmol), and THF (10 mL) was refluxed under nitrogen for 15 min to activate the
magnesium. To the mixture, anhydrous zinc chloride (0.6 g, 4.1mmol) in THF (60 mL)
was added and reflux was continued for another 15 min. 2-chloro-1,3-butadiene (4.9 mL,
25 mmol) (density 0.915 g/mL, 50 % in xylene) and 1,2-dibromoethane (0.95 g, 5 mmol)
in THF (30 mL) were added dropwise over a period of 30 min. This addition was

65
controlled so as to bring the mixture into a gentle reflux. The color of the contents
changed gradually from grayish white to greenish black. The mixture was heated to
reflux for an additional 30 min after completion of the addition. The Grignard reagent
thus obtained was immediately added dropwise to a solution of trimethoxyborane (4.25
mL, 38.5 mmol) in THF (25 mL) using a double-ended needle. The addition was
controlled in such a way that the internal temperature of the mixture was maintained
below –60 °C all the time. After completion of the addition, the solution was allowed to
warm to room temperature quickly. The cloudy grey colored reaction mixture was stirred
for 1 h . To the resulting mixture at room temperature, 0.5 M HCl solution (100 mL) was
added slowly. The reaction mixture was extracted with Et2O (2×75 mL). The combined
colorless clear organic layers were dried over MgSO4, and the volatiles were removed by
rotovap (30 °C, 20 mm) to yield diene boronic acid.
Preparation of 1,3-Butadiene-(2-propane-1,3-diol)- borate 2.36
General procedure 2 was followed to get diene boronic acid. The dried diene boronic
acid was added at once to a solution of propane-1,3-diol (0.7eq, 17.5 mmol, 1.33g)
dissolved in THF (100 mL). Sodium hydride (1.5 eq, 37.5 mmol, 0.86g) was added
slowly to the mixture at room temperature. The solvent was removed by rotovap after 2
hours. Then dry THF (3×100 mL) was added to dissolve and wash the solid. The solid
was removed by filtration and all the solution was removed by rotovap to get the white
solid product 2.36 (1.62g, 11.73mmol, 67%).

66
1H NMR (300 MHz, DMSO) 6.32 (dd, J =17.3, 10.5 Hz, 1H), 5.50 (dd, J=17.3,3.8 Hz,
1H), 5.06 (dd, J=17.3, 5.8Hz, 2H), 4.70 (dd, J=10.5, 3.8Hz, 1H), 3.62 (ddd, J=11.1, 4.3,
2.2Hz, 2H), 3.50 (td, J= 2.6, 11.0, 11.0Hz, 2H), 1.61 (m, 1H), 1.04 (dt, J=2.6, 2.6, 12.2Hz,
1H) 13
C NMR (300, MHz, DMSO) 145.5, 113.1, 112.4, 60.4, 30. HRMS: calcd for
C7H11BO2: 138.0852, found 138.08522.
Preparation of 1,3-Butadiene-(2-pinacol)- borate 2.37
General procedure 2 was followed to get diene boronic acid. The dried diene boronic acid
was added at once to a solution of pinacol (0.7eq, 17.5 mmol, 2.07g) dissolved in THF
(100 mL). Sodium hydride (1.5 eq, 37.5 mmol, 0.86 g) was added slowly to the mixture
at room temperature. The solvent was removed by rotovap after 2 hours. Then dry THF
(2×100 mL) followed by dry diethyl ether (2×100 mL) was added to dissolve and wash
the solid. The solid was removed by filtration and all the solution was removed by
rotovap to get the white solid product 2.37(1.83g, 10.15mmol, 58%).
1H NMR (300 MHz, DMSO) 6.28 (dd, J =17.5, 10.6 Hz, 1H), 5.40 (dd, J=17.5,
3.4Hz,1H), 5.08(d,J= 6.3Hz, 1H), 4.81 (d, J= 6.3, Hz, 1H), 4.71 (dd, J=10.6, 3.4Hz, 1H),
1.00 (s, 6H), 0.86 (s, 6H) 13
C NMR (300, MHz, DMSO) 145.7, 116.5, 112.9, 76.5, 26.7,
25.3 HRMS: calcd for C10H17BO2: 180.1322, found: 180.13217
Preparation of 1,3-Butadiene-(2-(2’,2’-dimethyl-propane-1,3-diol))- borate 2.38

67
General procedure 2 was followed to get diene boronic acid. The dried diene boronic
acid was added at once to a solution of 2,2-dimethylpropane-1,3-diol (0.7eq, 17.5 mmol,
1.82g) dissolved in THF (100 mL). Sodium hydride (1.5 eq, 37.5 mmol, 0.86 g) was
added slowly to the mixture at room temperature. The solvent was removed by rotovap
after 2 hours. Then dry THF (2×100 mL) followed by dry diethyl ether (2×100 mL) was
added to dissolve and wash the solid. The solid was removed by filtration and all the
solution was removed by rotovap to get the white solid product 2.38 (1.92 g, 11.55 mmol,
66%).
1H NMR (300 MHz, DMSO) 6.30 (dd, J =17.5, 10.6 Hz, 1H), 5.40 (dd, J=17.5,
3.7Hz,1H), 5.02(m,2H), 4.66 (dd, 10.6, 3.8Hz, 1H), 3.13(s, 4H), 0.91 (s, 3H), 0.54(s, 3H)
13C NMR (300, MHz, DMSO) 145.6, 119.0, 112.3, 32.1, 24.0, 22.7 HRMS: calcd for
C9H15BO2 : 166.1165, found: 166.11652
Preparation of sodium 2-(buta-1,3-dien-2-yl)-6-methyl-1,3,2-dioxaborocan-6-olate 2.39
General procedure 2 was followed to get diene boronic acid. The dried diene boronic
acid was added at once to a solution of 3-methylpentane-1,3,5-triol (0.7 eq, 2.34 g,17.5
mmol,) dissolved in THF (100 mL). Sodium hydride (1.5 eq, 37.5 mmol, 0.86 g) was

68
added slowly to the mixture at room temperature. The solvent was removed by rotovap
after 2 hours. Then dry acetone (2×100 mL) was added to dissolve and wash the solid.
The solid was removed by filtration and all the solution was removed by rotovap to get
the white solid product 2.39 (2.15 g, 11.03 mmol, 63%).
1H NMR (300 MHz, DMSO) 6.21 (dd, J =17.6, 10.7 Hz, 1H), 5.45 (dd, J=17.6,
3.9Hz,1H), 4.97(d,J= 6.4Hz, 1H), 4.82 (d, J= 6.4, Hz, 1H), 4.64 (dd, 10.7, 3.9Hz, 1H),
3.70 (m, 2H), 3.39 (m, 2H), 1.38(t, J=5.7, 4H), 0.97 (s, 3H) 13
C NMR (300, MHz, DMSO)
145.2, 116.5, 112.3, 64.3, 56.3, 41.4, 34.2 HRMS: calcd for C10H16BNaO3: 218.1090-
Na=195.1192, found: 195.1192
Preparation of sodium 1-(buta-1,3-dien-2-yl)-4-methyl-2,6,7-trioxa-1-borabicyclo(2,2,2)
octan-1-uide 2.40
General procedure 2 was followed to get diene boronic acid. The dried diene boronic
acid was added at once to a solution of 2-(hydroxymethyl)-2-methylpropane-1,3diol (0.7
eq, 2.10 g,17.5 mmol,) dissolved in THF (100 mL). Sodium hydride (1.5 eq, 37.5 mmol,
0.86 g) was added slowly to the mixture at room temperature. The solvent was removed
by rotovap after 2 hours. Dry acetone (3×100 mL) was added to dissolve and wash the
solid. The solid was removed by filtration and all the solution was removed by rotovap to
get the white solid product 2.40 (2.68 g, 13.13 mmol, 75%).
1H NMR (300 MHz, DMSO) 6.18 (dd, J =17.1, 10.5 Hz, 1H), 5.40 (dd, J=17.1,
3.8Hz,1H), 4.90(dd,J=15.2, 6.0Hz, 1H), 4.65 (dd, J= 10.5, 3.8Hz, 1H), 3.49 (s, 6H), 0.43

69
(s, 3H) 13
C NMR (300, MHz, DMSO) 144.5, 118.0, 113.0, 73.1, 34.4, 16.3 HRMS:
calcd for C9H14BNaO3: 204.0934+H=205.1012, found: M+H=205.1012

70
CHAPTER 3 Diels-Alder Reactions of 2-Boron Substituted-
1,3-Dienes
3.1 Results and Discussion
3.1.1 Diels-Alder Reactions
All the 2-boron substituted dienes were tried in Diels-Alder reactions to test their
reactivity with dienophiles. (Table III) Among these dienes, the diethanolamine borate
diene (2.4, entry 1) has proven to be significantly more reactive and more regioselective
in Diels-Alder reactions compared to its BF3 diene counterpart (Scheme 3.1).122, 123
Qualitatively, we initially noticed that whereas the BF3 diene required 16 h of heating at
95-100oC in a sealed tube in toluene with N-phenylmaleimide to obtain >90% yield of
cycloadduct, the diethanolamine borate diene (2.4) reacted with this same dienophile to
afford a 98% isolated yield of cyclaoadduct 3.1 in Table III after only 15 minutes at
25oC!
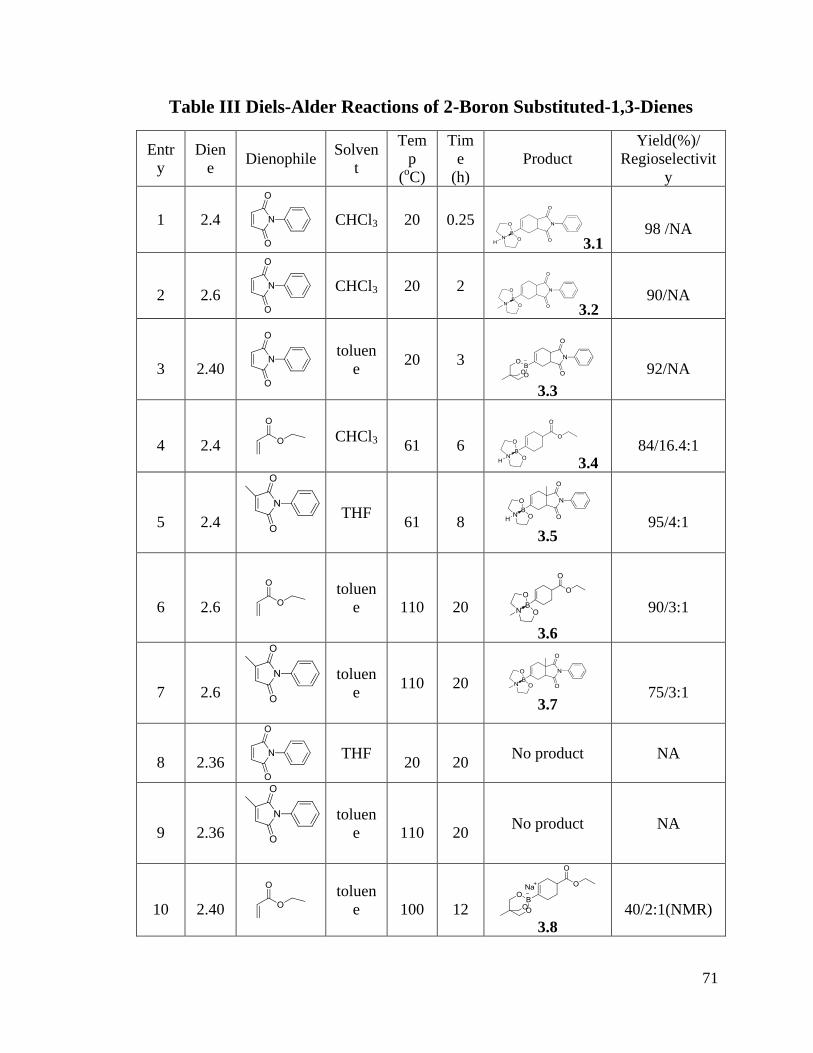
71
Table III Diels-Alder Reactions of 2-Boron Substituted-1,3-Dienes
Entr
y
Dien
e Dienophile
Solven
t
Tem
p
(oC)
Tim
e
(h)
Product
Yield(%)/
Regioselectivit
y
1 2.4
CHCl3 20
0.25
3.1
98 /NA
2
2.6
CHCl3 20 2
3.2
90/NA
3
2.40
toluen
e 20 3
3.3
92/NA
4
2.4 CHCl3
61
6
3.4
84/16.4:1
5
2.4
THF
61
8 3.5
95/4:1
6
2.6
toluen
e
110
20
3.6
90/3:1
7
2.6
toluen
e 110 20
3.7
75/3:1
8
2.36
THF
20
20 No product NA
9
2.36
toluen
e
110
20 No product NA
10
2.40
toluen
e
100
12 3.8
40/2:1(NMR)
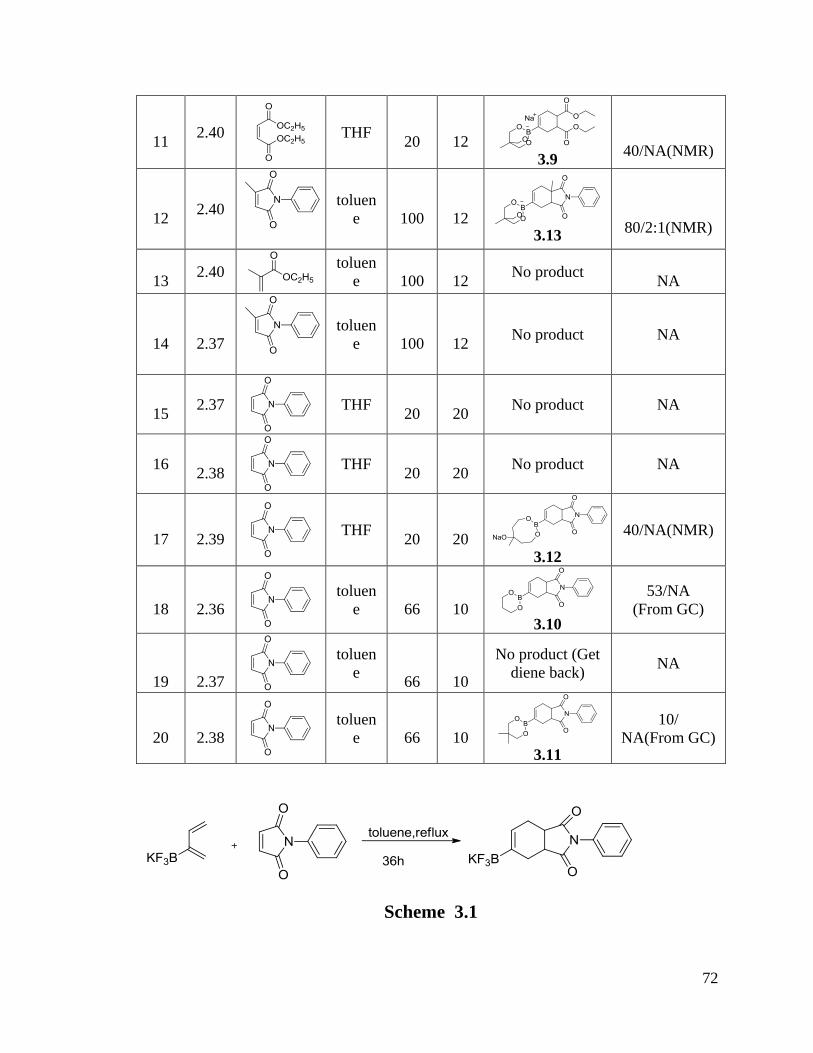
72
11 2.40
THF
20
12 3.9
40/NA(NMR)
12 2.40
toluen
e
100
12 3.13
80/2:1(NMR)
13 2.40
toluen
e
100
12 No product
NA
14
2.37
toluen
e
100
12 No product NA
15 2.37
THF
20
20 No product NA
16
2.38
THF
20
20 No product NA
17
2.39
THF
20
20 3.12
40/NA(NMR)
18
2.36
toluen
e
66
10 3.10
53/NA
(From GC)
19
2.37
toluen
e
66
10
No product (Get
diene back) NA
20
2.38
toluen
e
66
10 3.11
10/
NA(From GC)
Scheme 3.1

73
We tried to get more quantitative rate constant data about this Diels-Alder reaction via
NMR spectroscopy but when we tried to perform this reaction under pseudo first order
conditions at -10oC, we could only confirm that t1/2 is less than 4 minutes. Attempts to
get more accurate kinetic data using NMR at -40oC resulted instead in diene precipitation.
So, the diene (2.4) is by far the most reactive main group element substituted diene we
have made in the boron or silicon substituted series to date. What is perhaps even more
surprising to us is that in our group, Marcus Wright prepared a cobaloxime substituted
diene with a very short half life (t1/2=9.2 min) in Diels-Alder reactions at room
temperature. This diethanolamineborate diene is even more reactive than it124
and those
cobaloxime dienes consistently favored the s-cis conformation in the solid state (Scheme
3.2).
Scheme 3.2
Diene (2.4) is in the s-trans conformation in the solid state (Figure 3.1) but in this case
we suspect that the preference for the s-trans conformer is due to intermolecular
hydrogen bonding between the N-H and one of the adjacent molecule’s borate oxygens.
This hydrogen bonding would make a C(2)-C(3) dihedral angle of 50-60o on the order of
those we observed in cobaloxime diene solid state structures unfavorable. At 25oC in
CDCl3, we saw no evidence for the s-cis conformer by NOESY (Figure 3.2).
The boron substituted diene 2.4 thus obtained has C1 (5.23 vs 5.04, 4.96 (d6-DMSO)
and C3 (6.31 vs. 6.19) H’s which are significantly more deshielded than the BF3
substituted diene. In the solid state (Figure 3.1), C(1)-C(2) and C(2)-C(3) bond lengths

74
were virtually identical in both dienes whereas B-C(2) (1.609(5)Å vs 1.576(13)Å) and
C(3)-C(4) (1.308(6)Å vs 1.279(13)Å) were significantly longer in the diethanolamine
borate diene 2.4.
In an effort to further understand this reactivity difference, geometry-optimization with
DFT using the B3LYP functional and a 6-31G(d) basis set followed by population
analysis was performed using Gaussian 03 on 2-diethanolaminoborate-1,3-butadiene (2.4)
and its BF3 diene counterpart.
Figure 3.1 Crystal Structures of BF3 Substituted Diene and
Diethanolamineborate-1,3-Butadiene 2.4
2-Diethanolaminoborate-1,3-butadiene (2.4) has a HOMO energy of -6.00 eV, whereas
its BF3 diene counterpart has a HOMO energy of -12.58 eV. These energies are
consistent with our observations that 2-diethanolaminoborate-1,3-butadiene (2.4) is more
reactive than its BF3 diene counterpart. Furthermore, a Mulliken population analysis
indicates a build-up of electron density on carbons C1 and C4 of 0.15e and 0.14e,
respectively, in 2-diethanolaminoborate-1,3-butadiene compared to its BF3 diene
counterpart, which is also consistent with our observations.
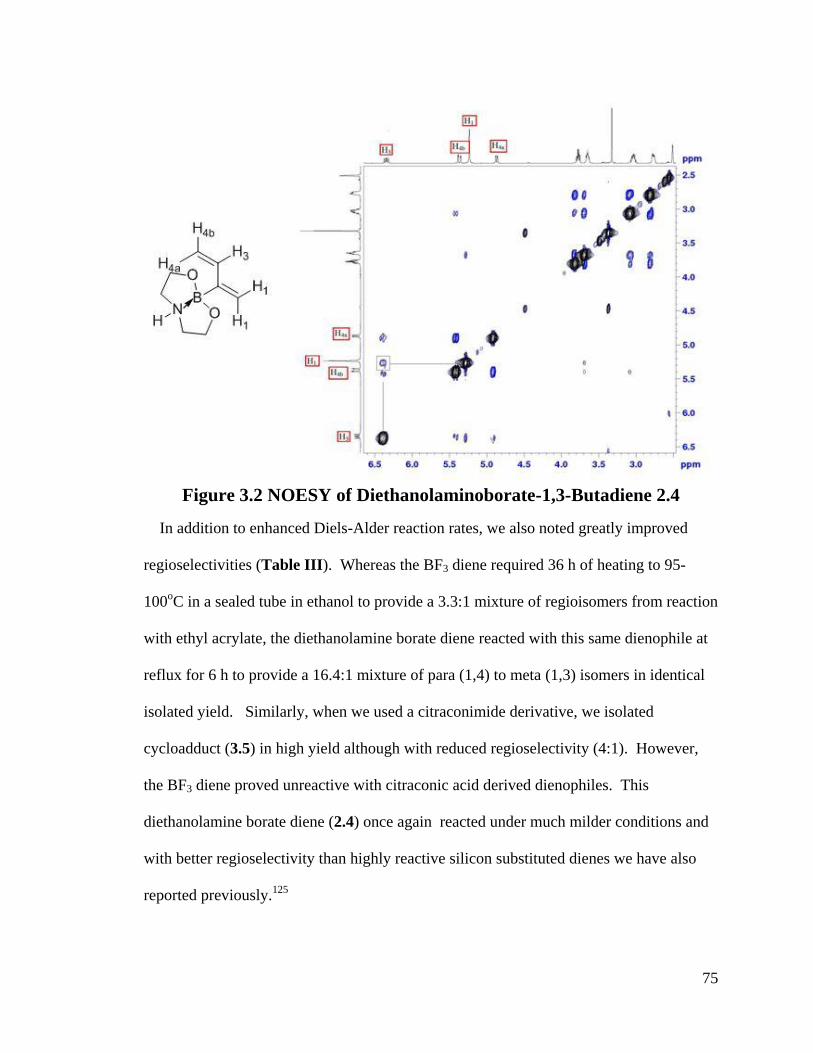
75
Figure 3.2 NOESY of Diethanolaminoborate-1,3-Butadiene 2.4
In addition to enhanced Diels-Alder reaction rates, we also noted greatly improved
regioselectivities (Table III). Whereas the BF3 diene required 36 h of heating to 95-
100oC in a sealed tube in ethanol to provide a 3.3:1 mixture of regioisomers from reaction
with ethyl acrylate, the diethanolamine borate diene reacted with this same dienophile at
reflux for 6 h to provide a 16.4:1 mixture of para (1,4) to meta (1,3) isomers in identical
isolated yield. Similarly, when we used a citraconimide derivative, we isolated
cycloadduct (3.5) in high yield although with reduced regioselectivity (4:1). However,
the BF3 diene proved unreactive with citraconic acid derived dienophiles. This
diethanolamine borate diene (2.4) once again reacted under much milder conditions and
with better regioselectivity than highly reactive silicon substituted dienes we have also
reported previously.125

76
Other D-A reactions was tried with dienes 2.6-2.40. Diene 2.6 has a similar structure to
2.4 and it was found to be a reactive diene in D-A reactions with N-phenylmaleimide too
(Table III, Entry 2). After 2 hours, D-A product was formed with a high yield of 90% in
CHCl3. For diene 2.6, CH2Cl2 was not a good solvent since it has a low boiling point in
the D-A reactions with di- and tri- substituted dienophiles, ethyl acrylate and 2-methyl N-
phenylmaleimide (Entry 6, 7). Diene 2.6 was not reactive in CH2Cl2 after refluxing for
16 hours but was reactive in CHCl3 after 20 hours with a yield of 80%. Toluene is the
best solvent in the reaction (90%) compared with CHCl3 because it has a high boiling
point and a good solubility for all dienes. In this reaction, regioselectivity (3:1) was found
to be lower than diene 2.4.
Diene 2.40 is also a reactive diene in a D-A reaction with N-phenylmaleimide. We can
get a complete cycloadduct with a yield of 92% after 3 hours (entry 3) at room
temperature. When diene 2.40 reacted with ethyl acrylate and diethyl fumarate, only a 40%
yield of product was found from NMR and GC without separation. Cycloproduct was
found with 80% yield when it reacted with sterically hindered dienophiles 2-methyl-N-
phenylmaleimide and ethyl methacrylate.
Diene 2.36 (entry 8, 18) can react with N-phenylmaleimide to give a 53% yield at high
temperature (66oC) and diene 2.38 gives a yield of 10% under the same conditions (entry
20). But for less reactive dienophiles, no D-A products were found (entry 9,14). Dienes
2.37 and 2.39 were found to have no reactivity in D-A reactions (entry 14, 15, 17, 19 ).
According to the results above we can conclude that tetra-coordinate boron substituted
dienes are more reactive than tri-coordinate boron substituted dienes in D-A reactions.
When tetra-coordinate boron substituted dienes react with N-phenylmaleimide, fast

77
reaction rates and high yields were observed. Tri-coordinate boron substituted dienes are
less reactive mainly because tetra-coordinate dienes all have an electron donating group
or atom enriching the electron density of boron. It eventually enriches the electron
density of the whole diene system to promote the HOMO energy and accelerates the
reaction rate of D-A reactions.
3.1.2 Dimerization of Boron Substituted Dienes
When D-A reactions were conducted at high temperatures, we noticed that no dimers of
boron substituted dienes were observed in NMR. This interesting result is different from
previously reported work as discussed in the Introduction.64, 95,103
From Table IV, only a
trace of dimers were observed from GC. The results showed that after the formation of
monomer of dienes, they are very stable at high temperature. Compared with the
formation of dimers, tri-coordinated boron substituted dienes can be formed at low
temperature. The first postulation of dimerization is that monomers may be kinetic
products and dimers may be thermodynamic products. For diene 2.39 and 2.38, higher
temperatures were required to get TS2 than 2.36 and 2.37 from the experiments (Figure
3.3).
However, some monomers showed no dimerization at high temperature (Table 2.2).
Temperature is not the only factor in this reaction. Further research showed that Lewis
acid could also be responsible for the self D-A reaction (Scheme 2.3, 2.4, 2.5). All
dimerizations occur in the presence of CrCl2 , Mg2+
or H+ that could act as catalysts for
self D-A reactions. When these 2 factors exist in the reaction system, the monomer will
have a high propensity to become a dimer.
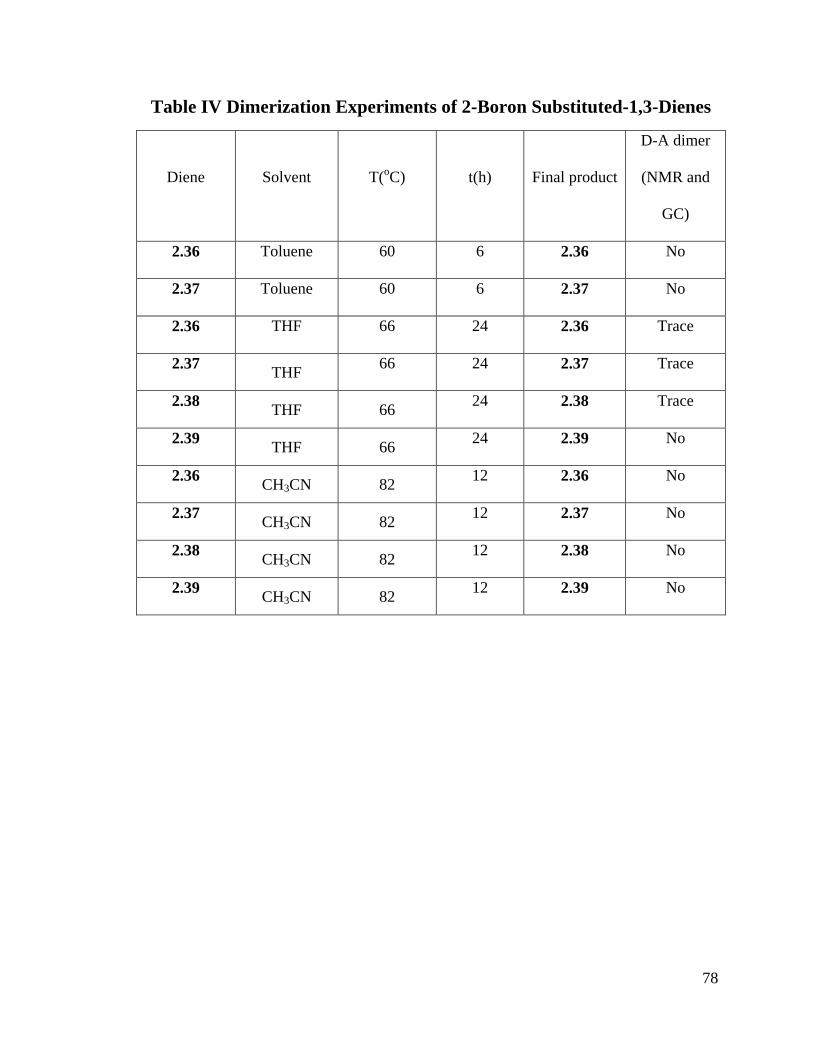
78
Table IV Dimerization Experiments of 2-Boron Substituted-1,3-Dienes
Diene Solvent T(oC) t(h) Final product
D-A dimer
(NMR and
GC)
2.36 Toluene 60 6 2.36 No
2.37 Toluene 60 6 2.37 No
2.36 THF 66 24 2.36 Trace
2.37 THF
66 24 2.37 Trace
2.38 THF 66
24 2.38 Trace
2.39 THF 66
24 2.39 No
2.36 CH3CN 82
12 2.36 No
2.37 CH3CN 82
12 2.37 No
2.38 CH3CN 82
12 2.38 No
2.39 CH3CN 82
12 2.39 No
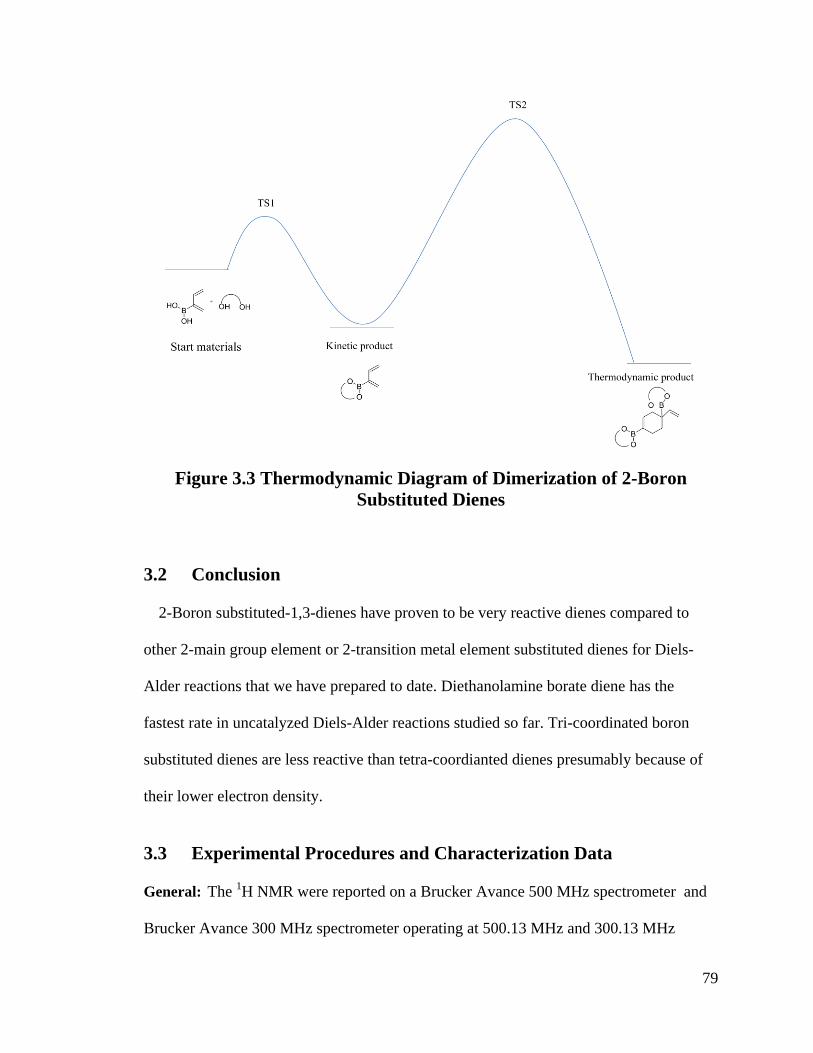
79
Figure 3.3 Thermodynamic Diagram of Dimerization of 2-Boron
Substituted Dienes
3.2 Conclusion
2-Boron substituted-1,3-dienes have proven to be very reactive dienes compared to
other 2-main group element or 2-transition metal element substituted dienes for Diels-
Alder reactions that we have prepared to date. Diethanolamine borate diene has the
fastest rate in uncatalyzed Diels-Alder reactions studied so far. Tri-coordinated boron
substituted dienes are less reactive than tetra-coordianted dienes presumably because of
their lower electron density.
3.3 Experimental Procedures and Characterization Data
General: The 1H NMR were reported on a Brucker Avance 500 MHz spectrometer and
Brucker Avance 300 MHz spectrometer operating at 500.13 MHz and 300.13 MHz

80
respectively. 13
C NMR were recorded on a Bruker Avance 300 MHz spectrometer and
Bruker Avance 500 MHz spectrometer operating at 75.48 MHz and 125.77 MHz
respectively. Chemical shifts were reported in parts per million () relative to
tetramethylsilane (TMS), or the residual proton resonances in the deuterated solvents:
chloroform (CDCl3). Coupling constants (J values) were reported in hertz (Hz).
All elemental analyses were performed by Atlantic Microlabs Inc., GA. High
resolution mass spectrometric (HRMS) analyses were performed at Caudill Laboratories
of University of North Carolina at Chapel Hill and Mass Spectrometry Lab, School of
Chemical Sciences, University of Illinois at Urbana-Champaign.
All reactions were carried out under an inert atmosphere unless otherwise noted. Flash
chromatography was performed using thick walled glass chromatography columns and
ultrapure silica gel. Ether and pentane were distilled over Na. Absolute ethanol and
methanol were used without further purification. THF was purchased from Fischer
Scientific in the form of solvent kegs and purified using a solvent dispensing system
developed by J.C. Meyer. Deuterated solvents were purchased from Cambridge Isotope
Laboratories and dried over molecular sieves. Magnesium sulfate, magnesium small
turnings, iodobenzene, 4-iodobenzotrifluoride, 2-iodoanisole, 4-iodoanisole, ethyl
acrylate, N-phenylmaleimide and N-phenyl methyl maleimide were purchased from
Aldrich Chemical Company and used as-received. 2-Chloro-1,3-diene, 50% in xylene
(Chloroprene) was purchased from Pfaltz & Bauer, Inc. and used as-received.
Isomer ratios were calculated by NMR (13
C NMR experiment with inverse gated 1H-
decoupling)126, 127

81
General Procedure for Diels-Alder Reactions:126
For dienes 2.4, 2.6 and 2.40
Diene and dienophile (1: 5) were dissolved in chloroform (2.4 and 2.6) or toluene (2.40)
in a round bottom flask at room temperature. The white product was precipitated with
pentane and obtained by vacuum filtration.
For dienes 2.36, 2.37, 2.38 and 2.39
Diene and dienophile (1: 5) were dissolved in THF (or toluene) in a round bottom flask at
room temperature. In refluxing reactions, toluene was used as a solvent. The white
product was precipitated with pentane and obtained by vacuum filtration.
Preparation of 3.1
Diene 2.4 (0.167 g, 1 mmol) and dienophile N-phenylmaleimide (0.865 g, 5 mmol) were
dissolved in chloroform (15 mL) in a round bottom flask at room temperature. After
stirring for 15 min., the product 3.1 was obtained by precipitation as a white powder
(0.330g, 0.97mmol, 98%) following addition of pentane (50 mL), vacuum filtration, and
drying under vacuum.
1H NMR (300 MHz, CDCl3) 7.49 -7.40 (m, 3H), 7.15-7.11(m, 2H), 6.26 (m, 1H), 4.78
(s, 1H), 4.03-3.94 (m, 3H), 3.61(dt, J =9.70, 3.20, 1H), 3.37-3.27 (m, 2H), 3.20 (m, 1H),
2.95 (ddd, J =12.3, 12.1, 6.2 Hz, 1H), 2.82-2.54 (m, 4H), 2.31(m, 1H), 2.17 (m, 1H).

82
13C NMR (300 MHz, CDCl3) 183.7, 180.0, 132.0, 129.8, 129.4, 128.9, 126.3, 63.6, 63.0,
52.4, 51.0, 41.4, 40.3, 26.0, 25.8. The signal of the vinyl carbon next to tetravalent boron
was not observed due to quadrupolar broadening.128
HRMS calcd for C18H21BN2O4 [M+H]+
= 341.1672, found [M+H]+= 341.1673
Preparation of 3.2
Diene 2.6 (0.181 g, 1 mmol) and dienophile N-phenylmaleimide (0.865 g, 5 mmol) were
dissolved in chloroform (15 mL) in a round bottom flask at room temperature. After
stirring for 2 hours, the product 3.2 was obtained by precipitation as a white powder
(0.318 g, 0.90 mmol, 90%) following addition of pentane (50 mL), vacuum filtration, and
drying under vacuum.
1H NMR (300 MHz, CDCl3) 7.43(t, J =7.6Hz, 2H), 7.35 (t, J=7.4Hz,1H), 7.23 (d,J=
7.6Hz, 2H), 6.35 (m, 1H), 3.98 (m, 4H), 3.21 (m, 2H), 3.05 (m, 2H), 2.81 (m, 2H), 2.69
(m, 2H), 2.44(s, 3H), 2.38 (m, 2H). 13
C NMR (300, MHz, CDCl3) 180.4, 179.8, 132.5,
132.2, 129.0, 128.8, 126.2, 62.0, 61.7, 60.9, 60.4, 46.4, 39.7,39.5, 26.5, 24.8.
EA: calcd: C 64.43; H 6.54; Found : C 63.73, H 6.43
Preparation of 3.3

83
Diene 2.40 (0.204 g, 1 mmol) and dienophile N-phenylmaleimide (0.865 g, 5 mmol) were
dissolved in toluene (15 mL) in a round bottom flask at room temperature. After stirring
for 2 hours, the product 3.3 was obtained by precipitation as a white powder (0.347 g,
0.92 mmol, 92%) by addition of pentane (50 mL), vacuum filtration, and drying under
high vacuum.
1H NMR (300 MHz, DMSO) 7.43 (m, 2H), 7.37 (m, 1H), 7.26 (m, 2H), 5.64(m, J=1H),
3.49 (s, 6H), 3.03 (m, 1H), 2.92 (m, 1H), 2.19(m, 4H), 0.44(s, 3H). 13
C NMR (300, MHz,
DMSO) 179.8, 179.7, 132.8, 128.6, 128.8, 127.9, 127.2, 73.3, 72.6, 34.5, 26.4, 23.6,
16.3 HRMS: calcd: 377.1410-Na+H=355.1597 found 355.1591
Preparation of 3.4
Diene 2.4 (0.167 g, 1 mmol) and dienophile ethyl acrylate (0.700g, 7 mmol) were
dissolved in chloroform (15 mL) in a round bottom flask and refluxed for 6 h. The white
product was precipitated with pentane (150 mL) and obtained by vacuum filtration
followed by drying under high vacuum, (0.224 g, 0.84 mmol, 84%).
1H NMR (300 MHz, CDCl3) 5.91 (m, 1H), 4.86 (s, 1H), 4.12 (q, J = 7.25, 2H), 3.97 (m,
2H), 2.89 (m, 2H), 3.224 (m, 2H), 2.79 (m, 2H), 2.48 (m, 1H), 2.23 (m, 2H), 2.11(m, 2H),
1.99 (m, 1H), 1.76 (s, 1H), 1.25 (t, J = 7.25, 3H). 13
C NMR (300 MHz, CDCl3) Major
isomer: 176.7, 139.9 (vinyl carbon next to the boron), 126.9, 62.85, 62.81, 60.0, 51.2,
40.1, 39.8, 28.6, 26.4, 25.9, 14.0. Minor isomer selected resonances: 176.2, 127.6, 24.7,
24.6 Major isomer : minor isomer = 16.4 : 1

84
Elemental anal. calcd. for C13H22BNO4: C, 58.45; H, 8.30. Found: 58.17, 8.32.
Preparation of 3.5
Diene 2.4 (0.167 g, 1 mmol) and dienophile 2-methyl-N-phenylmaleimide (0.16 g, 1.6
mmol) were dissolved in chloroform (10 mL) in a round bottom flask and refluxed for 6 h.
The white product was precipitated with pentane (150 mL) and obtained by vacuum
filtration followed by drying under high vacuum (0.336 g, 0.95 mmol, 95%).
1H NMR (300 MHz, CDCl3) 7.48-7.37 (m, 3H), 7.14 (s, 1H), 7.12 (s, 1H), 6.26 (m, 1H),
4.88 (s, 1H), 3.99 (m, 3H), 3.61 (m, 1H), 3.21(m, 1H), 2.92 (m, 2H), 2.79 (m, 1H), 2.68
(m, 2H), 2.57 (m, 1H), 2.17 (d, J = 15.0 Hz, 1H), 1.97 (d, J = 15.0 Hz, 1H), 1.47 (s, 3H).
13C NMR (300 MHz, CDCl3) Major isomer : 183.0, 182.9, 132.3, 131.0, 129.6, 129.2,
126.6, 63.9, 63.3, 52.6, 51.8, 49.4, 45.4, 35.7, 26.0, 23.4.
The signal of the vinyl carbon next to tetravalent boron was not observed due to
quadrupolar broadening. 128
Minor isomer found: 179.8, 130.1, 63.3, 53.0, 48.4, 46.9, 34.7, 27.2, 24.7
Major isomer : minor isomer = 4:1
HRMS calcd for C19H23BN2O4 [M+H]+=355.1830, found [M+H]
+= 355.1825
Preparation of 3.6

85
Diene 2.6 (0.181 g, 1 mmol) and dienophile ethyl acrylate (0.500g, 5 mmol) were
dissolved in chloroform (10 mL) in a round bottom flask and refluxed for 6 h. The white
product was precipitated with pentane (150 mL) and obtained by vacuum filtration
followed by drying under high vacuum (0.252 g, 0.90 mmol, 90%).
1H NMR (300 MHz, CDCl3) 6.05 (m, 1H), 4.07 (q, J=7.2 Hz, 2H), 3.97 (m, 2H), 3.90
(m, 2H), 3.01(m, 3H), 2.67 (m, 2H), 2,55 (s, 3H), 2.44 (m, 1H), 2.24 (m, 1H), 2.09 (m,
2H), 1.93 (m, 1H), 1.57 (m, 1H), 1.25 (t, 7.2 Hz, 3H)
13C NMR (300, MHz, CDCl3) Major isomer: 176.6, 130.2, 62.0, 60.3, 59.8, 46.3, 39.7,
28.7, 27.0, 25.9, and 14.1
Minor isomer’s peaks found: 177.0, 131.4, 59.9, 59.1, 59.0, 46.4, 40.1, 30.2, 29.1, 25.6,
and 25.2
HRMS calcd for C14H24BNO4: 281.1798 +Na=304.1696, found: M+Na=304.1696
Preparation of 3.7
Diene 2.6 (0.181 g, 1 mmol) and dienophile 2-methyl-N-phenylmaleimide (0.500 g, 5
mmol) were dissolved in chloroform (10 mL) in a round bottom flask and refluxed for 6h.
The white product was precipitated with pentane (150 mL) and obtained by vacuum
filtration followed by drying under high vacuum (0.276 g, 0.75 mmol, 75%).
1H NMR (300 MHz, CDCl3) Major isomer: 7.42 (t, J =7.8 Hz, 2H), 7.36 (t, J=7.2
Hz,1H), 7.22 (d,J= 7.8 Hz, 2H), 6.33 (m, 1H), 3.97 (m, 4H), 3.04 (m, 2H), 2.87 (m, 2H),
2.78 (m, 3H), 2.40 (s, 3H), 2.39 (m, 1H), 2.05 (dt, J=15, 7 Hz, 1H), 1.46 (s, 3H)

86
Minor isomer found: 2.41(s), 1.43 (s)
13C NMR (300, MHz, CDCl3) Major isomer: 182.6, 179.0, 132.6, 132.3, 129.0, 128.2,
126.1, 61.8, 60.9, 60.4, 47.3, 46.6, 44.2, 35.5, 26.3, 25.3, 25.2
Minor isomer found: 182.5, 179.6, 125.9, 61.8, 47.6, 44.3, 33.7, 26.9, and 25.0
HRMS calcd for C20H25BN2O4: calcd: 368.1907, found: 368.19075
Preparation of 3.13
Diene 2.40 (0.102 g, 0.5 mmol) and dienophile 2-methyl-N-phenylmaleimide (0.935 g,
5 mmol) were dissolved in toluene (10 mL) in a sealed tube. After refluxing for 18 hours,
the product 3.13 was observed from NMR and MS. No further separation was conducted.
The crude product was used in Suzuki cross coupling reactions which will be described in
the next chapter.

87
CHAPTER 4 Suzuki Cross Coupling Reactions
4.1 Results and Discussion
4.1.1 Suzuki Cross Coupling Reactions
In order to prove that 2-boron substituted dienes (2.4) can serve as a synthon for a host
of other organic dienes, we took cycloadducts (3.1-3.7) and proved that they could be
cross coupled efficiently to iodobenzene, 4-trifluoromethyl-1-iodobenzene, and 4-
iodoanisole.
Based on the initial D-A reactions, a series of Suzuki cross coupling reactions were
performed and conditions were optimized (Table V). The optimization conditions were
achieved by the Suzuki coupling reactions of boron cycloadduct (3.1) and iodobenzene
compounds shown in Table V.
We found in the solvent CH3CN, Pd2(dba)3 proved optimal in cross coupling reactions
(Table V, entry 3) with a high yield of 75%. Later we determined the best solvent for this
reaction is a mixture of CH3CN and C2H5OH (5:1).
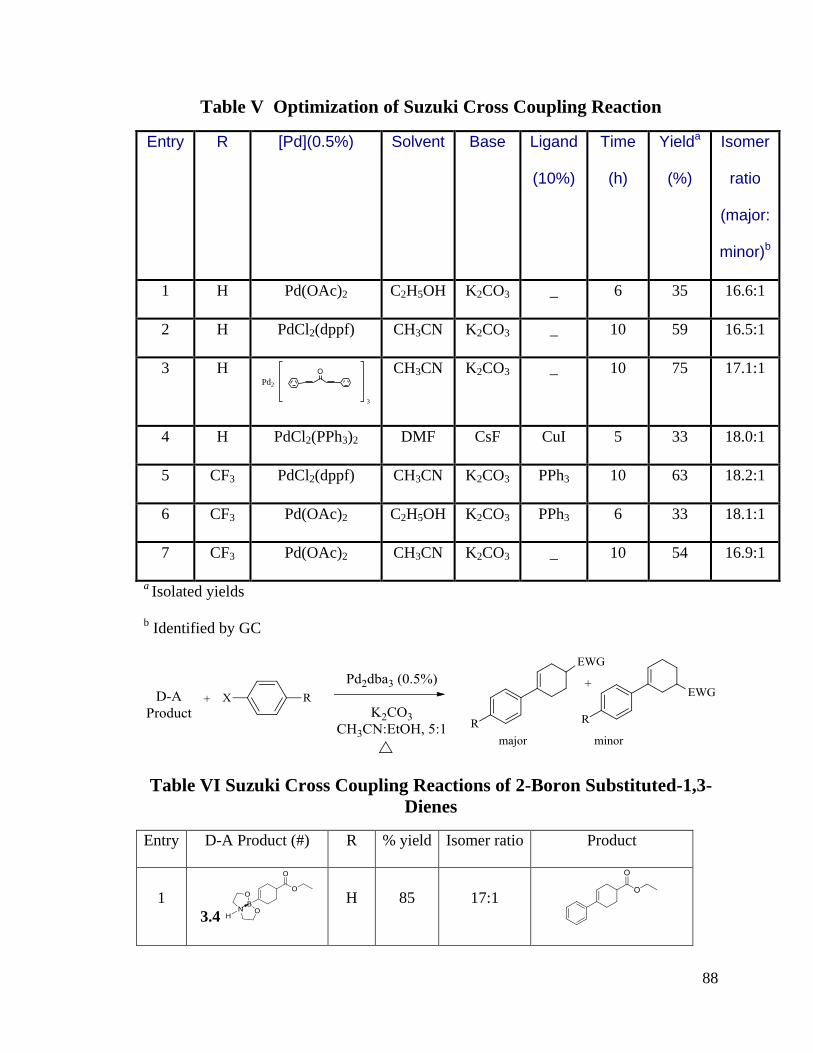
88
Table V Optimization of Suzuki Cross Coupling Reaction
Entry R [Pd](0.5%) Solvent Base Ligand
(10%)
Time
(h)
Yielda
(%)
Isomer
ratio
(major:
minor)b
1 H Pd(OAc)2 C2H5OH K2CO3 _ 6 35 16.6:1
2 H PdCl2(dppf) CH3CN K2CO3 _ 10 59 16.5:1
3 H O
3
Pd2
CH3CN K2CO3 _ 10 75 17.1:1
4 H PdCl2(PPh3)2 DMF CsF CuI 5 33 18.0:1
5 CF3 PdCl2(dppf) CH3CN K2CO3 PPh3 10 63 18.2:1
6 CF3 Pd(OAc)2 C2H5OH K2CO3 PPh3 6 33 18.1:1
7 CF3 Pd(OAc)2 CH3CN K2CO3 _ 10 54 16.9:1
a Isolated yields
b Identified by GC
Table VI Suzuki Cross Coupling Reactions of 2-Boron Substituted-1,3-
Dienes
Entry D-A Product (#) R % yield Isomer ratio Product
1
3.4
H 85 17:1
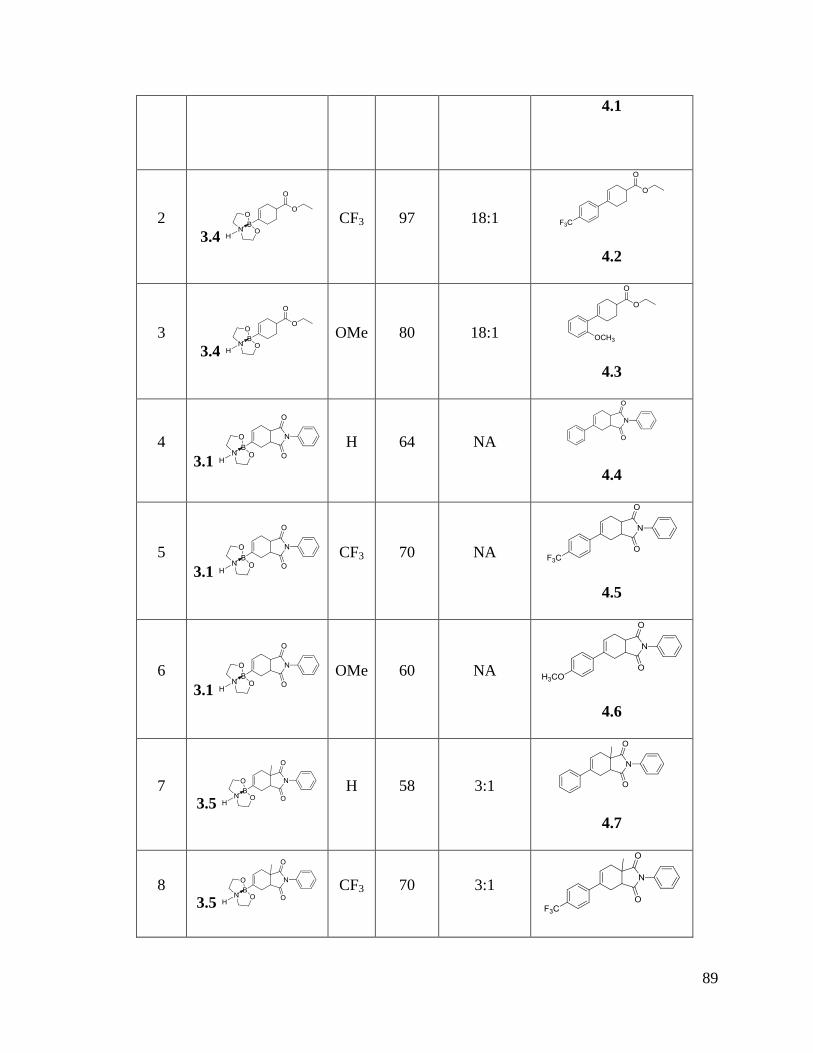
89
4.1
2
3.4
CF3 97 18:1
4.2
3
3.4
OMe 80 18:1
4.3
4
3.1
H 64 NA
4.4
5
3.1
CF3 70 NA
4.5
6
3.1
OMe 60 NA
4.6
7 3.5
H 58 3:1
4.7
8 3.5
CF3 70 3:1
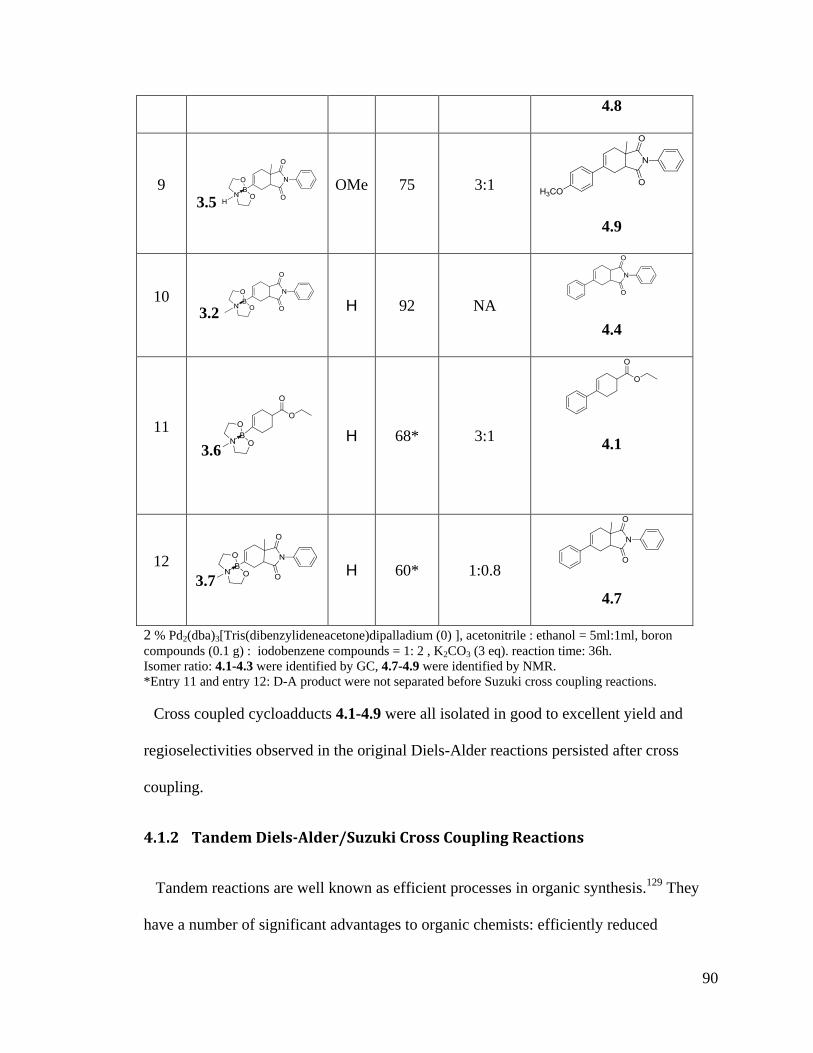
90
4.8
9 3.5
OMe 75 3:1
4.9
10 3.2
H 92 NA
4.4
11
3.6 H 68* 3:1
4.1
12
3.7 H 60* 1:0.8
4.7
2 % Pd2(dba)3[Tris(dibenzylideneacetone)dipalladium (0) ], acetonitrile : ethanol = 5ml:1ml, boron
compounds (0.1 g) : iodobenzene compounds = 1: 2 , K2CO3 (3 eq). reaction time: 36h.
Isomer ratio: 4.1-4.3 were identified by GC, 4.7-4.9 were identified by NMR.
*Entry 11 and entry 12: D-A product were not separated before Suzuki cross coupling reactions.
Cross coupled cycloadducts 4.1-4.9 were all isolated in good to excellent yield and
regioselectivities observed in the original Diels-Alder reactions persisted after cross
coupling.
4.1.2 Tandem Diels-Alder/Suzuki Cross Coupling Reactions
Tandem reactions are well known as efficient processes in organic synthesis.
129 They
have a number of significant advantages to organic chemists: efficiently reduced
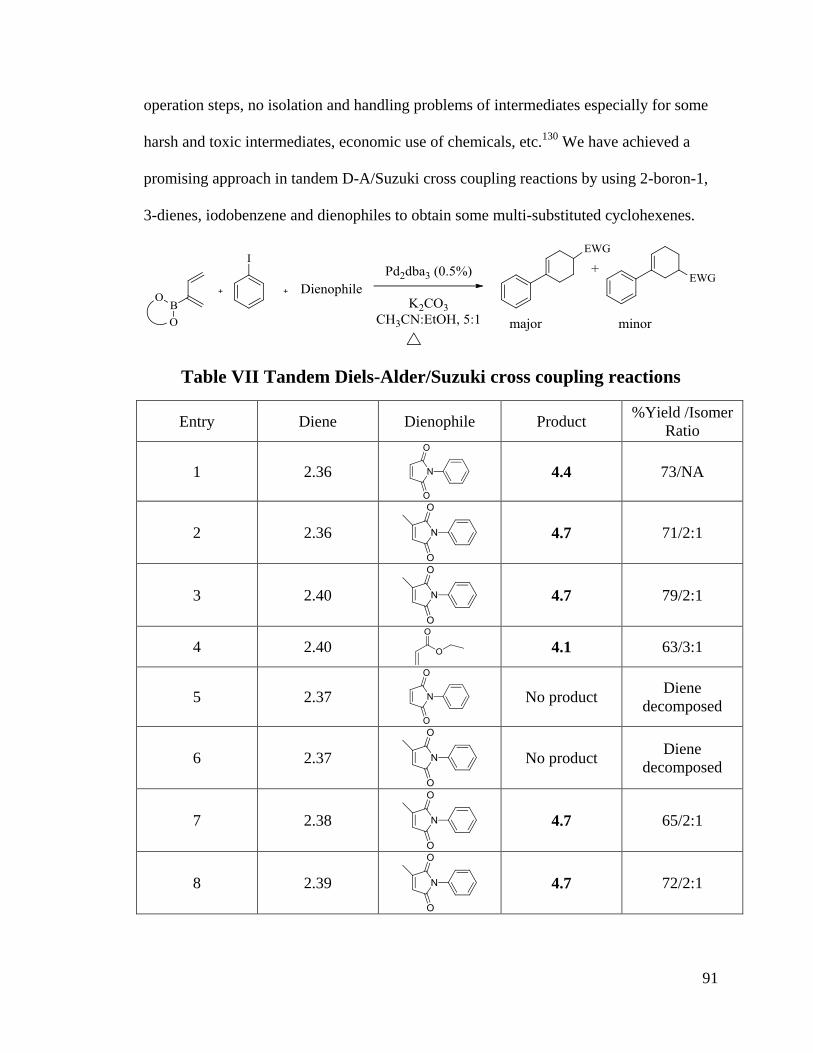
91
operation steps, no isolation and handling problems of intermediates especially for some
harsh and toxic intermediates, economic use of chemicals, etc.130
We have achieved a
promising approach in tandem D-A/Suzuki cross coupling reactions by using 2-boron-1,
3-dienes, iodobenzene and dienophiles to obtain some multi-substituted cyclohexenes.
Table VII Tandem Diels-Alder/Suzuki cross coupling reactions
Entry Diene Dienophile Product %Yield /Isomer
Ratio
1 2.36
4.4 73/NA
2 2.36
4.7 71/2:1
3 2.40
4.7 79/2:1
4 2.40
4.1 63/3:1
5 2.37
No product Diene
decomposed
6 2.37
No product Diene
decomposed
7 2.38
4.7 65/2:1
8 2.39
4.7 72/2:1

92
9 2.4
No product
Diene
decomposed
10 2.6
No product
Diene
decomposed
According to Table VII, dienes 2.36, 2.38, 2.40 and 2.39 can react in tandem D-A/
Suzuki cross coupling reactions with good yields. For the most reactive dienes 2.4 and
2.6 in Diels-Alder reactions, no positive results were observed. We observed diene
decomposition, perhaps because the amine groups in 2-boron-1,3-butadienes are reactive
in tandem conditions eventually causing the decomposition of dienes. Diene 2.37 is the
most unreactive diene in this group since it has a room temperature solution phase s-trans
structure (Appendix B), which also explains the reactivity in tandem D-A/Suzuki cross
coupling reactions. We have extensively investigated the mechanisms of tandem D-
A/Suzuki cross coupling reactions as shown in Scheme 4.1.
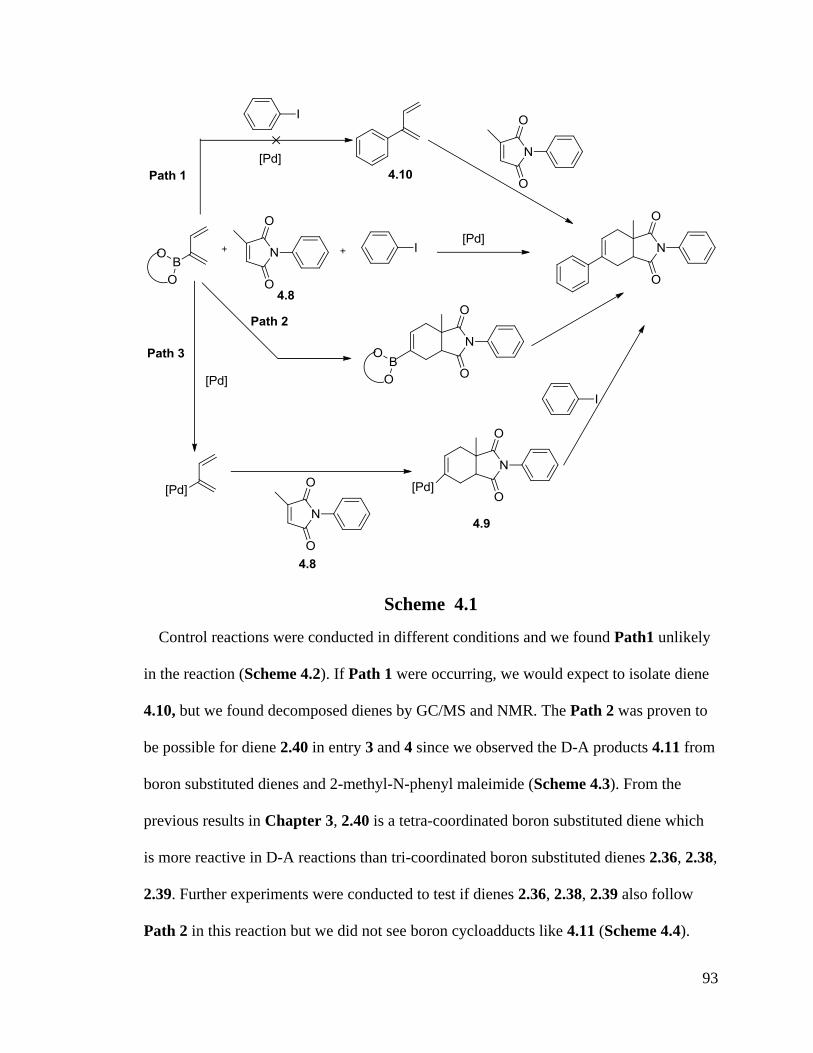
93
Scheme 4.1
Control reactions were conducted in different conditions and we found Path1 unlikely
in the reaction (Scheme 4.2). If Path 1 were occurring, we would expect to isolate diene
4.10, but we found decomposed dienes by GC/MS and NMR. The Path 2 was proven to
be possible for diene 2.40 in entry 3 and 4 since we observed the D-A products 4.11 from
boron substituted dienes and 2-methyl-N-phenyl maleimide (Scheme 4.3). From the
previous results in Chapter 3, 2.40 is a tetra-coordinated boron substituted diene which
is more reactive in D-A reactions than tri-coordinated boron substituted dienes 2.36, 2.38,
2.39. Further experiments were conducted to test if dienes 2.36, 2.38, 2.39 also follow
Path 2 in this reaction but we did not see boron cycloadducts like 4.11 (Scheme 4.4).
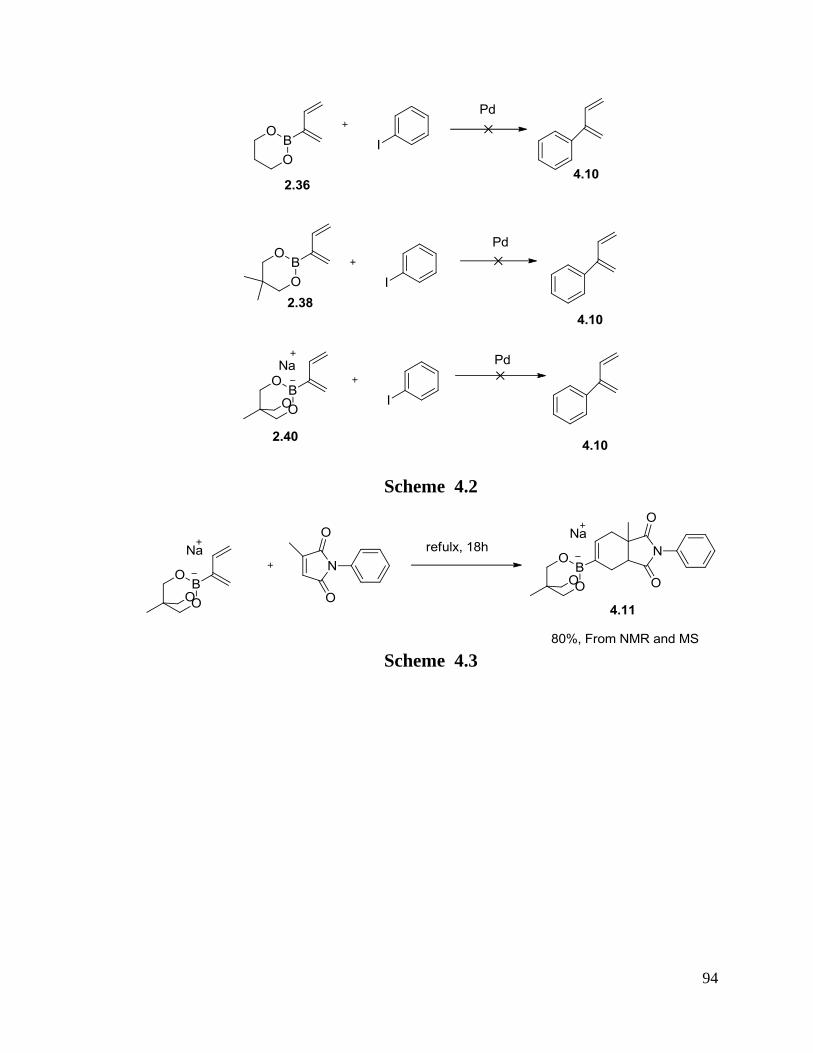
94
Scheme 4.2
Scheme 4.3
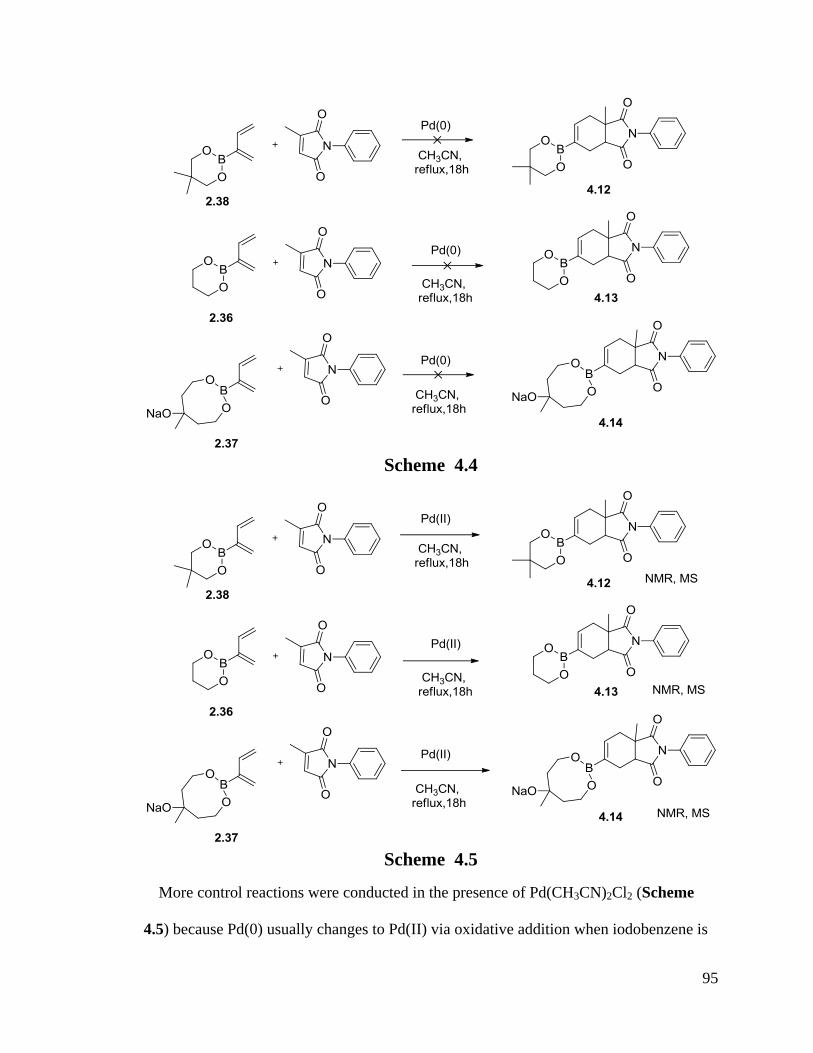
95
Scheme 4.4
Scheme 4.5
More control reactions were conducted in the presence of Pd(CH3CN)2Cl2 (Scheme
4.5) because Pd(0) usually changes to Pd(II) via oxidative addition when iodobenzene is

96
added to the system (Figure 1.6). We found Pd (II) plays an important role in the reaction
to catalyze D-A reactions between boron substituted dienes and dienophiles. So the Path
2 is the plausible mechanism for tandem D-A/Suzuki cross coupling reactions.
Also, we investigated Path 3 via a palladium substituted diene as an intermediate.
From the NMR data, when palladium was added to diene 2.36, some changes were found
after 30 minutes at room temperature. When we heated the solution to 50 ˚C for 3 h, the
transmetallation happened very fast and what we postulate as a palladium substituted
diene 4.8 was observed (Scheme 4.6) and (Figure 4.1). Isolation and separation of this
diene was tried by filtration but this failed. This diene 4.8 was only stable in acetonitrile
solution.
Scheme 4.6
Figure 4.1 Comparison of NMR Chemical Shifts of Boron Substituted
Diene and Palladium Substituted Diene in CD3CN

97
Palladium catalysts with different oxidation states and ligand sets were tried in order to
evaluate the transmetallation with diene 2.36 (Table VIII). Before Pd(dba)3 was added
to diene 2.36 in deuterated acetonitrile, it has to be oxidized by air to change to Pd(II).
When N2 was used to degas the solvent, only a small amount of 4.8 was observed. Pd(II)
was proven to be more active than Pd (0) in this transmetallation reaction (Table VIII,
entry 2 and 3). It only takes a few seconds to 1 minute for Pd(II) to transmetallate to
boron substituted diene to give 4.8 (entry 2 and 3) at room temperature but 3 hours at
50˚C or 60˚C for Pd (0) (entry 1 and 4).
The palladium substituted diene possibly has a ligand of CH3CN which coordinates
with Pd to stabilize the whole structure. From Table VIII, the ligands also are important.
DBA and pincer are better ligands (entry1 and 4) than PPh3 and CH3CN (entry 2 and 3).
The palladium substituted diene with DBA and pincer can be stable for 3 days (entry1
and 4) in a sealed tube but only 3 hours with PPh3 and CH3CN.
Table VIII Palladium Catalysts and 2-Palladium Substituted 1,3-Diene
Entry
[Pd] Solvent T(˚C)
Time Stability of
Palladium
Diene
1 Pd2(dba)3 CD3CN 50 3 h 3d
2 Pd(CH3CN)2Cl2 CD3CN 25 1 min 3h
3 Pd(PPh3)2Cl2 CD3CN 25 1 min 12h
4 Pd(Pincer) CD3CN
60 12 h 2d
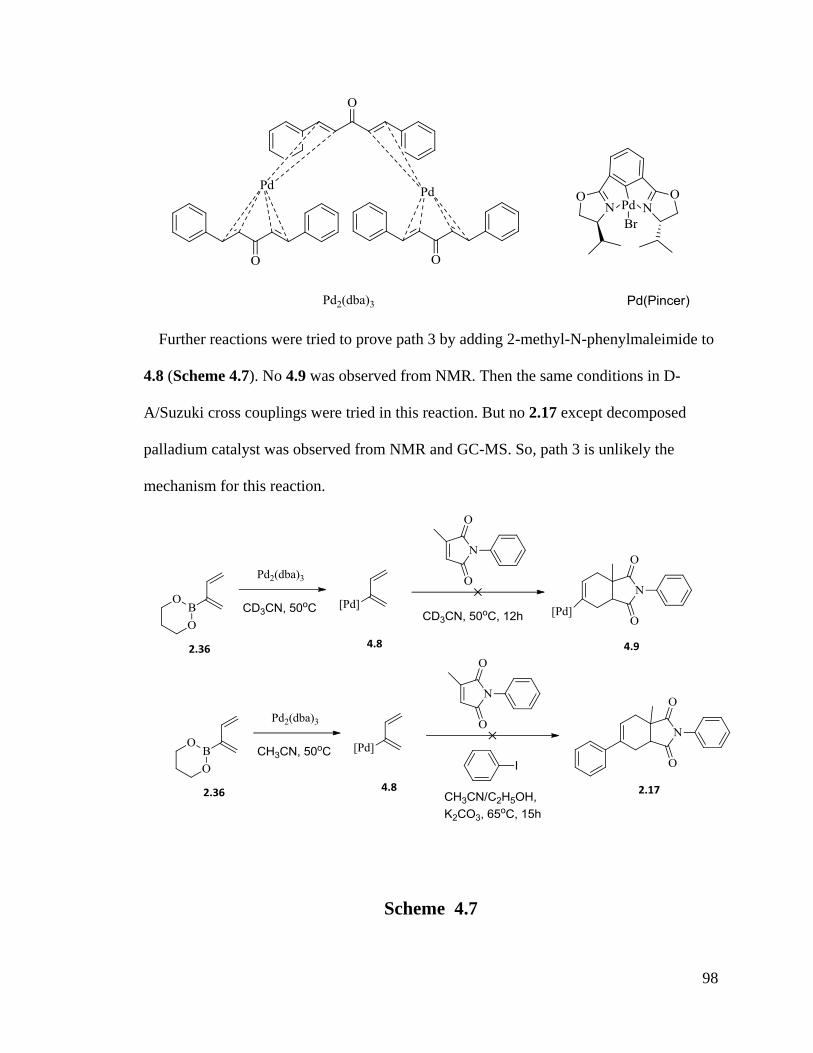
98
Further reactions were tried to prove path 3 by adding 2-methyl-N-phenylmaleimide to
4.8 (Scheme 4.7). No 4.9 was observed from NMR. Then the same conditions in D-
A/Suzuki cross couplings were tried in this reaction. But no 2.17 except decomposed
palladium catalyst was observed from NMR and GC-MS. So, path 3 is unlikely the
mechanism for this reaction.
Scheme 4.7

99
4.2 Conclusions
2-boron substituted dienes have demonstrated good reactivities and high
regioslectivities in stepwise and tandem D-A/Suzuki cross coupling reactions, which can
serve as excellent synthons for a host of organic dienes via cross coupling. The
mechanism research disclosed that Pd(II) could act as a Lewis acid catalyst to catalyze D-
A reaction of boron substituted dienes. Also, a possible palladium substituted-1,3-diene
as a new intermediate was observed in the reactions. Reactions showed that Pd(0) has to
be oxidized to Pd(II) to realize transmetallation to the boron substituted diene. The
reactivity of the possible palladium substituted diene is awaiting further investigation.
4.3 Experimental and Characterization Data
General procedure for stepwise Suzuki cross coupling reactions:
Boron compounds and iodoaromatic compounds were added to a N2 flushed flask with
Pd2(dba)3 and K2CO3 in acetonitrile and ethanol (30 mL). The mixture was refluxed for
36 h and cooled to room temperature. The solution was filtered through silica gel to
remove catalysts. The filtrate was quenched with water (50 mL) and extracted with Et2O
(4×50 mL). The combined organic layers were dried over MgSO4 and volatiles were
removed by rotary evaporation. The resulting cross-coupled cycloadduct residue was
purified by flash chromatography (ethyl ether: hexane=1:1).
Optimization of conditions: 2% Pd2(dba)3 [Tris(dibenzylideneacetone)dipalladium (0) ],
acetonitrile : ethanol = 5:1, boron cycloadduct : iodoaromatic compounds = 1: 2 , K2CO3
(3 equiv). reaction time: 36 h.

100
General procedures for Tandem Diels-Alder/ Suzuki cross coupling reactions
Diene and dienophile (1:4) were added to a N2 flushed high pressure resistance thick
wall tube with Pd2(dba)3 and K2CO3 in acetonitrile and ethanol (5 mL). Iodobenzene was
added. The mixture was sealed and refluxed for 24 h and cooled to room temperature.
The solution was filtered through silica gel to remove catalysts. The filtrate was quenched
with water (50 mL) and extracted with Et2O (4×50 mL). The combined organic layers
were dried over MgSO4 and volatiles were removed by rotary evaporation. The final
product was purified by flash chromatography (ethyl acetate: hexane=6:1).
Preparation of ethyl-4-phenyl cyclohex-3-enecarboxylate 4.1
Following the general procedure, iodobenzene (0.204 g, 1 mmol) and 3.4 (0.133 g, 0.5
mmol ) were added along with Pd2(dba)3 (10 mg, 0.01mmol ) and K2CO3 (0.207 g, 1.5
mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask was heated and
refluxed for 36 h and worked up as described in the general procedure. The resulting
brown oily crude product mixture was subjected to flash chromatography to yield the
cross-coupled product 4.1 as a light yellow oil (0.098 g, 0.43 mmol, 85%). 1H NMR data
was identical to that previously reported.131
1H NMR (300 Hz, CDCl3) 7.39–7.28 (m, 4H), 7.22 (m, 1H), 6.11–6.08 (m, 1H), 4.169
(q, J = 7.3 Hz, 2H), 2.60 (m, 1H), 2.54–2.42 (m, 4H), 2.25–2.14 (m, 1H), 1.84 (m, 1H),
1.27 (t, J = 7.3 Hz, 3H).

101
Major isomer: minor isomer = 17.5:1
Preparation of ethyl 4-[4-(trifluoromethyl)-phenyl]cyclohex-3-enecarboxylate 4.2
Following the general procedure, 4-iodobenzotrifluoride (0.272 g, 1 mmol) and 3.4
(0.133 g, 0.5mmol ) were added along with Pd2(dba)3 (10 mg, 0.01mmol) and
K2CO3(0.207 g, 1.5 mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask
was heated and refluxed for 36 h and worked up as described in the general procedure.
The resulting brown oily crude product mixture was subjected to flash chromatography to
yield the cross-coupled product 4.2 as a light yellow oil (0.144 g, 0.484 mmol, 97%). 1H
NMR data was identical to that previously reported.131
1H NMR (300 MHz, CDCl3) 7.48 (d, J =8.4 Hz, 1H), 7.38 (d, J = 8.4 Hz, 1H), 6.11 (m,
1H), 4.10 (q, J = 7.3 Hz, 2H), 2.55 (m, 1H), 2.47–2.39 (m, 4H), 2.13 (m, 1H), 1.79 (m,
1H), 1.209 (t, J = 7.3 Hz, 3H).
Major isomer: minor isomer = 18:1
Preparation of ethyl-4-(2-methoxyphenyl)-cyclohex-3-enecarboxylate 4.3

102
Following the general procedure, 2-iodoanisole (0.234 g, 1 mmol) and 3.4 (0.133 g, 0.5
mmol ) were added along with Pd2(dba)3 (10 mg, 0.01mmol) and K2CO3 (0.207 g, 1.5
mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask was heated and
refluxed for 36 h and worked up as described in the general procedure. The resulting
brown oily crude product mixture was subjected to flash chromatography to yield the
cross-coupled product 4.3 as a light yellow oil (0.104 g, 0.40 mmol, 80%). 1H NMR data
was identical to that previously reported. 131
1H NMR (300 Hz, CDCl3) 7.21 (ddd, J = 8.6, 7.4, 1.8 Hz, 1H), 7.103 (dd, J = 7.4, 1.8
Hz, 1H), 6.90 (ddd, J = 8.6, 7.4, 1.0 Hz, 1H), 6.85 (d, J = 8.6 Hz, 1H), 5.75 (m, 1H),
4.166 (q, J = 7.3 Hz, 2H), 3.80 (s, 1H), 2.64 (m, 1H), 2.55–2.37 (m, 4H), 2.10 (m, 1H),
1.82 (m, 1H), 1.28 (t, J = 7.3 Hz, 3H)
Major isomer: minor isomer = 21.3:1
Preparation of 2,5-diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione 4.4
Following the general procedure, iodobenzene (0.204 g, 1 mmol) and 3.1 (0.170 g,
0.5 mmol) were added along with Pd2(dba)3 (10 mg, 0.01 mmol) and K2CO3 (0.207 g, 1.5
mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask was heated and
refluxed for 36 h and worked up as described in the general procedure. The resulting
brown oily crude product mixture was subjected to flash chromatography to yield the

103
cross-coupled product 4.4 as a white solid (0.097 g, 0.32 mmol, 64%). 1H NMR data was
identical to that previously reported. 50
1H NMR (300 Hz, CDCl3) 7.28–7.45 (m, 8H), 7.10–7.20 (m, 2H), 6.15–6.27 (m, 1H),
3.44 (ddd, J = 9.5, 6.9, 2.5 Hz, 1H), 3.35 (ddd, J = 9.5, 6.9 2.5 Hz, 1H), 3.26 (dd, J = 15.5,
2.5 Hz, 1H), 2.95 (ddd, J = 15.5, 6.9, 2.5Hz, 1H), 2.64 (ddt, J = 15.5, 6.9, 2.5 Hz, 1H),
2.40–2.50 (m, 1H).
Preparation of 2-phenyl-5-[4-(trifluoromethyl)phenyl]-3a,4,7,7a-tetrahydo-1H-isoindole-
1,3(2H)-dione 4.5
Following the general procedure, 4-iodobenzotrifluoride (0.272 g, 1 mmol) and 3.1
(0.170 g, 0.5 mmol ) were added along with Pd2(dba)3 (10 mg, 0.01 mmol) and K2CO3
(0.207 g, 1.5 mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask was
heated and refluxed for 36 h and worked up as described in the general procedure. The
resulting brown oily crude product mixture was subjected to flash chromatography to
yield the cross-coupled product 4.5 as a white solid (0.112 g, 0.3 mmol, 60%).
1H NMR (300 MHz, CDCl3) 7.58 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 7.42 (t,
J =8.0 Hz, 2H), 7.3 (t, J = 8.0 Hz, 1H), 7.14 (d, J = 8.0 Hz, 2H), 6.31 (m, 1H), 3.48 (ddd,
J = 9.1, 7.4, 2.5 Hz, 1H), 3.38 (ddd, J = 9.1, 7.4, 2.5 Hz, 1H), 3.26 (dd, J = 15.5, 2.7 Hz,
1H), 2.98 (ddd, J = 15.5, 7.1, 2.7 Hz, 1H), 2.65 (ddt, J = 15.5, 7.1, 2.7 Hz, 1H), 2.43–2.50
(m, 1H). 13
C NMR (300 MHz, CDCl3) 179.2, 179.0, 144.0, 139.5, 132.1, 129.9 ( 2JC–F =

104
32.6 Hz), 129.5, 129.1, 126.7,126.3 ( 1JC–F = 273.1 Hz), 126.2, 125.9(
3JC–F = 3.8 Hz),
125.8, 40.4, 39.7, 27.8, 25.7.
HRMS calcd for C21H16F3NO2 [M+H]+ = 372.1211, found [M+H]
+ = 372.1211.
Preparation of 5-(4-methoxyphenyl)-2-phenyl-3a,4,7,7a-tetrahydro-1H-isoindole-
1,3(2H)-dione 4.6
Following the general procedure, 4-iodoanisole (0.234 g, 1 mmol) and 3.1(0.170 g, 0.5
mmol ) were added along with Pd2(dba)3 (10 mg, 0.01mmol) and K2CO3 (0.207 g, 1.5
mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask was heated and
refluxed for 36 h. The resulting brown oily crude product mixture was subjected to flash
chromatography to yield the cross-coupled product 4.6 as a white solid (0.099 g,
0.3 mmol, 60%). 1H NMR data was identical to that previously reported.
50
1H NMR (300 Hz, CDCl3) 7.43–7.28 (m, 5H), 7.15 (d, J = 7.6 Hz, 2H), 6.86 (d, J = 8.7
Hz, 2H), 6.12 (m, 1H), 3.80 (s, 3H), 3.42 (ddd, J = 9.3, 7.0, 2.6 Hz, 1H), 3.33 (ddd, J =
9.3, 7.0, 2.5 Hz,1H), 3.26 (dd, J = 15.5, 2.5 Hz, 1H), 2.95 (ddd, J = 15.5, 6.9, 2.5 Hz, 1H),
2.64 (ddt, J = 15.5, 6.9, 2.5 Hz, 1H), 2.40−2.50 (m, 1H).
Preparation of 3a-methyl-2,6-diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione
4.7

105
Followed the general procedure, 4-iodobenzene (0.204 g, 1 mmol) and 3.5 (0.178 g,
0.5 mmol ) were added along with Pd2(dba)3 (10 mg, 0.1mmol) and K2CO3 (0.207 g, 1.5
mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask was heated and
refluxed for 36 h and worked up as described in the general procedure. The resulting
brown oily crude product mixture was subjected to flash chromatography to yield the
cross-coupled product 4.7 as a white solid (0.0887 g, 0.28 mmol, 58%).
1H NMR (300 MHz, CDCl3) Major isomer: 7.42–7.23 (m, 8H), 7.16 (m, 1H), 7.13 (m,
1H), 6.19 (m, 1H), 3.28 (ddd, J = 15.3, 2.4 Hz, 1H), 3.01 (dd, J = 6.6, 2.4 Hz, 1H), 2.89
(dd, J = 6.6 Hz, 1H), 2.65 (ddt, J = 15.3, 6.6, 2.4 Hz, 1H), 2.12 (ddd, J = 15.3, 3.7, 2.4 Hz,
1H), 1.53 (s, 3H).
Minor isomer found: 3.18 (d, J = 15.3), 2.46 (d, J = 15.3), 2.34 (d, J = 15.3)
13C NMR (300 MHz, CDCl3) Major isomer: 182.2, 178.4, 140.2, 140.0, 132.3, 129.4,
128.9, 128.6, 127.5, 126.4, 125.6, 123.5, 48.3, 45.5, 34.4, 28.3, 25.9.
Minor isomer found: 181.6, 178.3, 140.6, 140.3, 128.5, 125.5, 47.5, 45.1, 36.3, 25.4
HRMS calcd for C21H19NO2 [M+H]+= 318.1494, found [M+H]
+ = 318.1494.
Major isomer: minor isomer = 3:1
Preparation of 3a-methyl-2-phenyl-6-[4-(trifluoromethyl)phenyl]-3a,4,7,7a-tetrahydro-
1H-isoindole-1,3(2H)-dione 4.8

106
Following the general procedure, 4-iodobenzotrifluoride (0.272 g, 1 mmol) and 3.5
(0.177 g, 0.5 mmol ) were added along with Pd2(dba)3 (10 mg, 0.01 mmol) and K2CO3
(0.207 g, 1.5 mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask was
heated and refluxed for 36 h and worked up as described in the general procedure . The
resulting brown oily crude product mixture was subjected to flash chromatography to
yield the cross-coupled product 4.8 as a white solid (0.135 g, 0.35 mmol, 70%).
1H NMR (300 MHz, CDCl3) Major isomer: 7.58 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0
Hz, 2H), 7.44–7.32 (m, 3H), 7.14–7.11 (m, 2H), 6.29 (m, 1H), 3.29 (dd, J = 15.2, 2.5 Hz,
1H), 3.05 (dd, J = 6.50, 2.5 Hz, 1H), 2.93 (m, 1H), 2.68 (ddt, J = 15.2, 6.5, 2.5 Hz, 1H),
2.15 (m, 1H), 1.56 (s, 3H); Minor isomer found: 3.188 (d, J = 15.2), 2.35 (dt, J = 15.2,
2.5 Hz)
13C NMR APT (300 MHz, CDCl3) Major isomer: 181.9, 178.4, 143.9, 139.5, 132.2,
129.8, 129.6, 129.4 ( 2JC–F = 33.3 Hz) , 126.7, 126.3, 126.1, 125.9(
1JC–F = 273.3 Hz),
125.5 ( 3JC–F =4Hz), 48.2, 44.9, 34.5, 28.1, 24.7 Minor isomer found: 180.0, 176.5,
140.0, 47.5, 45.6, 36.5, 30.6, 26.1.
HRMS calcd for C22H18F3NO2 [M+H]+= 386.1368, found [M+H]
+= 386.1368.
Major isomer: minor isomer = 3:1
Preparation of 6-(4-methoxyphenyl)-3a-methyl-2-phenyl-3a,4,7,7a-tetrahydro-1H-
isoindole-1,3(2H)-dione 4.9

107
Following the general procedure, 4-iodoanisole (0.234 g, 1 mmol) and 3.5 (0.178 g,
0.5 mmol) were added along with Pd2(dba)3 (10 mg, 0.01mmol) and K2CO3 (0.207 g, 1.5
mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask was heated and
refluxed for 36 h and worked up as described in the general procedure. The resulting
brown oily crude product mixture was subjected to flash chromatography to yield the
cross-coupled product 4.9 as a white solid (0.134 g, 0.39 mmol, 78%).
1H NMR (300 MHz, CDCl3) Major isomer: 7.38 (d, J = 7.5 Hz, 2H), 7.31 (d, J = 8.7
Hz, 2H), 7.26 (m, 1H), 7.13 (d, J = 7.5 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 6.1 (m, 1H),
3.80 (s, 3H), 3.25 (dd, J = 15.4, 2.4 Hz, 1H), 2.99 (dd, J = 6.5, 2.4 Hz, 1H), 2.86 (dd, J =
15.4, 6.5 Hz, 1H), 2.61 (ddt, J = 15.4, 6.5, 2.4 Hz, 1H), 2.16 (dd, J = 15.4, 2.4 Hz, 1H),
1.50 (s, 3H); Minor isomer selected resonances: 3.15 (d, J = 15.4), 2.44 (m), 2.30 (m).
13C NMR (300 MHz, CDCl3) 182.3, 178.5, 159.5, 139.8, 133.1, 132.4, 129.4, 128.8,
127.0, 126.8, 122.0, 114.3, 55.6, 48.4, 45.1, 36.7, 30.6, 25.9.
Elemental anal. calcd for C22H21NO3: C, 76.06; H, 6.09. Found: 76.34, 6.31.
Major isomer: minor isomer = 3:1
Preparation of 2,5-diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione 4.4 from
3.2 (Table VI, entry 10)

108
Following the general procedure, iodobenzene (0.204 g, 1 mmol) and 3.2 (0.177 g,
0.5 mmol) were added along with Pd2(dba)3 (10 mg, 0.01 mmol) and K2CO3 (0.207 g, 1.5
mmol) to a flask under N2 (30 mL acetonitrile and ethanol). The flask was heated and
refluxed for 36 h and worked up as described in the general procedure. The resulting
brown oily crude product mixture was subjected to flash chromatography to yield the
cross-coupled product 4.4 as a white solid (0.139 g, 0.46 mmol, 92%). 1H NMR data was
identical to that previously reported.50
1H NMR (300 Hz, CDCl3) 7.28–7.45 (m, 8H), 7.10–7.20 (m, 2H), 6.15–6.27 (m, 1H),
3.44 (ddd, J = 9.5, 6.9, 2.5 Hz, 1H), 3.35 (ddd, J = 9.5, 6.9 2.5 Hz, 1H), 3.26 (dd, J = 15.5,
2.5 Hz, 1H), 2.95 (ddd, J = 15.5, 6.9, 2.5Hz, 1H), 2.64 (ddt, J = 15.5, 6.9, 2.5 Hz, 1H),
2.40–2.50 (m, 1H).
Preparation of ethyl-4-phenyl cyclohex-3-enecarboxylate 4.1from 3.6 (Table VI, entry
11)
Diene 2.6 (0.090 g, 0.5 mmol) and dienophile ethyl acrylate (0.250g, 2.5 mmol) were
dissolved in chloroform (5 mL) in a round bottom flask and refluxed for 6 h. The flask
was cooled down to room temperature. Iodobenzene (0.204 g, 1mmol) was added along
with Pd2(dba)3 (10 mg, 0.01mmol ) and K2CO3 (0.207 g, 1.5 mmol) to the crude product
in the flask under N2 (30 mL acetonitrile and ethanol). The flask was heated and refluxed
for 36 h and worked up as described in the general procedure. The resulting brown oily

109
crude product mixture was subjected to flash chromatography to yield the cross-coupled
product 4.1 as a light yellow oil (0.082 g, 0.34 mmol, 68%). 1H NMR data was identical
to that previously reported.131
1H NMR (300 Hz, CDCl3) 7.39–7.28 (m, 4H), 7.22 (m, 1H), 6.11–6.08 (m, 1H), 4.169
(q, J = 7.3 Hz, 2H), 2.60 (m, 1H), 2.54–2.42 (m, 4H), 2.25–2.14 (m, 1H), 1.84 (m, 1H),
1.27 (t, J = 7.3 Hz, 3H).
Major isomer: minor isomer = 3:1
Preparation of 3a-methyl-2,6-diphenyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione
4.7 from 3.7 (Table VI, entry 12)
Diene 2.6 (0.090 g, 0.5 mmol) and dienophile 2-methyl-N-phenylmaleimide (0.250g, 2.5
mmol) were dissolved in chloroform (5 mL) in a round bottom flask and refluxed for 6h.
The flask was cooled down to room temperature. 4-Iodobenzene (0.204 g, 1 mmol) was
added along with Pd2(dba)3 (10 mg, 0.1mmol) and K2CO3 (0.207 g, 1.5 mmol) to a flask
under N2 (30 mL acetonitrile and ethanol). The flask was heated and refluxed for 36 h
and worked up as described in the general procedure. The resulting brown oily crude
product mixture was subjected to flash chromatography to yield the cross-coupled
product 4.7 as a white solid (0.095 g, 0.3 mmol, 60%). 1H NMR data was identical to that
previously reported.123

110
1H NMR (300 MHz, CDCl3) 7.42–7.23 (m, 8H), 7.16 (m, 1H), 7.13 (m, 1H), 6.19 (m,
1H), 3.28 (ddd, J = 15.3, 2.4 Hz, 1H), 3.01 (dd, J = 6.6, 2.4 Hz, 1H), 2.89 (dd, J = 6.6 Hz,
1H), 2.65 (ddt, J = 15.3, 6.6, 2.4 Hz, 1H), 2.12 (ddd, J = 15.3, 3.7, 2.4 Hz, 1H), 1.53 (s,
3H).
Major isomer: minor isomer =1:0.8
Tandem Diels-Alder/Suzuki cross coupling reactions:
Entry 1 of Table VII: preparation of 4.4
Diene 2.36 (0.10g, 0.72mmol) and N-phenylmaleimide (0.50g, 2.88mmol) (1:4) were
added to a N2 flushed high pressure resistance thick wall tube with Pd2(dba)3 and K2CO3
in acetonitrile (10 mL). Iodobenzene (0.59 g, 2.88 mmol) (diene:iodobenzene=1:4) was
added to the mixture. The tube was sealed and refluxed for 24 h and cooled to room
temperature. The solution was filtered through silica gel to remove catalysts. The filtrate
was quenched with water (50 mL) and extracted with Et2O (4×50 mL). The combined
organic layers were dried over MgSO4 and volatiles were removed by rotary evaporation.
The final product was purified by flash chromatography (ethyl acetate: hexane=4:1). 4.4
was obtained as a white powder (0.16 g, 0.52 mmol, 73%) 1H NMR data was identical to
that previously reported.50
1H NMR (300 Hz, CDCl3) 7.28–7.45 (m, 8H), 7.10–7.20 (m, 2H), 6.15–6.27 (m, 1H),
3.44 (ddd, J = 9.5, 6.9, 2.5 Hz, 1H), 3.35 (ddd, J = 9.5, 6.9 2.5 Hz, 1H), 3.26 (dd, J = 15.5,

111
2.5 Hz, 1H), 2.95 (ddd, J = 15.5, 6.9, 2.5Hz, 1H), 2.64 (ddt, J = 15.5, 6.9, 2.5 Hz, 1H),
2.40–2.50 (m, 1H).
Entry 2 of Table VII: preparation of 4.7
Diene 2.36 (0.10 g, 0.72 mmol) and 2-methyl-N-phenylmaleimide (0.54 g , 2.88 mmol)
(1:4) were added to a N2 flushed high pressure resistance thick wall tube with Pd2(dba)3
and K2CO3 in acetonitrile and ethanol (10 mL). Iodobenzene (0.29 g, 1.44 mmol)
(diene:iodobenzene=1:2) was added to the mixture. The tube was sealed and refluxed for
24h and cooled to room temperature. The solution was filtered through silica gel to
remove catalysts. The filtrate was quenched with water (50 mL) and extracted with Et2O
(4×50 mL). The combined organic layers were dried over MgSO4 and volatiles were
removed by rotary evaporation. The final product was purified by flash chromatography
(ethyl acetate: hexane=6:1). 4.7 was obtained as a white powder (0.19 g, 0.52 mmol,
71%)
1H NMR (300 MHz, CDCl3) Major isomer: 7.42–7.23 (m, 8H), 7.16 (m, 1H), 7.13 (m,
1H), 6.19 (m, 1H), 3.28 (ddd, J = 15.3, 2.4 Hz, 1H), 3.01 (dd, J = 6.6, 2.4 Hz, 1H), 2.89
(dd, J = 6.6 Hz, 1H), 2.65 (ddt, J = 15.3, 6.6, 2.4 Hz, 1H), 2.12 (ddd, J = 15.3, 3.7, 2.4 Hz,
1H), 1.53 (s, 3H).Minor isomer found: 3.18 (d, J = 15.3), 2.46 (d, J = 15.3), 2.34 (d, J =
15.3)

112
13C NMR (300 MHz, CDCl3) Major isomer: 182.2, 178.4, 140.2, 140.0, 132.3, 129.4,
128.9, 128.6, 127.5, 126.4, 125.6, 123.5, 48.3, 45.5, 34.4, 28.3, 25.9.
Minor isomer found: 181.6, 178.3, 140.6, 140.3, 128.5, 125.5, 47.5, 45.1, 36.3, 25.4
HRMS calcd for C21H19NO2 [M+H]+= 318.1494, found [M+H]
+ = 318.1494.
Major isomer: minor isomer = 2:1
Entry3 of Table VII: preparation of 4.7
Diene 2.40 (0.10 g, 0.49 mmol) and 2-methyl-N-phenylmaleimide (0.37 g , 1.96 mmol)
(1:4) were added to a N2 flushed high pressure resistance thick wall tube with Pd2(dba)3
and K2CO3 in acetonitrile and ethanol (10 mL). Iodobenzene (0.20 g, 0.98 mmol)
(diene:iodobenzene=1:2) was added to the mixture. The tube was sealed and refluxed
for 24h and cooled to room temperature. The solution was filtered through silica gel to
remove catalysts. The filtrate was quenched with water (50 mL) and extracted with Et2O
(4×50 mL). The combined organic layers were dried over MgSO4 and volatiles were
removed by rotary evaporation. The final product was purified by flash chromatography
(ethyl acetate: hexane=6:1). 4.7 was obtained as a white powder (0.12 g, 0.39 mmol,
79%). 1H NMR was identical to that previously reported.
123
1H NMR (300 MHz, CDCl3) 7.42–7.23 (m, 8H), 7.16 (m, 1H), 7.13 (m, 1H), 6.19 (m,
1H), 3.28 (ddd, J = 15.3, 2.4 Hz, 1H), 3.01 (dd, J = 6.6, 2.4 Hz, 1H), 2.89 (dd, J = 6.6 Hz,

113
1H), 2.65 (ddt, J = 15.3, 6.6, 2.4 Hz, 1H), 2.12 (ddd, J = 15.3, 3.7, 2.4 Hz, 1H), 1.53 (s,
3H).
Entry 4 of Table VII: preparation of 4.1
Diene 2.40 (0.10 g, 0.49 mmol) and ethyl acrylate (0.098 g , 0.98 mmol) (1:2) were
added to a N2 flushed high pressure resistance thick wall tube with Pd2(dba)3 and K2CO3
in acetonitrile and ethanol (10 mL). Iodobenzene (0.20 g, 0.98 mmol)
(diene:iodobenzene=1:2) was added to the mixture. The tube was sealed and refluxed
for 24h and cooled to room temperature. The solution was filtered through silica gel to
remove catalysts. The filtrate was quenched with water (50 mL) and extracted with Et2O
(4×50 mL). The combined organic layers were dried over MgSO4 and volatiles were
removed by rotary evaporation. The final product was purified by flash chromatography
(ethyl ether: hexane=1:1). 4.1 was obtained as a light yellow liquid. (0.071 g, 0.31 mmol,
63%).
1H NMR data was identical to that previously reported.
131
1H NMR (300 Hz, CDCl3) 7.39–7.28 (m, 4H), 7.22 (m, 1H), 6.11–6.08 (m, 1H), 4.169
(q, J = 7.3 Hz, 2H), 2.60 (m, 1H), 2.54–2.42 (m, 4H), 2.25–2.14 (m, 1H), 1.84 (m, 1H),
1.27 (t, J = 7.3 Hz, 3H).
Major isomer: minor isomer = 3:1

114
Entry 7 of Table VII: preparation of 4.7
Diene 2.38 (0.10g, 0.60mmol) and 2-methyl-N-phenylmaleimide (0.45 g , 2.40 mmol)
(1:4) were added to a N2 flushed high pressure resistance thick wall tube with Pd2(dba)3
and K2CO3 in acetonitrile and ethanol (10 mL). Iodobenzene (0.50 g, 2.4 mmol)
(diene:iodobenzene=1:4) was added to the mixture. The tube was sealed and refluxed
for 24h and cooled to room temperature. The solution was filtered through silica gel to
remove catalysts. The filtrate was quenched with water (50 mL) and extracted with Et2O
(4×50 mL). The combined organic layers were dried over MgSO4 and volatiles were
removed by rotary evaporation. The final product was purified by flash chromatography
(ethyl acetate: hexane=6:1). 4.7 was obtained as a white powder (0.12 g, 0.39 mmol,
65%). 1H NMR was identical to that previously reported.
123
1H NMR (300 MHz, CDCl3) 7.42–7.23 (m, 8H), 7.16 (m, 1H), 7.13 (m, 1H), 6.19 (m,
1H), 3.28 (ddd, J = 15.3, 2.4 Hz, 1H), 3.01 (dd, J = 6.6, 2.4 Hz, 1H), 2.89 (dd, J = 6.6 Hz,
1H), 2.65 (ddt, J = 15.3, 6.6, 2.4 Hz, 1H), 2.12 (ddd, J = 15.3, 3.7, 2.4 Hz, 1H), 1.53 (s,
3H).
Entry 8 of Table VII preparation of 4.7
Diene 2.39 (0.10g, 0.46mmol) and 2-methyl-N-phenylmaleimide (0.34 g, 1.83 mmol)
(1:4) were added to a N2 flushed high pressure resistance thick wall tube with Pd2(dba)3
and K2CO3 in acetonitrile and ethanol (10mL). Iodobenzene (0.37g, 1.83mmol)
(diene:iodobenzene=1:4) was added to the mixture. The tube was sealed and refluxed
for 24h and cooled to room temperature. The solution was filtered through silica gel to
remove catalysts. The filtrate was quenched with water (50 mL) and extracted with Et2O

115
(4×50 mL). The combined organic layers were dried over MgSO4 and volatiles were
removed by rotary evaporation. The final product was purified by flash chromatography
(ethyl acetate: hexane=6:1). 4.7 was obtained as a white powder (0.10g, 0.33 mmol, 72%).
1H NMR was identical to that previously reported.
123
1H NMR (300 MHz, CDCl3) 7.42–7.23 (m, 8H), 7.16 (m, 1H), 7.13 (m, 1H), 6.19 (m,
1H), 3.28 (ddd, J = 15.3, 2.4 Hz, 1H), 3.01 (dd, J = 6.6, 2.4 Hz, 1H), 2.89 (dd, J = 6.6 Hz,
1H), 2.65 (ddt, J = 15.3, 6.6, 2.4 Hz, 1H), 2.12 (ddd, J = 15.3, 3.7, 2.4 Hz, 1H), 1.53 (s,
3H).
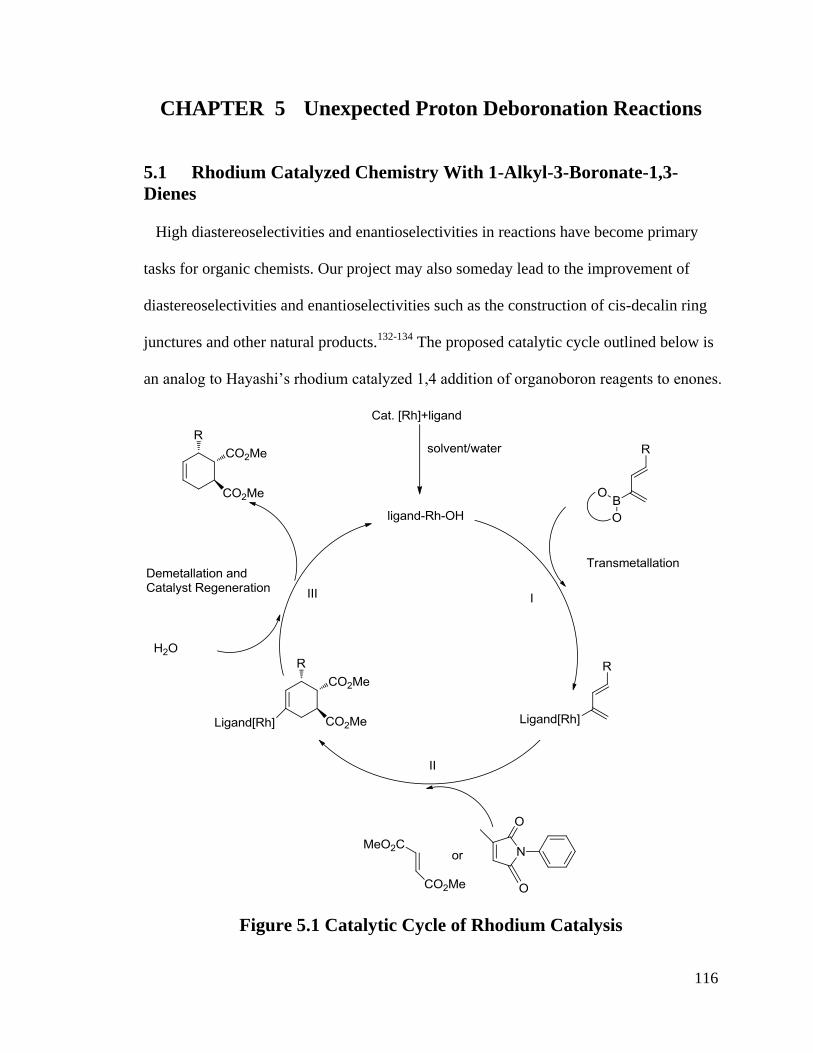
116
CHAPTER 5 Unexpected Proton Deboronation Reactions
5.1 Rhodium Catalyzed Chemistry With 1-Alkyl-3-Boronate-1,3-
Dienes
High diastereoselectivities and enantioselectivities in reactions have become primary
tasks for organic chemists. Our project may also someday lead to the improvement of
diastereoselectivities and enantioselectivities such as the construction of cis-decalin ring
junctures and other natural products.132-134
The proposed catalytic cycle outlined below is
an analog to Hayashi’s rhodium catalyzed 1,4 addition of organoboron reagents to enones.
Figure 5.1 Catalytic Cycle of Rhodium Catalysis

117
In the first step, the catalytic cycle starts with 1-alkyl-2-boronate-1,3-diene where the
transmetallation occurs with rhodium catalysts.106
There would be an option to attach
optically active ligands such as (R) or (S)- BINAP to induce controlled chirality into the
product as demonstrated in various examples of rhodium catalyzed asymmetric 1,4
addition reactions.135-137
The next step of the cycle is the Diels–Alder reaction between a
rhodium dienyl complex with a dienophile. It is also anticipated that the rhodium dienyl
complex would be in a conformation close to s-cis and would react rapidly with a variety
of dienophiles through exo transition states similar to the reported cobalt dienes.106, 138
The success of the overall scheme depends upon two factors106
(i) faster reactivity of the
rhodium dienyl complex in [4 + 2] cycloadditions than the corresponding boron dienyl
and (ii) its preferential reactivity towards [4 + 2] cycloaddition rather than 1,4-addition
with dienophiles. The final step in the Hayashi reaction is demetallation where the
rhodium carbon bond is cleaved via hydrolysis and regeneration of the catalyst occurs.
We have tried some rhodium catalysis of diethanolamine borate diene 2.4. Because this
diene is very reactive in Diels-Alder reaction with N-phenyl maleimide, we chose the
ethyl acrylate and 2-methyl –N-phenyl maleimide to do rhodium catalysis after the
control experiments showed negative results. Other boronate dienes and dienophiles also
have been tested in this reaction, with the results summarized in Table IX.
Table IX Rhodium Catalyzed Reactions
Entry Diene Dienophile Solvent/H2O Catalyst T(0C) Ligand Base T(h) Product
1 2.4
Toluene/ Rh(COD)2OTf
rt 19 no
118
H2O (5:1)
2 2.4
Toluene/
H2O (5:1) Rh(COD)2OTf
40 20 no
3 2.4
Toluene Rh(COD)2OTf
50 20 D-A
4 2.4
Toluene Rh(COD)2OTf
50
20 trace
5 2.4
Toluene/
H2O (5:1) Rh(COD)2OTf
50
20 no
6 2.4
Toluene/
H2O (5:1) Rh(COD)2OTf
rt 20 no
7 2.4
Toluene/
H2O (5:1) Rh(COD)2OTf
50 20 no
8 2
Toluene/
H2O (5:1) Rh(COD)2OTf
rt BINAP 20 no
9 2
Toluene/
H2O (5:1) Rh(COD)2OTf
50 BINAP 20 no
10 2
Toluene/
H2O (5:1) Rh(COD)2OTf
100 BINAP 20 no
11 2
Toluene/
H2O (5:1) Rh(COD)2OTf
rt BINAP
KOH 17 no
12 2
Toluene/
H2O (5:1) Rh(COD)2OTf
50 BINAP
KOH 17 no
13 2
Dioxane/
CH3OH
(5:1)
Rh(COD)2OTf 50
BINAP KOH 17 no
14 1
THF/
H2O(10:1)
Rh(COD)2OTf 55 Et3N 36 trace
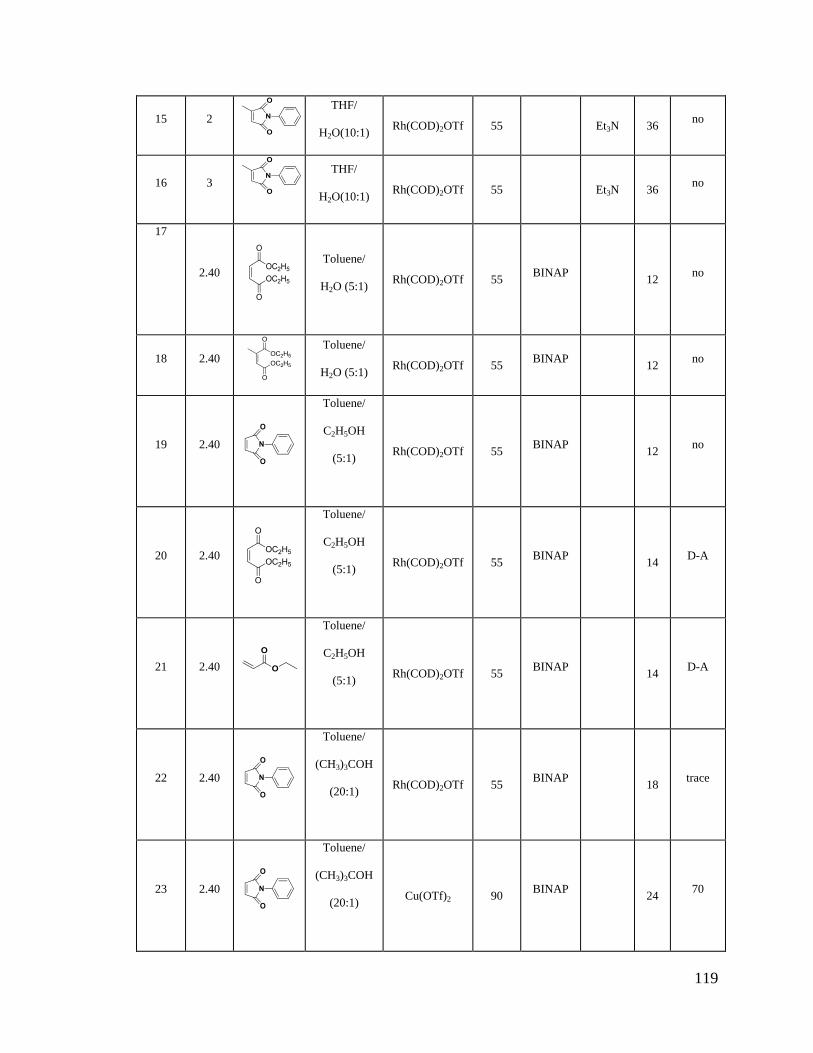
119
15 2
THF/
H2O(10:1) Rh(COD)2OTf 55
Et3N 36
no
16 3
THF/
H2O(10:1) Rh(COD)2OTf 55
Et3N 36
no
17
2.40
Toluene/
H2O (5:1) Rh(COD)2OTf 55
BINAP 12
no
18 2.40
Toluene/
H2O (5:1) Rh(COD)2OTf 55
BINAP 12
no
19 2.40
Toluene/
C2H5OH
(5:1)
Rh(COD)2OTf 55 BINAP
12 no
20 2.40
Toluene/
C2H5OH
(5:1)
Rh(COD)2OTf 55 BINAP
14 D-A
21 2.40
Toluene/
C2H5OH
(5:1)
Rh(COD)2OTf 55 BINAP
14 D-A
22 2.40
Toluene/
(CH3)3COH
(20:1)
Rh(COD)2OTf 55 BINAP
18 trace
23 2.40
Toluene/
(CH3)3COH
(20:1)
Cu(OTf)2 90 BINAP
24 70
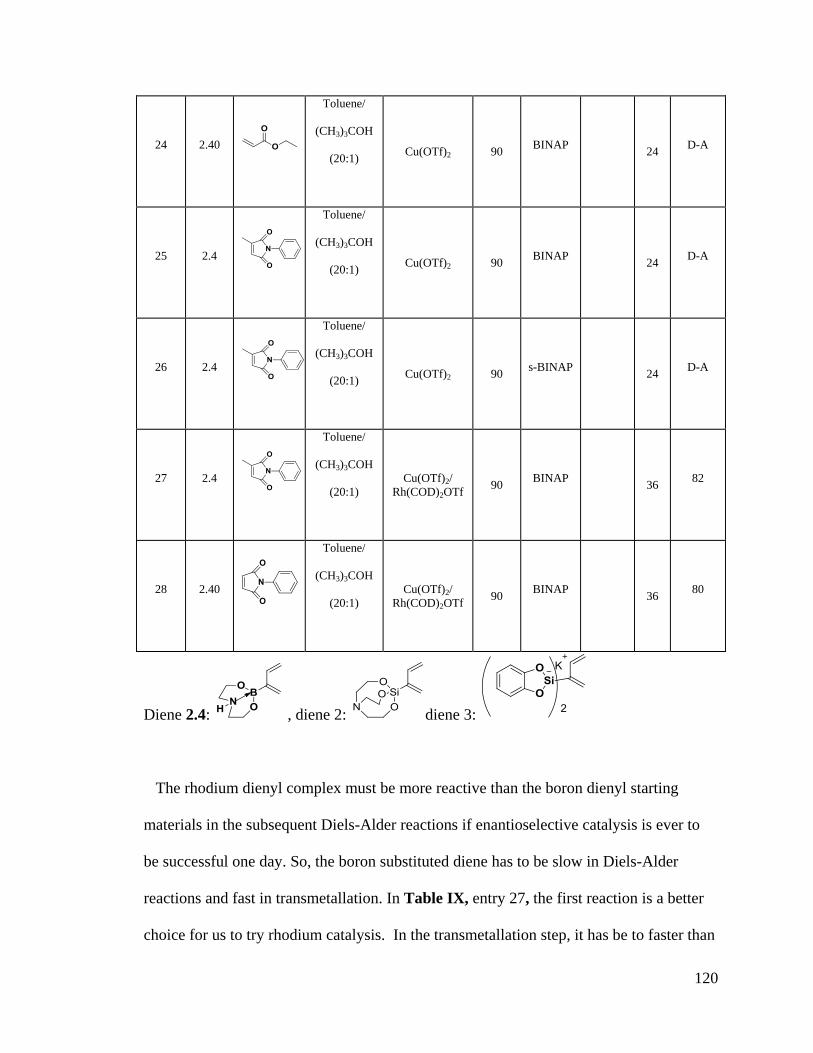
120
24 2.40
Toluene/
(CH3)3COH
(20:1)
Cu(OTf)2 90 BINAP
24 D-A
25 2.4
Toluene/
(CH3)3COH
(20:1)
Cu(OTf)2 90 BINAP
24 D-A
26 2.4
Toluene/
(CH3)3COH
(20:1)
Cu(OTf)2 90 s-BINAP
24 D-A
27 2.4
Toluene/
(CH3)3COH
(20:1)
Cu(OTf)2/
Rh(COD)2OTf 90
BINAP 36
82
28 2.40
Toluene/
(CH3)3COH
(20:1)
Cu(OTf)2/
Rh(COD)2OTf 90
BINAP 36
80
Diene 2.4: , diene 2: diene 3:
The rhodium dienyl complex must be more reactive than the boron dienyl starting
materials in the subsequent Diels-Alder reactions if enantioselective catalysis is ever to
be successful one day. So, the boron substituted diene has to be slow in Diels-Alder
reactions and fast in transmetallation. In Table IX, entry 27, the first reaction is a better
choice for us to try rhodium catalysis. In the transmetallation step, it has be to faster than

121
hydrolysis of the carbon-rhodium bond to finish the cycle. Different catalysts and
different solvents have been tried. We started with the solvents that have been used by
other chemists and former students:139
toluene/H2O, dioxane, toluene /t-butanol. Also,
different temperatures: room temperature, 50ºC and refluxing. Toluene/H2O and dioxane
have been tried and we found water decomposed the dienes. Toluene /t-butanol showed
good stability and reactivity in the reaction.
In Table IX, entries 1,2,5-7, diene 2.4 decomposed in the reaction , and we only got
dienophile back. The reason might be that water is not a good proton provider for dienes
or, nitrogen in diene 2.4 coordinated with rhodium catalyst and deactivated diene 2.4. In
entry 4, after we used water to separate the product, trace product was seen by GC-MS.
When the water was used again in entry 5, no product was found. Two silane substituted
dienes were also tried in this reaction, but no product was found (entries 8-16). In entry
14 we also saw trace product by GC but we could not improve the yield with this diene.
So, we suspect that dienes 2.4 and 2.6 are not good diene partners in rhodium catalysis, or
else the dienophile (2-methyl-N-phenylmalemide) is not a good choice. Then other
possible boronate dienes and dienophiles were also tested in this reaction. In entries 17-
19, diene 2.40 was used but no products were found. In entries 17-19, we found ethanol
can decompose N-phenyl maleimide as shown in Scheme 5.1. From COSY, HMBC and
HMQC, these two structures, 5.1 and 5.2 have similar signals and no further attempts
were made to assign a product structure.
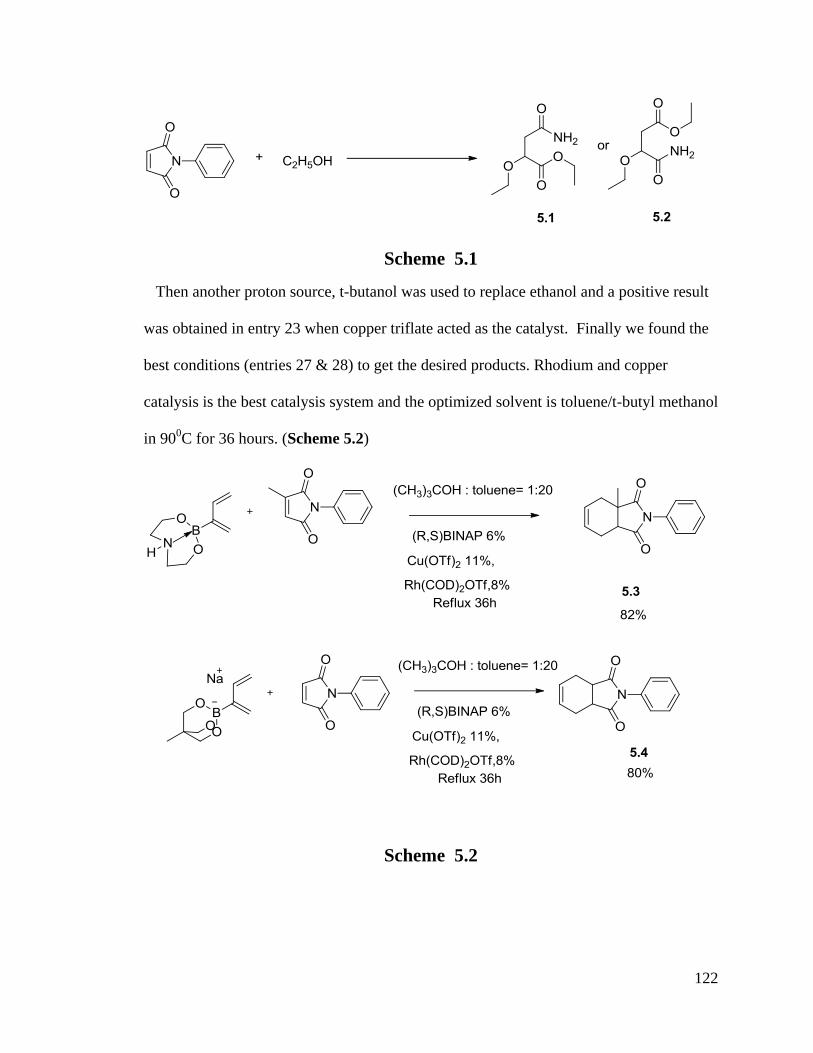
122
Scheme 5.1
Then another proton source, t-butanol was used to replace ethanol and a positive result
was obtained in entry 23 when copper triflate acted as the catalyst. Finally we found the
best conditions (entries 27 & 28) to get the desired products. Rhodium and copper
catalysis is the best catalysis system and the optimized solvent is toluene/t-butyl methanol
in 900C for 36 hours. (Scheme 5.2)
Scheme 5.2
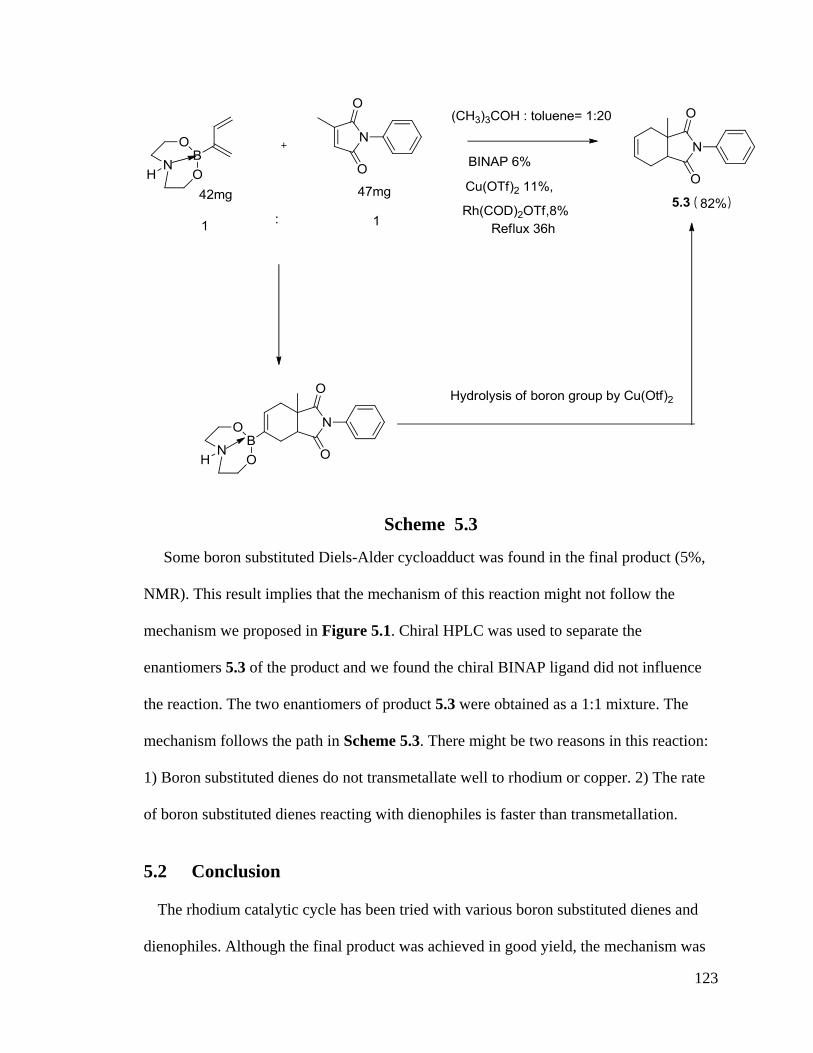
123
Scheme 5.3
Some boron substituted Diels-Alder cycloadduct was found in the final product (5%,
NMR). This result implies that the mechanism of this reaction might not follow the
mechanism we proposed in Figure 5.1. Chiral HPLC was used to separate the
enantiomers 5.3 of the product and we found the chiral BINAP ligand did not influence
the reaction. The two enantiomers of product 5.3 were obtained as a 1:1 mixture. The
mechanism follows the path in Scheme 5.3. There might be two reasons in this reaction:
1) Boron substituted dienes do not transmetallate well to rhodium or copper. 2) The rate
of boron substituted dienes reacting with dienophiles is faster than transmetallation.
5.2 Conclusion
The rhodium catalytic cycle has been tried with various boron substituted dienes and
dienophiles. Although the final product was achieved in good yield, the mechanism was

124
ddifferent from the original proposal. This catalytic system is not a good choice for
rhodium catalysis through transmetallation. However, from the result of the tandem D-
A/Suzuki cross coupling reactions, an intermediate palladium substituted diene was
observed in NMR tube (Scheme 4.5). We can expect the replacement of palladium for
rhodium may be a good future choice in this catalytic cycle (Figure 5.2).
Figure 5.2 Catalytic Cycle of Palladium Catalysis
5.3 Experimental and Characterization Data
Preparation of 3a-methyl-2-phenyl-3a, 4, 7, 7a-tetrahydro-1H-isoindole-1,3(2H)-dione
5.3 (Table IX, Entry 27)

125
Diene 2.4 (83.5 mg, 0.5 mmol) was added to a N2 flushed thick wall tube with
Rh(COD)2OTf (18.7 mg, 0.04 mmol, 8%) , Cu(OTf)2 (19.91 mg,0.055 mmol, 11%) and
(S)-BINAP (20 mg, 0.03 mmol, 6%) in toluene (10 mL) at rt for 30 mins. 2-Methyl-N-
phenyl-maleimide (93.5 mg, 0.5 mmol) was added to the mixture followed by t-butanol
(0.5 mL). The tube was sealed and heated up to reflux for 36 h then cooled to room
temperature. The solution was filtered through a silica gel pad to remove catalysts. The
filtrate was quenched with water (30 ml) and extracted with Et2O (3×50 mL). The
combined organic layers were dried over MgSO4 and volatiles were removed by rotary
evaporation. The resulting residue was purified by chromatography (ethyl ether: hexane =
1:4). The final product was obtained as a white powder (0.099 g, 0.41mmol, 82%).
1H NMR (300 MHz, CDCl3) 7.45(m, 3H), 7.24(m, 2H), 5.98(m, 2H), 2.84(m, 1H),
2.72(m, 1H), 2.66 (m, 1H), 2.33(m, 1H), 2.00(m, 1H), 1.46 (s, 3H).
13C NMR (300 MHz, CDCl3) 161.9, 176.3, 132.1, 129.0, 126.5, 126.1, 127.7, 127.5,
126.7, 47.4, 44.3, 32.7, 24.5, 23.9. HRMS calcd for C15H15NO2 =241.1103, found
241.1103.
Preparation of 2-phenyl-3a, 4, 7, 7a-tetrahydro-1H-isoindole-1,3(2H)-dione 5.4 (Table
IX, Entry 28)

126
Diene 2.40 (102 mg, 0.5 mmol) was added to a N2 flushed thick wall tube with
Rh(COD)2OTf (18.7 mg, 0.04 mmol, 8%) , Cu(OTf)2 (19.91 mg,0.055 mmol, 11%) and
(S)-BINAP (20 mg, 0.03 mmol, 6%) in toluene (10 mL) in rt for 30 mins. N-phenyl-
maleimide (87 mg, 0.5 mmol) was added to the mixture followed by t-butanol (0.5 mL).
The tube was sealed and heated up to reflux for 36 h then cooled to room temperature.
The solution was filtered through a silica gel pad to remove catalysts. The filtrate was
quenched with water (30ml) and extracted with Et2O (3×50 mL). The combined organic
layers were dried over MgSO4 and volatiles were removed by rotary evaporation. The
resulting residue was purified by chromatography (ethyl ether: hexane = 1:4). The final
product was obtained as a white powder (0.090 g, 0.40 mmol, 80%).1H NMR data was
identical to that previously reported.50
1H NMR (300 MHz, CDCl3) 7.32–7.47 (m, 3H), 7.22 (m, 2H), 5.98 (m, 2H), 3.25 (m,
2H), 2.74 (dq, J = 2.2, 2.2, 2.0, 4.0 Hz, 1H), 2.69 (dq, J = 2.2, 2.2, 2.0, 4.0 Hz, 1H), 2.34
(m, 1H), 2.28 (m, 1H). 13
C NMR (300 MHz, CDCl3) 179.1, 132.0, 129. 1, 128.5, 127. 3,
126.4, 39.2, 23.7
Preparation of 2-phenyl-3a, 4, 7, 7a-tetrahydro-1H-isoindole-1,3(2H)-dione 5.4 (Table
IX, Entry 23)
Diene 2.40 (102 mg, 0.5 mmol) was added to a N2 flushed thick wall tube with
Cu(OTf)2 (19.91 mg,0.055 mmol, 11%) and (S)-BINAP (20 mg, 0.03 mmol, 6%) in
toluene (10 mL) at rt for 30 mins. Then N-phenyl-maleimide (87 mg, 0.5 mmol) was

127
added to the mixture followed by t-butanol (0.5 mL). The tube was sealed and heated up
to 55˚C for 24 h then cooled to room temperature. The solution was filtered through a
silica gel pad to remove catalysts. The filtrate was quenched with water (30 mL) and
extracted with Et2O (3×50 mL). The combined organic layers were dried over MgSO4
and volatiles were removed by rotary evaporation. The resulting residue was purified by
chromatography (etheyl ether: hexane = 1:4). The final product was obtained as a white
powder (0.079 g, 0.35 mmol, 70%). 1H NMR data was identical to that previously
reported.
1H NMR (300 MHz, CDCl3) 7.32–7.47 (m, 3H), 7.22 (m, 2H), 5.98 (m, 2H), 3.25 (m,
2H), 2.74 (dq, J = 2.2, 2.2, 2.0, 4.0 Hz, 1H), 2.69 (dq, J = 2.2, 2.2, 2.0, 4.0 Hz, 1H), 2.34
(m, 1H), 2.28 (m, 1H). 13
C NMR (300 MHz, CDCl3) 179.1, 132.0, 129. 1, 128.5, 127. 3,
126.4, 39.2, 23.7

128
CHAPTER 6 CONCLUSIONS
The dimerization of boron substituted dienes were resolved. A series of monomers of
tri- and tetra coordinated boron substituted dienes were prepared at low temperature.
Tetra- coordinated boron substituted dienes were proven to be more active than tri-
coordinated complexes. High yields and regioselectivities were found in the D-A
reactions and maintained in Suzuki Miyaura cross coupling reactions. Boron substituted
dienes are good synthons for a host of organic dienes via D-A/cross coupling reactions. A
possible new intermediate palladium substituted diene was observed in mechanism
research. The stability varies with the different ligands coordinated with Pd.

129
References:
1. Hilt, G.; Hess, W.; Harms, K., Asymmetric Cobalt-Catalyzed Diels Alder
Reactions of a Boron-Functionalized 1,3-Diene with Alkynes and Allylboration with
Aldehydes. Organic Letters 2006, 8, 3287-3290.
2. Mukherjee, S.; Corey, E. J., Highly Enantioselective Diels Alder Reactions of
Maleimides Catalyzed by Activated Chiral Oxazaborolidines. Organic Letters 2010, 12,
632-635.
3. Borosy, A.; Frater, G.; Müller, U.; Schröder, F., Endo-selective Diels-Alder
reaction of methacrylonitrile: application to the synthesis of Georgywood. Tetrahedron
2009, 65, 10495-10505.
4. Douglas, C.; Overman, J.; Larry E., Catalytic asymmetric synthesis of all-carbon
quaternary stereocenters. Proceedings of the National Academy of Sciences of the United
States of America 2004, 101, 5363-5367.
5. Kano, T.; Tanaka, Y.; Maruoka, K., exo-Selective Asymmetric Diels-Alder
Reaction Catalyzed by Diamine Salts as Organocatalysts. Chemistry - An Asian Journal
2007, 2, 1161-1165.
6. Kawamura, M.; Kudo, K., Exo-selective asymmetric diels-alder reaction of
acrylate ester. Chirality 2002, 14, 727-730.
7. Houk, K. N.; Lin, Y. T.; Brown, F. K., Evidence for the concerted mechanism of
the Diels-Alder reaction of butadiene with ethylene. Journal of the American Chemical
Society 1986, 108, 554-556.
8. Fringuelli, F.; Taticchi, A., Dienes in The Diels Alder Reaction. Wiley, New York:
1990; p 30-34.

130
9. Geerlings, P.; De Proft, F.; Langenaeker, W., Conceptual Density Functional
Theory. Chemical Reviews 2003, 103, 1793-1874.
10. Dannenberg, J. J., Using Perturbation and Frontier Molecular Orbital Theory To
Predict Diastereofacial Selectivity. Chemical Reviews 1999, 99, 1225-1242.
11. Cirills Chmidts , S., Iths Chmdtan and D. K. Taylor, Substituent Effects on the
Orientation of Diels-Alder Reactions. I1. Canadian Journal of Chemistry 1971, 49, 371-
375.
12. Li, P.; Yamamoto, H., Lewis Acid Catalyzed Inverse-Electron-Demand
Diels−Alder Reaction of Tropones. Journal of the American Chemical Society 2009, 131,
16628-16629.
13. Boger, D. L., Modern Organic Synthesis. TSRI Press: La Jolla, CA, 1999, 213-
238.
14. Kurt, A.; Gerhard, S., The course of the diene synthesis. Angewandte Chemie
1937, 50, 510-519.
15. Martin, J. G. H., Richard K. , Stereochemistry of the Diels-Alder reaction. .
Chemical Reviews 1961, 61 537-562.
16. Sustmann, R.; Sicking, W.; Lamy-Schelkens, H.; Ghosez, L., endo/exo selectivity
in cycloadditions of 2,6-bis-silyloxy-3,4-dihydropyridine with methylacrylate.
Tetrahedron Letters 1991, 32, 1401-1404.
17. Fringuelli, F. M., Lucio; Radics, Lajos; Taticchi, Aldo; Wenkert, Ernest. , Diels-
Alder reactions of cycloalkenones. 13. Reactions of 2-cyclohexenones with (E)-1-
methoxy-1,3-butadiene. . Journal of Organic Chemistry 1988, 53, 4607-4610.

131
18. Oppolzer, W.; Chapuis, C.; Dao, G. M.; Reichlin, D.; Godel, T., High asymmetric
induction in diels-alder additions of cyclopentadiene to acrylates derived from isoborneol.
Tetrahedron Letters 1982, 23, 4781-4784.
19. Trost, B. M.; Chupak, L. S.; Lübbers, T., Short Preparation of (S)-(E)-1-(O-
Methylmandeloxy)butadiene. The Journal of Organic Chemistry 1997, 62, 736-736.
20. Trost, B. M.; O'Krongly, D.; Belletire, J. L., A model for asymmetric induction in
the Diels-Alder reaction. Journal of the American Chemical Society 1980, 102, 7595-
7596.
21. Bednarski, M.; Danishefsky, S., Interactivity of chiral catalysts and chiral
auxiliaries in the cycloaddition of activated dienes with aldehydes: a synthesis of L-
glucose. Journal of the American Chemical Society 1986, 108, 7060-7067.
22. Sarotti, A. M.; Spanevello, R. A.; Suliez, A. G., Second generation
levoglucosenone-derived chiral auxiliaries. Scope and application in asymmetric Diels-
Alder reactions. Tetrahedron 2009, 65, 3502-3508.
23. Jorgensen, K. A., Catalytic asymmetric hetero-Diels-Alder reactions of carbonyl
compounds and imines. Angewandte Chemie-International Edition 2000, 39, 3558-3588.
24. Douglas, C. J.; Overman, L. E., Catalytic asymmetric synthesis of all-carbon
quaternary stereocenters. Proc Natl Acad Sci U S A 2004, 101, 5363-7.
25. Furuta, K.; Shimizu, S.; Miwa, Y.; Yamamoto, H., Chiral (acyloxy)borane (CAB):
a powerful and practical catalyst for asymmetric Diels-Alder reactions. The Journal of
Organic Chemistry 2002, 54, 1481-1483.

132
26. Dieter, K.; Roland, B., Eine Borat-Propellerverbindung als chiraler Katalysator
einer asymmetrisch induzierten Diels-Alder-Reaktion. Angewandte Chemie 1990, 102,
568-569.
27. Ishihara, K.; Kurihara, H.; Matsumoto, M.; Yamamoto, H., Design of Bronsted
Acid-Assisted Chiral Lewis Acid (BLA) Catalysts for Highly Enantioselective Diels-
Alder Reactions. Journal of the American Chemical Society 1998, 120, 6920-6930.
28. Corey, E. J.; Sarshar, S.; Bordner, J., X-ray crystallographic and NMR studies on
the origins of high enantioselectivity in Diels-Alder reactions catalyzed by a chiral
diazaaluminolidine. Journal of the American Chemical Society 2002, 114, 7938-7939.
29. Evans, D. A.; Lectka, T.; Miller, S. J., Bis(imine)-copper(II) complexes as chiral
lewis acid catalysts for the Diels-Alder reaction. Tetrahedron Letters 1993, 34, 7027-
7030.
30. Corey, E. J.; Ishihara, K., Highly enantioselective catalytic Diels-Alder addition
promoted by a chiral bis(oxazoline)-magnesium complex. Tetrahedron Letters 1992, 33,
6807-6810.
31. Maruoka, K.; Murase, N.; Yamamoto, H., Chiral helical Lewis acids for
asymmetric Diels-Alder catalysts. The Journal of Organic Chemistry 2002, 58, 2938-
2939.
32. Narasaka, K.; Iwasawa, N.; Inoue, M.; Yamada, T.; Nakashima, M.; Sugimori, J.,
Asymmetric Diels-Alder reaction catalyzed by a chiral titanium reagent. Journal of the
American Chemical Society 2002, 111, 5340-5345.

133
33. Corey, E. J., Catalytic enantioselective Diels-Alder reactions: Methods,
mechanistic fundamentals, pathways, and applications. Angewandte Chemie-
International Edition 2002, 41, 1650-1667.
34. Wang, Y.; Li, H.; Wang, Y.-Q.; Liu, Y.; Foxman, B. M.; Deng, L., Asymmetric
Diels−Alder Reactions of 2-Pyrones with a Bifunctional Organic Catalyst. Journal of the
American Chemical Society 2007, 129, 6364-6365.
35. Ma, Y.; Zhang, Y. J.; Jin, S.; Li, Q.; Li, C.; Lee, J.; Zhang, W., C2-Symmetric
bipyrrolidines as organocatalysts for asymmetric Diels–Alder reactions. Tetrahedron
Letters 2009, 50, 7388-7391.
36. Fukuzawa, S.-i.; Yahara, Y.; Kamiyama, A.; Hara, M.; Kikuchi, S.,
Stereoselective Pinacol Coupling of Chiral Formylferrocene Using Divalent Samarium
Triflate: Preparation of a New Chiral Bisferrocenyl Oxazoline Ligand and Its Application
to Asymmetric Diels-Alder Reactions. Organic Letters 2005, 7, 5809-5812.
37. Brown, H. C.; Rangaishenvi, M. V., A simple procedure for the synthesis of
three-carbon homologated boronate esters and terminal alkenes via nucleopbilic
displacement in [alpha]-haloallylboronate ester. Tetrahedron Letters 1990, 31, 7115-7118.
38. Ramachandran, P.; Brown, H., Organometallics in Synthesis. Pergamon Press,
Oxford, NY 1994, 35-36.
39. Schlosser, M.; L.Lipshutz; H, N., Keith Smith, Organometallics in synthesis : a
manual In Wiley, New York: 2002; pp 465-505.
40. Komiya, S., Synthesis of Organometallic Compounds: A Practical Guide Press,
Oxford University, NY 1996, 345-362.

134
41. Roush, W. R. A., Kaori; Powers, Daniel B.; Palkowitz, Alan D.; Halterman,
Ronald L, Asymmetric synthesis using diisopropyl tartrate modified (E)- and (Z)-
crotylboronates: preparation of the chiral crotylboronates and reactions with achiral
aldehydes. . Journal of the American Chemical Society 1990, 112, 6339-48.
42. Miyaura, N.; Suzuki, A., Palladium-Catalyzed Cross-Coupling Reactions of
Organoboron Compounds. Chemical Reviews 1995, 95, 2457-2483.
43. Herbert C. Brown, N. G. B., and vishwanatha Somayaji, Convenient Procedures
for the Synthesis of Alkyl- And Alkenyl Boronic Acids and Esters. Organometallics
1983, 2, 1311-1316.
44. Pickin, K. A.; Kindy, J. M.; Day, C. S.; Welker, M. E., Simple preparation of
cobaloxime dienyl complexes and their exo selective Diels-Alder cycloadducts: Progress
toward transition metal-mediated Diels-Alder reactions which are catalytic in metal
dienyl complex. Journal of Organometallic Chemistry 2003, 681, 120-133.
45. Welker, M. E., Organocobalt complexes in organic synthesis Current Organic
Chemistry 2001, 5, 785-807.
46. Pidaparthi, R. R.; Welker, M. E.; Day, C. S., [6 + 4] and [4 + 2] Cycloaddition
Reactions of Cobaloxime 1,3-Dienyl Complexes and Tropones. Organometallics 2006,
25, 974-981.
47. Graham, P. M.; Delafuente, D. A.; Liu, W.; Myers, W. H.; Sabat, M.; Harman, W.
D., Facile Diels-Alder Reactions with Pyridines Promoted by Tungsten. Journal of the
American Chemical Society 2005, 127, 10568-10572.

135
48. Liu, W.; You, F.; Mocella, C. J.; Harman, W. D., A New Approach to Promoting
Sluggish Diels Alder Reactions:Dihapto-Coordination of the Diene. Journal of the
American Chemical Society 2006, 128, 1426-1427.
49. De, S.; Welker, M. E., Preparation of 2-BF3-Substituted 1,3-Dienes and Their
Diels Alder/Cross-Coupling Reactions. Organic Letters 2005, 7, 2481-2484.
50. Pidaparthi, R. R.; Welker, M. E.; Day, C. S.; Wright, M. W., Preparation of 2-
Trialkylsiloxy- Substituted 1,3-Dienes and Their Diels- Alder / Cross-Coupling Reactions.
Organic Letters 2007, 9, 1623-1626.
51. Pidaparthi, R. R.; Welker, M. E.; Day, C. S.; Wright, M. W., Preparation of 2-
Trialkylsiloxy- Substituted 1,3-Dienes and Their Diels−Alder/Cross-Coupling
Reactions. Organic Letters 2007, 9, 1623-1626.
52. Pidaparthi, R. R.; Welker, M. E., Preparation of siloxacyclopentene containing
1,3-dienes and their Diels-Alder reactions. Tetrahedron Letters 2007, 48, 7853-7856.
53. Welker, M. E., Recent advances in syntheses and reaction chemistry of boron and
silicon substituted 1,3-dienes. Tetrahedron 2008, 64, 11529-11539.
54. Garnier, L.; Plunian, B.; Mortier, J.; Vaultier, M., Diels-Alder reactions of 1,3-
dienylborate salts with activated dienophiles. Tetrahedron Letters 1996, 37, 6699-6700.
55. Renaud, J.; Oberer, C.-D. G. L., Ruthenium-Catalyzed Enyne Metathesis of
Acetylenic Boronates: A Concise Route for the Construction of Cyclic 1,3-Dienylboronic
Esters. Angewandte Chemie 2000, 39, 3101-3104.
56. Coleman, R. S.; Walczak, M. C., Tandem Stille/Suzuki Miyaura Coupling of a
Hetero-Bis-metalated Diene. Rapid, One-Pot Assembly of Polyene Systems. Organic
Letters 2005, 7, 2289-2291.

136
57. Lallemand, J.-Y.; Ricard, Y. S. L., A Concise Synthesis of an Advanced Clerodin
Intermediate through a Vaultier Tandem Reaction. European Journal of Organic
Chemistry 2002, 2002, 503-513.
58. Ishiyama, T.; Miyaura, N., Chemistry of Group 13 element-transition metal
linkage -- the platinum- and palladium-catalyzed reactions of (alkoxo)diborons. Journal
of Organometallic Chemistry 2000, 611, 392-402.
59. Babudri, F.; Farinola, G. M.; Fiandanese, V.; Mazzone, L.; Naso, F., A
straightforward route to polyenylsilanes by palladium- or nickel-catalyzed cross-coupling
reactions. Tetrahedron 1998, 54, 1085-1094.
60. Trost, B. M., Comprehensive Organic Synthesis. Elseiver, Oxford, UK, 1991; Vol.
5, p 336.
61. Barrett, A. G. M.; Bennett, A. J.; Menzer, S.; Smith, M. L.; White, A. J. P.;
Williams, D. J., Applications of Crotonyldiisopinocampheylboranes in Synthesis:
Total Synthesis of Restrictinol. The Journal of Organic Chemistry 1999, 64, 162-171.
62. Balma Tivola, P.; Deagostino, A.; Prandi, C.; Venturello, P., A New Synthesis of
Butadienyl- and Styrylboronic Esters: Highly Reactive Intermediates for Suzuki
Cross-Coupling. Organic Letters 2002, 4, 1275-1277.
63. Singidi, R. R.; RajanBabu, T. V., Borostannylation of Alkynes and Enynes. Scope
and Limitations of the Reaction and Utility of the Adducts. Organic Letters 2010, 12,
2622-2625.
64. Carreaux, F.; Possémé, F.; Carboni, B.; Arrieta, A.; Lecea, B.; Cossío, F. P., [4+3]
versus [4+2] Mechanisms in the Dimerization of 2-Boryl-1,3-butadienes. A Theoretical
and Experimental Study. The Journal of Organic Chemistry 2002, 67, 9153-9161.

137
65. Wrackmeyer, B.; Tok, O.; Klimkina, E.; Bubnov, Y. N., Reactivity of mono-1-
alkynyltin and -germanium compounds towards triallylborane. Inorganica Chimica Acta
2000, 300-302, 169-174.
66. De, S. D., Cynthia S.; Welker, Mark E.. , An unusual oligomerization/oxidation
reaction of a 3-boron-substituted 1-phenylbuta-1,3-diene produces 6,9,16,19-tetraphenyl-
5,15-distyryl-3,13,25,26-tetraoxa-2,12-
diborapentacyclo[16.2.2.28,11.12,5.112,15]hexacosa-1(20),7,10,17-tetraene Acta
Crystallographica, Section C: Crystal Structure Communications 2007, C63, o729-o730.
67. Geny, A.; Leboeliguf, D.; Rouquié, G.; Vollhardt, K. Peter C.; Malacria, M.;
Gandon, V.; Aubert, C., Cobalt(I)-Mediated Preparation of Polyborylated
Cyclohexadienes: Scope, Limitations, and Mechanistic Insight. Chemistry - A European
Journal 2007, 13, 5408-5425.
68. Morita, R.; Shirakawa, E.; Tsuchimoto, T.; Kawakami, Y., Synthesis of
multisubstituted 1,3-butadienes using the ruthenium-catalyzed double addition of
trimethylsilyldiazomethane to alkynylboronates Organic & Biomolecular Chemistry
2005, 3, 1263.
69. Kim, M.; Lee, D., Boron-Directed Regio- and Stereoselective Enyne Cross
Metathesis: Efficient Synthesis of Vinyl Boronate Containing 1,3-Dienes.
Organic Letters 2005, 7, 1865-1868.
70. Gonzaliez, J. R.; Gonzaliez, A. Z.; Soderquist, J. A., (E)-2-Boryl-1,3-butadiene
Derivatives of the 10-TMS-9-BBDs: Highly Selective Reagents for the Asymmetric
Synthesis of anti-1,2-Disubstituted 3,4-Pentadien-1-ols. Journal of the American
Chemical Society 2009, 131, 9924-9925.

138
71. Sherry, R. C.; Dirk, T.; Samuel, J. D., The B-Alkyl Suzuki-Miyaura Cross-
Coupling Reaction: Development, Mechanistic Study, and Applications in Natural
Product Synthesis13. Angewandte Chemie International Edition 2001, 40, 4544-4568.
72. Martin, R.; Buchwald, S. L., Palladium-Catalyzed Suzuki Miyaura Cross-
Coupling Reactions Employing Dialkylbiaryl Phosphine Ligands. Accounts of Chemical
Research 2008, 41, 1461-1473.
73. Suzuki, A., Recent advances in the cross-coupling reactions of organoboron
derivatives with organic electrophiles, 1995-1998. Journal of Organometallic Chemistry
1999, 576, 147-168.
74. Dennis, J. M. S.; Koten, G. v.; Robertus, J. M. K. G., Hexacationic Dendriphos
Ligands in the Pd-Catalyzed Suzuki-Miyaura Cross-Coupling Reaction: Scope and
Mechanistic Studies. Journal of the American Chemical Society 2009, 131, 11407-11416.
75. Erwan, G.; Shailesh, R.; John, M. B.; Andrew, C.; King Kuok, H.; Anny, J.,
Profound Steric Control of Reactivity in Aryl Halide Addition to Bisphosphane
Palladium(0) Complexes13. Angewandte Chemie International Edition 2002, 41, 1760-
1763.
76. Braga, A. A. C.; Morgon, N. H.; Ujaque, G.; Maseras, F., Computational
Characterization of the Role of the Base in the Suzuki Miyaura Cross-Coupling Reaction.
Journal of the American Chemical Society 2005, 127, 9298-9307.
77. Miyaura, N., Cross-coupling reaction of organoboron compounds via base-
assisted transmetalation to palladium(II) complexes. Journal of Organometallic
Chemistry 2002, 653, 54-57.

139
78. Ute, C.; Ramón, V., Monoligated Palladium Species as Catalysts in Cross-
Coupling Reactions. Angewandte Chemie International Edition 2005, 44, 366-374.
79. Ozawa, F. K., Kunihiko; Fujimori, Mizue; Hidaka, Takahiro; Toyoshima, Tsukasa;
Yamamoto, Akio. , Mechanism of the cross-coupling reaction of phenyl iodide and
methylmagnesium iodide catalyzed by trans-PdPh(I)(PEt2Ph)2 Organometallics 1989, 8,
180-8.
80. Takaya, Y.; Ogasawara, M.; Hayashi, T.; Sakai, M.; Miyaura, N., Rhodium-
Catalyzed Asymmetric 1,4-Addition of Aryl- and Alkenylboronic Acids to Enones. J. Am.
Chem. Soc. 1998, 120, 5579-5580.
81. Sakai, M.; Hayashi, H.; Miyaura, N., Rhodium-Catalyzed Conjugate Addition of
Aryl- or 1-Alkenylboronic Acids to Enones. Organometallics 1997, 16, 4229-4231.
82. Duan, W. L.; Iwamura, H.; Shintani, R.; Hayashi, T., Chiral Phosphine-Olefin
Ligands in the Rhodium-Catalyzed Asymmetric 1,4-Addition Reactions. J. Am. Chem.
Soc. 2007, 129, 2130-2138.
83. Hayashi, T.; Yamasaki, K., Rhodium-Catalyzed Asymmetric 1,4-Addition and Its
Related Asymmetric Reactions. Chem. Rev. 2003, 103, 2829-2844.
84. Shintani, R.; Duan, W. L.; Hayashi, T., Rhodium-Catalyzed Asymmetric
Construction of Quaternary Carbon Stereocenters: Ligand-Dependent Regiocontrol in the
1,4-Addition to Substituted Maleimides. J. Am. Chem. Soc. 2006, 128, 5628-5629.
85. Mediavilla Urbaneja, L.; Krause, N., Rhodium-catalyzed enantioselective 1,4-
additions of arylboronic acids to substituted enones. Tetrahedron: Asymmetry 2006, 17,
494-496.

140
86. Hayashi, T.; Takahashi, M.; Takaya, Y.; Ogasawara, M., Catalytic Cycle of
Rhodium-Catalyzed Asymmetric 1,4-Addition of Organoboronic Acids. Arylrhodium,
Oxa--allylrhodium, and Hydroxorhodium Intermediates. J. Am. Chem. Soc. 2002, 124,
5052-5058.
87. Fagnou, K.; Lautens, M., Rhodium-Catalyzed Carbon-Carbon Bond Forming
Reactions of Organometallic Compounds. Chem. Rev. 2003, 103, 169-196.
88. Ogasawara, M.; Yoshida, K.; Hayashi, T., 2,2'-Bis(diphenylphosphino)-1,1'-
biphenyl: New Entry of Bidentate Triarylphosphine Ligand to Transition Metal Catalysts.
Organometallics 2000, 19, 1567-1571.
89. Kasak, P.; Arion, V. B.; Widhalm, M., A chiral phosphepine-olefin rhodium
complex as an efficient catalyst for the asymmetric conjugate addition. Tetrahedron:
Asymmetry 2006, 17, 3084-3090.
90. Chen, G.; Tokunaga, N.; Hayashi, T., Rhodium-Catalyzed Asymmetric 1,4-
Addition of Arylboronic Acids to Coumarins: Asymmetric Synthesis of (<i>R</i>)-
Tolterodine. Org. Lett. 2005, 7, 2285-2288.
91. Duursma, A.; Boiteau, J.-G.; Lefort, L.; Boogers, J. A. F.; de Vries, A. H. M.; de
Vries, J. G.; Minnaard, A. J.; Feringa, B. L., Highly Enantioselective Conjugate
Additions of Potassium Organotrifluoroborates to Enones by Use of Monodentate
Phosphoramidite Ligands. The Journal of Organic Chemistry 2004, 69, 8045-8052.
92. Källström, S.; Jagt, R.; Sillanpää, R.; Feringa, B. L.; Minnaard, A. J.; Leino, R.,
Highly Enantio- and Diastereoselective One-Pot Reactions in Aqueous Media: Combined
Asymmetric Rh-Catalyzed Conjugate Addition/Metal-Mediated Allylation. European
Journal of Organic Chemistry 2006, 2006, 3826-3833.

141
93. Defieber, C.; Paquin, J.-F.; Serna, S.; Carreira, E. M., Chiral [2.2.2] Dienes as
Ligands for Rh(I) in Conjugate Additions of Boronic Acids to a Wide Range of
Acceptors. Organic Letters 2004, 6, 3873-3876.
94. Norihito, T.; Tamio, H., Asymmetric 1,4-Addition of Organoboron Reagents to
Quinone Monoketals Catalyzed by a Chiral Diene/Rhodium Complex: A New Synthetic
Route to Enantioenriched 2-Aryltetralones. Advanced Synthesis & Catalysis 2007, 349,
513-516.
95. de la Herrán, G.; Murcia, C.; Csákÿ, A. G., Rhodium -Catalyzed Reaction of Aryl-
and Alkenylboronic Acids with 2,4-Dienoate Esters: Conjugate Addition and Heck
Reaction Products†. Organic Letters 2005, 7, 5629-5632.
96. Chochois, H.; Sauthier, M.; Maerten, E.; Castanet, Y.; Mortreux, A., 1,4-
Carbonylative addition of arylboronic acids to methyl vinyl ketone: a new synthetic tool
for rapid furan and pyrrole synthesis. Tetrahedron 2006, 62, 11740-11746.
97. Shintani, R.; Okamoto, K.; Hayashi, T., Carbon-Carbon Bond-Forming
Enantioselective Synthesis of Chiral Organosilicon Compounds by Rhodium/Chiral
Diene-Catalyzed Asymmetric 1,4-Addition Reaction. Org. Lett. 2005, 7, 4757-4759.
98. Paquin, J. F.; Defieber, C.; Stephenson, C. R. J.; Carreira, E. M., Asymmetric
Synthesis of 3,3-Diarylpropanals with Chiral Diene-Rhodium Catalysts. J. Am. Chem.
Soc. 2005, 127, 10850-10851.
99. Genin, E.; Michelet, V.; Genet, J.-P., Rh-catalyzed addition of boronic acids to
alkynes for the synthesis of trisubstituted alkenes in a biphasic system - Mechanistic
study and recycling of the Rh/m-TPPTC catalyst. Journal of Organometallic Chemistry
2004, 689, 3820-3830.

142
100. Uozumi, Y.; Nakazono, M., Amphiphilic Resin-Supported Rhodium-Phosphine
Catalysts for C-C Bond Forming Reactions in Water. Advanced Synthesis & Catalysis
2002, 344, 274-277.
101. Sakuma, S.; Miyaura, N., Rhodium(I)-Catalyzed Asymmetric 1,4-Addition of
Arylboronic Acids to Unsaturated Amides. J. Org. Chem. 2001, 66, 8944-8946.
102. Ueda, M.; Miyaura, N., A Large Accelerating Effect of Tri(tert-butyl)phosphine
in the Rhodium-Catalyzed Addition of Arylboronic Acids to Aldehydes. J. Org. Chem.
2000, 65, 4450-4452.
103. Duan, H. F.; Xie, J. H.; Shi, W. J.; Zhang, Q.; Zhou, Q. L., Enantioselective
Rhodium-Catalyzed Addition of Arylboronic Acids to Aldehydes Using Chiral Spiro
Monophosphite Ligands. Org. Lett. 2006, 8, 1479-1481.
104. Hayes, B. L.; Adams, T. A.; Pickin, K. A.; Day, C. S.; Welker, M. E., Preparation
of Cobaloxime-Substituted Unsaturated Carbonyl Compounds and Their Subsequent
Conversion into 1-Cobaloxime-Substituted 1,3-Dienyl Complexes. Organometallics 2000,
19, 2730-2740.
105. Kamabuchi, A.; Miyaura, N.; Suzuki, A., Synthesis and cycloaddition of 2-
(dialkoxyboryl)-1,3-butadiene. Tetrahedron Letters 1993, 34, 4827-4828.
106. De, S., Preparation and Tandem Reactions of 2-Trifluoroborate Substituted
Dienes. Thesis 2006, 59.
107. Diaz, M.; Jaballas, J.; Tran, D.; Lee, H.; Arias, J.; Onak, T., Interaction of
Trimethylamine and closo-1,6-C2B7H9. Evidence for an “Open” Cage C2B7H9/Amine
Adduct. Inorganic Chemistry 1996, 35, 4536-4540.

143
108. Robertson, A. P. M.; Haddow, M. F.; Manners, I., Synthesis and the Thermal and
Catalytic Dehydrogenation Reactions of Amine-Thioboranes. Inorganic Chemistry 2012.
109. Hermanek, S., Boron-11 NMR spectra of boranes, main-group heteroboranes, and
substituted derivatives. Factors influencing chemical shifts of skeletal atoms. Chemical
Reviews 1992, 92, 325-362.
110. Poly, W.; Schomburg, D.; Hoffmann, H. M. R., Stereoselective generation and
facile dimerization of (E)-2-methylene-3-alkenoic acid esters. The Journal of Organic
Chemistry 1988, 53, 3701-3710.
111. Spino, C.; Crawford, J.; Cui, Y.; Gugelchuk, M., The dimerisation of 2-
methoxycarbonylbuta-1,3-diene: the importance of paralocalisation energy in assessing
diene reactivity. J. Chem. Soc., Perkin Trans. 2 1998, 1499-1507.
112. Akula, M. R.; Yao, M.-L.; Kabalka, G. W., Triolborates: water-soluble complexes
of arylboronic acids as precursors to iodoarenes. Tetrahedron Letters 2010, 51, 1170-
1171.
113. Yamamoto, Y., Cyclic Triolborates: Air- and Water-Stable Ate Complexes of
Organoboronic Acids. Angew. Chem. It. Ed. 2008, 47, 928-931.
114. Kelvin L. Billingsley, Timothy E. B. Stephen L. B., Palladium-Catalyzed
Borylation of Aryl Chlorides: Scope, Applications, and Computational Studies13.
Angewandte Chemie International Edition 2007, 46, 5359-5363.
115. Morrill, C.; Funk, T. W.; Grubbs, R. H., Synthesis of tri-substituted vinyl
boronates via ruthenium-catalyzed olefin cross-metathesis. Tetrahedron Letters 2004, 45,
7733-7736.

144
116. Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H., A General Model
for Selectivity in Olefin Cross Metathesis. Journal of the American Chemical Society
2003, 125, 11360-11370.
117. Harmata, M.; Lee, D. R.; Barnes, C. L., Stereospecific Synthesis of Dienones via
a Torquoselective Retro-Nazarov Reaction. Organic Letters 2005, 7, 1881-1883.
118. Hong, S. H.; Grubbs, R. H., Highly Active Water-Soluble Olefin Metathesis
Catalyst. Journal of the American Chemical Society 2006, 128, 3508-3509.
119. Ritter, T.; Day, M. W.; Grubbs, R. H., Rate Acceleration in Olefin Metathesis
through a Fluorine Ruthenium Interaction. Journal of the American Chemical Society
2006, 128, 11768-11769.
120. Blackwell, H. E.; O'Leary, D. J.; Chatterjee, A. K.; Washenfelder, R. A.;
Bussmann, D. A.; Grubbs, R. H., New Approaches to Olefin Cross-Metathesis. Journal
of the American Chemical Society 2000, 122, 58-71.
121. Darses, S. M., Guillaume; Genet, Jean-Pierre., Potassium organotrifluoroborates.
New partners in palladium-catalyzed cross-coupling reactions. . European Journal of
Organic Chemistry 1998, 8, 1875-1883.
122. De, S.; Welker, M. E., Preparation of 2-BF3-substituted 1,3-dienes and their
Diels-Alder/cross-coupling reactions. Org Lett 2005, 7, 2481-2484.
123. Wang, L.; Day, C. S.; Wright, M. W.; Welker, M. E., Preparation and Diels-
Alder/Cross Coupling Reactions of a 2-Diethanolaminoboron-Substituted 1,3-Diene.
Beilstein Journal of Organic Chemistry 2009, 5, 45.
124. Wright, M. W.; Smalley, T. L.; Welker, M. E.; Rheingold, A. L., Synthesis of
Cobalt-Substituted 1,3-Diene Complexes with Unusual Structures and Their Exo-

145
Selective Diels-Alder Reactions. Journal of the American Chemical Society 1994, 116,
6777-6791.
125. Pidaparthi, R. R.; Welker, M. E.; Day, C. S.; Wright, M. W., Preparation of 2-
trialkylsiloxy-substituted 1,3-dienes and their Diels-Alder/cross-coupling reactions. Org
Lett 2007, 9, 1623-1626.
126. Berger, S.; Braun, S., 200 and More NMR Experiments. . Wiley-VCH, Weinheim
1998, 318-320.
127. Silverstein, R. M.; Webster, F. X.; Kiemle, D. J., Spectrometric Identification of
Organic Compounds,Seventh edition. John Wilery & Sons, Hoboken, NJ: 2005; p 213-
214.
128. Darses, S.; Michaud, G.; Genêt, J.-P., Potassium Organotrifluoroborates: New
Partners in Palladium-Catalysed Cross-Coupling Reactions. European Journal of
Organic Chemistry 1999, 1999, 1875-1883.
129. Lebel, H.; Ladjel, C.; Bréthous, L., Palladium-Catalyzed Cross-Coupling
Reactions in One-Pot Multicatalytic Processes. Journal of the American Chemical
Society 2007, 129, 13321-13326.
130. Hussain, M. M.; Walsh, P. J., Tandem Reactions for Streamlining Synthesis:
Enantio- and Diastereoselective One-Pot Generation of Functionalized Epoxy Alcohols.
Accounts of Chemical Research 2008, 41, 883-893.
131. De, S., Preparation and Tandem Reactions of 2-Trifluoroborate Substituted
Dienes. Ph.D Dissertation of Wake Forest University Graduate School of Arts &
Sciences 2007.

146
132. Gossinger, E.; Schwartz, A.; Sereinig, N., Synthesis of the decalin subunit of
coloradocin. Tetrahedron 2000, 56, 2007-2014.
133. Gebbinck, E. A. K.; Jansen, B. J. M.; de Groot, A., Insect antifeedant activity of
clerodane diterpenes and related model compounds. Phytochemistry 2002, 61, 737-770.
134. Hanson, J. R., Steroids: reactions and partial synthesis. Natural Product Reports
2003, 20, 318-326.
135. Hayashi, T., Rhodium-catalyzed asymmetric 1,4-addition of organoboronic acids
and their derivatives to electron deficient olefins. Synlett 2001, 879-887.
136. Navarre, L.; Darses, S.; Genet, J. P., Tandem 1,4-addition/enantioselective
protonation catalyzed by rhodium complexes: Efficient access to alpha-amino acids.
Angewandte Chemie-International Edition 2004, 43, 719-723.
137. Hayashi, T., Rhodium-catalyzed asymmetric addition of aryl- and alkenylboron
reagents to electron-deficient olefins. Pure and Applied Chemistry 2004, 76, 465-475.
138. Welker, M. E., Organocobalt complexes in organic synthesis. Current Organic
Chemistry 2001, 5, 785-807.
139. De, S.; Solano, J. M.; Wang, L.; Welker., M. E., Rhodium catalyzed tandem
Diels-Alder/hydrolysis reactions of 2-boron-substituted 1,3-dienes. Journal of
Organometallic Chemistry 2009, 694, 2295-2298.

147
APPENDIX A Crystal Structure Data of Diene 2.4
Structure a22n / C8H14NO2B – CHCl3
X-Ray Crystallographic Comments
X-ray crystallographic data has been deposited with the CCDC and allocated dposition
number CCDC 736603. The asymmetric unit in crystalline C8H14NO2B – CHCl3
contains one C8H14NO2B molecule and one chloroform molecule of crystallization. All
displacement ellipsoids are drawn at the 50% probability level.
Brief Experimental Description to be Included as Text or as a Footnote at Time of
Publication
Colorless plate-shaped crystals of C8H14NO2B – CHCl3 are, at 203(2) K,
orthorhombic, space group Pna21 – C9
2v (No. 33) (1) with a = 19.587(3) Å, b = 7.742(1)
Å, c = 9.104(1) Å, V = 1380.6(3) Å3 and Z = 4 formula units {dcalcd = 1.378 g/cm
3;
a(MoK ) = 0.648 mm-1
}. A full hemisphere of diffracted intensities (1968 30-second
frames with a scan width of 0.30) was measured for a single domain specimen using
graphite-monochromated MoK radiation (= 0.71073 Å) on a Bruker SMART APEX
CCD Single Crystal Diffraction System (2). X-rays were provided by a fine-focus sealed
x-ray tube operated at 50kV and 30mA.
Lattice constants were determined with the Bruker SAINT software package
using peak centers for 1895 reflections having 8.07°≤ 2 ≤ 40.42°. A total of 10091

148
integrated absorption-corrected reflection intensities having 2((MoK )< 49.98 were
produced using the Bruker program SAINT(3); 2411 of these were unique and gave Rint =
0.040 with a coverage which was 99.5% complete. The Bruker software package
SHELXTL was used to solve the structure using “direct methods” techniques. All stages
of weighted full-matrix least-squares refinement were conducted using Fo2 data with the
SHELXTL Version 6.12 software package(4).
The final structural model incorporated anisotropic thermal parameters for
all nonhydrogen atoms and isotropic thermal parameters for all hydrogen atoms.
All hydrogen atoms on the diene (H1A, H1B, H3, H4A and H4B) as well as the
amine hydrogen atom (H1N) were located in a difference Fourier and included in
the structural model as independent isotropic atoms whose parameters were
allowed to vary in least-squares refinement cycles. The remaining hydrogen atoms
were included in the structural model as fixed atoms (using idealized sp3-hybridized
geometry and C-H bond lengths of 0.98 – 0.99 Å) "riding" on their respective carbon
atoms. The isotropic thermal parameters for these hydrogen atoms were fixed at a
value 1.2 times the equivalent isotropic thermal parameter of the carbon atom to
which they are covalently bonded. A total of 169 parameters were refined using
one restraint and 2411 data. Final agreement factors at convergence are:
R1(unweighted, based on F) = 0.045 for 2083 independent “observed” reflections
having 2(MoK )< 49.98 and I>2(I); R1(unweighted, based on F) = 0.053
and wR2(weighted, based on F2) = 0.096 for all 2411 independent reflections
having 2(MoK )< 49.98. The largest shift/s.u. was 0.000 in the final
refinement cycle. The final difference map had maxima and minima of 0.258 and

149
-0.167 e-/Å
3, respectively. The absolute structure was determined by refinement
of the Flack parameter x (5) ; x refined to a final value of 0.03(8) .
Acknowledgment
The authors thank the National Science Foundation (grant CHE-0234489) for
funds to purchase the x-ray instrument and computers.
References
(1) International Tables for Crystallography, Vol A, 4th
ed., Kluwer Academic
Publishers: Boston (1996).
(2) Data Collection: SMART (Version 5.628) (2002). Bruker-AXS, 5465 E. Cheryl
Parkway, Madison, WI 53711-5373 USA.
(3) Data Reduction: SAINT (Version 6.45) (2003). Bruker-AXS, 5465 E. Cheryl
Parkway, Madison, WI 53711-5373, USA and Sheldrick, G. M. (2006). SADABS
(Version 2006/3). University of G öttingen, Germany..
(4) G. M. Sheldrick (2001). SHELXTL (Version 6.12). Bruker-AXS, 5465 E. Cheryl
Parkway, Madison, WI 53711-5373 USA.
(5) Flack, H. D. (1983). Acta Cryst. A39, 876-881.
Table 1. Crystal data and structure refinement for C8H14NO2B – CHCl3
Identification code a22nfinal
Empirical formula C9 H15 B Cl3 N O2

150
Formula weight 286.38
Temperature 203(2) K
Wavelength 0.71073 Å
Crystal system Orthorhombic
Space group Pna21 - C9
2v (No. 33)
Unit cell dimensions a = 19.587(3) Å
b = 7.742(1) Å
c = 9.104(1) Å
Volume 1380.6(3) Å3
Z 4
Density (calculated) 1.378 g/cm3
Absorption coefficient 0.648 mm-1
F(000) 592
Crystal size 0.30 x 0.09 x 0.04 mm3
Theta range for data collection 4.03 to 24.99°
Index ranges -23≤h≤23, -9≤k≤9, -10≤l≤10
Reflections collected 10091
Independent reflections 2411 [R(int) = 0.0396]
Completeness to theta = 24.99° 99.5 %
Absorption correction Multi-scan (SADABS)
Max. and min. transmission 0.8383 and 0.6862
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 2411 / 1 / 169

151
Goodness-of-fit on F2 1.105
Final R indices [I>2σ(I)] R1 = 0.0446, wR2 = 0.0924
R indices (all data) R1 = 0.0534, wR2 = 0.0961
Absolute structure parameter 0.03(8)
Largest diff. peak and hole 0.258 and -0.167 e-/Å3
------------------------------------------------------------------------------------------------------------
---------
R1 = ||Fo| - |Fc|| / |Fo|
Replace
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters
(Å2x 103) for C8H14NO2B – CHCl3. U(eq) is defined as one third of the trace of the
orthogonalized Uij tensor.
________________________________________________________________________
______
x y z U(eq)
________________________________________________________________________
______
Cl(1) 2882(1) 2158(2) 2449(1) 83(1)
Cl(2) 2277(1) 2273(2) 5318(2) 88(1)
Cl(3) 2845(1) -875(1) 4234(1) 79(1)
C(9) 2923(2) 1369(4) 4236(4) 46(1)

152
B(1) 4744(2) 3757(5) 6048(4) 33(1)
N(1) 4623(1) 5484(3) 7112(3) 33(1)
O(1) 5329(1) 4375(3) 5169(2) 39(1)
O(2) 4119(1) 3711(3) 5208(2) 38(1)
C(1) 5536(2) 1348(5) 6878(6) 64(1)
C(2) 4910(2) 2020(4) 6950(3) 38(1)
C(3) 4414(2) 1167(4) 7904(4) 50(1)
C(4) 3760(2) 1477(6) 8032(5) 60(1)
C(5) 5713(2) 5560(4) 6025(4) 40(1)
C(6) 5189(2) 6688(4) 6739(4) 41(1)
C(7) 3834(2) 5403(4) 5161(4) 44(1)
C(8) 3932(2) 6101(4) 6696(4) 44(1)
________________________________________________________________________
______
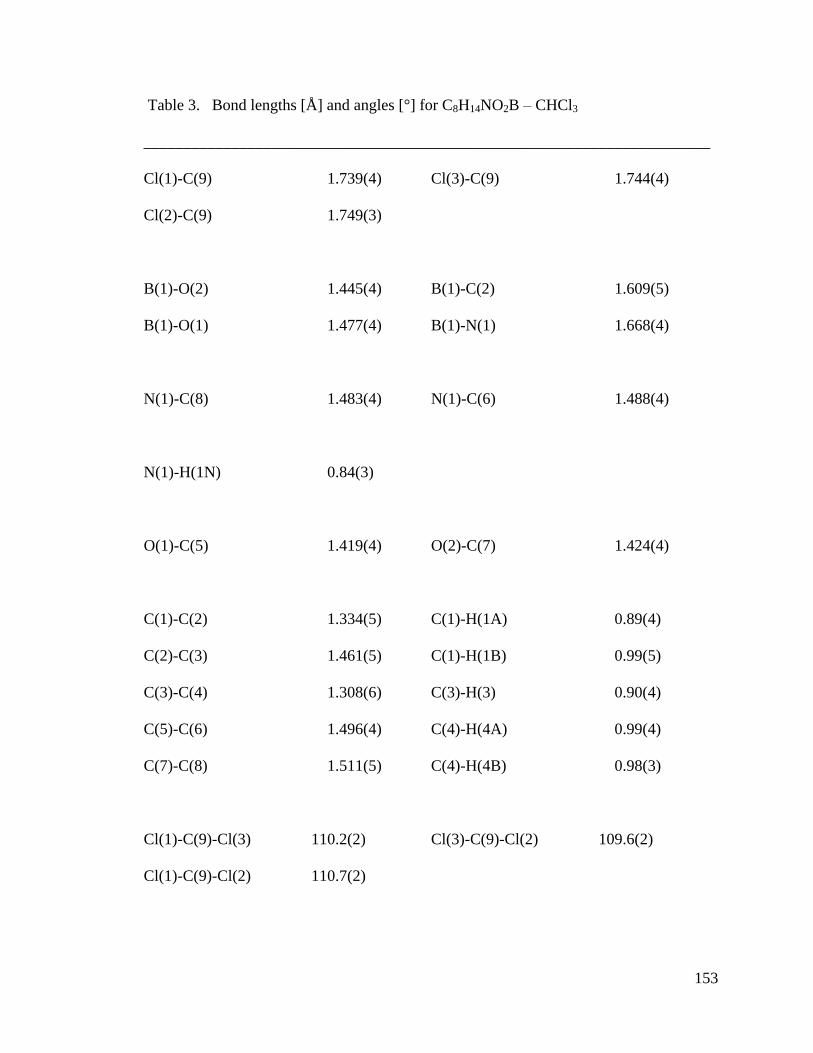
153
Table 3. Bond lengths [Å] and angles [°] for C8H14NO2B – CHCl3
_______________________________________________________________________
Cl(1)-C(9) 1.739(4)
Cl(2)-C(9) 1.749(3)
Cl(3)-C(9) 1.744(4)
B(1)-O(2) 1.445(4)
B(1)-O(1) 1.477(4)
B(1)-C(2) 1.609(5)
B(1)-N(1) 1.668(4)
N(1)-C(8) 1.483(4) N(1)-C(6) 1.488(4)
N(1)-H(1N) 0.84(3)
O(1)-C(5) 1.419(4) O(2)-C(7) 1.424(4)
C(1)-C(2) 1.334(5)
C(2)-C(3) 1.461(5)
C(3)-C(4) 1.308(6)
C(5)-C(6) 1.496(4)
C(7)-C(8) 1.511(5)
C(1)-H(1A) 0.89(4)
C(1)-H(1B) 0.99(5)
C(3)-H(3) 0.90(4)
C(4)-H(4A) 0.99(4)
C(4)-H(4B) 0.98(3)
Cl(1)-C(9)-Cl(3) 110.2(2)
Cl(1)-C(9)-Cl(2) 110.7(2)
Cl(3)-C(9)-Cl(2) 109.6(2)
154
O(2)-B(1)-O(1) 112.3(3)
O(2)-B(1)-C(2) 114.9(3)
O(1)-B(1)-C(2) 113.0(2)
O(2)-B(1)-N(1) 101.9(2)
O(1)-B(1)-N(1) 99.5(2)
C(2)-B(1)-N(1) 113.7(2)
C(8)-N(1)-C(6) 114.8(2)
C(8)-N(1)-B(1) 103.9(2)
C(6)-N(1)-B(1) 105.3(2)
C(8)-N(1)-H(1N) 109.4(19)
C(6)-N(1)-H(1N) 109.2(19)
B(1)-N(1)-H(1N) 114(2)
C(5)-O(1)-B(1) 108.8(2) C(7)-O(2)-B(1) 109.0(2)
C(2)-C(1)-H(1A) 118(3)
C(2)-C(1)-H(1B) 115(2)
H(1A)-C(1)-H(1B) 125(4)
C(1)-C(2)-C(3) 117.7(4)
C(1)-C(2)-B(1) 119.1(3)
C(3)-C(2)-B(1) 123.2(3)
C(4)-C(3)-C(2) 128.4(4)
C(4)-C(3)-H(3) 111(2)
C(2)-C(3)-H(3) 120(3)
C(3)-C(4)-H(4A) 119(2)
C(3)-C(4)-H(4B) 124(2)
H(4A)-C(4)-H(4B) 117(3)
O(1)-C(5)-C(6) 104.7(2)
N(1)-C(6)-C(5) 104.1(2)
O(2)-C(7)-C(8) 104.6(3)
N(1)-C(8)-C(7) 103.7(2)

155
________________________________________________________________________
____
Table 4. Anisotropic displacement parameters (Å2x 103) for C8H14NO2B – CHCl3.
The anisotropic displacement factor exponent takes the form: -2π2[ h2 a*2U11 + ... + 2
h k a* b* U12 ]
________________________________________________________________________
______
U11 U22 U33 U23 U13 U12
________________________________________________________________________
______
Cl(1) 68(1) 116(1) 65(1) 39(1) -13(1) -34(1)
Cl(2) 55(1) 103(1) 105(1) -23(1) 21(1) 8(1)
Cl(3) 96(1) 56(1) 83(1) 7(1) 3(1) -4(1)
C(9) 32(2) 60(2) 45(2) 1(2) -1(2) -6(2)
B(1) 34(2) 40(2) 24(2) -4(2) 1(1) -3(2)
N(1) 44(2) 33(1) 22(1) 1(1) 0(1) -3(1)
O(1) 38(1) 52(1) 27(1) -6(1) 3(1) -11(1)
O(2) 37(1) 42(1) 35(1) -7(1) -5(1) -5(1)
C(1) 56(2) 36(2) 99(4) -3(2) -18(3) 3(2)
C(2) 44(2) 31(2) 40(2) -9(1) -7(2) -5(1)
C(3) 78(3) 28(2) 43(2) 2(2) 1(2) -4(2)
C(4) 75(3) 50(2) 55(2) 0(2) 18(2) -16(2)

156
C(5) 45(2) 45(2) 31(2) 6(2) -2(1) -17(2)
C(6) 58(2) 34(2) 29(2) 5(1) -3(2) -13(2)
C(7) 38(2) 49(2) 45(2) 2(2) -10(2) 0(1)
C(8) 49(2) 39(2) 43(2) -1(2) 3(2) 9(2)
________________________________________________________________________
______

157
Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2x 10
3)
for C8H14NO2B – CHCl3
________________________________________________________________________
______
x y z U(eq)
________________________________________________________________________
______
H(9) 3371 1682 4663 55
H(1N) 4632(13) 5280(40) 8010(40) 26(8)
H(1A) 5620(20) 360(60) 7360(50) 67(13)
H(1B) 5830(20) 1820(60) 6080(50) 84(15)
H(3) 4543(19) 280(50) 8480(50) 63(12)
H(4A) 3490(20) 800(50) 8730(50) 76(13)
H(4B) 3523(16) 2410(40) 7500(40) 49(10)
H(5A) 6019 6244 5402 48
H(5B) 5986 4952 6764 48
H(6A) 5373 7234 7626 49
H(6B) 5033 7591 6063 49
H(7A) 3348 5360 4905 53
H(7B) 4073 6123 4440 53
H(8A) 3911 7366 6704 52

158
H(8B) 3585 5645 7367 52
________________________________________________________________________
______
159
Table 6. Torsion angles [°] for C8H14NO2B – CHCl3
________________________________________________________________
O(2)-B(1)-N(1)-C(8) 1.0(3)
O(1)-B(1)-N(1)-C(8) 116.4(2)
C(2)-B(1)-N(1)-C(8) -123.2(3)
O(2)-B(1)-N(1)-C(6) -120.0(2)
O(1)-B(1)-N(1)-C(6) -4.7(3)
C(2)-B(1)-N(1)-C(6) 115.7(3)
O(2)-B(1)-O(1)-C(5) 136.3(3)
C(2)-B(1)-O(1)-C(5) -91.8(3)
N(1)-B(1)-O(1)-C(5) 29.2(3)
O(1)-B(1)-O(2)-C(7) -81.2(3)
C(2)-B(1)-O(2)-C(7) 147.8(3)
N(1)-B(1)-O(2)-C(7) 24.4(3)
O(2)-B(1)-C(2)-C(1) 132.9(3)
O(1)-B(1)-C(2)-C(1) 2.3(4)
N(1)-B(1)-C(2)-C(1) -110.2(3)
O(2)-B(1)-C(2)-C(3) -49.7(4)
O(1)-B(1)-C(2)-C(3) 179.7(3)
N(1)-B(1)-C(2)-C(3) 67.2(4)
C(1)-C(2)-C(3)-C(4) -172.2(4)
B(1)-C(2)-C(3)-C(4) 10.4(5)
B(1)-O(1)-C(5)-C(6) -43.9(3)

160
C(8)-N(1)-C(6)-C(5) -133.0(3)
B(1)-N(1)-C(6)-C(5) -19.3(3)
O(1)-C(5)-C(6)-N(1) 38.3(3)
B(1)-O(2)-C(7)-C(8) -41.2(3)
C(6)-N(1)-C(8)-C(7) 90.6(3)
B(1)-N(1)-C(8)-C(7) -23.9(3)
O(2)-C(7)-C(8)-N(1) 40.0(3)
________________________________________________________________

161
Table 7. Hydrogen bonds for C8H14NO2B – CHCl3 [Å and °].
________________________________________________________________________
____
D-H...A d(D-H) d(H...A) d(D...A) <(DHA)
________________________________________________________________________
____
N(1)-H(1N)...O(1)#A 0.84(3) 1.98(3) 2.787(3) 161(3)
________________________________________________________________________
____
Symmetry transformations used to generate equivalent atoms:
#A -x+1,-y+1,z+1/2

162
Least-squares planes (x,y,z in crystal coordinates) and deviations from them
(* indicates atom used to define plane)
Plane 1
5.0842 (0.0326) x + 4.2998 (0.0122) y + 7.1928 (0.0087) z = 8.3678 (0.0147)
* -0.0260 (0.0017) C1
* -0.0035 (0.0027) C2
* 0.0632 (0.0029) C3
* -0.0437 (0.0020) C4
* 0.0101 (0.0009) B1
-0.0590 (0.0048) O1
-0.9320 (0.0049) O2
1.4560 (0.0051) N1
Rms deviation of fitted atoms = 0.0366
Plane 2
5.7480 (0.0488) x + 4.3356 (0.0097) y + 7.0537 (0.0071) z = 8.6119 (0.0210)
Angle to previous plane (with approximate esd) = 2.15 ( 0.24 )

163
* -0.0056 (0.0010) O1
* 0.0102 (0.0019) B1
* -0.0112 (0.0021) C2
* 0.0065 (0.0012) C1
0.0063 (0.0055) C3
-0.1446 (0.0079) C4
Rms deviation of fitted atoms = 0.0087
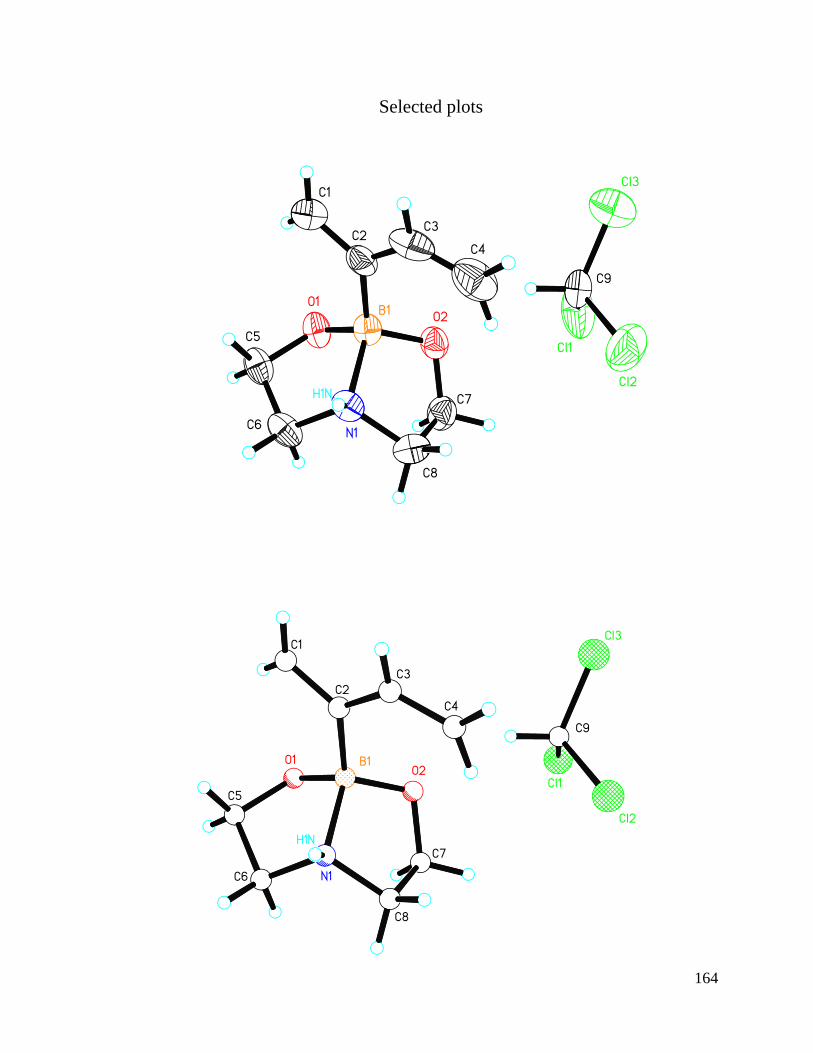
164
Selected plots
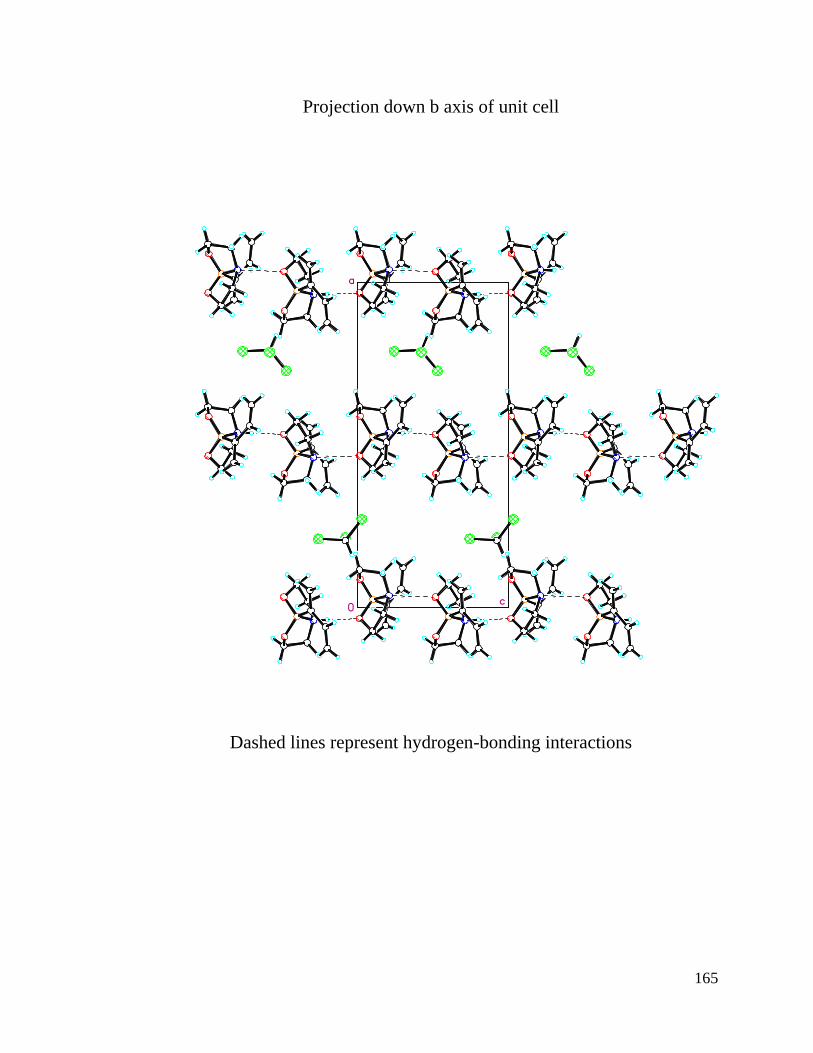
165
Projection down b axis of unit cell
Dashed lines represent hydrogen-bonding interactions
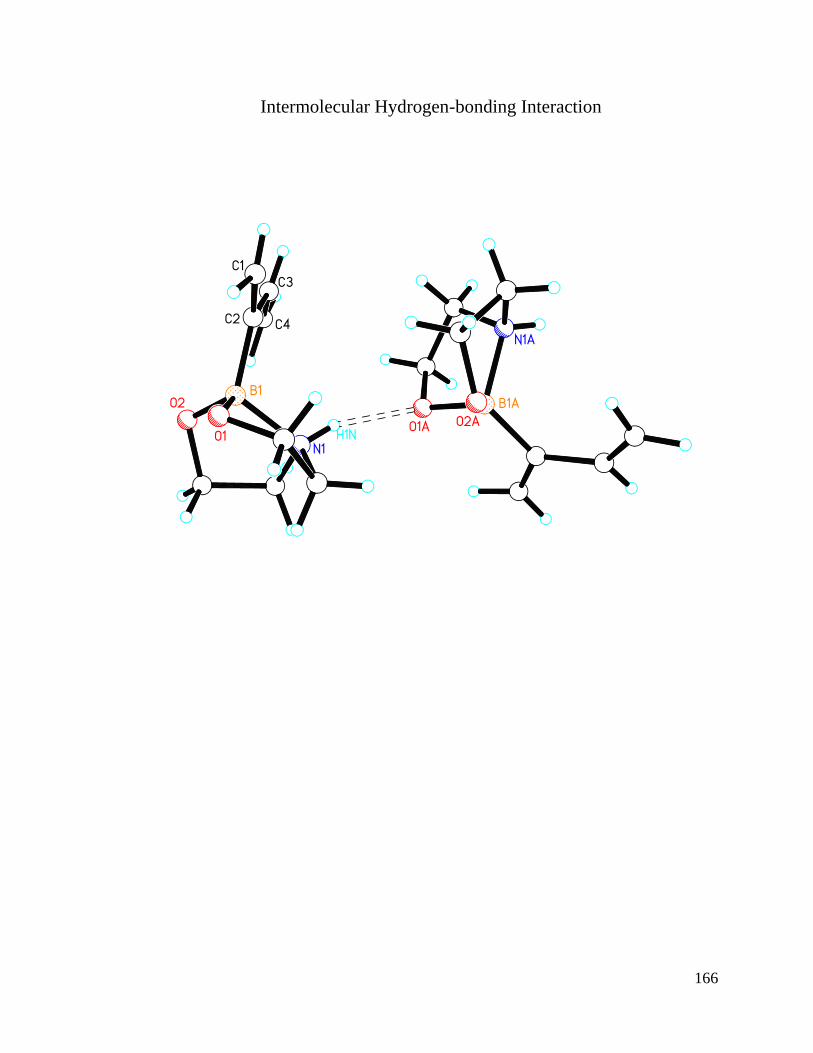
166
Intermolecular Hydrogen-bonding Interaction
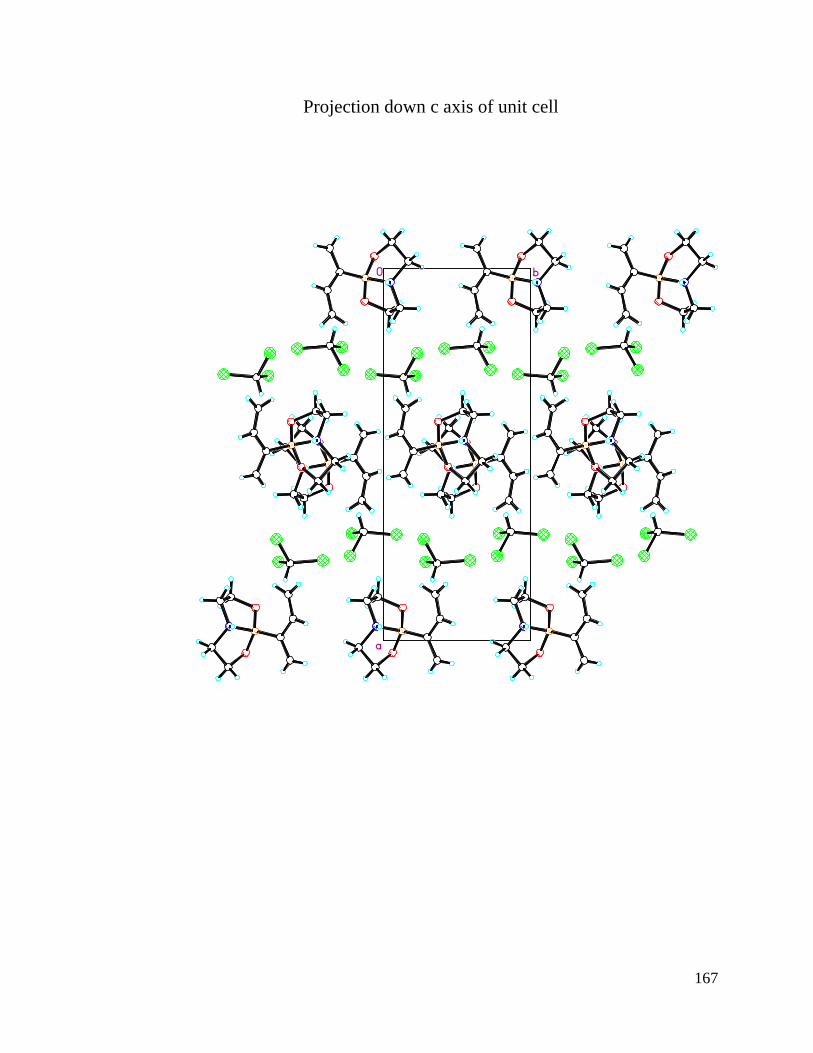
167
Projection down c axis of unit cell
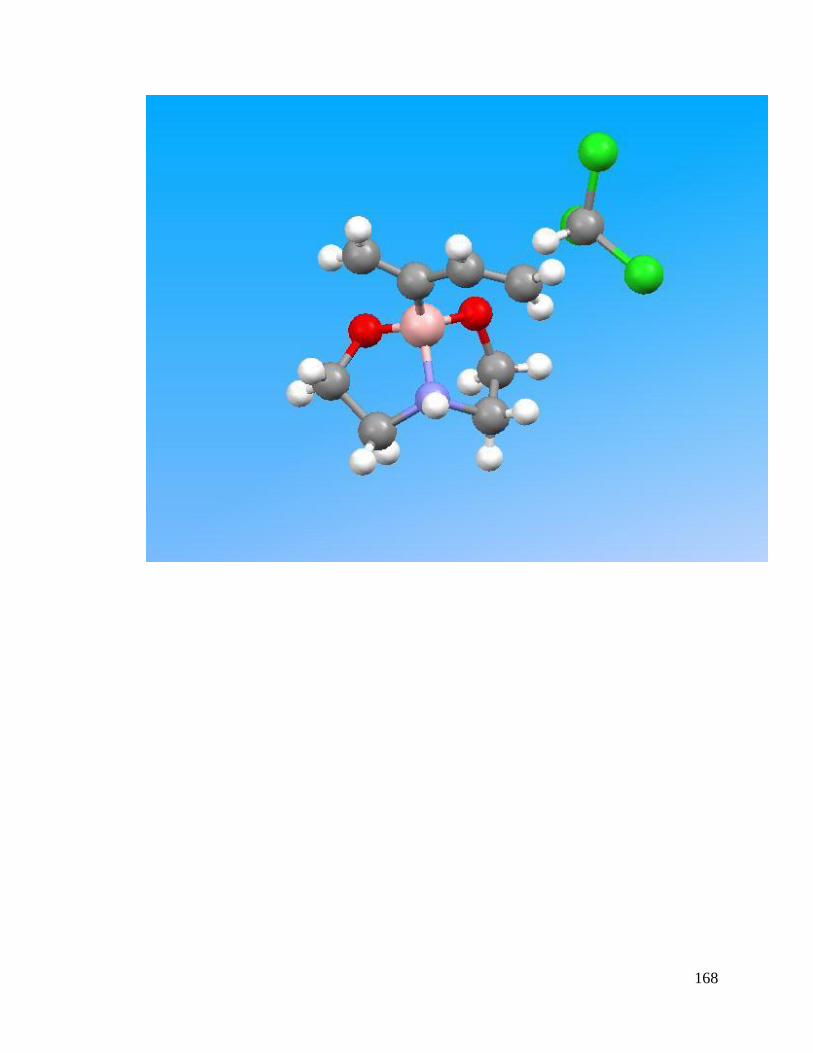
168
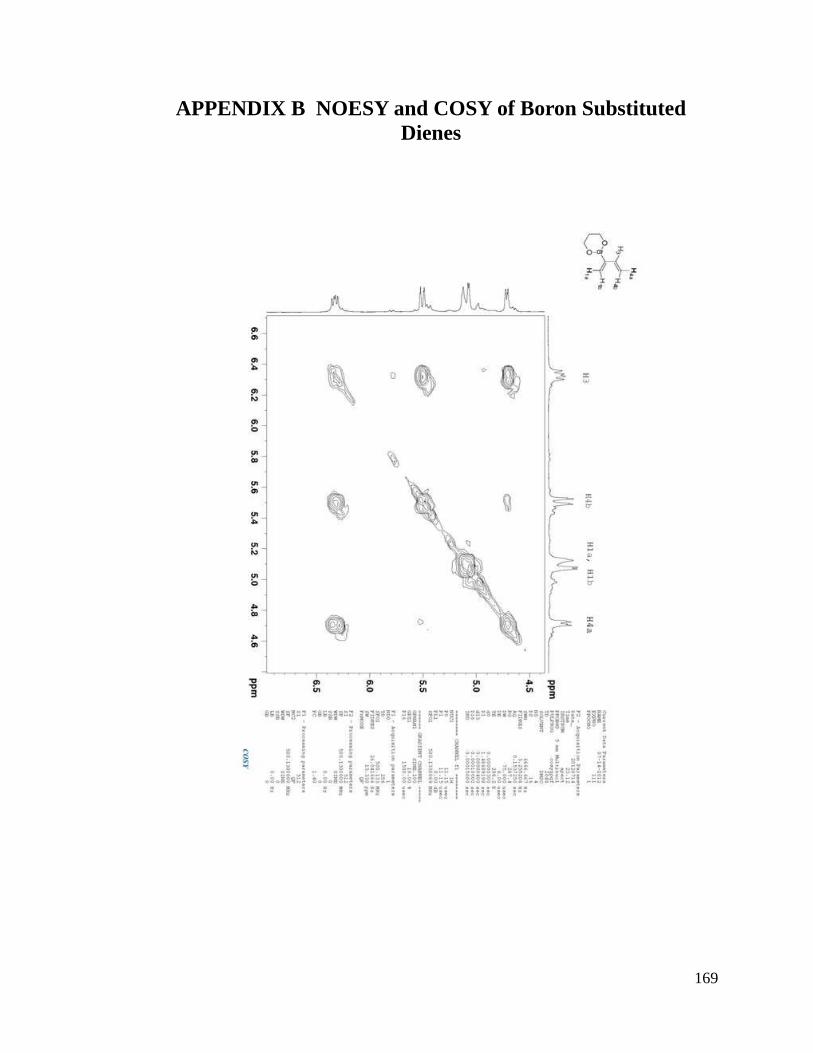
169
APPENDIX B NOESY and COSY of Boron Substituted
Dienes
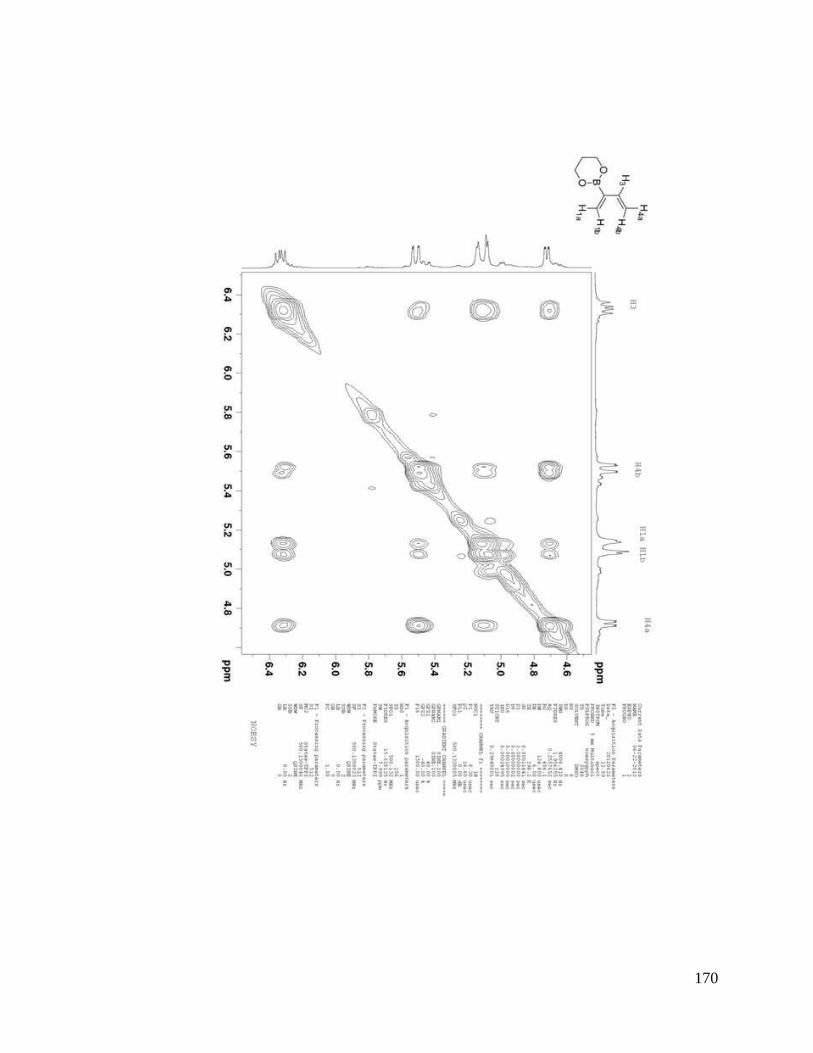
170
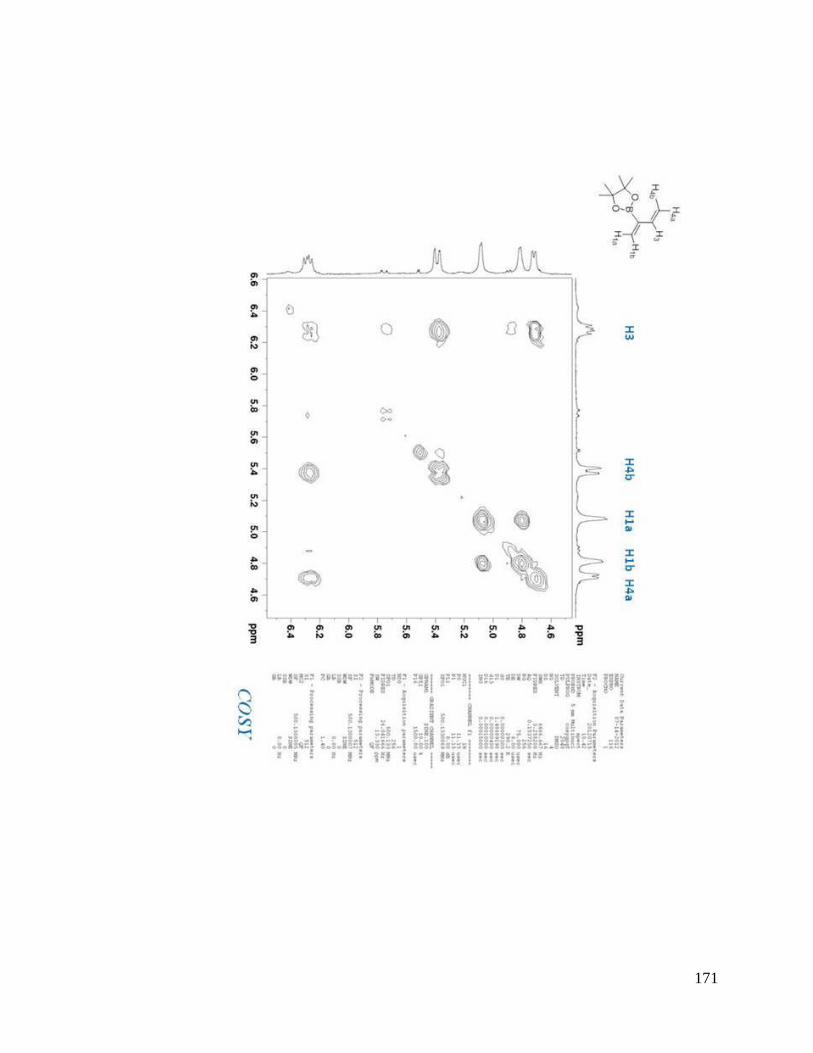
171

172

173
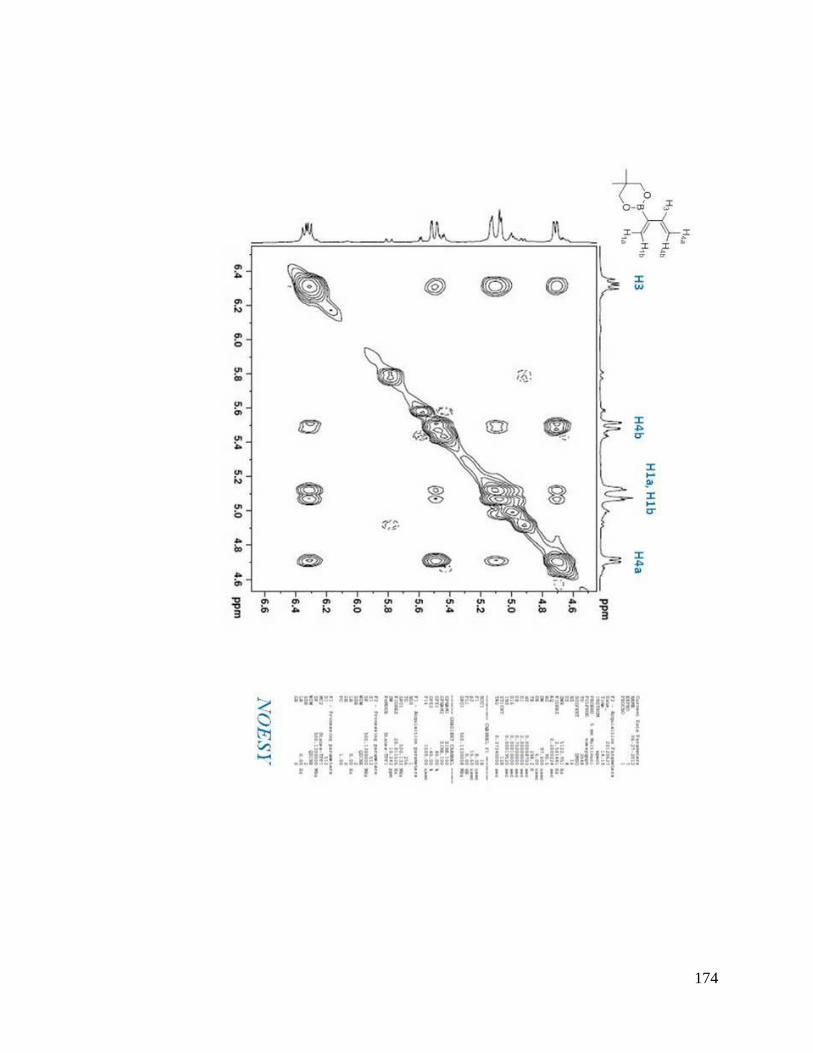
174
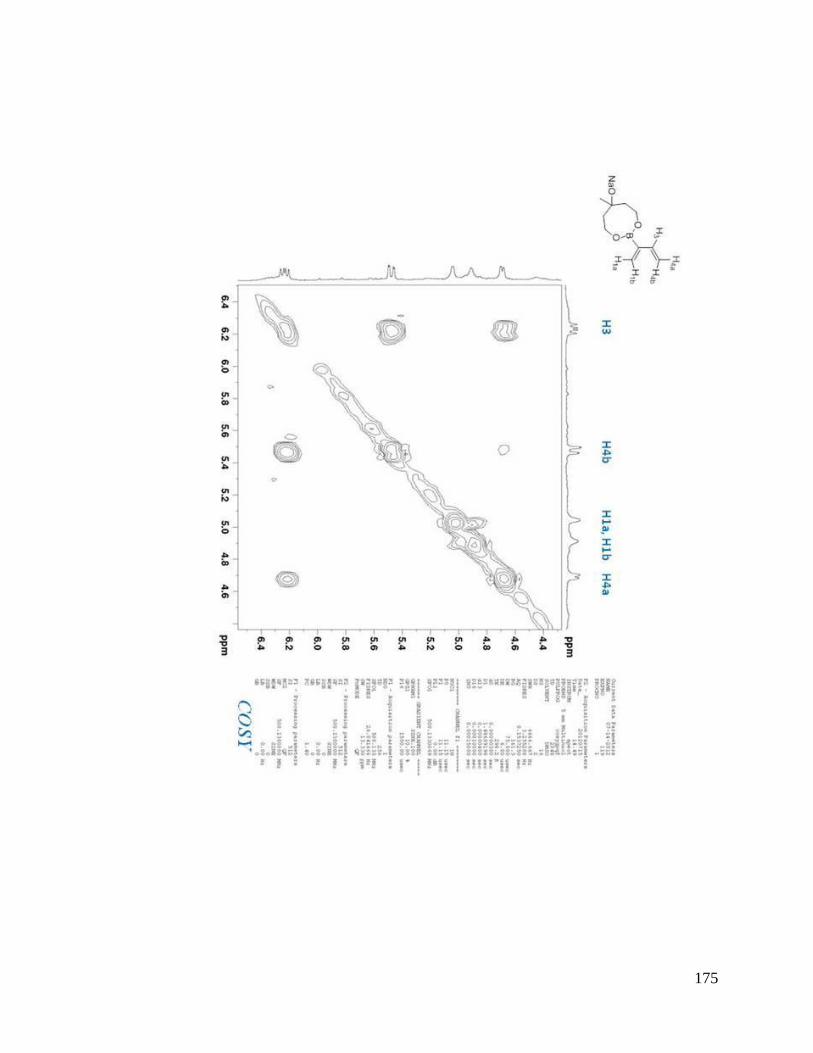
175
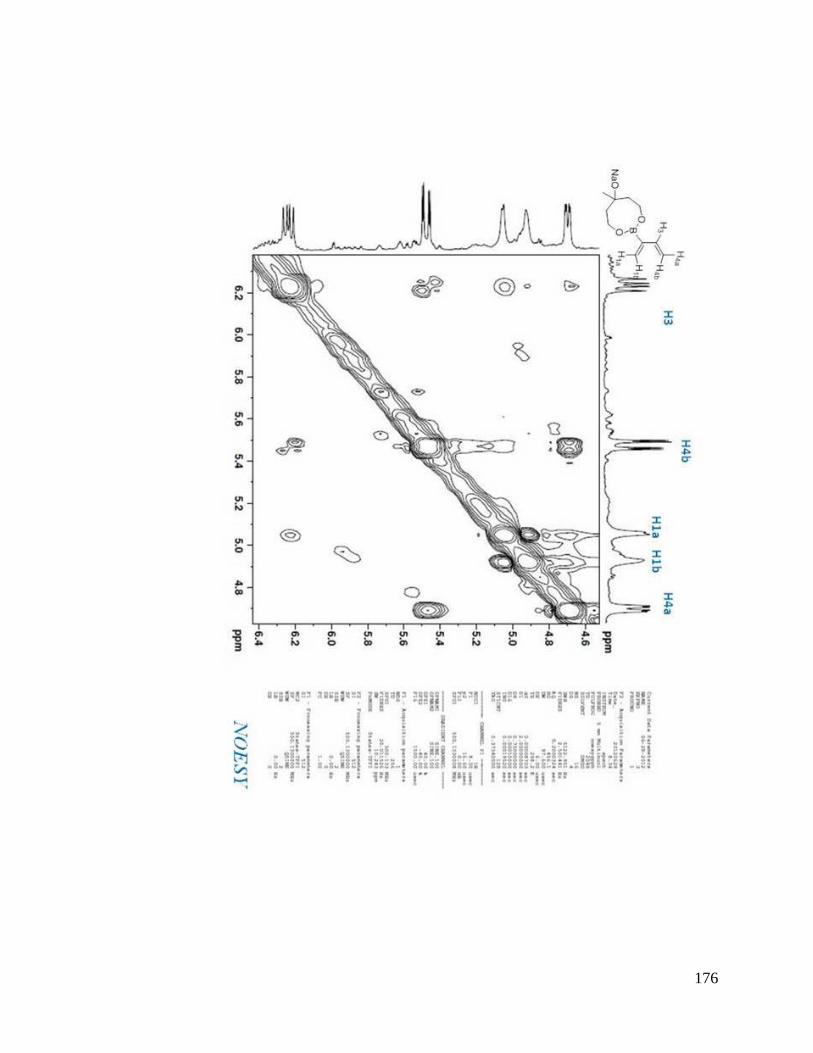
176
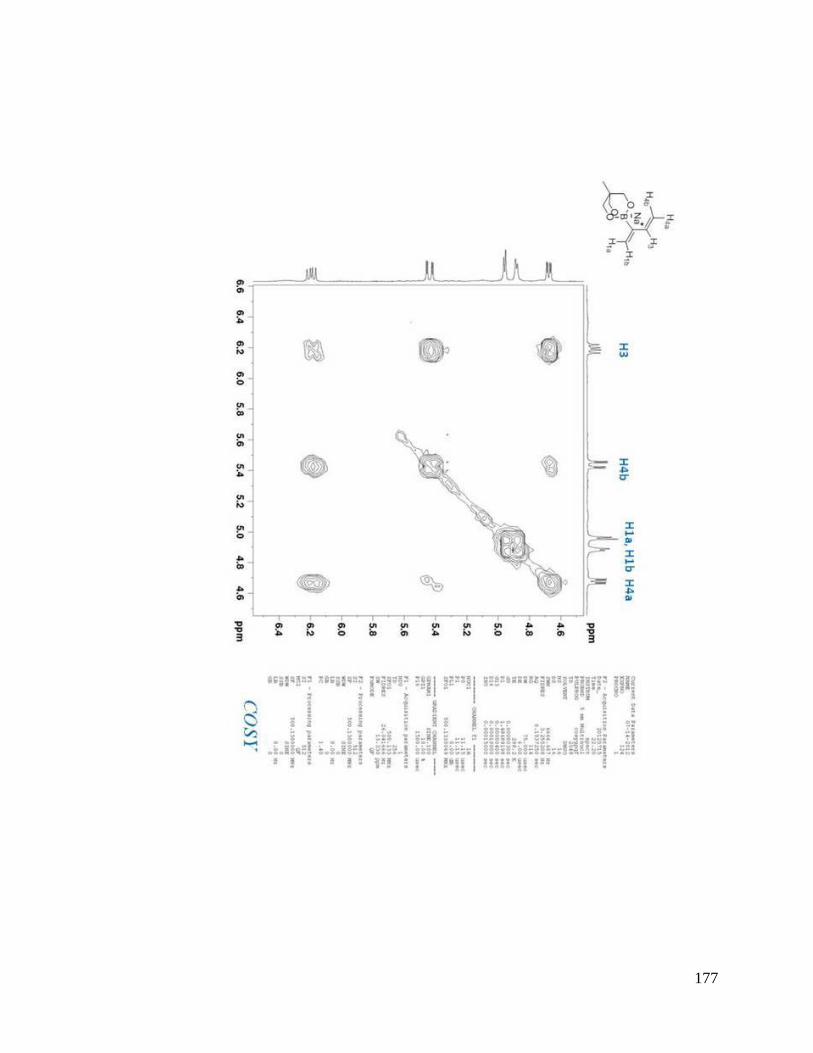
177
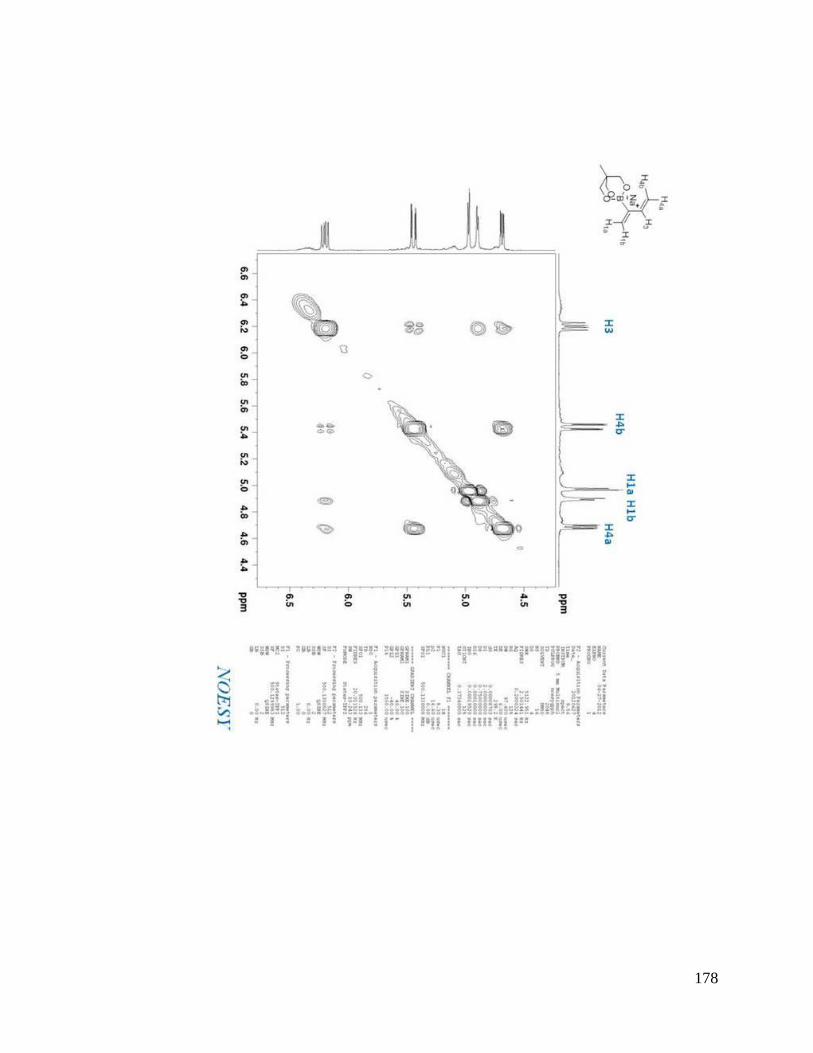
178

179
CURRICULUM VITAE
LIQIONG WANG
Born April 12th
, 1975, Yunnan, China
UNDERGRADUATE
STUDY(B.S.)
Yunnan Normal University, Kunming,
Yunnan, China, 1996
GRADUATE STUDY: (M.S.)
Huazhong University of Science and
Technology, Wuhan, Hubei, China, 2003
GRADUATE STUDY: (Ph.D.)
Wake Forest University
Winston Salem, North Carolina, USA
2006-present
SCHOLASTIC AND PROFESSIONAL EXPERIENCE:
Qujing Normal University – Teacher 2003-2006
Wake Forest University – Teaching Assistant 2006 – 2007
Wake Forest University – Research Assistant 2007-2012
HONORS AND AWARDS
Alumni Travel Award, Wake Forest University, 2009
Alumni Travel Award, Wake Forest University, 2010
Alumni Travel Award, Wake Forest University, 2011
PROFESSIONAL SOCIETIES
American Chemical Society, August 2007 to present, Organic Chemistry Division

180
PUBLICATIONS
Liqiong Wang, Mark E. Welker. Tandem Diels-Alder/Suzuki reactions of 2-boron
substituted 1,3-dienes. Manuscript under preparation.
Liqiong Wang, Cynthia S. Day, Marcus W. Wright, Mark E. Welker. Preparation and
Diels-Alder/cross coupling reactions of a 2-diethanolaminoboron-substituted 1,3-diene.
Beilstein Journal of Organic Chemistry, 2009, 5, No. 45.
Subhasis De; John M. Solano, Liqiong Wang, Mark E. Welker. Rhodium catalyzed
tandem Diels-Alder/hydrolysis reactions of 2-boron-substituted 1,3-dienes. Journal of
Organometallic Chemistry 2009, 694(15), 2295-2298.
PRESENTATIONS
Liqiong Wang; Mark E. Welker. Palladium Catalyzed Boron Substituted Dienes. (Oral)
Abstracts of papers, 63rd Southeast Regional Meeting of the American Chemical Society,
Richmond, VA, United States, October 26-29, 2011
Liqiong Wang; Mark E. Welker; Marcus W. Wright, Cynthia Day. Synthesis and fast
Diels-Alder/cross coupling reactions of boron substituted 1,3-dienes. (Oral) Abstracts of
papers, 240th ACS National Meeting, Boston, MA, United States, August 22-26, 2010
Liqiong Wang; Mark E. Welker; Marcus W. Wright, Cynthia Day. Preparation and
Diels-Alder/Cross Coupling Reactions of Boron Substituted 1,3-Dienes. (Oral) Abstracts
of papers, 61st Southeast Regional Meeting of the American Chemical Society, San Juan,
Puerto Rico, October 21-24 ,2009