synthesis of acetylenes, allenes and cumulenes || procedures and equipment
TRANSCRIPT

[13.1.2004–10:00pm] [1–10] [Page No. 1]
E:/Archive files/4188-Brandsma/Printer-Files/4188-Chapter-01.3d
1Procedures and Equipment
1.1 GENERAL
For most of the procedures described in this book a round-bottomed, three-
necked flask equipped with a combination of a dropping funnel and an inlet
for inert gas, a mechanical stirrer and a combination of a thermometer and
an outlet is recommended (Figure 1.1). If during the performance of the pro-
cedure no gases are evolved from the reaction mixture, the inlet for inert gas
may be omitted and the outlet connected to a balloon or relatively big flask
filled with inert gas. Depending upon the required efficiency of stirring a simple
glass rod with a flattened end, a chromium-plated paddle (Figure 1.2) or other
types may be used. A flask having both of the outer necks in a non-vertical
position is impractical since it is difficult to place the thermometer or gas inlet
tube such that contact with the stirrer is avoided. Instead of the usual mercury,
alcohol or pentane thermometer an electronic thermometer may be used.
If relatively small volumes of reagents have to be added over a short period,
the combination of a syringe and a rubber septum may be more convenient
than the dropping funnel.
Magnetic stirring may be carried out if the volume of the reaction mixture is
limited, not much suspended material is present and continuous control of the
temperature by using a cooling bath is not very essential.
1.2 REACTIONS IN LIQUID AMMONIA
Anhydrous liquid ammonia is an excellent solvent for many reactions involving
acetylenic compounds. The conversions can be carried out under atmospheric
pressure at the boiling point (�33 �C) or if necessary, at lower temperatures.
Depending upon the conditions of the procedure, the standard set-up
(Figure 1.1) or a variant may be used. Liquid ammonia of good quality
(water content less than 0.1%, absence of rust particles) can be drawn from
cylinders with or without a dip tube (cf. A. I. Vogel’s Textbook of Practical
1

[13.1.2004–10:00pm] [1–10] [Page No. 2]
E:/Archive files/4188-Brandsma/Printer-Files/4188-Chapter-01.3d
Organic Chemistry, 4th edn., Longmans, p. 98) and transferred through a
plastic tube into the reaction flask. Plugs of cotton wool are temporarily
placed on the necks, after which the flask is equipped as desired.
However, in many laboratories no ammonia of good quality is available and
purification by distillation may be necessary. Small pieces of sodium are added
with manual swirling to �0.6 litre of ammonia in a 1-litre round-bottomed
flask. After the blue colour has persisted, an additional amount (�2 g) of
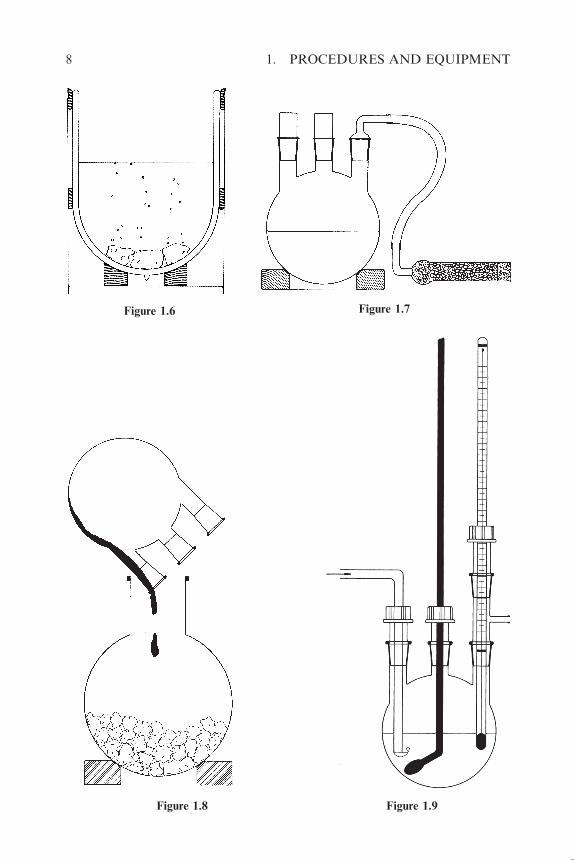
sodium is introduced and connection is made with the flask as shown in
Figure 1.9. The flask containing the solution of sodium is occasionally
placed in a bath at �35 �C, while condensation of the vapour is achieved by
(occasionally) cooling the flask (Figure 1.9) in liquid nitrogen. The distillation
can be completed in 1 to 1.5 h. Disposal of the sodium residue by addition of
ethanol should be carried out immediately after termination of the distillation.
Many reactions in liquid ammonia can be carried out at its boiling point.
Evaporation can be limited by insulating the flask in cotton wool.
If a volatile compound is to be prepared or a volatile or ammonia-sensitive
reagent (e.g. methyl iodide) is added, it is desirable to keep the reaction mixture
below the boiling point of ammonia (�33 �C). This can be done by occasional
cooling with liquid nitrogen or a mixture of dry ice and acetone in a Dewar
vessel (Figure 1.6).
Strong evaporation of ammonia during exothermic reactions can be avoided
by cooling the reaction mixture below the boiling point of ammonia.
For slow reactions, which require several hours and which can be carried out
without stirring, a flask with an evacuated space between the walls (Figure 1.4)
is recommended. If the flask is covered with aluminium foil, the rate of eva-
poration of ammonia is very low. An example is the reaction between lithium
acetylide and oxirane (Chapter 4, exp. 4.5.16).
If a reaction in ammonia is very fast and work-up has to be carried out imme-
diately, it may be more convenient to use a wide-necked round-bottomed flask
with the stirrer placed centrally (Figure 1.5). An example is described in
Chapter 3, exp. 3.9.28.
Note
The complicated equipment prescribed for performing reactions in liquid
ammonia,* even in some Organic Synthesis procedures (use of a reflux
*Some examples: A. L. Henne and K. W. Greenlee, J. Am. Chem. Soc. 67, 484 (1945);
Inorg. Synth. 2, 128 (1946); K. E. Schulte and K. P. Reiss, Chem. Ber. 86, 777 (1953); 87,
964 (1954); R. F. Parcell and C. B. Pollard, J. Am. Chem. Soc. 72, 2385 (1950); R. W.
Bradshaw, A .C. Day, E. R. H. Jones, C. B. Page, V. Thaller and R. A. V. Hodge,
J. Chem. Soc. [C], 1156 (1971).
2 1. PROCEDURES AND EQUIPMENT

[13.1.2004–10:00pm] [1–10] [Page No. 3]
E:/Archive files/4188-Brandsma/Printer-Files/4188-Chapter-01.3d
condenser, cooled with dry ice, placed on the reaction flask) may be an impor-
tant reason to prefer organic solvents. It should be emphasised, however, that
it is possible to carry out most reactions in ammonia without using a
reflux condenser. If strong evaporation is expected, for example during
dehydrohalogenations, the reaction flask may be insulated in cotton wool or
be placed in a bath with a dry ice–acetone mixture or liquid nitrogen as
described above.
1.3 SOME PRACTICAL HINTS
During some conversions in liquid ammonia, frothing may occur, especially
in the case of very thick suspensions. This can result in the loss of part of
the reaction mixture. The reaction flask therefore should never be more
than half-filled. Frothing may effectively be suppressed by adding small
amounts of diethyl ether (if the product to be isolated is not volatile), by
lifting the stirring motor so that the paddle rotates just below the surface
of the reaction mixture or by shortly cooling the flask in a bath with
liquid nitrogen.
If the product of a reaction in liquid ammonia is not very volatile (bp>110 �C
at atmospheric pressure), the ammonia may be allowed to evaporate
overnight or during the weekend. After removal of the usual equipment, the
flask is connected with a tube filled with cotton wool placed on the level of
the bottom of the flask so that a protecting atmosphere of ammonia remains
in the flask (Figure 1.7). Alternatively, the ammonia may be quickly removed
by placing the flask in a bath at 34–40 �C.
In the case of volatile products (bp<110 �C), removal of the ammonia
by evaporation gives rise to considerable losses. These can be considerably
minimised by adding a high-boiling solvent (e.g. a petroleum ether
fraction with bp>170 �C) to the reaction mixture after the reaction
has finished. Subsequently the mixture is poured on to a relatively
large amount of finely crushed ice in a large wide-necked round-bottomed
flask (Figure 1.8) or in a large beaker. The reaction is carried out at tempera-
tures below the boiling point of ammonia (cooling in dry ice–acetone or
liquid nitrogen).
In some elimination reactions in liquid ammonia an alkali acetylide
is formed. In some cases, for example in the preparation of methoxy-
and ethoxyacetylene, it is for reasons of safety essential to hydrolyse the
alkynylide before all ammonia has evaporated. This can be done by
successively adding the extraction solvent and a large amount of crushed
1.3 SOME PRACTICAL HINTS 3

[13.1.2004–10:00pm] [1–10] [Page No. 4]
E:/Archive files/4188-Brandsma/Printer-Files/4188-Chapter-01.3d
ice over a very short period (cf. Chapter 3, exp. 3.9.28). For this reason
the reaction is carried out in a large one-necked round-bottomed flask
(Figure 1.5).
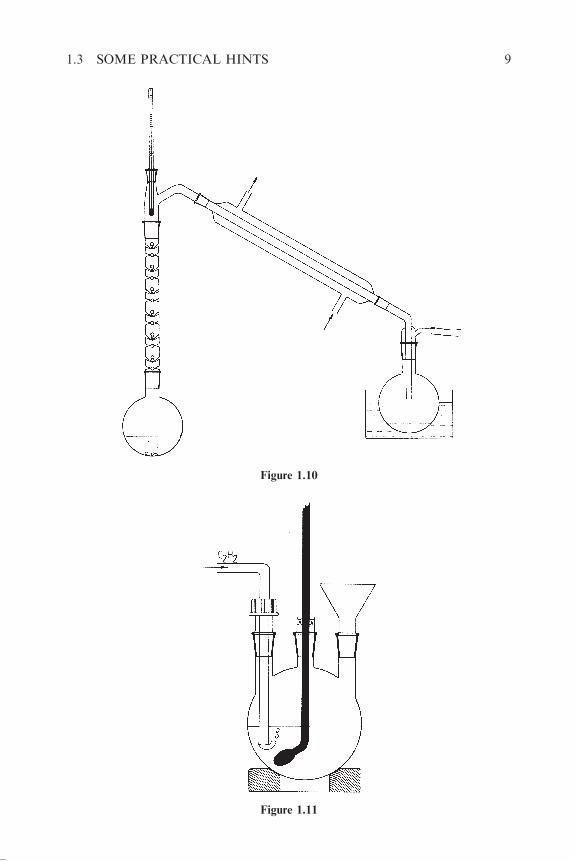
For the isolation of volatile or unstable products from the solution in a high-
boiling solvent the set-up of Figure 1.10 is used. After evacuation (between <1
and 25 Torr) the solution is warmed until a small amount of the extraction
solvent has passed over. The volatile product is collected in a strongly
cooled (bath with dry ice–acetone, �78 �C) receiver. If a water aspirator
is used, and entrance of moisture is undesired (for example in Chapter 3,
exp. 3.9.48), a tube filled with a drying agent should be placed between the
receiver and the aspirator.
Distillation can be accompanied by foaming especially in the case of com-
pounds with a long carbon chain. Foaming can be suppressed by completely
immersing the distillation flask in the liquid of the heating bath or by
adding a few drops of some anti-foaming liquid prior to carrying out the
distillation. Although special columns can be used for preventing the
foam passing over, it is, in general, advisable to use a relatively big distilla-
tion flask in such cases. Recovery by distillation of acetylenes or allenes that
have been stored for a long period may involve the risk of an explosive
decomposition of the residue. This danger can be minimised by adding a
certain amount (�20ml for the distillation of 50ml of product) of paraffin
oil prior to the distillation. The residue will not remain as a compact mass
but as an emulsion or dispersion in the oil after termination of the
distillation.
Concentrations of reactants in the procedures described in this book
generally are between 0.5 and 1mol/litre of solvent. A good estimate of the
time necessary for �100% conversion requires much experience and knowledge.
For the lithiation of a 1-alkyne with butyllithium, for example, one has to
know that the deprotonation of acetylenes with such very strongly basic
reagents is an extremely fast reaction. This means that the heat of this depro-
tonation is liberated over a very short period. Metallation of allenes, com-
pounds which are much less strongly acidic than 1-alkynes, proceeds less
fast. Nevertheless, one can make a rather good estimate of the time necessary
for (almost) complete lithiation of, for example methoxyallene,
H2C¼C¼CHOMe, by performing the following experiment. A solution of
0.05mol of n-BuLi in 33ml of hexane (the usual 1.6mol/litre concentration
of the commercially available reragent) and 40ml of tetrahydrofuran is
cooled down to �70 �C, then the cooling bath is removed and the flask is insu-
lated in cotton wool. The increase of the temperature of the slowly stirred solu-
tion is observed during a few minutes (an electronic thermometer is more
convenient than the usual one). Usually, rising of the temperature will
amount by a few degrees only. The temperature of the solution is adjusted
4 1. PROCEDURES AND EQUIPMENT

[13.1.2004–10:00pm] [1–10] [Page No. 5]
E:/Archive files/4188-Brandsma/Printer-Files/4188-Chapter-01.3d
again at �70 �C by dipping the reaction flask for a few seconds in the cooling
bath. Methoxyallene (0.05mol) is then added in one portion. This will result in
a rising of the temperature by several (at least 20) degrees over less than 1 min.
Even if smaller amounts of the allenic ether are added in one portion, the effect
is clearly visible. One may carry out a blank experiment by adding at �70 �C
the same volume of THF. In this case the temperature rises by a few (<5)
degrees only. Of course, for completion of the lithiation the reaction
mixture has to be stirred for a (limited) additional period at a temperature
higher than �70 �C. Reactions between strongly basic anionic species and
aldehydes or ketones usually proceed almost instantly, even at temperatures
far below 0 �C in solvents of low polarity. Silylations (with R3SiCl) and sulphe-
nylations (with disulphides, RSSR or thiocyanates, RSC�N) of organoalkali
compounds are other examples of very fast reactions, which are easy to
follow by temperature observation. Reactions in boiling liquid ammonia
(bp �33 �C) may be followed by keeping the end of a small slip of paper 1 cm
from the outlet. When there is no or a very slow reaction (e.g. when the reac-
tion has subsided) the flow of escaping ammonia is relatively weak and the
position of the slip remains vertical. In the case of very fast reactions much
ammonia may evaporate in a short time and the vigorous flow from the
outlet causes the slip to move off from the outlet. Dehydrohalogenations as
mentioned in Chapter 3, Section 3.3 are extremely fast and exothermic so
that addition of small portions of the halogen compound causes a vigorous
outward flow of ammonia. For reactions carried out at temperatures below
the boiling point of ammonia the method described above for methoxyallene
may be used.
It should be pointed out that heating effects can also be made visible by
performing the reaction on a very small (1mmolar) scale in a reagent tube
(wrapped in cotton wool) using an electronic thermometer.
Some reactions, if carried out on a preparative scale and with ‘preparative’
concentrations of the reactants, can be followed by observing the temperature
in the solution boiling under constant intensity of the reflux stirred at a
constant rate. A good example is the Pd/Cu-catalysed cross-coupling
between ethynyl(trimethyl)silane (bp �53 �C) and 1-bromo-1-cyclooctene in
piperidine (bp 106 �C) described in Chapter 16, exp. 16.7.15. The temperature
of the reaction mixture, in the beginning �90 �C, reaches a maximum
(�110 �C, somewhat higher than the boiling point of piperidine, due to the
presence of dissolved HBr salt) when most of the volatile acetylene
has reacted with bromocyclooctene. The isomerisation of methyl
propargyl ether (bp 61 �C) to methoxyallene (bp 51 �C) in the presence of
potassium t-butoxide in a small amount of DMSO (Chapter 17, exp. 17.2.8)
can be followed by looking at the thermometer in the refluxing solution.
1.3 SOME PRACTICAL HINTS 5

[13.1.2004–10:00pm] [1–10] [Page No. 6]
E:/Archive files/4188-Brandsma/Printer-Files/4188-Chapter-01.3d
The temperature drops as a result of the formation of a more volatile
compound.
In other procedures, carried out with high concentrations of starting com-
pounds, the refractive index of the solution can be measured, for example
in the isomerisation of HC�CCH(Br)C6H13 to BrCH¼C¼CHC6H13 under
the catalytic influence of lithium dibromocuprate, CuBr �LiBr (Chapter 12,
exp. 12.4.14), and in the bromination of 1,3-diynes, RC�CC�CH, with potas-
sium hypobromite (Chapter 9, exp. 9.2.3).
In a single case the only necessary instruments are the eyes. The conversion
of propargyl bromide into the corresponding iodide, using a hot (�70 �C) solu-
tion of sodium iodide in absolute ethanol (Chapter 20, exp. 20.1.11) is accom-
panied by precipitation of sodium bromide. The reaction is finished when salt
crystals are no longer being formed in the supernatant layer (stirring is not
necessary).
Particularly in the cases of hydroxyacetylenes, the question ‘How many
extractions with diethyl ether still have to be carried out?’ may arise. The follow-
ing practical tip may be useful. Take a small (0.5ml) sample from the extract
with a Pasteur pipette and allow the solution to flow along a ground-glass
stopper. Further extraction is no longer necessary when after evaporation of
the ether no residue is visible on the ground glass.
A quick and extremely simple method to investigate whether volatile
polymerisable liquids such as oxirane, ethyl vinyl ether and vinyl bromide
contain polymers is to put � 1 ml on a watch glass and to blow air over
the liquid. If not any oil-like residue is visible on the glass, purification is
not necessary.
The following generally valid advice with respect to the work-up of reac-
tion mixtures resulting from syntheses with Grignard reagents should be
given. If water or aqueous ammonium chloride is added at too fast a
rate to the reaction mixture, much heat will be evolved and as a result
part of the reaction mixture will be lost, especially if diethyl ether is
used as a solvent. Therefore, the reaction mixture always should be (cau-
tiously) poured into water or into aqueous ammonium chloride. If the pro-
duct is not acid-sensitive, dilute mineral acid can be used. Alternatively,
water or the aqueous solution of ammonium chloride or acid may be
added slowly to the efficiently stirred reaction mixture, preferably under
cooling.
6 1. PROCEDURES AND EQUIPMENT

[13.1.2004–10:00pm] [1–10] [Page No. 7]
E:/Archive files/4188-Brandsma/Printer-Files/4188-Chapter-01.3d
Figure 1.1 Figure 1.2 Figure 1.3
Figure 1.4 Figure 1.5
1.3 SOME PRACTICAL HINTS 7
[13.1.2004–10:00pm] [1–10] [Page No. 8]
E:/Archive files/4188-Brandsma/Printer-Files/4188-Chapter-01.3d
Figure 1.6 Figure 1.7
Figure 1.8 Figure 1.9
8 1. PROCEDURES AND EQUIPMENT
[13.1.2004–10:00pm] [1–10] [Page No. 9]
E:/Archive files/4188-Brandsma/Printer-Files/4188-Chapter-01.3d
Figure 1.10
Figure 1.11
1.3 SOME PRACTICAL HINTS 9

[13.1.2004–10:00pm] [1–10] [Page No. 10]
E:/Archive files/4188-Brandsma/Printer-Files/4188-Chapter-01.3d
ABBREVIATIONS
Et2O diethyl ether
THF tetrahydrofuran
DMSO dimethylsulphoxide
HMPT hexamethylphosphoric triamide
(Me3N)3P¼O
LDA lithium diisopropylamide
DMF N,N-dimethylformamide
TMEDA N1,N1,N2,N2-tetramethylethanediamine
rt (in experimental procedures) room temperature
BuLi n-butyllithium
t-BuOK potassium tert-butoxide
(the commercially available
non-complexed base)
TMS trimethylsilyl group
c-C6H11 cyclohexyl group
Me CH3 group
Et C2H5 group
n-Pr n-C3H7 group
n-Bu n-C4H9 group
t-Bu (CH3)3C group
(CH2)5C cyclohexane ring, in tables
10 1. PROCEDURES AND EQUIPMENT