synthesis of reduced metabolites of isoflavonoids, and their
TRANSCRIPT

Synthesis of Reduced Metabolites of Isoflavonoids, and their Enantiomeric Forms
Tuija Jokela
Laboratory of Organic Chemistry Department of Chemistry
Faculty of Science University of Helsinki
Finland
Academic Dissertation
To be presented with the permission of the Faculty of Science of the University of Helsinki for public examination in Auditorium A110 in the Department of Chemistry,
November 26th, at 12 o’clock.
Helsinki 2011

� ��
Supervisor Professor Kristiina Wähälä Laboratory of Organic Chemistry Department of Chemistry University of Helsinki Finland Reviewers Professor Harri Lönnberg Laboratory of Organic Chemistry Department of Chemistry University of Turku Finland Professor Victor A. P. de Freitas Laboratory of Organic Chemistry Department of Chemistry University of Porto Portugal Opponent Professor Michael Orfanopoulos Department of Chemistry University of Crete Greece ISBN 978-952-10-7328-1 (paperback) ISBN 978-952-10-7329-8 (PDF) http://ethesis.helsinki.fi Helsinki University Printing House Helsinki 2011


� ��
ABSTRACT Isoflavonoids are naturally occurring plant derived biochemicals, which act as phytoalexins. Isoflavonoids are of interest due to their estrogenic and other potential physiological properties, particularly in mammals that typically consume isoflavonoid rich nutrients such as soy and red clover. The literature review of this thesis mainly focuses on the reduced metabolites of hydroxy and/or methoxy substituted isoflavones with four groups: isoflavan-4-ols, isoflav-3-enes, isoflavans and �-methyldeoxybenzoins (1,2-diarylpropan-1-ones), which are all reduced metabolites of food derived isoflavones in mammals. Related isoflavan-4-ones are briefly discussed. Results of an extensive survey of the literature concerning the synthesis of polyhydroxy- or methoxysubstituted isoflavonoids and especially asymmetric approaches are discussed. The experimental section describes new synthetic methods to prepare polyphenolic reduced isoflavonoid structures such as isoflav-3-enes, isoflavan-4-ones, cis- and trans-isoflavan-4-ols, 1,2-diarylpropan-1-ones and isoflavans by various hydride reagents and hydrogenations. The specific reactivity differences of various hydride reagents toward isoflavonoids are discussed. The first enantioselective synthesis of natural (S)-(-)-equol and the opposite enantiomer (R)-(+)-equol is also described by the asymmetric iridium PHOX catalysed hydrogenation of isoflav-3-enes. Both of these equol enantiomers are found to possess biological activity in mammals due to estrogen receptor binding activity. The natural enantiomer prefers estrogen receptor � and the R-enantiomer prefers the estrogen receptor �. Also the precursor, isoflav-3-ene, is found to possess positive biological effects on mammals. In connection with the synthetic work, the (S)-(-)-equol was discovered from serum of ewes after isoflavone rich red clover feeding. The chiral HPLC method was developed to identify natural equol enantiomer for the first time in this species. The first synthesis of natural isoflavonoid (R)-(-)-angolensin and its enantiomer (S)-(+)-angolensin is desribed by the use of recyclable chiral auxiliaries (chiral pseudoephedrines). The method offers a general approach also to other natural polyphenolic 1,2-diarylpropan-1-ones and to further study isoflavonoid metabolism in human and other mammals. The absolute configurations of these new chiral isoflavonoid metabolites were determined by X-ray spectroscopy. Also thorough NMR and MS analysis of synthesised structures are presented.

� ��
ACKNOWLEDGEMENTS This study was carried out in the Laboratory of Organic Chemistry, Department of Chemistry, University of Helsinki. Funding from the Emil Aaltonen Foundation, The Graduate School of Bioorganic Chemistry, the Chancellor of the University of Helsinki and the University of Helsinki Research Fund are gratefully acknowledged. I wish to offer my sincere thanks to a number of people, for a great range of support. My supervisor, professor Kristiina Wähälä, introduced me to the field of isoflavonoid chemistry and gave her continuous support, encouragement, advice and confidence during the entire project. Also, Professor Emeritus Tapio Hase, who accepted me as a graduate student in his laboratory, offered his invaluable advice on numerous chemical problems. Both have placed the research facilities of the laboratory at my disposal. Dr. Auli Salakka provided pleasant cooperation concerning isoflavonoid synthesis research. Professor Hannu Saloniemi, Dr. Juhani Taponen, Mrs. Eeva Mustonen, and Mr. Ilkka Saastamoinen offered fruitful collaboration in our multidisciplinary research concerning isoflavonoids and animal health. Dr. Johanna Lampe and other coworkers contributed to the isoflavonoid metabolism study, and Professor Ian Rowland and coworkers participated in our multidisciplinary research project. Dr. Kalle Nättinen performed X-ray measurements, and did analysis of diiodo-(S)-equol, chiral PHOX-ligand and catalyst. Dr. Cristine Cardin performed the X-ray analysis of (R)-angolensin. Professor Kalevi Pihlaja and his group collaborated on the computational NMR. The personnel at the Laboratory of Organic Chemistry always provided me with advice whenever I needed it. Dr. Jorma Matikainen assisted me greatly with HRMS measurements and taught me how to measure LRMS spectra. Mr. Seppo Kaltia and Dr. Markku Mesilaakso introduced me to NMR measurements. I also thank all of the former and present members of our Phyto-Syn research group whom are gratefully acknowledged. Special thanks go to Eija, Barbara and Sari for being such good friends both on and off duty. I also want to thank the “unofficial” Phyto-Syn member Nina for sharing the interest in higher education teaching and our ECTN cooperation. Antti and Satu-Maarit gave valuable input on isoflavonoid metabolism research and provided pleasant companionship at the Soy Conference in San Diego. Special thanks go to Kauko for maintaining a full service chemical warehouse and Matti Keinänen for technical support especially concerning hydrogenation equipment. Professor Harri Lönnberg and Professor Victor A. P. de Freitas have kindly reviewed the manuscript of the thesis and gave extremely helpful comments, and Dr. Jennifer Rowland revised the language of the manuscript.

� ��
My present supervisors at Fermion, Arne and Lefa, have provided outstanding encouragement and given me the study leave to finish this manuscript. Also, other colleagues at Fermion have created an inspiring atmosphere. My sincerest thanks go to my parents for their unconditional love, support and trust in every aspect of my life. I’m especially grateful to your gentle caretaking of the boys. I would also like to thank Aino and Aukki for your kind support. I am especially grateful to my dear brother, who always kept the positive thinking no matter what. Finally, I would like to thank my Supermen, Mika, Roope and Lauri, for your love and support. I am most sincerely grateful for your endless patience and understanding during my late nights in our basement finishing up this manuscript. Vantaa, October 2011 Tuija Jokela

� ��
CONTENT ABSTRACT 2
ACKNOWLEDGEMENTS 3
LIST OF ORIGINAL PUBLICATIONS 6
LIST OF ABBREVIATIONS 7
1. INTRODUCTION 9
1.1. Isoflavonoid sources in nutrition, mammalian metabolism and biological activities 11 1.2. Synthesis of isoflavonoid metabolites ����������� ����� ��������������� � � � �� 1.2.2 Synthesis of isoflav-3-enes and isoflav-2-enes 18
1.2.2.1 2-O-Substituted or 2-N-substituted isoflav-3-enes 24 1.2.3 Synthesis of isoflavans 26 1.2.3.1 Catalytic hydrogenation of isoflavones 27 1.2.3.2 Multistep total syntheses 31 1.2.3.3 Synthesis of enantiopure isoflavans 35 1.2.4 Synthesis of ��-methyldeoxybenzoins 41 1.2.4.1 Synthesis of racemic �-methyldeoxybenzoins
(1,2-diarylpropan-1-ones) 41 1.2.4.2 Asymmetric synthesis of 1,2-diarylpropan-1-ones 45 2. AIMS OF THE STUDY 48 3. RESULTS AND DISCUSSION 49 3.1. Isoflavone reductions by hydride reagents and hydrogenations 49 3.2. Synthesis of trans- and cis-isoflavan-4-ols 51 3.3. Synthesis of isoflav-3-enes 52 3.4. Synthesis of isoflavans 52 3.4.1. Racemic polyphenolic isoflavans 52
3.4.2. Enantiopure polyphenolic isoflavans 53 3.5. Synthesis of �-methyldeoxybenzoins 58 3.5.1. Polyphenolic �-methyldeoxybenzoins 58 3.5.2. Enantiopure �-methyldeoxybenzoins 59 3.6. Isoflavonoids in biological studies 62 3.6.1. S-equol determination from serum of ewes 62
3.6.2. ODMA and equol in human isoflavone metabolism studies 62 3.6.3. Biological activity of S-equol vs. rac-equol in vitro 63 3.6.4 Equol quantitation in organic skimmed Finnish cow milk 63 4. CONCLUSIONS 64 5. EXPERIMENTAL 64 REFERENCES 70 ORIGINAL PUBLICATIONS 77

� ��
LIST OF ORIGINAL PUBLICATIONS This thesis is based on the following original publications, which are referred to in the text by their Roman numerals (I-VI):
I. Auli K. Salakka, Tuija H. Jokela, Kristiina Wähälä. Multiple hydride reduction pathways in isoflavonoids. Beilstein J. Org. Chem. 2, 2006, No. 16.
II. Pihlaja, K., Tähtinen, P., Klika, K.D., Jokela, T., Salakka, A.; Wähälä. K. Experimental and DFT 1H NMR Study of Conformational Equilibria in trans-4’,7-Dihydroxyisoflavan-4-ol and trans-Isoflavan-4-ol, J. Org. Chem. 68, 2003, 6864.
III. Mustonen, Eeva; Jokela, Tuija; Saastamoinen, Ilkka; Taponen, Juhani; Taponen, Suvi, Saloniemi, Hannu; Wähälä, Kristiina. High serum S-equol content in red clover fed ewes: the classical endocrine disruptor is a single enantiomer. Environ. Chem. Lett., 3, 2006, 154-159.
IV. Wähälä, K.; Jokela, T. Preparation of isoflavonoids. Finn. 2009, 20 pp. CODEN: FIXXAP FI 120584 B1 20091215 Patent. (Priority date 21.10.2005)
V. Magee, P. J.; Raschke, M.; Steiner, C.; Duffin, J. G.; Pool-Zobel, B. L.; Jokela, T.; Wähälä, K.; Rowland, I. R. Equol: A comparison of the effects of the racemic compound with that of the purified S-enantiomer on the growth, invasion, and DNA integrity of breast and prostate cells in vitro, Nutr. Cancer. 54 (2), 2006, 232.
VI. Frankenfeld, C. L.; Atkinson, C.; Thomas, W. K.; Goode, E. L.; Gonzalez, A.; Jokela, T.; Wähälä, K.; Schwartz, S. M.; Li, S. S.; Lampe, J. W. Familial correlations, segregation analysis, and nongenetic correlates of soy isoflavone-metabolizing phenotypes, Exp. Biol. Med. 229, 2004, 902.
Data published and discussed here for the first time are referred to as VII.

� �
LIST OF ABBREVIATIONS Ac acetyl Ad adamantyl BARF tetrakis[3,5-bis(trifluoromethyl)phenyl]borate aq. aqueous 9-BBN 9-borabicyclo[3.3.1]nonane BF3OEt2 borontrifluoride diethyletherate Bn benzyl n-BuPAd2 n-butyldi(1-adamantyl)phosphine BTED bis-tert-butylthioethane diborane ca. circa CD circular dichroism COD 1,5-cyclooctadienyl COSY correlated spectroscopy DAD diode array detector/ detection DBU 1,8-diazabicyclo[5.4.0]undec-7-ene DCC dicyclohexylcarbodiimide DCM dichloromethane DDQ 2,3-dichloro-5,6-dicyanobenzoquinone de diastereomeric excess DEAD diethyl azodicarboxylate DFT density functional theory DHE dehydroequol DIBAH, DIBAL-H diisobutylaluminum hydride DMAP 4-dimethylaminopyridine DMF N,N-dimethylformamide DMSO dimethylsulfoxide DT-DFT time-dependent density functional theory ECD electronic circular dichroism ee enantiomeric excess EI electron impact, electron ionisation ER estrogen receptor ERR estrogen-related receptor EtOAc ethyl acetate F-C Friedel-Crafts Fig. Figure FL fluorescence GC gas chromatography HMBC hetero multiple bond correlation HPLC high performance liquid chromatography HSQC hetero single quantum correlation IR infrared LC liquid chromatography LDA lithium diisopropylamide LHMDS lithium hexamethyl disilazide LICA lithium cyclohexylisopropylamide Me methyl MEM �-methoxyethoxymethyl

� �
MEK methylethylketone mg/d milligrams per day MOM methoxymethyl MS mass spectroscopy MTPPB methyl triphenyl phosphonium bromide MW microwave NaHMDS sodium bis(trimethylsilyl)amide; ((CH3)3Si)2NNa; sodium
hexamethyl disilazide NMR nuclear magnetic resonance PEG polyethylene glycol Ph phenyl PHOX phosphine oxazoline PPA polyphosphoric acid PPh3 triphenylphosphine quant. quantitative yield RP reversed phase rr rearrangement TBDMS tert-butyldimethylsilyl TBDPSCl tert-butylchlorodiphenylsilane TBS tert-butylsilyl TBSCl tert-butylsilylchloride THF tetrahydrofuran TLC thin layer chromatography TPPO triphenylphosphine oxide UV ultraviolet VCD vibrational circular dichroism vs. versus Xc chiral auxiliary

� ��
1. INTRODUCTION Isoflavonoids are naturally occurring plant derived biochemicals, which act as phytoalexins against bacteria, viruses and insect pests in flora. Isoflavonoids 1- 13 are a subclass of the taxonomically more widely occurring phytochemicals, flavonoids (flavone 14), both posessing a phenylchroman skeleton of 15 carbon atoms (Fig. 1). Isoflavonoids differ from flavonoids by the position of phenyl ring B, which is in the C-3 position instead of C-2 as in flavonoids. Isoflavonoids are of interest due to their estrogenic and other potential physiological properties, particularly in mammals that typically consume isoflavonoid rich nutrients. The work described in this thesis was entirely focused on the isoflavonoids, especially the reduced metabolites of hydroxy and/or methoxy substituted isoflavone 1 with four groups: isoflavan-4-ols 3, isoflav-3-enes 5, isoflavans 4 and �-methyldeoxybenzoins (1,2-diarylpropan-1-ones) 7, which are all reduced metabolites of food derived isoflavones in mammals (Fig. 1). Related isoflavan-4-ones 2 are briefly discussed. Results of an extensive survey of the literature concerning the synthesis of polyhydroxy- or methoxysubstituted isoflavonoids and especially asymmetric approaches are discussed. The motivation for this work came from the lack of expedient synthesis methods to prepare polyphenolic isoflavonoids. Particularly, the need for enantiopure or diastereopure reference compounds for use in clinical studies and biological activity studies gave reason to study isoflavonoid chemistry further. Isoflavones 1 (3-phenyl-4H-1-benzopyran-4-ones or 3-phenylchroman-4-ones) are the biggest subgroup in the isoflavonoid family in plants. Isoflavones occur mainly in the Leguminosae family, soy being the most important plant in human nutrition.1
Fig. 1. Isoflavonoid 1-13 and flavone 14 skeletons and numbering of rings A, B and C according to Dewick.1

���
In recent times, an enormous increase has been observed regarding the study of human metabolism of food derived isoflavones into reduced isoflavonoid substructures such as isoflavan 4 or isoflav-3-ene 5. Isoflavonoid clinical studies have focused on isoflavan equol 15 in particular, which is a reduced metabolite of one of the most abundant natural isoflavones, daidzein 16. For example in 2010, the biannual international symposium on the role of soy in preventing and treating chronic disease dedicated one day just for equol research.2 One of the topics was efficacy and safety of the natural S-equol supplementation for menopausal healthcare. This clearly demonstrates a trend in the study of isoflavone metabolism associated with the claims that isoflavones are beneficial to health. The first reduced isoflavone metabolite identified in human urine after consumption of an isoflavone rich diet is equol:3,4 this is consistent with equol being studied in the biggest number of publications among the reduced metabolites (Fig. 2).
Fig. 2 Frequency of publications concerning equol and isoflavan or isoflavene in general from 1990 to 2010 searched in SciFinder.
During the 21st century the number of patent applications concerning the potential daidzein metabolite dehydroequol 17 (7,4’-dihydroxyisoflav-3-ene)5-19 together with equol 1520 have increased dramatically. Their value and interest as patentable compounds, in particular, has increased due to their use as therapeutic agents in different diseases, as food supplements to improve human health, or as ingredients in cosmetic products against skin aging.19,21 Rising numbers of patent applications concerning either their preparation methods or their use as therapeutic agents demonstrate the rising interest of pharmaceutical and health oriented companies towards these reduced metabolites of natural isoflavones.
��
���
���
���
��
����
����
����� ����� ����� ����� �����
���������������
�������������
����������������������
������������������������

� ���
1.1. Isoflavonoid sources in nutrition, mammalian metabolism and biological activities
Isoflavones are the biggest subgroup of isoflavonoids, which are secondary metabolites formed in plants to act as phytoalexins. They are formed biosynthetically by the phenylpropanoid pathway from flavonoids by isoflavone synthase (IFS).22 These plant defensive compounds have raised special attention in medicinal and biological disciplines due to their various physiological effects also in mammals. For example, the effects of dietary flavonoids and isoflavonoids on xenobiotic and carcinogen metabolism have been under study.23
Isoflavonoid sources in human and other mammal nutrition Soy beans contain the phytoestrogenic isoflavones daidzein 16 and genistein 18.1 Soy is the most important isoflavone source in human nutrition. The typical isoflavone intake in Western cultures is less than 1 mg/d, whilst in Asian countries it is as high as 20-50 mg/d due to high soy consumption.25
Red clover (Trifolium pratens), which is used in organic farming as cow feed in Finland, also incorporates isoflavones abundantly, mostly formononetin 19 and biochanin A 20, but also their 4’-O-demethylated forms daidzein and genistein. Red clover isoflavones and their reduced metabolites are potential components also in human nutrition for example, if organic cow milk is consumed.26 Red clover extracts are also used as food supplements in human nutrition due to anticipated beneficial effects of isoflavones to human health. In plants, isoflavones occur mostly as glycosides having a sugar moiety. For example the precursor of daidzein, daidzin 21 (daidzein-7-O-�-glycoside) has a glycoside group connected to its 7-O- position. Isoflavonoid metabolism studies The known isoflavone metabolite equol 15 (7,4’-dihydroxyisoflavan) was first identified in the urine of pregnant mares and was found to be estrogenic.27 Equol itself does not occur in any plant. It has been isolated as a mammalian metabolite of isoflavones present in food. Isoflavone metabolism studies received more attention in the 1940’s, when subterranean clover grazing ovines were found to suffer from an infertility syndrome in Western Australia: from whence feed derived isoflavonoids were implied as a cause for hormonal disorders.28 The red clover isoflavones formononetin 19 and biochanin A 20 are first converted into the main soy isoflavones daidzein 16 and genistein 18 by mammalian gut microflora and further metabolised to various mostly unknown reduced structures. There seem to be notable differences between species and even individuals in their ability to metabolise food isoflavones. According to a study by our group and our collaborators, only 30 - 50% of the people studied were able to convert daidzein to equol 15 and 80-90% to ODMA 22.VI Gut microflora have an important role in isoflavone metabolism. For example germ free rats did not convert soy isoflavones to

� ���
equol,2 and newly born babies are not able to produce equol after soy milk consumption due to the lack of intestinal bacteria.30,31 Specific enzymes involved in the metabolism of dietary isoflavones, including daidzein, in man are currently unknown.32 In man, the metabolic pathways discovered thus far produce reduced metabolic structures such as equol 15 and O-DMA 22.33-35 Some minor hydroxylation36-38 of isoflavones 1 and/or their reduced derivatives has also been reported,39,40 however the concentrations of these oxidised forms are only fractional compared to the reduced metabolites. Isoflavone metabolism in man has only been studied to some extent thus far; predominantly in vitro studies, and some qualitative GC-MS analysis have been reported.41-43 Our group identified an isomer of equol, 3’,7-dihydroxyisoflavan 23 from human urine (Fig. 3).44 The same compound has also been discovered from cow’s milk.44 Our group also identified 6´-OH-ODMA 24 as the second human metabolic �-methyldeoxybenzoin derived from genistein.45 Isoflavone metabolism studies have also suffered from incorrect interpretations of GC-MS results due to the lack of authentic reference compounds. For example, one proposed daidzein metabolite from human urine was claimed to be 2,3-dehydro-O-DMA 25 and another the enol tautomer 27 of dihydrodaidzein 26 (Fig. 3).46,47
However, our group has shown that these compounds are just artefacts arising in the silylation of dihydrodaidzein during sample preparation and they are thus not true metabolites of daidzein.45

� ���
Fig. 3. Isoflavonoids discussed in the metabolism section.
The proposed main metabolic pathway for the soy isoflavone daidzin 21 in man is presented in Scheme 1. Biological activities or absolute configurations of most of the chiral metabolites have not yet been clarified in great detail in any species. Our group has identified the daidzein metabolite 7,4’-dihydroxyisoflavan-4-ol 28 to be cis-isomer, by applying the synthetic diastereopure reference compound.45 In contrast to this result, Kim and Han and coworkers claimed that this natural metabolite is the trans-isomer according to their semi-empirical CD studies with theoretical simulation of the chiroptical transitions.48-50 In the two later publications they reported that their initial absolute configurational determinations by used spectroscopic methods (CD, ECD) were not reliable (discussed in more detail in section 1.2.1). Also reported chemical structures were incorrectly drawn. The absolute configuration of the mammalian metabolite equol 15 was not determined before this thesis work due to lack of enantiopure reference compound.

� ���
Scheme 1. The proposed main catabolic pathways of natural soy isoflavone daidzin based on the urinary isoflavonoid metabolites found in human urine45,51 Dotted arrows indicate only tentative identification.
Biological effects of isoflavonoids
The most abundant soy isoflavones, daidzein 16 and genistein 18, are able to bind to estrogen receptors (ER) in mammals due to their structural similarity to natural hormone estradiol. 52 Isoflavone metabolites, such as the isoflavan equol 15, bind with even higher affinity and thus have stronger biological effects than the parent

� ���
compound daidzein 16.53 Equol is also a potent inhibitor of the Na-K-Cl cotransport system.54 Our group recently participated in a study of equol’s ligand binding capacity for estrogen-related receptor � (ERR �).55 Equol 15 was demonstrated to act as an ERR� agonist and is therefore capable of enhancing the growth inhibitory effect of ERR� on the prostate cancer PC-3 cells. Chiral equol is capable of binding to both estrogen receptors alpha and beta.56 The S and R enantiomers of equol (29, 30 respectively), obtained by chiral HPLC of the racemic mixture, have been shown to have different binding affinities to the estrogen receptors � and �; the R enantiomer preferring the ER� and the S enantiomer preferring the ER�. The S-enantiomer of equol 29 is also a potent antagonist for dihydrotestosterone, a growth stimulant for the prostate gland.57
According to in vitro studies, isoflavonoids may also prevent osteoporosis.58-60 Our group studied the antitumor activities of the reduced metabolites of daidzein trans- and cis- tetrahydrodaidzein 31 and 28 in an in vitro study. They were shown to be comparable to genistein 18 against human prostate cancer cells.61 The same compounds are also known potent antioxidants.62 The first identified human metabolic � methyldeoxybenzoin (1,2-diarylpropan-1-one), the triphenolic O-DMA 22, has shown equal binding activity to that of endogenous estradiol (E2) binding to estrogen receptor � in an in vitro experiment.63 In contrast, isoflavones genistein 18 and daidzein 16 showed weaker binding activity than O-DMA 22.
1.2. Synthesis of isoflavonoid metabolites Prior to this work, the absolute configurations of natural chiral isoflavonoids in man and other mammals were not determined using authentic chiral reference compounds. Synthetic isoflavonoids related to this thesis were also needed in our collaborative studies to measure their biological activities.V A thorough review of the literature suggested the lack of suitable synthesis methods to prepare polyphenolic isoflavonoids in general and especially the lack of asymmetric approaches was clear. 1.2.1 Synthesis of Isoflavan-4-ols To date, there are no reports about the synthesis of the single enantiomers of isoflavan-4-ols 3. In general, only unsubstituted or alkoxysubstituted isoflavone 1a-b reductions by nucleophilic hydrides such as NaBH4 to prepare mixtures of diastereomeric isoflavan-4-ols (cis- and trans-32 a-b) were described prior to commencement of this work (Scheme 2.). 64-66

� ���
Scheme 2. Reduction of methoxy- or unsubstituted isoflavones with sodium borohydride. The reduction of nonphenolic isoflavan-4-ones 2 by LiBH4, NaBH4, L-Selectride or Li(t-BuO)3AlH has also been used to produce mixtures of trans- and cis-isoflavan-4-ol enantiomer pairs cis-32, trans-32 (cis/trans ratio 4:5 – 4:3). 67,68
Unsubstituted or methoxysubstituted cis-isoflavan-4-ols cis-32 a-b have been produced from isoflavan-4-ones 2 by the use of large excess (80 eq) of electrophilic hydrides (borane-THF, BTED).68 Furthermore, a synthesis of methoxy or unsubstituted trans-isoflavan-4-ols 33a-b by hydroboration of the corresponding 3-aryl-4-hydroxycoumarins 34a-b in 42-54% yields was performed (Scheme 3).
Scheme 3. Trans-isoflavan-4-ols by a hydroboration of 3-phenyl-4-hydroxycoumarins. 68
The first papers describing the synthesis of diastereomeric mixtures of trans- or cis-isoflavan-4-ols 32 had no NMR information and there was no reliable data available for structural assignments.
An approach to synthesise the 2-aryl substituted isoflavan-4-ol analogue 35 was published by Venkati and Krupadanam69 who reported the synthesis of 2,3-trans-3,4-trans-2-aryl-3- arylchroman-4-ol 35 starting with the Knoevenagel reaction of aldehyde 36 and 7-O-methoxydeoxybenzoin 37 (Scheme 4) with an unknown total yield.
O
OH
R O
R
R
1) BH3
2) NaOH, H2O2
O
OH
R
R
R42-52%
34a R=H34b R=OMe
33a R=H33b R=OMe

� ��
Scheme 4. Synthesis of 2-aryl substituted analogue of isoflavan-4-ol 35 starting from p-methylbenzaldehyde 36 and 2’-hydroxy-4’-O-methyldeoxybenzoin 37.69
There is an isolated study about the chiral HPLC separation of a racemic mixture of unsubstituted isoflavan-4-ol stereoisomers:48 the semi-empirical analysis about the absolute configurations by CD and optical rotation of the isoflavan-4-ol stereoisomers is also reported: however, these results were reported to be unreliable more recently due to the spectroscopic methods used.49,50 To correct the unreliable configurational results, ECD spectroscopy with theoretical TD-DFT computations were used. The observed experimental ECD spectra resulted in a poor simulation, and thus inconclusive absolute configurational assignments of isoflavan-4-ol stereoisomers were again obtained. The correct absolute configurations of the four stereoisomers were determined by yet another method. DFT- assisted VCD spectroscopic analyses confirmed the absolute configurations of the isoflavan-4-ols 3. The absolute configurations of the isoflavan-4-ols were further confirmed by a modified Mosher method, yielding results opposite to their initial assignments. However, reported chemical structures were incorrectly drawn and thus this data remains unclear. Without synthetic enantiopure reference compounds, the elucidation of the correct stereochemistry of the natural metabolite is thus complicated and time consuming requiring various spectroscopic methods and theoretical calculations. Only single crystal X-ray crystallography will give absolute configuration most reliably, if good quality crystals are available.

� ��
1.2.2 Synthesis of isoflav-3-enes and isoflav-2-enes Synthesis of isoflav-3-enes 5 are only rarely described in the chemical literature. Even fewer papers describe the synthesis of isoflav-2-enes 6. The promising anticancer70 isoflavonoid dehydroequol 17 (other names for 17 according to SciFinder are 4’,7-dihydroxyisoflav-3-ene, 3-(4-hydroxyphenyl)-2H-1-benzopyran-7-ol, phenoxodiol, haginin E or idronoxil) has been isolated from the plant Lezpedeza homoloba.71 It has been suggested as a human metabolite of the isoflavone daidzein, and has raised interest also as an antioxidative71 and antiinflammatory72 agent. Dehydroequol and related structures have also been suggested as potent synthetic estrogens or antiestrogens.73-77 Dehydroequol 17 was found to prevent immune suppression induced by UV radiation and thus may have a future role as sun-protective cosmetic ingredient. It is also suggested to be a novel cardioprotective therapeutic together with other tested isoflavonoids dihydrodaidzein 26, equol 15, cis- and trans-tetrahydrodaidzein 28, 31.78 Detailed studies on the dehydroequol 17 and other phenolic isoflav-3-enes have been made difficult by the lack of synthesis methods reported in the literature. Just recently natural phytoisoflavonoid, 2’,3’-dimethoxydehydroequol (Haginin A) 38 was isolated from the branch of Lespedeza cyrtobotrya.79 Haginin A exhibits a strong hypopigmentary effect in Melan-A cells and significantly inhibits melanin synthesis according to Kim et al.79
There are only a few papers describing the synthesis of this class of compounds, which have low yields, are unselective and contain no hydroxysubstitution. The first approach to synthesise nonphenolic isoflav-3-enes 5 was the acid-catalysed dehydration of isoflavan-4-ols 3 prepared by hydride reductions of isoflavone 1 or isoflavan-4-one 2.80-82 The intermediate isoflavan-4-ols were usually not isolated but were dehydrated directly to the isoflav-3-enes. Also one patent application about the multistep synthesis of polyphenolic isoflav-3-enes by dehydration of isoflavan-4-ols in unspecified total yield has been published.83 In our experience this reduction dehydration sequence does not work for phenolic compounds due to the mixtures of products obtained and target isoflav-3-enes are formed only as minor products if at all.VII The synthesis of 1:1 mixtures of isoflav-2-enes 39 and isoflav-3-enes 40 in 3-10% yields from isoflavylium salts 41 has been reported (Scheme 5),84 whilst other publications describe nonphenolic isoflavylium salts as intermediates in the synthesis of isoflav-3-enes and /or isoflav-2-enes.85-87

� ���
Scheme 5. Reduction of isoflavylium salt 41 with sodium cyanoborohydride to the 1:1 mixture of corresponding isoflav-2-ene or isoflav-3-ene in 3-10% yields.84
There are also isolated papers describing the syntheses of unsubstituted or alkoxy or halosubstituted isoflav-3-enes and /or isoflav-2-enes. One method involves the reduction of isoflavones by the Clemmensen reaction (Scheme 6).88
Scheme 6. Synthesis of isoflavenes by the Clemmensen reduction, in which 4-OH products are not intermediates.88
A low-yielding non-reductive method to produce isoflav-2-enes 42 has also been reported, involving a palladium-catalysed tandem cyclisation-hydride capture process starting from bromobenzoacetophenone 44 and iodophenol derivative.89 The multistep route produces protected isoflav-2-ene 42 together with benzofuran 43 unselectively (Scheme 7).

� ���
Scheme 7. Preparation of isoflav-2-enes 42 together with 3-aryl-2,3-dihydrobenzo-furans 43.89
Another non-reductive route to produce the monobenzyloxy isoflav-3-ene 45 was introduced by Simas et al.90 Production of 3,4-chromene 46 after conjugate addition of phenyl lithium and desulfonation leads to isoflavan 47, isoflav-3-ene 45 or isoflavan-4-one 48 (Scheme 8).

� ���
Scheme 8. Synthesis of isoflav-3-ene 45, isoflavan 47 and isoflavan-4-one 48 structures through 4-chromenesulphone.90 Only recently, Li et al. (2009) published the total synthesis of dehydroequol 17 (Haginin E) and equol 15 starting from resorcinol 49 via the protected isoflavene intermediate 50 (Scheme 9).91 This multistep approach seemed complicated and time consuming. It contains a two step formylation of resorcinol 49 to corresponding benzaldehyde 51, which was further protected by monobenzylation to 4-benzyloxysalicylaldehyde 52. Then a Wittig reaction, O-alkylation, and another Wittig reaction produces the diene 53, transformed into an intermediate isoflav-3-ene 50 by a ring-closing metathesis using Grubbs’ catalyst (2nd generation). Finally, after deprotection with BCl3 under varying conditions the target isoflav-3-enes, Haginin E 17 or O-methyl derivative 54, were derived to give approximately 26% yields. Also 4’-O-methylequol 55 or equol 17 could be obtained from the intermediate isoflav-3-ene 50 by Pd(OH)2 in EtOH and cyclohexene reflux. Alternatively, the corresponding 7-O-benzyl-4’-O-methylisoflavan-4-ol 56 could be obtained in moderate 20% total yield by BH3-SMe2 in THF followed by treatment with water, 10% NaOH and 37% hydrogen peroxide. The protected isoflavan-4-ol 56 was used as starting material to prepare the protected isoflavone 57 and after demethylation or debenzylation gave isoflavones daidzein 16 and formononetin 19.

� ���
The total synthesis required several steps and chromatographic purifications, the first one already in the preparation of the starting material 4-benzyloxysalicylaldehyde 52. The overall yield of dehydroequol 17 was 26% and for daidzein 16 less than 15%.
Scheme 9. Synthesis of Haginin E 17, equol 15 and other isoflavonoids starting from resorcinol (Li et al.).91

� ���
Gupta and Ray presented a synthesis of dehydroequol 17 through a Friedel-Crafts (F-C) acylation reaction of 3-methoxyphenol 59 and 4-hydroxyphenylacetic acid 60 (Scheme 10).92 By the F-C reaction they obtained a mixture of two deoxybenzoins, 4”-O-Et substituted 62 and 4”-OH substituted 63. After chromatographic separation, the 4”-ethoxydeoxybenzoin 62 was reacted by cross aldol condensation with paraformaldehyde in an aqueous solution of sodium hydroxide to yield the corresponding isoflavan-4-one 61. The reduction of the carbonyl group by NaBH4 in methanol gave the isoflavan-4-ol 64, which was dehydrated into protected dehydroequol 65 by hydrogen chloride (HCl) in ethanol and then underwent deprotection by pyridinium hydrochloride to give dehydroequol 17 in a total yield of 27%. The route contained two chromatographic purifications of the intermediates and a third for the final product purification, thus was not a very efficient procedure. Furthermore, the use of ethoxylated deoxybenzoin 62 instead of free phenolic 63 was quite unexpected. The ethoxylated deoxybenzoin 62 is a side product formed from decomposed boron trifluoride etherate (BF3OEt2) if freshly distilled BF3-etherate is not used and is not normally formed.93
Scheme 10. Synthesis of dehydroequol 17 starting by Friedel-Crafts acylation of protected resorcinol 59.92
Recently, Erhardt and coworkers have published two articles and one patent application regarding the synthesis of isoflav-3-enes 66, 67 as key intermediates for the first total synthesis of racemic and natural enantiopure pterocarpans, (-)-glyceollin 68 and its derivatives.94-96 They used a Wittig reaction to produce the isoflav-3-ene substructures with appropriate MOM/Bn/TBDMS-protected substitution patterns with a yield of less than 12% overall as shown in Scheme 11.

� ���
Scheme 11. Synthetic route used to prepare protected DHE 67 as starting material for racemic and natural glyceollin 68 and derivatives.94-96
1.2.2.1 2-O-Substituted or 2-N-substituted isoflav-3-enes There are also some examples of the synthesis of 2-O- or 2-N-substituted isoflav-3-ene analogues, which are not true isoflavonoid derivatives. The preparation of isoflav-3-ene analogues with 2-O-substitution 70, 71 has been performed by reduction of 3-arylcoumarins 69 (Scheme 12).77,80,97-99 Synthesis of 2-OH-isoflav-3-ene 72 from protected salicylaldehyde 73 and arylglycidate salt 74 has also been reported (Scheme 13).86

� ���
Scheme 12. Synthesis of a 2-O-substituted isoflav-3-ene analogue 71 starting from corresponding 3-arylcoumarin 69.77
Scheme 13. Synthesis of a 2-OH-isoflav-3-ene 72 from protected salicylaldehyde 73 and arylglycidate salt 74.86 Varma and coworkers reported a method to produce nonphenolic 2-morpholinoisoflav-3-enes 75 with no other substitution or monomethoxy substitution via the condensation of N-styrylamines 76 with ortho-hydroxyarylaldehydes 77 to give 46-55% yields (Scheme 14).100-102 Later, they prepared other 2-N-substituted (morpholino, piperidino or pyrrolidino) derivatives of isoflav-3-enes under solvent free conditions using microwave irradiation in the presence of ammonium acetate as a catalyst to give 73-80% yields.103

� ���
Scheme 14. A solvent-free preparation of 2-morpholino-substituted isoflav-3-enes 75a and 75b using microwave irradiation (Varma and !�"�#�).103�
The formation of the 2-N-isoflav-3-ene derivatives 75 through the dehydration of the intermediate 2-N-isoflavan-4-ols was explained by the mechanism shown in Scheme 15.
Scheme 15. Proposed mechanism for the formation of 2-N-isoflav-3-ene derivatives 75.103�
1.2.3 Synthesis of isoflavans The preparations of isoflavans 4 reported in the literature are mainly based on the catalytic hydrogenation of nonphenolic isoflavones 1. Hydrogenation reactions have been achieved by palladium catalysis on a variety of supporting materials such as charcoal, calcium carbonate (CaCO3), aluminium oxide (AlO2) or barium sulfate (BaSO4) in different reaction media with varying results. The yields and product ratios are dependent on the concentration, pH, solvent, protection groups and the reducing reagent used. Hydrogenation of isoflavones 1 is reported to give mixtures of isoflavans 4, isoflavan-4-ones 2, isoflavan-4-ols 3, �-methyldeoxybenzoins 7, deoxybenzoins 78, pterocarpans 8, 3-cyclohexyl-3,4-dihydro-2H-1-benzopyrans 79, 3-phenyloctahydro-2H-chromenes 80, isoflav-3-enes 5 or isoflav-2-en-4-ols 81 (Fig. 1, Fig. 4).67,80,104,106-113

� ��
Fig. 4. Structures of the reported hydrogenation products of isoflavones discussed in section 1.2.3. Additionally, the isolated total syntheses of racemic isoflavans have been reported, but mostly in quite low yields with time consuming procedures. At the time of this thesis work, no method to prepare enantiopure polyphenolic isaflavans (such as (S)-equol 29 or (R)-equol 30) had been reported in the literature. Even to date, only two papers and one patent application describing the asymmetric multistep total syntheses of chiral phenolic isoflavans have been published (Heemstra et al. 2006 , Takashima et al. 2010, Setchell and Sorokin 2010; in more detail section 1.2.3.3).114-116
1.2.3.1 Catalytic hydrogenation of isoflavones
The most commonly used hydrogenation catalyst for isoflavones is palladium on charcoal (5 or 10% w/w). Different solvents and pH conditions have been used to try to optimise the reactions or improve the selectivity. Table 1 provides a summary of variable results of the Pd/C catalysed hydrogenation reactions of a number of isoflavones under different conditions collected from the literature. As can be seen in Table 1, results vary from one study to another depending on the Pd:isoflavone ratio, solvent, hydrogen consumption, reaction temperature, catalyst type or supplier used. Szabo et al. (1973, 1976) studied the effects of pH, different solvents and the amount of H2 consumed.110,111 Four different reduced structures were claimed to be produced in different reaction media. Chang et al. (1995) also reported product variability derived from the different solvents used.104 In previous experiments performed in our laboratory, no other products than isoflavans 4 were produced, regardless of which solvent was used.117
The identification of the products have also been uncertain in several cases. Chang et al. reported that the hydrogenation of daidzein 16 over 5% Pd/BaSO4 in EtOH produced the corresponding isoflavan-4-one (dihydrodaidzein) 26 as the major product.104 The identification of their synthetic product was said to be based on the comparison of 1H NMR shifts with published values not given in the paper. In one report, Pd on aluminium oxide and on calcium carbonate were claimed to have low selectivities and activities, but no product data was given.111 One report claimed isoflavan-4-ones 2 as reduction products with palladium on CaCO3 in ethanol, but identification of the compounds was based only on MS.118 A more recent paper (2009)119 reported a variety of products to be formed from the reduction of daidzein with different Pd/C catalyst types used in AcOH-EtOH (1:9 to 1:99). The solvent ratio was reported to have a significant effect on the reaction rate and products formed. A

� ��
new isoflavonoid structure was also claimed to be formed, namely 7,4’-dihydroxyisoflav-2-en-4-ol 82 from the reduction of the corresponding isoflavone daidzein 16, but no spectral data to confirm the structure or other physical properties were given.
In general, isoflav-2-en-4-ols 81 have only rarely been mentioned in the chemical literature and no reliable spectral data have been reported so far.120 The first mention was made by Thakar et al.121 when they claimed to have prepared unsubstituted isoflav-2-en-4-ol 81 in 75% yield by a hydride reduction with NaBH4 of the corresponding isoflavone 1 in ethanol, whereas isoflavan-4-ol 3 (88%) was obtained in diglyme. Product characterisation relied on the elemental analysis (C15H12O2) only. It should be kept in mind that the same elemental composition fits also to isoflavan-4-ones 2 (enol tautomer isoflav-3-en-4-ols 83), which is also a potential hydride reduction product of isoflavones 1 (Fig. 1).122,I Only recently (2010), a patent application was published concerning a selective reduction of isoflavone 4-keto group to 4-hydroxy, without the reduction of the 2,3-double bond.123 This may have occured via enzymatic or hydride reduction, various hydride reduction reagents were suggested; however no identification data such as spectral or crystallographic data or melting points were given for isoflav-2-en-4-ols 81 and thus this data is not entirely reliable.
Fig. 5. The elemental composition of the hypothetical isoflavonoid metabolite 81 is shared with the isoflavan-4-ones 2.

29
Tab
le 1
. Sum
mar
y of
Pd/
C c
atal
ysed
hyd
roge
natio
ns o
f is
ofla
vone
s in
the
liter
atur
e. R
eact
ion
tem
pera
ture
s an
d pr
essu
res
are
ambi
ent,
if
not
spe
cifi
ed.
Is
ofla
vone
sub
stit
utio
n P
d:is
ofla
vone
rat
io
(mol
. eq)
.
Sol
vent
, tem
pera
ture
Is
ofla
vono
id
prod
uct (
subc
lass
num
ber*
*) (
1-8,
79-
81),
yi
eld
%
Ref
.
7,3’
-diO
H-8
,2’,
4’-t
riO
Me-
*
HO
Ac
4, *
K
uros
awa
et
al.12
4 2’
,4’,
5’,6
’,7-
pent
aOM
e-
1:6
EtO
H +
2.5%
HO
Ac
4, 8
0%
Pelt
er&
A
men
echi
125
3’,4
’,5,
7-te
traO
Me-
1:
2.33
(P
dCl 2
/C)
HO
Ac,
100
o C
4 an
d oc
casi
onal
ly o
nly
1, 6
3%
Rob
erts
on e
t al
.126
No
subs
titu
ents
1:
5.8
HO
Ac
4,
61%
K
uros
awa
et
al.12
7 3’
,7-d
iOH
-4’,
8-di
OM
e-
1:5.
8 A
cOH
4,
80
%
Kur
osaw
a et
al
.128
1) 2
’,4’
,7-t
riO
CH
2OM
e-
2) 2
’,4’
,7-
triO
CH
2OC
H2C
H2O
Me-
1)-2
)1:1
ac
eton
e 1)
4, 6
3%
2) 4
, 92%
, 2 3
%
Suss
e67
No
subs
titu
ents
1:
4.8
1)
dio
xane
, 1 m
ol e
q H
2 2)
eth
anol
, 1 m
ol e
q H
2 3)
gla
cial
ace
tic
acid
, 3 m
ol e
q H
2 4)
Bri
tton
-Rob
inso
n bu
ffer
,pH
11-
12, 2
mol
eq
H2
5) B
-R b
uffe
r, p
H 3
-9, 2
mol
eq
H2
6) B
-R b
uffe
r, p
H 9
-10,
1 m
ol e
q H
2 7)
NaO
H, p
H 1
1-12
, 0.8
-2 m
ol e
q H
2 8)
eth
anol
, 3 m
ol e
q H
2
1) 2
, 95%
2)
3, 9
0%
3) 4
, 90%
4)
7, 6
0%
5) 3
, 90%
6)
2, 9
0%
7) 7
, 20-
60%
8)
4, 9
5%
Szab
o et
al
.110,
111
1)-2
) 7,
4’-d
iOH
- 3)
5,7
,4’-
triO
H-
4) 5
,7,4
’-T
riO
H-
* 1)
HO
Ac
2) e
than
ol
3) e
than
ol
4) H
OA
c
1) 4
, 46.
7%
2) 3
, 60%
3)
2, 6
7%
4) 4
, 67%
2, 2
7%
Cha
ng10
4
7,4
’-di
OH
- 1:
1.6
or
1.6:
1 aq
. eth
anol
4,
68-
99%
W
ähäl
ä117
5-O
H-
10%
Pd/
Cm
ol-e
q? +
0.1%
tri
ethy
l am
ine
as c
at.
aq. e
than
ol
2, *
B
ulut
80
1)-2
) 7,
4’-d
iOA
c 3)
5,7
,4’-
triO
Ac
4) 5
,7,4
’-tr
iOH
1) P
d/C
pre
oxyg
enat
ed f
or 2
-3
days
1.4
:1,
2) 1
:6.3
, 3)
-4)
1:5.
4
1) g
laci
al a
ceti
c ac
id
2) A
cOH
, 689
0kP
a, 9
5o C
3)
AcO
H, 9
990
kPa
,95o C
4)
AcO
H, 6
890
kPa,
95o C
1) 4
, 64%
2)
1, 7
9, 8
0, 4
3)
4, 6
2%
4) m
ainl
y 2,
but
mix
ture
of
seve
ral
com
poun
ds
Lam
bert
on10
9
1) a
cety
late
d is
ofla
vone
2)
hyd
roxy
subs
titu
ted
isof
lavo
ne
1)-2
) P
d/C
: iso
flav
one
rati
o no
t gi
ven
1)
-2)
HO
Ac
1) 4
, *
2) m
ixtu
re, *
A
ntus
107

30
1)-3
) tr
iben
zylo
xy
isof
lavo
ne
1) 1
:2.6
2)
1:2
.3
3) 1
: 6.6
5
1)ac
eton
e 2)
ace
tone
+ a
q. H
Cl
3) a
ceto
ne
1) 4
, 32%
8, 3
8%
2) 1
, 2, 8
*
3) m
ixtu
re o
f 5
prod
ucts
; *
Vis
ser10
6
4’-O
H-2
’-O
Me-
7-m
etho
xy-
etho
xy-
1:3.
5 H
OA
c 4,
28%
A
ntus
et a
l.129
2’
-OH
-4’-
OM
e-7-
OC
H2O
Me-
1:3.
4 E
tOH
:AcO
H 9
:1, 3
atm
4,
62%
V
an
Hee
rden
112
7-O
H-4
’-O
Me-
*
* 4,
93%
B
reyt
enba
ch
et a
l.130
1) 7
-OH
-4’-
OM
e-
2) 3
’,7-
diO
H-
1) 1
:22
2) 1
:14
glac
ial H
OA
c 1)
4, 9
2%
2) 4
, 86%
L
uk, e
t al.13
1
7,4’
-diO
H-
Pd/
C *
*
2 an
d 4,
*
Krä
mer
et
al.13
2 7,
2’-d
iOH
-4’-
OM
e-is
ofla
vone
ca
taly
tic
hydr
ogen
atio
n,
no
reag
ent o
r eq
uiva
lent
s w
ere
give
n 1)
ace
tone
2)
HO
Ac
1) 2
, *
2) 4
, *
Fark
as e
t al.13
3
2-be
nzyl
oxy-
1:
16
AcO
H
4, 9
0%
Fw
u et
al.13
4
7,2’
,4’-
triO
H-
1:4.
25
HO
Ac,
3 a
tm
4, 1
3%
Woo
dwar
d 11
3
7,4’
-diO
H-
1) 5
% P
d/C
(N
.E. C
hem
cat,
type
S
TD
) 2)
10%
Pd/
C (
Wak
o)
3) 5
% P
d/C
, wet
(50
% w
ater
, N
.E. C
hem
cat,
type
ST
D)
4) 5
% P
d/C
, wet
(50
% w
ater
, N
.E. C
hem
cat,
type
ST
D)
1)-3
) H
OA
c-E
tOH
1:9
4)
HO
Ac-
EtO
H 1
:99
1) m
ixtu
re o
f 2,
isof
lav-
3-en
e 5,
mai
nly
isof
lav-
2-en
-4-o
l 81,
trac
e of
4
2) m
ixtu
re o
f 2,
isof
lav-
3-en
e 5,
(is
ofla
v-2-
en-4
-ol)
81,
trac
e of
4
3) o
nly
4
isof
lava
n 4)
2, 8
1
(iso
flav
-2-e
n-4-
ol)
and
trac
es o
f 5
and
4,
but m
ainl
y un
reac
ted
1 w
as r
ecov
ered
Got
o et
al.11
9
*Not
spe
cifi
ed
** I
sofl
avon
e 1,
isof
lava
n-4-
one
2, is
ofla
van-
4-ol
3, i
sofl
avan
4, i
sofl
av-3
-ene
5, �
-met
hyld
eoxy
benz
oin
7, p
tero
carp
an 8
, 3-c
yclo
hexy
l-3,
4-di
hydr
o-2H
-1-b
enzo
pyra
n 79
, 4-
perh
ydro
-1-b
enzo
pyra
n-3-
yls
80, i
sofl
av-2
-en-
4-ol
81

� ���
1.2.3.2 Multistep total syntheses For the preparation of isoflavans with unsaturated substituents, other methods than the reduction of isoflavones have to be used. For example, prenyl substituted isoflavan 5-O-methyllicoricidin 84 (Scheme 16)135 was prepared by a complex 15-step route containing Wittig olefination and allylic oxidation by selenium dioxide, two Mitsunobu reactions, Claisen rearrangement, selective hydroboration and finally nuclear alkylation of the second intramolecular Mitsunobu reaction product 91, which had produced the isoflavan moiety. A major drawback in the route was the need of chromatographic flash purification of the acid sensitive Claisen rearrangement intermediate 88. The Claisen rearrangement also yielded 2-methyl-2-phenyl-2,3-dihydrobenzofuran 89 as reaction byproduct. The overall yield was less than 5 %.
Scheme 16. Total synthesis of 5-O-methyllicoricidin 84.135
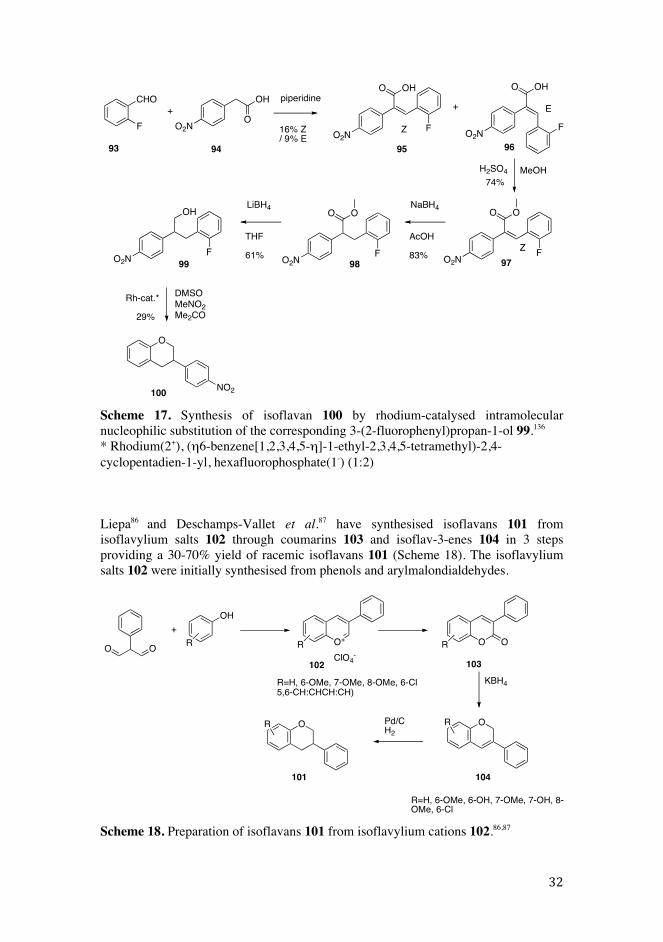
Methoxy-, nitro- and unsubstituted isoflavans, such as 100, have been prepared in 16-29% yields by rhodium-catalysed intramolecular nucleophilic substitution of the corresponding 3-(2-fluorophenyl)propan-1-ol 99 (Scheme 17).136 The overall yields of isoflavans were less than 2%.
OHO
O
+
OH
ORRO
PPh3-DEAD
O
O
O
RO OR
RO OR
O
O
OR
RO OR
O
O
OH OH
OHHO OH
O
O
O
(AcO)2ONaOAc, 210oC
Borane or9-BBN1. PPh3-DEAD
2. H2SO4MeOH
1. Et3NTBDMS2. n-BuLi, prenyl bromide
HO O
O
O
O
Si
HO OH
O
O
OHF, pyridine
R=CH2OCH2CH2Si(CH3)3
THF
R=H, -Me+
O
OMe
MeO
HO
OR
85 86
87
88
89
9091
92 84
� ���
Scheme 17. Synthesis of isoflavan 100 by rhodium-catalysed intramolecular nucleophilic substitution of the corresponding 3-(2-fluorophenyl)propan-1-ol 99.136
* Rhodium(2+), (�6-benzene[1,2,3,4,5-�]-1-ethyl-2,3,4,5-tetramethyl)-2,4-cyclopentadien-1-yl, hexafluorophosphate(1-) (1:2)
Liepa86 and Deschamps-Vallet et al.87 have synthesised isoflavans 101 from isoflavylium salts 102 through coumarins 103 and isoflav-3-enes 104 in 3 steps providing a 30-70% yield of racemic isoflavans 101 (Scheme 18). The isoflavylium salts 102 were initially synthesised from phenols and arylmalondialdehydes.
Scheme 18. Preparation of isoflavans 101 from isoflavylium cations 102.86,87

� ���
Usse et al. prepared unsubstituted 105 or monomethoxyisoflavans 106 in 3 steps starting from chromanone 107, which was converted to a triflate 108 and coupled with phenylboronic acid and finally hydrogenated with Pd/C in MeOH/CH2Cl2 to yield isoflavans 105-106 in unspecified total yields (Scheme 19).137
Scheme 19. Isoflavans 105-106 by Suzuki coupling of triflates 108 and phenyl boronic acids starting from chromanones 107 by Usse et al.137
Isoflavans 4 have also been prepared via isoflav-3-enes 5138-141 with multistep reactions and moderate yields by catalytic hydrogenations. Conti et al.139 and Burali et al.140 prepared isoflavans 109 in four steps starting from ortho-hydroxybenzyl alcohols 110 and �-bromoacetophenones 111 to yield 1-aryl-2-(o-hydroxymethyl)phenoxyethanones 112, which were converted into the corresponding triphenylphosphonium bromides 113. After the Wittig intramolecular cyclisation of triphenylphosphonium bromides 113 to isoflav-3-enes 114, Pd/C-catalysed hydrogenations gave isoflavans 109 as shown in Scheme 20. The reaction was not useful for chloro- or bromosubstituted derivatives, because partial dehalogenation occurred producing a product mixture.
Scheme 20. Synthesis of isoflavans 109a-b through the Wittig intramolecular cyclisation. Chloro- or bromo- substitution lead to partial dehalogenation in the last step and gave lower yields due to product mixtures.140

� ���
Recently, Gharpure et al. presented a multistep ortho-quinone methide 117 based approach to racemic mixtures of natural isoflavans equol 15, 3’-hydroxyequol 115 and vestitol 116 (Scheme 21).142 O-Methyl- or O-benzyl protected o-quinone methides 117 and aryl-substituted enol ethers 118 reacted in a Diels-Alder reaction followed by reductive cleavage of the methoxy acetal group of 119 using a Lewis acid and trialkylsilane. Then demethylation by pyridinium hydrochloride or debenzylation of 120 by H2/10% Pd/C in EtOAc gave hydroxyisoflavans in moderate yields.
Scheme 21. Synthesis of racemic isoflavans 15, 115, 116 by an ortho-quinone methide 117 based approach.142
Gupta and Ray presented a synthesis of rac-equol 15 through the Friedel-Crafts acylation reaction of 3-methoxyphenol 121 with 4-hydroxyphenylacetic acid 122. By modifying the reaction dehydroequol 17 could also be obtained as described earlier in

� ���
the previous section (Scheme 10).92 The formed isoflavan-4-one 123 was reduced to the corresponding isoflavan 124 by Pd/C hydrogenation, further converted to equol 15 by dealkylation with pyridinium hydrochloride in a 22% overall yield as shown in Scheme 22. The route contained two chromatographic purifications of intermediates. The final product also required chromatographic purification, thus causing low yields and numerous tedious steps.
Scheme 22. Synthesis of racemic equol 15 starting by a Friedel-Crafts acylation of the protected resorcinol 121.92
1.2.3.3 Synthesis of enantiopure isoflavans
There are only a few reports in the literature regarding the synthesis of enantiopure isoflavans. Natural isolated enantiopure pterocarpans 8 have been used to prepare chiral isoflavans 2 by Pd-catalysed hydrogenations to give approximately 70% yields144 or unspecified yields.144 This method however required a tedious chromatographic separation of natural starting materials at the beginning.
Versteeg et al.145,146 prepared non-phenolic isoflavans 128 via the �-alkylation of phenylacetate derivatives 129. The total yield starting from the synthesis of suitable benzyl bromide 130 was 15-21%, however this decreased when a more highly substituted benzyl bromide was used as a starting material. Various amounts of unwanted isomeric compounds, 2-benzyldihydrobenzo[b]furans 131, were formed as byproducts in the last cyclisation reaction.
Modification of the method by using a chiral auxiliary (imidazolidin-2-ones 132, 133), gave the enantiopure methoxy substituted isoflavan 128.147,148 The reaction started by stereoselective �-benzylation of phenylacetic acid derivatives 129149-153 (diastereomeric excess, de > 95%) and in the last step a Mitsunobu reaction was used to close the heterocyclic ring C. After reductive removal of the chiral auxiliary and cyclization, methoxysubstituted chiral isoflavan 128 was prepared in a total of 6-7 steps in a 35% yield (enantiomeric excess, ee > 96%) in a total of 6-7 steps (Scheme 23). However, the cleavage of the methoxy substitution was not published at the time, and was not even attempted for the production of chiral hydroxy substituted natural isoflavan (S)-equol 29.

� ���
Scheme 23. First asymmetric route to methoxysubstituted isoflavan 128.147,148
No asymmetric synthesis route to (S)-equol 29 or other free phenolic isoflavans were available at the time of this work. At the same time as we filed our own patent applicationIV about the asymmetric hydrogenation of dehydroequol derivative to prepare (S)-equol 29 and (R)-equol 30 (Scheme 46, section 3.4.2 Results and discussion), Setchell and Sorokin had filed their patent application (2005).116 Their approach was longer than ours, but they also used asymmetric hydrogenation of chromenes as a key step to prepare enantiopure isoflavan (S)-equol 29 (Scheme 24). The overall yield in their 6 step route to (S)-equol was 38.9%.
Scheme 24. Setchell’s and Sorokin’s asymmetric hydrogenation of the protected chromene 137 to prepare S-equol 29.116

� ���
They prepared the key intermediate isoflav-3-ene 137 by previously known reductive sequences. Protected isoflavone 134 was reduced to isoflavan-4-one 135, which was reduced to the corresponding protected isoflavan-4-ol 136 and further dehydrated to isoflav-3-ene 137 by methanesulfonic anhydride and triethyl amine in THF at room temperature. Shortcomings of the method of preparing 29 were the several steps, and the need to use energy consuming cryo chemistry and chromatographic purifications with toxic hexane as eluent in several steps. It should be mentioned that the melting point of their synthetic claimed enantiopure (enantiomeric excess, ee 100%) (S)-equol was considerably lower, only 162 oC, compared to previously reported natural isolated enantiopure equol (191 – 193 oC).154
After this thesis work and filing the patent applications, the second asymmetric synthesis of (S)-equol 29 by a total synthesis was reported (Scheme 25).114 The stereocenter was formed by Evans alkylation using a chiral auxiliary 138 ((S)-4-benzyloxazolidin-2-one). The key step to create the chiral center by Evans alkylation was very similar compared to the previously presented Versteeg et al. approach described in scheme 23. Intramolecular Buchwald etherification formed the chroman ring to give the isoflavan moiety 141, whereas the Mitsunobu reaction was used in Versteeg et al.’s approach. The method was only used for the enantiopure equol 29/30 production and no other chiral isoflavans were reported. Demethylation of protected isoflavan 141 was actually the only major difference in this approach compared to the Versteeg et al. procedure as they produced the natural phenolic chiral isoflavans 29 and 30.

� ��
Scheme 25. Total synthesis of enantiopure (S)-equol 29 by an asymmetric Evans alkylation to create the chiral center in 139.114
The most recent asymmetric synthesis by Takashima et al. (2010) produced (S)-equol 29 by a 15 step procedure.115,155 This approach also used allylic substitution as a key step to produce chirality (Scheme 26).

� �$�
Scheme 26. Enantioselective total synthesis of (S)-equol 29 by allylic substitution as the key step that gives a yield of approximately 24%.115,155
The method was only applicable to prepare chiral equol 29/30 and no other natural isoflavans ((R)-sativan 143 or (R)-vestitol 116) could be prepared selectively. When tested for the synthesis of natural (R)-sativan by the same procedure, 2-(2,4-dimethoxybenzyl)-2,3-dihydrobenzofuran-6-ol 144 was obtained as a 1:1 sideproduct along with the desired isoflavan (R)-sativan 143 from (R)-3-(2,4-bis(benzyloxy)phenyl)-2-(2,4-dimethoxyphenyl)propan-1-ol 145 (Scheme 27). The sideproduct was explained to be formed from the bromide isomer 146, which was a product of the 2,4-(OMe)2C6H3 group migration in 147 after bromination by CBr4 and PPh3. Other unidentified sideproducts were also formed in the bromination step. Because of these sideproducts, the previously known Mitsunobu reaction (Scheme 28) had to be applied after debenzylation to construct the benzopyran ring135,145,146,148 of isoflavans other than equol. 2,4-Dimethoxy substitution in the B-aromatic ring in 147 caused bromide group migration and formation of the sideproduct 146.

� ��
Scheme 27. 2-(2,4-Dimethoxybenzyl)-2,3-dihydrobenzofuran-6-ol 144 as sideproduct in benzopyran ring formation after unstable bromination product by Takashima et al.115,155
Scheme 28. Preparation of (R)-sativan 143 by using the Mitsunobu reaction in the final step.115,155 The route from Takashima et al.115,155 suffered from not being a generally applicable and robust route to variously substituted isoflavans without the Mitsunobu reaction in the formation of the heterocyclic ring C. The procedure contained also energy consuming cryo chemistry in many steps and expensive reagents in larger scale.

� ���
1.2.4 Synthesis of ��-methyldeoxybenzoins No synthesis methods for free polyphenolic �-methyldeoxybenzoins 7 were available prior to our work or even to date. The lack of asymmetric synthetic methods of polyphenolic �-methyldeoxybenzoins was especially obvious. 1.2.4.1 Synthesis of racemic ������������� �� � (1,2-diarylpropan-1-ones)
Human urinary metabolites, O-DMA 22 and angolensin 168, have been prepared by multistep routes with protection groups. First angolensin 168 was prepared by a Friedel-Crafts reaction of resorcinol dibenzyl ether and 2-(4-methoxyphenyl)propionic acid chloride.156 After debenzylation, racemic angolensin was obtained in a 13% yield. Corresponding 4-O-methyl ether of angolensin was produced from 1-benzyloxy-3-methoxybenzene in a 29% yield. The method was modified in our laboratory to prepare O-DMA 22 by using 1,3-dimethoxybenzene 169 and 2-(4-methoxyphenyl)propionic acid 170 in polyphosphoric acid (PPA). After demethylation of 171 with 1M boron tribromide (BBr3 ) in dichloromethane (DCM, CH2Cl2), the product 22 was obtained in an 85% overall yield (Scheme 29).35
Scheme 29. First synthesis of O-DMA 22 in our laboratory.35 Angolensin 168 has also been prepared to give an 11% yield by 2-methylation and deprotection of 1-[2,4-bis(benzyloxy)phenyl]-2-(4-methoxyphenyl)propan-1-one.157 In another report, angolensin was prepared by a base catalysed �-C-methylation of deoxybenzoin 172, O-methylation being prevented by benzyl protection (Scheme 30).158 After acidic debenzylation, angolensin was produced in a yield of about 60%. The preparation of these starting materials 172 and 173 clearly required further synthetic steps which lower the total yield.
Scheme 30. Synthesis of angolensin 168 by a C-methylation of deoxybenzoin to give a yield of about 60%.158

� ���
2,4-Dimethoxy-4’-hydroxy-�-methyldeoxybenzoin 174 has been prepared by a multistep route of eight steps.159 In this route, the starting tosylated dihydrochalcone 175 was obtained in five steps, starting from 2,4-dimethoxyacetophenone 176 and anisaldehyde 177. Irradiation in dioxan at 350 nm gave the rearranged product 178, which was reduced with sodium cyanoborohydride (NaBH3CN) to the target product 174 (Scheme 31).
Scheme 31. Photochemical route to 2,4-dimethoxy-4’-hydroxy-�-methyldeoxybenzoin 174.159
As noted previously (Table 1), catalytic hydrogenation of isoflavones 1 with Pd/C at pH 11-12 has also been claimed to produce �-methyldeoxybenzoins 7.110,111 Previously, in our laboratory, unprotected natural �-methyldeoxybenzoins 22 and 168 have been prepared from corresponding phenolic isoflavones 16 and 19 with lithium aluminium hydride (LAH, LiAlH4) in tetrahydrofuran (THF) both in 42% yields (Scheme 32).160 By the same method, genistein metabolite 6’-OH-O-DMA 24 has also been prepared in a 66% yield.161 The method provided OH-substituted �-methyldeoxybenzoins easily except when there was no hydroxy substituent at the isoflavone 7-position. In such cases, prop-2-en-1-ols 179 were the main products.
Scheme 32. Reduction of natural isoflavones with LiAlH4. 160,161
In contrast to some earlier proposals,111 it is assumed that the initial step in these reductions is a 1,4-addition to the enone system, driven by the 4-oxypyrylium contributor I to the isoflavone structure (Scheme 33).160,I This is followed by ring opening to a prop-2-en-1-one II, reduced by another 1,4-addition to give the deoxybenzoin enolates III, IV, or by an alternative 1,2-addition to give propenols VI.

� ���
It was suggested that the effect of the 7-hydroxy substituent is to provide a further resonance contributor IV where the propenone carbonyl is lacking, thus favouring conjugate hydride attack at the propene terminus.
Scheme 33. Proposed alternative reaction pathways for the LAH reduction of isoflavones with or without 7-OH-substitution by our group.160, I
Regioselective methylations of O-DMA 22 were carried out during the course of this thesis (Scheme 34).162
Scheme 34. Regioselective methylation of O-DMA. 162

� ���
Regioselective methylation of O-DMA 22 offers an expedient route to mono- or dimethylated O-ether derivatives by appropriately adjusting the substrate : base : electrophile ratios according to pKa differences between these three phenolic groups. 4’’-O-alkylation occurs via triphenolate anion formation by one equivalent of MeI in DMF to give angolensin 168 in a yield of 63%. In contrast, the 4’-O-alkylation to 180 occurs by using only one equivalent of base and MeI due to the more acidic nature of the C-4’ site than the C-4’’. Bis(methyl ether) 181 can be prepared by using excess base and MeI in a 70% yield. Chelation effects obviously obstructed O-alkylation at C-2’ thus favouring O-alkylation at C-4’.
The green chemistry approach by using microwave (MW) reactor and ionic liquid as co-solvent in Friedel-Crafts (F-C) acylation to prepare polyphenolic deoxybenzoins has also been studied in our laboratory.163 In case of �-methyldeoxybenzoins such as O-DMA 22 (4’,4’’-dihydroxy-�-methyldeoxybenzoin), it did not crystallise, but had to be extracted by organic solvent and then purified by flash chromatography in a 55% yield. In general, MW reaction times were short, and other deoxybenzoins than �-methylated were easily crystallised from ionic liquids. The alkyl substituted �-methyldeoxybenzoin 185 has been prepared by the palladium-(II) acetate/n-BuPAd2 catalysed �-arylation of ketone 186 with a non-activated chloroarene 187 (Scheme 35).164 More expensive Cs2CO3 was reported to be needed as base instead of K3PO4 to improve selectivity.
Scheme 35. Preparation of alkyl substituted �-methyldeoxybenzoin 185. 164
More recently, the methoxy substituted �-methyldeoxybenzoin 188 was prepared by a related Pd(II) catalysed �-arylation of the ketone 189 by 4-OMe-chlorobenzene 190 (Scheme 36).165,166 Aryl bromides gave twice as fast reactions, but yields were about 8-10% lower.

� ���
Scheme 36. Preparation of methoxy-substituted �-methyldeoxybenzoin 188 by �-arylation of the enolisable ketone 189. 165,166
Recently, monohydroxy-�-methyldeoxybenzoins 191-193 and deoxybenzoin 194 were prepared by the spontaneous Truce-Smiles rearrangement starting from propiophenones 195 and 1-fluoro-2-nitrobenzenes 196 (Scheme 37).167 The intramolecular nucleophilic aromatic substitution was improved by the additional electron withdrawing groups, such as NO2, in the B ring. Methoxy-substitution in the para-position of acyl ring A prohibited the Truce-Smiles rearrangement and intermediate diaryl ether 197a, not deoxybenzoin 194, was the main product isolated in a yield of 65 % due to electronic effects. This indicates that the procedure is not valuable in producing natural methoxy substituted or polyphenolic derivatives such as O-DMA 22 and angolensin 168.
Scheme 37. �-Arylation of aryl ketones by the Truce-Smiles rearrangement.167
1.2.4.2 Asymmetric synthesis of 1,2-diarylpropan-1-ones
No reports about the asymmetric synthesis of natural polyphenolic �-methyldeoxybenzoins existed prior to our work. The asymmetric synthesis of a

� ���
tetramethoxy substituted �-alkyldeoxybenzoin has been published,168 reporting the use of chiral pseudoephedrine auxiliary 199 (Scheme 38). The pathway consisted of the asymmetric alkylation of an arylacetic acid 200 via its (S,S)-pseudoephedrine amide 201. After a hydrolysis of 202 to remove the chiral auxiliary, followed by a Friedel-Crafts acylation, the chiral non-racemic methoxy-deoxybenzoin, with the appropriate substitution at the benzylic carbon, was obtained.
Scheme 38. Synthesis of enantiopure methoxy-1,2-diarylpropan-1-one 198.168
(S,S)-Pseudoephedrine 199 was first introduced by Myers et al. (1994) as a practical chiral auxiliary for the synthesis of highly enantiomerically enriched �-alkylated carboxylic acids, alcohols, aldehydes and ketones.169,170 The diastereoselective alkylation of pseudoephedrine amides was suggested to be derived from the
electrophilic attack on the putative (Z)-enolate 205 (R syn to the enolate oxygen) from the same face (1,4-syn) as the carbon-bound methyl group of the pseudoephedrine auxiliary when the enolate is drawn in a planar, extended conformation (see Fig. 6).
Fig. 6. Mnemonic for pseudoephedrine amide enolate 205 alkylation.169,170

� ���
Vicario et al.168 suggested that a more rigid structure than 206 for the reaction intermediate should be required in order to explain the high degree of stereoselection obtained. It was concluded that lithium ions, from LiCl, must be coordinated to the enolate oxygen and lithium alkoxide in the pseudoephedrine moiety through a bridging species present in the reaction media, such as the solvent or a chlorine anion from LiCl as drawn in 207 (Fig. 7).
Fig. 7. Proposed mechanism in the asymmetric alkylation of pseudoephedrine derived amide enolate 207. X denotes solvent, Cl or (iPr)2NH molecules.168

� ��
2. AIMS OF THE STUDY The objective of this study was to synthesise and identify plant derived isoflavone metabolites formed in the intestinal track in human, and other mammals such as ruminants, consuming a phytoestrogen rich diet. Interest in isoflavone metabolism derives from the anticipated beneficial biological effects of phytoestrogen metabolites on human and animal health. Possible adverse effects also require further research. The isoflavonoids synthesised in this thesis were used as authentic reference compounds in clinical chemistry studies to identify and quantify the metabolites in body fluids and tissues. The synthesis of these isoflavonoids was also important for the biological activity studies. The main goal in the synthetic work was to develop atom economical and expedient routes to produce free hydroxy substituted or phenolic isoflavonoids, which were unknown, according to chemical literature, prior to this work. For example, no published methods were available for reducing hydroxysubstituted isoflavones with hydride reducing agents in general. In particular, there were no methods to prepare enantiopure synthetic reference compounds for the characterisation of new chiral metabolites.

� �$�
3. RESULTS AND DISCUSSION
3.1. Isoflavone reductions by hydride reagents and hydrogenationsI, II, IV
An expedient one-pot synthesis of various polyhydroxy isoflavones 1 from an appropriate phenol and aryl acetic acid had been developed earlier in our group.93 The availability of these various polyphenolic isoflavones as starting materials offered a practical approach to study hydride reductions and hydrogenations by different available reagents in producing reduced isoflavonoid structures 2-5 and 7 (for the summary of the results see Scheme 39 below).
Scheme 39. Summary of the selective isoflavone reductions achieved in this study.
In general, sodium borohydride (NaBH4) is the most used reagent in reducing isoflavone 1 or isoflavan-4-one 2 skeletons according to the chemical literature. However, published results covered only unsubstituted or protected starting materials with variable product combinations formed (isoflavan-4-one 2 or cis- and/or trans-isoflavan-4-ols 3 or sometimes isoflav-2-en-4-ol 81 (Fig. 5)). In addition, the product characterisations were contradictory from one study to another as already discussed in section 1.2.1. This study describes the conformational equilibria of diastereomeric pairs of isoflavan-4-ols determined by NMR and clarifies the isoflavone hydride reductions in comparison to the prior ambiguous results reported in the literature.I,II,IV

� ��
The hydride reduction of �,�-unsaturated ketones, such as the isoflavone ring C, can take place by two alternative pathways. Addition to the conjugated double bond proceeds by 1,4-addition to first give an isoflavan-4-one 2, which is further reduced to an isoflavan-4-ol 3. Alternatively, the reduction can follow the 1,2-addition pathway to the carbonyl group. For example, the claimed isoflav-2-en-4-ols 81 were reported in the literature to be 1,2-addition products. During the experimental work, we discovered that the addition pathway of various hydride reagents to isoflavones 1 mostly involves the 1,4-pathway in contrast to the ordinary non-flavonoid �-alkoxy-�,�-unsaturated ketones, which favour 1,2-addition. Various products to be formed were dependent on the following factors: 1) the hydride reagent (electrophilic/nucleophilic, steric effects), 2) the substrate, (free OH or not; especially 7-position phenolic group was important in LAH reductions and determined whether a 1,4- or 1,2-addition took place) or 3) the pH of the reaction media. Under basic conditions with NaBH4 in EtOH 1-(2-hydroxyaryl)-2-arylprop-2-en-1-ols 211 were formed as sideproducts among isoflavan-4-ols 212, 213 (Scheme 40). The addition of boric acid blocked the sideproduct formation.
Scheme 40. Reduction of non-phenolic isoflavones with sodium borohydride.
The hydride addition reactions tested followed mainly the initial 1,4-addition route to give the isoflavan-4-one enolate. In a hydroxylic solvent, or even on workup under basic conditions, the ketone is generated, and reduced further to the saturated alcohol (NaBH4). The presence and number of phenolic or alkoxy groups in the substrates does not have a major effect on these reductions except in the case of NaBH4 reduction, which fails completely presumably due to solubility reasons, and LAH reduction where the outcome depends on the presence or absence of an OH substituent at C-7. �-Methyldeoxybenzoins could be prepared selectively by the 1,4-addition if starting isoflavone had the 7-hydroxy substitution.I In the case of 7-methoxy or hydrogen substitution in the starting isoflavone, 1-(2-hydroxyaryl)-2-arylprop-2-en-1-ols 211 were the main products observed after 1,2-addition (Scheme 40 above). A rare exception for the typical 1,4-addition by borohydrides was observed with the bulky 9-BBN. It was found to react by 1,2-addition to the carbonyl group to give isoflav-3-ene and not the 1,4-addition product isoflavan-4-one with both protected and free phenolic isoflavones as described in section 3.3 Scheme 43. IV

� ���
The previously claimed potential 1,2-addition product isoflav-2-en-4-ol 81 was not formed in any hydride addition reaction.I In addition, the dehydration of polyphenolic isoflavan-4-ols 3 by acidic treatment was not selective in producing isoflav-3-enes 5 as previously claimed by others.VII
Isoflavans 4 were only obtained by Pd catalysed hydrogenations (more details in section 3.4.1), not as hydride reduction products. 3.2. Synthesis of trans- and cis-isoflavan-4-olsI, II
As no diastereoselective syntheses of polyphenolic trans isomers of isoflavan-4-ols 3 were available or discovered by us, we developed an RP HPLC method to separate the two diastereomers from each other.II Diastereopure reference compounds are needed for the identification of natural metabolites as biological activities are determined to various degrees by the adopted conformations of the substrates.171 Trans-isoflavan-4-ols 215, 216 and cis-isoflavan-4-ols 217, 218 were separated from the prepared diastereomixtures by HPLC or column chromatography in order to study their conformational equilibria. Intermediate isoflavan-4-ones 219, 26 were prepared from corresponding isoflavones 1, 16 by catalytic transfer hydrogenation reactions as described in scheme 41.
Scheme 41. The structures of the isoflavones, isoflavanones and the isoflavanols (trans- 215, 216 and cis- 217, 218). Reaction conditions: (a) Pd/C, NH4OCHO, MeOH, reflux; (b) Li(t-BuO)3AlH (10 eq.), THF, 25-30oC (219 �� 215, 217). (c) LiBH4, THF, 0oC (26 � 216, 218).
Scheme 42. The generalized half-chair conformations adopted by the isoflavan-4-ols looking across the heterocyclic ring towards the fused aromatic ring. Cis isoflavan-4-ols can be anancomeric with only one conformer predominating61 whilst the trans isoflavan-4-ols are an equilibrium mixture of diequatorial and diaxial half-chair conformations.II

� ���
3.3. Synthesis of Isoflav-3-enesIV
7,4’-Dihydroxyisoflav-3-ene 17 (dehydroequol) was expediently prepared from readily available daidzein 16 by reduction with 9-BBN in THF (Scheme 43) in one step. Dehydroequol has previously been prepared either by multistep routes or unselectively together with corresponding isoflav-2-ene by the Clemmensen reaction (Scheme 6).
Scheme 43. Approach to the synthesis of dehydroequol 17 and O,O'-dibenzyldehydroequol 58. Reaction conditions: i. 9-BBN, THF, rt, 15h, 50-60%; ii. K2CO3, BnCl, DMF, refl., 90%.IV
Isoflav-3-enes 17 and 58 were useful substrates for the asymmetric hydrogenation experiments to prepare enantiopure polyphenolic isoflavans. The chiral iridium-PHOX precatalysts 231, 232 (Scheme 45) and 235, 236 (Fig. 8) were found to be highly enantioselective (ee 80-93 %) towards hydrogenations of isoflav-3-enes tested (see Section 3.4.2). 3.4. Synthesis of IsoflavansIV, VII
3.4.1 Racemic polyphenolic isoflavansVII
Due to problems caused by Pd on charcoal (varying reactivity between different batches, partly because of variable amounts of HCl traces left)172, another reagent was used in this work. Palladium on barium sulfate proved to be an excellent reagent for the heterogenous hydrogenation of isoflavones to produce polyphenolic isoflavans (Scheme 44). Ethanol (96%) was already proved to be a superior solvent for the reaction over AcOH or other solvents.117 Starting isoflavones are poorly soluble in EtOH, but product isoflavans are soluble, thus offering an easy separation of traces of starting material from the raw product by filtration.

� ���
Scheme 44. Preparation of isoflavans from unprotected isoflavones.VII
The most used method to prepare racemic isoflavans has been the hydrogenation of isoflavones with Pd/C. The problems with catalytic hydrogenation reactions of isoflavones have been in many cases the need to use protection of phenolic hydroxy groups, the unselectivity, moderate yields and many reaction steps. In this study, the objective was to try to optimise the Pd-catalysed hydrogenation of unprotected isoflavones, which are easily obtained by the one-pot Friedel-Crafts acylation developed in our laboratory.93 In a previous study to prepare isoflavans in our laboratory, it was concluded that excess Pd on charcoal (isoflavone:Pd metal ratio, 1 mol-eq : 1.6 mol-eq) produced the best yields.117 The Pd/C was not an optimal reagent though, because results varied when different batches or suppliers of the reagent were used. For example, it has been discovered in our laboratory that certain commercial batches of the Pd/C actually contained large quantities of PdCl2 which is reduced to Pd and HCl.172 Thus, a better carrier for the palladium metal was needed. 3.4.2. Enantiopure polyphenolic isoflavansIV
At the time of this thesis work, there were no prior reports about the synthesis of enantiopure polyphenolic isoflavans. The main objective was to develop an enantioselective synthesis of isoflavans, especially natural S-equol and its nonnatural enantiomer R-equol, for biological studies. The existing asymmetric approach with chiral auxiliaries developed by Versteeg et al.145,147,148 (Scheme 23) was not valuable in producing free hydroxy substitution through demethylation since known methods were not described at that time and would have added even more steps to an already long route. Only recently, a total synthesis of enantiopure equol was reported to give a less than 10% overall yield,114 There is also a recent study,173 where a certain human anaerobic bacteria strain was used to convert dihydrodaidzein 26, a known isoflavone metabolite itself, to (S)-equol 29. Another approach was taken in this study, namely the asymmetric organometal catalysed hydrogenation of isoflav-3-enes 5, which are conveniently prepared from readily available isoflavones 1 as shown in the previous section (Scheme 46).

� ���
Asymmetric hydrogenation of 58 with 3 mol% of iridium PHOX-catalysts 231 or 232 (preparation of 231 and 232 in Scheme 45) in dichloromethane (DCM) yielded dibenzyloxyequols 233 or 234 successfully, in yields of approximately 90%. Alternatively, the commercial threonine derived iridium PHOX complex 235 yielded 233 in a 90% yield. Given the limited solubility of even dibenzyloxydehydroequol 58 in DCM, catalyst loading was, to some extent, higher than in some earlier asymmetric olefinic hydrogenations with similar Ir-PHOX catalysts (0.3-3 mol%). With 58, 3-4 mol% precatalyst loadings were required to achieve good yields (ca. 90%) and enantioselectivities (ee 92-93%) at ambient temperature. Almost full conversions were also achieved by lower catalyst loadings, but ee values decreased to 40%. (S)-(-)-equol 29 and (R)-(+)-equol 30 were obtained after debenzylation of protected equol enantiomers 233 and 234. 7,4’-Dihydroxyisoflav-3-ene 17 (dehydroequol) was not soluble in DCM at a practical synthetic level, thus the asymmetric hydrogenation of 17 was not possible under similar conditions as for 58. The addition of co-solvent such as acetone repressed the catalysis as indicated by calculated results174 although the solubility of 17 would have been much better.
Two commercially available threonine derived iridium PHOX catalysts 235 and 236 were also tested. Both gave (R)-(+)-equol 30 with good yields and ee 80 - 93% starting from 58 (Table 3).IV
Scheme 45. Synthesis of iridium-BARF complexes 231 and 232 via corresponding chiral PHOX ligands.IV ORTEP plot for 231 in Fig. 9.

� ���
Fig. 8. Commercial threonine based iridium PHOX BARF-catalysts 235 and 236.
Fig. 9. X-ray crystal structure of the synthesised (S)-PHOX -based iridium BARF complex 231. ORTEP plot.

� ���
Table 3. Enantioselective hydrogenation experiments of 58 with use of different Iridium PHOX catalysts 231, 232, 235 and 236. All experiments gave yields about 90%.
experiment starting material
solvent catalyst T / oC
P / bar
reaction time / h
mol% catalyst
eea abs. conf.
1 58 DCM 231 rt 50 42 3 92 (R)
2 58 DCM 231 120 50 15 2 40 (R)
3 58 DCM 232 rt 50 15 3 92 (S)
4 58 DCM 232 rt 50 42 3 92 (S)
5 58 DCM 235 rt 50 15 4 93 (R)
6 58 DCM 236 rt 50 15 3 80 (R)
aThe ee values were determined by chiral HPLCIV and the absolute configuration was determined by single crystal XRD analysis (Fig. 10, Scheme 47).
Scheme 46. Synthesis of (S)-(-)-equol 29 and (R)-(+)-equol 30 from 7-O,4’-O-dibenzyldehydroequol 58.IV

� ���
The absolute configuration of the synthesised (S)-(-)-equol 29 was determined by a single crystal XRD measurement of the diiodo-(S)-equol derivative 29b (Fig. 10, Scheme 47), prepared from enantiopure (S)-(-)-equol 29 by the selective diiodination developed during this thesis to improve the accuracy of the XRD analysis.
Fig. 10. X-ray crystal structure of the synthesised (S)-(-)-6,8-diiodoequol 29b. ORTEP plot. �
�
������� ����!���� ��� ����� ��� synthetic (S)-(-)-equol 29 to (S)-(-)-6,8-diiodoequol 29b to be used in the single crystal XRD analysis in determining the absolute configuration of the prepared parent enantiopure (S)-(-)-equol 29 (more details in Experimental section).
In comparison with other published methods, this asymmetric hydrogenation of dehydroequol derivatives offers a competitive method for the preparation of bioactive chiral isoflavans.
OHO
OH
OHO
OH29b
N-iodosuccinimide
DMF, 0oC to rt49%29
I
I

� ��
3.5. Synthesis of ��-methyldeoxybenzoinsVII
No asymmetric synthesis of polyphenolic �-methyldeoxybenzoins 7 was available prior to this work. However, the reaction of chiral 2-arylpropionic acids 241, 242 with recorcinol 49 offered an expedient route to chiral polyphenolic �-methyldeoxybenzoins by the Friedel-Crafts acylation procedure (more details in section 3.5.2).VII A number of racemic polyphenolic �����"#�����#���������������
����������� �����#��"��� ��� ���� �"� VII
3.5.1. Polyphenolic �-methyldeoxybenzoinsVII
Previously, O-DMA 22 has been prepared from 1,3-dimethoxybenzene 169 and 2-(4-methoxyphenyl)propionic acid 170 in PPA giving an 85% yield as described in section 1.2.4.1 (Scheme 29) or by LAH reduction in THF of daidzein in 42% yield (Scheme 32). By adapting the boron trifluoride etherate (BF3
.OEt2) catalysed Friedel-Crafts acylation, developed for the one-pot synthesis of unprotected polyphenolic isoflavones previously in our laboratory, also a number of different polyphenolic �-methyldeoxybenzoins 22, 243-248 from the corresponding 2-arylpropanoic acids 256 a-d and polyphenols could be prepared only in one step and in good yields (ca. 70-90%) (Scheme 48, for more details, see section 3.5.1).VII �
R1, R2, R3=H 256a R4,R5=H 22 R1,R2,R3,R4,R5=H R1=OH, R2,R3=H 256b R4=OH, R5=H 243 R1,R2,R3,R4=H, R5=Me R1=H, R2=Me, R3=OMe 256c R4=H, R5=Me 244 R1,R2,R3,R5=H, R4=OH 256d R4=OMe, R5=Me 245 R1=OH, R2,R3, R4,R5=H 246 R1,R4=OH, R2,R3,R5=H 247 R1,R4=H, R3=OMe,R2,R5=Me 248 R1=H, R3,R4=OMe, R2,R5=Me
Scheme 48. Preparation of polyphenolic O-DMA derivatives by a direct F-C acylation without protection of phenolic groups. VII
Only recently, Goto et al. (2009) used an identical approach to prepare a number of different OMe- or OH-substituted �-methyldeoxybenzoins for use in radical-scavenging activity studies.119

� �$�
3.5.2. Enantiopure �-methyldeoxybenzoins [1,2-(diaryl)propan-1-ones]VII The asymmetric synthesis of tetramethoxy substituted �-methyldeoxybenzoins (no substitution in the C-2 position in ring A as in natural O-DMA derivatives) has been published,168 where the use of chiral pseudoephedrines was also introduced. However, the methoxy-substitution is not really effective in producing polyphenolic O-DMA derivatives owing to the partial racemisation in the deprotection step as demonstrated in this study by the demethylation of diphenolic (R)-(-)-angolensin 249 (described in more detail at the end of this section).VII
In this study, a natural diphenolic (R)-(-)-angolensin 253 and its enantiomer (S)-(+)-angolensin 255 were prepared for the first time by adapting the approach already described in the previous section, using the one-step BF3-etherate catalysed Friedel-Crafts acylation between resorcinol 49 and a chiral acid 241/242. The chiral acids 241/242 were first prepared by using pseudoephedrine 199 and its enantiomer 210 as chiral auxilliaries (XC) (Scheme 49, Table 4; for more details see section 5. Experimental). The pseudoephedrine amide enolates 251 and 252 were prepared from commercially available 4-methoxyphenylacetic acid (253) after in situ acid chloride formation and by using (R,R)-(-)- or (S,S)-(+)-pseudoephedrines as chiral auxiliaries.168,VII The diastereoselective methylation of the formed pseudoephedrine amide enolates was readily performed by electrophilic attack of MeI after dianion formation with LDA in the presence of LiCl in THF at –78oC (2h) and at 0oC for 6 hours to produce diastereopure �-methylated pseudoephedrine amides 254 and 255 (d.e. >95% by 1H NMR). Chiral auxiliaries were cleanly removed by acid hydrolysis yielding 241 and 242 in a yield of approximately 70% after a standard aqueous acid-base extraction and recrystallisation from aq. MeOH.
The chiral acid 242 has been previously prepared by the enzymatic hydrolysis of racemic �-methylarylacetonitrile using Rhodococcus sp. CGMCC 0497 in an unspecified yield,175 but the other chiral acid 242 has not been prepared before. The enantiopure angolensins 249 and 250 were prepared by the F-C acylation reaction of commercially available resorcinol 49 and the chiral acid 241 or 242. The reaction conditions were equal to already described method for the preparation of racemic O-DMA derivatives in the previous section. The overall yields of (R)- and (S)-angolensins 249 and 250 were 44% and ee 97-98%. The absolute configuration of the prepared (-)-angolensin 249 was determined to be R by a single crystal X-ray (XRD) measurement (Fig. 11). The opposite enantiomer was concluded to be (S)-(+)-angolensin 250. (R)-(-)-angolensin has also been isolated from the plant Pterocarpus angolensis, but no x-ray data was reported then.176,177
Demethylation of the first prepared diphenolic (R)-(-)-angolensin 249 or (S)-(+)-angolensin 250 (Scheme 49) by BBr3 in DCM, as described for racemic angolensin in Scheme 29, led to partial racemisation according to our experience (ee decreased from

� ��
98% to 53%)VII and therefore this approach was not useful for the preparation of enantiopure triphenolic (R)-O-DMA or the other enantiomer (S)-O-DMA.VII
Scheme 49. First synthesis of enantiopure (R)-(-)-angolensin 249 and (S)-(+)-angolensin 250 developed during this work. Reagents and conditions: i) SOCl2, toluene, refl.; ii) (R,R)-(-)-/ (S,S)-(+)-pseudoephedrine, Et3N, THF, -20oC, ~85%; iii) LDA, LiCl, THF, -78oC; iv) MeI, THF, 0oC, ~88%; v) dioxane, 4M H2SO4, refl., 69-70%; vi) resorcinol, BF3OEt2, 79-81% (Table 4 yields and de and ee in more detail).

���
Fig. 11. X-Ray crystal structure of the synthesised (R)-(-)-angolensin 249. ORTEP Plot.
Table 4. Stereoselectivities (diastereomeric excess, de or enantiomeric excess, ee) and yields obtained in the asymmetric synthesis of the enantiopure angolensins 249, 250.
product yield
(%)d
de
(%)a
product yield
(%)
product
yield
(%)d
ee
(%)b
tR(min)
254c 87 >95 241c 69 249 R-(-)-angolensin
79 98 13.5
255c 88 >95 242 70 250c S-(+)-angolensin
81 97 27.5
a Determined by 1H NMR. b Determined by chiral HPLC; Ultron ES-OVM for 249 and 250; c new compound. dafter column chromatography purification. Analysis conditions described in section 5. Experimental.

� ���
3.6. Isoflavonoids in biological studies Table 5. Isoflavonoid metabolites identified from mammalian species in conjunction with the synthesis research project and their biomedical studies. Isoflavonoid
/ reference
Body fluids found
/ reference
Biomedical studies / reference
(S)-equol (29)IV Serum of ewesIII Receptor binding activity of S-enantiomer vs. rac-mixtureV
Rac-O-DMA (22)VII Human urineVI
Cow milk26 ODMA producing ability in manVI
Rac-equol (15)VII Human urineVI
Cow milk26 Equol producing ability in manVI
Trans-7,4’-dihydroxyisoflavan-4-ol I, II
Not found Antitumor activity against human prostate cancer cells in vitro were comparable to genistein and cis-isomer.
61 cis-7,4’-dihydroxyisoflavan-4-ol (28)II
Human urine45 Antitumor activity against human prostate cancer cells in vitro were comparable to genistein and trans-isomer.61
3.6.1. S-equol 29 determination from serum of ewesIII
In this study (S)-(-)-equol 29 has been identified from the serum of ewes after red clover feeding for the first time using the enantiopure reference compound.III In the same study O-DMA 22 was also identified. Coinciding with the publication of this paper, a report about X-ray single-crystal analysis of (-)-S-equol 29 isolated from rat faeces in vivo was published.154 Simultaneously, human intestinal bacteria was reported to solely produce the S-enantiomer of equol. 178
Quantitative HPLC-DAD determination of isoflavonoids from red clover fed ewe serum showed that equol 15 was the main metabolite present (7.7 mg mL-1). Thus the chiral HPLC analysis was developed to determine absolute configuration of the natural enantiomer by the aid of prepared chiral reference compounds.III, IV Thus, the natural equol was proved to be solely the S-enantiomer 29. 3.6.2. ODMA 22 and equol 15 in human isoflavone metabolism studies Only about half of the human population are able to convert isoflavones to further metabolites such as equol 15and 80-90% are able to produce O-DMA 22.179-184 For example, rats, chimpanzees and mice are proved to be equol producers and no interindividual differences are found.185 Bovines are also able to convert daidzein to equol and excrete it in their milk.105 Our interdisciplinary study186 about the concordance within an individual for the equol-producer and O-DMA producer

� ���
phenotypes were measured at two time points (T1, T2), 1-3 years apart. Phenotypes were ascertained by analyzing equol and O-DMA using GC-MS in a spot urine sample collected after 3 days of soya (source of daidzein) supplementation. In ninety-two individuals, 41% were equol producers at T1 and 45% were equol producers at T2, and 90% were O-DMA producers at T1 and 95% were O-DMA producers at T2. These results suggest that these phenotypes are stable in most individuals over time, providing a useful biomarker for evaluating disease risk associated with harboring particular intestinal bacteria responsible for the metabolism of the soy isoflavone daidzein. Another study from our group187 revealed the differences between equol and O-DMA producing abilities among Caucasian and Korean American women and girls. The Caucasian population had a lower equol-producing prevalence, but a higher O-DMA producing ability compared to Korean Americans. These findings support the reports that, compared with Western populations, Asian populations have a higher equol-producer prevalence, which may be in association with a lower mortality due to hormonal cancers. Synthetic equol has also been used in studies about serum steroid hormones, sex hormone-binding globulin concentrations, and urinary hydroxylated estrogen metabolites in post-menopausal women in relation to daidzein-metabolizing phenotypes. VI,188 3.6.3. Biological activity of S-equol 29 vs. rac-equol 15 in vitroV A comparison study of the effects of the racemic equol 29 with that of S-enantiomer 29 on the growth, invasion and DNA integrity of breast and prostate cells in vitro was performed. Both racemic and pure S-equol inhibited the growth of the breast cancer cell line MDA-MB-231 (>10 mM) and the prostate cancer cell lines LNCap (>5 mM) and LAPC-4 (>2.5 mM). The compounds also showed equipotent effects in inhibiting the invasion of MDA-MB-231 and PC-3 cancer cells through matrigel. S-equol (1, 10, 30 mM) was unable to prevent DNA damage in MCF-7 or MCF-10A breast cancer cells following exposure to 2-hydroxy-4-nonenal, menadione or benzo(a)pyrene-7,8-dihydrodiol-9,10-epoxide. On the contrary, racemic equol 15 (10, 30 mM) prevented DNA damage in MDF-10A cells following exposure to 2-hydroxy-4-nonenal or menadione. These findings indicate that racemic equol has strong antigenotoxic activity in contrast to the single S-enantiomer implicating the R-, rather than metabolic S-enantiomer as being responsible for the antioxidant effects of equol. These results may have implications on the in vivo chemoprotective properties attributed to equol.
3.6.4 Equol quantitation in organic skimmed Finnish cow milk26
The isoflavonoids, equol rac-15, formononetin 19, daidzein 16, genistein 18, biochanin A 20 and O-DMA 22, were analyzed from commercial cartons of skimmed Finnish cow milk by HPLC- (DAD)-FL. Identification was confirmed by comparing the UV-spectra of the eluting peaks with authentic reference compounds. Flavone 14 was used as a standard to calculate recovery, which was 80% +/- 6%. According to analysis 411 +/- 65 ng/mL of equol 15 and traces of formononetin 19 and daidzein 16

� ���
were detected in organic skimmed milk whereas conventionally produced milk contained only 62 +/- 16 ng/mL of equol and no formononetin or daidzein. Results indicate that isoflavone rich red clover feed used in Finnish organic farming is responsible for high levels of equol in cow’s milk. Discovery indicates that more studies about the health effects of organic products should be conducted due to growing consumption of these products. 4. CONCLUSIONS In this thesis work, a direct and highly enantioselective route toward enantiopure (R)-equol 30 and (S)-equol 29 was developed. Both of these equol enantiomers are found to have positive biological activity in mammals. In addition, the first direct and efficient synthesis of dehydroequol 17, a potent anticancer drug, by a one step synthesis from readily available daidzein 16, was presented. The first reported enantioselective synthesis of enantiopure angolensins (249, 250) has also been described. The method also offers a synthesis route towards enantiopure O-DMA 22 by modifying the 4’’-O-protection group in chiral acids to, for example, silyl or benzyl which are more easily cleaved than methyl. Synthesis of chiral (R)-O-DMA and its enantiomer are under further development to be used in the identification of the metabolic eutomer. Other polyhydroxy isoflavonoids have also been synthesized to be used as standards in analytical and metabolic studies of these natural nutrition based metabolites. In this study, the isoflavone hydride reductions were clarified in comparison to the prior ambiguous results reported in the literature. The discovery of (S)-equol 29 from the serum of ewes by using authentic reference compound clarified the metabolism track of daidzein 16 and formononetin 19 in these production animals. The described metabolic findings suggested that more studies about the biological effects of these metabolites are needed, since for example cows’ milk, and also blood, are used in the human food industry as ingredients, and they are direct sources of reduced isoflavonoids in human nutrition. Effects and biologically safe dosages of (S)-equol 29 and other mammalian metabolites to human health are still to be discovered.
5. EXPERIMENTAL The experimental part of the work that has not been included in the publications I-VI is described in this section. General Experimental Procedures NMR spectra were recorded on a Bruker 500 Avance or a Varian 300 Mercury Plus spectrometer. 1H-NMR (500 MHz) and 13C-NMR (125 MHz or 75 MHz) spectra were referenced to tetramethylsilane (TMS) at 0.00 ppm. J values are reported in Hz. Low and high resolution mass analyses were recorded on a JEOL JMS SX102 mass spectrometer operating at 70 eV. TLC was conducted on Merck silica gel 60 F254

� ���
plates, and Merck silica 60 (0.040-0.063 mm, 230-400 mesh) was used for flash chromatography. Chiral HPLC analysis for ee determinations were performed on a Waters model 996 HPLC instrument with Photodiode Array detector and Waters 717 plus autosampler. Measurement conditions: Ultron ES-OVM, 250 x 4.6mm, ID., 10 mm particle size, MeOH-phosphate buffer (pH 4.65), 25:75 as eluent flow rate 1mLmin-1 for (R)- and (S)-angolensin. Cyclobond I 2000 RSP column, 250x10 mm, 10 mm, Astec guard column system 4 mm ID, 2 cm lenght, 30:70 ACN:0.1% formic acid 3.5 mLmin-1 for (R)- and (S)-O-DMA. Melting points were determined in open capillary tubes, with an Electrothermal apparatus, and are uncorrected. Specific optical rotations were measured with a Jasco DIP-1000 Digital Polarimeter. THF was purified and dried by refluxing in continuous distillation apparatus with sodium and benzophenone as an indicator prior to use. Triethyl amine, toluene and dichloromethane were distilled over CaH2.
5.1 Preparation of (S)-6,8-diiodo-7,4’-dihydroxyisoflavan 29b
Chiral (S)-equol 29 (0.024g, 0.10 mmol) and sodium iodosuccinimide (0.023g, 0.10 mmol) were dissolved in dry DMF (1 mL) under argon atm and stirred at 0oC for 17h. The reaction was quenched by adding water followed by extraction with diethyl ether and evaporation to dryness. The raw product was a light yellowish oily prepicipitate and was flash chromatographed in silica gel by eluting with DCM. The combined DCM fractions were evaporated to dryness to give 0.024g (0.049 mmol, 49.0%) of 29b, which was recrystallised from hexane/diethyl ether to give white crystals, which could be used in the single crystal XRD measurement of the absolute configuration of the parent (S)-equol 29 (Fig. 10)
� (300 MHz, d6-acetone) 4.08 (dd, J 10.5, 10.5, H-2 ax), 4.37 (ddd, J 10.5, 3.6, 2.1, H-2 eq), 3.11 (dddd, J 10.5, 10.6, 5.7, 3.6, H-3 ax), 2.96 (ddd, J 15.6, 10.6, 1.0, H-4 ax), 2.91 (ddd, J 15.6, 5.7, 2.0, H-4 eq), 7.50 (s, H-5), 7.17 (d, J 8.6, H-2’,6’) 6.83 (d, J 8.5, H-3’,5’), 7.69 (br s, OH), 8.28 (br s, OH); (EI) m/z (rel. int): M+ 493.9 (100%), 368.0 (50%). HRMS C15H12O3I2 requires 493.8876, observed 493.8879.
5.2 Preparation of chiral 2-(4-methoxyphenyl)-propionic acids 241 and 242.
Commercial 4-methoxyphenyl acetic acid (5.9 g, 0.030 mol) was converted to it’s corresponding acid chloride by refluxing with SOCl2 (0.044mol, 5.5mL) in toluene (78mL) and further to the amides 251 and 252 by using either (S,S)-(+)- or (R,R)-(-)-pseudoephedrine (4.95g, 0.030mol) and Et3N (5.0mL) in THF (150mL) at -20oC for 4.5 hours in 88% yield. Amides 251 and 252 were asymmetrically methylated using LDA (0.053mol), LiCl (6.70g, 0.158mol) and MeI (0.029mol, 1.82mL) in THF (70mL) in –78oC to 0oC to produce 254 and 255 with a yield of 89%. Chiral auxiliaries were cleanly removed by refluxing with 4M HCl in dioxane yielding 241 and 242 in a yield of approximately 70%.
(-)-(1R,2R)- and (+)-(1S,2S)-N-(2-Hydroxymethyl-2-phenylethyl)-N-methyl (4-methoxyphenyl) acetamide 251 and 252. White plates, 88%, mp 131oC (toluene) lit.:189 130-132oC. [a]D
28=-108.0 (c. 0.2, CH2Cl2) 251, [a]D27=+108.0 (c. 0.2, CH2Cl2)
252. 1H and 13C NMR and IR as reported.189 (EI) m/z (rel. int): 313 (M+, 0.5), 295 ([M-

� ���
H2O]+, 100), 206 (55), 148 (42), 121 (58); (M–H2O)+ C19H21O2N requires 295.1572 observed 295.1570.
(-)-(1R,2R)- and (+)-(1S,2S)-N-(2-Hydroxymethyl-2-phenylethyl)-N-methyl-2-(4-methoxyphenyl) propionamide 254 and 255. Colourless oil 89%, [a]D
29=-92.0 (c. 0.2, CH2Cl2) 254 and [a]D
29=+92.0 (c. 0.2, CH2Cl2) 255. 1H NMR d (2.75:1 rotamer ratio, * denotes minor rotamer peaks, 300 MHz CDCl3) 0.52* (d, J=7.0, 3H), 1.10 (d, J=7.0, 3H), 1.37 d, J=7.0, 3H), 2.67 (s, 3H), 2.90* (s, 3H), 3.67-3.72 (m, 3H), 3.73* (s, 3H), 3.77 (s, 3H), 4.0-4.58 (m, 4H), 6.78-6.85 (m, 2H), 7.09-7.16 (m, 2H), 7.24-7.40 (m, 5H); 13C NMR d (3:1 rotamer ratio, * denotes minor rotamer peaks, CDCl3, 75 MHz ) 14.1, 14.6*, 20.8, 21.0*, 42.7*, 43.3, 55.3, 57.8, 75.5*, 76.4, 114.3, 126.4, 126.8, 127.3*, 127.6, 128.2, 128.7*,133.5, 134.5*, 141.5*, 142.3, 158.2*, 158.4, 171.2, 175.1*, 176.2; (EI) m/z (rel. int): 327 (M+, 1%), 309 ([M-H2O]+, 100), 221(98), 220(98); (M–H2O)+ C20H23O2N requires 309.1729 obsvd 309.1743.
(R)-(-)- and (S)-(+)-2-(4-methoxyphenyl)propanoic acid 241 and 242.
White solid, mp 74oC (MeOH) lit.:190 73-74oC. [a]D27=-86.2 (c. 0.2, CH2Cl2) lit.:190
[�]D20=-59 (c. 0.3, EtOH) 241, [�]D
27=+86.2 (c. 0.2, CH2Cl2) 242. 1H NMR, IR and MS as reported for corr. (S)-(+)-acid.175
5.3 Preparation of enantiopure polyhydroxy ��-methyldeoxybenzoins by the one step Friedel-Crafts acylation
Resorcinol (0.012g, 0.12 mmol) and enantiopure 2-aryl propionic acid (0.022g, 0.12 mmol for 241) and BF3OEt2 (1 mL) were stirred at 70oC under argon atmosphere, until no starting material was observed on TLC-monitoring, and quenched with 2M HCl. The mixture was extracted with diethyl ether, washed carefully with water and dried with Na2SO4, filtrated and solvent evaporated to yield (R)- or (S)-1,2-diarylpropan-1-ones as yellowish oils. After short column chromatography, recrystallisation yielded purified crystalline �-methyldeoxybenzoins ee 97 to 98% as confirmed by chiral HPLC analysis (Table 4.).
[2R]-(-)- and [2S]-(+)-1-(2,4-dihydroxyphenyl)-2-(4-methoxyphenyl)propan-1-one 249 and 250.
Flash chromatography ((7:1)CH2Cl2/EtOAC) yielded 249 (81%) and 250 (79%) as colourless oil, recrystallisation from aq. EtOH yielded light yellowish crystals; mp 121-122oC (lit.:177 120-122oC, lit.:160 84oC for racemic angolensin). [�]D
26=-115.0 (c. 1.0, CH2Cl2) lit.:
177 [�]D19=-115.0 (EtOH) 249, [�]D
26=+115.0 (c. 1.0, CH2Cl2) 250; 1H NMR, 13C NMR, MS, IR and UV as reported for racemic angolensin.160

� ���
5.4 Preparation of racemic polyhydroxy-��-methyldeoxybenzoins 244-246
Racemic 2-(aryl)propionic acids 256a-d were prepared by the procedure previously described.35,191 The general procedure to prepare rac-polyhydroxy-�-methyldeoxybenzoins 244-246 by a Friedel-Crafts acylation was as described for enantiopure derivatives in section 5.3.
1-(2,4,-Dihydroxyphenyl)-2-(3,4-dihydroxyphenyl)-propan-1-one 244
Flash chromatography (EtOAc) yielded 244 (80%) as a yellowish oil, recrystallization from aq. EtOH yielded light yellowish crystals; mp 90oC; d (500 MHz, d6-acetone) 1.41 (3H, d, J 6.8, C(3)Me), 4.68 (1H, q, J 6.8, C(2)H), 6.29 (1H, d, J 2.4, C(3’)H), 6.34 (1H, dd, J 8.6, 2.4, C(5’)H), 6.70 (1H, ddd, J 8.6, 2.2, 0.35, C(5”)H), 6.75 (1H, d, J 8.1, C(6”)H), 6.82 (1H, d, J 2.2, C(2”)H), 7.88 (1H, d, J 9.0, C(6’)H), 8.61 (1H, s, C(4’)OH), 12.9 (1H, s, C(2’)OH); d (125 MHz, d6-acetone) 19.4 (C3), 46.7 (C2), 103.5 (C3’), 108.6 (C5’), 113.1 (C1’), 115.3 (2”), 116.4 (C6”), 119.8 (C5”), 134.1 (C6’), 134.5 (C1”), 144.8 (C3”),146.1 (C4”), 165.3 (C2’), 166.6 (C4’), 204.8 (C1); m/z (EI) 274 (M+, 11%), 137(100); C15H14O5 requires 274.0841 obsvd 274.0853.
1-(2,3,4-Trihydroxyphenyl)-2-(4-hydroxyphenyl)-propan-1-one 245
Flash chromatography (EtOAc) yielded 245 (90%) as a yellowish oil, recrystallization from benzene yielded light yellowis crystals; mp 202oC; d (500 MHz, d6-acetone) 1.43 (3H, d, J 6.8, C(3)Me), 4.78 (1H, q, J 6.8, C(2)H), 6.40 (1H, d, J 9.0, C(5’)H), 6.77 (1H, d, J 8.6 C(3”)H), 6.77 (1H, d, J 8.6 C(5”)H), 7.19 (1H, d, J 8.6, C(2”)H), 7.19 (1H, d, J 8.6, C(6”)H), 7.50 (1H, d, J 9.0, C(6’)H), 7.74 (1H, s, C(3’)OH), 8.61 (1H, s, C(4’)OH), 12.9 (1H, s, C(2’)OH); d (125 MHz, d6-acetone) 19.4 (C3), 46.0 (C2), 108.1 (C5’), 113.2 (C1’), 115.7 (C3”,5”), 123.7 (C6’), 129.3 (C2”,6”), 133.1 (C4’), 133.6 (C1”), 152.3 (C3’), 153.6 (C2’), 157.1 (C4”), 206.8 (C1); m/z (EI) 274 (M+, 29%), 153(100), 121 (34); C15H14O5 requires 274.0841 obsvd 274.0826.
1-(2,3,4-Trihydroxyphenyl)-2-(3,4-dihydroxyphenyl)-propan-1-one 246
Flash chromatography (EtOAc) yielded 246 (75%) as a yellowish oil, recrystallization from chloroform/EtOAc yielded off-white crystals; mp 113-115oC; d (500 MHz, d6-acetone) 1.41 (3H, d, J 6.8, C(3)Me), 4.71 (1H, q, J 6.8, C(2)H), 6.40 (1H, d, J 9.0, C(5’)H), 6.70 (1H, ddd, J 8.6, 2.2, 0.35, C(5”)H), 6.75 (1H, d, J 8.1, C(6”)H), 6.82 (1H, d, J 2.2, C(2”)H), 7.49 (1H, d, J 9.0, C(6’)H), 7.74 (1H, s, C(3’)OH), 8.61 (1H, s, C(4’)OH), 12.9 (1H, s, C(2’)OH); d (125 MHz, d6-acetone) 19.2 (C3), 46.8 (C2), 103.9 (C3’), 111.8 (C1’), 115.4 (2”),116.0 (C6”), 116.3 (C6’), 119.7 (C5”),134.8 (C1”), 138.5 (C5’), 144.9 (C3”),146.2 (C4”), 154.8 (C4’), 160.7 (C2’), 206.4 (C1); m/z (EI) 290 (M+, 20%), 153 (100), 137 (17); C15H14O6 requires 290.0790, obsvd 290.0783.

� ��
5.5 General procedure for isoflavans
Isoflavans 4, 15, 226-230 were synthesised by hydrogenation of the corresponding isoflavones over palladium on barium sulfate with vigorous stirring at room temperature. Isoflavones (1 g) were dissolved in 96% ethanol (50 mL). Hydrogenation in the presence of 10% Pd/BaSO4 (1.6:1 [mol-eq.] isoflavone:Pd) was continued until no more hydrogen was consumed. Filtration and evaporation of the solvent yielded yellowish oils, which were crystallised from aq. EtOH. 7-Hydroxy-4’-methoxyisoflavan 226 White crystals 85% mp 153-155oC (from aq. ethanol) (lit.192 mp 160oC) � (500 MHz, d6-acetone) 3.96 (dd, J 10.4, 10.5, H-2 ax), 4.21 (ddd, J 10.5, 3.6, 2.0, H-2 eq), 3.12 (dddd, J 10.5, 10.6, 5.7, 3.6, H-3 ax), 2.91 (ddd, J 15.6, 10.6, 1.0, H-4 ax), 2.86 (ddd, J 15.6, 5.7, 2.0, H-4 eq), 6.90 (d, J 8.2, H-5), 6.37 (dd, J 8.2, 2.5, H-6), 6.29 (d, J 2.4, H-8), 7.26 (d, J 8.4, H-2’,6’) 6.91 (d, J 8.8, H-3’,5’), 8.12 (s, 7-OH), 3.79 (s, 4’-OMe); � (125 MHz, d6-acetone) 71.5 (C-2), 38.8 (C-3), 32.6 (C-4), 131.0 (C-5), 108.9 (C-6), 157.6 (C-7), 103.7 (C-8), 114.0 (C-4a), 156.0 (C-8a), 134.7 (C-1’), 129.3 (C-2’,6’), 114.9 (C-3’,5’), 159.6 (C-4’), 55.5 (4’-OCH3); m/z (EI) 256 (M+, 85%), 149 (13), 134 (100), 121 (20), 91 (10),77 (5), 65 (3), 51 (2); HRMS calcd for C16H16O3
256.1099, found 256.1109. UV and IR as reported.88
5,7-Dihydroxy-4’-methoxyisoflavan 227
White crystals 84% mp 229-230oC (from aq. ethanol) (lit.84 mp 235-237oC); � (500 MHz, d6-acetone) 3.92 (dd, J 10.4, 10.4, H-2 ax), 4.17 (ddd, J 10.4, 3.5, 2.1, H-2 eq), 3.06 (dddd, J 10.4, 10.4, 5.5, 3.5, H-3 ax), 2.63 (dd, J 16.1, 10.4, H-4 ax), 2.91 (ddd, J 16.1, 5.5, 2.0, H-4 eq), 5.88 (d, J 2.3, H-6), 6.29 (d, J 2.3, H-8), 7.27 (d, J 8.4, H-2’,6’) 6.91 (d, J 8.8, H-3’,5’), 7.97 (s, 7-OH), 8.18 (s, 5-OH), 3.79 (s, 4’-OMe); � (125 MHz, d6-acetone) 71.2 (C-2), 38.5 (C-3), 27.6 (C-4), 157.1 (C-5), 95.6 (C-6), 157.6 (C-7), 96.0 (C-8), 102.1 (C-4a), 156.9 (C-8a), 135.1 (C-1’), 129.3 (C-2’,6’), 114.8 (C-3’,5’), 159.6 (C-4’), 55.5 (4’-OCH3); m/z (EI) 272 (M+, 98%), 163 (30), 151 (70), 134 (100), 121 (85), 119 (65), 91 (55); HRMS calcd for C16H16O4 272.1049, found 272.1057; � cm-1 3361, 1626, 1606, 1513, 1474, 1286, 1227, 1144, 1095, 1024, 955.
5,7,4’-Trihydroxyisoflavan 228 White crystals 75% mp 213-214oC (from aq. ethanol) (lit.,104 mp 209-211oC); � (300 MHz, d6-acetone) 3.89 (dd, J 10.5, 10.4, H-2 ax), 4.16 (ddd, J 10.4, 3.5, 2.1, H-2 eq), 3.03 (dddd, J 10.5, 10.7, 5.4, 3.5, H-3 ax), 2.61 (dd, J 16.1, 10.7, H-4 ax), 2.90 (ddd, J 16.1, 5.4, 2.0, H-4 eq), 5.88 (d, J 2.3, H-6), 6.03 (d, J 2.3, H-8), 7.17 (d, J 8.4, H-2’,6’) 6.83 (d, J 8.5, H-3’,5’), 7.97 (s, 7-OH), 8.16 (s, 5-OH), 8.23 (s, 4’-OH); � (75 MHz, d6-acetone) 71.3 (C-2), 38.5 (C-3), 27.6 (C-4), 157.1 (C-5), 95.6 (C-6), 157.5 (C-7), 95.8 (C-8), 102.1 (C-4a), 156.8 (C-8a), 133.9 (C-1’), 129.3 (C-2’,6’), 116.2 (C-3’,5’), 157.1 (C-4’); m/z (EI) 258 (M+, 83%), 246 (15), 163 (20), 139 (100), 120 (80), 107 (15), 91 (10), 69 (7); HRMS calcd for C15H14O4 258.0892, found 258.0903; � cm-1 3373, 1611, 1514, 1471, 1380, 1220, 1095, 1043, 1020, 962.

� �$�
7,4’-Dihydroxyisoflavan 15 White needles 88% mp 160oC (from benzene) (lit.,193158oC); � (500 MHz, d6-acetone) 3.90 (dd, J 10.5, 10.5, H-2 ax), 4.18 (ddd, J 10.5, 3.6, 2.1, H-2 eq), 3.07 (dddd, J 10.5, 10.6, 5.7, 3.6, H-3 ax), 2.89 (ddd, J 15.6, 10.6, 1.0, H-4 ax), 2.84 (ddd, J 15.6, 5.7, 2.0, H-4 eq), 6.89 (d, J 8.2, H-5), 6.37 (dd, J 8.2, 2.4, H-6), 6.29 (d, J 2.4, H-8), 7.16 (d, J 8.6, H-2’,6’) 6.82 (d, J 8.5, H-3’,5’), 8.07 (s, 7-OH), 8.21 (s, 4’-OH); � (125 MHz, d6-acetone) 71.6 (C-2), 38.8 (C-3), 32.7 (C-4), 131.0 (C-5), 108.9 (C-6), 157.6 (C-7), 103.7 (C-8), 114.1 (C-4a), 156.0 (C-8a), 133.5 (C-1’), 129.2 (C-2’,6’), 116.3 (C-3’,5’), 157.2 (C-4’); MS as reported.194; � cm-1 3348, 1614, 1599, 1513, 1454, 1242, 1120, 1021, 827.
Isoflavan 4 White powder 80% mp 55-56oC (from aq. ethanol) as previously reported.195 � (500 MHz, d6-acetone) 4.06 (dd, J 10.5, 10.5, H-2 ax), 4.30 (ddd, J 10.5, 3.7, 2.0, H-2 eq), 3.22 (dddd, J 10.5, 10.3, 5.6, 3.7, H-3 ax), 3.08 (dd, J 16.1, 10.3, H-4 ax), 2.98 (ddd, J 16.1, 5.6, 1.7, H-4 eq), 7.09 (m, H-5), 6.85 (td, J 7.1, 1.2, H-6), 6.79 (dd, J 8.7, 1.2 H-8), 7.38 (m, H-2’,5’, 6’), 7.35 (m, H-3’,5’), 7.28 (m, H-4’); � (125 MHz, d6-acetone) 71.2 (C-2), 39.3 (C-3), 33.0 (C-4), 130.7 (C-5), 121.0 (C-6), 127.8 (C-7), 117.1 (C-8), 123.0 (C-4a), 155.3 (C-8a), 142.6 (C-1’), 129.5 (C-2’,6’), 128.3 (C-3’,5’), 128.0 (C-4’); IR as reported.193 m/z (EI) 210 (M+, 100%), 149 (12), 119 (76), 104 (56), 91 (28); HRMS calcd for C15H14O 210.1045 , found 210.1029.
5,7,3’,4’-Tetrahydroxyisoflavan 229 White crystals 70% mp 220oC (from aq. ethanol); � (300 MHz, d6-acetone) 3.87 (dd, J 10.3, 10.5, H-2 ax), 4.14 (ddd, J 10.3, 3.2, 2.0, H-2 eq), 3.00 (dddd, J 10.5, 10.8, 5.4, 3.2, H-3 ax), 2.58 (dd, J 16.1, 10.8, H-4 ax), 2.88 (ddd, J 16.1, 5.4, 1.5, H-4 eq), 5.87 (d, J 2.2, H-6), 6.02 (d, J 2.0, H-8), 6.82 (s, H-2’), 6.63 (dd, J 8.1, 2.1, H-5’), 6.80 (d, J 8.1, H-6’), 8.17 (s, 5-OH), 7.97 (s, 7-OH), 7.79 (s, 3’-OH), 7.84 (s, 4’-OH); � (75 MHz, d6-acetone) 71.4 (C-2), 38.7 (C-3), 27.6 (C-4), 157.1 (C-5), 95.6 (C-6), 157.5 (C-7), 95.9 (C-8), 102.2 (C-4a), 156.9 (C-8a), 134.8 (C-1’), 115.3 (C-2’,6’), 144.8 (C-3’), 146.0 (C-4’), 119.5 (C-5’), 116.2 (C-6’); m/z (EI) 258 (M+, 83%), 246 (15), 163 (20), 139 (100), 120 (80), 107 (15), 91 (10), 69 (7); m/z (EI) 274 (M+,55%), 167 (10), 139 (100), 123 (15); HRMS calcd for C15H14O5 274.0841, found 274.0853.
5,7-Dihydroxy-3’,4’-dimethoxyisoflavan 230 White crystals 75% mp 208-209oC (from aq. ethanol); � (500 MHz, d6-acetone) 3.93 (dd, J 10.4, 10.4, H-2 ax), 4.19 (ddd, J 10.4, 3.6, 2.1, H-2 eq), 3.05 (dddd, J 10.4, 11.1, 5.5, 3.6, H-3 ax), 2.65 (dd, J 16.1, 11.1, H-4 ax), 2.91 (ddd, J 16.1, 5.5, 2.0, H-4 eq), 5.88 (d, J 2.3, H-6), 6.03 (d, J 2.3, H-8), 6.98 (d, J 2.0, H-2’) 6.86 (d, J 8.2, 2.0, H-3’,5’), 6.91 (d, J 8.2, H-6’ ), 7.95 (s, 7-OH), 8.16 (s, 5-OH), 3.79 (s, 3’-OMe), 3.82 (s, 4’-OMe); � (125 MHz, d6-acetone) 71.2 (C-2), 38.9 (C-3), 27.6 (C-4), 157.1 (C-5), 95.6 (C-6), 157.6 (C-7), 95.9 (C-8), 102.1 (C-4a), 156.9 (C-8a), 135.8 (C-1’), 112.5 (C-2’), 149.3 (C-3’), 150.5 (C-4’), 120.2 (C-5’), 113.1 (C-6’), 56.1 (3’, 4’-OCH3); m/z (EI) 302 (M+, 65%), 164 (100), 151 (34), 121 (10); HRMS calcd for C17H18O5
302.1154, found 302.1140.

� ��
REFERENCES 1. Dewick, P. M.; in The Flavonoids, Advances in Research Since 1986, ed.
Harborne, J.B., Chapman and Hall, London, 1993, 117. 2. 9th International symposium on the role of soy in health promotion and chronic
disease prevention and treatment, October 16-19, 2010, Capital Hilton, Washington DC, USA.
3. Axelson, M.; Kirk, D. N.; Farrant, R. D.; Cooley, G.; Lawson, A. M.; Setchell, K. D. R. Biochem. J. 1982, 201, 353.
4. Adlercreutz, H.; Fotsis, T.; Heikkinen, R.; Dwyer, J. T.; Woods, M.; Goldin, B. R.; Gorbach, S. L. Lancet 1982, 2, 1295.
5. Kelly, G. E.; Brown, D. “Combination radiotherapy and chemotherapy compositions using isoflavone compounds, and methods for the treatment of cancer”. US2006167037 and WO2005049008.
6. Kelly, G. E. “Therapeutic methods and compositions involving isoflav-3-ene and isoflavan structures”. US2006167083.
7. Heaton, Andrew.; Husband, A. J. (Novogen Research Pty Ltd., Australia). “Preparation of substituted chroman derivatives for use in pharmaceutical compositions as anti-cancer agents”. WO2006032085.
8. Heaton, A.; Husband, A. J. “Preparation of chroman derivatives for use in pharmaceutical compositions for the treatment of cancer”. WO2006032086.
9. Merchiers, P. G.; Koenraad, F. F. (Galapagos Genomics N.V., Belg.) “Methods, compositions and compound assays for inhibiting �-amyloid protein production by inhibiting sphingosine kinase (SPHK), and anti-Alzheimer’s uses”. WO2005103692.
10. Heaton, A.; Jeoffreys, G. (Novogen Res. Pty Ltd). “Improved process for the synthesis of isoflavene and uses of a new hydrogenation catalyst”. WO2005103025.
11. Heaton, A.; Kelly, G. E. (Novogen Res. Pty Ltd). “Isoflavonoid conjugates, compositions thereof and therapeutic methods involving same”. WO2005049627.
12. Kelly, G. E.; Brown, D. (Novogen Res. Ltd). “Combinational radiotherapy and chemotherapy compositions and methods”. WO2005049008.
13. Kelly, G. E. (Novogen Res. Ltd). “Synergistic anticancer compositions containing platinum-isoflavonoids”. WO2004030662.
14. Kelly, G. E.; Husband, A.; Walker, C. (Novogen Res., Ltd). “Skin photoageing and actinic damage treatment with equol and related compounds”. WO2004026274.
15. Kelly, G. E.; Husband, A.; Walker, C. (Novogen Res., Ltd). “Use of equol and related compounds for the repair of DNA mutagenic damage”. WO2004022023.
16. Kelly, G. E.; Husband, A. J. (Novogen Res., Ltd). “Therapeutic methods and compositions using isoflav-3-ene and isoflavan compounds”. WO2003086386.
17. Heaton, A.; Kelly, G. E. (Novogen Res., Ltd). “Isoflavonoid conjugates, compositions thereof and therapeutic methods involving same”. WO2003051864.
18. Galliant, S.; Venzke, K.; Herpens, A.; Biergiesser, H.; Schoenrock, U.; Staeb, F. (Beiersdorf AG, Germany). “Use of isoflavonoids in cosmetic or dermatological preparations for the prophylaxis or treatment of sensitive skin”. DE10122342.
19. Kelly, G. E.; Husband, A. J. (Novogen Res., Ltd). “Compositions and method for protecting skin from UV-induced immunosuppression and skin damage”. WO9936050.

� ���
20. For example Adlercreutz, H.; Murphy, C.; Fotsis, T.; Al-Maharik, N.; Heinonen, S-M. “Isoflavan and isoflavene compounds and their use as angiogenesis inhibitors”. WO2007/096471.
21. Patent applications for therapeutic uses: Oe, K.; Kimura, T. (Unitika Ltd., Japan) “Aldonic acids for promoting the production of equol”. WO 2007148631, JP 2008001608.; Constantinou, A. Method for enhancing tamoxifen efficasy as a cancer therapeutic. US 2007166414.; Hyatt, J. A. “Preparation of isoflavonoid compounds”. US2007149788.; Kimura, H.; Yamauchi, T.; Ueno, T.; Suzuki, T.; Tadano, K.; Sato, I.; Uchiyama, S.; Oono, M.; Mizuno, M. (Otsuka Pharmaceutical Co.; Ltd., Japan). WO2007066655.; Pan, Y. (Nestec S.A., Switz.). “Isoflavone compositions for reducing accumulation of body fat in male mammals, and methods for their use”. WO2007051629.; Lephart, E. D.; Lund, T. D.; Setchell, K. D. R.; Handa, R. J. “Use of equol for ameliorating or preventing neuropsychiatric and neurodegerative diseases or disorders”. US2007043108.
22. Dixon, R. A.; Achnine, L.; Kota, P.; Liu, C-J.; Reddy, S. M.S.; Wang, L. Review. Mol. Plant. Path. 2002, 3, 371.
23. Moon, Y. J.; Wang, X.; Morris, M. Toxicol. in Vitro 2006, 20, 187. 24. Hosny, M.; Rosazza, J. P. N. J. Nat. Prod. 2002, 65, 805. 25. Mazur, W., Adlercreutz, H. Nutr. 2000, 16, 654. 26. Hoikkala, A., Mustonen, E., Saastamoinen, I., Jokela, T., Taponen, J., Saloniemi,
H., Wähälä, K. Mol. Nutr. Food Res. 2007, 51(7), 782. 27. Marrian, G. F.; Haslewood, G. A. D. Biochem. J. 1932, 26, 1226. 28. Bennetts, H. W.; Underwood, E.J.; Shier, F. L. Aust. Vet. J. 1946, 22, 2. 29. Bowey, E.; Adlercreutz, H.; Rowland, I. Food Chem. Toxicol., 2003, 41, 631. 30. Setchell, K. D. R.; Zimmer-Nechemias, L.; Cai, J.; Heubi, J. E. Lancet, 1997, 350,
23. 31. Setchell, K. D. R.; Zimmer-Nechemias, L.; Cai, J.; Heubi, J. E. Am. J. Clin. Nutr.,
1998, 68, 1453S. 32. Kim, M.; Marsh, E. N. G.; Kim, S-U.; Han, J. Biochemistry, 2010, 49, 5582. 33. Atkinson, C.; Frankenfeld, C. L.; Lampe, J. W. Exp. Biol. Med. 2005, 230, 155. 34. Wang, J.; Han, L.; Zhu, L.; Gao, Y. Fenxi Huaxue 2006, 34(4), 569. Only abstract
in English. 35. Bannwart, C.; Adlercreutz, H.; Fotsis, T.; Wähälä, K.; Hase, T.; Brunow, G. Finn.
Chem. Lett. 1984, 4-5, 120. 36. Roberts-Kirchhoff, E. S.; Crowley, J. R.; Hollenberg, P. F.; Kim, H. Chem. Res.
Toxicol. 1999, 12, 610. 37. Kulling, S. E.; Honig, D. M.; Metzler, M. J. Agric. Food Chem., 2001, 49, 3024. 38. Kulling, S. E.; Honig, D. M.; Simat, T. J.; Metzler, M. J. Agric. Food Chem.,
2000, 48, 4963. 39. Maatooq, G. T.; Rosazza, J. P. N. Phytochemistry 2005, 66, 1007. 40. Heinonen, S-M,; Wähälä, K.; Adlercreutz, H. Phytochemistry Reviews 2002, 1,
175. 41. Heinonen, S-M.; Wähälä, K.; Adlercreutz, H. J. Agric. Food Chem. 2004, 52(22),
6802. 42. Heinonen, S-M.; Hoikkala, A.; Wähälä, K.; Adlercreutz, H. J. Steroid Biochem.
Mol. Biol. 2003, 87(4-5), 285.

� ���
43. Heinonen, S-M.; Wähälä, K.; Liukkonen, K-H.; Aura, A-M.; Poutanen, K.; Adlercreutz, H. J. Agric. Food Chem. 2004, 52(9), 2640.
44. Bannwart, C.; Adlercreutz, H.; Wähälä, K.; Kotiaho, T.; Hesso, A.; Brunow, G.; Hase, T. Biomed. Environ. Mass Spectrom. 1988, 17, 1.
45. Heinonen, S-M.; Wähälä, K.; Adlercreutz, H. Anal. Biochem. 1999 274, 211. 46. Kelly, G. E.; Nelson, C.; Waring, M. A.; Joannou, G. E.; Reeder, A. Y. Clin.
Chim. Acta, 1993, 223, 9. 47. Joannou, G. E.; Kelly, G. E.; Reeder, A. Y.; Waring, M.; Nelson, C. J. Steroid
Biochem. Molec. Biol. 1995, 54, 167. 48. Won, D.; Shin, B-K.; Kang, S.; Hur, H-G.; Kim, M.; Han, J. Bioorg. Med. Chem.
Lett. 2008, 18, 1952. 49. Kim, M.; Won, D.; Han, J. Bioorg. Med. Chem. Lett. 2010, 20, 4337. 50. Kim, M.; Marsh, E. N. G.; Kim, S-U.; Han, J. Biochemistry, 2010, 49, 5582. 51. Yuan, J-P.; Wang, J-H.; Liu, X. Mol. Nutr. Food Res. 2007 51(7), 765. 52. Miksicek, R. J. Proc. Soc. Exp. Biol. Med. 1995, 208, 44. 53. Fujioka, M.; Uehara, M.; Wu, J.; Adlercreutz, H.; Suzuki, K.; Kanazawa, K.;
Takeda, K.; Yamada, K.; Ishimi, Y. J. Nutr. 2004, 134, 2623. 54. Alda, J. O.; Mayoral, J. A.; Lou, M.; Gimenez, Y.; Martinez, R. M.; Garay, R. P.;
Biochem. Biophys. Res. Commun. 1996, 221, 279. 55. Hirvonen, J.; Rajalin, A-M.; Wohlfahrt, G.; Adlercreutz, H.; Wähälä, K.;
Aarnisalo, P. J. Steroid Biochem. Mol. Biol. 2011, 123, 46. 56. Muthyala, R. S.; Ju, Y. H.; Sheng, S.; Williams, L. D.; Doerge, D. R.;
Katzenellenbogen, B. S.; Helferich, W. G.; Katzenellenbogen, J. A. Bioorg. Med. Chem. 2004, 12, 1559.
57. Kuiper, G.G.J.M.; Carlsson, B.; Grandien, K.; Enmark, E.; Häggblad, J.; Nilsson, S.; Gustaffson, J.Å.; Endocrinology, 1997, 138, 863.
58. Lieberherr, M.; Cournot, G.; Robins, S. P. British J. Nutr. 2003, 89 suppl. 1., S59. 59. Coxam, V. British J. Nutr. 2003, 89 suppl. 1, S75-S85. 60. Valtuena, S.; Cashman, K.; Robins, S. P.; Cassidy, A.; Kardinaal, A.; Branca, F.
British J. Nutr. 2003, 89 suppl. 1, S87. 61. Wähälä, K.; Koskimies, J. K.; Mesilaakso, M.; Salakka, A. K.; Leino, T. K.;
Adlercreutz, H. J. Org. Chem. 1997, 62, 7690. 62. Arora, A.; Nair, M. G.; Strasburg, G. M. Arch. Biochem. Biophys. 1998, 356, 133. 63. Kinjo, J.; Tsuchihashi, R.; Morito, K.; Hirose, T.; Aomori, T.; Nagao, T.; Okabe,
H.; Nohara, T.; Masamune, Y. Biol. Pharm. Bull. 2004, 27(2), 185. 64. Yamaguchi, S.; Ito, S.; Nakamura, A.; Inoue, N. Bull. Chem. Soc. Jpn. 1965, 38,
2187. 65. Anjaneyulu, A. S. R.; Sri Krishna, C.; Ramachandra, Row, L. Bull. Nat. Inst. Sci.
India, 1965, 31, 118. 66. Anjaneyulu, A. S. R.; Sri Krishna, C.; Ramachandra, Row, L. Tetrahedron 1965,
21, 2677. 67. Susse, M.; Johne, S.; Hesse, M. Helv. Chim. Acta 1992 75, 457. 68. Chidiak, H.; Kirkiacharian, S. Arm. Khim. Zh. 1996, 49, 94. 69. Venkati, M.; Krupadanam, G. L. D. Ind. J. Chem. Sect. B: Org. Chem. Including
Med. Chem. 2005, 44B(3), 618. 70. Constantinou, A. I.; Mehta, R.; Husband, A. Eur. J. Cancer, 2003, 39, 1012. 71. Miyase, T.; Sano, M.; Nakai, H.; Muraoka, M.; Nakazawa, M.; Suzuki, M.;
Yoshino, K.; Nishihara, Y.; Tanai, J. Phytochemistry, 1999, 52(2), 303. 72. Widyarini, S.; Spinks, N.; Husband, A. J.; Reeve, V. E. Photochem. Photobiol.,
2001, 74(3), 465.

� ���
73. Lawson, W. J. Chem. Soc. 1954, 4448. 74. Micheli, R. A.; Booth, A. N.; Livingstone, A. L.; Bickoff, E. M. J. Med. Chem.
1962, 5, 321. 75. Durani, S.; Agarwal, A. K.; Saxena, R.; Setty, B. S.; Gupta, R. C.; Kole, P. L.;
Ray, S.; Anand, N. J. Steroid Biochem. 1979, 11(1A), 67. 76. Miksicek, R. J. Proc. Soc. Exp. Biol. Med. 1995, 208(1), 44. 77. Grese, T. A.; Pennington, L. D. Tetrahedron Lett. 1995, 36, 8913. 78. Chin-Dusting JPF, Fisher LJ, Lewis TV, Piekarska A, Nestel PJ, Husband A.
British J. Pharm. 2001, 133, 595. 79. Kim, J. H.; Baek, S. H.; Kim, D. H.; Choi, T. Y.; Yoon, T. J.; Hwang, J. S.; Kim,
M. R.; Kwon, H. J.; Lee, C. H.. J. Investig. Dermatol. 2008, 128, 1227. 80. Bulut, M. Chim. Acta Turc. 1991, 19, 17. 81. Major, A.; Nogradi, M.; Vermes, B.; Kajtar-Peredy, M. Liebigs Ann. Chem. 1988,
555. 82. Shoukry, M. M.; Darwish, N. A.; Morsi, M. A. Gass. Chim. Ital. 1982, 112, 289. 83. Heaton, A.; Kumar, N. Patent Application publication US 2005/0143588 A1, Jun.
30, 2005. 84. Liepa, A. J. Aust. J. Chem., 1981, 34, 2647. 85. Bouvier, P.; Adrieux, J.; Cunha, H.; Molho, D. Bull. Soc. Chim. Fr. 1977, 1187. 86. Liepa, A. J. Aust. J. Chem. 1984, 37, 2545. 87. Deschamps-Vallet, C.; Ilotse, J-B.; Meyer-Dayan, M. Tetrahedron lett. 1983,
24(37), 3993. 88. Dudley, K. H.; Miller, H. W.; Corley, R. C.; Wall, M. E. J. Org. Chem. 1967, 32,
2317. 89. Diaz, P.; Gendre, F.; Stella, L.; Charpentier, B. Tetrahedron 1998, 54, 4579. 90. Simas, A. B. C.; Furtado, L. F. O.; Costa, P. R. R. Tetrahedron Lett. 2002, 43,
6893. 91. Li, S-R.; Chen, P-Y.; Chen, L-Y.; Lo, Y-F.; Tsai, I-L.; Wang, E-C. Tetrahedron
Lett. 2009, 50, 2121. 92. Gupta, A.; Ray, S. Synthesis, 2008, 23, 3783. 93. Wähälä, K.; Hase, T. J. Chem. Soc., Perkin Trans 1. 1991, 3005. 94. Khupse, R. S.; Erhardt, P. W. Org. Lett. 2008, 10(21) 5007. 95. Luniwal, A.; Khupse, R. S.; Reese, M.; Fang, L.; Erhardt, P. W. J. Nat. Prod.
2009, 72, 2072. 96. Erhardt, P. W.; Khupse, R. S.; Sarver, J. G.; Cleveland, T. E.; Boue, S. M.; Wiese,
T. E.; Burow, M. E.; McLachlan, J. A. PCT Int. Appl. (2009), WO 2009111428 A2 20090911.
97. Verma, P.; Singh, S.; Dikshit, D. K.; Ray, S. Synthesis, 1988, 68. 98. Cook, C. E.; Twine, C. E.; J. Chem. Soc., Chem. Commun. 1968, 791. 99. Cook, C. E.; Corley, R. C.; Wall, M. E. J. Org. Chem. 1965, 30, 4114. 100. Dean, F. M.; Varma, R. S.; J. Chem. Soc., Perkin Trans 1 1982, 1193. 101. Dean, F. M.; Varma, M.; Varma, R. S. J. Chem. Soc., Perkin Trans 1 1982,
2771. 102. Dean, F. M.; Varma, R. S. Tetrahedron Lett. 1981, 22, 2113. 103. Varma, R. S.; Dahiya, R. J. Org. Chem. 1998, 63, 8038. 104. Chang, Y-C.; Nair, M. G.; Nitiss, J. L. J. Nat. Prod. 1995, 58(12), 1901. 105. Mustonen, E. A.; Tuori, M.; Saastamoinen, I.; Taponen, J.; Wähälä, K.;
Saloniemi, H.; Vanhatalo, A. British J. Nutr. 2009, 102, 1552. 106. Visser, F. R.; Lane, G. A. Aust. J. Chem. 1987, 40, 1705.

� ���
107. Antus, S.; Gottsegen, A.; Kolonits, P.; Nagy, Z.; Nogradi, M.; Vermes, B. J. Chem. Soc., Perkin Trans 1, 1982, 1389.
108. McMurry, T. H. B.; Martin, E.; Donnelly, D. M. X.; Thompson, J. C. Phytochemistry, 1972, 11, 3283.
109. Lamberton, J. A.; Suares, H.; Watson, K. G. Aust. J. Chem., 1978 31, 455. 110. Szabo, V.; Antal, E. Tetrahedron Lett. 1973 19, 1659. 111. Szabo, V.; Antal, E. Acta Chim. Acad. Sci. Hung. 1976 90(4), 381. 112. van Heerden, F. R.; Brandt, E. V.; Roux, D. G. J. Chem. Soc., Perkin Trans 1,
1980 2463. 113. Woodward, M. D. Phytochemistry, 1980 19, 921. 114. Heemstra, J. M.; Kerrigan, S. A.; Doerge, D. R.; Helferich, W. G.; Boulanger,
W. A. Org. Lett. 2006, 8(24), 5441. 115. Takashima, Y.; Kaneko, Y.; Kobayashi, Y. Tetrahedron 2010, 66, 197. 116. Setchell, K. D. R.; Sorokin, V. D. Patent Application Publication US
2010/0069653 A1 (Continuation of application No. 11/194,280, filed on Aug. 1, 2005, now Pat. No. 7,528,267)
117. K. Wähälä, “Synthesis of Isoflavonoid Phytoestrogens, their Precursors and Metabolites”, doctoral thesis, J-Paino, Helsinki, p. 50-53, 1992.
118. Joannoau, G. E.; Kelly, G. E.; Reeder, A. Y.; Waring, M.; Nelson, C. J. Steroid Biochem. Molec. Biol., 1995, 54, 167.
119. Goto, H.; Terao, Y.; Akai, S. Chem. Pharm. Bull. 2009 57(4), 346. 120. Tsukayama, M.; Kawamura, Y.; Tamaki, H.; Kubo, T.; Horie, T. Bull. Chem.
Soc. Jpn, 62, 1989, 826. 121. Thakar, G. P.; Janaki, N.; Subba Rao, B. C. Indian J. Chem. 3, 1965, 74. 122. Eguchi, T.; Hoshino, Y. Bull. Chem. Soc. Jpn, 74, 2001, 967. 123. Steffan, B. WO 2010/018199 A1. 124. Kurosawa, K.; Ollis, W. D.; Redman, B. T.; Sutherland, I. O. J. Chem.
Soc.,Chem. Commun., 1968, 1263. 125. Pelter, A.; Amenechi, P. I. J. Chem. Soc. (C), 1969, 887. 126. Robertson, A.; Suckling, C. W.; Whalley, W. B. J. Chem. Soc., 1949, 1571. 127. Kurosawa, K.; Ollis, W. D.; Redman, B. T.; Sutherland, I. O.; Alves, H. M.;
Gottlieb, O. R. Phytochemistry, 1978, 17, 1423. 128. Kurosawa, K.; Ollis, W. D.; Sutherland, I. O.; Gottlieb, O. R.; de Oliveiras, A.
B. Phytochemistry, 1978, 17, 1405. 129. Antus, S.; Gottsegen, A.; Kolonits, P.; Nogradi, M. Liebigs Ann Chem. 1986,
2179. 130. Breytenbach, J. C.; van Zyl, J. J.; van der Merwe, P. J.; Rall, G. J. H.; Roux, D.
G. J Chem Soc, Perkin Trans 1, 1981, 2684. 131. Luk, K.-C.; Stern, L.; Weigele, M. J.Nat. Prod. 1983, 46, 852. 132. Krämer, R. P.; Hindorf, H.; Jha, H. C.; Tsotsonos, P.; Egge, H. Phytochemistry
1984, 23(10), 2203. 133. Farkas, L.; Gottsegen, A.; Nogradi, M. J. Chem. Soc., Perkin trans 1, 1974, 305. 134. Fwu, S-Y.; Chang, C-Y.; Huang, L-J.; Teng, C-M.; Wang, J-P.; Chen, S-C.;
Kuo, S-C. Chin. Pharm. J. 1999, 51(4), 255. 135. Shih, T. L.; Wyratt, M. J.; Mrozik, H. J. Org. Chem., 1987, 52, 2029. 136. Houghton, R. P.; Shervington, L. A. J. Chem. Res. S., 1989, 239. 137. Usse, S.; Guillaumet, G.; Viaud, M-C. Tetrahedron Lett. 1997, 38(31), 5501. 138. Liepa, A. Aust. J. Chem., 1984, 37, 2545. 139. Conti, C.; Desideri, N.; Orsi, N.; Sestili, I.; Steir, M. L. Eur. J. Med. Chem.,
1990, 25, 725.

� ���
140. Burali, C.; Desideri, N.; Stein, M. C.; Conti, C.; Orsi, N. Eur. J. Med. Chem., 1987, 22(2), 119.
141. Heaton, A.; Kumar, N. Production of isoflavone derivatives. US 2005/0143588 A1. June 30, 2005.
142. Gharpure, S. J.; Sathiyanarayanan, A. M.; Jonnalagadda, P. Tetrahedron Lett. 2008, 49, 2974.
143. Spencer, G. F.; Jones, B. E.; Plattner, R. D.; Barnekow, D. E.; Brinen, L. S.; Clardy, J. Phytochemistry, 1991, 30(12), 4147.
144. Goda, Y.; Kiuchi, F.; Shibuya, M.; Sankawa, U. Chem. Pharm. Bull. 1992, 40(9), 2452.
145. Versteeg, M.; Bezuidenhoudt, B. C. B.; Ferreira, D. Heterocycles 1993, 36(8), 1743.
146. Versteeg, M.; Bezuidenhoudt, B. C. B.; Ferreira, D. Heterocycles 1998, 48(7), 1373.
147. Versteeg, M.; Bezuidenthoudt, B. C. B.; Ferreira, D.; Swart, K. J. J. Chem. Soc., Chem. Commun. 1995, 1317.
148. Versteeg, M.; Bezuidenhoudt, B. C. B.; Ferreira, D. Tetrahedron, 1999, 55, 3365.
149. Close, W. J. J. Org. Chem. 1950, 15, 1131. 150. Roder, H.; Helmchen, G.; Peters, E. M.; Peters, K.; Von Schnering, H. G.
Angew. Chem. 1984, 96, 895. 151. Evans, D. A.; Britton, T. C.; Ellman, J. A. Tetrahedron Lett. 1987, 28, 6141. 152. Cardillo, G.; D’Amico, A.; Orena, M.; Sandri, S. J. Org. Chem. 1988, 53, 2354. 153. Drewes, S. E.; Malissar, D. G. S.; Roos, G. H. P.; Chem. Ber. 1993, 126, 2663. 154. Liang, G.; Zhang, T.; Wang, J.; Chen, B.; Cheng, K.; Hu, C.
Chemistry&Biodiversity 2005, 2, 959. 155. Takashima, Y.; Kobayashi, Y. Tetrahedron Lett. 2008, 49, 5156. 156. Gupta, V. N.; Seshadri, T. R. Proc. Ind. Acad. Sci. A 1956, 44, 223. 157. Aggarwal, S. K.; Grover, S. K.; Seshadri, T. R. Indian J. Chem. 1970, 8, 478. 158. Jain, A. C.; Paliwar, P. Indian J. Chem. 1988, 27B, 985. 159. Bezuidenhoudt, B. C. B.; Brandt, E. V.; Roux, D. G. J. Chem. Soc., Perkin
Trans 1 1981, 263. 160. Salakka, A.; Wähälä, K. J. Chem. Soc., Perkin Trans 1 1999, 2601. 161. Wähälä, K.; Salakka, A.; Adlercreutz, H. Proc. Soc. Exp. Biol. Med. 1998, 217,
293. 162. Wähälä, K.; Jokela, T.; Salakka, A.; Kaltia, S.; Mesilaakso, M. J. Chem. Soc.,
Perkin Trans 1 2001, 642. 163. Hakala, U.; Wähälä, K., Tetrahedron Lett., 2006, 47(47), 8375. 164. Ehrentraut, A.; Zapf, A.; Beller, M. Adv. Synth. Catal. 2002, 344, 209. 165. Navarro, O.; Marion, N.; Oonishi, Y.; Kelly III, R. A.; Nolan, S. P. J. Org.
Chem. 2006, 71, 685. 166. Marion, N.; Ecarnot, E. C.; Navarro, O.; Amoroso, D.; Bell, A.; Nolan, S. P. J.
Org. Chem. 2006, 71, 3816. 167. Snape, T. J. Synlett, 2008, 2689. 168. Vicario, J. L.; Badia, D.; Dominguez, E.; Carrillo, L. J. Org. Chem. 1999, 64,
4610. 169. Myers, A. G.; Yang, B. H.; Chen, H.; Gleason, J. L. J. Am. Chem. Soc. 1994,
116, 9361. 170. Myers, A. G.; Yang, B. H.; Chen, H.; McKinstry, L.; Kopecky, D. J.; Gleason, J.
L.; J. Am. Chem. Soc. 1997, 119, 6496.

� ���
171. James, L. C.; Roversi, P.; Tawfik, D. S. Science 2003, 299, 1362. 172. Kaisalo, L. H.; Hase, T. A. Tetrahedron Lett. 2001, 42, 7699. 173. Wang, X-L.; Hur, H-G.; Lee, J. H.; Kim, K. T.; Kim, S-I. Apl. Environ.
Microbiol. 2005, 71, 214. 174. Brandt, P.; Hedberg, C.; Andersson, P. G. Chem. Eur. J. 2003, 9, 339. 175. Wu, Z-L.; Li, Z-Y. Tetrahedron: Asymm. 2001, 12, 3305. 176. Bezuidenhoudt, B.C.B.; Brandt, E.V.; Roux, D.G. J. Chem. Soc., Perkin Trans 1,
1980, 2179. 177. Ollis, W.D.; Ramsay, M.V.J.; Sutherland, I.O. Aust. J. Chem. 1965, 18, 1787. 178. Secthell, K.D.R.; Clerici, C.; Lephart, E. D.; Cole, S. J.; Heenan, C.; Castellani,
D.; Wolfe, B. E.; Nechemias-Zimmer, L.; Brown, N. M.; Lund, T. D.; Handa, R. J.; Heubi, J. E. Am. J. Clin. Nutr. 2005, 81, 1072.
179. Setchell, K. D. R.; Borriello, S. P.; Hulme, P.; Kirk, D. N.; Axelson, M. Am. J. Clin. Nutr. 1984, 40, 569.
180. Kelly, G. E.; Nelson, C.; Waring, M. A.; Joannou, G. E.; Reeder, A. Y. Clin Chim Acta 1993, 223, 9.
181. Sathyamoorthy, N.; Wang, T. T. Y. Eur. J. Cancer 1997, 33, 2384. 182. Lampe, J. W.; Karr, S. C.; Hutchins, A. M.; Slavin, J. L. Proc. Soc. Exp. Biol.
Med. 1998, 217, 335. 183. Arai, Y.; Uehara, M.; Sato, Y.; Kimira, M.; Eboshida, A.; Adlercreutz, H.;
Watanabe, S. J. Epidemiol. 2000, 10, 127. 184. Rowland, I. R.; Wiseman, H.; Sanders, T. A. B.; Adlercreutz, H.; Bowey, E. A.;
Nutr. Cancer 2000, 36, 27. 185. Setchell, K. D.; Brown, N. M.; Lydeking-Olsen, E. J. Nutr. 2002, 132, 3577. 186. Frankenfeld, C.; Atkinson, C.; Thomas, W.; Gonzalez, A.; Jokela, T.; Wähälä,
K.; Schwartz, S.; Li, S.; Lampe, J. British Journal of Nutrition 2005, 94, 873. 187. Song, K. B.; Atkinson, C.; Frankenfeld, C. L.; Jokela, T.; Wähälä, K.; Thomas,
W. K.; Lampe, J. W. J. Nutr. 2006, 136(5), 1347. 188. Frankenfeld, C. L.; McTiernan, A.; Tworoger, S. S.; Atkinson, C.; Thomas, W.
K.; Stanczyk, F. Z.; Marcovina, S. M.; Weigle, D. S.; Weiss, N. S.; Holt, V. L.; Schwartz, S. M.; Lampe, J. W. J. Steroid. Biochem. Mol. Biol. 2004, 88, 399.
189. Smitrovich, J. H.; Boice, G. N.; Qu, C.; DiMichele, L.; Nelson, T. D.; Huffman, M. A.; Murry, J.; McNamara, J.; Reider, P. J. Org. Lett. 2002, 4(11), 1963.
190. Clark-Lewis, J.W.; Jemison, R. W. Aust. J. Chem. 1965, 18, 1791. 191. Fujii, K.; Nakao, K.; Yamauchi, T. Synthesis 1982, 456. 192. Wessely, F.; Prillinger, F. Monatshefte für Chemie 1938, 72, 197. 193. Wessely, F.; Prillinger, F. Chem. Ber. 1939, 72, 629. 194. Hayashi, Y.; Shirato, T.; Sakurai, K.; Takahashi, T. Mokuzai Gakkaishi, 1978,
24, 898. 195. Szabo, V.; Borbely, J.; Antus, E. Acta Chim. Acad. Sci. Hung. 1979, 102, 51.