synthetic approaches to ionophore antibiotics a thesis
TRANSCRIPT

SYNTHETIC APPROACHES TO IONOPHORE ANTIBIOTICS
a thesis presented by
ANNETTE MARIAN DOHERTY
in partial fulfilment of the requirements
for the award of the degree of
DOCTOR OF PHILOSOPHY
OF THE
UNIVERSITY OF LONDON
WHIFFEN LABORATORY
CHEMISTRY DEPARTMENT
IMPERIAL COLLEGE
LONDON SW7 2AY SEPTEMBER 1985

2
ABSTRACT
This thesis begins with a review of the Sharpless asymmetric
epoxidation with particular emphasis on its application to natural
product synthesis.
The major part of the thesis involves synthetic studies of the
novel acyltetronic acid ionophore M139603. The synthesis of some
interesting model compounds has been described incorporating as the key
step, the stereospecific Julia-sulphone coupling to introduce the
required trans-disubstituted double bond. The biological activity is
presently being investigated.
Degradation studies of the naturally-occurring ionophore M139603
have been described. The first synthesis of the optically pure
tetrahydrofuran fragment was accomplished from (5)-(+)-methyl-3-
hydroxy-2-methylpropionate in 16 steps and comparison made with auth
entic material derived from degradation of natural M139603.
Thus, the [2+2] cycloaddition of (J?)-(Z)-1-t-buty1 diphenyl silyI -
oxy-2-methylpent-3-ene with dichioroketene, followed by reduction and
Baeyer-Vi11iger oxidation afforded a 1:1 mixture of diastereoisomeric
lactones. After separation 4-(i?)-[(2-t-butyldiphenylsilyloxy-l-(5)-
methyl)ethyl]-3-(s')-methylbutan-4-olide, possessing three of the required
five chiral centres was further elaborated to the key allylic alcohol
(S’)-[55, 6/?, 75]-8-1-butyl diphenyl si lyloxy-6-hydroxy-5,7-dimethyloct-2-
en-l-ol in 3 steps (61% overall yield). The Sharpless asymmetric
epoxidation under modified conditions was accompanied by simultaneous
ring closure to the tetrahydrofuran, thus introducing the two remaining
chiral centres. After further elaboration, the synthetic compound was
found to be identical in all respects to that derived from degradation,
thus unambiguously establishing the absolute configuration of the natural
product.

3
Derivatisation of this right-hand fragment and coupling studies
with the left-hand aldehyde of M139603 obtained from degradation and
other model aldehydes are discussed.
The final part of the thesis describes model studies towards the
pyrrole-containing ionophore indanomycin (X-14547A). This work
culminated in the synthesis of a novel model compound whose biological
activity is presently being investigated.

4
LIST OF CONTENTS
Page
ABSTRACT 2
ACKNOWLEDGEMENTS 5
ABBREVIATIONS 7
REVIEW 9
The Use of the Sharpless Asymmetric Epoxidation
in Natural Product Synthesis
REVIEW REFERENCES 56
SYNTHETIC APPROACHES TO IONOPHORE ANTIBIOTICS
INTRODUCTION 61
1. The Ionophores 62
2. The Polyether Ionophore M139603 66
RESULTS AND DISCUSSION
3. Model Studies related to the Ionophore M139603 73
4. Degradation Studies of M139603 104
5. Synthetic Studies towards the Tetrahydrofuranyl 113
Portion of M139603
6. Coupling Studies 158
7. Model Studies related to the Ionophore 173
Antibiotic Indanomycin (X-14547A)
APPENDIX : SPECTRAL DATA 192
EXPERIMENTAL 211
REFERENCES 348

5
ACKNOWLEDGEMENTS
I wish to express my deepest thanks to Prof. S.V. Ley for his
guidance, encouragement and friendship throughout the past three years.
I also thank Dr. D.J. Williams for providing the X-ray structure
determinations, Mr. K.I. Jones and his staff for the microanalytical
service, Mr. J. Bilton and Mrs. J. Challis for the mass spectra and
Dr. D. Sheppard for the high field n.m.r. spectra. Special thanks are
due to Biddy for her kindness and friendship.
I would like to express my sincere thanks to all Whiffenites and
Perkinites past and present whom I will never forget for their friend
ship and help. I am especially grateful to the patient and invaluable
proof-readers : Don, Neville, Phil, Gary and Howard.
Thanks are also due to Mrs. E. Pinn for her excellent typing of
this manuscript.
I gratefully acknowledge the supply of M139603 from ICI Ltd.,
Macclesfield, Cheshire, and Coopers Animal Health Ltd., Berkhamsted,
and also the SERC for their financial support.
Last, but not least, I would like to thank my parents for their
continual support and encouragement throughout my career.
ANNETTE DOHERTY

TO MY PARENTS
WITH GRATITUDE AND AFFECTION

7
ABBREVIATIONS
nBuLi - n-Butyl-lithium
CSA - Camphor sulphonic acid
DBU - Diazobicyclo[5.4.0]undec-7-ene
DCC - Di cyclohexylcarbodi imide
DET - Diethyl tartrate
DHP - Dihydropyran
DIBAL - Diisobutylaluminium hydride
DIPT - Diisopropyl tartrate
DMAP - N,N-Dimethyl-4-ami nopyridine
DME - 1,2-Dimethoxyethane
DMF - Dimethylformamide
DMPU - l,3-Dimethyl-3,4,5,6-tetrahydro-2(lH)-pyrimidinone
DMSO - Dimethylsulphoxide
eq - Equivalents
HMPA - Hexamethylphosphorami de
h.p.1.c. - High pressure liquid chromatography
LDA - Lithium diisopropylamide
mCPBA - w-Chloroperbenzoic acid
MeLi - Methyl-1ithium
Ms - Methanesulphony1 (mesyl)
Na-Hg - Sodium-Mercury amalgam.
N-PSP - N-Phenylselenophthalimide
n.m.r. - Nuclear magnetic resonance
PCC - Pyridinium chiorochromate
PDC - Pyridinium dichromate
r. t. - Room temperature
TBAF - Tetra-n-butylammonium fluoride
TBDMS - t-Butyldimethylsilyl

TBDPS t-Butyldiphenyl silyl
TBHP t-Butylhydroperoxide
THF Tetrahydrofuran
THP Tetrahydropyranyl
t.l.c. Thin layer chromatography
TMS Trimethyl silyl
Ts p-toluenesulphonyl (tosyl)
VO(acac)2 Vanadyl acetylacetonate

9
PRODUCT SYNTHESIS
INTRODUCTION
In recent years, asymmetric synthesis has become increasingly
important in the construction of naturally occurring substances. The
stereochemical complexity of many natural products necessitates the
use of highly stereoselective chemical reactions for their successful
synthesis. The intense research activity in this area has led to the
discovery of a wide range of new synthetic methods which have enabled
the synthesis of an impressive array of organic structures.
The epoxide functional group is one of the most useful inter
mediates in organic synthesis. Its importance arises from the
existence of regio- and stereoselective methods for its construction2-5and for controlling its subsequent reactions.
6 7The discovery by Katsuki and Sharpless ’ in 1980, that it was
possible to synthesize enantiomerically pure epoxides from achiral
olefins has been one of the most significant advances in the field of
asymmetric synthesis. Indeed since the initial report of theg
asymmetric Sharpless epoxidation reaction, it has rapidly become
accepted as one of the best means for synthesizing a great variety of
optically active substances.
The now well-known process involves the treatment of an allylic
alcohol with tert-butyl hydroperoxide (TBHP) (1.5 - 4.0 eq),8 titanium (IV)
isopropoxide (1.2 eq) and either (+)- or (-)-dialkyl tartrate (1.5 eq)
to furnish an epoxy alcohol of high optical purity (generally > 90%
e.e. and usually > 95% e.e.), the absolute configuration of which cang
be predicted (Scheme 1).
REVIEW : THE USE OF THE SHARPLESS ASYMMETRIC EPOXIDATION IN NATURAL

10
D.-0-diethyl tartrate*0 ’ (unnatural)
O ’
JL.-Q-diethyl tartrate (natural) 70-90 %
>90 % e.e.
4*These conditions are often known as the 'standard' Sharpless conditions and will be referred to as such throughout this review.
Thus the epoxide oxygen is delivered from a specific enantioface
of the olefin depending on the chirality of the tartrate diester
employed. Indeed since both enantiomers of tartaric acid of high
optical purity are commercially available, either enantiomer of the
epoxy alcohol can be prepared.
One of the most surprising features of this asymmetric reaction is
its generality with regard to substrate; almost every type of allylic
alcohol substitution pattern can be epoxidised with high enantio-
selectivity. The remarkable combination of substrate generality and
high selectivity exhibited by this titanium alkoxide-tartrate

11
epoxidation catalyst is without precedent among either man-made or
enzyme catalysts.
In view of the considerable contribution that this reaction has
made in the past five years to synthetic organic chemistry, it was
deemed interesting to provide a short review of its wide range of
applications to natural product synthesis. It is obviously not
possible to cover the area completely and indeed several useful
reviews and reports have already appeared on the mechanistic^’ and 3 5 8 9synthetic aspects ’ ’ ’ of the reaction. The purpose at present is
merely to demonstrate the importance of this discovery and the impact
it has had on natural product synthesis.
SECTION 1 : KINETIC RESOLUTION
As might have been expected, the titanium alkoxide - tartrate
epoxidation catalyst was found to be sensitive to pre-existing
chirality in the allylic alcohol substrate. Indeed, shortly after its
discovery in 1980, Sharpless and co-workers reported that with racemic
secondary allylic alcohols as substrates, the relative rates of epoxida
tion for enantiomeric pairs were surprisingly high (ranging from 15 to12140 in measured cases). Thus the slow reacting enantiomer of the
racemic substrate could often be re-isolated with high optical
activity (> 90% e.e.). Moreover, as for the epoxidation of prochiral 6 13allylic alcohols, ’ the stereochemical outcome of these kinetic
resolutions was found to be highly predictable. Thus, in general,
the L-(+)-tartrates strongly favour the erythvo product (Scheme 2).
Experimentally the (s)-enantiomer of (£)-cyclohexylpropenylcarbinol
reacts ea. 104 times faster than the (i?)-enantiomer.

12
Scheme 2
L -H -D IP TSlow
Thus the usual oxygen-delivery from the underside (as shown on these
diagrams) by the L-(+)-tartrate catalyst is consonant with the eryth.ro
selectivity in the (S)-enantiomer giving rise to a high erythro : threo
product ratio, while in the (i?)-enantiomer these two effects are
opposed, leading to a much lower ratio.
This kinetic resolution process is remarkably general and is
potentially an extremely useful route to a variety of optically pure
allylic alcohols.
14Roush and Brown have employed a kinetic resolution in their
stereoselective synthesis of two monosaccharides, 2,6-dideoxy-D-arab'ino-

13
hexose (1) (olivose) which is a component of a number of natural
products including olivomycin A (2), and 2,6-dideoxy-£-r£2?0-hexose (3)
(digitoxose) which is a component of the cardiac glycosides.
Thus when the racemic secondary allylic alcohol (4AB) was treated
with Z7-(-)-diisopropyl tartrate ((-)-DIPT) (1.5 eq), titanium (IV) iso-
propoxide (1.0 eq) and tert-butyl hydroperoxide (TBHP) (0.4 eq) in
dichioromethane at -20°C, the fast reacting (f?)-enantiomer (4A)
afforded the product of erythvo upper-face attack (as indicated on the
diagram) (5) in greater than 95% enantiomeric excess (e.e.). The
slow reacting (S')-enantiomer (4B) was re-isolated with 72% e.e.
(Scheme 3).

14
Scheme 3
(5) Major
The epoxy alcohol (5) was then converted via a sequence of simple
steps to olivose (1) (Scheme 4).
Scheme 4
olivose

15
This synthesis of (+)-(l) proceeds in six steps from crotonaldehyde and
ally! bromide and is applicable to the preparation of the (-)-enantiomer.
By comparison, the shortest synthesis of (+)-(l) from S-glucose proceeds 15ain eight steps; the (-)-enantiomer is available in five steps from
L-rhamnose.
The slow reacting (S')-enantiomer (4B) was re-isolated and epoxidised
under ‘standard1 Sharpless conditions to afford the epoxy alcohol (6)
which was easily converted to digitoxose (3) (Scheme 5).
Scheme 5
L -fl-P IPTTi(0‘Pr)4TBHP-20°C
aOH
(6)
H O a - z O
HO
(3)
digitoxose

16
These two syntheses of monosaccharides from non-carbohydrate
precursors are short and highly stereoselective featuring asymmetric
epoxidation-kinetic resolution and highly regioselective epoxide ring
opening reactions.
Another example of the kinetic resolution process is illustrated
by the preparation of the (i?)- and {S)-enantiomers of ipsdienol (7)
(which are both insect pheromones).
> =HC
The resolution of racemic (7) which is commercially available was per
formed by treatment with the appropriate tartrate diester [(+)-DIPT
R ; (-)-DIPT 5],3
Cava and Dominguez employed the Sharpless kinetic asymmetric1 fiepoxidation to establish a route to (+)-4-demethoxydaunomycinone (8A)
which can easily be converted to (+)-4-demethoxydaunomycin (8B).
Structure-activity relationships have indicated that (8B) possesses
antitumour activity ca. 10 times greater than either natural
daunomycin (9B) or adriamycin (10B).
(8A)
(SB)
(9B)
(10B)
R1
H
H
H
OH
R2
H
H
OMe
OMe
OH
Thus kinetic resolution of (±)-(ll) was carried out by treatment with
titanium (IV) isopropoxide, (+)-DIPT and TBHP (0.6 eq) at -20°C for

17
17 h to afford a mixture of (-)-(12) and (i?)-( + )-(ll) (slow reacting
enantiomer) in 34% and 23% yields respectively (Scheme 6).
Scheme 6
Chromic acid oxidation of this mixture gave the epoxy ketone
(-)-(13) (major product) and the enone (14) (minor product). Sodium
dithionite reduction of (-)-(13) gave (-)-4-demethoxy-7-deoxydaunomycin
(15) (Scheme 7) which can be converted to (+)-4-demethoxydaunomycinone
by literature methods.

18
Scheme 7
and
(15)
Work closely related to that of Cava (described above) has been
reported more recently by Rama Rao and co-workers.
Mori and co-workers have employed the Sharpless asymmetric
epoxidation-kinetic resolution process in their enantioselective
synthesis of (-)-pestalotin (16) which is a gibberellin synergist
isolated from culture filtrate of a phytopathogenic fungus Pestalotia
cryptomeriaecola Sawada and also independently from an unidentified18penicillin species. Thus treatment of (±)-l-hepten-3-ol with
Z?-(-)-DIPT, titanium (IV) isopropoxide and TBHP at -20°C for 40 h
afforded (2S, 3i?)-(17) in 74% yield (94% e.e.). This epoxy alcohol
(17) could be converted to natural (-)-pestalotin (16) (Scheme 8) and
also to the stereoisomer (-)-epipestalotin (18) (Scheme 9).

19
Scheme 8
Mitsunobu
conditions *
H
RO H
EE = C H 2C H 2O C H 2CH 3
He Ph3P ,Et02CN=NC02Et,
no2
°2*\ X > - C 0 2H
Scheme 9
(18)

20
The kinetic resolution process (KR) is not only restricted to
secondary allylic alcohols in which chirality resides at the carbinol3carbon and some examples are shown in Scheme 10.
Scheme 10
Racemic Substrate Recovered Substrate
V Kinetic0H Resolution
(KR)
I1
Ph^Sf^OH95% e-e-
80% e.e.
In addition to the kinetic resolution of secondary allylic
alcohols. Sharpless has recently reported the highly successful
oxidative kinetic resolution of 3-hydroxyamines employing a 2:119titanium : tartrate ligand ratio. This new process is also highly
predictable in terms of stereochemical outcome. An example of the
use of this method is illustrated in the synthesis of the N-benzyl
derivative of (S)-bevantolol (19) (Scheme 11) which is a precursor to19the cardioselective (3-blocker antagonists.

21
Scheme 11
(19)
85 % e • e .
(r)-N- oxide
Thus the N-oxide product which results from the oxidation of the
fast-reacting (-ff)-enantiomer can easily be separated from the (£)-
enantiomer (19) because of the dramatic solubility difference.
Many other examples of the Sharpless epoxidation - kinetic20-24resolution process have been reported and although this is only
a brief review, it clearly indicates the wide-ranging potential in
natural product synthesis.
SECTION 2 : CHIRAL 2,3-EPOXY ALCOHOLS AS USEFUL INTERMEDIATES IN
SYNTHESIS
The asymmetric epoxidation of prochiral allylic alcohols in con
junction with selective epoxide-cleavage reactions shows great potential
as a versatile approach to a wide variety of useful substrates.
Indeed since the discovery of the asymmetric epoxidation, the major

22
consideration has been to increase the synthetic utility of the1 S PR
O l - o n n w s l r f t h n l cresulting 2,3-epoxy alcohols.
The opening of epoxides with nucleophiles occurs under an extremely
wide range of reaction conditions. The regio- and stereoselectivity
of an epoxide-cleavage reaction are related to the mechanism and
therefore dependent on the conditions employed. In principle there
are three reactive sites for nucleophilic substitution in a 2,3-epoxy
alcohol (20). Thus attack at the C-2 and C-3 positions is immediately
apparent and is controlled by both electronic and steric factors and
also by the nature of the nucleophile.
When R is an electron-withdrawing group, attack at C-3 is disfavoured
because of the well-known rate deceleration such groups cause on S^2
reactions. In addition, when R is sterically demanding nucleo
philic attack occurs preferentially at C-2. Thus, combination of
these two factors can lead to high regioselectivity in the ring opening
of 2,3-epoxy alcohols to afford the corresponding 1,3-diols.
Regioselective C-2 ring opening by the azide anion is illustrated
in Kishi's synthesis of the precursors to four diastereoisomeric 2-27amino-2-deoxy-Z^-pentitols (Scheme 12). Thus the bulky nature of the
acetonide moiety at C-3 and its additional electron-withdrawing effect
presumably control the regioselectivity of nucleophilic attack.
O
OH
(20)

23
Scheme 12
OAc NHAc
OAc OAc
a) NaN3, NH^Cl, MeOCH2CH2OH, A .b) 1 atm.. H2, Pd/C, MeOH, r.t.
c) Ac20, pyr-
d) Dowex 50X8-200 acidic resin, aq. MeOH, r.t
In addition, some nucleophilic reagents can be delivered intramolecu-
larly by coordination to the hydroxy group giving rise to predominant
28 29attack at C-2. ’ One example of this type of reaction involves
the reduction of 2,3-epoxy alcohols with sodium bis(2-methoxyethoxy)-
aluminium hydride (Red-Al ) affording exclusively the 1,3-diol products

24
30This useful reaction has been used recently by Takano and co-workers
as the key step in the first enantioselective synthesis of the bicyclic
ant venom alkaloid (-)-[3£-(3p, 5p, 8a)]-3-heptyl-5-methylpyrrolizidine
( 21 ) .
Thus epoxidation of the furanyl-substituted allylic alcohol (22)
under 'standard' Sharpless conditions followed by regioselective
cleavage of the resulting 2,3-epoxy alcohol with Red-Al afforded the
1,3-diol (23A) in 86% overall yield. The secondary hydroxyl was then
substituted by phthalimide with inversion of configuration by employing
31Mitsunobu's conditions. Debenzoylation followed by treatment with
hydrazine hydrate afforded the amino alcohol (24).
Hydrolysis of the compound (24) afforded the pyrroline derivative
(25), which after reduction with sodium cyanoborohydride, gave a mixture
of pyrrolizidines (26) and (27) (1:1). Further elaboration to the
natural product (21) proceeded without difficulty (Scheme 13).
Thus the Sharpless epoxidation and regioselective ring-opening of
the resulting 2,3-epoxy alcohol introduced the first chiral centre
which served as a control for the incorporation of the two remaining
centres of asymmetry.
Many other examples of this controlled C-2 epoxide ring opening
32-34reaction have been reported.
An example of regioselective C-3 attack was illustrated by the
reaction of the trityl-protected epoxy alcohol (28A) with 1-trimethyl-
si lylvinylmagnesium bromide in the presence of catalytic copper (I)
35iodide to afford the alcohol (29A) as the sole product (Scheme 14).

Scheme 14

26
The corresponding enantiomer (29B) was also synthesized (using D-(-)-
DIPT in the Sharpless epoxidation). Deprotection of (29A) and (29B)
followed by periodate cleavage afforded the (i?)- and (S')-aldehydes
(30A) and (30B) (Scheme 15).
Scheme 15
Me,Si
X l^OCPh,1'c h c i2c o 2hMe3Si
- y cho^ (30A)1 (29A) 2‘
Nal04
Me3Si 9H.OCPh3as _Me3Si
Jv cho11l above
(29 B) (30 B)
These aldehydes (30A) and (30B) were then converted to syn- and
antf-p-methylhomoallyl alcohols via addition of a Grignard reagent;
such compounds are potentially very useful intermediates in the synthe
sis of macrolide and ionophore antibiotics.
The regioselective C-3 ring opening of 2,3-epoxy alcohols has
also been usefully employed for the stereoselective introduction ofQC
the hydroxy groups in the side chain of a variety of steroids.
Since the biological activity of various physiologically active
steroids has been found to depend on the configuration of the
functional groups present in the side chain, it is particularly
important for any synthesis to be highly stereoselective.

27
Treatment of the allylic alcohol (31) with (+)- and (-)-diethyl
tartrate (DET) separately, afforded the epoxy alcohols (32) and (33)
respectively. Protection of the alcohol, C-3 ring opening by treat
ment with lithium aluminium hydride and deprotection afforded the diols
(34) and (35) which possess the correct stereochemistry for (25R)- and
(25S)-25,26-dihydroxyvitamin D3 (Scheme 16) respectively.
Scheme 16

28
Sharpless has recently reported that titanium (IV) isopropoxide
mediates the nucleophilic opening of 2,3-epoxy alcohols,acids and 37 bamides. Thus, a variety of substituted epoxides were ring-opened
with high regioselectivity for C-3 by amines, thiophenol and many other
nucleophiles in the presence of a stoichiometric amount of titanium
(IV) isopropoxide. When the titanium catalyst was not present
reaction was very sluggish and under more forcing conditions, the
regioselectivity was low (often slightly in favour of the C-2 product).
The reaction of tpans-2,3-epoxydecanoic acid with diethylamine in the
presence of Ti(01Pr)i, (1.1 eq) afforded predominantly 3-(diethylamino)-
2-hydroxydecanoic acid (36) i.e. the product resulting from attack at
C-3. In the absence of the metal catalyst, only 10% conversion
occurred even after 4 days at room temperature and the ratio of C-2
to C-3 products was 2.2:1. (Scheme 17). The C-3 selectivity in the
presence of Ti(01Pr)t+ has been found to be quite general for a range of
epoxy alcohols, acids and amides, but its magnitude appears to be
nucleophile dependent.
Scheme 17
NEt2
C - 3 C -2
With Ti(0 PrK >20 : 1
Without Ti(0 Pr)^ l : 2.2

29
It has been suggested that these reactions proceed via an inter
mediate titanium complex such as (37) and (38).
Enhanced C-3 selectivity may be a result of the bond between C-3
and oxygen being better able to overlap with an empty d-orbital on
titanium, than the corresponding C-2 oxygen bond which lies almost in
the plane of the five-membered ring. This new titanium-mediated
nucleophilic opening procedure should extend the utility of 2,3-epoxy
alcohols for the synthesis of polyfunctional homochiral organic
molecules.
3 5 38-40Although there are many more examples ’ ’ of impressive
regiochemical control in the ring-opening of 2,3-epoxy alcohols, it is
not possible to cover them all in this review; moreover several3 5reviews of the subject have already been published. ’
Having discussed the nucleophilic reactions at C-2 and C-3 of the
2,3-epoxy alcohol moiety and their application to natural product
synthesis, it now remains to illustrate how reactions at C-l contribute
to the usefulness of these intermediates.
The hydroxyl group can be replaced by a variety of other functional
groups via conversion to a good leaving group such as a tosylate or
mesylate, followed by S^2 displacement with a range of nucleophiles
(Scheme 18).^

30
Scheme 18
R
A simple example of this type of reaction was provided recently 41by Katsuki in the synthesis of (-)-propanolol (39) which is a
(3-adrenergic receptor antagonist (Scheme 19).
Scheme 19
Me3SL ^ /O HMe3Si^ ^ ^ 0H P-f-VDIPT^ CH3SO?CI ^
TBHP Et3NTi(0*Pr) >95% e.e.
(39)

31
Mori and Ebata converted the 2,3-epoxy tosylates (40) and (41)
to the optically active pheromones (+)-disparlure (42) and (z,Z)-3,6-
e£s-9,10-epoxyheneicosadiene (43) respectively (the latter is the
saltmarsh caterpillar moth pheromone) (Scheme 20).
Scheme 20
42
1. L -H -P E T ,
T B H P , Ti(0'Pr)
2- TsC I4 9
TsO
(C9H19)CuLi
If
C10H21
(42) 0
"OHI. L - H - p e t ,
TBHP ,Ti(o6Pr)42. TsCI
Our own research (see Results and Discussion) has revealed that
it is possible for certain epoxy alcohols to be directly converted to
the corresponding epoxy selenides via treatment with two equivalents
of both N-phenylselenophthalimide (N-PSP) and tri-n-butylphosphine in
THF at -20°C (Scheme 21).

32
Scheme 21
ON-PSP,*B u 3P fTHF
-20°C
A useful modification to the latent reactivity at C-l was provided
by Mori and Ueda in their synthesis of 2,6-dimethyl-l,5-heptadien-3-ol43acetate, the pheromone of the comstock mealybug. Thus conversion of
the epoxy alcohols (44) and (45) (obtained from the allylic alcohol
(46) via Sharpless asymmetric epoxidation employing £-(-)- or
L-(+)-tartrate respectively) to the corresponding iodides (47) and
(48) followed by Zn-AcOH reduction gave after acetylation the {R)~ and
is)- pheromones (49) and (50) (Scheme 22).
Scheme 22

33
Certain 2,3-epoxy alcohols can be selectively substituted at C-l44via the Payne rearrangement. This involves a rearrangement in basic
medium to the isomeric 1,2-epoxy-3-ol followed by nucleophilic attack
at C-l affording a 1,2-diol (Scheme 23).
Scheme 23
A recent example of this reaction is incorporated into the45synthesis of (2S, 3S')-4-amino-2,3-dihydroxy-3-methylbutyric acid
(51) which is a degradation product from carzinophi1 in, an antitumour
antibiotic which selectively inhibits the synthesis of cellular DNA.
Thus enantioselective epoxidation of (52) according to the Sharpless
procedure afforded the epoxy alcohol (53) (e.e. 90%). Heating a
mixture of (53) and excess sodium azide in dioxane - 1M sodium
hydroxide (1:1) containing cetyl trimethyl ammonium bromide (0.2 eq,
phase-transfer catalyst) at 100°C led to the expected Payne rearrange-
ment/epoxide opening and gave the desired azide-diol (54) in 52%
yield. Compound (54) was converted to the target (51) via a sequence
of four simple steps (Scheme 24).

34
Scheme 24
PhCH D -f-)- DIPTTi (0^PrJ4 TBHP
-15° C
PhCH20.
4(53)
NaN3OH NaOH
CTAB
OBOC =*BuOC
The Payne rearrangement of 2,3-epoxy alcohols has been extensively46 47investigated by Sharpless and Masamune * over the last few years
with numerous applications to the synthesis of sugars and related
natural products and several excellent reviews have already been3 5 48published related to this useful methodology. ’ ’
This brief review concerned with the utility of the 2,3-epoxy
alcohol intermediate has illustrated its wide applicability and
potential in natural product synthesis.

35
SECTION 3 : SELECTED SYNTHESES EMPLOYING THE SHARPLESS ASYMMETRIC
EPOXIDATION
Since the discovery in August 1980 of the enantioselective
Sharpless epoxidation, the reaction has been used in the syntheses of
many different types of natural products and it is obviously not
possible to provide a complete review of them. The intent of this
section is to describe some selected examples from the literature in
order to illustrate the wide-ranging success and efficiency of this
relatively new synthetic method.
The extraordinary antitumour activity of the macrocycle maytansine
(55) and related derivatives has led to considerable activity by
various groups in their efforts to achieve a total synthesis. In
1983, Meyers and co-workers reported the first enantioselective49synthesis of (-)-maysine (56).
The north-eastern zone of the molecule possesses four of six
chiral centres and its synthesis was derived from an enantiomerically
pure starting material (5)-(+)-3-hydroxy-2-methylpropionic acid. A
number of simple synthetic steps converted this compound to the tri-
substituted allylic alcohol (57). Sharpless epoxidation of (57)

36
proceeded with greater than 99% enantioselectivity to introduce two of
the required chiral centres (Scheme 25).
Scheme 25
COjH
several steps
HO
The key epoxy alcohol (58) was further elaborated to the fragment (59)
which was coupled to the lithio derivative (60) (Scheme 26).
Scheme 26
OH

37
Finally, cyclization using the intramolecular Horner-Emmons Wadsworth
procedure and functional group manipulation afforded the desired
product (-)-maysine (56).
In recent years, the construction of C-glycopyranosides has been50accomplished with varying degrees of success. Masamune and co-workers
have reported an improved procedure for the introduction of the C-2
centre of the tetrahydropyran system using the titanium-catalyzed
asymmetric epoxidation under modified conditions (5 mol DET, 3.6 mol
Ti(01Pr)i+ and 2-4 mol TBHP per mole of the substrate). Thus treatment
of the allylic alcohol (61) with either (+)- or (-)-DET led to the
corresponding epoxy alcohols (62) and (63). It is interesting to note
here that the triethylsilyl alcohol protecting group was stable to the
Lewis acidic Sharpless conditions. Deprotection of the epoxy alcohols
(62) and (63) with tetra-n-butyI ammonium fluoride followed by treatment
with sodium hydride in DMF afforded the tetrahydropyranyl derivatives
(64) and (65) (Scheme 27) respectively.
Scheme 27
R = O CH 2Ph

38
Periodate cleavage of the 1,2-diols followed by reduction with sodium
borohydride afforded the alcohols (66) and (67). Finally protection
as the allyl ether derivatives, reductive ring opening of the
benzylidene group and acetylation afforded compounds (68) and (69)
which are potentially useful fragments for the synthesis of the51complex natural product palytoxin (Scheme 28).
Scheme 28
Palytoxin, the toxic principle isolated from marine soft corals of
the genus Palythoa is the most poisonous substance known to date except
for a few polypeptides and proteins found in bacteria and plants.
Kishi and co-workers have completely assigned the stereochemistry of 51palytoxin, primarily on the basis of organic synthesis. One of the
degradation products (70) possessed two unassigned asymmetric centres
(in R2).

39
* chiral centres
In order to determine the configuration of these asymmetric centres
unambiguously, all the possible stereoisomers of compound (71) were
synthesized [(71A) and (71B) from (72); (71C) and (71D) from inter
mediates used in the transformation of (72) to (71A) and (71B)].
The assignment of the absolute configuration of these compounds
depended upon Sharpless asymmetric epoxidation. Compound (71D) was
found to possess the same stereochemistry as present in palytoxin
itself. The stereochemical predictability of the Sharpless procedure

40
was used to establish the absolute configuration of several other51degradation products from palytoxin.
The Sharpless asymmetric epoxidation has been employed in the13 23 42 43 52-54synthesis of several chiral insect pheromones. ’ ’ ’ ’55Recently Johnston and Oehlschlager have reported an extremely
efficient route to multi gram quantities of both enantiomers of
frontal in (81) which has been shown to possess aggregation pheromone
activity in several species of Dendvootonus bark beetles. Although
there are many existing syntheses of this compound, several are not
amenable to large-scale work and some are only applicable to the
production of one of the two possible enantiomers. Incorporation of
the Sharpless procedure has enabled both enantiomers in greater than
90% e.e. to be readily available. The synthesis is shown in
Scheme 29.
Thus ring-opening of the 2,3-epoxy alcohols at C-3 by treatment
with lithium aluminium hydride afforded the 1,3-diols (73A) and (73B)
which were easily converted in high yield to the two enantiomers of
frontalin (74A) and (74B).
The recent synthesis of (-)-swainsonine (75) by Sharpless and 48co-workers employs the methodology of the Sharpless/Masamune
47 56approach to polyhydroxylated natural products. ’ This indolizidine
alkaloid is known to be an effective inhibitor of both lysosomal
a-mannosidase, an enzyme involved in the cellular degradation of
polysaccharides, and mannosidase II, a key enzyme in the processing of
asparagine-linked glycoproteins. Three syntheses of this biologically
important compound have appeared in the literature in the past year,
each starting from a glucose or mannose derivative. This most
recent synthesis of swainsonine is a totally different approach
starting from an achiral compound.

41
Scheme 29
MgBr
CuBr
T
OH

42
Thus Sharpless asymmetric epoxidation of the allylic alcohol (76)
obtained in three steps from trans-1,4-dichioro-2-butene, afforded
the crystalline epoxy alcohol (77) in 95% e.e. Payne rearrangement- 44epoxide cleavage (Section 2) was effected by treatment of (77) with
thiophenol (1.2 eq) in tevt-butanol and 0.5M sodium hydroxide at
85°C to afford, after benzylation, the sulphide (78) (Scheme 30).
Scheme 30
.OHTsN'
k 6Ph (77)
PhSHNaOH*BuOH,85°C
(78)
Oxidation to the sulphoxide followed by Pummerer rearrangement in
the presence of acetic anhydride, trifluoroacetic acid and 2,6-lutidine
afforded the acetoxy sulphide (79). Reduction with lithium aluminium
hydride followed by Swern oxidation afforded aldehyde (80). Homologa
tion with triethyl phosphonoacetate gave the ester (81). Reduction
followed by Sharpless epoxidation ((-)-DIPT) resulted in the desired

43
epoxy alcohol (82) homogeneous by h.p.l.c. (> oa. 300:1) (Scheme 31).
Scheme 31
OBn
OBn(78)
OBn
1. U A IH 42. rC0Cl)2
DM SO -60°C
BnTsN^
OBn
T CHO OBn
(80)
O 0 Na©(EtOlP-CHCOzEt^
OBn
I.DIBAL
OBn
2-D-Q-DIPTrTBHP, Ti(o*Pr)4 ,
- 20°C
OBn(82)
Bn=CH 2Ph
The epoxy-alcohol (82) was oxidised under the Pfitzner-Moffatt
conditions followed by homologation with carbethoxymethylidenetriphenyl -
phosphorane and diimide reduction of the resultant a,p-unsaturated
ester to afford the epoxy ester (83) in 74% yield for the three steps.
Removal of the tosyl protecting group with sodium naphthalide followed
by immediate protection afforded the silyl ether (84) (Scheme 32).

44
Scheme 32
OBn
(83)
severalsteps
(a) D C C , D M S 0 ,C 5H5N H 0 S 0 2CF3
PhjP = C H C 0 2E t ;
K02CN=NC02K , AcOH .^BuMe2S i0 S 0 2CF3 ;
DIBAL .
Compound (84) was converted to (-)-swainsonine (75) after ester reduc
tion, primary alcohol mesylation and final deprotection of the
intermediate bicyclic quaternary ammonium salts (ois and trans-fused).
This synthesis of swainsonine (75) is an extremely elegant
example of the use of the enantioselective Sharpless epoxidation and
highly regioselective epoxide-cleavage reactions in natural product
synthesis.
The asymmetric epoxidation has also found application in the57synthesis of polyether ionophores. The complex ionophore ionomycin
(85) isolated from Streptomyoes oonglobatus (ATCC 31005) is unique in
that it chelates metal ions as the dibasic acid and has much greater
Ca2+ selectivity than does A-23187 (calcimycin), the other major
calcium-selective ionophore.

45
(85)
lonomycin
The synthesis of the left-hand portion (86) of ionomycin (85) has been5reported by Wilts and co-workers starting from geraniol acetate to
establish the carbon skeleton and utilizes the Sharpless asymmetric
epoxidation to introduce the required chirality. Thus selenium
dioxide allylic oxidation of geraniol acetate introduced the function
ality necessary for asymmetric epoxidation and for the subsequent
introduction of the remaining three carbon atoms (Scheme 33).
Four of the five chiral centres of the furanoid fragment were thus
secured and the fifth centre may be fixed after coupling of the right-
hand portion to the bisfuranoid fragment.
The last example in this section is an extremely elegant use of
the Sharpless asymmetric epoxidation and kinetic resolution processes.24Dolle and Nicolaou have reported the total synthesis of aurodox (87)
and efrotomycin (88), narrow-spectrum antibiotics belonging to the
newly-discovered class of compounds known as the elfamycins.
Aurodox (87)

46
Scheme 33
OAc
D-0- DET
T ifc m l TBHP 4
-23°C
PhNCOEt3N
AcO
H CI04
DihydropyranH®
'OAc2-Dowex
H©
HOi n n
TBHP V ^ O ' T i ^ OT iln^D ^l < HTi(o*Pr) - 23° C
4 OH
severalsteps
BuPh.Si O

47
Efrotomycin (88)
The stereocontrol led synthesis of the c^s-tetrahydrofuran system
was achieved via a new tandem methodology based on oligoepoxide
openings (Scheme 34).
Scheme 34
The synthesis of this fragment began with a kinetic resolution of the
secondary allylic alcohol (89) (0.6 eq t-BuOOH, 1.0 eq (-)-DET, 1.0
eq Ti(01Pr)1+) to afford after protection of the alcohol, compound
(90). Addition of sodium phenyl selenide (from NaBH4 and PhSeSePh)
followed by oxidation and elimination of the resulting selenoxide
afforded the allylic alcohol (91). Sharpless epoxidation, deprotec
tion of the silyl ether and partial hydrogenation of the alkyne
afforded the epoxy diol (92) (Scheme 35).

48
Scheme 35
Ph'1. TBHP,Ti(b‘Pr)
g-n-D ET /2. Protection
OSiPh^Bu
"O
1. PhSeSePh
NaBH4 ^2. H20 2
Ph'
O SiPh2* Bu
OH
(91)
TBHP,Ti(o"Pr)4
D -0- DET*"~
-20° C
Per-acid epoxidation, dio1 protection, deprotection of the benzyl
ether followed by oxidation (RuO^-NalO^) and reaction with diazomethane
afforded the di-epoxide (93). Treatment with dimsyl potassium
(KCH2S(0)CH3) gave the c-is-fused tetrahydrofuran (94) (Scheme 35) after
hydroxyl protection.
Scheme 36
(93) (94)
OSiMe/Bu

49
The asymmetric synthesis of the key intermediate (95) which is a
stable degradation product of aurodox was also achieved employing the
Sharpless epoxidation procedure to introduce two of the required
chiral centres. Thus treatment of the allylic alcohol (96) with
Z?-(-)-DET (2 eq), TBHP (2 eq) and titanium (IV) isopropoxide (1 eq)
afforded the epoxy alcohol (97A). Conversion to (97B) followed by
ring opening of the epoxide in the presence of aluminium chloride gave
the secondary alcohol (98) which was further elaborated to goldinono-
lactone (95) (Scheme 37).
Scheme 37
The syntheses described briefly in this section and many other
examples employing the Sharpless asymmetric e p o x i d a t i o n ^ illustrate
the importance of this reaction to the synthetic chemist.

50
SECTION 4 : FURTHER APPLICATIONS
The purpose of this section is to discuss some recent develop
ments of the Sharpless asymmetric epoxidation reaction.
Although this titanium-tartrate-mediated epoxidation has been
shown to be effective with allylic alcohols of widely varying sub
stitution patterns, it was recently reported to be very sensitive to64steric bulk in the substrate. Thus allylic alcohols with tert-
butyl groups at each one of four possible positions (R1-R1+) (Scheme 38)
were employed in the Sharpless epoxidation.
Scheme 38
V ! -Q - D E TR2 / R1
j < > HT B H P ^
r 4 HT i(0 ‘ Pr)4 K
R 4 H
For R1, R2, R3 = ^Bu separately, the face selection of the sub-g
strates was in the normal direction but the enantioselectivity was
much lower (oa. 40-80% e.e.). When R4 = ^Bu the corresponding racemic
secondary allylic alcohols did not show any useful kinetic resolution.
These results suggest that attempts to asymmetrically epoxidize or
kinetically resolve allylic alcohols with tertiary groups are not
likely to produce the high enantioselectivity normally exhibited by
this asymmetric epoxidation procedure.
Sharpless has recently reported the results of the asymmetric65epoxidation of various homoallylic alcohols. Interestingly the
enantiofacial selection is opposite to that observed for allylic
alcohols, but the enantiomeric purities of the products range from only
23-55%. Although this method for the production of 3,4-epoxy alcohols

51
is inefficient, it may prove to be of some value due to its ability to
furnish a wide variety of chiral compounds predictably.
Oehlschlager and Czyzewska and others have applied the Sharpless
asymmetric epoxidation to allylic alcohols containing a conjugated 66 67alkyne. ’ Reaction of the resulting alkynyl epoxy alcohols with
dialkyl cuprates gave dihydroxy allenes of high optical purity 66(Scheme 39). The stereochemistry of the reaction was shown to be
exclusively anti when sulphide was present. Organocuprates cause
isomerization of allenes probably via electron transfer; dimethyl
sulphide was found to retard this process.
Scheme 39
C — CH20HHCeC —
(-)-DET
Ti(01Pr)„ TBHP
/R2CuMgBr.Me2S R. . C ^ C ^ O H----------- Z = C = C ' " n0H
One of the major factors establishing the Sharpless epoxidation
procedure as an extremely useful reaction in organic synthesis has been
the compatibility of the reaction conditions to the presence of a range
of functionality within the allylic alcohol substrate. Pridgen andCQ
co-workers have recently reported the presence of the 4,5-diphenyl-
oxazoyl moiety as a masked ester, to be fully compatible with the
Sharpless conditions. Since this moiety has been found to be a
versatile carboxyl equivalent, the result should further extend the

52
this application by the synthesis of (-)-methyl (5£), (6S)-oxido-7-
acetoxyheptanoate (99) (Scheme 40,n=l) which is a widely used inter-finmediate in the construction of optically active leukotrienes A-E.
Scheme 40
utility of the epoxidation procedure. These workers have illustrated
Ph
Phii^ OH L-0-DET,
Ph
T B H P , Ph
Tl(0<,pr)4 *-23° C
ry v c ^ ° *H
H
1. Ac20pyridine
2. 10 2Ph
OA H
o o
OAcTsOHMeOHA
o M
H
(99)
Certain epoxy alcohols, notably the 2-alkyl series (100) are
sensitive to ring opening under the standard asymmetric epoxidation
conditions. Although the use of TitO^BuK is a partial solution
to this problem it was thought that intentional and selective opening
of these epoxy alcohols (100) might serve to trap the chiral inter-69mediates in a useful form.
OH
(100)R
OH
(101)

53
Substitution of Ti(01Pr)i+ with TiCl2(01Pr)2 was successful
in trapping the intermediate epoxides as 3-chloro-l,2-diols.
Subsequent treatment with base effected ring closure to the epoxide
with opposite chirality to that normally obtained under the 'standard'g
asymmetric epoxidation conditions (Scheme 41).
Scheme 41
Cl
29 14
0H 2 TiC I2(ofPrj2 U ° % H1 l -Q -d e t h c ^
nOr* 29 14TBH P, 0UC
OH ©
H2SC14
OH
This same result was obtained employing a 2:1 TiCl2(01Pr)2 : tartrate
ratio and the reversed face selection was observed for a range of
allylic alcohols. In addition to this result the use of 2:1 ratio
of Ti(01Pr)lt. : tartramide has also been found to effect epoxidation
with opposite enantiofacial selectivity to that of the standard69
catalyst (Scheme 42).

54
Scheme 42
Ph
PhOH
2Ti(o^Pr)4TBH P2.4(102)
Ph
Ph'
OHO. 1
V ^ ^ N H C H 2Ph. * L / N H C H 2Ph
HO" if O
(102)
This discovery enables access to either enantioface of an allylic
alcohol employing the appropriate derivative of the inexpensive
£-(+)-tartaric acid. It should be noted, however, that the 2:1
system does appear to be rather substrate dependent. Many questions
regarding the mechanism of both 2:1 and 2:2 systems remain to be
answered and the current ideas have recently been publ ished. 10»H>69
Nevertheless, these new inverse induction systems are valuable
additions to the already popular parent asymmetric epoxidation
process.

55
Although this review has been selective rather than exhaustive, it
is clear that the Sharpless asymmetric epoxidation is an extremely
important discovery and there is no doubt that its applications in
organic synthesis will continue to increase particularly as the
mechanism of the process becomes more fully understood.

56
REVIEW REFERENCES
1. (a) J.D. Morrison, "Asymmetric Synthesis", Academic Press, Inc.,
London, 1983-1985,Vols. 1-5.
(b) S. Masamune, Heterocycles, 1984, 21_, 107.
2. A.S. Rao, S.K. Paknikar, and J.G. Kirtane, Tetrahedron, 1983,
39, 2323, and references therein.
3. K.B. Sharpless, C.H. Behrens, T. Katsuki, A.W.M. Lee, V.S. Martin,
M. Kakatani, S.M. Viti, F.J. Walker, and S.S. Woodward, Pure and
Appl. Chem. , 1983, 55_, 589.
4. D. Seebach, B. Weidmann, and L. Wilder, "Modern Synthetic Methods
1983", ed. R. Scheffold, Otto Salle Verlag, Frankfurt, 1983, p.323.
5. C.H. Behrens and K.B. Sharpless, Aldrichimica Acta, 1983, J6.j 67,
and references therein.
6. T. Katsuki and K.B. Sharpless, J. Am. Chem. Soc. , 1980, 102, 5974.
7. K.B. Sharpless, "Discovery of the titanium-catalyzed asymmetric
epoxidation - a personal account", Proa. Robert A. Welch Found. Conf.
Chem. Res. , 1984, 27., 59.
8. J.G. Hill, B.E. Rossiter, and K.B. Sharpless, J. Org. Chem., 1983,
48, 3607.
9. J.G. Hill, K.B. Sharpless, C.M. Exon, and R. Regenye, Org. Synth.
1985, 63, in press.
10. K.B. Sharpless, S.S. Woodard, and M.G. Finn, Pure and Appl. Chem. ,
1983, 55, 1823.
11. I.D. Williams, S.F. Pedersen, K.B. Sharpless, and S.J. Lippard,
J. Am. Chem. Soc., 1984, 106, 6430.

57
12. V.S. Martin, S.S. Woodard, T. Katsuki, Y. Yamada, M. Ikeda, and
K.B. Sharpless, J. Am. Chem. Soc. , 1981, 103, 6237.
13. B.E. Rossiter, T. Katsuki, and K.B. Sharpless, J. Am. Chem. Soc.,
1981, 103, 464.
14. W.R. Roush and R.J. Brown, J. Org. Chem., 1982, £7, 1371.
15. (a) P.L. Durette, Synthesis3 1980, 1037.
(b) B. Iselin and T. Reichstein, Helv. Chim. Acta, 1944, 2J_, 1146.
16. D. Dominguez and M.P. Cava, J. Org. Chem., 1983, £8, 2820.
17. A.V. Rama Rao, J.S. Yadav, K. Bal Reddy, and A.R. Mehendale,
J. Chem. Soc. 3 Chem. Commun. , 1984, 453.
18. K. Mori, T. Otsuka, and M. Oda, Tetrahedron, 1984, £0, 2929.
19. S. Miyano, L.D.-L. Lu, S.M. Viti, and K.B. Sharpless, J. Org. Chem.,
1983, £8, 3608.
20. M. Bessodes, E. Abushanab, and K. Antonakis, Tetrahedron Lett.,
1984, 5899.
21. J.D. White, M. Kang, and B.G. Sheldon, Tetrahedron Lett., 1983,
4539.
22. J.A. Marshall and R.C. Andrews, J. Org. Chem. , 1985, 50, 1602.
23. K. Mori and T. Otsuka, Tetrahedron, 1983, 39, 3267.
24. R.E. Dolle and K.C. Nicolaou, J. Am. Chem. Soc., 1985, 107, 1691.
25. S. Masamune and W. Choy, Aldrichimica Acta, 1982, 15, 47.
26. (a) M.G. Finn and K.B. Sharpless "Asymmetric Synthesis", ed.
J.D. Morrison, Academic Press, Inc., London, 1985, Vol. 5,
Chapter 8.
(b) B.E. Rossiter, "Asymmetric Synthesis", ed.J.D. Morrison,
Academic Press, Inc., London, 1985, Vol. 5, Chapter 7.

58
27. N. Minami, S.S. Ko, and Y. Kishi, J. Am. Chem. Soa. , 1982, 104,
1109.
28. J.M. Finan and Y. Kishi, Tetrahedron Lett., 1982, 2719.
29. S.M. Viti, Tetrahedron Lett., 1982, 4541.
30. S. Takano, S. Otaki, and K. Ogasawara, J. Chem. Soo.3 Chem. Commun. ,
1983, 1172.
31. 0. Mitsunobu, Synthesis, 1981, 1.
32. W. Pickenhagan and H. Bronner-Schindler, Helv. Chim. Acta, 1984,
67, 947.
33. K. Prasad and 0. Repic, Tetrahedron Lett., 1984, 3391.
34. P. Ma, V.S. Martin, S. Masamune, K.B. Sharpless, and S.M. Viti,
J. Org. Chem. , 1982, 47, 1378.
35. Y. Kobayashi, Y. Kitano, and F. Sato, J. Chem. Soc.3 Chem. Commun.,
1984, 1329.
36. N. Koizumi, M. Ishiguro, M. Yasuda, and N. Ikekawa, J. Chem. Soo. 3
Perkin Trans. I, 1983, 1401.
37. (a) M. Caron and K.B. Sharpless, J. Org. Chem., 1985, 50, 1557.
(b) J.M. Chong and K.B. Sharpless, J. Org. Chem., 1985, 50, 1560.
38. P. Sundararaman, G.Barth, and C. Djerassi, J. Am. Chem. Soo., 1981,
103, 5004.
39. P.G. Gassmann and R.S. Gremban, Tetrahedron Lett., 1984, 3259.
40. D. Gani, D.O'Hagan, K. Reynolds, and J.A. Robinson, J. Chem. Soo.3
Chem. Commun. , 1985, 1002.
41. T. Katsuki, Tetrahedron Lett., 1984, 2821.
42. K. Mori and T. Ebata, Tetrahedron Lett., 1981, 4281.

59
43. K. Mori and H. Ueda, Tetrahedron, 1981, _37, 2581.
44. G.B. Payne, J. Org. Chem. , 1962, 27, 3819.
45. P. Garner, J.M. Park, and V. Rotello, Tetrahedron Lett. , 1985, 3299.
46. A.W.M. Lee, V.S. Martin, S. Masamune, K.B. Sharpless, and F.J. Walker,
J. Am. Chem. Soe. , 1982, 104, 3515.
47. T. Katsuki, A.W.M. Lee, P. Ma, V.S. Martin, S. Masamune, K.B. Sharpless,
D. Tuddenham, and F.J. Walker, J. Org. Chem., 1982, 47, 1373.
48. C.E. Adams, F.J. Walker, and K.B. Sharpless, J. Org. Chem., 1985,
50, 420.
49. A.I. Meyers, K.A. Babiak, A.L. Campbell, D.L. Comins, M.P. Fleming,
R. Henning, M. Heuschmann, J.P. Hudspeth, J.M. Kane, P.J. Reider,
D.M. Roland, K. Shimizu, K. Tomioka, and R.D. Walkup, J. Am. Chem.
Soe., 1983, 105, 5015.
50. L.A. Reed, III, Y. Ito, S. Masamune, and K.B. Sharpless, J. Am.
Chem. Soe. , 1982, 104, 6468.
51. (a) L.L. Klein, W.W. McWhorter, Jr., S.S. Ko, K.-P. Pfaff, and
Y. Kishi, J. Am. Chem. Soe., 1982, 104, 7362.
(b) S.S. Ko, J.M. Finan, M. Yonaga, Y. Kishi, D. Uemura, and
Y. Hirata, J. Am. Chem. Soe., 1982, 104, 7364.
(c) H. Fujioka, W.J. Christ, J.K. Cha, J. Leder, Y. Kishi,
D. Uemura, and Y. Hirata, J. Am. Chem. Soe., 1982, 104, 7367.
52. L. Guo-quiang, X. Hai-jian, W.Bi-chi, G. Guong-zhong, and
Z. Wei-shan, Tetrahedron Lett., 1985, 1233.
53. N. Nakagawa and K. Mori, Agrie. Biol. Chem., 1984, 48, 2505.
54. B.D. Johnston and A.C. Oehlschlager, J. Org. Chem., 1982, 47,
5384.

60
55. B.D. Johnston and A.C. Oehlschlager, Can. J. Chem. , 1984, 62, 2148.
56. S.Y. Ko, A.W.M. Lee, S. Masamune, L.A. Reed, III, K.B. Sharpless,
and F.J. Walker, Sotenee (Washington3 D.C.), 1983, 220, 949.
57. P.G.M. Wuts, R.D'Costa, and W.Butler, J. Org. Chem., 1984, 49,
2582.
58. W.H. Rastetter and J. Adams, Tetrahedron Lett. , 1982, 1319.
59. E.J. Corey, A. Marfat, and B.C. Laguzza, Tetrahedron Lett. ,
1981, 3339.
60. E.J. Corey, S. Hashimoto, and A.E. Barton, J. Am. Chem. Soo. ,
1981, 103, 721.
61. W.R. Roush and T.A. Blizzard, J. Org. Chem., 1984, 49, 4332.
62. S.R. Baker, J.R. Boot, and S.E. Morgan, Tetrahedron Lett. ,
1983, 24, 4469.
63. A.M. Doherty and S.V. Ley, submitted for publication.
64. M.J. Schweiter and K.B. Sharpless, Tetrahedron Lett., 1985, 2543.
65. B.E. Rossiter and K.B. Sharpless, J. Org. Chem., 1984, 49, 3707.
66. A.C. Oehlschlager and E. Czyzewska, Tetrahedron Lett., 1983, 5587.
67. B. Bernet and A. Vasella, Tetrahedron Lett., 1983, 5491.
68. L.N. Pridgen, S.C. Shilcrat, and I. Lantos, Tetrahedron Lett. ,
1984, 2835.
69. L.D.-L.Lu, R.A. Johnson, M.G. Finn, and K.B. Sharpless, J. Org.
Chem., 1984, 49, 728.

61
RESULTS AND DISCUSSION
INTRODUCTION
In 1981, the isolation of a novel antibacterial agent designated
M139603 (1), obtained from the aerobic fermentation of Streptomyces
longispovoflavus NCIB 11426, was first reported in the literature.*
This compound belongs to the general class of polyether ionopnores,2-4one which has received increasing attention over the past few years.
It is easy to understand the reasons for the intense research activity
in this area. Besides the important biological properties of polyether
ionophores, the complexity of their structures presents a formidable
challenge to the synthetic chemist.
We became interested in the synthesis of this novel ionophore and
of model compounds which might mimic its biological activity. Before
embarking upon a discussion of the work presented in this thesis, a
very brief introduction into this class of ionophores is given,
followed by a more detailed discussion of the literature related co
M139603 itself.
29 15 27
34M139603 (l) M = Na , H

62
SECTION 1 : THE IQNOPHQRES
As the name suggests, an ionophore is an ion-carrier, a molecule
capable of complexing with an ion and enabling its transport through a
lipophilic medium. The diversity of structure within this class of
molecules is considerable, but a common feature is the presence of
heteroatoms capable of acting as ligands for an ion. The ionophore
antibiotics are naturally occurring compounds produced by micro
organisms, however they are often extremely toxic to, or have strong
physiological effects on higher organisms and this can limit their
antibiotic applicability. In addition to the naturally occurring
lonophores, there exist several examples of synthetic analogues which
possess many of the same properties, for example, the crown ethers and
cryptates.
One of the most recent entries into the field of ionophores is
the polyether group. These are characterized by having a linear
carbon backbone, containing tetrahydrofurans and -pyrans. They
possess a wide range of heteroatom functional groups for cation
liganding and they all possess many centres of asymmetry. In addition,
most of the polyether ionophores characterized to date have a structure
terminating in a carboxylic acid and hence this group of compounds has
often been termed the 'carboxylic acid ionophores'.
The structures of two ionophore antibiotics, monensin and indano-
mycin (X-14547A), are illustrated below. The former ionophore was the
first to have its structure fully elucidated and has been synthesized 6 7by two groups. ’ Indanomycin is a novel pyrrole-containing lonophore
owhich transports divalent cations and it has recently been synthesized
by our group^ ana two others.

63
Figure 1
MO NENSIN
Figure 2 : (see Section 7 for a stereopair representation of the
conformation of X-14547A in the crystalline state)
(X-14547A) INDANOMYCIN

64
An essential feature of the ionophoric activity of the polyethers
originates from the capability of the ionophore complexes to adopt a
conformation in which all of the liganding heteroatoms orient towards
interior of the complex, thus exposing a relatively nonpolar exterior
(Figure 1). The lipid bilayer in biological membranes presents a
barrier to the free transport of cations since the hydrophilic hydrated
cations cannot cross the central hydrophobic region. In facilitated
diffusion, the cation can cross this barrier in association with an
ionophore.
BIOLOGICAL ACTIVITY
The polyether antibiotics generally exhibit activity against
Gram-positive bacilli, cocci and filamentous organisms and it has been
suggested that this microbial inhibition is probably due to losses of
essential monovalent cations, since all polyethers are capable of
transporting such cations.
It was first reported in 1968 that monensin and a variety of other12polyether ionophores exhibited coccidiostat activity, that is, they
were effective in controlling infection in coccidia, a parasite which
affects the internal tract of birds and mammals. Monensin was the
first polyether to be marketed as a coccidiostat in 1971.
Several polyethers have also been found to improve ruminant feed
utilization by increasing the amount of propionic acid produced in the
rumen of animals such as cows, sheep and goats.
Ionophores have also been of interest in the field of pharmacology.
Since the discovery that lasalocid (X-537A) and calcimycin (A-23187). 14could mobilize Ca2+ ions, these two polyether antibiotics have
received much attention. They have been found to exert a profound

65
positive effect upon the contractility of heart muscle and, lasalocid15in in vivo experiments caused dramatic increases in coronary flow
and cardiac output. The potential pharmacological applications are
still under study.
Although this is a very brief and simplified summary of the various
biological properties that these ionophores possess, it clearly indi
cates their importance and the immense interest they have stimulated.

66
SECTION 2 : THE POLYETHER IONOPHORE M1396031 1 fiThe novel ionophore M139603 (1) ’ possesses the
characteristic tetrahydrofuran and tetrahydropyran groups common
to polyether ionophores. However, it also possesses an unusual
six-membered carbocyclic ring and a biosynthetically rare acyltetronic
acid moiety. In fact, this was the first example of a polyether iono
phore with the terminal carboxylic acid group replaced by a tetronic
acid functionality.
M139603 and a number of its derivatives (prepared by the ICI group)
have been found to be effective in reducing the proportion of methane
produced by ruminal fermentation and in increasing the proportion of
propionic acid in rumen fluid and are therefore believed to possess
growth promoting properties in ruminants.'*'
Shortly after the structure of M139603 had been reported, a Swiss
group‘d isolated a closely related ionophore antibiotic known as
tetronomycin (2), from a culture of a new strain of Streptomyces sp.
nov (S 53161/A). This antibiotic differs structurally by the absence
of methyl groups on C-20 and C-22 and by the presence of a methylene
group at C-34 (numbered according to the convention proposed by3Westley ). In addition, the absolute configuration was, somewhat
surprisingly, reported to be opposite to that of M139603 at all of
the comparable chiral centres.

67
29 15 27
TETRONOMYCIN (2)
The ICI research team published* the X-ray crystal structure of
the 4-bromo-3,5-dinitrobenzoyl derivative of M139603 and were able to
show that in the crystalline state, five of the oxygen atoms in the
molecule were acting as ligands for the metal ion along with the
oxygen from a molecule of water. These six ligands then formed a
distorted octahedral array around the metal ion (Figure 3a).
The lipid solubility of the resulting complex is thus explained by
the shielding of the polar interior, which delocalizes the cation
charge, and by the simultaneous exposure of the nonpolar groups on the
exterior.

Figure 3a
Crystal structure of the mono-0- acetyltetronomycin silver salt.The upper inset view shows the silver coordination more clearly.

69
An X-ray crystal structure of the mono-O-acetyltetronomycin silver
salt^ showed that the metal ion was complexed by the acidic diketone,
by the tetrahydropyran and tetrahydrofuran oxygen atoms, by the terminal
ether oxygen and, interestingly by the 018 double bond (Fig. 3b).
One may question whether these solid-state structures are really
representative of the conformation adopted in solution. Using COSY1945 and 90 n.m.r. techniques, Grandjean and Laszlo have established
that the solution geometry is closely representative of the solid-state
structure. In addition, the conformation of the free acid and sodium
salt are found to be almost identical for both ionophores. These
workers have also studied the binding properties with a number of
different cations and have shown that both display a selectivity for
Na+ and Ca2+, two cations with very similar ionic radii. In addi
tion they display a marked positive cooperativity in the transport of19Pr3+ across lipid bilayers when used jointly with lasalocid.
Biosynthetic studies of the antibiotic M139603 have been reported2 0 - 2 2recently by Staunton and co-workers. From n.m.r. labelling
techniques they have shown that the sequence of chain assembly and
resulting stereochemistry at the various chiral centres are not consis
tent with either of the stereochemical prototypes recently proposed by 23Cane et at. and thus suggested that M139603 might belong to a new
stereochemical class of polyether ionophores. The carbon skeleton is
derived from a linear combination of seven acetate units, six propionate
units and a C2 unit of unknown origin as illustrated (Figure 4). The
two carbon atoms of the tetronic acid ring remain uniabelled in all of
their experiments.

70
Figure 4
The unusual carbocyclic ring could be formed by an aldol reaction
between a carbanion at C-10 and a carbonyl group at C-5, followed by
dehydration and reduction, or by a Michael reaction of the carbanion at
C-10 onto a double bond at C-4 - C-5 (Figure 5).
Figure 5
The nucleophilic reactivity at C-10 could be achieved from a variety of
structures (A-D).

71
Structure C can be eliminated because biosynthetic studies indicate
that C-30 is hydroxylated after the carbocyclic ring closure. With
regard to the formation of the tetrahydropyran unit, 180 labelling
experiments indicate that the oxygen from acetate at C-17 is retained
in the biosynthetic precursor. An attractive proposal for the con
certed generation of both the tetrahydropyran and cyclohexane rings
can therefore be considered (Figure 6) where both rings would be formed
in chair conformations with all groups equatorial.
Figure 6
M e O ‘

72
It is possible that this proposed polyene cyclisation could occur
without enzymatic catalysis, providing a suitable cation could hold the
structure in the required conformation. This proposal seems very
likely since models indicate that the conformation required is close to
that adopted by the ionophore itself.
180 Labelling studies on the top right-hand portion of M139603
have shown a retention of oxygen from propionate at C-21 but a lack of
retention of acetate at C-25 in the biosynthetic precursor. This seems
to indicate a precursor of type (3) with oxygen absent at C-25.
OMe
(3)

73
SECTION 3 : MODEL STUDIES RELATED TO THE IONOPHORE M139603
INTRODUCTION
Since methodology for the synthesis of the acyltetronic acid por-24tion of the molecule had already been established in our group, we
were interested in developing a route to the right-hand portion of the
molecule. In addition to the importance of setting up the required
chiral centres, it was also necessary to be able to ensure the trans-
stereochemistry of both double bonds in any coupling strategy.
Focusing attention on the disubstituted double bond of the iono-
phore, it was hoped to couple a tetrahydrofuranyl fragment (5) (where
X is some suitable anion-stabilizing group) with the tetrahydropyranyl
aldehyde (4) (Scheme 1).
Scheme 1
An alternative strategy (Scheme 2) involving the addition of (6)
to the tetrahydrofuranyl aldehyde (7) seemed less favourable since our 25 26group and others have found that systems similar to (6) are prone
to ring opening via (3-oxyelimination (Scheme 3).

74
Scheme 2
Scheme 3
Although our major interest at this time was to develop a route
towards the synthesis of the naturally occurring system, we considered
that it would be both useful and interesting to design and construct
an unnatural system which might possess similar properties.
Relatively little work has been reported on model compounds for
these complex ionophores although the importance of discovering a
biologically active model compound cannot be underestimated.
27Wierenga and co-workers have synthesized the model compound (8)
which has been shown to possess transport properties which compare
very favourably with the best known Ca2+ ionophores, calcimycin
and lasalocid.

75
(«)
28Hackler has also reported the synthesis of a very simple model
ionophore from the reaction of diol (9) with potassium hydroxide. The
diol was easily obtained from the permanganate oxidation of geranyl
acetate (Scheme 4).
Scheme 4
AcO
The presence of the water molecule was confirmed by n.m.r. and
elemental combustion analysis and was explained by its ability to
confer maximum steric and electrostatic stability upon the complex.
The acetate group apparently functions in a similar manner to the
carboxyl group in the carboxylic acid ionophores.

76
RESULTS
Shortly after the X-ray crystal structure of the 3-bromo-2,4-
dinitrobenzoyl derivative of M139603 was reported,* we obtained a
sample of this compound from ICI and also recrystallized it from
methanol. Our X-ray crystal structure determination (Figure 7)
showed that the terminal methoxy group was no longer acting as a ligand
for the metal ion in the crystalline state and had been replaced by a
molecule of methanol. The absence of the liganding water molecule in
our system gave rise to a complex involving only five ligands compared
to six in the ICI crystal structure. Thus it seemed that the free
rotating side-chain possessing the methoxy substituent could easily be
substituted for an external ligand. It might be suggested that this
result may be due merely to the purity of the methanol used, although
since we made no effort to dry the solvent prior to its use, this does
not seem to be an adequate explanation. Indeed, the initial report*
stated that all the oxygen-metal distances were within a normal range
except for the distance between the metal and the methyl ether oxygen
which was too short, perhaps indicating some strain present. Moreover
it should be noted that the conformation of the ionophore did change
during this ligand rearrangement, possibly resulting in a slightly
more favourable energy state.
With this result in mind, and our interest in model compounds
which might possess ionophoric oroperties, we decided to begin our
studies by synthesizing compounds of the type (10) with the side-chain
absent and where R could be variable.
R ^ H O'
C«)

o
Figure 7
Br

78
Synthetic approaches towards (10) must ensure (a) cis-stereo-
chemistry about the tetrahydropyran ring and (b) introduction of the
^-double bond stereoselectively, By our earlier consideration of
the carbon-carbon double bond formation, we anticipated coupling a
fragment of type (11), where X is some anion-stabi1izing substituent,
with aldehydes of type (12).
■ < r
(11) (12)
There are several synthetic methods available for such a coupling29 30reaction, but the Julia aldehyde-sulphone method ’ where X would
be -S02Ph seemed to be the most attractive in terms of maximizing the
transiois double bond ratio. In addition, it is known that when
branching occurs adjacent to the newly formed double bond, much higher30trans-stereoselectivity is observed. In the naturally occurring
system, of course, there is a methyl group present at position 20
adjacent to the double bond (13).
(13)
We were therefore confident that this route would give a high degree of
trans-stereoselectivity. Hence, our initial targets were to synthesize

79
compounds of type (11) and (12). The tetrahydrofuran portion was
easily obtained in high yield from furan using standard chemistry
(Scheme 5).
Scheme 5
7 V''O ''(14)
HO) h 2 ^^ 5%Rh/AI203
HO
(15)
NP$
- B u3P
PhSmCPBA,
SO-Ph
(16) O’)
NPS = N -(phenylthio)succinimide
31Thus quenching of 2-1ithiofuran with ethylene oxide followed by
hydrogenation over rhodium on alumina^a gave the 2-substituted tetra
hydrofuran (15) in 74% overall yield. Conversion to the sulphide (16)
using N-phenylthiosuccinimide and tri-n-butylphosphine at room tempera- 32ture, followed by oxidation with two equivalents of mCPBA afforded
the crystalline sulphone (17) in high yield.
The advantage of this route was that it enabled us to synthesize
a variety of other coupling fragments from alcohol (15) (Scheme 6).
Scheme 6
O' V OH (15)
Ph3P, imidazol^
l2 , CH3CN O
Ph,P , A
PhCH3I"
PPh3
3N NaOH
A

80
With the right-hand portion now available, we turned our attention
to the synthesis of the aldehyde fragment. We decided to form the33tetrahydropyran ring via an acid-catalysed closure onto an epoxide
which after oxidation would give the required aldehyde for coupling
(Scheme 7). The epoxy-alcohol where R = CH2C02Me was synthesized
as shown in Scheme 8.
34Thus quenching of the Weiler dianion of ethylacetoacetate at
the more reactive site with 4-bromobut-l-ene gave the p-ketoester
adduct (18) in 84% yield. Sodium borohydride reduction followed by
epoxidation of the double bond gave the required epoxy-alcohol (20) as
an inseparable mixture of diastereoisomers. Acid-catalysed cyclisa- 35tion (CSA) then proceeded to afford a 1:1 mixture of tetrahydro-
pyranyl alcohols (21A) and (21B). Although these alcohols could be
separated by h.p.l.c., we actually found that the aldehydes obtained
after oxidation were easily separated by conventional flash chromato
graphy. Although this route is inefficient in that it produces a 1:1
mixture of ois- and trans-tetrahydropyrans. this was deliberate, since
the trans-compound was required for model studies on another system
[see Section 6 for the use of trans-compound].
Oxidation of the diastereoisomeric mixture of alcohols proved to
be more difficult than we had anticipated. Chromium-based (PDC ,37PCC ) oxidations were slow and low-yielding (even with the addition
of molecular sieves and with or without a buffer present). Swern 38oxidation employing trifluoroacetic anhydride and dimethyl 39asulphoxide was also unsatisfactory in terms of yield. However,
39b cthe oxalyl chloride - dimethyl sulphoxide ’ reagent was successful
and gave optimised yields of ca. 65% of the unstable aldehydes,
although it was necessary to use two equivalents of the oxidant to
achieve this. 1H and 13C N.m.r. spectroscopy enabled the

81
Scheme 7
Scheme 8
O O1. NaH
OMe 2V'Bul\3. Butenyl
bromide(18)
CCXJVIe OH
(21A) (21B)
oxalyl chloride D M SO , -60°C
. nH w H CHO
COzMe
(22B)

82
unequivocal assignment of the stereochemistry about the tetrahydrofuran
ring. The two aldehyde proton resonances for the ois- and trans
isomers (22A) and (22B), appeared at 9.58 and 9.87 p.p.m. respectively.
This is in accord with the reported data of cyclohexane derivatives
where the signal due to the axial CHO proton always appears downfield
stereoisomer the CHO is equatorial and would therefore be expected
to resonate upfield from the CHO in the trans-stereoisomer as was
indeed observed.
In the 13C n.m.r. spectrum the 6 value for a cyclohexane ring
carbon atom bearing an equatorial group is expected to be higher than
that for the corresponding compound with an axial group (Figure 8).
Figure 8
9.58 ppm
(22B)
40from the signal due to the equatorial CHO proton. In the ois-
R
13C resonance downfield when
R is equatorial
Also in -CHX groups attached to cyclohexanes the carbon resonance
shifts upfield when CHX is equatorial (Figure 9).

83
Figure 9
Application of these rules
aldehydes further corroborated
Figure 10
X = 0
resonates upfield when CHX is
equatorial.
the 13C n.m.r. data of the two
initial assignments (Figure 10).
13C n.m.r. Data (22.5 MHz)
(22A) (Spectrum 1)
6 22.50 (C-3') 25.67 {C-4')30.61 ((7-5' ) 40.99 ((7H2C02Me)51.61 (0(7H3) 74.31 ((7-21 ) 80.63 ((7-6' )171.29 ((702Me) 201.56 ((7H0)
(22B) (Spectrum 2)
5 19.51 ((7-31 ) 23.48 ((7-4* ) 30.01 ((7-5' ) 40.57 ((7ri2C02Me) 51.61 (0(7Hj) 71.38 ((7-2' ) 78.84 ((7-6' ) 171.17 ((702Me) 204.80 ((7H0)
In order to confirm unambiguously that our assignments were
correct the X-ray crystal structure determination of the p-nitrobenzoyl
derivative of the c^s-compound (23) (from our assignments) (obtained

sp e c tru m 113.22.5 MHz C n.m.r
lk™#A wiiwutaf nL 1 » « » __I__L
3LX5 OoO
_i__ I— i----> > . 1 -L
n s i*so
J__I__1 1_t 1__Lia.5 ioo
I ‘ ‘ ■__1 * ■■ A.
I S 60
j __ ..
zsa__.----L
o00

sp e c tru m 2

after sodium borohydride reduction of the corresponding alcohol) was
obtained (Figure 11). Clearly both substituents are on the same face
of the tetrahydropyran ring.
O
(23)
Initially,in order to establish the optimum conditions for the
a-sulphone anion formation, its reaction, and quenching of the result
ing adduct, we chose a few simple electrophiles as reaction partners.
Thus deprotonation of the sulphone (17) in THF with nBuLi at -78°C
gave rise to a pale yellow solution of the anion which was quenched
with TMSC1, ally1 bromide and benzaldehyde. In the last example,
the initially-formed adduct was quenched with benzoyl chloride prior to
work-up (see Experimental section). The results are shown in Table 1.
Table 1
Electrophi1e Product Yield
TMS-C1-----> TM S
83%
70%
PhCHOOCOPh
(24)
85%(diastereoisomeric
mixture)

87
Figure 11

88
The benzaldehyde adduct (24) which was present as a diastereoisomeric
mixture, was then treated with 6% sodium amalgam in THF/MeOH (3:1) at
-20°C to afford the corresponding olefin (25) as a 90:10 E:Z
mixture of double bond isomers in 67% yield (56% overall yield from
the sulphone) (Scheme 9).
Scheme 9
S 0 2Ph
Ph
OCOPh
(24)
6% Na-Hg ,
THFj MeOH
-20° C
OPh
Now familiar with the reaction conditions, we attempted to repeat
the experiment with the cfs-aldehyde (22A) as the electrophile.
Unfortunately, on a small scale (less than 1 mmol) the reaction was
not successful, resulting in substantial recovery of the starting
sulphone and total disappearance of the aldehyde. Even treatment of
the crude reaction mixture with sodium amalgam gave none of the
required olefin. Various modifications including change of solvent,
addition of HMPA , quenching with acetic anhydride etc. were equally
unsatisfactory. In view of this result we thought that perhaps the
1ithio-sulphone anion, being strongly basic (pK ca. 29) was causingCithe aldehyde to enolise and/or causing some deprotonation a to the
ester group. Obviously with benzaldehyde as the electrophile, neither29 b 41of these possibilities existed. Lythgoe et al. ’ have reported
that for aldehydes that are readily enolised the sulphone is best
deprotonated with a Grignard reagent thereby giving rise to a softer

89
anion. However, all attempts to deprotonate the sulphone with ethyl-
magnesium bromide were unsuccessful; there was no reaction with any of
the previously used electrophiles.
When the reaction of the 1ithio-sulphone anion with (22A) was
attempted on a larger scale, followed by direct treatment of the crude
product with sodium amalgam, a low yield (38%) of a 95:5 E:Z
olefinic mixture was obtained (26) (Scheme 10).
Scheme 10
SO zPh
( 1 7 )
1. nBuLi, -78°C2 . ( 22A)
3. C6H5C0C14. Na-Hg, -20°C
THF-MeOH
It has been mentioned above that these tetrahydropyranyl aldehydes were
found to be unstable; indeed Ireland and coworkers have recently
reported some closely related aldehydes to be prone to hydration and
decomposition. In addition, purification of these compounds to
analytical standards proved to be difficult, even by flash chromato
graphy of samples immediately prior to analysis. Eventually distil
lation followed by immediate combustion analysis was successful. These
findings may partly indicate why the coupling reaction was unsuccessful
on a small scale, resulting only in recovery of starting material.
Presumably, the aldehyde absorbed moisture during handling which simply
quenched the sulphone anion. This cannot however be the only explana
tion because as will be seen later, other anions, notably Wittig and
Grignard reagents can be coupled in much higher yield to the aldehydes
(22A) and (22B).

90
Although the Julia aldehyde-sulphone coupling gave high trans-
stereoselectivity, the yield was not satisfactory and so we decided
to turn our attention to Wittig methodology.
Treatment of the aldehyde (22A) with propylidenetriphenyl-
phosphorane at -78°C (generated by treatment of propyltriphenyl-
phosphonium iodide with nBuLi at 0°C) followed by rapid warming to
room temperature, gave as expected only the Z-olefin (27) in 53% yield
(Scheme 11).
Scheme 11
There has been much study on the mechanism and stereoselectivity of
the Wittig reaction. It is known that under totally "salt-free"
conditions saturated aliphatic non-stabi1ized triphenylphosphonium
ylids react with aliphatic aldehydes in nonpolar solvents to afford44the Z-olefin almost exclusively. In nonpolar solvents, the
stereochemistry of the olefin product is dependent on the nature of
the inorganic salt present. It has been postulated that in the
presence of lithium salts, the initial evythvo 'betaine' adduct is
partially converted to the thermodynamically more stable thveo
'betaine' via reversion to the starting aldehyde and ylid, resulting
in an increase in the proportion of the E -isomer in the olefin45adduct. Schlosser and coworkers developed a highly stereo
selective trans-olefin synthesis whereby the primary Wittig inter
mediate, with predominantly the erythro configuration is a-metallated
by addition of a second equivalent of an alkyl or aryl lithium

91
reagent and the resulting intermediate “betaine ylid" is reprotonated
to generate the threo Wittig intermediate which then gives the E-
olefin. However, this was obviously not compatible with the presence
of the ester group in aldehydes (22A) and (22B).
In an attempt to overcome this limitation, the Wittig intermediate
was treated with potassium hydride (inverse-addition) at -78°C and
allowed to warm to -30°C, but no equilibration occurred and the yield
of the Z-olefin was low. A similar result has been obtained by
Anderson and Henrick, who isolated some p-ketoester condensation product46indicating partial removal of the proton a to the ester group.
Schlosser was the first to discover that alcohols can promote
equilibration of the Wittig intermediate and hence increase the propor-45ation of E -isomer in the product. More recently Anderson and
Henrick have reported that addition of non-hindered alcohols such as
methanol and ethanol can increase the proportion of S'-isomer quite 46substantially.
Thus treatment of propyltriphenylphosphonium iodide with nBuLi in
THF at -78°C followed by addition of aldehyde (22A) and warming to
-40°C presumably leads initially to the erythro intermediate. Excess
dry methanol was then added and the solution stirred for oa. 4 h at this
temperature prior to quenching and work-up. This procedure gave a
22:78 Z ; E mixture of the olefinic adduct [(27) and (28)] in 53%
yield (Scheme 12).

92
Scheme 12
1. nBuLi, -78°C
2 . ( 22A)C3H7PPh3 r -----------
3. MeOH (xs), -40°C
4 h
4. H20
Although this ratio was not as high as we would require, we were hopeful
that the higher degree of substitution present in the naturally
occurring system would increase it to an acceptable level.
In order to assess the effect of the ester functionality on these
coupling reactions, we decided to replace this group for one which
would not possess any acidic protons (29).
(27) and (28)
(29) P = protecting group
Thus protection of the aldehyde (22B) as the dimethyl acetal
47(30B) followed by lithium aluminium hydride reduction of the ester
gave (31B) in high yield {trccns-series used in an attempt to establish
satisfactory reaction conditions).

93
Unexpectedly, attempted deprotection at this stage proved to be
difficult. Silica gel doped with various acids of increasing strength
in THF either caused no deprotection or, in the case of the stronger
acids, resulted in total decomposition of the compound. Trifluoro-49acetic acid in chloroform was also unsuccessful. Interestingly,
10% aqueous oxalic acid on silica gel in dichloromethane under the48reported conditions gave only the dimeric product (32).
48
Increasing the dilution resulted in no deprotection or formation of
dimer. With the alcohol protected as either the TBDMS (33) or
TBDPS (34) ether, successful deprotection of the acetal group still
proved to be an elusive process.
One interesting result however, was the successful (93%) deprotec
tion of the dimethyl acetal (30B) with two equivalents of boron
tribromide in dichloromethane at -78°C (Scheme 13).
Scheme 13
BBr3/CH2Cl2y
-78°Cr « -CO,Me
fr'CHO
(22 b)

94
However, when we attempted to repeat this reaction on (33) and (34),
deprotection of the alcohol occurred.
Having become rather pessimistic about the prospects of finding suitable
conditions to deprotect this remarkably stubborn acetal, we chose 1,3-
dithiolane as an alternative protecting group for the aldehyde because
it may be removed under a variety of neutral conditions. Thus treatment
of the aldehydes (22A) and (22B) with 1,2-ethanedithiol and boron tri- 50fluoride gave the dithiolanes (35A) and (35B), which after lithium
aluminium hydride reduction and protection of the resulting alcohols
(36A) and (36B), gave compounds (37A) and (37B) (Scheme 14).
Scheme 14
(33) (34)
trans -series
(3 6 A)(37A)

95
Again however, we had considerable difficulty in the deprotection
of these compounds (37A) and (37B). Thallium trifluoroacetate under51buffered conditions was slow and low-yielding. Indeed in order to
force the reaction to completion it was necessary to use large excesses
of the thallium reagent which caused problems in the work-up for large-
scale reactions. The only successful conditions that were found
involved the use of methyl iodide in wet acetonitrile with excess sodium52carbonate as a buffer. These conditions gave the aldehydes (38A) and
(38B) in a maximum yield of 61%, which although not particularly
satisfactory did provide us with sufficient quantities of the aldehydes
required for the coupling reaction.
Our attempts to synthesize model aldehydes with a longer side-chain on
the tetrahydropyran ring were thwarted by the low-yielding deprotection
of compounds (42A) and (43A) (Scheme 15) under a variety of different
conditions.

96
Scheme 15
CH
Nss
(43A)
Treatment of the lithio-anion of sulphone (17) as before with (38A)
gave, after quenching with benzoyl chloride, a diastereoisomeric mixture
of benzoyloxy-sulphones (44A) and a small amount of the more polar
hydroxy-sulphone (45A). Reduction of the benzoyloxy-sulphones (44A)
with 6% sodium amalgam at -20°C gave a separable mixture of the olefins
(46A) in an oa. 85:15 tvans\ois ratio as the major products, along with
variable amounts of vinyl sulphone (47A) (Scheme 16). This minor
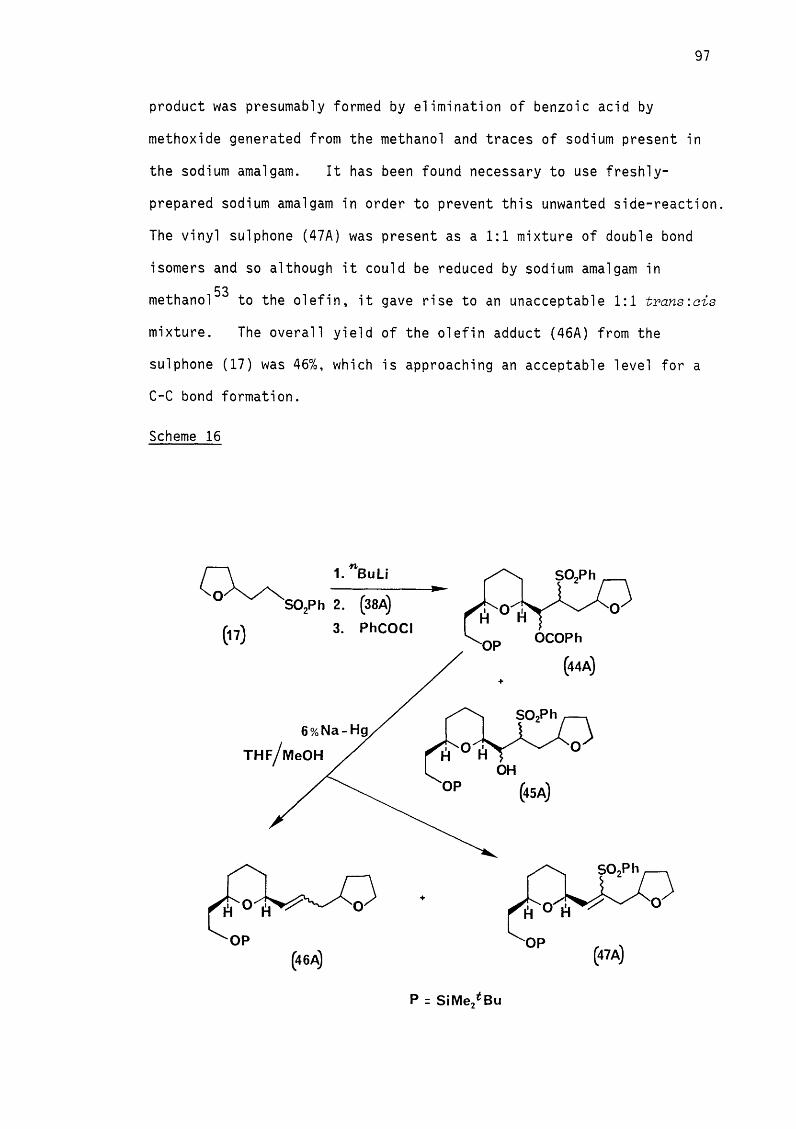
97
product was presumably formed by elimination of benzoic acid by
methoxide generated from the methanol and traces of sodium present in
the sodium amalgam. It has been found necessary to use freshly-
prepared sodium amalgam in order to prevent this unwanted side-reaction.
The vinyl sulphone (47A) was present as a 1:1 mixture of double bond
isomers and so although it could be reduced by sodium amalgam in 53methanol to the olefin, it gave rise to an unacceptable 1:1 trans:ois
mixture. The overall yield of the olefin adduct (46A) from the
sulphone (17) was 46%, which is approaching an acceptable level for a
C-C bond formation.
Scheme 16
P = SiM e2*Bu

98
Since we were also interested in comparing the sulphone and Wittig
methodology, we attempted the Schlosser-Wittig modification to the
reaction between propylidenetriphenylphosphorane and aldehyde (38B),
which was now viable in the absence of the ester group. Thus, after
addition of the aldehyde to the orange-red solution of the phosphorane
at -78°C, a further equivalent of nBuLi was added, followed by
potassium tert-butoxide (1.5 eq) and tevt-butanol (1.5 eq). After
stirring at -40°C for 1 h the mixture was warmed to 25°C. However,
equilibration of the Wittig intermediate was much less effective than
under the modified Wittig conditions used previously and we were only
able to achieve a 40:60 cis\tvans ratio of double bond isomers (48B).
1. nBuLi, -78°CC3H7PPh3 I ---------------
2. (38B)
3. nBuLi
4. K0tBu, tBuOH
Thus it seemed that for our system, addition of a non-hindered
alcohol in the Wittig reaction was definitely the method of choice.
The sulphone methodology was however more acceptable in terms of the
ratio of double bond isomers obtained.
Having now established a route to introduce trorcs-stereochemistry
about the double bond, we were interested in modifying our adducts, so
that they would resemble the natural ionophore M139603 more closely.
Therefore deprotection of pure tvans (46A) with 40% aqueous HF 54in acetonitrile (5:95) gave the corresponding alcohol (49A) in 98%
yield. Swern oxidation to the aldehyde (50A) followed by homologation

99
with the stabilized phosphorane carbethoxymethylidenetriphenyl-
phosphorane gave the separable a,$-unsaturated esters (51AT) and (51AC)
in 89 and 4% yields respectively (Scheme 17).
Scheme 17
The trans-aldehyde (38B) was subjected to the same sequence of
reactions as (38A) for the purposes of providing greater quantities of
material. Most of the reactions were therefore tested on the trans
series first to find the optimum conditions, thereby preventing wastage
of the required ois-series.

100
Scheme 18
X (T V
O7)
1. *BuLi , -78°C
SOjPh 2. (38B)
3. PhCOCI
4. 6%Na-Hg
T H F /M e O H -20° C
several steps
It was found that the mixture of C-10 double oond isomers was more
easily separated at the final step in this Scheme; the a,3~unsaturated
esters (51BT) and (52) were separable by flash chromatography (N.B.
only the £ 0 2 double bond was produced in the phosphorane reaction)
(Scheme 18).
The reader will recall that in the 'real' system M139603, the C-ll
double bond is one position closer to the tetrahydropyran ring than in
our model compounds (51AT) and (51BT) (Figure 12).
Figure 12
M 139603

101
We thought that it would be interesting to see whether the double bond
in our model compounds could be transposed. Thus addition of the
ester (51BT) to LDA (1 eq) and HMPA (1 eq) in THF at -78°C
followed by quenching with glacial acetic acid gave the required decon-
jugated product (53) as an inseparable Z:E (1:2) mixture of
double bond isomers (Scheme 19).
Scheme 19
It is interesting to note that although the anion formed in this reac
tion prior to quenching could undergo ring opening via £-oxyelimination,
total tvccns-stereochemistry about the tetrahydropyran ring was retained;
if ring opening and closure were occurring we would have expected to
have isolated some of the more thermodynamically stable diequatorial
product (Scheme 20).

102
Scheme 20
55Kende and Toder have systematically examined the stereochemical out
come of the deconjugative discharge of the lithium dienolates of a
series of unsaturated esters. Their results indicate that the
electrophilic discharge of the Li dienolate from the ester of an
(£’)-2-alkenoate (disubstituted double bond) leads stereospecifically to
the (Z)-3-alkenoate ester unless the 0 4 carbon bears a substituent
larger than methyl, beyond which the reaction becomes increasingly
stereorandom. In our system the 0 4 carbon substituent is considerably
larger than a methyl and so we would not have expected to be able to
predict the double bond isomer ratio. Indeed there are many examples
in the literature which show little stereospecificity when the 0 450substituent is large.

103
Recalling the characteristic features of the polyether ionophores
(Section 1), we considered that the carboxylic acids derived from our
a,p-unsaturated esters might possess biological activity. Thus mild
basic hydrolysis of the ester (51AT) (using 1M LiOH) gave the corres
ponding acid (54) (see Appendix for 250 MHz n.m.r. spectrum) which has
been sent to ICI for biological evaluation.
At this point, we felt that enough time had been spent on the model
work and were eager to embark upon synthetic studies towards the ’real'
system.
We had established a coupling strategy which was satisfactory for
introducing the trans-double bond between the tetrahydropyran and
-furan rings and in addition, by varying the side-chain on the tetra
hydropyran ring had synthesized some interesting model compounds.

104
SECTION 4 : DEGRADATION STUDIES OF M1396Q3
Having established in our model studies (Section 3) that the trans-
disubstituted double bond could be introduced via the coupling of a
suitably substituted tetrahydrofuran with a tetrahydropyranyl aldehyde,
we wanted to apply this methodology to the 'real' system M139603.
It was our hope that degradation studies would provide useful
information for an eventual total synthesis. Indeed slow ozonolysis
of the sodium salt of M139603 (la) at -78°C followed by dimethyl sulphide
decomposition of the ozonide gave the aldehyde (7) (Scheme 21).
Scheme 21
0
However no other aldehydes could be isolated from this reaction.
Initially it was thought that perhaps the C-ll - C-12 trisubstituted
double bond was being attacked, but that the allylic alcohol was inter
fering in some way with the intermediate ozonide. We therefore hoped
that protection of the free alcohol might enable us to isolate fragments
from the left-hand side of the molecule.

105
Thus treatment of M139603 Na+ salt (la) with acetic anhydride and
pyridine^ gave the mono-O-acetyl derivative (55) which was found to be
slightly more polar, perhaps as a result of conformational effects.
Treatment of (55) with ozone at -78°C followed by dimethyl sulphide as
before, gave the right-hand side aldehyde (7) and a very polar left-hand
side aldehyde (56) (from this point on, 'right-hand' and 'left-hand'
refer to these portions of M139603) which from n.m.r. still possessed
the trisubstituted double bond. Thus it seems that ozone is not
attacking this double bond at -78°C (Scheme 22).
Scheme 22
In fact, it was later found that the trisubstituted double bond is
only cleaved at oa. -20°C. We believe that this stability to ozone is
due to its hindered nature. Indeed later work provided further evidence42for this explanation. The polar, unstable aldehyde (56) decomposed

on attempted flash chromatography using silica gel, alumina or Florisil
and was observed to decompose in solution, making recrystallization very
difficult. In an attempt to decrease the polarity of the left-hand
side fragment and make the compound easier to handle, we decided to
protect the tetronic acid moiety of M139603 as its methyl ether. Thus
treatment of (55) with 20% aqueous phosphoric acid in ethanol/acetone
for 48 h gave the free acid derivative (57). The reported conditions+ 16for the conversion of the tetronomycin Na salt to its free acid
indicate rapid reaction with 10% phosphoric acid (10 min). However,
we found that these conditions were not satisfactory for M139603.
Even after 24 h (extraction of an aliquot from the reaction followed by
work-up), the infra-red spectrum still showed the presence of some
sodium salt (la) (v (CHC13) 3300, 1725, and 1645 cm"1). 20%max.Phosphoric acid over a 48 h period was therefore generally used to force
the reaction to completion. Treatment of (57) with diazomethane then
gave the methyl tetronic acid derivative (58) (Scheme 23), which is
itself more polar than (1), (55) or (57)!
Scheme 23
106

107
Although it is possible to change the sequence of steps in protect
ing M139603, it was found that such modified routes were less clean and
lower-yielding.
i . e .
Na+ salt (la) — s- free acid
or Na+ salt (la) — ► free acid
(lb) — > (57) — > (58)
(lb) — ► methyl ether — ► (58)
Because of the tautomerism which exists between the various isomers
of the acyltetronic acid moiety, we might have expected isomeric
alkylated products to be formed in the diazomethane methylation 57(Scheme 24). However, the 250 MHz n.m.r. spectrum indicated the
presence of only one isomer (58) (see Appendix).
Scheme 24
R = Alkyl

108
Ozonolysis of derivative (58) followed by dimethyl sulphide or
triphenylphosphine work-up gave the two aldehydes (7) and (59).
This left-hand side aldehyde (59) was clearly unstable but seemed
to be easier to handle than the sodium salt derivative (56). However
it was still not stable to chromatography and needed to be stored at
low temperature in the solid state. The separation of (59) and (7)
was therefore usually performed by repeated trituration of the crude
product from the ozonolysis reaction with petrol, since (59) is
insoluble in this solvent while (7) is readily soluble, being much less
polar. Use of dimethyl sulphide in the work-up generates dimethyl
sulphoxide which is generally removed by washing with water. However,42Ireland and Norbeck have recently reported several aldehydes that
were inductively destabilized towards hydration and decomposition by
strongly electronegative substituents. Ln view of this recent report
we now prefer not to use the dimethyl sulphide work-up. Moreover, use
of oa. 0.9 eq of triphenylphosphine causes very clean and rapid decompo
sition of the ozonide and we believe that after separation of the two
aldehydes by trituration with petrol as described, the left-hand side
aldehyde should be used crude (contaminated with triphenylphosphine
oxide) in any future coupling work.

109
The unusual order of polarity among the M139603 derivatives is
worth mentioning (Scheme 25). Indeed the order is opposite to that
which we might have predicted and did cause some confusion at the outset!
We believe that these results are due to conformational changes. For
example, presumably the aldehydes (55) and (59) which were found be very
much more polar than any of the other derivatives, can no longer adopt
a conformation in which most of the nonpolar groups are orientated to
the exterior.
Scheme 25
Polarity
increasing
ltfF)
Aldehydes (56) and (59)
Mono-O-acetyl M139603 methyl ether (58)
Mono-O-acetyl M139603 Na+ salt (55) and free acid (57)
M139603 Na+ salt (la) and free acid (lb)
In any synthesis of M139603, it will obviously be necessary to prove
that both the allylic acetate and methyl ether protecting groups may be
removed easily and without any loss of optical activity.
Removal of the methyl ether was readily accomplished by treatment
with 3M aqueous HC1 in THF. As expected, no cleavage of the
terminal methoxyl occurred under these conditions. The allylic acetate
proved to be somewhat more stable than was initially anticipated.
Indeed, 3M sodium hydroxide in THF/water was required for its removal
and even so the reaction was slow, taking over a week to reach
completion. Allylic acetates are usually easily cleaved with potassium58carbonate in aqueous methanol but these conditions were ineffective
for our system. Presumably the hydroxide nucleophile has to approach
a sterically hindered part of the molecule and this is in keeping with

110
the remarkable resistance of the trisubstituted double bond to undergo
ozonolytic cleavage (see above).
However, both these deprotection steps were clean and high-yielding
and the optical activity of the re-isolated M139603 Na+ salt (la) was
identical to that reported in the literature. Moreover, the treatment
with 3M sodium hydroxide not only removes the acetate but also
converts the free acid (lb) to the crystalline sodium salt (la)
(Scheme 26).
Scheme 26
3M HC1 3M NaOH(58) ------- ► (57) -------► M139603 Na salt
THF-H20 THF-H20(la)
As was mentioned earlier, it is also possible to cleave the trisub-
stituted double bond by treatment with ozone at ca. -20°C to afford (60)
presumably via (61). However, we were not able to isolate any of the
tetronic acid portion from the reaction and it is thought that under
these vigorous conditions it too may be undergoing some ozonolytic
cleavage (Scheme 27).
Scheme 27
M (61)

Ill
Although from our model studies we had shown that an ester
functionality would be compatible with some of the coupling conditions,
we also realised that it might be necessary to replace the acetate
with some other suitable protecting group. It was possible to protect
the allylic alcohol as the TBDMS ether (62) using the imidazole/DMF 59route. Surprisingly however, no reaction was observed under the
fintriethylamine/DMAP/CH2Cl2 conditions. Perhaps DMF was required
as the solvent in order to coordinate to the cation and produce a more
reactive anion. The allylic alcohol could also be protected as the
3-(trimethylsilyl)ethoxymethyl ether (SEM ether) (63) using diisopropyl-61amine (4 eq) and SEM-C1 (ea. 3 eq) in dichioromethane (followed by
protection of the tetronic acid as before). However, this compound was
very unstable and difficult to handle, undergoing decomposition on
attempted chromatography.
6 (250 MHz) 0.04 (9H, s, SiMe3), 0.71 (3H, d, J 6 Hz, Me), 0.91 (3H, d,
J 6 Hz, Me), 0.96 - 2.02 (29H, m), 2.24 (3H, m, 10-H, 20-H, and 22-H),
3.17 (1H, d, J 9.5 Hz, 13-H), 3.32 (1H, m, CffOMe), 3.35 (3H, s, OMe),
3.43 - 3.70 (5H, m, 4-H, 17-H, 21-H, CH20C772CH2SiMe3), 3.94 (3H, s,
=C~0Me), 3.99 (2H, s, =CCff20SEM), 3.95 - 4.03 (1H, m, 24-H), 4.60 (1H,
d, J 9.5 Hz, Cfl20C=0), 4.64 (1H, d, J 9.5 Hz, Cff20C=0), 4.68 (2H, s,
=CCH20Cff20), 5.46 (1H, d, J 10 Hz, Cff=CCH20SEM), 5.48 (1H, dd, J 16 and
6 Hz, 0CHC&CH), and 5.67 (1H, dd, J 16 and 7 Hz, 0CHCH=Cff).

112
We had established a suitable protection-deprotection strategy for
M139603 and shown that it was possible to selectively degrade the
ionophore to smaller fragments. The next section is concerned with
synthetic efforts towards the tetrahydrofuranyl fragment obtained from
degradation.

113
SECTION 5 : SYNTHETIC STUDIES TOWARDS THE TETRAHYDRQFURANYL PORTIONOF M139603
5.1 Enantioselective construction of the trares-tetrahydrofuran system
OHC
OMe
( 6 4 )
Having obtained the right-hand aldehyde (7) from the degradation
of M139603, we sought to synthesize the corresponding alcohol (64).
which we hoped to convert to a variety of coupling fragments, and to
compare it directly with the naturally derived material obtained from
reduction of (7).
The alcohol (64) is a trans-tetrahydrofuranyl system possessing
five chiral centres. One of our major considerations at the outset
was the problem of introducing the trans-stereochemistry across the
ring, between C-21 and C-51.
Prior to the recent biosynthetic studies of M139603 we anticipated
introducing the chiral centres at C-5' and C-l" via an acid-catalyzed
closure of an alcohol on to a chiral epoxide (Scheme 28) (P is some
suitable protecting group).
Scheme 28

114
Gratifyingly, this is the route that labelling studies have indi
cated probably occurs during the biosynthesis of this right-hand 22fragment (Section 2). We hoped to be able to prepare the correct
62chiral epoxide via the Sharpless asymmetric epoxidation of an allylic
alcohol (65), which led us to consider the route shown in Scheme 29.
Scheme 29
The frans-tetrahydrofuran stereochemistry would be ensured by the
correct choice of tartrate-ester used in the Sharpless epoxidation.
The (2?)- double bond in the allylic alcohol is required to ensure the
correct stereochemical relationship between C-51 and the side-chain
diol unit formed in (66).
5.2 The Methoxy Side-chain
Our next consideration was the problem of converting the diol unit
in (66) to the side-chain present in the natural compound. The primary
alcohol was obviously no longer needed, having been introduced solely
for the purpose of controlling the epoxidation of the double bond. We
therefore required a synthetic method to convert the CH20H to a methyl
group. Some of our model studies had revealed that it was possible to
convert an epoxy-alcohol to an epoxy-selenide via two routes, (A) and
(B) (Scheme 30).

115
Scheme 30(A)
The direct route (A) requires two equivalents of tri-n-butyl-n 63phosphine ( Bu3P) and N-phenylselenophthal imide (N-PSP), indeed use
of only one equivalent of each reagent resulted in no reaction at all.
We believe that the initial complex formed between N-PSP and nBu3P
is coordinating in some way to the epoxide oxygen, preventing conver
sion of the primary alcohol to the selenide. The two-step route 64(B), although longer, is high-yielding and extremely easy to perform.
Simple epoxy-selenides (R = alkyl), were found to be unstable to
chromatography on silica gel, and generally Florisil was used for their
purification.
65Attempts to reduce the selenide function in some simple model
epoxy-selenides were unsuccessful, resulting in simultaneous ring
opening (with both Raney nickel in ether and tri-n-butyltin hydride
methods).
Combination of this idea with our proposed strategy for the tetra-
hydrofuran ring formation, led us to conclude that we would need to
reduce the selenide group after the acid-catalyzed ring closure
{i.e. in the absence of the epoxide) (Scheme 31). It might also be2necessary to protect the secondary alcohol in this strategy (P ).

116
Scheme 31
Conversion to (64) would be effected by methylation of the secondary
alcohol and deprotection of the primary alcohol.
5.3.1 The Carbon Framework
Having planned the construction of the tetrahydrofuran ring and
the methoxy side-chain, our new target was a compound of type (67).
OHP1,P 2 = protecting groups

117
We anticipated introducing the trans double bond via the addition of a
stabilized phosphorane to an aldehyde and therefore proposed the route
shown in Scheme 32.
Scheme 32
The lactol (68) could easily be obtained from the corresponding
lactone (69) and this molecule became the new target.
Initially we had hoped to be able to find conditions to introduce
the correct chirality in the side-chain of this lactone via a conjugate
addition into an a,p-unsaturated sulphone of type (70) or (71).

118
This would then have led eventually to the sulphone coupling partner
provided our proposed strategy was successful. Recently however66Isobe and co-workers have reported the diastereoselective introduction
of a methyl group into the vicinity of a secondary alcohol completely
in the syn-orientation (Scheme 33).
Scheme 33
OH SOzPh
1. MeLi
2. KF
Me
v SOH S 0 2Ph
They have extended this methodology to the addition of nucleophiles to
the pyranosyl heteroolefin (72) (Scheme 34).
Scheme 34
They explain this remarkable stereoselectivity as being the result
of lithium coordination between the two oxygen atoms present, thus
directing the nucleophile in the syn manner. If these results can be
applied to our system (70) it can be seen that the methyl would be
introduced with the wrong stereochemistry. We thought however that it
might be possible to reverse the stereoselectivity by addition of some
suitable coordinating species prior to the addition of the methyl cuprate
reagent (methyl-lithium could not be used in the presence of the lactone
functionality). Failing this, we hoped to use the results of Isobe 66and co-workers to introduce a hydride reagent stereoselectively to

119
(71), which if successful would give us the methyl in the correct
orientation.
Our proposed route to (70) is shown in Scheme 35 (analogous route
for (71)).
Scheme 35
Thus the cis-stereochemistry of the lactone would be ensured by
the stereospecific [2 + 2] cycloaddition between dichloroketene and
olefin (73). An obvious drawback with this route is its racemic
nature, however at the outset we were interested in rapid access to
the sulphone (70) so that we could examine the conjugate addition
methodology.

120
5.3.2 Synthetic efforts towards the sulphone (70)
Hydrogenation of alkynes is the standard method for the prepara
tion of cis-olefins and Lindlar's palladium catalyst is widely used for
. 67this purpose.
Thus protection of 2-butyn-l-ol as the tert-butyldiphenyl silyl
60ether (74) followed by hydrogenation with Lindlar's catalyst gave the
c-^s-olefin (75) in 98% overall yield (Scheme 36).
Scheme 36
Me— =DSi^BuPh,
Me— = H,
(74)Lindlar’scatalyst
OSi*BuPh 2
It was found necessary to stop the hydrogenation as soon as all of the
alkyne had been consumed to prevent overreduction to the alkane. In
addition we generally used 10% or less of the Lindlar's reagent per
weight of the alkyne. Larger quantities of catalyst also caused
problems of overreduction.
There are two common methods used for the preparation of ketenes
69(a) the triethylamine dehydrohalogenation of an acid halide and
(b) the dehalogenation of an a-haloacid with activated zinc.^ Attempts
to use method (a) to generate dichloroketene in situ and its reaction
with olefin (75) were unsuccessful, resulting in substantial recovery
of starting material and polymerization of the ketene. Method (b)
however was successful. Thus slow addition of a solution of distilled
trichloroacetyl chloride in diethyl ether to freshly prepared zinc-
copper couple71 and the olefin (75) followed by refluxing for 2 h
gave the unstable dichloroadduct as a mixture of regioisomers (76A) and

121
and (76B). The product was found to be unstable both to chromato
graphy and distillation and was therefore reduced directly with zinc
72and glacial acetic acid at 40°C for 3 h to afford the crystalline
cyclobutanones (77A) and (77B) in ca. 3:1 ratio (71% overall yield
from (75)) (Scheme 37).
Scheme 37
c l c o c i ; z
OP
ci L sci-Cl \ — Z c
S Zn-Cu , Et20 A r ^ °
*
OSi*BuPh2 OP
(75) (76A) (76B)
P = Si*BuPh2
AcOH ,Zn , 40°C
OP
r ^ °OP
(77A) (77B)
The cycloaddition of ketenes with olefins is usually considered
to be a concerted ^2$ + ^2a process possessing some degree of charge
73build-up in the transition state. By consideration of the substi
tuents best able to stabilize the developing charge, it is often
possible to predict the orientation of the addition. Thus C2 of the
ketene becomes bonded to the carbon atom of the olefin that is best
able to stabilize positive charge (Scheme 38).

122
Scheme 38
C l - ^
Cl
nucleophilic e ectrophilic
R----- CH- —CH;
f
5+:CH,
c r
R
t\ ____ ^ 0
R = alkyl , a ry l, etc.
With our disubstituted olefin (75), there is little difference
between the ability of the double bond carbon atoms to stabilize
positive charge. We were hoping however that the bulky nature of the
-CH20Si^BuPh2 substituent would control the regiochemistry of the
cycloaddition to produce mainly the adduct (76A). As can be seen
from our product ratio, this prediction turned out to be correct. Thus
in the transition state leading to the major product (76A), the two
large chlorine atoms are positioned remote from the bulky -CH20SitBuPh2
substituent (Scheme 39).
Scheme 39
MeC H 2OSi*BuPh2
(76A)

123
Since zinc salts are produced in the reaction of trichioroacetyl
chloride and zinc-copper couple, we also thought that the cycloaddition
might be partially controlled by coordination of the metal between the
two oxygen atoms (Scheme 40).
Scheme 40
Cl
The major cyclobutanone (77A) was then oxidised with 30% aqueous72hydrogen peroxide in glacial acetic acid at aa. 5°C to afford the
lactone (78) in 86% yield. Deprotection with tetra-n-butylammonium
fluoride gave the alcohol (79).
It is now easy to understand why this silicon protecting group was
chosen for the alcohol. Acid stability was necessarily required for
the reduction of the dichiorocyclobutanones and for the Baeyer-Vi11iger
oxidation. In addition, its bulky nature was required to control the
regiochemistry in the cycloaddition step. Lastly, we needed to be
able to remove it easily in the presence of the lactone functionality.

124
Having obtained the alcohol (79) we now wanted to oxidise it to
the aldehyde. We had considerable difficulty with this oxidation.36PDC (with or without crushed molecular sieves), oxalyl chloride-39b c 75DMSO ’ modified Collins procedure and sulphur trioxide-pyridine
were all unsuccessful, affording either none of the aldehyde or very76low yields. The Pfitzner-Moffatt oxidation has been used on systems
similar to (79);^ indeed treatment of the alcohol (79) with DCC and
pyridinium trifluoroacetate in benzene and DMSO gave the unstable
aldehyde (80) in a poor 24% yield. Attempts to improve this yield
were unsuccessful. It was thought that the instability of this 42aldehyde was the reason for the low yield. Although this step was
obviously not satisfactory, it did enable access to a small quantity
of the required aldehyde (80) and we therefore decided to press on in
order to try some of the conjugate addition reactions discussed
earlier.
It was hoped to obtain the required vinylic sulphone using78methodology already developed in our laboratories. Thus addition
of the aldehyde (80) to the anion generated from treatment of phenyl-79 nsulphonyltrimethylsilylmethane with BuLi (1.1 eq) in 1,2-dimethoxy-
ethane at -78°C, gave after work-up, a low yield of a separable oa. 4:1
mixture of cis- and trans-\jinylic sulphones, (81A) and (81B)
respectively (Scheme 41).

125
Scheme 41
Disappointingly, all attempts to react a methyl cuprate reagent
with either (81A) or (81B) were unsuccessful. Initially the cuprate
was generated from methyl-lithium and copper bromide-dimethyl sulphide 80complex but after addition of the vinyl sulphone it was not possible
to isolate any product or recovered starting material. Our first
thought was that perhaps this was not the best type of methyl cuprate81reagent to use and consequently the reaction with the Lipshutz mixed
cyanocuprate reagent Me2Cu(CN)Li2 was attempted. However, the
same disappointing result was obtained.
Not wanting to abandon this route just yet, it was decided to
protect the lactone as the 0-methyl-lactol hoping that the yield of the
oxidation step would also improve. Thus treatment of the lactone (78)
with DIBAL in toluene at -78°C gave the lactol (82) in high yield. It
is important in the work-up of this reaction to use the correct pro
portions of glacial acetic acid, water and solid sodium bicarbonate

126
(see Experimental section) to prevent formation of the 0-acetyl-lactol
(83) (although (83) may be easily converted to (82) by treatment with58potassium carbonate in aqueous methanol at 40°C) (Scheme 42).
Scheme ^2
K 2 C O 3 1
MeOH / H20 40° C
< ^ c t " oac
OSi*BuPh2
(83)
Protection of the lactol (82) using methanol and CSA gave the
tetrahydrofuranyl ether (84) in 75% yield. It is important to
chromatograph compound (84) prior to its deprotection to (85) with
tetra-n-butylammoniurn fluoride; failure to do so results in a substan
tial yield of the tetrahydropyranyl ether (86), presumably formed by
the mechanism shown in Scheme 43.

127
Scheme 43
6 (250 MHz) 0.96 (0.3H, d, J 6.5 Hz, Me, minor anomer), 1.02 (2.7H, d,
J 6.5 Hz, major anomer), 1.40 (1H, m, 3-H), 1.73 - 1.94 (3H, m, 3-H,
4-H, and OH), 3.30 (1H, m, 5-H), 3.35 (0.3H, s, OMe, minor anomer),
3.36 (2.7H, s, OMe, major anomer), 3.40 (1H, dd, J 19.5 and 10.5 Hz,
6-H), 3.63 (1H, dd, J 10.5 and 4.5 Hz, 6-H), 4.62 (0.9H, dd, J 4 and
1 Hz, 2-H, major anomer), and 4.82 (0.1H, dd, J 12.5 and 3.5 Hz, minor
anomer).
The ether (86) existed as a mixture of anomers in an oa. 90:10
ratio. The major anomer was assumed to possess the methoxy group at820 2 in the axial orientation as a result of the anomeric effect.
This assumption is in keeping with the observed spectral data (Figs. 13
and 14). Thus in the major anomer the coupling constants J^g and
, which are between protons with a di-equatorial and equatorial-
axial relationship respectively,were expected to be relatively small
(J 4 and 1 Hz observed). In the minor anomer J which is a coupling

128
between di-axial protons was expected to be large as observed (J^
12.5 Hz).
Figure 13
Figure 14
JQF 19.5 Hz
JQE 4.5 Hz
JAB’ °AC 4 and 1 HZ
Minor
Oxidation of (86) to (87) under the Pfitzner-Moffatt conditions further
corroborated our assignments.
O ^ T O M e
(87)
vmax (film) 2935’ 1725’ 1178’ 1053’ and 956 cm’1’*6 (90 MHz) 1.08 (2.70H, d, J 6.5 Hz, Me, major
anomer), 1.10 (0.30H, d, J 6.5 Hz, minor anomer),
1.45-3.00 (3H, m, 3-H2 and 4-H), 3.40 (2.70H, s,
OMe, major anomer), 3.48 (0.30H, s, OMe, minor
anomer), 3.48 (0.30H, s, OMe, minor anomer), 3.86
(0.10H, d, J 14 Hz, 6-H, minor anomer), 3.88 (0.90H
d, J 17 Hz, 6-H, major anomer), 4.18 (0.10H, d
J 14 Hz, 6-H, minor anomer), 4.20 (0.90H, d, J
17 Hz, 6-H, major anomer), and 4.95 (1H, m, 2-H);
m/z 143 (M+-H), 111 (M+-Me0H-H),
83 (CH2=CH-CH(Me)C0+), and 57 (C3H50+).

Pfitzner-Moffatt oxidation of (85) as before gave a more satisfac
tory yield (49%) of the reasonably stable aldehyde (88). Although
this yield is still not as high as we would like, since we were not
sure whether our conjugate addition route would work, it was decided
not to spend any time optimising the conditions at this point.
Reaction of the aldehyde (88) as before with the lithio-anion of
phenylsulphonyltrimethylsilylmethane gave a 62% yield of the vinyl
sulphones (89A) and (89B) as an inseparable 1:1 mixture (Scheme 44).
Scheme 44
(89A) and (89b) 1 1
62%
In all of these tetrahydrofuranyl compounds, there was one major
anomer present which we assumed for steric reasons to be the compound
with the methoxy on the opposite side of the ring to the two other
substi tuents.

130
major anomer
The unusually high differences between the 6-values of the proton
at C-4 (adjacent to the double bond) of the ois- and trans-vinyl sul-
phones is worth noting (this effect also exists in the p-protons of
the vinyl sulphone side-chain). The difference is present between
compounds (79A) and (79B) and also (89A) and (89B) (Scheme 45).
Scheme 45 (See Appendix for full (250 MHz) spectral data)
6-24 6.29
(89A)(81A)
6-926-90
(89B)(81B)

131
Molecular models indicate that the ois-\jinyl sulphones probably
take up a conformation where the sulphone oxygen atoms are much closer
to the proton at C-4 than in the trans-\jinyl sulphones (Scheme 46).
Scheme 46
9 ok
Thus the unusually high 6-value for the C-4 proton in the ois
compounds may be a result of the deshielding effect of the sulphur-
oxygen bonds.

132
Having now prepared compounds (89A) and (89B) we were ready to
try the cuprate addition. Again however, no success was realised
with either of the methyl cuprate reagents tried previously. At this
point we were beginning to lose hope that our strategy of the addition
of a methyl cuprate would ever be successful. The reader will recall
that at the beginning of this discussion, two possible routes were
proposed to introduce the correct chirality for the side-chain methyl
group. Since we had a small quantity of the sulphones (89A) and (89B)
available,it was decided to attempt a hydride addition with these
compounds to ensure that this second route would be feasible. Thus
treatment of the sulphones (89A) and (89B) with lithium triethyl-
borohydride ('Super-Hydride') (1.85 eq) gave, after work-up, the
saturated sulphone (90) in 96% yield (Scheme 47).
Scheme 47
Although we could have gone back and synthesized the required
sulphone (71) for the hydride addition via a similar sequence of reac
tions to those described in the preceding discussion (Scheme 48),
there was no guarantee that the conjugate addition would be successful
in terms of stereochemical control. In addition, the racemic nature
of the route would have necessitated resolution at some stage. y

133
Scheme 48
Me-----= — H + C H 3CHO
In view of these factors, it was decided to embark upon a route
starting from a chiral compound with the side-chain methyl present in
its correct absolute configuration. As will be seen in the next sub
section, the [2 + 2] cycloaddition strategy has still been used to set
up the cis relationship between the substituents on the tetrahydrofuran
ring.
Thus, although the sulphone-mediated approach was eventually
abandoned, it did provide useful insights which enabled us to find an
alternative and successful route to our target.
5.3.2 Chiral route to lactone (69)
In order to synthesize the lactone (69) with the side-chain methyl
in its correct configuration, we needed to obtain the eTs-alkene (91)
for the cycloaddition route.
P = protecting group

134
Compound (91) can be synthesized easily in four steps from the83commercially available hydroxy-ester (S)-(+)-methyl-3-hydroxy-2-
methylpropionate (92).
C02Me C02Me
OH
(92)
^OSi*BuPh2
(93)
Thus treatment of (92) with £er£-butyldiphenyl silyl chloride,60triethyl amine and DMAP gave the protected derivative (93) in high
yield. Since we were planning to introduce the ois double bond via a
Wittig reaction with an unstabilized phosphorane,we required the
aldehyde (94). Attempts to obtain (94) directly from the ester by
reduction with DIBAL (1 eq) were problematic, resulting in mixtures of
the aldehyde and the overreduced product (95).
TsCHO
OSi*BuPh2
(94)
It was therefore decided to deliberately reduce the ester down to the
alcohol (95) which could then be oxidised to the required aldehyde.
Surprisingly treatment of compound (93) with excess lithium aluminium
hydride resulted in immediate deprotection even at low temperatures
(-40°C). We therefore used an excess of DIBAL in toluene to obtain
the alcohol (95) in 75% yield. Although a range of oxidation

135
conditions were tried, it was found that the Swern oxalyl chloride -
DMSO route^9*3’0 was the most efficient, affording the aldehyde in 72%
yield. Treatment of ethyltriphenylphosphonium iodide with nBuLi at
_78°C followed by addition of the aldehyde (941 and rapid warming to
room temperature gave after work-up, the required olefin (91)
(P = tBuPH2Si) (>95% ois) in 86% yield. With this olefin in hand, we
were now ready to carry out the cycloaddition with dichloroketene.
Thus slow addition, as described previously (Section 5.3.1) of
trichloroacetyl chloride to a refluxing mixture of the olefin (91) and
excess freshly prepared zinc-copper couple in diethyl ether,
followed by refluxing for a further 2 h, gave after work-up, the
unstable dichloroadducts (96AB) and (97AB). It was necessary to use
oa. three equivalents of trichloroacetyl chloride in order to force the
reaction to completion. Again the dichloroadducts were found to be
unstable to both chromatography and distillation and were therefore
used in the reduction step without purification. It is worth noting
here that the work-up conditions for this cycloaddition greatly affect
the yield obtained. Literature experimental procedures'*3’ often
involve aqueous work-ups. However, we found that any addition of
water to the crude reaction mixture resulted in the formation of hydro
chloric acid (presumably from the hydrolysis of any traces of trichloro
acetyl chloride still remaining) which then caused removal of the
silicon protecting group. Even though reported procedures often
involve washing with aqueous sodium bicarbonate solution, we did not
find that this completely solved the problem. A modified non-aqueous
work-up (see Experimental section) resulted in far superior yields
emphasizing the need for rigorously dried reagents and solvents in this
reaction.

136
Treatment of the crude dichloro-cycloadducts with zinc and glacial 72acetic acid at 40°C as described previously (Section 5.3.1) resulted
in incomplete reduction even after prolonged reaction times. Indeed,
under these conditions, we obtained substantial quantities of the
monochloro-adducts. In order to effect complete reduction, heating
to ca. 90°C was necessary but unfortunately, the silicon protecting
group was not stable to these conditions.
Recently Liebeskind and Baysdon^ have reported the use of zinc
and solid ammonium chloride at room temperature for the reduction of
dichlorocyclobutanones. Use of these mild conditions gave a high
yield of the cyclobutanones (98) and (99) as a separable 6:1 mixture in
71% overall yield (from olefin (91)) (Scheme 49). Compounds (98) and
(99) were each present as a 1:1 diastereoisomeric mixture (designated
A and B).
The regioisomeric ratio is even higher than that obtained when
olefin (75) was used in the cycloaddition. Thus the bulky nature of
the silicon protecting group and the methyl substitution present in
olefin (91) both seem to be controlling the way in which dichloroketene
approaches the double bond (Scheme 50).
As part of an attempt to discover the extent to which this silicon
protecting group was controlling the regiochemistry in the cyclo
addition we thought that it would be interesting to replace it with a
less bulky group. We chose to replace it with the thiophenyl group
since the products obtained would be useful in our projected synthesis
of the right-hand portion of M139603 by virtue of the simple conversion
of -SPh to -S02Ph. Thus treatment of (92) with diphenyl disulphide86and tri-n-butylphosphine gave the sulphide (100) in 88% yield.
Reduction of (100) with lithium aluminium hydride in diethyl ether gave
the alcohol (101) which was oxidised to the corresponding aldehyde (102)

137
Scheme 49
(98AB) (99AB)
(96A)
Scheme 50
rs/ie

138
using the oxalyl chloride - DMSO Swern conditions. ’ Treatment of
(102) with the phosphorane generated from ethyltriphenylphosphonium
iodide and nBuLi as before, gave the alkene (103) (>95% ois) in 48%
overall yield from the alcohol (102). Reaction with dichloroketene
formed in situ as before, gave an unstable mixture of dichloro-QC
adducts (104) which after Zn/NH^Cl reduction gave a 2:1 inseparable
mixture of regioisomeric cyclobutanones (105AB) and (106AB) in 45%
overall yield from the olefin (103) (Scheme 51).
Scheme 51
39b c
(92)
.C02Me"Bu,P Y
PhSSPhOH THF
(100)
LiAlH4Et20
COH oxalyl chloride>D M SO , -60°C
a r h
(101)
CHO
^SPh
(102)
Ph,P=CHCH3
THF
c i 9c = c = o
Et20
Ph
(103)
Dichloro-adducts (104)
Zn,NH4CI(s)
THF, MeOH
2 1

139
Comparing this result with that for olefin (91), we see a marked
reduction in the regioselectivity when -SPh replaces -0SitBuPh2.
Thus, it seems that our earlier explanations for the high regioselec
tivity in the presence of the bulky silicon group are further
corroborated. These results are summarized in Table 2.
Table 2
Major regioisomer (after reduction)
JY _ ^
r^ O S i*8 u P h 2 ^ O S i* B u P h 2
(91)
Y __
Y p^ S P h ^ S P h
(103)
RegioisomericRatio
oa . 6:1
ca. 2:1
72Baeyer-Vi11iger oxidation of the major cyclobutanone (98AB)
gave a 1:1 mixture of diastereoisomeric lactones (107A) and
(107B). These could easily be separated by high pressure liquid
chromatography on high resolution silica gel.

140
At the outset we were hoping that the methyl at C-2 in (91)
would cause the olefin to exhibit at least a modest ir-facial selec
tivity in favour of the required diastereoisomer (96A) (Scheme 49).
Of course this hope was based on the assumption that the conformation
adopted by the olefin (91) would be as indicated in Scheme 52.
However, the free rotating nature of the side-chain makes it very
difficult to predict the lowest energy conformation and indeed
energy calculations would really be required for this purpose.
However, we did feel that variation of the reaction conditions,
particularly the temperature, should affect the ir-facial selectivity.
The cycloaddition did proceed at room temperature but predictably
took longer to reach completion {oa. 10 h). However, the same 1:1
ratio of (107A) and (107B) was obtained after reduction and Baeyer-
Villiger oxidation. Lowering the temperature still further to 0°C
caused the cycloaddition to proceed extremely slowly and in fact was
not practical, since the dichloroketene polymerized faster than it
reacted with the olefin (91) at this temperature. Changing the rate
of formation of dichloroketene, controlled by the addition of tri-
chloroacetyl chloride using a syringe pump at various speeds did not
affect the diastereoisomeric ratio of products. Addition of a Lewis

141
acid such as phosphorus oxychloride^^’ or change of solvent were
similarly unsuccessful in affecting the ir-facial selectivity.
Scheme 52
After separation of the lactones (107A) and (107B) by high
pressure liquid chromatography, we obviously needed to be able to
assign the stereochemistry to each. Unfortunately the 250 MHz 1H
n.m.r. data was not very helpful in this respect; indeed the spectral
data was very similar for both compounds. Molecular models indicated
that the unwanted diastereoisomer (107B) could take up a reasonable
conformation where the protons at C-2' were fairly close to the methyl
ring protons. In the required diastereoisomer (107A),the distance
between these two sets of protons was greater in any of its possible
conformations. Thus we decided that a nuclear Overhauser experiment
might distinguish between the two lactones. The results for the
irradiation at the 0 4 and C-2' protons are shown (see n.O.e. spectra).

142

J A
Nuclear Overhauser Experiment
X
C-3 Me
2'-HL
J*—r—i—?—i—r—r—r— \—r—j—»—r-»—r— r~
•+ 3---- 1-----1---- 1— t----- <----- 1-----1-----r — r r p
z 1 GO

144
The major difference between the two sets of data is the nuclear
Overhauser enhancement of the ring methyl doublet upon irradiation of
the C-2‘ protons as predicted. In one diastereoisomer there is no
signal for the ring methyl indicating that it is not positioned close
in space to the C-2‘ protons; we assumed that this corresponded to
the required lactone (107A). The spectral data for the other dia
stereoisomer indicates a considerable nuclear Overhauser enhancement
for the ring methyl on irradiation of the C-21 protons indicating
their proximity; this presumably corresponds to the lactone (107B).
Although we were fairly confident of our assignments we really
wanted to prove unambiguously that they were correct before proceeding
any further on our synthetic path.
It was decided therefore to convert each lactone separately to a
compound where comparison of their spectral data would inequivocally
differentiate between them. Thus reduction of (107A) and (107B)
separately with 1M DIBAL in toluene at -78°C gave the corresponding
lactols (108A) and (108B). Treatment of these lactols with tetra-n-
butylammonium fluoride affected their deprotection to afford compounds
(109A) and (109B). These lactols were then ring-opened by the
addition of the stabilized phosphorane carbethoxymethylidenetriphenyl-
phosphorane to afford the a,p-unsaturated esters (110A) and (HOB)
both as inseparable mixtures of cis- and trans-isomers {ca. 5:1 in
each case). These diols were then treated without purification with87CSA and anhydrous copper sulphate in acetone to afford the acetonides
(111AC), (111AT), (112BC) and (112BT). At this stage the cis- and
trans-\ somers of each pair of acetonides were separable by conventional
column chromatography on silica gel (Scheme 53).

145
Scheme 53
PO
HO
(107S)
P= *BuPh,$i
C S A ,C uS 0 4

146
It was found necessary to deprotect the compounds (108A) and
(108B) prior to the phosphorane homologation reaction. Reversing
the order of these steps simply caused ring closure via the Michael
addition of the alkoxide onto the double bond of the a,j3-unsaturated
ester (Scheme 54).
Scheme 54
Comparison of compounds (111AT) and (111BT) in their chair con
formations indicates that the coupling constant between the protons
at C-6 and C-7 (H^ and Hg respectively) should be significantly
different (Scheme 55).
Scheme 55

147
In compound (111AT), 6-H and 7-H are diaxially disposed and should
therefore exhibit a large coupling constant. This indeed turned out
to be the case (Jpo 9.6 Hz). In compound (111BT), 6-H and 7-H have AB
an axial-equatorial relationship and as expected J D was muchAd
smaller (J^g 2.3 Hz). These results unamoiguously confirmed the
original stereochemical assignments.
Although our route to the lactone does give rise to a 1:1 mixture
of diastereoisomers we felt that it was worth pursuing, in view of the
fact that separation could be easily effected. In addition, the route
is short and easy to perform, providing a compound with three chiral
centres in only seven steps from a commercially available starting
material.
5.4 Conversion of the lactone (107A) to the right-hand portion of
M1396Q3
Having successfully completed the synthesis of the required lactone
(107A) we were now ready to convert it to the allylic alcohol (67) whichc?
was required for the asymmetric Sharpless epoxidation (Section 5.3.1).
It will be recalled that in the previous section compound (111AT) was
synthesized in order to confirm the lactone stereochemistry.
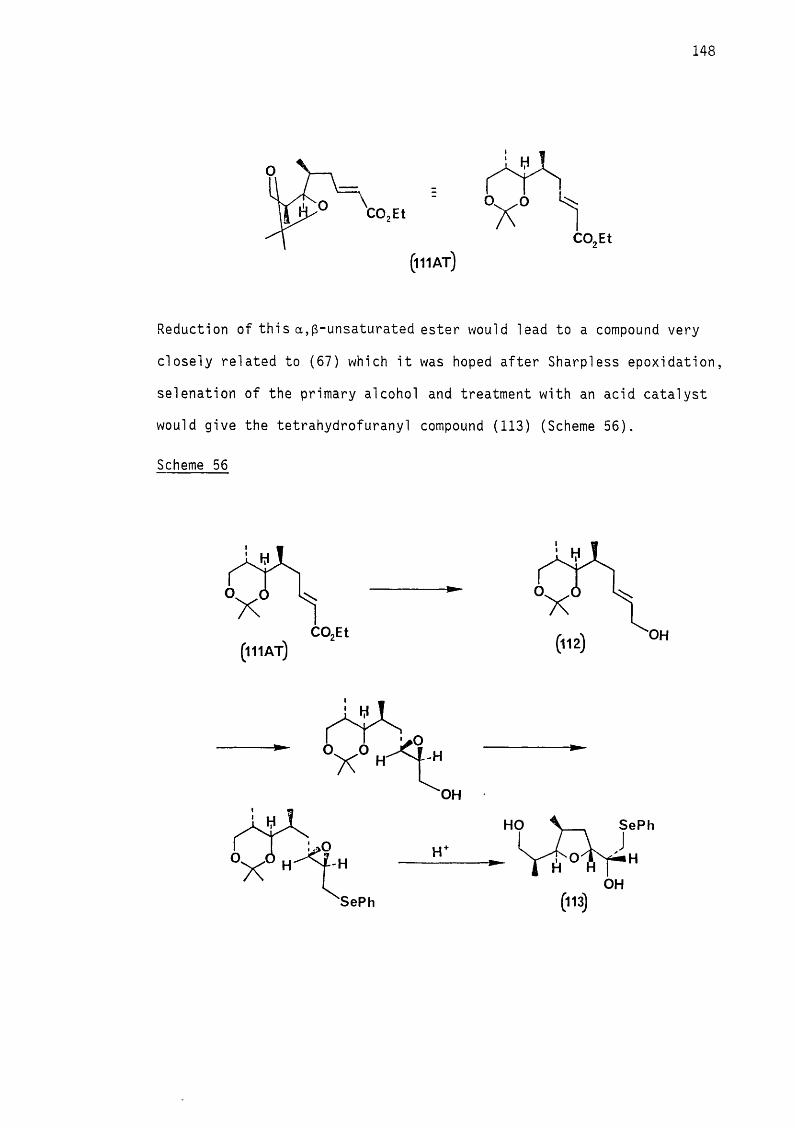
148
Reduction of this a,£-unsaturated ester would lead to a compound very
closely related to (67) which it was hoped after Sharpless epoxidation,
selenation of the primary alcohol and treatment with an acid catalyst
would give the tetrahydrofuranyl compound (113) (Scheme 56).
Scheme 56
SePh

149
Thus DIBAL reduction of (111AT) in toluene proceeded uneventfully to
afford the allylic alcohol (112) in 80% yield. At this point we88decided to use the non-asymmetric Sharpless epoxidation first to tes
the viability of the method. Thus epoxidation of (112) with ^BuOOH
and V0(acac)2 in dicnloromethane gave an inseparable 1:1 mixture of
diastereoisomeric epoxy-alcohols (114) in 89% yield. Conversion to
the primary selenide using the previously described conditions of tri-
n-butylphosphine (2 eq) and N-phenylselenophthalimide (2 eq) in THF
at -20°C (Section 5.2) afforded a 1:1 diastereoisomeric mixture of
epoxy-selenides (70%) (115) (Scheme 57).
As mentioned previously, the epoxy-selenide was found to be
unstable to flash chromatography on silica gel and was purified by
rapid chromatography using Florisil. To our disappointment, all
attempts to deprotect the acetonide in the presence of this extremely
sensitive epoxide were unsuccessful. Indeed, ring opening always
occurred prior to the acetonide cleavage. Since acid-catalyzed ring
opening of epoxides involves a carbocationic intermediate, loss of
stereochemistry at the epoxide carbon atoms in the chiral series would
obviously lead to a mixture of cis- and trans-tetrahydrofurans.
Scheme 57
SePh

150
Thus our hopes of synthesizing the required tozns-tetrahydrofuran
(113) via this route were destroyed. Instead, it was decided to
adopt an approach in which the silyl ether in lactone (107A) was left
intact. The lactone (107A) was therefore reduced with DIBAL to the
lactol (108A) as described in Section 5.3.2. Treatment with
carbethoxymethylidenetriphenylphospnorane in dichloromethane gave the
ois- and trans-a, (3-unsaturated esters (116) and (117), in 12 and 75%
yields respectively. The reaction was relatively slow, taking about
48 h to reach completion and variation of the solvent did not seem to
affect the rate of reaction significantly. Treatment of the trans
isomer (117) with 1.5M DIBAL in toluene at -78°C gave a disappointing
yield (50%) of the allylic alcohol (118), the other product from the
reaction being the tetrahydrofuran (119) as a mixture of diastereo-
isomers (Scheme 58).
Scheme 58
'BuPHSiO v
(107A)
DIBALPhCH3-78°C
^BuPKSiO -
-(108A)
Ph,P=CHCQ2Ef

151
We initially thought that in order to prevent this Michael reaction,
it would be necessary to protect the secondary alcohol. From earlier
considerations, we required the protecting group to be easily removable
under mild conditions i.e. in the presence of an epoxide. In addition,
it would be necessary to be able to remove the secondary alcohol protec
tion in the presence of the tert-butyldiphenylsilyl group. Lastly, the
protecting group chosen would have to be stable to the conditions
employed in the Sharpless reaction (Lewis acidic). These criteria
certainly narrowed the choice of suitable protecting groups quite89considerably. Recently, Guindon and co-workers reported the tert-
butylmethoxyphenylsilyl ether protecting group to be both selective and
stable whilst at the same time exhibiting enhanced lability towards
fluoride thus enabling its selective cleavage in the presence of a TBDPS
ether. In addition, it would be expected to survive the Sharpless
conditions. To this end, compound (118) was treated with tert-butyl-89methoxyphenylsilyl bromide (prepared as reported ) and triethylamine in
DMF at room temperature. Although starting material was slowly con
sumed, the only product isolated no longer possessed the double bond;
presumably ring closure to the tetrahydrofuran was occurring prior to
the reaction of the alkoxy-anion with the silicon reagent. Although
reaction conditions were varied (different base and solvent) we were
never successful in obtaining the protected secondary alcohol.
We were now faced with the possibility of having to be content with
a poor 50% yield of the required allylic alcohol from the DIBAL reduction.
It was felt however that it should be possible to increase this yield by
variation of the reaction conditions. Indeed, treatment of compound
(117) in THF at -78°C with DIBAL (3 eq of 1.5M solution in THF or hexane)
gave the required allylic alcohol (118) as the only product in a grati
fying 84% yield.

152
This interesting observation may be the result of the fact that
toluene, being a nonpolar solvent may increase the rate of the ring
closure reaction to form the less polar tetrahydrofuranyl product. THF
may coordinate, perhaps via hydrogen bonding, to the free alcohol thus
reducing the basicity of the lone pair of electrons on the oxygen which
may decrease the rate of ring closure.
The next step in the projected synthesis was the Sharpless62asymmetric epoxidation of the allylic alcohol (118). Recalling our
original thoughts on the strategy of this synthesis (Scheme 31), we had
proposed to perform the epoxidation upon the allylic alcohol (57) with
the secondary alcohol protected. It was then hoped that after epoxi
dation, the primary alcohol could be converted to the corresponding
selenide followed by acid-catalyzed ring closure to the tetrahydrofuran.
By performing the Sharpless epoxidation on the allylic alcohol (118)
where the secondary alcohol was unprotected, we considerably altered the
ensuing steps. Indeed, it was hoped that it would not be necessary to
isolate the intermediate epoxy-alcohol but that it would undergo ring
closure to the tetrahydrofuran under the Lewis acidic conditions of the90Sharpless reaction. Several attempts to epoxidise the allylic
alcohol (118) under the 'standard' Sharpless reaction conditions were
unsuccessful. Because it was believed that the failure of the allylic
alcohol to undergo epoxidation was due to coordination between the91initial titanium-tartrate complex and our substrate, it was decided to
attempt the reaction using larger excesses of the reagents. Sharpless
epoxidation employing these modified conditions (see Experimental92section) followed by the "NaF" work-up procedure afforded a 75% yield
of the trans-fused tetrahydrofuran (120) (Scheme 59). The work-up used
here has been recommended by Sharpless for isolating water-soluble and/or
sensitive products. Indeed compound (120) was found to be considerably
water-soluble.

153
Scheme 59
^ B u Ph jS i
OH
Sharplessepoxidation
^BuPI^Si
OH
JOH
(118) (120)
75%
93Masamune et at. recently reported the use of larger excesses of
reagents in the epoxidation of their polyoxygenated substrates.
Having obtained the tetrahydrofuran (120) it remained to convert
the diol side-chain to that present in the natural compound (Scheme 60).
Scheme 60
Attempted conversion of the primary alcohol to the selenide using
ful even when more than one equivalent of each reagent was employed.
We therefore decided to attempt the same conversion via the less direct
route (Section 5.2, Route B). Since we wanted to mesylate the primary
alcohol in the presence of the secondary alcohol we decided to employ
the hindered base N,N-diisopropylethylamine (Hunig's base) instead of94the more usual triethylamine and also to perform the reaction at low
the direct route employing N-PSP and tri-n-butylphosphine63 was unsuccess-
temperature. Thus treatment of the diol (120) in dichioromethane at
-20°C with methanesulphonyl chloride (1.1 eq) followed by Hunig's base
(1.0 eq) gave after aqueous work-up, the primary mesylate (121) with

154
only traces of the dimesylate. Attempts to convert (121) to the
selenide (122) were unsuccessful using the diphenyl diselenide-sodium95borohydride conditions. Since these conditions produce the phenyl-
selenide anionic species in its mildest nucleophilic form, we decided to
generate a more reactive anion. Recently in our laboratories a novel
method of generating sodium phenyl selenide has been developed. This
procedure involves the treatment of diphenyl diselenide with sodium
metal in THF under ultrasonic conditions (see Experimental section).
Treatment of the primary mesylate (121) at 0°C with this form of the
phenyl selenide anion gave the primary selenide (122) in 51% overall
yield from the diol (Scheme 61).
Scheme 61
Reduction of the selenide was easily effected by treatment with
Raney nickel in diethyl ether under an atmosphere of hydrogen to afford
compound (123) in quantitative yield. Methylation of the secondary
hydroxyl may be achieved under the classical Purdie conditions using
methyl iodide and freshly prepared silver oxide in DMF at room
temperature. However, this reaction is relatively slow and on a
larger scale it has been found more convenient to treat (123) with

155
sodium hydride in THF/DMPU followed by methyl iodide to afford the
methyl ether (124) in 77% yield (Scheme 62).
Scheme 62
93
Because we only had a small quantity of (124) in hand we decided to com
pare it directly with the material obtained from degradation after
oxidation and protection (Scheme 63).
Scheme 63
Mono-O-acetyl
M139603 methyl ether
The synthetic material (124) was identical in all respects (1H
n.m.r., i.r., mass spectrum, rotation, t.l.c.) to the material obtained
from degradation (125).

156
On obtaining larger quantities of (124) we were able to show that
it could be deprotected using anhydrous 2M tetra-n-butylammonium
fluoride in THF to afford the alcohol (64) (Scheme 64).
Scheme 64
Thus the synthesis of the tetrahydrofuranyl portion (64) of M139603
has been achieved in 16 steps from (S)-(+)-methyl-3-hydroxy-2-methyl
propionate (Scheme 65). In addition this chemical synthesis of (124)
unambiguously established the absolute configuration of M139603 of which
there was some doubt.^
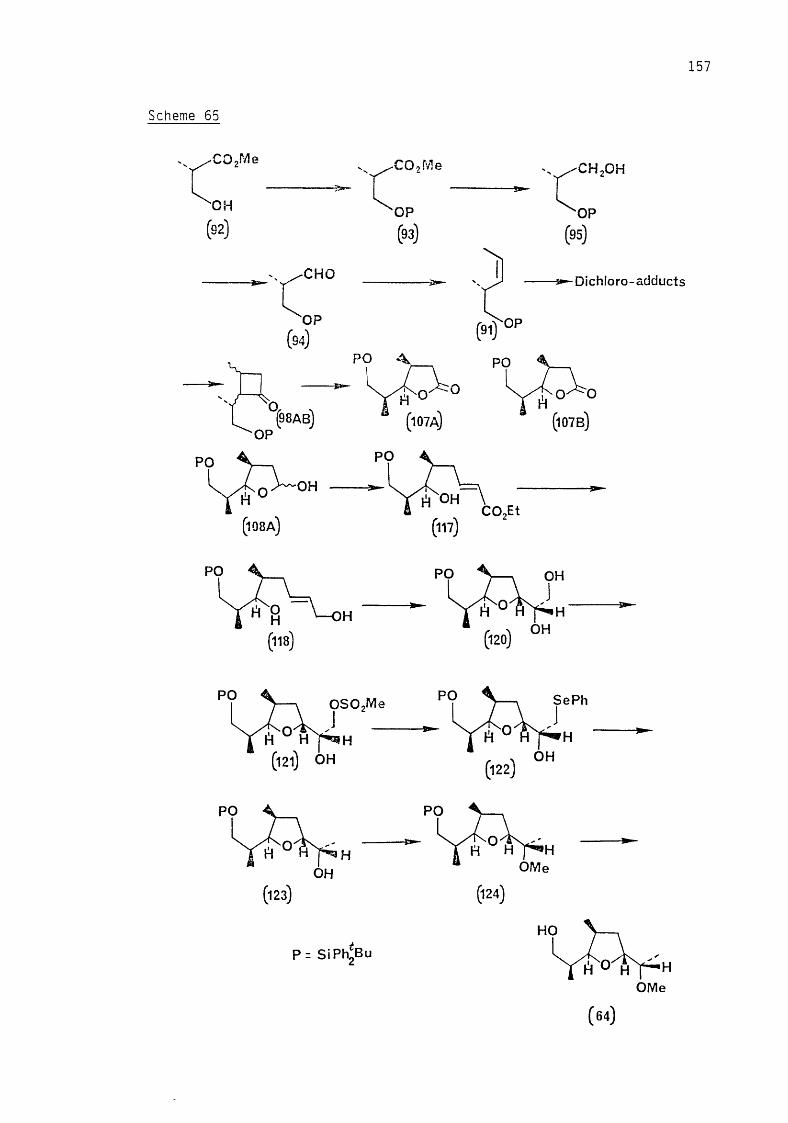
157
Scheme 65
^•CQ 2Me CO 2 Me ' ^ C H 2OH------- !»-
OP ^ O P
(93) (95)
■*» C H O -5»~Dich!oro-adducts
-9**
OP
(S4)PO
(91) 0 P
PO
^ 0 ^ °
(107a)
• 0 ^ 0
(107B)
PO PO
(64)

158
SECTION 6 : COUPLING STUDIES
Having obtained the right-hand portion of M139603 by total syn
thesis and the left-hand portion from degradation, our next goal was
the coupling of these two fragments (Scheme 66). In our model studies
(Section 3) we had established that it was possible to introduce the
trans-disubstituted double bond via either sulphone or Wittig method
ology, the former being more successful in terms of the double bond
isomer ratio obtained.
Scheme 66
X = anion - stabilizing group
We therefore decided to begin by converting alcohol (64) to the
corresponding sulphone and phosphonium salt in an attempt to repeat
these coupling reactions for the naturally-occurring system. Thus the
alcohol (64) was treated with diphenyl disulphide and tri-n-butyl- 86phosphine to afford the sulphide (126) in 95% yield. Oxidation with
two equivalents of mCPBA in dichloromethane afforded the crystalline
sulphone (127) in 90% yield.
Conversion of the alcohol (64) to the corresponding iodide (128)
using triphenylphosphine, imidazole and iodine in acetonitrile-diethyl
ether (1:3) at room temperature was easily effected. Treatment of

159
(123) with triphenylphosphine in refluxing toluene for 24 hours
afforded the phosphonium salt (129) in 79% yield (Scheme 67). The
corresponding bromide reached much more sluggishly with triphenyl-
phosphine.
Scheme 67
HO V
OMe
Ph3P,
12 *,, imidazole
o v □ i'
(128) OMe
To our disappointment, all attempts to couple the 1ithio-sulphone
(generated from nBuLi and sulphone (127)) with the left-hand side
aldehyde (59) obtained from degradation were unsuccessful, resulting in
substantial recovery of the starting sulphone (127) and total dis
appearance of the aldehyde. Even treatment of the crude reaction
mixture (after quenching with benzoyl chloride) with sodium amalgam did
not lead to the required olefin.

160
Deprotonation of the phosohonium salt with nBuLi at -73°C
followed by addition of aldehyde (59) was similarly unsuccessful. At
this point, we decided to attempt the addition of some other coupling
fragments to the aldehyde (59).
Thus the iodide (128) was heated with redistilled triethylohosohite
(Arbusov reaction^) at ca. 120°C for 48 h to afford the phosphonate 101ester (130) in 70% yield. The phosphine oxide (131) could be syn
thesized from the phosphonium salt by a simple 30 min reflux with 3M102aqueous sodium hydroxide. Alternatively, we have found that it is
more convenient to treat the iodide (128) with sodium diphenylphosphide
(obtained from the treatment of diphenylphosphine with sodium metal) to
obtain, after air oxidation, the phosphine oxide (131) in 93% yield
(Scheme 68).
Scheme 68

161
Since neither of tnese types of reagent had been used before in
coupling reactions with the model aldehydes, we decided co attempt: the
addition of the phosphine oxide (121) to aldehyde (22A) in a Horner-
Wittig reaction before applying it to the naturally occurring system.
Thus treatment of the phosphine oxide (131) with nBuLi in THF at
-78°C followed by the addition of the aldehyde (22A) afforded after
work-up,a diastereoisomeric mixture of hydroxyphosphine oxides (132)
(40%) and reisolated phosphine oxide (40%). The isolation of starting
phosphine oxide from the reaction seemed to indicate that the anion
was causing some deprotonation of the aldehyde. Treatment of the
purified hydroxyphosphine oxides (132) with freshly sublimed potassium
tert-butoxide (1 eq) in THF at 0°C caused rapid elimination to afford
the trans-olefin (133) in 81% yield with a trace of the less polar cis-
olefin (Scheme 69). Thus although the overall yield of this two-step
procedure was low (32%) it did afford mainly the trans-olefin and we
were therefore encouraged to attempt it with the aldehyde (59). Since
the aldehyde (22A) was racemic and the phosphine oxide optically pure,
the olefin adduct (133) would have been expected to be a diastereo
isomeric mixture. The 400 MHz n.m.r. spectrum only indicated one set
of signals and it was therefore assumed that the resonances of the
diastereoisomeric protons were coincident (see Appendix).

162
Scheme 69
yO
Unfortunately all attempts to repeat this coupling reaction with
aldehyde (59) have been unsuccessful. Similarly we could not obtain
the required olefin from the attempted addition of the phosphonate 101ester anion to aldehyde (59).
In all of these coupling reactions, the right-hand partner was re
isolated from the reaction in varying yield (30-70%) despite rigorous
attempts to ensure that all the reagents and solvents were dry. We
feel that the failure of these coupling reactions arises mainly from
the sensitivity of aldehyde (59) and its tendency to undergo hydration

163
and decomposition. As mentioned previously the separation of the
right- and left-hand aldehydes obtained from the ozonolysis reaction
was carried out by repeated trituration with petrol. The left-hand
aldehyde was then washed once with water to remove the DMSO generated
in the czonolysis work-up, dried, and then kept under high vacuum for
24 h. No other form of purification being possible, we had simply
used the aldehyde after this procedure. As mentioned previously, we
now feel that it is not advisable to use the dimethyl sulphide work-up
since the aldehyde may be undergoing extensive hydration in our
attempts to remove the DMSO.
Although we felt that the instability of the aldehyde was contri
buting to the failure of these coupling reactions, we also thought
that lithium might not be the best choice of counter-ion since the
intermediate 1ithio-species obtained after the addition of the aldehyde
to any of our right-hand partners might be expected to possess con
siderable reactivity. We considered it possible therefore that trap
ping of this reactive intermediate was not proceeding efficiently and
that side-reactions were predominating. In order to test whether this
was the case it was decided to attempt the addition of a Grignard
reagent to the left-hand side aldehyde (59).
Thus addition of phenethylmagnesiurn bromide to aldehyde (59) at
-30°C followed by warming to room temperature afforded a low yield of
the adduct (134) (Scheme 70) as a 1:1 mixture of diastereoisomers.
42

164
Scheme 70
With this pleasing result, we decided to return to our model aldehyde
(22B) (we only had a very small quantity of the cis-aldehyde (22A) in
hand) in order to establish a route from a hydroxy-adduct such as (134)
to the trans-olefin.
Thus treatment of (22B) with phenethylmagnesium bromide (1.5 eq)
afforded a diastereoisomeric mixture of hydroxy-adducts (135A) and
(135B) in a gratifying 84% yield. This was the highest yield that we
had obtained in any coupling with the model aldehydes and it therefore
seemed that the magnesium cation was superior to the lithium cation in
these reactions (Scheme 71).

165
Scheme 71
1. P h C H 2C H ,M g 8 r
2. H20
Obviously elimination of water from (135A) and (135B) could occur in
two directions, either towards or away from the ring. If elimination
occurred towards the ring, the enol ether formed (136) would be expected
to isomerise readily to afford compound (137) (Scheme 72).
Scheme 72

166
Our task was therefore to find a suitable route which would cause
elimination away from the ring to afford the olefin (138), which
involves removal of the less acidic proton Hg rather than H .
Of the methods available for the direct elimination of alcohols104we considered that the use of 3urgess' salt , where a bulky polar
intermediate is formed,might effect selective removal of the less
hindered proton Hg. Thus treatment of the mixture of (135A) and
(135B) in THF with methyl(carboxysulphamoyl)triethylammonium hydroxide
inner salt (139) (Burgess' salt, prepared according to the literature
procedure"^0*) at room temperature for 24 h caused the conversion of
the alcohols to a more polar product, assumed to be the initially
formed salt (140). However, no elimination had occurred at room
temperature and therefore the solution was heated to reflux. After
6 h, there was still no further reaction and therefore the THF was
evaporated in a stream of argon and replaced with toluene. By increas
ing the temperature gradually, elimination began to occur at oa. 80°C.
After complete conversion of (140) to a much less polar product the
reaction mixture was evaporated and chromatographed to afford mainly
the enol ether (137) with only a trace of the required olefin (138)
(Scheme 73).

167
Scheme 73
Ph
51%
Ph
trans
At this point we decided to try to find milder conditions for the
elimination, since we considered that the high temperature required in
the Burgess' salt elimination might be having a detrimental effect on
the observed regioselectivity. Thus conversion of the alcohols (135A)
and (135B) to the corresponding mesylates (141AB) was effected by treat-94ment with methanesulphonyl chloride and triethylamine.
Ph
(141AB)

168
Attempted elimination with DBU was unsuccessful even after
heating. Some mild conditions reported for the elimination of
mesylates involve their treatment with activated alumina in dichloro- 106methane. This method afforded both products (137) and (138) in
50 and 35% yields respectively.
In view of our failure to produce (138) selectively we decided to
attempt the conversion of the secondary alcohol to the corresponding
selenide since it is known that elimination of selenoxides in related
systems proceeds away from oxygen.^ Treatment of (135AB) with108phenylselenocyanide and tri-n-butylphosphine,which is reported to
be superior to the N-PSP-nBu3P route^ for the conversion of secondary
alcohols to their corresponding selenide derivatives, was low-yielding.
Surprisingly treatment of the alcohols (135AB) with N”PSP and tri-n-
butylphosphine (2 eq of each) afforded the secondary selenide (142) in
62% yield. Oxidation with 30% aqueous hydrogen peroxide in aceto- 109nitrile afforded the trans-olefin almost exclusively (Scheme 74).
Scheme 74
105
C02Me
(138)

169
Consi deration of the two possible modes of sz/n-el i mi nation
indicates that tpons-sterc-oselsctivity is expected where the two
largest groups R1 and R2 are orientated with minimum steric inter-
action (Scheme 75).
Scheme 75
Disappointingly, we were unable to convert the alcohol (134) to
the required selenide. Not wanting to abandon this Grignard route,
it was decided to attempt the addition of the right-hand side Grignard

170
partner to aldehyde (59). Unfortunately all attempts to form the
Grignard reagent from the iodide (123) were unsuccessful even at elevated
temperatures. It was therefore decided to convert the alcohol (64) to°4the corresponding bromide (144) via the mesylate derivative^ (143) in
the hope that it would be possible to form the bromo-Grignard reagent
(Scheme 76).
Scheme 76
Again however we had no success in our attempts to form (145).
There are three basic modifications of the general procedure for the
direct synthesis of difficulty formed Grignard reagents, namely
(a) solvent variation, and (b) use of high reaction temperatures, and
(c) activation of the magnesium metal. Since our attempts at (a) and
(b) had failed,we decided to use activated magnesium as a last resort to
generate (145). Addition of the bromide (144) to Rieke magnesium"^ at
-50°C followed by the aldehyde (59) (from the triphenylphosphine work-up)

171
and warming to 0°C did not afford any of the required adduct. Indeed,
in addition to the Grignard quenched product (146), we obtained a sub
stantial quantity of the dimeric compound (147).
6 (90 MHz) 0.80 (3H, d, J 6 Hz,
Me), 0.90 (3H, d, J 6 Hz, Me),
1.00 (3H, d, J 6.5 Hz, Me), 1.12
(3H, d, J 6.5 Hz, Me), 1.48 - 2.40
(4H, m), 3.20 - 3.70 (5H, m con
taining s, OMe at 3.38), and 3.92
(1H, ddd, J 9.5, 7.5, and 5 Hz.
5-H).
6 (90 MHz) 0.80 (6H, d, J 6 Hz,
Me x 2), 0.90 (6H, d, J 6 Hz,
Me x 2), 1.08 (6H, d, J 6 Hz,
Me x 2). 1.40 - 2.60 (12H, m),
3.18 - 3.50 (10H, m, containing s,
OMe at 3.38), and 3.90 (2H, m,
5-H x 2); m/z 371 (MH+), 311
(M+ - CH(Me)OMe), 141, 85
(-CHCH2CH(Me)CH20-+), and 59
(CH(Me)0Me+); (Found: M+-
CH(Me)0Me, 311.2596. C22Htt20(+
requires M+-CH(Me)0Me, 311.2586).
Conclusi on
The synthesis of a variety of possible right-hand side coupling
fragments for M139603 has been achieved and it has been possible to show
that the trans-disubstituted double bond can be introduced via the coupling

172
of the phosphine oxide derivative (131) with the model aldehyde (22A).
Although lack of time has prevented further investigation, coupling
studies are presently being continued in our laboratories.

173SECTION 7 : MODEL STUDIES RELATED TO THE IONOPHORE ANTIBIOTIC
INDANOMYCIN (X-14547A)
In 1978, the naturally occurring ionophore indanomycin (X-14547A)
(148) was isolated from a strain of Streptomyces ant-ibioticus 112NRRL 8167. This structurally unique carboxylic acid ionophore,
possesses the ability to transport divalent as well as monovalent
cations. It also exhibits antibacterial, antitumour and antihyperten-112sive activity and promotes feed utilization in ruminants.
The structure and absolute configuration of indanomycin was
determined by Westley et at. by X-ray analysis of a crystal of the
derived (ff)-(+)-I-amino-l-(4-bromophenyl)ethane salt which had an
unusual antibiotic to amine molecular ratio of 2:1.
Indanomycin possesses the characteristic tetrahydropyran ring and
terminal carboxylic acid moiety common to ionophore antibiotics
(Section 1). However, in addition it possesses an unusual trans-fused
tetrahydroindan and a pyrrolylcarbonyl functionality. The latter is113common to only a few other ionophores of the calcimycin type. It is
interesting that these molecules also show a particular specificity and
transport ability for divalent cations. The (Iff),(3ff)-butadienyl unit
between the tetrahydropyran and tetrahydroindan ring systems is unique
to this ionophore antibiotic.
The novel structure and important biological properties of indano
mycin have stimulated considerable interest, culminating in its totalQ-1 1synthesis by our group and two others as noted previously (Section 1).
(148)

174
Because of our interest in synthesizing simple model compounds for
these complex naturally occurring ionophores, we decided to construct
a model system for indanomycin, with the hope that it would possess
similar biological properties.
112bThe dimeric nature of the salt complex reported by Westley
(see above) involves two antibiotic molecules forming a jaw-like struc
ture within which the ammonium salt is bound (Scheme 77 shows a stereopair representation of the complex).
Scheme 77
Structure of antibiotic X-14547A (148) and conformation in the crystalline state of the salt complex consisting of two antibiotic molecules and one molecule of i?-( + )-1-amino-l-(4-bromophenyl)ethane.

175
The amine is held within the dimer by three hydrogen bonds, to 0-3 and
0-4 of one antibiotic molecule and the carboxyl ate 0-1' of the second
(prime) molecule. The two antibiotic molecules are held together by
hydrogen bonds between 0-1' and NH, 0-1 and NH', and 0-2 and
0-2' of the carboxylic acid groups. The 2:1 stoichiometry of the
complex arises from the fact that only one of the indanomycin molecules
is ionized, allowing the two 0-2 oxygens to be hydrogen bound via the
proton on the nonionized carboxyl OH.
Consideration of this structure suggests that the carboxylic acid,
tetrahydropyran and pyrrolylcarbonyl groups are utilized in cation
binding. The trans-butadienyl unit may stabilize this configuration\
by reducing the conformational flexibility of the ionophore. With
these factors in mind it was decided to attempt the synthesis of model
compound (149) which would hopefully possess all the necessary features
for biological activity.
(149)
We proposed two possible routes (A) and (B) to this model compound
(149) (Scheme 78).gOur group's total synthesis of indanomycin involved introduction
29 30of the diene portion via the Julia aldehyde-sulphone coupling ’
between (153) and (154), followed by elaboration to the natural product
{i.e. Route B) (Scheme 79).

Scheme 78
Scheme 79
II
Indanomycin

177
Micolaou's synthesis^ involved the coupling of the sulphone (155)
with the allylic bromide (156) followed by elimination of phenyl-
sulphinic acid to introduce the diene moiety.
M
Br
Roush'*''*' has used a Wadsworth-Emmons*^ reaction between the aldehyde
(153) and a phosphonate ester (157).
O
CHO
Clearly, all of these syntheses involved the introduction of the
butadienyl moiety via a route of type B.
We thought it would be interesting to construct our model system
using the alternative pathway, Route k,via a Julia aldehyde-sulphone
coupling i.e. X = S02Ph. The synthesis of the fcrans-tetrahydrofuran
(22B) has already been discussed in the model studies for M139603
(Section 3).
Our synthetic plan for the required allylic sulphone (150) is
shown in Scheme 80.

Scheme 80
Thus treatment of a suitably protected 2-1ithio-pyrrole anion with
phthalide was expected to afford the pyrrolylcarbonyl alcohol (158)
which could be further elaborated to the required allylic sulphone
(150).
It was decided to incorporate the previously used pyrrole protect
ing group (3-trimethylsilylethoxymethyl (SEN), as it was shown in the

179
indanomycin synthesis to be readily cleaved by treatment with anhydrous
tetra-n-butylanr.monium fluoride s o l u t i o n . T h u s treatment of9SEM-pyrrole (prepared as reported previously ) at 0°C in 1,2-dimethcxy-
ethane with nBuLi followed by quenching with phthaliae (0.5 eq)
afforded the expected product alcohol (158) (where P = SEM) in an
optimised 64% yield (based on recovered SEM-pyrrole). Oxidation of
(158) to the aldehyde (159) was easily performed by treatment with115activated manganese dioxide in acetonitrile (72%). It was
anticipated that the required trans double bond could be introduced
via the addition of a stabilized phosphorane to aldehyde (159).
Consequently, treatment of (159) with carbethoxymethylidenetriphenyl -
phosphorane in dichioromethane for ea. 6 h afforded the trans-a,p-
unsaturated ester (160) (Scheme 81) in 95% yield.
Scheme 81

180
As expected, selective reduction of the ester in the presence of
the pyrrolylcarbonyl moiety was not possible. Therefore (160) was
treated with excess DIBAL (3 eq) in toluene at -78°C to afford the
diol (161) in 56% yield.
O H
0 6 0Attempts to improve this yield were unsuccessful; indeed the major
side-reaction involved Michael addition of the alcohol oxygen onto the
a,3-unsaturated ester to afford the tetrahydrofuranyl compound (162)
(Scheme 82).
Scheme 82
M (162)
(162) v (film) 2955, 1728, and 1069 cnT^; 6 (250 MHz) 0.00 (9H, s, max •SiMe3), 0.92 (2H, m, D72SiMe2), 1.27 (3H, t, J 7 Hz, C02CH2Ctf3), 3.10
(1H, dd, J 16 and 8 Hz, Ctf2C02Et), 3.30 (1H, dd, J 16 and 4 Hz,
C#2C02Et), 3.51 (2H, m, NCH20Ci72), 3.84 (1H, dd, J 8 and 4 Hz,
CffCH2C02Et), 4.18 (2H, m, -C02Ctf2CH3), 4.90 (1H, s, OCtfCH-N), 5.34 (1H,
d, J 10.5 Hz, NCtf20), 5.48 (1H, d, J 10.5 Hz, NC£20), 5.68 (1H, dd,
J 3.5 and 1.5 Hz, 3'-H pyrrole), 5.98 (1H, dd, J 3.5 and 3 Hz, 41-H
pyrrole), 6.78 (1H, dd. J 3 and 1.5 Hz, 5‘-H pyrrole), and 7.30 (4H, m, Ph).

181
In order to convert the diol (161) to the reauired allylic sulphon
(150), we anticipated conversion of the primary alcohol to the tosylats
or mesylate, followed by oxidation of the secondary alcohol and then
displacement with sodium phenylsulphinate (Scheme 83).
Scheme 83
(l50) (164)
The allylic tosylate (164) could be attacked by a nucleophile in an
S^2 or S^2' approach; however we were hoping that with a suitable
choice of conditions the former would predominate to afford the
required allylic sulphone (150).
Unfortunately attempts to synthesize the primary mesylate or
tosylate were unsuccessful. In the former case the diol (161) was
treated with triethylamine followed by methanesulphonyl chloride at 94-10°C, but an immediate deep red colour was noted upon addition of
the latter reagent. T.l.c. indicated total disappearance of starting
diol to base-line material. Treatment of the diol with p-toluene-116sulphonyl chloride and pyridine in dichloromethane afforded only a

182
very low yield of the required tosylate (163), the remainder of the
material being of much greater polarity as observed in the attempted
mesylate formation. It was thought that the failure of these reaction
might have been due to the instability of the product (mesylate or
tosylate) under the reaction conditions (Scheme 84 shows some
possibi1ities).
Scheme 84
Having had no success with this route to the sulphone, it was
decided to attempt selective protection of the primary alcohol as a
bulky silyl ether.
Fortunately treatment of the diol with triisopropylsilyl chloride 59and imidazole in DMF afforded the alcohol (165) (60-80%) accompanied
by variable quantities (0-20%) of the oxidised material (166)
(Scheme 85).

183
Scheme 85
(166}
Presumably the alcohol (165) was prone to air oxidation and the
pyrrolylcarbonyl compound (166) may have been produced on work-up.
The alcohol (165) could be oxidised to the pyrrolylcarbonyl derivative
(166) by treatment with activated with manganese dioxide in aceto- 115nitrile. Removal of the triisopropyl silyl group was easily
effected by treatment with 40% aqueous HF in acetonitrile, conditions
which interestingly did not remove the SEM-ether protecting group.
This triisopropylsilyl group could also be removed using 0.01M aqueous59HC1 in ethanol at 90°C but the former procedure was generally faster
and higher-yielding. The resulting alcohol (167) was converted to
the corresponding sulphide (168) in 95% yield using N-ohenylthio-
succinimide and tri-n-butylphosphine (1.8 eq of each). The presence
of a double bond in the sulphide (168) precluded the use of many of
the common oxidising agents for its conversion to the sulphone. However
use of diphenyl diselenide and 30% aqueous hydrogen peroxide afforded
the desired sulphone (169) in 83% yield (Scheme 86).

184
Scheme 86
Having now obtained the required trans-tetrahydropyranyl aldehyde (22B)
(Section 3) and the allylic sulphone (169), we were ready to attempt the
Julia aldehyde-sulphone coupling.
Thus treatment of the sulphone (169) in THF at -78°C with nBuLi
afforded a deep red-black solution which gradually lightened in colour
after addition of the trans-aldehyde (22B). The reaction mixture was
quenched with benzoyl chloride prior to work-up to afford a diastereo-
isomeric mixture of benzoyloxy-sulphones (170). When the crude adduct
(170) was treated with 6% sodium amalgam in THF/MeOH at -20°C for 1 h
the only product observed was the alcohol (171) (Scheme 87).
Reduction of the diene (172), the expected product from this
reaction, was presumably very rapid, which is perhaps not altogether
surprising in view of its highly conjugated nature (Scheme 88).

185
Scheme 87
(171): v (film) 3455, 3154, 2977, 2811, 1733, 1646, and 1111 cm"1; max •6 (250 MHz) 0.00 (9H, s, SiMe3), 0.94 (2H, m, C#2SiMe3), 1.20 - 1.44
(2H, m, 4-H2), 1.54 - 1.80 (4H, m, 3-H2 and 5-H2), 2.11 (1H, m, l'-H),
2.33 (1H, m, l'-H), 2.40 (1H. dd, J 15 and 5 Hz, Ctf2C02Me), 2.61 (1H,
dd, J 15 and 7.5 Hz, Ctf2C02Me), 3.08 - 3.35 (2H. m, Ctf2Ph), 3.40 - 3.75
(4H, m, OH, 6-H and 0C#2CH2SiMe3), 3.61 (3H, s (two close), OMe), 4.18
(1H, m, 2-H), 5.32 (2H, s (two close), 0CH2N), 5.28 - 5.65 (2H, m, CH=CH),
5.62 (1H, dd, J 3 and 1.5 Hz, 3"'-H pyrrole), 5.98 (1H, t, J 3 Hz,
4m -H pyrrole) . 6.16 UH, d, J 1 Hz, CtfOH), 6.73 (1H, dd, J 3 and
1.5 Hz, 51,1 -H pyrrole), 7.12 - 7.36 (3H, m, Ph), and 7.78 (1H, m, Ph).

186
Scheme 88
In an attempt to suppress this overreduction, the reaction
temperature was lowered to ca. -45°C and the sodium amalgam was added
periodically in small portions. Under these conditions it was
possible to isolate some of the keto-compound (173) accompanied by
(171) as before, and the methoxy-adduct (174) {ca. 1.5 : 1 : 2
respectively, 60% overall yield) (Scheme 89). The diene (172) was
never isolated from these reactions.

187
Scheme 89
A possible mechanism for the formation of (174) is shown in Scheme 90.
The structure of (174) has been proved unambiguously by the 250 MHz
COSY spectrum (vide infra).

188
Scheme 90

Scheme 90 (Contd)
5''/
v (film) 2947, 1737. 1628, and 1097 cm”1; 5 (250 MHz) 0.00 (6H, s,max.SiMe3), 0.90 (2H, m, Ctf2SiMe3), 1.50 - 1.70 (2H, m, 4*-H2), 2.46 (1H,
dd, J 14.5 and 7 Hz. Ctf2C02Me), 2.66 (1H, dd, J 14.5 and 7.5 Hz,
Ctf2C02Me), 3.17 (3H, s, OMe), 3.48 - 3.70 (4H, m, 6-H, 31-H, and
NCH20C#2CH2SiMe3), 3.65 (3H, s, C02Me), 4.25 (1H, m, 2-H (from
decoupling)), 5.37 (1H, oa. dd, J 15.5 and 7.5 Hz, 11-H), 5.80 (1H, m,
2‘-H), 5.88 (2H, s, NCtf20), 6.19 (1H, dd, J 4 and 2.5 Hz, 5m-H pyrrole),
6.53 (1H, dd, J 4 and 2 Hz, 4m -H pyrrole), 7.IS (1H, dd, J 2.5 and
2 Hz, 3"'-H pyrrole), and 7.32 (4H, m, Ph); m/z 541 (M+), 509 (M+-MeOH),
and 392 (M+-MeOH-OCH2CH2SiMe3).
Thus the COSY spectrum shows the coupling between the proton at
C-2 and the Ctf2C02Me protons. Also the two protons at oa. 3.55 p.p.m.
[6-H and 3'-H] are each coupled to one of the double bond protons;
however there is no coupling between the protons adjacent to oxygen. These
observations strongly suggest an arrangement of the type shown in Figure 15.

Correlated shift 2-D n.m.r. spectrum (recorded on WM-250 machine).

191
Although we were not able to obtain the required diene it was
decided to continue with the synthesis as planned. Thus the alcohol
(171) was easily oxidised to (173) with activated manganese dioxide in 115acetonitrile. Removal of the SEM-ether protecting group was
effected by treatment with anhydrous 3M tetra-n-butylammoniurn fluoride
to afford the free pyrrolyl compound (175). Lastly mild basic
hydrolysis using 1M lithium hydroxide in THF/water afforded the acid
(176) (Scheme 91) in an overall yield of 44% from (171).
Scheme 91
This acid (176) is presently undergoing biological evaluation at ICI.
Thus in our model studies towards the ionophore indanomycin, we
have shown that it is possible to couple an allylic sulphone with a
trons-tetrahydropyranyl aldehyde. The problem of overreduction could
possibly be eliminated by coupling the SEM-pyrrole lithio-anion with a
saturated lactone or, alternatively, by reducing the keto-group prior
to coupling, thereby lowering the reduction potential of the resulting
adduct. Further studies in this area are currently being continued
in our laboratories.

192
APPENDIX
400 MHz and 250 MHz LH n.m.r. spectra

400 MHz H n.m.r.
193

250 MHz n.m.r.
ID

195

250 MHz *H n.m.r.
196

400 MHz H n.m.r.
10

198

250 MHz H n.m.r.
w L>.
o
199

200
Ck^o-

o O M eH KJ
250 MHz H n.m.r.
201

(l2o)
T1 T s
T-
250 MHz H n.m.r.
’ r 2.
202

250 MHz 1H n.m.r.
(synthetic)
ui * t i ' ' ' ' ' ’ ' ' ' r * 1 ' ' ' ■ ' ' 1 i ' ' ’ ' ' ' ' ' ' rn to s h
~ i ' ’ 1 ' ' ' ' ' ' i ' ■ ■ ■ ■ ' ■ - ' i ' ’ ' ' 1 ' ' 1 r
3 2. I 0 203

204

250 MHz 1H n.m.r.
205

250 MHz 1H n.m.r.
<\~T1
~r5 ' r
206

400 MHz V
° ' '-^ rx
C02IVIe
(133)
r6
m m AT5 207

203

250 MHz 1H n.m.r.
209

5' i ' ' ' ■ ' 1 ’ ' ' i ' 1 1 1 ' ■ • ■ ■ i .................................................................................i ..................................................................... * * ,
^ 3 2 » O
o

211
EXPERIMENTAL
Melting points were determined using a Kofler hot-stage apparatus
and are uncorrected. Optical rotation measurements were conducted
using an Optical Activity AA-1000 polarimeter at 25°C. Infrared
spectra were recorded on Perkin Elmer 298 and 983G grating infrared
spectrophotometers using a thin film or as a solution in chloroform.
1H n.m.r. spectra were recorded at 60 MHz on a Varian EM-360A, at
90 MHz on a Jeol FX 90Q, at 250 MHz on a Bruker WM-250, and at 400 MHz
on a Bruker WH-400 machine, and are quoted for solutions in deuterio-
chloroform with tetramethylsilane as an internal standard. Mass
spectra were determined with a VG micromass 7070 B instrument.
Elemental microanalyses were performed in the Imperial College
Chemistry Department microanalytical laboratory.
Analytical thin layer chromatography was performed on precoated
aluminium- or glass-backed plates (Merck Kieselgel 60 ^ 5 4 ) anc*
preparative chromatography was conducted under low pressure using either
MN Kieselgel 60 (230-400 mesh) or BDH Florisil (200-300 mesh). Silica
gel refs to the Kieselgel. The high pressure liquid chromatographic
separation of the diastereoisomeric lactones [Exp 72] was conducted
with a Gilson 302-303 machine on a DYNAMAX Macro-h.p.1.c. column (21.4 cm
internal diameter, 25 cm length) employing 8 pm silica gel with 7.5%
isopropanol/92.5% hexane as eluant. The flow rate was 12 ml/min and a
UV detector at 254 nm was employed.
Petrol refers to light petroleum ether with b.p. 40-60°C and was
redistilled before use. Diethyl ether, 1,2-dimethoxyethane and tetra-
hydrofuran were dried by reflux over sodium/benzophenone and distilled
before use. Dichloromethane was dried by reflux over phosphorus
pentoxide and distilled before use. Dimethylformamide and dimethyl-osulphoxide were dried by prolonged storage over 4A molecular sieves

212
followed by distillation under reduced pressure onto 4A molecular
sieves. Benzene and toluene were dried over sodium wire and aceto
nitrile dried over calcium hydride and all were distilled before use.
Pyridine, diisopropylamine and triethylamine were distilled from
calcium hydride and stored over potassium hydroxide pellets. Pyrrole
was redistilled from calcium hydride at slightly reduced pressure
immediately prior to use. All other solvents and reagents were
purified by standard techniques. Solutions were dried over anhydrous
sodium sulphate and evaporated with a rotary evaporator followed by
static evaporation with an oil pump.
Various authors have used different numbering systems for1o pQ-pp pn-pp
M139603. ’ The numbering used by Staunton and co-workers
has been employed throughout the text and the Experimental and is
shown in the diagram below.
29 15 27
Na®salt

213
1. Preparation of 2-(2-Furyl)ethanol (14)
To THF (100 ml) at -25°C was added nBuLi (42.0 ml of a 1.20M
solution in hexane, 50.4 mmol) followed by freshly distilled furan
(3.64 ml, 50.0 mmol). The solution was stirred for 6 h at room
temperature and then cooled to -20°C before addition of excess ethylene
oxide (5 ml, condensed with a Dry ice-acetone bath and transferred via
a precooled gas-tight syringe into THF). The resulting solution was
allowed to warm to room temperature and stirred overnight. The reac
tion mixture was poured onto ice (lOg) and solid sodium chloride (lOg)
was added. The aqueous layer was repeatedly extracted with diethyl
ether (5 * 50 ml) and the combined organic extracts washed with
saturated aqueous sodium chloride (1 x 50 ml). After drying (Na2S0,J ,
tne solvent was removed under reduced pressure and either chromato
graphed (diethyl ether) or distilled (b.p. 80° at 12 mm Hg; to afford
2-(2-furyl )ethanol (14) (3.41g, 61%) as a colourless oil, 6 (60 MHz')
2.80 (2H, t, 2-H2, J 6.5 Hz), 3.10 (1H, m, -OH), 3.55 - 3.90 (2H, m,
1-H2), 6.00 (1H, d, J 3 Hz, 31-H), 6.20 (1H, dd, J 3 and 1.5 Hz, 41-H),
and 7.17 (1H, d. J 1.5 Hz, 51-H); identical to the previously118reported compound.

214
2. Preparation of 2-(2-Tetrahydrofur,yl )ethanol (15)
2-(2-Furyl)ethanol (14) (2.36g, 21.1 mmol) in anhydrous methanol
(40 ml) was hydrogenated over 5% rhodium on alumina at atmospheric 31apressure. After 96 h the catalyst was filtered off through a pad
of silica gel and the filtrate evaporated to afford 2-(2-tetrahydro-
furyljethanol (15) (2.44g, 100%) as a colourless oil, v (film) 3400,max *2940, and 1060 cm"1; <5 (60 MHz) 1.30 - 2.30 (7H, m, 2-H2, 3'-H2,
41-H2 and OH), and 3.10 - 4.30 (5H, m, 1-H2, 2 1-H and 5'-H2); m/z
+ 11998 (M - H20); identical to the previously reported compound.
3. Preparation of 2-(2-Phenylthioethyl )tetrah,ydrofuran (16)
HO
(15)
4___ 3 PhS
'2'
To tri-n-butylpnosphine (1.42 ml, 5.72 mmol) in dry THF (10 ml)
under argon was added recrystallized N-phenylthiosuccimide (1.18g,
5.72 mmol) in THF (5 ml) at room temperature. After stirring for
5 min, 2-(2-tetrahydrofuryl)ethanol (15) (332 mg, 2.86 mmol) in THF
(2 ml) was added. The solution was stirred for 2 h followed by
addition of water (10 ml) and diethyl ether (30 ml). The aqueous
layer was extracted with diethyl ether (3 * 20 ml) and the combined

215
ethereal extracts washed with water (1 x 10 ml). Drying (Na^O^) and con
centration under reduced pressure gave a brown residue which was purified
by chromatography (gradient elution, petrol 10% diethyl ether-petrol
to afford 2-{2-phenylthioethyl)tetrahydrofuran (16) (506 mg, 85%) as a
colourless oil, v (film) 3090, 2950, 2890, 1075, 760, and 705 cm”1; max.6 (90 MHz) 1.25 - 2.10 (6H, m, l'-H2, 3-H2, and 4-H2), 3.00 (2H, m,
2‘-H2), 3.80 (3H, m, 2-H and 5-H2). and 1.22 (5H, m, Ph); m/z 208 (M+),
123 (CH2SPh+), 109 (PhS+), 99 (M+-SPh), and 71 (M+-CH2CH2SPh);
(Found: M+, 208.0926. C12H160S requires M+, 208.0922); (Found:
C, 69.24; H, 7.63; S, 15.20. C12H160S requires C, 69.19; H, 7.74;
S, 15.39%).
4. Preparation of 2-(2-Phenylsulphonylethy1)tetrahydrofuran (17)
To a stirred solution of the sulphide (16) (2.40g, 11.5 mmol) in
dry dichloromethane (10 ml) was added mCPBA (4.59g, 90% purity, 24.0
mmol) in dichloromethane (50 ml) at 0°C. After 15 min, the reaction
was allowed to warm to room temperature and stirred for a further 2 h.
The solution was then diluted with diethyl ether (200 ml) and washed
with 1M aqueous sodium hydroxide solution (2 x 30 ml) and water (1 x
30 ml) respectively. Drying (Na2S0tt) and concentration under reduced
pressure followed by chromatography (45% diethyl ether-petrol afforded
2-(2- phenytsulphonylethyZ)tetrahydrofuran (17) (2.69g, 97%) as a low-
melting crystalline solid (m.p. 28°C), v (CHC13) 3060, 2950, 2870,max •

216
1310, 1230, and 1060 cm"1; 6 (250 MHz) 1.45 (1H, m), 1.78 - 2.08 (5H,
m), 3.14 (1H, ddd, J 14, 11.5, and 5 Hz, Cff2S02Ph), 3.30 (1H, ddd,
J 14, 11.5, and 4 Hz, Ctf2S02Ph), 3.60 - 3.90 (3H, m, 2-H and 5-H2),
7.52 - 7.70 (3H, m, Ph), and 7.92 (2H, m, Ph); m/z 240 (M+) and 163
(M+-Ph); (Found: M+, 240.0814. C12H1602S requires M+, 240.0820);
(Found: C, 59.71; H, 6.67; S, 13.43. C12H1602S requires C, 59.98;
H, 6.71; S, 13.34%).
5. Preparation of Methyl 3-hydroxyoct-7-enoate (19)
To a rapidly stirred solution of methyl 3-oxo-oct-7-enoate (18-)120prepared by the literature procedure ) (ll.OOg, 64.6 mmol) in dry
methanol (50 ml) at -10°C was added excess sodium borohydride in por
tions until no starting material was observed by t.l.c. The solution
was then poured into saturated aqueous ammonium chloride (50 ml) over
laid with diethyl ether (200 ml). The aqueous layer was extracted
with further diethyl ether (3 x 100 ml) and the combined organic
extracts washed with water (1 x 50 ml) and brine (1 x 50 ml). After
drying (Na2S01+) and concentration under reduced pressure the crude
residue was chromatographed (35% diethyl ether-petrol) to afford
methyl 3-hydroxyoct-7-enoate (19) (ll.OOg, 99%) as a colourless oil, vm3V (film) 3400, 2900, 2720, 1725, 1640, and 1020 cm"1; 6 (250 MHz)
1.40 - 1.64 (4H, m, 4-H2 and 5-H2), 2.04 - 2.14 (2H, m, 6-H2), 2.42
(1H, dd, J 16.5 and 8.5 Hz, Cff2C02Me), 2.52 (1H, dd, J 16.5 and 3.5 Hz,

217
6. Preparation of Methyl 7,8-epoxy-3-hydroxyoctanoate (20)
OH O
OMe 4,
51
1 T>Me8
(19) (20)
To the alcohol (19) (5.50g, 31.9 mmol) in dry dichloromethane
(100 ml) at 0°C was added dropwise a solution of mCPBA (6.88g, 90% purity,
36.0 mmol) in dry dichloromethane (120 ml). After 2 h, the reaction
was allowed to warm to room temperature and then stirred overnight.
The precipitated benzoic acid was filtered off and washed with a small
quantity of diethyl ether. The combined filtrate was treated with
excess dimethyl sulphide to remove any remaining mCPBA and then washed
with 5% aqueous sodium bicarbonate solution (2 x 30 ml). The aqueous
layer was extracted with diethyl ether (2 x 30 ml) and the combined
organic solution dried (Na2S0iJ and concentrated under reduced pressure.
Column chromatography of the residue (gradient elution, 50% diethyl
ether-petrol diethyl ether) afforded a 1:1 diastereoisomeric mixture
of methyl l , 8 - e p o x y -2-hydroxyoctanoate (20) (4.60g, 77%) as a colour
less oil, v (film) 3470, 2950, 1730, 1200, 1090, 915, and 845 cm"1; max.6 (60 MHz) 1.20 - 1.90 (6H. m, 4-H2, 5-H2, and 6-H2), 2.45 (2H, m,
C#2C02Me), 2.75 (1H, m, 7-H), 3.38 (2H, m, 8-H2), 3.68 (1.5H. s, OMe),
3.69 (1.5H, s, OMe), 3.96 (1H, m, CtfOH), and 4.68 (1H, br s, OH);

218
m/z 170 (M+-H20), 157 (M+-0Me), and 115 (M+-CH2C02Me); (Found:
C, 57.24; H, 8.84. C9H1604 requires C, 57.43; H, 8.57%).
7. Preparation of Methyl e-£s-6“(methylenehydroxy )tetrahydropyran-2-
ylacetate (21a) and Methyl trans-6-(methylenehydroxy)tetrahydro-
pyran-2-ylacetate (21B)
To the diastereoisomeric mixture of epoxy-alcohols (20) (4.00g,
21.3 mmol) in dry dichloromethane (100 ml) was added CSA (247 mg,
1.07 mmol) at room temperature. The solution was stirred for 48 h
and then filtered through a small silica gel pad. After concentra
tion under reduced pressure, the residue was purified by chromatography
(70% diethyl ether-petrol) to afford a 1:1 inseparable mixture of
methyl cis-6-(methylenehydroxy)tetrahydropyran-2-ylacetate (21A) and
methyl tra.r\s-6-(methylenehydroxy)tetrahydropyran-2-ylacetate (21B)
(4.00g, 100%) as a colourless oil, \> v (film) 3500, 2950, 2890, 1735.
and 1040 cm"1; 6 (250 MHz) 1.08 - 1.82 (6H, m), 2.27 - 2.64 (2H, m,
Ctf2C02Me), 3.28 - 3.49 (2H, m, Ctf20H), 3.58 (1.5H, s. C02Me), 3.60
(1.5H, s, C02Me), 3.65 - 4.18 (3H, m, OH, 2-H, and 6-H): m/z 188 (M+,
170 (M+-H20), 157 (M+-CH20H), 156 (M+-Me0H), 129 (M+-C02Me), 125 (M+-
CH20H-Me0H), and 115 (M+-CH2C02Me); Found: C, 57.17; H, 8.68.
C9H160t+ requires C, 57.43; H, 8.57%).

219
(22A) and Methyl trcms-6-formyl tetrah.ydropyran-2-yl acetate (22B)
8. Preparation of Methyl efs-6-formyltetrahydropyran-2-ylacetate
To a solution of oxalyl chloride (3.84 ml, 44.0 mmol) in dichloro-
methane (100 ml) at -60°C under argon was added dropwise DMSO (6.24 ml,
88.0 mmol) in dichloromethane (20 ml). After 5 min, the diastereo-
isomeric mixture of alcohols (21A) and (21B) (4.14g, 22.0 mmol) in
dichloromethane (45 ml) was added dropwise, maintaining the temperature
between -60 and -55°C. The resulting solution was stirred for a
further 20 min before addition of triethylamine (30.4 ml, 0.22 mol)
and subsequent warming to room temperature. Water (50 ml) was
added and the aqueous layer extracted with dicnloromethane (3 x 50 ml).
The combined organic extracts were washed with brine (1 x 50 ml) and
dried (Na2S0i+). The solution was then concentrated under reduced
pressure to ca. 30 ml and filtered through a small silica gel pad (with
diethyl ether). The filtrate was evaporated and the residue chromato
graphed (gradient elution, 40 50% diethyl ether-petrol) to afford
methyl trans-6-fovmyltetvahydropyrccn-2.-ylacetate (22B) (i?p 0.38, 40%
diethyl ether-petrol) (1.30g, 32%) and methyl cis-6-formyltetrahydro-
pyran-2-ylaeetate (22A) (i?p 0.23, 40% diethyl ether-petrol) (1 -30g, 32%)
both as slightly yellow oils, trans-isomer (22B): v (film) 2940,max.2870, 1735, 1210, 1160, 1100, and 1050 cm"1; 6 (250 MHz) 1.20 - 1.80
(5H, m), 2.04 (1H, m), 2.46 (1H, dd, J 15 and 5 Hz, Ctf2C02Me), 2.64
(1H, dd, J 15 and 8 Hz, C#2C02Me), 3.72 (3H, s, OMe), 4.08 (1H, m,
2'-H), 4.18 (1H, dd, J 5.5 and 3 Hz, 6‘-H), and 9.87 (1H, d, J 1 Hz,

220
CHO); m / z 186 (M+), 157 (M+-CH0), 125 (M+-CH0-Me0H), 113 (M+-CH2C02Me), and 97 (M+-CH0-CH3C02H); (Found: C, 57.92; H, 7.86. C9H1It0,* requires
C, 58.05; H, 7.58%).
cfs-i somer (22A): v (film) 2945, 2870, 1735, 1200, 1165, 1110, andmax.1040 cm"1; 6 (250 MHz) 1.20 - 2.05 (6H, m), 2.47 (1H, dd, J 15.5 and
5 Hz, C#2C02Me), 2.66 (1H, dd, J 15.5 and 8 Hz, Ctf2C02Me), 3.70 (3H, s,
OMe) ,3.80 - 3.95 (2H, m, 2'-H and 6'-H), and 9.58 (1H, s, CHO); m / z
186 (M+), 157 (M+-CH0), 125 (M+-CH0-Me0H), 113 (M+-CH2C02Me), and 97
(M+-CH0-CH3C02H); (Found: M+, 186.0896. C9H1(f0,* requires M+, 186.0892).
9. Preparation of Methyl efs-6-(p-nitrobenzoyloxymethyl)tetrahydro-
pyran-2-ylacetate (23)
To the cfs-alcohol (21A) (54 mg, 0.29 mmol) in dry pyridine
(1.5 ml) was added p-nitrobenzoyl chloride (110 mg, 0.58 mmol). After
stirring at room temperature overnight the pyridine was removed under
reduced pressure and the residue taken up in diethyl ether (5 ml)
followed by filtration through a small pad of silica gel. Concentration
under reduced pressure and chromatography (50% diethyl ether-petrol)
gave the p-nitrobenzoate derivative m e t h y l c i s - 6 - { p - n i t r o b e n z o y l o x y -
m e t h y l ) t e t r a h y d r o p y r a n - 2 - y l a c e t a t e (23) as a semi-solid (73 mg, 75%). Crystallization from diethyl ether gave crystals of sufficient quality
for an X-ray crystal structure determination (m.p. 62°C), \>max (CHC13)
2940, 2860, 1725, 1600, 1525, 1440, 1350, 1230, 1200, and 1100 cm"1:

221
6 (250 MHz) 1.20 - 2.00 (6H, m), 2.43 (1H, dd, J 15 and 5 Hz, Ctf2C02Me),
2.58 (1H, dd, J 15 and 8 Hz, Ctf2C02Me), 3.62 (3H, s, C02Me), 3.72-3.92
(2H, m, 21-H and 61-H), 4.32 (2H, d, J 5.5 Hz, Ci720C0Ar), and 8.25 (4H,
m, Ph); m/z 337 (M+), 306 (M+-0Me), 111 (M+-MeC02H), 264 (M+-CH2C02Me),
and 170 (M+-ArC02H); (Found: C, 56.69; H, 5.60; N, 4.06.
Ci6Hi907N requires C, 56.97; H, 5.68; N, 4.15%).
10. Preparation of (E)-2-(3-Phenyl-prop-2-enyl)tetrahydrofuran (25)
(25)
To the sulphone (17) (63 mg, 0.26 mmol) in THF (3 ml) at -78°C
was added nBuLi (0.17 ml of a 1.50M solution in hexane, 0.26 mmol)
dropwise with stirring. After 10 min, benzaldehyde (26 mg, 0.26 mmol)
in THF (0.5 ml) was added dropwise to the yellow solution. The
yellow colour immediately disappeared and after a further 10 min,
benzoyl chloride (60 pi, 0.52 mmol) was added in one portion and the
solution allowed to warm to room temperature over 2 h. 3-N,N-
Dimethylaminopropyl-1-amine (66 pi, 0.52 mmol) was then added to
destroy the excess benzoyl chloride and the solution was diluted with
diethyl ether (10 ml) and water (5 ml). The aqueous layer was

222
extracted with diethyl ether (3 x 5 ml) and the combined ethereal
extracts washed with saturated sodium chloride solution (2 x 5 ml)
before drying (Na2S0it). Removal of the solvent under reduced pressure
and chromatography (50% diethyl ether-petrol) gave a diastereoisomeric
mixture of benzoyloxy-sulphones (24) (100 mg, 85%) as a white foam,
v a (CHC1 3) 3060, 2980, 2770, 1730, 1150, and 1080 cm"1; 6 (60 MHz)max.1.30 - 2.40 (6H, m), 3.10 - 4.20 (4H, m), 6.30 - 6.80 (1H, m, CffOBz),
and 7.10 - 7.80 (15H, m, Ph).
The mixture of benzoyloxy-sulphones (24) (100 mg, 0.22 mmol) was
dissolved in methanol (0.5 ml) and THF (1.5 ml) and the resulting
solution cooled to -20°C. 6% Sodium amalgam (0.3g, oa. 6.0 eq) was
added to the solution at -20°C. Further small portions of the amalgam
were added periodically until all starting material had been consumed
(t.l.c.). The solution was then poured into petrol (100 ml) and water
(10 ml) and the aqueous layer extracted with diethyl ether (2 x 20 ml).
The combined organic extracts were washed with saturated aqueous
sodium chloride solution (2 x 10 ml) and after drying (Na2S0i+), the
solvent was removed under reduced pressure. The resulting yellow oil
was purified by chromatography (10% diethyl ether-petrol) to give the
olefinic adduct (£)-2-(3-phenyl-prop-2-enyl)tetrahydrofuran (25)
(28 mg, 66%) {oa. 10:90, Z:E mixture of double bond isomers) as a
colourless oil, (film) 2970, 2870, 1445, 1350, and 1105 cm"1;
6 (250 MHz) (^-isomer) 1.20 - 2.08 (4H, m), 2.45 (2H, m, Ctf2CH=CH),
3.74 (1H, m, 2-H), 3.92 (2H, m, 5-H2), 6.23 (1H, dt, J 16 and 7 Hz,
2'-H), 6.46 (1H, d, J 16 Hz, 3'-H), and 7.20 - 7.40 (5H, m, Ph);
m/z 188 (M+), 111 (M+-Ph) , 85 (M+-CH=CH-Ph), 77 (Ph+), and
71 (M+-CH2CH=CHPh).

223
11. Preparation of Methyl <gfs-6-r3-i tetrahydrofuran-2-yl )prop-l-(ff ,Z )-
en.yl 1tetranydropyran-2-yl acetate (26)
To the sulphone (2.20g, 9.12 mmol) in dry THF (20 ml) at -78°C
was added nBuLi (7.32 ml of a 1.20M solution in hexane, 10.0 mmol)
dropwise with stirring. After 10 min, the cfs-aldehyde (22A) (1.70g,
9.12 mmol) in THF (5 ml) was added dropwise to the yellow solution.
After a further 15 min, benzoyl chloride (2.12 ml, 18.24 mmol) was
aaded in one portion and the solution allowed to warm to room tempera
ture over 2 h. 3-N,N-Dimethylaminopropyl-l-amine (2.30 ml, 18.24
mmol) was then added to destroy the excess benzoyl chloride and the
reaction worked-up as described previously (Experiment 10). The crude
residue in THF/methanol (20:5 ml) was treated directly witn 6% sodium
amalgam. After work-up as before (Experiment 10) and chromatography
(60% diethyl ether-petrol) m e t h y l z \ s - § J L $ - { t e t T d h y d r o f u ¥ a n - 2 - y l ) i p v o ' p - l -
(E, Z ) - e n y l ] t e t r a h y d r o p y r a n - 2 - y l a c e t a t e (26) was obtained (934 mg, 38%) as an inseparable c i s i t r a n s mixture { c a . 5:95) as a colourless oil,
(film) 2970, 2870, 1740, 1197. and 1068 cm-1; E-isomer: 6 (250
MHz) 1.18 - 2.00 (10H, m), 2.14 - 2.45 (2H, m, CE2-CH=), 2.41 (1H, dd,
J 15 and 5 Hz, CE2C02Me), 2.62 (1H, dd, J 15 and 8 Hz, CE2C02Me), 3.68
(3H. s, OMe), 3.70 - 3.92 (5H, m, 2-H, 6-H, 2"-H, and 5"-H2), and
5.48 - 5.70 (2H, m, J 16 Hz, CH=CH); m/z 268 (M+), 237 (M+-0Me), 209
(M+-C02Me), 195 (M+-CH2C02Me), and 71 (C,ri70+); (Found: C, 66.94;

224
H, 8.81. Ci5H2£f0tf requires C, 67.14; H, 9.01%).
12. Preparation of Methyl <?Ts-6-[(z)-but-l-en,y1]tetrahydrop,yran-2-
ylacetate (27)
121To a suspension of propyltriphenylphosphonium iodide (130 mg,
0.30 mmol) in dry THF (3 ml) at 0°C was added nBuLi (0.23 ml of a 1.18M
solution in hexane, 0.27 mmol) and the solution stirred for a further
30 min at 0°C. After precooling to -78°C, the cis-aldehyde (22A)
(50 mg, 0.27 mmol) in THF (0.3 ml) was added dropwise to the orange-
red solution. After stirring for 5 min at -78°C and subsequent
warming to room temperature over 30 min, the mixture was poured into
saturated aqueous ammonium chloride solution (10 ml) and diethyl
ether (50 ml). The aqueous layer was extracted with diethyl ether
(2 x 20 ml) and the combined organic extracts washed with water
1 x 20 ml) and dried (Na2S0tf). Concentration under reduced pressure,
followed by chromatography (30% diethyl ether-petrol) gave methyl cis
6-[(Z)-but-l-enyl]tetrahydropyran-2-ylacetate (27) (32 mg, 56%) as a
colourless oil; vmax (CHC13) 2930, 2860, 1730, 1640, 1200, and 1050
cm"1; 6 (250 MHz) 0.98 (3H, t, J 8 Hz, CH2CH2), 1.15 - 1.90 (6H, m),
2.08 (2H, m, CH=CH-Ctf2), 2.40 (1H, dd, J 15 and 6 Hz, Ctf2C02Me), 2.59
(1H, dd, J 15 and 7 Hz, Ctf2C02Me), 3.67 (3H, s, OMe), 3.83 (1H, m, 2-H),
4.14 (1H, m, 6-H), 5.26 (1H, ddt, J 11, 8 and 1 Hz, l'-H), and 5.39
(1H, ddt, J 11, 7, and 1 Hz, 2'-H); m/z 181 (M+-0Me) and 55 (CH=CH-Et)+.

225
13. Preparation of Methyl cis-6-[(E,Z)-but-l-enylltetrah.ydropyran-2-
ylacetate (27) and (28)
C2H5CH = PPh3
+
O H'CHO C 0 2Me
(22A)(27) and (28)
To a stirred suspension of the phosphonium salt (260 mg, 0.60 mmol
in dry THF (2 ml) at 0°C was added nBuLi (0.46 ml of a 1.30M solution
in hexane, 0.60 mmol) dropwise. After stirring for 30 min, the orange
red solution was precooled to -40°C and the cfs-aldehyde (22A) (93 mg,
0.50 mmol) in dry THF (0.5 ml) was added dropwise. After 90 min at
-40°C, dry methanol (2.5 ml) was slowly added and the solution main
tained between -45 and -40°C for 4 h. Water (1 ml) was then added
and the reaction allowed to warm to room temperature. After 1 h, the
mixture was poured into diethyl ether (15 ml) and the organic layer
was washed with brine (1 x 5 ml), dried (Na2S04) and concentrated under
reduced pressure to afford the crude adduct as a yellow oil.
Purification by chromatography (30% diethyl ether-petrol) afforded
methyl cis-6-[(2J£')-but-l-enyl]tetrahydropyran-2-ylacetate (27) and
(28) (22:78; Z:E) (56 mg, 53%) as a colourless oil, v (CHC13) 2930,
2860, 1730, 1600, 1200, and 1030 cm"1; tf-isomer: 6 (250 MHz) 0.99 (3H,
t, J 8 Hz, CH3), 1.15 - 1.92 (6H, m), 2.14 (2H, m, Ctf2-CH=CH), 2.42
(1H, dd, J 15 and 6 Hz, C#2C02Me), 2.64 (1H, dd, J 15 and 7 Hz,
C#2C02Me), 3.68 (3H, s, OMe), 3.82 (2H, m, 2-H and 6-H), 5.45 (1H, ddt,
J 15, 6, and 1 Hz, Ctf=CHEt), and 5.69 (1H, ddt, J 15, 6.5, and 1 Hz,

226
=CffEt); m / z 181 (M+-OMe) and 55 (CH=CHEt)+; Z-isomer; data as
reported previously.
14. Preparation of Methyl trares-6 -dimethoxymethyltetrahydropyran-2-
,yl acetate (30B)
K-10 MontmorilIonite clay (lg) was stirred with trimethylortho
formate (3 ml) for 5 min and then filtered. The resultant wet filter-
cake was used directly in the reaction. To the t r a n s - aldehyde (22B)
( 2 0 0 mg, 1.08 mmol) in hexane-diethyl ether (1 :1 , 1 0 ml) was added a
small portion of the filter-cake prepared. After rapid stirring for
30 min at room temperature, the product was isolated by filtration
through a small silica gel pad, followed by washing with 5% aqueous
sodium bicarbonate solution (5 ml) and water (5 ml), drying (Na2S04)
and concentration under reduced pressure to afford m e t h y l trans-6 -
d i m e t h o x y m e t h y l t e t r a h y d r o ' p y r a n - 2 - y l a c e t a t e (30B) (210 mg, 84%) as a
colourless oil, v (film) 2940, 2860, 1735, 1200, and 1080 cm"1;
6 (60 MHz) 1.35 - 2.00 (6 H, ml, 2.54 (2H, m, C#2C02Me), 3.35 (6 H, s.
CH(0Me)2), 3.70 (3H, s, C02Me), 3.70 - 4.30 (2H, m, 2-H and 6 -H), and
4.38 (1H, d, J 6 Hz, C£(0Me)2): m / z 201 (M+-0Me), 169 (M+-0Me-Me0H),159 (M+-CH2C02Me), 157 (M+-CH(0Me)2), 127 (M+-CH2C02Me-Me0H), 95 (M+-
CH2C02Me-2Me0H), and 75 (CH(0Me)2)+; (Found: M+-0Me, 201.1120.
C1 1 H2 0O5 requires M+-0Me, 201.1127); (Found: C, 57.06; H, 8.93.
C1 1 H2 0O5 requires C, 56.88; H, 8 .6 8%).

227
15. Preparation of 2-(trans-6-Dimethoxymethyltetrahydropyran-2-yl)-
ethanol (31B)
The dimethyl acetal (30B) (210 mg, 0.91 mmol) in dry diethyl ether
(2 ml) was added dropwise to a stirred slurry of lithium aluminium
hydride (27 mg, 0.71 mmol) at 0°C. The solution was allowed to warm
to room temperature and stirred for a further 20 min. After recool
ing to 0°C, water (1 ml) was added dropwise, followed by saturated
aqueous ammonium chloride solution (5 ml) and diethyl ether (15 ml).
Extraction of the aqueous layer with diethyl ether (3 x 10 ml),
washing of the organic extracts with brine (1 x 10 ml), drying ^Na2S0^)
and concentration under reduced pressure, gave a colourless oil which
was purified by chromatography (diethyl ether) to afford 2-(trans-6-
d - i m e t h o x y m e t h y l t e t r a h y d r o p y r a n - Z - y l ) e t h a n o l (31B) (180 mg, 97%) as a colourless oil, v (film) 3500, 2940, 2860, 1200, and 1080 cm-1;
6 (60 MHz) 1.15 - 2.20 (8H, m), 3.38 (6H, s, Ctf(0Me)2), 3.70 - 3.90
(4H. m, 1-H2, 21 -H and 6'-H), 4.48 (1H, d, J 7 Hz, Ctf(0Me)2), and 6.00
(1H, br s, OH); m / z 204 (M+ j, 189 (M+-Me), 173 ^M+-0Me), ana 141 (M+- MeOH-OMe); (Found: C, 58.61; H, 10.17. C10H2oO«f requires C, 58.80;
H, 9.87%).

228
16. Preparation of the dimer (32)
10% Aqueous oxalic acid (4 drops) was added with continuous
magnetic stirring to a suspension of silica gel (600 mg) in dichloro-
methane (2 ml). After 2-3 min, the water phase had disappeared and
the acetal (31B) (150 mg, 0.74 mmol) was added at room temperature.i
After 4 h, diethyl ether (10 ml) was added and the suspension filtered
through a small pad of silica gel to afford the dimer (32) as a mixture
of diastereoisomers (90 mg. 60%) as a colourless oil, v (film) 293.0,
2880, 1120, 1065, 1025, and 975 cm”1; 5 (250 MHz) 1.20 - 2.05 (16H, m),
3.40 (6H, s, -OMe. (several close singlets)), 3.45 - 4.03 (8H, m), 4.18
(0.30H, d, J 6.6 Hz, CtfOMe), 4.35 (0.35H, d, J 6.5 Hz, CtfOMe), 4.38
(0.35H, d, J 6.5 Hz, CffOMe), and 4.58 (1H, m, CffOMe); m/z (FAB) 363
(M+ + H + H20), 313 (M+-0Me), and 311(M+-Me0H-H); (Found: C, 62.39;
H, 9.74. C18H3206 requires C, 62.77; H, 9.36%).
17. Preparation of trares-2-(2-t-Butyldimethylsilyloxyethyl1-6-di
me t hoxyme t hy 1tetrahydropyran (33)

229
A mixture of the alcohol (190 mg, 0.93 mmol), triethylamine
(0.16 ml, 1.12 mmol), t-butyldimethyl silyl chloride (154 mg, 1.02 mmol)
and DMAP (a few crystals) in dry dichioromethane (10 ml) was stirred
at room temperature for 1 h. After dilution with water (10 ml) and
dichioromethane (30 ml) the aqueous layer was extracted with further
dichioromethane (2 x 20 ml) and the combined organic extracts washed
with brine (1 * 20 ml ). Drying (Na2S0I+) and concentration underv
reduced pressure followed by chromatography (10% diethyl ether-petrol
afforded tvans-2-(2-t-butyldimethyl silyloxyethyl)-6-dimethoxymethyl-
tetrahydropyran (33) (228 mg, 77%) as a colourless oil, v (film)
2933, 2857. 1094, and 836 cm"1; 6 (90 MHz) 0.00 (6H, s, SiMe2), 0.87
(9H, s, tBu), 1.20 - 2.14 (8H, m), 3.38 (6H, s, CH(0 Me) 2) , 3.55 - 3.80
(3H, m, C7/20TBDMS and 2-H), 3.95 (1H, m, 6-H), and 4.32 (1H, d, J 6.5
Hz, C#(0Me)2); m/z 287 (M+-0Me), 261 (M+-tBu), 243 (M+-CH(0Me)2), 229
(M+-tBu-Me0H), 198 (M+-tBu-0Me-Me0H), and 155 (M+-0TBDMS-Me0H).
18. Preparation of Methyl cfs-6-(l,3-dithiolan-2-,yl )tetrah.ydropyran-
2-ylacetate (35A)
To the cis-aldehyde (22A) (180 mg, 0.97 mmol) in dry dichloro-
methane (5 ml) at room temperature under argon was added redistilled
l,2-ethanedithiol (0.12 ml, 1.45 mmol) followed by boron trifluoride

230
etherate (24 pil, 0.20 mmol). After stirring at room temperature
overnight, 5% aqueous sodium hydroxide solution (1.5 ml) was added
and the organic layer diluted with dichioromethane (10 ml) and washed
with water (5 ml), dried (Na2S0lt) and concentrated under reduced
pressure. Column chromatography (35% diethyl ether-petrol) gave pure
methyl c£s-6-(l,3-dithiolan-2-yl)tetrahydropyran-2-ylacetate (35A)
(231 mg, 91%) as a pale yellow oil, v (film) 2950, 2863, 1730, 1195,max.
and 1070 cm-1; 6 (60 MHz) 1.25 - 2.00 (6H, m), 2.40 - 2.88 (2H, m,
C#2CO2Me}, 3.18 (4H, s, S(CH2)2S), 3.28 - 3.58 (1H, m, 6-H), 3.68 (3H,
s, C02Me), 3.78 (1H, m, 2-H), and 4.38 (1H, d, J 7.5 Hz, C#(SCH2CH2S));
m/z 262 (M+ ), 157 (M+-CH(SCH2CH2S)), 125 (M+-CH(SCH2CH2S)-MeOH), 97 (M+-
CH(SCH2CH2S)-MeC02H ), 83 (M+-CH(SCH2CH2S)-CH3C02Me), and 61 (HSCH2CH2S)+ .
19. Preparation 2-{ c - i s-6-( 1,3-Dithiolan-2-yl )tetrah,ydrop.yran-2-y1 )-
ethanol (36A)
Following the procedure of Experiment 15, the ester (35A)(210 mg,
0.91 mmol) was reduced with lithium aluminium hydride and after work-up
and chromatography (60% diethyl ether-petrol) afforded 2-(cis-6-(l,3-
dith.i-o'lan~2-yZ)tetvahydvo'pyvan-2-yZ) ethanol (36A) (167 mg, 89%) as a
colourless oil, v (film) 3620, 3480, 2938, 2880, and 1040 cm"1:
6 (60 MHz) 1.10 - 2.10 (8H, m) 2.85 - 4.00 (5H, m, 1-H2, 2'-H, 6 1-H,
and OH), 3.18 (4H, s, S(CH2)2S), and 4.32 (1H, d, J 7.5 Hz, Ctf(SCH2CH2S))
m/z 234 (M+ ), 129 (M+-CH(SCH2CH2S)), 111 (M+-H20-CH(SCH2CH2S)),

231
105 (CH(SCH2CH2S))+ , 93 (HSCH2CH2S)+ ; (Found: C, 51.38; H, 7.94;
S, 27.58. C10H1802S2 requires C, 51.25; H, 7.74; S, 27.36%).
20. Preparation of gis-2-(2-t-Butylaimethylsilyloxyethyl)-6-(1,3-
dithiolan-2-yl)tetrahydropyran (37A)
Following the procedure of Experiment 17, the alcohol (36A) (264 mg,
1.13 mmol) was protected as the TBDMS ether. Chromatography (10% di
ethyl ether-petrol) afforded z \ s - 2 - ( 2 - t - b u t y t d i m e t ' h y l s i t y Z o x y e t h y ' l ) - § -
{ 1 , 2 > - d i t h i o l a n - 2 - y l ) t e t v a h y d r o p y r a n (37A) (361 mg, 92%) as a colourless
oil, vmax (film) 2940, 2870, 1260, 1075, 840, and 660 cm-1; 6 (60 MHz)
0.00 (6H, s, SiMe2 ), 0.90 (9H, s, tBu), 1.15 - 2.00 (8H, m), 3.10 (4H,
s, S(CH2)2S), 3.25 - 3.85 (4H, m, 2-H, 6-H, and C7/20TBDMS), and 4.30
(1H, d, J 8 Hz, Cff(S(CH2)2S)); m / z 291 (M+-tBu), 243 (M+-CH(SCH2CH2S)), 171 (M+-tBu-Me-CH(SCH2CH2S)), 131 (0TBDMS)+ , and 105 (CH(SCH2CH2S)+ );
(Found: C, 55.27; H, 9.53; S, 18.60. C16H3202S2Si requires
C, 55.12; H, 9.25; S, 18.39%).

232
21. Preparation of c^s-6-(2-t-But.yldimethylsil.yloxyethyl)-2-tetra-
hydrop.yrancarbaldehyde (38A).
To the dithioacetal (37A) (670 mg, 1.93 mmol) in acetonitrile
(30 ml) and water (1 ml) was added solid sodium carbonate (4g, c a .
20 eq) and methyl iodide (1.27 ml, 19.3 mmol). The mixture was stirred
rapidly at room temperature until t.l.c. indicated that the reaction
was complete { c a . 72 h) and then diluted with dichloromethane (50 ml)
and water (20 ml). The aqueous layer was extracted with further
dichioromethane (3 * 30 ml) and the combined organic extracts washed
with water (1 x 20 ml) and brine (1 x 20 ml). Drying (Na2S0,J and
concentration under reduced pressure, followed by chromatography (10%
diethyl ether-petrol) gave c \ s - ^ > - { Z - t - b u t y l d i m e t h y Z s ' L Z y l o x y e t h y l ) - Z -
t e t r a h y d r o p y r a n c a r b a Z d e h y d e (38A) (318 mg, 61%) as a colourless oil,
vmax 2960’ 2870' 1740’ 1260’ 1080’ and 830 cm"1: $(250 MHz)
0.02 (6H. s, SiMe2), 0.85 (9H, s, tBu), 1.10 - 1.96 (8H, m), 3.52 (1H,
m, 6-H), 3.62 - 3.84 (3H, m, C7/20TRDMS and 2-H), and 9.58 (1H, s, CH0);
m / z 257 (M+-Me), 243 (M+-CH0), and 215 (M+-tBu); (Found: C, 61.68;
H, 10.57. Cli+H280^Si requires C, 61.72; H, 10.36%).

233
22. Preparation of trans-2-(2-t-Butyldimethyl si1,y1oxyethyl)-6-(1,3-
dithiolan-2-yl)tetrahydropyran (37B)
C02Me
(22B)
Following the procedures of Experiments 18-20, the trans-olefin
(22B) (500 mg, 2.69 mmol) was converted to t r & x \ s - 2 - ( 2 - t - b u t y l d i m e t h y l
s % l y l o x y e t h y l ) - § - ( l , 2 - d i t h i o l a n - 2 - y l ) t e t v a h y d v o p y v a n (37B) (702 mg,
75% overall yield) which was a colourless oil, v (film) 2940, 2860,
1255, 1080, and 840 cm"1; 6 (250 MHz) 0.05 (6H, s, SiMe2), 0.88 (9H,
s, tBu), 1.30 - 2.05 (8H, m), 3.10 - 3.23 (4H, m, S(CH2)2S), 3.52 (1H,
ddd, J 8.5, 8, and 3.5 Hz, 6-H), 3.73 (2H, m, C7/20TBDMS), 3.94 (1H,
m, 2-H), and 4.68 (1H, d, J 8.5 Hz, Ctf(SCH2CH2S)); m/z 291 (M+-tBu),
243 (M+-CH(SCH2CH2S)). 171 (M+-tBu-Me-CH(SCH2CH2S)), 131 (0TBDMS+ ),
and 105 (CH(SCH2CH2S)+ ); (Found: C, 54.96; H, 9.38; S, 18.18.
Ci6H3202S2Si C, 55.12; H, 9.25; S, 18.39%).
23. Preparation of trans-6-(2-t-Butyldimethyl si 1yloxyethyl)-2-tetra-
hydropyrancarbaldehvde (38B1

234
To the dithioacetal (37B) (500 mg, 1.44 mmol) in acetonitrile
(25 ml) and water (1 ml) was added solid sodium carbonate (4g) and
methyl iodide (1.10 ml, 16.7 mmol). After stirring at room temperature
for ca. 72 h the reaction was worked-up following the procedure in
Experiment 21. Chromatography (10% diethyl ether-petrol) afforded
trans-6- (2-t-butyldimethyls'ilyloxyethyl)-2-tetrahydropyrancarbaldehyde
(3SB) (211 mg, 54%) as a colourless oil, v (film) 2960, 2870, 1745,max.
1260, 1080, and 820 cm"1; 6 (250 MHz) 0.00 (6H, s, SiMe2), 0.88 (9H,
s, tBu), 1.10 - 2.03 (8H, m), 3.74 (3H, m, Ctf20TBDMS and 6-H), 4.13
(1H, m, 2-H), and 9.83 (1H, d, J ^ 1 Hz, CHO); m/z 257 (M+-Me), 243
(M+-CH0), and 215 (M+-tBu); (Found: C, 61.48; H, 10.49.
Cii+H2 803Si requires C, 61.72; H, 10.36%).
24. Preparation of cfs-6-(l,3-Dithiolan-2-yl)-2-tetrahydropyran-
acetaldehyde (39A)
To oxalyl chloride (0.24 ml, 2.74 mmol) in dry dichioromethane
(4 ml) was added DMS0 (0.39 ml, 5.48 mmol) dropwise at -60°C. After
stirring for 10 min at this temperature, the alcohol (36A) (310 mg,
1.37 mmol) in dich1oromethane (10 ml) was added dropwise maintaining
the temperature at ca. -60°C. After 15 min, triethyl amine (1.43 ml,
10.28 mmol) was added rapidly and the solution allowed to warm to room
temperature over 45 min. Water (5 ml) and dichloromethane (20 ml)
were added and the aqueous layer extracted with further dichloromethane

235
(2 x 10 ml). The combined organic extracts were washed with water
(1 x 10 ml) and brine (1 x 10 ml), before drying and removal of the
solvent under reduced pressure. Chromatography (55% diethyl ether-
petrol) afforded the aldehyde (39A)(226 mg, 74%) as an off-white
solid, v (film) 2938, 2851, 1718, 1198, and 1071 cm"1;
6 (90 MHz) 1.10 - 2.00 (6H, m), 2.55 (2H, m, C#2CH0), 3.00 - 3.50
(1H, m, 6-H), 3.20 (4H, s, SCH2CH2$), 3.90 (1H, m, 2-H), 4.38 (1H,
d, J 7.5 Hz, C#(SCH2CH2S)), and 9.80 (1H, t, J 1 Hz, CHO); m/z 233
(MH+ ), 232 (M+ ), 204 (MH+-CH0), 127 iM+-CH(SCH2CH2S)), and 105
(CH(SCH2CH2S)+ ).
25. Preparation of (ff)-Eth,yl-4-(gfs-6-(l,3-dithiolan-2-y1)tetrahydro-
pyran-2-yl)but-2-enoate (4QA)
To the aldehyde (39A) (220 mg, 0.95 mmol) in dichioromethane
(10 ml) at room temperature was added carbethoxymethylidenetriphenyl-
1 2 2phosphorane (364 mg, 1.05 mmol) in dichioromethane (5 ml) and the
resulting mixture stirred for oa. 5 h. The solution was then concen
trated under reduced pressure and chromatographed (50% diethyl ether-
petrol) to afford (E)- e t h y l - 4 - {cis-6-{l d - i t h b o l a n - 2-y l ) t e t r a h y d r o -
p y r a n - 2 - y l ) b u t - 2 - e n o a t e (40A) (272 mg, 95%) as a colourless oil,
v v (f^m) 2940, 2880, 1718, 1658, and 1040 cm"1; 6 (250 MHz) 1.28
(3H, t, J 7 Hz, CH2Ctf3), 1.45 - 1.90 (6H, m). 2.26 - 2.52 (2H, m, 4-H2),

236
3.18 (4H, m, SCH2CH2S), 3.32 (1H, m, ddd, J 11, 7.5, and 2 Hz, 6'-H),
3.47 (1H, m, 2' -H), 4.19 (2H, q, J 7 Hz, C02C/72CH3). 4.41 (1H, d,
J 7.5 Hz, C#(SCH2CH2S)), 5.90 (1H, dt, J 15.5 and 1.5 Hz, 2-H), and
6.99 (1H, dt, J 15.5 and 7 Hz, 3-H); m/z 302 (M+ ), 243 (M+-C02Et),
197 (M+-CH(SCH2CH2S)), and 105 (CH(SCH2CH2S))+ ; (Found: C, 55.39;
H, 7.41. C1I+H2203S2 requires C, 55.60; H, 7.33%).
26. Preparation of (ff)-4-(gfs-6-(l,3-Dithiolan-2-yl)tetrahydropyran-
2-yl)but-2-en-l-ol (41A)
To a stirred solution of the ester (40A) (196 mg, 0.65 mmol) in
dry toluene (3 ml) at -78°C was added DIBAL (1.0 ml of a 1.50M solu
tion in toluene, 1.50 mmol) dropwise. After one hour, t.l.c. indi
cated complete reduction and the reaction was quenched at -78°C by
dropwise addition of water (0.5 ml). After warming to room tempera
ture the reaction was diluted with ethyl acetate (20 ml). Solid
sodium bicarbonate (2g) was added and the slurry shaken vigorously
and then allowed to stand for 10 min. The supernatant liquid was
filtered through a short pad of silica gel and the remaining solid
extracted several times with further portions of ethyl acetate
(5 x 20 ml), these extracts being filtered through the same silica
gel pad. The combined organic extracts werp concentrated under
reduced pressure and the residue chromatographed (75% diethyl

237
ether-petrol) to afford (E)-4-(cis-6-(l, 2 - d i t h i o l a . n - 2 . - y l ) t e t v a h y d r o p y r a n -
2 - y l ) b u t - 2 - e n - l - o l (41A) (160 mg, 95%) as a colourless oil, v (film)
3390, 2931, 2854, 1668, 1194, 1083, and 974 cm"1; 6 (90 MHz) 1.10 - 2.05
(7H, m), 2.25 (2H, m, 4-H2), 3.10 - 3.50 (2H, m, 2‘-H and 6'-H), 3.20
(4H, s, SCH2CH2S). 4.10 (2H, br d, J 2.5 Hz, Ctf20H), 4.40 (1H, d, J 7.5
Hz, Ctf(SCH2CH2S)), and 5.70 (2H, m, CH=CH); m/z 260 (M+ ), 242 (M+-H20),
189 (|v|+-CH2CH=CHCH20H), 155 (Mf-CH(SCH2CH2S)), and 105 (CH(SCH2CH2S)) + ;
(Found: C, 55.10; H, 7.66; S, 24.41. C12H2002S2 requires C, 55.35;
H, 7.74; S, 24.62%).
27. Preparation of (ff )-c^s-2 -(4-* t-Butyl dimethyl si 1yloxybut-2-enyl)-6-
(1,3-dithiolan-2-yl)tetrahydropyran (42A)
4
A mixture of the alcohol (41A) (140 mg, 0.54 mmol), triethyl amine
(90 pi, 0.65 mmol), t e r t -butyl dimethyl silyl chloride (90 mg, 0.59 mmol)
and DMAP (a few crystals) in dry dichioromethane (6 ml) was stirred at
room temperature f o r l h. Work-up as described previously (Experi
ment 17) and chromatography (15% diethyl ether-petrol) afforded (E)-cis-
2- { i\ - t - b u t y Z d ' i m e t h y Z s ' Z Z y Z o x y b u t - 2 - e n y Z ) -6 - (1, 2 - d - i t h i - o Z a n ~ 2 - y Z 1t e t v a ~
h y d r o p y v a n (42A) (187 mg, 93%) as a colourless oil, v (film) 2930,max.
2855, 1254, 1098, 1050, 973. and 836 cm"1; 6 (90 MHz) 0.05 (6H, s,
SiMe2), 0.90 (9H. s, tBu), 1.10 - 2.00 (6H, m), 2.20 (2H, m, l'-H2),
3.10 - 3.50 (2H, m, 2-H and 6-H), 3.20 (4H, s, SCH2CH2S), 4.11 (2H,

238
br d, CE,OTBDMS), 4.40 (1H, d, J 7.5 Hz, C#(SCH2CH2S)), and 5.64 (2H,
m, CH=CH); m/z 374 (M+ ), 359 (M+-Me), 317 (M+-tBu), 269 (M+-CH(SCH2CH2S)),
and 105 (CH(SCH2CH2S) + ); (Found: C, 57.76; H, 9.42. C18H3^ O ^ S i
requires C, 57.70; H, 9.15%).
28. Preparation of (E) - c i s - 2 -(4-Benzyloxybut-2-enyl )-6-(l,3-dithio1an-
2-y1jtetrahydropyran (43A)
4
To sodium hydride (22 mg, 50% dispersion in oil), prewashed with
sodium-dried 30-40° b.p. petrol (2 x 2 ml) in dry THF (2 ml) at room
temperature was added the allylic alcohol (41A) (100 mg, 0.39 mmol)
in THF (2 ml) dropwise over 5 min. The resulting orange solution was
stirred at room temperature for 2 h before addition of freshly distilled
benzyl bromide (70 pi, 0.59 mmol). After 1 h, the reaction mixture
was cooled to 0°C and quenched by dropwise addition of water (0.5 ml).
The solution was diluted with diethyl ether (20 ml) and water (5 ml),
the aqueous layer extracted with diethyl ether (2 * 10 ml) and the
combined organic extracts washed with water (1 x 5 ml) and brine
( 1 x 5 ml). Drying (Na2S0lf) and concentrateon under reduced pressure.
followed by chromatography (45% diethyl ether-petrol) afforded the
benzyl ether (43A) (104 mg, 77%) as a colourless oil, v (film) 3027,max.
2929, 2850, 1668, 1071, 974, and 737 cm-1; 6 (90 MHz) 1.10 - 1.95
(6H, m), 2.15 - 2.35 (2H, m, l'-H2), 3.20 - 3.55 (2H, m, 2-H and 6-H),

239
3.20 (4H, s, SCH2CH2S), 4.00 (2H, d, J 4.5 Hz, Ctf20Bz), 4.40 (1H, d,
J 7.5 Hz, CH(SCH2CH2S)), 4.52 (2H. s, 0C#2Ph), 5.70 (2H, m, CH=CH), and
7.28 - 7.42 (5H, m, Ph); m/z 350 (M+ ), 259 (M+-CH2Ph), 245 (M+-
CH(SCH2CH2S)), 189 (M+-CH2CH=CHCH20CH2Ph), 105 (CH(SCH2CH2S)+ ), and
91 (CH2Ph+ ); (Found: M+-CH(SCH2CH2S), 245.1540. C lgH2602S2 requires
M+-CH(SCH2CH2S), 245.1546).
29. Preparation of (ff)-cds-2-(2-t-Butyldimeth.y1si1y'loxyethyl )-6-f3-
(tetrahydrofuran-2-yl)prop-l-en.y1]tetrahydropyran (46A)
To the sulphone (17) (77 mg, 0.32 mmol) in dry THF (2 ml) at -78°C
was added nBuLi (0.24 ml of 1.35M solution in hexane, 0.32 mmol) dropwise
over 5 min. After stirring for 10 min at this temperature the ois-
aldehyde (38A) (79 mg, 0.29 mmol) in dry THF (0.5 ml) was added drop-
wise and the mixture stirred for a further 15 min before addition of
freshly distilled benzoyl chloride (67 pi, 0.58 mmol) in one portion.
The reaction was then allowed to warm to room temperature over 2 h
followed by addition of 3-N,N-dimethy1ami nopropyl amine (73 pi, 0.58 mmol).

240
After dilution with diethyl ether (20 ml) and water (10 ml), the aqueous
layer was extracted with diethyl ether (3 x 10 ml) and the combined
organic extracts washed with water (1 x 5 ml) and brine (1 x 5 ml).
Drying (Na2S0t+) and concentration under reduced pressure gave a yellow
oil which after chromatography (gradient elution, 10 — 50% diethyl
ether-petrol) afforded a diastereoisomeric mixture of benzoyloxy-
sulphones (44A) (107 mg, 60%) and a slightly more polar product corres
ponding to a diastereoisomeric mixture of hydroxy-sulphones (45A) (25
mg, 17%); (44A) : \> (film) 2940, 2860, 1720, 1270. 1150, 1075, andmax.
830 cm-1; 6 (60 MHz) 0.05 (6H, s, (several close singlets), SiMe2),
0.95 (9H. s (several close singlets), ^Bu), 1.10 - 2.15 (14H, m),
3.00 - 4.00 (8H, m), 5.30 (1H, m, CtfOCOPh), and 7.35 - 8.00 (10H, m, Ph)
m / z 455 (MH+-C0Ph-tBu), 353 (M+-PhC02H-S02Ph), 296 (M+-tBu-PhC02H-S02Ph)
145 (CH20TBDMS+ ), 131 (0TBDMS+ ), 105 (PhC0+ ), and 71 (C,H70+ ); (45A):
v (film) 3540, 3350, 2940, 2860, 1260, 1150, 1085, and 840 cm"1; max.
6 (250 MHz) 0.05 (6H, s (several close singlets), SiMe2), 0.88 (9H,
s (several close singlets), ^Bu), 1.00 - 2.12 (14H, m), 3.15 - 4.05
(10H, m), 7.34 - 7.80 (3H, m, Ph), and 7.92 (2H, m, Ph); m / z 455 (M+- tBu), 352 (M+-H20-PhS02H), and 71 (C^H70+ ).

241
(b)
4
To the diastereoisomeric mixture of benzoyloxy-sulphones (44A)
(119 mg, 0.19 mmole) in dry THF (1 ml) and dry MeOH (0.25 ml) was
added solid Na2HP0t, as a buffer (250 mg, c a - 10 eq). After cooling
to -20°C, freshly-ground 6% sodium amalgam (0.2g) was added and the
mixture stirred rapidly for 1 h. Further small portions of the amalgam
were added periodically until t.l.c. indicated no remaining starting
material. The reaction was diluted with 50% diethyl ether-petrol
(20 ml) and washed with water (2 * 5 ml). The aqueous layer was
extracted with diethyl ether (2 * 5 ml) and the combined organic
extracts washed once with water (10 ml) and dried (Na2S0tt). Evapora
tion of the solvent under reduced pressure followed by chromatography
(25% diethyl ether-petro 1 ) afforded ( 2 ) - z \ s - 2 - { 2 - t - b u t y l d i m e t h y I s i l y l -
o x y e t h y l ) - 6 - {3-(t e t r a h y d r o f u r a n - 2 - y l ) p r o p ~ l ~ e n y l } t e t r a h y d r o p y r a n (46A) (43 mg, 65%) and the corresponding (Z)-isomer (8 mg, 12%) and variable
quantities of the vinyl sulphone (47A) (0-15%) all as colourless oils,

242
olefin (46A): v (film) 2933. 2857, 1603, 1093, 1040, and 834 cm-1; max.5 (250 MHz) 0.05 (6H, s, SiMe2/), 0.88 (9H, s, tBu), 1.16 - 2.00 (12H, m),
2.12 - 2.40 (2H, m, CH=CH-C/72). 3.45 - 4.02 (7H, m). 5.55 (1H, ddt,
J 15.5, 5.1, and 1 Hz, 1"-H), and 5.66 (1H, dt, J 15.5 and 6.5 Hz,
2"-H); m/z 354 (M+), 297 (M+-t3u), and 71 (C,H70+); (Found: C, 67.43;
H, 10.68. C20H3803Si requires C, 67.74; H, 10.80%).
Vinyl sulphones (47A): v v (film) 2950, 2870, 1260, 1150, 1075, andmax *835 cm-1; 6 (250 MHz) 0.00 (2.5H, s (two close singlets), SiMe2),
0.05 (3.5H, s (two close singlets), SiMe2), 0.82 (3.7H, s (two close
singlets), ^Bu), 0.87 (5.3H, s (two close singlets), ^Bu), 1.20 - 1.98
(14H. m), 3.25 - 4.22 (7H, m), 6.87 (0.58H, d, J 8 Hz, CH=), 6.89 (0.42H,
d, J 7.5 Hz, CH=), 7.45 - 7.64 (3H, m, Ph), and 7.82 - 7.88 (2H, m, Ph);
m/z 437 (M+-tBu), 353 (M+-S02Ph), 297 (MH+-S02Ph-tBu), and 71 (C,H70+).
30. Preparation of (ff,Z)-trans-2-(2-t-Butyldimethylsilyloxyethy1)-6-
[3-(tetrahydrofuran-2-yl)prop-l-eny1Itetrahydropyran (46B)
4
The trans-aldehyde (38B) (90 mg, 0.33 mmol) was converted to (E,Z)-
trans-2-(2-£-butyIdimethytsilyloxyethy£)-6-[3-(tetvahyarofuran~2-yl )-
prop-l-enyl]tetrahydropyran (46B) (57 mg, 49%) (ca. 85:15, E:Z mixture)
via the procedure described in Experiment 29, v (film) 2934, 2858,
1664, 1093, 1040, and 837 cm'1; 6 (250 MHz) 0.04 (0.9H, s, SiMe2,
(Z)-isomer), 0.07 (5.1H, s, SiMe2, (E)-isomer), 0.86 (1.35H, s, tBu,

243
(Z)-isomer), 0.88 (7.65H, s, Bu, {E)-isomer), 1.30 - 2.00 (12H, m),
2.16 - 2.42 (2H, m, CH=CH-Ctf2), 3.64 - 3.92 (6H, m), 4.28 (0.85H. br s,
6-H, {E )-i sorrier), 4.53 (0.15H, br s, 6-H, (Z)-isomer), and 5.62 (2H. m,
CH=CH); m/z 354 (M+), 297 (M+-tBu), 209 (M+-CH2CH20TBDMS), 187, and
71 (CuH;0+); (Found: C, 67.69: H, 10.92. C20H3803Si requires
C, 67.74; H, 10.80%).
31. Preparation of {E,Z)-trans-2-(But-l-eny 1 )-6-(2-t-butyldimeth,yl-
si1yloxyethyl)tetrahydropyran (48B)
4
To a stirred suspension of propy1triphenylphosphonium iodide
(130 mg, 0.30 mmol) in dry THF (3 ml) at 0°C was added nBuLi (0.23 ml
of a 1.18M solution in hexane, 0.27 mmol) dropwise. After stirring for
30 min, the orange-red solution was precooled to -78°C before dropwise
addition of the trans-aldehyde (38B) (73 mg, 0.27 mmol) in THF (0.3 ml).
The orange-red colour gradually disappeared and after 5 min, the solu
tion was allowed to warm to -40°C over 10 min, then recooled to -78°C
before dropwise addition of nBuLi (0.23 ml of a 1.18M solution in
hexane,0.27 mmol). After a further 5 min at -78°C, potassium

244
t-butoxide (52 mg, 0.41 mmol) and t-butanol (39 pi, 0.41 mmol) were
added followed by aqueous hydrochloric acid (0.27 ml of a IN solution,
0.27 mmol) and the resulting solution stirred at ca. -45°C for 4 h.
The reaction was then allowed to warm to room temperature over 1 h and
quenched with saturated aqueous ammonium chloride solution (1 ml).
Dilution with water (5 ml) and diethyl ether (20 ml), extraction of
the aqueous layer with ether (3 x 10 ml), washing of the combined organic
extracts with water (1 x 10 ml), drying (Na2S01+) and concentration under
reduced pressure gave the crude product as a yellow oil. Purification
by chromatography (10% diethyl ether-petrol) afforded(E,Z)-trans-2-(&wt-
l-enyl)-§-(-2-t-butyldimethylsilyloxyethyl)tetrahydropyvan (48B) (45 mg,
56%) (60:40, E:Z mixture, homogeneous by t.l.c.) as a colourless oil,
(film) 2950, 1600, 1260, 1080, and 840 cm"1; 6 (250 NHz) 0.04
(2.4H, s, SiMe2, (Z)-isomer), 0.05 (3.6H, s, SiMe2, (E)-isomer), 0.89
(3.6H, s, tBu, (Z)-isomer), 0.90 (5.4H, s, tBu, (El-isomer), 0.98 (1.2H,
t, J 7.5 Hz, Me, (Z)-isomer), 1.00 (1.8H, t, J 7.5 Hz, Me, (E)-isomer),
1.20 - 1.95 (8H, m), 2.00 - 2.18 (2H, m, CH=CH-CE2), 3.64 - 3.72 (2H. m,
CE20TBDMS), 3.88 (1H, m, 6-H), 4.24 (0.6H, m, 2-H, (E)-isomer), 4.54
(0.4H, m, 2-H, (z)-isomer), and 5.45 - 5.73 (2H, m, CH=CH, (EZ)-mixture);
m/z 241 (N+-tBu), 183 (M+-TBDMS), and 131 (0TBDMS+); (Found: C, 68.10;
H, 11.70. C17H3l+02Si requires C, 68.40; H, 11.48%).
32. Preparation of (E)-2-{gfs-6-f3-(Tetrahydrofuran-2-yl)prop-l-en,yl1-
tetrahydropyran-2-yllethanol (49A)

245
To the silyl ether (46A) (2.00g, 5.65 mmol) in acetonitrile (20 ml)
was added a 95:5 mixture of acetonitrile and 40% aqueous HF solution
(1 ml) at room temperature. After stirring for 10 min the reaction
was diluted with water (10 ml) and chloroform (50 ml). The aqueous
layer was extracted with chloroform (3 x 20 ml) and the combined organic
extracts washed with water (1 x 10 ml) and brine (1 x 10 ml). Drying
(Na2S0(+) and concentration under reduced pressure, followed by filtra
tion through a short silica gel pad afforded the alcohol (49A) (1.33g,
98%) as a colourless oil, v (film) 3435, 2934, 2858, 1650, and 1068
cm"1; 6 (250 MHz) 1.20 - 2.00 (12H, m), 2.21 (1H, m, CH=CHC£2), 2.28
(1H, m, CH=CHC7/2), 3.00 (1H, br s, OH), 3.58 - 3.90 (7H, m), 5.46 (1H,
ddt, J 15.5, 5.5, and 1 Hz, 1"-H), and 5.65 (1H, dt, J 15.5 and 6.5 Hz,
2"-H); m/z 240 (M+), 222 (M+-H20), 170 (MH+-C„H70), and 71 (C4H70+).
33. Preparation of (E£)-2-{trans-6-[3-(Tetrahydrofuran-2-yl)prop-l-eny 1-
tetrahydropyran-2-yljethanol (49B)
The silyl ether (46B) (2.00g, 5.65 mmol) was deprotected with HF :
acetonitrile as described in Experiment 32 to afford (E,Z)-2-{trans-6-
[3-(tetrahydrofuran-2-yl)prop-1-enyl]tetrahydropyran-2-yl}ethanol (49B )
(85:15, E:Z, homogeneous by t.l.c.) (1.30g, 96%) as a colourless oil,
v (film) 3438, 2936, 2867, 1663, and 1063 cm"1; 6 (60 MHz)max.1.20 - 2.10 (12H, m), 2.20 - 2.40 (2H, m, CH=CH-Cff2), 2.70 (1H, br s, OH),

246
3.50 - 4.20 (6H, m), 4.35 (1H, m, 6'-H), and 5.50 - 5.70 (2H, m, CH=CH);
m/z 240 (M+), 222 (M+-H2Q), 195 (M+-CH2CH20H), 170 (MH+-ClfH70), ana 71
(C^O"1"); (Found: C, 69.81; H, 10.38. C11+H2it03 requires C, 69.96;
H, 10.06%).
34. Preparation of (g)-o^s-6-{3-(Tetrahydrofuran-2-yl)prop-l-enyl}-2-
tetrahydropyranacetaldehyde (50A)
To oxalyl chloride (46 pi, 0.53 mmol) in dry dichioromethane
(1.5 ml) was added DMSO (78 pi, 1.10 mmol) dropwise at -60°C. After
stirring for 10 min at this temperature, the alcohol (49A) (84 mg,
0.35 mmol) in dichioromethane (2 ml) was added dropwise, maintaining
the temperature between -60 and -50°C. After 15 min, triethylamine
(0.36 ml, 2.60 mmol) was added rapidly and the solution allowed to
warm to room temperature over 45 min. Water (5 ml) and dichioromethane
(10 ml) were added and the aqueous layer extracted with further
dichioromethane (2 x 10 ml). The combined organic extracts were
washed with water (1 x 10 ml) and brine (lx 10 ml) before drying
(Na2S01+) and removal of the solvent under reduced pressure.
Chromatography (75% diethyl ether-petrol) afforded the aldehyde (50A)
(67 mg, 80%) as a colourless oil, v (film) 2941, 2863, 1721, 1198,UlaX •
1063, 974, and 751 cm"1; 6 (90 MHz) 1.00 - 2.00 (10H, m), 2.20 (2H,
m, CH=CH-CH2), 2.45 (2H, m, Ctf2CH0), 3.60 - 4.00 (5H, m, 2-H, 6'-H,
2"-H, and 5m-H2), 5.35 - 5.80 (2H, m, CH=CH), and 9.86 (1H, t, 0 2 Hz,

247
CHO); m/z 239 (MH+), 238 (M+), 210 (MH+-CH0), 125 (M+-CH2CHO-C4H70),
85 (C5H90+), and 71 (C„H70+).
35. Preparation of (£ ,Z)-frro?2s-6-{3-(Tetrahydrofuran-2-yl )prop-l-enyl }-
2-tetrahydropyranacetaldehyde (50B)
The alcohol (49B) (100 mg, 0.42 mmol) was oxidised by the Swern
procedure described in Experiment 34 to afford after flash chromatography
(75% diethyl ether-petrol) the aldehyde (50B) (80 mg, 80%) (85:15,
E\Zy homogeneous by t.l.c.) as a colourless oil, v (film) 2934,max •2858, 2727, 1727, 1198, 1069, and 973 cm-1; 6 (250 MHz) 1.20 - 2.00
(10H, m), 2.18 - 2.45 (2H, m, CH=CHCtf2), 2.40 - 2.75 (2H, m, Ctf2CH0),
3.68 - 3.97 (4H, m, 2-H, 2"-H, and 5"-H2), 4.17 (0.15H, m, 6-H, (Z)-
isomer), 4.34 (0.85H, m, 6-H, (£)-isomer), 5.50 - 5.84 (2H, m, CH=CH),
9.78 (0.15H, t, J 2 Hz, CHO, (Z)-isomer), and 9.79 (0.85H, t, J 2 Hz,
CHO. (E)-isomer); m/z 238 (M+), 210 (MH+-CH0), 195 (M+-CH2CH0), 167
(M+-C4H70), and 71 (CttH70+).
(50B)

248
36. Preparation of (ff,ff)-Ethyl-4-{cTs-6-r3-(tetrahydrofuran-2-yl )prop-
1-enyl 1tetrahydropyran-2-yl}but-2-enoate (51AT)
To the aldehyde (50A) (94 mg, 0.39 mmol) in dry dichioromethane
(2 ml) at room temperature was added carbethoxymethylidenetriphenyl -
phosphorane (150 mg, 0.43 mmol) in dry dichioromethane (2 ml) and the
resulting mixture was stirred for 5 h. The reaction solution was
concentrated under reduced pressure and chromatographed directly (35%
diethyl ether-petrol) to afford {£,£■)-ethyl-4-{c-\s-6-[3-tetrahydro furcra-
2-yl)prop-l-enyl']tetrahydropyran-Z-yl}but~2.-enoate (51 AT) (109 mg, 89%)
and the (Z,E) - i s o m e v (51AC) (5 mg, 4%) as colourless oils; more polar
(E,E)-isomer (51AT): v (film) 2935, 2857, 1718, 1654, 1267, 1180,max.1068, and 1041 cm"1; 6 (90 MHz) 1.28 (3H, t, J 7 Hz, CH2Ctf3), 1.20 -
2.05 (10H, m), 2.15 - 2.50 (4H, m, 4-H2 and 3M-H2), 3.30 - 4.00 (5H, m,
2'-H, 61-H, 2*"-H, and 5m -H2), 4.20 (2H, q, J 7 Hz, C02CH2), 5.40 - 5.87
(2H, m, ,CH=CH), 5.85 (1H, dt, J 16 and 1 Hz, =CtfC02Et), and 6.98 (1H,
dt, J 16 and 7.5 Hz, Ctf=CHC02Et); m/z 308 (M+), 263 (M+-0Et), 195 (M+-
CH2CH=CHC02Et), and 71 (C4H70+); (Found: C, 69.92; H, 9.33. C18H280,

249
requires C, 70.10; H, 9.15%).
Less polar (Z,E)-isomer (51AC): v (film) 2934, 2356, 1718, 1644.max.1181, 1069, and 1035 cm"1; 6 (250 MHz) 1.15 - 2.00 (10H, m), 1.26 (3H,
t, J 7 Hz, CH2CH3), 2.10 - 2.38 (2H, m, 3"-H2), 2.75 (1H, m, 4-H),
2.98 (1H, m, 4-H), 3.42 (1H, m, 2'-H), 3.62 - 3.90 (4H, m, 6'-H,
2m -H, and 5"'-H2), 4.14 (2H, q, J 7 Hz, C02CH2), 5.57 (1H, ddt, J 15.5,
5.5, and 1 Hz, 0CHCH=CH), 5.68 (1H, dt, J 15.5 and 6.5 Hz, 0CHCH=C£),
5.82 (1H, dt, J 11.5 and 2 Hz, CH=CtfC02Et), and 6.41 (1H, dt, J 11.5
and 7 Hz, C£=CHC02Et); m/z 308 (M+), 263 (M+-0Et), 195 (M+-
CH2CH=CHC02Et), and 71 (C„H70+); (Found: C, 69.90; H, 9.24.
Ci8H2804 requires C, 70.10; H, 9.15%).
37. Preparation of (IT,ff)-Ethyl-4-(trans-6-[3-(tetrahydrofuran-2-y1)-
prop-l-enyl ]tetrahydrop.yran-2-yl }but-2-enoate (51BT)
The {E,Z)-mixture of aldehydes (50B) (150 mg, 0.62 mmol) was homol
ogated as described in Experiment 36 to afford {E,E)-ethyl-4-{£rans-6-
[3-(tetrahydrofuran-2-yl)prop-l-enyl]tetrahydropyran-2-yl }but-2-enoate

250
(51BT) (155 mg, 79%) and the {E, Z)-isomer (52) (27 mg, 14%) as colour
less oils;
Less polar {E,E)~isomer (51BT): v (film) 2978, 2940, 2862, 1708,max.
and 1 Hz, 0CHCtf=CH), 5.68 (1H, dt, J 15.5 and 6.5 Hz, 0CHCH=C#CH2),
5.87 (1H, dt, J 16 and 1 Hz, CH=CtfC02Et), and 6.98 (1H, dt, J 16 and
7.5 Hz, C£=CHC02Et); m/z 308 (M+), 263 (M+-0Et), 195 (M+-CH2CH=CHC02Et),
and 71 (C,H70+).
More polar {E,Z)-isomer (52): v (film) 2938, 2867, 1708, 1654, 1035,max.and 981 cm"1; 6 (250 MHz) 1.28 (3H, t, J 7 Hz, CH2C//3), 1.45 - 2.00
(10H, m), 2.20 - 2.55 (4H, m, 4-H2 and 3"-H2), 3.73 (1H, m, 2'-H),
3.78 - 3.92 (3H, m, 2"'-H and 5"'-H2), 4.18 (2H, q, J 7 Hz, C02CH2),
4.30 (1H, br s, 61-H), 5.64 (2H, m, CH=CH), 5.87 (1H, td, J 16 and
1 Hz, CH=C#C02Et), and 6.87 (1H, dt (5 lines), J 16 and 7 Hz, Ctf=CHC02Et)
m/z 308 (M+), 263 (M+-0Et), 195 (M+-CH2CH=CHC02Et), and 71 (CuH70+).
38. Preparation of (E,E) and (Z.ff)-Ethyl-4-(trans-6-f~3-(tetrah,ydro-
furan-2-yl )prop-l-enyl]tetrahydropyran-2-y1)but-3-enoate (53)
1654, 1186, 1062, and 980 cm"1; 6' (250 MHz) 1.27 (3H, t, J 7.5 Hz,
CH2Ctf3), 1.20 - 2.00 (10H, m), 2.15 - 2.53 (4H, m, 4-H2 and 3M-H2),
3.46 (1H, m, 2'-H), 3.72 (1H, m, 6'-H), 3.70 - 3.92 (3H, m, 2"'-H and
5,m-H2), 4.18 (2H, q, J 7.5 Hz, C02CH2), 5.56 (1H, ddt, J 15.5, 5.5,

251
To dry diisopropylamine (0.22 ml, 1.58 mmol) under argon at -2G°C
was added nBuLi (1.20 ml of a 1.50M solution in hexane, 1.80 mmol)
dropwise. The resulting solution was allowed to warm to -10°C over
10 min. Ory THF (0.5 ml) was added to the white slurry and the solu
tion precooled to -30°C before transferring, via a gas-tight syringe,
an aliquot (0.19 ml of LDA solution, 0.16 mmol) to a 1 ml flask under
argon. The solution of LDA (0.16 mmol) was precooled to -78°C and
dry HMPA (0.10 ml, 0.58 mmol) was added, followed by the (£’,£’)-a,(3-
unsaturated ester (51BT) (45 mg, 0.15 mmole) in THF (0.5 ml). The
yellow solution was stirred at -78°C for 5 min before addition of
glacial acetic acid (0.2 ml) in dry THF (0.3 ml) followed by saturated
aqueous sodium bicarbonate solution (0.3 ml). The mixture was
diluted with diethyl ether (10 ml) and washed with saturated aqueous
sodium bicarbonate solution (2 * 2 ml), water (2 * 2 ml) and brine
(2 x 1 ml), before drying (Na2S0t+) and removal of the solvent under
reduced pressure. Filtration through a short silica gel pad gave the
deconjugated ester (53) (41 mg, 91%) (ca. 2:1 (E,E)- : (Z,E)-isomers,
homogeneous by t.l.c.) as a colourless oil, v (film) 2959, 2935,
2871, 1727, 1028, and 972 cm"1; 6 (250 MHz) 1.26 (2H, t, J 7.5 Hz,
CH2Ctf3, (£’,£’)-isomer), 1.27 (1H, t, J 7.5 Hz, CH2C#3, (Z,E)-isomer),
l. 30 - 2.00 (10H, m), 2.22 - 2.40 (2H, m, 3"-H), 3.05 (1.33H, d,
J 6.5 Hz, C/Z2C02Et, (Z?,2?)-isomer), 3.16 (0.67H, d, J 6.5 Hz,
CZZ2C02Et, ( Z,£)-isomer), 3.74 (1H, m, 2'-H), 3.70 - 3.92 (4H, m, 6'-H,
2 m -H, and 5m-H2), 4.13 (0.67H, q, J 7.5 Hz, C02C HZs (Z,£)-isomer),
4.15 (1.33H, q, J 7.5 Hz, C02CHz, (£,£)-isomer), 5.46 - 5.72 (3.33H,
m, CH=CH and Ctf=CHC02Et), and 5.80 (0.67H, ddt (12 lines), J 16, 6.5,
and 1 Hz, =C#C02Et, (E,E)-isomer); m/z 308 (M+), 263 (M+-0Et), 238
(MH+-C„H70), 220 (M+-CH3C02Et), and 71 (Ct+H70+); (Found: C, 70.24;
H, 9.24. Ci8H280i+ requires C, 70.10; H, 9.15%).

252
39. Preparation of (E J2 )-4-{cis -5-f3-fTetrahydrofuran-2-yl )prop-l-gnyl ~j-
tetrahydropyran-2-yl}but-2-snoic acid (54)
To the ester (51AT) (52 mg, 0.17 mmol) in THF (2 ml) was added
LiOH (1.72 ml of a 1M aqueous solution, 1.72 mmol) at room temperature.
The mixture was stirred rapidly for 8 h. The solution was then acidi
fied with 5% aqueous hydrochloric acid solution and after dilution
with diethyl ether (20 ml) and water (5 ml), the aqueous layer was
extracted several times with diethyl ether (3 x 10 ml). The combined
ethereal extracts were washed with 5% aqueous hydrochloric acid solution
and water (1 x 10 ml ) followed by drying (Na2S0i+) and concentration
under reduced pressure. The resulting yellow oil was passed through
a short silica gel pad (diethyl ether) to afford (E,E)-4-{cis-6-[3-
tetrahydrofuran-2-yt)prop-1-enyl]tetrahydropyran-2-yl}but-2-enoic acid
(54) (26 mg, 55%) as a colourless oil, v (film) 3426, 3080, 2935,
1697, 1654, 1070, and 978 cm"1; 6 (250 MHz) 1.20 - 2.00 (10H, m),
2.12 - 2.56 (4H, 4-H2 and 3"-H2), 3.49 (1H, m, 2‘-H) , 3.68 - 3.94 (4H,
m, 61 -H. 2 "‘-H, and 5m -H2), 5.50 (1H, ddt, J 15.5, 5.5, and 1 Hz,
0CHC#=CH), 5.68 (1H, dt, J 15.5 and 6.5 Hz, 0CHCH=C7/), 5.87 (1H, dt,
J 16 and 1.5 Hz, CH=CtfC02H), 7.09 (1H, m, (5 lines), J 16 and 7 Hz,
Ctf=CHC02H), and (C02H, diffuse); m/z 280 (M+), 195 (M+-CH2CH=CHC02H),
and 71 (C^HyO*); Found: C, 68.74; H, 8.93. C16H2<+01+ requires

253
C, 58.55; ri, 8.53%).
40. Preparation of Mono-Q-acetyl M1396Q3 sodium salt (55)
A mixture of M139603 Na+ salt"'’ (la) (l.OOg, 1.60 mmol) in acetic
anhydride (10 ml) and pyridine (10 ml) was stirred at room temperature
for 6 h. The residue obtained after evaporation under reduced
pressure was taken up in dichioromethane (50 ml) and washed with water
(1 x 10 ml) followed by 1M aqueous sodium bicarbonate solution
(1 x 10 ml ). After drying (Na2S0t+), the organic solution was
evaporated to afford the mono-O-acetyl M139603 sodium salt (55) as a white foam (l.OOg, 97%). Chromatography (diethyl ether) provided a
sample of microanalytical purity, v (CHC13) 2920, 1738, 1640, 1440,
1268, 1242, 1050, and 1002 cm'1; 5 (90 MHz) 0.65 (3H, d, J 6 Hz, Me),
0.80 - 2.50 (33H, m), 2.00 (1H. s, 0C0CH3), 2.07 (2H, s, 0C0CH3), 3.30
(3H, s, OMe), 3.18 - 4.00 (5H, m, 4-H, 13-H, 17-H, 21-H, and 25-H),
4.00 - 4.80 (5H, m, CH20C=0, Cfl20Ac, and 24-H), 5.25 (1H, d, J 10 Hz,
CH= CCH20Ac), 5.40 (1H, dd, J 15 and 9 Hz, 0CHCH=C7/CH(Me)), and 5.98
(1H, dd, J 15 and 10 Hz, OCHC//=CHCH(Me)); (Found: C, 66.69; H, 8.50.
^HssOgNa requires C, 66.65; H, 8.31%).

254
41. Preparation of Mono-Q-acetyl M1396Q3 free acid (57)
Mono-O-acetyl M139603 sodium salt (55) (5.00g, 7.51 mmol) in
ethanol (50 ml) and acetone (50 ml) was acidified with phosphoric acid
(10 ml of a 20% aqueous solution) under vigorous stirring for 48 h.
After concentration under reduced pressure, the remaining aqueous
solution was extracted with dich1oromethane (3 x 70 ml) and the organic
phase washed with water (1 x 50 ml). Evaporation of the solvent under
reduced pressure afforded the free acid monoacetate (57) (4.74g, 98%)
as a white foam, v (CHCU) 3402, 2926, 1764, 1736, 1687, 1648,
1229, and 1011 cm"1; 6 (90 MHz) 0.60 (3H, d, J 6 Hz, Me), 0.80 - 2.50
(33H, m), 1.98 (1H, s, 0C0CH3), 2.07 (2H, s, 0C0CH3), 3.30 (3H, s, OMe),
3.18 - 4.00 (5H, m, 4-H, 13-H, 17-H, 21-H, and 25-H), 4.00 - 4.70 (5H,
m, CH20C=0, CH20(\c , and 24-H), 5.10 - 5.58 (2H, m, Ctf=CCH20Ac and
0CHCH=C7/CH(Me)), and 5.98 (1H, dd, J 15 and 10 Hz, 0CHCtf=CHCH(Me)),
and C02 H, diffuse.

255
42. Preparation of Mono-O-acetyl M139603 methyl tetronic acid (58)
A flow of diazomethane (from potassium hydroxide and N-methyl-N-123nitroso-p-toluenesulphonamide(Diazald) ) was passed through a solu
tion of mono-O-acetyl M139603 free acid (4.00g, 6.21 mmol) in diethyl ether (100 ml) until all of the acid had been consumed (t.l.c.)
Evaporation under reduced pressure followed by chromatography (diethyl
ether, short column) or recrystallization (ethyl acetate-petrol)
afforded mono-O-acetyl M139603 methyl tetronic acid (58) (3.47g, 85%)
as a white solid, v (CHC13) 2927, 1762, 1737, 1664, 1626, 1244, 1119,
and 1063 cm'1; 6 (250 MHz) 0.70 (3H, d, J 6 Hz, Me), 0.92 (3H, d,
J 6 Hz, Me), 0.93 (3H, d, J 6 Hz, Me), 1.00 (3H, d, J 6 Hz, Me), 1.08
(6H, t, J 6 Hz, Me x 2), 1.12 - 2.07 (15H, m), 2.00 (3H, s, 0C0CH3),
2.13 - 2.30 (3H, m, 10-H, 20-H, and 22-H), 3.22 (1H, d, J 9.5 Hz, 13-H),
3.32 (1H, m, 25-H), 3.37 (3H, s, OMe), 3.51 (1H, dd, J 10 and 4 Hz,
21-H), 3.56 - 3.74 (2H, m, 4-H and 17-H), 3.98 (3H, s, =C-0Me), 3.99
(1H, m, 24-H), 4.55 (1H, d, J 12 Hz, CH20C=0), 4.60 (1H, d, J 12 Hz,
CH20C=0), 4.76 (2H, s, Ctf20Ac), 5.42 - 5.58 (2H, m, Cff=CCH20Ac and
0CHCH=C//CH(Me)), and 5.68 (1H, dd, 0 15 and 7 Hz, 0CHCtf=CHCH(Me));

256
(Found: C, 69.39; H, 9.07. C38H5809 requires C, 69.27; H, 8.87%).
43. Hydrolysis of M1396Q3 sodium salt (la)
A solution of M139603 sodium salt'*' (la) (l.OOg, 1.60 mmol) in
acetone (20 ml) and ethanol (20 ml) was acidified with phosphoric acid
(10 ml of a 20% aqueous solution) under vigorous stirring for 48 h.
After dilution with water (20 ml) and concentration under reduced
pressure, the remaining aqueous solution was extracted with dichloro-
methane (3 * 70 ml). The organic extracts were washed with water
(1 x 30 ml), dried (Na2S0lt) and evaporated to afford M139603 free
acid (lb) (945 mg, 98%) as a slightly yellow oil, v (film) 3500,max.1765, 1685, and 1650 cm-1; identical to the previously reported
1compound.

257
44. Ozonolysis of Mono-O-acetyl M1396Q3 methyl tetronic acid.
O M e
COMono-O-acetyl M139603 methyl tetronic acid (58) (2.00g, 3.04 mmol)
in dichioromethane (150 ml) at -78°C was ozonized slowly over a period+of oa. 8 h. Excess dimethyl sulphide (0.67 ml, 9.12 mmol) was then
added and the solution allowed to warm to room temperature and stirred
for a further 2 h. After concentration under reduced pressure, the
residue was taken up in diethyl ether (150 ml) and washed with water
(2 x 20 ml). Drying (Na2S0H), followed by evaporation of the solvent
gave a mixture of the left- and right-hand side aldehydes (59) and (7).
Separation was effected by repeated trituration with petrol. The
petrol solution was evaporated under reduced pressure and after
chromatography (15% diethyl ether-petrol) afforded the right-hand side
aldehyde (7) (878 mg, 95%) as a colourless oil. The remaining solid
from the trituration procedure was dried in vaeuo for 24 h to afford the unstable left-hand side aldehyde (59) (1.27g, 85%) as a white foam.

258
(7): v v (film) 2970, 1720, and 1085 cm'1; 6 (400 MHz) 0.97 (3H, d,
J 7.0 Hz, ring methyl), 1.02 (3H, d, J 7.0 Hz, OHCCH(Ate)), 1.10 (3H, d,
J 6.5 Hz, CH(Afe)OMe), 1.70 (1H, ddd, J 12.5, 7.0, and 1 Hz, 4'-H),
2.03 (1H. ddd, J 12.5, 9.0, and 6.8 Hz, 4'-H), 2.35 (1H, m, 3'-H),
2.48 (1H, m, 2-H), 3.37 (1H, m, CHOMe), 3.38 (3H, s, OMe), 3.89 (1H,
dd, J 10.0 and 4.0 Hz, 2'-H), 4.40 (1H, ddd, J 9.0, 7.0, and 4.5 Hz,
51-H), and 9.81 (1H, d, J 2.0 Hz, CHO); m/z 200 (M+), 157 (M+-C2H30),
141 (M+-CH(Me)0Me), 111 (M+-CH(Me)CHO-MeOH), 85, 83 (M+-CH(Me)CH0-
CH(Me)OMe), and 59 (CH(Me)0Me+); (Found: M+ , 200.1410. C:XH2 003
requires M+, 200.1412); (Found: C, 65.75; H, 10.16. C11H2003
requires C, 65.97; H, 10.07%).
(59): v (film) 2927, 1745, 1735, 1679, 1619, 1244, 1066, 962, and
911 cm-1; 6 (400 MHz) 0.72 (3H, d, J 7.5 Hz, Me), 1.00 (3H, d, J 7.5 Hz,
Me), 1.07 (3H, d, J 7.5 Hz, Me), 0.94 - 1.98 (13H, m), 2.02 (3H, s,
0C0CH3), 2.20 - 2.38 (1H, m, 10-H), 3.38 (1H, d, J 10.0 Hz, 13-H),
3.51 (1H, m, 4-H), 3.77 (1H, d, J 12.0 Hz, CtfCHO), 3.99 (3H, s, OMe),
4.60 (1H, d, J 12.0 Hz, CH20C=0), 4.70 (1H, d, J 12.0 Hz, CH20C=0),
4.78 (2H, s, Ctf20Ac), 5.40 (1H, d, J 10.5 Hz, C7=CCH2OAc), and 9.60
(1H, s, CHO); m/z 490 (M+), 461 (M+-CHO), 430 (M+-AcOH), 289 (M+-AcOH-O H
C^O141), 261 (M+-Ac0-170), 260 (M+-Ac0H-170), 170 ^
O
141 and 59 (0C0CH3+).
An alternative work-up procedure involves the addition of triphenyl-
phosphine (0.9 eq) to the crude reaction mixture at -78°C and subsequent
warming to room temperature. This method eliminates the necessity for
an aqueous work-up.

259
45. Ozonolysis of the left-hand side aldehyde (59)
To a solution of the left-hand side aldehyde (59) (100 mg,
0.20 mmol) in dichioromethane at -20°C was passed a flow of ozone (in
oxygen). After 4 h excess dimethyl sulphide (0.20 ml) was added and
the solution allowed to warm to room temperature. After a further
2 h, the solvent was evaporated under reduced pressure, the residue
taken up in diethyl ether (30 ml) and washed with water (2 x 10 ml).
The aqueous phase was extracted with diethyl ether (2 * 20 ml) and
the combined ethereal extracts washed with water (1 x 10 ml) and brine
(1 x 10 ml ). Drying (Na2S0i+) and concentration under reduced pressure
followed by chromatography (5% ethanol-chioroform) afforded the lactol
(60) (20 mg, 46%) as a colourless oil, vm (CHC13) 3411, 2926, 1736,
1234, and 1070 cm'1; 6 (90 MHz) 0.90 (1.2H, d, J 6.5 Hz, Me, minor
anomer), 0.92 (1.8H, d, 0 6.5 Hz, Me, major anomer), 1.20 - 1.90 (5H.
m), 2.17 (1.2H, s, OMe, minor anomer), 2.18 (1.8H, s, OMe, major anomer),
3.52 - 3.90 (1H, m, 2-H), 4.98 (3H, m, 6' -H and C/720Ac), and 7.52 (1H,
m, OH); m/z 199 (M+-OH), 127, 115 (M+-C0CH20Ac), and 43 (C0CH3+);
(Found; M+-OH, 199.0979. CloH1605 requires M+-OH, 199.0970).

260
46. Preparation of the t-Butyldimethyl si 1yl derivative of M1396Q3
sodium salt (62)
A solution of J4139603 sodium salt (la) (200 mg, 0.32 mmol),
imidazole (44 mg, 0.64 mmol) and t-butyldimethyl si]y1 chloride (53 mg,
0.35 mmol) in dry DMF (6 ml) was stirred at room temperature for 24 h.
The reaction was diluted with diethyl ether (30 ml) and washed with
water (2 x 10 ml). The aqueous phase was extracted with diethyl ether
(2 x 20 ml) and the combined ethereal extracts washed with water
(1 x 10 ml ) and brine (1 x 10 ml). Drying (Na2S0tt) and concentration
under reduced pressure followed by chromatography on Florisil (diethyl
ether) afforded the TBDMS derivative of M139603 sodium salt (62)
(168 mg, 71%) as a white foam, vmav (CHC13) 2926, 1717, 1646, and
1054 cm-1; 6 (400 MHz) 0.09 (3H, s, SiMe2), 1.00 (3H, s, SiMe2), 0.65
(3H, d, J 6.0 Hz, Me), 0.89 (9H, s, tBu), 0.90 - 1.90 (31H, m), 2.20
(1H, m, 22-H), 2.37 (2H, m, 10-H and 20-H), 3.22 (1H, d, J 10.0 Hz,
13-H), 3.29 (3H, s, OMe), 3.30 (1H, m, CtfOMe), 3.69 (1H, dd, J 10.0
and 4.0 Hz, 21-H), 3.73 (1H, m, 4-H), 3.86 (1H, m, 17-H), 3.97 (1H,
d, J 10.0 Hz, Cff20TBDMS), 4.15 (1H, d, 0 10.0 Hz, C#20TBDMS), 4.22 (1H,
d, J 14.5 Hz, CH20C=0), 4.23 (1H, m, 24-H), 4.33 (1H, d, J 14.5 Hz,
CH20C=0), 5.11 (1H, d, 0 10.0 Hz, C#=CCH20TBDMS), 5.48 (1H, dd, J 15.5
and 8.8 Hz, 0CHCff=CH), and 5.96 (1H, dd, J 15.5 and 10.5 Hz, 0CHCH=Cff);
m/z 738 (M+); (Found: M+, 738.4489. C41H6708SiNa requires M+,
738.4496).

261
47. Preparation of 1-t-Butyldiphenyl silyloxy-but-2-yne (74)
4 3 o *Me — =:— \ Me — = —OH 0SitBuPh2
(74)
1 24To 2-butyne-l-ol (2.32g, 33.1 mmol) in dry dichioromethane
(40 ml) was added triethylamine (5.49 ml, 39.6 mmol) followed by t-butyl-
diphenylsilyl chloride (10.Og, 9.44 ml, 36.4 mmol) and DMAP (160 mg,
1.30 mmol) at room temperature. After stirring for 15 min, reaction
was complete (t.l.c.) and was diluted with dichioromethane (30 ml) and
saturated aqueous ammonium chloride solution (20 ml). The aqueous
layer was extracted with dichioromethane (3 * 30 ml) and the combined
organic extracts washed with brine (1 * 20 ml) and dried (Na2SOtt).
Concentration under reduced pressure followed by chromatography (3%
diethyl ether-petrol) afforded 1-t-butyldiphenylsilyloxy-but-2-yne (74) (10.Ig, 99%) as a colourless oil, v (film) 3071, 3050, 2931, 2894,
2858, 2238, 1148, 1112, and 1072 cm’1; 6 (90 MHz) 1.10 (9H, s, tBu),
l. 82 (3H, t, J 2 Hz, Me), 4.30 (2H, q, J 2 Hz, CH2), 7.30 - 7.50 (6H,
m, Ph), and 7.65 - 7.80 (4H, m, Ph); m/z no M+, 251 (M+-tBu);
(Found: C, 77.63; H, 7.95. C20H2t*OSi requires C, 77.87; H, 7.84%).
48. Preparation of (Z)-1-t-Butyldiphenyl silyloxy-but-2-ene (75)
Me — = — ^ —0SitBuPh2
(74)
To a rapidly stirred suspension of Lindlar's catalyst67 (330 mg,
10% per weight of substrate) in THF (60 ml) under a hydrogen atmosphere

262
was added the alkyne (74) (3.30g. 10.6 mmol) in THF (10 ml) in one
portion. The reaction was carefully monitored by t.l.c. and
terminated just as all the starting alkyne had been consumed. The
catalyst was filtered off through a small pad of silica gel and washed
several times with diethyl ether. Evaporation of the solvent under
reduced pressure gave (D-l-t-butyldiiphenyls'ilyloxy -but-2-ene (75) as a
colourless oi I (329 mg, 99%). Chromatography (3% diethyl ether-petrol) gave a
sample of microanalytical purity, v (film) 3071, 3024, 2931, 2858,max •
1658, 1112, and 1092 cm’1; 6 (250 MHz) 1.06 (9H, s, ^u), 1.46 (3H, d,
J 7 Hz, Me), 4.28 (2H, d, J 6 Hz, CH2), 5.42 - 5.70 (2H, m, CH=CH),
7.30 - 7.45 (6H, m, Ph), and 7.65 - 7.74 (4H, m, Ph); m/z no M+ , 255
(CSitBuPh2) + , 253 (M+-tBu), and 55 (CH2-CH=CH-Me)+ ; (Found: C, 77.36;
H, 8.61. C2oH26OSi requires C, 77.36; H, 8.44%).
49. Preparation cf cis-2-t-Butyldiphenyl silyloxymethyl-4,4-dichi or0-3-
methyl cycl obutanone (76A) and gfs-3-t-Butyldiphen,ylsilylox,ymethy1-
2,2-dichloro-4-mettiy1cyclobUtanone (76B)
} -
O Si^BuPh
O S iP h 2 Bu
(75) (76 A] (76 B)

263
In a two-necked flask fitted with a reflux condensor and dropping
funnel was placed freshly prepared zinc-copper couple"^ (450 mg) the
ois-olefin (75) (760 mg, 2.45 mmol) and dry diethyl ether (6 ml) under
argon. Dry redistilled trichioroacetyl chloride (0.82 ml, 7.35 mmol)
in diethyl ether (10 ml) was added dropwise to a rapidly stirred
refluxing solution of the olefin over a period of 4 h. After a
further hour at reflux, the solution was cooled to room temperature and
poured into a flask containing solid sodium bicarbonate (5g) and petrol
(50 ml). After shaking and allowing to stand until there was no
further effervescence, the supernatant liquid was filtered rapidly
through a small pad of silica gel and the resulting solid washed with
10% diethyl ether-petrol, these organic washings being filtered through
the same silica gel pad. After concentration under reduced pressure
to ca. 5 ml, petrol (50 ml) and solid sodium bicarbonate (2g) were
added. After no further effervescence was noted, the supernatant
liquid was again filtered rapidly through a fresh pad of silica gel,
the solid being repeatedly washed with further petrol. This procedure
removed the precipitated zinc salts and prevented any loss of the silyl
protecting group. Removal of the solvent under reduced pressure gave
an unstable mixture of dichioroadducts (76A) and (76B) (3:1) as a
yellow oil (l.OOg, 97%) which was used directly in the next step,
v (film) 3080, 2964, 2830, 1803, 1115, and 702 cm-1; 6 (250 MHz)max.
l. 04 (6.75H, s, tBu), 1.05 (2.25H, s, tBu), 1.35 (2.25H, d, J 7.5 Hz,
Me), 1.39 (0.75H, d, J 7.5 Hz, Me), 3.22 (1H, m, CHC=0), 3.84 - 4.07
(3H, m, CHCC12 and Ctf20TBDPS), 7.35 - 7.48 (6H, m, Ph), and 7.70 (4H,
m, Ph); m/z no M+ , 365 (M+(37C1) - tBu), and 363 (M+ (35C1) - tBu).

264
50. Preparation of cfs-2-t-Butyidiphenylsi 1yloxymethyl-3-methylcyclo-
butanone (77A) and gfs-3-t-Butyldiphenyl si 1y 1oxymethyl-2-methyl -
cyclobutanone (77B)
JZ\l— d^CXL
fO
O S iP h , Bu
(76A)
O S iP h , Bu
Cl3 ^Cl
o
(76B)
O S iP h ^B uj 1
V ? . 4 V 3 4
r ?O S iP h /B u
(77A) (77 b )
To the crude mixture of dichiorocyclobutanones (76A) and (76B)
(3.30g, 7.84 mmol) in glacial acetic acid (15 ml) at room temperature
was added excess powdered zinc (2g). After 15 min, the mixture was
warmed to 40°C and stirred for o a . 4 h (or until t.l.c. indicated
completion). The acetic acid was then removed under reduced pressure
(taking care to keep the temperature below 50°C) and the residue
diluted with petrol (100 ml) before filtration through a short pad of
silica gel. Chromatography (gradient elution, 5 10% diethyl ether-
petrol ) afforded c \ s - 2 - t - b u t y l d ' i p h e n y l s ' L l y l o x y m e t h y l - 7 > - m e t h y ' l c y o l o -
b u t a n o n e ( 7 1 A ) (1.36g, 56%) and c i s - 2 > - t - b u t y l d i p h e n y l s - i l y l o o c y m e t h y l - 2 -
m e t h y I c y c l o b u t a n o n e (77B) (420 mg. 17%) both as white crystalline solids
(m.p. 54°C), (77A): v (film) 3069, 2926, 2855, 1783, 1111, and 704max.
cm-1; 6 (250 MHz) 1.06 (9H, s, tBu), 1.32 (3H, d, J 7.5 Hz, Me), 2.57
(1H, ddd, J 16. 6, and 2 Hz, 4-H), 2.65 (1H, m, 3-H), 3.15 (1H, ddd,
J 16, 9, and 3.5 Hz, 4-H), 3.44 (1H, m, 2-H), 3.83 (1H, dd, J 11 and

265
4 Hz, C^OTBDPS), 3.92 (1H, dd, J 11 and 7 Hz, Ctf20TBDPS), 7.38 (6H, m,
Ph), and 7.68 (4H, m, Ph); m/z 352 (M+ ), 295 (M+-tBu), and 253 (M+-tBu-
CH2C0); (Found: C, 75.12; H, 8.06. C22H2802Si requires C, 74.95;
H, 8.01%);
(77B): (film) 3050, 2931, 1773, 1105, and 706 cm'1; 6 (250 MHz)max •
I. 05 (9H, s, tBu), 1.18 (3H, d, J 7.5 Hz, Me), 2.63 (1H, m, 3-H), 2.73
(1H, ddd, J 17, 4, and 2 Hz, 4-H), 3.09 (1H, ddd, J 17, 9, and 3 Hz, 4-H),
3.49 (1H, m, 2-H), 3.78 (1H, dd, J 11 and 5 Hz, Ctf20TBDPS), 3.84 (1H, dd,
J 11 and 6 Hz, Ctf20TBDPS), 7.40 (6H, m, Ph), and 7.68 (4H, m, Ph);
352 (M+ ), 295 (M+-tBu), and 253 (M+-tBu-CH2C0); (Found: C, 74.88;
H, 7.99. C22H2802Si requires C, 74.95: H, 8.01%).
51. Preparation of c i s -4-t-8uty1dipheny1 si 1yloxymethy1-3-methy1butan-4-
olide (78)
V4j
O S iP h 2 Bu
(77A)
->»■
O S i*B u P h 2
(78)
To a solution of the major cyclobutanone (77A) (256 mg, 0.73 mmol)
in glacial acetic acid (1.5 ml) at ca. 5°C was added 30% aqueous hydrogen
peroxide solution (0.23 ml, 2.20 mmol). The solution was stirred at
this temperature for a a . 12 h. The acetic acid was then removed under
reduced pressure and the resulting semi-solid dissolved in a minimum
quantity of dichioromethane and filtered through a short pad of silica
gel. Evaporation of the solvent under reduced pressure followed by

266
chromatography (50 -* 75% dichioromethane-petrol) gave cis-4-f-£>ufz/7-
diyhenylsilyloxymethyt-Z-methylbutan-‘\-ol'Lde (73) (230 mg, 36%) as a
wh^e solid, v (CHC1 3) 3070, 2926, 2857, 1779, 1171, 1110, and 708
cm"1; 6 (250 MHz) 1.08 (9H, s, tBu), 1.22 (3H, d, J 7 Hz, Me), 2.49
(1H, dd, J 17 and 10 Hz, 2-H), 2.60 (1H, dd# J 17 and 8 Hz, 2-H), 2.77
(1H. m, 3-H), 3.78 (1H, dd, J 11.5 and 3 Hz, O/20TBDPS}; 3.87 (1H, dd,
J 11.5 and 3.5 Hz, Ctf.OTBDPS), 4.44 (1H. dt, J 7.5, 3.5, and 3 Hz, 4-H),
7 42 (6H, m, Ph), and 7.68 (AH, m, Ph); m/z no M+ , 311 (M+-tBu), 281
(M+-CH(Me)CH2C00H), 259 (CH20TBDPS+ ), and 69 (CH2=CH-CH(Me)CH2+ );
(Found: C, 71.75; H, 7.73. C22H2803Si requires C, 71.70; H, 7.66%).
52. Preparation of cis -4-(Hydroxymethyl)-3-methylbutan-4-olide (79)
To the lactone (78)(1.08g, 2.93 mmol) in dry THF (20 ml) was
added tetra-n-butylammoniurn fluoride (2.93 ml of a 1M solution in THF
2.93 mmol) at room temperature. After stirring for 30 min, the
solvent was removed under reduced pressure and the residue chromato
graphed (60% ethyl acetate-petrol) to afford cfs-4-(hydroxymethyl)-
3-methylbutan-4-olide (79) (323 mg, 85%) as a colourless oil, v (film)max.
3424, 2970, 2884, 1763, 1176, and 1038 cm"1; <5 (250 MHz) 1.15 (3H,
d, J 7.5 Hz, Me), 2.38 (1H, dd, J 17 and 8 Hz, 2-H). 2.62 (1H, dd,
J 17 and 8.5 Hz, 2-H). 2.75 (1H, m, 3-H), 3.43 ( 1H, br s, OH), 3.85
(2H, m, C7/20H), and 4.54 (1H, ddd, J 7.5, 4, and 3.5 Hz, 4-H); m/z
130 (M+ ), 112 (M+-H2Q), and 99 (M+-CH20H); identical to the previously
125reported compound.

267
53. Preparation of cfs-4-Formyl-3-methylbutan-4-olide (80)
To the alcohol (79) (83 mg, 0.64 mmol) in dry DMSO (0.60 ml) and
benzene (1.0 ml) was added pyridine (52 pi, 0.64 mmol) and trifluoro-
acetic acid (25 pi, 0.32 mmol). To the resulting solution was added
redistilled dicyclohexylcarbodiimide (400 mg, 1.93 mmol) in DMSO (1 ml)
at room temperature. After stirring overnight, oxalic acid (173 mg,
1.93 mmol) in methanol (1 ml) was added and when gas evolution had
ceased (ca. 30 min), water (10 ml) was added followed' by filtration
of the precipitated dicyclohexylurea. The aqueous layer was extracted
with dichioromethane (3 * 20 ml) and the combined organic extracts
washed once with water (5 ml) and dried (Na2S0<J. Removal of the
solvent under reduced pressure followed by chromatography (gradient
elution, 30 75% ethyl acetate-petrol) gave the unstable aldehyde
(80) (19.6 mg, 24%) as a colourless oil, 6 (250 MHz) 1.15 (3H, d,
J 7.5 Hz, Me), 2.28 (1H, dd, J 17 and 5 Hz, 2-H), 2.78 (1H, dd, 0 17
and 8.5 Hz, 2-H), 2.98 (1H, m, 3-H), 4.76 (1H, dd, J 7.5 and 2 Hz,
4-H), and 9.76 (1H, d, J 2 Hz, CHO); m/z no M+ , 99 (M+-CH0), and 71
(M+-CH0-C0).

268
54. Preparation of (ff)-efs-3-Methyl-4-(2-phenylsulphonylethenyl)butan-
4-olide (81B) and ( Z)-ois-3-Methyl-4-(2-phenylsulphonylethenyl )-
butan-4-olide (81A)
79To phenyl sulphonyltrimethylsilylmethane (240 mg, 1.05 mmol) in
dry 1,2-dimethoxyethane (6 ml) at -78°C under argon was added nBuLi
(0.74 ml of a 1.56M solution in hexane, 1.16 mmol) dropwise. After
10 min, the aldehyde (80) (190 mg, 1.48 mmol) in 1,2-dimethoxyethane
(2 ml) was added dropwise to the pale yellow solution* Stirring was
continued at -78°C for 10 min before allowing to warm to room tempera
ture over 30 min. The solution was then diluted with water (5 ml)
and diethyl ether (20 ml). The aqueous layer was extracted with
diethyl ether (2 * 10 ml) and the combined ethereal extracts washed
with water (1 * 10 ml) and brine (1 * 10 ml). Drying (Na2S0„) and
concentration under reduced pressure followed by chromatography (gradient
elution, 50 80% diethyl ether-petrol) gave the (Z)-vinyl sulphone
(81A) (67 mg, 24%) and the (E)-vinyl sulphone (81B) (26 mg, 9.3%) both
as white sol ids;
Less polar (Z)-isomer (81A): \> v (CHC13) 3060, 2970, 1783, 1631, 1149,
and 1085 cm-1; 6 (250 MHz) 1.02 (3H, d, J 8 Hz, CH3), 2.25 (1H, dd,
0 17 and 3 Hz, 2-H), 2.82 (1H, dd, J 17 and 8 Hz, 2-H), 3.06 (1H, m,
3-H), 6.10 (1H, ddd (7 lines), J 8, 6.5, and 1 Hz, 4-H), 6.24 (1H, dd,

269
J 11.5 and 8 Hz, Cff=CHS02Ph), 6.42 (1H, dd, J 11.5 and 1 Hz, CH=CffS02Ph),
7.60 (3H, m, Ph), and 7.90 (2H, m, Ph); m/z 266 (M+ ), 224 (M+-CH2C0),
125 (M+-S02Ph), and 77 (Ph+ ).
More polar (£’)-isomer (81B): v (CHC13) 3059, 2927, 1782, 1629, 1147,
and 1086 cm”1; 6 (250 MHz) 1.02 (3H, d, J 8 Hz, CH3), 2.20 (1H, dd,
J 17 and 5 Hz, 2-H), 2.71 (1H, dd, J 17 and 8 Hz, 2-H), 2.85 (1H, m,
3-H), 5.12 (1H. ddd, J 6.5, 3.5, and 2 Hz, 4-H), 6.55 (1H, dd, J 15 and
2 Hz, CH=CtfS02Ph), 6.90 (1H, dd, J 15 and 3.5 Hz, Ctf=CHS02Ph), 7.60 (3H,
m, Ph), and 7.88 (2H, m, Ph); m/z 266 (M+ ), 224 (M+-CH2C0), 125 (M+-
S02Ph), 99 (M+-CH=CHS02Ph), and 77 (Ph+ ); (Found: M+ , 266.0605.
CiaHi.O.S requires M+ , 266.0613); (Found: C, 58.54; H, 5.68.
Ci3H11+01+S requires C, 58.63; H, 5.30%).
55. Preparation of (±)-2-(7?)-t-Buty1diphenyl silyloxymethyl-5-hydroxy-
3-( 5)-methyltetrahydrofuran (82)
To the lactone (500 mg, 1.36 mmol) in dry toluene (1C ml) at -78°C
was added DIBAL (1.70 ml of an oa. 1.50M solution in toluene, 2.55 mmol)
dropwise. After 45 min, reaction was complete and was quenched by
dropwise addition of glacial acetic acid (1.56 ml) followed by warming
to room temperature. Water (0.5 ml) and solid sodium bicarbonate
(3.14g) were added sequentially and the resulting solid extracted with

270
ethyl acetate (4 x 30 ml). The solution was concentrated under
reduced pressure to o a . 10 ml and diethyl ether (50 ml) was added
followed by filtration through a small pad of Florisil to afford after
evaporation of the solvent the lactol (82) (458 mg, 91%) as a viscous,
colourless oil, v (film) 3409, 3070, 2933, 2858, 1112, 1012, and max.703 cm-1; 6 (90 MHz) 1.08 (6.3H, s, ^Bu, major anomer), 1.10 (2.7H,
s, Bu, minor anomer), 1.20 (3H, t, J 7 Hz, Me), 1.78 - 2.30 (2H, m,
4-H2), 2.60 (1H, m, 3-H), 3.60 - 3.80 (3H, m, C£20TBDPS and OH),
3.90 - 4.34 (1H, m, 2-H), 5.42 (0.3H, m, 5-H, minor anomer), 5.56 (0.7H,
m, 5-H, major anomer), 7.35 (6H, m, Ph), and 7.70 (4H, m, Ph); m / z 353
(M+-0H), 313 (M+-tBu), 295 (M+-H20-tBu), and 269 (CH20TBDPS+).
56. Preparation of (±)-2-(fl)-t-Butyl diphenylsilyloxymethyl-5-methoxy-
3-(s)-methy1tetrahydrofuran (84).
The lactol (450 mg, 1.22 mmol) was dissolved in methanol (10 ml)
and CSA (14 mg, 0.06 mmol) was added. After stirring at room tempera
ture for o a . 4 h, the methanol was removed under reduced pressure and the residue chromatographed (15% diethyl ether-petrol) to afford (±)-2-
(R) -t - b u t y l d i - p h e n y l s % l y l o x y m e t h y ' l - 5 - m e t h o x y-3- (S) - m e t h y l t e t r a h y d r o f u r a n
(84) (350 mg, 75%) as a colourless oil, v (film) 3070, 2931, 1112,max.and 703 cm'1; 5 (60 MHz) 1.10 (12H, m, tBu and Me), 1.80 - 2.70 (3H, m,
3-H and 4-H2), 3.30 (0.9H, s, OMe, minor anomer), 3.35 (2.1H, s, OMe,
(82) (84)

271
major anomer), 3.72 (2H, m, Cff20TBDPS), 4.10 (1H, m, 2-H), 4.95 (1H, m,
5-H), 7.38 (6H, m, Ph), and 7.70 (4H, m, Ph); m/z 353 (M+-0Me), 327
(M+-tBu), and 295 (M+-tBu-MeOH); (Found: M+-tBu, 327.1412.
C23H3203Si requires M+-^Bu, 327.1416); (Found: C, 72.05; H, 8.68.
C23H3203Si requires C, 71.83; H, 8.39%).
57. Preparation of (±)-5-Methoxy-(3)-(s)-methyltetrah,ydrofuran-2-(R)-
ylmethanol (85)
To the silyl ether (84) (630 mg, 1.64 mmol) in dry THF (10 ml)
under argon was added tetra-n-butylammonium fluoride (1.64 mis of a 1.0M
solution in THF, 1.64 mmol) at room temperature. After stirring for
1 h, the solution was concentrated under reduced pressure and chromato
graphed directly (75% diethyl ether-petrol) to afford (±)-5-methoxy-(3)-
{S)-methyltetrahydrofuran-2-{R)-ylmethanol (85) (195 mg, 83%) as a
colourless oil, vmav (film) 3430, 2931, 1112, and 703 cm"1; 6 (250 MHz)
1.00 (3H, d, J 7.5 Hz, Me), 1.71 (1H, ddd (7 lines), J 12.5, 8, and
5.5 Hz, 41-H), 2.03 (1H, ddd, J 12.5, 7, and 1 Hz, 4'-H), 2.56 (1H, m,
31-H), 2.59 (1H, br s, OH), 3.35 (3H, s, OMe), 3.61 (2H, m, Ctf20H),
4.15 (1H, ddd (6 lines), J 7.5, 7, and 4 Hz, 2'-H), and 5.03 (1H, dd,
J 5.5 and 1 Hz, 51 -H); m/z 146 (M+), 128 (M+-H20), 115 ((M+-0Me) and
(M+-CH20H)), 97 (M+-H20-0Me), and 71 (Ci+H70+); (Found: C, 57.71;
H, 9.72. C7Hlt+03 requires C, 57.51; H, 9.65%).

272
58. Preparation of (±)-5-Methoxy-3-(5)-methyl-2-{r )-tetrahvdrofuran-
carbaldehyde (88)
The alcohol (85) (538 mg, 3.74 mmol) was oxidised following the
procedure of Experiment 53 and after chromatography (70% diethyl ether-
petrol) afforded the aldehyde (88) (260 mg, 49%) as a colourless oil,
v (film) 2938, 2833, 1730, 1100, 897 , and 842 cm"1; 6 (250 MHz)
1.04 (2.55H, d, J 7.5 Hz, Me, major anomer), 1.12 (0.45H, d, J 7.5 Hz,
Me, minor anomer), 1.72 (1H, ddd (7 lines), J 13.5. 8.5, and 5.5 Hz,
4-H), 2.12 (1H, ddd, J 13.5, 7.5, and 1.5 Hz, 4-H), 2.68 (0.15H, m,
3-H, minor anomer), 2.85 (0.85H, m, 3-H, major anomer), 3.39 (2.55H,
s, OMe, major anomer), 3.48 (0.45H, s, OMe, minor anomer), 4.26 (0.15H,
dd, J 8.5 and 3 Hz, 2-H, minor anomer), 4.40 (0.85H, dd, J 8 and
2 Hz, 2-H, major anomer), 5.18 (0.15H, dd, J 5.5 and 2 Hz, 5-H, minor
anomer), 5.22 (0.85H, dd, 0 5.5 and 1.5 Hz, 5-H, major anomer), 9.70
(0.85H, d, J 2 Hz, CH0, major anomer), and 9.77 (0.15H, d, J 3 Hz, CH0,
minor anomer); m/z 144 (M+), 115 (M+-CH0), and 83 (M+-CH0-Me0H);
(Found: M+, 144.0780. C7H1203 requires M+, 144.0786).

273
59. Preparation of (g, z)-(t)-5-Methoxy-3-(g)-2-,(/?)-(2,-phenyl sulphonyl -
ethen.yl )tetrahydrofuran (89AB)
79To a solution of phenyl sulphonyltrimethylsilylmethane (250 mg,
1.10 mmol) in dry THF (7 ml) at -78°C was added nBuLi dropwise (0.86 ml
of a 1.34M solution in hexane, 1.15 mmol). The pale yellow solution
was stirred for 20 min at -78°C before addition of the aldehyde (88)
(150 mg, 1.04 mmol) in dry THF (3 ml). After 15 min, the reaction was
warmed to room temperature and worked-up following the procedure
described in Experiment 54. Chromatography (35% diethyl ether-petrol)
afforded an inseparable mixture (1:1) of double bond isomers (89A) and
(89B) (182 mg, 62%) as a colourless oil, v (film) 3160, 2964, 1629,max.1203, 1149, 1086, 1057, and 826 cm-1; 6 (250 MHz) 0.92 (1.5H, d,
J 7 Hz, Me), 0.93 (1.5H, d, J 7 Hz, Me), 1.65 (0.5H, ddd (7 lines),
J 13, 7.5,and 5.5 Hz, 4'-H, (E)-isomer), 1.76 (0.5H, ddd, J 13, 5.5,
and 1 Hz, 4'-H, (Z)-isomer), 2.03 (0.5H, ddd, J 13, 7.5, and 1.5 Hz,
41-H, (E)-isomer), 2.14 (0.5H, ddd, J 13, 7.5, and 2 Hz, 41-H,(Z)-isomer),
2.68 (0.5H, m, 3'-H, (E)-isomer), 2.82 (0.5H, m, 31-H, (z)-isomer),
3.32 (1.5H, s, OMe), 3.35 (1.5H, s, OMe), 4.75 (0.5H, ddd, J 7.5, 4,
and 2 Hz, 21-H, (E)-isomer), 5.03 (0.5H, dd, J 5.5 and 1.5 Hz, 5'-H,
(E)-isomer), 5.07 (0.5H, dd, J 5.5 and 2 Hz, 51-H, (Z)-isomer), 5.66
(0.5H, ddd (7 lines), J 8.5, 7, and 1 Hz, 21-H, (Z)-isomer), 6.29
(0.5H, dd, J 11.5 and 8.5 Hz, Ctf=CHS02Ph, (Z)-isomer), 6.45 (0.5H, dd,

274
J 11.5 and 1 Hz, CH=CF/S02Ph, (Z)-isomer), 6.59 (0.5H, dd, J 15 and 2 Hz,
CH=C#S02Ph, (£)-isomer), 6.92 (0.5H. dd, J 15 and 4 Hz, Ctf=CHS02Ph,
(S’)-isomer), 7.50 - 7.70 (3H, m, Ph), and 7.85 - 7.98 (2H, m, Ph);
m/z 251 (M+-0Me), 250 (M+-MeOH), 224 (M+-CH2CH0Me), 141 (M+-S02Ph), 109
(M+-S02Ph-Me0H) , and 77 (Ph+); (Found: M+-0Me. 251.0738. C^H^O^S
requires M+-0Me, 251.0742).
60. Preparation of (±)-5-Methoxy 3-(S)-methyl-2-(/?)-(2-phenyl sulphonyl-
ethyl)tetrahydrofuran (90)
To a solution of the vinyl sulphones (89AB) (120 mg, 0.27 mmol) in
THF (1 ml) under argon was added LiBHEt3 (’Super-Hydride1) (0.50 ml of a
1.0M solution in THF, 0.50 mmol) dropwise. After stirring for 2 h at
room temperature, the reaction was quenched with water (5 ml) and
diluted with diethyl ether (20 ml). The aqueous layer was extracted
into diethyl ether (3 x 10 ml) and the combined organic extracts
dried (Na2S0O and concentrated under reduced pressure. Chromatography
(35% diethyl ether-petrol) gave {±)- 5 - m e t h o x y - 2 - { S ) - m e t h y l - 2 - { R ) ~ ( 2 -
p h e n y l s u l p h o n y l e t h y l ) t e t r a h y d r o f u r a n (90) (166 mg, 96%) as a white
solid (m.p. 53°C), v (film) 2962, 1148, 1088, 1065, and 1031 cm-1;max.6 (90 MHz) 0.90 (3H, d, J 7 Hz, Me), 1.50 - 2.50 (5H, m, 4-H2, 3-H,
l‘-H2), 3.05 - 3.45 (2H, m, Ctf2S02Ph), 3.25 (3H, s, OMe), 3.96 (1H, m,
2-H), 4.90 (1H, dd, J 5 and 2 Hz, 5-H), 7.60 (3H. m, Ph), and 7.95 (2H,

275
m, Ph); m/z 284 (M+, weak), 253 (M+-OMe), 252 (M+-MeOH), 226 (M+-
CH2CH0Me), 143 (M+-S02Ph), 115 (M+-CH2CH2S02Ph), 110 (M+-Me0H-PhS02H),
and 86 (CH(Me)CH2CH0Me+); (Found: M+-0Me, 253.0900. C11>H2O0,,S
61. Preparation of (5)-Methyl 3-t-butyldiphen.ylsi 1yloxy-2-methyl-
propionate (93)
(+)-S-Methyl 3-hydroxy-2-methylpropionate (8.00g, 67.8 mmol) was
stirred with t-butyldiphenylsilyl chloride (18.5 ml, 71.1 mmol),
triethyl amine (10.4 ml, 75.0 mmol) and DMAP (415 mg, 3.4 mmol) in dry
dichioromethane (100 ml) for oa . 6 h at room temperature. After
dilution with further dichioromethane (100 ml) and water (50 ml), the
organic layer was combined (3 * 100 ml) extract of the aqueous layer,
before washing with water (1 * 50 ml) and brine (1 x 50 ml). The
solution was dried (Na2S0t+) and the solvent removed under reduced
pressure to give a pale yellow oil which was purified by chromatography
(5% diethyl ether-petrol) to afford (%)-methyl-^-t-butyldiphenylsilyloxy-
Z-methylpvopionate (93) (22.0g, 94%) as a colourless oil, M q 2 5 + 23°
{c 7.8, MeOH); v (film) 3071, 2934, 2858, 1741, 1200, 1112, and 823 max.cm'1; 6 (60 MHz) 1.05 (9H, s, tBu), 1.20 (3H, d, J 6.5 Hz, Me), 2.70
requires M+-0Me, 253.0898): (Found: C, 58.85; H, 7.19; S, 11.32.
Cit+H2001+S requires C, 59.13; H, 7.09; S, 11.27%).
(92) (93)
(1H, m, 2-H), 3.60 (3H, s, C02CH3), 3.60 - 3.85 (2H, m, 3-H2), 7.28 (6H,

276
m, Ph), and 7.60 (4H, m, Ph); m / z 325 (M+-0Me), 299 (M+-tBu), 269 (M+- CH(Me)C02Me), 213, and 183; (Found: C, 70,87; H, 8.07. C21H2303Si
requires C, 70.74; H, 7.92%).
62. Preparation of (ff)-3-t-Butyl diphenyl silylox;y-2-methyl propan-l-ol
(95)
/ C 0 2MeTsk .OSrBuPh,(93)
To the ester (93) (32.Og, 89.8 mmol) in dry toluene (100 ml) at
-78°C under argon was added DIBAL (150 ml of al.50M solution in toluene,
0.23 mol). The mixture was stirred at this temperature for 1 h
followed by dropwise addition of water (10 ml) and subsequent warming
to room temperature. The solution was then poured into a flask con
taining ethyl acetate (300 ml) and solid sodium bicarbonate (15g).
After shaking this mixture for 10 min, it was allowed to stand at
room temperature for 20 min. The supernatant liquid was filtered
through a pad of silica gel (under pressure) and the solid residue
repeatedly extracted with further ethyl acetate (5 x 100 ml) and the
organic solution filtered through the same silica gel pad. Removal of
the solvent under reduced pressure followed by chromatography (35%
diethyl ether-petrol) afforded ( R ) - ? > - t - b u t y l d i p h e n y l s i l y l o x y - Z - m e t h y l -
p r o p a n - l - o l (95) (22.Og, 75%) as a colourless oil, v (film) 3370,ITlaX •
3070, 2931, 2859, 1112, 1035, and 823 cm'1; 6 (60 MHz) 0.85 (3H, d,
J 7 Hz, Me), 1.10 (9H, s, tBu), 1.95 (1H, m, 2-H), 2.55 (1H, br s, OH),

277
3.40 - 3.80 (4H, m, 1-H2 and 3-H2), 7.30 (6H, m, Ph), and 7.70 (4H, m,
Ph); rn/z 271 (M+-tBu) and 199; (Found; C, 72.98; H, 8.78.
C2oH2802Si requires C, 73.12; H, 8.59%).
63. Preparation of (g)-3-t-Buty1diphenyl silyloxy-2-methylpropan-l-al
1941
To oxalyl chloride (4.89 ml, 56.1 mmol) in dry dichioromethane
(120 ml) at -60°C was added DMS0 (7.93 ml, 0.11 mol) in dichioromethane
(30 ml) dropwise. After stirring for 5 min the alcohol (95) (10.Og,
30.5 mmol) in dry dichloromethane (35 ml) was added dropwise and the
reaction stirred at -60°C for a further 20 min. Triethylamine (38.8
ml, 0.28 mol) was then added and the reaction mixture allowed to warm
slowly to room temperature. After dilution with further dichloro
methane (100 ml) and water (70 ml), the aqueous layer was extracted
with dichloromethane (3 * 100 ml). The combined organic extracts
were washed with brine (100 ml) and dried (Na2S0J. Concentration
under reduced pressure followed by chromatography (25% diethyl ether-
petrol ) afforded (s)-3-t-buty1diphenylsilyloxy-2-methylpropan-l-al
(95) (94)
(94) (7.20g, 72%) as a white solid, (m.p. 60°C); [a]^25 + 2.1 (o 5.7,
CHC13); v (CHC13) 3071, 2933, 2858, 1735, 1112, 1035, and 823 cm"1; max.6 (250 MHz) 1.05 (9H, s, tBu), 1.12 (3H, d, J 7 Hz), 2.58 (1H, m, 2-H),
3.85 (1H, dd, J 10 and 6.5 Hz, 3-H), 3.92 (1H, dd, J 10 and 5 Hz, 3-H),

278
7.35 - 7.48 (6H. m, Ph), 7.63 - 7.69 (4H, m, Ph), and 9.79 (1H, d,
J 2 Hz. CHO); m/z 269 (M+-tBu) *md 199; (Found: C, 73.52; H, 8.34.
C2oH2s02Si requires C, 73.57; H, 8.03%).
64. Preparati on of (fl)-f Z)-l-t-Butv1diphenyl si 1yloxy-2-methylpent-3-
To ethyltriphenylphosphomum iodide (4.10g, 9.80 mmol) suspended
in dry THF (100 ml) at 0°C was added nBuLi (6.45 ml of a 1.52M solution
in hexane, 9.80 mmol) dropwise with stirring. After 30 min, the
resultant red solution was precooled to -78°C and the aldehyde (94)
(3.20g, 9.80 mmol) in dry THF (10 ml) was added dropwise over 3 min.
The red colour immediately disappeared and after stirring for 5 min at
-78°C, the solution was allowed to warm to room temperature over 30 min.
The mixture was poured into saturated aqueous ammonium chloride solu
tion (50 ml) and diethyl ether (200 ml). The aqueous phase was
extracted with diethyl ether (2 * 70 ml) and the combined ethereal
extracts were washed with water (50 ml) and brine (50 ml). Drying
(Na2S01+) and concentration under reduced pressure, followed by flash
chromatography (3% diethyl ether-petrol) afforded (R)-(Z)-1-t-butyl-
diphenylsilyloxy-Z-methyl-pent-Z-ene (91) [>95% (Z)] (2.85g, 86%) as a
colourless oil, [a]25 - 18.3° [o 10.2, CHC13); v (film) 3070,u [ l i e * X •
ene (91)
OSieB u P h 2
(94)

279
2958, 2930, 1558, 1112, and 823 cm’1; 6 (250 MHz) 1.00 (3H, d, J 7 Hz,
0 2 methyl), 1.06 (9H, s, tBu), 1.57 (3H, d, J 7 Hz, =CHMe), 2.71 (1H,
m, 4-H), 3.47 (2H, m, Cff20TBDPS), 5.19 (1H, m, J 10.5 Hz, 4-H), 5.43
(1H, m, J 10.5 Hz, 3-H), 7.38 (6H, m, Ph), and 7.68 (4H, m, Ph); m/z
281 (M+-rBu) and 239 (M^Bu-CH^CHMe); (Found; C, 77.83; H, 9.16.
C22H30OSi requires C. 78.05; H, 8.93%).
65. Preparation of 2-(S)-[(2-t-Butyldiphenyl si 1yioxy-1-(R)-methyl)-
e thy 11-4,4-d i ch 1 or o-3-(7?)-methyl cycl obutanone and 2-(i?)-[(2-t-
Butyl di phenyl si 1 yi oxy-1-(7?) -methyl)ethyl 1-4,4-di chi oro-3-(S)-
methy1cyclobutanone (96AB) and 3-(7?)-[(2-t-Butyl diphenyl si 1 yloxy-
1- (7?) -methyl ) ethyl ]-2,2-dich1 oro-4-(ff)-methylcyclobutanone and
3- (S) -f (2-1-Butyl di phenyl si 1 yl ox.y-l- (R) -methyl ) ethyl 1-2,2-dichloro-
4- (R)-methylcyclobutanone (97AB)
In a two-necked flask fitted with a reflux condensor and dropping
funnel was placed the olefin (lO.OOg, 29.6 mmol), freshly-prepared zinc-
copper couple (lOg) and dry diethyl ether (500 ml) under argon.
Redistilled trichioroacetyl chloride (9.91 ml, 88.8 mmol) in diethyl
ether (90 ml) was added dropwise to a rapidly stirred refluxing solu
tion of the olefin over a period of oa. 4 h. After a further 2 h

280
reflux, the reaction was cooled to room temperature and poured into a
flask containing solid sodium bicarbonate (20g) and petrol (200 ml).
After shaking and allowing to stand until no further bubbling occurred,
the supernatant liquid was filtered rapidly through a small pad of
silica gel and the resulting solid washed with 10% diethyl ether-petrol,
these organic washings being filtered through the same silica gel pad.
After concentration under reduced pressure to oa. 30 ml, petrol (200 ml)
and solid sodium bicarbonate (lOg) were added. After no further
effervescence was seen, the supernatant liquid was again filtered
rapidly through a fresh silica gel pad; the solid repeatedly washed
with further petrol and filtered through the same silica pad. This
procedure is necessary to prevent loss of the silyl protecting group.
Removal of the solvent under reduced pressure gave an unstable mixture
of dich1oroadducts (96AB) and (97AB) (12.50g, 94% crude yield) as a
viscous yellow oil which was used directly in the next step (unstable
to column chromatography or distillation), v _v (film) 3071, 2858,
1803, 1113, 1045, 823, and 803 cm"1; 5 (60 MHz) 1.00 - 1.70 (15H. m),
2.00 - 2.50 (1H, m, l'-H), 2.70 - 4.10 (4H, m, Ctf20TBDPS and ring
protons), 7.30 (6H, m, Ph), and 7.60 (4H, m, Ph); m/z 448 [M+(3 5 C1)j,
413 [M+(35C1)-HC1], 391 [M+(35Cl)-tBu], 338 (M+-C0CC12), 323 (M+-C0CC12-
Me), 281, 217, 199, and 135.

281
66. Preparation of 2-(fl)-C(2-t-Butyldiphenylsi lyloxy-l-W^-methyl )-
ethyl1-3-(ff)-methylcycl obutanone and 2-(£)-[(2-t-Butyldiphenyl -
si1y1oxy-l-(i?)-methyl) e thy 1 ]-3-(7?)-me thy lcycl obutanone (98AB) and
3- {R) -f (2-t-Butyl di phenyl si 1 y 1 oxy-1- (R) -methyl ) ethyl "j-2-(di
methyl cycl obutanone and 3-(S)-[~(2-t-But.yl diphenyl si 1yloxy-l-(di
methyl )ethyl]-2-( R)-methylcyclobutanone (99AB)
B u P h ,S iO
rBuPh,SiOCl Cl
Cl/> C I
(96AB) (?7Ab )B u P h ^ iO
V 3 4
BuPh-SiO
X •
2 1X Q
(98AB)
O(99AB)
To a stirred solution of the crude mixture of dichioroadducts
(96AB) and (97A8) (12.50g, 27.8 mmol) in dry THF/methanol (100 ml :
25 ml) at 0°C was added powdered ammonium chloride (lOg), followed by
zinc powder (lOg). After 20 min, the mixture was allowed to warm to
room temperature and stirred for 24 h. The solution was then diluted
with petrol (200 ml) to precipitate the zinc salts and filtered through
a short pad of silica gel. The solid was washed several times with
further petrol. Removal of the solvent under reduced pressure
followed by chromatography of the residue (10% diethyl ether-petrol)
gave each of the v e g i o i s o m e v i o c y c l o b u t a n o n e s (98AB) (6.84g, 60.9% from
alkene (91)) and (99AB) (1.13g, 10.1%) both a white crystalline solids,
less polar (98AB) : v (CHC13) 3070, 2960, 1172, 1112, and 823 cm’1;
6 (250 MHz) 0.99 (1.5H, d, J 6.5 Hz, Me), 1.00 (1.5H, d, J 6.5 Hz, Me),
1.06 (9H, s, tBu), 1.12 (1.5H, d, J 6.5 Hz, Me), 1.22 (1.5H, d, J 6.5
Hz, Me), 2.03 (1H, m, 11-H), 2.27 (1H, ddd, J 16.5, 3.5, and 2 Hz,

282
4-H), 2.42 (1H, m, 3-H). 3.15 (1H, ddd, J 16.5, 9, and 2.5 Hz, 4-H),
3.20 (1H, m, 2-H), 3.40 (1H, dd, J 10 and 6 Hz, 2'-H), 3.49 (1H, dd,
J 10 and 4 Hz, 2'-H), 7.40 (6H. m, Ph), and 7.76 (4H, m, Ph); m/z 338
(M+-CH2C0), 323 (M+-tBu), 281 (M+-tBu-C0CH2), and 253 (M+-tBu-CH(Me)-
CH2C0); (Found: C, 75.56; H, 8.56. C2ttH3 202Si requires C, 75.57;
H, 8.47%).
67. Preparation of Methyl 2-(/?)-methyl-3-pheny1 thiopropionate (100)
.C02Me
X)H
(92)
To a stirred solution of the alcohol (92) (5.00g, 42.4 mmol)
in dry THF (100 ml) at room temperature was added tri-n-butylphosphine
(15.80 ml, 63.4 mmol) followed by diphenyl disulphide (13.90g, 63.8
mmol) in THF (50 ml). After stirring overnight diethyl ether (100 ml)
and water (50 ml) were added followed by extraction of the aqueous
layer with diethyl ether (2 x 50 ml). The combined organic extracts
were washed once with water (1 x 50 ml) and then dried (Na2S0£+).
Concentration under reduced pressure followed by chromatography
(gradient elution, petrol ■+ 10% diethyl ether-petrol) afforded m e t h y l
2-(R)-m e t h y l - 3 - p h e n y l t h - i o p r o p i o n a t e (100) (7.83g, 88%) as a colourless
oil, [a]n 2 5 + 57.2° {c 6.7, CHC1 3); \> (film) 3058, 2976, 1737,
1212, 1166, 741, and 692 cm"1; 6 (90 MHz) 1.28 (3H, d, J 7 Hz, Me),
2.30 - 3.35 (3H, m, 2-H and Ctf2SPh), 3.65 (3H, s, OMe), and 7.32 (5H,
m. Ph); m/z 210 (M+), 179 (M+-0Me), 151 (M+-C02Me), 150 (M+-MeC02H),

283
123 (PhSCH2+), 109 (PhS+), and 77 (Ph+); (Found: M+ , 210.0703.
C1:Hit+02S requires M+, 210.0715); (Found: C, 62.64; H, 6.78.
C n H ltf02S requires C. 62.83: H, 6.71%).
68. Preparation of 2-(7?)-Methyl-3-ohenythiopropan-l-ol (101)
^/C02Me
SPh
(100)
OH
I2 1
3 ,SPh
M
To a suspension of lithium aluminium hydride (720 mg, 18.9 mmol)
in dry diethyl ether (50 ml) at 0°C under argon was added the ester
(100) (5.20g, 24.8 mmol) in diethyl ether (20 ml) dropwise. After
addition was complete (ca. 15 min) the solution was allowed to warm
to room temperature and stirred for a further 30 min. The reaction
was recooled to 0°C and quenched by dropwise addition of wet ethyl
acetate (5 ml). Saturated aqueous ammonium chloride solution (20 ml)
and diethyl ether (50 ml) were added followed by extraction of the
aqueous layer with diethyl ether (3 * 50 ml). The combined organic
extracts were washed with water (1 x 20 ml), brine (1 x 20 ml) and
dried (Na2S0,J. Concentration under reduced pressure and chromato
graphy (50% diethyl ether-petrol) of the residue gave {R)-2-methyl-2-
phenylthiopropan-l-ol (101) (4.00g, 89%) as a colourless oil, [a]^25
- 20.1 {c 14.5, CHC13); v (film) 3371, 3058, 2958, 2925, 1028,
739, and 691 cm"1; 6 (60 MHz) 0.98 (3H, d, J 7 Hz, Me), 2.00 (1H, m,
2-H), 2.10 (1H, br s, OH), 2.88 (2H, m, C#2SPh), 3.52 (2H, d, J 5 Hz,
CH20H), and 7.18 (5H, m, Ph); m/z 182 (M+), 123 (M+-CH(Me)CH20H),

284
110 (PhSH+), 109 (PhS+), and 77 (Ph+); (Found: M+, 182.0782.
Ci0Hi4OS requires M+, 182.0765); (Found: C, 66.03; H, 7.97.
CioHiz+OS requires C, 65.89; H, 7.74%).
69. Preparation of 2-(R)-Methyl-3-phenylthiopropan-l-al (102)
To oxalyl chloride (1.92 ml, 22.0 mmol) in dichioromethane
(60 ml) under argon was added DMSO (3.12 ml, 44.1 mmol) in dichloro-
methane (20 ml) dropwise maintaining the temperature at ca. -60°C.
After addition was complete, the solution was stirred for a further
5 min, before addition of the alcohol (101) (2.00g, 11.0 mmol) in
dichioromethane (20 ml). After 15 min at ca. -60°C, triethyl amine
(12.20 ml, 88.0 mmol) was added and the reaction mixture warmed to
room temperature over 30 min. Work-up as described in Experiment 63
and chromatography (20% diethyl ether-petrol) gave 2-(R)- m e t h y7-3-
v h e n y t t h ' i o ' p r o v a n ~ ^ ~ a t (102) (1.22g, 62%) as a colourless oil, vmax.(film) 3058, 2971, 2931, 1724, 741, and 690 cm'1; 6 (60 MHz) 1.20
(3H, d, J 7 Hz, Me), 2.50 - 3.50 (3H, m, 2-H and C/72SPh), 7.22 (5H,
m, Ph), and 9.55 (1H, d, J 1 Hz, CH0); m/z 180 (M+), 123 (PhSCH2+),
110 (PhSH+), and 77 (Ph+); (Found: C, 66.30; H, 6.69. C10H12OS
(101}
requires C, 66.63; H, 6.71%).

285
70. Preparation of (z)-(ff)-l-Phenylthio-2-methy1pent-3-ene (103)
5
bPh0°2) (103)
To ethyltriphenylphosphonium iodide (2.97g, 7.11 mmol) suspended
in THF (70 ml) at 0°C was added nBuLi (4.75 ml of a 1.50M solution in
hexane, 7.13 mmol) dropwise with rapid stirring. After 30 min, the
resultant red solution was precooled to -78°C and the aldehyde (102)
(1.22g, 6.78 mmol) in dry THF (10 ml) added dropwise. After stirring
for 5 min at -78°C, the reaction was allowed to warm to room temperature
over 30 min. Work-up as described in Experiment 64 and chromatography
(5% diethyl ether-petrol) afforded the (Z)-olefin (103) (l.Olg, 78%)
as a colourless oil, vmax (film) 3059, 3008, 2961, 1654, 1092, 1026.
738, and 690 cm"1; 5 (250 MHz) 1.08 (3H, d, J 6 Hz, Me), 1.57 (3H, dd,
J 7 and 2 Hz, =CHAfe). 2.76 (1H, m, 2-H), 2.86 (2H, m, Ctf2SPh), 5.23
(1H, m, J 10.5 Hz, 3-H), 5.46 (1H, qdd, J 10.5, 6 and 1 Hz, 4-H), and
7.10 - 7.35 (5H, m, Ph).

236
71. Preparation of 2-{r )-\ {1-(i?)-Methyl-2-phenvlthio)ethyl]-3-(di
methyl cycl obutanone and \ZS, 3i?1 isomer (105AB) and 2-(5)-Methyl-
3-(ff)-(l-(fl)-meth,yi-2-pheny1thio)ethy1cyc1obutanone and [2.?, 37?1
isomer (106AB)
X r~ i2" t c
2
• A * / 4 1 ^ 0
'SPh
(105AB) (106AB)2 1
By the procedure reported in Experiment 65, the (Z)-olefin (103)
(390 mg, 2.03 mmol) in diethyl ether (50 ml) was treated with dichloro-
ketene in situ, generated as before by the dropwise addition of tri-
chloroacetyl chloride (0.68 ml, 6.10 mmol) to zinc-copper couple (0.5g)
and the olefin at reflux. Work-up as oefore gave an unstable mixture
of dichioroadducts (104) which was reduced directly with zinc as
described in Experiment 66 to afford an inseparable mixture of cyclo-
butanones (105AB) and (106AB) (ca. 2:1) [216 mg, 45% from (103)J as a
colourless oil. v (film) 3058, 2961, 2925, 1768, 1089. 1025, 740,
and 692 cm-1; 6 (250 MHz) 1.02 (2H, t, J 7 Hz, Me, major regioisomer,
two diastereoisomers), 1.10 (1H, d, J 7 Hz, Me, minor regioisomer,
two diastereoisomers), 1.14 (l‘H, d, J 7 Hz, Me, major regioisomer),
1.25 (1H, d, J 7 Hz, Me, major regioisomer), 1.33 (0.5H, d, J 7 Hz,
Mering, minor regioisomer), 1.36 (0.5H, d, J 7 Hz, Mer- , minor
regioisomer), 2.00 - 2.32 (2H, m), 2.48 - 3.76 (5H, m), and 7.10 -
7.40 (5H, m, Ph); m/z 234 (M+), 192 (M+-CH2C0), 124 (M+-PhSH), 123
(PhSCH2+), 110 (PhSH+), and 109 (PhS+); (Found: M+, 234.1083.
Cit*Hi80S requires M+, 234.1078).

287
72. Preparation of 4-(fl)-r(2-t-Butyldiphenylslly’]ox,y-l-(£)-methyl )-
ethyl]-3-(ff)-methylbutan-4-olide (107A) and 4-(£,)-r(2-t-Butyl-
diphenylsilyloxy-1-(g)-methyl)ethy!1-3-(r )-methylbutan-4-olide
(1Q7B)
To the diastereoisomeric mixture of cyclobutanones (6.00g, 15.8
mmol) in glacial acetic acid (20 ml) at ca. 5°C was added hydrogen
peroxide (5.40 ml of a 30% aqueous solution, 47.6 mmol) and the mixture
stirred for 8 h. The excess peroxide was destroyed by addition of
dimethyl sulphide (1 ml) followed by removal of the solvent under
reduced pressure. The resulting residue was chromatographed (25%
diethyl ether-petrol) to afford an inseparable mixture of 4-(R)-[(2-t-
butyldiphenylstlyloxy-l- (S) -methyl) ethyl] - 3- (S) -methylbutan-Q-olide
(107A) and 4-(S)-[(2-t-butyIdiphenyIsdlyloxy-1-(S)-methyl)ethyl]-2-(R)-
methylbutan-4-ol-ide (1078) (6.00g, 96%) as a semi-solid. The lactones
were separated by high pressure liquid chromatography using high resolu
tion silica gel (7.5%isopropanol-hexane) to afford (107A) (2.95g. 47%)
as a white crystalline solid (m.p. 77°C) and (107B) (2.95g, 47%) as a
viscous colourless oil, more polar (107A) M q 25 - 20.3° (e 7.7. CHC13)
vm=v (CHC13) 3070, 2931. 2858, 1780, and 1113 cm"1; 6 (250 MHz) 0.98
(3H. d, J 7 Hz, C-l* methyl), 1.03 (3H, d, J 6.5 Hz, C-3* methyl), 1.08
(9H, s, tBu), 1.90 (1H, m, l'-H), 2.20 (1H, d, J 16.5 Hz, 2-H), 2.54
(1H, m, 3-H), 2.75 (1H, dd, J 16.5 and 7.5 Hz, 2-H), 3.76 (1H, dd,

288
J 10 and 5 Hz, 2'-H), 3.82 (1H, dd, J 10 and 3.5 Hz, 2'-H), 4.30 (1H,
dd. J 10.5 and 4.5 Hz, 4-H), 7.40 (6H, m, Ph), and 7.76 (4H, m, Ph);
m/z 339 (M+-tBu) and 309 (M+-CH(Me)CH2C02H); (Found: Fl+-tBu, 339.1421.
C2 ^ 3 203Si requires M+-tBu, 339.1416); (Found: C, 72.91: H, 8.23.
C21+H3 203 Si requires C. 72.68; H, 8.13%).
Less polar (107B): [a]n25 + 24° {a 5.7, CHC13); v v (film) 3040, 2932,u m3 X •1778, and 1112 cm-1: 5 (250 MHz) 0.93 (3H, d, J 7 Hz, C-3 methyl),
1.07 (9H, s, tBu), 1.11 (3H, d, J 6.5 Hz, C-l' methyl), 2.02 (1H, m,
l'-H), 2.17 (1H, dd, J 17 and 2.5 Hz, 2-H), 2.47 (1H, m, 3-H). 2.68
(1H, dd. J 17 and 7.5 Hz, 2'-H), 3.53 (1H, dd, J 10 and 5 Hz, 2'-H),
3.59 (1H, dd, J 10 and 5.5 Hz, 2'-H), 4.37 (1H, dd, J 8.5 and 5 Hz,
4-H), 7.40 (6H, m, Ph), and 7.76 (4H, m, Ph); m/z 339 (M+-tBu) and
309 (M+-CH(Me)CH2C02H); (Found: M+-tBu, 339.1421. C2l+H3 203Si
requires M+-tBu, 339.1516); (Found: C, 72.49; H, 8.42. C21+H3 203Si
requires C, 72.68; H, 8.13%).
73. Preparation of 2-(i?)-(2-t-Butyldi phenyl si 1yloxy-1-[S)-methyl)ethyl -
5-h,ydroxy-3-(5)-methyl tetrahydrofuran (108A)
To a stirred solution of the lactone (107A) (2.13g, 5.38 mmol) in
dry toluene (40 ml) at -78°C was added DIBAL (4.67 ml of a 1.5M solution
in toluene, 7.00 mmol) dropwise. After stirring at -78°C for 1 h,
glacial acetic acid (1.2 ml) was added dropwise followed by warming to

289
room temperature. Water (1.7 ml) was then added and the solution
poured into a flask containing ethyl acetate (50 ml) and solid sodium
bicarbonate (5g). After shaking and then allowing to stand for 10 min,
the supernatant liquid was filtered through a short silica gel pad.
The remaining solid was extracted with further ethyl acetate (4 x 50 ml),
these extracts being filtered through the same silica gel pad. The combined
organic solution was evaporated under reduced pressure and the crude
residue chromatograohed (35% diethyl ether-petrol) to afford 2-(R)-(2-
t-butylchiphenylszlyloxy-1- (S)-methyl) ethyl-b-hydroxy -3-(S)-methyltetvar
hydrofuran (108A) (2.10g, 97%) as a colourless oil, M q25 - 5.7° (a 9.4,
CHC13); v (film) 3410, 3070, 2930, 1112, 1064, 997, and 823 cm-1;
6 (90 MHz) 0.80 (3H, d, J 7 Hz, Me), 1.05 (9H, s, tBu), 1.10 (3H, d,
J 7 Hz, Me), 1.70 - 2.30 (4H, m, 4-H2, 3-H, and l'-H), 3.20 - 3.80 (3H,
m, 2'-H2 and OH), 3.96 (1H, dd, J 8 and 5 Hz, 2-H), 5.50 (1H, m, 5-H),
7.40 (6H. m, Ph), and 7.65 (4H, m, Ph); m/z 341 (M+-tBu) and 323
(M+-tBu-H20); (Found: C, 72.57; H, 9.04. C2t+H3t+03Si requires
C, 72.32; H, 8.60%).
74 . Preparation of 2-( S)-(5-Hydroxy-3-(ff)-meth.y1tetrah.ydrofuran-2-(fl)-
yl )propan-l-ol (109A)
To the lactol (108A) (422 mg, 1.06 mmol) in THF (10 ml) at room
temperature was added tetra-n-butylammoniurn fluoride (1.17 ml of a 1.0M

290
solution in THF, 1.17 mmol). The solution was stirred until reaction
was complete (t.l.c.) {oa. 1 h) and the solvent then removed under
reduced pressure. Chromatograohy (gradient elution, 50% ethyl acetate-
petrol -» ethyl acetate) afforded 2-{S)-{5-hydroxy-2>-{$)-methyltetva-
hydrofuran-2-{R)“yl)propan-l-ol (109A) (151 mg, 89%) as a viscous
colourless oil, v „ (film) 3378, 2966, 2878, 1059, 994. and 896 cm-1;
6 (90 MHz) 0.85 (3H, d, J 7 Hz, Me), 1.70 - 2.45 (4H, m, 4'-H2, 3'-H,
and 2-H), 3.40 - 3.90 (4H, m, C H 20H and OH), 3.94 (1H. dd, J 10.5
and 5 Hz, 2'-H), and 5.40 - 5.70 (1H. m, 5 1 -H ); m/z 142 (M+-H20) and
101 (M+-CH(Me)CH20H); (Found: C, 59.54; H, 10.20. C8H1603 requires
C, 59.98; H, 10.07%).
75. Preparation of 2-(ff)-(5-Hydroxy-3-(i?)-methyltetranydrofuran-2-(5)-
yl )propan-l-o1 (109B)
The lactol (108B) (177 mg, 0.45 mmol) (obtained from the reduction
of lactone (107B) following the procedure of Experiment 73) was treated
with tetra-n-butylammoniurn fluoride (0.49 ml of a 1.0M solution in THF,
0.49 mmol). Work-up as described in Experiment 74 followed by
chromatography (gradient elution, 50% ethyl acetate-petrol -> ethyl
acetate) afforded 2-(S)-(5-hydroxy-2-(R)-methyltetvahydrofuran-2-($)-
yl) propan-1-ol (109B) (62 mg, 87%) as a viscous colourless oil, vmax.
(film) 3404, 2966, 2880, 1034, 992, and 897 cm"1; 6 (90 MHz) 0.98 (3H,

291
d, J 7 Hz, Me), 1.02 (3H, d, J 6.5 Hz, Me), 1.58 - 2.65 (6H, m, 4'-H2,
3' -H, 2-H, and OH (X2) ), 3.38 - 3.98 (2H, m, Ctf20H). 4.12 (1H, t,
J 6.5 Hz, 21-H), and 5.38 - 5.62 (1H, m, 5'-H); m/z 142 (M+-H20) and
101 (M+-CH(Me)CH2OH); (Found: C, 59.94; H, 10.29. C8H1503
requires C, 59.98; H, 10.07%).
76. Preparation of (27)- Ethyl (55, 67?. 7S)-6,8-dioxy-6.8-0-1sopropy1 -
idene-5,7-dimethyloct-2-enoate (1 H A T )
OH
To carbethoxymethylidenetriphenylphosphorane (175 mg, 0.50 mmol)
in dichioromethane (5 ml) at room temperature was added the lactol
(109A) (74 mg, 0.46 mmol) in dichioromethane (5 ml). After stirring
for ca. 48 h reaction was complete (t.l.c.) and the solution was con
centrated under reduced pressure. Chromatography of the residue
(diethyl ether) afforded the a, fl-unsaturated ester (110A) (95 mg, 90%)
(oa . 5:1, E: Z mixture) as a colourless oil. The diol-ester (110A)
(95 mg, 0.40 mmol) was immediately taken up in acetone followed by
addition of anhydrous copper sulphate (200 mg) and a trace of CSA.

292
The reaction was stirred at room temperature for 30 min and then
filtered through a short pad of silica gel. After concentration
under reduced pressure, the residue was chromatographed (10% diethyl
ether-petrol) to afford (Z)-ethyl (5S, 6R, 7S)-6,8-dioxy-6,8-0-iso-
propylidene-5,7-dimethyloct-2-enoate (111AC) (15 mg, 13%) and (£’)-
ethyl (55, 65, 75)-6.8-dioxy-6,8-0-isopropylidene-5,7-dimethyloct-2-
enoate (73 mg, 66%) (111AT) both as colourless oils, less polar (Z)-
isomer (111AC): v v (film) 2964, 1718, 1639, 1383, and 1181 cm"1;
6 (250 MHz) 0.71 (3H, d, J 7 Hz, Me), 0.92 (3H, d, J 7 Hz, Me), 1.29
(3H, t, J 7 Hz, C02CH2C53), 1.35 (3H, s, acetonide Me), 1.37 (3H,
s, acetonide Me), 1.86 - 2.05 (2H, m, 5-H and 7-H), 2.68 (2H, m, 4-H2),
3.43 (1H. dd, J 9.5 and 2.5 Hz, 6-H), 3.48 (1H, t, J 11.5 Hz, 8-H ),S X •
3.69 (1H, dd, J 11.5 and 5 Hz, 8-H ), 4.17 (2H, q, J 7 Hz, C02C52CH3),
5.80 (1H, dt, J 11.5 and 1.5 Hz, =C5C02Et), and 6.27 (1H, dt, J 11.5
and 7.5 Hz, C5=CHC02Et); m/z 270 (M+, weak), 255 (M+-Me), 171 (M+-
CH=CHC02Et), and 129 (M+-CH(Me)CH2CH=CHC02Et); (Found: M+-Me, 255.1600.
Ci5H2 601+ requires M+-Me, 255.1596).
More polar (5)-isomer (1 H A T ): v (film) 2969, 1718, 1650, 1380,max.1199, 1181, and 1117 cm"1; 6 (250 MHz) 0.70 (3H, d, J 7 Hz, Me), 0.91
(3H, d, J 7 Hz, Me), 1.29 (3H, t, J 7 Hz, C02CH2C53), 1.37 (3H, s,
acetonide Me), 1.38 (3H, s, acetonide Me), 1.75 - 1.94 (2H, m, 5-H and
7-H), 2.10 - 2.36 (2H, m, 4-H2), 3.30 (1H, dd, J 12 and 2.5 Hz, 6-H),
3.38 (1H, t, J 11.5 Hz, 8-H ), 3.69 (1H, dd, J 11.5 and 5 Hz, 8-H ),eq. ax.4.09 (2H, q, J 7 Hz, C02Ctf2CH3), 5.82 (1H, dt, J 15.5 and 1.5 Hz,
=C#C02Et), and 6.94 (1H, ddd (7 lines), J 15.5, 8, and 7.5 Hz,
C/7=CHC02Et); m/z 270 (M+ , weak), 255 (M+-Me), 226 (M+-Me-Et), 209
(M+-Me-EtOH), 171 (M+-CH=CHC02Et), and 129 (M+-CH(Me)CH2CH=CHC02Et);
(Found: M+-Me, 255.1600. C15H2601+ requires M+-Me, 255.1596).

293
77 . Preparation of (ff,Z)-Ethyl [5/?, 6 S. 751-6.8-di hydroxy-5,7-
dimethyloct-2-enoate (110B)
The did (109B) (43 mg, 0.27 mmol) was treated with carbethoxy-
methylidenetriphenylphosphorane (103 mg, 0.30 mmol) in dichloro-
methane (5 ml). After oa. 6 h and work-up as described previously
in Experiment 76, chromatography (diethyl ether) afforded the a,(3-
unsaturated ester (HOB) (51 mg, 83%) {oa. 5:1, (E)\{Z), homogeneous
by t.l.c.) as a colourless oil, v (film) 3390, 2929, 1701, 1648,max.1180, and 981 cm-1; 6 (90 MHz) 0.98 (3H, d, J 7 Hz, Me), 1.02 (3H, d,
J 7 Hz, Me), 1.28 (3H, t, J 7 Hz, C02CH2Ctf3), 1.65 - 2.70 (6H, m, 4-H2,
5-H, 7-H, and OH (x2)), 3.40 - 3.78 (3H, m, 6-H and 8-H2), 4.18 (2H,
q, J 7 Hz, C02C#2CH3), 5.45 (0.17H, m, =C#C02Et, (Z)-isomer), 5.82
(0.83H, d, J 15.5 Hz, =CffC02Et, {E)-isomer), 6.25 (0.17H, m, Ctf=CHC02Et,
(Z)-isomer), and 6.92 (0.83H, dt (5 lines), J 15.5 and 7.5 Hz,
Ci7=CHC02Et, (E)-isomer); m/z 212 (M+-H20), 171 (M+-CH(Me)CH20H), 142
(M+-CH(Me)CH20H-Et), and 125 (M+-CH(Me)CH20H-Et0H); (Found: M+-
CH(Me)CH20H, 171.1022. C12H220u requires M+-CH(Me)CH20H, 171.1021).
HO
(l09B) (h o b )

294
78. Preparation of (ff)-Ethyl (5E, 65. 7<S')-6,8-dioxy-6.8-0-isopropy1-
idene-5,7-dimethyloct-2-enoate (111BT)
The diol-ester (HOB) (25 mg, 0.11 mmol) was protected as the
acetonide in an analogous manner to the procedure described in
Experiment 76. Chromatography (10% diethyl ether-petrol) of the
residue after work-up afforded the (Z)-a,£-unsaturated ester acetonide
(111BC) (3 mg, 10%) and corresponding (E)-isomer (111BT) (19 mg, 63%)
both as colourless oils, less polar (Z)-isomer: v (CHC1,) 2988,
1717, 1641, 1270, and 1178 cm-1; 6 (250 MHz) 0.92 (3H, d, 0 6.5 Hz,
Me), 1.06 (3H, d, J 7 Hz, Me), 1.28 (3H, t, J 7 Hz, C02CH2CH 3 ) , 1.38
(3H, s, acetonide Me), 1.39 (3H, s, acetonide Me), 1.52 - 1.74 (2H, m,
5- H and 7-H), 2.52 - 2.62 (2H, m, 4-H2), 3.52 (1H, dd, J 11 and 2.5 Hz,
6- H). 3.58 (1H, dd, J 11.5 and 2 Hz, 8-H ), 4.04 (1H. dd, J 11.5 andecj.
2.5 Hz, 8-H ), 4.14 (2H, q, J 7 Hz, C02CE2CH3), 5.82 (1H, dt, J 11.5d X i
and 2 Hz, =CEC02Et), and 6.20 (1H, ddd, J 11.5, 8, and 7 Hz,
CE=CHC02Et); 777/3 270 (M+ , weak), 255 (M+-Me), 209 (M+-Me-Et0H), 171
(M+-CH=CHC02Et), and 129 (M+-CH(Me)CH2CH=CHC02Et); (Found: M+-Me,
C i5H260h requires M+-Me, 255.1596).255.1600.

295
Mors polar (^)-isomer: v (film) 2988, 1718, 1650, 1270, and 1178ITlclX .
cm-1; 6 (250 MHz) 0.94 (3H. d, J 6.5 Hz, Me), 1.07 (3H, d, J 7 Hz, Me),
1.29 (3H, t, J 7 Hz, C02CH2Ctf3), 1.39 (3H. s, acetonide Me), 1.40 (3H,
s, acetonide Me), 1.52 - 1.62 (1H, m), 1.73 (1H, m), 1.91 (1H, dddd
(12 lines), J 13.5, 8, 8, and 1 Hz, 4-H), 2.28 (1H. m, J 13.5 Hz, 4-H),
3.51 (1H, dd, J 9.5 and 2.5 Hz, 6-H), 3.59 (1H, dd, J 11.5 and 1.5 Hz,
8-H^v ), 4.07 (1H, dd, J 11.5 and 3 Hz, 8-H ), 4.19 (2H, q, J 7 Hz,ax. eq.
C02Ctf2CH3), 5.84 (1H, dt, J 15.5 and 1 Hz, =C£C02Et), and 6.92 (1H,
ddd (7 lines), J 15.5, 9, and 6.5 Hz, Ctf=CHC02Et); m/z 270 (M^, weak),
255 (M+-Me), 226 (M+-Me-Et), 209 (M+-Me-Et0H), 171 (M+-CH=CHC02Et),
and 129 (M+-CH(Me)CH2CH=CHC02Et); (Found: M+-Me, 255.1600.
Ci5H2604 requires M+-Me, 255.1596).
79 . Preparation of (ff)-(55, 6R, IS)-6,8-Diox,y-6,8-0-isopropyl idene-
5, 7-dimethyloct-2-en-l-ol (112)
To the (£)- a, (3-unsaturated ester (1 H A T ) (40 mg, 0.15 mmol) in
toluene (2 ml) at -78°C was added DIBAL (0.24 ml of a 1.50M solution
in toluene, 0.36 mmole) dropwise. The resulting solution was stirred
for 30 min after which time reaction was complete (t.l.c.). Work-up
as before (Experiment 62) and chromatography (35% diethyl ether-petrol)
afforded the (E)-allylic alcohol (112) (27 mg, 80%) as a colourless
oil, v (film) 3394, 2964, 1668, 1383, and 1200 cm-1; 6 (250 MHz)max.
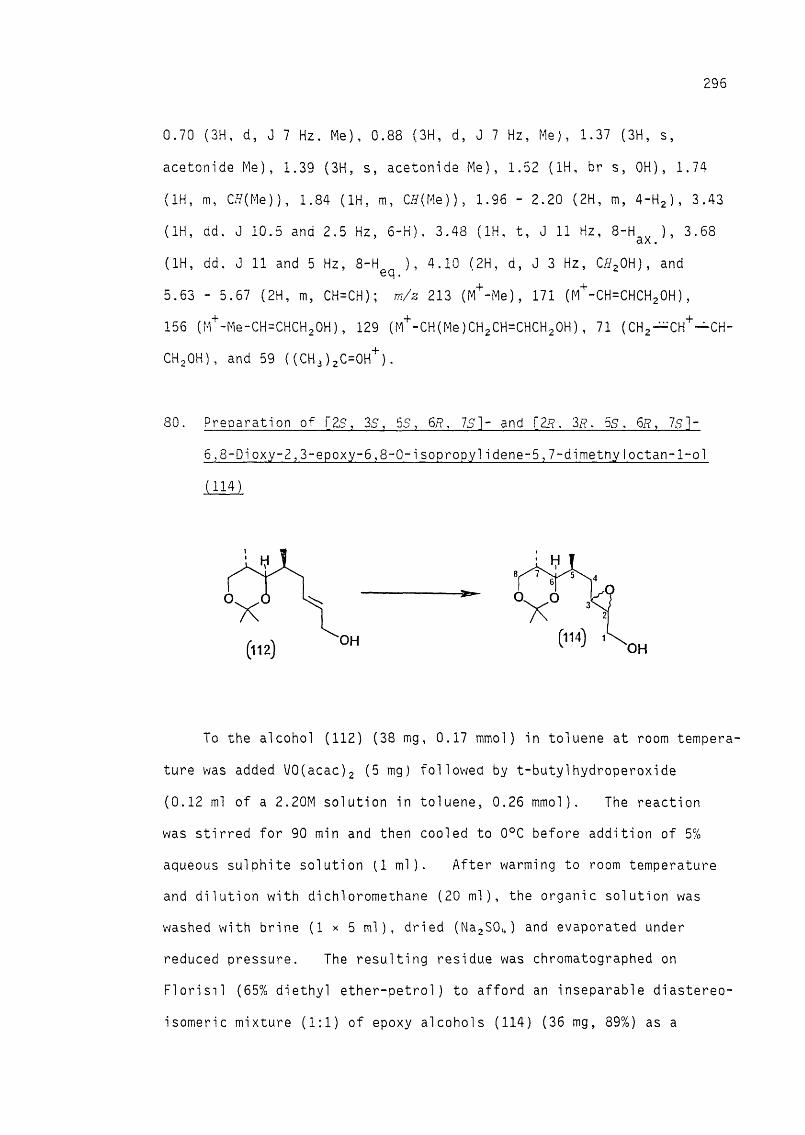
296
0.70 (3H, d, J 7 Hz. Me), 0.88 (3H, d, J 7 Hz, Me), 1.37 (3H, s,
acetonide Me), 1.39 (3H, s, acetonide Me), 1.52 (1H, br s, OH), 1.74
(1H, m, Ctf (Me)), 1.84 (1H, m, Ctf(Me)), 1.96 - 2.20 (2H, m, 4-H2), 3.43
(1H, dd, J 10.5 and 2.5 Hz, 6-H), 3.48 (1H. t, J 11 Hz, 8-H v ), 3.68
(1H, dd, J 11 and 5 Hz, 8-H ), 4.10 (2H, d, J 3 Hz, Ctf20H), andecj.
5.63 - 5.67 (2H, m, CH=CH); m/z 213 (M+-Me), 171 (M+-CH=CHCH20H),
156 (M+-Me-CH=CHCH20H), 129 (M+-CH(Me)CH2CH=CHCH20H), 71 (CH2— CH+— CH-
CH20H), and 59 ((CH3)2C=0H+ ).
80. Preparation of [25, 35, 5g, 6/?, 75]- and 1~2R. 3i?. 5S. 6R, 7g]-
6 ,8-Di oxy-2,3-epoxy-6,8-0-i sopropy1i dene-5,7-dimethy 1octan-l-ol
(114)
To the alcohol (112) (38 mg, 0.17 mmol) in toluene at room tempera
ture was added V0(acac)2 (5 mg) followed by t-butylhydroperoxide
(0.12 ml of a 2.20M solution in toluene, 0.26 mmol). The reaction
was stirred for 90 min and then cooled to 0°C before addition of 5%
aqueous sulphite solution (1 ml). After warming to room temperature
and dilution with dichioromethane (20 ml), the organic solution was
washed with brine (1 * 5 ml ) , dried (Na2S01+) and evaporated under
reduced pressure. The resulting residue was chromatographed on
FI ori si 1 (65% diethyl ether-petrol) to afford an inseparable diastereo-
isomeric mixture (1:1) of epoxy alcohols (114) (36 mg, 89%) as a

297
colourless oil (7?p 0.38, 75% diethyl ether-petrol), vmax (CHC13) 3435,
2970, 1384, 1257, 1236, 1200, and 1171 cm’1; 6 (250 MHz) 0.73 (3H, d,
J 7.5 Hz, Me), 0.95 (3H, d, J 7.5 Hz, Me), 1.35 (1.5H, s, acetonide
Me), 1.35 (1.5H, s, acetonide Me), 1.40 (1.5H. s, acetonide Me), 1.41
1.5H, s, acetonide Me), 1.48 - 1.76 (2H. m, 5-H and 7-H), 1.78 - 2.01
(2H, m, 4-H2), 2.08 (1H, br t, OH), 2.92 (1H, m, 3-H), 3.00 (1H, m,
2-H), and 3.42 - 3.94 (5H, m, Ctf20H, 6-H, and 8-H2): m/z 229 (M+-Me),
129 (M+-CH(Me)CH2CH(0)CHCH20H), 87 (CH2CH(0)CHCH20H), and 59 (CH3C02+ );
(Found: M+-Me, 229.1437. Ci3H21+0„ requires M+-Me, 229.1434).
81. Preparation of [2 H, 3 S. 55, 6 R, IS]- and \Z S, 31?, 5S, 6/?, 75j-
6.8-Dioxy-2,3-epoxy-6 ,8-0-i sopropyli dene-5,7-dimethyl octyl phenyl
selemde (115)
To the diastereoisomeric mixture of epoxy alcohols (114) (21 mg,
0.09 mmol) in THF (1 ml) at -20°C was added tri-n-butylphosphine (42 pi,
63a0.18 mmol) followed by N-phenylselenophthalimide (52 mg, 0.18 mmol)
in THF (1 ml). After stirring for 10 min, the mixture was diluted
with diethyl ether (15 ml) and water (5 ml). The aqueous layer was
extracted with diethyl ether (3 x 10 ml) and the combined organic
extracts washed with water (1x5 ml). Drying (Na2S0£+) and concentra
tion under reduced pressure gave a yellow oil which was purified by
chromatography on Florisil (gradient elution, petrol 15% diethyl

298
ether-petrol) to afford an inseparable diastereoisomeric mixture (1:1)
of epoxy selenides (115) (24 mg, 70%) as a colourless oil, v (film)max.
2928, 2852, 1265, 1235, 1198, 1059, 1010, and 737 cm"1; 6 (250 MHz)
0.68 (3H. d, J 7 Hz, Me), 0.85 (i.5H, d, J 7 Hz, Me), 0.88 (1.5H, d,
J 7 Hz, Me), 1.23 (1.5H, s, acetomde Me), 1.29 (1.5H, s, acetonide Me),
l. 30 (1.5H, s, acetonide Me), 1.37 (1.5H, s, acetonide Me), 1.42 (1H,
m, C#(Me)), 1.55 (1H, m, Ctf(Me)), 1.80 (2H, m, 4-H2), 2.68 (1H, m, 3-H),
2.80 (1H, dt, J 14 and 8 Hz, Ctf2SePh), 2.92 (1H, m, 2-H), 3.12 (1H,
ddd (7 lines), J 14, 9, and 5 Hz, C#2SePh), 3.35 - 3.50 (2H, m, 6-H
and 8-H), 3.65 (1H, dd, J 11.5 and 5 Hz, 8-H), 7.25 (3H, m, Ph), and
7.52 (2H. m, Ph); m/z 384 (M+ ), 369 (M+-Me), 213 (M+-CH28°SePh),
157 (Ph80Se+ ). and 129 (M+ - CH(Me)CH2CH(0)CHCH280SePh); (Found:
M+ . 384.1210. C19H2 303 80Se requires M+ , 384.1204).
82. Preparation of (ff)-Ethyl [55, 67?, 751-8-t-butyldiphenyl silyloxy-
6-hydroxy-5,7-dimethyloct-2-enoate (117)
The lactol (108A) (1.26g, 3.20 mmol) and carbethoxymethylidenetri-
phenylphosphorane (1.22g, 3.50 mmol) in dry dichioromethane (30 ml)
under argon were stirred at room temperature for gcl. 48 h. After con
centration under reduced pressure, the residue was chromatographed (25%
diethyl ether-petrol) to afford (E)-e t h y l [5S. 6R, 1 ^ 1 ~ S - t - b u t y l d i p h e n y l ~
s ' C l y ' l o x y - & - h y d x > o x y ~ ii ,7 ~ d i m e t h y t o o t ~ 2 . - e n o a t e (117) (880 mg, 75%) and the

299
corresponding {l)-isomev (116) (slightly less polar) (140 mg, 12%) both
as colourless oils, less polar (z)-isomer (116): M q 25 “ 11.7°
{o 2.2, CHC13); v (film) 3498. 3071, 2962, 2858, 1700, 1639, 1213,
1183, 1112, 910, and 823 cm-1; 6 (250 MHz) 0.84 (3H, d, J 7 Hz, Me),
0.96 (3H, d, J 7 Hz, Me), 1.05 (9H, s, tBu), 1.26 (3H, t, J 7 Hz,
C02CH2C7/3), 1.79 (2H, m, 5-H and 7-H), 2.41 (1H, m, from decoupling,
J 14 and 6.5 Hz, 4-H), 2.94 (1H, m, from decoupling, J 14 and 9 Hz, 4-H),
3.52 (1H, dt, J 9 and 3 Hz, CtfOH), 3.69 (1H, dd, J 9.5 and 6 Hz, 8-H),
3.69 (1H, d, J 4 Hz, OH), 3.82 (1H, dd, J 9.5 and 4 Hz, 8-H), 4.16 (2H,
q, J 7 Hz, C02C/72CH3), 5.85 (1H. dd, J 11.5 and 1 Hz, =CtfC02Et), 6.28
(1H, ddd, J 11.5, 9, and 7.5 Hz, C77=CHC02Et), 7.40 (6H, m, Ph), and
7.68 (4H, m, Ph); m/z 411 (M+-tBu), 381 (M+-CH2C02Et), 365 (M+-tBu-
EtOH), and 199 (M+-CH20SitBuPh2); (Found: M+-tBu, 411.2001.
C2sHu0OaSi requires M+-tBu, 411.1991); (Found: C, 71.97; H, 8.83.
^ a ^ o ^ S i requires C, 71.75; H, 8.60%).
More polar (iTJ-isomer: [a]D25 + 16.5° {o 9.1, CHC13); (film)
3498, 3071, 2961, 1717, 1648, 1215, 1176, 1113, and 823 cm"1;
6 (250 MHz) 0.71 (3H, d, J 7 Hz, Me), 0.94 (3H, d, J 7 Hz, Me), 1.05
(9H, s, tBu), 1.30 (3H, t, J 7 Hz, C02CH2C773), 1.70 - 1.93 (2H, m, 5-H
and 7-H), 2.24 (1H, m, 4-H). 2.39 (1H, m, 4-H), 3.51 (1H, dt, J 9 and
2.5 Hz), 3.64 (1H, dd, J 10.5 and 8 Hz, 8-H), 3.74 (1H, dd, J 10.5 and
4 Hz, 8-H), 3.77 (1H, d, J 1.5 Hz, OH), 4.19 (2H. q, J 7 Hz, C02C^2CH3),
5.88 (1H, dt, J 15.5 and 1 Hz, =C#C02Et), 6.98 (1H. dt (5 lines),
J 15.5 and 7 Hz, Ctf=CHC02Et), 7.42 (6H, m, Ph), and 7.68 (4H, m, Ph);
m/z 411 (M+-tBu), 381 (M+-CH2C02Et), 365 (M+-tBu-EtOH), and 199 (M+-
CH20Si^BuPh); (Found: C, 71.69; H, 8.74. CjsH^oO^Si requires
C, 71.75; H, 8.60%).

300
83. Preparation of (ff)-[55, 6i?, 75]-8-t-Butyldiphenyl si 1yloxy-6-
hydroxy-5,7-dimethyloct-2-en-l-ol (118)
To a solution of the (£}-a,p-unsaturated ester (117) (653 mg,
l. 40 mmol) in dry THF (10 ml) at -78°C under argon was added DIBAL
(2.82 ml of a 1.50M solution in hexane, 4.20 mmol) dropwise. After
stirring for 1 h the reaction was quenched with water (3 ml) and
allowed to reach room temperature. The solution was diluted with
ethyl acetate (20 ml) and then poured into a flask containing ethyl
acetate (100 ml) and solid sodium Dicarbonate (5g). After shaking
and allowing to stand for 10 min, the supernatant liquid was filtered
through a short pad of silica gel and the residual solid washed with
further portions of ethyl acetate (4 * 70 ml); these extracts were
filtered through the same silica gel pad. The combined organic
extracts were evaporated under reduced pressure and the residue
chromatographed (65% diethyl ether-petrol ) to afford (5)-[55, 6#, 75]-
8-1-butyldiphenyl si 1yloxy-6-hydroxy-5,7-dimethyloct-2-en-l-ol (118)
(500 mg, 84%) as a colourless oil, [a]^25 + 16.3° (o 3.0, CHC13);
vm3V (film) 3455, 2963, 2932, 2860, 1589, 1113, 1006. and 909 cm"1;
6 (250 MHz) 0.72 (3H, d, J 7 Hz, Me), 0.90 (3H, d, J 6.5 Hz, Me), 1.06
(9H, s, tBu), 1.60 - 1.95 (3H, m, 5-H, 7-H, and OH), 2.02 - 2.28 (2H,
m, 4-H2), 3.52 (1H, dd, J 9.5 and 2.5 Hz, 6-H), 3.63 (1H, br s, OH),
3.66 (1H. dd, J 10 and 7.5 Hz, 8-H), 3.75 (1H, dd, J 10 and 4.5 Hz,

301
8-H), 4.08 (2H, d, 0 3.5 Hz, C/72OH), 5.70 (2H, m, CH=CH), 7.42 (6H. m,
Ph), and 7.68 (4H, m, Ph); m/z 351 (M+-tBu-H20), 269 (Ph2tBuSi0CH2^),
239 (Ph2tBuSi+ ), and 199; (Found: M+-tBu-H20, 351.1770. C26H3803Si
requires M+-tBu-H20, 351.1780).
84. Preparation of 1-(ff)-(5-(i?)-[(2-t-Butyl diphenyl silyloxy-1-(di
methyl )ethyl]-4-(5)-methyltetrahydrofuran-2-(/?)-yl )-l, 2-ethane-
diol (120)
To a stirred solution of titanium (IV) isopropoxide (0.71 ml, 2.38
mmol) in dry dichioromethane (15 ml) at -78°C under argon was added
diethyl (2R, 3 i?)-tartrate (590 mg, 2.86 mmol) (dried by stirring witho
4A molecular sieves at 1 mm Hg followed by bulb to bulb distillation
(Kugelruhr) in dichioromethane (3 ml) followed by the allylic alcohol
(118) (500 mg, 1.17 mmol) in dichioromethane (3 ml). After 5 min
anhydrous t-butylhydroperoxide (TBHP) (1.95 ml of a 3.0M solution
in toluene, 5.85 mmol, precooled to -20°C) was added to the reaction
mixture and the solution allowed to reach -20°C over a 2 h period.
After a further 6 h at -20°C dry acetonitrile (10 ml) and saturated
92aqueous sodium fluoride solution (10 ml) were added and the mixture
stirred vigorously for 2 h at room temperature. After extraction
with dichioromethane (3 * 30 ml), the organic layer was dried and
concentrated to afford the crude diol {120) along with remaining
tartrate diester and TBHP.

302
An equally successful work-up procedure involved addition of
dry diethyl ether (10 ml) followed by saturated sodium sulphate solution
(6 ml) to the reaction at -20°C. After rapid stirring at room tempera
ture for 2 h, the slurry was filtered through a Celite pad and the
resulting orange-yellow paste washed with several portions of
anhydrous diethyl ether until the paste became somewhat granular. The
orange-yellow layer was scraped off the Celite pad into an Erlenmeyer
flask. Ethyl acetate was added and the resulting suspension stirred
vigorously for 5 min in boiling ethyl acetate. The slurry was filtered
through the same Celite pad and the orange-yellow solid washed twice
with hot ethyl acetate. The combined filtrates were concentrated to
afford the crude product along with tartrate diester and any TBHP. In
both work-up procedures the latter may be removed azeotropically by
repeated addition of dry toluene and evaporation.
The tartrate diester may be removed by chromatography (gradient
elution, 50 -> 75% diethyl ether-petrol) to afford l-(S)-{5-(R)-[(2-t-
butyldiy>henylsilyloxy-\ - { S) -methyl) ethyV\-<\ - (S )-methyltstrahydrofuran-
2-{R)-yl}-l,2-ethaneddol (120) (385 mg, 75%) as a white crystalline
solid, (m.p. 67 °C); [a]n25 + 16.1° (o 1.4, CHC13); v (CHCU) 3385,
2923, 2855, 1113, and 1073 cm"1; 6 (250 MHz) 0.91 (3H, d, J 7 Hz, C-4'
methyl), 0.99 (3H, d, J 7 Hz, C-l" methyl), 1.08 (9H, s, rBu), 1.65 -
1.78 (2H, br dd, J 13 and 6.5 Hz, 3'-H and 1"-H), 2.03 (1H, ddd, J 13,
8.5, and 6.5 Hz, 3 1-H), 2.12 - 2.45 (3H, m, 4'-H and 0H(x2)), 3.50 -
3.80 (6H, m, C/720H, 1-H, 5'-H, and Ctf20TBDPS), 4.01 (1H, ddd (6 lines),
J 10, 6.5,and 6 Hz, 2 1-H) , 7.40 (6H, m, Ph), and 7.70 (4H, m, Ph);
m/z 385 (M+-tBu), 381 (M+-CH(0H)CH20H), 307, 199, and 126 (M+-H20-
CH2 (Me )CH20Si tBuPh2 ); (Found: M+-tBu, 385.1831. C25H380l4Si requires
M+-tBu, 385.1835); (Found: C, 70.38; H, 8.72. C 26H 3S0 kSi requires
C, 70.55; H, 8.65%).
127

303
85. Preparation of 1-{S)-\5-(fl)-f(2-t-Butyldiphenyl si 1y1oxy-l-(S )-
methyl)ethyl]-4-(S)-methyl tetrahydrofuran-2-(i?)-yl )-2-(meth,y1 -
sulphony]oxy)ethano1 (121)
To the diol (120) (58 mg, 0.13 mmol) in dry dich1oromethane (1 ml)
at -30°C was added methanesulphonyl chloride (10 pi. 0.13 mmol)
followed by di isopropyl ethyl amine (Hiinig's base) (25 pi, 0.14 mmol).
After stirring for 30 min, the solution was diluted with ice-water
(2 ml) and dichioromethane (10 ml). The aqueous layer was extracted
with further dichioromethane (2 * 5 ml) and the combined organic
extracts washed with brine (1 x 5 ml) and dried (Na2S0H). Chromato
graphy (75% diethyl ether-petrol) afforded l-(S)-{5-{R)-[{Z-t-butyl-
diphenyIsilyloxy-1 - (S) -methy7 ) e t % Z- ] - 4 - (S) -methyltetrahydrofuran- 2 - (R) -
yl)-2-{methylsulphonyloxy)ethanol (121) (48 mg, 70%) as a viscous,
colourless oil, [a]n2S + 4.7° {a 1.8, CHC13); v (film) 3437 ,
2962, 2933, 1428, 1356, 1176, 1113, and 1074 cm-1; 6 (250 MHz) 0.91
(3H, d, J 7 Hz, C-41 methyl), 1.01 (3H, d, J 6.5 Hz, C-l" methyl),
1.06 (9H, s, tBu), 1.71 (1H. m, OH), 1.77 (1H, dd, J 13 and 7 Hz, 1"-H),
2.05 (1H, ddd, J 14, 8.5, and 6.5 Hz, 3'-H), 2.26 (1H, d, J 5 Hz,
4'-H), 2.32 (1H, m, 3'-H), 2.98 (3H, s, 0S02Me), 3.70 - 3.87 (4H, m,
1-H, 5' -H, and Cff20TBDPS), 3.99 (1H, m, 2'-H), 4.16 (1H, dd, J 11 and
6.5 Hz, Ctf20S02Me), 4.30 (1H, dd, J 11 and 3 Hz, C/720S02Me), 7.40
(6H, m, Ph), and 7.68 (4H, m, Ph); m/z 463 (M+-tBu), 381 (M+-CH(0H)-
CH20S02Me), and 367 (M+-tBu-H0S02Me); (Found: M+-tBu, 463.1601.

304
^yH^oOeSSi requires M+-^Bu, 463.1611); (Found: C, 62.32; H, 7.91;
S, 5.90. C27Ht+006SSi requires C, 62.27; H, 7.74; S, 6.16%).
86. Preparation of !-(/?)-(5-(7?) -[(2-1-Butyl diphenyl si 1 yl oxy-1-(S) -
methyl )ethyll-4-(6')-methy1 tetrahydrofuran-2-(^)-yl }-2-phenyl-
seleno-ethanol (122)
To a flask containing diphenyl diselenide (1.73g, 0.55 mmol) in
dry THF (20 ml) in an ultrasonic bath (under argon) was added sodium
metal in small pieces (0.28g, 1.20 mmol). After 16 h, all the sodium
had been consumed and a fine off-white suspension of sodium phenyl
selenide had formed.
To the primary mesylate (121) (200 mg, 0.38 mmol) in THF (5 ml)
at 0°C was added sodium phenyl selenide (0.76 ml of the 0.55M solution
prepared above, 0.42 mmol). The cooling bath was removed and the
reaction stirred at room temperature for 30 min. The mixture was
then diluted with diethyl ether (20 ml) and washed with water (5 ml).
The aqueous layer was extracted with further diethyl ether (2 x 10 ml)
and the combined organic extracts washed with brine (1 * 5 ml) before
drying (Na2S04) and concentration under reduced pressure. Chromato
graphy of the residue (15% diethyl ether-petrol) afforded l-(R)-{5-(R)-
[ Z - t - b u t y Z d ' i p h e n y Z s ' i Z y Z o x y - l - (S) - m e t h y Z ) e t h y Z ~ \ ~ a>~ (S ) - m e t h y Z t e t r a h y a r o -
f u r a n - 2 - { R ) - y Z } - Z - p h e n y Z s e Z e n o ~ e t h a n o Z (122) (150 mg, 65%) as a
OH(121) (122)

305
colourless oil (ftp 0.55, 35% diethyl ether-petrol), [aj^25 - 4.5°
{o 1.4, CHC13); vmav (film) 3446, 2961, 2857, 1112, 1073, and 702
cm"1; 6 (400 MHz) 0.90 (3H, d, J 7.2 Hz, C-4' methyl), 1.00 (3H, d,
J 7.2 Hz, C-l" methyl), 1.06 (9H, s, tBu), 1.68 (1H, dd, J 12.8 and
7.2 Hz. 31-H), 1.70 (1H, m, 1"-H), 2.05 (1H, ddd, J 12.8, 7.2, and
6.4 Hz, 31-H), 2.28 (2H, m, 41-H and OH), 2.92 (1H, dd, J 12.0 and
8.0 Hz, C.72SePh), 3.11 (1H, dd, J 12.0 and 3.5 Hz, Ctf2SePh), 3.68 -
3.80 (4H, m, 1-H, 51-H, and Ctf20TBDPS), 4.02 (1H, ddd (6 lines),
J 12.0, 5.5, and 5.0 Hz, 2'-H), 7.22 (3H, m, SePh), 7.40 (6H, m, Ph),
7.50 (2H, m, SePh), and 7.68 (4H, m, Ph); m/z 582 (M+(80Se)), 525
(M+(80Se)-tBu), 411 (M+(80Se)-CH2SePh), 367 (M+(8GSe)-SePh-tBu-H),
and 199; (Found: M+(80Se)-tBu, 525.1353. C32H„Q03 Si80Se requires
M+(8JSe)-tBu, 525.1364); (Found: C, 66.12; H, 7.58. C32H,203SiSe
requires C, 66.07; H, 7.28%).
87 . Preparation of l-(g')-(5-(/?)-[(2-t-Butyldiphenyl si lyloxy-l-(ff)-
methyl)ethyl ]-4-(S) -methyl tetrah,ydrofuran-2-(i?)-yl}ethanol (123)
-To Raney nickel in diethyl ether (3 ml) under a hydrogen atmosphere
was added the selenide (122) (78 mg, 0.13 mmol) in diethyl ether (2 ml).
After 10 min, reduction was complete and the solution was filtered
through a short pad of silica gel. The catalyst was washed several
times with further portions of diethyl ether. Concentration of the

306
combined organic solution under reduced pressure afforded l-(S)-{5-(R)-
[(2-* -butytdi'phenyZs'iZy'loxy-l- (S) -methyl )ethyV\-(\ - (S) -methyltetrahydro- furan-2-(R)-yl}ethanol (123) (56 mg, 100%) as a colourless oil.
Chromatography (30% diethyl ether-petrol) gave a sample of micro-
analytical purity (i?p 0.37, 35% diethyl ether-petrol), [a]p25 + 15.1°
{o 4.4, CHC13); v v (film) 3437, 2962, 2930, 1112, and 1075 cm"1;
6 (250 MHz) 0.75 (3H, d, J 7 Hz, C-4' methyl), 0.98 (9H, s, tBu), 1.04
(6H, t, J 7 Hz, C-2 methyl and C-l" methyl), 1.50 (1H, br s, OH),
1.75 - 2.15 (4H, m, 3'-H2, 4'-H, and 1"-H), 3.45 - 3.60 (4H, m, 1-H,
51-H, and Ctf20TBDPS), 3.90 (1H, m, 21-H), 7.34 (6H, m, Ph), and 7.60
(4H, m, Ph); m/z 381 (M+-CH(Me)0H), 369 (M+-tBu), and 255 (tBuPh2SiO+);
(Found: M+-tBu, 369.1877. C26H3803Si requires M+-tBu, 369.1886);
(Found: C, 73.21; H, 9.08. C26H3803Si requires C, 73.19; H, 8.98%).
88. Preparation of 2-(fl)-[(2-t-Buty1diphen,ylsilyloxy-l-(S)-methyl)-
ethyl]-5-(fl)-(1-(S)-methoxyethyl)-3-(S)-methyltetrahydrofuran
(124)
To a stirred suspension of sodium hydride (10 mg, 0.20 mmol, 50%
dispersion in oil) prewashed with sodium-dried 30-40° b.p. petrol
(2 x 3 ml), in dry THF (2 ml) at 0°C under argon was added a solution
of the alcohol (123) (44 mg, 0.10 mmol) in THF (3 ml). After 15 min,
DMPU (31 pi, 0.25 mmol) was added, stirring continued for a further

307
10 min, and then methyl iodide (38 pi, 0.60 mmol) was added. The mixture
was stirred at room temperature for 3 h followed by dilution with diethyl
ether (10 ml) and water (5 ml). The aqueous layer was extracted with
diethyl ether (2 * 10 ml) and the combined ethereal extracts dried
(Na2S0,J and evaporated under reduced pressure to afford the crude methyliether.' Purification by chromatography (8% diethyl ether-petrol)
afforded 2-(R)-[{2-t-butyldiphenylsilyloxy-l-{$)-methyl)et/zyZ]-5-(R)-(l-
(S)-methoxyethyl)-3-(S)-methyltetrahydrofuran (124) (35 mg, 77%) as a
colourless oil (Z?p 0.33, 10% diethyl ether-petrol), M q25 + 20.8°
[a 0.4, CHC13); vm=v (CHC13 ) 3070, 2963, 2931, 2858, and 1112 cm-1;
6 (400 MHz) 0.90 (3H, d, J 7.2 Hz, C-3 methyl), 0.99 (3H, d, J 7.2 Hz,
CH(Afe)CH20TBDPS), 1.05 (9H, s, tBu), 1.11 (3H, d, J 6.5 Hz, CH(Me)OMe),
1.71 (2H, br dd, J 12.5 and 6.8 Hz, 4-Ha and l'-H), 2.00 (1H, ddd, J 12.5,9
and 6.5 Hz, 4-H ), 2.26 (1H, m, from decoupling, J 7.2, 6.5, and 4.0 Hz,
3-H), 3.31 (1H, m, Ctf(Me)OMe), 3.34 (3H, s, OMe), 3.73 (1H, dd, J 9.5
and 3.0 Hz, C/720TBDPS), 3.76 (1H, dd, J 10.0 and 4.0 Hz, 2-H), 3.83 (1H,
dd, J 9.5 and 5.0 Hz, Ctf20TBDPS), 3.94 (1H, ddd, J 9.0, 6.8, and 5.2 Hz,
5-H), 7.39 (6H, m, Ph), and 7.70 (4H, m, Ph); m/z 383 (M+-tBu), 269
(0TBDPS+), 253, 235, 209, 199, 169, 143 (M+-CH(Me)CH20TBDPS), 85 (MH+-
CH(Me)0Me-CH(Me)CH20TBDPS), and 59 (CH(Me)0Me+); (Found: M+-tBu,
383.2047. C27Hl+003Si requires M+-tBu, 383.2042); (Found: C, 73.52;
H, 9.27. C27Hl+ 003Si requires C, 73.59; H, 9.15%), identical to the
compound from the degradation of the natural product (see Experiment 90).

308
vervsnMiUr ^
11241
COUPLING CONSTANTS
The methylation could also be effected by treatment of the alcohol
(123) in DMF with freshly prepared silver (I) oxide (5 eq ) and methyl
iodide (5 eq ) at room temperature. After stirring for ca. 48 h all
starting material had been consumed (t.l.c.) and the solution was
diluted with diethyl ether (20 ml). After washing with water (3 x 5 ml),
the aqueous layer was extracted with diethyl ether (2 x 10 ml) and the
combined organic extracts washed with water (1 x 10 ml) and brine
(1 x 10 ml). Drying and concentration under reduced pressure followed
by chromatography as before gave the methyl ether (124) (31 mg,68%)
identical to the previously reported compound.

309
89. Preparation of 2-(S)-{~5-(i?)-(l-fSl-Methoxyethyl )-3-(ff)-methyltetra-
hydrofuran-2-(i?)-yl1 propan-l-ol (64)
To sodium borohydride (386 mg, 11.2 mmol) in dry methanol (10 ml)
at -20°C was added the aldehyde (7) (obtained from degradation of the
natural product) (678 mg, 3.40 mmol) in dry methanol (5 ml). After
stirring for 15 min, reduction was complete and the reaction was
quenched by careful addition of water (2 ml) followed by dilution with
diethyl ether (70 ml) and saturated aqueous ammonium chloride solution
(10 ml). The aqueous layer was extracted with diethyl ether (2 * 20 ml)
and the combined ethereal extracts washed with water (1 * 10 ml) and
brine (1 * 10 ml). Drying (Na2S0«J and evaporation of the solvent
under reduced pressure followed by chromatography (60% diethyl ether-
petrol ) afforded 2-{Z)-[5-{R)-{l-{Z)-methoxyethyl)-2-{R)-methyltetr>a-
hydrofuran-2.-{R)-yV\propan-l-ol (64) (500 mg, 73%) as a viscous,
colourless oil (ftp 0.23, 50% diethyl ether-petrol), [a.]q25 + 38.8°
{a 5.0, CHC13); \> (film) 3450, 2960, 1190, 1140, 1080, 1030, and
1005 cm-1; 5 (250 MHz) 0.75 (3H, d, J 7 Hz, CH(Me)CH20H), 0.90 (3H, d,
J 7 Hz, C-31 methyl), 1.08 (3H, d, J 6.5 Hz, CH(Me)OMe), 1.63 (1H, dd,
J 12.5 and 6.5 Hz, 4-H ), 1.83 (1H, m, Ctf(Me)CH20H), 1.95 (1H, ddd,
J 12.5, 9.5, and 7 Hz, 4-H^), 2.29 (1H, m, from decoupling, J 7, 6.5,
and 4 Hz, 3’-H), 3.33 (1H, m, Ctf(Me)OMe), 3.36 (3H, s, OMe), 3.50 - 3.67
(4H, m, CH20H and 2'-H), and 3.98 (1H, ddd, J 9.5, 6.5, and 5 Hz, 51-H);

310
m/z 202 (M+), 184 (M+-H20), 170 (M+-MeOH), 143 (M+-CH(Me)0Me and M+- CH(Me)CH20H), 85 (MH+-CH(Me)0Me-CH(Me)CH20H), and 59 (CH(Me)0Me+ and
CH(Me)CH20H+); (Found: M+, 202.1554. ClxH2203 requires M+, 202.1569);
(Found: C, 65.02; H, 10.75. C11H2203 requires C, 65.31; H, 10.96%).
90. Preparation of 2-(7?)-[(2-t-Butyldiphenylsil yl oxy-1-(-S')-methyl )-
ethyl ]-5-(.??)-(1-(S)-methoxyethyl)-3-(5)-methyltetrahydrofuran
11211
!BuPlv,SiO
(125)OMe
To the alcohol (64) (14 mg, 0.07 mmol) in dry dichioromethane
(1 ml) was added triethylamine (11 pi, 0.08 mmol), t-butyldiphenyl si 1yl
chloride (20 pi, 0.08 mmol) and DMAP (1 crystal). After stirring for
2 h at room temperature the solution was diluted with dich1oromethane
(15 ml) and water (5 ml). The aqueous layer was extracted with
dichioromethane (2 * 10 ml) and the combined organic extracts washed
with brine (1 x 5 ml) before drying (Na2S01+) and concentration under
reduced pressure. Chromatography (10% diethyl ether-petrol) afforded
the silyl ether (125) (25 mg, 82%) as a colourless oil, M q 25 + 18.7°
{c 1.3, CHC13) identical to the synthetic material (124) (t.l.c.,
XH n.m.r., i.r., mass spectrum).

311
91. Preparation of 2-(S)-[5-(i?)-( l-(S)-Methoxyethyl )-3-(i?)-methyl-
tetrahydrofuran-2-(/?)-yl 1 propan-l-o1 (64)
To the silyl ether (124) (20 mg, 0.05 mmol) in dry THF (1 ml) at
room temperature was added anhydrous tetra-n-butylammoniurn fluoride
(24 pi of a 2.5M solution in THF, 0.06 mmol) and the reaction stirred
for 24 h. The solution was diluted with diethyl ether (10 ml) and
water (4 ml). Extraction of the aqueous layer with diethyl ether
(3 x 10 ml), followed by washing of the combined ethereal extracts with
brine (1 * 5 ml ), drying (Na2S01+) and concentration under reduced
pressure gave a crude yellow oil. Chromatography (60% diethyl ether-
petrol afforded the alcohol (64) (8.6 mg, 94%) with spectroscopic
data as reported in Experiment 89.
92. Preparation of 5-(/?)-(l-(5'J-Methoxyethyl) -3-{S) -methyl-2-(/?)-[(!-
(R)-methyl - 2-phenyl thi o)ethyl ]tetrah,ydrof uran (126)
2‘

312
To tri-n-butylphosphine (0.60 ml.- 2.40 mmol) in dry THF (3 ml) at
room temperature was added diphenyl disulphide (520 mg, 2.40 mmol) in
THF (3 ml) followed by the alcohol (64) (242 mg, 1.20 mmol) in THF
(6 ml). After stirring for 2 h at room temperature the solution was
poured into a flask containing water (10 ml) overlaid with diethyl ether
(50 ml). The aqueous layer was extracted with diethyl ether (2 x 20 ml)
and the combined ethereal extracts washed with water (1 x 10 ml) and
brine (1 x 10 ml). Drying (Na^SOiJ and removal of the solvent under
reduced pressure followed by chromatography (gradient elution,
petrol -+ 10% diethyl ether-oetrol ) afforded 5-{R)-(l-{S)-methoxyethyl)-
3 - (S) -mevhyl-2- (R)-[(1~(R) -methyl-2~y>henylthio )ethyl]tetrahydrofuran
(126) (336 mg, 95%) as a colourless oil (7?p 0.24, 10% diethyl ether-
petrol), v (film) 3060, 2960, 2920, 1090, 1005, 735, and 690 cm'1;
6 (90 MHz) 0.88 (3H, d, J 7 Hz, C-3 methyl), 0.98 (3H, d, J 7 Hz,
CH(Me)CH2SPh), 1.12 (3H, d, J 6.5 Hz, CH(Me)OMe), 1.40 - 2.39 (4H, m,
C7/(Me)CH2SPh, 3-H, and 4-H2), 2.72 (1H, dd, J 12.5 and 9 Hz, Ctf2SPh),
3.15 (3H, s, OMe), 3.26 - 3.68 (3H, m, Ctf2SPh, Ctf(Me)0Me, and 2-H),
3.95 (1H, ddd, J 9, 6.5, and 5 Hz, 5-H). and 7.10 - 7.48 (5H, m, Ph);
m/z 294 (M+), 263 (M+-0Me), 235 (M+-CH(Me)0Me), 123 (PhSCH2+), 85 (MH+-
CH(Me)OMe-CH(Me)CH2SPh), and 59 (CH(Me)0Me+); (Found: M+, 294.1661.
C17H2602S requires M+, 294.1653); (Found: C, 69.55; H, 9.12.
C17H2502S requires C, 69.34; H, 8.90%).

313
93 . Preparation of 5-(/?)-(l-(g)-Methoxyethyl)-3-{S) -methyl -2-(i?)-1~ (1-
{R)-methyl-2-phenylsulphonyl)ethyl1tetrahydrofuran (127)
To the sulphide (127) (336 mg, 1.14 mmol) in dry dichioromethane
(10 ml) at 0°C was added mCPBA (550 mg, 90% purity, 2.85 mmol). The
reaction was warmed to room temperature and stirred for 1 h. After
dilution with dichioromethane (60 ml), the solution was washed with
0.5M aqueous sodium hydroxide solution (2 x 15 ml), followed by water
(1 x 10 ml ) and brine (1 x 10 ml ). Drying (Na2S0t+) and evaporation
of the solvent under reduced pressure gave the crude sulphone as a
yellow oil. Chromatography (60% diethyl ether-petroi ) afforded 5-(R)-
(1-(S)-methoxyethyl)-3-(S)-methyl-2-{R)-[(l—(R)-methyl-2-phenylsulphonyl)-
ethy V] tetrahydrofuran (127) (334 mg, 90%) as a white crystalline solid,
(m.p. 43-44°C); [<x]n25 + 10.3° (c3.9, CHC13); v v (CHCT 3) 3062,
2964, 2880, 1448, 1377, 1205, 1150, 1093, and 745 cm-1; 6 (250 MHz)
0.84 (3H, d, J 7 Hz, C-3 methyl), 1.03 (3H, d, J 6.5 Hz, CH(Me)OMe),
1.12 (3H, d, J 7 Hz, CH(Me)CH2S02Ph), 1.65 (1H, ddd. J 12.5, 6.5, and
0.5 Hz, 4-H ), 1.97 (1H, ddd, J 12.5, 9.5, and 6.5 Hz, 4-H1, 2.10 - 2.30Ct p
(2H, m, 3-H and Ctf(Me)CH2S02Ph), 2.98 (1H, dd, J 14 and 10.5 Hz,
Ctf2S02Ph), 3.25 (1H. dq, J 6.5 and 4.5 Hz, Ctf(Me)OMe), 3.32 (3H, s, OMe),
3.43 (1H, dd. J 10.5 and 4 Hz, 2-H), 3.77 (1H, dd, J 14 and 2 Hz,
C#2S02Ph), 3.85 (1H, ddd, J 9.5, 6.5, and 4.5 Hz, 5-H), 7.60 (3H, m, Ph),
and 7.94 (2H, m, Ph); m/z 327 (MH+), 294 (M+-Me0H), 267 (M+-CH(Me)0Me),

314
143 (M+-CH(Me)CH2S02Ph), 125, 107, 97, 85 (MH+-CH(Me)CH2S02Ph-CH(Me)0Me),
and 59 (CH(Me)0Me+): (Found: C, 62.32; H, 7.99; S, 9.69. C^H^O^S
requires C, 62.55; H, 8.03; S, 9.82%).
94. Preparation of 2-(i?)-[(2-Iodo-l-(i?)-methyl)ethyl ]-5-(j?)-(l-(g)-
methoxyethyl)-3-(ff)-methyltetrahydrofuran (128)
To a rapidly stirred solution of the alcohol (64) (255 mg, 1.26
mmol) in acetonitrile-diethyl ether (4 ml : 12 ml) was added triphenyl-
phosphine (495 mg, 1.89 mmol), imidazole (129 mg, 1.89 mmol) and
finally resublimed iodine (480 mg, 1.89 mmol) at room temperature (the
reaction was exothermic and larger scale reactions required cooling).
After 5 min, reaction was complete and the solution was diluted with
diethyl ether (50 ml) followed by washing with 5% aqueous sodium thio
sulphate solution (1 x 10 ml) and water (1 x ml). After concentration
under reduced pressure to oa. 5 ml and dilution with petrol (15 ml),
the solution was filtered through a short pad of silica gel to remove
the triphenylphosphine oxide. Evaporation of the solvent under
reduced pressure followed by chromatography (5% diethyl ether-petrol) afforded 2 - {R)-[(2-£odo-l-{R)-methyl)ethyl]-5-{R)-{1-(S)-methoxyethyl)-
2-{S)-methyltetrahijdrofuran (128) (332 mg, 84%) as a colourless oil
[7?p 0.45, 10% diethyl ether-petrol), M q25 " 5.5° {o 11.7, CHC13);
vmax 2966’ 2930, 2877, 1199> and 1093 cm"1; 6 (25° MHz) °-92

315
(6H, d, J 7 Hz, C-3 methyl and CH(Me)CH2I), 1.12 (3H, d, J 6.5 Hz,
CH(Me)OMe), 1.40 (1H, m, C#(Me)CH2I), 1.70 (1H, dd, J 12.5 and 7 Hz,
4-H ), 2.00 (1H, ddd, J 12.5, 9, and 6.5 Hz, 4-HJ, 2.26 (1H, m, 3-H),a p
3.30 - 3.54 (4H, m, Ctf(Me)OMe, 2-H, and C#2I), 3.40 (3H, s, OMe), and
3.96 (1H, ddd, J 9, 7, and 4.5 Hz, 5-H); m/z 312 (M+), 253 (M+-
CH(Me)OMe), 209 (M+-CH2CH(0H)CH(Me)0Me), 169 (CH(Me)CH2I+), 143 (M+-
CH(Me)CH2I), and 59 (CH(Me)0Me+); (Found: M+-CH(Me)OMe, 253.0071.
C n H210 2I requires M+-CH(Me)OMe, 253.0089); (Found: C. 42.62; H, 7.01.
CiiH2i02I requires C, 42.32; H, 6.78%).
95. Preparation of 2-(fl)-[5-(/?)-(l-(g)-Methoxyethyl )-3-(g)-methyl-
tetrahydrofuran-2-(i?)-.yl]prop-l-yltriphenylphosphoniurn iodide
(129)
To the iodide (128) (158 mg, 0.51 mmol) in dry toluene (5 ml) was
added recrystallized triphenylphosphine (147 mg, 0.56 mmol) and the
mixture refluxed for oa. 48 h. The toluene was removed under reduced
pressure and the phosphonium salt purified by repeated addition of
ethyl acetate and separation of the supernatant liquid (phosphonium
salt insoluble in ethyl acetate) to afford after drying in vaouo the
phosphonium iodide (129) (230 mg, 79%) as a white foam, v (CHC13)
2967, 1111, 997, 725, and 690 cm-1; 6 (250 MHz) 0.69 (3H, d, 0 7 Hz,
Me), 0.80 (3H, d, J 6.5 Hz, Me), 1.09 (3H, d, J 6.5 Hz, CH(Me)OMe),

316
1.71 (1H, dd, J 12.5 and 6.5 Hz. 4'-H ), 1.95 - 2.13 (2H, m, 4'-H0 anda p
C7/(Me )CH2PPh3+I~), 2.25 (1H, m, 31 -H), 3.25 - 3.80 (4H, m, Ctf2PPh3V ,
21-H, and Ctf(Me)OMe), 3.35 (3H, s, OMe), 3.86 (1H, dt, J 10.5 and
4 Hz, 5'-H), and 7.68 - 7.95 (15H, m, Ph); m/z 448 (MH+-I), 447 (M+-I),
185 (M+-PPh3I), and 85 (MH+-CH(Me)OMe-CH(Me)CH2PPh3I).
96. Preparation of Diethyl 2-(1?)-f5-(/?) -(1-( S)-metnoxyethy 1 )-3-(S)-
methyl tetrahydrofuran-2-( /?)-,yl ]prop-l-y1 phosphonate (130)
To the iodide (150 mg, 0.48 mmol) was added redistilled triethyl-
phosphite (3 ml) and the mixture heated at 120°C for 24 h. The tri
ethyl phosphite was then removed under high pressure and the residue
chromatographed (ethyl acetate) to afford the phosphonate ester (130)
(116 mg, 75%) as a colourless oil, v (film) 2969, 1247, 1095, 1060,
1028, and 959 cm'1; 6 (250 MHz) 0.91 (3H, d, J 7 Hz, Me), 1.03 (3H, d,
J 7 Hz, Me), 1.09 (3H, d, 0 6.5 Hz, CH(Me)OMe), 1.32 (6H, t, 0 7 Hz,
P0(0CH2C7/3) 2), 1.47 (1H, ddd, J 17.5, 15.5, and 11 Hz, C£2P0(0Et) 2),
1.69 (1H, dd, J 12.5 and 7 Hz, 4'-H ), 1.85 - 2.08 (1H, m, Ctf(Me)CH2-
P0(0Et)2), 1.99 (1H, ddd, J 12.5, 9. and 6.5 Hz, 4'-H ), 2.26 (1H, m,
3' -H) , 2.40 (1H, ddd, J 19, 15.5, and 2.5 Hz, C/72P0(0Et)2), 3.32 (1H,
dq, J 6.5 and 5 Hz, Ctf(Me)OMe), 3.37 (3H, s, OMe), 3.41 (1H, dd, J 10
and 4 Hz, 21-H), 3.93 (1H, ddd, J 9, 7, and 5 Hz, 5'-H), and 4.01 - 0II + , +4.18 (4H, m, P(0Ctf2CH3)2); m / z 323 (MH ), 290 (M -MeOH) ,

317
263 (M+-CH(Me)OMe), 180 (CH(Me)CH2P(OEt)2), 179 (CH(Me)CH2P0(0Et)2+),
152 (CH2=fj>(0Et)2 + ), 138 (HOP(OEt)2+), and 59 (CH(Me)OMe+); (Found:OH
MH+, 323.1984. C15H3105P requires MH + , 323.1987).
97 . Preparation of 2-(#)-[5-(i?)-( l-(S)-Methoxyeth,yl )-3-(S)-methy1-
tetrahydrofuran-2-(i?)-yl]prop-1-yldiphenyl phosphine oxide (131)
The phosphomum salt (129) (200 mg, 0.35 mmol) was refluxed in 3M
aqueous sodium hydroxide solution (5 ml) for 30 min. After cooling
to room temperature, chloroform (30 ml) and water (10 ml) were added
and the aqueous layer extracted with chloroform (3 x 20 ml). The
combined organic extracts were washed with water (1 x 10 ml) and dried
(Na2S0„). Evaporation of the solvent under reduced pressure followed
by chromatography (ethyl acetate) gave the phosphine oxide (131)
(125 mg, 93%) as a viscous, colourless oil, v (film) 2926, 1735,max •1638, 1437, 1376, 1170, 1120, and 1096 cm"1; 5 (250 MHz) 0.77 (3H, d,
J 7 Hz, Me), 1.01 (3H, d, J 6.5 Hz, Me), 1.12 (3H, d, J 6.5 Hz, Me),
1.67 (1H, dd, J 12.5 and 6.5 Hz, 4'-Ha), 1.86 - 2.12 (3H, m, 4'-H ,
1-H, and 2-H), 2.22 (1H, m, 3‘-H), 2.98 (1H, ddd, J 14.5, 10, and
0.5 Hz, 1-H), 3.31 (1H, dq, J 6.5 and 5 Hz, Ctf(Me)OMe), 3.38 (3H, s,
OMe), 3.49 (1H, dd, J 10 and 4 Hz, 2'-H), 3.90 (1H, ddd, J 9.5, 6.5,
and 5 Hz, 5'-H), 7.46 (6H, m, Ph), 7.72 (2H, m, Ph), and 7.84 (2H, m,
Ph); m/z 387 (MH+, weak), 386 (M+, weak), 354 (M+-Me0H),

318
327 (M+-CH(Me)OMe), 243 (CH(Me)CH2P(0)Ph2+), 215 (Ph2P(0)CH2+), and
201 (Ph2P=0+); (Found: M+, 386.2011. C s a ^ ^ P requires M+,
386.2011).
98. Preparation of 2-(j?)-[5-(i?)-( l-(S)-Methoxyethy1 )-3-(5)-methyl-
tetrahydrofuran-2-(i?)-,y11 pr op-1-yl diphenyl phosphine oxide (131)
O
Diphenylphosphine (101 pi, 0.58 mmol) was added to a flask fitted
with a reflux condensor containing sodium metal (12 mg, 0.53 mmol,
washed with 30-40° b.p. petrol (2 * 5 ml) to remove oil) in dry THF
(5 ml) under argon and the solution refluxed for oa. 12 h. The
resulting orange slurry was cooled to 0°C and the iodide (150 mg,
0.48 mmol) added in dry THF (2 ml) with rapid stirring. The orange
colour immediately disappeared and-the solution was allowed to stir
/ at 0°C for a further hour before warming to room temperature.
Dilution with ethyl acetate (20 ml), washing with water (1 x 5 ml),
drying (Na2S01+) and concentration under reduced pressure gave the
crude phosphine oxide as a yellow oil. Chromatography (ethyl
acetate) followed by drying at 50°C at 0.10 mm Hg afforded the pure
phosphine oxide (131) (167 mg, 90%) as a viscous, colourless oil
identical to the previously prepared compound (Experiment 97).

319
99. Preparation of Methyl 6-(S)-{3-(fl)-[5(i?)-( l-(S)-methox,yethyl )-3-
(S)-methyltetrahydrofuran-2-(/?)-yV}-3-(fl)-methylprop-l-(ff)-enyl}-
tetrahydropyran-2-(#)-yl acetate and [ 6 3 'R, 5"R, l111 S, 311 5. 2S\-
isomer (133) via the hydroxyphosphine oxides (132)
To a stirred solution of the phosphine oxide (131) (145 mg, 0.38
mmol) in dry THF (1.5 ml) at -78°C was added nBuLi (0.31 ml of a 1.29M
solution in hexane, 0.40 mmol) dropwise. After 1 h at -78°C, the
c-fs-aldehyde (22A) (70 mg, 0.38 mmol) in dry THF (1 ml) was added to
the red solution and stirring continued for 1 h before warming to room
temperature. The pale yellow mixture was diluted with dichloro-
methane (15 ml) and water (5 ml). The aqueous layer was extracted
with further dichioromethane (2 x 10 ml) and the combined organic
extracts washed with water (1 x 5 ml) and brine (1 x 5 ml). Drying
(Na2S0t+) and evaporation of the solvent under reduced pressure gave a
yellow oil which by t.l.c. contained no remaining aldehyde (22A), but
some starting phosphine oxide (131) and a mixture of diastereoisomeric
hydroxyphosphine oxide adducts (132) (slightly less polar than (131)).
Chromatography (90% ethyl acetate-petrol) afforded a diastereoisomeric

320
mixture of hydroxyphosphine oxides (132) (85 mg, 40%) as a white foam
and phosphine oxide (131) (58 mg, 40%) as a colourless oil, (132):
v v (CHC13) 3345, 2930, 1735, 1436, 1161, and 1100 cm"1; 6 (250 MHz)
(diascereoisomeric resonances coincident) 0.28 (3H, d, J 7 Hz, Me),
1.11 (3H, d, J 7 Hz, Me). 1.43 (3H, d, J 6.5 Hz, Me), 1.20 - 2.20
(10H, m), 1.99 (1H, dd, J 15.5 and 7.5 Hz, Ctf2C02Me), 2.27 (1H, dd,
J 15.5 and 5 Hz, Cn72C02Me), 3.20 - 4.05 (6H, m), 3.36 (3H, s, OMe),
3.79 (3H, s, C02Me), 4.33 (1H, m), 5.67 (1H, d, J 8.5 Hz, CtfOH), 7.42
(6H, m, Ph), 7.73 (2H, m, Ph), and 7.93 (2H, m, Ph); m/z 573 (MH+),
572 (M+), 571 (M+-H), 541 (M+-0Me), 513 (M+-CH(Me)0Me), 415 (M+-(C5H80)-
CH2C02Me), 354 (M+-Ph2P(0)0H), 245, 243, 201, 157 ((C5H80)CH2C02Me+),
and 143 ((CsH80)CH(Me)0Me+); (Found: M+, 572.2891. C32Hk507P
requires M+, 572.2903).
( b )
To the diastereoisomeric mixture of hydroxyphosphine oxides (60 mg,
0.10 mmol) in dry THF (2 ml) at 0°C was added freshly sublimed potassium
t-butoxide (12 mg, 0.10 mmol) with stirring. After 1 h, the orange-
yellow solution was diluted with water (5 ml) and diethyl ether (15 ml).
The aqueous layer was extracted with diethyl ether (2 x 10 ml) and the
combined ethereal extracts washed with brine (5 ml) and dried (Na2S0lt).
Evaporation of the solvent under reduced pressure followed by chromato
graphy (gradient elution, 30 -> 50% diethyl ether-petrol) afforded the

321
(2?)-olefin (133) (30 mg, 81%) and a trace of the (z)-olefin (slightly
less polar) as colourless oils, (E)-olefin: 6 (400 MHz) 0.94 (3H, d,
J 7 Hz, Me), 0.98 (3H, d, J 7 Hz, Me), 1.03 (3H, d, J 6.5 Hz, Me),
1.10 - 1.80 (7H, m), 1.89 (1H, ddd, J 12.5, 9.2, and 7 Hz, 4"-HJ,p2.18 (2H, m, =CHC7/(Me)CH(0)C#(Me)), 2.33 (1H, dd, J 15 and 6.5 Hz,
Ctf2C02Me), 2.53 (1H, dd, J 15 and 6.8 Hz, Ctf2C02Me), 3.27 (1H, m,
Ctf(Me)OMe), 3.31 (3H, s, OMe), 3.44 (1H, dd, J 9.5 and 4 Hz, 2"-H),
3.60 (3H, s, C02Me), 3.69 - 3.82 (2H, m, 2-H and 6-H), 3.88 (1H, ddd,
J 9, 7.5, and 5 Hz, 5"-H), 5.45 (1H, dd, J 16 and 5.5 Hz, OCHCH=), and
5.71 (1H, dd, J 16 and 6.2 Hz, 0CHCH=C#); m/z 354 (M+), 295 (M+-
CH(Me)OMe), 143 ((C5H 80)CH(Me)0Me+), 111 (143-MeOH). 85 (C5H90+), and
43 (CH3C0+); (Found: M+, 354.2396. C20H3405 requires M+, 354.2406).
100. Preparation of 5-(/?)-(l-(S)-Methoxyethyl)-2-(/?)-r(2-methyl-
sulphonylox.y-1-(S)-methyl)ethyl]-3-(S)-methyltetrahydrofuran
m ix
To the alcohol (64) (270 mg, 1.34 mmol) in dry dichioromethane
(10 ml) at ca. -5°C was added triethyl amine (0.22 ml, 1.60 mmol)
followed by methanesulphonyl chloride (0.13 ml, 1.47 mmol). After
stirring for 15 min the solution was diluted with ice-water (10 ml)
and dichioromethane (20 ml). The aqueous layer was extracted with
further dich1oromethane (2 * 20 ml) and the combined organic extracts

322
washed with brine (1 x 10 ml) before drying (Na2S0l+). Chromatography
(50% diethyl ether-petrol) afforded ^{^)-{\-{%)-methoxyethyl)-2-{^)-{(2-
methy lsulphonyloxy-l-{ S) -methyl) ethyl'\~'l- (S) -methyltetrdhydrofuran (143)
(344 mg, 94%) as a colourless oil, v (film) 2959, 1354, 1175, 1090,max.and 943 cm"1; 5 (60 MHz) 0.90 (3H, d, J 7 Hz, Me), 0.95 (3H, d, J 7 Hz,
Me), 1.12 (3H, d, J 7 Hz, Me ), 1.40 - 2.60 (4H, m, 3-H, 4-H2, and
Ctf(Me)CH20S02Me), 3.05 (3H, s, 0S02Me), 3.38 (3H, s, OMe), 3.25 - 4.25
(3H, m, 2-H, 5-H, and Ctf(Me)OMe), and 4.35 (2H, m, C/720S02Me); m/z
281 (MH+). 249 (M+-0Me), 248 (M+-MeOH), 221 (M+-CH(Me)0Me), 143 (M+-
CH(Me)CH20S02Me), 125 (M+-CH(Me)0Me-MeS03H), 111 (M+-CH(Me)CH20S02Me-
MeOH), 107, and 59 (CH(Me)0Me+); (Found: M+-CH(Me)0Me, 221.0850.
Ci2H2l+05S requires M+-CH(Me)0Me, 221.0848); (Found: C, 51.49;
H, 8.83; S, 11.54. C12H21+05S requires C, 51.41; H, 8.63; S, 11.43%).
101. Preparati on of 2-(j?)-f(2-Bromo-l-(R)-methy I)ethyl]-5-(R)-(1-(S)-
methoxyethy1)-3-(S)-methyltetrahydrofuran (144)
Anhydrous lithium bromide (2.14g, 24.6 mmol) was dissolved in a
minimum quantity of THF. To the resulting slurry was added the
mesylate (143) (344 mg, 1.23 mmol) in THF (2 ml) and the mixture
stirred at room temperature for ca. 48 h. The slurry was then diluted
with water (20 ml) and diethyl ether (50 ml) followed by extraction
of the aqueous layer with diethyl ether (2 x 30 ml). The combined
ethereal extracts were washed with water (1 x 10 ml) and brine

323
(1 x 10 ml) and dried (Na2S04). Concentration under reduced pressure
followed by chromatography (8% diethyl ether-petrol) afforded 2-(R)-
[[Z-bromo-1-{R)-methyl) ethy t]-5-(R)-(1-(S)-methoxyethyl)~3-(S)~methyl~
tetrahyarofuran (144) (307 mg, 95%) as a colourless oil, M q25 " 4-3°
(c 4.1, CHC13); v av (film) 2967, 2932, 2879, 1359, 1093, 1010, and
862 cm-1; 6 (400 MHz) 0.84 (3H, d, J 7.5 Hz, C-3 methyl), 0.91 (3H, d,
J 6.5 Hz, CH(Me)CH2Br), 1.05 (3H, d, J 6.5 Hz, CH(Me)OMe), 1.63 (1H, dd,
J 12.5 and 6.6 Hz, 4-H ), 1.77 (1H, m, from decoupling, J 9.6, 6.4, and
3 Hz, Cff(Me)CH2Br), 1.93 (1H, ddd, J 12.5, 9.5, and 6.5 Hz, 4-H ),
2.19 (1H, m, from decoupling, J 7.5, 6.5, and 4 Hz, 3-H), 3.26 (1H, dq,
J 6.5 and 4.5 Hz, Ctf(Me)OMe), 3.32 (3H, s, OMe), 3.47 (1H, dd, J 9.8
and 6.4 Hz, C772Br), 3.50 (1H, dd, J 9.6 and 4 Hz, 2-H), 3.62 (1H, dd,
J 9.8 and 3 Hz, C/72Br), and 3.90 (1H, ddd, J 9.5, 6.6, and 4.5 Hz, 5-H);
m/z 266 (M+(slBr), weak), 264 (M+(79Br), weak), 207 (M+-(81Br)-
CH(Me)OMe), 205 (M+(79Br)-CH(Me)0Me), 163 (M+(8xBr)-C2H1+0-CH(Me)0Me),
161 (M+( /9Br )-C2H1+0-CH(Me)0Me), 143 (M+-CH(Me)CH2Br), 111 (M+-
CH(Me)CH2Br-Me0H), and 59 (CH(Me)0Me+); (Found: (M+(79Br)-CH(Me)0Me),
205.0231. CilH2102Br requires (M+(79Br)-CH(Me)0Me), 205.0228 ;
(Found: C, 49.99; H, 7.92. CliH2102Br requires C, 49.82; H, 7.98%).
102. Addition of Pheneth.ylmagnesiurn bromide to the left-hand side
aldehyde (59) obtained from degradation
MeO

324
To a two-necked flask fitted with a reflux condensor containing
dry magnesium turnings (240 mg, 10.0 mmol) and diethyl ether (5 ml)
was added freshly distilled (2-bromoethy1)benzene (1.37 ml, 10.0 mmol)
in diethyl ether (5 ml) (a few drops of neat bromide were added
initially and the solution briefly heated in order to start formation
of the Grignard reagent). To a stirred solution of the aldehyde
(59) (490 mg, 1.00 mmol) in dry THF (10 ml) at -30°C was added phen-
ethylmagnesium bromide (1.50 ml of the 1.0M solution prepared as
above, 1.30 mmol, transferred via a gas-tight syringe). After allow
ing to warm to 0°C over 30 min with stirring, the reaction was quenched
by dropwise addition of saturated aqueous ammonium chloride solution
(1 ml). After dilution with further ammonium chloride solution
(10 ml) and diethyl ether (50 ml), the aqueous layer was extracted
with diethyl ether (2 x 20 ml) and the combined organic extracts washed
with water (1 x 10 ml) and dried (Na2S0<J. Concentration under reduced
pressure followed by chromatography (gradient elution, 50% ethyl acetate-
petrol -* ethyl acetate) gave two diastereoisomeric alcohols (134) (120
mg, 19%) as a yellow oil, v (CHC13) 3403, 2926, 1726, 1629, 1250,
1049, 1020, 910, and 733 cm"1; 6 (90 MHz), 0.60 - 2.30 (27H, m),
1.97 (1.5H, s, 0C0CH3), 1.98 (1.5H, s, 0C0CH3), 3.25 - 4.00 (4H, m),
4.02 (3H, s, OMe), 4.05 - 4.98 (5H, m), 5.20 (1H, d, J 10 Hz, CH=),
and 7.20 (5H, m, Ph); m/z 537 (M+-CH3C02), 536 (M+-CH3C03H), 518 (M+-
H20-CH3C02H), 461 (M+-CH(0H)CH2CH2Ph), 401 (M+-CH(0H)CH2CH2Ph-CH3C02H),
367 (M+-170-CH3C02), 231 (M+-170-CH(0H)CH2CH2Ph-CH3C02H), 171, 170

325
103. Preparation of Methyl (±)-6-(i?)-[(1-(i?ff)-hydroxy-3-phenyl )propy1 ]
tetrahydropyran-2-(fl)-ylacetate.
To a stirred solution of the trans-aldehyde (22B) (190 mg, 1.00
mmol) in THF (10 ml) at -30°C was added phenethylmagnesiurn bromide
(1.50 ml of a 1.0M solution, 1.50 mmol, prepared as in Experiment 102).
After allowing the reaction mixture to warm to 0°C over 30 min, work-up
as before (Experiment 102) followed by chromatography (40% diethyl
ether-petrol) afforded two diastereoisomeric alcohols (135AB) (244 mg,
84% combined yield) (1:1 mixture) both as colourless oils, (135A, less
polar): v (film) 3491, 3025, 2938, 1733, 1207, 1047, and 911 cm'1;max.6 (90 MHz) 1.20 - 2.00 (8H, m), 2.20 - 2.95 (4H, m, C#2C02Me and C#2Ph),
3.15 (1H, br s, OH), 3.30 - 3.95 (2H, m, CtfOH and Ctf-0), 3.58 (3H, s,
OMe), 4.08 (1H, m, Ctf-0), and 7.15 (5H, m, Ph); m/z 292 (M+, weak),
274 (M+-H20), 219 (M+-CH2C02Me), 183 (M+-CH2Ph-H20), 170, 157 (M+-
CH(0H)CH2CH2Ph), 125 (M+-CH(OH)CH2CH2Ph-MeOH), and 105 (PhCH2CH2+);
(Found: M+, 292.1652. C17H2h0w requires M+, 292.1675).
(135B, more polar): v (film) 3446, 3025, 2938, 1736, 1208, 1043,max.and 912 cm'1; 6 (90 MHz) 1.20 - 1.90 (8H, m), 2.00 (1H, br s, OH),
2.25 - 2.90 (4H, m, C#2C02Me and Ctf2Ph), 3.30 - 3.75 (2H, m, CtfOH and
C.7-0), 3.62 (3H, s, OMe), 4.20 (1H, m, Cff-0), and 7.15 (5H, m, Ph);
m/z 292 (M+, weak), 274 (M+-H20), 219 (M+-CH2C02Me), 183 (M+-CH2Ph-H20),
170, 157 (M+-CH(0H)CH2CH2Ph), 125 (M+-CH(OH)CH2CH2Ph-MeOH), and 105
(PhCH2CH2+); (Found: M+, 292.1652. C17H21+01+ requires M+, 292.1675).

326
104. Attempted Elimination of Alcohols (135AB) using Burgess1 salt
(139)
To methyl (carboxysulphamoyl)triethylammonium hydroxide inner
salt^^0* (Burgess' salt, 81 mg, 0.34 mmol) in THF (3 ml) under argon
was added the mixture of alcohols (135 AB) (67 mg, 0.23 mmol) in THF
(2 ml) and the resulting solution stirred overnight at room temperature.
The solvent was then evaporated in a stream of argon and replaced with
toluene (5 ml). The solution was gradually heated until elimination
began to occur {ca. 80°C) and maintained at this temperature until
reaction was complete (t.l.c.). Removal of the solvent under
reduced pressure followed by chromatography (15% diethyl ether-petrol)
gave methyl 6-(3-phenylpropyl)-2^3-dlhydro-(\\\-pyran-2-ylacetate (137)
(32 mg, 51%) and a slightly more polar product (£,Z)-methyl trccns-6-
(3-phenylprop-l-enyl)tetrahydropyran-2-ylacetate (138) as ca. 6:1
E:Z mixture (6 mg, 9%) both as colourless oils, (137): v (film)max.2927, 1732, 1675, 1287, 1153, and 1058 cm”1; 6 (250 MHz) 1.40 - 1.80
(4H, m), 1.90 - 2.05 (4H, m, allylics), 2.41 (1H, dd, J 15 and 6 Hz,
C#2C02Me), 2.52 (2H, t, J 8 Hz, Ctf2Ph), 2.41 (1H, dd, J 15 and 7.5 Hz,
Ctf2C02Me), 3.61 (3H, s, OMe), 4.19 (1H, m, 2-H), 4.42 (1H, m, CH=),
and 7.15 (5H, m, Ph); m/z 275 (MH+), 274 (M+), 170 (MH+-CH2CH2Ph),
155 (M+-CH2CH2CH2Ph), and 96 (M+-CH2CH2CH2Ph-C02Me); (Found: C, 74.15;

327
H, 8.35. Ci7H:203 requires C, 74.42; H, 8.08%).
(138): x> (CHC13) 2942, 1731, 1601, 1098, 1033, and 974 cm'1;
6 (250 MHz) 1.40 - 1.80 (6H, m), 2.36 (1H, dd, J 14 and 5.5 Hz,
Cii2C02Me), 2.54 (1H, dd, J 14 and 8 Hz, Ctf2C02Me), 3.34 (2H, d,
J 7 Hz, Ctf2Ph), 3.60 (3H, s, OMe), 4.16 (1H, m, 2-H), 4.30 (1H, m, 6-H),
5.55 (1H, ddt, J 15.5, 5, and 1 Hz, l'-H), 5.72 (1H, m, J 15.5 Hz,
21-H), and 7.18 (5H, m, Ph); m/z 274 (M+) and 183 (M+-CH2Ph).
105. Preparation of (t)-Methyl 6-(i?)-r(l-(flS)-methylsulphonyloxy-3-
phenyl )propy11tetrah,vdrop,yran-2-(-fi>)-,y1acetate (141AB)
To the diastereoisomeric mixture of alcohols (135AB) (103 mg,
0.35 mmol) in dichioromethane (3 ml) at -5°C was added triethyl amine
(58 pi, 0.42 mmol) followed by methanesulphonyl chloride (30 pi, 0.39
mmol). After stirring for 15 min, the solution was diluted with ice-
water (5 ml) and dichioromethane (15 ml). The aqueous layer was
extracted with dichioromethane (2 * 10 ml) and the combined organic
extracts washed with brine (1 x 5 ml) and dried (Na2S0<J. Chromato
graphy (40% diethyl ether-petrol) afforded a diastereoisomeric mixture
(1:1) of mesylates (141AB) (127 mg, 97%) as a colourless oil, vmax.(film) 3026, 2942, 1734, 1171, 1094, and 922 cm'1; 5 (90 MHz) 1.30 -
2.10 (8H, m), 2.20 - 2.90 (4H, m, Ctf2C02Me and Ctf2Ph), 3.02 (3H, s,
0S02Me), 3.60 (1.5H, s, OMe), 3.62 (1.5H, s, OMe), 3.70 (1H, m, 2-H),
4.28 (0.5H, m, 6-H), 4.34 (0.5H, m, 6-H), 4.55 (0.5H, dd, J 11.5 and

328
6.5 Hz, C#0S02Me), 4.78 (0.5H, dd, J 11.5 and 6.5 Hz, C£0S02Me), and
7.20 (5H, s, Ph); m/z 274 (M+-MeS03H), 183 (M+-CH2Ph-MeS03H), 170,
157 (M+-CH(OMs)CH2CH2Ph), 125 (M^-CHfOMs)CH2CH2Ph-MeOH), and 91
(PhCH2+); (Found: M+-MeS03H, 274.1576. C18H2606S requires M+-
MeS03H, 274.1569).
106. Attempted Elimination of the Mesylates (141AB)
To neutral alumina (400 mg, activated by heating at 200°C at
0.1 mm Hg for 24 h) in dry dichioromethane (2 ml) was added the
diastereoisomeric mixture of mesylates (141AB) (30 mg, 0.08 mmol) in
dichioromethane (1 ml) at room temperature. After 48 h, the alumina
was filtered off and washed several times with dichioromethane.
After concentration under reduced pressure and chromatography (15%
diethyl ether-petrol) a mixture of the enol ether (137) (11 mg, 50%)
and the required olefin (138) (7.7 mg, 35%, E : Z mixture, 6:1) were obtained with spectral properties as reported previously (Experiment
104).

329
107. Preparation of (i)-Methyl 6-(7?)-|~(3-phenYl -l-(/?S)-selenophenvl )-
propyl ]tetrahydropyran-2-(ff)-yl acetate (142)
To a diastereoisomeric mixture (65:35) of alcohols (135AB) (47 mg,
0.16 mmol) in THF (4 ml) at room temperature was added tri-n-butyl-
phosphine (80 pi, 0.32 mmol) and N-phenylselenophthalimide (72 mg,
0.24 mmol) in THF (1 ml). After stirring at room temperature for
30 min, the reaction was diluted with water (5 ml) and diethyl ether
(15 ml) and the aqueous layer extracted with further ether (2 * 10 ml).
The combined ethereal extracts were washed with brine (1 x 10 ml),
dried (Na2S0it) and evaporated under reduced pressure. The resulting
residue was chromatographed (20% diethyl ether-petrol) to afford the
secondary selenide (142) (43 mg, 62%) as a slightly yellow oil,
vm^v (film) 2938, 1735, 1169, and 1036 cm-1; 5 (250 MHz) 1.40 - 2.20
(8H, m), 2.32 - 3.05 (4H, m, C#2C02Me and Ctf2Ph), 3.18 (0.65H, m,
CffSePh), 3.33 (0.35H, m, CtfSePh), 3.58 (1.05H, s, OMe), 3.62 (1.95H,
s, OMe), 3.80 (1H. m, 2-H), 4.10 (0.35H, m, 6-H), 4.43 (0.65H, m, 6-H),
7.25 (8H, m, Ph and SePh), and 7.54 (2H, m, SePh); m/z 432 (M+), 275
(PhCH2CH2CH80SePh+), 183 (C2H280SePh+), 157 (PhSe+ and (C5H80)CH2C02Me+),
and 91 (PhCH2 + ); (Found: M+, 432.1214. C^HjsOaSe requires M+,
432.1204).
(135AB) (142)

330
108. Elimination of the selenide (142) to afford ( 7)~(±j-Methyl 6-(.?)-
(3-phenylorop-l-enyl)tetrahydrooyran-2-(i?)-y1acetate (138)
H H }
C O M e SePh
(142)
Ph
To a stirred solution of the selenide (142) (40 mg, 0.09 mmol)
in acetonitrile (3 ml) was added 30% aqueous hydrogen peroxide solution
(50 pi, 0.45 mmol) at 0°C. After 1 h the solution was taken up in
diethyl ether (20 ml) and washed with 5% aqueous sodium thiosulphate
solution (1 x 7 ml) and water (1 x 5 ml). Drying (Na2S0,J, concen
tration under reduced pressure and chromatography (15% diethyl ether-
petrol) afforded solely the (£)-olefin (138) (19 mg, 74%) as a colour
less oil and with spectral properties as reported in Experiment 104.
109. Preparation of o-r2-N-(g-Trimeth.ylsilylethox.vmeth.yl )p,yrrolyl-
carbonylIbenzyl alcohol (158)
O
To a stirred solution of N-(p-t
(3.12g, 15.84 mmol) in dry DME (15 m
nBuLi (11.30 ml of a 1.56M solution
5 '
"imethylsi1ylethoxymethyl)pyrrole
1) at 0°C under argon was added
in hexane, 17.63 mmol) dropwise
1

331
over 5 min. After 10 min at 0°C, phthalide (1.07g, 8.00 mmol) in
dry DME (10 ml) was added rapidly in one portion to the yellow
solution. The reaction was allowed to warm to room temperature over
10 min before pouring into saturated aqueous ammonium chloride
solution (50 ml) and diethyl ether (100 ml). The aqueous layer was
extracted with diethyl ether (2 x 50 ml) and the combined ethereal
extracts washed with water (1 x 20 ml) and brine (1 x 20 ml). After
drying (Na2S0u), the solvent was removed under reduced pressure and
the orange-brown residue chromatographed (gradient elution, petrol^
50% diethyl ether-petrol) to afford recovered SEM-pyrrole (2.10g,
67%) (i?p 0.33, 6% diethyl ether-petrol) and the product alcohol (158)
(1.10g, 64% based on recovered SEM-pyrrole) (i?p 0.40, 50% diethyl
ether-petrol) as a yellow oil, v (film) 3605, 2950, 2890, 1770,FTiaX .
1605, 1400, and 1240 cm-1; 6 (250 MHz) 0.00 (9H, s, SiMe3), 0.92 (2H,
m, Ctf2SiMe3), 3.62 (2H, t, J 8 Hz, 0C//2CH2SiMe3), 4.54 (1H, s, OH),
4.78 (2H, br s, CH20H), 5.88 (2H, s, 0C#2N), 6.22 (1H, dd, J 4 and
2.5 Hz, 41 -H pyrrole), 6.66 (1H, dd, J 4 and 1.5 Hz, 5'-H pyrrole),
7.22 (1H, dd, J 2.5 and 1.5 Hz, 3'-H pyrrole), and 7.32 - 7.71 (4H, m,
Ph); m/z 331 (M+), 313 (M+-H20), 303, 285 (M+-CH20H-Me), 213 (M+-
Me3SiCH2CH20H), 105 (PhC0+), and 73 (SiMe3+); (Found: M+, 331.1606.
Ci8H25N03Si requires M+ , 331.1603).

332
110. Preparation of o-[2-N-(S-Trimetnylsi 1ylethoxymethyl)pyrrolvl-
carbonylIbenzaidehyde (159)
To the alcohol (158) (1.10g, 3.32 mmol) in dry acetonitrile
(20 ml) was added excess activated manganese dioxide at room temperature.
The reaction was stirred until t.l.c. indicated consumption of all
starting material (c a . 48 h) and then filtered through a small pad of
silica gel, the solid being washed with diethyl ether several times.
Concentration under reduced pressure followed by chromatography of the
residue (35% diethyl ether-petrol) gave o-[2-N-(&-tr'imethylsilylethoxy-
methyl)pyrrolylcarbonyl'\benzaldehyde (159) (790 mg, 72%) as a slightly
yellow oil, v av (film) 3110, 2960, 2900, 1710, 1640, 1420, 1250. and
1090 cm-1; 6 (250 MHz) 0.00 (9H, s, SiMe3), 0.98 (2H, t, J 8 Hz,
Ctf2SiMe3), 3.67 (2H, t, J 8 Hz, 0Ci72CH2SiMe3), 5.88 (2H, s, 0CH2N),
6.19 (1H, dd, J 4 and 2.5 Hz, 4'-H pyrrole), 6.50 (1H, dd, J 4 and 1.5
Hz, 5'-H pyrrole), 7.23 (1H, dd, J 2.5 and 1.5 Hz, 31-H pyrrole),
7.55 - 7.68 (3H, m, Ph), 7.99 (1H, m, Ph), and 10.08 (1H, s, CH0);
m/z 329 (M+), 313, 256 (M+-SiMe3), 242 (M+-CH2SiMe3), 213 (M+-CH0-
CH2SiMe3), 211 (M+-Me3SiCH2CH20H), 191, 182 (M+-CH0-Me3SiCH2CH20H),
and 73 (SiMe3+); (Found: C, 65.68; H, 7.08; N, 4.34. Ci8H23N03Si
requires C, 65.62; H. 7.04; N, 4.25%).

333
111. Preparation of (ff)-Ethyl 3-{2-f2-N-(S-trimethy1 si 1ylethoxymethyl)-
pyrrolylcarbonyllphen-1-yl)prop-2-enoate (160)
To carbethoxymethylidenetriphenylphosphorane (840 mg, 2.41 mmol)
in dichioromethane (15 ml) under argon was added the aldehyde (159)
(790 mg, 2.40 mmol) in dichioromethane (5 ml) at room temperature.
After stirring for ca. 5 h, the solvent was evaporated under reduced
pressure and the residue chromatographed (35% diethyl ether-petrol)
to afford (E)-et^yZ.-3-{2-[2-A7-(p - tvirr.e thylsilyle thoxyme thyl) pyrro lyl-
carbonyl]pherrl~yl}prop-2-enoate (160) (910 mg, 95% as a colourless oil,
v (film) 3050, 2960, 1710, 1630. 1595, 1175, and 1075 cm-1; max.6 (250 MHz) 0.00 (9H, s, SiMe3), 0.98 (2H, t, J 8 Hz, C/72SiMe3), 1.28
(3H, t, J 7.5 Hz, C02CH2C#3), 3.68 (2H, t, J 8 Hz, 0C#2CH2SiMe3), 4.20
(2H, q, J 7.5 Hz, C02Ctf2CH3), 5.87 (2H, s, 0CH2N), 6.18 (1H, dd,
J 4 and 2.5 Hz, 4"-H pyrrole), 6.40 (1H, d, J 16 Hz, =CtfC02Et), 6.52
(1H, dd, J 4 and 1.5 Hz, 5"-H pyrrole), 7.22 (1H, dd, J 2.5 and 1.5 Hz,
3"-H pyrrole), 7.38 - 7.52 (3H, m, Ph), 7.70 (1H, d, J 8 Hz, Ph), and
7.84 (1H, d, J 16 Hz, PhC#=); m/z 354 (M+-0Et), 326 (M+-C02Et and
M+-SiMe3), 298 (M+-CH2CH2SiMe3), 281 (M+-Me3SiCH2CH20H), 268 (M+-
CH20CH2CH2SiMe3), 227, and 215; (Found: C, 66.04; H, 7.61; N, 3.54.
C22H29N04Si requires C, 66.13; H, 7.32; N, 3.51%).

334
112. Preparation of (ff)-o-(3-Hydroxyproo-l-eny1)-g-[2-N-(g-trimethyl-
silylethoxymethyl)pyrrolylIbenzyl alcohol (161)
To the (£')-a,p-unsaturated ester (160) (700 mg, 1.75 mmol) in
toluene (10 ml) at -78°C under argon was added excess DIBAL (3.63 ml
of a 1.50M solution in toluene, 5.45 mmol) with stirring. After 1 h
the reaction was quenched by dropwise addition of glacial acetic acid
(1 ml) followed by warming to room temperature. The solution was
then poured into ethyl acetate (150 ml) and the liquid filtered
rapidly through a short pad of silica gel. Extraction of the solid
alum with further ethyl acetate, filtration through the same silica
gel pad and concentration under reduced pressure gave a yellow oil
which after chromatography (50% diethyl ether-petrol) afforded (E)-o-
(3- hydroxypvopr1- enyl) -a-[2-/7- (3 - tvimethylsi lylethoxymethyl) pyrrolyt] -
benzyl alcohol (161) (353 mg, 56%) as a colourless oil, v (film)max.3600, 3460, 3040, 2950, 1605, 1280, 1260, 1190, and 1070 cm-1;
6 (250 MHz) 0.00 (9H, s, SiMe3), 0.92 (2H, m, C/72SiMe3), 1.80 (2H,
br s, OH (x2) ), 3.50 (2H, m, 0C/72CH2SiMe3), 4.12 (2H, br s, C/720H),
5.25 (2H, s, OCH2N), 5.68 (1H, dd, J 3.5 and 1.5 Hz, 3"-H pyrrole),
5.98 (1H, t, J 3.5 Hz, 4"-H pyrrole), 6.07 (1H, dt, J 15.5 and 5.5 Hz,
=C/7C02Et), 6.13 (1H, d, J 4 Hz, CHQH), 6.67 (1H, d, J 15.5 Hz, PhC#=),
6.72 (1H, dd, J 3.5 and 1.5 Hz, 5"-H pyrrole), 7.22 - 7.42 (3H, m, Ph),

335
and 7.65 (1H, m, Ph); m/z 359 (M+), 341 (M+-H20), 331, 310 (M+-H20-
CH2OH), 234, 223 (M+-Me3SiCH2CH20H-H20), and 210 (M+-Me3SiCH2CH20H-
CH20H); (Found: C, 66.67; H, 8.26; N, 3.63. C20H29N03Si requires
C, 66.80; H, 8.13; N, 3.91%).
113. Preparation of (g)-o-(3-Triisopropylsi 1yloxyprop-l-enyl)-a-f2-N-
(6-trimethyi si 1 vl ethoxymethy 1 )pyrro 1 y 1 ]benzyl alcohol (165")r
To the diol (392 mg, 1.10 mmol) in DMF (8 ml) was added imidazole
(150 mg, 2.20 mmol) in DMF (2 ml) and tnisopropylsi 1yl chloride
(0.24 ml, 1.10 mmol) at room temperature. After stirring overnight,
the mixture was poured into diethyl ether (60 ml) and water (20 ml).
The aqueous layer was extracted with diethyl ether (3 * 40 ml) and the
combined ethereal extracts washed with water (1 x 20 ml) and brine
(1 x 20 ml). After drying (Na2S04) and removal of the solvent under
reduced pressure, the resulting residue was chromatographed (gradient
elution, 20 40% diethyl ether-petrol) to afford (E)-o-(3- t r i i s o -
p r o p y l s i l y l o x y p r o p -1-e n y l ) -a- [2-N - ( $ - t r i m e t h y l s i l y l e t h o x y m e t h y l ) -
p y v r o l y t ] b e n z y l a l c o h o l ( 165) (375 mg, 67%) and some of the o x i d i s e d
m a t e r i a l (166) (See Experiment 114) (132 mg, 23%) both as colourless
oils, (165) :v (film) 3427, 3064, 3029, 2945, 1653, 1601, 1571,ITlaX .
1067, and 836 cm-1; 6 (60 MHz) 0.00 (9H, s, SiMe3), 0.90 - 2.00 (23H,
m), 3.48 (3H, m, OH and 0Ctf2CH2SiMe3), 4.25 (2H, dd, J 4 and 1.5 Hz,

336
C#20Si(1 Pr)3)r 5.21 (2H, m, 0CH2N), 5.55 (1H, m,
3"-H pyrrole), 5.90 - 6.20 (3H, m, =C£C02Et, CtfOH, and 4"-H pyrrole),
6.55 - 6.85 (2H, m, PhC#= and 5"-H pyrrole), and 7.22 - 7.65 (4H, m,
Ph); m/z 500 (M+-Me), 497 (M+-H20), 397 (M+-Me3 SiCH2CH20H), 380 (M+-
0H-Me3 SiCH2CH20H), 342 (M+-0Si(1Pr)3), and 73 (SiMe3+); (Found:
C, 67.31; H, 9.80: N, 2.72. C^H^MOaSi requires C, 67.52; H, 9.57;
N, 2.72%).
114. Preparation of (ff)-(2-N-(3-Trimethy1si1ylethoxymethylpyrrolyl)-
carbonyl)-o-(3-triisopropyl silyloxyprop-l-enylIbenzene (166)
To the hydroxy-compound (165) (132 mg, 0.26 mmol) in acetonitrile
(10 ml) was added excess activated manganese dioxide (500 mg). The
reaction was stirred until t.l.c. indicated no remaining starting
material {ca. 1 h) and then filtered through a small silica gel pad.
Concentration under reduced pressure followed by chromatography (20%
diethyl ether-petrol) afforded (t)-[2-i'l-{$-tr%methyls'Llyle-bhoxymethyl)-
p y r r o l y l o a r b o n y t ] -o- (3-t r i i s o p r o p y l s i l y l o x y p r o p - 1-e n y l ) b e n z e n e (166)(102 mg, 78%) as a colourless oil, v (film) 2947, 2865, 1634, 1409,max •1245, and 1084 cm"1; 6 (60 MHz) 0.00 (9H, s, SiMe3), 0.70 - 1.80 (23H,
m), 3.40 -3.80 (2H, m, 0Cfl2CH2 SiMe3), 4.34 (2H, dd, J 4.5 and 1.5 Hz,
Ctf20 ( 1 Pr)3), 5.85 (2H, s, 0CH2N), 6.05 - 6.80 (4H, m, CH=CH, 4"-H
and 5"-H pyrrole), and 6.85 - 7.80 (5H, m, Ph and 3"-H pyrrole);

337
m/z 514 (MH+), 499 (MH+-Me), 471 (MH+-’lPr), 413 (MH+-CH2CH2SiMe3), 396
115. Preparation of (E)-3-{2-F2-N-(S-Trimethylsi 1ylethoxymethyl)-
pyrroly 1 carbonyl ]phen-l-,y1 }prop-2 -en-l-ol (167)
To the silyl ether (166) (560 mg, 1.09 mmol) in acetonitrile
(10 ml) was added 40% aqueous HF in acetonitrile (1:9) (0.5 ml) drop-
wise at room temperature. After stirring for 1 h the solution was
diluted with chloroform (50 ml) and water (10 ml). The aqueous layer
was extracted with chloroform (3 * 20 ml) and the combined organic
extracts washed with water and brine (1 * 20 ml). After drying
(Na2S0,J and concentration under reduced pressure, the resulting yellow
oil was purified by chromatography (75% diethyl ether-petrol) to
afford (E)-3-{2-[2-77-(3 -trimethyIsilylethoxymethyl)pyrrolyIcarbonyl]-
phen-l-yl}prop-2~en-l-ol (167) (385 mg, 99%) as a colourless oil.
v (film) 3430, 3108, 3060, 2960, 1628, 1565, 1527, 1250, 1085, and max.840 cm"1; 6 (250 MHz) 0.00 (9H, s, SiMe3), 0.95 (2H. m, C/72SiMe3),
1.63 (1H, br s, OH), 3.62 (2H, m, 0C772CH2SiMe3), 4.21 (2H, d, J 5.5 Hz,
CT/20H), 5.84 (2H, s, 0CH2N), 6.16 (1H, dd, J 4 and 2.5 Hz, 4“-H
pyrrole), 6.31 (1H, dt, J 16 and 5.5 Hz, =C77CH2OH), 6.51 (1H, dd, J 4
and 1.5 Hz, 5M-H pyrrole), 6.74 (1H, d, J 16 Hz, PhCtf=), 7.17 (1H, dd,
N, 2.74. C2 9H1+vN02Si requires C, 67.78; H, 9.22; N, 2.73%).

338
J 2.5 and 1.5 Hz, 3"-H pyrrole), 7.22 - 7.44 (3H, m, Ph);and 7.60 (1H,
d, J 8 Hz, Ph); m/z 357 (M+, weak), 339 (M+-H20), 327 (MH+-CH20H),
268,and 73 (SiMe3+); (Found: C, 67.34; H, 7.76; N, 4.03.
C2oH2 7NO3Si requires C, 67.19; H, 7.61; N, 3.92%).
116. Preparation of {E)~ l-(3-Phenyl thioprop-l-enyl )-2-(~2-N-(6-tri-
methylsi 1v1ethoxymethyl)pyrrolylcarbonylIbenzene (168)
32To N-phenylthiosuccinimide (400 mg, 1.93 mmol) in dry THF
(3 ml) was added redistilled tri-n-butylphosphine (0.48 ml, 1.90 mmol).
After stirring for 5 min at room temperature, the alcohol (167) (385
mg, 1.08 mmol) in THF (3 ml) was added to the pink solution with
rapid stirring. After 5 min, reaction was complete and the solution
was diluted with diethyl ether (20 ml) and water (10 ml). Extraction
of the aqueous layer with diethyl ether (3 x 30 ml) followed by washing
of the combined ethereal extracts with water ( 1 x 2 0 ml), drying
(Na2S01+) and concentration under reduced pressure gave a yellow oil
which after chromatography (gradient elution, petrol -> 15% diethyl
ether-petrol ) afforded {E)-l-{3-phenylthioprop-l-enyl)-2-[2-N-{$-
tr-imethyls'ilylethoxymethyl)pyrrolylcarbonyl]benzene (168) (470 mg, 95%)
as a colourless oil, v (film) 3153, 3065, 2926, 1628, 1584, 1567,max.1527, 1250, and 1080 cm-1; 6 (90 NHz) 0.00 (9H, s, SiMe3), 1.02 (2H,
t, J 8 Hz, Ctf2SiMe3), 3.65 - 3.80 (4H, m, Ctf2SPh and 0Ctf2CH2SiMe3),
5.92 (2H, s, 0CH2N), 6.25 (1H, dd, J 4 and 2.5 Hz, 4"-H pyrrole),

339
6.27 (1H, dt, J 16 and 7 Hz, =C#CH2SPh), 6.55 (1H, dd, J 4 and 1.5 Hz,
5"-H pyrrole), 6.75 (1H, dt, J 16 and 1 Hz, PhC#=), and 7.15 - 7.70
(10H, m, Ph and 3"-H pyrrole); m/z 449 (M+), 434 (M+-CH3), 340 (M+-
SPh), and 73 (SiMe3+); (Found: C, 69.30; H, 7.06; N, 3.27.
C2 6H3 iN02SSi requires C, 69.45; H, 6.95; N, 3.11%).
117. Preparation ot (g)-l-(3-Phenylsulphonylprop-1-enyl)-2-[2 -N-(p-
trimethylsilylethoxymethyl)pyrroly1carbonylIbenzene (169)
5"
To the sulphide (168) (200 mg, 0.44 mmol) in dichloromethane
(0.50 ml)/diethyl ether (4 ml) and diphenyl diselenide (140 mg,
0.44 mmol) at 0°C was added 30% aqueous hydrogen peroxide solution
(0.23 ml, 2.03 mmol) dropwise over 3 min. After stirring for 1 h at
0°C, the solution was allowed to reach room temperature and stirred
for a further 6 h. Work-up involved addition of dichloromethane
(50 ml) followed by washing with saturated aqueous sodium bicarbonate
solution (2 x 5 ml). 5% aqueous sodium metabisulphite solution
(1 x 10 ml) and brine (1 x 10 ml) respectively. The organic solu
tion was dried (Na2S0£+), 5% aqueous sodium metabisulphite solution
(1 x 10 ml) and brine (1 x 10 ml)respectively. The organic solution
was dried (Na2S04) and the solvent evaporated under reduced pressure.
Chromatography (60% diethyl ether-petrol) afforded (E)-l-(3- p h e n y l -
s u l p h o n y I p r o p - 1 - e n y Z)-2-[2-iV-(3- t r i m e t h y l - s i l y Z e t h o x y m e t h y 1 ) p y r r o l y l ~

340
carbonyllbenzene (169) (178 mg, 83%) as a white crystalline
solid, v (CHC13} 3027. 2955, 1628, 1596, 1567, 1250, 1178,
1155, and 1082 cm'1; 5 (250 MHz) 0.00 (9H, s, SiMe3), 0.95 (2H, t,
J 8 Hz, C^2Si Me3), 3.60 (2H, t, J 8 Hz, 0Cff2CH2SiMe3), 3.91 (1H, d,
J 7 Hz, C#2S02Ph), 3.92 (1H, d, J 7 Hz, Ctf2S02Ph), 5.78 (2H, s,
0CH2N), 6.09 (1H, dt, J 16 and 7 Hz, =Ci7CH2S02Ph), 6.18 (1H, dd,
J 4 and 2.5 Hz, 4"-H pyrrole), 6.46 (1H, dd, J 4 and 2 Hz, 5“-H
pyrrole), 6.60 (1H, d, J 16 Hz, PhCtf=), 7.18 (1H, dd, J 2.5 and 2 Hz,
3"-H pyrrole), 7.28 - 7.58 (7H, m, Ph), and 7.82 (2H, m, Ph);
m/z 466 (M+-CH3), 364 (M+-0CH2CH2SiMe3), 340 (M+-S02Ph), 282, and 73
(SiMe3 +); (Found: C, 65.01; H, 6.57; N, 2.92. C2 6H3 iNO^SSi
requires C, 64.83; H, 6.49; N, 2.91%).
118. Preparation of (ff)-(±)-Methyl 6-(i?)-{4-[2-(2-N-(B-trimethylsi 1y1 -
ethoxymethvl)pyrro1ylhydroxymethylene)phen-l-yllbut-2-enyl}tetra-
hydropyran-2 -(i?)-yl acetate (171)

341
(a) To the sulphorte (169) (100 mg, 0.21 mmol) in THF (1.5 ml) at -78°C
was added nBuLi (0.16 ml of a 1.46M solution in hexane, 0.23 mmol)
dropwise over 2 min. The deep red solution was stirred for 10 min
at -78°C before addition of the aldehyde (22B) (35 mg, 0.19 mmol) in
THF (1 ml) dropwise. After 20 min, benzoyl chloride (44 pi, 0.42 mmol)
was added in one portion and the solution allowed to warm to room
temperature over a 2 h period. Water (5 ml) and diethyl ether (15 ml)
were added and the aqueous layer extracted with diethyl ether
(3 x 10 ml). The combined organic extracts were washed with water
(1 x 10 ml) and brine (1 x 5 ml). Drying (Na2S0(+) and concentration
under reduced pressure afforded a diastereoisomeric mixture of benzoyl-
oxy-sulphones (170) (177 mg, 82%) as a yellow foam, \> (CHC13) 3062,
2950, 2871, 1731, 1629, 1585, 1526, 1177, 1149, 1047, and 1000 cm-1;
6 (60 MHz) 0.00 (9H, s, SiMe3), 0.80 - 2.85 (10H, m), 3.20 - 4.70
(8 H, m), 5.60 - 6.80 (7H, m), and 7.00 - 8.30 (15H, m, Ph and 3"'-H
pyrrole).
(b) To the crude mixture of benzoyloxy-sulphones (170) in dry THF/MeOH
(3 ml/1 ml) was added anhydrous solid Na2HP01+ (540 mg) as a buffer and
6% sodium amalgam (210 mg) at -20°C.* After rapid stirring for 1 h a
further portion ( 2 1 0 mg) of sodium amalgam was added and stirring
continued for 2 h, before pouring into petrol (20 ml) and water (5 ml).
The aqueous layer was extracted with diethyl ether (3 x 10 ml) and the
combined organic extracts washed with water ( 1 x 1 0 ml) and dried
(Na2S0t+). Removal of the solvent under reduced pressure followed by
chromatography (40% diethyl ether-petrol) afforded the (fi’J-olefin (171)
(1:1 diastereoisomeric mixture) (60 mg, 77%) as a colourless oil,
vmav (film) 3456, 3154, 2977, 2811, 1733, 1646, and 1111 cm"1;fTlaX .
6 (250 MHz) 0.00 (9H, s, SiMe3), 0.94 (2H, m, C//2SiMe3), 1.20 - 1.44
(2H, m, 4-H2 ), 1.54 - 1.80 (4H, m, 3-H2 and 5-H2), 2.11 (1H, m, l'-H),

342
2.33 (1H, m, i'-H), 2.40 (1H, dd, J 15 and 5 Hz, C#2 C02Me), 2.61 (1H,
dd, J 15 and 7.5 Hz, Ctf2C02Me), 3.08 - 3.35 (2H, m, C#2Ph), 3.40 -
3.75 (4H, m, OH, 6 -H, and OCtf2CH2SiMe3), 3.61 (3H, s, (two v. close),
OMe), 4.18 (1H, m, 2-H), 5.32 (2H, s (two v. close), 0CH2N), 5.28 -
5.65 (2H, m, CH=CH), 5.62 (1H, dd, J 3 and 1.5 Hz, 3"'-H pyrrole),
5.98 (1H, t, J 3 Hz, 4“'-H pyrrole), 6.16 (1H, d, J 1 Hz, CtfOH), 6.73
(1H, dd, J 3 and 1.5 Hz, 5m -H pyrrole), 7.12 - 7.36 (3H, m, Ph), and
7.78 (1H, m, Ph); m/z 509 (M+-2H2), 196 (N-SEM-pyrrole*+), 157 (M+-
CH2CH=CHCH2Ar), 101 (Me3 SiCH2CH2+), and 73 (Me3 Si+ and Me02CCH2+).
i When the reduction was performed at -45°C with periodic addition of
small portions of sodium amalgam it was possible to isolate the
pyrrolylcarbonyl adduct (173) (see Experiment 118) in 20% yield in
addition to the reduced compound (171) (13%) and the methoxy-adduct
(174) (30%) (see Results and Discussion section and the Appendix for
spectral data). 1
119. Preparation of (ff)-(±)-Methyl 6-(/?)-{4-[2-(2-N-(g-trimethy1silyl~
ethoxymethyl)pyrrol,y1 carbonyl)phen-l-yl]but-2 -eny!jtetrahydro-
pyran-2-(7?)-ylacetate (173)
5"'
To the alcohol (171) (77 mg, 0.15 mmol) in acetonitrile (2 ml) was
added excess activated manganese dioxide (300 mg) at room temperature.
The reaction was stirred until t.l.c. indicated completion and then

343
filtered through a short pad of silica gel. Evaporation under
reduced pressure followed by chromatography (30% diethyl ether-petrol)
afforded the pure pyrrolylcarbonyl compound (173) (64 mg, 84%) as a
colourless oil, (CHC13) 3153, 2952, 1733, 1628, 1527, 1100, and
858 cm-1; 6 (250 MHz) 0.00 (9H, s, SiMe3), 0.92 (2H, m, Ctf2SiMe3),
1.20 - 1.40 (2H, m, 4-H2), 1.50 - 1.80 (4H, m, 3-H2 and 5-H2), 2.12
(1H, m, I'-H), 2.28 (1H, m, l'-H), 2.40 (1H, dd, J 14.5 and 5.5 Hz,
Ctf2C02Me), 2.63 (1H, dd, J 14.5 and 8 Hz, Ctf2C02Me), 3.41 (2H, d,
J 7 Hz, Ctf2Ph), 3.58 - 3.65 (3H, m, 6-H and 0Ctf2CH2SiMe3), 3.66 (3H, s,
C02Me), 4.19 (1H, m, 2-H), 5t. 38 (1H, m, J 15 Hz, 2 ‘ -H), 5.54 (1H, m,
J 15 Hz, =C#CH2Ph), 5.87 (2H, s, 0CH2N), 6.17 (1H, dd, J 4 and 2.5 Hz,
4"'-H pyrrole), 6.49 (1H, dd, J 4 and 2 Hz, 5'" pyrrole), 7.17 (1H, dd,
J 2.5 and 2 Hz, 3"'-H pyrrole), and 7.20 - 7.42 (4H, m, Ph); m/z
511 (M+), 393 (M+-Me3SiCH2CH20H), 314 (M+-N-SEM-pyrrole), 157 (M+-
CH2CH=CHCH2Ar), and 73 (Me3 Si+ and Me02CCH2+); (Found: M+, 511.2754.
C3 2H3 9N20 2Si requires M+, 511.2781).
120. Preparation of ( E’)-(±)-Methyl 6-(i?)-{4-[2-(2-p.yrrolyl carbonyl )-
phen-l-yl ]but-2-enyl }tetrahydropyran-2-(i?)-ylacetate (175)
5 " '
To the N-SEM-pyrrole protected adduct (173) (21 mg, 0.04 mmol) in
THF (1 ml) was added anhydrous tetra-n-butylammoniurn fluoride (15 pi1 Pf)of a 3M solution in THF , 0.045 mmol) and the solution stirred at

344
40°C for 16 h. After dilution with diethyl ether (10 ml) and water
(2 ml), the aqueous layer was extracted with diethyl ether (2 x 4 ml)
and the combined organic extracts washed with brine (1 x 5 ml).
Drying and concentration under reduced pressure afforded the free
pyrrolyl adduct (175) as a yellow oil. Chromatography (50% diethyl
ether-petrol) afforded the pure compound (175) (10 mg, 64%) as a
colourless oil, (CHC13) 3449, 3153, 2930, 1731, 1622, 1570, 1540,
1085, 1039, and 873 cm-1; 5 (250 MHz) 1.20 - 1.40 (2H, m, 4-H2),
l. 45 - 1.80 (4H, m, 3-H2 and 5-H2), 2.04 (1H, m, l'-H), 2.26 (1H, m,
l'-H), 2.40 (1H, dd, J 14.5 and 5.5 Hz, C/72C02Me), 2.63 (1H, dd,
J 14.5 and 8 Hz, Ctf2C02Me), 3.48 (2H, d, J 7 Hz, Ctf2Ph), 3.60 (1H,
m, 6 -H), 3.68 (3H, s, C02Me), 4.18 (1H, m, 2-H), 5.35 (1H, m, J 15 Hz,
2 1 -H), 5.54 (1H, m, J 15 Hz, =CtfCH2Ph), 6.28 (1H, m, 4"'-H pyrrole),
6.59 (1H, m, 5m -H pyrrole), 7.12 (1H, m, 3“'-H pyrrole), 7.22 - 7.40
(4H. m, Ph), and 9.59 (1H, br s, NH); m/z 381 (M+), 350 (M+-0Me),
315 (M+-Ct,Hi+N), 314 (M+-CitH5N), 308 (M+-CH2C02Me), 196 (M+-CH2Ar),
157 (M+-CH2CH=CHCH2Ar), and 125 (M+-CH2CH=CHCH2Ar-MeOH); (Found:
M+, 381.1943. C^H^NO* requires M+, 381.1940).
121. Preparation of (£)-(t)-6 -(/?)-{4-f2-(2-Pyrrolylcarbonyl)phen-l-
yl lbut-2-enyl}tetrah,ydropyran-2-(i?)-ylacetic acid (176)
hi
C v 2,t,c c o 2h
(175) (l76)

345
Tc the ester (175) (10 mg, 0-025 mmol) in THF (0.50 ml) was
added 1M aqueous lithium hydroxide solution (0.26 ml, 0.26 mmol) and
the resulting solution stirred at room temperature for ca. 5 h (t.l.c.
indicated completion). 5% Aqueous hydrochloric acid solution (2 ml)
was added and the aqueous layer extracted with diethyl ether (5 x 5 ml).
The combined ethereal extracts were washed with water (1 x 5 ml) and
dried (Na2S0u). Concentration under reduced pressure followed by
purification by filtration through a small pad of silica gel (in a
pasteur pipette using diethyl ether as eluant) afforded the pure acid
(176) ( 8 mg, 83%) (i?p 0.20, diethyl ether) as a white semi-solid,
v (CHC13) 3393, 2926, 1710, 1619, and 1118 cm"1; 6 (250 MHz)
1.20 - 1.80 (6H, m, 3-H2, 4-H2, and 5-H2), 1.98 (1H, m, l'-H), 1.30
(1H, m, l'-H), 2.38 (1H, dd, J 14.5 and 3.5 Hz, Ctf2C02H), 2.66 (1H, dd,
J 14.5 and 10 Hz, Ctf2C02H), 3.44 (2H, d, J 5 Hz, Ctf2Ph), 3.72 (1H, m,
6 -H), 4.17 (1H, m, 2-H), 5.36 (1H, m, 2 1 -H), 5.56 (1H, m, = 0 0 2Ph),
6.28 (1H, m, 4I,'-H pyrrole), 6.60 (1H, m, 5"'-H pyrrole), 7.16 (1H, m,
3"'-H pyrrole), 7.22 - 7.48 (4H, m, Ph), 10.10 (1H, br s, NH), and
C02H, diffuse; m/z 350 (M+-0H), 322 (M+-C00H), 308 (M+-CH2C00H), 256
[(M+-0H-C5HuN0) and (M+-C00H-CuHuN)], and 67 (C„HSN+).

346
X-Ray Crystallographic Data for the 3-Bromo-2,4-dimtrobenzoy 1
derivative of M139603 (Figure 7, Section 3)
Crystals of the 3-bromo-2,4-dinitrobenzoyl derivative of M139603
Clt2H5 4BrN2 0 1 3Na.CH3 0 H are monoclinic, cl - 10.418(2), b - 10.954(3),c = 20.720(5)A, p = 91.84(2)°, U = 2363A3, space group P2lf Z = 2,
M = 929.8, D = 1.31 gem"3, p(Cu-K ) = 18 cm"1, F(ooo) = 974.
3175 independent observed reflections [|F | > 3 a (|F |), % $ 58°]
were measured on a Nicolet R3m diffractometer with Cu-X radiationa
(graphite monochromator) and using w-scans. An empirical absorption
correction based upon 338 azimuthal measurements was applied. The
structure was solved by the heavy-atom method and the non-hydrogen
atoms refined anisotropically. The position of the hydroxy hydrogen
atom and the orientation of the methyl group hydrogen atoms in the
MeOH molecule were obtained from a AF map. The hydroxy hydrogen
atom was refined isotropically. The positions of all the othero
hydrogen atoms were idealised (C-H = 0.96A) assigned isotropic thermal
parameters, U(H) = 1.2 £/ (C), and allowed to ride on their parenteH
carbon atoms. The methyl groups were refined as rigid bodies. The
absolute configuration was determined both by the refinement of a
free variable n which multiplies all f" and by an F-factor test.
n refined to a value of 1.44(4) and the values of r and r _ were
0.0394 and 0.0427 respectively, and R , and R were 0.0437 andw+ w-0.0485 respectively, [w“1 = o2(F) + 0.00071 F2]. Maximum residual

347
electron density was 0.33eA“3 and the mean and maximum shift/error in
the final refinement were 0.07 and 0.59 respectively.
X-Ray Crystallographic Data for (23) (Figure 11, Section 3)
o
(2 3 )
Crystals of (23) C16H19N07 are monoclinic, a = 19.669(3),
b = 5.552(1), e = 18.736(3)A, $ = 124.35(1)°, U = 1689A3 space group Cq3
2 = 4, M = 337.3, D = 1.33 gem-3, \i{Cu-K ) = 8 cm"1, F(ooo) = 712.
1135 independent observed reflections [|F J > 3 o(|F |), 3 $ 53°] were
measured on a Nicolet R3m diffractometer with Cu-#a radiation (graphite
monochromator) and using w-scans. The structure was solved by direct
methods and the non-hydrogen atoms refined anisotropically. All theo
hydrogen atom positions were idealised (C-H = 0.96A, assigned isotropic
thermal parameters, £/(H) = 1.2 U (C), and allowed to ride on theirc \ - l
parent carbon atoms. The methyl group was refined as a rigid body.
Refinement converged to give R = 0.031, R = 0.037, [w"1 = a2(F) +w0.00067 F2]. Maximum residual electron density was 0.13eA“3 and the
mean shift/error in the final refinement were 0.09 and 0.43
respectively.

348
REFERENCES
1. U.K. Pat. Appl. 2, 027, 013 A; U.K. Pat. Appl. 0, 070, 622 A1;
D.H. Davies, E.W. Snape, P.J. Suter, T.J. King, and C.P. Falshaw,
J. Chem. Soc., Chem. Cornmun. , 1981, 1073.
2. B.C. Pressman, Ann. Review Biochem. , 1976, _45, 925.
3. J.W. Westley, Adv. Appl. Microbiol. , 1977, 22_, 177.
4. U. Graefe, R. Schlegel, and M. Bergholz, Rharmazie, 1984, 39, 661.
5. A. Agtarap, J.W. Chamberlin, M. Pinkerton, and L.K. Steinrauf,
J. Am. Chem. Soc. , 1967, 89_, 5737.
6. T. Fukuyama, K. Akasaka, D.S. Karanewsky, C.-L.J. Wang, G. Schmid,
and Y. Kishi, J. Am. Chem. Soc., 1979, 101, 262.
7. D.B. Collum, J.H. McDonald, III, and W.C. Still, J. Am. Chem. Soc.,
1980, J02, 2117.
8. J.W. Westley, R.H. Evans, Jr., C.-M. Liu, T. Hermann, and
J.F. Blount, J. Am. Chem. Soc. , 1978, 100, 6784.
9. M.P. Edwards, S.V. Ley, S.G. Lister, B.D. Palmer, and D.J. Williams,
J. Org. Chem. , 1984, 4-9, 3503.
10. K.C. Nicolaou, D.P. Papahatjis, D.A. Claremon, R.L. Magolda, and
R.E. Dolle, J. Org. Chem., 1985, J50, 1440.
11. W.R. Roush, S.M. Peseckis, and A.E. Walts, J. Org. Chem., 1984,
49, 3429.
12. R.F. Shumard and M.E. Callender, Antimicrob. Agents Chemother. ,
1968, 359.
13. A.P. Raun, U.S. Patent 3, 937, 866 (1976).
14. (a) B.C. Pressman, 'The Role of Membranes in Metabolic Regulation',
eds. M.A. Mehlem and R.W. Hanson, Academic Press, New York,
1972, p.149.

34914. (b) P. Reed and H.A. Lardy, J. Biol. Chem. , 1972, 247, 6970.
15. (a) N.T. de Guzman, B.C. Pressman, K. Lasseter, and R. Palmer,
Clin. Res. , 1973, 21_, 413.
(b) A. Schwartz, R.M. Lewis, H.G. Hanley, R.G. Munson, R.D. Dial,
and M.Y. Ray, Circ. Res. , 1974, 34, 102.
16. A.M. Doherty and S.V. Ley, submitted for publication.
17. C. Keller - Juslen, H.D. King, M. Kuhn, H.-R. Loosli, V. Pache,
T.J. Petcher, H.P. Weber, and A.von Wartburg, J. Antibiot. ,
1982, 35, 142.
18. J. Grandjean and P. Laszlo, Tetrahedron Lett., 1983, 3319.
19. J. Grandjean and P. Laszlo, J. Am. Chem. Soc. , 1984, 1_06, 1472.
20. J.M. Bulsing, E.D. Laue, F.J. Leeper, J. Staunton, D.H. Davies,
G.A.F. Ritchie, A. Davies, A.B. Davies, and R.P. Mabelis,
J. Chem. Soo., Chem. Commun. , 1984, 1301.
21. D.M. Doddrell, E.D. Laue, F.J. Leeper, J. Staunton, A. Davies,
A.B. Davies, and G.A.F. Ritchie, J. Chem. Soc., Chem. Commun.,
1984, 1302.
22. A.K. Demetriadou, E.D. Laue, J. Staunton, G.A.F. Ritchie, A. Davies,
and A.B. Davies, J. Chem. Soc., Chem. Commun. , 1985, 408.
23. D.E. Cane, W.D. Celmer, and J.W. Westley, J. Am. Chem. Soc. ,
1983, 105, 3594.
24. P.M. Booth, C.M.J. Fox, and S.V. Ley, Tetrahedron Lett., 1983, 5143.
25. B.D. Palmer, D.I.C. Thesis, 1983, University of London.
26. (a) D. Seebach and M. Pohmakotr, Helv. Chim. Acta, 1979, 62 , 843.
(b) D. Seebach, M. Pohmakotr, C. Schregenberger, B. Weidmann,
R.S. Mali, and S. Pohmakotr, Helv. Chim. Acta, 1982, 65, 419.

350
27. W. Wierenga, B.R. Evans, and J.A. Woltersom, J. Am. Chem. Soc. ,
1979, T01_, 1334.
28. R.E. Hackler, J. Org. Chem. , 1975, 40, 2978.
29. (a) M. Julia and J.-M. Paris, Tetrahedron Lett., 1973, 4833.
(b) P.J. Kocienski, B. Lythgoe, and S. Ruston, J. Chem. Soc.,
Perk-in Trccns. I, 1978, 829.
30. P.J. Kocienski, B. Lythgoe, and I. Waterhouse, J. Chem. Soc.,
Perkin Trans. I, 1980, 1045.
31. (a) M.J. Arco, M.H. Trammell, and J.D. White, J. Org. Chem.,
1976, 41_, 2075.
(b) E.R. Marinelli and A.B. Levy, Tetrahedron Lett., 1979, 2313.
(c) V. Ramanathan and R. Levine, J. Org. Chem. , 1962, 27, 1216.
32. K.A.M. Walker, Tetrahedron Lett., 1977, 4475.
33. P.-T. Ho, Can. J. Chem. , 1982, 6_0, 90.
34. S.N. Huckin and L. Weiler, J. Am. Chem. Soc. , 1974, 9(5, 1082.
35. K.C. Nicolaou,. D.P. Papahatjis, D.A. Clareman, and R.E. Dolle, III,
J. Am. Chem. Soc., 1981 , 103, 6967.
36. (a) W.M. Coates and J.R. Corrigan, Chem. and Ind. , 1969, 1594.
(b) E.T. Corey and G. Schmidt, Tetrahedron Lett. , 1979,399.
(c) S. Czernecki, C. Georgoulis, C.L. Stevens, and K. Vijayakumaran,Tetrahedron Lett. , 1985, 1699.
37. (a) E.J. Corey and J.W. Suggs, Tetrahedron Lett., 1975, 2647.
(b) G. Piancatelli, A. Scettri, and M.D'Auria, Synthesis, 1982, 245.

37. (c) J. Herscovici, M.-J. Egron, and K. Antonakis, J. Chem. Soc. 3
Perkin Trans. I, 1982, 1967.
38. A.J. Mancuso and D. Swern, Synthesis , 1981, 165.
39. (a) S.L. Huang, K. Omura, and D. Swern, J. Org. Chem. , 1976, 41,
3329.
(b) M. Marx and T.T. Tidwell, J. Org. Chem., 1984, 4_9, 788.
(c) A.J. Mancuso, D.S. Brownfain, and D. Swern, J. Org. Chem.,
1979, 44, 4148.
40. (a) T.J. King and J.P. Yardley, J. Chem. Soc. , 1961, 4308.
(b) J.M. Coxon, M.P. Hartshorn, and D.N. Kirk, Tetrahedron, 1967,
23, 3511.
(c) B.N. Blackett, J.M. Coxon, M.P. Hartshorn, B.L.J. Jackson, and
C.N. Muir, Tetrahedron, 1969, 25, 1479.
41. J.W. Morzycki, H.K. Schnoes, and H.F. DeLuca, J. Org. Chem.,
1984, 49, 2148.
42. R.E. Ireland and D.W. Norbeck, J. Org. Chem. , 1985, 50, 2198.
43. H.J. Bestmann and 0. Vostrowsky, Topics Curr. Chem., 1983, 109,
85 and 165.
44. (a) M. Schlosser and K-F. Christmann, Angew. Chem., Int. Ed. Engl.,
1965, 4, 689.;
(b) M. Schlosser, Top. Stereochem. , 1970, 5_, 1.
(c) M. Schlosser, K.-F. Christmann, and A. Piskala, Chem. Ber. ,
1970, T03, 2814.
(d) L.D. Bergelson, L.I. Barsukov, and M.M. Shemyakin, Tetrahedron,
1967, 23_, 2709, and references cited therein.
45. (a) M. Schlosser and K.-F. Christmann, Justus Liebigs Ann. Chem.,
1967, 708, 1.
351

352
45. (b) M. Schlosser and K.-F. Christmann, Angew Chem. , Ind. Ed. Engl. ,
1965, 5, 126.(c) E.J. Corey and H. Yamamoto, J. Am. Chem. Soc. , 1970, 92, 226.(d) M. Schlosser and K.-F. Christmann, Synthesis, 1969, 38.(e) D.J. Faulkner, Synthesis , 1971, 175.
46. R.J. Anderson and C.A. Henrick, J. Am. Chem. Soc. , 1975, 97_, 4327.
47. E.C. Taylor and C.-S. Chiang, Synthesis, 1977, 467.
48. F. Huet, A. Lechevallier, M. Pellet, and J.M. Conia, Synthesis,
1978, 63.
49. R.A. Ellison, E.R. Lukenbach, and C.-W. Chiu, Tetrahedron Lett. ,
1975, 499.
50. R.P. Hatch, J. Shringarpure, and S.M. Weinreb, J. Org. Chem., 1978,43, 4172.
51. T.-L. Ho and C.M. Wong, Can. J. Chem., 1972, 50, 3740.
52. S. Takano, S. Hatakeyama, and K. Ogasawara, J. Chem. Soc.j Chem.
Commun., 1977, 68.
53. B.M. Trost, H.C. Arndt, P.E. Strege, and T.R. Verhoeven, Tetrahedron Lett. , 1976, 3477.
54. R.F. Newton, D.P. Reynolds, M.A.W. Finch, D.R. Kelly, and S.M. Roberts,Tetrahedron Lett., 1979, 3981.
55. A.S. Kende and B.H. Toder, J. Org. Chem., 1982, 47, 163.
56. (a) M. Hayashi, Y. Arai, H. Wakatsuka, M. Kawamura, Y. Konishi,T. Tsuda, and K. Matsumoto, J. Med. Chem., 1980, 23_, 525.(b) P.W. Pfeffer and L.D. Silbert, J. Org. Chem., 1971 , 36, 3290.
57. L.J. Haynes and J.R. Plimmer, Quart. Rev. , 1960, 14, 292.

58.
353
J.J. Plattner, R.D. Gless, and H. Rapoport, J. Am. Chem. Soc. ,
1972, 94, 8513.
59. K.K. Ogilvie, E.A. Thompson, M.A. Quilliam, and J.B. Westmore,Tetrahedron Lett. , 1974, 2865.
50. S.K. Chaudhary and 0. Hernandez, Tetrahedron Lett. , 1979, 99.
61. B.H. Lipshutz and J.J. Pegram, Tetrahedron Lett., 1980, 3343.
62. T. Katsuki and K.B. Sharpless, J. Am. Chem. Soe. , 1980, 102,5974.
63. (a) K.C. Nicolaou, D.A. Claremon, W.E. Barnette, and S.P. Seitz J. Am. Chem. Soc. , 1979, 101 , 3704.(b) P.A. Grieco, J.Y. Jaw, D.A. Claremon, and K.C. Nicolaou,J. Org. Chem., 1981 , 4(5, 1215.
64. K.B. Sharpless, C.H. Behrens, T. Katsuki, A.W.M. Lee, V.S. Martin, M. Takatani, S.M. Viti, F.J. Walker, and S.S. Woodward, Pure and Appl. Chem. , 1983, 55_, 589.
65. (a) D.L.J. Clive, G.J. Chittattu, V. Farina, W.A. Kiel,S.M. Menchen, C.G. Russell, A. Singh, C.K. Wong, and N.J. Curtis,J. Am. Chem. Soc. , 1980, 102, 4438.(b) M. Sevrin, D. Van Ende, and A. Krief, Tetrahedron Lett.,1976, 2643.
66. (a) M. Isobe, Y. Funabashi, Y. Ichikawa, S. Mio, and T. Goto,Tetrahedron Lett., 1984, 2021.(b) M. Kitamura, M. Isobe, Y. Ichikawa, and T. Goto, J. Am. Chem.
Soc. , 1984, 106, 3252.
67. (a) H. Lindlar, Helv. Chim. Acta, 1952, _35, 446.(b) H. Lindlar and R. Dubuis, Org. Synth. Coll. Vol. V, 1973, 880.

354
68. VI.T. Brady, Tetrahedron, 1981 , 37_, 2949.
69. (a) J.C. Sauer, J. Am. Chem. Soe. , 1947, _69, 2444.(b) E.C. Taylor, A. McKillop, and G.H. Hawks, Ora. Synth.,1972, 52, 36.(c) ' W.T. Brady and G.A. Scherubel, J. Org. Chem., 1974, 3 9 ,3790.
70. (a) H. Staudinger, Chem. Ber. , 1905, 38, 1735.(b) C.W. Smith and D.G. Norton, Org. Synth. Coll. Vol. IV,
1963, 348.(c) C.C. McCarney and R.S. Ward, J. Chem. Soc. , Verkin Trans. I,
1975, 1600.
71. (a) D.A. Bak and W.T. Brady, J. Org. Chem., 1979, 4 4, 107.(b) L.R. Krepski and A. Hassner, J. Org. Chem., 1978, 43, 2879.
72. E.J. Corey, Z. Arnold, and J. Hutton, Tetrahedron Lett., 1970, 307.
73. (a) R.W. Holder, J. Chem. Ed., 1976, _53_, 81.(b) R. Montaigne and L. Ghosez, Angew. Chem., Int. Ed. Engl.,
1968, 7, 221.
74. R. Ratcliffe and R. Rodehorst, J. Org. Chem., 1970, _35, 4000.
75. (a) J.R. Parikh and W. von. E. Doering, J. Am. Chem. Soo. , 1967, 89, 5505.(b) J.M. Landesberg and J. Sieczkowski, J. Am. Chem. Soo., 1971, 93, 972.
76. K.E. Pfitzner and J.G. Moffatt, J. Am. Chem. Soc., 1963, 85, 3027;K.E. Pfitzner and J.G. Moffatt, J. Am. Chem. Soc., 1965, 817, 5661 ,5670.
77. (a) I. Tomoskozi , L. Gruber, G. Kovacs, I. Szekely, andV. Simonidesz, Tetrahedron Lett., 1976, 4639.

77.
355
(b) E.J. Corey and C.U. Kim, J. Org. Chen. , 1973, 38, 1233.(c) N.A. Nelson and R.W. Jackson, Tetrahedron Lett., 1975,3275.
78. (a) S.V. Ley and N.S. Simpkins, J. Chem. Soo., Chem. Commun.,1983, 1281.(b) D. Craig, S.V. Ley, N.S. Simpkins, G.H. Whitham, and M.J. Prior, J. Chem. Soo. , Perkin Trans. I, 1985, in press.
79. (a) D.J. Ager, J. Chem. Soo., Chem Commun., 1984, 485.(b) G.D. Cooper, J. Am. Chem. Soo., 1954, _76, 3713.
80. H.O. House, C.-Y. Chu, J.M. Wilkins, and M.J. Umen, J. Org. Chem.,
1975, 40, 1460.
81. B.H. Lipshutz, R.S. Wilhelm, and D.M. Floyd, J. Am. Chem. Soo.,
1981, 103, 7672.
82. (a) R.U. Lemieux and N.J. Chii, Abstracts of Papers; Am. Chem. Soo. ,
1958, 133, 31N .(b) P. Deslongchamps, 'Stereoelectronic Effects in Organic Chemistry1 Pergamon Press, Oxford, 1983.
83. Aldrich Chemical Co. Ltd.
84. A.E. Greene, M.-J. Luche, and J .- P . Depres, J. Am. Chem. Soo.,
1983, J05, 2435.
85. L.S. Liebeskind and S.L. Baysdon, Tetrahedron Lett., 1984, 1747.
86. I. Nakagawa and T. Hata, Tetrahedron Lett., 1975, 1409.
87. P.A. Grieco, Y. Ohfune, Y. Yokoyama, and W. Owens, J. Am. Chem. Soo.,
1979, ] 0]_, 4749.
88. S. Tanaka, H. Yamamoto, H. Nozaki, K.B. Sharpless, R.C. Michaelson, and J.D. Cutting, J. Am. Chem. Soc. , 1974, 96, 5254.

89. Y. Guindon, R. Fortin, C. Yoakim, and J.W. Gil lard, Tetrahedron Lett. , 1984, 4717.
90. P.G.M. Wuts, R.D'Costa, and W. Butler, J. Org. Chem. , 1984, 4 9 ,2582.
91. (a) K.B. Sharpless, S.S. Woodward, and M.G. Finn, Pure and Appl. Chem. , 1983, _55_, 1823.(b) I.D. Williams, S.F. Pedersen, K.B. Sharpless, and S.J. Lippard,J. Am. Chem. Soa. , 1984, 106, 6430.
92. (a) B.E. Rossiter, T. Katsuki, and K.B. Sharpless, J. Am. Chem.
Soc. , 1981, 103, 464.(b) T. Katsuki, Tetrahedron Lett., 1984, 2821.
93. L.A. Reed, III, Y. Ito, S. Masamune, and K.B. Sharpless, J. Am.Chem. Soc. , 1982, 1 0 4 , 6468.
94. R.K. Crossland and K.L. Servis, J. Org. Chem., 1970, 35, 3195.
95. D.L.J. Clive, Tetrahedron3 1978, _34, 1049.
96. S.V. Ley and I.A. O'Neil, unpublished results.
97. T. Purdie and J.C. Irvine, J. Chem. Soc., 1903, 83_, 1021.
98. T. Mukhopadhyay and D. Seebach, Eelv. Chim. Acta. , 1982, 6J5, 385.
99. (a) P.d. Garegg and B. Samuelsson, J. Chem. Soc., Perki-n Trans. I,
1980, 2866.(b) E.J. Corey, S.G. Pyne, and W. Su, Tetrahedron Lett., 1983,4883.
100. B.A. Arbusov, Pure and Appl. Chem., 1964, 9_, 307.
101. W.S. Wadsworth, Jr. and W.D. Emmons, J. Am. Chem. Soc., 1961,83, 1733.
356

357
102. S. Trippett, J. Chem. Soc. , 1961, 2813.
103. (a) T.A.M. van Schaik, A.V. Henzen, and A. van der Gen,Tetrahedron Lett. , 1983, 1303.(b) J. Boutagy and R. Thomas, Chem. Rev., 1974, 74, 87.
104. (a) E.M. Burgess, H.R. Penton, Jr., and E.A. Taylor, J. Am. Chem.
Soc. , 1970, 92, 5224.(b) E.M. Burgess, H.R. Penton, Jr., and E. A. Taylor, J. Org.
Chem. , 1973, 38_, 26.(c) P. Crabbe and C. Leon, J. Org. Chem. , 1970, 35_, 2594.(d) E.M. Burgess, H.R. Penton, Jr., E.A. Taylor, and W.M. Williams, Org. Synth. , 1977, 56_, 40.
105. H. Oediger, F. Moller, and K. Eiter, Synthesis, 1972, _56_, 591.
106. L.A. Paquette, W.B. Farnham, and S.V. Ley, J. Am. Chem. Soc. ,
1975, 97, 7273.
107. K.C. Nicolaou, Tetrahedron, 1981, _37, 4097.
108. (a) P.A. Grieco, S. Gilman, and M. Nishizawa, J. Org. Chem. ,
1976, 41_, 1485.(b) T.G. Back and D.J. McPhee, J. Org. Chem. , 1984, 49_, 3842.
109. K.B. Sharpless, M.W. Young, and R.F. Lauer, Tetrahedron Lett.,1973, 1979.
110. E.C. Ashby and J.N. Argyropoulos, Tetrahedron Lett., 1984, 7.
111. (a) R.D. Rieke and S.E. Bales, J. Am. Chem. Soc., 1974, _96, 1775. (b) R.D. Rieke, S.E. Bales, P.M. Hudnall, and G.S. Poindexter,Org. Synth., ed. R.M. Coates, Wiley, New York, 1980, 59, 85.
112. (a) J.W. Westley and C.-M. Liu, U.S. Patent, 4, 100, 171, 1978.,J.W. Westley, R.H. Evans, Jr., C.-M. Liu, T.E. Hermann, andJ.F. Blount, J. Am. Chem. Soc. , 1978, 100, 6784.

358
112. (b) C.-M. Liu, T.E. Hermann, M. Liu, D.N. Bull, N.J. Palleroni,
B. L.T. Prosser, J.W. Westley, and P.A. Miller, J. Antibiot. ,
1979, 32, 95.
(c) J.W. Westley, R.H. Evans, Jr., L.H. Sello, N. Troupe,
C. -M. Liu, and J.F. Blount, J. Antibiot, , 1979, 32, 100.
113. (a) M.O. Chaney, P.V. Demarco, N.D. Jones, and J.L. Occolowitz,
J. Am. Chem. Soo. , 1974, _96, 1932.
(b) L. David and A. Kergomard, J. Antibiot., 1982, 1409.
114. J.M. Muchowski and D.R. Solas, J. Org. Chem., 1984, 49, 203.
115. (a) A.J. Fatiadi, Synthesis, 1976, 133.
(b) G.R. Martinez, P.A. Grieco, and C.V. Srinivasan, J. Org. Chem.,
1981, 46, 3760.
116. (a) L.F. Fieser and M. Fieser, "Reagents for Organic Synthesis",
Wiley, New York, 1967, p. 1179.
(b) W.S. Johnson, J.C. Collins, Jr., R. Rappo, M.B. Rubin,
P.J. Kropp, W.F. Johns, T.E.Pike, and W. Bartmann, J. Am. Chem. Soo.,
1963, 85, 1409.
117. (a) K.C. Nicolaou, D.A. Claremon, D.P. Papahatjis, and R.L. Magolda,
J. Am. Chem. Soc. , 1981, 103, 6969.
(b) K.C. Nicolaou, R.L. Magolda, W.J. Sipio, W.E. Barnette,
Z. Lysenko, and M.M. Jouillie, ibid., 1980, 102, 3784.
(c) H.J. Reich, F. Chow, and S.L. Peake, Synthesis, 1978, 299.
(d) P.A. Grieco, Y. Yokoyama, S. Gilman, and M. Nishizawa,
J. Org. Chem. , 1977, 42_, 2034.
118. D.J. Chadwick, J. Chambers, G.D. Meakins, and R.L. Snowdon,
J. Chem. Soc., Perkin Trans. I, 1975, 523.

359
119. H. Merz and K. Stockhaus, J. Med. Chem. , 1979, 22, 1475.
120. S.V. Ley, B. Lygo, and H. Molines, J. Chem. Soe. , Perkin Trans. I ,
1984, 2403.
121. J.B. Hendrickson, M.L. Maddox, J.J. Sims, and H.D. Kaesz,
Tetrahedron, 1964, 20, 449.
122. 0. Isler, H. Gutmann, M. Montavon, R. Ruegg, G. Ryser, and P. Zeller,
Helv. Chim. Acta, 1957, 4C), 1242.
123. H. Schlenk and J.L. Gellerman, Anal. Chem. , 1960, .32, 1412.
124. Aldrich Chemical Co. Ltd.
125. D.B. Coll urn, J.H. McDonald, III, and W. Clark Still, J. Am. Chem.
Soc. , 1980, 102, 2218.
126. J.G. Hill, E.B. Rossiter, and K.B. Sharpless, J. Org. Chem.,
1983, 48, 3607.
127. J.G. Hill, K.B. Sharpless, C.M. Exon, and R. Regenye, Org. Synth.,
1985, 63_, in press.
128. M.P. Edwards, Ph.D. Thesis, 1982, University of London.
NOTE:-
Ireland and co-workers have recently reported synthetic studies towards
the polyether ionophore monensin (See Refs. 6 and 7 for previous
syntheses).
R.E. Ireland, D. Habich, and D.W. Norbeck, J. Am. Chem. Soc., 1985, 107,
3271 ; R.E. Ireland and D.W. Norbeck,ibid. , 1985, 107, 3279; R.E. IrelandD.W. Norbeck, G.S. Mandel , and N.S. Mandel , ibid., 1985, 107, 3285.