synthetic and computational studies of metal-ligand ... · synthetic and computational studies of...
TRANSCRIPT

Synthetic and Computational Studies of Metal-Ligand Cooperationwith Iron Group Complexes for Water Splitting and Ketone
Hydrogenation
by
Demyan Evan Prokopchuk
A thesis submitted in conformity with the requirementsfor the degree of Doctor of PhilosophyGraduate Department of Chemistry
University of Toronto
c© Copyright 2015 by Demyan Evan Prokopchuk

Abstract
Synthetic and Computational Studies of Metal-Ligand Cooperation with Iron Group Complexes for
Water Splitting and Ketone Hydrogenation
Demyan Evan Prokopchuk
Doctor of Philosophy
Graduate Department of Chemistry
University of Toronto
2015
The paradigm of metal-ligand cooperation pervades the fields of organometallic chemistry and catalysis.
In its simplest form, metal-ligand cooperation can be viewed as a strategic partnership between the
metal and its surrounding ligand system to achieve new modes of chemical reactivity. Classic cases of
metal-ligand cooperation involve elaborate chiral catalysts containing noble metals for enantioselective
hydrogenation reactions, which are able to generate chiral compounds of great value to the pharma-
ceutical, agrochemical, and flavour industries. In recent years, creative design innovations have allowed
chemists to create new molecules that use inexpensive iron-based catalysts for the same enantioselective
transformations. Furthermore, metal-ligand cooperation has been recently employed in catalyst design
pertaining to renewable/green energy applications. In particular, the activation of benign small molecules
such as water, carbon dioxide, and methanol can be performed with well-defined molecular catalysts ex-
hibiting metal-ligand cooperativity. This thesis encompasses two areas in which metal-ligand cooperation
plays a vital role in the overall reactivity. First, new ruthenium complexes containing bidentate, triden-
tate, and tetradentate ligand systems are synthesized and investigated for applications in water splitting
(2 H2O→2 H2 + O2). Second, computational investigations are performed to elucidate the mechanism
of ketone hydrogenation using three generations of iron hydrogenation catalysts containing tetradentate
PNNP ligand frameworks.
ii

Acknowledgements
I am indebted to many people who have supported my endeavours throughout the last 5.5 years. Firstly,
thank you to Bob for his guidance and wisdom; he is always willing to talk chemistry with his students
(especially reaction mechanisms!), discuss new ideas, and always encouraged me to pursue my own ideas.
His “hands off, but always open door” policy gave me the independence I needed with my research
projects while always being available in times of need. Next, thank you to all present and past Morris
group members: Vivian, Alex, Wylie, Alba, Brian, Peter, Kanghee, Jessica, Nils, Kai, Sam, Karl, Molly,
Heiko, Florian; you kept my spirits up during my many, many hours spent in lab and motivated me to
keep trying when results were discouraging (and of course, there’s always time for a coffee break and
chat, no matter how busy we are). Also, thank you to my PhD advisory committee members, Datong
and Ulrich, for their valuable input throughout the duration of my studies.
I will deeply miss all the talented and friendly support/research staff at the University of Toronto.
Thanks to Alan and his decades long crystallography experience, the X-ray lab is one of the finest in
the world. Alan was always ready to strike up a conversation with me about current events and the
outdoors while I anxiously waited for the unit cell data collection to finish. In the NMR lab, many
thanks to Darcy, Tim, Dmitry, and Joel. You are extremely friendly, helpful, and were always available
to answer my (silly) NMR questions. Thanks to all the other staff members in the department for
their generous help and support through the years: Ken in chem stores, Matt in ANALEST, Jack in
glassblowing, John in the machine shop, and Jack/Patrick/Violeta/Frank in the electronics shop. A
special and hearty thanks to the Graduate Office, especially Anna Liza, Stefanie, Penny, and Denise for
their outstanding support and cheerful personality.
I had the honour of spending five months at ETH Zurich working in the Grutzmacher lab. Thank
you to Hansjorg for accepting me into his lab for this short time, it was an unforgettable experience.
Thanks to all group members (the “Grutzis”) for their support and friendship, especially to my lab
mates Bruno, Monica, and Xiuxiu.
I would not have been able to spend as much time doing research without scholarships from the
province of Ontario and NSERC, who I deeply thank for appreciating the importance of my scientific
research.
I wish to thank all my family and friends, near and far, for their support and encouragement during
my studies. I especially want to thank my parents (“Mama and Tato”) and sister Oksana for always
supporting my interests and passions. Finally, thank you to my wife Natalia - her love, patience, and
kindness are unparalleled.
iii

Contents
List of Abbreviations xiv
1 Introduction 1
1.1 Metals and Ligands Working Together: “Bifunctional,” “Cooperative”, and “Non-Innocent”
Behaviour . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.2 Proximal Metal-Ligand Cooperation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2.1 Applications of MLC in Ketone Hydrogenation . . . . . . . . . . . . . . . . . . . . 4
1.3 Distal Metal-Ligand Cooperation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.3.1 Applications of MLC in Homogeneous Water Splitting . . . . . . . . . . . . . . . . 8
1.4 Thesis Outline . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
2 Structural Properties of Trans Hydrido-Hydroxo Complexes and their Proton Ex-
change Behaviour with Water in Solution 15
2.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.2 Results and Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.4 Experimental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.4.1 General Comments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.4.2 trans-Ru(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2 (51) . . . . . . . . . . . . . . . . . 24
2.4.3 trans-Ru(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2·H2O (52) . . . . . . . . . . . . . . 24
2.4.4 Thermolysis/photolysis of 52 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
2.4.5 OsH(NHCMe2CMe2NH2)(PPh3)2 (54) nH2O(n = 1, 2). . . . . . . . . . . . . . . . 25
2.5 Supplementary Information . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
2.5.1 X-ray Structural and NMR Data . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
3 From Amine to Ruthenaaziridine to Azaallyl – Unusual Transformation of Di-(2-
pyridylmethyl)amine on Ruthenium 29
3.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
3.2 Results and Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
3.2.1 Synthesis of Ruthenium Di-(2-pyridylmethyl)amine and Ruthenaaziridine Com-
plexes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
3.2.2 Synthesis of a Ruthenium Azaallyl Complex. . . . . . . . . . . . . . . . . . . . . . 32
3.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
3.4 Experimental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
iv

3.4.1 General Comments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
3.4.2 [RuH{κ3N-fac-1,3-di-(2-pyridylmethyl)amine}(PPh3)2]Cl (60). . . . . . . . . . . . 37
3.4.3 RuH{κ3C alkNN py-1,3-di-(2-pyridylmethyl)amine}(PPh3)2 (61). . . . . . . . . . . 37
3.4.4 RuH(κ3N -1,3-di-(2-pyridyl)-2-azaallyl)(PPh3)2 (62). . . . . . . . . . . . . . . . . . 38
4 Intramolecular C-H/O-H Bond Cleavage With Water and Alcohol Using a Phosphine-
Free Ruthenium Carbene NCN Pincer Complex 39
4.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
4.2 Results and Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
4.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
4.4 Experimental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
4.4.1 General Comments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
4.4.2 1-(2-methylpyridyl)-4,5-diphenylimidazole. . . . . . . . . . . . . . . . . . . . . . . . 49
4.4.3 1,3-di(2-methylpyridyl)-4,5-imidazolium bromide (63). . . . . . . . . . . . . . . . . 49
4.4.4 Ag(NCN)Br (64). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
4.4.5 [Ru(NCN)(NCCH3)3][PF6]2 (65). . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
4.4.6 [Ru(NCN)(tBu2bpy)(NCCH3)][PF6]2 (66). . . . . . . . . . . . . . . . . . . . . . . 50
4.4.7 [RuBr(NCN)(tBu2bpy)][PF6] (67). . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
4.4.8 Ru(OtBu)(NCN*)(
tBu2bpy) (68). . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
4.4.9 Ru(NCN**)(PPh3)(tBu2bpy) (70). . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
4.4.10 Ru(OH)(NCN*)(tBu2bpy) (71). . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
4.5 Supplementary Information . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
4.5.1 NMR Peak Numbering and Spectra. . . . . . . . . . . . . . . . . . . . . . . . . . . 54
4.5.2 X-Ray crystal structure and refinement data. . . . . . . . . . . . . . . . . . . . . . 64
4.5.3 DFT Energy Diagram . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
5 Phosphine-Free Ruthenium Complexes Bearing Tetradentate Amino-Olefin Ligands 69
5.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
5.2 Results and Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
5.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
5.4 Experimental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
5.4.1 General Comments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
5.4.2 [K(db18c6)][RuH(trop2dad)] (78). . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
5.4.3 [Bu4N][RuH(trop2dad)] (79). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
5.4.4 (±)-trans-N ,N -bis(5H-dibenzo[a,d]cyclohepten-5-yl)-1,2- diaminocyclohexane (trop2dach,
80). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
5.4.5 Synthesis of Ru2Cl4(trop2dach)(PPh3)2 (81). . . . . . . . . . . . . . . . . . . . . . 76
5.4.6 Ru0(trop2dach) (82). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
5.4.7 [K]2[Ru–II
(trop2dach)] (83). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
6 First Generation Iron PNNP [6.5.6] Complexes for the Transfer Hydrogenation of
Ketones: Mechanistic Insights from DFT Calculations 80
6.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
6.2 Results and Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
v

6.2.1 Summary of Catalysis Investigations by Spectroscopic Methods . . . . . . . . . . . 82
6.2.2 Calculated Mechanism for Formation of Complexes 86, 87, and Fe(0) Species 91 . 85
6.2.3 Using a CO Scale Factor to Support the Proposed Structures of 87 and 91 . . . . 88
6.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
6.4 Experimental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
6.4.1 Computational Details . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
6.4.2 General . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
6.5 Supplementary Information . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
6.5.1 Calculated Complex 85, and Scale Factor Data . . . . . . . . . . . . . . . . . . . . 90
7 Second and Third Generation Iron PNNP [5.5.5] Complexes: The Calculated Mech-
anism of Ketone Hydrogenation 93
7.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
7.2 Results and Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
7.2.1 Activation Period with Model Bis(eneamido) Complex During ATH . . . . . . . . 97
7.2.2 Catalytic Cycle for ATH with Model Amido-eneamido Complex . . . . . . . . . . 101
7.2.3 KIE Calculations for Activation and Catalysis . . . . . . . . . . . . . . . . . . . . 106
7.2.4 Formation of a Bis(amido) PNNP Complex and Modelling its ATH Catalytic Activity108
7.2.5 Calculated Activity and Enantioselectivity of the Third Generation Full Catalyst
System 97Ph During ATH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 110
7.2.6 Third-Generation FePNNP [5.5.5] Complexes are Also Moderately Active AH Cat-
alysts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111
7.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
7.4 Computational Details . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
7.4.1 General . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
7.5 Supporting Information . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
7.5.1 Transfer Hydrogenation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
7.5.2 Direct Hydrogenation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
7.5.3 Cartesian Coordinates , Free Energies, and Enthalpies of Optimized Structures . . 124
8 Conclusions and Future Work 132
Bibliography 135
vi

List of Tables
2.1 Chemical shifts of interest (ppm) in the 1H NMR spectra (400 MHz, toluene-d8) of 52 at
-80 ◦C and 55 at -60 ◦C. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.2 Selected Bond Lengths (A) and Bond Angles (◦) for 52 and 55. . . . . . . . . . . . . . . . 22
2.3 X-Ray crystal structure and refinement data for complexes 52 and 55. . . . . . . . . . . . 26
3.1 Selected bond lengths (A) and angles (◦) for complexes 60, 61, and 62. . . . . . . . . . . 35
3.2 X-Ray crystal structure and refinement data for complexes 60, 61, and 62. . . . . . . . . 36
4.1 Experimental and calculated activation parameters for intramolecular proton transfer
(68→69). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
4.2 Selected X-Ray crystal structure and refinement data for complexes 1-(2-methylpyridyl)-
4,5-diphenylimidazole (Im), 65, 68, and 70. . . . . . . . . . . . . . . . . . . . . . . . . . . 65
5.1 Selected NMR data for complexes 81, 82 and 83. . . . . . . . . . . . . . . . . . . . . . . . 74
5.2 Selected X-Ray crystal structure and refinement data for complexes 81 and 82. . . . . . . 79
6.1 Comparison of Bond Lengths (A) and Angles (◦) for Compounds 86 and 86DFT. . . . . . 84
6.2 Comparison of Bond Lengths (A) and Angles (◦) for Compounds 85 and 85DFT. . . . . . 91
6.3 Experimental and calculated carbonyl stretches of compounds 85, 86, 92,309 and 93.295 . 91
7.1 Experimental302 and Calculated KIE Values (1 atm, 29 ◦C) for Hydride Transfer During
the Activation Period and Catalytic Cycle Using iPrOD-d1 and iPrOD-d8. . . . . . . . . . 107
7.2 Comparison of Experimental46 and Calculated (from DFT) Activity and Enantioselectiv-
ity Parameters During the ATH of AcPh. . . . . . . . . . . . . . . . . . . . . . . . . . . . 111
7.3 Comparison of Experimental and Calculated (from DFT) Activation Parameters for Hy-
dride Transfer to Acetophenone in ATH Relative to 122 (Figure 7.10), and H2 Splitting
in AH Relative to 100 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
7.4 Electronic energies of key transition states using various density functionals. . . . . . . . . 118
vii

List of Figures
1.1 Three conceptual forms of ligands working/interacting together with metals. M = tran-
sition metal, Ln = multidentate ligand, m = number of electrons, E = H, OH, OR, NHR. 2
1.2 A non-exhaustive collection of stable and well-defined complexes exhibiting proximal
MLC, arranged in approximate chronological order of publication. . . . . . . . . . . . . . 3
1.3 The dramatic difference in ATH activity between catalysts 17 and 18. . . . . . . . . . . . 5
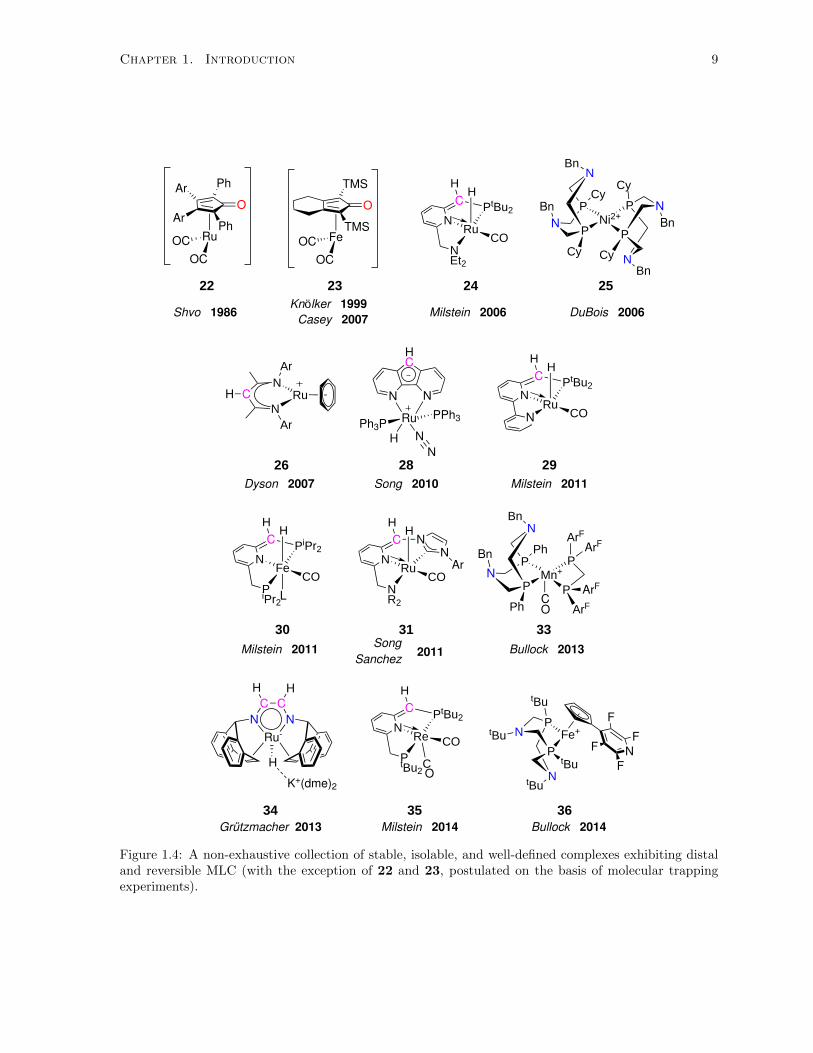
1.4 A non-exhaustive collection of stable, isolable, and well-defined complexes exhibiting distal
and reversible MLC (with the exception of 22 and 23, postulated on the basis of molecular
trapping experiments). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
1.5 Top: Half-cell reactions for the production of H2 and O2 from H2O. Bottom: recent
examples of well-defined catalysts for each half-reaction. . . . . . . . . . . . . . . . . . . . 10
1.6 Four different ligand systems reported in this thesis for applications in water splitting. . . 13
1.7 Three generations of iron hydrogenation catalysts examined in this thesis. . . . . . . . . . 14
2.1 (a) Overlaid 1H NMR spectra (400 MHz) of 51 at 25 ◦C (black) and -60 ◦C (green). (b)
Overlaid 1H NMR spectra (400 MHz) of 52 at 25 ◦C (black), -40 ◦C (green), and -80 ◦C
(blue). Residual solvent signal (CD3) for toluene-d8 appears at 2.09 ppm. Only the inset
spectrum on the bottom left was obtained using a 600 MHz NMR spectrometer to increase
the resolution of the H2O signal. The aromatic resonances appear from 8.0–6.8 ppm and
some have been omitted and some spectra have been magnified on the y-axis for clarity. . 18
2.2 Left: Asymmetric unit of 52 depicted with thermal ellipsoids at the 30% probability
level. Hydrogen atoms on aromatic rings and methyl groups have been omitted for clarity.
Selected bond distances and angles are listed in Table 2.2. Right: Crystal packing of 52
as a water bridged dimer depicted with thermal ellipsoids at the 30% probability level.
Phenyl carbons/hydrogens and methyl hydrogens have been omitted for clarity. . . . . . . 22
2.3 1H NMR spectrum (400 MHz) of 51 at -60 ◦C in toluene-d8. . . . . . . . . . . . . . . . . 27
2.4 1H NMR spectrum (400 MHz) of 52 at -80 ◦C in toluene-d8. . . . . . . . . . . . . . . . . 27
2.5 1H NMR spectrum (400 MHz) of 54 + H2O at -40 ◦C in toluene-d8. . . . . . . . . . . . . 28
2.6 1H NMR spectrum (400 MHz) of 54 + 2 H2O at -60 ◦C in toluene-d8. . . . . . . . . . . . 28
3.1 Examples of ligand systems containing anionic amido ligand donors. . . . . . . . . . . . . 30
3.2 Molecular structure of 60 depicted with thermal ellipsoids at the 30% probability level.
Hydrogen atoms on aromatic rings and methylene carbons have been omitted for clarity. . 31
3.3 Molecular structure of 61 depicted with thermal ellipsoids at the 30% probability level.
Hydrogen atoms on the aromatic rings have been omitted for clarity. . . . . . . . . . . . . 32
viii

3.4 Molecular structure of 62 depicted with thermal ellipsoids at the 30% probability level.
Hydrogen atoms on the phenyl rings have been omitted for clarity. . . . . . . . . . . . . . 34
4.1 Molecular structure of 68 with ellipsoids at the 30 % probability level. Phenyl and
methyl hydrogens have been omitted for clarity. Distances (A) and angles (◦): Ru1–
O1 2.111(2), Ru1–C1 1.955(3), Ru1–N3 2.101(2), Ru1–N4 2.116(2), Ru1–N5 2.041(2),
Ru1–N6 2.102(2), C22–C23 1.377(4), O1· · ·H16A 2.341; N5-Ru1-N6 78.04(8), N3-Ru1-C1
87.13(10), N4-Ru-C1 88.92(10), O1-Ru1-C1 93.30(10). . . . . . . . . . . . . . . . . . . . . 42
4.2 1H−1H ROESY spectrum (THF-d8, 0 ◦C, 600 ms mixing time, 1 s relaxation delay)
of 68. The coloured asterisks (matched with Scheme 4.4) indicate the protons on the
pincer framework undergoing two-site chemical exchange. The exchange cross peak at
δ = (7.3, 7.4) belongs to the phenyl rings attached to the NHC. . . . . . . . . . . . . . . . 43
4.3 Molecular structure of 70 with ellipsoids at the 30 % probability level. Only one of the
two crystallographically independent complexes is shown. Hydrogens have been omitted
for clarity. Distances (A) and angles (◦): Ru1A–P1A 2.3187(14), Ru1A–C1A 1.966(7),
Ru1A–N3A 2.121(5), Ru1A–N4A 2.131(5), Ru1A–N5A 2.104(4), Ru1A–N6A 2.155(6),
C16A–C17A 1.366(10), C22A–C23A 1.348(10); N5A-Ru1A-N6A 76.58(19), N3A-Ru1A-
C1A 90.3(2), N4A-RuA-C1A 87.1(2), C1A-N1A-C16A-C17A -3.98, C1A-N2A-C22A- C23A
-23.46. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
4.4 Selected chemical shift regions of experimental 1H NMR spectra of 68 (left, THF-d8, 600
MHz) at various temperatures and simulated NMR spectra (right) with rate constants
(s−1). Arrows indicate the exchange peaks of interest that were modeled in the simulation. 45
4.5 Eyring Plot of Proton Transfer 68→69. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
4.6 Transition state geometry of TS68,69 (ν = 1160i). Distances are in A. . . . . . . . . . . . 46
4.7 NMR peak numbering for all complexes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
4.8 1H NMR spectrum of 1-(2-methylpyridyl)-4,5-diphenylimidazole. . . . . . . . . . . . . . . 55
4.9 13C NMR spectrum of 1-(2-methylpyridyl)-4,5-diphenylimidazole. . . . . . . . . . . . . . . 55
4.10 1H NMR spectrum of 63. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
4.11 13C NMR spectrum of 63. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
4.12 1H NMR spectrum of 65. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
4.13 13C NMR spectrum of 65. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
4.14 1H NMR and 1D NOESY spectrum (red line, 600 ms mixing time, 4 s relaxation delay,
irradiation at δ = 8.15) of 66. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
4.15 13C NMR spectrum of 66. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
4.16 1H NMR and 1D NOESY spectrum (red line, 600 ms mixing time, 4 s relaxation delay,
irradiation at δ = 8.14) of 67. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
4.17 13C NMR spectrum of 67. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
4.18 1H NMR spectrum of 68. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
4.19 13C NMR spectrum of 68. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
4.20 Stacked 1H VT-NMR spectrum of 68. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
4.21 1H NMR spectrum of 70. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
4.22 13C NMR spectrum of 70. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
4.23 31P NMR spectrum of 70. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
4.24 1H NMR spectrum of 71. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
ix

4.25 13C NMR spectrum of 71. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
4.26 1H-1H ROESY spectrum (THF−d8, 0 ◦C, 600 ms mixing time, 1 s relaxation delay) of 71. 64
4.27 Molecular structure of 1-(2-methylpyridyl)-4,5-diphenylimidazole with 30% probability
ellipsoids. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
4.28 Molecular structure of 65 with 30% probability ellipsoids. Hydrogen atoms and couter-
anions omitted for clarity. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
4.29 Free energy (G◦, kcal/mol) diagram of 68, TS68,69, and 69. Enthalpies (H◦, kcal/mol)
are given in parentheses. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
5.1 Left: The “trop” moiety as part of a chelating ligand for transition metals. Right: Low
valent metal complexes with tetradentate amino/amido olefin ligands. . . . . . . . . . . . 70
5.2 Molecular structure of (S,S )-81 with ellipsoids at the 30 % probability level. Hydrogen
atoms have been omitted for clarity. Distances (A), angles (◦), and torsions (◦): Ru1–P1
2.282(2), Ru1–P2A 2.23(1), Ru1–Cl1 2.421(1), Ru1–Cl2 2.434(1), Ru1–Cl3 2.504(1), Ru1–
Cl4 2.477(1), Ru2–Cl3 2.460(1), Ru2–Cl4 2.432(1), Cl2· · ·H2 2.213, Ru2–N1 2.123(5),
Ru2–N2 2.135(5), Ru2–ct1 2.127(4), Ru2–ct2 2.136(6), C4–C5 1.412(7), C19–C20 1.373(6);
P1-Ru1-P2A 95.5(4), Cl1-Ru1-Cl2 168.96(5), N1-Ru2-N2 80.1(2), ct1-Ru2-ct2 99.0(2);
C4-C5-C19-C20 -74.8(5). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73
5.3 Molecular structure of 82 with ellipsoids at the 30 % probability level. Hydrogen atoms
have been omitted for clarity. Distances (A) and angles (◦): Ru1–N1 2.131(4), Ru1–
N2 2.129(3), Ru1–ct1 1.980(4), Ru1–ct2 1.973(5), C4–C5 1.423(7), C19–C20 1.435(7),
O1· · ·H1 2.100; N1-Ru1-N2 82.0(2), ct1-Ru1-ct2 98.30(2), N1-Ru1-ct2 92.18(2), N2-Ru1-
ct1 92.18(2), N1-Ru1-ct1 163.51(2), N2-Ru1-ct2 162.37(2). . . . . . . . . . . . . . . . . . . 74
5.4 Drawings of structurally characterized 84,279 72,266 and proposed solution state structure
of 83. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
6.1 Iron precatalysts for the TH of ketones with [6.5.6] metallacycles prepared by our group. . 81
6.2 Molecular structure of 2 (top row, ellipsoids at 30 % probability level) and calculated gas-
phase structural model of 86 (bottom row). Phenyl hydrogens and counteranions have
been omitted for clarity. Selected bond distances and angles are presented in Table 6.1. . 84
6.3 Calculated geometries and selected bond lengths (A, left to right, top to bottom): 88,
TS88,89AcMe(−726i cm−1), TS90,87iPrOH
(−1049i cm−1), TS90,91iPrOH(−1055i cm−1),
TS87,91 (−319i cm−1), and 91. Phenyl hydrogens have been omitted for clarity. . . . . . 87
6.4 Free energy profile for the compounds shown in Scheme 6.3 with energies of free iPrO–,iPrOH, and AcMe used where appropriate. Energies are all calculated relative to 85 and
2 equivalents iPrO– in iPrOH solvent continuum. . . . . . . . . . . . . . . . . . . . . . . . 88
6.5 Experimental, calculated, and scaled carbonyl stretches for proposed complexes 87 and 91. 89
6.6 Calculated structure of 85, gas phase. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
6.7 Molecules 85, 86, 92,309 and 93295 and their calculated models. . . . . . . . . . . . . . . 92
6.8 Calculation scale factor λ.306 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
7.1 First-generation Fe(II) [6.5.6] (94), second-generation Fe(II) [5.5.5] complexes (95), and
third-generation Fe(II) [5.5.5] complexes (96) for the TH of ketones. . . . . . . . . . . . . 94
7.2 HOMO (left) and LUMO (right) for complex 102. . . . . . . . . . . . . . . . . . . . . . . 98
x

7.3 Energy profile for the catalyst activation process and enolate side reactions. All energies
are relative to 102, 3iPrOH, and AcPh. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100
7.4 Optimized structures and selected bond lengths (A, left to right, top to bottom): 102,
TS102AcPh,103 (1120i cm−1), TS102AcPh,104 (1269i cm−1), TS102iPrOH,105 (1409i cm−1),
TS102iPrOH,106 (1649i cm−1), 106, TS106,107 (869i cm−1), and 107. . . . . . . . . . . . 102
7.5 Energy profile of hydrogen transfer from iPrOH to AcPh via amido-eneamido complex
107 (continued from Figure 7.3). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
7.6 Optimized structures and selected bond lengths (A, left to right, top to bottom): TS107aiPrOH,108
(1143i cm−1), 108, TS108,109AcMe(430i cm−1), 109, TS109AcPh,110 (407i cm−1), 110,
TS110,107PE(1096i cm−1), an TS107biPrOH,111 (483i cm−1). . . . . . . . . . . . . . . . . 105
7.7 Transition states marked with potential deuteration sites for KIE calculations. . . . . . . 107
7.8 Energy profile to reduce the eneamido ligand portion of complex 107 (continued from
Figure 7.5). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
7.9 Energy profile of hydrogen transfer from iPrOH to AcPh via bis(amido) complex 116
(continued from Figure 7.8). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
7.10 Calculated structures relative to full amido-eneamido complex 100 . . . . . . . . . . . . . 110
7.11 Optimized structure and selected bond lengths (A) for TS124,109(ν = 1074i). . . . . . . . 112
7.12 Optimized structures and selected bond lengths (A) for 126 (top), 101 (middle), and
TS127,101(ν = 1089i, bottom). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 114
7.13 Energy profile for alcohol assisted proton transfer. All energies are relative to 102. . . . . 119
7.14 Optimized structures and selected bond lengths (A) of TS128,129 (1733i cm−1) and 129. 119
7.15 Energy profile for other considered transfer hydrogenation mechanisms. All energies are
relative to 102. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
7.16 Optimized structures and selected bond lengths (A, left to right, top to bottom): 131
(79i cm−1), TS131,132 (466i cm−1), TS134,135 (1455i cm−1), 135, and TS135,136 (419i cm−1).122
xi

List of Schemes
1.1 Reaction of Ir(I) PNP complex 1 with H2. . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 Ruthenium-catalysts for the AH (top) and ATH (bottom) of valuable chiral ketones. . . . 5
1.3 Mechanistic work performed with 7 and 19 in AH. . . . . . . . . . . . . . . . . . . . . . . 6
1.4 Proposed mechanisms for the hydrogenation of ketones, from experimental and calculated
evidence, using RuH2(diamine)(diphosphine) catalysts.64 . . . . . . . . . . . . . . . . . . 7
1.5 Thermal and photochemical production of H2 and O2 with a transition metal Ru-PNN
pincer complex. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
1.6 Organometallic systems which display unique reactivity relevant to water splitting. . . . . 12
1.7 Other known complexes that react with water via MLC. . . . . . . . . . . . . . . . . . . . 12
2.1 Reaction of 50 with H2O to generate 51. . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
2.2 Hydrogen bonding equilibria in solution when reacting 50 with two equivalents of H2O
to generate 52. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
2.3 Reactions of 54 with H2O. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
3.1 Synthesis of complex 60. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
3.2 Synthesis of 61. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
3.3 Summary of the reactivity of complexes 60 and 61. . . . . . . . . . . . . . . . . . . . . . . 33
3.4 Azaallyl complexes prepared by Wolczanski and co-workers. . . . . . . . . . . . . . . . . . 33
4.1 General deprotonation behavior and O–H activation using pyridine-centered Ru-PNL pin-
cer complexes, where L = N or P with the curved line representing various linkers. . . . . 40
4.2 Synthesis of compounds 63-67. Conditions: Step i) Ag2O, CHCl3, 3 A sieves, 89 %;
step ii) [RuCl2(NBD)]n, CH3CN/CHCl3, 60 ◦C, then MPF6 (M = Na, K), 60 %; step iii)tBu2bpy, (CH3)2CO, reflux, 89 %; step iv) KBr, (CH3)2SO, 110 ◦C, 89 %. . . . . . . . . . 41
4.3 Synthesis of 68. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
4.4 Top: intramolecular proton exchange behavior of 68 in solution with proposed inter-
mediate 69; the colour-matched asterisks indicate the protons on the pincer framework
undergoing two-site chemical exchange while the red proton is being transferred. Bottom:
trapping experiments with PPh3, generating doubly dearomatized complex 70. . . . . . . 44
4.5 Reaction of 68 with H2O or D2O. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
5.1 Left: dehydrogenation of methanol/water mixtures catalyzed by 76. Right: stoichiometric
reactivity studies of 76, generating Ru(0) complex 75 or 77. dme = 1,2-dimethoxyethane. 71
5.2 Reaction of 76 with db18c6 or NBu4Br and subsequent reaction with H2O. . . . . . . . . 72
xii

5.3 Synthesis of 81. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72
5.4 Synthesis of neutral Ru complex 82 and dianionic complex 83. HMDS = hexamethyldis-
ilazane. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73
6.1 Reaction Scheme for TH of Ketones Using 85 As a Precatalyst in the Presence of Excess
Base. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
6.2 Summary of the observed reactivity of Fe PNNP [6.5.6] complexes. . . . . . . . . . . . . . 82
6.3 Proposed Mechanism for Formation of 86, 87, and Fe(0) Species 91. . . . . . . . . . . . . 86
7.1 General Equations for Inner Sphere Hydrogenation, ATH, and AH of Prochiral Ketones
Catalyzed by Metal Complexes with Ancillary Ligands L and Amine Ligand NH2R. . . . 96
7.2 General Reaction Scheme for the TH of Ketones Catalyzed by Iron PNNP Complexes. . . 96
7.3 Diimine Precatalysts 95 and Their Reactivity with Strong Base to Generate 97. . . . . . 97
7.4 Reaction of Third-generation Fe(II) PNNP [5.5.5] Complex 98 with Base and Hydrogen
Sources. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
7.5 Proposed Mechanism Responsible for the Activation Period and Enolate Side Reactions. . 99
7.6 Proposed Catalytic Cycle for the Transfer Hydrogenation of Ketones via Amido-Eneamido
complex 107 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
7.7 Reaction of Bis(amino) Complex 113302 with Base, Which Likely Generates Bis(amido)
Complex 114 in Situ. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 108
7.8 Calculated Structures Starting with Simplified amido-eneamido Complex 107 (1 atm, 298
K) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
7.9 Calculated Structures Starting with Amido-eneamido Complex 99 or 100 . . . . . . . . . 113
7.10 General Mechanism for the ATH (right loop, clockwise from active isomer) and AH (left
loop, counter-clockwise from active isomer) of Prochiral Ketones Using Fe(II) 5.5.5 PNNP
Complexes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 116
7.11 Proton transfer to ligand with the assistance of a second equivalent of iPrOH. . . . . . . . 119
7.12 Other considered mechanisms for the transfer hydrogenation of ketones. . . . . . . . . . . 120
7.13 Mechanisms considered in the main text and alternate conformers/complexes. . . . . . . . 123
8.1 Potential water splitting cycle with RuNCN pincer complexes. . . . . . . . . . . . . . . . . 133
xiii

List of Abbreviations
∆ change, difference.
δ chemical shift.
ε extinction coefficient.
λ wavelength.
µL microlitre.
ν frequency.
◦ degrees.
‡ transition state.
A Angstrom.
ArF fluoroaryl.
iPrOH 2-propanol (isopropanol).
iPr isopropyl.
tBu tert-butyl.
trop2dach N,N-bis(5H-dibenzo[a,d]- cyclohepten-5-yl)-1,2-diaminocyclohexane.
trop2dpen N,N-bis(5H-dibenzo[a,d]- cyclohepten-5-yl)-1,2-diphenyl-1,2-ethylenediamine.
G Gibbs free energy.
H enthalpy.
J coupling constant.
S entropy.
h Planck’s constant.
i imaginary unit.
k rate constant.
k b Boltzmann’s constant.
xiv

1D one-dimensional.
2.2.2-crypt 4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane.
2D two-dimensional.
AcMe acetone.
AcPh acetophenone.
AH asymmetric H2 hydrogenation.
alk alkyl.
Ar aryl.
ATH asymmetric transfer hydrogenation.
BINAP 2,2’-bis(diphenylphosphino)-1,1’-binaphthyl.
Bn benzyl.
bpy 2,2’-bipyridine.
BSSE basis set superposition error.
Bu butyl.
C Celsius.
cal calorie.
calcd calculated.
CCD charge-coupled device.
cm centimetre.
coord coordinated.
Cp η5-cyclopentadienyl.
CP/MAS cross-polarization/magic angle spinning.
Cy cyclohexyl.
dad 1,4-diazabuta-1,3-diene.
dae 1,2-diaminoethane.
DART direct analysis in real time.
db18c6 2,3,11,12-Dibenzo-1,4,7,10,13,16-hexaoxacyclooctadeca-2,11-diene (dibenzo-18-crown-6).
DFT density functional theory.
xv

dme 1,2-dimethoxyethane.
DMPE 1,2-bis(dimethylphosphino)ethane.
DNMR dynamic nuclear magnetic resonance.
DOSY diffusion ordered spectroscopy.
dpen 1,2-diphenyl-1,2-ethylenediamine.
DSSC dye-sensitized solar cell.
e exponential function.
EA elemental analysis.
ee enantiomeric excess.
ESI electrospray ionization.
Et ethyl.
exptl experimental.
EXSY exchange spectroscopy.
FT Fourier transform.
HMDS hexamethyldisilazane.
HOMO highest occupied molecular orbital.
Hz Hertz.
IEF-PCM integral equation formalism polarizable continuum model.
IMes 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene.
IR infrared.
IRC intrinsic reaction coordinate.
K Kelvin.
KHMDS potassium hexamethyldisilazide.
KIE kinetic isotope effect.
ln natural logarithm.
LUMO lowest unoccupied molecular orbital.
M molarity.
xvi

Me methyl.
mg milligram.
MHz megahertz.
min. minutes.
MLC metal-ligand cooperation.
mm millimetre.
mmol millimole.
MO molecular orbital.
mol mole.
MS mass spectrometry.
ms millisecond.
nacnac 1,3-diketimine.
NBD bicyclo[2.2.1]hepta-2,5-diene.
NHC N-heterocyclic carbene.
nm nanometre.
NMR nuclear magnetic resonance.
NOESY nuclear Overhauser effect spectroscopy.
olef olefin.
PE 1-phenylethanol.
Ph phenyl.
ppm parts per million.
py pyridine.
quat quaternary.
R gas constant.
ROESY rotating-frame nuclear Overhauser effect spectroscopy.
s second.
T temperature.
xvii

TH transfer hydrogenation.
THF tetrahydrofuran.
tmen 1,1,2,2-tetramethylethylenediamine.
TMS trimethylsilyl.
TOF turnover frequency.
TON turnover number.
trop 5H-dibenzo[a,d]-cycloheptenyl.
TS transition state.
uncoord uncoordinated.
UV ultraviolet.
VT variable temperature.
Xyl xylyl.
xviii

Chapter 1
Introduction
1.1 Metals and Ligands Working Together: “Bifunctional,” “Co-
operative”, and “Non-Innocent” Behaviour
The principles of homogeneous transition metal catalysis classically treats ligands as “spectators,” pro-
viding ancillary services to the action occurring at the metal center. Many of the elementary processes
in homogeneous catalysis either involve direct reaction of a substrate/ligand with the metal (association,
oxidative addition, insertion) or release into the bulk solution (dissociation, reductive elimination), often
accompanied by changes in the metal’s oxidation state.1 In the last few decades, however, catalyst design
has shifted towards the incorporation of “hybrid” multidentate ligand systems that remain bonded to the
metal complex while actively participating in the formation and cleavage of chemical bonds (Figure 1.1).
In one case, the transition metal can be thought to have Lewis acidic character while the multiden-
tate ligand has Lewis basic character, with additional electron donation from the ligand to the metal.
(A). Cleavage of E−H across the metal-ligand scaffold often leaves the metal’s formal oxidation state
unperturbed, with a concomitant change in the ligand-to-metal electron donor properties. The second
case can be perceived as a donor-acceptor interaction between metal and ligand, respectively, where
an electron accepting Lewis acidic ligand in combination with an electron rich transition metal cleaves
chemical bonds (B). This often results in a formal oxidation at the metal and reduction at the ligand,
creating an M−E−L binding motif.2 The final conceptual form is somewhere in between – aptly called
“non-innocent” – where metal-based and ligand-based redox orbitals are similar in energy, resulting in
ambiguous assignment of formal oxidation states (C). This can lead to the redox and bond cleavage
events occurring at the ligand or metal.3–7
Thus, the concept of a metal working together with a ligand can mean many things, depending on the
choice of transition metal and ligand(s). Cases A and B have been coined metal-ligand bifunctional,8,9
metal-ligand cooperative,10 or chemically non-innocent11 behaviour. Case C is often differentiated
from the former examples by the more specific term “redox non-innocence.”7 The concept described
above in case A, which is the focus of this thesis, will be consistently referred to as “metal-ligand
cooperation” (MLC). There have been many reviews written on the topic of MLC in recent years,2,6,11–29
and samples have been chosen to highlight some of the common design elements without exhaustively
reviewing all known compounds, particularly focusing on monometallic MLC with applications in ketone
hydrogenation and water splitting. The MLC concept will the divided into two broad categories: (1)
1

Chapter 1. Introduction 2
Mx [Ln] Mx [Ln]
E Hδ+E H+
E H-
δ−
Mx [Ln] Mx+2 [Ln]
H Eδ−H E+
H E-
δ+
Mx [Ln ] Mx+m [Ln]m
Acidic Metal,Basic Ligand
RedoxNon-Innocence
Acidic Ligand,Basic Metal
A
B
C
Figure 1.1: Three conceptual forms of ligands working/interacting together with metals. M = transitionmetal, Ln = multidentate ligand, m = number of electrons, E = H, OH, OR, NHR.
those which cleave chemical bonds across a multidentate ligand fragment directly bonded to the metal
(proximal MLC) and (2) those which cleave chemical bonds across a multidentate ligand fragment not
directly bonded to the metal (distal MLC).
1.2 Proximal Metal-Ligand Cooperation
The origins of explicitly observed MLC in the primary coordination sphere were initially discovered by
Fryzuk and co-workers, showing that the coordinatively unsaturated Ir(I)-PNP complex 1 containing an
anionic amido donor oxidatively adds H2 to generate 2 and then heterolytically cleaves H2 to obtain a
mixture of the mer- and fac- the hydrido-amino Ir(III) complexes 3 and 4 (Scheme 1.1).30 The coor-
dination chemistry and reactivity with commonly available ligands was investigated (CO, PR3, olefins,
MeI)31,32 and other transition metals were explored,33–35 however applications in catalysis remained
restricted to simple olefin hydrogenations with complex 1.31 Thus, the potential for exploiting the MLC
properties of metal-amido moieties for other catalytic applications remained dormant for many years.
IrN PPh2
PPh2
Si
Si
IrN PPh2
HPPh2
Si
Si
H
HH
IrN
PPh2
HPPh2
Si
Si
H
HH
IrN PPh2
HPPh2
Si
SiHH2 H2
1 2 3 4
Scheme 1.1: Reaction of Ir(I) PNP complex 1 with H2.
A non-exhaustive collection of well-characterized MLC ligand frameworks are shown in Figure 1.2.
All compounds are structurally characterized and/or have been isolated and fully characterized by NMR
spectroscopy. Most of these coordinatively unsaturated compounds are all synthesized by salt elimination
reactions in the presence of strong base. The arrow drawn from the blue amido nitrogen to the metal
emphasizes that there is additional π-donor stabilization from the ligand to the metal, with the exception
of 14 which contains a central π-donor carbene ligand coloured in pink. In all cases, the “hybrid” Lewis
basic fragment on the ligand and Lewis acidic metal center reversibly and heterolytically cleaves various
H−X bonds.
Many of the compounds in Figure 1.2 are active catalysts in a variety of reactions. Compound
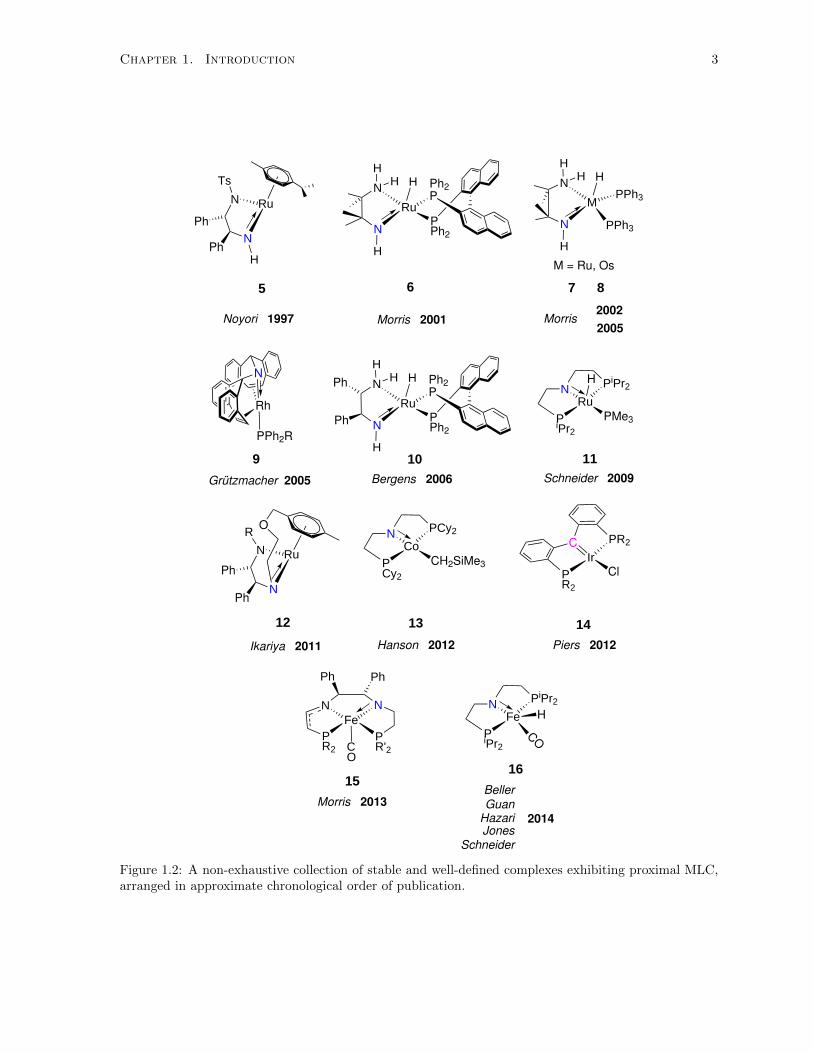
Chapter 1. Introduction 3
PPh2
Rh
N
PPh2R
RuN PiPr2
PMe3PiPr2
Schneider 2009
Jones
IrC PR2
ClPR2
Grützmacher 2005
M
H
PPh3
N
PPh3N
H
H
H
2002
2005
RuN
NPh
PhH
Ts
Noyori 1997
RuN
N
Ph
Ph
RO
Ikariya 2011
M = Ru, Os
FeN N
PR2
PR'2C
O
Ph Ph
Morris 2013
FeN PiPr2
H
PiPr2
CO
Beller
Schneider
Guan
2014
Piers 2012
H
CoN PCy2
CH2SiMe3PCy2
Hanson 2012
Morris
Bergens 2006
Hazari
Ru
HN
N
Ph
Ph
H
H
HPh2P
PPh2
Ru
HN
N
H
H
HPh2P
Morris 2001
5 6 7 8
9 10 11
12 13 14
1516
Figure 1.2: A non-exhaustive collection of stable and well-defined complexes exhibiting proximal MLC,arranged in approximate chronological order of publication.

Chapter 1. Introduction 4
58,9 is an active catalyst for the asymmetric transfer hydrogenation of ketones and imines, while 6,36
7,37 8,38 9,39 and 1040,41 are active for the (asymmetric) H2 hydrogenation of ketones (Section 1.2.1).
Compound 1142,43 is an extremely active catalyst for the dehydrogenation of ammonia-borane, while
complexes 1244 and 1545,46 are enantioselective catalysts for both transfer and H2 hydrogenation. The
paramagnetic Co(II)PNP complex 1347,48 is a versatile H2 hydrogenation catalyst of alkenes, ketones,
and imines under very mild conditions. The iridium carbene complex 1449 reversibly splits H2 across the
Ir−−C moiety. Finally, complex 16 was reported around the same time by many groups, with applications
in acceptorless (de)hydrogenation of N-heterocycles,50 acceptorless (de)hydrogenation of alcohols and
ketones,51 hydrogenation of esters to alcohols,52,53 and the hydrogenation of aliphatic and aromatic
nitriles.54
Some common features appear among the above described complexes. The Lewis basic nitrogen
atom is flanked by at least one sp3-hybridized atom, with the exception of 14, increasing the basicity
at nitrogen to furnish heterolytic bond cleavage. In addition, the above ligand designs have creatively
circumvented β-hydride elimination, with the exception of 11, where the PNP ligand can selectively
dehydrogenate and rehydrogenate itself.42 Finally, the above complexes are neutral, decreasing Lewis
acidity at the metal and in turn preventing irreversible addition of the heterolytically cleaved molecules.
These and other design elements have emerged in the development of MLC catalysts for the various
applications described above.
1.2.1 Applications of MLC in Ketone Hydrogenation
The asymmetric H2 hydrogenation (AH) of ketones is performed in the pharmaceutical, agrochemical
and fragrance industries to produce valuable enantiopure alcohols and amines. For example, specially
designed ruthenium catalysts containing elaborate enantiodirecting diphosphine and diamine ligands
are important for catalyzing the addition of hydrogen gas to inexpensive aryl ketones in order to obtain
(S )- or (R)-alcohol products for perfumes or pharmaceutical intermediates. Scheme 1.2 (top) shows the
AH of 3-(dimethylamino)propiophenone to the (R)-enantioenriched alcohol using a ruthenium complex
developed by Noyori and coworkers.55 This alcohol is later used to prepare (R)-fluoxetine, a potent
serotonin-uptake inhibitor used for treating depression. Another useful reduction process is asymmetric
transfer hydrogenation (ATH) where the hydrogen is transferred from the solvent, often isopropanol,
to the ketone function to produce the enantiopure alcohol. For example, Baratta and co-workers56
made ruthenium complexes containing the (R,S )-Xyliphos ligand to reduce a simple ketone to (S )-1-(3-
trifluoromethylphenyl)ethanol, used in the synthesis of the fungicide (S)-MA20565 (Scheme 1.2, bottom).
In these examples, the ligands have been carefully crafted for optimum turn over frequency (TOF) and
turn over number (TON).
Gao, Ikariya, and Noyori synthesized (S,S )-trans-RuCl2(PNNP) (17) and
(S,S )-trans-RuCl2(PNHNHP) (18), which are both precatalysts for the asymmetric transfer hydro-
genation of aromatic ketones in basic isopropanol (Figure 1.3).57,58 However, complex 17 was poorly
active/selective for the ATH of acetophenone (3 % yield, 18 % ee after 48 h where ee is enantiomeric
excess) while 18 had excellent activity and selectivity (91 % yield, 97 % ee after 25 h). It was later
discovered by our lab that the chiral hydride-chloride analog of 18 was highly active for both ATH and
AH when treated with base; however, the activity and enantioselectivity were not the same as above.59
This suggested that different catalysts or mixtures of catalysts were forming in solution depending on
the reaction conditions, specifically the amount of added base and the nature of the reductant.

Chapter 1. Introduction 5
Fe
Ru
ClH2N
NH2
PXyl2
Xyl2P
catalyst (0.01 mol%)
Cl
MeO
MeO
O
NMe2
OH
NMe2KOtBu
H2 (8 atm)iPrOH
25 oC
H
N
NH
RuPh2P
PXyl2
Cl
catalyst (0.01 mol%)
NaOiPr
60 oC
O
H
H
O
CF3
OH
CF3
H
(excess)
Scheme 1.2: Ruthenium-catalysts for the AH (top) and ATH (bottom) of valuable chiral ketones.
The dramatic difference in activity for diamine precatalysts versus diimines can be rationalized by
MLC and the so-called “NH effect.”8,9,12,13,15 Base was required during catalysis and it was postulated
that its role was to deprotonate the amine (NH) functionality and generate a reactive metal-amido
moiety. This reacts with dihydrogen and then transfers a proton and hydride equivalent in the outer
coordination sphere to ketones in a “bifunctional” (MLC) manner, initially proposed to be going through
a six-membered pericyclic transition state (Figure 1.3, right).
Ru
N
PPh2
N
PPh2
Cl
Cl
Ru
N
PPh2
N
PPh2
Cl
Cl
H
H
poor activity and selectivitygood activity
high enantioselectivity
Ru N
H H
C O
proposedtransition state
δ+
δ−
δ+
δ+
δ−
δ−
17 18
Figure 1.3: The dramatic difference in ATH activity between catalysts 17 and 18.
A key discovery by our laboratory was that neutral, structurally characterized metal-amido complexes
such as 7 could cleave H2 heterolytically to yield the trans-dihydride complex 19, and that these are cru-
cial intermediates during catalysis (Scheme 1.3).37,60,61 Once the NH−RuH moiety is in place, proton and
hydride can then be transferred to the substrate. Using the “tmen” (1,1,2,2-tetramethylethylenediamine)
ligand, which has methyl groups instead of hydrogens on carbons α to the amido group (β to the ruthe-

Chapter 1. Introduction 6
nium) was important to allow the isolation of an amido species. If hydrogens are present, the ruthenium
amido complexes tend to undergo β-hydride elimination to catalytically inactive imine complexes.
Ru PPh3
PPh3
H2N
NH
H
Ru
PPh3
PPh3
H2N
N
H
HH2
ketonealcohol
Ru N
HH
δ+
δ−δ+
δ−
benzene HH
7 19
Scheme 1.3: Mechanistic work performed with 7 and 19 in AH.
Rigorous mechanistic investigations in recent years have shown that the nature of proton and hydride
transfer is likely to be a two-step process as opposed to a concerted one in many cases, and this is a subject
of ongoing experimental and computational investigation, which is briefly summarized in Scheme 1.4.
The achiral RuH2(tmen)(PPh3)2 (19) generates the fully characterized amido complex 7 and free alcohol
in the presence of a ketone substrate (top), which could transfer a proton and hydride in a concerted
manner as originally proposed by Noyori and co-workers.37,60,61 The chiral trans-dihydride complex
RuH2((R,R)−dpen)((R)−BINAP) (20), on the other hand, does not generate the free amido species;
instead, an alkoxide complex 21 is observed and characterized, leading to the proposal of a concerted
transition state and rearrangement to an alkoxide (bottom).41,62–64 Finally, computational investigations
have indicated a stepwise reaction mechanism, first transferring a hydride and then forming a transient
metal-alkoxide ion pair (middle).65–67 From this ion pair, either 7 or 21 can be accessed, depending on
whether proton transfer or alkoxide rearrangement occurs. In light of these recent computational studies,
which include higher levels of theory and solvent continuum effects, the ion pair intermediate mechanism
is becoming more widely accepted, as opposed to the originally proposed bifunctional/concerted proton-
hydride transfer.
We were motivated to explore analogous iron-based systems; however early attempts by our group
to synthesize iron diamine-diphosphine analogues akin to dihydrides 7 and 19 were unsuccessful. The
catalytic potential of such iron-based hydrogenation catalysts has been investigated using Density Func-
tional Theory (DFT).68 The low cost of iron, its high abundance, and its reduced toxicity are all desirable
catalyst features for the pharmaceutical, fragrance, and agrochemical industries. If active, well-defined,
homogeneous iron catalysts could be developed, experimental and computational mechanistic analyses
would be conducted in parallel.
1.3 Distal Metal-Ligand Cooperation
Another subset of compounds also exhibit reversible MLC, which also involve the secondary, or distal,
ligand coordination sphere. The distinction made between the compounds in Figure 1.4 and those
presented above in Figure 1.2 is that a Lewis basic ligand site accepting hydrogen is not directly bonded to
the metal. In many of these systems, the driving force for reversible H−X addition to the coordinatively
unsaturated metal and ligand involves the protonation-deprotonation of a distal carbon atom (coloured

Chapter 1. Introduction 7
Ru NH
ORR'
HH
M NH
ORR'
HH
Ru
H
PPh3
N
PPh3N
H
H
H
Ru
HPN
PNH
H2
H2
onlyhydride transfer
+Ru NH
O-RR'
HH
Metal-alkoxide ion pairadduct
(calculated)
H+ transfer
+ ORR'
HHRu NH
ORR'
HH
concertedproton-hydride transfer
and alcohol release
Rearrangement
O
R' R
Hconcerted
proton-hydride transferand rearrangement
Observed:
Observed:
RuH
2(tm
en)(
PP
h 3) 2
RuH
2(dp
en)(
BIN
AP
)
PPh2
Ru
HN
N
Ph
Ph
H
H
HPh2P
H
197
20
21
Scheme 1.4: Proposed mechanisms for the hydrogenation of ketones, from experimental and calculatedevidence, using RuH2(diamine)(diphosphine) catalysts.64

Chapter 1. Introduction 8
in pink) on the multidentate ligand which aromatizes-dearomatizes the ligand, with the exception of
25, 33, and 36, where protonation occurs at a distal nitrogen atom. The sp2 carbon atoms involved in
distal MLC are converted to an sp3 hybridized state upon protonation or electrophilic attack, with the
exception of 22 and 23, where the ketone oxygen is converted to an alcohol.
Many of these compounds are also widely used in homogeneous catalysis. Intermediates 2269,70 and
2371,72 contain a zero-valent unsaturated metal complex in its most active form, and are active hydro-
genation catalysts. The electrophilic cyclopentadienone oxygen atom accepts a proton, aromatizing the
ring to an anionic “Cp” moiety, while the electrophilic metal accepts a hydride and the formal oxidation
state at the metal increases by two. The dearomatized Ru-PNN pincer complex 24,73 which is useful
in a huge variety of chemical transformations, contains a dearomatized pyridyl moiety which rearom-
atizes upon addition of H−X compounds. Other neutral pincer compounds such as 29,74 30,75 and
31,76,77 have been developed in recent years, and are active catalysts for an impressive array of hydro-
genation/dehydrogenation reactions. Complexes 25,78 33,79 and 3680 reversibly bind H2 using basic
pendant amine moieties installed on a bidentate phosphine ligand, carefully constructed for applications
in either electrocatalytic H2 oxidation or H+ reduction. Compound 2681 possesses a bidentate “nacnac”
ligand with a distal β-carbon atom that can cooperate with the metal to invoke reversible cycloaddition
reactions with ethylene and acetylene. Ruthenium complex 2882,83 can not only reversibly cleave H2
across the metal and ligand, but also reversibly insert CO2 at the distal carbon atom of the diazaflu-
orene ring. The anionic hydride complex 3484 contains a planar conjugated metallacycle, where both
distal carbon and proximal nitrogen atoms can be hydrogenated, and is an active catalyst for methanol
dehydrogenation in the presence of water. Complex 3585,86 reversibly binds CO2, H2, and nitriles, and
is an active formic acid dehydrogenation catalyst.
1.3.1 Applications of MLC in Homogeneous Water Splitting
One of the key challenges facing society in the future is a source of renewable energy, where a carbon-
neutral or carbon-free energy conversion cycle is realized.87 Water splitting, or the conversion of H2O
to H2 and O2, is a carbon neutral, environmentally benign candidate for this application in which
organometallic chemistry and catalysis can play a vital role.87–91 Nature is our champion water splitter,
with the Mn4O4 cluster in plant leaves’ photosystem II92 converting 2 H2O → O2 + 4 H+ + 4 e– and
the hydrogenase93–95 family of enzymes converting 2 H+ + 2 e– → H2 (Figure 1.5, top). The overall
redox reaction is thermodynamically uphill, with the most difficult step being O2 production, requiring
a half-cell potential of 1.23 V (vs. NHE).
One approach in catalyst development for this application is designing separate molecular catalysts
for the reduction (cathodic) and oxidation (anodic) half-reactions, which could then be embedded into
a solar-powered fuel cell.87 For example, Ru(II) polypyridyl complexes such as 37 can catalyze the
production of O2 in the presence of a sacrificial oxidant in acidic aqueous solutions.91,96–101 Rigorous
mechanistic analysis98,102 of 37 and related compounds have revealed that the mechanism of O2 produc-
tion goes through high valent intermediates (formally Ru(II) to Ru(V)), with O−−O formation being the
rate-limiting step. Although catalyst design has historically been dominated by ruthenium polypyridyl
complexes, other molecular catalysts acting as structural and/or functional mimics to nature’s photo-
system II have been designed using Mn, Fe, Co, Ir, and Cu complexes.91 At the other (cathodic) end,
proton reduction catalysts such as 25 have been developed, which use MLC principles (Figure 1.4) to
combine protons and electrons at the metal-ligand interface to eletrocatalytically generate H2 in aqueous
Chapter 1. Introduction 9
Ni2+
PP
PP
NBn
Cy
Cy
Cy
NBn
Cy
N
Fe+
P
P
tBu
NtBu
tBu
tBu
Ru
NEt2
NPtBu2
CO
CH
H
Ru
NR2
N
N
CO
CH
H
NAr
Song
Sanchez
Milstein 2006 DuBois 2006
Bullock 2014
N
HC
N
RuPh3PPPh3
H N
N
Song 2010
N
F
F
F
F
Mn+
PP
PP
NBn
Ph
Ph
ArF
ArF
ArF
ArF
N
Bn
CO
Bullock 2013
Ph
Ph
O
RuOC
OC
Ar
Ar
2011
Shvo 1986
Re
PtBu2
NPtBu2
CO
C
C
H
O
RuN
PtBu2
CO
CH
H
N
Milstein 2011
Milstein 2014
TMS
TMS
O
FeOC
OC
Knölker 1999
Casey 2007
RuN
NAr
Ar
CH
Dyson 2007
N
N
Bn
Bn
K+(dme)2
N N
C C
Ru-
H
Grützmacher 2013
H H
Fe
PiPr2
NPiPr2
CO
CH
H
Milstein 2011
L
22 23 24 25
26 28 29
30 31 33
34 35 36
Figure 1.4: A non-exhaustive collection of stable, isolable, and well-defined complexes exhibiting distaland reversible MLC (with the exception of 22 and 23, postulated on the basis of molecular trappingexperiments).

Chapter 1. Introduction 10
acidic media.78,103–105
N
NN
N
NN
N Ru
H2O2+
(4Ce4+ + 2 H2O 4Ce3+ + O2 + 4H+)
Ni2+
PP
PP
NBn
Cy
Cy
Cy
NBn
Cy
N
N
Bn
Bn
2H2O O2 + 4H+ + 4e- Eanode = 1.23 V
4H+ + 4e- 2H2 Ecathode = 0 V
2H2O 2H2 + O2Erxn = -1.23 V
O2 production catalyst H2 production catalyst
(vs. NHE)
electrocatalysis in aqueous acidic media
using a sacrificial oxidant
37 25
Figure 1.5: Top: Half-cell reactions for the production of H2 and O2 from H2O. Bottom: recent examplesof well-defined catalysts for each half-reaction.
Such homogeneous oxidation and reduction catalysts could then be used in dye sensitized solar cells
(DSSCs), which use a light harvesting organometallic complex (a dye/chromophore, such as [Ru(bpy)3]2+
)
attached to a heterogeneous semiconductor (such as TiO2) to drive the oxidation half-reaction forward
(instead of a sacrificial reductant as shown above).106 The ejected electrons and protons are then recom-
bined in the presence of a proton reduction catalyst to generate H2. Photocatalytic molecular systems
for H2 production have also been intensely studied.107,108 This is an oversimplified interpretation to
highlight the essence of the reaction using well-defined molecular catalysts, and there are still many
challenges to overcome in achieving efficient catalytic turnovers with minimal energy loss and maxi-
mal efficiency.91 Complete, standalone photoelectrochemical water splitting devices (“artificial leaves”)
are indeed known, which generate H2 and O2 using water and light.109 These systems are based on
nanostructured materials instead of molecular components; a recent example is Nocera’s water splitting
catalyst system, which uses a triple-junction Si photovoltaic light harvesting device that is modified with
an electrodeposited Ni-Mo-Zn alloy for H2 evolution and cobalt-borate catalyst for O2 evolution.110
A new concept for water splitting catalyst design appeared in 2009 when Milstein and co-workers
discovered that the ruthenium pincer complex 24 stoichiometrically splits water in solution to gener-
ate one equivalent H2 and a half equivalent O2 in two discrete steps by using, heat, light, and MLC
(Scheme 1.5).111–113 First, the dearomatized Ru-PNN complex 24 quantitatively and reversibly cleaves
H2O to the generate hydrido-hydroxo complex 38 in 45 % yield. Next, complex 38 is refluxed for three
days, liberating H2 and forming the structurally characterized bis(hydroxo) complex 39, presumably
through intermediate A. Finally, 39 is irradiated with UV light under a flow of argon, complex 38 is
regenerated in 49 % yield, with formation of O2. The detection of O2 was quantified using multiple
detection techniques by analyzing the gaseous headspace from sealed reaction vessels and determined to
be 23 % on average. The mixed isotopomer of 39 was also prepared (Scheme 1.5, bottom left), which
suggests that the O2 formation process is intramolecular due to the predominant formation of 34O2 after
UV irradiation. The transient Ru(0) intermediate B is proposed to form, along with formation of H2O2,

Chapter 1. Introduction 11
which then disproportionates to evolve O2 and oxidatively add H2O to regenerate 38. Other analytical
and chemical techniques were also conducted to rule out the presence of ·OH radicals being the source
of O2 formed in the reaction, and additional computational studies have been conducted to elucidate
more intimate mechanistic details regarding H2 and O2 production.114–118
Ru
NEt2
NPtBu2
CO
16OH
18OHhν
32O2 : 34O2 : 36O2
3.8 : 16.2 : 1
Ru
NEt2
NPtBu2
CO
H2O
OH
-H2
Ru
NEt2
NPtBu2
OH
H CO
OH
H2O"HOOH"0.5 O2
Ru
NEt2
NPtBu2
CO
H
+H2O
-H2O
hν
H H H H H
Ru
NEt2
NPtBu2
CO
OH
H
Ru0
NEt2
NPtBu2
CO
H H
+
A
B
24 38 39
Scheme 1.5: Thermal and photochemical production of H2 and O2 with a transition metal Ru-PNNpincer complex.
To date, there is still no other system that has performed a water splitting cycle via MLC, either
stoichiometrically or catalytically. However, there have been some interesting reactions of relevance
to organometallic water splitting (Scheme 1.6). In the presence of aqueous acid, osmocene (40) can
be irradiated with visible light to generate [Cp2Os(IV)(H2O)2]2+
(41) and H2. In an independent
reaction, the osmocene hydroxide cation 42 reacts with UV light to generate osmocene, acid, and O2
gas.119 A recent theoretical investigation has shown that Cp ring slippage probably occurs en route to
O2 formation, suggesting that the “ancillary” ligand works together with the metal in some capacity
to facilitate the reaction.120 The Pt(IV)(hydroxo)(hydroperoxo) species 43 photoeliminates dihydrogen
trioxide and forms 44 at low temperatures, which has been investigated in detail by molecular trapping
experiments.121 Finally, the disilane-linked titanocene (bis)hydroxide complex 45 was shown to cleanly
photoeliminate a ·OH radical at low temperatures by using a radical trapping agent, subsequently forming
Ti(III) complex 46.122
Aside from hydroxide complex 38, there are only two other known complexes that react with water
via MLC (Scheme 1.7). The hydrido-amido complex 10 reacts with trace amounts of water at low
temperatures, and the trans-RuH(OH)((R,R)−dpen)((R)−BINAP) complex is characterized by 1H and13C NMR spectroscopy.41 The other example is using nickel carbene pincer complex 48, which reacts
with various H−X molecules, including water, to irreversibly generate 49.123
1.4 Thesis Outline
The work described in the first part of this thesis (Chapters 2–5) was inspired by the water splitting chem-
istry exhibited with Ru-PNN pincer complex 24. We sought to design MLC systems for organometallic
water splitting reactions (Figure 1.6). Chapter 2 investigates the known ruthenium (7) and osmium (8)
amido complexes using ligand A, and their reactivity with H2O is explored (Prokopchuk, D. E.; Collado,

Chapter 1. Introduction 12
PtHO
Et3P
PEt3
Cl
TiIV
CF3
OOH
hν (380 nm)
-78 oCPt
Et3P
PEt3
Cl
CF3
+ HOOOH
16-20 %
OH
OH
Si
Si
hν (visible)TiIII OH
Si
Si-23 oC+ OH
Os OsIV
OH2
OH2
hν (visible)
2+
+ H2+ H2O + 2H+
OsIV OH
+
hν (350 nm)+ H+ 0.5 O2+Os
40
40 41
42
43 44
45 46
Scheme 1.6: Organometallic systems which display unique reactivity relevant to water splitting.
NiC PiPr2
PPh3PiPr2
NiC PiPr2
OHPiPr2
H
H2O
characterized at -60 oC by NMR
THF
H2O(trace)P
Ph2
Ru
HN
N
Ph
Ph
H
H
HPh2P
PPh2
Ru
HN
N
Ph
Ph
H
H
HPh2P
OHH
10 47
48 49
Scheme 1.7: Other known complexes that react with water via MLC.

Chapter 1. Introduction 13
A.; Lough, A. J.; Morris, R. H. Dalton Transactions 2013, 42, 10214–10220).∗ In this work, I synthe-
sized and characterized new Ru/Os complexes, and wrote the manuscript; Alba Collado synthesized the
Os complex 8 as part of a graduate student research exchange project and investigated its reactivity
with water; Dr. Alan Lough carried out all work related to single crystal X-ray diffraction; Professor
Morris directed the research and prepared the manuscript for submission.
Chapter 3 investigates ruthenium complexes containing tridentate ligand B, with hopes that the cen-
tral nitrogen atom can participate in proximal MLC and subsequent water splitting chemistry
(Prokopchuk, D. E.; Lough, A. J.; Morris, R. H. Dalton Transactions 2011, 40, 10603–10608).† In
this work, I synthesized, characterized all the described compounds and wrote the manuscript; Dr. Alan
Lough carried out all work related to single crystal X-ray diffraction; Professor Morris directed the
research and prepared the manuscript for submission.
Chapter 4 explores the reactivity of N-heterocyclic carbene C, which contains flanking methylpyridyl
ligands that could potentially be deprotonated/dearomatized for distal MLC water splitting applica-
tions (Prokopchuk, D. E.; Tsui, B. T. H.; Lough, A. J.; Morris, R. H. Chemistry – A European Journal
2014, 20, 16960–16968).‡ I synthesized and characterized most compounds, performed VT-NMR ex-
periments/simulations and DFT calculations, and wrote the manuscript; Brian Tsui synthesized and
characterized some ruthenium complexes as part of an undergraduate summer research project; Dr.
Alan Lough carried out all work related to single crystal X-ray diffraction; Professor Morris directed the
research and prepared the manuscript for submission.
Chapter 5 investigates the coordination chemistry with tetradentate amino-olefin ligand D, which
could potentially utilize the proximal amine atoms and distal carbon backbone in MLC water splitting,
as seen with complex 34. I synthesized and characterized all new ruthenium complexes in the laboratory
of Prof. Hansjorg Grutzmacher (ETH Zurich) in 2014 as part of a research exchange program; crystal
mounting and data collection for the first structurally reported complex in Chapter 5 was performed
by Rafael Rodrıguez-Lugo during his PhD studies at ETH Zurich; I carried out all other work related
to single crystal X-ray diffraction with the assistance of Bruno Pribanic (PhD student, ETH Zurich)
and Michael Worle (Head, Small Molecule Crystallography Center, ETH Zurich). This is an ongoing
collaborative project between Professors Morris and Grutzmacher.
N
N
NH2N NH2N N
CH2H2C
N N
HNH HN
C C
=
H H
A B C D
Figure 1.6: Four different ligand systems reported in this thesis for applications in water splitting.
Throughout the last seven years, our lab has pioneered three generations of enantioselective iron
catalysts containing PNNP ligands for the asymmetric hydrogenation of ketones and imines.124 Driven
by the desire to determine their mechanism of action by both experiment and theory, the second part of
∗Reproduced with permission from The Royal Society of Chemistry.†Reproduced with permission from The Royal Society of Chemistry.‡Reprinted with permission. Copyright c© 2014 John Wiley and Sons.

Chapter 1. Introduction 14
this thesis (Chapters 6 and 7) deal with the DFT calculated mechanisms involving our first, second, and
third generation iron catalysts (Figure 1.7). In particular, Chapter 6 examines the mechanistic insights
gained from DFT calculations on our first generation system (Prokopchuk, D. E.; Sonnenberg, J. F.;
Meyer, N.; Zimmer-De Iuliis, M.; Lough, A. J.; Morris, R. H. Organometallics 2012, 31, 3056–3064).∗ I
performed most DFT calculations and wrote the DFT portions of the manuscript, in addition to editing
revised versions of the entire manuscript; Jessica Sonnenberg synthesized/characterized new compounds
and performed NMR/catalysis experiments; Dr. Nils Meyer synthesized one complex and grew crystals
suitable for X-ray diffraction; Marco Zimmer-De Iuliis performed preliminary DFT calculations; Dr.
Alan Lough carried out all work related to single crystal X-ray diffraction; Professor Morris directed the
research and prepared the manuscript for submission.
Chapter 7 first explores the mechanism of transfer hydrogenation with our homogeneous second
generation catalysts (Prokopchuk, D. E.; Morris, R. H. Organometallics 2012, 31, 7375–7385),† for which
I performed all DFT calculations and wrote the manuscript. Next, the third generation catalysts, which
are the most ATH catalysts in existence,46,125 are reported to be both ATH and DH catalysts (Zuo, W.;
Tauer, S.; Prokopchuk, D. E.; Morris, R. H. Organometallics 2014, 33, 5791–5801).‡ In this manuscript,
I performed all DFT calculations and wrote the DFT portion of the manuscript; Dr. Weiwei Zuo wrote
all other parts of the manuscript and performed most synthesis and catalysis experiments; Sebastian
Tauer performed some synthesis and catalysis experiments as part of an undergraduate student research
exchange project; Professor Morris directed the research and prepared the manuscripts for submission.
Previously unpublished work in this chapter includes preliminary results comparing the calculated ATH
activity and enantioselectivity with experimental values.
FeN
PPh2
2+
N
PPh2
MeCN
CO
R = aryl; R' = H, aryl
2+
FeN N
PR2
PR2C
O
R' R'
MeCN
+
FeN N
PR2
PR'2C
O
Ph PhX
R, R' = arylX = Cl, Br
H
1st generation 2nd generation 3rd generation
Figure 1.7: Three generations of iron hydrogenation catalysts examined in this thesis.
∗Adapted with permission. Copyright c© 2012 American Chemical Society.†Adapted with permission. Copyright c© 2012 American Chemical Society.‡Adapted with permission. Copyright c© 2014 American Chemical Society.

Chapter 2
Structural Properties of Trans
Hydrido-Hydroxo Complexes and
their Proton Exchange Behaviour
with Water in Solution
Abstract
We report the synthesis of Ru(II) and Os(II) trans hydrido-hydroxo complexes by reacting
the unsaturated amido complexes MH(NHCMe2CMe2NH2)(PPh3)2 (M = Ru, Os) with sto-
ichiometric amounts of water. Proton exchange is rapid at room temperature between the
amine/water/hydroxide moieties leading to signal averaging of the NMR properties, which
can be slowed at low temperature in order to see resonances of separate complexes. These
compounds can also be cleanly converted back to their starting complexes by dehydration
in the presence of 3 A molecular sieves. X-ray crystal structures of these Ru(II) and Os(II)
trans hydrido-hydroxo complexes reveal that the unit cell contains an additional molecule
of water trapped in the crystal lattice which hydrogen bonds with a neighbouring hydroxo
ligand, forming a water bridged dimer in the solid state. Although there are many cases
of oxidative addition of water to transition metal complexes, relatively few cases are known
where water addition occurs via metal-ligand cooperation (bifunctional addition) without
altering the oxidation state of the metal center.
2.1 Introduction
Water is a very attractive chemical feedstock to produce hydrogen as an energy resource due to its vast
abundance on Earth. Therefore, the reactivity of transition metal complexes with water has important
implications in developing well-defined catalysts that can convert H2O into H2 and/or
O2.87–89,96,97,99,112,113,126 In general, the intramolecular splitting of a water O-H bond at a well-defined
metal complex can be facilitated in two ways: (a) water can be oxidatively added, therefore increasing
15

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 16
the metal’s oxidation state by two and creating new M-H and M-OH bonds;127–130 (b) water can add
in a cooperative metal-ligand fashion where the hydroxide ion binds to the metal center and the lig-
and system is protonated, leaving the metal’s oxidation state unchanged. In the latter case, this can
be achieved using monodentate oxo ligands on high oxidation state transition metal complexes,131–134
a bidentate amino-amido ligand,41 or a tridentate PNN-type pincer ligand.111 While there have been
many reports of water adding to transition metal complexes via oxidative addition or using high-valent
metal-oxo compounds, there are relatively few studies of intramolecular metal-ligand water addition
using multidentate bifunctional ligands.41,111 Furthermore, NMR characterization can be challenging
because the resultant complexes may rapidly exchange protons on the NMR time scale which broadens
signals of interest.135,136
In our efforts to understand how bifunctional metal-amido containing complexes react with water, we
report the 1H NMR, 31P NMR, and structural characterization when the five-coordinate ruthenium(II)
and osmium(II) metal-amido complexes of the general formula MH(NHCMe2CMe2NH2)(PPh3)2 (M =
Ru, Os) are exposed to stoichiometric amounts of H2O. These Ru and Os amido complexes have been
previously studied by our group as catalysts for the hydrogenation of polar bonds14,36–38,61,137 and for
Michael additions.137 Both complexes reversibly cleave the O-H bond of H2O across the metal-amido
fragment to produce trans-MH(OH)(NH2CMe2CMe2NH2)(PPh3)2· nH2O (M = Ru, n = 0, 1; M = Os,
n = 1) which are only clearly observable at low temperatures by 1H NMR spectroscopy. The addition
of stoichiometric equivalents of water changes the appearance of the 1H NMR spectra and provides
information on proton exchange equilibria occurring in solution. Furthermore, we also report molecular
structures of these products which contain an additional water molecule in the crystal lattice that acts
as a hydrogen bonding bridge between neighbouring metal complexes.
2.2 Results and Discussion
We mixed the highly reactive five-coordinate ruthenium hydrido-amido complex
RuH(NHCMe2CMe2NH2)(PPh3)2 (50) with one equivalent of H2O in an NMR tube in toluene-d8
(Scheme 2.1). The 1H NMR spectrum at 25 ◦C (aromatic region excluded) shows broad signals at
2.88 and 0.88 ppm and a triplet at -17.5 ppm (Figure 2.1a, black line) with no detectable amount of 50.
These three signals integrate to a 5:12:1 ratio, respectively, and we assign them as follows: the broad
signal at 2.88 ppm is a combination of NH and OH protons; the signal at 0.88 ppm belongs the methyl
groups on the diamine backbone; the triplet at -17.5 ppm is the metal hydride with a 2JHP coupling
constant of 25 Hz, consistent with being cis to both PPh3 ligands.
Cooling the sample to -60 ◦C significantly changes the appearance of the signals in Figure 2.1a (green
line). The signal originally at 2.88 ppm separates into two distinct peaks at 3.73 and 1.95 ppm (labelled
NHa and NHb, respectively), each accounting for two protons. Based on the chemical shift of these four
protons as compared to similar RuHX(NH2CMe2CMe2NH2)(PPh3)2 complexes,137 we assign these to
be the NH protons belonging to the compound trans-Ru(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2 (51).
The methyl resonance has resolved into two distinct signals at 0.89 and 0.74 ppm and the hydride signal
has shifted upfield to -16.9 ppm. The two CH3 signals at -60 ◦C are also consistent with the presence
of unique chemical environments above and below the plane of the Ru-diamine metallacycle. At room
temperature, there is signal averaging over all four methyl groups as a result of the rapid interconversion
between their axial and equatorial positions,137 whereas at low temperatures these methyl groups resolve

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 17
Ru
H
PPh3
N
PPh3N
H
H
H
H2O
3 Å sieves
Ru
HPPh3N
PPh3NH
OHH
H H
50 51
Scheme 2.1: Reaction of 50 with H2O to generate 51.
into two discrete signals. A new signal appears at -1.51 ppm accounting for one more proton which
we assign to be the Ru-OH moiety, correlating well with other known monomeric trans-Ru(H)(OH)L4
complexes.41,111,135,138 A very broad and weak signal centered at -21.1 ppm is detectable at -60 ◦C which
is characteristic of the starting complex 50. Therefore, we conclude that at room temperature there
is rapid exchange between NH and OH protons while at low temperatures the observed compound is
predominantly 51. The hydride signal position at room temperature (-17.5 ppm) can then be considered
a weighted average of the fraction of 50 and 51 in solution whose signals can be separated at low
temperatures. Taking the chemical shift of the product complex (-16.9 ppm) and the starting material
(-21.1 ppm) with their weighted average found at -17.5 ppm at room temperature means that there is
about 14% 50 and 86% 51 at 25 ◦C. Moreover, an intermediate amido-aqua complex is not observed
at temperatures between 25 ◦C and -80 ◦C. If 3 A molecular sieves are added to the reaction mixture
at room temperature, the major product is the starting material 50, demonstrating the labile nature of
the water molecule. Reacting 50 with one equivalent of D2O followed by probing the reaction solution
at -60 ◦C via 1H NMR resulted in a decrease of signal intensity for peaks NHa, NHb and the Ru-OH,
leaving the hydride, methyl, and phenyl protons unchanged. This supports the notion that rapid proton
exchange is localized to the nitrogen and oxygen atoms.
If two equivalents of water are added to an NMR tube filled with 50 in toluene-d8, the orange reaction
solution turns yellow after a few seconds of shaking and variable temperature 1H NMR experiments were
performed (Figure 2.1b). At room temperature (black line), we rationalize that the broad five-proton
signal at 2.74 ppm is the signal averaging of three groups of protons: two diamine protons NHa, H2O, and
a Ru-OH proton. The remaining two diamine protons, NHb, can be clearly seen at 1.94 ppm at 25 ◦C.
Resolution in the methyl region (0.95 and 0.74 ppm) at room temperature indicates unique chemical
environments above and below the diamine metallacycle. The Ru-H signal appears upfield at -17.41
ppm as a triplet, maintaining its 25 Hz coupling constant due to neighbouring cis phosphines.
When the temperature is decreased to -40 ◦C, the broad five-proton signal at 2.74 ppm disappears
and a new signal appears at 3.75 ppm belonging to NHa (Figure 2.1b, green line). The two-proton signal
formerly at 1.94 ppm shifts slightly to 1.89 ppm. However, there are three protons, belonging to the
ruthenium hydroxide and water moieties, which are not visible at -40 ◦C. Cooling the NMR sample down
to -80 ◦C (Figure 2.1b, blue line) reveals a broad signal at -1.24 ppm belonging to the Ru-OH moiety. In
addition, a very broad signal becomes visible at 6.48 ppm (inset, Figure 2.1b), which partly overlaps with

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 18
Figure 2.1: (a) Overlaid 1H NMR spectra (400 MHz) of 51 at 25 ◦C (black) and -60 ◦C (green). (b)Overlaid 1H NMR spectra (400 MHz) of 52 at 25 ◦C (black), -40 ◦C (green), and -80 ◦C (blue). Residualsolvent signal (CD3) for toluene-d8 appears at 2.09 ppm. Only the inset spectrum on the bottom leftwas obtained using a 600 MHz NMR spectrometer to increase the resolution of the H2O signal. Thearomatic resonances appear from 8.0–6.8 ppm and some have been omitted and some spectra have beenmagnified on the y-axis for clarity.

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 19
the phenyl protons of PPh3. We assign this to be the second water molecule which reveals the presence
of trans-Ru(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2·H2O (52) in solution. The severe broadening of the
H2O and OH peaks when they become visible at -80 ◦C suggests that proton exchange between these
two moieties is much more rapid than the NH-OH proton exchange. As with 51, 52 can be dehydrated
to produce 50 in high yield in the presence of 3 A molecular sieves. Signals belonging to 50 could not
be detected at any temperature between 25 ◦C and -80 ◦C, suggesting that the equilibrium lies much
further toward the product side.
The diamine protons NHb which appear at about 1.8 ppm at low temperatures in Figure 2.1 are
syn-coplanar to the metal hydride (Scheme 2.2). As the diamine conformation changes in solution, Hb is
either in an axial position (syn-coplanar to the hydride) or equatorial which renders it unavailable to H-
bond with water. Ha, on the other hand, is either equatorial or anti-coplanar to the hydride, which makes
it available to form a six-membered ring with the water and hydroxide moieties; as a result, these protons
become de-shielded and appear at about 3.8 ppm. This behaviour was also observed in a previous study
where 50 was reacted with weak acids HX, where X contains an oxygen atom available to participate in
hydrogen bonding.137 Furthermore, the same study showed that the reaction of 50 with two equivalents
of phenol crystallizes to form RuH(OPh)(NH2CMe2CMe2NH2)(PPh3)2·HOPh, in which the OH group
on phenol forms a six-membered H-bonding ring by situating itself between a coordinated phenoxide
oxygen and an amine proton. Our attempts to spectroscopically detect the selective proton exchange
behaviour in the reaction of 52 using low temperature 1H−1H EXSY experiments were unsuccessful due
to the overly broad amine and hydroxide resonances.
Ru
N N
P P
H
OHHa
Hb
Hb
Ha
HO
H
Ru
NN
PP
H
OHHa
Hb
Hb
Ha
HO
H
Scheme 2.2: Hydrogen bonding equilibria in solution when reacting 50 with two equivalents of H2O togenerate 52.
In a related reaction, Bergman and co-workers reacted Ru(DMPE)2(C2H4)
(DMPE = 1,2-bis(dimethylphosphino)ethane) with excess H2O at 90 ◦C followed by crystallization to
obtain [trans-RuH(OH)(DMPE)2(µ−H2O)]2 (53) which is a water bridged dimer in the solid state.135
Variable temperature NMR experiments were also performed on complex 53, and at room temperature,
a broad signal was detected at 2.30 ppm in THF-d8, as compared to 2.90 ppm for 52 in toluene-d8.
Upon cooling 53 to -92 ◦C, the water protons were assigned as a broad signal at 5.70 ppm and the broad
hydroxide proton appeared at -4.55 ppm, as compared to 6.48 and -1.24 ppm for 52, respectively. The
similarities between our findings and those of Bergman and co-workers further support our peak assign-
ments. Furthermore, Piers and co-workers recently observed rapid and temperature dependent proton
exchange between cis aqua-hydroxo ligands on a sterically bulky Pd(OH)(H2O)(α−diimine) complex.136
Given the temperature dependent spectral data of 53, Bergman and co-workers were unable to deter-
mine its structure in solution, presumably uncertain whether the compound was monomeric or dimeric
since the molecular structure afforded a water-bridged dimer. We tried to determine whether our com-

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 20
plex 52 was monomeric or dimeric in solution by running Diffusion Ordered Spectroscopy (DOSY) NMR
experiments at room temperature and comparing the self-diffusion coefficients of the amido complex 50
and 52. By probing the hydride resonances, which were well-resolved at room temperature, we deter-
mined that the self-diffusion coefficient of 50 is 5.5 ± 0.5 ×10−10 m2/s and 5.3 ± 0.2 ×10−10 m2/s for
52, indicating that the overall difference in size of the individual molecules in solution is indistinguish-
ably small, and therefore 52 remains monomeric in solution. Due to the temperature dependent spectral
similarities and similar crystal packing of 52 and 53 (vide infra), we expect that 53 is also monomeric
in solution.
Given the recent successes in consecutive thermal and photochemical evolution of H2 and O2 by a
Ru-PNN pincer complex,111 we were interested to determine whether 52 could evolve H2 by releasing a
proton from the diamine ligand and hydride from the metal center, followed by addition of the second
equivalent of H2O across the metal-amido bond to generate a bis(hydroxo) complex. NMR scale reactions
in toluene-d8 were carried out after injection of two equivalents of H2O to 50 where either (a) the sample
was heated up to 90 ◦C or (b) the sample was irradiated with ultraviolet light at room temperature.
Both reaction solutions gradually changed from yellow to dark orange and were monitored by 31P and 1H
NMR. In both the thermolysis and photolysis reactions, decomposition occurred by dissociation of the
phosphine and diamine ligands which was accompanied by the formation of triphenylphosphine oxide
(PPh3−−O). After several days, the only detectable phosphorus-containing product in the photolysis
reaction was PPh3−−O. Oxidation of the phosphine by water might be accompanied by reduction of the
metal centre, which has been demonstrated in one case for palladium-catalyzed aryl halide activation.139
Furthermore, no detectable amounts of free H2 were identified in the 1H NMR spectra. Attempts to
synthesize a bis(hydroxo) complex by using N2O to transform the metal hydride into a metal hydroxide
moiety138 led to the immediate formation of PPh3−−O and subsequent decomposition of the metal
complex.
The difference in metal-amido bond strength between the analogous ruthenium and osmium com-
plexes is apparent when 54 is reacted with one or two equivalents of H2O. With only one equivalent of
H2O present, the complex trans-Os(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2 is not observed at low tem-
peratures; instead, there is a 1:1 ratio of 54 and trans-Os(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2·H2O
(55) in solution (Scheme 2.3). This equilibrium is rationalized by the fact that 5d metals tend to prefer
metal-ligand multiple bonds over 4d metals,140 especially in the case of 50 versus 54.38 When a second
equivalent of water is added, 54 is present in lower quantities and the major product at low temperatures
is 55. Based on the relative integrations of the hydride and NH regions of 54 and 55 at -60 ◦C, we
estimate that there is about 25% 54 and 75% 55 in solution. The spectral features of 55 are similar to
those of 52 and a summary of key peak assignments is reported in Table 2.1. 1H NMR spectra for these
reactions are provided in the Supporting Information.
We were able to obtain molecular structures of 52 and 55 by growing crystals from a concentrated
solution of each respective metal-amido complex and 2 equivalents of H2O. Both ruthenium and osmium
compounds share strikingly similar metrical parameters (Table 2.2) and only the structure of 52 will
be discussed. Complex 52 conforms to an octahedral geometry with an acute diamine bite angle (N1–
Ru1–N2) of 74.4(1) degrees and an obtuse P1–Ru1–P2 angle of 96.68(3) degrees (Figure 2.2, left). The
oxygen atom bonded to ruthenium is angled towards the diamine ligand (N1–Ru–O1 and N2–Ru–O1
angles of 80.77(9) and 80.9(1) degrees, respectively), with the distance between the closest two NH
hydrogens and oxygen atom being less than the sum of their Van der Waals radii (O1· · ·H1A = 2.504 A

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 21
Table 2.1: Chemical shifts of interest (ppm) in the 1H NMR spectra (400 MHz, toluene-d8) of 52 at-80 ◦C and 55 at -60 ◦C.
Peak AssignmentChemical Shift (ppm)
52 55
H2O 6.48 6.10
NHa 3.88 4.06
NHb 1.86 2.45
M−OH -1.17 -0.71
M−H -17.63 -19.01
Os
H
PPh3
N
PPh3N
H
H
H
H2O
3 Å sieves
Os
HPPh3N
PPh3NH
OHH
H H
. H2O
54 55
Scheme 2.3: Reactions of 54 with H2O.
and O1· · ·H2B=2.649 A, respectively). The oxygen of the hydroxo ligand acts as an H-bond acceptor
while a neighbouring water molecule is the H-bond donor (O1· · ·O2 = 2.692(4) A). Pertinent crystal
structure and refinement data are provided in the Supporting Information.
The crystal packing of 52 reveals a hydrogen bonded dimer in which two water molecules act as
H-bond donors for two hydroxo ligands (Figure 2.2, right). The second molecule of 52 is related to the
first by an inversion centre located in the middle of the O· · ·O· · ·O· · ·O ring. The protons belonging
to the hydroxo ligands do not participate in hydrogen bonding and are positioned between two phenyl
rings. This unusual type of crystal packing motif has been reported by Bergman and co-workers (53, as
mentioned above).135
To our knowledge, the only structurally characterized monomeric Ru(H)(OH) complexes are those
of Bergman and co-workers (53), Milstein and co-workers’ trans-Ru(H)(OH)(CO)(PNN)·nH2O (PNN =
(2-(di-tert-butylphosphinomethyl)-6-(diethylaminomethyl)pyridine)),111 and Whittlesey and co-workers’
cis-Ru(H)(OH)(CO)2(IMes)2 and cis-Ru(H)(OH)(CO)(IPr)2 (IMes = 1,3-bis(2,4,6-trimethylphenyl)
imidazol-2-ylidene, IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene).141,142 The Ru-O bond dis-
tances for these four compounds fall between 1.995(2) A and 2.230(2) A, with the longest Ru-O distances
occurring when the hydroxide is trans to the powerful σ-donating hydride ligand. The metal-oxygen
bond for 52 is 2.190(2) A which falls within this range and only slightly shorter than the 2.230(2) A
Ru-O bond distance in Bergman and co-workers’ compound (53).135 Also, the O· · ·O distance between
water and hydroxide is shorter for 52 (2.692(4) A) than in 53 (2.758(3) A) which indicates a stronger
hydrogen bond in our system.
As for structurally characterized Os(H)(OH) complexes, there are two molecular structures reported
in the literature: Caulton and co-workers’ five coordinate Os(H)(OH)(CO)(PtBu2Me)2
143 and Esteruelas

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 22
Figure 2.2: Left: Asymmetric unit of 52 depicted with thermal ellipsoids at the 30% probability level.Hydrogen atoms on aromatic rings and methyl groups have been omitted for clarity. Selected bonddistances and angles are listed in Table 2.2. Right: Crystal packing of 52 as a water bridged dimerdepicted with thermal ellipsoids at the 30% probability level. Phenyl carbons/hydrogens and methylhydrogens have been omitted for clarity.
Table 2.2: Selected Bond Lengths (A) and Bond Angles (◦) for 52 and 55.
52 55
Ru1–N1 2.182(3) Os1–N1 2.185(4)
Ru1–N2 2.185(3) Os1–N2 2.186(5)
Ru1–P1 2.2485(9) Os1–P1 2.241(1)
Ru1–P2 2.2506(8) Os1–P2 2.250(1)
Ru1–H1Ru 1.61(3) Os1–H1Os 1.57(5)
Ru1–O1 2.190(2) Os1–O1 2.192(3)
O1· · ·O2 2.692(4) O1· · ·O2 2.709(6)
N1–Ru1–N2 74.4(1) N1–Os1–N2 74.5(2)
P1–Ru1–P2 96.68(3) P1–Os1–P2 98.82(5)
N1–Ru1–O1 80.77(9) N1–Os1–O1 79.6(1)
N2–Ru1–O1 80.9(1) N2–Os1–O1 80.2(1)
O1–Ru1–H1Ru 170(1) O1–Os1–H1Os 163(2)

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 23
and co-workers’ Os(H)(OH)(CO)(η2−CH2−−CHCO2CH3)(PiPr3)2.144 In both cases, the hydride and
hydroxide ligands are cis to one another with Os-O bond lengths of 2.022(5) and 2.111(3) A, respectively,
expectedly shorter than the Os-O bond distance of 2.192(3) A for 55 since it is trans to a hydride.
2.3 Conclusion
In summary, we have reacted five coordinate complexes of the general formula
MH(NHCMe2CMe2NH2)(PPh3)2 (M = Ru, Os) with stoichiometric amounts of water and examined the
proton exchange behaviour of the resultant solutions via variable temperature NMR. At low temperatures
we were able to observe and spectroscopically characterize the compounds
trans-M(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2·nH2O (M = Ru, n = 0, 1; M = Os, n = 1) and obtain
molecular structures when n = 1. Interestingly, the additional molecule of water serves as a hydrogen
bonding bridge to a neighbouring metal complex in the solid state, forming a dimer in the crystal lattice
for both Ru and Os. We expect that the proton exchange behaviour observed in these systems would
also be relevant for other unsaturated transition metal complexes containing a metal-amido linkage and
stoichiometric reactions with water could have implications in characterizing inorganic/organometallic
complexes capable of water splitting.
2.4 Experimental
2.4.1 General Comments
All experiments were carried out under argon using standard Schlenk and glove box techniques. Com-
pounds RuH(NHCMe2CMe2NH2)(PPh3)2 (50)37 and OsH(NHCMe2CMe2NH2)(PPh3)2 (54)38 were
synthesized according to known literature procedures. Oxygen-free H2O was prepared by pouring dis-
tilled water into a Schlenk flask, applying high vacuum for about one minute and then sonicating for
10 minutes to remove dissolved gases. The vacuum/sonication steps were repeated twice more after
which the vessel was filled with argon. Toluene-d8 was purchased from Aldrich in individually packaged
ampoules and stored in an argon filled glove box over 3 A molecular sieves before use. D2O was pur-
chased from Aldrich and degassed prior to use according to the procedure described above and stored
under argon. Due to the oxygen sensitivity of the observed products and labile nature of the OH/H2O
moiety, mass spectrometric and elemental analyses were not performed. All NMR spectroscopic data
were collected using a Varian 400 MHz NMR system (operating at 400 MHz for 1H or 162 MHz for 31P)
or an Agilent DD2 600 MHz spectrometer with an Agilent OneNMR probe (operating at 600 MHz for1H). All 1H NMR samples were referenced to their respective residual solvent methyl peak of toluene-d8
(δ = 2.08) and 31P NMR were referenced to an external standard of 85% aqueous H3PO4 at δ = 0.00.
Coupling constant J values are given in Hz. For DOSY NMR experiments, the Bipolar Pulse Pair
Stimulated Echo with Convection Compensation NMR experiment was used with a diffusion delay of
100 ms, diffusion-encoding pulse-width of 2 ms, and a diffusion-encoding pulse strength of 0.001966
gauss/cm.145 All reactions and spectroscopic experiments were carried out in toluene-d8 and variable
temperature NMR studies were conducted immediately after reacting the metal complexes with water;
prolonged exposure of the reaction solutions to low temperatures led to product precipitation or crystal-
lization. Ultraviolet irradiation was conducted using a 450 W Hanovia medium-pressure mercury lamp.

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 24
Single-crystal X-ray diffraction data were collected at 150 K using a Nonius Kappa-CCD diffractometer
with Mo Kα radiation (λ = 0.71073 A). The CCD data were integrated and scaled using the Denzo-
SMN package.146 The structures were solved and refined using SHELXTL V6.1.147 Refinement was by
full-matrix least-squares on F 2 using all data.
2.4.2 trans-Ru(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2 (51)
In an argon filled glove box, 50 (20 mg, 0.027 mmol) was placed into an NMR tube and then dissolved
in toluene-d8 (0.8 mL). The tube was sealed with a tight fitting rubber septum which was then wrapped
with Parafilm-M R©. The NMR tube was removed from the glove box and distilled/degassed H2O (0.50
µL, 0.027 mmol) was injected with a microsyringe. Shaking the tube for a few seconds allowed the droplet
of water deposited at the top to react with the 50 solution below and a subsequent change of colour from
orange to yellow-orange was observed. The solution was immediately analyzed by NMR spectroscopy.
At -40 ◦C, trans-Ru(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2 (51) is observed. X-ray quality crystals of
52 (vide infra) can be obtained in a few days by leaving the solution sealed and undisturbed at room
temperature. Addition of 3 A molecular sieves at room temperature (before crystallization) followed by
vigorous stirring regenerates 50 in high yield. Spectral data for 51: 1H NMR (400 MHz; -60 ◦C): δH
7.84 (12 H, m, PPh3), 6.99 (18 H, m, PPh3), 3.73 (2 H, br, NHa), 1.95 (2 H, br, NHb), 0.89 (6 H, s,
CH3), 0.74 (6 H, s, CH3), -1.51 (1 H, br s, RuOH), -16.88 (1 H, br, RuH). 31P{1H} NMR (162 MHz;
-60 ◦C): δP 73.93 (s).
2.4.3 trans-Ru(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2·H2O (52)
The same procedure was used as above except two equivalents of distilled/degassed H2O (1.0 µL, 0.054
mmol) were added to 50 (20 mg, 0.027 mmol) in toluene-d8 (1.6 mL). The product became yellow
after shaking for a few seconds. At -80 ◦C, trans-Ru(H)(OH)(NH2CMe2CMe2NH2)(PPh3)2·H2O (52)
is observed. X-ray quality crystals of 52 can be obtained overnight by leaving the solution sealed
and undisturbed at room temperature. Addition of 3 A molecular sieves at room temperature (before
crystallization) followed by vigorous stirring regenerates 50 in high yield. Spectral data for 52: 1H NMR
(400 MHz; -80 ◦C): δH 7.94–7.77 (12 H, m, PPh3), 7.07–6.99 (18 H, m, PPh3), 6.48 (2 H, br, partly
buried under Ph), H2O), 3.85 (2 H, br, NHa), 1.83 (2 H, br, NHb), 0.94–0.54 (1 2 H, br, CH3), -1.24 (1
H, br s, Ru-OH), -17.43 (1 H, br, RuH). 31P{1H} NMR (162 MHz; -80 ◦C): δP 66.70 (s).
2.4.4 Thermolysis/photolysis of 52
A solution of 52 was prepared as described above. The sample was taken into an argon-filled glove box
and transferred via syringe to a J. Young tube. In two independent experiments, the tube was either
immersed in a 90 ◦C oil bath or exposed to ultraviolet light. The reaction solutions were periodically
analyzed by 1H and 31P{1H} NMR. After three days of photolysis, the only observable product by31P{1H} NMR is PPh3−−O; after five days of thermolysis, the only observable products by 31P{1H}NMR are 52, PPh3, and PPh3−−O.

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 25
2.4.5 OsH(NHCMe2CMe2NH2)(PPh3)2 (54) nH2O(n = 1, 2).
The same procedure was used as above. When one equivalent of distilled/degassed H2O (0.5 µL, 0.027
mmol) was to 54 (22 mg, 0.027 mmol) in toluene-d8 (1.1 mL), the solution turned yellow-orange after
shaking. At -40 ◦C, both 54 and 55 are observed a 1:1 ratio. The chemical shifts of 54 in toluene-
d8 are nearly identical to those already reported elsewhere in benzene-d6.38 When two equivalents of
distilled/degassed H2O (1.0 µL, 0.054 mmol) were added to 54 (22 mg, 0.027 mmol) in toluene-d8 (2.2
mL), the solution turned yellow after shaking. The major observed product by 1H NMR and 31P{1H}NMR is 55. Addition of 3 A molecular sieves at room temperature (before crystallization) followed by
vigorous stirring regenerates 54 in high yield. Spectral data for 55: 1H NMR (400 MHz; -60 ◦C): δH
7.78 (12 H, m, PPh3), 7.06 -6.94 (18 H, m, PPh3), 6.09 (2 H, br, H2O), 4.04 (2 H, br, NHa), 2.43 (2
H, br, NHb), 0.83-0.70 (12 H, m, CH3), -0.72 (1 H, br s, OsOH), -19.02 (1 H, br, OsH). 31P{1H} NMR
(162 MHz; -60 ◦C): δP 22.22 (s).

Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 26
2.5 Supplementary Information
2.5.1 X-ray Structural and NMR Data
Table 2.3: X-Ray crystal structure and refinement data for complexes 52 and 55.
Compounds 52 55
Empirical formula C42H50N2O2P2Ru C42H50N2O2P2Os
Formula mass 777.85 866.98
Temperature (K) 150(2) 150(1)
Wavelength (A) 0.71073 0.71073
Crystal system Monoclinic Monoclinic
Space group P21/n P21/n
a (A) 13.9422(4) 13.9210(3)
b (A) 15.7847(2) 15.8226(4)
c (A) 16.9683(4) 16.9605(6)
α (◦) 90 90
β (◦) 94.2440(9) 94.033(2)
γ (◦) 90 90
Volume (A3) 3724.0(2) 3726.5(2)
Z 4 4
Density (calculated, g/cm3) 1.387 1.545
Absorption Coefficient (mm−1) 0.545 3.546
F(000) 1624 1752
Crystal Size (mm3) 0.14 × 0.12 × 0.06 0.10 × 0.10 × 0.10
Theta range for data collection (◦) 2.73–27.56 2.57–27.55
Reflections collected 20367 34052
Independent reflections (R(int)) 8073 (0.0473) 8502 (0.0843)
Completeness to θ = 25.00◦ (%) 98.0 98.9
Absorption correction Multi-scan Multi-scan
Max. and min. transmission 0.969, 0.882 0.712, 0.577
Refinement methodFull-matrixleast-squares on F 2
Full-matrixleast-squares on F 2
Data / restraints / parameters 8073/0/449 8502/0/449
Goodness-of-fit on F2 1.040 1.054
R1(I > 2σ(I))a 0.0461 0.0428
wR2 (all data)b 0.1268 0.0974
a R1 =Σ(Fo − Fc)
Σ(Fo)
b wR2 =
√Σ[w(F 2
o − F 2c )2]
Σ[w(F 2o )2]
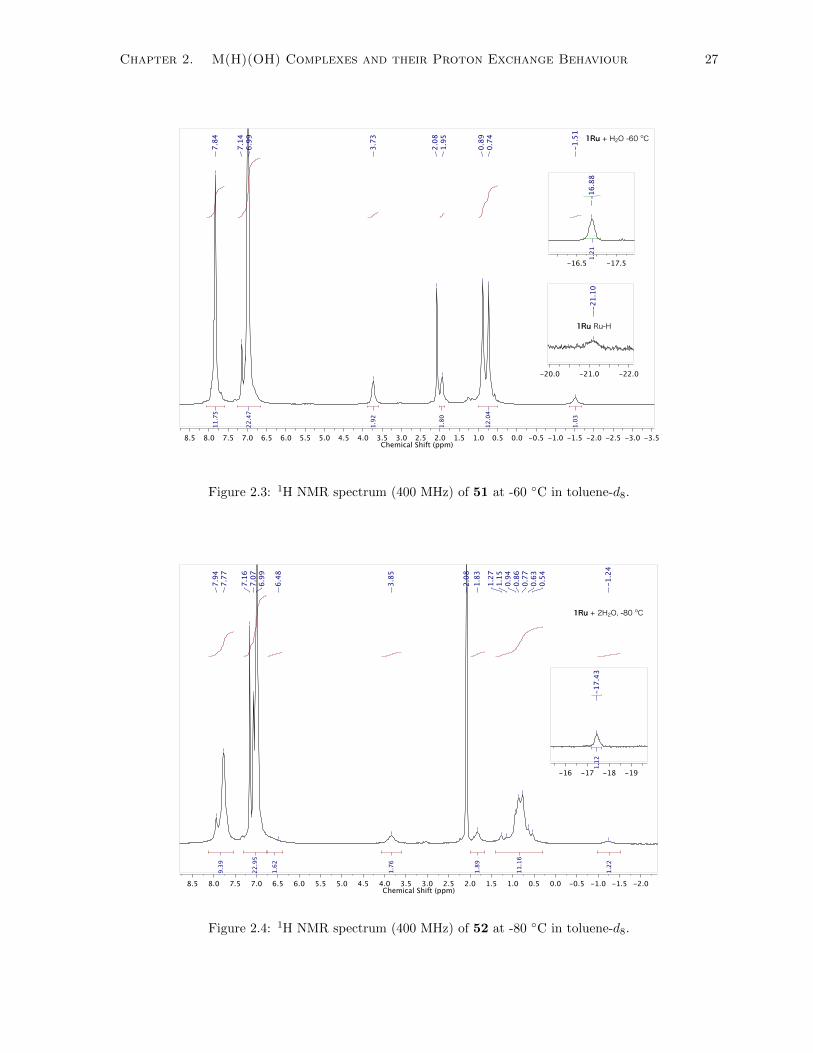
Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 27
-3.5-3.0-2.5-2.0-1.5-1.0-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5Chemical Shift (ppm)
1.03
12.04
1.80
1.92
22.47
11.75
-1.51
0.74
0.89
1.95
2.08
3.73
6.99
7.14
7.84
-17.5-16.5 1.21
-16.8
8
-22.0-21.0-20.0
-21.1
0
Figure 2.3: 1H NMR spectrum (400 MHz) of 51 at -60 ◦C in toluene-d8.
-2.0-1.5-1.0-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5Chemical Shift (ppm)
1.22
11.16
1.89
1.76
1.62
22.95
9.39
-1.24
0.54
0.63
0.77
0.86
0.94
1.15
1.27
1.83
2.08
3.85
6.48
6.99
7.07
7.16
7.77
7.94
-19-18-17-16 1.12
-17.4
3
Figure 2.4: 1H NMR spectrum (400 MHz) of 52 at -80 ◦C in toluene-d8.
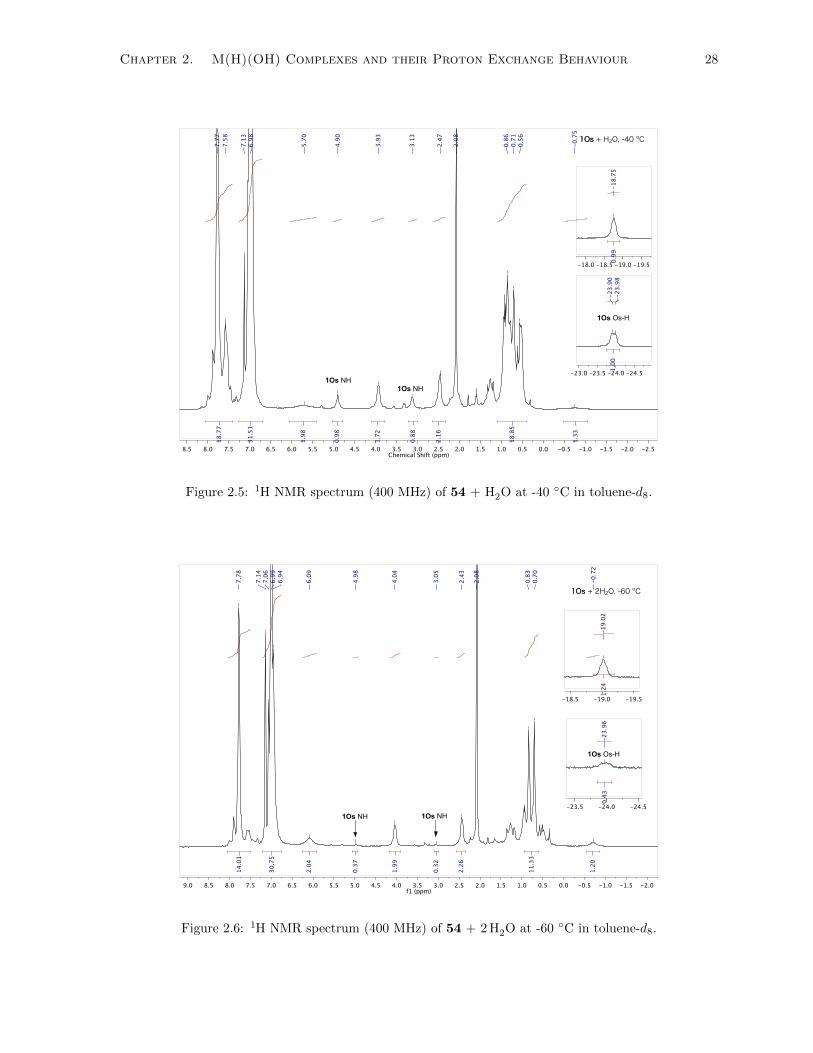
Chapter 2. M(H)(OH) Complexes and their Proton Exchange Behaviour 28
-2.5-2.0-1.5-1.0-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5Chemical Shift (ppm)
1.33
18.85
2.16
0.88
1.72
0.98
1.98
31.51
18.77
-0.75
0.56
0.71
0.86
2.08
2.47
3.13
3.93
4.90
5.70
6.98
7.13
7.58
7.77
-19.5-19.0-18.5-18.0 0.99
-18.7
5
-24.5-24.0-23.5-23.0 1.00
-23.9
8-2
3.90
1Os NH1Os NH
1Os Os-H
Figure 2.5: 1H NMR spectrum (400 MHz) of 54 + H2O at -40 ◦C in toluene-d8.
-2.0-1.5-1.0-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.0f1 (ppm)
1.20
11.33
2.26
0.32
1.99
0.37
2.04
30.75
14.01
-0.72
0.70
0.83
2.08
2.43
3.05
4.04
4.98
6.09
6.94
6.99
7.06
7.14
7.78
-19.5-19.0-18.5 1.24
-19.0
2
-24.5-24.0-23.5 0.43
-23.9
6
1Os Os-H
1Os NH 1Os NH
Figure 2.6: 1H NMR spectrum (400 MHz) of 54 + 2 H2O at -60 ◦C in toluene-d8.

Chapter 3
From Amine to Ruthenaaziridine to
Azaallyl – Unusual Transformation
of Di-(2-pyridylmethyl)amine on
Ruthenium
Abstract
The complexation of di-(2-pyridylmethyl)amine to RuHCl(PPh3)3 affords the salt [RuH{κ3N-fac-1,3-di-(2-pyridylmethyl)amine}(PPh3)2]Cl. Reaction with potassium tert-butoxide at
room temperature yields the unusual ruthenaaziridine complex RuH{κ3C alkNN py-1,3-di-(2-
pyridylmethyl)amine}(PPh3)2, where the central nitrogen atom, adjacent alkyl carbon, and
pyridine arm coordinate to the metal, leaving the second pyridine arm uncoordinated. Sur-
prisingly, heating of this ruthenaaziridine complex with concomitant H2 formation affords
the ruthenium azaallyl complex RuH(κ3N -1,3-di-(2-pyridyl)-2-azaallyl)(PPh3)2. This is a
rare example of a 4d metal complex containing the azaallyl ligand. X-ray crystal structures
and NMR characterization of all three compounds are presented herein.
3.1 Introduction
Tridentate ligands containing mixed nitrogen and phosphorus donors have recently received much atten-
tion, affording tunable and robust ligand systems for late transition metals.11,21,32,148–151 For example,
the highly reactive five-coordinate Ru-PNP complex 56, which contains a central secondary nitrogen,
reversibly reacts with H2152 and is a highly active ammonia-borane dehydrogenation catalyst (Fig-
ure 3.1).42 Moreover, Ru-PNN complex 57 which contains a central pyridine ring that is dearomatized
also reversibly reacts with H2.21 It efficiently catalyzes the formation of alcohols into esters153 and
splits water into H2 and O2.111 Ruthenium compounds with tridentate PNP and PNN ligands have
also been utilized in the catalytic reduction of ketones.154–157 The nucleophilic nitrogen atom of 56 and
29

Chapter 3. Transformation of Di-(2-pyridylmethyl)amine on Ruthenium 30
vinylic carbon atom next to phosphorus in 57 are thought to assist in the activation of substrates during
bifunctional/cooperative catalysis.12,14–16,18–20,23,158,159
As a simple starting point, we chose to use the parent compound di-(2-pyridylmethyl)amine, which
contains two pendant methylpyridine “arms” and a central secondary amine donor. It is easily syn-
thesized in high yields160 and is cost-effective, both desirable properties of any ligand system. Some
iron-group complexes containing tridentate nitrogen donor ligands (NNN) have been examined for use
in catalysis,161–175 and iron complexes of amine-functionalized di-(2-pyridylmethyl)amines have been
intensely studied as analogues of active sites of iron-containing oxidases.176–178 The parent ligand can
also be deprotonated twice to generate the neutral amido iridium complex 58 and anionic, dearomatized
diamido species 59 (Figure 3.1).179,180 However, there are only a few reports of ruthenium compounds
containing this ligand.181–185 We decided to investigate its reactivity with the common ruthenium hy-
dride precursor RuHCl(PPh3)3, rationalizing that a combination of a secondary amine proton and metal
hydride in the presence of base may offer a promising metal-ligand bifunctional system for catalytic trans-
formations, such as H2 hydrogenation and/or transfer hydrogenation.14,18–20,23,36,37,61,155,156,159,186–188
RuN PiPr2
PMe3PiPr2
H
Ru
N PtBu2
CONEt2
H
IrN
N
NIr
NN
N
-
56 57 58 59
Figure 3.1: Examples of ligand systems containing anionic amido ligand donors.
3.2 Results and Discussion
3.2.1 Synthesis of Ruthenium Di-(2-pyridylmethyl)amine and Ruthenaaziri-
dine Complexes.
Our investigations began by reacting RuHCl(PPh3)3 with di-(2-pyridylmethyl)amine, expecting that re-
moval of two phosphines would lead to coordination of the tridentate ligand. However, stirring for several
minutes in tetrahydrofuran (THF) afforded the salt
[RuH{κ3N-fac-1,3-di-(2-pyridylmethyl)amine}(PPh3)2]Cl (60) in excellent yield (Scheme 3.1). Com-
plex 60 is freely soluble in protic solvents and was fully characterized by NMR studies in methanol-d4.
This product resulted from the displacement of the chloride anion and removal of only one phosphine.
The 1H NMR shows that the NH proton on the di-(2-pyridylmethyl)amine ligand is shifted significantly
downfield (6.2 ppm) compared to the methylene protons (3.8-3.6 ppm). The hydride resonance appears
at -14.7 ppm as a triplet (2JHP = 29 Hz), given the Cs symmetry of the cation.
An X-ray crystal structure of complex 60 was also obtained (Figure 3.2, Table 3.1 and Table 3.2).
In the solid state, 60 adopts a distorted octahedral geometry with the chloride counteranion hydrogen
bonded to the amine proton. The bite angles between the central nitrogen atom and pendant pyridine
arms are 77.4(2)◦ and 83.7(2)◦ for the N1-Ru-N2 and N1-Ru-N3 angles, respectively (Table 3.1). This
is complemented by the larger angle of 95.70(7)◦ between the phosphine ligands. The Ru-N2 bond

Chapter 3. Transformation of Di-(2-pyridylmethyl)amine on Ruthenium 31
RuN
H
PPh3
PPh3N
NH
+Cl-
NN N
H
RuHCl(PPh3)3 +THF
10 min., 25 oC-PPh3
60
Scheme 3.1: Synthesis of complex 60.
is longer for 60 than in the related complexes trans,fac-[Ru{di-(2-pyridylmethyl)amine)}(CO)2Cl][PF6]
and cis,fac-[Ru{di-(2-pyridylmethyl)amine}(CO)2(MeCN)][PF6]2 due to the presence of a hydride trans-
to the central nitrogen atom.182
Figure 3.2: Molecular structure of 60 depicted with thermal ellipsoids at the 30% probability level.Hydrogen atoms on aromatic rings and methylene carbons have been omitted for clarity.
When 60 is reacted with one equivalent of potassium tert-butoxide, we expected rapid and se-
lective deprotonation of the central nitrogen atom at room temperature to afford RuH{fac-1,3-di-(2-
pyridylmethyl)amide}(PPh3)2. However, when monitoring the course of the reaction by NMR spec-
troscopy, we observed a complex mixture of products as indicated by multiple metal hydride, ligand,
and phosphorus signals. The mixture was allowed to stir for 24 hours at room temperature and these
coalesced into one major product. The 1H NMR spectrum of this product contained a hydride signal
coupled to two inequivalent cis- phosphines (-15.8 ppm, 2JHP = 29 and 25 Hz). When trying to assign
the proton signals in the 1H NMR spectrum, we noticed that one of the protons on the CNC back-
bone of the ligand, appearing as a broad multiplet, was shifted far downfield (at 5.1 ppm) compared
to three other protons, which have well-resolved couplings (at 3.5-3.1 ppm). This is reminiscent of the
NH proton in 60, which was also far downfield with respect to the methylene protons. Indeed, IR
analysis reveals an NH stretching mode at 3271 cm−1. Based on complete 1D/2D NMR spectral char-
acterization and a crystal structure, we determined the compound to be RuH{κ3C alkNN py-1,3-di-(2-

Chapter 3. Transformation of Di-(2-pyridylmethyl)amine on Ruthenium 32
pyridylmethyl)amine}(PPh3)2 which contains an anionic ruthenaaziridine moiety and a decoordinated
pyridine ring. (61, Scheme 3.2, Figure 3.3, Table 3.1 and Table 3.2). The methine hydrogen of the alkyl
in complex 61 appears at 3.1 ppm.
KOtBuRuN
H
PPh3
PPh3N
NH
+Cl-
THF24 h, 25 oC
RuN
PPh3
PPh3
HN
NH
H
60 61
Scheme 3.2: Synthesis of 61.
Figure 3.3: Molecular structure of 61 depicted with thermal ellipsoids at the 30% probability level.Hydrogen atoms on the aromatic rings have been omitted for clarity.
Crystals of 61 were obtained via diffusion of pentane into a concentrated solution of benzene. Se-
lected bond lengths and angles are given in Table 3.1. The complex is severely distorted from an ideal
octahedron with large P1-Ru-C7 and P2-Ru-N2 angles of 103.5(1)◦ and 113.19(8)◦, respectively, and
a very acute N2-Ru-C7 angle of 39.9(1)◦. The hydride ligand is trans- to the coordinated pyridine
arm. The N2-C7 bond length is 1.452(4) A, which is indicative of a sp3-hybridized carbon atom and
is comparable with the slightly longer N2-C7 distance of 1.487(8) A in 60. The only other two other
crystallographically defined compounds that contain the ruthenaaziridine motif are prepared by react-
ing the carbonyl cluster Ru3(CO)12 with 1,4-diazabutadienes (α-diimines).189,190 The products in these
cases were dimers while 61 remains monomeric, possibly due to the steric bulk of the phosphines.
3.2.2 Synthesis of a Ruthenium Azaallyl Complex.
In the preparation of complex 61 by reaction of base with complex 60 at room temperature, a deep
emerald green solid also forms in low yield as a byproduct. Its presence is signaled by a hydride triplet

Chapter 3. Transformation of Di-(2-pyridylmethyl)amine on Ruthenium 33
at -8.6 ppm (2JHP = 29 Hz) in crude samples of 61. When ruthenium salt 60 is heated in THF with
potassium tert-butoxide, an intensely coloured emerald green solution formed, which we thought to be
(RuH{fac-1,3-di-(pyridylmethyl)amide}(PPh3)2). The NMR spectrum showed that 61 was replaced by
a highly symmetrical molecule containing a triplet at -8.6 ppm (2JHP = 29 Hz) and a singlet phosphorus
peak (proton decoupled) at 50.9 ppm appeared. Surprisingly, it was identified by use of single crystal
X-ray diffraction to be a rare azaallyl complex191–195 with trans-PPh3 ligands (Scheme 3.3, Figure 3.4).
Selected metrical parameters for 62 are presented in Table 3.1. The bite angle between the pyridine
nitrogens is 157.96(3)◦, while the central Ru-N bond is slightly shorter (Ru-N2 = 2.055(2) A) than the
pyridine Ru-N bonds (Ru-N1 = 2.107(3), Ru-N3 = 2.113(2) A). The azaallyl N-C bonds are identical
(N2-C6 = 1.335(4) A, N2-C7 = 1.334(4) A), which suggests delocalization of negative charge about all
three atoms. These bond lengths and angles are similar to a previously reported iron 1,3-di-(2-pyridyl)-
2-azallyl complex.193
The symmetrical structure of 62 explains the triplet pattern of the hydride resonance in the 1H
NMR spectrum and the singlet phosphorus peak. Not only was the central amine proton removed
from di-(2-pyridylmethyl)amine, but a proton and hydride equivalent were also eliminated to form the
tridentate anionic azaallyl ligand. The trans-arrangement of the PPh3 ligands suggests that some ligand
rearrangement must have occurred during the formation of 62, possibly involving phosphine or pyridine
arm dissociation.
KOtBuRu
N
H
PPh3
PPh3N
NH
+Cl-
RuN
PPh3
PPh3
HN
NH
∆
-H2Ru
N N
H
PPh3
N
PPh3
H
-KCl-HOtBu
H
H
60 61 62
Scheme 3.3: Summary of the reactivity of complexes 60 and 61.
Initially discovered as a degradation product attached to zinc,192 the azaallyl ligand has attracted
attention more recently due to the intense colours it imparts on a variety of 3d metal precursors. Wol-
czanski and co-workers have synthesized similar complexes by reacting the lithium dipyridylazaallyl salt
with a variety of metals (Scheme 3.4).193,196 These molecules have two intraligand transitions193 in the
visible region with extinction coefficients ranging from 20000 – 50000 M−1cm−1. Azaallyl compound 62
also has two intraligand UV-vis absorption bands, with broad absorbances centered at 697 nm (ε = 8500
M−1cm−1) and 405 nm (ε = 32000 M−1cm−1).
NNN
[M] = FeBr2(THF)2, CoCl2, NiCl2(dme) HH
NNN
HH
M[M] NNN
HH
- Li+
+
Scheme 3.4: Azaallyl complexes prepared by Wolczanski and co-workers.

Chapter 3. Transformation of Di-(2-pyridylmethyl)amine on Ruthenium 34
There is a minor byproduct that forms with 62 which contains a hydride triplet 0.1 ppm upfield and
a phosphorus singlet 2 ppm downfield from the signals in 62, respectively. We were unable to identify or
isolate this electronically similar product, but its presence can be minimized if two equivalents of base
are used to synthesize complex 62 instead of one.∗
The azaallyl complex 62 can also be synthesized directly from 61 by refluxing in THF with no added
base (Scheme 3.3). This shows that 61 is indeed a reaction intermediate. However, the azaallyl complex
62 can be synthesized in higher yields and greater purity if two equivalents of base are used when starting
from 60 (vide supra). Interestingly, the amine proton and hydride of 61 are cis- to one another in the
solid state (Figure 3.3), which suggests that H2 loss occurs in a bifunctional manner.37
Figure 3.4: Molecular structure of 62 depicted with thermal ellipsoids at the 30% probability level.Hydrogen atoms on the phenyl rings have been omitted for clarity.
∗The electronically similar product may be a dimer. Wolczanski and co-workers found that one of their monomericiron(II) azaallyl complexes was in equilibrium with a dimerized product connected through the azaallyl carbons. Seereference 193.

Chapter 3. Transformation of Di-(2-pyridylmethyl)amine on Ruthenium 35
Table 3.1: Selected bond lengths (A) and angles (◦) for complexes 60, 61, and 62.
Parameter 60 61 62
Ru-H1 1.74(7) 1.63(3) 1.61(3)
Ru-N1 2.128(6) 2.175(3) 2.107(3)
Ru-N2 2.234(5) 2.131(3) 2.055(2)
Ru-N3 2.111(6) - 2.113(2)
Ru-C7 - 2.121(3) -
Ru-P1 2.293(2) 2.244(1) 2.3002(7)
Ru-P2 2.284(2) 2.276(1) 2.3081(7)
N2-C6 1.492(8) 1.463(5) 1.335(4)
N2-C7 1.487(8) 1.452(4) 1.334(4)
N1-Ru-N2 77.4(2) 76.7(1) 78.8(1)
N1-Ru-N3 83.7(2) - 157.1(1)
N2-Ru-N3 77.0(2) - 78.5(1)
P1-Ru-P2 95.70(7) 103.37(4) 157.96(3)
P2-Ru-N1 172.1(2) 94.39(7) 88.24(7)
P1-Ru-N3 172.6(2) - 90.84(7)
3.3 Conclusion
We have shown that a simple ligand precursor, di-(2-methylpyridyl)amine, can be manipulated into
adopting three different coordination modes. Firstly, the salt
[RuH{κ3N-fac-1,3-di-(2-pyridylmethyl)amine}(PPh3)2]Cl is synthesized where it binds as a neutral tri-
dentate ligand. Upon addition of base, one of the alkyl carbons becomes an anionic two-electron donor
and an unusual ruthenaaziridine complex (61) is formed. Finally, refluxing 60 with base or 61 with-
out base leads to the novel ruthenium hydrido azaallyl complex 62 with concomitant H2 production.
This deprotonation/dehydrogenation reaction pathway is unusual, and mechanistic studies are currently
under way.
3.4 Experimental
3.4.1 General Comments
All experiments were carried out under argon using standard Schlenk and glove box techniques. Tetrahy-
drofuran (THF) was distilled over Na/benzophenone prior to use. RuHCl(PPh3)3 was prepared accord-
ing to a known literature procedure.197 Di-(2-picolyl)amine, sublimed potassium tert-butoxide, and
methanol-d4 (ampules) were used as received (Aldrich). Air and moisture free benzene-d6 was prepared
by stirring with Na/benzophenone for 2 days, followed by three freeze-pump-thaw cycles and vacuum
transfer. All NMR spectroscopic data were collected using a Varian 400 MHz or a Bruker 400 MHz
NMR system operating at 400 MHz for 1H, 101 MHz for 13C, or 162 MHz for 31P. All 1H and 13C
NMR samples were referenced to their respective residual solvent peaks. Spectroscopic data for 31P
NMR were referenced to an external standard of 85% aqueous H3PO4 at δ = 0.00. Single-crystal X-ray
diffraction data were collected at 150 K using a Nonius Kappa-CCD diffractometer with Mo Kα radia-
tion (λ = 0.71073 A). The CCD data were integrated and scaled using the Denzo-SMN package.146 The
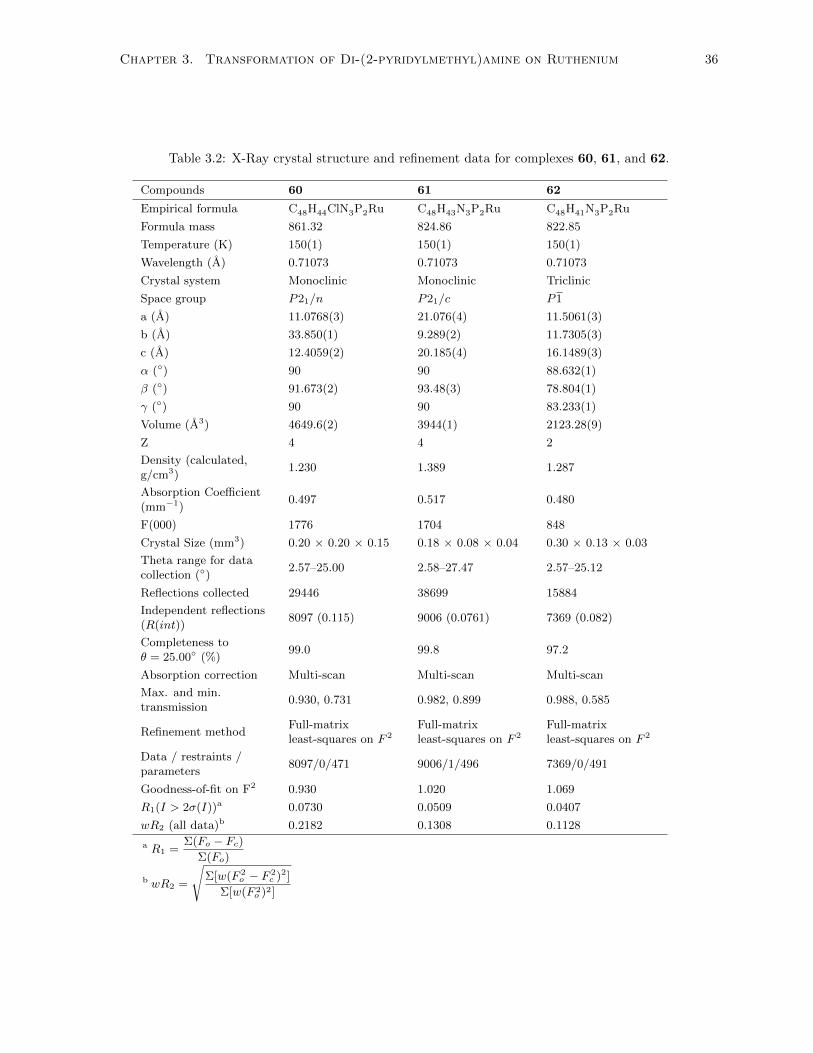
Chapter 3. Transformation of Di-(2-pyridylmethyl)amine on Ruthenium 36
Table 3.2: X-Ray crystal structure and refinement data for complexes 60, 61, and 62.
Compounds 60 61 62
Empirical formula C48H44ClN3P2Ru C48H43N3P2Ru C48H41N3P2Ru
Formula mass 861.32 824.86 822.85
Temperature (K) 150(1) 150(1) 150(1)
Wavelength (A) 0.71073 0.71073 0.71073
Crystal system Monoclinic Monoclinic Triclinic
Space group P21/n P21/c P1
a (A) 11.0768(3) 21.076(4) 11.5061(3)
b (A) 33.850(1) 9.289(2) 11.7305(3)
c (A) 12.4059(2) 20.185(4) 16.1489(3)
α (◦) 90 90 88.632(1)
β (◦) 91.673(2) 93.48(3) 78.804(1)
γ (◦) 90 90 83.233(1)
Volume (A3) 4649.6(2) 3944(1) 2123.28(9)
Z 4 4 2
Density (calculated,g/cm3)
1.230 1.389 1.287
Absorption Coefficient(mm−1)
0.497 0.517 0.480
F(000) 1776 1704 848
Crystal Size (mm3) 0.20 × 0.20 × 0.15 0.18 × 0.08 × 0.04 0.30 × 0.13 × 0.03
Theta range for datacollection (◦)
2.57–25.00 2.58–27.47 2.57–25.12
Reflections collected 29446 38699 15884
Independent reflections(R(int))
8097 (0.115) 9006 (0.0761) 7369 (0.082)
Completeness toθ = 25.00◦ (%)
99.0 99.8 97.2
Absorption correction Multi-scan Multi-scan Multi-scan
Max. and min.transmission
0.930, 0.731 0.982, 0.899 0.988, 0.585
Refinement methodFull-matrixleast-squares on F 2
Full-matrixleast-squares on F 2
Full-matrixleast-squares on F 2
Data / restraints /parameters
8097/0/471 9006/1/496 7369/0/491
Goodness-of-fit on F2 0.930 1.020 1.069
R1(I > 2σ(I))a 0.0730 0.0509 0.0407
wR2 (all data)b 0.2182 0.1308 0.1128
a R1 =Σ(Fo − Fc)
Σ(Fo)
b wR2 =
√Σ[w(F 2
o − F 2c )2]
Σ[w(F 2o )2]

Chapter 3. Transformation of Di-(2-pyridylmethyl)amine on Ruthenium 37
structures were solved and refined using SHELXTL V6.1.147 Refinement was by full-matrix least-squares
on F 2 using all data. All IR spectra were prepared as KBr pellets and carried out on a Perkin-Elmer
Spectrum One FT-IR spectrometer. All UV-vis spectra were recorded on a Hewlett-Packard Agilent
8453 UV-vis spectrophotometer. Elemental Analyses were performed on a Perkin-Elmer 2400 CHN
elemental analyzer. For compounds 61 and 62, elemental analyses were attempted several times and
consistently produced low carbon percentages despite being pure by all other spectroscopic means.
3.4.2 [RuH{κ3N-fac-1,3-di-(2-pyridylmethyl)amine}(PPh3)2]Cl (60).
A suspension of RuHCl(PPh3)3 (1.00 g, 1.08 mmol) in THF (15 mL) was placed in a glass vial charged
with a teflon-coated stir bar. Di-(2-pyridylmethyl)amine (237 mg, 1.19 mmol) was added and after 10
minutes of stirring the solution became cloudy yellow. This was immediately filtered over a medium-pore
glass frit and washed with THF (2 × 1 mL). The yellow precipitate on the frit was washed through with
methanol (10 mL) and the yellow filtrate was collected. Excess solvent was removed in vacuo to afford
a bright yellow microcrystalline solid (857 mg, 92%). Crystals suitable for X-ray diffraction were grown
via slow diffusion of diethyl ether into a concentrated methanol solution at 25 ◦C. EA: Found C 66.4,
H 5.3, N 4.9. Calc. for C48H44N3P2ClRu: C 66.9, H 5.15, N 4.9%. UV-vis: λmax(CH3CN)/nm 345
(ε/M−1cm−1 14000). IR: νmax/cm−1 3273w (NH), 1956s (RuH). 1H NMR (CD3OD): δ 8.67 (2H, d,3J = 6 Hz, 6-pyH), 7.33-7.23 (14H, m, 4-pyH and C6H5), 7.09 (6H, m, C6H5), 6.95 (12H, m, C6H5),
6.89 (2H, d, 3J = 8 Hz, 3-pyH), 6.61 (2H, m, 5-pyH), 6.16 (1H, m, NH), 3.83 (2H, d, 2J = 16 Hz,
CH2), 3.64 (2H, dd, 2J = 16 Hz, CH2), -14.66 (1H, t, 2JHP = 29 Hz, RuH). 13C {1H} NMR: δ 161.55
(2-pyC), 156.61 (6-pyC), 138-139 (C6H5) 137.24 (4-pyC), 135.09 (C6H5), 129.76 (C6H5), 128.56 (C6H5),
124.88 (3-pyC), 122.89 (5-pyC), 63.06 (CH2). 31P {1H} NMR: δ 67.40 (s, RuP). MS (ESI): Found: m/z
826.1961. Calc. for [C48H44N3P2Ru]+
826.2048.
3.4.3 RuH{κ3C alkNN py-1,3-di-(2-pyridylmethyl)amine}(PPh3)2 (61).
A suspension of 60 (28 mg, 0.033 mmol) in tetrahydrofuran (2 mL) was placed in a glass vial charged
with a teflon-coated stir bar. Potassium tert-butoxide (4 mg, 0.033 mmol, 0.085 M in THF) was added
(excess base is not detrimental). The solution darkened immediately after addition of base and was
stirred for 24 hours at room temperature, during which the colour changed to green-brown. This green-
brown solution was filtered through Celite, dried in vacuo, then redissolved in hexanes (3 mL) and stirred
overnight. Finally, the solution was filtered over a medium-pore glass frit and the solid was washed with
hexanes (about 3 mL) until the outgoing filtrate was colourless. The remaining solid was dried in vacuo
to afford a yellow-brown powder (14 mg, 52%). Crystals suitable for X-ray diffraction were grown via
slow diffusion of pentane into a concentrated benzene solution at 25 ◦C. EA: Found: 63.7, 5.1, 4.9. Calc.
for C48H43N3P2Ru: C 69.9, H 5.25, N 5.1%. IR: νmax/cm−1 3271w (NH), 1953s (RuH). 1H NMR (C6D6,
coord = coordinated, uncoord = uncoordinated): δ 8.50 (1H, d, 2J = 5 Hz, 6-pyHuncoord), 7.80 (1H, d,2J = 5 Hz, 6-pyHcoord), 7.55 (12H, m, C6H5), 7.02-6.94 (18H, m, C6H5), 6.87 (1H, m, 4-pyHuncoord),
6.58 (1H, m, 5-pyHuncoord), 6.56 (1H, m, 4-pyHcoord), 6.11 (1H, d, 2J = 7 Hz, 3-pyHcoord), 5.94 (1H,
d, 2J = 7 Hz, 3-pyHuncoord), 5.86 (1H, m, 5-pyHcoord), 5.10 (1H, m, NH), 3.42 (1H, dd, 2J = 16 Hz,
CH2), 3.26 (1H, dd, 2J = 16 Hz, CH2), 3.11 (1H, m, CHNH), -15.81 (1H, dd, 2JHP = 29 Hz, 2JHP = 25
Hz, RuH). 13C {1H} NMR: δ 169.65 (2-pyCuncoord), 158.52 (2-pyCcoord), 154.41 (6-pyCcoord), 149.26 (6-
pyCuncoord), 143.86-142.71 (C6H5), 134.91 (4-pyCuncoord), 134.64-133.86 (C6H5), 131.57 (4-pyCcoord),

Chapter 3. Transformation of Di-(2-pyridylmethyl)amine on Ruthenium 38
127.84 (C6H5), 121.97 (5-pyCcoord), 120.56 (3-pyCcoord), 119.13 (3-pyCuncoord), 116.27 (5-pyCuncoord),
59.54 (CHNH), 56.10 (CH2). 31P {1H} NMR: δ 77.39 (m, RuP), 61.73 (m, RuP). MS (DART): Found:
m/z 824.2. Calc. for [C48H40N3P2Ru]+
(loss of H–): 824.2.
3.4.4 RuH(κ3N -1,3-di-(2-pyridyl)-2-azaallyl)(PPh3)2 (62).
A suspension of 60 (28 mg, 0.033 mmol) in THF (2 mL) was placed in a Schlenk flask charged with a
teflon-coated stir bar. Potassium tert-butoxide (8 mg, 0.067 mmol, 0.085 M in THF) was added. The
solution turned dark immediately after addition of base and was heated at 60 ◦C overnight. The resulting
deep emerald green solution was filtered through Celite and dried in vacuo. Pentane was added (1 mL),
the solution was stirred for 1 hour, then filtered over a medium-pore glass frit (the product is soluble
in pentane so it must be used sparingly). Solvent was removed in vacuo to afford a dark green powder
(20 mg 75%). Crystals suitable for X-ray diffraction were grown via slow diffusion of pentanes into a
concentrated ethereal solution at -30 ◦C. EA: Found: C 68.0, H 5.2, N 5.1. Calc. for C48H41N3P2Ru:
C 70.1, H 5.0, N 5.1%. UV-vis: λmax(C6H6)/nm 697 (ε/M−1cm−1 8500), 405 (32000). IR: νmax/cm−1
1818m (RuH). 1H NMR (C6D6): δ 7.87-7.83 (12H, m, C6H5), 7.07-6.99 (18H, m, C6H5), 6.39 (2H, d,2J = 6 Hz, 6-pyH), 6.38 (2H, s, CHazaallyl), 6.25 (2H, m, 4-pyH), 5.88 (2H, d, 2J = 7 Hz, 3-pyH), 5.16
(2H, m, 5-pyH), -8.59 (1H, t, 2JHP = 29 Hz, RuH). 13C {1H} NMR: δ 166.89 (2-pyC), 156.53 (6-pyC),
137.07 (C6H5), 134.56 (C6H5), 128.75-128.00 (C6H5 buried under solvent), 115.58 (Cazaallyl), 113.82
(5-pyC), 113.67 (3-pyC). 31P {1H} NMR: δ 50.94 (s, RuP). MS (DART): Found: m/z 822.2. Calc. for
[C48H40N3P2Ru]+
(loss of H–): 822.2.

Chapter 4
Intramolecular C-H/O-H Bond
Cleavage With Water and Alcohol
Using a Phosphine-Free Ruthenium
Carbene NCN Pincer Complex
Abstract
Transition metal complexes that exhibit metal-ligand cooperative (MLC) reactivity could
be suitable candidates for applications in water splitting. Ideally, the ligands around the
metal should not contain oxidizable donor atoms, such as phosphines. With this goal in
mind, we report new phosphine-free ruthenium NCN pincer complexes with a central N-
heterocyclic carbene donor and methylpyridyl N-donors. Reaction with base generates a
neutral, dearomatized alkoxo-amido complex, which has been structurally and spectroscopi-
cally characterized. The tert-butoxide ligand facilitates regioselective, intramolecular proton
transfer through a C–H/O–H bond cleavage process occurring at room temperature. Kinetic
and thermodynamic data have been obtained by VT NMR experiments; DFT calculations
support the observed behavior. Isolation and structural characterization of a doubly dearom-
atized phosphine complex also strongly supports our mechanistic proposal. The alkoxo-amido
complex reacts with water to form a dearomatized ruthenium hydroxide complex, a first step
towards phosphine-free MLC water splitting.
4.1 Introduction
Metal-ligand cooperation (MLC) can occur in transition metal complexes containing a Lewis basic lig-
and site that works together with a Lewis acidic metal center to cleave chemical bonds. In recent
years, several groups have isolated and characterized reactive transition metal species that highlight
the utility and importance of this design principle for applications in small molecule activation and
catalysis.6,25,27,28,84,123 In particular, PNN pincer ligand systems incorporating a functionalized pyri-
39

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 40
dine ring in the central ligating position are active catalysts for a variety of chemical transformations
(Scheme 4.1).21,24,74,198,199 Active catalysts are generally prepared and/or isolated by deprotonation
with a bulky external base, which dearomatizes the methylpyridyl fragment of the pincer ligand. With
reactions involving alcohols, a key proposed step is O–H splitting to generate a neutral alkoxide com-
plex with subsequent rearomatization of the heterocycle.114,116,117,200–205 Other systems replacing the
phosphine ligand arm with an N-heterocyclic carbene have also been reported.76,77,206
N
PR2
Ru
HH
-HB, -Cl-
L'
COH
Cl
N
PR2
Ru
H
L'
COH
N
PR2
Ru
HH
L'
COH
RO
+ROH
R = H, alkyl
-ROH
B-
Scheme 4.1: General deprotonation behavior and O–H activation using pyridine-centered Ru-PNL pincercomplexes, where L = N or P with the curved line representing various linkers.
Understanding the mechanism of MLC O–H splitting, especially with water, is relevant to renew-
able energy applications and the environment.87–89,109,110,113,207–211 In 2009, Milstein and co-workers
reported a novel series of reactions in which a structurally characterized [RuH(OH)(PNN)(CO)] pincer
complex split water into H2 and O2 in two discrete steps using only heat and light (PNN = 2-(di-tert-
butylphosphinomethyl-6-diethylaminomethyl)pyridine).111,112,212 This is a first step forward in designing
well-defined systems to catalytically generate H2 and O2 in a clean, atom-efficient manner.
Ideally, suitable catalyst candidates for water splitting do not contain oxidizable donor atoms, such
as phosphines, that might readily react with H2O139,213 or O2214–217 (2 H2O→2 H2 +O2; Erxn = 1.23 V
vs. SHE). Although the standard oxidation potential of pyridine is +1.6 V (versus 1 M Ag/AgNO3),218
pyridine-containing chelates are widely used for the catalytic oxidation of H2O to O2 by using a sacrificial
reductant.98–101,219 Furthermore, research into the oxidation of N-heterocyclic carbenes (NHCs) is lim-
ited, with a few known examples involving free NHCs220–222 and some NHC ligands bound to Cu223–225
and Rh226 capable of being oxidized in the presence of O2. In our efforts to design a phosphine-free
MLC system that could be suitable for water splitting, we present a carbene-centered NCN pincer ligand
platform that undergoes regioselective, intramolecular C–H/O–H proton exchange at room temperature
by aromatization/dearomatization of the two outer pyridine rings. This is facilitated by a coordinated
tert-butoxide ion, demonstrating that the bulky alkoxide base plays an important role in the coordina-
tion sphere of the metal by acting as an “intraligand” proton shuttle, which has not been previously
observed in other MLC systems. A rare dearomatized ruthenium tert-butoxide complex has been spec-
troscopically and structurally characterized, while NMR kinetic studies and DFT calculations strongly
support the proposed proton shuttle mechanism. A phosphine trapping experiment further validates
our mechanistic proposal, allowing us to structurally characterize the first example of a neutral, doubly
dearomatized Ru-NCN pincer complex. Initial experiments show that the alkoxo-amido complex reacts
with water to form a dearomatized ruthenium hydroxide complex, a first step towards designing new
MLC systems for water splitting.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 41
4.2 Results and Discussion
The synthesis begins by preparing the imidazolium salt 1,3- di(2-methylpyridyl)-4,5-diphenylimidazolium
bromide (64; Scheme 4.2) in two steps using commercially available reagents.227,228 Next, 63 is treated
with silver oxide in the presence of activated 3 A molecular sieves to generate the silver carbene transfer
agent 64 in good yield. It is important to note that the yield of 2 is low and/or inconsistent if 3 A molec-
ular sieves are omitted or insufficiently dried. Water released during the reaction with silver oxide deacti-
vates the imidazolium salt and dry solvent in combination with activated molecular sieves must be used.
The monosubstituted carbene configuration of 64, as opposed to being a bis(carbene) silver salt,229–231
was confirmed by elemental analysis. Installation of the NCN pincer ligand onto ruthenium was achieved
by transmetalation from 64 using [RuCl2(NBD)]n (NBD = bicyclo[2.2.1]hepta-2,5-diene), followed by
salt metathesis using sodium or potassium hexafluorophosphate to obtain the tris-acetonitrile complex
[Ru(NCN)(NCCH3)3][PF6]2 (65). Two of the acetonitrile ligands are easily displaced by refluxing in the
presence of tBu2bpy (4,4-di-tert-butyl-2,2-dipyridyl) to produce [Ru(NCN)(tBu2bpy)(NCCH3)][PF6]2
(66). To remove the third acetonitrile ligand, heating 66 in the presence of excess potassium bromide
followed by alumina column purification yields the deep red-orange solid [RuBr(NCN)(tBu2bpy)][PF6]
(67). Complex 67 is deeply coloured and absorbs strongly between 300 and 700 nm, with broad signals
centered at 444 nm (ε = 7000m−1cm−1) and 352 nm (ε = 10000m−1cm−1). The syntheses of 64-67 are
performed under air-free and moisture-free conditions, but the final products are conveniently stable in
air/moisture. Using a suite of 1D and 2D NMR spectroscopic techniques, complexes 63-67 were fully
characterized in solution. In particular, 66 and 67 exhibit characteristic 1H NOESY signals, as indicated
in Scheme 4.2. The molecular structures of 63 and 65 have also been obtained by single crystal X-ray
diffraction (see the Supporting Information).232
NN
N
NRu
N
N
Br
+
PF6-
NN
N
NRu
N
N
N
2+
2PF6-
NN
N
NRu
N
N
N
2+
2PF6-
N N
N NAg
Br
N N
N N
+
Br-
i
iii
iv
ii
H
H
H
H
H
H1H NOESY1H NOESY
63 64 65
67 66
Scheme 4.2: Synthesis of compounds 63-67. Conditions: Step i) Ag2O, CHCl3, 3 A sieves, 89 %; step ii)[RuCl2(NBD)]n, CH3CN/CHCl3, 60 ◦C, then MPF6 (M = Na, K), 60 %; step iii) tBu2bpy, (CH3)2CO,reflux, 89 %; step iv) KBr, (CH3)2SO, 110 ◦C, 89 %.
Next, complex 67 was treated with potassium tert-butoxide under an argon atmosphere to determine

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 42
if deprotonation at the methylene position was possible, resulting in dearomatization of the pyridine ring.
The addition of two equivalents of potassium tert-butoxide to a suspension of deep red-orange 67 in
THF generates a deep maroon-purple solution in high yield (95 %) that is sensitive to air and moisture.
Crystals suitable for X-ray diffraction revealed the compound to be the neutral, dearomatized, alkoxo-
amido complex Ru(OtBu)(NCN*)(
tBu2bpy) (68; Scheme 4.3 and Figure 4.1). Similar to 67, complex
68 is also deeply coloured, possessing broad and intense absorbance signals at 487 nm (ε = 7000 M−1
cm−1) and 378 nm (ε = 10000m−1 cm−1).
NN
N
NRu
N
N
O HH
HNN
N
NRu
N
N
Br
+
PF6-
2KOtBu
THF
quantitative by 1H NMR
-HOtBu
-KBr, -KPF6
67 68
Scheme 4.3: Synthesis of 68.
The molecular structure of 68 has some notable features. A tert-butoxide ligand occupies the apical
position, possessing an unusually long Ru–O bond length of 2.111(2) A.233–235 Tert-butoxide bound
ruthenium complexes are rare and have only been structurally characterized in distorted tetrahedral
complexes containing one or more phosphine ligands.233–235 One of the methylene protons has indeed
been deprotonated, leading to dearomatization of the pyridine moiety and shortening of bonds C22–
C23 (1.377(4) A), C24–25 (1.348(4) A), and C26–27 (1.357(4) A) (Figure 4.1). Finally, there is a strong
interaction between the oxygen atom and a methylene hydrogen (O1· · ·H16A = 2.341 A), situated about
0.4 A closer together than the sum of their van der Waals radii.
Figure 4.1: Molecular structure of 68 with ellipsoids at the 30 % probability level. Phenyl and methyl hy-drogens have been omitted for clarity. Distances (A) and angles (◦): Ru1–O1 2.111(2), Ru1–C1 1.955(3),Ru1–N3 2.101(2), Ru1–N4 2.116(2), Ru1–N5 2.041(2), Ru1–N6 2.102(2), C22–C23 1.377(4), O1· · ·H16A2.341; N5-Ru1-N6 78.04(8), N3-Ru1-C1 87.13(10), N4-Ru-C1 88.92(10), O1-Ru1-C1 93.30(10).

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 43
Complex 68 was also analyzed in detail by solution-state NMR spectroscopy. At 25 ◦C in THF-d8,1H NMR signals belonging to the pincer ligand are broad whereas the tBu2bpy proton resonances are
sharp. Cooling the sample to 0 ◦C sharpens and resolves the pincer ligand signals, allowing for complete
1D and 2D NMR characterization of 68 (see the Supporting Information).232 Diagnostic 1H NMR signals
include the vinylic CH singlet at 5.06 ppm (attached to C22; Figure 4.1) corresponding to the exocyclic
proton of the pincer ligand, and the diastereotopic methylene doublets appearing at 4.74 and 6.47 ppm
(attached to C16; Figure 4.1).
The 1H−1H NOESY pulse sequence at 0 ◦C reveals unique chemical exchange (EXSY) cross peaks,
whereby each proton on the pincer ligand framework is undergoing two-site chemical exchange; a 1H−1H
ROESY experiment confirms that the cross-peaks in phase with the diagonal were indeed due to chemical
exchange (Figure 4.2). The observed two-site chemical exchange is due to regioselective C–H/O–H
hydrogen transfer, where the tert-butoxide ligand acts as a proton shuttle between both sides of the
pincer ligand (Scheme 4.4). The absence of an EXSY cross peak for the CH2 doublet at 6.47 ppm
suggests that this is the proton undergoing intramolecular exchange (Scheme 4.4, red H atom). This
process could occur through a doubly deprotonated/dearomatized intermediate 69, which we were unable
to detect at elevated temperatures.∗ Instead, a phosphine trapping experiment and DFT calculations
were performed (see below).
Figure 4.2: 1H−1H ROESY spectrum (THF-d8, 0 ◦C, 600 ms mixing time, 1 s relaxation delay) of68. The coloured asterisks (matched with Scheme 4.4) indicate the protons on the pincer frameworkundergoing two-site chemical exchange. The exchange cross peak at δ = (7.3, 7.4) belongs to the phenylrings attached to the NHC.
We conducted variable temperature (VT) 1H NMR spectroscopy in THF-d8 (0-50 ◦C) and observed
selective broadening of protons on the pincer ligand while all the signals on tBu2bpy remained sharp (see
the Supporting Information).232 VT-NMR experiments of the crude reaction mixture in THF-d8, which
contains free tert-butanol and potassium tert-butoxide, reveals nearly identical proton chemical shifts
∗We were unable to observe any peak coalescence at elevated temperatures due to product loss above 70 ◦C in toluene-d8.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 44
NN
N
NRu
N
N
O
H
H
HN
N
N
NRu
N
N
O HH
H NN
N
NRu
N
N
OH H
H
****
*
****
* ** *
* ** *
*
* *
PPh3
NN
N
NRu
N
N
PPh3
H
H
-tBuOH
> 2KOtBu
PPh3
68 69 68′
67
70
Scheme 4.4: Top: intramolecular proton exchange behavior of 68 in solution with proposed intermediate69; the colour-matched asterisks indicate the protons on the pincer framework undergoing two-sitechemical exchange while the red proton is being transferred. Bottom: trapping experiments with PPh3,generating doubly dearomatized complex 70.
and peak broadening behavior when compared to a pure sample of 68, suggesting that proton transfer
is intramolecular. To support the presence of doubly deprotonated intermediate 69, we performed a
phosphine trapping experiment either by addition of PPh3 to 68, or addition of PPh3 to 67 with excess
base (Scheme 4.4). A new deep orange compound forms with a characteristic 31P NMR singlet at 59.5
ppm and a vinylic 1H NMR singlet at 4.53 ppm. NMR spectroscopic and X-ray crystallographic char-
acterization unambiguously confirm that this complex is indeed the doubly deprotonated/dearomatized
complex Ru(NCN**)(PPh3)(tBu2bpy) (70; Figure 4.3).
According to the NMR spectroscopic data, 70 is Cs symmetric in solution, however the molecular
structure reveals that one of the pincer arms is tilted significantly out of plane with respect to the
N-heterocyclic carbene, with a dihedral angle of C1A-N2A-C22A-C23A = -23.468 (Figure 4.3). The
bond lengths of each pincer ligand arm of 70 resemble that of the monodearomatized portion of 68.
Double deprotonation of related Re,236 Rh,237 Pd/Pt,238 and Ni239 PNP pincer systems with a central
2,6-dimethylpyridyl moiety have been described elsewhere; to our knowledge, this is the first reported
pincer complex that contains two independently dearomatized pyridine rings.
The regiospecific chemical exchange of 68 was modeled using dynamic NMR (DNMR) simulations
(Figure 4.4). The CH2 protons and CH proton on the pincer ligand system, attached to carbons C16 and
C22 (Figure 4.1), were modeled as a two-site ABC↔BAC mutual exchange system using the DNMR3240
simulation package.241 Excellent line fitting was achieved, allowing us to use the rate constants to obtain
activation parameters by Eyring plot analysis (Table 4.1 and Figure 4.5). The small entropy contribution
(∆S‡ = −1.5 ± 1.7 cal·mol−1K−1), is consistent with this being an intramolecular chemical exchange
process.242,243
Density functional theory (DFT) calculations were per formed to determine if the energy barriers

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 45
Figure 4.3: Molecular structure of 70 with ellipsoids at the 30 % probability level. Only one of the twocrystallographically independent complexes is shown. Hydrogens have been omitted for clarity. Distances(A) and angles (◦): Ru1A–P1A 2.3187(14), Ru1A–C1A 1.966(7), Ru1A–N3A 2.121(5), Ru1A–N4A2.131(5), Ru1A–N5A 2.104(4), Ru1A–N6A 2.155(6), C16A–C17A 1.366(10), C22A–C23A 1.348(10);N5A-Ru1A-N6A 76.58(19), N3A-Ru1A-C1A 90.3(2), N4A-RuA-C1A 87.1(2), C1A-N1A-C16A-C17A -3.98, C1A-N2A-C22A- C23A -23.46.
Figure 4.4: Selected chemical shift regions of experimental 1H NMR spectra of 68 (left, THF-d8, 600MHz) at various temperatures and simulated NMR spectra (right) with rate constants (s−1). Arrowsindicate the exchange peaks of interest that were modeled in the simulation.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 46
between 68 and the proposed doubly dearomatized complex 69 corresponded with our experimental
observations. We chose to use the M06-L244 DFT functional combined with the TZVP245/TZVPFit
basis sets on all atoms (except QZVP246 for Ru) since accurate thermochemical parameters were re-
cently obtained for other organometallic reactions.247 Ground-state structures for the alkoxide (68) and
doubly dearomatized alcohol adduct (69) were found, along with a transition state structure (TS68,69)
(Figure 4.6). The calculated ground state free energy of 69 is 11.5 kcal·mol−1 higher in energy than 68,
close to that of TS68,69 (∆G‡ = 15.7 kcal·mol−1, see the Supporting Information).232
Table 4.1: Experimental and calculated activation parameters for intramolecular proton transfer(68→69).
Sample ∆H‡ (kcal·mol−1) ∆S‡ (cal·mol−1K−1) ∆G‡298 (kcal·mol−1)
68→69 (exptl) 14± 1 −1.5± 1.7 15± 1
68→69 (calcd) 15.8 0.2 15.7
y = -‐7241.5x + 21.888 R² = 0.99515
-‐5.0 -‐4.5 -‐4.0 -‐3.5 -‐3.0 -‐2.5 -‐2.0 -‐1.5 -‐1.0 -‐0.5 0.0
3.0E-‐03 3.1E-‐03 3.2E-‐03 3.3E-‐03 3.4E-‐03 3.5E-‐03 3.6E-‐03 3.7E-‐03
ln(k/T)
1/T (K-‐1)
Figure 4.5: Eyring Plot of Proton Transfer 68→69.
Figure 4.6: Transition state geometry of TS68,69 (ν = 1160i). Distances are in A.
When 68 is exposed to water, the deep maroon-purple solution turns deep maroon-orange, replac-
ing the tert-butoxide moiety with hydroxide to generate the compound Ru(OH)(NCN*)(tBu2bpy) (71;

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 47
Scheme 4.5). The NMR signals corresponding to 71 are similar to that of 68, revealing intramolecular
chemical exchange by 1H−1H ROESY and a broad Ru–OH signal at -2.4 ppm (0 ◦C) in the 1H spectrum
(see the Supporting Information).232 Reaction of 68 with D2O reveals deuterium incorporation at ex-
actly four sites, with the affected 1H NMR signals integrating to about 50 % of their original value. This
suggests that the substitution of tert-butanol for hydroxide is not stereoselective, whereas with other
pincer systems D2O splitting only deuterates one ligand site.111
NN
N
NRu
N
N
O HH
HTHF-d8
NN
N
NRu
N
N
O
D/H
D/HD/H
D/HH2O/D2O
-HOtBu
40 oC, 5 min
NN
N
NRu
N
N
O
D/H
D/H
D/H
D/HNN
N
NRu N
N
H
H
6868 7171′
Scheme 4.5: Reaction of 68 with H2O or D2O.
4.3 Conclusion
The dearomatized complexes 68 and 71 exhibit unprecedented intramolecular proton-transfer behavior.
This exchange may also be operative in other pincer-type systems in which alkoxide bases, alcohols, or
water are employed. VT-NMR spectroscopy, DNMR simulations, phosphine trapping experiments, and
DFT calculations of the proton shuttle mechanism of 68 are all are in excellent agreement with one
another. The deeply coloured nature of these compounds is encouraging for future use in photochemical
reactions. We believe that compounds 68 and 71 are promising entry points into well-defined systems
capable of water splitting, which is currently under investigation in our laboratory.
4.4 Experimental
4.4.1 General Comments
All reactions containing silver or ruthenium reagents were carried out in an argon atmosphere (5.0
grade, ultrahigh purity) using standard Schlenk and glove box techniques using air and moisture free
solvents/reagents unless otherwise stated. Di-µ-chloro-(η4-bicyclo[2.2.1]hepta-2,5-diene)ruthenium(II)
polymer was synthesized according to known literature procedures.248 Sublimed potassium tert-butoxide
was purchased from Aldrich and stored in an argon-filled glove box. Alumina (activated, neutral, Brock-
mann I) was purchased from Aldrich and used as received. Silver(I) oxide was purchased from Aldrich
and stored in an argon-filled glove box protected from light. Dichloromethane and acetonitrile were
refluxed over calcium hydride, distilled, and stored under argon. Acetone was refluxed over phosphorus
pentoxide, distilled, and stored under argon. Air and moisture free dimethylsulfoxide was purchased
from Aldrich and stored under argon. Pentane was refluxed over Na/benzophenone, distilled, and stored
under argon. Tetrahydrofuran was refluxed over sodium/benzophenone, distilled, and stored under ar-
gon with activated 3 A molecular sieves (20% w/v). Toluene was refluxed over sodium, distilled, and
stored under argon. Degassed H2O/D2O was prepared by pouring distilled/deionized water (or D2O

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 48
purchased from Cambridge Isotope Laboratories) into a Schlenk flask, applying high vacuum for about
one minute, and then sonicating for 5 minutes to remove dissolved gases. The vacuum/sonication steps
were repeated twice more after which the vessel was filled with argon. Molecular sieves were acti-
vated by heating at 300 ◦C under high vacuum for 2-3 days and were stored in an argon-filled glove
box. Acetonitrile-d3 and chloroform-d were purchased from Cambridge Isotope Laboratories and used
as received. Tetrahydrofuran-d8 and toluene-d8 were purchased in individually packaged ampoules from
Aldrich and dried in an argon-filled glove box over activated 3 A molecular sieves (20% w/v). All other
solvents/reagents were used as received without further purification. NMR spectra with compound
numbering schemes, X-ray crystal structure and refinement data, and DFT Cartesian coordinates are
provided in the Supporting Information.232
All NMR spectroscopic data were collected using an Agilent DD2 500 MHz spectrometer with an
Agilent HC 5 mm XSens cryogenically cooled probe (operating at 500 MHz for 1H, 126 MHz for 13C),
Agilent DD2 600 MHz spectrometer with an Agilent 5-mm OneNMR Probe (operating at 600 MHz
for 1H, 151 MHz for 13C, 243 MHz for 31P), or Bruker Avance III 400 spectrometer (operating at 400
MHz for 1H, 101 MHz for 13C). All 1H and 13C NMR samples were referenced to their respective
residual solvent peaks249 and data was collected at 298 K unless stated otherwise. Coupling constant
J values are given in Hz. Spectral data was processed using MNova 9.0. Variable temperature 1H
NMR analysis of 68 was collected at spectrometer frequency of 600 MHZ between 0 ◦C and 50 ◦C.
The probe temperature was calibrated using a 100% ethylene glycol standard250 in a flame sealed NMR
tube supplied by Cambridge Isotope Laboratories. VT airflow was set to 10 L/min and deviation from
desired temperatures was no more than 0.25 ◦C. Dynamic NMR simulations were performed using the
DNMR3240 utility in Spinworks 4.0.3.241 The protons in 6 labelled as H3b (A), H28 (B), and H3a (C)
were modeled as an ABC↔BAC spin system with J(A,B) = 0 Hz and J(A,C) = J(B,C) = 13.2 Hz.
Simulations were modelled at 600 MHz with a spectral window of 9615 Hz, lowest frequency of -2321.8
Hz, and 65536 points. Rate constants k were obtained by systematic trial and error, modifying rate
constant (k) and relaxation (T2) parameters at until a best fit was achieved for each experimental NMR
spectrum at a given temperature.
UV-vis spectra were recorded on a Hewlett-Packard Agilent 8453 UV-vis spectrophotometer. Elemen-
tal analyses were performed on a Perkin-Elmer 2400 or LECO TruSpec Micro CHN elemental analyzer.
Single-crystal X-ray diffraction data were collected at 150 K using a Nonius Kappa-CCD diffractometer
with Mo Kα radiation (λ = 0.71073 A). The CCD data were integrated and scaled using the Denzo-
SMN package.146 The structures were solved and refined using SHELXTL V6.1.147 Refinement was by
full-matrix least-squares on F 2 using all data.
All DFT calculations were performed using Gaussian 09.251 The M06-L244 density functional with
the TZVP245 (also known as def-TZVP) basis set with TZVPFit density fitting basis set for C, H, N, O
atoms was used and QZVP246 (also known as def2-QZVP) basis set was used for Ru. Normal convergence
criteria were used for all optimizations and a pruned (99,590) integration grid was used throughout
(grid=ultrafine). Optimizations were performed in tetrahydrofuran (THF) solvent using the integral
equation formalism polarizable continuum model (IEF-PCM)252,253 with radii and non-electrostatic
terms from the SMD254 solvation model (scrf=smd). Full vibrational and thermochemical analyses (1
atm, 298 K) were performed on optimized structures to obtain solvent-corrected free energies (G◦solv)
and enthalpies (H◦solv). Optimized ground states were found to have zero imaginary frequencies while
transition states had exactly one imaginary frequency. Three-dimensional visualizations of calculated

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 49
structures were generated by ChemCraft.255
4.4.2 1-(2-methylpyridyl)-4,5-diphenylimidazole.
This procedure has been adapted from a synthetic protocol for a similar compound.256
4,5-diphenylimidazole (31.6 mmol, 6.97 g), 2-(bromomethyl)pyridine hydrobromide (31.6 mmol, 8.00
g), KOH (127 mmol, 7.1 g), and a Teflon coated stir bar were added to a round bottom flask, followed
addition of tetrahydrofuran (250 mL). The solution was refluxed for 2 days. The solvent was removed,
followed by addition of dichloromethane (200 mL), and then the solution was washed with water (2
× 200 mL) in a separatory funnel. The organic layer was dried with magnesium sulfate, filtered, and
solvent was removed. The solid was redissolved in tetrahydrofuran (about 20 mL) and pentane was
added (600 mL) to precipitate a white solid. The product was collected on a medium-pore glass frit,
washed with pentane (30 mL), and dried under high vacuum (8.61 g, 87%). EA: Found: C 80.57, H
5.71, N 13.25. Calc. for C21H17N3: C 81.00, H 5.50, N 13.49%. 1H NMR (400 MHz; CDCl3): δ =
8.50 (m, 1H; H8), 7.73 (s, 1H; H1), 7.57 (m, 1H; H6), 7.50-7.47 (2H; Ph), 7.38-7.32 (3H; Ph), 7.22-7.10
(5H; Ph), 7.17 (m, 1H; H7 (buried under Ph)), 6.76 (m, 1H; H5), 5.09 (s, 2H; H3). 13C {1H} NMR:
δ = 156.39 (C4), 149.56 (C8), 138.35 (C9), 137.44 (C1), 136.95 (C6), 134.44-128.74 (Ph), 128.67 (C2),
128.08-126.34 (Ph), 122.66 (C7), 120.94 (C5), 50.33 (C3).
4.4.3 1,3-di(2-methylpyridyl)-4,5-imidazolium bromide (63).
1,3-di(2-methylpyridyl)-4,5-diphenylimidazole (27.6 mmol, 8.61 g), 2-(bromomethyl)pyridine hydrobro-
mide (27.6 mmol, 6.99 g), NaHCO3 (55 mmol, 4.6 g), and a Teflon-coated stir bar were added to a round
bottom flask, followed by addition of acetonitrile (600 mL). The solution was refluxed for 2 days. The
deep red solution was dried and redissolved in 50 mL dichloromethane. The solids were filtered off and
the filtrate was collected and dried under high vacuum. About 600 mL tetrahydrofuran was added and
the suspension was stirred vigorously for several hours until the lumpy solid became a fine powder. The
solid was isolated on a medium-pore glass frit, washed with tetrahydrofuran (30 mL), pentane (30 mL),
and dried under vacuum to afford a pale tan-pink powder (9.42 g, 70%). Crystals suitable for X-ray
diffraction were grown via bi-layer diffusion (diethyl ether/acetonitrile). EA: Found: C 66.67, H 5.15,
N 11.66. Calc. for C27H23BrN4: C 67.08, H 4.80, N 11.59%. 1H NMR (500 MHz; CDCl3): δ = 10.37
(s, 1H; H1), 8.50 (m, 2H; H8), 7.68 (m, 2H; H6), 7.42 (m, 2H; H5), 7.36 (m, 2H; Ph), 7.28 (m, 4H;
Ph), 7.24-7.21 (m, 4H; Ph), 7.21 (m, 2H; H7 (buried under Ph)), 5.64 (s, 4H; H3). 13C {1H} NMR:
δ = 152.75 (C4), 149.70 (C8), 138.67 (C1), 137.64 (C6), 132.37 (C2), 130.95-124.91 (Ph), 123.72 (C7),
123.15 (C5) 52.31 (C3).
4.4.4 Ag(NCN)Br (64).
Compound 63 (18.6 mmol, 9.00 g) and a Teflon coated stir bar were added to a Schlenk flask and placed
under an inert atmosphere. Under argon, Ag2O (9.31 mmol, 2.16 g) and activated 3 A molecular sieves
(10 g) were added, followed by dichloromethane (120 mL). The solution was vigorously stirred in the
dark for 4 hours at room temperature. In air, about 250 mL chloroform was added to the solution, stirred
for 15 minutes, then filtered through a pad of Celite. The Celite pad was washed with dichloromethane
(20 mL) and then the solvent was removed from the brown solution. The residue was redissolved in 50
mL chloroform and about 600 mL diethyl ether was added to precipitate the product from the vigorously

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 50
stirred solution. The solid was isolated on a medium-pore glass frit, washed with diethyl ether (20 mL),
and dried under high vacuum to obtain a pale tan-pink solid (9.79 g, 89%). The hygroscopic product
is air and moisture stable. EA: Found: C 53.93, H 3.68 N 9.39. Calc. for C27H22N4AgBr · (CHCl3)0.1:
C 54.05, H 3.70, N 9.30%. After multiple EA results from independent batches of product, carbon was
consistently 1% lower than expected, leading us to conclude that about 10% CHCl3 remains with the
product. 1H NMR (400 MHz; CDCl3): δ = 8.50 (m, 2H; H8), 7.60 (m, 2H, H6), 7.28-7.11 (10H; Ph),
7.17 (m, 2H; H7 (buried under Ph), 7.08 (m, 2H, H5), 5.39 (s, 4H; H3). 13C {1H} NMR: δ = 183.72
(C1), 155.78 (C4), 149.84 (C8), 137.09 (C6), 132.11 (C2), 130.76-127.78 (Ph), 122.99 (C7), 122.07 (C5),
54.76 (C3).
4.4.5 [Ru(NCN)(NCCH3)3][PF6]2 (65).
Ruthenium(II) dichloride norbornadiene polymer ([RuCl2(NBD)]n, 10.2 mmol, 2.68 g), 64 (10.2 mmol,
6.00 g), and a Teflon coated stir bar were added to a Schlenk flask and placed under an inert atmosphere.
Chloroform (80 mL) and acetonitrile (80 mL) were added and the solution was stirred for 24 h at 60 ◦C.
The solution was cooled to room temperature and MPF6 was added (20.3 mmol, 2 equiv. with respect to
[RuCl2(NBD)]n, M = Na or K). The solution was stirred for 1 h at room temperature. In air, the solution
was filtered through a pad of Celite which was washed with acetonitrile until the outgoing liquid was
colourless. The yellow-orange solution was dried, and then 1,2-dimethoxyethane (500 mL) was added.
This solution was vigorously stirred for several hours until a fine powder formed. The solid was collected
on a medium-pore glass frit, washed with pentane (20 mL) and dried under high vacuum to afford a pale
tan powder (5.58 g, 60%). The product is air and moisture stable. Crystals suitable for X-ray diffraction
were grown via bi-layer diffusion (diethyl ether/acetonitrile). The labile acetonitrile ligand trans to the
N-heterocyclic carbene begins exchanging with deuterated acetonitrile when the sample is dissolved for
NMR analysis. EA: Found: C 43.88, H 3.74, N 11.10. Calc. for C33H31F12N7P2Ru·0.5(CH3CN): C
43.57, H 3.50, N 11.21%. 1H NMR (600 MHz; CD3CN): δ = 9.10 (m, 2H; H8), 7.92 (m, 2H; H6), 7.52
(m, 2H; H5), 7.50 (m. 2H; H7), 7.49-7.48 (m, 6H; Ph), 7.39-7.37 (m, 4H; Ph), 5.28 (s, 4H; H3), 2.24
(s, 6H; H10), 1.96 (s, 3H; H12). 13C {1H} NMR: δ = 185.25 (C1), 158.17 (C8), 157.97 (C4), 139.35
(C6), 132.68 (C2), 131.84-128.37 (Ph), 128.13 (C9), 127.01 (C5), 126.04 (C7), 128.35 (C11 (overlaps
with CD3CN)), 52.18 (C3), 4.61 (C10), 1.77 (C12, overlaps with CD3CN)).
4.4.6 [Ru(NCN)(tBu2bpy)(NCCH3)][PF6]2 (66).
Complex 65 (2.18 mmol, 2.00 g), 4,4’-di-tert-butyl-2,2’-bipyridine (3.27 mmol, 0.876 g), and a Teflon
coated stir bar were added to a Schlenk flask and placed under an inert atmosphere. Acetone (15 mL)
was added and the solution was refluxed for 2 hours, after which the dark orange solution was cooled
to room temperature. In air, diethyl ether (80 mL) was added to the solution and a bright orange
precipitate formed. This was stirred vigorously for a few hours until it became a fine orange powder.
The solid was isolated on a medium-pore glass frit, washed with diethyl ether (20 mL), and dried under
high vacuum. (2.14 g, 89%). The product is air and moisture stable. EA: Found: C 51.16, H 5.22,
N 8.97. Calc. for C47H49F12N7P2Ru·0.5(C4H10O): C 51.63, H 4.77, N 8.60%. 1H NMR (600 MHz;
CD3CN): δ = 9.44 (d, 3J(H,H) = 5.7 Hz, 1H; H18), 8.56 (d, 4J(H,H) = 1.8 Hz, 1H; H15), 8.36 (d,4J(H,H) = 2.0 Hz, 1H; H12), 8.15 (d, 3J(H,H) = 6.1 Hz, 1H; H9), 8.03 (m, 1H; H17), 7.69 (m, 2H; H6),
7.66 (br, 2H; H8), 7.55-7.48 (m, 10H; Ph), 7.40 (d, 3J(H,H) = 7.0 Hz, 2H; H5), 7.25 (dd, 3J(H,H) =

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 51
6.1 Hz, 4J(H,H) = 2.1 Hz, 1H; H10), 7.02 (m, 2H; H7), 5.31 (d, 2J(H,H) = 16.2 Hz, 2H; 3Ha), 5.14
(d, 2J(H,H) = 16.2 Hz, 2H; 3Hb), 2.38 (s, 3H; H24), 1.57 (s, 9H; H22) 1.36 (s, 9H; H20). 13C {1H}NMR: δ = 189.94 (C1), 164.56 (C16), 163.41 (C11), 159.35 (C13), 158.58 (C4), 156.79 (C14), 155.72
(C9), 155.61 (C8), 151.83 (C18), 138.89 (C6), 133.06 (C2), 131.94 (Ph), 130.58 (C23), 130.34-128.59
(Ph), 127.01 (C5), 126.12 (C17), 125.80 (C7), 124.55 (C10), 122.81 (C12), 122.27 (C15), 52.14 (C3),
36.59 (C21), 36.14 (C19), 30.65 (C22), 30.39 (C20), 5.14 (C24).
4.4.7 [RuBr(NCN)(tBu2bpy)][PF6] (67).
Compound 66 (1.81 mmol, 2.00 g), potassium bromide (5.44 mmol, 0.647 g), and a Teflon coated stir bar
were added to a Schlenk flask and placed under an inert atmosphere. Dimethyl sulfoxide (10 mL) was
added and the deep orange solution was heated at 110 ◦C for 2 hours. In air, the solvent was removed
by vacuum distillation at 100 ◦C. The deep red-orange residue was washed with dichloromethane (10
mL) and dried with heat and high vacuum to remove more dimethyl sulfoxide. The solid was redissolved
in acetonitrile (50 mL) and the salts were filtered off with a medium pore glass frit. The salts were
washed with acetonitrile until the outgoing liquid was colourless. The deep red-orange solution was
concentrated, passed through a short path alumina column (acetonitrile), and the leading red-orange
fraction was collected. After solvent removal, the residue was redissolved in about 15 mL acetonitrile
and precipitated by adding diethyl ether (500 mL) while vigorously stirring to afford a red-orange
powder. The product was isolated on a medium-pore glass frit and dried under high vacuum (1.61
g, 89%). The product is air and moisture stable. EA: Found: C 54.58, H 5.15, N 8.39. Calc. for
C45H46BrF6N6PRu·0.5(C4H10O): C 54.60, H 4.97, N 8.13%. 1H NMR (600 MHz; CD3CN): δ = 10.18
(d, 3J(H,H) = 5.8 Hz, 1H; H18), 8.49 (d, 4J(H,H) = 1.9 Hz, 1H; H15), 8.28 (d, 4J(H,H) = 2.1 Hz, 1H;
H12), 8.14 (d, 3J(H,H) = 6.2 Hz, 1H; H9), 8.03 (dd, 3J(H,H) = 5.8 Hz, 4J(H,H) = 2.0 Hz, 1H; H17),
7.76 (br, 2H; H8), 7.54 (m, 2H; H6), 7.51-7.45 (m, 10H; Ph), 7.26 (d, 3J(H,H) = 7.0 Hz, 2H; H5), 7.10
(ddd, 3J(H,H) = 6.2 Hz, 4J(H,H) = 2.1 Hz, 1H; H10), 6.87 (m, 2H; H7), 5.69 (br d, 2H; 3Ha), 5.07 (d,2J(H,H) = 15.6 Hz, 2H; 3Hb), 1.55 (s, 9H; H22) 1.33 (s, 9H; H20). 13C {1H} NMR: δ = 195.50(C1),
163.17 (C16), 161.21 (C11), 160.57 (C13), 159.45 (C4), 157.22 (C14), 156.67 (C8), 156.42 (C9), 152.57
(C18), 137.57 (C6), 132.63 (C2), 131.87-129.01 (Ph), 125.99 (C5), 125.70 (C17), 124.78 (C7), 123.92
(C10), 122.44 (C12), 121.33 (C15), 52.48 (C3), 36.40 (C21), 35.89 (C19), 30.73 (C22), 30.41 (C20).
UV/Vis (CH3CN): λmax(ε, M−1cm−1)= 444 (7000), 352 (10000).
4.4.8 Ru(OtBu)(NCN*)(tBu2bpy) (68).
Compound 67 (0.10 mmol, 100 mg) and potassium tert-butoxide (0.22 mmol, 25 mg), and a Teflon coated
stir bar are combined in a vial inside an argon-filled glove box. Tetrahydrofuran (2 mL) was added and
the solution immediately turned deep maroon-orange and was stirred for 2 hours at room temperature.
After 2 hours, the deep maroon-purple solution was dried under high vacuum and redissolved in toluene
(2 mL). The solution was filtered through a pad of Celite and dried. Trituration with pentane and drying
under high vacuum afforded deep maroon-purple solid 6 (80 mg, 95%). Crystallization by slow vapour
diffusion of pentane into a concentrated toluene solution yields deep purple-black crystals (71%), which
were suitable for X-ray diffraction. Compound 68 is freely soluble in toluene, tetrahydrofuran, diethyl
ether, and partly soluble in pentane. Due to the highly sensitive nature of 68, a powder or crystalline
sample did not yield successful elemental analysis data after several attempts, despite being pure by

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 52
NMR spectroscopy. 1H NMR (600 MHz; THF−d8, 0 ◦C): δ = 10.13 (d, 3J(H,H) = 5.8 Hz, 1H; H18),
8.34 (d, 3J(H,H) = 6.3 Hz, 1H; H9), 8.29 (d, 1H; H15), 8.09 (d, 1H; H12), 7.73 (dd, 3J(H,H) = 5.8 Hz,
1H; H17), 7.41-7.33 (m, 6H; Ph), 7.32 (m, 1H; H6), 7.28-7.25 (m, 4H; Ph), 7.16 (m, 1H; H5), 7.00 (d,3J(H,H) = 5.7 Hz, 1H; H8), 6.77 (dd, 3J(H,H) = 6.3 Hz, 1H; H10), 6.50 (m, 1H; H7), 6.47 (d, 2J(H,H)
= 13.2 Hz, 2H; 3Ha), 5.89 (d, 3J(H,H) = 6.4 Hz, 1H; H23), 5.55 (m, 1H; H25), 5.06 (s, 1H; H28), 5.04
(d, 3J(H,H) = 8.8 Hz, 1H; H26), 4.74 (d, 2J(H,H) = 13.2 Hz, 2H; 3Hb), 4.20 (m, 1H; H24), 1.52 (s, 9H;
H22) 1.29 (s, 9H; H20), 0.99 (s, 9H; H31). 13C {1H} NMR: δ = 190.74 (C1), 162.00 (C4), 160.18 (C13),
158.22 (C16), 156.77 (C14), 154.39 (C9), 153.33 (C11), 153.28 (C8), 152.03 (C23), 150.32 (C18), 145.72
(C27), 133.80 (C6), 131.68-130.63 (Ph), 130.36 (C2), 129.13-127.69 (Ph), 125.87 (C29), 125.81 (C25),
123.41 (C5), 122.54 (C7), 121.90 (C17), 121.15 (C10), 119.88 (C12), 117.93 (C15), 114.79 (C26), 98.48
(C24), 86.99 (C28), 71.36 (C30), 51.22 (C3), 35.70 (C21), 35.04 (C31), 34.97 (C19), 30.81 (C22), 30.40
(C20). UV/Vis (THF): λmax(ε, M−1cm−1)= 487 (7000), 378 (10000).
4.4.9 Ru(NCN**)(PPh3)(tBu2bpy) (70).
Compound 67 (0.040 mmol, 40 mg), PPh3, (0.040 mmol, 11 mg), KOtBu (0.12 mmol, 14 mg), and
a Teflon-coated stir bar were combined in a vial inside an argon- filled glove box. Tetrahydrofuran
(3 mL) was added and the solution was stirred overnight at room temperature. The deep red-orange
solution was dried under high vacuum and redissolved in toluene (3 mL), then filtered through a pad
of Celite and dried. The solid was washed and suspended by vigorously stirring with hexane (3 mL),
collected on a medium-pore glass frit, and dried under high vacuum (30 mg, 73 %). Crystals suitable
for X-ray diffraction were grown by slow vapor diffusion of pentane into a concentrated toluene solution.
Formation of 70 was also detected by 1H NMR spectroscopy when one equivalent of PPh3 was added
to 68 in a vigorously stirred THF solution in an argon-filled glove box. EA: Found: C 72.96, H 6.04, N
7.80. Calc. for C63H59N6PRu: C 73.31, H 5.76, N 8.14%. 1H NMR (500 MHz; THF−d8): δ = 9.11 (d,3J(H,H) = 5.9 Hz, 1H; H18), 8.69 (dd, 3J(H,H) = 5.9 Hz, 4J(H,H) = 2.9 Hz, 1H; H9), 8.30 (d, 4J(H,H)
= 2.0 Hz, 1H; H15), 8.27 (m, 1H; H12), 7.72-7.69 (m, 6H; Ph), 7.48 (m, 1H; H10), 7.31 (dd, 3J(H,H) =
5.9 Hz, 4J(H,H) = 2.0 Hz, 1H; H17), 7.22-7.09 (m, 19H; Ph), 5.50 (m, 2H; H6), 4.97 (m, 2H; H8), 4.79
(m, 2H; H5), 4.53 (s, 2H; H3), 4.08 (m, 2H; H7), 1.41 (s, 9H; H20), 1.39 (s, 9 H ; H22). 13C {1H} NMR:
δ = 174.19 (d, 2J(C,P) = 17.9 Hz); C1), 160.38 (C11), 160.37 (C16), 158.33 (C14), 157.35 (C13), 154.18
(C18), 153.05 (C9), 150.91 (C8), 147.78 (C4), 139.67127.16 (Ph), 126.23 (C6), 125.59 (C2), 123.33 (C17),
123.20 (C10), 120.60 (C15), 119.21 (C12), 115.15 (C5), 99.89 (C7), 88.06 (C3), 35.67 (C21), 35.59 (C19),
30.46 (C20), 30.35 (C22). 31P {1H} NMR: δ = 57.69 (s).
4.4.10 Ru(OH)(NCN*)(tBu2bpy) (71).
Compound 68 (0.019 mmol, 16 mg) was placed into an NMR tube and then dissolved in THF-d8. The
tube was sealed with a tight fitting rubber septum which was then wrapped with Parafilm-M R©. The
NMR tube was removed from the glove box and distilled/degassed H2O (0.50 µL, 0.028 mmol) was
injected with a microsyringe. Shaking the tube for a few seconds allowed the droplet deposited at the
top to dissolve into the solution below. The solution was heated at 40 ◦C for 5 minutes, changing
from deep maroon-purple to deep maroon-orange in colour and was immediately analyzed by NMR
spectroscopy (0 ◦C). The same procedure was used when reacted with D2O (0.50 µL, 0.025 mmol). 1H
NMR (600 MHz; THF−d8, 0 ◦C): δ = 9.87 (d, 3J(H,H) = 5.8 Hz, 1H; H18), 8.32 (m, 1H; H9), 8.27 (d,

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 53
3J(H,H) = 6.2 Hz, 1H; H15), 8.09 (m, 1H; H12), 7.69 (m, 1H; H17), 7.40-7.26 (m, 10H; Ph), 7.32 (m,
1H; H6), 7.22 (m, 1H; H5), 6.80 (m, 1H; H8), 6.73 (m, 1H; H10), 6.49 (m, 1H; H7), 6.41 (d, 2J(H,H) =
14 Hz, 2H; 3Ha), 6.09 (d, 3J(H,H) = 6.3 Hz, 1H; H23), 5.62 (m, 1H; H25), 5.15 (d, 3J(H,H) = 8.9 Hz,
1H; H26), 5.02 (s, 1H; H28), 4.92 (d, 2J(H,H) = 14 Hz, 2H; 3Hb), 4.36 (m, 1H; H24), 3.33 (br, H2O
+ (CH3)3COH), 1.51 (s, 9H; H22) 1.29 (s, 9H; H20), 1.07 (s, 9H; (CH3)3COH), -2.35 (br, 1H; H30).13C {1H} NMR: δ = 188.41 (C1), 162.86 (C4), 159.31 (C13), 157.70 (C16), 156.67 (C14), 154.59 (C9),
153.61 (C11), 153.56 (C8), 150.51 (C23), 150.37 (C18), 146.21 (C27), 133.63 (C6), 131.37-130.52 (Ph),
130.19 (C2), 129.19-127.92 (Ph), 126.11 (C29), 125.99 (C25), 124.06 (C5), 122.94 (C7), 122.18 (C17),
120.78 (C10), 119.80 (C12), 118.20 (C15), 114.29 (C26), 99.37 (C24), 87.63 (C28), 66.72 ((CH3)3COH),
51.38 (C3), 35.67 (C21), 35.00 (C19), 31.80 ((CH3)3COH), 30.78 (C22), 30.41 (C20).

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 54
4.5 Supplementary Information
4.5.1 NMR Peak Numbering and Spectra.
1
2
3
45
6
7 8
N N
N
9
1
2
3
45
6
7 8
N N
N N
+
Br-
NN
N
NRu
N
N
N
2+
2PF6-
1
2
34
5
67
8
9
10
11
12
1D NOESY
NN
N
NRu
N
N
N
2+
2PF6-
1
2
34
5
67
8
9
1011
12
1314 15
16
1718
19
21
20
22
23
24
Ha
Hb
H
NN
N
NRu
N
N
Br
+
PF6-
1D NOESY
1
2
34
5
67
8
9
1011
12
1314 15
16
1718
19
21
20
22H
Ha
Hb
NN
N
NRu
N
N
O H
Hb
1
2
34
5
67
8
9
1011
12
1314 15
16
1718
19
21
20
22
2324
252629
30
2728
31
Ha
N N
N NAg
Br
1
2
3
45
6
7 8
NN
N
NRu
N
N
O
H
H
Hb
1
2
34
5
67
8
9
1011
12
1314 15
16
1718
19
21
20
22
2324
252629
30
2728
HaNN
N
NRu
N
N
PPh3
1
2
3
45
67
8
9
1011
12
1314
1516
1718
19
21
20
22H
63 64
65 66 67
68 70 71
Figure 4.7: NMR peak numbering for all complexes.
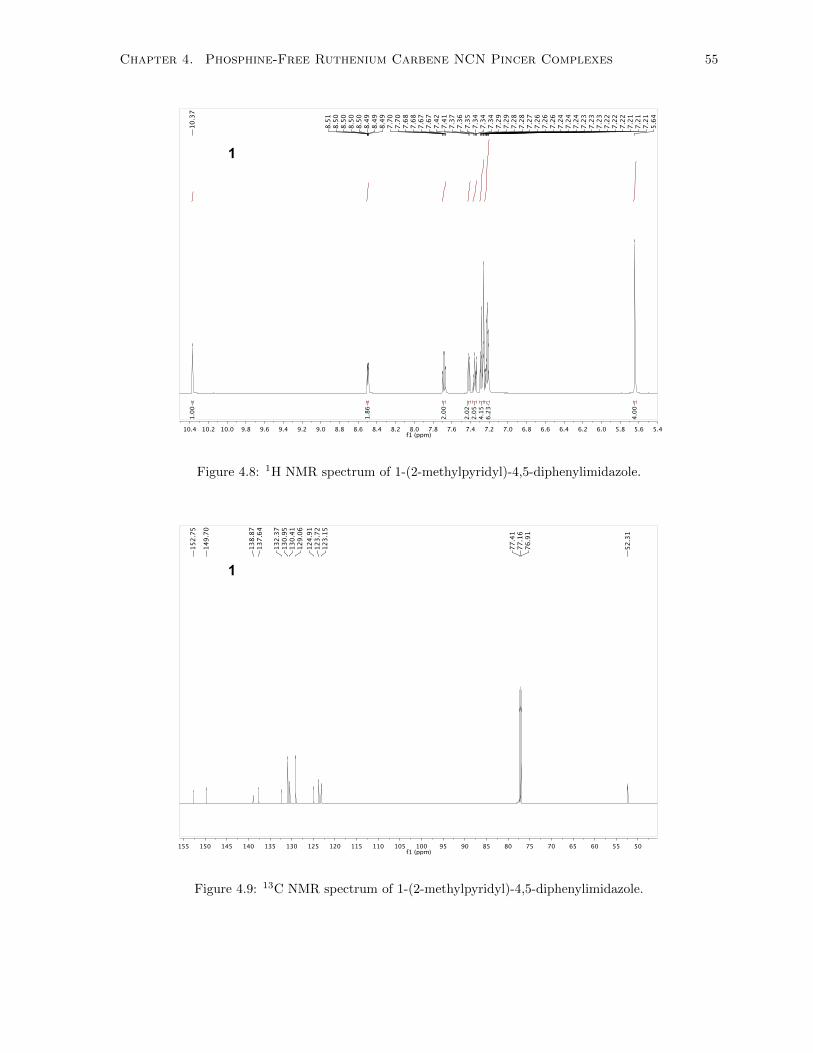
Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 55
Figure 4.8: 1H NMR spectrum of 1-(2-methylpyridyl)-4,5-diphenylimidazole.
Figure 4.9: 13C NMR spectrum of 1-(2-methylpyridyl)-4,5-diphenylimidazole.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 56
Figure 4.10: 1H NMR spectrum of 63.
Figure 4.11: 13C NMR spectrum of 63.
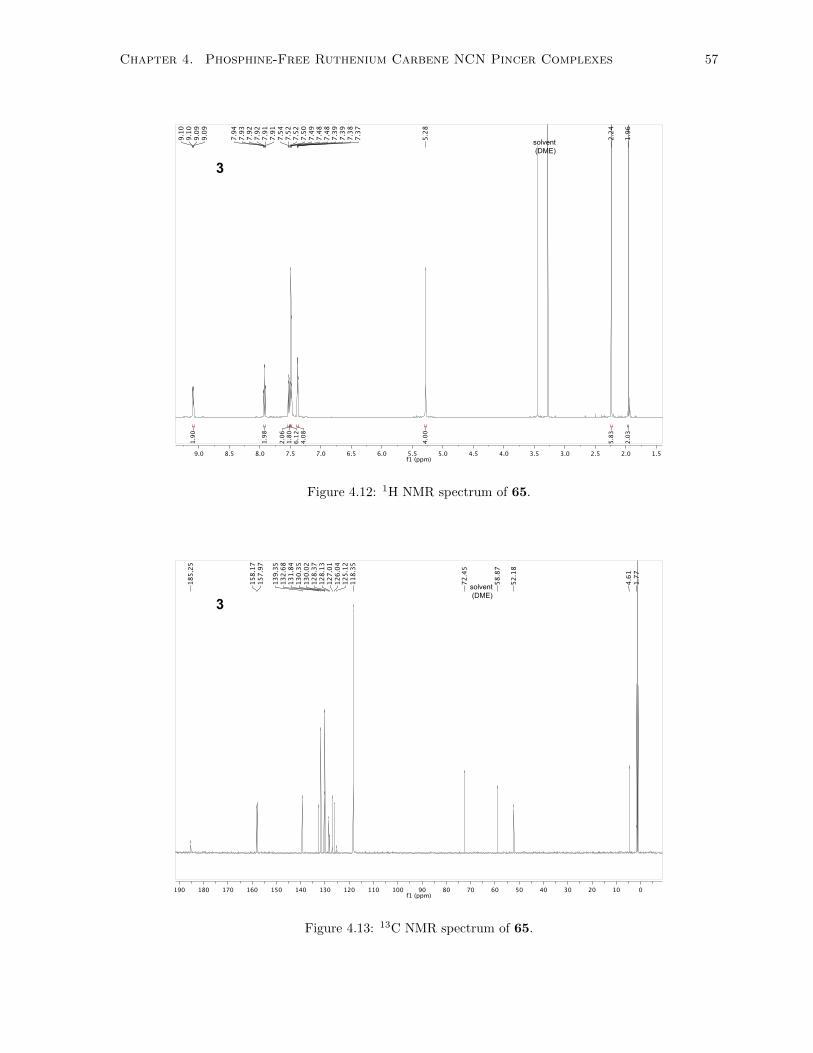
Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 57
Figure 4.12: 1H NMR spectrum of 65.
Figure 4.13: 13C NMR spectrum of 65.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 58
Figure 4.14: 1H NMR and 1D NOESY spectrum (red line, 600 ms mixing time, 4 s relaxation delay,irradiation at δ = 8.15) of 66.
Figure 4.15: 13C NMR spectrum of 66.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 59
Figure 4.16: 1H NMR and 1D NOESY spectrum (red line, 600 ms mixing time, 4 s relaxation delay,irradiation at δ = 8.14) of 67.
Figure 4.17: 13C NMR spectrum of 67.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 60
Figure 4.18: 1H NMR spectrum of 68.
Figure 4.19: 13C NMR spectrum of 68.
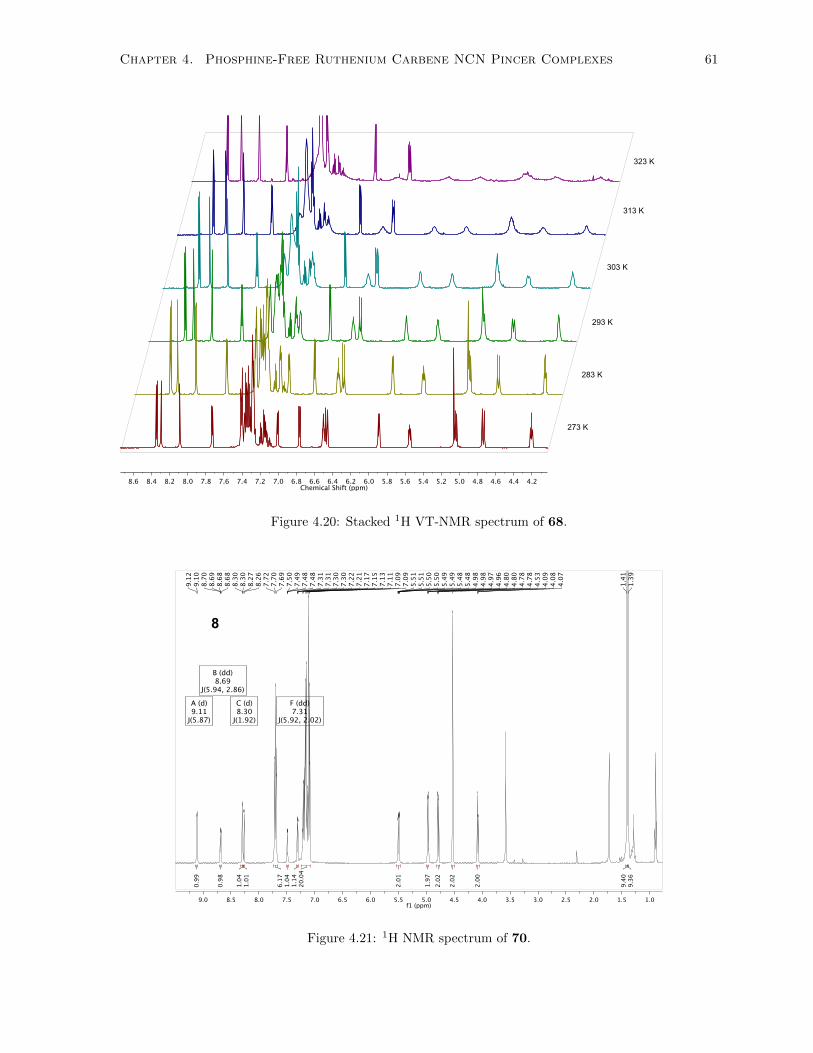
Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 61
Figure 4.20: Stacked 1H VT-NMR spectrum of 68.
Figure 4.21: 1H NMR spectrum of 70.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 62
Figure 4.22: 13C NMR spectrum of 70.
Figure 4.23: 31P NMR spectrum of 70.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 63
Figure 4.24: 1H NMR spectrum of 71.
Figure 4.25: 13C NMR spectrum of 71.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 64
Figure 4.26: 1H-1H ROESY spectrum (THF−d8, 0 ◦C, 600 ms mixing time, 1 s relaxation delay) of 71.
4.5.2 X-Ray crystal structure and refinement data.

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 65
Table 4.2: Selected X-Ray crystal structure and refinement data for complexes 1-(2-methylpyridyl)-4,5-diphenylimidazole (Im), 65, 68, and 70.
Compound Im·(H2O)2 65·CH2Cl2 68·C5H12
Empirical formula C27H27BrN4O2 C34H33Cl2F12N7P2RuC54H66N6ORu
Formula mass 519.43 1001.58 916.19
Temp. (K) 147(2) 147(2) 147(2)
Crystal system Triclinic Triclinic Monoclinic
Space group P1 P1 P21/n
a (A) 10.0240(19) 11.4038(9) 21.480(3)
b (A) 10.447(2) 13.1369(10) 10.0850(12)
c (A) 12.072(2) 14.5455(11) 22.272(3)
α (◦) 78.011(5) 75.854(2) 90
β (◦) 88.339(4) 72.083(2) 90.234(3)
γ (◦) 78.932(4) 85.274(2) 90
Volume (A3) 1213.5(4) 2010.5(3) 4824.6(10)
Z 2 2 4
Calc. density (g/cm3) 1.422 1.655 1.261
Absorption coeff.(mm−1)
1.726 0.695 0.369
F(000) 536 1004 1936
Crystal size (mm3)0.280 × 0.200 ×0.100
0.400 × 0.260 ×0.130
0.290 × 0.120 ×0.030
θ range collected (◦) 1.725–27.530 1.512–27.538 1.314–27.531
Reflections collected 35658 33752 60817
Independent reflectionsR(int)
5546 (0.0220) 9128 (0.0220) 11081 (0.0610)
Completeness toθ = 27.52◦ (%)
99.8 99.2 99.9
Absorption correctionSemi-empirical fromequivalents
Semi-empirical fromequivalents
Semi-empirical fromequivalents
Max. and min.transmission
0.7456, 0.6286 0.7455, 0.6656 0.7456, 0.6796
Data / restraints /parameters
5546/0/323 9128/91/548 11081/0/570
Goodness-of-fit on F2 1.054 1.032 1.036
R1(I > 2σ(I))a 0.0228 0.0433 0.0423
wR2 (all data)b 0.0623 0.1176 0.1014
a R1 =Σ(Fo − Fc)
Σ(Fo)
b wR2 =
√Σ[w(F 2
o − F 2c )2]
Σ[w(F 2o )2]

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 66
Compound 70·(C7H8)0.75·(C5H12)0.25Empirical formula C69.5H68N6PRu
Formula mass 1119.33
Temp. (K) 147(2)
Crystal system Triclinic
Space group P1
a (A) 18.1397(7)
b (A) 18.7592(9)
c (A) 20.7229(9)
α (◦) 89.894(3)
β (◦) 72.527(2)
γ (◦) 67.912(3)
volume (A3) 6183.3(5)
Z 4
Calc. density (g/cm3) 1.202
Absorption coeff. (mm−1) 2.634
F(000) 2344
Crystal size (mm3) 0.080 × 0.050 × 0.050
θ range collected (◦) 2.253–68.241
Reflections collected 135915
Independent reflections R(int) 21971 (0.2308)
Completeness to θ = 27.52◦ (%) 98.1
Absorption correctionSemi-empirical fromequivalents
Max. and min. transmission 0.8796, 0.8170
Data / restraints / parameters 21971/23/1337
Goodness-of-fit on F2 0.977
R1(I > 2σ(I))a 0.0741
wR2 (all data)b 0.2258
a R1 =Σ(Fo − Fc)
Σ(Fo)
b wR2 =
√Σ[w(F 2
o − F 2c )2]
Σ[w(F 2o )2]

Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 67
Figure 4.27: Molecular structure of 1-(2-methylpyridyl)-4,5-diphenylimidazole with 30% probability el-lipsoids.
Figure 4.28: Molecular structure of 65 with 30% probability ellipsoids. Hydrogen atoms and couter-anions omitted for clarity.
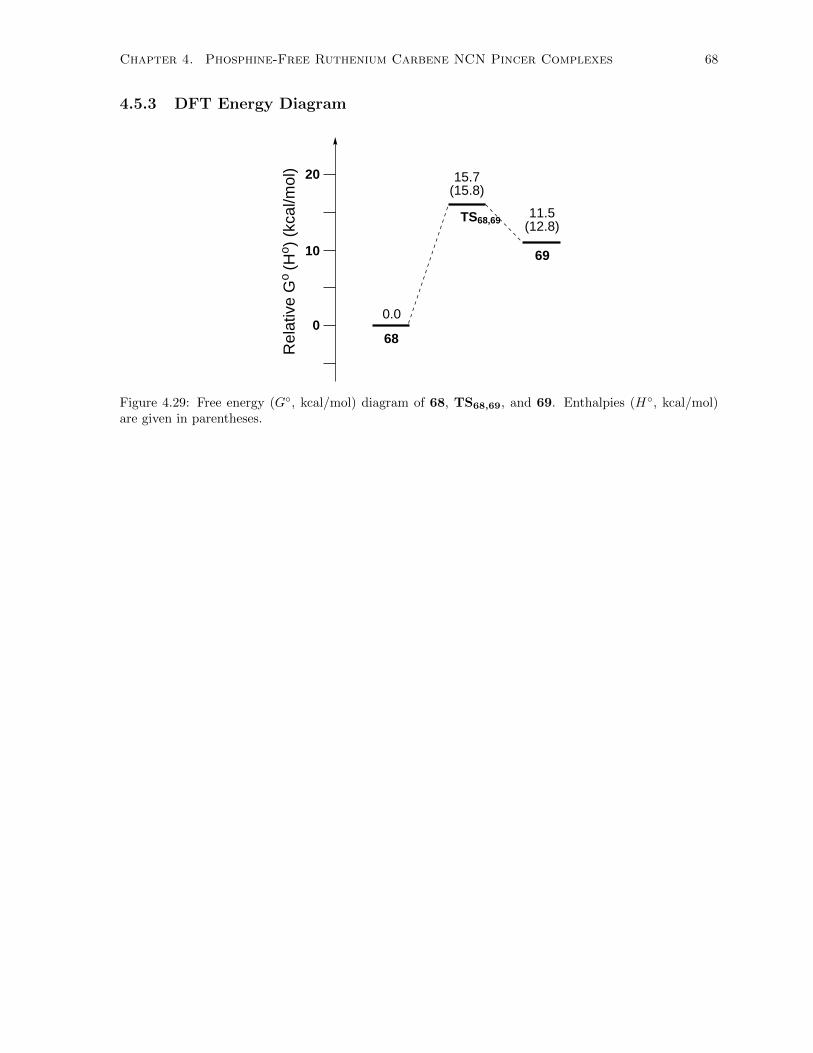
Chapter 4. Phosphine-Free Ruthenium Carbene NCN Pincer Complexes 68
4.5.3 DFT Energy Diagram
0
10
20
11.5(12.8)
0.0
15.7(15.8)
Rel
ativ
e G
o (H
o ) (
kcal
/mol
)
68
TS68,69
69
Figure 4.29: Free energy (G◦, kcal/mol) diagram of 68, TS68,69, and 69. Enthalpies (H◦, kcal/mol)are given in parentheses.

Chapter 5
Phosphine-Free Ruthenium
Complexes Bearing Tetradentate
Amino-Olefin Ligands
Abstract
New ruthenium complexes with tetradentate amino-olefin ligands including the “trop” olefinic
ligand donor framework (trop = 5H-dibenzo[a,d]-cycloheptenyl) were synthesized. Recently,
it was shown that an anionic ruthenium hydride complex [K][RuH(trop2dad)] containing the
tetradentate ligand 1,4-bis(5H-dibenzo[a,d]cyclohepten-5-yl)-1,4-diazabuta-1,3-diene
(trop2dad) which readily liberates H2 in the presence of water and is an active methanol
dehydrogenation catalyst in methanol/water reaction mixtures (Nature Chemistry, 2013,
5, 342–347). In our quest to synthesize phosphine-free ruthenium complexes capable of
water splitting, we investigated the reactivity of this anionic metal-hydride complex with
water, and also the effect of the potassium ion by encapsulation with a crown ether or re-
placement with a tetrabutylammonium ion. Finally, the synthesis and reactivity of new
ruthenium complexes with the tetradentate ligand N,N -bis(5H-dibenzo[a,d]- cyclohepten-5-
yl)-1,2-diaminocyclohexane (trop2dach) were performed, resulting the synthesis of an electron
rich four-coordinate Ru(0) complex, Ru0(trop2dach), which has been structurally character-
ized. Double deprotonation at the amine NH positions results in the synthesis of a rare
Ru(-II) complex, [K2][Ru–II
(trop2dach)], which has been characterized by 1D and 2D NMR
spectroscopy.
5.1 Introduction
In organometallic chemistry, the ubiquitous alkene binding motif is commonly associated with hydrocar-
bon based π-donor haptic modes such as η2 (ethylene, cycloolefins), η3 (allyl, indenyl), η4 (cyclobutadi-
enyl), η5 (cyclopentadienyl), or η6 (benzene). An unique variation on this motif is the η2-5H-dibenzo[a,d]-
cycloheptenyl (η2-trop) moiety, which serves as a two-electron π-donor that can be functionalized with
69

Chapter 5. Ru Complexes With Tetradentate Amino-Olefin Ligands 70
nitrogen or phosphorus donor atoms at the sp3 carbon within the seven-membered ring (Figure 5.1,
left).39,84,257–266 Functional groups can be introduced at the olefin, making these complexes enantiose-
lective hydrogenation catalysts when coordinated to transition metals.267–269 The amine-functionalized
tetradentate trop ligands trop2dpen (dpen = (S,S )-1,2-diphenylethylenediamine), trop2dach (dach =
(R,R)-1,2-diaminocyclohexane), and trop2dae (dae = 1,2-diaminoethane) have been shown to stabilize
electron rich rhodium261,266 (72 and 73), iridium262 (74), and ruthenium84 (75) compounds, respec-
tively, much owing to the robust π-accepting nature of the olefin ligands (Figure 5.1, right). In addition,
X-ray crystallographic measurements and NMR chemical shifts of the olefinic proton and carbon nuclei
are very sensitive to changes in their electronic environment, making them useful probes into the overall
electronic structure.
L
ML'n
=
trop
L = P; R, R' = trop, Ph, alkylL = N; R = H, trop, alkyl; R' = H, alkyl
R
R'
N N
Ru0
M = Ir
M = Rh
N N
Rh-
N N
M-
H
H
Ph PhK+ K+
72 7374
75
Figure 5.1: Left: The “trop” moiety as part of a chelating ligand for transition metals. Right: Lowvalent metal complexes with tetradentate amino/amido olefin ligands.
Recently, the structurally characterized anionic ruthenium hydride complex 76 was found to be an
active catalyst for the formation CO2/H2 from 1:1 methanol:water mixtures (Scheme 5.1, left).84 Based
on stoichiometric reactivity studies, three elementary reaction steps are proposed during catalysis: (1)
conversion of methanol to formaldehyde and hydrogen; (2) the reaction of formaldehyde and water to
generate formic acid and hydrogen; (3) the dehydrogenation of formic acid to generate carbon dioxide
and hydrogen. These reactions are facilitated by several hydride/proton metal-ligand cooperation steps
involving the metal center, nitrogen atoms, and the ethylene backbone. In a stoichiometric reaction
with alcohol and water, the formally Ru(0) complex 75 has been isolated and structurally characterized.
Complex 75 can be converted back to hydride complex 76 in the presence of strong base, liberating
H2 (Scheme 5.1, right). Furthermore, in the absence of alcohol, the complex Ru(trop2dad) (77, dad =
1,4-diazadiene) can be isolated. When ethanol is subsequently reacted with 77 in the presence of KOH,
complex 75 forms by reducing the ligand framework and oxidizing ethanol, forming potassium acetate.
We were curious to know how hydride complex [K][RuH(trop2dad)] would react in the absence of
alcohol for applications in water splitting, since hydrogen is readily liberated from 76. Based on previous
work by Milstein, the next logical step after hydrogen production is to attempt synthesizing ruthenium
hydroxide complexes for O2 formation.111 We also wanted to know what role the potassium ion played
in the reactivity of 76∗ and whether new ruthenium complexes with functionalized tetradentate ligand
frameworks (trop2dach, trop2dpen) could be synthesized. The reactivity of 76 with water under various
conditions will be presented, along with the synthesis and characterization of new ruthenium complexes
∗Potassium ions have been shown to accelerate ketone hydrogenation catalysis.67,270

Chapter 5. Ru Complexes With Tetradentate Amino-Olefin Ligands 71
N N
Ru0
H
H
(1) CH3OH
(2) H2C=O + H2O
(3) HCOOH
H2C=O + H2
HCOOH + H2
CO2 + H2
CH3OH + H2O0.5 mol %
CO2 + 3 H2
H2O/EtOH
-H2, -MeCOOK
KOtBu
-H2, -tBuOH
65 oC
90 oC K+(dme)2
N N
Ru-
H
H2O
-H2, -KOH
N N
Ru
EtOH-MeCOOK
KOH
76
76
75
77
Scheme 5.1: Left: dehydrogenation of methanol/water mixtures catalyzed by 76. Right: stoichiometricreactivity studies of 76, generating Ru(0) complex 75 or 77. dme = 1,2-dimethoxyethane.
bearing the trop2dach ligand framework.
5.2 Results and Discussion
Three different molecules were synthesized: (1) 76,84 (2) 78, encapsulating the potassium ion with
db18c6 (dibenzo-18-crown-6), and (3) 79, replacing the potassium ion with a NBu+4 (tetrabutylammo-
nium) ion. Reacting 76 with db18c6 is trivial, forming complex 78 in quantitative yield when db18c6 is
added (Scheme 5.2). Reacting 76 with NBu4Br is also trivial; stirring the starting materials in THF and
then removing KBr by filtration yields 79 in high purity. Successful reactions are indicated by the large
change in chemical shift of db18c6 methylene protons for 78, precipitation of KBr in the case of 79, and
the downfield hydride chemical shift change from -10.25 ppm to -9.00 (78) and -9.43 (79). The change
in hydride chemical shift likely indicates decreased ion pairing between the hydride/trop ligands and
potassium ion since the molecular structure of 7684 and other anionic [K][(trop(diamide)trop)M] com-
plexes mentioned earlier (72–74)261,262,266 reveals the formation of electrostatically enforced host-guest
complexes, where the potassium ion is π-bound to the a trop arene ring in solid-state molecular struc-
tures. Reacting 76 with 2.2.2-crypt leads to complete precipitation of a substance which we were unable
to redissolve in hydrocarbon or ethereal solvents for characterization. Although the target complex
[K(2.2.2-crypt)][RuH(trop2dad)] may have been synthesized, it is also expected that cryptand ligands
will completely separate the ions, leading to low solubility in hydrocarbon solvents.271
Compounds 76–79 were reacted in THF-H2O mixtures using up to 10 equivalents H2O and heat.
Unfortunately, after prolonged heating under various conditions, the complex 77 predominantly formed,
as observed in earlier studies,84 with eventual precipitation of a poorly soluble multimetallic ruthenium
species.∗ Substitution of the counter ion or encapsulation of the potassium ion only slowed this process
down (Scheme 5.2), however the eventual fate of the reaction was the same.
Our next approach was to explore the synthesis of other trop2(diamine) ligands with ruthenium,
motivated by the fact that electron rich rhodium and iridium diamido complexes 72–74 were eas-
ily synthesized in earlier reports.261,262,266 We hoped that a functionalized diamine backbone would
lead to more desirable reactivity. Upon reaction of one equivalent RuCl2(PPh3)3 with (±)-trans-
∗H. Grutzmacher, M. Trincado, unpublished results.

Chapter 5. Ru Complexes With Tetradentate Amino-Olefin Ligands 72
δRuH = -10.25 (s)
Y+ = K+(db18c6), δRuH = -9.00 (s)
K+(dme)2
N N
Ru-
H
Y+
N N
Ru
H
Y+ = Bu4N+, δRuH = -9.43 (s)
db18c6
or
Bu4NBr
N N
Ru
- H2, - YOH
H2O, heat
multimetallicproduct
-
76 78
79
77
Scheme 5.2: Reaction of 76 with db18c6 or NBu4Br and subsequent reaction with H2O.
N,N -bis(5H-dibenzo[a,d]cyclohepten-5-yl)-1,2-diaminocyclohexane (trop2dach, 80), the chloro-bridged
bimetallic species 81 precipitates from solution (Scheme 5.3). Doubling the amount of ruthenium at the
outset of the reaction affords 81 in 88 % yield.
RuNH HN
2 RuCl2(PPh3)3 Cl
Cl
Ru
Cl
Cl
PPh3
PPh3
N
N
H
H
THF, reflux
=
80 81
Scheme 5.3: Synthesis of 81.
The molecular structure of (S,S )-81 is shown in Figure 5.2 along with selected metrical parameters.
The ruthenium atoms are bridged by two chloride ligands and both metals conform to a distorted
octahedral geometry. The bridging chlorine atoms, Cl1 and Cl2, respectively, are slightly closer to
Ru2 (2.460(1) and 2.432(1) A) versus Ru1 (2.504(1) and 2.477(1) A). Atom Cl2 is canted towards
the H atom connected to N2 (Cl2· · ·H2 = 2.213 A) making the Cl1-Ru1-Cl2 angle acute (168.96(5)◦).
This hydrogen bonding motif might be useful in the rationally synthesizing other bridged dimer species
containing trop2(diamine) ligands – other structurally characterized ruthenium dimers are known which
contain such NH· · ·Cl interactions between multidentate amine ligands and terminal halides.151,272–275
The trop2dach ligand is bonded in a tetradentate fashion to the metal and possesses unusually
long Ru-centroid bonds (Ru2–ct1 = 2.127(4), Ru2–ct2 = 2.136(6) A) when compared to other known
RR’Ntrop ligands, which typically contain metal-centroid distances of around 2.0 A. The longer bonds
in (S,S )-81 may be due to the strained non-planar coordination geometry. In fact, this is the first
instance of a structurally characterized trop2(diamine) complex where its tetradentate binding mode
is not planar. Typically, trop2(diamine) ligands can also bind in a κ3 mode, leaving one trop ligand
uncoordinated.84,260 The dihedral angle generated between the olefin ligands (C4-C5 and C19-C20) are
approaching orthogonality (-74.8(5)◦).
Due to the insoluble nature of 81 in most organic solvents, 81 was analyzed by CP/MAS solid-state

Chapter 5. Ru Complexes With Tetradentate Amino-Olefin Ligands 73
Figure 5.2: Molecular structure of (S,S )-81 with ellipsoids at the 30 % probability level. Hydrogen atomshave been omitted for clarity. Distances (A), angles (◦), and torsions (◦): Ru1–P1 2.282(2), Ru1–P2A2.23(1), Ru1–Cl1 2.421(1), Ru1–Cl2 2.434(1), Ru1–Cl3 2.504(1), Ru1–Cl4 2.477(1), Ru2–Cl3 2.460(1),Ru2–Cl4 2.432(1), Cl2· · ·H2 2.213, Ru2–N1 2.123(5), Ru2–N2 2.135(5), Ru2–ct1 2.127(4), Ru2–ct22.136(6), C4–C5 1.412(7), C19–C20 1.373(6); P1-Ru1-P2A 95.5(4), Cl1-Ru1-Cl2 168.96(5), N1-Ru2-N280.1(2), ct1-Ru2-ct2 99.0(2); C4-C5-C19-C20 -74.8(5).
13C and 31P{1H} NMR. The 31P spectrum reveals an unresolved multiplet 53.84 ppm while the 13C
spetrum reveals broad signals in the aromatic region (141.43–127.86 ppm) and further upfield (92.36–
61.91 ppm). When the dimer 81 is heated in the presence of four equivalents strong base and ex-
cess hydrogen gas, the pale pink suspension turns deep orange with formation of a white precipitate
(Scheme 5.4). Analysis of the crude product in situ by 1H and 31P NMR under H2 indicates the pres-
ence of two new major products, one whose hydride chemical shift matches the known polyhydride
complex with the general formula RuH6(PPh3)2.276–278 Presumably, the potassium ions from KHMDS
(potassium hexamethyldisilazide) abstract the chloride ions and base deprotonates H2, generating a
polyhydride compound as previously synthesized by protonation of the salt [K][RuH5(PPh3)2]278 or
reaction of Ru(η2−styrene)2(PPh3)2 with H2.276,277
RuN N
Cl
Cl
Ru
Cl
Cl
PPh3
PPh3
N
N
H
H
-4 HMDS, -4 KCl
4 KHMDS, H2 (xs)
THF, 65 oC
+
Ru0
H
HRuxHy(PPh3)z
2 KHMDS-2 HMDSN N
Ru
2-
2K+
81 82
83
Scheme 5.4: Synthesis of neutral Ru complex 82 and dianionic complex 83. HMDS = hexamethyldisi-lazane.
The second product is the complex Ru0(trop2dach) (82), which was fully characterized by 1D/2D

Chapter 5. Ru Complexes With Tetradentate Amino-Olefin Ligands 74
NMR spectroscopy and single crystal X-ray diffraction (Figure 5.3). The complex resides in a distorted
square planar geometry, with one molecule of THF hydrogen bonded to an amine proton (O1· · ·H1
2.100 A). The Ru-centroid bond distances (Ru1–ct1 = 1.980(4), Ru1–ct2 = 1.973(5) A) are significantly
shorter than in 81 (Ru2–ct1 = 2.127(4), Ru2–ct2 = 2.136(6) A). In comparison with the known Ru(0)
complex 75 (Figure 5.1 and Scheme 5.1), the metrical parameters are quite similar.84
Figure 5.3: Molecular structure of 82 with ellipsoids at the 30 % probability level. Hydrogen atoms havebeen omitted for clarity. Distances (A) and angles (◦): Ru1–N1 2.131(4), Ru1–N2 2.129(3), Ru1–ct11.980(4), Ru1–ct2 1.973(5), C4–C5 1.423(7), C19–C20 1.435(7), O1· · ·H1 2.100; N1-Ru1-N2 82.0(2),ct1-Ru1-ct2 98.30(2), N1-Ru1-ct2 92.18(2), N2-Ru1-ct1 92.18(2), N1-Ru1-ct1 163.51(2), N2-Ru1-ct2162.37(2).
Reacting 82 in THF−d8 in a J. Young tube with two equivalents strong base generates the new
doubly deprotonated complex [K]2[Ru–II
(trop2dach)] (83) that has been fully characterized by 1D/2D
NMR spectroscopy. Unlike 75, which undergoes dehydrogenation of the diamine backbone to form hy-
dride complex [K][RuH(trop2dad)], we observe that the backbone remains intact, locating the tertiary
CHdach signals at 2.09 ppm (1H) and 74.14 ppm (13C). The NH signals of 82 (4.70 ppm in THF−d8)
also disappear. We propose that the solution state structure contains two intimate ion pairs, similar
to the structurally characterized 84279 and 72266 (Figure 5.4), which could possibly prevent backbone
dehydrogenation. Confirmation of our hypothesis by X-ray crystallography would be crucial and crys-
tallization attempts are currently under way. Table 5.1 summarizes the change in olefinic chemical shifts
for compounds 82, and 83, revealing the increasing electron richness as the formal charge at ruthenium
decreases.
Table 5.1: Selected NMR data for complexes 81, 82 and 83.
Parameter 82 83
CH olef (ppm)3.18 2.25
2.21 1.32
CHolef (ppm)60.17 55.85
50.09 51.47

Chapter 5. Ru Complexes With Tetradentate Amino-Olefin Ligands 75
N
K+
N
Ru2-
K+
(THF)n
(THF)n
NN
Ph Ph
Rh-
K+
(THF)3
Ru2-P
P
P
P
Li+
Li+
(THF)3
(THF)3
84 72 83
Figure 5.4: Drawings of structurally characterized 84,279 72,266 and proposed solution state structureof 83.
5.3 Conclusion
New ruthenium complexes featuring the trop2dad framework have been synthesized. Although our efforts
in reacting [Y][RuH(trop2dad)] complexes (Y = K+, NBu+4 ) with water were unsuccessful, three new
compounds with the trop2dach ligand were synthesized, two of which were structurally characterized.
The trop2dach ligand has been shown to stabilize low valent oxidation states of ruthenium, both Ru(0)
(82) and Ru(-II) (83) complexes. We continue to work on structurally characterizing the novel dianionic
complex 83 and investigate these low valent Ru compounds for applications in water splitting reactions.
5.4 Experimental
5.4.1 General Comments
All reactions were carried out in an argon atmosphere using standard Schlenk and glove box techniques
using air and moisture free solvents/reagents unless otherwise stated. RuCl2(PPh3)3280 and 7684 were
synthesized according to known literature procedures. Celite, KHMDS, db18c6, and NBu4Br were stored
in an argon-filled glove box prior to use. THF-d8 was dried in an argon-filled glove box over activated 3
A molecular sieves. THF, DME, and hexanes were dried using a Solvent Purification System (Innovative
Technology) and stored under argon. All other solvents/reagents were used as received without further
purification or handling precautions. Solution state NMR spectroscopic data were collected using a
Bruker 400 MHz spectrometer (operating at 400 MHz for 1H, 101 MHz for 13C), or a Bruker 500 MHz
spectrometer (operating at 500 MHz for 1H, 126 MHz for 13C). 1H and 13C NMR samples were referenced
to their respective residual solvent peaks249 and data was collected at 298 K unless stated otherwise.
Coupling constant J values are given in Hz.
CP/MAS solid-state NMR spectra were collected on a 16.45 T Agilent DD2 700 spectrometer (op-
erating at 283 MHz for 31P, 176 MHz for 13C at MAS rates of 15 or 18 kHz) equipped with a 1.6 mm
Double Resonance T3-HX MAS Solids Balun Probe. 31P chemical shifts were referenced to external
phosphoric acid at 0 ppm and 13C chemical shifts were externally referenced to 43.67 ppm using glycine
powder. The sample was ground to a fine powder and packed into 1.6 mm OD zirconia rotor. Spectral
data was processed using MNova 9.0.
Single-crystal X-ray diffraction data were collected at 100 K using a Bruker SMART APEX or a
Bruker APEX II diffractometer with Mo Kα radiation (λ = 0.71073 A). The structures were solved and

Chapter 5. Ru Complexes With Tetradentate Amino-Olefin Ligands 76
refined using ShelXL-2013147 in the Olex2 software package.281 Refinement was by full-matrix least-
squares on F 2 using all data.
5.4.2 [K(db18c6)][RuH(trop2dad)] (78).
In an argon-filled glove box, compound 76 (0.017 mmol, 13 mg) and dibenzo-18-crown-6 (0.017 mmol,
6 mg) were combined in an NMR tube. The mixture was dissolved in THF−d8 and turned deep
yellow-brown after shaking for a few seconds. The sample was then investigated by NMR and indicated
quantitative conversion to complex 78. The product was not isolated for further analysis. 1H NMR
(500 MHz; THF−d8): δ = 7.23 (d, 3J(H,H) = 7.6 Hz, 2H; CHar), 7.14 (d, 3J(H,H) = 7.6 Hz, 2H;
CHar), 6.94–6.89 (m, 8H; CHar K(db18c6)), 6.87 (m, 4H; CHar), 6.77 (m, 2H; CHar), 6.72 (m, 2H;
CHar), 6.57 (s, 2H; CH−−CH), 6.52 (m, 2H; CHar), 6.45 (m, 2H; CHar), 5.05 (s, 2H; CHbenzyl), 3.77 (m,
8H; CH2 K(db18c6)), 3.37 (m, 8H; CH2 K(db18c6)), 3.06 (d, 3J(H,H) = 8.7 Hz, 2H; CHolef), 2.96 (d,3J(H,H) = 8.7 Hz, 2H; CHolef), -9.00 (s, 1H; RuH). 13C {1H} NMR(126 MHz; THF−d8): δ = 150.12
(Cquat), 148.70 (Cquat), 147.89 (Cquat K(db18c6)), 147.47 (Cquat), 145.20 (Cquat), 127.28 (Car), 126.57
(Car), 126.18 (Car), 125.91 (Car), 125.77 (CH−−CH), 125.24, (Car), 121.71 (Car K(db18c6)), 121.47 (Car),
119.91 (Car), 112.15 (Car K(db18c6)), 78.41 (CHbenzyl), 69.29 CH2 K(db18c6)), 67.96 CH2 K(db18c6)),
64.75 (Colef), 53.83 (Colef).
5.4.3 [Bu4N][RuH(trop2dad)] (79).
Compound 76 (0.026 mmol, 20 mg), NBu4Br (0.026 mmol, 9 mg), and a Teflon coated stir bar were
combined in an argon-filled glove box. About 3 mL THF was added and the solution was stirred for 3
hours at room temperature. The solution was filtered through a pad of Celite and dried. The sample was
then investigated by 1H NMR (THF−d8) and indicated clean conversion to complex 79. The product
was not isolated for further analysis. 1H NMR (200 MHz; THF−d8): δ = 7.22 (m, 2H; CHar), 7.13 (m,
2H; CHar), 6.94–6.73 (m, 8H; CHar), 6.57 (m, 4H; CHar), 6.51 (s, 2H; CH−−CH), 5.05 (s, 2H; CHbenzyl),
2.63–2.51 (m, 12H; CHolef + CH2 TBAB), 1.30 (m, 8H; CH2 TBAB), 1.12 (m, 8H; CH2 TBAB), 0.97
(t, 3J(H,H) = 7.1 Hz, 12H; CH3 TBAB), -9.43 (s, 1H; RuH).
5.4.4 (±)-trans-N ,N-bis(5H-dibenzo[a,d]cyclohepten-5-yl)-1,2- diaminocyclo-
hexane (trop2dach, 80).
The compound was synthesized using virtually the same procedure as for synthesizing the enantiopure
(R,R)-trop2dach ligand,260 however instead the free diamine (±)-trans-1,2-Diaminocyclohexane was
used.
5.4.5 Synthesis of Ru2Cl4(trop2dach)(PPh3)2 (81).
Compound 80 (0.30 mmol, 150 mg), RuCl2(PPh3)3 (0.61 mmol, 581 mg), and a Teflon coated stir bar
were added to a Schlenk flask and placed under an inert atmosphere. About 20 mL DME was added
and the solution was refluxed for 12 h. In air, the solution was filtered through a glass frit and the
solid was washed with diethyl ether (10 mL) and dried under high vacuum to afford a pale pink solid,
81 (363 mg, 88%). Complex 81 is insoluble in halogenated, hydrocarbon, and ethereal solvents. X-ray
quality crystals were obtained by reacting (S,S )-80260 with RuCl2(PPh3)2 in DME in an NMR tube

Chapter 5. Ru Complexes With Tetradentate Amino-Olefin Ligands 77
under argon without stirring at 60 ◦C, after which pink needle-shaped crystals deposited on the side of
the NMR tube. EA: Found: C 62.97, H 3.33, N 2.42. Calc. for C72H64Cl4N2P2Ru2: C 63.44, H 4.73, N
2.05%. Solid-state CP/MAS 13C NMR (MAS rate = 18 kHz): δ = 141.43–127.86, 92.36, 80.33, 77.68,
67.55, 63.67, 61.91. Solid-state CP/MAS 31P{1H} NMR (MAS rate = 18 kHz): δ = 53.84 (m). Mp:
275–277 ◦C (dec).
5.4.6 Ru0(trop2dach) (82).
Complex 81 (0.22 mmol, 300 mg), KHMDS (0.88 mmol, 176 mg), and a Teflon coated stir bar were added
to a 100 mL Schlenk tube with a one-way Teflon vacuum valve and placed under an inert atmosphere.
This mixture was suspended in 15 mL THF and the argon atmosphere was replaced with H2 (about
1.5 bar) by one freeze-pump-thaw cycle. The tube was sealed and heated at 65 ◦C for 2 hours, after
which the suspension became a deep brown-orange colour. The solution was taken into an argon-filled
glove box, filtered through a pad of Celite, and dried. The brown solid was suspended in 50 mL hexane
and vigorously stirred overnight. The brown solid was isolated on a glass frit, and the outgoing orange
filtrate was discarded. The solid was washed with more hexane (about 30 mL) until the outgoing filtrate
was colourless and dried under vacuum. The resulting brown solid (120 mg) is predominantly complex
82 with varying amounts of minor RuxHy(PPh3)z impurities, depending on the reaction batch. Single
crystals suitable for X-ray diffraction were grown by slow evaporation from a concentrated THF solution.
Due to the difficulties in completely separating 82 from the RuxHy(PPh3)z impurities, the product was
insufficiently pure for elemental analysis. 1H NMR (500 MHz; THF−d8): δ = 7.44 (m, 2H; CHar), 7.35
(m, 2H; CHar), 7.06 (m, 2H; CHar), 7.02 (m, 2H; CHar), 6.93 (m, 4H; CHar), 6.85 (m, 4H; CHar), 4.83 (s,
2H; CHbenzyl), 4.70 (d, 3J(H,H) = 11.2 Hz, 2H; NH), 2.59 (d, 3J(H,H) = 8.0 Hz, 2H; CHolef), 2.51 (m,
2H; CH2), 1.99 (m, 2H; CHdach), 1.69 (m, 2H; CH2), 1.60 (d, 3J(H,H) = 8.0 Hz, 2H; CHolef), 1.10-1.00
(m, 4H; CH2). 1H NMR (500 MHz; C6D6): δ = 7.83 (m, 2H; CHar), 7.64 (m, 2H; CHar), 7.21 (m, 2H;
CHar), 7.04-6.83 (m, 8H; CHar), 6.72 (m, 2H; CHar), 4.46 (s, 2H; CHbenzyl), 3.18 (d, 3J(H,H) = 8.0
Hz, 2H; CHolef), 2.49 (d, 3J(H,H) = 11.9 Hz, 2H; NH), 2.21 (d, 3J(H,H) = 8.0 Hz, 2H; CHolef), 1.74
(m, 2H; CH2), 1.24 (m, 2H; CHdach), 1.13 (m, 2H; CH2), 0.42 (m, 2H; CH2), -0.55 (m, 2H; CH2). 13C
{1H} NMR(126 MHz; THF−d8): δ = 150.64 (Cquat), 148.49 (Cquat), 136.85 (Cquat), 136.62 (Cquat),
130.05 (Car), 128.97 (Car), 128.63 (Car), 128.27 (Car), 128.22 (Car), 127.18 (Car), 122.84 (Car), 121.70
(Car), 65.26 (CHbenzyl), 61.92 (CHdach), 60.17 (Colef), 50.09 (Colef), 30.24 (CH2), 25.63 (CH2). 13C {1H}NMR(126 MHz; C6D6): δ = 150.27 (Cquat), 148.04 (Cquat), 135.70 (Cquat), 135.38 (Cquat), 129.94 (Car),
129.69 (Car), 129.16 (Car), 129.04 (Car), 128.47 (Car), 127.79 (Car), 123.13 (Car), 121.91 (Car), 65.48
(CHbenzyl), 60.81 CHdach), 60.40 (Colef), 50.27 (Colef), 29.74 (CH2), 24.36 (CH2).
5.4.7 [K]2[Ru–II(trop2dach)] (83).
In an argon-filled glove box, compound 82 (0.017 mmol, 10 mg) and KHMDS (0.034 mmol, 6 mg) were
combined in an NMR tube. The mixture was dissolved in THF−d8 and then investigated by NMR
spectroscopy. The product was not isolated for further analysis. 1H NMR (400 MHz; THF−d8): δ =
7.5-6.5 (m, Car), 4.83 (s, 2H; CHbenzyl), 2.49 (m, 2H; CH2), 2.25 (d, 3J(H,H) = 8.0 Hz, 2H; CHolef), 2.09
(m, 2H; CHdach), 1.59 (m, 2H; CH2), 1.32 (d, 3J(H,H) = 8.0 Hz, 2H; CHolef), 1.12 (m, 2H; CH2), 0.65
(m, 2H; CH2). 13C {1H} NMR(400 MHz; THF−d8): δ = 152.74 (Cquat), 152.32 (Cquat), 148.17 (Cquat),
144.47 (Cquat), 127.40 (Car), 127.29 (Car), 126.35 (Car), 126.02 (Car), 125.94 (Car), 123.68 (Car), 119.47

Chapter 5. Ru Complexes With Tetradentate Amino-Olefin Ligands 78
(Car), 118.86 (Car), 74.14 CHdach), 70.32 (CHbenzyl), 55.85 (Colef), 51.47 (Colef), 32.35 (CH2), 27.07
(CH2).

Chapter 5. Ru Complexes With Tetradentate Amino-Olefin Ligands 79
Table 5.2: Selected X-Ray crystal structure and refinement data for complexes 81 and 82.
Compound 81 82
Empirical formula C72H64Cl4N2P2Ru2 C40H42N2ORu
Formula mass 1363.13 667.82
Temp. (K) 100 100
Crystal system Triclinic Monoclinic
Space group P1 P21/c
a (A) 10.3736(3) 9.121(2)
b (A) 13.2432(4) 15.475(3)
c (A) 13.7718(4) 21.720(5)
α (◦) 99.178(2) 90
β (◦) 107.6360(10) 92.615(5)
γ (◦) 97.263(2) 90
Volume (A3) 1748.98(9) 3062.6(12)
Z 1 4
Calc. density (g/cm3) 1.294 1.448
Absorption coeff.(mm−1)
0.670 0.548
F(000) 696 1392
Crystal size (mm3)0.230 × 0.130 ×0.060
0.250 × 0.040 ×0.020
2Θ range collected (◦) 3.17–63.852 3.232–52.746
Reflections collected 34265 18434
Independent reflectionsR(int)
20631 (0.0396) 6258 (0.1228)
Completeness toθ = 26.00◦ (%)
100.0 100.0
Absorption correctionSemi-empirical fromequivalents
Semi-empirical fromequivalents
Max. and min.transmission
0.9583, 0.8097 0.8623, 0.7891
Data / restraints /parameters
20631/3/771 6258/0/397
Goodness-of-fit on F2 0.983 0.985
R1(I > 2σ(I))a 0.0429 0.0558
wR2 (all data)b 0.0981 0.1213
a R1 =Σ(Fo − Fc)
Σ(Fo)
b wR2 =
√Σ[w(F 2
o − F 2c )2]
Σ[w(F 2o )2]

Chapter 6
First Generation Iron PNNP [6.5.6]
Complexes for the Transfer
Hydrogenation of Ketones:
Mechanistic Insights from DFT
Calculations
Abstract
Iron complex trans-[Fe(CO)(MeCN)(PPh2C6H4CH−−NCH2−)2−κ4P,N,N, P ][BF4]2 (85),
when reacted with KOiPr in benzene, produced the unusual complex
[Fe(CO)(PPh2C6H4CH−−NCH2CH2NHCHC6H4PPh2)−κ5P,N,C,N, P ][BF4] (86), which
has been characterized by spectroscopy and by single-crystal X-ray diffraction. The C–N
bond length in this complex indicates that it is best viewed as an iron(II) ligand-folded
ferraaziridine-κ2C,N complex instead of an iron(0) η2-iminium complex. Density functional
theory (DFT) calculations have been employed on simplified structural models to support a
mechanism of formation of this complex via the transfer of a hydride from an iron isopropoxide
complex to an imine carbon on the ligand to produce an iron imino-amido complex followed by
liberation of acetone. Two energetically similar pathways have been proposed in which depro-
tonation of a methylene carbon on the PNNP ligand
by strong base produces an experimentally observed ferraaziridinido
complex Fe(CO)(PPh2C6H4CH−−NCH2CH2NCHC6H4PPh2)−κ5P,N,C,N, P ) (87) or
a square-pyramidal Fe(0) complex. Protonation of 87 by free isopropyl alcohol produces
the structurally characterized ferraaziridine complex 86. NMR and IR spectroscopy data
show that during the transfer hydrogenation of acetophenone catalyzed by 85 in basic iso-
propyl alcohol, free ligand is observed along with one major iron-containing species identified
as 87. On the basis of our calculations of relative free energies and a CO scale factor, we
80

Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 81
predict that ferraaziridine 86 is easily deprotonated to form the electron-rich iron complex
87 and a square-pyramidal Fe(0) complex, which are responsible for the two observed CO
stretches below 1900 cm−1 in catalytic mixtures. Mass balance studies indicate that the
catalytically active species is not observable by NMR. Although 86 and 87 are poor trans-
fer hydrogenation catalysts, we present experimental and theoretical evidence that ligand
folding/distortion is feasible.
6.1 Introduction
The hydrogenation of carbonyl compounds to alcohols is a vital process in the pharmaceutical, fine
chemical, and perfume industries.16,282–288 The alcohols may be prepared via direct hydrogenation,
hydrosilylation, or transfer hydrogenation (TH) reactions, usually utilizing catalysts based on expensive
and sometimes toxic platinum metals.289 The first studies into the asymmetric iron-catalyzed TH of
ketones were carried out by Vancheesan et al.290,291 and Gao et al.,292 and we recently reported highly
efficient iron catalysts for the asymmetric TH of ketones to the corresponding alcohols using isopropyl
alcohol as a hydrogen source.293–295 As a common structural feature in these catalysts, an iron(II)
atom is coordinated by a tetradentate diimine ligand, a carbonyl or isonitrile, and an acetonitrile or
bromide ligand, as depicted in Figure 6.1. The catalysts work at room temperature with excellent
turnover frequencies (TOFs) and enantioselectivities (TOF up to 28 000 h −1, ee up to 99 %). While
the mechanism of ruthenium-catalyzed TH has been extensively studied,14,296 little was known about
the mechanism of iron-catalyzed TH prior to the publication of this work.297 A detailed knowledge
of the mechanism will be essential to optimize the design of our systems to make more efficient and
enantioselective iron catalysts, to make them more competitive with the best platinum metal based
systems. Hence, this is currently under extensive study in our laboratory.
FeN
PPh2
2+
N
PPh2
MeCN
L
R = H, Ph; R---R = -(CH2)4-
L = CO, CNtBu
R R
5
6 6
* *
Figure 6.1: Iron precatalysts for the TH of ketones with [6.5.6] metallacycles prepared by our group.
In this chapter, we discuss the species observable by NMR and IR spectroscopy during the TH of ace-
tophenone (AcPh) to 1-phenylethanol (PE) using the iron complex
trans-[Fe(CO)(MeCN)(PPh2C6H4CH−−NCH2−)2−κ4P,N,N, P ][BF4]2 (85)293 as the precatalyst, iso-
propyl alcohol (iPrOH) as the hydrogen source, and potassium tert-butoxide (KOtBu) as the base,
generating acetone (AcMe) as a byproduct (Scheme 6.1). To gain further insight into the species formed
during catalysis, we investigated the reaction of precatalyst 85 with stoichiometric amounts of base and
performed density functional theory (DFT) calculations on simplified models of the reactive species to
further support our results.

Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 82
Ph
O HO H
Excess KOtBuiPrOH
O
Ph
HO H+ +
26 oCiPrOH AcMeAcPh PE
*
FeN
PPh2
2+
N
PPh2
MeCN
CO
2BF4-
85
Scheme 6.1: Reaction Scheme for TH of Ketones Using 85 As a Precatalyst in the Presence of ExcessBase.a
a8 equivalents base with respect to [Fe].
6.2 Results and Discussion
6.2.1 Summary of Catalysis Investigations by Spectroscopic Methods
Scheme 6.2 summarizes the observed reactivity with Fe PNNP [6.5.6] complexes.∗ When 85 is dissolved
in a solution of acetophenone in basic iPrOH, the solution immediately becomes clear and dark brown.
The activated catalyst mixture is highly air sensitive, and exposure to air results in an immediate
colour change to pale yellow, rendering it inactive for TH. When the reaction mixture of a typical TH
experiment was analyzed via NMR and IR techniques by using 85 as a precatalyst with relatively high
catalyst loadings (Scheme 6.2, bottom arrow), a mixture of compounds is observed by 31P NMR, with
the major signals belonging to the 87, possessing two doublets at 84.1 and 68.7 ppm (2JPP = 29 Hz).
When synthesized independently via 86, compound 87 was found to be a poorly active catalyst.
N
Fe
H
CO
N
PPh2
PPh2
KOtBuNH
Fe
H
CO
N
PPh2
PPh2
BF4-
Fe
N
PPh2
CO
N
PPh2
MeCN
NaOiPr
excess KOtBu, iPrOH, evaluate by 31P NMR
active catalyst in basic isopropanol
poor catalytic activity,structurally characterized
poor catalytic activity,
2+
2BF4-
+
detected by 31P NMR
iPrOHC6H6 or iPrOH
2JPP = 43 Hz (d)2JPP = 29 Hz (d)
85 86 87
Scheme 6.2: Summary of the observed reactivity of Fe PNNP [6.5.6] complexes.
The IR spectrum of the crude catalytic mixture revealed weak, broad absorptions for CO vibrations
at 1960 and 1946 cm−1, and intense broad absorptions from 1835 to 1890 cm−1, with distinct maxima
at 1870 and 1846 cm−1, indicating that a carbonyl ligand is still coordinated to a metal center during
catalysis, possibly to the active species. The broadness of the absorption below 1900 cm−1 is likely due
∗The synthetic work was performed by the other authors of this manuscript.297

Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 83
to the presence of multiple, similar species in solution, with two major species giving rise to peaks at 1870
and 1846 cm−1. While 1960 and 1946 cm−1 are typical values for a monocarbonyl iron(II) compound,
signals below 1900 cm−1 are usually indicative of iron(0) complexes or bridging carbonyl ligands.298,299
Assignment of these two peaks was done in conjunction with DFT calculations (vide infra).
Furthermore, mass balance 31P {1H} NMR experiments were performed on catalytic mixtures using
a PPh3−−O internal standard, which concluded that about 25 % of the phosphorus signals observed
belonged to a known species that is poorly active for TH.297 This is significant, because it shows that a
maximum of 75 % of the iron is either catalytically active and NMR inactive or is catalytically active
and found in extremely low concentrations.∗
With the goal of isolating the intermediates observed by 31P NMR and IR spectroscopy, we inves-
tigated the reaction of precatalyst 85 with 2 equiv of sodium isopropoxide in neat benzene without AcPh
present. Surprisingly, we did not observe the formation of an octahedral
iron alkoxide complex; instead, the cationic ferraaziridine complex
[Fe(CO)(PPh2C6H4CH−−NCH2CH2NHCHC6H4PPh2)−κ5P,N,C,N, P ][BF4](86) was isolated in 65 %
yield (Scheme 6.2). Since we were able to measure clean and well-resolved NMR spectra without contact-
shifted signals, we expect that the electronic state of the iron is low-spin iron(II) and the complex is
diamagnetic (this is supported by unrestricted DFT calculations on a related Fe(II) complex, which will
be discussed later). The IR spectrum shows a characteristic CO vibration band at 1940 cm−1, lower
than that for the starting material 85 (2002 cm−1), as expected for a reduction in positive charge and
increase in π back-bonding from Fe to CO on going from 85 to 86.
The single-crystal X-ray diffraction structure (Figure 6.2) revealed a distorted geometry where the
C(10) carbon of the ferraaziridine moiety is bound approximately trans to the carbonyl ligand, with
a C(10)–Fe–C(17) angle of 158.3(2)◦. The complex appears to adopt a distorted-octahedral geometry,
with a strained C(10)–Fe–N(2) angle of 42.0(1)◦ and obtuse P(1)–Fe–C(10) and C(17)–Fe–N(2) angles
of 106.1(1) and 116.6(1)◦, respectively. The complex is best viewed as an iron(II) κ2-ferraaziridine
complex instead of an iron(0) η2-iminium complex, given the N(2)–C(10) bond distance of 1.434(5)
A, characteristic for an N–C single bond. The bond distances from iron to the imine, amine, and alkyl
functions are 1.980(3) (Fe–N(1)), 1.988(3) (Fe–N(2)), and 2.012(2) A (FeC(10)), respectively (Table 6.1).
The metal-carbonyl distance Fe–C(17) (1.781(4) A) is typical for an iron-carbonyl bond and is similar to
that of complex 85.293 We have also calculated the ground-state structures of 85 and 86 in the gas phase
using DFT (simplified structural models, phenyls replaced with hydrogens). Both structures are in good
agreement with those of 85 (see the Supporting Information)297 and 86 (Figure 6.2, Table 6.1). The
gas-phase metrical parameters for 85 and 86 are nearly identical with those calculated with solvation
effects (vide infra).
Complex 86 displays certain spectral features similar to those of the TH reaction mixture (Scheme 6.2,
bottom arrow). It has an IR absorption at 1940 cm−1 and doublets in the 31P {1H} NMR spectrum
at 84 and 69 ppm. However, the signal of the latter spectrum has a 2JPP coupling constant of 43 Hz
while the doublets of the TH solution have 2JPP = 29 Hz, thus indicating that the complexes are not
the same. Complex 86 does not catalyze the TH of acetophenone in neat iPrOH without base at 26 ◦C.
On the basis of the coupling constant mismatch between 86 and the TH mixture, it is not the species
present during TH.
∗We reported elsewhere that the true identity of the active species is 4 nm iron(0) nanoparticles coated with the chiralligand.300
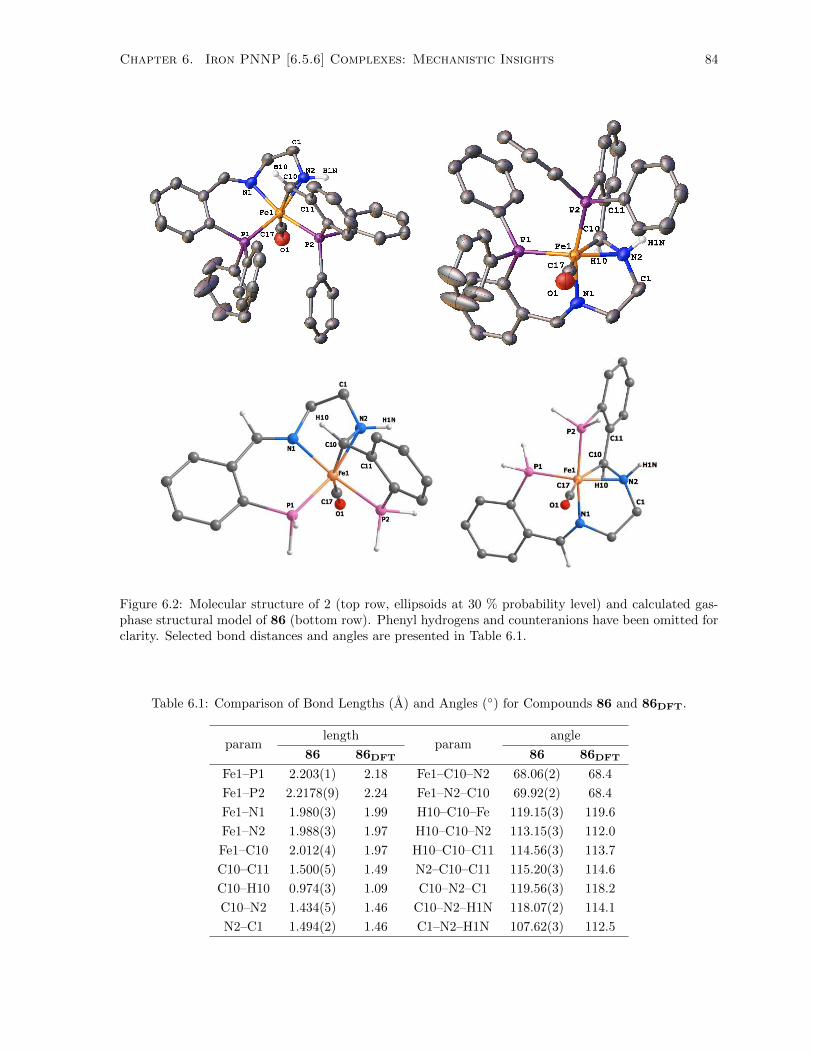
Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 84
Figure 6.2: Molecular structure of 2 (top row, ellipsoids at 30 % probability level) and calculated gas-phase structural model of 86 (bottom row). Phenyl hydrogens and counteranions have been omitted forclarity. Selected bond distances and angles are presented in Table 6.1.
Table 6.1: Comparison of Bond Lengths (A) and Angles (◦) for Compounds 86 and 86DFT.
paramlength
paramangle
86 86DFT 86 86DFT
Fe1–P1 2.203(1) 2.18 Fe1–C10–N2 68.06(2) 68.4
Fe1–P2 2.2178(9) 2.24 Fe1–N2–C10 69.92(2) 68.4
Fe1–N1 1.980(3) 1.99 H10–C10–Fe 119.15(3) 119.6
Fe1–N2 1.988(3) 1.97 H10–C10–N2 113.15(3) 112.0
Fe1–C10 2.012(4) 1.97 H10–C10–C11 114.56(3) 113.7
C10–C11 1.500(5) 1.49 N2–C10–C11 115.20(3) 114.6
C10–H10 0.974(3) 1.09 C10–N2–C1 119.56(3) 118.2
C10–N2 1.434(5) 1.46 C10–N2–H1N 118.07(2) 114.1
N2–C1 1.494(2) 1.46 C1–N2–H1N 107.62(3) 112.5

Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 85
An isolated, pure sample of 86 was reacted with a stoichiometric amount
of KOtBu in iPrOH to afford the postulated ferraaziridinido complex
Fe(CO)(PPh2C6H4CH−−NCH2CH2NCHC6H4PPh2)−κ5P,N,C,N, P ) (87; Scheme 6.2), which only dif-
fers from 86 in that the ferraaziridine nitrogen atom is deprotonated. This structure is proposed on
the basis of NMR spectra, IR spectra, and DFT calculations (vide infra). Complex 87 could not be
isolated as a pure product, and therefore analysis was done on the crude solution after removal of excess
base. The 31P {1H} NMR spectrum of the resulting solution shows two doublets at 84.1 and 68.7 ppm
(2JPP = 29 Hz) (along with singlets at 12 and 13.4 ppm for various isomers of the free ligand), which
now matches the chemical shift and coupling constant of the phosphorus-containing complex in the TH
mixture. The IR spectrum shows absorptions associated with carbonyl ligands at 1862 and 1870 cm−1,
as well as broad peaks at 1934 and 1957 cm−1. From the similarities of the 31P {1H} NMR and IR
spectra between this reaction mixture and the TH solutions, we reason that 87 is present during TH.
As mentioned previously, the crude mixture of 87 in iPrOH is only minimally catalytically active (0.5
% conversion of 3.85 mmol of acetophenone to 1-phenethanol at 26 ◦C in 1 h, 40 % in 24 h); therefore,
it is also not the catalytically active species present during TH.
This chemically “non-innocent” behavior of the ligand is reminiscent of the PNP systems designed
by Milstein24 and Schneider42 and PNNP complexes developed in our group,46,301,302 whereby the
ligand exhibits reversible protonation-deprotonation behavior. However, this system is flexible and
preferentially adopts a folded/distorted geometry.
6.2.2 Calculated Mechanism for Formation of Complexes 86, 87, and Fe(0)
Species 91
We have used DFT calculations to rationalize the formation of the experimentally observed complexes
86 and 87 and also propose the energetically favorable reduction to a five-coordinate Fe(0) species via
two energetically similar pathways (Scheme 6.3). In order to reduce computational cost, the phenyl
substituents on phosphorus have been truncated to hydrogen atoms in all calculated structures. In
addition, solvent effects have been taken into account and all calculations have been performed usingiPrOH as the solvent (see Computational Details).
Starting with the dicationic TH precatalyst 85, the labile acetonitrile ligand is replaced by an in-
coming isopropoxide anion, which is exergonic by 16.3 kcal/mol to generate 88 (Figure 6.4). Next,
hydride transfer from the tertiary carbon on the alkoxido ligand reduces the imine moiety, generating
the cationic Fe(II) imino-amido species 89AcMe, which is endergonic by 7.3 kcal/mol and has a transi-
tion state barrier of 20.1 kcal/mol to overcome (TS88,89AcMe; Figure 6.3 and Figure 6.4). Dissociation
of weakly coordinating acetone from 89AcMe produces 89, a five-coordinate imino-amido species, which
reacts with another one equivalent of alkoxide, yielding the neutral octahedral complex 90, which is 24.9
kcal/mol lower in energy than 85.
The exergonic process of going from 89AcMe to 90 is followed by two energetically similar pathways:
simultaneous ligand folding/distortion and deprotonation to generate a neutral Fe(II) ferraaziridinido
complex (87iPrOH) with a barrier of 22.2 kcal/mol (TS90,87iPrOH); alternatively, deprotonation without
generation of an ironalkyl bond reduces the metal center to form a distorted-square-pyramidal Fe(0)
complex (91iPrOH) with a calculated barrier of 21.9 kcal/mol (TS90,91iPrOH). The activation energy for
these transitions differs by only 0.3 kcal/mol, and the free energies of products 87iPrOH and 91iPrOH are
lower than that of 90 by 8.6 and 5.8 kcal/mol, respectively. The transition-state geometries TS90,87iPrOH
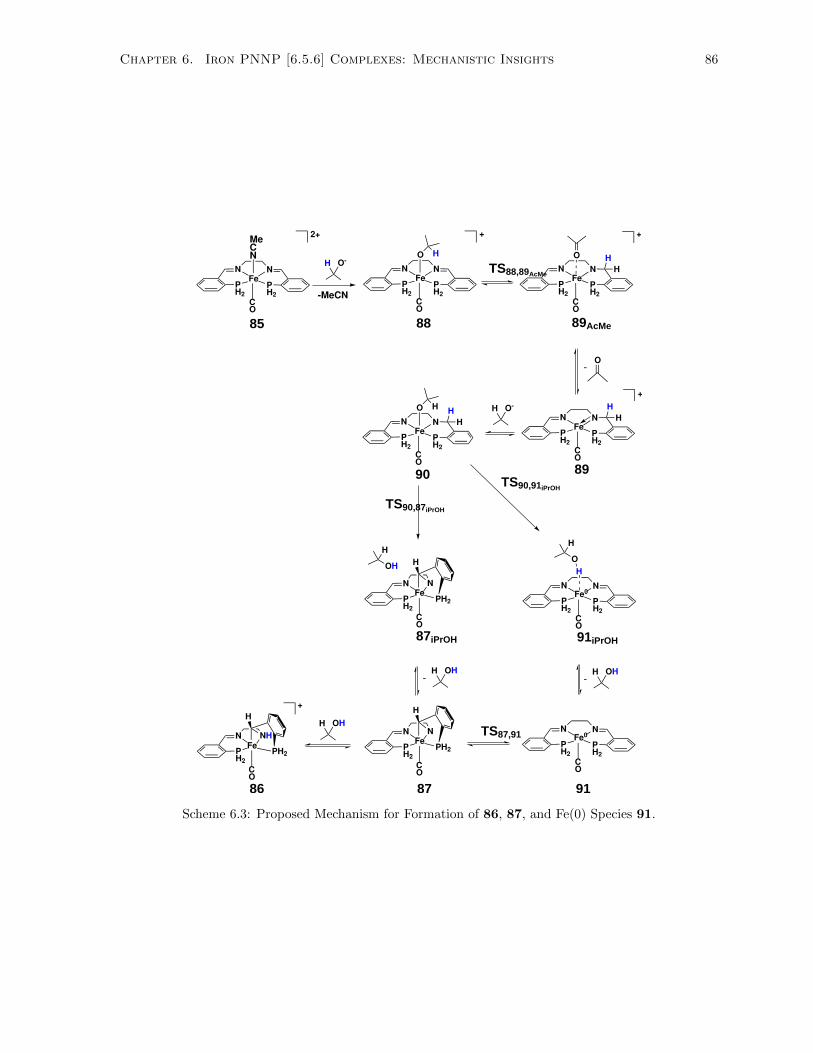
Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 86
FeN
PH2
CO
N
PH2
O H
+
NFe
H
CO
N
PH2
PH2
FeN
PH2
CO
N
PH2
H
H
+
O
+
FeN
PH2
CO
N
PH2
H
H
FeN
PH2
CO
N
PH2
O HH
H
Fe0N
PH2
CO
N
PH2
O
H
HOH
H
NFe
H
CO
N
PH2
PH2
Fe0N
PH2
CO
N
PH2
FeNH
PH2
H
CO
N
PH2
+
OHH-
OHH-
OHH
FeN
PH2
CO
N
PH2
MeCN
-MeCN
2+
O-
O-H
O-H
85 88
TS88,89AcMe
89AcMe
90 89
TS90,87iPrOH
TS90,91iPrOH
87iPrOH 91iPrOH
86 87
TS87,91
91
Scheme 6.3: Proposed Mechanism for Formation of 86, 87, and Fe(0) Species 91.

Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 87
and TS90,91iPrOHare shown in Figure 6.3. The major difference between these transition states is the
iron-alkoxide distance and the degree of ligand distortion during deprotonation. For TS90,87iPrOH, the
alkoxide is further away from iron (Fe· · ·O = 3.05 A) than in TS90,91iPrOH, where the Fe–O distance is
close (Fe–O = 2.05 A) and the alkoxide remains trans to the carbonyl (O–Fe–C = 175.8◦). Tetradentate
PNNP ligands on ruthenium303 and iron304 are known to fold to produce cis-octahedral complexes.
The Fe(0) complex 91, with or without isopropyl alcohol, is proposed on the basis of the low carbonyl
stretches in the IR spectrum of the catalytic mixture (vide supra), which will be discussed in the following
section.
Figure 6.3: Calculated geometries and selected bond lengths (A, left to right, top to bottom): 88,TS88,89AcMe
(−726i cm−1), TS90,87iPrOH(−1049i cm−1), TS90,91iPrOH
(−1055i cm−1), TS87,91 (−319icm−1), and 91. Phenyl hydrogens have been omitted for clarity.
Dissociation of iPrOH from the deprotonated ferraziridine complex 87iPrOH leads to 87 + acetone
+ iPrOH (Scheme 6.3), which has the lowest relative free energy of all the compounds calculated (−37.6
kcal/mol relative to 85 + 2 iPrO–) and has been experimentally observed in the presence of more than

Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 88
2 equivalents of base (Scheme 6.2). The metrical parameters of 87 do not differ much from those of
the calculated 86, but the electronics change dramatically. The relative carbonyl stretch of 86 is 83
cm−1 higher than that of 87 in the gas phase, which suggests that iron is very electron rich when the
ferraaziridine nitrogen is deprotonated (the difference is 86 cm−1 when solvation effects are incorporated).
We rationalize that 86 is formed by protonation of the tetrahedral nitrogen atom by free iPrOH, given
a modest energy difference of 4.8 kcal/mol between 87 and 86.
-30
-20
-10
0
10
0.0
Rel
ativ
e G
o (k
cal/m
ol)
+ 2iPrO-
-40
-16.3
3.8
-9.0
-25.7 -24.9
-3.0
-2.7
-30.7
-33.5
-37.6
-23.0
-32.8
-39.4
+ iPrO-
+ iPrO-
+ iPrO-
+ AcMe+ AcMe
+ AcMe
+ AcMe
+ iPrOH+ AcMe
+ iPrOH+ AcMe
+ iPrO-
+ AcMe
85
88
TS88,89AcMe
89AcMe
90 89
TS90,87iPrOH
TS90,91iPrOH
87iPrOH
91iPrOH
86
87
TS87,91
91
Figure 6.4: Free energy profile for the compounds shown in Scheme 6.3 with energies of free iPrO–,iPrOH, and AcMe used where appropriate. Energies are all calculated relative to 85 and 2 equivalentsiPrO– in iPrOH solvent continuum.
Reduction to the square-pyramidal Fe(0) complex 91 can also be accomplished by ligand “unfolding”
of 87 with a barrier of 14.6 kcal/mol (TS87,91, Figure 6.3 and Figure 6.4). The relative free energy
of this electron-rich Fe(0) complex is similar to that of 87 (1.8 kcal/mol lower) and can also form by
simultaneous deprotonation/reduction of 90 (vide supra).
All calculations were performed on low-spin iron complexes in the singlet state (S = 0). To account
for the possibility of odd-electron intermediates, we also optimized an intermediate spin analogue of
88 (S = 1, two unpaired electrons; see the Supporting Information).297 Intermediate-spin 88 retains
a distorted-octahedral geometry but is 10.6 kcal/mol higher in energy than low-spin 88. Attempts at
calculating a high-spin analogue of 88 resulted in immediate CO loss from the coordination sphere. Since
the metal-carbonyl bond is clearly intact in compound 86 and its synthesis is reproducible in open and
closed systems, we do not think CO dissociation plays a role in its formation. However, decomposition
of the metal complex cannot be ruled out if iron adopts a high-spin electron configuration.
6.2.3 Using a CO Scale Factor to Support the Proposed Structures of 87
and 91
Observed metal-carbonyl stretches in our Fe-PNNP infrared spectra are easily identifiable and sen-
sitive to changes in the overall electronic structure.305,306 Although there have been studies report-
ing DFT scale factors for metal-carbonyl complexes,307,308 they do not report scale factors using the

Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 89
mPW1PW91 density functional. Furthermore, the theoretical models of 85 and 86 are not the same
as their experimental analogues. A new CO scale factor was determined using simplified models
of 85 and 86, along with simplified models of two other fully characterized iron(II) PNNP com-
plexes, trans-[Fe(CO)(NCMe)(PPh2CH2CHN((R, R)−C(Ph)HC(Ph)H)NCHCH2PPh2)][BPh4]2 (92)309
and trans-[Fe(CO)(NCMe)(PPh2C6H4CHN((R, R)−C6H10)NCHC6H4PPh2)][BF4]2 (93)295 (see the Sup-
porting Information).297 As for all other complexes, phenyl groups were truncated to hydrogen atoms
and anions were removed to reduce computational cost. The experimental carbonyl stretches were then
compared with the calculated carbonyl stretches, which resulted in a calculated306 CO scale factor of
0.917± 0.002.
The square-pyramidal complex 91 has a calculated carbonyl stretching frequency of 2020 cm−1,
which is only 8 cm−1 lower than the calculated CO stretching frequency of complex 87 (2028 cm−1),
indicating electron-rich iron centers in both cases. Applying the CO scale factor to 87 and 91 results
in predicted experimental CO stretches of 1860 ± 4 and 1852 ± 4 cm−1, respectively (Figure 6.5). In
comparison with the observed transfer hydrogenation CO stretches of 1870 and 1862 cm−1, we predict
that complexes 87 and 91 are responsible for the two experimentally observed carbonyl stretches below
1900 cm−1 in TH mixtures.
NFe
H
CO
N
PH2
PH2
Fe0N
PH2
CO
N
PH2
νCO(exp) = 1870 cm-1
νCO(calc) = 2028 cm-1
νCO(scaled) = 1860 cm-1
1862 cm-1
2020 cm-1
1852 cm-1
87 91
Figure 6.5: Experimental, calculated, and scaled carbonyl stretches for proposed complexes 87 and 91.
6.3 Conclusion
We investigated the mechanism of TH of AcPh to PE utilizing
trans-[Fe(CO)(MeCN)(PPh2C6H4CH−−NCH2−)2−κ4P,N,N, P ][BF4]2 (85) as a precatalyst and basiciPrOH as a hydrogen source. Spectroscopic and computational investigations were performed to deter-
mine the structures of any intermediates involved in catalysis. NMR data showed that, during catalysis,
free ligand is observed along with an iron-containing species thought to be the neutral ferraaziridinido
complex Fe(CO)(PPh2C6H4CH−−NCH2CH2NCHC6H4PPh2)−κ5P,N,C,N, P ) (87). This is suggested
on the basis of the synthesis, characterization, and reactivity of the unusual ligand-folded ferraaziri-
dine complex [Fe(CO)(PPh2C6H4CH−−NCH2CH2NHCHC6H4PPh2)−κ5P,N,C,N, P ][BF4] (86). Both
of these complexes are mediocre catalysts in comparison with precatalyst 85. Using DFT, we proposed
thermodynamically favorable mechanisms for the formation of 86 and 87 that involve simultaneous
deprotonation/folding of the ligand arm or deprotonation without formation of an iron-alkyl bond. IR
spectra showed that during catalysis, two electron-rich iron species form with CO stretches below 1900
cm−1. On the basis of a calculated CO scale factor and relative free energies, we predict that 86 is easily

Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 90
deprotonated in the presence of excess base to form electron-rich iron complexes 87 and the square-
pyramidal Fe(0) complex 91, which are responsible for the two observed CO stretches below 1900 cm−1
in catalytic mixtures. Elsewhere, evidence is provided that the actual catalyst in the system is 4 nm
Fe(0) nanoparticles coated with chiral ligand.300
6.4 Experimental
6.4.1 Computational Details
All calculations were performed using Gaussian09251 or Gaussian03.310 The mPW1PW91 density func-
tional was used for all calculations.311,312 Iron was treated with the SDD basis set to include relativistic
effective core potentials.313 Atoms C, H, N, O, and P were treated with the 6-31++G(d,p) basis set,
which includes diffuse and polarization functions.314,315 Unrestricted open-shell calculations of 88 showed
no evidence of significant spin contamination; therefore, only closed-shell structures were considered. The
phenyl substituents on phosphorus atoms were replaced with hydrogen atoms to reduce computational
cost. Such simplifications were shown in previous computational studies on iron and ruthenium sys-
tems to have no significant effect on the core structures.294,316 Lynch and Truhlar have reported that
the mPW1PW91 functional is better for the prediction of transition states and energy barriers.317 All
structures were optimized with solvent correction (2-propanol) using the integral equation IEF-PCM
protocol.252,253 Radii and nonelectrostatic terms from the SMD solvation model were also used, which
have been reported by Truhlar and co- workers to be more accurate in calculating the solvation free
energy of bare ions such as isopropoxide.254 Ground-state structures of 85, 86, 87, 91, 92, and 93 were
performed in the gas phase for CO scale factor calculations. Optimized ground states were found to
have zero imaginary frequencies, while optimized transition states were found to have one imaginary fre-
quency. Three-dimensional visualizations of the calculated structures were generated by ChemCraft.255
Three-dimensional coordinates for all calculated compounds have been published elsewhere.297
6.4.2 General
Detailed experimental procedures and characterization were performed by other authors of this published
work.297
6.5 Supplementary Information
6.5.1 Calculated Complex 85, and Scale Factor Data

Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 91
Figure 6.6: Calculated structure of 85, gas phase.
Table 6.2: Comparison of Bond Lengths (A) and Angles (◦) for Compounds 85 and 85DFT.
paramlength
paramangle
85 85DFT 85 85DFT
Fe1–P1 2.288(1) 2.25 P1–Fe1–P2 102.98(4) 94.2
Fe1–P2 2.81(1) 2.25 P1–Fe1–N2 87.29(9) 89.6
Fe1–N1 2.020(3) 2.00 P2–Fe1–N1 86.93(9) 91.6
Fe1–N2 2.020(3) 2.01 C17–Fe1–N3 174.5(2) 179.3
Fe1–N3 1.959(3) 1.97 Fe1–N1–C10 132.6(3) 133.5
Fe1–C17 1.775(4) 1.80 Fe1–N1–C1 109.0(2) 109.3
N1–C10 1.276(5) 1.29
N1–C1 1.487(5) 1.47
N3–C18 1.143(5) 1.16
O1–C17 1.143(5) 1.14
Table 6.3: Experimental and calculated carbonyl stretches of compounds 85, 86, 92,309 and 93.295
Compound νexpCO (cm−1) νcalcCO (cm−1)
85 2002 2185
86 1940 2110
92 2001 2188
93 1999 2180

Chapter 6. Iron PNNP [6.5.6] Complexes: Mechanistic Insights 92
NH
Fe
H
CO
N
PPh2
PPh2
FeN
PPh2
CO
N
PPh2
MeCN
FeN
PPh2
CO
N
PPh2
MeCN PhPh
FeN
PPh2
CO
N
PPh2
MeCN
FeN
PH2
CO
N
PH2
MeCN
2+
FeN
PH2
CO
N
PH2
MeCN
2+
NH
Fe
H
CO
N
PH2
PH2
FeN
PH2
CO
N
PH2
MeCN
2+
2+
[BF4]2
[BF4]2
[BPh4]2
[BF4]2
85
86
92
93
Figure 6.7: Molecules 85, 86, 92,309 and 93295 and their calculated models.
λ =ΣνexpCO
ΣνcalcCO
= 0.917± 0.002
Figure 6.8: Calculation scale factor λ.306

Chapter 7
Second and Third Generation Iron
PNNP [5.5.5] Complexes: The
Calculated Mechanism of Ketone
Hydrogenation
Abstract
We have studied the mechanism of asymmetric transfer hydrogenation (ATH) and asymmet-
ric hydrogenation (AH) in silico using Density Functional Theory (DFT) with Fe(II) PNNP
bis(eneamido) and amido-eneamido complexes. The general formula for these compounds is
Fe(CO)(R2PCHCHN(S, S−CH(R′)CH(R′))NCHCHPR2) and
Fe(CO)(R2PCH2CH2N(S, S−CH(R′)CH(R′))NCHCHPR2) (R, R′ = alkyl, aryl), respec-
tively, and calculations on model complexes and the full structures have been conducted.
For the bis(eneamido) species, an initial activation period involving one equivalent of iso-
propyl alcohol reduces the bis(eneamido) ligand by a stepwise inner-sphere mechanism. This
activation step is proposed to be slow because of the relatively high barrier calculated for the
inner-sphere transfer of hydride to an imine carbon on the ligand to produce the proposed
active species, an unsymmetrical amido-eneamido complex which was later confirmed exper-
imentally to be the active species during catalysis. The ATH catalytic cycle propagates by
addition of isopropyl alcohol over Fe–amido part of the ligand to generate an FeH–NH unit.
In contrast to the activation step, a stepwise outer-sphere mechanism has been calculated to
transfer the proton and hydride in two discrete steps which are connected through a ground
state involving an NH-stabilized alkoxide ion. During ATH with the bis(eneamido) systems,
the highest calculated barrier is hydride transfer in the activation period, while the highest
barrier in the catalytic cycle involves hydride transfer to/from iron. The resting states dur-
ing catalysis are either an Fe(alkoxide)–NH or FeH–NH complex on the basis of their low
relative free energies. The high H2 splitting barrier across the Fe–amido portion of the active
amido-eneamido complex explains why it is only a moderately active AH catalyst. Calcula-
93

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 94
tions on the full structures confirms the observed catalytic behavior for ATH and AH, with
preliminary results showing that the calculated ATH activity and enantioselectivity are in
agreement with experimental values. This work leads to a deeper understanding of how this
family of highly active “green” catalysts operates under catalytic conditions.
7.1 Introduction
The asymmetric hydrogenation (AH) and asymmetric transfer hydrogenation (ATH) of prochiral ketones
and imines are valuable transformations because they allow the production of enantiopure alcohols and
amines for the use in the pharmaceutical, fragrance, food, and materials industries.16,282–288 Previously,
the predominant catalysts of these asymmetric transformations were based on rare and expensive metals
such as ruthenium and rhodium. Replacing these precious metals with common metals such as iron has
been the subject of significant investigations,29,318–324 as iron is cheaper and potentially less toxic and
more environmentally friendly.
Our group discovered the first well-defined Fe(II) complexes with chiral tetradentate PNNP lig-
ands can catalyze the ATH of ketones.293–295 These ATH catalysts have the general formula trans-
[Fe(L)(NCMe)(PPh2C6H4CHN((S, S)−C(R)H−C(R)H)NCHC6H4PPh2)]2+
(R = alkyl, aryl; L = CO,
CNtBu) and contain a tetradentate [6.5.6] PNNP ligand with o-phenylene groups in the linkers be-
tween the diamine nitrogen and phosphorus atoms, forming two six-membered metallacycles and a
five-membered metallacycle around the diamine backbone (94; Figure 7.1). These complexes exhibit
high conversion, good enantiomeric excess (ee), and moderate turnover frequencies (TOFs) for ATH
(>99% conversion, up to 96% ee, TOF up to 2600 h−1). Our second generation of Fe(II) complexes
with the general formula trans-[Fe(CO)(L)(PR2CH2CHN((S, S)−C(R′)HC(R′)H)NCHCH2PR2)]n+
(95;
R, R′ = alkyl, aryl; L = CH3CN, Br–; n =2, 1 respectively) contain a planar tetradentate [5.5.5] PNNP
ligand without an o-phenylene linker and are highly active and enantioselective ATH catalysts (up to
98% conversion, up to 99% ee, TOF up to 28000 h−1).309,325–328 The smaller metallacycles lead to a
more rigid ligand, as opposed to the conformationally flexible [6.5.6] PNNP ligand, which can distort
itself to form a pentadentate ferraaziridine complex in the presence of base.297
FeN
PPh2
2+
N
PPh2
MeCN
L
R = H, Ph; R---R = -(CH2)4-
L = CO, CNtBu
R R
5
6 6
R = alkyl, aryl; R' = H, aryl; R---R = -(CH2)4- L = CH3CN, n = 2 L = Br-, n = 1
n+
* *
555
FeN N
PR2
PR2C
O
R' R'L* *
+
555
FeN N
PR2
PR'2C
O
Ph PhX* *
R, R' = arylX = Cl, Br
H
94 95 96
Figure 7.1: First-generation Fe(II) [6.5.6] (94), second-generation Fe(II) [5.5.5] complexes (95), andthird-generation Fe(II) [5.5.5] complexes (96) for the TH of ketones.
Our third generation catalysts with the general formula
trans-[Fe(CO)(X)(PR2CH2CHN((S, S)−C(Ph)HC(Ph)H)NHCH2CH2PR′2)]+
(R, R′ = aryl; X = Cl,
Br) are the most efficient asymmetric transfer hydrogenation of ketones to date (96, Figure 7.1), which

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 95
contain a variety of amineiminediphosphine hybrid ligands (P-NH-N- P).45,46,125 Upon activation with
2–8 equivalents of base, these imineamine iron complexes showed exceptionally high catalytic activity in
the hydrogenation of ketone substrates by transferring hydrogen from the 2-propanol solvent, with the
highest TOF being greater than 200 s−1 (720000 h−1). The ee of the final chiral alcohols obtained from
these iron catalysts are >99% for some substrates.
The mechanism of transition-metal-catalyzed ketone hydrogenation has been thoroughly studied by
experimental14,16,37,329–331 and computational61,66,67,158,202,332–338 means. It is widely accepted that
hydrogen transfer, by using either H2 or a sacrificial hydrogen donor such as isopropyl alcohol, can
occur by an inner-sphere or outer-sphere mechanism and is usually classified by the hydride transfer
step. Inner-sphere hydrogenation requires a vacant coordination site, with a metal-bound hydride or
a sacrificial hydrogen donor transferring an H– equivalent to the metal-bound substrate (Scheme 7.1;
A).339–342 Proton transfer to the substrate can be facilitated by the ancillary ligand, protic solvent, or
by the heterolytic splitting of H2 (if it is used as a reductant).
During an outer sphere hydrogenation mechanism, a vacant coordination site is not required and a
coordinated metal hydride is transferred from the metal complex directly to the substrate while a proton
is acquired from an external protic solvent or the ancillary ligand. In particular, the orchestrated outer-
sphere motion of proton and hydride to the ancillary ligand and metal, respectively, is generally referred
to as “bifunctional” or “cooperative” catalysis.18–21,28,343 In the case of bifunctional ATH (Scheme 7.1;
B), a proton is transferred to a basic site on the ligand, such as an amide, and a hydride is transferred to
the acidic metal center. With H2 gas (C), the molecule is split heterolytically across the metal-amide to
generate the same MH-NH intermediate as in ATH. Once this intermediate is formed, the proton/hydride
are then transferred to the ketone substrate in the same manner during ATH and AH (D).
A typical ATH reaction catalyzed by iron PNNP complexes is shown in Scheme 7.2. Depending
on the iron precatalyst used in the TH of acetophenone (AcPh) to 1-phenylethanol (PE), there is an
activation period up to 10 minutes long before rapid catalysis.302,326,328 When the strong base KOtBu
is added, isopropanol (iPrOH) is the reaction solvent and thus iPrO– is the base and reductant in the
system, generating acetone (AcMe) as a byproduct. If the reaction of the iron precursors with iPrO–
were to directly produce the active species, the activation period would be negligible and rapid catalysis
would occur without delay. In attempts to isolate the active species, the diimine precatalyst 95 was
reacted with iPrO– or tBuO– salts to form coordinatively unsaturated bis(eneamido) complexes of the
type Fe(CO)(R2PCHCHN(S, S−CH(R′)CH(R′))NCHCHPR2)(97; Scheme 7.3).301,302 These electron-
rich complexes have been spectroscopically characterized and, in one case, structurally characterized.
Once isolated, base-free catalysis can be achieved using a three-component mixture which consists of
AcPh as the substrate, iPrOH as the reductant/solvent, and 97 as the precatalyst at room temperature
under an inert atmosphere. However, the order of addition and delay time in adding AcPh to 97 iniPrOH caused dramatic changes in activity.∗ For the highly active systems of type 97 where R,R′ = Ph
(97Ph), the activation period could be avoided only after reacting 97Ph with iPrOH for 12 minutes prior
to adding AcPh.302 Therefore, a reaction between 97Ph and iPrOH must occur before rapid catalysis
takes place, which is responsible for the activation period.
We recently reported that our first generation iron system 94 involves the formation of an Fe(0)
∗No activation period has been detected for iron PNNP precatalysts with ethyl substituents on phosphorus, albeitcatalysis occurs with much lower turnover frequency. The elevated temperature required for catalysis compared to thesystem with phenyl groups on phosphorus may be sufficient to allow the rapid transformation of the precatalyst into theactive species.301

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 96
H
M NHR
O
H
RR'
H
M NHR
H
OR
R'Both Outer Sphere
ATH and AH:
Ln Ln
M NHR
O
H
H3CH3C
H
M NHR
H H
OH3C
H3C
LnLn
Outer Sphere ATH:
M NHR
H H
M NHR
H H
LnLn
Outer Sphere AH:
Inner Sphere ATH:
M
O
H3C CH3
H
LnO
RR'
M
O
H3CCH3
H
LnO
R
R'
A
B
C
D
M
H
Ln
O
RR'
M
H
LnO
R
R'
Scheme 7.1: General Equations for Inner Sphere Hydrogenation, ATH, and AH of Prochiral KetonesCatalyzed by Metal Complexes with Ancillary Ligands L and Amine Ligand NH2R.
Ph
O HO HExcess tBuOK
2-propanol O
Ph
HO H+ +
28 oC
iPrOH AcMeAcPh PE
[Fe]
*
Scheme 7.2: General Reaction Scheme for the TH of Ketones Catalyzed by Iron PNNP Complexes.a
a8 equivalents base with respect to [Fe].

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 97
complex297 and Fe nanoparticles300 during ATH, but the complexes of this current study do not show
any signs of nanoparticle formation.302 Thus, the mechanism was investigated on the basis of evidence
that it is a homogeneous process.
FeN N
PR2
PR2C
O
R' R'
FeN N
PR2
PR2C
O
R' R'n+
L2 tBuOK
- 2 tBuOH
95 97
Scheme 7.3: Diimine Precatalysts 95 and Their Reactivity with Strong Base to Generate 97.
It was later established that reacting the third-generation amine-enamine complex 98 with two equiv-
alents base produces two amido-eneamido isomers, 99 and 100 (Scheme 7.4).45,46 When this mixture
of isomers is exposed to either iPrOH or H2, only isomer 100 reacts to generate the FeH–NH complex
101 which is within the catalytic cycle for both ATH and AH.
+
BF4-
2 KOtBu
active isomerinactive isomer
+
FeN N
PPh2
PPh2C
O
Ph PhClH
FeN N
PPh2
PPh2C
O
Ph Ph
FeN N
PPh2
PPh2C
O
Ph Ph or
H2 (20 atm)Fe
N N
PPh2
PPh2C
O
Ph PhHH
O
H H
98 99 100 101
Scheme 7.4: Reaction of Third-generation Fe(II) PNNP [5.5.5] Complex 98 with Base and HydrogenSources.
We present a detailed DFT study using the model bis(eneamido) complex
Fe(CO)(H2PCHCHNCH2CH2NCHCHPH2) and full amido-eneamido complex
Fe(CO)(Ph2PCH2CH2N(S, S−CH(Ph)CH(Ph))NCHCHPPh2) to elucidate the mechanism of ATH and
AH of ketones using Fe(II) PNNP catalysts. Although the simple model complex is not suitable for ad-
dressing features governing enantioselectivity, it allows us to establish the fundamental mode of action of
the catalyst with minimal computational cost. Once the lowest-energy catalytic pathway is found, we are
able to use full catalyst structures to compare activation energies, rate-limiting steps, and enantiodeter-
mining steps with our experimental data. On the basis of the relative free energies of reactants, products,
and their transition states, plausible mechanisms are presented that match well with our experimental
observations.
7.2 Results and Discussion
7.2.1 Activation Period with Model Bis(eneamido) Complex During ATH
We have based our studies on the initial presence of three species in solution during catalysis: AcPh,iPrOH (solvent and reductant), and the square pyramidal achiral bis(eneamido) model complex 102

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 98
(Scheme 7.5); thus, all free energies are compared relative to 102, 3 equivalents of iPrOH, and AcPh.
Analysis of the MOs of 102 reveals that the HOMO is mainly a ligand-based nonbonding π-type orbital
with electronic contributions from the eneamido nitrogen atoms and carbon atoms adjacent to phospho-
rus. The electronic contribution of the LUMO predominantly consists of an empty dz2-type orbital on
iron (Figure 7.2). Low-lying bonding MOs in 102 reveal delocalization across the planar PNNP ligand
plane, which distributes electron density about the metal center and eneamido atoms. This motif is
represented by curved lines in Scheme 7.5 and other schemes throughout. For subsequent octahedral
complexes that contain the eneamido moiety, low-lying orbitals delocalize electrons throughout the ene
carbons and amido nitrogen but not the iron centre.
Figure 7.2: HOMO (left) and LUMO (right) for complex 102.
From this point, four reaction pathways were considered: enolate formation by which AcPh transfers
a proton to nitrogen (102→103), enolate formation by which AcPh transfers a proton to the carbon
atom adjacent to phosphorus (102→104), alkoxide formation by which iPrOH transfers a proton to
nitrogen (102→105), and alkoxide formation by which iPrOH transfers a proton to the carbon atom
adjacent to phosphorus (102→106). Increasing the concentration of AcPh has been experimentally
shown to increase the duration of the activation period, presumably due to the formation of metal-
bound enolates.302
Calculations clearly show that the kinetic product is the formation of alkoxido-NH complex 105
(G◦‡(TS102iPrOH,105) = 11.2 kcal/mol, G◦(105) = 9.2 kcal/mol) while the thermodynamic product
is the alkoxido–CH2 complex 106 (G◦‡(TS102iPrOH,106) = 16.8 kcal/mol, G◦(106) = −2.4 kcal/mol)
(Figure 7.3). Reaction of AcPh with 102 is also feasible, resulting in the formation of enolate complexes
103 and 104; however, the energy required for their formation is higher (G◦‡(TS102AcPh,103) = 22.0
kcal/mol and G◦‡(TS102AcPh,104) = 25.9 kcal/mol, respectively). Nonetheless, formation of iron enolate
complexes is feasible and could prolong the activation period. Selected ground state and transition state
geometries are shown in Figure 7.4.
Alcohol-assisted proton transfer during ruthenium-188,335,344 and iron-catalyzed345 ketone hydro-
genation has been calculated to lower the reaction barrier. We also investigated a “proton shuttle”
mechanism, where an additional molecule of iPrOH assists in the proton transfer step going from 102 to
106 (see the Supporting Information).346 The calculated free energy barrier for alcohol-assisted proton
transfer toward the formation of 106 was found to be 18.9 kcal/mol, which is only 2.1 kcal/mol higher
than TS102iPrOH,106. This small energy difference also makes solvent-assisted proton transfer feasible
on going from compound 102→106; probing the role of hydrogen-bonding solvent molecules such asiPrOH in this proton transfer event using explicit solvent modeling could uncover more information on
the assistance of solvent in a “proton shuttle” type mechanism.332
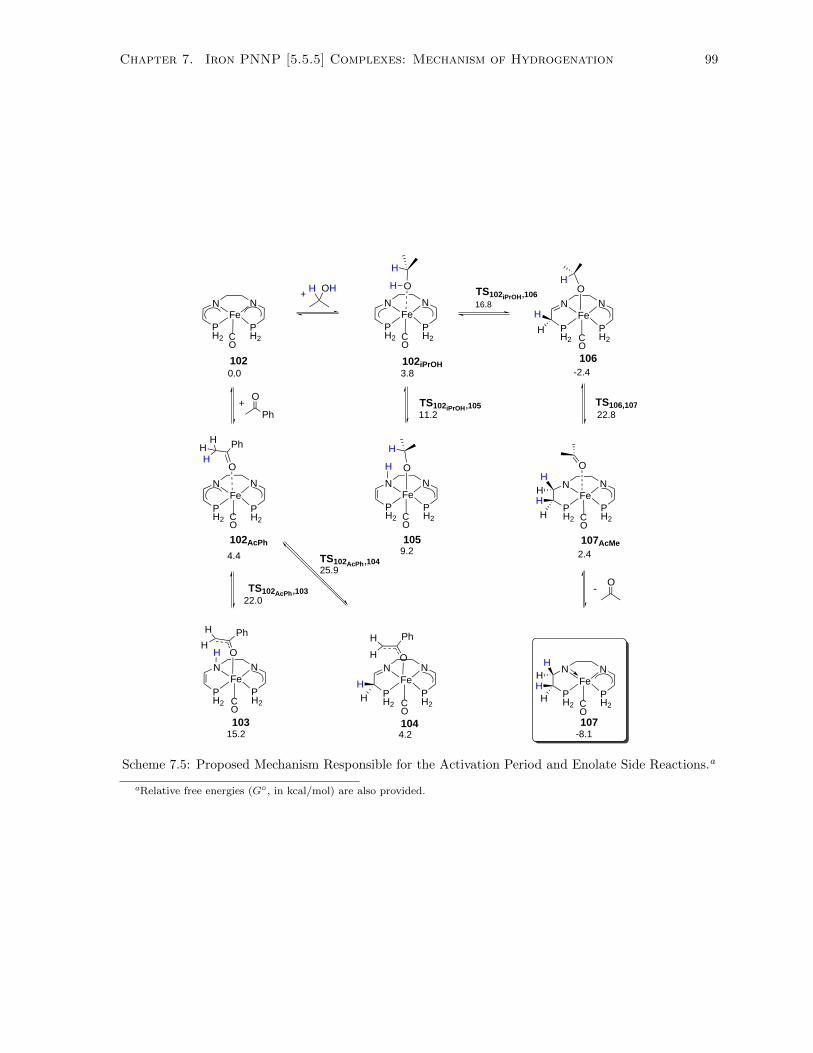
Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 99
FeN N
PH2
PH2
O
H
HH
H
FeN N
PH2
PH2
O
H
H
H
CO
CO
FeN N
PH2
PH2C
O
O-
OH+
FeN N
PH2
PH2
H
HH
H
CO
H
FeN N
PH2
PH2C
O
O
Ph
HH
H
FeN N
PH2
PH2
O
HH
CO
PhH
H
O
Ph+
FeN N
PH2
PH2
OH
CO
H
FeN N
PH2
PH2
OH
CO
H
FeN N
PH2
PH2
O
CO
PhH
HH
0.0 3.8
22.0
25.9
11.2
16.8
-2.4
22.8
9.24.4
15.2 4.2
2.4
-8.1
102 102iPrOH
102AcPh
103
TS102AcPh,103
104
TS102AcPh,104
105
TS102iPrOH,105
106
TS102iPrOH,106
107
TS106,107
107AcMe
Scheme 7.5: Proposed Mechanism Responsible for the Activation Period and Enolate Side Reactions.a
aRelative free energies (G◦, in kcal/mol) are also provided.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 100
-10
0
10
20
3.8
11.2
0.0
4.4
16.8
25.9
22.0
15.2
9.2
4.2
-2.4
22.8
2.4
-8.1
Rel
ativ
e G
o (k
cal/m
ol)
FeN N
PH2
PH2
O
HH
H
CO
FeN N
PH2
PH2
H
HH
H
CO
FeN N
PH2
PH2
OH
CO
PhH
H
FeN N
PH2
PH2
OH
HH
H
CO
FeN N
PH2
PH2
OH
CO
PhH
H
FeN N
PH2
PH2
OH
CO
H
FeN N
PH2
PH2
OH
CO
H
FeN N
PH2
PH2C
O
102102iPrOH
102AcPh
103
TS102AcPh,103
104
TS102AcPh,104
105TS102iPrOH,105
106
TS102iPrOH ,106
107
TS106,107
107AcMe
Figure 7.3: Energy profile for the catalyst activation process and enolate side reactions. All energies arerelative to 102, 3iPrOH, and AcPh.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 101
Of the four scenarios presented above, compound 106 can react further by transferring a hydride
equivalent to the imine carbon (G◦‡(TS106,107) = 22.8 kcal/mol), which is an overall exergonic reaction
by 8.1 kcal/mol after dissociation of AcMe (relative to 102). The product of this reaction is the amido-
eneamido complex 107 which is within the catalytic cycle (vide infra). This is the rate-limiting step
in the reduction of 102 to 107 by iPrOH; we propose that the activation period seen during catalysis
is due to the barrier associated with the hydride transfer during inner-sphere reduction of the PNNP
ligand to generate 107, a mixed amido-eneamido complex.
7.2.2 Catalytic Cycle for ATH with Model Amido-eneamido Complex
Our mechanistic studies regarding the catalytic cycle begin with 107, the structure reached after the
activation period (G◦(107) = −8.1 kcal/mol with respect to 102; Scheme 7.6 and Figure 7.5). As seen
for 102, low-lying MOs in square-pyramidal 107 delocalize the electrons throughout the metal center
and eneamido atoms. One equivalent of iPrOH approaches 107 with the O−H hydrogen atom above
the amido nitrogen atom and the C−H atom pointing towards iron (G◦(107aiPrOH) = −3.9 kcal/mol).
Next, a transition state was found (TS107aiPrOH,108) in which proton transfer to the amido nitrogen
produces 108 (G◦ = −1.9 kcal/mol), a ground state ion pair adduct in which the formally anionic oxygen
atom is stabilized by the newly formed N−H moiety (N−H = 1.09 A, O−−−H = 1.51 A, Figure 7.6). In
addition, the C−H bond pointing towards iron has been elongated (1.10 A vs. 1.21 A for 107aiPrOH)
and the Fe−H distance is short (1.81 A). The bonding mode between iron and hydrogen can be classified
as agostic but with an obtuse Fe−H−C angle of 145◦ and a strong electrostatic interaction between iron
and hydrogen.347 The subsequent hydride transfer is exergonic and proceeds with only a 1.3 kcal/mol
free energy barrier (TS108,109AcMe) to form the ketone adduct 109AcMe. Hydride transfer from the
NH-stabilized isopropoxide to 109 has been calculated to be the rate-limiting step (G◦‡(TS108,109AcMe)
= −0.6 kcal/mol) but is only 1.2 kcal/mol higher in energy than the hydride transfer barrier from 109
to AcPh (vide infra). This difference is small and may not accurately predict which of these two steps
is indeed rate-limiting, considering that the model complex being used does not account for metal–
substrate steric/electronic interactions and different density functionals report higher or lower electronic
barriers (see Section 7.4). Regardless of which step is rate limiting in reality, these barriers are much
lower in energy than the 22.8 kcal/mol required for hydride transfer to the imine carbon in the activation
period (TS106,107, Figure 7.3).∗ After AcMe dissociation, the free energy of octahedral amino-hydrido
complex 109 is −13.4 kcal/mol with respect to 102 plus all relevant small molecules.
Complex 109 can now perform a stepwise outer-sphere proton/hydride transfer to AcPh through a
sequence of events reverse of that described for iPrOH. First, hydride transfer from iron to AcPh occurs to
generate an NH-stabilized ion pair adduct (109AcPh→TS109AcPh,110→110); second, barrierless proton
transfer from nitrogen to oxygen (110→TS110,107PE→107PE) produces PE, regenerating intermediate
107 after product dissociation. The metrical parameters of the intermediates and transition states for
hydrogen transfer from 109 to PE are very close to those calculated for iPrOH→107 hydrogen transfer
(Figure 7.6).
Very similar stepwise proton-hydride transfer sequences containing NH-stabilized ion pair adducts
have been calculated for transition-metal complexes with tridentate ligands incorporating a central amido
donor. Grutzmacher and co-workers have developed the catalyst Rh(trop2N)(PPh3), a Rh(I) complex
utilizing a tridentate diolefin amido ligand, and calculated the mechanism by which it catalyzes the TH
∗Section 7.2.5 presents data on the calculated full catalyst structure 97Ph.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 102
Figure 7.4: Optimized structures and selected bond lengths (A, left to right, top to bottom):102, TS102AcPh,103 (1120i cm−1), TS102AcPh,104 (1269i cm−1), TS102iPrOH,105 (1409i cm−1),TS102iPrOH,106 (1649i cm−1), 106, TS106,107 (869i cm−1), and 107.
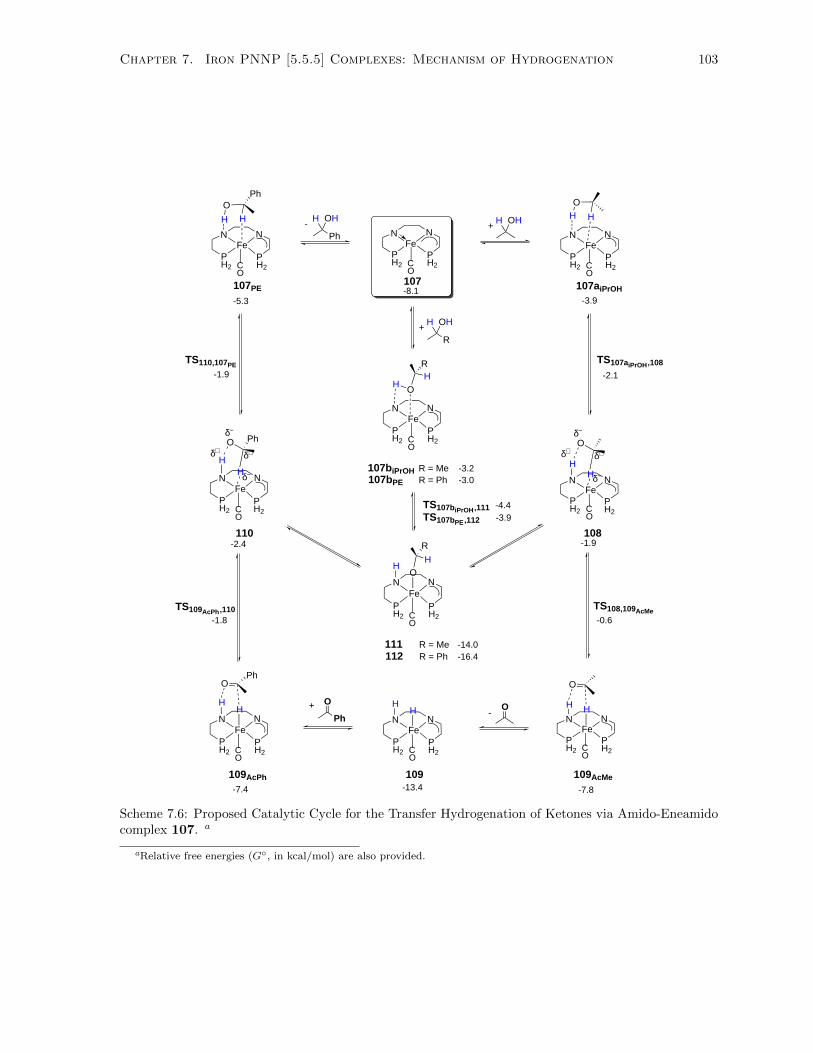
Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 103
FeN N
PH2
PH2C
O
O
HH
FeN N
PH2
PH2
H H
O
CO
FeN N
PH2
PH2
HH
OPh
CO
FeN N
PH2
PH2
OPh
CO
HH
FeN N
PH2
PH2C
O
O
HH
FeN N
PH2
PH2C
O
O Ph
HH
O-
+ O
Ph
δ+
δ−
δ+
δ−
δ+
δ−
δ+
δ−
FeN N
PH2
PH2C
O
FeN N
PH2
PH2
HH
CO
OH+
HOH
Ph-
H
OH
R+
H
R = Me
FeN N
PH2
PH2C
O
O
RH
H
FeN N
PH2
PH2C
O
O
R
HH
R = Ph
-8.1-3.9
-2.1
-3.2-3.0
-0.6
-7.8
-14.0-16.4
-13.4-7.4
-1.8
-4.4-3.9
-2.4 -1.9
-1.9
-5.3
R = MeR = Ph
107 107aiPrOH
108
TS107aiPrOH ,108
109 109AcMe
TS108,109AcMe
109AcPh
110
TS109AcPh,110
107PE
TS110,107PE
107biPrOH
111
TS107biPrOH ,111
107bPE
112
TS107bPE,112
Scheme 7.6: Proposed Catalytic Cycle for the Transfer Hydrogenation of Ketones via Amido-Eneamidocomplex 107. a
aRelative free energies (G◦, in kcal/mol) are also provided.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 104
-20
-10
0
10
20
-16.4
-3.9 -3.0
-14.0
-4.4 -3.2
-7.6
-8.1
-3.9
-5.3
-1.9
-2.1
-1.9
-2.4
-0.6
-1.8
-7.4
-7.8
-13.4
22.8R
elat
ive
Go
(kca
l/mol
)
+ PE
+ iPrOH
intramolecularrearrangement
+ AcMe
FeN N
PH2
PH2C
O
FeN N
PH2
PH2C
O
O
RH
H
FeN N
PH2
PH2C
O
O
RH
H
FeN N
PH2
PH2
HH
CO
FeN N
PH2
PH2C
O
O R
HH
FeN N
PH2
PH2C
O
O R
HH
O+
O
Ph
R = Me
R = Ph
107
107
107aiPrOH
108
TS107aiPrOH ,108
109
109AcMe
TS108,109AcMe
109AcPh110 TS109AcPh,110
107PE
TS110,107PE
107biPrOH
111TS107biPrOH,111
107bPE
112
TS107bPE,112
TS106,107
Figure 7.5: Energy profile of hydrogen transfer from iPrOH to AcPh via amido-eneamido complex 107(continued from Figure 7.3).
of ketones263 and the dehydrogenative coupling of primary alcohols with water, methanol, or amines.264
Bi and co-workers have calculated348 the mechanism of the transfer hydrogenation of ketones for the
PNP iridium catalyst IrH3[(iPr2PC2H4)2NH]. which was originally developed by Abdur-Rashid and co-
workers.349 Gusev and co-workers have calculated NH-stabilized alkoxo adducts for the dehydrogenation
of alcohols using the osmium catalysts OsH3[N(C2H4PiPr2)2] and OsH(CO)[N(C2H4PiPr2)2], which also
incorporate PNP-type ligands.350,351 Of note, an OH-stabilized ion pair adduct has been identified using
ab initio molecular dynamics simulations and an explicit solvent model for the Ru-catalyzed hydrogen
transfer to formaldehyde using RuH(η6−C6H6)(OCH2CH2NH2).332
Metal-bound alkoxides also play an important role as resting states in the proposed catalytic cycle.
The formation of 107 (Scheme 7.6 and Figure 7.5) makes it possible for the iPrOH O−H bond to add
across the Fe−Namido bond, producing the alkoxido complex 111, which has a free energy of −14.0
kcal/mol, very similar to that of 109 (G◦ = −13.4 kcal/mol). iPrOH coordinates to 107 such that
the O−H bond sits atop the Fe−Namido bond (107biPrOH), which then goes through a barrierless
transition state (TS107biPrOH,111) to generate 111. Presumably, either 111 or 109 are resting states
during catalysis, which means that the activation energy for the entire catalytic process is approximately
13 kcal/mol if the rate-limiting step of transfer hydrogenation is either hydride transfer to iron fromiPrOH (TS108,109AcMe
) or from iron to AcPh (TS109AcPh,110). Given our simplified DFT models, this
value is close to the activation free energy of 16.1 kcal/mol for the transfer of H– from iPrOH to iron,
which is obtained from the estimated rate constants based on experimentally determined equilibrium
constants and kinetic simulations.302 For the transfer of H– from iron to AcPh (TS109AcPh,110), the
free energy barrier is about 12 kcal/mol if 111 and/or 109 are indeed resting states, also close to the
experimentally derived value of 14.3 kcal/mol. Calculations performed using the full catalyst system
reveal that stereoelectronic parameters (i.e., steric repulsion between the substrate and phenyl rings on
catalyst) increase the calculated free energy barriers and lead to better agreement with experiment.∗
∗See Section 7.2.5.
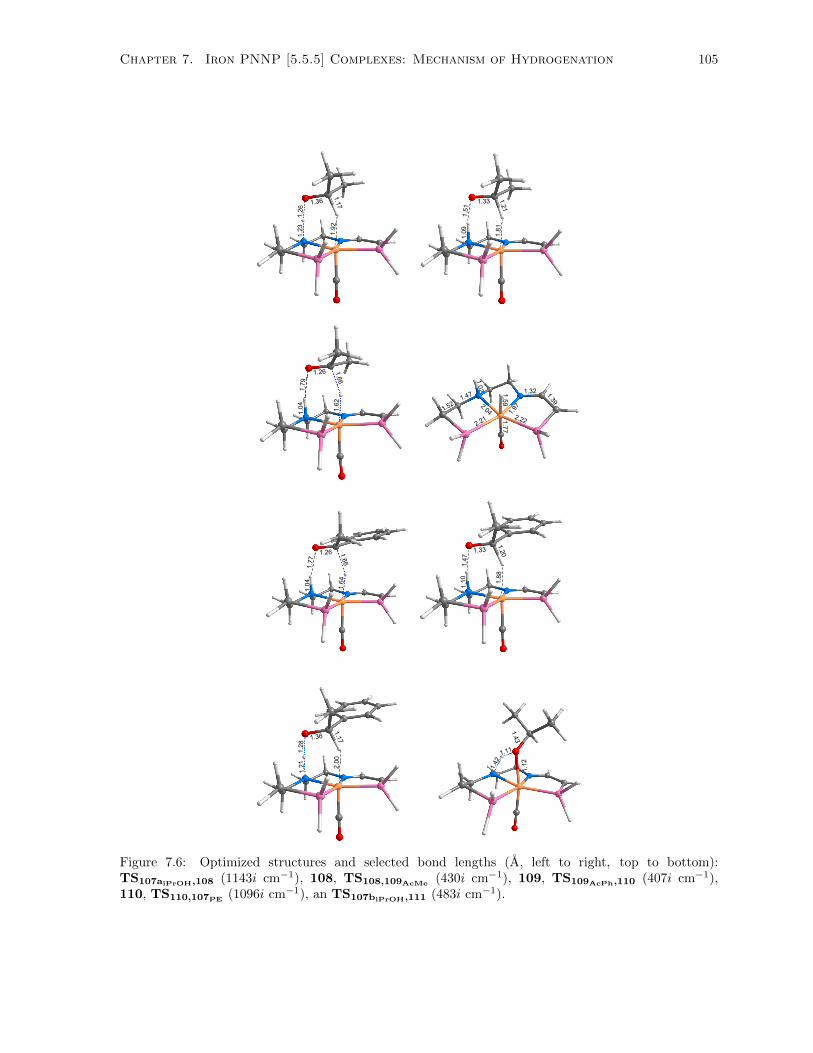
Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 105
Figure 7.6: Optimized structures and selected bond lengths (A, left to right, top to bottom):TS107aiPrOH,108 (1143i cm−1), 108, TS108,109AcMe
(430i cm−1), 109, TS109AcPh,110 (407i cm−1),110, TS110,107PE
(1096i cm−1), an TS107biPrOH,111 (483i cm−1).

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 106
The TOF of this catalytic cycle can be estimated from the calculated activation free energy barrier
using conventional transition state theory.352 The calculated overall turnover-limiting step is hydride
transfer from isopropoxide to iron (TS108,109AcMe) and two possible resting states are 111 and 109,
giving an activation free energy between 12.8 and 13.4 kcal/mol (vide supra). This translates to a
calculated TOF range (29 ◦C) of 1.1× 107–4.2× 106 h−1, which is 2–3 orders of magnitude higher than
the experimentally observed maximum TOF of 5.5 × 104 h−1 at 29 ◦C.302 Although this discrepancy
may seem dramatic, the calculated free energy barrier for the experimentally observed maximum TOF
is 16 kcal/mol, only about 3 kcal/mol higher than the calculated activation free energy.
A previous study has shown that when additional AcPh is added during catalysis as the rate decreases
and the reaction progresses toward equilibrium, the rate increases again and rapid catalysis occurs for a
brief period of time.326 An increased concentration of the product alcohol (PE) after prolonged catalysis
would also make it possible to generate the alkoxido complex 112 in solution (Scheme 7.6 and Figure 7.5),
which is even more stable than its isopropoxide analogue (G◦(112) = −16.4 kcal/mol, G◦(111) = −14.0
kcal/mol). Thus, it follows that an increased concentration of PE would also slow down catalysis, due
to the larger activation barrier that is required (about 16 kcal/mol) to achieve productive catalytic
turnovers. This argument is verified when comparing relative energies with the full catalyst system
97Ph(Section 7.2.5).
The formation of metal-bound alkoxides has experimental relevance in the field of ruthenium-catalyzed
H2 hydrogenation catalysis.41 62 Bergens and co-workers have recently performed low-temperature in-
tramolecular trapping experiments to investigate the reactions between a free
RuH2(diamine)(diphosphine) complex and ketone to generate RuH(alkoxide)(diamine)(diphosphine).63
They conclude that bonding between the incoming ketone and Ru dihydride complex persists throughout
the course of the reaction and formation of a free Ru amide complex and the product alcohol does not
occur. On the basis of these findings, it may also be possible for alkoxide complexes 111 and 112 to
undergo reversible intramolecular rearrangement to form the ion pair adducts 108/110. Although we
have no computational evidence for this type of rearrangement, we believe that the overall low ther-
modynamic barriers during catalysis also make this possible. It is clear that our DFT studies classify
hydride transfer as occurring in the outer sphere via an NH-stabilized ion pair adduct; however, the ther-
modynamically favourable formation of Fe alkoxide complexes must be taken into account. A detailed
mechanistic DFT study concerning low barrier intramolecular rearrangement from an NH-stabilized ion
pair adduct to form a Ru alkoxide during H2 hydrogenation is reported elsewhere.65
7.2.3 KIE Calculations for Activation and Catalysis
The presence or absence of a KIE can provide useful information to the experimentalist and theorist about
the mechanism of transition-metal catalyzed reactions.353 In a typical transfer hydrogenation reaction
using the diimine precatalyst 95Ph (R, R′ = phenyl; Scheme 7.3), the solvent/reductant iPrOH was
replaced with monodeuterated iPrOD-d1 ((CH3)2CHOD) or fully deuterated iPrOD-d8 ((CD3)2CDOD)
followed by calculation of the KIEs at 29 ◦C.302 A primary KIE value of 1.3 ± 0.1 is obtained wheniPrOD-d1 is used, while a primary KIE value of 2.5± 0.1 is obtained with iPrOD-d8. Since concentrations
of precatalyst and active species are constantly changing throughout catalysis, calculating separate KIEs
during the activation period and rapid catalysis allows us to obtain more information about the origin
of the experimental KIEs.
To calculate the KIE for the activation period, we examined the rate-limiting hydride transfer step

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 107
106→TS106,107. Selective deuteration of one hydrogen atom adjacent to phosphorus simulates a re-
action with iPrOD-d1 and deuterating seven additional hydrogen atoms on the isopropoxide moiety
simulates a reaction with iPrOD-d8 (Figure 7.7; TS106,107). The calculated KIE using iPrOD-d1 is
0.9, suggesting that there is no significant isotope contribution (Table 7.1). However, a calculated KIE
value of 2.6 was found when iPrOD-d8 was used, suggesting that the rate-limiting hydride transfer step
contributes significantly and correlates with the observed value of 2.5±0.1. These findings are consistent
with the qualitative observation of a longer activation period when iPrOD-d8 is used.302
FeN N
PR2
PR2C
O
O CH3(D3)
CH3(D3)
H(D)(D)H
FeN N
PR2
PR2
O
(D3)H3C
H
(D)H
(D)H
CO
CH3(D3)
(D)HH
H
(D)H
FeN N
PR2
PR2C
O
O Ph
CH3
H(D)(D)H
(D)HH
H
(D)H
TS106,107 TS108,109AcMe TS109AcPh,110
Figure 7.7: Transition states marked with potential deuteration sites for KIE calculations.
To calculate the KIE during catalysis, we examined the transfer of a hydride equivalent from the
NH-stabilized ion pair adduct to iron (108→TS108,109AcMe) and transfer of a hydride equivalent from
the FeH-NH complex to AcPh to generate the phenethoxide adduct (109AcPh→TS109AcPh,110). In
order to simulate the use of monodeuterated iPrOD-d1, only two sites were deuterated: the hydrogen
atom adjacent to phosphorus (which is deuterated during the activation period) and hydrogen attached
to nitrogen (Figure 7.7; TS108,109AcMeand TS109AcPh,110). To simulate the reaction with iPrOD-d8,
eight additional sites are deuterated when a hydride equivalent is transferred to iron: the hydrogen atom
adjacent to nitrogen (which is deuterated during the activation period) along with all hydrogen atoms on
the isopropoxide moiety. When a hydride equivalent is transferred from iron to AcPh using iPrOD-d8,
only four sites are deuterated, as shown in Figure 7.7 (TS109AcPh,110). Calculations using iPrOD-d1
for hydride transfer to/from iron reveal no significant kinetic isotope effects (0.9 and 1.1), while usingiPrOD-d8 generated KIE values of 1.9 and 1.5 (Table 7.1).
Table 7.1: Experimental302 and Calculated KIE Values (1 atm, 29 ◦C) for Hydride Transfer During theActivation Period and Catalytic Cycle Using iPrOD-d1 and iPrOD-d8.
stepkH/kD
(CH3)2CHOD (CD3)2CDOD
exptl302 1.3± 0.1 2.5± 0.1
106→TS106,107 0.9 2.6
108→TS108,109AcMe0.9 1.9
109AcPh→TS109AcPh,110 1.1 1.5
Bearing in mind that simple model complexes were used for KIE analysis, the calculated trends are
in rough agreement with the experimental values. Using iPrOD-d1 reveals a minimal KIE during the
activation period and rapid catalysis while catalysis with iPrOD-d8 reveals KIE values greater than 1.
On the basis of these results, we predict that the dominant contribution to the experimentally observed
KIE when using iPrOD-d8 is hydride transfer during the activation period, which is also the largest
surmountable barrier toward generating the active catalytic species.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 108
7.2.4 Formation of a Bis(amido) PNNP Complex and Modelling its ATH
Catalytic Activity
Our group recently synthesized the chiral Fe(II) PNNP diamine complex
[Fe(CO)(Br)(PPh2CH2CH2NH((S, S)−C(Ph)HC(Ph)H)NHCH2CH2PPh2)][BF4] (113; Scheme 7.7), a
saturated analog of the diimine precatalyst 95 whose general formula is shown in Scheme 7.3.302 When
complex 113 is used under standard catalytic conditions, the presence of an 8-fold excess of base presum-
ably deprotonates both nitrogen atoms to generate the bis(amido) catalyst
[Fe(CO)(PPh2CH2CH2N((S, S)−C(Ph)HC(Ph)H)NCH2CH2PPh2) (114). This complex can then ac-
cept a proton/hydride equivalent from iPrOH and transfer it to AcPh, presumably across the Fe–N
bond of the catalyst as described above for the amido-eneamido compounds. Bis(amino) complex 113 is
a precatalyst for the TH of AcPh to PE; however, its activity is poor (10% conversion, 2 h, 82% ee, cat-
alyst:base:substrate ratio of 1:8:6000)302 versus the analogous diimine precatalyst (90% conversion, 30
min, 82% ee, catalyst:base:substrate ratio of 1:8:2000),309 but the enantioselectivity remains unchanged.
FeN N
PPh2
PPh2C
O
Ph PhH H
+
BPh4-
FeN N
PPh2
PPh2C
O
Ph PhtBuOK
(excess)
Cl
113 114
Scheme 7.7: Reaction of Bis(amino) Complex 113302 with Base, Which Likely Generates Bis(amido)Complex 114 in Situ.
Since reduction of the PNNP ligand of bis(eneamido) complex 102 is thermodynamically favorable,
we extended our calculations to include stepwise inner-sphere reduction of the eneamido portion of
mixed amido-eneamido complex 107, yielding the bis(amido) complex 116 (Figure 7.8). The reduction
proceeds in a way similar to that described above for 102. First, iPrOH coordination and proton transfer
generates the alkoxido complex 115 (107biPrOH→115, G◦‡ = 16.0 kcal/mol) followed by hydride
transfer and AcMe dissociation to afford the Fe(II) bis(amido) complex 116 (115→116, G◦‡ = 18.1
kcal/mol). Note that the hydride transfer free energy of TS115,116AcMeis 4.7 kcal/mol lower in energy
than TS106,107 (G◦‡ = 22.8 kcal/mol) in the activation period (vide supra) and 116 is exergonic by
10.6 kcal/mol with respect to 102. This provides evidence that complete reduction of the ligand system
is thermodynamically favorable and that “deactivation” or “decomposition” of precatalyst 95 after
prolonged catalysis309 may in fact be the gradual formation of bis(amido) complexes such as 116 in
solution.
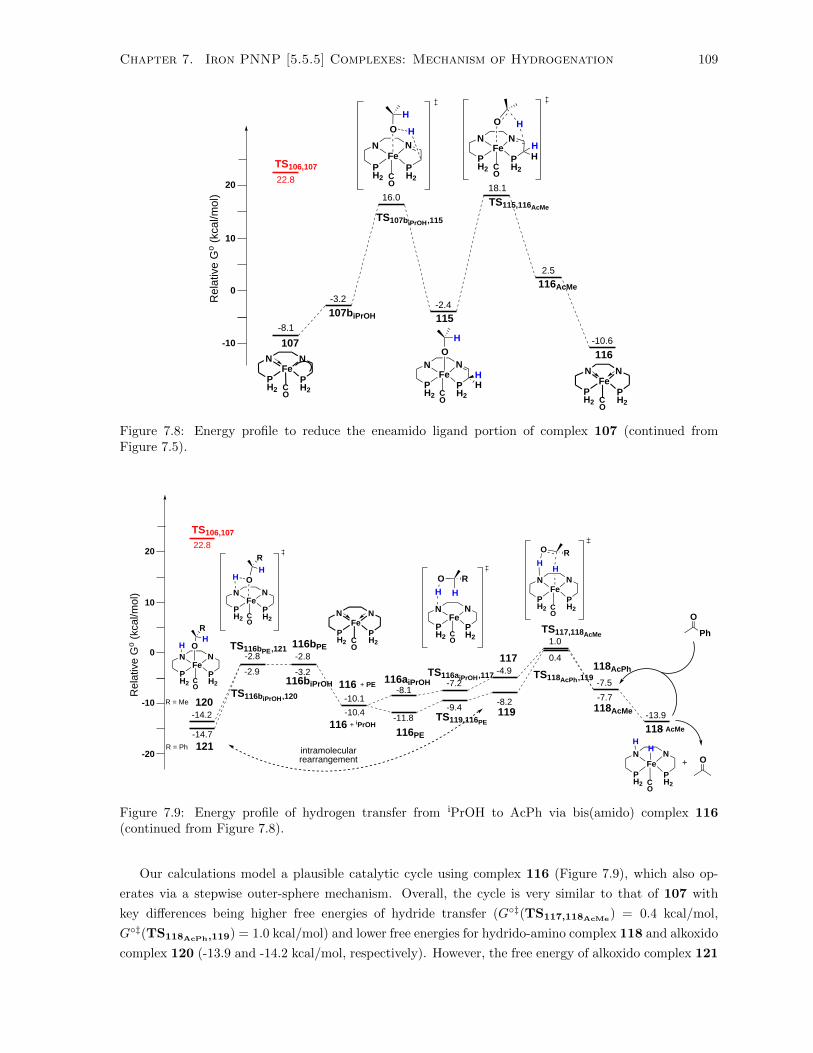
Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 109
-10
0
10
20
-8.1
-3.2
16.0
-2.4
18.1
2.5
-10.6
Rel
ativ
e G
o (k
cal/m
ol)
22.8
FeN N
PH2
PH2
HCO
O
H
H
FeN N
PH2
PH2
HCO
H
HO
FeN N
PH2
PH2C
O
O H
H
FeN N
PH2
PH2C
O
FeN N
PH2
PH2C
O
107
107biPrOH 115
TS107biPrOH,115
116
116AcMe
TS115,116AcMe
TS106,107
Figure 7.8: Energy profile to reduce the eneamido ligand portion of complex 107 (continued fromFigure 7.5).
-20
-10
0
10
20
-14.7
-2.8 -2.8
-14.2
-2.9 -3.2
-10.1
-10.4
-8.1
-11.8
-7.2
-9.4
-4.9
-8.2
1.0
0.4
-7.5
-7.7
-13.9
22.8
Rel
ativ
e G
o (k
cal/m
ol)
+ PE
+ iPrOH
intramolecularrearrangement
+ AcMe
FeN N
PH2
PH2C
O
FeN N
PH2
PH2C
O
O
RH
H
FeN N
PH2
PH2C
O
O
RH
H
FeN N
PH2
PH2
HH
CO
FeN N
PH2
PH2C
O
O R
HH
FeN N
PH2
PH2C
O
O R
HH
O+
O
Ph
R = Me
R = Ph
116
116 116aiPrOH
117TS116aiPrOH ,117
118
118AcMe
TS117,118AcMe
118AcPh
119
TS118AcPh,119
116PE
TS119,116PE
116biPrOH
120TS116biPrOH,120
116bPE
121
TS116bPE,121
TS106,107
Figure 7.9: Energy profile of hydrogen transfer from iPrOH to AcPh via bis(amido) complex 116(continued from Figure 7.8).
Our calculations model a plausible catalytic cycle using complex 116 (Figure 7.9), which also op-
erates via a stepwise outer-sphere mechanism. Overall, the cycle is very similar to that of 107 with
key differences being higher free energies of hydride transfer (G◦‡(TS117,118AcMe) = 0.4 kcal/mol,
G◦‡(TS118AcPh,119) = 1.0 kcal/mol) and lower free energies for hydrido-amino complex 118 and alkoxido
complex 120 (-13.9 and -14.2 kcal/mol, respectively). However, the free energy of alkoxido complex 121

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 110
is 1.7 kcal/mol higher than that of the analogous eneamido-alkoxido complex 112. If the resting states
are indeed complexes 118 and 120/121, the activation free energy will be about 15–16 kcal/mol, which
is consistent with the lower experimentally observed activity with 114 in comparison to an estimated
activation energy of 12–13 kcal/mol for amido-eneamido catalyst 107. All intermediate and transition
state geometries closely resemble those already discussed above.
7.2.5 Calculated Activity and Enantioselectivity of the Third Generation
Full Catalyst System 97Ph During ATH
The activation period observed during catalysis with second-generation catalysts has been verified by
experiment and theory, leading to the active amido-eneamido species by reduction of an imine group
on the ligand by iPrOH. Our group recently developed a novel synthetic method to synthesize unsym-
metrical third generation amine-imine complexes as mentioned above (Figure 7.1), which are the most
active TH catalysts to date when activated by strong base.46 Calculating the thermodynamic properties
of full catalyst system 100 by incorporating phenyl substituents on the phosphines and diamine allows
us obtain more accurate energy values for the above proposed resting states 109, 111, and 112. Fur-
thermore, the enantioselectivity can be evaluated by comparing the energies of the transition states that
lead to formation of R or S alcohols. Preliminary results are presented below.
Figure 7.10 shows the six structures that were calculated along with their free energies, all relative to
100 plus relevant small molecules under catalytic conditions.46 The general trends are in agreement with
calculations on the model system; FeH–NH complex 101 and tert-butoxide complex 122 have similar
free energies (-5.8 and -4.5 kcal/mol, respectively) while product alkoxide complex 123 is lower in energy
(-8.5 kcal/mol). The pro-R and proS enantiodetermining transition states are represented by TSR and
TSS respectively, and formation of the R product is lower in energy by 1.5 kcal/mol, also in agreement
with the predominant formation of R-phenylethanol during catalysis (88 % R).
FeN N
PPh2
PPh2C
O
Ph Ph
FeN N
PPh2
PPh2C
O
Ph PhHH
FeN N
PPh2
PPh2C
O
Ph PhHH
FeN N
PPh2
PPh2C
O
Ph PhOH
H
FeN N
PPh2
PPh2C
O
Ph PhOH
PhH
R - phenylethanol pro-R pro-S
O Ph
FeN N
PPh2
PPh2C
O
Ph PhHH
O
Ph
0.0 -5.8 -4.5
-8.5 10.7 12.2
100 101 122
123 TSR TSS
Figure 7.10: Calculated structures relative to full amido-eneamido complex 100 (1 atm, 301 K)a
aRelative free energies (G◦, in kcal/mol) are also provided.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 111
We have summarized our preliminary results of the comparison between observed and calculated
activity/enantioselectivity in Table 7.2. Using conventional transition state theory,352 we calculated the
TOF for each proposed transition state and compared it with the observed value of 119 s−1, which
corresponds to ∆G◦ = 14.8 kcal/mol. The calculations reveal that the most likely resting state during
catalysis is the isopropoxide complex 122, with a calculated TOF of 58 s−1 (∆G◦ = 15.2 kcal/mol).
Since iPrOH is the reductant and reaction solvent, the catalyst will predominantly be in this form during
initial catalytic turnovers. As the product alcohol concentration increases, the more stable alkoxide 123
will dominate in solution, reducing catalytic activity.∗ The free energy difference between the pro-S and
pro-R transition states is calculated to produce R-phenylethanol with an ee of 84 %, surprisingly close to
the observed ee of 88 %. We continue to perform additional calculations using different levels of theory
to further support these results.
Table 7.2: Comparison of Experimental46 and Calculateda (from DFT) Activity and EnantioselectivityParameters During the ATH of AcPh.
parameter value
exptl. TOF (s−1) 119
calcd.1 TOF (s−1), TSR−122 58
calcd.1 TOF (s−1), TSR−101 6.6
calcd.1 TOF (s−1), TSR−123 0.1
exptl. ee (%) 88
calcd.2 ee (%), TSS−TSR 84
1 rate =kbT
he
∆G◦RT
2 ee =e
∆∆G‡RT − 1
e∆∆G‡RT + 1
a1 atm, 301 K, iPrOH solvent continuum
7.2.6 Third-Generation FePNNP [5.5.5] Complexes are Also Moderately Ac-
tive AH Catalysts
We demonstrated that the mechanism of ATH using simplified models of 107 and 109 involves stepwise
proton/hydride transfer from FeH–NH complex 109 to acetophenone Scheme 7.6. On the basis of the
calculated relative free energies, this was postulated to be the turnover-limiting step when iPrOH is used
as the reductant. The hydride (FeH–NH) and alkoxide (Fe(OCHMePh)–NH) compounds were found to
be the lowest-energy species, acting as possible resting states in the catalytic cycle. Thus, once the
FeH–NH complex is formed in solution, we expect that the same mechanism is operational to reduce
ketonic substrates, with the only difference now being that H2 gas is responsible for regenerating the
FeHNH complex (Scheme 7.1). Since H2 splitting was found to be the turnover-limiting step during
catalysis, our focus will be on calculating H2 splitting barriers using a simplified version of complex 107
and comparing this with the calculated energies of the full system 107, which includes all of the phenyl
substituents.
∗As noted earlier, the formation of bis(amido) complexes such as 116 could also lead to reduced activity or deactivationof the catalyst.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 112
We have calculated the barrier for H2 splitting on the same simplified amido-eneamido system, using
the same level of theory as in our previous work346 and also using the M06-L functional with the
TZVP/TZVPfit basis set (see the Experimental Section).45 The latter functional/basis set combination
was shown to be an efficient and accurate method in comparing thermochemical values between theory
and experiment for other organometallic transformations.247 In addition, solvation effects were taken
into account by using a THF solvent continuum. Scheme 7.8 shows that the H2 splitting barriers
for both functional/basis set combinations are comparable (G◦‡(TS124,109) = 17.2 vs 18.0 kcal/mol),
with the relative free energies differing by only 0.8 kcal/mol relative to 107 plus all relevant small
molecules. Formation of the FeH–NH complex 109 is exergonic by 9.0 kcal/mol (M06/6-31++G(d,p))
and 7.1 kcal/mol (M06-L/TZVP/TZVPfit), respectively. The metrical parameters for TS124,109 are
very similar using both levels of theory, and the structure is presented in Figure 7.11.
H
H
FeNN
PPCO
H
H
H
H
H H
HHHH
H
H
FeNN
PPCO
H
H
H
H
H H
HHHH
H HH
H
FeNN
PPCO
H
H
H
H
H H
H
HH
H
H
H
0.0 12.3 -9.0
H2
17.2M06/6-31++G(d,p):
M06-L/TZVP/TZVPfit:
H2 splitting
0.0 12.9 -7.118.0
107 124 TS124,109 109
Scheme 7.8: Calculated Structures Starting with Simplified amido-eneamido Complex 107 (1 atm, 298K)a
aRelative free energies (G◦, in kcal/mol) are also provided.
We were also interested in optimizing structures and obtaining H2 splitting barriers using a full
structural analogue of 107, which included (S,S)-dpen and all phenyl substituents on phosphorus. For all
further calculations discussed, we use M06-L/TZVP/TZVPFit to optimize ground-state and transition-
state structures. When 98 (Scheme 7.4) reacts with 2 equivalents of base, two enantiomers form in
solution (99 and 100), where the carbonyl ligand is located on either side of the PNNP ligand plane.
Simplified complex 107, as discussed above, is structurally analogous to 100.
Figure 7.11: Optimized structure and selected bond lengths (A) for TS124,109(ν = 1074i).
Using molecule 99 as our free energy reference point, along with all relevant small molecules, we
wanted to investigate why H2 splitting selectively occurred with complex 100, leaving 99 untouched
(Scheme 7.9). First, coordination of H2 to 99 successfully leads to optimization of the η2−H2 complex

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 113
125, which is 13.8 kcal/ mol higher in energy than 99. Carefully inspecting the orientation of the amide
ligand of 125 reveals that the nitrogen lone pair is pointing away from the η2−H2 ligand (Figure 7.12),
similar to the orientation of the NH group in complex 98.
Our attempts at trying to find an H2 splitting transition state between 125 and the FeH–NH complex
126 were unsuccessful. In order for heterolytic H2 splitting to occur across the metalamide moiety, the
nitrogen atom must orient its electron pair toward the σ∗ molecular orbital of hydrogen. The pyramidal
inversion at nitrogen might be too energetically unfavorable, since the amine hydrogen and hydrogen
atoms on the alkyl groups attached to nitrogen are in “syn-axial” positions; pyramidal inversion would
then require the entire ligand system to reorient itself in order to relieve the axial strain (Scheme 4).
Calculations were performed where the ligand is initially reoriented to relieve these unfavorable “syn-
axial” interactions, but attempts to find both an η2−H2 ground-state structure and an H2 splitting
transition state were also unsuccessful (see the Supporting Information).45 Furthermore, the energy of
the FeH–NH complex 126 is substantially higher than the free energies of 100 and 101 (vide infra).
H
FeN N
P PCO
HH
H
HH
H
FeN N
P PCO
HH
HH
H
H
H HH2
1.4
FeN N
P PCO
HH
HH
H
H
HH
15.2 6.8
FeNN
PPCO
H
H
H
H
H
HFe
NN
PPCO
H
H
H
H
H
H HHFe
NN
PPCO
H
H
H
H
H
H
H
0.0 14.6 -6.9
H2 16.1
+
no TS found
x
"syn-axial" interactions unfavorable
amide lone pairpoints away from H2
99 125 126
100 127
TS127,101
101
Scheme 7.9: Calculated Structures Starting with Amido-eneamido Complex 99 or 100.a
a1 atm, 298 K, THF solvent continuum. Relative free energies (G◦, in kcal/mol) are also provided.
Starting with complex 100(G◦ = 1.4 kcal/mol relative to 99), an η2−H2 ground state was found
(127), which is only about 0.5 kcal/mol lower in energy than 125. However, in this case, the amide
lone pair is oriented on the same side as coordinated dihydrogen. Comparing bond length pairs in 125
and 127 (Figure 7.12) reveals that there is little difference between the two structures, but there is
obviously a dramatic difference in the orientation of the P–Namido portion of the ligand with respect to
coordinated dihydrogen. From 127, an H2 splitting transition state was found (G◦‡(TS127,101) = 17.5
kcal/mol), generating hydride complex 101, which is exergonic by 6.9 kcal/mol (when compared with
amido-eneamido compound 100). The calculated structure of TS127,101 is shown in Figure 7, and bond
lengths are strikingly similar to its simplified analogue TS124,109 (Figure 7.11).
We were also interested to know why complexes such as 98 does not exhibit the same activity and
enantioselectivity for ATH and AH. Table 7.3 lists the calculated and experimentally measured activation

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 114
Figure 7.12: Optimized structures and selected bond lengths (A) for 126 (top), 101 (middle), andTS127,101(ν = 1089i, bottom).

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 115
parameters (∆G◦‡, ∆H◦‡, ∆S◦‡) in both ATH and AH, relative to the free energy of amido-eneamido
complex 4bH for ATH and 4a for AH. The relative free energies (∆G◦) in ATH for the turnover-limiting
step (hydride transfer to acetophenone) were taken from our previous experimental302 study on the
mechanism of transfer hydrogenation using these amido-eneamido systems. Table 7.3 clearly shows
that the experimental and calculated H2 splitting free energy barrier for AH are both higher than the
turnover-limiting step for ATH and that there is good agreement between the experiment and theory for
the case of AH. Unfortunately, the enantioselectivity during the AH of acetophenone produces racemic
products, which is possibly due to side reactions of 100 and/or 101 during catalysis, likely resulting from
the harsher conditions required for catalysis (20 atm H2, 323 K). The products of these side reactions
are currently under investigation in our laboratory.
Table 7.3: Comparison of Experimental and Calculated (from DFT) Activation Parameters for HydrideTransfer to Acetophenone in ATH Relative to 122 (Figure 7.10), and H2 Splitting in AH Relative to100a
parameter ATHb exptl. ATH calcd.c AH exptl.d AH calcd.d
∆H◦‡ (kcal/mol) 6.0± 0.2 15.1 10.0± 0.2 7.0
∆S◦‡ (cal/mol·K) −28± 1 −0.1 −31.0± 0.5 −29.2
∆G◦‡ (kcal/mol) 14.3± 0.5 15.2 20.0± 0.5 16.4
aBoth ATH and AH are with M06-L/TZVP/TZVPFit level of theory, however ATH uses an iPrOH solvent continuumwhile AH uses a THF solvent continuum.
bReference 302. There are potentially large errors associated with these data due to the multiparameter kinetic modelinginvolved and the difficulty in separating the kinetics of catalyst activation from the kinetics of the catalytic cycle.
c1 atm, 301 K.d20 atm, 323 K.
7.3 Conclusion
With the aid of DFT, we have a comprehensive understanding of the mechanism of ATH and AH of
ketones using Fe PNNP [5.5.5] complexes, which corresponds well with experimental data. A general
mechanism is shown in Scheme 7.10. The largest calculated free energy barrier for ATH using our
second-generation Fe PNNP [5.5.5] catalysts is hydride transfer from an iron-bound isopropoxide to an
imine carbon on the ligand during a stepwise inner-sphere activation period. Overcoming this barrier
transforms the bis(eneamido) complex into a mixed amido-eneamido compound, which is the catalytically
active species. During ATH (right loop, clockwise from active isomer) and AH (left loop, counter-
clockwise from active isomer), the catalytic cycle operates via a stepwise outer-sphere mechanism where
an H+/H– pair is transferred across the amido nitrogen and iron atom, respectively. The turnover
limiting step in ATH is H– transfer from catalyst to substrate. Calculated kinetic isotope effect values
are also in rough agreement with the experimentally determined values, which supports the proposed
mechanism of catalysis. Preliminary calculations modelling the activity/enantioselectivity free energies
with the full catalyst structure also agree with experimental observations.
For our model amido-eneamido system, it is energetically feasible to reduce the ligand through another
stepwise inner-sphere reduction to generate the symmetrical bis(amido) compound
Fe(CO)(H2PCH2CH2NCH2−)2. This complex has been calculated to be active for the TH of ketones
and operates in a similar stepwise outer sphere fashion but has higher hydride transfer barriers and a
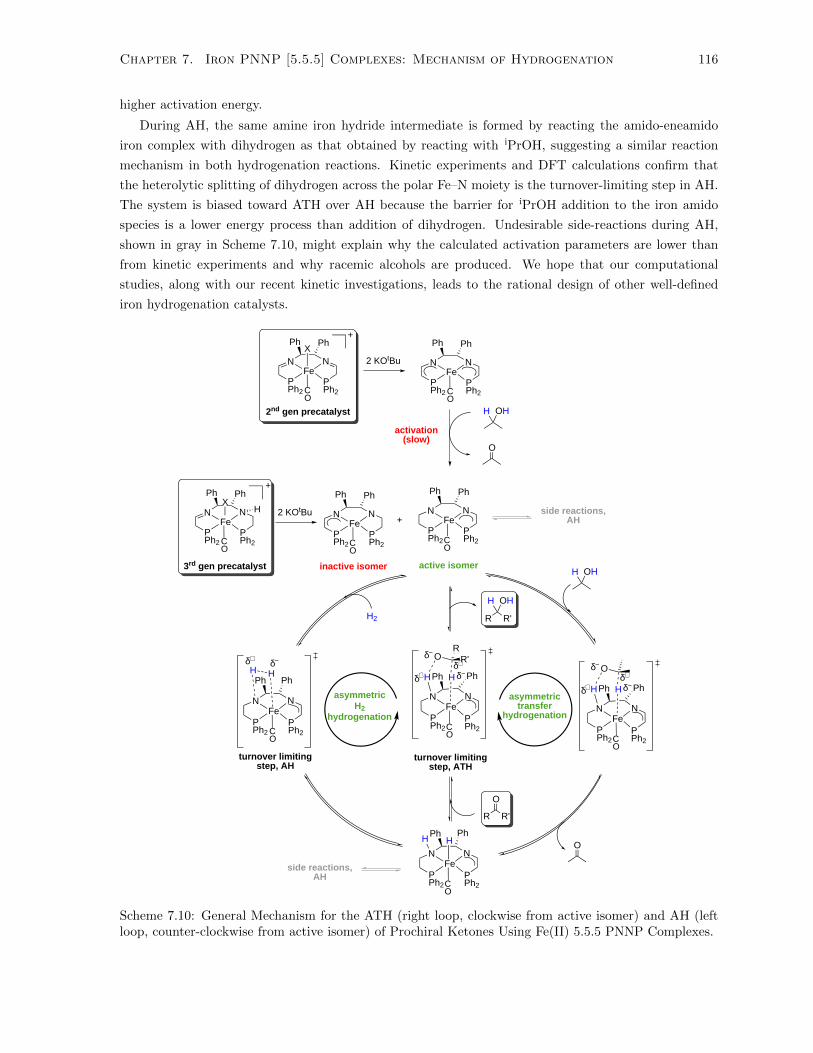
Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 116
higher activation energy.
During AH, the same amine iron hydride intermediate is formed by reacting the amido-eneamido
iron complex with dihydrogen as that obtained by reacting with iPrOH, suggesting a similar reaction
mechanism in both hydrogenation reactions. Kinetic experiments and DFT calculations confirm that
the heterolytic splitting of dihydrogen across the polar Fe–N moiety is the turnover-limiting step in AH.
The system is biased toward ATH over AH because the barrier for iPrOH addition to the iron amido
species is a lower energy process than addition of dihydrogen. Undesirable side-reactions during AH,
shown in gray in Scheme 7.10, might explain why the calculated activation parameters are lower than
from kinetic experiments and why racemic alcohols are produced. We hope that our computational
studies, along with our recent kinetic investigations, leads to the rational design of other well-defined
iron hydrogenation catalysts.
FeN N
PPh2
PPh2C
O
O R'R
HHδ+
δ−
δ+
δ−
OHH
FeN N
PPh2
PPh2C
O
Ph Ph+
HX
3rd gen precatalyst
2 KOtBuFe
N N
PPh2
PPh2C
O
Ph Ph
+ FeN N
PPh2
PPh2C
O
Ph Ph
inactive isomer active isomer
turnover limiting step, ATH
Ph Ph
FeN N
PPh2
PPh2
H H
CO
Ph Ph
FeN N
PPh2
PPh2
H H
CO
Ph Ph
δ−δ+
turnover limiting step, AH
H2
side reactions,AH
side reactions,AH
FeN N
PPh2
PPh2C
OOHH
Ph Ph
FeN N
PPh2
PPh2C
O
Ph Ph
2 KOtBu
+
O
2nd gen precatalyst
activation(slow)
R
OH
R'
H
R
O
R'
FeN N
PPh2
PPh2C
O
O
HHδ+
δ−
δ+
δ−Ph Phasymmetric
H2hydrogenation
asymmetrictransfer
hydrogenation
X
O
Scheme 7.10: General Mechanism for the ATH (right loop, clockwise from active isomer) and AH (leftloop, counter-clockwise from active isomer) of Prochiral Ketones Using Fe(II) 5.5.5 PNNP Complexes.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 117
7.4 Computational Details
7.4.1 General
DFT calculations were performed using Gaussian09.251 Simplified complexes 102–121 and 124 were
treated with the M06354 hybrid functional and 6-31++G(d,p)315,355–357 basis set while full/complete
complexes 99–101, 122–127, TSR, and TSS were treated with the M06-L244 functional and TZVP245
basis set with the TZVPFit density fitting basis set. Either normal (opt) or tight (opt = tight) conver-
gence criteria were used for all optimizations, and a pruned (99, 590) integration grid was used throughout
(grid = ultrafine). All NH-stabilized ion pair adducts of model complexes were connected to their TSs
by performing IRC calculations.358,359 Optimizations were performed in iPrOH (2-propanol) for ATH
calculations and THF (tetrahydrofuran) for AH calculations using the IEF-PCM252,253 with radii and
nonelectrostatic terms from the SMD254 solvation model. KIE values (1 atm, 29◦C) were calculated with
the freqchk utility (supplied by Gaussian) using conventional transition state theory,61,360 neglecting
tunnelling/recrossing effects and assuming that the geometry of the protio and deuterio transition state
remains the same. The freqchk utility was also used to calculate any non-STP thermochemical pa-
rameters for comparison with experimental values. Basis set superposition error (BSSE) was examined
by using the counterpoise method361,362 and computed to be 1–2 kcal/mol for weakly coordinating
ketone/alcohol adducts 107aiPrOH, 109AcMe, 109AcPh, and 107PE; thus, it is expected that the
BSSE for other ketone/alcohol adducts is within the same range. Open-shell triplet state optimizations
were performed on five-coordinate complexes 102, 107, and 116; in comparison to their singlet state
free energies, each structure was higher in energy by 6.4, 9.3, and 3.2 kcal/mol, respectively. Triplet
state electronic energies were calculated for transition states TS106,107, TS109AcPh,110, TS115,116AcMe,
and TS118AcPh,119 by using optimized singlet-state geometries and calculating single point energies;
in comparison to their singlet state electronic energies, each structure was higher in energy by 38, 40,
34, and 36 kcal/mol, respectively. Therefore, odd-electron species were not further considered. Full
vibrational and thermochemical analyses (1 atm, 298 K) were performed on optimized structures to
obtain solvent-corrected free energies (G◦) and enthalpies (H◦). Optimized ground states were found
to have zero imaginary frequencies, while transition states were found to have one imaginary frequency.
Three-dimensional coordinates not published in this thesis and additional calculations discussed in the
main text appear in the Supporting Information of their respective publications.45,346 Three-dimensional
visualizations of calculated structures were generated by ChemCraft.255
7.5 Supporting Information
7.5.1 Transfer Hydrogenation
Energy Evaluation of Model Complexes Using Other Density Functionals
To assess the validity of the calculated energies using the M06354 density functional, a variety of other
common density functionals were also tested with the 6-31++G(d,p) basis set (Table 7.4). Single-point
energies using M06/6-31++G(d,p) optimized structures were obtained for two key transition states: hy-
dride transfer to the imine carbon in the activation period (106→TS106,107, Figure 7.3) and hydride
transfer from iron to AcPh in the proposed catalytic cycle (109AcPh→TS109AcPh,110, Figure 7.5).
The following density functionals were chosen: pure M06L244 and TPSS,363 hybrid B3LYP,364,365

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 118
B3PW91,311,365–369 BMK,370 HSE06 (also known as HSEh1PBE),371–377 LC-ωPBE,378–381 MPW1k,382
mPW1PW91,312,366 PBE0 (also known as PBE1PBE),383 TPSSh,363 and ωB97X-D.384 With regard to
other research groups that have calculated NH-stabilized ion pair adducts, the MPW1k and PBE0 den-
sity functionals were used by Gusev,350,351 B3PW91 was used by Grutzmacher,263,264 and B3LYP was
used by Bi.348 Yang has recently reported a similar comparison of density functionals in conjunction
with mechanistic studies of an Fe(II) PNP pincer complex for the hydrogenation of ketones.202
Our results show that energies for the imine reduction step (E(TS106,107) − E(106)) deviate no
more than 4 kcal/mol from the barrier calculated using the M06 density functional (E = 26.7 kcal/mol),
with the exception of MPW1k (19.4 kcal/mol, ∆E = 7.3 kcal/mol). Hydride transfer barriers from
complex 109AcPh to AcPh are all within 3 kcal/mol from M06 (E = 5.7 kcal/mol). The LC-ωPBE
density functional predicts the barriers will be highest for these two steps, while MPW1k predicts the
lowest barriers. In general, we conclude that our choice of density functional is reasonable and does not
alter our discourse.∗
Table 7.4: Electronic energies of key transition states using various density functionals.
Density Functional E(TS106,107)− E(106) E(TS109AcPh,110)−E(109AcPh)
M06L 23.2 6.2
TPSS 22.8 5.4
B3LYP 26.2 7.6
B3PW91 23.5 6.3
BMK 28.9 6.0
HSE06 24.2 5.3
LC-ωPBE 30.3 8.4
M06 26.7 5.7
MPW1k 19.4 4.8
mPW1PW91 24.3 5.9
PBE0 23.9 5.4
TPSSh 24.3 5.8
ωB97X-D 28.3 5.6
Alcohol-assisted Proton Transfer in the Activation Period.
Two equivalents of iPrOH are added to 102 (Scheme 7.11). The free energy required for proton transfer
from iPrOH to the carbon atom is 18.9 kcal/mol (TS128,129, Figure 7.13). The bond lengths (Fig-
ure 7.14) reveal that a pure proton transfer to carbon occurs while the O−H bond above iron remains
intact. This suggests that a calculable intermediate and transition state exists between TS128,129 and
the alkoxide product 129. An IRC calculation of TS128,129 revealed that an alkoxide adduct should ex-
ist that is very similar in energy to TS128,129, but all attempts to minimize a ground state intermediate
were unsuccessful. In addition, attempts at finding a proton transfer transition state to generate 129
were unsuccessful. We expect both these structures to be similar to or lower in energy than TS128,129.
∗However, larger energy differences have been found between DFT methods (B3LYP) and ab initio techniques (MP2,MP4), which was beyond the scope of this study.158

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 119
OH2
H
FeN N
PH2
PH2
OH
CO
H
O
HH
FeN N
PH2
PH2
O
H
H
CO
H
O
H
H
102
128
TS128,129
129
Scheme 7.11: Proton transfer to ligand with the assistance of a second equivalent of iPrOH.
-10
0
10
20
0.0
8.8
18.9
5.0
Rel
ativ
e G
o (k
cal/m
ol)
16.8
102
128
TS128,129
129
TS102iPrOH,106
Figure 7.13: Energy profile for alcohol assisted proton transfer. All energies are relative to 102.
Figure 7.14: Optimized structures and selected bond lengths (A) of TS128,129 (1733i cm−1) and 129.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 120
Other Considered TH Mechanisms.
Starting with mixed amido-eneamido compound 107, two alternative possibilities for hydrogen transfer
were explored. The first case involves proton transfer to the eneamido nitrogen and hydride trans-
fer to iron (107→133, Scheme 7.12 and Figure 7.15). Based on our results, this is also a stepwise
proton-hydride transfer sequence, however our attempts to find a proton transfer transition state were
unsuccessful. Furthermore, the NH-stabilized ion pair adduct 131 is not a true ground state; a small
imaginary frequency exists (79i cm−1) and our attempts to optimize this structure using tighter con-
vergence criteria failed. However, the metrical parameters of 131 (Figure 7.16) closely match other
NH-stabilized ion pair adducts we have calculated and we expect that the proton transfer step is low
in energy, much like structures 108 and 117 discussed in the article. The hydride transfer free energy
(G◦‡(TS131,132) = 23.1 kcal/mol) is the same as for inner sphere hydride transfer during the proposed
activation period (TS106,107, 22.8 kcal/mol) and much higher in energy than hydride transfer to/fromiPrOH during the proposed catalytic cycle (G◦‡(TS108,109AcMe
) = −0.6 kcal/mol).
FeN N
PH2
PH2C
O
OH
H
FeN N
PH2
PH2C
O
O
H H
FeN N
PH2
PH2C
O
O
H H
FeN N
PH2
PH2C
O
O
HH
FeN N
PH2
PH2C
O
HHOH
+H O
-
FeN N
PH2
PH2
HCO
OH
H FeN N
PH2
PH2
HCO
O
HH Fe
N N
PH2
PH2
HCO
H
H
OH+
H O-
δ+
δ−δ+
δ−
δ+
δ−δ+
δ−
107
107
130 131
TS131,132
132 133
134
TS134,135
135
TS135,136
136 137,110
Scheme 7.12: Other considered mechanisms for the transfer hydrogenation of ketones.
The second possibility for hydrogen transfer is proton transfer to the eneamido carbon adjacent
to phosphorus and hydride transfer to iron (107→137, Scheme 7.12). Again, a stepwise proton-
hydride transfer sequence is also occurring with an NH-stabilized ion pair adduct as an intermediate
(135). However, the barrier for proton transfer has been calculated to be higher than hydride transfer
(G◦‡(TS134,135) = 13.8 kcal/mol vs. G◦‡(TS135,136) = 11.0 kcal/mol, Figure 7.15). An interaction
between a P−H hydrogen and alkoxide oxygen for structures 135 and TS135,136 (2.52 and 2.53 A,
respectively) may help lower the calculated energies (Figure 7.16).

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 121
-10
0
10
20
-8.8
-0.5
13.8
Rel
ativ
e G
o (k
cal/m
ol)
-0.6
22.8
-1.3
13.8
13.8
13.8
13.8
13.8
13.8
13.8
13.8
107
130
131
TS131,132
132
133
134
TS134,135
135 TS135,136
136
137
TS106,107
TS109AcPh,110
Figure 7.15: Energy profile for other considered transfer hydrogenation mechanisms. All energies arerelative to 102.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 122
Figure 7.16: Optimized structures and selected bond lengths (A, left to right, top to bottom): 131(79i cm−1), TS131,132 (466i cm−1), TS134,135 (1455i cm−1), 135, and TS135,136 (419i cm−1).

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 123
7.5.2 Direct Hydrogenation
H
FeN N
P PCO
HH
H
HH
H
FeN N
P PCO
HH
HH
H
H
H HH2
1.4
FeN N
P PCO
HH
HH
H
H
HH
15.2 6.8
FeNN
PPCO
H
H
H
H
HH
FeNN
PPCO
H
H
H
H
H
HFe
NN
PPCO
H
H
H
H
H
H HHFe
NN
PPCO
H
H
H
H
H
H
H
FeNN
PPCO
H
H
H
H
HH
FeNN
PPCO
H
H
H
H
HH
HH
14.6 -6.9
H2 17.5
4.0-0.2
H2
H
H
+
FeN N
P PCO
H
H
H
H
H H
HH
FeN N
P HPCO
H
H
H
H
H HH
H
2.8 7.3 0.1
FeN N
P PCO
H
H
H
H
H H
H2
no TS found
x
no TS found
x
6.5
0.0
"syn-axial" interactions unfavorable
"syn-axial" interactions unfavorable
99 125 126
100 127
TS127,101
101
Scheme 7.13: Mechanisms considered in the main text and alternate conformers/complexes.a
a1 atm, 298 K, THF solvent continuum. Relative free energies (G◦, in kcal/mol) are also provided.

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 124
7.5.3 Cartesian Coordinates (A), Free
Energies (G◦, Hartree), and En-
thalpies (H◦, Hartree) of Opti-
mized Structures∗
iPrOH
H◦ -194.291128
G◦ -194.324792
C 0.00260 0.03438 0.36276
H -0.01064 0.08668 1.46070
C 1.33435 -0.51028 -0.08819
H 2.15412 0.12418 0.24946
H 1.49682 -1.51151 0.30996
H 1.37657 -0.56879 -1.17775
C -1.16167 -0.81100 -0.10246
H -1.10031 -1.82070 0.30624
H -2.11275 -0.38249 0.21842
H -1.17376 -0.88550 -1.19179
O -0.09186 1.36929 -0.16410
H -0.94685 1.72521 0.10490
AcMe
H◦ -193.115521
G◦ -193.149197
C 0.00000 0.17857 0.00000
C -1.27170 -0.60888 -0.00329
H -2.13449 0.03240 -0.16508
H -1.24103 -1.39249 -0.76166
H -1.38181 -1.11751 0.95723
C 1.27170 -0.60888 0.00329
H 1.24103 -1.39249 0.76166
H 1.38181 -1.11751 -0.95723
H 2.13449 0.03240 0.16508
O -0.00000 1.39880 0.00000
AcPh
H◦ -384.832833
G◦ -384.875082
C -1.68877 -0.19759 0.00011
O -2.21080 -1.30508 0.00018
C -2.52412 1.04421 0.00013
H -2.30651 1.65855 0.87537
H -2.30659 1.65855 -0.87512
H -3.57926 0.78422 0.00018
C -0.20863 -0.06195 0.00001
C 0.42175 1.18463 -0.00014
C 0.57648 -1.21787 0.00006
C 1.80488 1.27065 -0.00022
∗1 atm, 298 K, iPrOH solvent continuum.
H -0.16673 2.09356 -0.00018
C 1.95624 -1.13092 -0.00002
H 0.08366 -2.18242 0.00017
C 2.57330 0.11486 -0.00016
H 2.28490 2.24140 -0.00033
H 2.55563 -2.03307 0.00003
H 3.65446 0.18375 -0.00022
PE
H◦ -386.005614
G◦ -386.04809
C -1.64834 -0.31264 -0.26673
H -1.84327 -1.15766 -0.94060
C -2.27253 -0.60585 1.07954
H -3.34848 -0.75253 0.98202
H -1.84335 -1.50742 1.51545
H -2.09788 0.22205 1.76929
O -2.30185 0.85616 -0.77035
H -1.92662 1.04891 -1.63817
C -0.15220 -0.13187 -0.16070
C 0.69868 -1.21661 -0.35647
C 0.39790 1.10063 0.18323
C 2.07149 -1.07502 -0.21230
H 0.27810 -2.17922 -0.62965
C 1.77069 1.24546 0.32408
H -0.25866 1.95034 0.33368
C 2.61164 0.15775 0.12870
H 2.72157 -1.92706 -0.37221
H 2.18589 2.21062 0.58959
H 3.68347 0.27148 0.23728
100
H◦ -3792.283705
G◦ -3792.406769
Fe -0.24763 0.07380 -0.20102
P 1.10453 1.66505 0.72316
P 1.11095 -1.62370 -0.71120
N -1.66644 1.19905 0.49840
N -1.62978 -1.20850 -0.32049
C -2.99076 0.60223 0.54040
C -2.94976 -0.58861 -0.43649
H -2.24721 3.01433 1.34742
C -1.40611 2.40946 1.00058
H -0.13080 -3.64472 -0.20469
C -0.11484 -2.97014 -1.06118
H 0.18619 -3.55637 -1.92923
C -0.17787 0.79936 -1.74302
O -0.26407 1.28842 -2.79893
C -1.47160 -2.32106 -1.25024

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 125
H -1.58734 -1.97321 -2.29109
H -2.25705 -3.07264 -1.09861
C -0.12656 2.90147 1.08591
H 0.09481 3.86731 1.52157
C 2.17108 -2.24142 0.65711
C 3.50664 -1.84051 0.72974
C 1.63090 -2.98102 1.70985
C 4.28631 -2.18202 1.82476
H 3.94243 -1.26201 -0.07797
C 2.41830 -3.33691 2.79504
H 0.58667 -3.27277 1.69516
C 3.74553 -2.93548 2.85797
H 5.32084 -1.86251 1.86717
H 1.98739 -3.91870 3.60106
H 4.35544 -3.20511 3.71161
C 2.27653 -1.60290 -2.12801
C 2.79263 -2.80157 -2.62559
C 2.70588 -0.39867 -2.67920
C 3.70971 -2.79135 -3.66385
H 2.48339 -3.74715 -2.19348
C 3.62979 -0.39006 -3.71645
H 2.32928 0.53997 -2.29095
C 4.12907 -1.58487 -4.21165
H 4.10171 -3.72679 -4.04435
H 3.95843 0.55463 -4.13286
H 4.84836 -1.57892 -5.02169
C 1.91752 1.27836 2.33066
C 2.92216 2.07503 2.88246
C 1.42842 0.20034 3.06844
C 3.43593 1.78592 4.13797
H 3.30443 2.92637 2.33013
C 1.93651 -0.08217 4.32822
H 0.64338 -0.42111 2.64604
C 2.94669 0.70610 4.86218
H 4.21916 2.40838 4.55427
H 1.54959 -0.92523 4.88841
H 3.35062 0.48148 5.84193
C 2.43350 2.40988 -0.29021
C 2.10246 3.37665 -1.23987
C 3.74187 1.92401 -0.24744
C 3.06011 3.85063 -2.12503
H 1.08582 3.75451 -1.28252
C 4.69854 2.40232 -1.13032
H 4.01431 1.16466 0.47747
C 4.35970 3.36449 -2.07278
H 2.79023 4.60304 -2.85647
H 5.71078 2.01815 -1.08531
H 5.10703 3.73556 -2.76367
H -3.17159 0.18480 1.54462
H -3.07160 -0.18456 -1.45936
C -4.13080 1.53366 0.21149
C -5.29418 1.52567 0.97537
C -4.06671 2.36909 -0.90405
C -6.37459 2.32882 0.63362
H -5.35236 0.87582 1.84307
C -5.14209 3.17443 -1.24581
H -3.16118 2.38639 -1.50291
C -6.30113 3.15497 -0.47851
H -7.27414 2.30961 1.23750
H -5.07806 3.81941 -2.11426
H -7.14095 3.78503 -0.74585
C -4.11080 -1.52166 -0.18275
C -5.22764 -1.50272 -1.01318
C -4.11071 -2.37732 0.91839
C -6.32383 -2.31416 -0.75054
H -5.23641 -0.83755 -1.87133
C -5.20106 -3.19295 1.18104
H -3.24025 -2.40436 1.56620
C -6.31338 -3.16156 0.34824
H -7.18621 -2.28496 -1.40616
H -5.18528 -3.85592 2.03831
H -7.16594 -3.79754 0.55427
101
H◦ -3793.467893
G◦ -3793.591660
Fe 0.17528 -0.01769 -0.15259
P -1.13823 1.76224 -0.50773
P -1.13561 -1.76880 0.22296
N 1.71711 -1.38328 -0.29796
N 1.65896 1.20425 -0.70005
C 2.97864 0.62549 -0.82895
C 2.97692 -0.66916 0.01503
H 2.20712 3.12685 -1.27713
C 1.38634 2.46580 -0.98648
H 0.13619 -3.51617 -0.89350
C 0.11277 -3.14948 0.13473
H -0.16226 -3.99042 0.77058
C 0.44591 0.37003 1.55529
O 0.64624 0.65779 2.66567
C 1.46887 -2.59189 0.51046
H 1.49829 -2.29279 1.56207
H 2.25405 -3.33903 0.35536
C 0.10093 2.96718 -0.91939
H -0.12285 4.00202 -1.14757
C -2.44886 -2.25399 -0.96998
C -3.79414 -2.04267 -0.66821
C -2.10948 -2.71660 -2.24244

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 126
C -4.77800 -2.30091 -1.61279
H -4.07950 -1.67288 0.31057
C -3.09441 -2.98498 -3.18060
H -1.06854 -2.85496 -2.51317
C -4.43174 -2.77704 -2.86881
H -5.81826 -2.12980 -1.36311
H -2.81485 -3.35000 -4.16167
H -5.19986 -2.98035 -3.60503
C -1.98608 -2.03223 1.83556
C -2.39716 -3.30611 2.23451
C -2.28360 -0.94327 2.65069
C -3.07369 -3.48366 3.43078
H -2.19934 -4.16582 1.60344
C -2.96882 -1.11990 3.84586
H -1.98950 0.05360 2.34477
C -3.35983 -2.39044 4.23968
H -3.38353 -4.47747 3.73100
H -3.19599 -0.26031 4.46525
H -3.89076 -2.53144 5.17341
C -2.42453 1.84934 -1.83441
C -3.20320 3.00404 -1.95960
C -2.59514 0.82490 -2.76051
C -4.12807 3.12547 -2.98425
H -3.08532 3.81531 -1.24865
C -3.52705 0.94333 -3.78438
H -1.98849 -0.06954 -2.68008
C -4.29559 2.09216 -3.89856
H -4.72104 4.02850 -3.06993
H -3.64908 0.13462 -4.49571
H -5.02165 2.18524 -4.69731
C -2.10485 2.39391 0.93239
C -1.46128 3.14347 1.91545
C -3.43448 2.01874 1.13630
C -2.12895 3.51224 3.07510
H -0.42645 3.43677 1.76462
C -4.10306 2.38863 2.29389
H -3.94954 1.42493 0.38777
C -3.45111 3.13424 3.26797
H -1.61588 4.09715 3.82950
H -5.13471 2.08967 2.43880
H -3.97324 3.42200 4.17257
H 3.16587 0.31621 -1.87269
H 2.90765 -0.38290 1.06930
C 4.11754 1.51763 -0.39365
C 5.27710 1.61827 -1.15575
C 4.04887 2.20141 0.82040
C 6.35173 2.37973 -0.71399
H 5.33729 1.08857 -2.10116
C 5.11907 2.96267 1.26292
H 3.14407 2.13482 1.41689
C 6.27574 3.05213 0.49719
H 7.24867 2.44876 -1.31805
H 5.05283 3.48840 2.20815
H 7.11172 3.64847 0.84233
C 4.22378 -1.48785 -0.18934
C 5.20405 -1.52645 0.79867
C 4.44623 -2.17130 -1.38386
C 6.38608 -2.22520 0.59693
H 5.03855 -0.99435 1.72986
C 5.62369 -2.87605 -1.58522
H 3.69322 -2.15264 -2.16560
C 6.59868 -2.90138 -0.59626
H 7.14148 -2.24244 1.37341
H 5.78280 -3.40426 -2.51773
H 7.51964 -3.44913 -0.75523
H -0.10476 -0.31624 -1.68288
H 1.74496 -1.66110 -1.27806
122
H◦ -3986.606279
G◦ -3986.738926
Fe 0.14586 -0.01958 -0.05435
P -1.22001 -1.47754 1.08624
P -1.11785 1.63773 -0.99692
N 1.64508 -1.34586 0.36526
N 1.65302 0.87598 -0.98549
H 2.90025 -0.00342 1.32061
C 2.93191 -0.61952 0.41567
H 3.25454 -0.30780 -1.67226
C 2.98154 0.32143 -0.80966
H 2.26247 2.40800 -2.25224
C 1.41693 1.93537 -1.74506
H -0.25579 -3.49574 2.16721
C 0.00761 -2.84300 1.33557
H -0.03004 -3.45036 0.42777
C 0.41452 0.92531 1.38687
O 0.65052 1.52939 2.35254
C -0.04399 -1.03869 -2.95636
C -1.19679 -1.71846 -3.68736
H -0.13228 0.04703 -3.13569
H -2.15801 -1.43195 -3.25673
H -1.20849 -1.46289 -4.75050
H -1.10946 -2.80695 -3.61099
C 0.15497 2.47008 -1.90702
H -0.00581 3.37684 -2.47560
C 1.39057 -2.25104 1.49705
H 2.14621 -3.04163 1.54476
H 1.46947 -1.66692 2.41906

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 127
O -0.10519 -1.32166 -1.58738
H 1.55781 -1.86566 -0.51582
C 4.14311 -1.51351 0.48249
C 4.31891 -2.54505 -0.43947
C 5.13191 -1.28847 1.43536
C 5.45888 -3.33353 -0.40664
H 3.55775 -2.73288 -1.19108
C 6.27732 -2.07248 1.46611
H 5.00274 -0.48474 2.15301
C 6.44304 -3.09674 0.54495
H 5.58219 -4.13285 -1.12744
H 7.04034 -1.88303 2.21149
H 7.33536 -3.71038 0.56858
C 4.08343 1.33995 -0.62601
C 5.30582 1.18413 -1.27225
C 3.91480 2.41732 0.24350
C 6.34358 2.08101 -1.05296
H 5.44339 0.34773 -1.95039
C 4.94811 3.31485 0.46270
H 2.96143 2.55099 0.74532
C 6.16717 3.14804 -0.18411
H 7.28939 1.94687 -1.56442
H 4.80386 4.14825 1.14008
H 6.97371 3.85104 -0.01396
C -2.69401 -2.35322 0.42507
C -3.94957 -2.24529 1.02526
C -2.56879 -3.09432 -0.75267
C -5.05318 -2.87046 0.46167
H -4.07330 -1.67111 1.93586
C -3.67551 -3.71370 -1.31329
H -1.60408 -3.15200 -1.24110
C -4.92017 -3.60584 -0.70796
H -6.02045 -2.78144 0.94172
H -3.56314 -4.27932 -2.23116
H -5.78397 -4.08985 -1.14753
C -1.76330 -0.95735 2.75875
C -1.66781 -1.76795 3.88792
C -2.33056 0.31236 2.88379
C -2.12268 -1.31224 5.11793
H -1.24238 -2.76186 3.81691
C -2.79757 0.76102 4.10918
H -2.40152 0.95571 2.01100
C -2.68977 -0.05080 5.23051
H -2.03632 -1.94837 5.99052
H -3.23929 1.74710 4.18928
H -3.04506 0.30044 6.19166
C -2.49150 1.15306 -2.11639
C -2.51075 1.53754 -3.45505
C -3.51771 0.34265 -1.63001
C -3.54135 1.12485 -4.28873
H -1.70821 2.15291 -3.84707
C -4.55174 -0.06391 -2.46077
H -3.50685 0.02576 -0.59252
C -4.56256 0.32456 -3.79432
H -3.54439 1.42687 -5.32946
H -5.34246 -0.69199 -2.06772
H -5.36403 0.00144 -4.44776
C -1.90362 2.90973 0.09229
C -1.05329 3.78602 0.77080
C -3.27667 3.00561 0.31790
C -1.55914 4.72119 1.66004
H 0.01622 3.73503 0.59241
C -3.78308 3.94499 1.20647
H -3.96404 2.35063 -0.20289
C -2.92763 4.80176 1.88348
H -0.88295 5.39219 2.17647
H -4.85311 4.00710 1.36609
H -3.32465 5.53309 2.57704
C 1.27944 -1.48834 -3.56372
H 1.31184 -1.31763 -4.64325
H 2.12170 -0.95547 -3.12207
H 1.43064 -2.55879 -3.39263
123
H◦ -4178.330667
G◦ -4178.471225
Fe -0.14871 -0.24957 -0.00916
P -1.58543 -0.53795 -1.77782
P -1.34187 0.24835 1.87885
N 1.35084 -0.44450 -1.40061
N 1.43329 0.02704 1.15847
H 2.28781 -2.01113 -0.41445
C 2.54386 -0.99153 -0.72094
H 3.13302 0.83223 0.21345
C 2.74322 -0.14064 0.55564
H 2.14582 0.47295 3.05957
C 1.25972 0.34007 2.43323
H -0.68912 -0.96999 -4.05470
C -0.33058 -0.49867 -3.14008
H -0.16170 0.55707 -3.36887
C -0.18167 -1.95421 0.36288
O -0.15049 -3.09375 0.59466
C 0.28380 2.74223 0.16488
C -0.52522 3.98226 -0.21193
H 0.09790 2.54260 1.23337
H -1.59418 3.78248 -0.10721
H -0.27330 4.83927 0.41835
H -0.33492 4.26235 -1.25205

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 128
C 0.00982 0.49381 2.99894
H -0.11358 0.68766 4.05652
C 0.95510 -1.12407 -2.64495
H 1.73997 -1.04940 -3.40401
H 0.82150 -2.18600 -2.41768
O -0.10891 1.66801 -0.63048
H 1.47948 0.55659 -1.58364
C 1.76595 3.05385 0.05643
C 2.52558 3.31877 1.19390
C 2.39946 3.08778 -1.18434
C 3.88466 3.58647 1.10105
H 2.04181 3.29292 2.16640
C 3.75590 3.36916 -1.28571
H 1.81858 2.88014 -2.07959
C 4.50620 3.61163 -0.14193
H 4.46303 3.77063 1.99952
H 4.23329 3.39111 -2.25927
H 5.56713 3.81843 -0.21806
C 3.79140 -1.04034 -1.56396
C 4.13103 0.01057 -2.41269
C 4.66921 -2.11530 -1.44387
C 5.32669 -0.00543 -3.11615
H 3.45283 0.84965 -2.52546
C 5.86706 -2.13316 -2.14401
H 4.41229 -2.93920 -0.78574
C 6.20060 -1.07588 -2.97963
H 5.57651 0.82081 -3.77150
H 6.54127 -2.97435 -2.03665
H 7.13540 -1.08857 -3.52673
C 3.78499 -0.76863 1.44850
C 5.07013 -0.23905 1.51197
C 3.49695 -1.92618 2.17180
C 6.05386 -0.85506 2.27498
H 5.29900 0.66119 0.94886
C 4.47635 -2.54191 2.93516
H 2.49380 -2.34087 2.13509
C 5.75939 -2.00899 2.98663
H 7.05075 -0.43221 2.31382
H 4.24058 -3.44098 3.49227
H 6.52421 -2.49023 3.58426
C -2.86407 0.65600 -2.33560
C -4.21396 0.30718 -2.40307
C -2.49052 1.97115 -2.62492
C -5.16708 1.25184 -2.75753
H -4.52889 -0.70455 -2.17586
C -3.44708 2.91112 -2.97706
H -1.44984 2.25874 -2.54687
C -4.78729 2.55527 -3.04547
H -6.21055 0.96479 -2.80943
H -3.14183 3.92820 -3.19460
H -5.53333 3.29145 -3.31940
C -2.43354 -2.15840 -1.90764
C -2.50274 -2.89698 -3.08747
C -3.06954 -2.64769 -0.76518
C -3.18861 -4.10333 -3.11975
H -2.02529 -2.53662 -3.99071
C -3.76563 -3.84589 -0.80198
H -3.01460 -2.08459 0.16223
C -3.82321 -4.57768 -1.98036
H -3.23008 -4.67164 -4.04108
H -4.25732 -4.21076 0.09192
H -4.35929 -5.51847 -2.01006
C -2.41633 1.74176 1.85560
C -2.20128 2.80117 2.73491
C -3.44271 1.84207 0.91619
C -3.00264 3.93342 2.68145
H -1.39350 2.74188 3.45652
C -4.24969 2.97011 0.86777
H -3.61266 1.03093 0.21617
C -4.02848 4.01967 1.74972
H -2.82342 4.75180 3.36878
H -5.04563 3.03108 0.13511
H -4.65149 4.90503 1.70780
C -2.43543 -1.03663 2.63363
C -1.81774 -2.15014 3.20766
C -3.82931 -0.98977 2.60274
C -2.57082 -3.19329 3.72363
H -0.73422 -2.19262 3.25348
C -4.58299 -2.03337 3.12334
H -4.33888 -0.13487 2.17575
C -3.95791 -3.13937 3.68077
H -2.07282 -4.04828 4.16537
H -5.66486 -1.97762 3.09512
H -4.54783 -3.95285 4.08544
TSR
H◦ -4178.299480
G◦ -4178.440637
Fe -0.00983 -0.26612 0.05395
P -1.45366 -1.60552 -1.08891
P -1.17419 1.23590 1.30438
N 1.44969 -1.17812 -1.04529
N 1.58307 0.66466 0.78190
H 2.50171 -1.88773 0.60818
C 2.69129 -1.21101 -0.23101
H 3.19861 0.82561 -0.54615
C 2.88069 0.21687 0.31310
H 2.32107 2.14953 2.03632
Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 129
C 1.42668 1.66074 1.64176
H -0.62044 -3.21590 -2.79260
C -0.26894 -2.29684 -2.32508
H -0.16351 -1.54929 -3.11643
C 0.07400 -1.39425 1.39129
O 0.21299 -2.12150 2.28519
C 0.67706 1.64014 -2.44635
C -0.58216 1.79212 -3.27239
H -0.00673 0.90530 -1.22750
H -0.88391 0.82086 -3.65941
H -1.41985 2.23727 -2.74079
H -0.34817 2.43435 -4.12669
C 0.18411 2.08714 2.06050
H 0.07321 2.87826 2.79056
C 1.06656 -2.47386 -1.63520
H 1.82500 -2.81600 -2.34619
H 1.00967 -3.21421 -0.83132
O 1.57129 0.86697 -2.90976
H 1.60122 -0.51023 -1.82268
C 1.13413 2.86569 -1.69011
C 0.23944 3.81314 -1.19233
C 2.49972 3.08926 -1.51854
C 0.69564 4.92638 -0.50231
H -0.82520 3.67764 -1.33122
C 2.95918 4.20000 -0.82754
H 3.20023 2.38211 -1.94636
C 2.05698 5.11990 -0.30904
H -0.01663 5.64480 -0.11318
H 4.02480 4.35281 -0.70004
H 2.41299 5.98763 0.23331
C -2.95817 -1.15880 -2.05211
C -4.15416 -0.96521 -1.35512
C -2.94677 -0.97326 -3.43304
C -5.30606 -0.58218 -2.02134
H -4.18297 -1.10806 -0.27953
C -4.10372 -0.58774 -4.09874
H -2.04046 -1.13162 -4.00360
C -5.28276 -0.38841 -3.39689
H -6.22396 -0.43349 -1.46539
H -4.07986 -0.44869 -5.17286
H -6.18241 -0.08727 -3.91948
C -2.09756 -3.06372 -0.16061
C -2.13882 -4.34352 -0.71208
C -2.61634 -2.86430 1.11911
C -2.67462 -5.40102 0.00963
H -1.75632 -4.52634 -1.70905
C -3.16609 -3.91823 1.83311
H -2.58565 -1.87472 1.56325
C -3.19166 -5.19094 1.28045
H -2.69224 -6.39236 -0.42689
H -3.56773 -3.74208 2.82396
H -3.61224 -6.01840 1.83884
C -2.31151 2.48017 0.56020
C -2.96682 2.21476 -0.63764
C -2.52155 3.70781 1.18941
C -3.80999 3.15991 -1.20874
H -2.79928 1.26927 -1.13903
C -3.35993 4.65333 0.61881
H -2.01759 3.93216 2.12324
C -4.00435 4.38247 -0.58303
H -4.30577 2.94224 -2.14794
H -3.50956 5.60668 1.11183
H -4.65497 5.12500 -1.02906
C -2.25626 0.56842 2.64350
C -1.67116 0.05884 3.80247
C -3.63791 0.45449 2.48044
C -2.44719 -0.55946 4.77201
H -0.59810 0.14977 3.94064
C -4.41352 -0.16569 3.45002
H -4.11290 0.85070 1.58912
C -3.81977 -0.67772 4.59594
H -1.97909 -0.94896 5.66835
H -5.48511 -0.24683 3.31027
H -4.42552 -1.16160 5.35262
C 3.90866 -1.69238 -0.97572
C 4.69343 -2.71453 -0.44984
C 4.29900 -1.08790 -2.17108
C 5.84971 -3.12499 -1.09982
H 4.39550 -3.18530 0.48136
C 5.45068 -1.50117 -2.82388
H 3.69326 -0.28984 -2.59032
C 6.23069 -2.51855 -2.28837
H 6.45262 -3.91978 -0.67719
H 5.74345 -1.02549 -3.75230
H 7.13155 -2.83849 -2.79781
C 3.98381 0.26325 1.34120
C 5.19031 0.89401 1.05423
C 3.82610 -0.35651 2.58092
C 6.22586 0.89807 1.98051
H 5.31565 1.38538 0.09401
C 4.85722 -0.35356 3.50681
H 2.88348 -0.83922 2.81958
C 6.06220 0.27213 3.20744
H 7.15976 1.39393 1.74383
H 4.72295 -0.84069 4.46542
H 6.86719 0.27535 3.93251
TSS
Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 130
H◦ -4178.297990
G◦ -4178.438209
Fe 0.27142 0.25308 -0.09636
P -1.04321 0.22286 1.74612
P -0.96481 0.92369 -1.87142
N 1.67483 -0.66893 1.05793
N 1.75041 0.12644 -1.41262
H 3.06340 0.86880 0.84153
C 3.00995 -0.19928 0.60802
H 3.12056 -1.43904 -1.12840
C 3.03061 -0.35842 -0.92904
H 2.38385 0.50206 -3.35743
C 1.54264 0.52236 -2.66012
H -0.27069 -0.91095 3.82843
C -0.02190 -0.93091 2.76780
H -0.22538 -1.94316 2.40485
C 0.80158 1.88972 0.22550
O 1.23202 2.95464 0.39490
C 0.26500 -2.87554 -0.81900
H -0.18147 -1.38233 -0.49212
C 0.31007 0.95717 -3.10203
H 0.16782 1.32245 -4.11072
C 1.43295 -0.58861 2.51002
H 2.09292 -1.27228 3.05291
H 1.66663 0.42817 2.84100
O 1.31344 -3.11507 -0.14544
H 1.58920 -1.65998 0.77096
C -2.78813 -0.33884 1.90972
C -3.82153 0.60199 1.89553
C -3.11438 -1.69362 1.96030
C -5.14665 0.19662 1.93330
H -3.59614 1.66110 1.85303
C -4.44254 -2.09452 2.01403
H -2.33556 -2.44630 1.96193
C -5.46183 -1.15361 1.99596
H -5.93404 0.94078 1.92075
H -4.67771 -3.15050 2.07005
H -6.49683 -1.47087 2.03274
C -1.09369 1.79435 2.70842
C -0.88633 1.83559 4.08624
C -1.39759 2.98111 2.03867
C -0.97000 3.03850 4.77409
H -0.66001 0.93108 4.63716
C -1.49639 4.17936 2.72875
H -1.55967 2.96741 0.96521
C -1.27759 4.21090 4.09910
H -0.79728 3.05596 5.84347
H -1.73823 5.08907 2.19215
H -1.34528 5.14731 4.63943
C -2.37699 -0.08887 -2.48917
C -3.43549 -0.42567 -1.64389
C -2.39686 -0.54945 -3.80525
C -4.50563 -1.17388 -2.11278
H -3.41832 -0.11443 -0.60651
C -3.45794 -1.31583 -4.26822
H -1.57072 -0.31477 -4.46704
C -4.51828 -1.62320 -3.42593
H -5.31984 -1.42386 -1.44252
H -3.45702 -1.67052 -5.29212
H -5.34669 -2.21939 -3.78952
C -1.70469 2.61671 -1.77675
C -0.86580 3.71306 -1.98359
C -3.02632 2.85008 -1.39359
C -1.33238 5.00704 -1.80518
H 0.16251 3.54441 -2.28708
C -3.49258 4.14553 -1.21613
H -3.70466 2.02155 -1.22998
C -2.64718 5.22768 -1.41724
H -0.66700 5.84564 -1.97333
H -4.52255 4.30784 -0.92063
H -3.01225 6.23800 -1.27786
C 4.17295 -0.88926 1.27000
C 5.18718 -0.13515 1.85399
C 4.28244 -2.27980 1.26702
C 6.29396 -0.75335 2.41953
H 5.10833 0.94735 1.85661
C 5.38477 -2.89862 1.83783
H 3.49910 -2.87725 0.81200
C 6.39493 -2.13736 2.41231
H 7.07716 -0.15327 2.86719
H 5.45807 -3.97969 1.83099
H 7.25662 -2.62238 2.85482
C 4.24666 0.32890 -1.50228
C 5.34645 -0.41480 -1.91936
C 4.31672 1.72094 -1.55687
C 6.49724 0.21553 -2.37520
H 5.29888 -1.49836 -1.87969
C 5.46326 2.35234 -2.01316
H 3.46093 2.30991 -1.24207
C 6.55883 1.60069 -2.42169
H 7.34579 -0.37728 -2.69564
H 5.50415 3.43461 -2.04996
H 7.45435 2.09450 -2.77923
C 0.42495 -2.69630 -2.31242
H 0.49055 -3.68892 -2.76758
H -0.39850 -2.16288 -2.78468
H 1.35093 -2.16638 -2.52096
C -1.00414 -3.56227 -0.37251

Chapter 7. Iron PNNP [5.5.5] Complexes: Mechanism of Hydrogenation 131
C -2.17914 -3.52680 -1.12371
C -0.97264 -4.34513 0.78162
C -3.29224 -4.25425 -0.72909
H -2.22737 -2.93234 -2.02840
C -2.08296 -5.07712 1.17469
H -0.05394 -4.38620 1.35428
C -3.24684 -5.03560 0.41811
H -4.19745 -4.21637 -1.32419
H -2.03840 -5.68622 2.07000
H -4.11446 -5.61024 0.71987

Chapter 8
Conclusions and Future Work
A series of synthetic and computational studies have been conducted which revolve around the general
theme of metal-ligand cooperation (MLC) for applications in water splitting and ketone hydrogenation,
respectively. In the Chapters 2–5, new ruthenium and osmium complexes have been synthesized, which
in most cases contain ligands that participate in proximal or distal MLC. In Chapters 6 and 7, the
mechanism of iron-catalyzed ATH and AH has been explored in depth with our first, second, and third
generation Fe-PNNP catalysts.
Chapter 2 has covered the synthesis of new ruthenium and osmium hydrido-hydroxo complexes
that exhibit unique intramolecular proton transfer behaviour, requiring VT-NMR studies for solution
state characterization.385 Molecular structures of two new ruthenium and osmium hydrido-hydroxo
compounds have also been reported, which exist as water-bridged H-bonding dimers in the solid state.
Unfortunately, thermal or photochemical excitation leads to the rapid and irreversible oxidation of
the PPh3 ligands, releasing free tmen ligand and rendering these systems inactive for water splitting.
Future work using the tmen ligand as a MLC ligand for water splitting could include substitution of the
monodentate phosphine ligands for a chelating diphosphine ligand, or even a second tmen ligand.386 A
chelating amino-carbene or pyridyl-carbene ligand could also be incorporated in the place of phosphines.
Since water splitting catalysts using earth-abundant metals would be a desirable goal in this field, tmen
coordination complexes using iron should also be explored.
Chapter 3 has explored the coordination chemistry of the di-(2-pyridylmethyl)amine donor ligand.
Three new metal complexes have been synthesized, and all of these compounds have been struc-
turally characterized.385 It was originally envisioned that the central NH donor atom of the di-(2-
pyridylmethyl)amine ligand could be deprotonated and act as a proximal MLC donor atom to het-
erolytically cleave water. Instead, we discovered that deprotonation with base leads to the formation
of a well-defined ruthenaaziridine complex, followed by evolution of H2 to form a very stable azaallyl
ruthenium species. Future work using this ligand system for MLC applications, or a modified derivative,
would begin by fully understanding the reaction mechanism on going from di-(2-pyridylmethyl)amine
→ ruthenaaziridine → azaallyl. The mechanism of reaction could be investigated by NMR or UV-vis
spectroscopy, since the reaction is rather slow (several hours or longer) and the resultant species are
all strongly absorbing in the visible spectrum. This investigation could also be accompanied by DFT
calculations to help identify key intermediates and transition states.
Chapter 4 has described the synthesis of new ruthenium NCN pincer complexes, which hold the great-
132

Chapter 8. Conclusions and Future Work 133
est promise for applications in water splitting.232 The rigid nature of the carbene-centered NCN ligand
enforces a meridional ligand coordination geometry while the absence of phosphine ligands in the coordi-
nation sphere holds promise for its survival in oxidizing chemical environments; ruthenium and iridium
carbene complexes are known be water oxidation catalysts in the presence of a sacrificial oxidant.387–389
Future work includes the irradiation of the deeply coloured hydroxo complex 71 with light, which might
promote the formation of hydrogen peroxide, ·OH radicals, and or even O2 (Scheme 1.6). Reaction of 69
with H2 would also be important, since well-defined ruthenium hydride compounds might subsequently
react with water to generate H2 and O2. A potential water splitting cycle is shown in Scheme 8.1 in-
volving MLC in the H2 and O2 formation steps. In addition, the modular syntheses of these complexes
also allows for the installation of other counter-ions in place of PF–6 and other multidentate ligands in
place of bipyridine.
NN
N
NRu
N
N
PhPh O
HH
H
H2O
-H2
NN
N
NRuII
N
N
PhPh
HH
H
H2O "HOOH"0.5 O2+
NN
N
N
Ru
N
N
PhPh H
H
H
H
H
O
H
NN
N
N
Ru
N
N
PhPh O
H
H
H
H
O
H
H
H2/H2O
- tBuOH hν
NN
N
NRu0
N
N
PhPh
HH
H
H?
H
H2O
+- tBuOHH2
69
Scheme 8.1: Potential water splitting cycle with RuNCN pincer complexes.
Chapter 5 has described the synthesis of phosphine-free ruthenium complexes using tetradentate
amino-olefin ligands. The π-accepting nature of the trop olefinic moieties allowed for the stabilization of
low valent ruthenium complexes, ranging from formally Ru(0) to Ru(-II) oxidation states. The previously
reported complex [K][RuH(trop2dad)], which is known to produce H2 in the presence of H2O, was
investigated under various reaction conditions relevant to water splitting. Unfortunately, the trop2dad
ligand framework on ruthenium did not lead to any desirable outcomes. However, the tetradentate
trop2dach ligand was successfully coordinated to ruthenium, stabilizing new four-coordinate Ru(0) and
Ru(-II) complexes. Dianionic four-coordinate ruthenium complexes are rare and may readily react with
H-X compounds such as H2O and/or H2. Future work includes obtaining a molecular structure of Ru(-II)
complex [K2][Ru–II
(trop2dach)] (83) and investigating its reactivity for applications in water splitting.
Additional work may also involve the incorporation of other functionalized trop2(diamine) ligands, such
as trop2dach or trop2tmen, onto ruthenium.
Chapter 6 has investigated the mechanism of reaction using our first generation Fe-PNNP [6.5.6]
complexes during ATH.297 Structural characterization of a κ5-ferraaziridine complex (86) indicated to
us that the large six-membered metallacycles are able to easily fold and distort themselves under ATH

Chapter 8. Conclusions and Future Work 134
reaction conditions. A mechanism of reaction was proposed, based on DFT calculations, to generate this
ferraaziridine complex. An additional mechanism was calculated that examined the thermodynamically
favourable formation of Fe(0) compounds during catalysis, which supported the experimentally observed
CO stretches in IR spectra. Furthermore, these pieces of evidence strongly supported experiments
which showed that our first generation Fe-PNNP [6.5.6] catalysts are in fact enantioselective 4 nm
Fe(0) nanoparticle catalysts coated with chiral ligands.300 Future work in this field could entail the
development of a modular and general method to access enantioselective iron nanoparticle catalysts for
the ATH of ketones.
Chapter 7 has dealt with detailed mechanistic investigations using our second and third generation
Fe-PNNP [5.5.5] catalysts. Using a combination of experiment and theory, our lab has developed a
detailed understanding of how these catalysts operate during ATH and AH. Key computational find-
ings,346 in collaboration with kinetic studies,302 showed that our second generation catalysts underwent
an activation period which selectively reduced an imine group on the PNNP ligand, generating the
true catalyst, an amido-eneamido iron species. This led to the rational development of third genera-
tion catalysts which contain an unsymmetrical PN(H)NP ligand and are able to immediately enter the
catalytic cycle after reaction with base to generate the most active ATH catalysts in existence.46 DFT
calculations also revealed that these third generation catalysts are poor AH catalysts due to the high,
rate-determining heterolytic H2 splitting barrier.45 Future work includes expanding calculations into the
origins of enantioselectivity to include different levels of theory and a wider substrate scope. This would
help us understand which regions of the catalyst to modify first in order to maximize selectivity during
the enantiodetermining step, saving time and resources in our quest for developing cheap, abundant, and
robust hydrogenation catalysts. It is not well understood why our third generation complexes are less
enantioselective for AH versus ATH, and theoretical investigations into side reaction or decomposition
pathways would be beneficial (Scheme 7.10).
Metal-ligand cooperation has left an indelible mark in the field of ligand design and catalyst de-
velopment. The possible variations of metal, oxidation state, donor/acceptor atoms, linkers, denticity,
and charge are virtually endless in the rational development of new catalyst systems. With the rapid
development of new pharmaceuticals, agrochemicals, and fragrances, it is certain that MLC elements
will continue to hold a privileged position in catalysis for industrial applications – without the need
for expensive and toxic metals if clever and creative ligands are employed. In a world with dwindling
petroleum supplies and rocketing CO2 levels, organometallic chemists hold the key to unlocking the true
potential of MLC for renewable energy applications, which would give humanity a new hope in securing
its energy future for generations to come.

Bibliography
[1] Halpern, J. Inorg. Chim. Acta 1981, 50, 11 – 19. (page 1)
[2] Devillard, M.; Bouhadir, G.; Bourissou, D. Angew. Chem. Int. Ed. 2015, 54, 730–732. (page 1)
[3] Ward, M. D.; McCleverty, J. A. J. Chem. Soc., Dalton Trans. 2002, 275–288. (page 1)
[4] Mukherjee, S.; Weyhermuller, T.; Bothe, E.; Wieghardt, K.; Chaudhuri, P. Dalton Trans. 2004,
3842–3853. (page 1)
[5] Hetterscheid, D. G. H.; Kaiser, J.; Reijerse, E.; Peters, T. P. J.; Thewissen, S.; Blok, A. N. J.;
Smits, J. M. M.; de Gelder, R.; de Bruin, B. J. Am. Chem. Soc. 2005, 127, 1895–1905. (page 1)
[6] Annibale, V. T.; Song, D. RSC Adv. 2013, 3, 11432–11449. (page 1, 39)
[7] Berben, L. A.; de Bruin, B.; Heyduk, A. F. Chem. Commun. 2015, 51, 1553–1554. (page 1)
[8] Noyori, R.; Hashiguchi, S. Acc. Chem. Res. 1997, 30, 97–102. (page 1, 4, 5)
[9] Haack, K.-J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R. Angew. Chem. Int. Ed. Engl. 1997,
36, 285–288. (page 1, 4, 5)
[10] Ben-Ari, E.; Leitus, G.; Shimon, L. J. W.; Milstein, D. J. Am. Chem. Soc. 2006, 128, 15390–15391.
(page 1)
[11] van der Vlugt, J.; Reek, J. Angew. Chem. Int. Ed. 2009, 48, 8832–8846. (page 1, 29)
[12] Noyori, R.; Ohkuma, T. Angew. Chem. Int. Ed. 2001, 40, 40–73. (page 1, 5, 30)
[13] Noyori, R.; Yamakawa, M.; Hashiguchi, S. J. Org. Chem. 2001, 66, 7931–7944. (page 1, 5)
[14] Clapham, S. E.; Hadzovic, A.; Morris, R. H. Coord. Chem. Rev. 2004, 248, 2201 – 2237. (page 1,
16, 30, 81, 95)
[15] Ikariya, T.; Murata, K.; Noyori, R. Org. Biomol. Chem. 2006, 4, 393–406. (page 1, 5, 30)
[16] Ikariya, T.; Blacker, A. J. Acc. Chem. Res. 2007, 40, 1300–1308. (page 1, 30, 81, 94, 95)
[17] Grutzmacher, H. Angew. Chem. Int. Ed. 2008, 47, 1814–1818. (page 1)
[18] Ikariya, T.; Gridnev, I. Top. Catal. 2010, 53, 894–901. (page 1, 30, 95)
[19] Kuwata, S.; Ikariya, T. Dalton Trans. 2010, 39, 2984–2992. (page 1, 30, 95)
135

BIBLIOGRAPHY 136
[20] Grotjahn, D. Top. Catal. 2010, 53, 1009–1014. (page 1, 30, 95)
[21] Milstein, D. Top. Catal. 2010, 53, 915–923. (page 1, 29, 40, 95)
[22] Gunanathan, C.; Milstein, D. Bond Activation by Metal-Ligand Cooperation: Design of “Green”
Catalytic Reactions Based on Aromatization-Dearomatization of Pincer Complexes. In Bifunc-
tional Molecular Catalysis; Ikariya, T., Shibasaki, M., Eds.; Springer Berlin / Heidelberg, 2011;
Vol. 37, pp 55–84. (page 1)
[23] Ikariya, T. Bull. Chem. Soc. Jpn. 2011, 84, 1–16. (page 1, 30)
[24] Gunanathan, C.; Milstein, D. Acc. Chem. Res. 2011, 44, 588–602. (page 1, 40, 85)
[25] van der Vlugt, J. I. Eur. J. Inorg. Chem. 2012, 2012, 363–375. (page 1, 39)
[26] Schneider, S.; Meiners, J.; Askevold, B. Eur. J. Inorg. Chem. 2012, 2012, 412–429. (page 1)
[27] Gelman, D.; Musa, S. ACS Catal. 2012, 2, 2456–2466. (page 1, 39)
[28] Askevold, B.; Roesky, H. W.; Schneider, S. ChemCatChem 2012, 4, 307–320. (page 1, 39, 95)
[29] Eisenstein, O.; Crabtree, R. H. New J. Chem. 2013, 37, 21–27. (page 1, 94)
[30] Fryzuk, M. D.; MacNeil, P. A. Organometallics 1983, 2, 682–684. (page 2)
[31] Fryzuk, M. D.; MacNeil, P. A. Organometallics 1983, 2, 355–356. (page 2)
[32] Fryzuk, M. D.; MacNeil, P. A.; Rettig, S. J. Organometallics 1985, 4, 1145–1147. (page 2, 29)
[33] Fryzuk, M. D.; MacNeil, P. A.; Rettig, S. J. Organometallics 1986, 5, 2469–2476. (page 2)
[34] Fryzuk, M. D.; MacNeil, P. A.; Rettig, S. J. J. Am. Chem. Soc. 1987, 109, 2803–2812. (page 2)
[35] Fryzuk, M. D.; Montgomery, C. D.; Rettig, S. J. Organometallics 1991, 10, 467–473. (page 2)
[36] Abdur-Rashid, K.; Faatz, M.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2001, 123, 7473–
7474. (page 4, 16, 30)
[37] Abdur-Rashid, K.; Clapham, S. E.; Hadzovic, A.; Harvey, J. N.; Lough, A. J.; Morris, R. H. J.
Am. Chem. Soc. 2002, 124, 15104–15118. (page 4, 5, 6, 16, 23, 30, 34, 95)
[38] Clapham, S. E.; Morris, R. H. Organometallics 2005, 24, 479–481. (page 4, 16, 20, 23, 25)
[39] Maire, P.; Buttner, T.; Breher, F.; Le Floch, P.; Grutzmacher, H. Angew. Chem. Int. Ed. 2005,
44, 6318–6323. (page 4, 70)
[40] Ohkuma, T.; Koizumi, M.; Muniz, K.; Hilt, G.; Kabuto, C.; Noyori, R. J. Am. Chem. Soc. 2002,
124, 6508–6509. (page 4)
[41] Hamilton, R. J.; Bergens, S. H. J. Am. Chem. Soc. 2006, 128, 13700–13701. (page 4, 6, 11, 16,
17, 106)
[42] Kaß, M.; Friedrich, A.; Drees, M.; Schneider, S. Angew. Chem. Int. Ed. 2009, 48, 905–907. (page
4, 29, 85)

BIBLIOGRAPHY 137
[43] Marziale, A. N.; Friedrich, A.; Klopsch, I.; Drees, M.; Celinski, V. R.; Schmedt auf der Gunne, J.;
Schneider, S. J. Am. Chem. Soc. 2013, 135, 13342–13355. (page 4)
[44] Touge, T.; Hakamata, T.; Nara, H.; Kobayashi, T.; Sayo, N.; Saito, T.; Kayaki, Y.; Ikariya, T. J.
Am. Chem. Soc. 2011, 133, 14960–14963. (page 4)
[45] Zuo, W.; Tauer, S.; Prokopchuk, D. E.; Morris, R. H. Organometallics 2014, 33, 5791–5801. (page
4, 95, 97, 112, 113, 117, 134)
[46] Zuo, W.; Lough, A. J.; Li, Y. F.; Morris, R. H. Science 2013, 342, 1080–1083. (page vii, 4, 14,
85, 95, 97, 110, 111, 134)
[47] Zhang, G.; Scott, B. L.; Hanson, S. K. Angew. Chem. Int. Ed. 2012, 51, 12102–12106. (page 4)
[48] Zhang, G.; Hanson, S. K. Chem. Commun. 2013, 49, 10151–10153. (page 4)
[49] Burford, R. J.; Piers, W. E.; Parvez, M. Organometallics 2012, 31, 2949–2952. (page 4)
[50] Chakraborty, S.; Brennessel, W. W.; Jones, W. D. J. Am. Chem. Soc. 2014, 136, 8564–8567.
(page 4)
[51] Chakraborty, S.; Lagaditis, P. O.; Forster, M.; Bielinski, E. A.; Hazari, N.; Holthausen, M. C.;
Jones, W. D.; Schneider, S. ACS Catal. 2014, 4, 3994–4003. (page 4)
[52] Chakraborty, S.; Dai, H.; Bhattacharya, P.; Fairweather, N. T.; Gibson, M. S.; Krause, J. A.;
Guan, H. J. Am. Chem. Soc. 2014, 136, 7869–7872. (page 4)
[53] Werkmeister, S.; Junge, K.; Wendt, B.; Alberico, E.; Jiao, H.; Baumann, W.; Junge, H.; Gallou, F.;
Beller, M. Angew. Chem. Int. Ed. 2014, 53, 8722–8726. (page 4)
[54] Bornschein, C.; Werkmeister, S.; Wendt, B.; Jiao, H.; Alberico, E.; Baumann, W.; Junge, H.;
Junge, K.; Beller, M. Nat. Commun. 2014, 5, 1–11. (page 4)
[55] Ohkuma, T.; Ishii, D.; Takeno, H.; Noyori, R. J. Am. Chem. Soc. 2000, 122, 6510–6511. (page 4)
[56] Baratta, W.; Chelucci, G.; Magnolia, S.; Siega, K.; Rigo, P. Chem. Eur. J. 2009, 15, 726–732.
(page 4)
[57] Gao, J.-X.; Ikariya, T.; Noyori, R. Organometallics 1996, 15, 1087–1089. (page 4)
[58] Gao, J.-X.; Zhang, H.; Yi, X.-D.; Xu, P.-P.; Tang, C.-L.; Wan, H.-L.; Tsai, K.-R.; Ikariya, T.
Chirality 2000, 12, 383–388. (page 4)
[59] Rautenstrauch, V.; Hoang-Cong, X.; Churlaud, R.; Abdur-Rashid, K.; Morris, R. H. Chem. Eur.
J. 2003, 9, 4954–4967. (page 4)
[60] Li, T.; Churlaud, R.; Lough, A. J.; Abdur-Rashid, K.; Morris, R. H. Organometallics 2004, 23,
6239–6247. (page 5, 6)
[61] Zimmer-De Iuliis, M.; Morris, R. H. J. Am. Chem. Soc. 2009, 131, 11263–11269. (page 5, 6, 16,
30, 95, 117)
[62] Hamilton, R. J.; Bergens, S. H. J. Am. Chem. Soc. 2008, 130, 11979–11987. (page 6, 106)

BIBLIOGRAPHY 138
[63] Takebayashi, S.; Dabral, N.; Miskolzie, M.; Bergens, S. H. J. Am. Chem. Soc. 2011, 133, 9666–
9669. (page 6, 106)
[64] John, J. M.; Takebayashi, S.; Dabral, N.; Miskolzie, M.; Bergens, S. H. J. Am. Chem. Soc. 2013,
135, 8578–8584. (page xii, 6, 7)
[65] Hasanayn, F.; Morris, R. H. Inorg. Chem. 2012, 51, 10808–10818. (page 6, 106)
[66] Dub, P. A.; Ikariya, T. J. Am. Chem. Soc. 2013, 135, 2604–2619. (page 6, 95)
[67] Dub, P. A.; Henson, N. J.; Martin, R. L.; Gordon, J. C. J. Am. Chem. Soc. 2014, 136, 3505–3521.
(page 6, 70, 95)
[68] Chen, H.-Y. T.; Di Tommaso, D.; Hogarth, G.; Catlow, C. R. A. Dalton Trans. 2011, 40, 402–412.
(page 6)
[69] Shvo, Y.; Czarkie, D.; Rahamim, Y.; Chodosh, D. F. J. Am. Chem. Soc. 1986, 108, 7400–7402.
(page 8)
[70] Conley, B. L.; Pennington-Boggio, M. K.; Boz, E.; Williams, T. J. Chem. Rev. 2010, 110, 2294–
2312. (page 8)
[71] Casey, C. P.; Guan, H. J. Am. Chem. Soc. 2007, 129, 5816–5817. (page 8)
[72] Casey, C. P.; Guan, H. J. Am. Chem. Soc. 2009, 131, 2499–2507. (page 8)
[73] Gunanathan, C.; Milstein, D. Chem. Rev. 2014, 114, 12024–12087. (page 8)
[74] Balaraman, E.; Gunanathan, C.; Zhang, J.; Shimon, L. J. W.; Milstein, D. Nat. Chem. 2011, 3,
609–614. (page 8, 40)
[75] Langer, R.; Leitus, G.; Ben-David, Y.; Milstein, D. Angew. Chem. Int. Ed. 2011, 50, 2120–2124.
(page 8)
[76] Sun, Y.; Koehler, C.; Tan, R.; Annibale, V. T.; Song, D. Chem. Commun. 2011, 47, 8349–8351.
(page 8, 40)
[77] del Pozo, C.; Iglesias, M.; Sanchez, F. Organometallics 2011, 30, 2180–2188. (page 8, 40)
[78] Wilson, A. D.; Newell, R. H.; McNevin, M. J.; Muckerman, J. T.; Rakowski DuBois, M.;
DuBois, D. L. J. Am. Chem. Soc. 2006, 128, 358–366. (page 8, 10)
[79] Hulley, E. B.; Helm, M. L.; Bullock, R. M. Chem. Sci. 2014, 5, 4729–4741. (page 8)
[80] Liu, T.; Wang, X.; Hoffmann, C.; DuBois, D. L.; Bullock, R. M. Angew. Chem. Int. Ed. 2014, 53,
5300–5304. (page 8)
[81] Phillips, A. D.; Laurenczy, G.; Scopelliti, R.; Dyson, P. J. Organometallics 2007, 26, 1120–1122.
(page 8)
[82] Stepowska, E.; Jiang, H.; Song, D. Chem. Commun. 2010, 46, 556–558. (page 8)
[83] Annibale, V. T.; Song, D. Chem. Commun. 2012, 48, 5416–5418. (page 8)

BIBLIOGRAPHY 139
[84] Rodrıguez-Lugo, R. E.; Trincado, M.; Vogt, M.; Tewes, F.; Santiso-Quinones, G.; Grutzmacher, H.
Nat. Chem. 2013, 5, 342–347. (page 8, 39, 70, 71, 72, 74, 75)
[85] Vogt, M.; Nerush, A.; Iron, M. A.; Leitus, G.; Diskin-Posner, Y.; Shimon, L. J. W.; Ben-David, Y.;
Milstein, D. J. Am. Chem. Soc. 2013, 135, 17004–17018. (page 8)
[86] Vogt, M.; Nerush, A.; Diskin-Posner, Y.; Ben-David, Y.; Milstein, D. Chem. Sci. 2014, 5, 2043–
2051. (page 8)
[87] Lewis, N. S.; Nocera, D. G. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15729–15735. (page 8, 15,
40)
[88] Crabtree, R. H. Organometallics 2011, 30, 17–19. (page 8, 15, 40)
[89] Piers, W. E. Organometallics 2011, 30, 13–16. (page 8, 15, 40)
[90] Klahn, M.; Beweries, T. Rev. Inorg. Chem. 2014, 34, 177–198. (page 8)
[91] Karkas, M. D.; Verho, O.; Johnston, E. V.; Akermark, B. Chem. Rev. 2014, 114, 11863–12001.
(page 8, 10)
[92] McEvoy, J. P.; Brudvig, G. W. Chem. Rev. 2006, 106, 4455–4483. (page 8)
[93] Darensbourg, M. Y.; Lyon, E. J.; Zhao, X.; Georgakaki, I. P. Proc. Natl. Acad. Sci. U. S. A. 2003,
100, 3683–3688. (page 8)
[94] Fontecilla-Camps, J. C.; Volbeda, A.; Cavazza, C.; Nicolet, Y. Chem. Rev. 2007, 107, 4273–4303.
(page 8)
[95] Tard, C.; Pickett, C. J. Chem. Rev. 2009, 109, 2245–2274. (page 8)
[96] Yagi, M.; Kaneko, M. Chem. Rev. 2001, 101, 21–36. (page 8, 15)
[97] Alstrum-Acevedo, J. H.; Brennaman, M. K.; Meyer, T. J. Inorg. Chem. 2005, 44, 6802–6827.
(page 8, 15)
[98] Concepcion, J. J.; Jurss, J. W.; Brennaman, M. K.; Hoertz, P. G.; Patrocinio, A. O. T.; Mu-
rakami Iha, N. Y.; Templeton, J. L.; Meyer, T. J. Acc. Chem. Res. 2009, 42, 1954–1965. (page 8,
40)
[99] Sala, X.; Romero, I.; Rodrıguez, M.; Escriche, L.; Llobet, A. Angew. Chem. Int. Ed. 2009, 48,
2842–2852. (page 8, 15, 40)
[100] Concepcion, J. J.; Tsai, M.-K.; Muckerman, J. T.; Meyer, T. J. J. Am. Chem. Soc. 2010, 132,
1545–1557. (page 8, 40)
[101] Liu, X.; Wang, F. Coord. Chem. Rev. 2012, 256, 1115 – 1136. (page 8, 40)
[102] Concepcion, J. J.; Jurss, J. W.; Templeton, J. L.; Meyer, T. J. J. Am. Chem. Soc. 2008, 130,
16462–16463. (page 8)
[103] Rakowski DuBois, M.; DuBois, D. L. Chem. Soc. Rev. 2009, 38, 62–72. (page 10)

BIBLIOGRAPHY 140
[104] Helm, M. L.; Stewart, M. P.; Bullock, R. M.; DuBois, M. R.; DuBois, D. L. Science 2011, 333,
863–866. (page 10)
[105] DuBois, D. L. Inorg. Chem. 2014, 53, 3935–3960. (page 10)
[106] Gratzel, M. Nature 2001, 414, 338–344. (page 10)
[107] Dempsey, J. L.; Brunschwig, B. S.; Winkler, J. R.; Gray, H. B. Acc. Chem. Res. 2009, 42, 1995–
2004. (page 10)
[108] Han, Z.; Eisenberg, R. Acc. Chem. Res. 2014, 47, 2537–2544. (page 10)
[109] Joya, K. S.; Joya, Y. F.; Ocakoglu, K.; van de Krol, R. Angew. Chem. Int. Ed. 2013, 52, 10426–
10437. (page 10, 40)
[110] Nocera, D. G. Acc. Chem. Res. 2012, 45, 767–776. (page 10, 40)
[111] Kohl, S. W.; Weiner, L.; Schwartsburd, L.; Konstantinovski, L.; Shimon, L. J. W.; Ben-David, Y.;
Iron, M. A.; Milstein, D. Science 2009, 324, 74–77. (page 10, 16, 17, 20, 21, 29, 40, 47, 70)
[112] Eisenberg, R. Science 2009, 324, 44–45. (page 10, 15, 40)
[113] Hetterscheid, D.; van der Vlugt, J.; de Bruin, B.; Reek, J. Angew. Chem. Int. Ed. 2009, 48,
8178–8181. (page 10, 15, 40)
[114] Li, J.; Shiota, Y.; Yoshizawa, K. J. Am. Chem. Soc. 2009, 131, 13584–13585. (page 11, 40)
[115] Chen, Y.; Fang, W.-H. J. Phys. Chem. A 2010, 114, 10334–10338. (page 11)
[116] Yang, X.; Hall, M. B. J. Am. Chem. Soc. 2010, 132, 120–130. (page 11, 40)
[117] Sandhya, K. S.; Suresh, C. H. Organometallics 2011, 30, 3888–3891. (page 11, 40)
[118] Ma, C.; Piccinin, S.; Fabris, S. ACS Catal. 2012, 2, 1500–1506. (page 11)
[119] Kunkely, H.; Vogler, A. Angew. Chem. Int. Ed. 2009, 48, 1685–1687. (page 11)
[120] Chen, Y.; Han, J.; Fang, W.-H. Inorg. Chem. 2012, 51, 4938–4946. (page 11)
[121] Wickramasinghe, L. A.; Sharp, P. R. J. Am. Chem. Soc. 2014, 136, 13979–13982. (page 11)
[122] Godemann, C.; Dura, L.; Hollmann, D.; Grabow, K.; Bentrup, U.; Jiao, H.; Schulz, A.; Bruck-
ner, A.; Beweries, T. Chem. Commun. 2015, 51, 3065–3068. (page 11)
[123] Gutsulyak, D. V.; Piers, W. E.; Borau-Garcia, J.; Parvez, M. J. Am. Chem. Soc. 2013, 135,
11776–11779. (page 11, 39)
[124] Sues, P. E.; Demmans, K. Z.; Morris, R. H. Dalton Trans. 2014, 43, 7650–7667. (page 13)
[125] Zuo, W.; Morris, R. H. Nat. Protoc. 2015, 10, 241–257. (page 14, 95)
[126] Cook, T. R.; Dogutan, D. K.; Reece, S. Y.; Surendranath, Y.; Teets, T. S.; Nocera, D. G. Chem.
Rev. 2010, 110, 6474–6502. (page 15)

BIBLIOGRAPHY 141
[127] Ozerov, O. V. Chem. Soc. Rev. 2009, 38, 83–88 and references therein. (page 16)
[128] Millard, M. D.; Moore, C. E.; Rheingold, A. L.; Figueroa, J. S. J. Am. Chem. Soc. 2010, 132,
8921–8923. (page 16)
[129] Klaring, P.; Pahl, S.; Braun, T.; Penner, A. Dalton Trans. 2011, 40, 6785–6791. (page 16)
[130] Kessler, M.; Hansen, S.; Hollmann, D.; Klahn, M.; Beweries, T.; Spannenberg, A.; Bruckner, A.;
Rosenthal, U. Eur. J. Inorg. Chem. 2011, 2011, 627–631. (page 16)
[131] Parkin, G.; Bercaw, J. E. Polyhedron 1988, 7, 2053 – 2082. (page 16)
[132] Jee, J.-E.; Comas-Vives, A.; Dinoi, C.; Ujaque, G.; van Eldik, R.; Lledos, A.; Poli, R. Inorg. Chem.
2007, 46, 4103–4113. (page 16)
[133] Poli, R. Coord. Chem. Rev. 2008, 252, 1592 – 1612. (page 16)
[134] Poverenov, E.; Efremenko, I.; Frenkel, A. I.; Ben-David, Y.; Shimon, L. J. W.; Leitus, G.; Kon-
stantinovski, L.; Martin, J. M. L.; Milstein, D. Nature 2008, 455, 1093–1096. (page 16)
[135] Burn, M. J.; Fickes, M. G.; Hartwig, J. F.; Hollander, F. J.; Bergman, R. G. J. Am. Chem. Soc.
1993, 115, 5875–5876. (page 16, 17, 19, 21)
[136] Lohr, T. L.; Piers, W. E.; Parvez, M. Inorg. Chem. 2012, 51, 4900–4902. (page 16, 19)
[137] Clapham, S. E.; Guo, R.; Zimmer-De Iuliis, M.; Rasool, N.; Lough, A.; Morris, R. H.
Organometallics 2006, 25, 5477–5486. (page 16, 19)
[138] Kaplan, A. W.; Bergman, R. G. Organometallics 1998, 17, 5072–5085. (page 17, 20)
[139] Grushin, V. V.; Marshall, W. J. J. Am. Chem. Soc. 2006, 128, 4632–4641. (page 20, 40)
[140] Nugent, W. A.; Mayer, J. M. Metal-ligand multiple bonds: the chemistry of transition metal com-
plexes containing oxo, nitrido, imido, alkylidene, or alkylidyne ligands; New York: Wiley, 1988.
(page 20)
[141] Jazzar, R. F. R.; Bhatia, P. H.; Mahon, M. F.; Whittlesey, M. K. Organometallics 2003, 22,
670–683. (page 21)
[142] Saker, O.; Mahon, M. F.; Warren, J. E.; Whittlesey, M. K. Organometallics 2009, 28, 1976–1979.
(page 21)
[143] Renkema, K. B.; Huffman, J. C.; Caulton, K. G. Polyhedron 1999, 18, 2575 – 2578. (page 21)
[144] Edwards, A. J.; Elipe, S.; Esteruelas, M. A.; Lahoz, F. J.; Oro, L. A.; Valero, C. Organometallics
1997, 16, 3828–3836. (page 23)
[145] Jerschow, A.; Muller, N. J. Magn. Reson. 1997, 125, 372 – 375. (page 23)
[146] Otwinowski, Z.; Minor, W. [20] Processing of X-ray diffraction data collected in oscillation mode.
In Macromolecular Crystallography Part A; Charles W. Carter, J., Ed.; Academic Press, 1997;
Vol. 276, pp 307 – 326. (page 24, 35, 48)

BIBLIOGRAPHY 142
[147] Sheldrick, G. M. Acta Cryst. 2008, 64, 112–122. (page 24, 37, 48, 76)
[148] Benito-Garagorri, D.; Kirchner, K. Acc. Chem. Res. 2008, 41, 201–213. (page 29)
[149] Liang, L.-C. Coord. Chem. Rev. 2006, 250, 1152 – 1177. (page 29)
[150] Tsvetkov, N.; Fan, H.; Caulton, K. G. Dalton Trans. 2011, 40, 1105–1110. (page 29)
[151] Friedrich, A.; Drees, M.; Kass, M.; Herdtweck, E.; Schneider, S. Inorg. Chem. 2010, 49, 5482–5494.
(page 29, 72)
[152] Friedrich, A.; Drees, M.; Schmedt auf der Gunne, J.; Schneider, S. J. Am. Chem. Soc. 2009, 131,
17552–17553. (page 29)
[153] Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D. J. Am. Chem. Soc. 2005, 127, 10840–10841.
(page 29)
[154] Del Zotto, A.; Baratta, W.; Ballico, M.; Herdtweck, E.; Rigo, P. Organometallics 2007, 26, 5636–
5642. (page 29)
[155] Clarke, M. L.; Dıaz-Valenzuela, M. B.; Slawin, A. M. Z. Organometallics 2007, 26, 16–19. (page
29, 30)
[156] Dıaz-Valenzuela, M.; Phillips, S.; France, M.; Gunn, M.; Clarke, M. Chem. Eur. J. 2009, 15,
1227–1232. (page 29, 30)
[157] Phillips, S.; Fuentes, J.; Clarke, M. Chem. Eur. J. 2010, 16, 8002–8005. (page 29)
[158] Yamakawa, M.; Ito, H.; Noyori, R. J. Am. Chem. Soc. 2000, 122, 1466–1478. (page 30, 95, 118)
[159] Ito, M.; Ikariya, T. Chem. Commun. 2007, 5134–5142. (page 30)
[160] Gruenwedel, D. W. Inorg. Chem. 1968, 7, 495–501. (page 30)
[161] Chavez-Gil, T. E.; Yasaka, M.; Senokuchi, T.; Sumimoto, M.; Kurosaki, H.; Goto, M. Chem.
Commun. 2001, 2388–2389. (page 30)
[162] Nishiyama, H.; Furuta, A. Chem. Commun. 2007, 760–762. (page 30)
[163] Inagaki, T.; Phong, L.; Furuta, A.; Ito, J.-i.; Nishiyama, H. Chem. Eur. J. 2010, 16, 3090–3096.
(page 30)
[164] Poater, A.; Mola, J.; Saliner, A. G.; Romero, I.; RodrIguez, M.; Llobet, A.; Sola, M. Chem. Phys.
Lett. 2008, 458, 200 – 204. (page 30)
[165] Britovsek, G. J. P.; England, J.; Spitzmesser, S. K.; White, A. J. P.; Williams, D. J. Dalton Trans.
2005, 945–955. (page 30)
[166] Knijnenburg, Q.; Gambarotta, S.; Budzelaar, P. H. M. Dalton Trans. 2006, 5442–5448. (page 30)
[167] Britovsek, G. J. P.; Gibson, V. C.; Hoarau, O. D.; Spitzmesser, S. K.; White, A. J. P.;
Williams, D. J. Inorg. Chem. 2003, 42, 3454–3465. (page 30)

BIBLIOGRAPHY 143
[168] Abbenhuis, R. A. T. M.; del Rıo, I.; Bergshoef, M. M.; Boersma, J.; Veldman, N.; Spek, A. L.;
van Koten, G. Inorg. Chem. 1998, 37, 1749–1758. (page 30)
[169] Mishra, H.; Patra, A. K.; Mukherjee, R. Inorg. Chim. Acta 2009, 362, 483 – 490. (page 30)
[170] Dayan, O.; Cetinkaya, B. J. Mol. Catal. A: Chem. 2007, 271, 134 – 141. (page 30)
[171] Small, B. L.; Brookhart, M. Macromolecules 1999, 32, 2120–2130. (page 30)
[172] Bhor, S.; Anilkumar, G.; Tse, M. K.; Klawonn, M.; Dobler, C.; Bitterlich, B.; Grotevendt, A.;
Beller, M. Org. Lett. 2005, 7, 3393–3396. (page 30)
[173] Kaul, F. A. R.; Puchta, G. T.; Frey, G. D.; Herdtweck, E.; Herrmann, W. A. Organometallics
2007, 26, 988–999. (page 30)
[174] Fukui, S.; Suzuki, N.; Wada, T.; Tanaka, K.; Nagao, H. Organometallics 2010, 29, 1534–1536.
(page 30)
[175] O’Reilly, R. K.; Gibson, V. C.; White, A. J.; Williams, D. J. Polyhedron 2004, 23, 2921 – 2928.
(page 30)
[176] Panda, M. K.; John, A.; Shaikh, M. M.; Ghosh, P. Inorg. Chem. 2008, 47, 11847–11856. (page
30)
[177] Fuchs, M. G. G.; Dechert, S.; Demeshko, S.; Ryde, U.; Meyer, F. Inorg. Chem. 2010, 49, 5853–
5858. (page 30)
[178] Visvaganesan, K.; Mayilmurugan, R.; Suresh, E.; Palaniandavar, M. Inorg. Chem. 2007, 46, 10294–
10306. (page 30)
[179] Tejel, C.; Ciriano, M. A.; del Rıo, M. P.; Hetterscheid, D. G.; Tsichlis i Spithas, N.; Smits, J.
M. M.; de Bruin, B. Chem. Eur. J. 2008, 14, 10932–10936. (page 30)
[180] Tejel, C.; del Rıo, M. P.; Ciriano, M. A.; Reijerse, E. J.; Hartl, F.; Zalis, S.; Hetterscheid, D. G.;
Tsichlis i Spithas, N.; de Bruin, B. Chem. Eur. J. 2009, 15, 11878–11889. (page 30)
[181] Sahni, S. K.; Drew, M. G. B.; Bell, T. W.; Brunschwig, B. S. J. Chem. Soc., Chem. Commun.
1993, 123–125. (page 30)
[182] Gibson, D. H.; Wu, J.; Mashuta, M. S. Inorg. Chim. Acta 2006, 359, 309 – 319. (page 30, 31)
[183] Soliman, A.; Khattab, M.; Ramadan, R. Transition Met. Chem. 2007, 32, 325–331. (page 30)
[184] Omar, M. J. Therm. Anal. Calorim. 2009, 96, 607–615. (page 30)
[185] Yogi, A.; Oh, J.-H.; Nishioka, T.; Tanaka, R.; Asato, E.; Kinoshita, I.; Takara, S. Polyhedron
2010, 29, 1508 – 1514. (page 30)
[186] Abdur-Rashid, K.; Abbel, R.; Hadzovic, A.; Lough, A. J.; Morris, R. H. Inorg. Chem. 2005, 44,
2483–2492. (page 30)
[187] Haque, F. N.; Lough, A. J.; Morris, R. H. Inorg. Chim. Acta 2008, 361, 3149 – 3158, Protagonists
in Chemistry: Robert J. Angelici. (page 30)

BIBLIOGRAPHY 144
[188] Hedberg, C.; Kallstrom, K.; Arvidsson, P. I.; Brandt, P.; Andersson, P. G. J. Am. Chem. Soc.
2005, 127, 15083–15090. (page 30, 98)
[189] Staal, L. H.; Polm, L. H.; Balk, R. W.; Van Koten, G.; Vrieze, K.; Brouwers, A. M. F. Inorg.
Chem. 1980, 19, 3343–3351. (page 32)
[190] Polm, L. H.; Elsevier, C. J.; Van Koten, G.; Ernsting, J. M.; Stufkens, D. J.; Vrieze, K.; An-
drea, R. R.; Stam, C. H. Organometallics 1987, 6, 1096–1104. (page 32)
[191] Frazier, B. A.; Wolczanski, P. T.; Lobkovsky, E. B. Inorg. Chem. 2009, 48, 11576–11585. (page
33)
[192] Westerhausen, M.; Kneifel, A. N. Inorg. Chem. Commun. 2004, 7, 763 – 766. (page 33)
[193] Frazier, B. A.; Wolczanski, P. T.; Lobkovsky, E. B.; Cundari, T. R. J. Am. Chem. Soc. 2009, 131,
3428–3429. (page 33, 34)
[194] Malassa, A.; Agthe, C.; Gorls, H.; Friedrich, M.; Westerhausen, M. J. Organomet. Chem. 2010,
695, 1641 – 1650. (page 33)
[195] Volpe, E. C.; Wolczanski, P. T.; Lobkovsky, E. B. Organometallics 2010, 29, 364–377. (page 33)
[196] Wolczanski, P. T.; Frazier, B. A. U.S. Pat. Appl. Publ. US 20100204473, CAN 153:302632,
2010. (page 33)
[197] Hallman, P. S.; McGarvey, B. R.; Wilkinson, G. J. Chem. Soc. A 1968, 3143–3150. (page 35)
[198] Gunanathan, C.; Milstein, D. Science 2013, 341, 249. (page 40)
[199] Balaraman, E.; Khaskin, E.; Leitus, G.; Milstein, D. Nat. Chem. 2013, 5, 122–125. (page 40)
[200] Gunanathan, C.; Ben-David, Y.; Milstein, D. Science 2007, 317, 790–792. (page 40)
[201] Li, H.; Wang, X.; Huang, F.; Lu, G.; Jiang, J.; Wang, Z.-X. Organometallics 2011, 30, 5233–5247.
(page 40)
[202] Yang, X. Inorg. Chem. 2011, 50, 12836–12843. (page 40, 95, 118)
[203] Li, H.; Wen, M.; Wang, Z.-X. Inorg. Chem. 2012, 51, 5716–5727. (page 40)
[204] Hasanayn, F.; Baroudi, A.; Bengali, A. A.; Goldman, A. S. Organometallics 2013, 32, 6969–6985.
(page 40)
[205] Li, H.; Hall, M. B. J. Am. Chem. Soc. 2014, 136, 383–395. (page 40)
[206] Fogler, E.; Balaraman, E.; Ben-David, Y.; Leitus, G.; Shimon, J. W., Linda; Milstein, D.
Organometallics 2011, 30, 3826–3833. (page 40)
[207] Tanaka, R.; Yamashita, M.; Chung, L. W.; Morokuma, K.; Nozaki, K. Organometallics 2011, 30,
6742–6750. (page 40)
[208] Pitman, C. L.; Miller, A. J. M. ACS Catal. 2014, 4, 2727–2733. (page 40)
[209] Tong, L.; Zong, R.; Thummel, R. P. J. Am. Chem. Soc. 2014, 136, 4881–4884. (page 40)

BIBLIOGRAPHY 145
[210] Stoll, T.; Gennari, M.; Fortage, J.; Castillo, C. E.; Rebarz, M.; Sliwa, M.; Poizat, O.; Odobel, F.;
Deronzier, A.; Collomb, M.-N. Angew. Chem. Int. Ed. 2014, 53, 1654–1658. (page 40)
[211] Mulyana, Y.; Keene, F. R.; Spiccia, L. Dalton Trans. 2014, 43, 6819–6827. (page 40)
[212] Hammarstrom, L.; Styring, S. Nat. Chem. 2009, 1, 185–186. (page 40)
[213] Prokopchuk, D. E.; Collado, A.; Lough, A. J.; Morris, R. H. Dalton Trans. 2013, 42, 10214–10220.
(page 40)
[214] Birk, J. P.; Halpern, J.; Pickard, A. L. J. Am. Chem. Soc. 1968, 90, 4491–4492. (page 40)
[215] Halpern, J.; Pickard, A. L. Inorg. Chem. 1970, 9, 2798–2800. (page 40)
[216] Sen, A.; Halpern, J. J. Am. Chem. Soc. 1977, 99, 8337–8339. (page 40)
[217] Kanan, M. W.; Nocera, D. G. Science 2008, 321, 1072–1075. (page 40)
[218] Turner, W. R.; Elving, P. J. Anal. Chem. 1965, 37, 467–469. (page 40)
[219] Concepcion, J. J.; Jurss, J. W.; Templeton, J. L.; Meyer, T. J. Proc. Natl. Acad. Sci. U. S. A.
2008, 105, pp. 17632–17635. (page 40)
[220] Tskhovrebov, A. G.; Solari, E.; Wodrich, M. D.; Scopelliti, R.; Severin, K. Angew. Chem. Int. Ed.
2012, 51, 232–234. (page 40)
[221] Legault, M. C.; McKay, C. S.; Moran, J.; Lafreniere, M. A.; Pezacki, J. P. Tetrahedron Lett. 2012,
53, 5663 – 5666. (page 40)
[222] Tskhovrebov, A. G.; Vuichoud, B.; Solari, E.; Scopelliti, R.; Severin, K. J. Am. Chem. Soc. 2013,
135, 9486–9492. (page 40)
[223] Liu, B.; Zhang, Y.; Xu, D.; Chen, W. Chem. Commun. 2011, 47, 2883–2885. (page 40)
[224] Slivarichova, M.; Ahmad, R.; Kuo, Y.-Y.; Nunn, J.; Haddow, M. F.; Othman, H.; Owen, G. R.
Organometallics 2011, 30, 4779–4787. (page 40)
[225] Liu, X.; Chen, W. Organometallics 2012, 31, 6614–6622. (page 40)
[226] Jeletic, M. S.; Ghiviriga, I.; Abboud, K. A.; Veige, A. S. Organometallics 2007, 26, 5267–5270.
(page 40)
[227] Magill, A. M.; McGuinness, D. S.; Cavell, K. J.; Britovsek, G. J.; Gibson, V. C.; White, A. J.;
Williams, D. J.; White, A. H.; Skelton, B. W. J. Organomet. Chem. 2001, 617-618, 546 – 560.
(page 41)
[228] Jahnke, M. C.; Pape, T.; Hahn, F. E. Eur. J. Inorg. Chem. 2009, 2009, 1960–1969. (page 41)
[229] Catalano, V. J.; Malwitz, M. A. Inorg. Chem. 2003, 42, 5483–5485. (page 41)
[230] Catalano, V. J.; Malwitz, M. A.; Etogo, A. O. Inorg. Chem. 2004, 43, 5714–5724. (page 41)
[231] Catalano, V. J.; Moore, A. L. Inorg. Chem. 2005, 44, 6558–6566. (page 41)

BIBLIOGRAPHY 146
[232] Prokopchuk, D. E.; Tsui, B. T. H.; Lough, A. J.; Morris, R. H. Chem. Eur. J. 2014, 20, 16960–
16968. (page 41, 43, 46, 47, 48, 133)
[233] Sanford, M.; Henling, L.; Day, M.; Grubbs, R. Angew. Chem. Int. Ed. 2000, 39, 3451–3453. (page
42)
[234] MacInnis, M. C.; McDonald, R.; Ferguson, M. J.; Tobisch, S.; Turculet, L. J. Am. Chem. Soc.
2011, 133, 13622–13633. (page 42)
[235] Lipke, M. C.; Tilley, T. D. J. Am. Chem. Soc. 2013, 135, 10298–10301. (page 42)
[236] Korstanje, T. J.; Lutz, M.; Jastrzebski, J. T. B. H.; Klein Gebbink, R. J. M. Organometallics
2014, 33, 2201–2209. (page 44)
[237] Hahn, C.; Spiegler, M.; Herdtweck, E.; Taube, R. Eur. J. Inorg. Chem. 1998, 1998, 1425–1432.
(page 44)
[238] Feller, M.; Ben-Ari, E.; Iron, M. A.; Diskin-Posner, Y.; Leitus, G.; Shimon, L. J. W.; Konstanti-
novski, L.; Milstein, D. Inorg. Chem. 2010, 49, 1615–1625. (page 44)
[239] Vogt, M.; Rivada-Wheelaghan, O.; Iron, M. A.; Leitus, G.; Diskin-Posner, Y.; Shimon, L. J. W.;
Ben-David, Y.; Milstein, D. Organometallics 2013, 32, 300–308. (page 44)
[240] Stephenson, D. S.; Binsch, G. J. Magn. Reson. 1978, 30, 625 – 626. (page 44, 48)
[241] Marat, K. SpinWorks 4.0.3, Copyright 2014. (page 44, 48)
[242] Perrin, C. L.; Dwyer, T. J. Chem. Rev. 1990, 90, 935–967. (page 44)
[243] Jessop, P. G.; Morris, R. H. Coord. Chem. Rev. 1992, 121, 155 – 284. (page 44)
[244] Zhao, Y.; Truhlar, D. G. J. Chem. Phys. 2006, 125, 194101. (page 46, 48, 117)
[245] Schafer, A.; Huber, C.; Ahlrichs, R. J. Chem. Phys. 1994, 100, 5829–5835. (page 46, 48, 117)
[246] Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. (page 46, 48)
[247] Gusev, D. G. Organometallics 2013, 32, 4239–4243. (page 46, 112)
[248] Albers, M. O.; Singleton, E.; Yates, J. E.; Mccormick, F. B. In Dinuclear Ruthenium(II) Carboxy-
late Complexes; John Wiley & Sons, Inc., 2007; Vol. 26, pp 249–258. (page 47)
[249] Fulmer, G. R.; Miller, A. J. M.; Sherden, N. H.; Gottlieb, H. E.; Nudelman, A.; Stoltz, B. M.;
Bercaw, J. E.; Goldberg, K. I. Organometallics 2010, 29, 2176–2179. (page 48, 75)
[250] Berger, S.; Braun, S. In 200 and More NMR Experiments; Wiley-VCH, 2004; p 145. (page 48)
[251] Frisch, M. J. et al. Gaussian 09 Revision B.01, 2009, Gaussian, Inc., Wallingford CT, 2010. (page
48, 90, 117)
[252] Tomasi, J.; Mennucci, B.; Cances, E. J. Mol. Struct. THEOCHEM 1999, 464, 211 – 226. (page
48, 90, 117)
[253] Tomasi, J.; Mennucci, B.; Cammi, R. Chem. Rev. 2005, 105, 2999–3094. (page 48, 90, 117)

BIBLIOGRAPHY 147
[254] Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. (page
48, 90, 117)
[255] ChemCraft, http://www.chemcraftprog.com. (page 49, 90, 117)
[256] Chiu, P. L.; Lai, C.-L.; Chang, C.-F.; Hu, C.-H.; Lee, H. M. Organometallics 2005, 24, 6169–6178.
(page 49)
[257] Schonberg, H.; Boulmaaz, S.; Worle, M.; Liesum, L.; Schweiger, A.; Grutzmacher, H. Angew.
Chem. Int. Ed. 1998, 37, 1423–1426. (page 70)
[258] Harmer, J.; Frison, G.; Rudolph, M.; Schonberg, H.; Deblon, S.; Maire, P.; Boulmaaz, S.; Bre-
her, F.; Bohler, C.; Ruegger, H.; Schweiger, A.; Grutzmacher, H. New perspectives for olefin
complexes: Synthesis and characterisation of stable rhodium(0) and iridium(0) complexes. In Per-
spectives in Organometallic Chemistry ; Screttas, C. G., Steele, B. R., Eds.; The Royal Society of
Chemistry, 2003; pp 222–239. (page 70)
[259] Buttner, T.; Breher, F.; Grutzmacher, H. Chem. Commun. 2004, 2820–2821. (page 70)
[260] Maire, P.; Breher, F.; Schonberg, H.; Grutzmacher, H. Organometallics 2005, 24, 3207–3218.
(page 70, 72, 76)
[261] Maire, P.; Konigsmann, M.; Sreekanth, A.; Harmer, J.; Schweiger, A.; Grutzmacher, H. J. Am.
Chem. Soc. 2006, 128, 6578–6580. (page 70, 71)
[262] Konigsmann, M.; Donati, N.; Stein, D.; Schonberg, H.; Harmer, J.; Sreekanth, A.; Grutzmacher, H.
Angew. Chem. Int. Ed. 2007, 46, 3567–3570. (page 70, 71)
[263] Zweifel, T.; Naubron, J.-V.; Buttner, T.; Ott, T.; Grutzmacher, H. Angew. Chem. Int. Ed. 2008,
47, 3245–3249. (page 70, 104, 118)
[264] Zweifel, T.; Naubron, J.-V.; Grutzmacher, H. Angew. Chem. Int. Ed. 2009, 48, 559–563. (page
70, 104, 118)
[265] Vogt, M.; de Bruin, B.; Berke, H.; Trincado, M.; Grutzmacher, H. Chem. Sci. 2011, 2, 723–727.
(page 70)
[266] Maire, P.; Breher, F.; Grutzmacher, H. Angew. Chem. Int. Ed. 2005, 44, 6325–6329. (page x, 70,
71, 74, 75)
[267] Maire, P.; Deblon, S.; Breher, F.; Geier, J.; Bohler, C.; Ruegger, H.; Schonberg, H.;
Grutzmacher, H. Chem. Eur. J. 2004, 10, 4198–4205. (page 70)
[268] Piras, E.; Lang, F.; Ruegger, H.; Stein, D.; Worle, M.; Grutzmacher, H. Chem. Eur. J. 2006, 12,
5849–5858. (page 70)
[269] Defieber, C.; Grutzmacher, H.; Carreira, E. Angew. Chem. Int. Ed. 2008, 47, 4482–4502. (page
70)
[270] Hartmann, R.; Chen, P. Angew. Chem. Int. Ed. 2001, 40, 3581–3585. (page 70)
[271] Gusev, D. G.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 1998, 120, 13138–13147. (page 71)

BIBLIOGRAPHY 148
[272] Ito, M.; Shibata, Y.; Watanabe, A.; Ikariya, T. Synlett 2009, 2009, 1621–1626. (page 72)
[273] Samanta, S.; Goswami, S. Inorg. Chem. 2011, 50, 3171–3173. (page 72)
[274] Kuroiwa, K.; Yoshida, M.; Masaoka, S.; Kaneko, K.; Sakai, K.; Kimizuka, N. Angew. Chem. Int.
Ed. 2012, 51, 656–659. (page 72)
[275] Sgro, M. J.; Stephan, D. W. Dalton Trans. 2013, 42, 10460–10472. (page 72)
[276] Chaudret, B.; Devillers, J.; Poilblanc, R. J. Chem. Soc., Chem. Commun. 1983, 641–643. (page
73)
[277] Chaudret, B.; Devillers, J.; Poilblanc, R. Organometallics 1985, 4, 1727–1732. (page 73)
[278] Abdur-Rashid, K.; Gusev, D. G.; Lough, A. J.; Morris, R. H. Organometallics 2000, 19, 1652–
1660. (page 73)
[279] Rosa, P.; Mezailles, N.; Ricard, L.; Mathey, F.; Le Floch, P.; Jean, Y. Angew. Chem. Int. Ed.
2001, 40, 1251–1253. (page x, 74, 75)
[280] Hallman, P. S.; Stephenson, T. A.; Wilkinson, G. In Tetrakis(triphenylphosphine)dichlororuthenium(II)
and Tris(triphenylphosphine)dichlororuthenium(II); John Wiley & Sons, Inc., 1970; pp 237–240.
(page 75)
[281] Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Cryst.
2009, 42, 339–341. (page 76)
[282] Noyori, R. Adv. Synth. Catal. 2003, 345, 15–32. (page 81, 94)
[283] Saudan, L. A. Acc. Chem. Res. 2007, 40, 1309–1319. (page 81, 94)
[284] Huang, Y.; Liu, N.; Wu, X.; Chen, Y. Curr. Org. Chem. 2010, 14, 1447–1460. (page 81, 94)
[285] Palmer, A. M.; Zanotti-Gerosa, A. Curr. Opin. Drug Discovery Dev. 2010, 13, 698—716. (page
81, 94)
[286] Blaser, H.-U. Top. Catal. 2010, 53, 997–1001. (page 81, 94)
[287] Magano, J.; Dunetz, J. R. Org. Process Res. Dev. 2012, 16, 1156–1184. (page 81, 94)
[288] Organometallics as Catalysts in the Fine Chemical Industry ; Beller, M., Blaser, H.-U., Eds.;
Springer Berlin Heidelberg, 2012; Vol. 42. (page 81, 94)
[289] Bolm, C.; Legros, J.; Le Paih, J.; Zani, L. Chem. Rev. 2004, 104, 6217–6254. (page 81)
[290] Jothimony, K.; Vancheesan, S.; Kuriacose, J. J. Mol. Catal. 1985, 32, 11 – 16. (page 81)
[291] Jothimony, K.; Vancheesan, S. J. Mol. Catal. 1989, 52, 301 – 304. (page 81)
[292] Chen, J.-S.; Chen, L.-L.; Xing, Y.; Chen, G.; Chen, W.-Y.; Dong, Z.-R.; Li, Y.-Y.; Gao, J.-X. Acta
Chim. Sinica 2004, 62, 1745. (page 81)
[293] Meyer, N.; Lough, A.; Morris, R. Chem. Eur. J. 2009, 15, 5605–5610. (page 81, 83, 94)

BIBLIOGRAPHY 149
[294] Sui-Seng, C.; Haque, F. N.; Hadzovic, A.; Putz, A.-M.; Reuss, V.; Meyer, N.; Lough, A. J.;
Zimmer-De Iuliis, M.; Morris, R. H. Inorg. Chem. 2009, 48, 735–743. (page 81, 90, 94)
[295] Sui-Seng, C.; Freutel, F.; Lough, A.; Morris, R. Angew. Chem. Int. Ed. 2008, 47, 940–943. (page
vii, x, 81, 89, 91, 92, 94)
[296] Samec, J. S. M.; Backvall, J.-E.; Andersson, P. G.; Brandt, P. Chem. Soc. Rev. 2006, 35, 237–248.
(page 81)
[297] Prokopchuk, D. E.; Sonnenberg, J. F.; Meyer, N.; Zimmer-De Iuliis, M.; Lough, A. J.; Morris, R. H.
Organometallics 2012, 31, 3056–3064. (page 81, 82, 83, 88, 89, 90, 94, 97, 133)
[298] Rybin, L.; Petrovskaya, E.; Struchkov, Y.; Batsanov, A.; Rybinskaya, M. J. Organomet. Chem.
1982, 226, 63 – 69. (page 83)
[299] van der Vlugt, J. I.; Rauchfuss, T. B.; Wilson, S. R. Chem. Eur. J. 2006, 12, 90–98. (page 83)
[300] Sonnenberg, J. F.; Coombs, N.; Dube, P. A.; Morris, R. H. J. Am. Chem. Soc. 2012, 134, 5893–
5899. (page 83, 90, 97, 134)
[301] Lagaditis, P. O.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2011, 133, 9662–9665. (page 85,
95)
[302] Mikhailine, A. A.; Maishan, M. I.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2012, 134,
12266–12280. (page vii, xiii, 85, 95, 97, 98, 104, 106, 107, 108, 115, 134)
[303] Mezzetti, A. Dalton Trans. 2010, 39, 7851–7869. (page 87)
[304] Bigler, R.; Otth, E.; Mezzetti, A. Organometallics 2014, 33, 4086–4099. (page 87)
[305] Rauhut, G.; Pulay, P. J. Phys. Chem. 1995, 99, 3093–3100. (page 88)
[306] Scott, A. P.; Radom, L. J. Phys. Chem. 1996, 100, 16502–16513. (page x, 88, 89, 92)
[307] Yu, L.; Srinivas, G. N.; Schwartz, M. J. Mol. Struct. THEOCHEM 2003, 625, 215 – 220. (page
88)
[308] Sierraalta, A.; Martorell, G.; Ehrmann, E.; Anez, R. Int. J. Quantum Chem. 2008, 108, 1036–1043.
(page 88)
[309] Mikhailine, A.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2009, 131, 1394–1395. (page vii,
x, 89, 91, 92, 94, 108)
[310] Frisch, M. J. et al. Gaussian 03 Revision E.01, 2003, Gaussian, Inc., Wallingford, CT, 2004. (page
90)
[311] Burke, K.; Perdew, J. P.; Yang, Y. In Electronic Density Functional Theory: Recent Progress and
New Directions; Dobson, J. F., Vignale, G., Das, M. P., Eds.; Plenum: New York, 1998; p 81.
(page 90, 118)
[312] Adamo, C.; Barone, V. J. Chem. Phys. 1998, 108, 664–675. (page 90, 118)
[313] Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. J. Chem. Phys. 1987, 86, 866–872. (page 90)

BIBLIOGRAPHY 150
[314] Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer, P. V. R. J. Comput. Chem. 1983, 4,
294–301. (page 90)
[315] Frisch, M. J.; Pople, J. A.; Binkley, J. S. J. Chem. Phys. 1984, 80, 3265–3269. (page 90, 117)
[316] Li, T.; Bergner, I.; Haque, F. N.; Zimmer-De Iuliis, M.; Song, D.; Morris, R. H. Organometallics
2007, 26, 5940–5949. (page 90)
[317] Lynch, B. J.; Truhlar, D. G. J. Phys. Chem. A 2001, 105, 2936–2941. (page 90)
[318] Morris, R. H. Chem. Soc. Rev. 2009, 38, 2282–2291. (page 94)
[319] Junge, K.; Schroder, K.; Beller, M. Chem. Commun. 2011, 47, 4849–4859. (page 94)
[320] Nakazawa, H.; Itazaki, M. Fe–H Complexes in Catalysis. In Iron Catalysis; Plietker, B., Ed.;
Springer Berlin / Heidelberg, 2011; Vol. 33, pp 27–81. (page 94)
[321] Darwish, M.; Wills, M. Catal. Sci. Technol. 2012, 2, 243–255. (page 94)
[322] Bezier, D.; Sortais, J.-B.; Darcel, C. Adv. Synth. Catal. 2013, 355, 19–33. (page 94)
[323] Bullock, R. M. Science 2013, 342, 1054–1055. (page 94)
[324] Gopalaiah, K. Chem. Rev. 2013, 113, 3248–3296. (page 94)
[325] Mikhailine, A. A.; Kim, E.; Dingels, C.; Lough, A. J.; Morris, R. H. Inorg. Chem. 2008, 47,
6587–6589. (page 94)
[326] Mikhailine, A. A.; Morris, R. H. Inorg. Chem. 2010, 49, 11039–11044. (page 94, 95, 106)
[327] Lagaditis, P. O.; Lough, A. J.; Morris, R. H. Inorg. Chem. 2010, 49, 10057–10066. (page 94)
[328] Sues, P. E.; Lough, A. J.; Morris, R. H. Organometallics 2011, 30, 4418–4431. (page 94, 95)
[329] Sandoval, C. A.; Ohkuma, T.; Muniz, K.; Noyori, R. J. Am. Chem. Soc. 2003, 125, 13490–13503.
(page 95)
[330] Baratta, W.; Rigo, P. Eur. J. Inorg. Chem. 2008, 2008, 4041–4053. (page 95)
[331] Perry, R. H.; Brownell, K. R.; Chingin, K.; Cahill, T. J.; Waymouth, R. M.; Zare, R. N. Proc.
Natl. Acad. Sci. U. S. A. 2012, 109, 2246–2250. (page 95)
[332] Handgraaf, J.-W.; Meijer, E. J. J. Am. Chem. Soc. 2007, 129, 3099–3103. (page 95, 98, 104)
[333] Chen, Y.; Liu, S.; Lei, M. J. Phys. Chem. C 2008, 112, 13524–13527. (page 95)
[334] Comas-Vives, A.; Ujaqu, G.; Lledos, A. Inner- and Outer-Sphere Hydrogenation Mechanisms: A
Computational Perspective. In Theoretical and Computational Inorganic Chemistry ; van Eldik, R.,
Harvey, J., Eds.; Academic Press, 2010; Vol. 62, pp 231 – 260. (page 95)
[335] Guo, X.; Tang, Y.; Zhang, X.; Lei, M. J. Phys. Chem. A 2011, 115, 12321–12330. (page 95, 98)
[336] Zhang, X.; Guo, X.; Chen, Y.; Tang, Y.; Lei, M.; Fang, W. Phys. Chem. Chem. Phys. 2012, 14,
6003–6012. (page 95)

BIBLIOGRAPHY 151
[337] O, W. W. N.; Lough, A. J.; Morris, R. H. Organometallics 2012, 31, 2137–2151. (page 95)
[338] O, W. W. N.; Lough, A. J.; Morris, R. H. Organometallics 2012, 31, 2152–2165. (page 95)
[339] Meerwein, H.; Schmidt, R. Justus Liebigs Ann. Chem. 1925, 444, 221–238. (page 95)
[340] Verley, A. Bull. Soc. Chim. Fr. 1925, 37, 537. (page 95)
[341] Ponndorf, W. Angew. Chem. 1926, 39, 138–143. (page 95)
[342] Cohen, R.; Graves, C. R.; Nguyen, S. T.; Martin, J. M. L.; Ratner, M. A. J. Am. Chem. Soc.
2004, 126, 14796–14803. (page 95)
[343] Bifunctional Molecular Catalysis; Ikariya, T., Shibasaki, M., Eds.; Springer Berlin / Heidelberg,
2011; Vol. 37. (page 95)
[344] Hadzovic, A.; Song, D.; MacLaughlin, C. M.; Morris, R. H. Organometallics 2007, 26, 5987–5999.
(page 98)
[345] Zhang, H.; Chen, D.; Zhang, Y.; Zhang, G.; Liu, J. Dalton Trans. 2010, 39, 1972–1978. (page 98)
[346] Prokopchuk, D. E.; Morris, R. H. Organometallics 2012, 31, 7375–7385. (page 98, 112, 117, 134)
[347] Brookhart, M.; Green, M. L. H.; Parkin, G. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6908–6914.
(page 101)
[348] Bi, S.; Xie, Q.; Zhao, X.; Zhao, Y.; Kong, X. J. Organomet. Chem. 2008, 693, 633 – 638. (page
104, 118)
[349] Clarke, Z. E.; Maragh, P. T.; Dasgupta, T. P.; Gusev, D. G.; Lough, A. J.; Abdur-Rashid, K.
Organometallics 2006, 25, 4113–4117. (page 104)
[350] Bertoli, M.; Choualeb, A.; Gusev, D. G.; Lough, A. J.; Major, Q.; Moore, B. Dalton Trans. 2011,
8941–8949. (page 104, 118)
[351] Bertoli, M.; Choualeb, A.; Lough, A. J.; Moore, B.; Spasyuk, D.; Gusev, D. G. Organometallics
2011, 30, 3479–3482. (page 104, 118)
[352] McQuarrie, D. A.; Simon, J. D. Physical Chemistry: A Molecular Approach, 6th ed.; Calif:
University Science Books, 1997. (page 106, 111)
[353] Gomez-Gallego, M.; Sierra, M. A. Chem. Rev. 2011, 111, 4857–4963. (page 106)
[354] (a) Zhao, Y.; Truhlar, D. Theor. Chem. Acc. 2008, 120, 215–241; (b) Zhao, Y.; Truhlar, D. G.
Acc. Chem. Res. 2008, 41, 157–167. (page 117)
[355] Hehre, W. J.; Ditchfield, R.; Pople, J. A. J. Chem. Phys. 1972, 56, 2257–2261. (page 117)
[356] Hariharan, P. C.; Pople, J. A. Theor. Chem. Acc. 1973, 28, 213–222. (page 117)
[357] Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650–654. (page
117)
[358] Fukui, K. Acc. Chem. Res. 1981, 14, 363–368. (page 117)

BIBLIOGRAPHY 152
[359] Hratchian, H. P.; Schlegel, H. B. Chapter 10 - Finding minima, transition states, and following
reaction pathways on ab initio potential energy surfaces. In Theory and Applications of Compu-
tational Chemistry ; Dykstra, C. E., Frenking, G., Kim, K. S., Scuseria, G. E., Eds.; Elsevier:
Amsterdam, 2005; pp 195 – 249. (page 117)
[360] Laidler, K. J. Chemical Kinetics, 3rd ed.; New York: Harper & Row, 1987. (page 117)
[361] Boys, S.; Bernardi, F. Mol. Phys. 1970, 19, 553–566. (page 117)
[362] Simon, S.; Duran, M.; Dannenberg, J. J. J. Chem. Phys. 1996, 105, 11024–11031. (page 117)
[363] Tao, J.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Phys. Rev. Lett. 2003, 91, 146401. (page
117, 118)
[364] Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. (page 117)
[365] Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. (page 117, 118)
[366] Perdew, J. P. In Electronic Structure of Solids ’91 ; Ziesche, P., Eschrig, H., Eds.; Akademie Verlag,
Berlin, 1991; p 11. (page 118)
[367] Perdew, J. P.; Chevary, J. A.; Vosko, S. H.; Jackson, K. A.; Pederson, M. R.; Singh, D. J.;
Fiolhais, C. Phys. Rev. B 1992, 46, 6671–6687. (page 118)
[368] Perdew, J. P.; Chevary, J. A.; Vosko, S. H.; Jackson, K. A.; Pederson, M. R.; Singh, D. J.;
Fiolhais, C. Phys. Rev. B 1993, 48, 4978–4978. (page 118)
[369] Perdew, J. P.; Burke, K.; Wang, Y. Phys. Rev. B 1996, 54, 16533–16539. (page 118)
[370] Boese, A. D.; Martin, J. M. L. J. Chem. Phys. 2004, 121, 3405–3416. (page 118)
[371] Heyd, J.; Scuseria, G. E. J. Chem. Phys. 2004, 121, 1187–1192. (page 118)
[372] Heyd, J.; Scuseria, G. E. J. Chem. Phys. 2004, 120, 7274–7280. (page 118)
[373] Heyd, J.; Peralta, J. E.; Scuseria, G. E.; Martin, R. L. J. Chem. Phys. 2005, 123, 174101. (page
118)
[374] Heyd, J.; Scuseria, G. E.; Ernzerhof, M. J. Chem. Phys. 2006, 124, 219906. (page 118)
[375] Izmaylov, A. F.; Scuseria, G. E.; Frisch, M. J. J. Chem. Phys. 2006, 125, 104103. (page 118)
[376] Krukau, A. V.; Vydrov, O. A.; Izmaylov, A. F.; Scuseria, G. E. J. Chem. Phys. 2006, 125, 224106.
(page 118)
[377] Henderson, T. M.; Izmaylov, A. F.; Scalmani, G.; Scuseria, G. E. J. Chem. Phys. 2009, 131,
044108. (page 118)
[378] Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. J. Chem. Phys. 2004, 120, 8425–
8433. (page 118)
[379] Vydrov, O. A.; Scuseria, G. E. J. Chem. Phys. 2006, 125, 234109. (page 118)

BIBLIOGRAPHY 153
[380] Vydrov, O. A.; Heyd, J.; Krukau, A. V.; Scuseria, G. E. J. Chem. Phys. 2006, 125, 074106. (page
118)
[381] Vydrov, O. A.; Scuseria, G. E.; Perdew, J. P. J. Chem. Phys. 2007, 126, 154109. (page 118)
[382] Lynch, B. J.; Fast, P. L.; Harris, M.; Truhlar, D. G. J. Phys. Chem. A 2000, 104, 4811–4815.
(page 118)
[383] Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158–6170. (page 118)
[384] Chai, J.-D.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. (page 118)
[385] Prokopchuk, D. E.; Lough, A. J.; Morris, R. H. Dalton Trans. 2011, 40, 10603–10608. (page 132)
[386] Chiu, W.-H.; Peng, S.-M.; Che, C.-M. Inorg. Chem. 1996, 35, 3369–3374. (page 132)
[387] Lalrempuia, R.; McDaniel, N. D.; Muller-Bunz, H.; Bernhard, S.; Albrecht, M. Angew. Chem. Int.
Ed. 2010, 49, 9765–9768. (page 133)
[388] Bernet, L.; Lalrempuia, R.; Ghattas, W.; Mueller-Bunz, H.; Vigara, L.; Llobet, A.; Albrecht, M.
Chem. Commun. 2011, 47, 8058–8060. (page 133)
[389] Petronilho, A.; Rahman, M.; Woods, J. A.; Al-Sayyed, H.; Muller-Bunz, H.; Don MacElroy, J. M.;
Bernhard, S.; Albrecht, M. Dalton Trans. 2012, 41, 13074–13080. (page 133)