synthesis to clinic – c studies in man a chemistry...
TRANSCRIPT

Synthesis to Clinic – 14C studies in man a Chemistry perspectiveperspective
Stephen LewintonManaging Director – Chemistry & MetabolismFeb 2013

“Synthesis-to-Clinic”
• Tightly integrated supply chain
�Single vendor
�Single project manager
14C API synthesis
Preclinical Data
Regulatory affairs
�Single project manager
�Integrated Quality process
�Continuity of the science
�Removal of management burden
• Relevant to all 14C containing study types• Microdose, microtracer, human ADME
IMP production
Clinical conduct
Sample analysis
Biometrics

GMP
The Regulatory FrameworkGeneral Provisions• European Clinical Trials Directive 2001/20/EC (EUCTD)• Medicines for Human Use (Clinical Trials) Regulations 2004 (SI 2004/1031)• UK MHRA• http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm• APIs and Drug Products for Use in Very Early Phase Studies
3
• APIs and Drug Products for Use in Very Early Phase StudiesGuidance for Industry, Investigators, and Reviewers – Exploratory IND Studies, January 2006
• cGMP for Phase 1 Investigational Drugs; Food and Drug Administration, July 2008
The Clinical Trials Authorisation (CTA) application• The Investigational Medicinal Product Dossier (IMPD)The Investigational New Drug application (IND)

GMP
GMP or not GMP?
• [14C]IMP (Drug Product) must be manufactured to GMP
• [14C]API (Drug Substance) for use in clinical trials is the starting material for manufacture of the [14C]IMP administered in the clinical trial
4
in the clinical trial
• It does not have to be prepared to GMP but……………appropriate GMP concepts should be applied

GMP
Appropriate GMP Concepts• Clinical radiosynthesis laboratory (minimise risk of
cross-contamination)• Quarantine and identity testing of raw materials
(BSE/TSE statements, CoAs)• Equipment maintained and calibrated• Clinical project glassware – washed and inspected
before use
5
• Analytical methods should be scientifically sound• Production data documented• QA review of analytical and production data• QA Batch Release
[14C]API for clinical trials prepared at Quotient released with a GMP CoA
The final scope of [14C]API project is agreed with the customer

GMP
Is GMP for 14C labelled API really GMP?
Not all GMP controls are appropriate for the manufacture of [14C]API for clinical trial use:• Process validation for [14C]API production is not required• Analytical methods may not be fully validated
6
The objective is to produce [14C]API using a procedure that:• satisfies regulatory requirements• allows the material to be accepted into a [14C]IMP manufacturing procedure
Compliance is not Science

Preparing 14C Labelled Compounds for Clinical Use
A Representative [ 14C]API Radiosynthesis
Synthesis of [14C]DSM
Analytical method transfer
7
Technical [14C]API synthesis1
Clinical / GMP [14C]API synthesis
IMPDSubmission
Storage stability study
1 = Technical batch is not mandatory
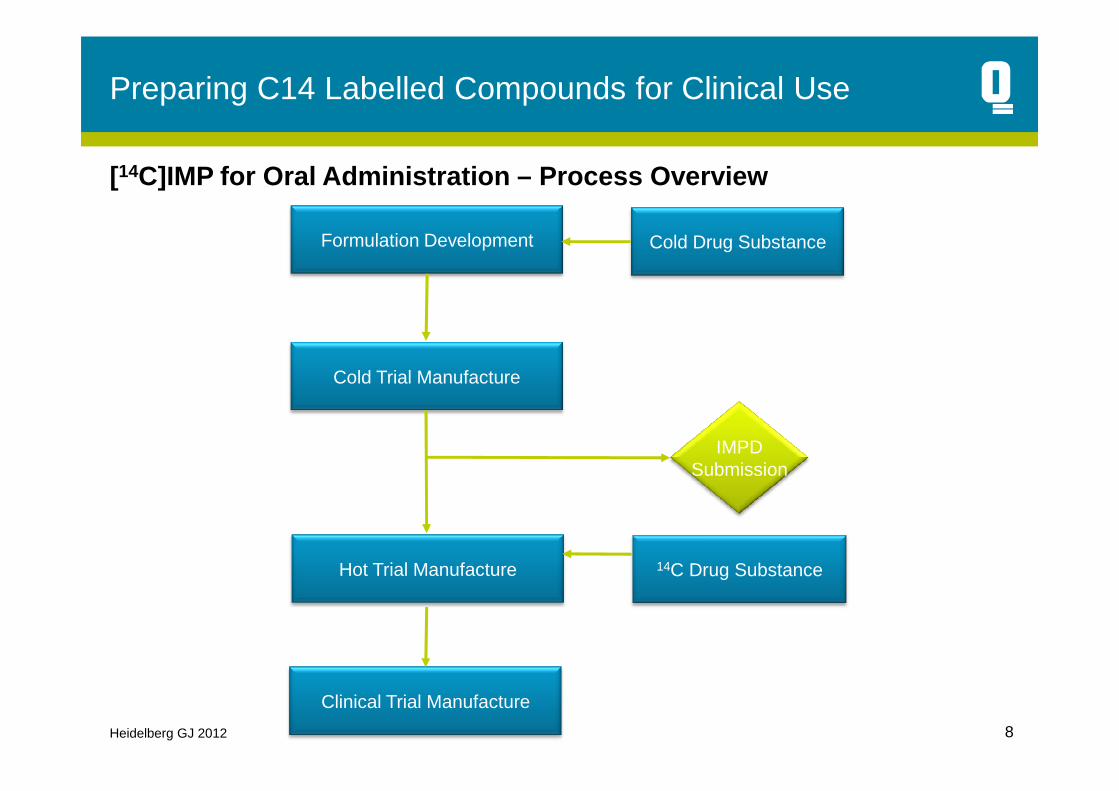
Preparing C14 Labelled Compounds for Clinical Use
Cold Trial Manufacture
Formulation Development Cold Drug Substance
[14C]IMP for Oral Administration – Process Overview
Heidelberg GJ 2012 8
Hot Trial Manufacture
Clinical Trial Manufacture
14C Drug Substance
IMPDSubmission

Some Key Considerations
• Analytical Release & Impurity profiles– Radiosynthesis route should mimic cold
route as far as practical.• Helps ensure toxicological coverage for
impurities
– Radiosynthesis scale typically <0.5g and so compound requirement for analytical release must factor in calculations of
9
Radiotrace
release must factor in calculations of required production scale.
– Cold analytical method must be adapted for radiodetector to allow determination of radiochemical purity.

Some Key Considerations
• Proposed drug product– Majority of these studies are undertaken using
oral solutions. Advantages include:• Concerns about form of drug substance avoided• Easy to demonstrate homogeneity of the Drug
Product• Ensures good bioavailability
– Key challenges with moving to clinical dosage
10
– Key challenges with moving to clinical dosage form (oral solid most commonly)
• Ensure physico form of radiolabelled matches clinical API. Most milling techniques require multi grams of material.
• Achieving homogeneity – co-crystallisation versus blending
– Program design requires early decision on whether to mimic clinical dosage form and the strategy for doing so.

Some key considerations
• Determination of Dose for Human AME
– Need to determine safe radioactive dose for Human AME study
• 28-day Quantitative Whole Body Autoradiography study (QWBA) in pigmented rats. Data used for human dosimetry calculation.
– Combining safe radioactive dose with
11
– Combining safe radioactive dose with the proposed clinical dose allows calculation of the specific activity of the Drug Product (IMP).
• Dose for microdose study determined from ICH M3
– Dosimetry is not required to support these studies
– Rapid determination of required specific activity possible

Some key considerations
• Why is the specific activity important?– Stability
• Stability often depends upon the specific activity and form of storage. • Stability can vary depending on label position.• Not possible to use cold data and structure to predict impact of labelling on
stability. • Stability studies need to be conducted at the appropriate or a higher specific
activity and in correct form.
12
activity and in correct form.• Stability data is important in allowing assignment of an expiry date.
– If the specific activity of the Drug product is high and so the level of cold dilution is low then importance of characterisation of the radiochemical API is increased.
• Early discussions around dose and degree of dilution are therefore important in planning

Stability Example
• Dominant effect of solution storage here lies in the suppression of formation of impurities
• Radiochemical purity specification of ≥ 97% both the -80°C solid and -80°C solution remain in specification
95.0
96.0
97.0
98.0
99.0
100.0
0 1 2 3 4 6
Analysis Time Points (weeks)
-80°C Solid Radiochemical purity %-80°C Solution Radiochemical Purity %
13
remain in specification• Radiochemical impurity
specification is ‘no single impurity >1.0%’ only the -80°C solution remains in specification after 4 weeks.
93
94
95
96
97
98
99
100
0 1 2 3 4
Analysis Time Points (weeks)
-80°C Solution Radiochemical Purity %-20°C Solution Radiochemical Purity %

QA
GMP[14C]API Synthesis
GM
P F
acili
ty
Com
mis
sion
ing
Stability Trials
Typical Time-lines for GMP[ 14C]API
Start Clinical Program
[14C]DSM Synthesis
Analytical Method TransferC
usto
mer
ap
prov
es B
atch
D
ocum
enta
tion[14C]
Technical Batch
Synthesis
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Week
Cus
tom
er
appr
oves
P
roto
col
14

Summary
• In undertaking studies using 14C the early planning of radiosynthesis plays a key role.
• Some of the key early decision include:– Proposed dose product– Radioactive and cold dose– Analytical release criteria– Understanding importance of physico form to
15
– Understanding importance of physico form to absorption
• In considering Chemistry requirements QA/QPs should consider for:– AME studies: Single dose, Clinic based, ~6
volunteers and normally healthy.– Requirements for exploratory studies for
Microdose studies
