tailoring nanoparticle designs to target cancer … · tailoring nanoparticle designs to target...
TRANSCRIPT

Tailoring nanoparticle designs to target cancer basedon tumor pathophysiologyEdward A. Sykesa, Qin Daia, Christopher D. Sarsonsb, Juan Chenc, Jonathan V. Rocheleaua,d, David M. Hwange,Gang Zhengc, David T. Crambf, Kristina D. Rinkerb, and Warren C. W. Chana,g,h,i,j,1
aInstitute of Biomaterials and Biomedical Engineering, University of Toronto, Toronto, ON, Canada M5S 3G9; bBiomedical Engineering, University ofCalgary, Calgary, AB, Canada T2N 1N4; cDepartment of Medical Biophysics, Princess Margaret Cancer Centre, University of Toronto, Toronto, ON, CanadaM5G 1L7; dToronto General Research Institute, University Health Network, Toronto, ON, Canada M5G 2M9; eDepartment of Pathology, University HealthNetwork, Toronto, ON, Canada M5G 2C4; fDepartment of Chemistry, University of Calgary, Calgary, AB, Canada T2N 1N4; gDonnelly Center for Cellular andBiomolecular Research, University of Toronto, Toronto, ON, Canada M5S 3E1; hDepartment of Chemistry, University of Toronto, ON, Canada M5S 3H6;iDepartment of Chemical Engineering, University of Toronto, ON, Canada M5S 3E5; and jDepartment of Materials Science and Engineering, University ofToronto, ON, Canada M5S 3E4
Edited by Mark E. Davis, California Institute of Technology, Pasadena, CA, and approved January 12, 2016 (received for review November 16, 2015)
Nanoparticles can provide significant improvements in the di-agnosis and treatment of cancer. How nanoparticle size, shape,and surface chemistry can affect their accumulation, retention, andpenetration in tumors remains heavily investigated, because suchfindings provide guiding principles for engineering optimal nano-systems for tumor targeting. Currently, the experimental focus hasbeen on particle design and not the biological system. Here, wevaried tumor volume to determine whether cancer pathophysiologycan influence tumor accumulation and penetration of different sizednanoparticles. Monte Carlo simulations were also used to model theprocess of nanoparticle accumulation. We discovered that changesin pathophysiology associated with tumor volume can selectivelychange tumor uptake of nanoparticles of varying size. We furtherdetermine that nanoparticle retention within tumors depends onthe frequency of interaction of particles with the perivascular ex-tracellular matrix for smaller nanoparticles, whereas transport oflarger nanomaterials is dominated by Brownian motion. Theseresults reveal that nanoparticles can potentially be personalizedaccording to a patient’s disease state to achieve optimal diagnosticand therapeutic outcomes.
cancer | nanoparticles | targeting | nano–bio interactions | tumor
Nanotechnology remains an emerging and important researchdiscipline for detecting and treating cancer. Nanomaterials
can be engineered with different sizes, shapes, and surfacechemistries, as well as assembled into hierarchical nanosystems(1). Nanomaterials can also be engineered with unique propertiessuch as emission of light for fluorescence detection (2), magnetismfor magnetic resonance imaging (3, 4), and thermal emission forablation of tumor cells (5). Despite the potential of nanomaterials,typically less than 5% of an administered dose reaches the tumorcompartment (6) because of poor retention within the tumorspace and uptake by the skin (7), spleen, and liver (8–10). Re-finements to the size, shape, and surface chemistry of nano-materials have improved their blood half-lives (11, 12) andinteractions with cancer cells (13–15). Unfortunately, clinicaltranslation of cancer nanomedicine remains stagnated by ad-herence to the ideology that nanoparticles and other agents canbe designed to “universally” detect and treat tumors independentof type or stage of cancer progression. Tumor growth leads tophysiological changes in their tissue composition (cell density,vascularity, necrosis, and stroma). If nanoparticles could be tai-lored according to the physiological state of each tumor, cancerdetection and treatment may be drastically improved. However,investigations into the effect of tumor pathophysiology on nano-particle accumulation and kinetics have been limited.Fundamental analysis of tumor pathophysiology has identified
unique cellular and structural properties associated with variousstages of cancer progression. We currently understand that theincreasing vascular tortuosity, inhomogeneity, and restricted
blood flow (and subsequent low blood pressure) associated withtumor growth prevents chemotherapeutic agents from reachingtheir target. This impairment of drug delivery may lead to poortherapeutic efficacy and cancer recurrence (16, 17). As we learnmore about the cellular, vascular, and compositional character-istics of tumors, it is increasingly evident that tailoring drug de-livery vehicles to the physiological state of a tumor may beinstrumental to improving treatment of this disease (18, 19).However, enabling clinicians to personalize patient care willrequire a deeper understanding of the implications of tumoranatomy and pathophysiology on the delivery and function ofmedicinal agents.Here, we determine whether the delivery of spherical gold
nanoparticles (AuNPs) can be affected by changes in tumorvolume—a surrogate of cancer progression. Specifically, we(i) characterize the changes in the physiological structures andmicroenvironment of tumors as they grow in a tumor xenograftmouse model, and (ii) explore how such changes impact uptake,permeation, and retention of polyethylene glycol (PEG)-coatedAuNPs. Understanding these variations will enable clinicians topersonalize cancer therapy by catering nanotherapeutic regimensaccording to tumor characteristics. As a proof of concept, wesuccessfully demonstrate that observable changes in tumor
Significance
Nanotechnology is a promising approach for improving cancerdiagnosis and treatment with reduced side effects. A keyquestion that has emerged is: What is the ideal nanoparticlesize, shape, or surface chemistry for targeting tumors? Here,we show that tumor pathophysiology and volume can signifi-cantly impact nanoparticle targeting. This finding presents aparadigm shift in nanomedicine away from identifying andusing a universal nanoparticle design for cancer detection andtreatment. Rather, our results suggest that future clinicians willbe capable of tailoring nanoparticle designs according to thepatient’s tumor characteristics. This concept of “personalizednanomedicine” was tested for detection of prostate tumorsand was successfully demonstrated to improve nanoparticletargeting by over 50%.
Author contributions: E.A.S., D.M.H., D.T.C., K.D.R., and W.C.W.C. designed research;C.D.S., D.T.C., and K.D.R. designed the collagen experiments; E.A.S., Q.D., and C.D.S. performedresearch; J.C., J.V.R., and G.Z. contributed new reagents/analytic tools; J.C. generated theprostate cancer model; J.V.R. set up second harmonic generation measurements and tissueimaging; G.Z. produced the prostate cancer mouse model; E.A.S., Q.D., and W.C.W.C. ana-lyzed data; and E.A.S. and W.C.W.C. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1521265113/-/DCSupplemental.
E1142–E1151 | PNAS | Published online February 16, 2016 www.pnas.org/cgi/doi/10.1073/pnas.1521265113

pathophysiology can be used in a decision matrix to rationallyselect AuNP designs according to desired function.
ResultsCharacterization of Tumors. Pathophysiological changes associatedwith tumor volume were studied to identify biological parame-ters that might impact AuNP targeting. The degree of vascu-larization, cell density, and extracellular matrix (ECM) contentof different-sized orthotopic human breast melanoma xenografttumors derived from MDA-MB-435 cells in CD1 nude athymicmouse models were characterized. These parameters were se-lected because they have been shown to individually impactnanoparticle uptake rate, accumulation, and retention (20–22).Histological sections stained with CD31 antibodies were used tocolorimetrically visualize tumor blood vessels, whereas Movat’sPentachrome staining was performed to highlight nuclei andECM components such as proteoglycans, mucopolysaccharides,and collagen. Vascular density was calculated by counting thenumber of vessels per tumor cross-section. We observed thatthe concentration of blood vessels increased with tumor volumebut plateaued at 44 ± 3 blood vessels/mm2 for tumor volumesexceeding 1.0 cm3 (Fig. 1A). Interestingly, the tumor vascula-ture was only uniformly distributed in small tumors. Tumorblood vessels became increasingly concentrated near necroticregions and at the tumor perimeter as tumors enlarged (SIAppendix, Fig. S1).Beyond tumor vascularization, the fraction of the tumor com-
posed of proteoglycans and mucopolysaccharides increased ata rate of 4.2 arbitrary units/cm3 (Fig. 1B), whereas tumor celldensity increased at a rate of 1.70 cells/cm3 (Fig. 1C) with tumorvolume. Unstained acellular space also proportionally decreasedwith tumor growth (Fig. 1D). These factors coincided with height-ened ECM production at regions surrounding tumor blood vesselsand necrotic tissue, whereas ECM content in regions of densetumor tissue around the core became reduced (SI Appendix, Fig.S2). A closer examination of ECM composition by Picrosiriusred staining (Fig. 2A) and second harmonic generation (SHG)imaging (Fig. 2B) identified that these regions contained type Icollagen with a density and structure that evolved with tumorgrowth. Picrosirius red-stained samples spectrally shifted from
deep red to pale pink (Fig. 2C), whereas SHG microscopy im-ages decreased in intensity (Fig. 2D) as tumors enlarged. There isa decrease of 9% per cm3 in Picrosirius red intensity and thespectral shift in SHG peak intensity was characteristic of a loss instructural ECM via reduction in collagen fiber thickness andlength (23–25).Together, these results indicate that as tumors mature through
growth, their tissue and vasculature become denser and morechaotic. In particular, the ECM appears to remodel during tu-mor enlargement, thus leading to a more amorphous phenotype.Given that ECM components were observed to encapsulate tu-mor blood vessels (SI Appendix, Fig. S3) and are known to bi-ologically function as a basal support for blood vessels thatinterfaces with the stroma, changes in ECM may be a primarymediator of nanoparticle entry into the tumor compartment.
Gold Nanoparticle Model System. Having characterized the evolu-tion of tumor tissues during growth, we sought to determinewhether these physiological changes could be used to tune thetumor targeting efficacy of nanoparticles. Because tumor uptakeis dependent on nanoparticle diameter (12, 26, 27), a library ofmethoxy-PEG–coated AuNPs of varying diameter were designedto examine the effect of tumor growth on particle delivery. Al-though clinical trials for AuNPs are limited, AuNPs were se-lected over more clinically appropriate polymeric nanomaterialsbecause AuNPs can be reproducibly and precisely synthesized ina broad range of sub-100-nm sizes. Furthermore, AuNPs providea nondeformable formulation for testing the effect of core di-ameter on tumor uptake, are easily surface modified, and can bequantified in tissues with high sensitivity. A schematic illustratingthe AuNP design used in this study is depicted in SI Appendix,Fig. S4A.Spherical AuNPs with core diameters of 15, 30, 45, 60, and
100 nm (SI Appendix, Fig. S4B) were synthesized using standardcitrate and hydroquinone reduction techniques (28). These sizeswere selected to systematically characterize how the tumormicroenvironment impacts a broad range of particle diameters.AuNP surfaces were modified with hetero-bifunctional 5-kDaPEG with methoxy and sulfhydryl termini as well as AlexaFluor 750-labeled 10-kDa sulfhydryl-PEG to respectively stabilize
Fig. 1. Summary of the pathophysiological changes in tumors during growth. The graphs depict the changes in vascular density (A), the proportion of tumorsoccupied by ECM components (B), acellular space (C), and cellular density (D) associated with tumor volume. All values were normalized to tumor cross-sectional area.
Sykes et al. PNAS | Published online February 16, 2016 | E1143
ENGINEE
RING
PNASPL
US

particles for blood transport and to fluorescently track particlesin vivo. Although it is difficult to use fluorescence as an absolutequantification technique, we have shown previously that fluo-rescence is an accurate modality for monitoring relative changesin nanoparticle biodistribution (26, 29). Surface modifications resul-ted in AuNPs with a PEG packing density of 0.3–1.5 ligands/nm2. Atthese densities, surface-bound PEG moieties were calculatedaccording to their Flory diameter to be in the brush layer con-formation, ensuring that the tested nanoparticles were suffi-ciently passivated (SI Appendix, Table S1). Surface modificationswere also found to increase nanoparticle hydrodynamic diame-ters by 20–40 nm (SI Appendix, Fig. S4C) and positively shiftnanoparticle zeta potentials by 20–30 mV (SI Appendix, Fig.S4D). Particle fluorescence was confirmed by the migration ofdistinct fluorescent bands during agarose gel electrophoresis (SIAppendix, Fig. S4E). AuNP fluorescence was shown to increaseproportionally with particle diameter (SI Appendix, Fig. S5).Fluorescent PEG groups were also confirmed to be stably boundto particle surfaces because the rate of desorption in the pres-ence of serum remained below 0.2 PEG molecules/h (SI Ap-pendix, Fig. S5D). In vivo pharmacokinetics of our functionalizedAuNPs was also characterized by analysis of blood plasma at 0, 2,4, 8, and 24 h posttail vein injection (HPI) in non–tumor-bearingCD1 nude athymic mice. Inductively coupled plasma atomicemission spectroscopy (ICP-AES) analysis of blood samplesrevealed that the blood half-lives of our AuNPs ranged from 2 to10 h. A complete characterization of our formulations is pre-sented in SI Appendix, Table S2.
Analysis of Nanoparticle Accumulation in Tumors. AuNP accumu-lation was evaluated via tail vein injection of formulations intoCD1 nude athymic mice bearing orthotopic MDA-MB-435 humanbreast melanoma tumors. Tumors volumes evaluated in thisstudy ranged from 0.05–3.00 cm3. AuNP delivery to the differentsized tumors was fluorescently profiled in mice to assess tumoraccumulation kinetics and to measure total AuNP exposure.
Fluorescent tracking was achieved by whole-animal imaging us-ing a Carestream In Vivo Imaging System at time points rangingfrom 0–24 HPI.Total area under the curve (AUC) was calculated from the
kinetic curves in SI Appendix, Fig. S6 as a metric for AuNP ac-cumulation within the tumor. Overall, AUC values increasedwith tumor volume (Fig. 3A). Accumulation for 15-, 30-, and45-nm AuNPs steadily increased with tumor volume from 490 ±70% to 720 ± 30% ID·h, 280 ± 50% to 750 ± 10% ID·h, and480 ± 70% to 960 ± 100% ID·h, respectively. Changes inaccumulation of larger formulations occurred as step increasesat discrete tumor volumes. There was an ∼1.5 times higheraccumulation for 60-nm formulations once tumors exceeded2.2 cm3, whereas 100-nm particles exhibited a ∼4.6 times increasein accumulation for volumes 0.5 cm3 and larger in comparisonto smaller tumors. These trends were confirmed by ICP-AESmeasurements of gold content in tumors at 24 HPI (Fig. 3B).The ICP-AES results indicated that by 24 HPI tumor uptakeof 15- and 30-nm particles were consistently higher than allother formulations and steadily increased from 0.39 ± 0.04%to 0.99 ± 0.18% ID and 0.28 ± 0.03% to 0.90 ± 0.18% ID,respectively (two-way ANOVA, P = 0.05), whereas largerparticles such as 60 nm trended higher (although statisticallynot significant) from 0.18 ± 0.02% to 0.26 ± 0.12% ID as tumorvolumes increased.In combination with our histological observations, these re-
sults suggest that the higher porosity of the ECM increasinglyaccommodates the entry of larger nanoparticles at later stages oftumor growth. This implies that a minimum tumor size must bereached to support entry of each AuNP diameter. An AuNPaccumulation threshold of 500% was selected to illustrate thispoint (Fig. 3A). This threshold was defined as the mean AUC of15-nm AuNPs in sub-0.5-cm3 tumors as particles in this sizerange would experience the least steric hindrance. AUC valuesfor each AuNP diameter were statistically compared with thethreshold (two-way ANOVA, P = 0.05); 15-nm AuNPs have this
Fig. 2. Structural changes to type I collagen associ-ated with tumor size. (A) Representative bright-fieldimages of Picrosirius red-stained sections that depictthe evolution of collagen with tumor size. (B) Rep-resentative SHGmicroscopy images of collagen (green)overlaid with DRAQ5-stained nuclei (blue). (C) Graphdelineating how Picrosirius red intensity fades withrising tumor volume as collagen fibrils convert tolesser-organized constructs. (D) Histograms of SHGintensity in collagen-enriched zones validates thattype I collagen becomes increasingly amorphous withtumor enlargement. (Scale bars, 50 μm.) DRAQ5 is1,5-bis{[2-(di-methylamino) ethyl]amino}-4, 8-dihy-droxyanthracene-9,10-dione.
E1144 | www.pnas.org/cgi/doi/10.1073/pnas.1521265113 Sykes et al.
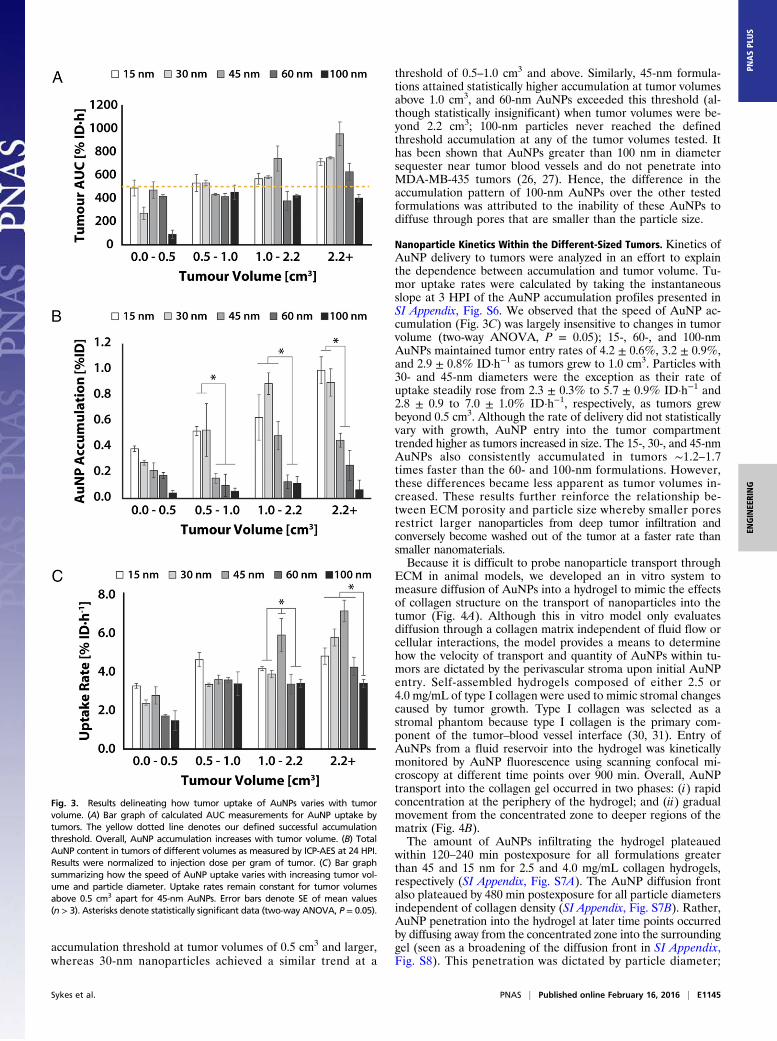
accumulation threshold at tumor volumes of 0.5 cm3 and larger,whereas 30-nm nanoparticles achieved a similar trend at a
threshold of 0.5–1.0 cm3 and above. Similarly, 45-nm formula-tions attained statistically higher accumulation at tumor volumesabove 1.0 cm3, and 60-nm AuNPs exceeded this threshold (al-though statistically insignificant) when tumor volumes were be-yond 2.2 cm3; 100-nm particles never reached the definedthreshold accumulation at any of the tumor volumes tested. Ithas been shown that AuNPs greater than 100 nm in diametersequester near tumor blood vessels and do not penetrate intoMDA-MB-435 tumors (26, 27). Hence, the difference in theaccumulation pattern of 100-nm AuNPs over the other testedformulations was attributed to the inability of these AuNPs todiffuse through pores that are smaller than the particle size.
Nanoparticle Kinetics Within the Different-Sized Tumors. Kinetics ofAuNP delivery to tumors were analyzed in an effort to explainthe dependence between accumulation and tumor volume. Tu-mor uptake rates were calculated by taking the instantaneousslope at 3 HPI of the AuNP accumulation profiles presented inSI Appendix, Fig. S6. We observed that the speed of AuNP ac-cumulation (Fig. 3C) was largely insensitive to changes in tumorvolume (two-way ANOVA, P = 0.05); 15-, 60-, and 100-nmAuNPs maintained tumor entry rates of 4.2 ± 0.6%, 3.2 ± 0.9%,and 2.9 ± 0.8% ID·h−1 as tumors grew to 1.0 cm3. Particles with30- and 45-nm diameters were the exception as their rate ofuptake steadily rose from 2.3 ± 0.3% to 5.7 ± 0.9% ID·h−1 and2.8 ± 0.9 to 7.0 ± 1.0% ID·h−1, respectively, as tumors grewbeyond 0.5 cm3. Although the rate of delivery did not statisticallyvary with growth, AuNP entry into the tumor compartmenttrended higher as tumors increased in size. The 15-, 30-, and 45-nmAuNPs also consistently accumulated in tumors ∼1.2–1.7times faster than the 60- and 100-nm formulations. However,these differences became less apparent as tumor volumes in-creased. These results further reinforce the relationship be-tween ECM porosity and particle size whereby smaller poresrestrict larger nanoparticles from deep tumor infiltration andconversely become washed out of the tumor at a faster rate thansmaller nanomaterials.Because it is difficult to probe nanoparticle transport through
ECM in animal models, we developed an in vitro system tomeasure diffusion of AuNPs into a hydrogel to mimic the effectsof collagen structure on the transport of nanoparticles into thetumor (Fig. 4A). Although this in vitro model only evaluatesdiffusion through a collagen matrix independent of fluid flow orcellular interactions, the model provides a means to determinehow the velocity of transport and quantity of AuNPs within tu-mors are dictated by the perivascular stroma upon initial AuNPentry. Self-assembled hydrogels composed of either 2.5 or4.0 mg/mL of type I collagen were used to mimic stromal changescaused by tumor growth. Type I collagen was selected as astromal phantom because type I collagen is the primary com-ponent of the tumor–blood vessel interface (30, 31). Entry ofAuNPs from a fluid reservoir into the hydrogel was kineticallymonitored by AuNP fluorescence using scanning confocal mi-croscopy at different time points over 900 min. Overall, AuNPtransport into the collagen gel occurred in two phases: (i) rapidconcentration at the periphery of the hydrogel; and (ii) gradualmovement from the concentrated zone to deeper regions of thematrix (Fig. 4B).The amount of AuNPs infiltrating the hydrogel plateaued
within 120–240 min postexposure for all formulations greaterthan 45 and 15 nm for 2.5 and 4.0 mg/mL collagen hydrogels,respectively (SI Appendix, Fig. S7A). The AuNP diffusion frontalso plateaued by 480 min postexposure for all particle diametersindependent of collagen density (SI Appendix, Fig. S7B). Rather,AuNP penetration into the hydrogel at later time points occurredby diffusing away from the concentrated zone into the surroundinggel (seen as a broadening of the diffusion front in SI Appendix,Fig. S8). This penetration was dictated by particle diameter;
Fig. 3. Results delineating how tumor uptake of AuNPs varies with tumorvolume. (A) Bar graph of calculated AUC measurements for AuNP uptake bytumors. The yellow dotted line denotes our defined successful accumulationthreshold. Overall, AuNP accumulation increases with tumor volume. (B) TotalAuNP content in tumors of different volumes as measured by ICP-AES at 24 HPI.Results were normalized to injection dose per gram of tumor. (C) Bar graphsummarizing how the speed of AuNP uptake varies with increasing tumor vol-ume and particle diameter. Uptake rates remain constant for tumor volumesabove 0.5 cm3 apart for 45-nm AuNPs. Error bars denote SE of mean values(n > 3). Asterisks denote statistically significant data (two-way ANOVA, P = 0.05).
Sykes et al. PNAS | Published online February 16, 2016 | E1145
ENGINEE
RING
PNASPL
US

45-nm AuNPs achieved the highest permeation at 17.0 ± 2.0 and13.2 ± 0.4 μm, whereas 100-nm AuNPs exhibited the poorestpenetration at 8.0 ± 1.0 and 5.4 ± 0.6 μm for collagen densitiesof 2.5 and 4.0 mg/mL, respectively (Fig. 4C). Although AuNPpermeation appeared to decrease with collagen concentration,differences were not statistically significant (two-way ANOVA,P > 0.05). These trends were consistent with the tumor-permeationresults at 24 HPI, where AuNP infiltration did not vary withtumor volume (Fig. 4D). Particle permeation also did not changestatistically between the tumor periphery, regions neighboringnecrotic zones, or within the core of the tumor tissue. Despite thelower collagen density, diffusion of 15-nm AuNPs into the2.5 mg/mL collagen gels was unexpectedly 2.0 and 1.3 timeslower than our 45-nm formulation in vitro and in vivo, re-spectively. These differences in diffusion were similar to previousstudies (26, 27) and were attributed to the speed of AuNP uptakeby and expulsion from the collagen matrix (Fig. 4 E and F). Incomparison to 4.0 mg/mL collagen gels, 15-nm formulationswere taken up by the 2.5 mg/mL gels 14% slower and expelled51% faster. This result leads to a lower overall AuNP concen-tration within the 2.5 mg/mL collagen gel and, accordingly,slower particle diffusion. Alternatively, because the uptake rateof AuNPs exceeding 45 nm does not vary with collagen density(two-way ANOVA, P = 0.05), their slower depletion from thecollagen matrix allows for greater nanoparticle retention andconsequently greater infiltration distances.
Computational Modeling of Nanoparticle Diffusion Through PorousMatrices. To help understand how nanoparticles interact withthe collagen matrix, Monte Carlo numerical simulations of AuNPdiffusion through collagen matrices were conducted; 2D modelswere used to examine the frequency of AuNP collisions withcollagen fibers within pores of the hydrogel matrix. The frequencyof such collisions can influence the retention and path of theAuNPs in the tumor; 3D simulations were also conducted tocompare AuNP permeation capacity through stroma with differentcollagen densities. These computational models were conducted inMatlab using custom algorithms to simulate AuNP interaction anddiffusion within collagen matrices. These simulations followedsimilar strategies used by Stylianopoulos et al. (32). AuNP motilitywas modeled as step-wise random walk obeying Einstein–Stokesdiffusion (Eq. 2), whereas particle–fiber interactions were modeledas elastic collisions. Fig. 5A provides an illustration delineating thepath of AuNP motion within a collagen pore. Obeying Brownianmotion, AuNPs move randomly and can collide with collagen fi-bers. For our 3D simulations, collagen fibers were approximatedas cylinders with radii between 0.05 and 0.50 μm. Representativeimages of the collagen matrices of varying collagen density simu-lated in Matlab are presented in Fig. 5B. The modeled radii werechosen according to measured thicknesses from scanning electronmicroscopy images of our 2.5 and 4.0 mg/mL collagen hydrogels(SI Appendix, Fig. S9). A detailed summary of our model and itsunderlying assumptions can be found in Methods.
Fig. 4. In vitro collagen hydrogel model of AuNPtransport through tumor ECM. (A) Schematic depict-ing the in vitro setup used to profile AuNP infiltrationinto type I collagen hydrogels. (B) Illustration of theobserved AuNP (red) infiltration process for the col-lagen hydrogels (green). AuNPs first concentrate atthe gel–reservoir interface dependent on particle sizeand collagen density. Once an equilibrium is reachedbetween AuNPs in the matrix and interface, the AuNPfront gradually diffuses deeper into the hydrogel.(C) Bar graph depicting the permeation of AuNPswithin the collagen hydrogels at 900 min postexposure.(D) Whisker plot depicting the cumulative results ofAuNP penetration from blood vessels into tumor tis-sues at 24 HPI. No differences were found betweentumor sizes. Bar graphs (E and F) summarize the dif-ferences in AuNP entry and exit from hydrogels basedon collagen density and AuNP diameter. Error barsdenote SEM for n = 3. Asterisks denote statisticallysignificant data (two-way ANOVA, P = 0.05).
E1146 | www.pnas.org/cgi/doi/10.1073/pnas.1521265113 Sykes et al.

AuNP movement in our 2D models for 1,000-particle replicateswas simulated in 0.005-, 0.020-, 0.108-, and 0.640-μm2 squarestromal pores for 10,000 steps at 0.1-s intervals. Our simulationsdetermined that AuNP–fiber collision rates increased with re-ducing pore size and decreasing AuNP diameter (Fig. 5C); 15-nmAuNPs achieved the highest frequency of interaction with col-lagen fibers at rates between 0.038–0.023 collisions/s (cps),whereas 100-nm formulations ranged from 0.016–0.001 cps forpore sizes between 0.005–0.640 μm2. Interestingly, collisionrates for 15-, 45-, and 60-nm AuNPs were statistically similarfor 0.005-μm2 pores (ANOVA, P = 0.05) but became increas-ingly dissimilar as pores enlarged. These simulations suggest thatimpact of particle size on Brownian motion is a primary mediatorof AuNP motility within the hydrogel over its frequency of col-lision with the ECM. This result suggests that the greater thenanoparticles interact with collagen, the longer they will be retainedwithin the tumor.Expanding on these results, AuNP diffusion was also modeled
in three dimensions to compare how AuNP diameter and collagendensity might impact stromal accumulation and infiltration. Stro-mal–ECM of increasing collagen density was modeled computa-tionally as 27,000-μm3 cubes containing anisotropically orientedcollagen fibers. The number of fibers were chosen to achievecollagen volume fractions (8.72–87.20%), reflective of conditionsfound within tumors (33, 34). Diffusion distance for 500 AuNPreplicates was tracked for 5,000 discrete steps at 1-s intervals. Oursimulations indicate that diffusion rates changed with AuNP di-ameter but did not change with collagen density (Fig. 5D); 15-nmAuNPs exhibited the greatest mobility at 1.95 ± 0.03 nm/s in thesimulated hydrogels, whereas 45-, 60-, and 100-nm particles dif-fused at rates of 0.78 ± 0.01, 0.60 ± 0.01, and 0.36 ± 0.01 nm/s,respectively. These findings support our AuNP-permeation obser-vations from histological tumor sections whereby AuNP diffusionaway from blood vessels (SI Appendix, Fig. S10) did not vary withtumor volume (Fig. 5D).
Together, these 2D and 3D models elucidate how the stromalmatrix is implicated in particle permeation. Although thesesimplified 2D and 3D models ignore the effect of fluid flow,oncotic pressure, and inelastic collagen–AuNP interactions, themodels provide a mechanism for our in vitro and in vivo per-meation observations. They suggest that AuNP permeation is thebalance between the effects of particle size on Brownian motionand the frequency of particle collision with the ECM. The in-creased mobility of smaller AuNPs afforded greater diffusion butwas also inhibitory because of the higher frequency of collisionwith the ECM. Conversely, AuNPs of larger diameters exhibitedslower motion but also a lower propensity to interact with thestroma. The volume fractions tested and simulated in this studyequate to ECM pore sizes ranging from 0.45 to 1.74 μm. Becausethese pores exceed the size of our AuNPs, differences in diffu-sivity associated with collagen density would be negligible for alltested particle diameters. Extended further, these computationalfindings demonstrate that AuNP transport within the tumor canbe distorted through collisions with ECM fibers. These collisionscan limit retention within the tumor compartment if AuNP volumeapproaches the porosity of the stromal ECM.
Nanoparticle Selection According to Tumor Maturity. Given thecomplex dependence of tumor AuNP uptake on both particlesize and tumor pathophysiology, we asked whether there was ameans to rationally select AuNP formulations according to tumorvolume. In our proof-of-concept work, we evaluated whether adecision matrix could be used to select nanomaterials for eithertumor detection (diagnostic) or drug delivery (treatment). AuNPformulations with rapid delivery and high tumor contrast weredefined as effective probes for delineating tumors, whereas AuNPscapable of high tumor retention and homogeneous tissue distribu-tion were anticipated to fare well as drug delivery vehicles.Relative measurements of AuNP fluorescence in vitro and in
vivo were used as an estimate of tumor contrast achievable by
Fig. 5. Monte Carlo models simulating the dynam-ics of AuNP transport through and interactions withcollagen matrices. (A) Pictorial representation ofAuNP randomwalk in two dimensions within collagenpores. Number of collisions with the pore was trackedas a measure of AuNP interactions with collagen ma-trices. (B) Representative images of simulated hydro-gels of varying collagen densities in three dimensions.Images were rendered in Matlab using the samealgorithms used for assessment of AuNP diffusionthrough collagen matrices in three dimensions. (C) Bargraph comparing the rate of AuNP collisions withcollagen matrices of varying pore size obtained from2D simulations. Collision frequency decreases withincreasing pore and AuNP size. Asterisks denotes thescenario whereby AuNP size exceeded the dimensionsof the pore. (D) Line graph depicting the simulatedchanges to AuNP-diffusion rate in collagen gels ascollagen density increases. Collagen density did notappear to impact AuNP diffusivity but was insteaddictated by AuNP size.
Sykes et al. PNAS | Published online February 16, 2016 | E1147
ENGINEE
RING
PNASPL
US

each formulation. Surface area-to-volume ratios were also cal-culated to approximate the drug-loading capacity of each AuNPsize (SI Appendix, Fig. S5C). These parameters in conjunctionwith tumor accumulation, uptake rate, and penetration capacitywere ranked from best (4) to worst (1) for each of the four AuNPsizes used in our experiments. Each parameter was also given amultiplier according to the parameter’s importance to a givenAuNP function. The weighted sum of these rankings was thencalculated for each AuNP design for the different tumor sizeranges. Eq. 1 is a summary of the scoring scheme, where μ is theimportance multiplier, β represents the ranking factor, and idenotes the ranked AuNP parameters. Fig. 6B highlights theseparameters and the associated values used to calculate the scoresfound in our decision matrices (Fig. 6 A and B).
ScoreAuNPjTumor =X
i
μi · βi. [1]
Overall, smaller (<45-nm) AuNPs were favored for both diag-nostic and therapeutic applications across all tumor sizes. Diam-eters in the 100-nm range were consistently predicted as poorcandidates for either application, whereas 15- and 45-nm parti-cles were both expected to be useful for detection and treatmentof large (>1.0-cm3) tumors. AuNPs in the 60-nm range werethe exception to these trends because these AuNPs were pre-dicted to be better for detection of small, early-stage tumors(<0.5 cm3). This exception for 60-nm nanoparticles was empiri-cally attributed to the statistical similarity in AUC values forparticles with diameters between 15 and 60 nm (Fig. 3A) as wellas the higher tumor contrast seen for 60-nm particles (SI Appen-dix, Fig. S5) in the 0.0- to 0.5-cm3 range. Because macrophageuptake of nanoparticles increases with particle diameter (35),the enhanced utility of 60-nm AuNPs may also be related tochanges in phagocytic capacity of tumor-associated macrophages(36) because the macrophages’ phenotypes evolve during tumorprogression (37).These results imply that passively targeted AuNPs with smaller
diameters would be more applicable for detection and drug de-livery when tumor size is unknown. However, 45-nm AuNPs may
be the more effective vehicle for later-staged tumors becausetheir larger surface area-to-volume ratio (SI Appendix, Fig. S5C)theoretically allows for 900% greater drug loading than 15-nmparticles with merely a drop to tumor accumulation by less than57.3%. Although these findings are specific to passively targetedAuNPs, the proposed decision matrix schema can be generalizedto provide a systematic method for assessing other particle types.A flowchart detailing a potential means of implementing thisstrategy is outlined in Fig. 6C.
Validation of the Decision Matrix for Personalized Targeting ofProstate Tumors. Toward validating our results, we evaluatedwhether our formulated decision matrices could be used to predictthe ideal AuNP design for other tumor models. A blinded studywas conducted in CD1 nude athymic mice bearing orthotopichuman tumors with PC3 prostate cancer cells to verify whetherour tumor-size dependent predictions were accurate; 15- and100-nm AuNPs were tail vein-injected into tumor-bearing miceto evaluate AuNP efficacy for tumor detection and accumula-tion. Both particle designs were effective at delineating the lo-cation of the tumor (Fig. 7A) but at varying efficacies. Tumordetection speed and contrast for 15-nm AuNPs were, respec-tively, 53.7% and 50.8% higher than 100-nm particles; 15 nmachieved greater tumor accumulation than 100-nm designs andtrended higher with increasing tumor size (Fig. 7 C–E). Thesefindings were consistent with our decision matrix, alluding to thepotential of our system for use on other particle formulations andtumor types.
DiscussionGiven the observed limitations of AuNP accumulation in tumors,it is clear that careful design of nanomaterials is necessary to achieveoptimal tumor delivery. Currently, interaction of nanoparticles withthe hepatic and renal systems can be manipulated by the nano-particle’s size, shape, and surface chemistry (38). However, toengineer an optimal nanoparticle delivery system for cancertargeting is more complicated and one needs to balance theparticular function (i.e., payload and signal intensity), tumorinteraction, and the tumor-competing organs for nanoparticle
Fig. 6. Proposed method of selecting AuNPsaccording to tumor maturity. (A) Pseudocolored heatmaps qualitatively depict the utility of each AuNPdiameter for therapeutic (Left) and diagnostic(Right) applications predicted by our proposed de-cision matrices. Rankings for particle utility for agiven tumor volume have been rated from high (red)to low (green). Tabular values were calculated bytaking the weighted sum of empirically ranked tu-mor accumulation potential, uptake rate, contrast,and permeation data according to AuNP diameterand tumor size. (B) Weighted importance (μ) of de-cision matrix parameters for application of nano-particles to tumor diagnosis and treatment. (C) Flowdiagram illustrating a proposed method of person-alizing AuNP selection in the clinic for cancer de-tection and treatment.
E1148 | www.pnas.org/cgi/doi/10.1073/pnas.1521265113 Sykes et al.

sequestration. Unfortunately, optimization of only the physico-chemical properties of nanomaterials has reached an impassewhereby tumor targeting efficiency remains stagnated at 5%(6, 39). In our work, we have alternatively approached tumordelivery from the biological perspective by characterizing theunique physiological changes that occur during tumor growth totailor nanoparticles.We determined that for MDA-MB-435 orthotopic human
tumor xenografts, malignant tissues become more disordered asthe tumors increase in volume. Starting from homogeneouslyvascularized tissues with minimal necrotic space, tumors transi-tion toward higher cell densities with vasculature that concen-trates at sparsely distributed regions. This disproportionatevascularization coincides with an increase in necrotic tissue andexpression of collagen and other ECM components that sur-round tumor blood vessels. Type I collagen in the tumor ECMwas found to convert from long filamentous fibers to shorter andmore amorphous structures as tumors increase in volume.Through use of an in vitro collagen hydrogel model, we ratio-nalized that these structural changes in the ECM are a primarymediator of passive tumor delivery of spherical AuNPs. Thiscollagenous basal membrane acts as a “sponge” for extravasatingAuNPs but can delay particle infiltration. The densely packedECM of early-stage tumors appears to sterically restrict AuNPentry based on particle diameter. In larger tumors, the moreporous and less rigid structure of type I collagen facilitates entryof larger AuNPs and enhances accumulation of smaller particlesby the stroma. Because AuNP infiltration depth did not changewith tumor size in vivo nor with variations to collagen density invitro, bulk tumor accumulation of particles likely depends (i) onthe capacity of ECM to interact with AuNPs and (ii) on thenumber of blood vessels available for AuNP entry for a giventumor volume.These tumor growth-associated changes highlight physiologi-
cal parameters that can be exploited for selecting and designingAuNPs for tumor targeting. The reduction of available inter-stitial volume and enhanced porosity of stroma caused bytumor growth hinder permeation of AuNPs but allow for higherAuNP extravasation into the tumor space. This finding suggests
that large AuNPs become more effective when tumors maturebut this improvement in accumulation comes at the expense ofdeep tissue permeation. While small AuNPs may have deeperpenetration they may not be ideal in many applications. Forexample, early diagnosis and treatment of cancer is associatedwith increased patient survival (40–42). Unfortunately, smallerAuNPs, which are best suited to target low-volume tumors, maybe less effective drug-delivery vehicles because the payloads ofthese AuNPs may be smaller than larger particles. This possi-bility illustrates the dichotomy of AuNP design for cancer tar-geting application, because a tradeoff must be made between theintended function of a nanomaterial and optimal tumor delivery.Toward personalized medicine, a simplified decision matrix
was developed to illustrate a means of personalizing the selectionof AuNPs according to tumor stage and desired AuNP function.Our proof-of-concept decision matrix facilitates the personali-zation of a nanomaterial according to the patient by providing anunbiased score of how well a formulation might fare based ontumor volume and the AuNP’s design parameters: tumor signal(fluorescence), accumulation, uptake rate, and permeation. Fig.6C presents a flowchart illustrating how such a decision matrixmight be used clinically to select nanotherapeutic regimens.Simulations established that for MDA-MB-435 tumors, pas-
sively targeted AuNPs with 60-nm diameters provide the bestcontrast for detecting early stages of tumor growth and sites ofmetastasis. Alternatively, particles in the 15- to 45-nm rangeappear to be more effective for diagnostics because tumors in-crease in size or in situations where tumor maturity and phe-notype are unknown. For therapeutic regimens, our work alsoidentifies that AuNPs with diameters between 15 and 45 nm arebest used for tumors exceeding 1.0 cm3, because these AuNPs’permeation distances exceed 60- to 100-nm AuNPs despitehaving lower loading capacities. These results imply that AuNPsmust be rationally designed according to the intended function.Formulations optimized for diagnostic applications may notnecessarily be effective designs for drug delivery or vice versa.Although we have shown that our AuNP tumor size trends werealso valid for a PC3 prostate tumor model, our results may notnecessarily be generalizable to all tumor types because the de-
Fig. 7. Blinded study assessing passive AuNP tar-geting of prostate tumors. (A) Whole-animal fluo-rescent images of mice bearing orthotopic prostatetumors. Bright regions highlight areas of AuNP ac-cumulation. (B) Magnetic resonance images used toconfirm the presence and size of prostate tumors inmice. Dotted circles demarcate the location of thetumor. Graphs C, D, and E, respectively, compare theICP-AES measured accumulation, tumor uptake rates,and tumor contrast of 15- and 100-nm AuNPs in smalland large tumors. Error bars in all graphs denote SEmean values for n = 3. Asterisks denote statisticallysignificant data (two-way ANOVA, P = 0.05).
Sykes et al. PNAS | Published online February 16, 2016 | E1149
ENGINEE
RING
PNASPL
US

cision matrix presented here was constructed from a single tumortype and nanoparticle design. However, because our proposeddecision matrix uses phenotypic parameters that are common tomalignant tissues, the proposed strategy could be easily adoptedby pathologists and researchers. With a concerted effort amongresearchers to elucidate how different nanoparticle physico-chemical properties and microarchitectures of different tumormodels and host species impact nanoparticle entry and retentionwithin tumors, a generalized decision matrix may be realized.Production of this large database may allow future clinicians to usestandard magnetic resonance, computer tomographic, and histo-logical imaging techniques to landmark and approximate the sizeof a tumor, which will guide the specifications for designing thenanoparticles. This approach would cater the design of nano-medicine to the patient.
ConclusionsTo improve cancer detection and therapy, researchers are nowinvestigating how the physicochemical properties of a nano-material mediate nanoparticle transport and function. Althoughit is clear that the synthetic properties of the nanoparticle arecritical to the nanoparticles’ biological interactions, how thephysiological characteristics of the tumor impact nanoparticle fateremains largely unexplored. Here, we show that tumor biology isequally important as nanoparticle size in dictating nanoparticletargeting efficacy. We further show that a thorough assessment oftumor composition can be used to develop a simple algorithm forrational selection of AuNPs according to cancer stage. Imple-mentation of nanomaterials in tandem with radiological imagingand tissue biopsies may be clinically useful to optimally detectnascent tumors and personalize therapeutic regimens. However,realization of this personalized approach to cancer nanomedicinewill require a greater understanding of the physical changes in thetumor microenvironment associated with cancer progression andthe microenvironment’s implications on nanoparticle function.The conclusions presented here have been formulated with
passively targeted AuNPs using an orthotopic MDA-MB-435tumor model. Although we successfully demonstrate that ourproposed decision matrix can predict AuNP targeting efficacy fororthotopic prostate tumors, ascertaining how tumor growth canaffect malignant tissues in other tumor models remains critical toensure that animal and nano-based research can be translated tohumans. It would also be prudent to study how other nano-particle types and targeting schemes may change nanoparticleinteractions with the host and tumor microenvironment. Forexample, analysis of how tumor pathophysiology influences ac-tive targeting may help to explain why the decoration of bio-recognition molecules on nanoparticle surfaces appear to onlyenhance tumor targeting for nanoparticles within the 60-nmrange (26). Further investigation on such topics will broaden ourunderstanding of nano–bio interactions and allow for the de-velopment of a fundamental framework for design of cancer-centric nanomaterials. Nevertheless, our results illustrate thattumor maturity is a critical parameter that both impacts the fateof a nanomaterial and can be exploited to rationally designbetter diagnostic probes and therapeutic vehicles in the future.
MethodsAnimal-handling protocols were approved by the Faculty of Medicine andPharmacy Animal Care Committee, University of Toronto.
Tumor Accumulation Measurements. Efficiency of AuNP delivery to tumors wasmeasured by ICP-AES. Tumors were harvested at 24 HPI and digested in 1 mLof aqua regia (1:3 vol/vol nitric acid to hydrochloric acid) supplemented with1 μg/mL yttrium for 2 h at 70 °C. Yttrium was used as an internal reference toaccount for sample loss during the digestion and purification process.Postdigestion, acidic solutions were diluted with 2 mL of double-distilledwater and filtered through 0.22-μm PVDF membranes to remove undigestedtissue. Volumes of the digested samples were then adjusted to achieve a
final volume of 4 mL via addition of double-distilled water. Gold and yttriumcontents in each sample were measured using a Perkin-Elmer Optima 3000.AuNP accumulation in tumors at 24 HPI was determined by normalizingmeasured gold concentrations to yttrium content and tumor mass.
Analysis of Nanoparticle Infiltration into Collagen Matrices. Synthesizednanoparticles were tested in vitro for their permeation capacity through typeI collagen hydrogels. Self-assembled hydrogels were first prepared by mixingpresolubilized rat tail type I collagen on ice with 10× PBS and 1 M sodiumbicarbonate at an 8:1:1 volumetric ratio, followed by dilution with double-distilled water to achieve final collagen concentrations of 2.5 and 4.0 mg/mLcollagen solutions that were then placed into gel molds and allowed to self-assemble at 37 °C for 3 h. Postpolymerization, hydrogels were equilibratedin double-distilled water for 2 h, followed by immediate water exchangeand introduction of AuNPs. AuNP infiltration into hydrogels was monitoredevery 30 min for 15 h via laser-scanning confocal microscopy using anOlympus Fluoview FV1000. A transillumination lamp was used to determinethe collagen edge, whereas differential interference contrast (DIC) andfluorescence were invoked to profile AuNP distribution within the hydrogel.AuNP permeation was profiled along the length of the collagen hydrogel byanalyzing confocal images of AuNP fluorescence in ImageJ. Fluorescent-intensity profiles were then placed into GraphPad Prism to calculate totalAuNP uptake and track the mean AuNP-infiltration distances. Calculatedvalues were used to determine AuNP-accumulation rates for the differenthydrogel densities by taking the slope of the linear regression curves seen inSI Appendix, Fig. S11A.
Analysis of Nanoparticle Expulsion from Collagen Matrices. Collagen hydrogelswere constructed using a similar pH-based self-assembly process as men-tioned in Analysis of Nanoparticle Infiltration into Collagen Matrices. Beforegelation, AuNPs equivalent to a total surface area of 30 cm2 were thor-oughly mixed with hydrogel solutions on ice. AuNP–collagen mixtures werethen allowed to set overnight at 37 °C, rinsed with PBS, and suspended in 1mL of PBS. At 0, 1, 2, 3, 5, 6, 8, and 24 h, the PBS suspension solution wassampled (90 μL) to track AuNP expulsion from the hydrogels. AuNP quantityin sample solutions was approximated by measurement of sample fluores-cence in 384 fluorescent well plates (Nunc 384-well optical well plates) usinga Carestream Multispectral MS Fx Pro in vivo imager (excitation/emission:750/830 nm) at an exposure time of 10 min. Fluorescent images were ana-lyzed by densitometry in ImageJ. AuNP expulsion rates were obtained bytaking the slope of the linear regression curves seen in SI Appendix,Fig. S11B.
Simulation of Nanoparticle Diffusion in Collagen Matrices. Two-dimensionaland 3D stochastic models of AuNP movement in collagen matrices wereprogrammed and simulated in Matlab; 2D models were used to study howAuNP diameter and differences in the available area fraction of collagenmatrices would affect the frequency of AuNP–collagen collisions; 3D simu-lations were conducted to investigate the impact of collagen density onAuNP diffusion distance. For both models, AuNP movement was taken asdiscrete random walk steps obeying Einstein–Stokes Brownian motion, asdictated by Eq. 2, where KB, η, T, and r denote the Boltzmann constant,solvent viscosity, temperature in kelvins, and AuNP radii, respectively:
D=κBT6πηr
. [2]
Particle movement was approximated as the mean square displacementaccording to Fick’s second law (Eq. 3), where δ and dt were taken as thediscrete distance and time interval between steps:
δ=ffiffiffiffiffiffiffiffiffiffiffiffi2Ddt
p. [3]
AuNPs were approximated as circles and spheres for two and three dimen-sions, respectively. AuNP–collagen fiber collisions were assumed to beelastic with collagen fibers approximated as immobile cylinders. Simula-tions were also conducted in the limit of dilute AuNP concentrationswhere AuNP–AuNP collisions could be neglected and particles could beindependently tracked.
For the 2D simulations, collagen matrices of differing available areafractions were approximated as square pores of varying size. Pore sizes wereselected based on empirically measured spaces between collagen fibers seenin scanning electron microscopy images of collagen hydrogels of varyingconcentration (SI Appendix, Fig. S9). Images were imported into ImageJ andthresholded to differentiate collagen fibers from pores. The size of each
E1150 | www.pnas.org/cgi/doi/10.1073/pnas.1521265113 Sykes et al.

pore was measured by calculating the rolling ball radius of each pore usingImageJ’s built-in algorithm. Initial AuNP positions were randomized withinthe collagen pores and were permitted to move stochastically within thesquare. Upon AuNP movement beyond the dimensions of the pore, collisionevents were counted and AuNP trajectories were elastically reflected. Par-ticles were tracked for 10,000 steps at 0.1-s intervals. AuNP–collagen colli-sions were tallied for each condition.
To simulate 3D collagen, matrices composed of 100, 300, 600, and 1,000cylinders ranging in length from 0 to 30 μm and radii ranging from 0.05 to0.50 μm were randomly distributed and oriented within 30 × 30 × 30 μmcubes to mimic hydrogels with collagen volume fractions between 8.72%and 87.20%. Collagen volume fractions were determined by calculating theratio of volume occupied by collagen fibers (approximated as cylinders)versus the total region of interest (30 × 30 × 30 μm cube). Volume fractionswere equated to empirical collagen pore sizes by taking a 2D projection ofthe generated tissue, followed by calculation using the same ImageJ processas mentioned for our 2D simulations. For particle-motility simulations,AuNPs were randomly placed in our 3D matrices, allowed to move freelywithin the confines of the cube, and reflect off collagen fibers. The direction
of AuNP motion was randomized with each step. AuNPs were tracked for5,000 steps at 0.5-s intervals. Displacement between start and end pointswere measured to determine AuNP diffusion distances.
Statistical Analysis. All statistical analysis comparing between groups wereperformed using one-way ANOVA (one variable per group) and two-wayANOVA (two variables per group) in GraphPad Prism.
Supporting Information. See the SI Appendix for details regarding materials,gold nanoparticle synthesis and characterization, tumor induction, nano-particle administration, tumor histology analysis, and whole-animal imaging.
ACKNOWLEDGMENTS. This work was supported by Natural Sciences andEngineering Research Council Grant RGPIN288231, Canadian Institute ofHealth Research Grants MOP130143 and COP126588, and Canadian ResearchChair Grant 950-223824. E.A.S. and C.D.S. acknowledge fellowships from theNatural Sciences and Engineering Research Council. C.D.S. acknowledges ascholarship from the Alberta Innovates Technology Futures.
1. Chou LYT, Zagorovsky K, Chan WCW (2014) DNA assembly of nanoparticle super-structures for controlled biological delivery and elimination. Nat Nanotechnol 9(2):148–155.
2. He X, Wang K, Cheng Z (2010) In vivo near-infrared fluorescence imaging of cancerwith nanoparticle-based probes.Wiley Interdiscip Rev Nanomed Nanobiotechnol 2(4):349–366.
3. Nam T, et al. (2010) Tumor targeting chitosan nanoparticles for dual-modality optical/MR cancer imaging. Bioconjug Chem 21(4):578–582.
4. Yang J, et al. (2007) Multifunctional magneto-polymeric nanohybrids for targeteddetection and synergistic therapeutic effects on breast cancer. Angew Chem Int EdEngl 46(46):8836–8839.
5. Cherukuri P, Glazer ES, Curley SA (2010) Targeted hyperthermia using metal nano-particles. Adv Drug Deliv Rev 62(3):339–345.
6. Bae YH, Park K (2011) Targeted drug delivery to tumors: Myths, reality and possibility.J Control Release 153(3):198–205.
7. Sykes EA, Dai Q, Tsoi KM, Hwang DM, Chan WCW (2014) Nanoparticle exposurein animals can be visualized in the skin and analysed via skin biopsy. Nat Commun5:3796.
8. De Jong WH, et al. (2008) Particle size-dependent organ distribution of gold nano-particles after intravenous administration. Biomaterials 29(12):1912–1919.
9. Cole AJ, David AE, Wang J, Galbán CJ, Yang VC (2011) Magnetic brain tumor tar-geting and biodistribution of long-circulating PEG-modified, cross-linked starch-coated iron oxide nanoparticles. Biomaterials 32(26):6291–6301.
10. Kumar R, et al. (2010) In vivo biodistribution and clearance studies using multimodalorganically modified silica nanoparticles. ACS Nano 4(2):699–708.
11. Moghimi SM, Hunter AC, Murray JC (2001) Long-circulating and target-specificnanoparticles: Theory to practice. Pharmacol Rev 53(2):283–318.
12. Yaehne K, et al. (2013) Nanoparticle accumulation in angiogenic tissues: Towardspredictable pharmacokinetics. Small 9(18):3118–3127.
13. Huang X, Teng X, Chen D, Tang F, He J (2010) The effect of the shape of mesoporoussilica nanoparticles on cellular uptake and cell function. Biomaterials 31(3):438–448.
14. Hauck TS, Ghazani AA, Chan WCW (2008) Assessing the effect of surface chemistry ongold nanorod uptake, toxicity, and gene expression in mammalian cells. Small 4(1):153–159.
15. Treuel L, Jiang X, Nienhaus GU (2013) New views on cellular uptake and trafficking ofmanufactured nanoparticles. J R Soc Interface 10(82):20120939.
16. Brown JM, Giaccia AJ (1998) The unique physiology of solid tumors: Opportunities(and problems) for cancer therapy. Cancer Res 58(7):1408–1416.
17. Junttila MR, de Sauvage FJ (2013) Influence of tumour micro-environment hetero-geneity on therapeutic response. Nature 501(7467):346–354.
18. Rosenblum D, Peer D (2014) Omics-based nanomedicine: The future of personalizedoncology. Cancer Lett 352(1):126–136.
19. McMillin DW, Negri JM, Mitsiades CS (2013) The role of tumour-stromal interactionsin modifying drug response: Challenges and opportunities. Nat Rev Drug Discov 12(3):217–228.
20. Thurber GM, Schmidt MM, Wittrup KD (2008) Factors determining antibody distri-bution in tumors. Trends Pharmacol Sci 29(2):57–61.
21. Stylianopoulos T, et al. (2010) Diffusion of particles in the extracellular matrix: Theeffect of repulsive electrostatic interactions. Biophys J 99(5):1342–1349.
22. Lu P, Weaver VM, Werb Z (2012) The extracellular matrix: A dynamic niche in cancerprogression. J Cell Biol 196(4):395–406.
23. Rich L, Whittaker P (2005) Collagen and picrosirius red staining: A polarized lightassessment of fibrillar hue and spatial distribution. Braz J Morphol Sci 22:97–104.
24. Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ (2012) Second harmonic gener-ation microscopy for quantitative analysis of collagen fibrillar structure. Nat Protoc7(4):654–669.
25. Brown E, et al. (2003) Dynamic imaging of collagen and its modulation in tumors invivo using second-harmonic generation. Nat Med 9(6):796–800.
26. Sykes EA, Chen J, Zheng G, Chan WCW (2014) Investigating the impact of nano-particle size on active and passive tumor targeting efficiency. ACS Nano 8(6):5696–5706.
27. Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW (2009) Mediating tumortargeting efficiency of nanoparticles through design. Nano Lett 9(5):1909–1915.
28. Perrault SD, Chan WCW (2009) Synthesis and surface modification of highly mono-dispersed, spherical gold nanoparticles of 50-200 nm. J Am Chem Soc 131(47):17042–17043.
29. Chou LYT, Chan WCW (2012) Fluorescence-tagged gold nanoparticles for rapidlycharacterizing the size-dependent biodistribution in tumor models. Adv HealthcMater 1(6):714–721.
30. Provenzano PP, et al. (2008) Collagen density promotes mammary tumor initiationand progression. BMC Med 6(1):11.
31. Dudley DT, et al. (2014) A 3D matrix platform for the rapid generation of therapeuticanti-human carcinoma monoclonal antibodies. Proc Natl Acad Sci USA 111(41):14882–14887.
32. Stylianopoulos T, Diop-Frimpong B, Munn LL, Jain RK (2010) Diffusion anisotropy incollagen gels and tumors: The effect of fiber network orientation. Biophys J 99(10):3119–3128.
33. Jakobsen I, Lyng H, Kaalhus O, Rofstad EK (1995) MRI of human tumor xenografts invivo: Proton relaxation times and extracellular tumor volume. Magn Reson Imaging13(5):693–700.
34. Krol A, Maresca J, Dewhirst MW, Yuan F (1999) Available volume fraction of mac-romolecules in the extravascular space of a fibrosarcoma: Implications for drug de-livery. Cancer Res 59(16):4136–4141.
35. Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW (2012) Nanoparticle size and surfacechemistry determine serum protein adsorption and macrophage uptake. J Am ChemSoc 134(4):2139–2147.
36. Varin A, Mukhopadhyay S, Herbein G, Gordon S (2010) Alternative activation ofmacrophages by IL-4 impairs phagocytosis of pathogens but potentiates microbial-induced signalling and cytokine secretion. Blood 115(2):353–362.
37. Wang B, et al. (2011) Transition of tumor-associated macrophages from MHC classII(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol 12(1):43.
38. Albanese A, Tang PS, Chan WCW (2012) The effect of nanoparticle size, shape, andsurface chemistry on biological systems. Annu Rev Biomed Eng 14:1–16.
39. Dawidczyk CM, Russell LM, Searson PC (2014) Nanomedicines for cancer therapy:State-of-the-art and limitations to pre-clinical studies that hinder future develop-ments. Front Chem 2:69.
40. Josefsson A, et al. (2005) Tumor size, vascular density and proliferation as prognosticmarkers in GS 6 and GS 7 prostate tumors in patients with long follow-up and non-curative treatment. Eur Urol 48(4):577–583.
41. Wisnivesky JP, Yankelevitz D, Henschke CI (2004) The effect of tumor size on cur-ability of stage I non-small cell lung cancers. Chest 126(3):761–765.
42. Yu K-D, et al. (2012) Effect of large tumor size on cancer-specific mortality in node-negative breast cancer. Mayo Clin Proc 87(12):1171–1180.
Sykes et al. PNAS | Published online February 16, 2016 | E1151
ENGINEE
RING
PNASPL
US