tara leanne knoll thesis submitted in partial fulfillment...
TRANSCRIPT

STUDIES ON THE MECHANISM AND INHIBITION OF SIALIDASES
Tara Leanne Knoll B.Sc., Kinesiology, Simon Fraser University, Burnaby, Canada 2000
THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
MASTER OF SCIENCE
In the Department of
Chemistry
O Tara Leanne Knoll, 2003
SIMON FRASER UNIVERSITY
December 2003
All rights reserved. This work may not be reproduced in whole or in part, by photocopy
or other means, without permission of the author.

APPROVAL
Name:
Degree:
Title of Thesis:
Examining Committee:
Chair:
Date Approved:
Tara Leanne Knoll
M.Sc.
Studies on the Mechanism and Inhibition of Sialidases.
Dr. N.R. Branda
Dr. A.J. Bennet, Professor, Senior Supervisor
( / " 6 c ' { L C , --,, - ,. , "/-,, Dr. E. Plettner, Assistant Professor, Committee Member
) / O l * " 'I -.
Dr. J.C. Clyburne, ~dsociate ~Gfessor, Committee Member
Dr. P.D. Wilson, A sis ant Professor, Internal Examiner E r

PARTIAL COPYRIGHT LICENCE
I hereby grant to Simon Fraser University the right to lend my thesis,
project or extended essay (the title of which is shown below) to users of the
Simon Fraser University Library, and to make partial or single copies only
for such users or in response to a request from the library of any other
university, or other educational institution, on its own behalf or for one of its
users. I further agree that permission for multiple copying of this work for
scholarly purposes may be granted by me or the Dean of Graduate
Studies. It is understood that copying or publication of this work for
financial gain shall not be allowed without my written permission.
Title of Thesis/Project/Extended Essay:
Studies on the Mechanism and Inhibition of Sialidases
Author: Y-v- Ud wry -. (signature)
Tara Leanne Knoll (name)
- - - - - - - (date)

ABSTRACT
Sialic acids (N-acetylneuraminic acids, NeuAc) are found in nature as the terminal
residue of oligosaccharide chains in glycoconjugates. Their biological roles include
stabilization of glycoconjugate conformations, cell adhesion, and molecular and cellular
recognition. Sialidases are enzymes that catalyze the hydrolysis of a-2+3; 2+6; and
2 4 linked sialosides. This family of enzymes is involved in the pathogenicity of many
diseases such as influenza and cholera.
As part of an ongoing investigation into the mechanisms of spontaneous and
enzyme-catalyzed hydrolysis reactions of sialosides, 3,4-dihydro-2H-pyrano[3,2-
clpyridinium sialoside (DHP NeuAc) was synthesized. Studies on the spontaneous
solvolysis of DHP NeuAc revealed that reaction occurred via an DN + AN (SN1)
mechanism with no intramolecular nucleophilic participation from the anomeric
carboxylate.
The rate of DHP NeuAc hydrolysis catalyzed by sialidases from Vibrio cholerae,
Clostridium perfringens, and Salmonella typhimurium was compared to that of other
pyridinium sialosides in order to determine how changing the leaving group affects
catalysis. Based on the data, it was concluded that for V. cholerae, rate-determining
sialylation occurs concurrently with a conformational change. While conformational
change was rate-limiting for S. typhimurium, sialylation was partially rate-limiting for C.
perfringens. In addition, DHP NeuAc was found to be a competitive inhibitor of the
Newcastle hemagglutinin-neuraminidase bifunctional enzyme with a K, of 58 pM.

Hydrolysis of DHP NeuAc catalyzed by the Micromonospora viridifaciens wild-
type sialidase was compared with that for by two active site mutants of this enzyme.
When the general acidbase aspartic acid residue was mutated to a glycine, there was a
slight increase in rate of hydrolysis which was attributed to making more room in the
active site for this conformationally restricted substrate. But, the rate of hydrolysis of
DHP NeuAc decreased significantly when the nucleophilic tyrosine residue was mutated
to a glycine. This indicates that in contrast to the p-nitrophenyl sialoside substrate, water
is not able to act as an efficient nucleophile to displace the leaving group.
Using this mechanistic information, a new class of sialidase inhibitors was
designed. Progress toward the synthesis of these inhibitors via two different routes is
outlined.

DEDICATION
This thesis is dedicated to my family, whose love has kept me going and whose humour has given me joy.

ACKNOWLEDGEMENTS
I would like to thank Dr. A. J. Bennet for giving me the opportunity to work in his
group. His enthusiasm, support and patience made him a wonderful "Boss."
Thank-you to Dr. Jacki Watson for all her advice regarding enzyme kinetics as
well for the use of all the sialidase enzymes. I am extremely grateful to Veedeeta
Dookhun for her help in the lab as well as for her friendship. I would also like to thank
my lab mate Ivan Hemeon who keeps us in good spirits.
I am grateful to Mrs. Marcy Tracey for running the numerous NMR spectra as
well as for introducing me to new extra-curricular activities. I also appreciate Dr. Alan
Tracey for his help with processing the 2D NMR spectra. I thank Dr. David McGillivray
for measuring the high resolution mass spectrum, and Mr. M. K. Yang for performing the
elemental analyses.
I would especially like to thank my family for their encouragement and love, I
couldn't have done it without them!

TABLE OF CONTENTS
.. Approval ............................................................................................................................. 11
... Abstract .......................................................................................................................... m
Dedication ........................................................................................................................... v
Acknowledgements ........................................................................................................... vi ..
Table of Contents ............................................................................................................. vll
List of Figures ................................................................................................................... ix
List of Tables ...................................................................................................................... x
List of Schemes .................................................................................................................. xi .. ............................................................................. List of Abbreviations and Acronyms xu
Chapter One: Introduction ............................................................................................. 1 Carbohydrates .................................................................................................................. 1 Sialic Acids ..................................................................................................................... 1 Spontaneous Hydrolysis of Carbohydrates ...................................................................... 2
Research Objective ....................................................................................................... 3
.................................................................................................................... Glycosidases 5 ......................................................................................................................... Sialidases 7
General Mechanism of Sialidase-Catalyzed Hydrolysis .................................................. 8 Bacterial and Viral Sialidases ........................................................................................ 1 1
Research Objective ..................................................................................................... 12
............................................................... The Micromonospora viridifaciens Sialidase 1 3 ................................................................................................ Research Objective 1 3
...................................................................................................................... Influenza -13 .......................................................... Mode of Influenza Infection and Drug Targets 15 ........................................................... Previous Work: Influenza Sialidase Inhibitors 16
..................................................................................................... Research Objective 19
Chapter Two: Enzymatic and Spontaneous Solvolysis of an a-D-N- ..................................................................... Acetylneuraminyl Pyridinium Zwitterion 20
....................................................................................................................... Overview 20
vii

Spontaneous Solvolysis of 3,4.dihydro.2H.pyrano[3, 2.~1 pyridinium N- acetylneuraminide ......................................................................................... 20
Results ................................................................................................................... 20 Discussion .................................................................................................................. 26 Experimental .............................................................................................................. 30 Kinetics ................................................................................................................... 36 Product Studies ........................................................................................................... 36
Hydrolysis of Pyridinium a-D-N-Acetylneuraminides Catalyzed by Bacterial and Viral Sialidases ....................................................................................... 37
................................................................................................................... Results 37 Discussion ............................................................................................................ 3 8 Experimental .............................................................................................................. 42
Hydrolysis of DHP NeuAc Catalyzed by the Micromonospora viridifaciens Neuraminidase .............................................................................................. -44
Results ................................................................................................................... 44 Dicussion .................................................................................................................. -46 Experimental .............................................................................................................. 48
Chapter Three: Progress Toward the Synthesis of a Novel Influenza Inhibitor ............................................................................................................................ 50
Overview ....................................................................................................................... 50
Proposed Synthesis 1 ..................................................................................................... 50 Progress ................................................................................................................... 52 Experimental .............................................................................................................. 54
..................................................................................................... Proposed Synthesis 2 57 Progress ................................................................................................................... 59 Experimental .............................................................................................................. 60
............................................................................................ Chapter Four: Conclusions 66
......................................................................................................................... References 68
... Vl l l

Figure 1 : Figure 2: Figure 3: Figure 4: Figure 5:
Figure 6: Figure 7:
Figure 8: Figure 9: Figure 10: Figure 1 1 : Figure 12: Figure 13:
LIST OF FIGURES
Naturally occurring sialic acids ......................................................................... 2 ............................ Generic a-aryl sialoside. P-CMP sialoside and DHP NeuAc 4
General mechanism for inverting glycosidases ................................................. 6 ................................................ General mechanism for a retaining glycosidase 6
General reaction for the catalyzed hydrolysis of sialic acid from glycoconjugate chains ..................................................................................... 7
..................................................................... General mechanism for sialidases 8
Trapping the sialosyl-enzyme intermediate using a 2, 3-difluorosialic acid ................................................................................................................... 10
. . Pyridinium N-acetylneurammides ................................................................ 1 1
................................................... The novel p yridinium N-acetylneuraminide 1 3 .................................................. Classification system for the influenza virus 14
* . ........................................................................... Influenza sralidase inhibitors 17 ................................................................................ Mannosidase inhibitors 1 8
3,4-Dihydro-2 H-pyrano[3 , 2-clpyridinium N-acetylneuraminide (DHP NeuAc) .......................................................................................................... 2 0
Figure 14: Plot of absorbance versus wavelength for pKa determination of DHPH+ ............................................................................................................. 22
Figure 15: DHP NeuAc (3) and P-D-glucopyranosyl4'-bromoisoquinolinium tetrafluoroborate salt (20) ................................................................................ 25
Figure 16: Plot of log(ko& versus log(kObs )zo for the aqueous solvolyses of 3 and 20 at T = 65'C .................................................................................................. 25
................................................................ Figure 17: Products of DHP NeuAc solvolysis 26
........................................................................ Figure 18: CMP-N-acetylneuraminate (2) 29 Figure 19: Glucopyranosyl cation (25) and N-acetylneuraminyl cation (26) .................... 29 Figure 20: Glycal formation via proton abstraction by the pyridine leaving group .......... 30 Figure 2 1 : Pyridinium a.D.N.acetylneuraminides ............................................................ 38 Figure 22: Plot of log(kCat) versus p ~ a ( ~ - ~ + ) for the NDV sialidase-catalyzed
hydrolysis of 27-30 and 3 ................................................................................ 41 Figure 23 : Substrates for M . viridifaciens neuraminidase-catalyzed hydrolysis ............... 44
............................................................................................. Figure 24: Target compound 50

LIST OF TABLES
Table 1 : Observed rate constants for the aqueous methanolysis of DHP NeuAc (3), T = 65 OC .................................................................................................... 24
Table 2: Observed products formed during the reactions of DHP NeuAc (3) in aqueous methanol (vlv) at 65 OC. ...................................................................... 26
Table 3: Michaelis-Menten parameters for the sialidase-catalyzed hydrolysis of DHP NeuAc at 37OC. ....................................................................................... .37
Table 4: Calculated PI, values for kcat and kCatlKm on the siladase-catalyzed ........................ hydrolyses of pyridinium a-D-N-acetylneuraminides at 37 OC. 38
Table 5: Kinetic parameters for the wild-type, D92G, and Y370G M. viridifaciens sialidase-catalyzed hydrolysis of pNP NeuAc and DHP NeuAc. ............................................................................................................. -44
Table 6: Rates of hydrolysis of DHP NeuAc (3). ............................................................. 45

LIST OF SCHEMES
Scheme 1: Scheme 2: Scheme 3: Scheme 4: Scheme 5: Scheme 6: Scheme 7: Scheme 8: Scheme 9:
Synthesis of 3.4.dihydro.2H.pyrano[3. 2.~1 pyridine ................................... 21 Synthesis of sialyl chloride ........................................................................... 23 Coupling between 18 and 16, and deprotection to yield DHP NeuAc ......... 23
............................................. Possible pathways for DHP NeuAc solvolysis 27 ..................................................................................... Proposed synthesis 1 51
................................................................... Proposed Synthesis 1 continued 52 Reaction of tosylate 33 ................................................................................ 5 4
..................................................................................... Proposed synthesis 2 58 Proposed synthesis 2 continued ................................................................... 59
............................................................. Scheme 10: Alternative to proposed synthesis 2 -60

LIST OF ABBREVIATIONS AND ACRONYMS
A Angstrom
Abs absorbance
AcCl
AcOH
AgBF4
aq
BF30Et2
Bn
b.p.
CDC13
CHC13
CH2C12
CMP
COSY
d
DHP
D20
DMSO
equiv
ESI
EtOH
acetyl chloride
acetic acid
silver tetrafluoroborate
aqueous
boron trifluoride diethyl etherate
benzyl
boiling point
deuterated chloroform
chloroform
dichloromethane
cytidine monophosphate
correlation spectroscopy
doublet
3,4-dihydro-2H-pyran[3,2-c] pyridine
deuterated water
dimethyl sulfoxide
equivalents
electron spray ionization
ethanol
xii

HC1
HETCOR
H2S04
HPLC
HRMS
J
K2co3
OAc
LiAlH4
m
MCPBA
Me
MeOH
MES
MgS04
min
m.p.
NaOH
NeuAc, Neu5Ac
NH3
NMR
Pd/C
P
hydrochloric acid
heteronuclear correlation
sulphuric acid
high performance liquid chromatography
high resolution mass spectrometry
coupling constant in Hz
potassium carbonate
acetate
lithium aluminium hydride
multiplet
3-chloroperoxybenzoic acid
methyl
methanol
4-morpholineethanesulfonic acid monohydrate
magnesium sulphate
minutes
melting point
sodium hydroxide
N-acetylneuraminic acid
ammonia
nuclear magnetic resonance
palladium on carbon
para
. . . X l l l

RNA
NaOMe
sat
S
t1/2
THF
TLC
TsCl
TsOH
UV
vis
v
ribonucleic acid
sodium methoxide
saturated
singlet
half-life
tetrahydrofuran
thin layer chromatography
p-toluenesulfonyl chloride
p-toluenesulfonic acid
ultraviolet
visible
volume
xiv

CHAPTER ONE: INTRODUCTION
Carbohydrates
The term "carbohydrate" literally means "carbon hydrate" and stems from the
general stoichiometric formula (cH~o),.' Carbohydrates are the most abundant class of
biological molecules and play many roles, such as in energy storage, in metabolism and
in cell-cell signalling.* The group of carbohydrates that will be discussed in detail in this
thesis is the sialic acids.
Sialic Acids
The term "sialic acid" is often used interchangeably with "neuraminic acid" but is
now defined as the family comprised of derivatives of neuraminic acids.3 In 1936, the
first neuraminic acid (5-amino-3,5-dideoxy-~-glycero-~-galacto-nonulosonic acid)
species were crystallized by Blix yet the full scope of their distribution and function still
remains to be completely determined.4
Neuraminic acids are found in mammals, viruses, bacteria, and many other
organisms.5 In general, they occupy the terminal position of oligosaccharides in
glycoconjugates, and are usually linked a-(2,3) or a-(2,6) to D-galactose, a-(2,6) to N-
acetyl-D-galactosamine residues3, or a-(2,8) to other sialic acid m~iet ies .~ Because of
their position at the termini of glycoconjugate chains, sialic acids are important in
molecular and cellular recognition event^.^
Some of the naturally occurring derivatives of neuraminic acid are shown below
(Figure 1). The amine of the parent neuraminic acid compound can be substituted with

either an acetyl or a glycoloyl group. Other sialyl species include derivatization of one or
more of the alcohol groups as methyl ethers or as acetyl, lactyl, sulfonyl, or phosphoryl
For example, 9-OAc-NeuSAc (neuraminic acid that is acetylated at positions 5
and 9) was found a-(2,8)-linked to a Neu5Ac (neuraminic acid acetylated at position 5) in
gangliosides of human melanoma cells.8
Figure 1: Naturally occurring sialic acids.
Spontaneous Hydrolysis of Carbohydrates
The mechanisms of hydrolysis for carbohydrate derivatives have been of interest
for the past thirty years, because of the importance of these compounds in biological
systems. In 1977, Young and Jencks used acetophenone acetals as models for glucosyl
pyranosides in order to estimate the lifetime of solvent-equilibrated oxacarbenium ion
intermediates in water.9 Based upon these trapping experiments they were able to

estimate that the lifetime of the methoxymethyl oxacarbenium ion (CH30C~2f ) in water
was too short to form a solvent-equilibrated oxacarbenium ion;9 therefore the reactions of
these acetals occur via an ANDN (SN2) mechanism (concerted association of the
nucleophile and dissociation of the n~cleofu~e). '~ '"
Compared to the methoxyrnethyl oxacarbenium ion, aldopyranosyl cations were
expected to be even more unstable and hence, aldopyranosides were predicted to
hydrolyze via concerted mechanisms. If this were the case, one would expect complete
inversion of configuration at the anomeric carbon. However, product studies of the
solvolysis of both anomers of various glucopyranosyl derivatives showed that a mixture
of a- and P-products was formed, and that for two anomeric substrates different product
ratios were formed. To reconcile these results with the above prediction, Sinnott and
Jencks proposed that the reaction transition states possessed weak interactions between
the departing nucleofuge and the incoming solvent m~lecules. '~
In agreement with these results, the lifetime in aqueous solution of the
glucopyranosylium ion has been estimated to be around (1 .O-2.5) x 1 0-l2 s, 13*14 a time
interval that is too short to allow solvent equilibration.15 Indeed, the only glycosidic
hydrolysis reactions in which the aglycon departs as an anion have been shown to occur
via ANDN (SN2) mechanism^.'^-^^
Research Objective
In contrast to the numerous studies performed on glycopyranosides, little research
has been performed on the solvolysis of sialosides. The key differences between
sialosides and glucosides are that sialosides have a carboxylate group attached to the
anomeric carbon while glucosides do not, and that sialosides have also have a CH2 group

adjacent to the anomeric carbon while most glycosides have a hydroxyl group on the
corresponding carbon atom. An effect of deoxy generation, that is removal of an
electron-withdrawing group, is to increase the stability of the oxacarbenium ion
intermediate.13 The effect of adding an anomeric carboxylate on the mechanism of
hydrolysis is a subject of debate. For instance, Ashwell et al. proposed that the
carboxylate formed a transient a-lactone and provided nucleophilic assistance to
departure of a phenolate group during hydrolysis of aryl a-sialosides (1) (Figure 2).19 In
contrast, Horenstein and Bruner studied the hydrolysis of P-CMP (cytidine
monophosphate) sialoside 2 and these authors argued that the carboxylate is coplanar
with the oxacarbenium ion intermediate and they suggested that its role was to assist the
attacking water molecule via electrostatic interaction^.^' Chou et al. also argued against
nucleophilic participation by the carboxylate and they proposed that their pyridinium
sialosides hydrolyzed via a dissociative DN + AN (SNl) me~han i sm.~~
Figure 2: Generic a-aryl sialoside, P-CMP sialoside, and DHP NeuAc.
~5 0
H &a AcH HO - 'X H o ~ ~ ~ ~ AcH HO OH OH
a-aryl sialoside 1 P-CMP sialoside 2
DHP NeuAc 3

It should be noted that sialic acid, like other glycosidases, undergoes rapid
mutarotation in water, which in this case gives an equilibrium mixture that favors the P-
anomer by a factor of about 2 0 . ~ ~ AS a result, it is impossible to determine the
stereochemistry of sialoside hydrolysis reactions. For this reason, one of the research
objectives of this thesis was to examine the aqueous methanolysis of 3,4-dihydro-2H-
pyrano[3,2-clpyridinium a-D-N-acetylneuraminoate (DHP NeuAc, 3). Results and
discussion of these experiments are outlined in Chapter 2.
Glycosidases
Glycosidases are enzymes that catalyze the hydrolysis of glycosidic linkages in
carbohydrates. They can be divided into two major groups: those that hydrolyze
glycosides with retention of configuration and those that do so with inversion of
configuration.23 The general mechanism for an inverting glycosidase is pictured below
(Figure 3). In this mechanism two active site carboxyl groups act in concert as a general
acid-general base pair to catalyze the displacement of the aglycon with a water molecule.
The distance between the two carboxyl groups is 9.0 - 9.5 A, a distance that allows room
for direct attack of a water molecule on the substrate.23324

Figure 3: General mechanism for inverting glycosidases.23
General Acid A.
P
General Base
+ ROH
In constrast, retaining glycosidases, an enzyme class that acts by retaining the
configuration at the anomeric carbon, contain a pair of carboxylate groups that are
approximately 4.5 - 5.5 A apart.23'24 During the enzyme-catalyzed reaction one
carboxylate acts as a general acidhase while the other acts as a nucleophile to form an
enzyme-bound intermediate (Figure 4).
Figure 4: General mechanism for a retaining glycosidase.23
General Acid
Nucleophile I
General T""
Enzyme-bound Intermediate

Sialidases
Sialidases (EC 3.2.1.18: N-acetylneuraminosyl glycohydrolases or
ne~raminidases)~~ are a family of retaining glycosidases that catalyze the removal of
terminal sialic acids from oligosaccharide chains found in glycoconjugates as shown in
Figure 5.26 These enzymes are found in mammals, viruses, bacteria, fungi, and other
species. As such, they are important in regulating the displayed neuraminic acids on cell
surfacesz7 and these enzymes are also important determinants of pathogenicity.
The mechanism of sialidase-catalyzed hydrolysis can be deduced using a variety
of methods. One method that will be discussed in this report is measuring the kinetic
parameters of sialoside hydrolysis catalyzed by different sialidases as well as for mutants
of one sialidase. Also, knowledge concerning the spontaneous hydrolysis of sialosides
can provide insight into the intrinsic reactivity of these compounds. This allows
comparisons to be made between different substrate classes for their enzyme-catalyzed
hydrolysis reactions.
Figure 5: General reaction for the catalyzed hydrolysis of sialic acid from glycoconjugate chains.
sialidase HO
H20/-ROH AcH N AcHN

Gen era1 Mechanism of Sialidase-Catalyzed Hydrolysis
Sialidases cleave a-linked sialic acid with retention of configuration at the
anorneric X-ray crystallographic studies have revealed that the reaction
product, sialic acid, binds to the influenza sialidase in a boat c o n f ~ r m a t i o n . ~ ~ ' ~ ~ This
information combined with a kinetic isotope effect study on the catalyzed hydrolysis of
p-nitrophenyl N-acetylneuraminide led Sinnott and co-workers to postulate that the
reaction proceeds via an oxacarbenium ion-like transition state (Figure 6).34


Only recently has it been shown that the role of the conserved tyrosine residue is
to act as a nucleophile, presumably in conjunction with the conserved glutamate acting as
a general base, to displace the aglycon and form a sialosyl-enzyme intermediate. Work
by Watson et al. highlighted the importance of the tyrosine residue by characterizing
three tyrosine mutants of the Micromonospora viridifaciens (M. viridifaciens) sialidase.
It was found that with these tyrosine mutants, the sialic acid product had an inverted
anomeric configuration. These authors concluded that in this case, a hole was created
where a water molecule could bind and then act nucleophilically to displace the leaving
In addition, Watts et al. were able to isolate a sialosyl-enzyme intermediate
using a fluoro-sugar to trap the nucleophilic tyrosine residue (Figure 7). It should be
noted that in this work a trans-sialidase (an enzyme that transfers sialic acid from one
glycosidic linkage to another) rather than a sialidase was used.36
Figure 7: Trapping the sialosyl-enzyme intermediate using a 2,3-difluorosialic acid.
Once the sialosyl-enzyme intermediate is formed, the conserved aspartate can act
as a general base to aid water in its attack at the anomeric carbon, while the protonated
glutamate residue assists the breakdown of the sialosyl-enzyme intermediate by donating
10

a proton to the tyrosine leaving group (Figure 6). The result of which is to yield a-sialic
acid as the first formed carbohydrate product.
Bacterial and Viral Sialidases
The general mechanism described above can have variations specific to different
sialidases. For example, substrates can bind in different conformations, and the rate-
limiting step can depend on both the sialidase and the substrate.
A Brarnsted plot is one way in which to visualize the effect of leaving group
ability on the rate of a reaction. That is, a plot of the pK, of the conjugate acid of the
leaving group versus the log of the reaction rate constant. The gradient of such a plot is
known as PI,, and is a measure of the reaction rate's sensitivity to the leaving group
ability. As a result, if a PI, value of zero, or close to it, is observed in a Brmsted plot for
the hydrolysis of pyridinium N-acetylneuraminides (Figure 8), then cleavage of the
leaving group is not the rate-determining step for the reaction.
Figure 8: Pyridinium N-acetylneuraminides.
R , = R 2 = H HO R, =Me,R2 =H
/ R, = H , R Z = M e R , = R 2 = M e
HNAc
In order to determine the effect of leaving group on influenza type A sialidase-
catalyzed hydrolysis, a series of pyridinium N-acetylneuraminides was synthesized by D.
Chow (Figure 8). Such substrates by virtue of their positive charge do not require acid
catalysis for departure of the leaving group, thereby simplifying the hydrolysis kinetics.
11

A Brmnsted plot for the hydrolysis of the pyridinium N-acetylneuraminides was then
constructed and the Slg value for k,,lK, was determined to be 0.00 k 0.12 at pH 9.5.
Thus, the authors concluded that C-N cleavage was not the slow step of the reaction and
that the evidence was consistent with a rate-determining conformational change.21
Subsequently, Watson et al. tested these pyridinium N-acetylneuraminides against
a panel of bacterial and viral sialidases. They found that the sialidases tested were able to
hydrolyze the pyridinium sialosides, therefore Brmsted plots were constructed for the
enzymes derived from Clostridium perfringen (A99 strain), Salmonella typhimurium
(LT2 strain), Vibrio cholerae (395 strain) and Newcastle disease virus (Hitchner B1
Research Objective
In such studies, a wide range of pKa values should ideally be used when
constructing Brmnsted plots. However, the pyridinium N-acetylneuraminides synthesized
by chou2' covered a pKa range of only 1.5 units and the data points measured for the
various sialidases were "well" scattered about the best-fit line. As a result, the calculated
PI, values were associated with a sizeable error. Therefore, our objective was to
synthesize the novel pyridinium N-acetylneuraminide shown in Figure 9, and use it as a
substrate in measuring the kinetic parameters for the bacterial and viral sialidases tested
by Watson et ~ 1 . ~ ~ The results and discussion of these experiments can be found in
Chapter 2.

Figure 9: The novel pyridinium N-acetylneuraminide.
The Micromonospora viridifaciens Sialidase
Research Objective
In addition to examining the mechanistic differences between sialidases from
different origins, much can be learned by kinetically characterizing mutated forms of an
enzyme. Previous work already described by Watson et aE. with tyrosine mutants of
Micromonospora viridifaciens included kinetic characterization of these mutants withp-
nitrophenyl N-acetylneuraminic acid as well as with the two natural substrates a-(2,3)
sialyl-lactose and a-(2,6) sialyl-lactose.35 It would be interesting to know how mutation
affects this sialidase's ability to hydrolyze a representative member of the pyridinium N-
acetylneuraminides. The results from these experiments are discussed in Chapter 2 of
this thesis.
Having knowledge of the mechanism of action of sialidases is useful as an aid in
the design of inhibitors. Such compounds could be used to help control the action of
these sialidases, for example the influenza sialidase.
Influenza
Influenza is a respiratory illness that has afflicted man throughout history and
continues to cause significant mortality. For example, in 1989 an outbreak of influenza
13

caused the deaths of approximately 26,000 individuals in England and Even
worse than this, 20 million people died due to the influenza pandemic in 19 18- 19 19.
This toll surpasses that of the number of casualties in World War
There are two main influenza variants that cause disease, namely A and B. In
terms of virulence, influenza A affects pigs, horses, seals, whales, birds, and humans. It
is this type that is responsible for the human pandemics while influenza B causes regional
epidemics and affects only humans." A third type of influenza, type C, also exists
however it does not result in disease. The classification of influenza subtypes is shown in
Figure 10. The initial letter denotes the type, which is followed by the origin of the
strain, the particular strain sequence number and the year of isolation. The nomenclature
for type A influenza also identifies the subtypes of the two surface proteins as there are
15 known hemagglutinin (HA) subtypes and 9 known neuraminidase (NA) subtypes.
Figure 10: Classification system for the influenza virus.
Neuraminidase
Hemagglutinin
Attempts at preventing human infection by the influenza virus have been mainly
through the use of vaccines. However, due to the variability in the virus' surface
antigens, there is no universal influenza vaccine. Each year, the World Health

Organization predicts which strains are going to be circulating, and they prepare the
yearly vaccine accordingly.40
As an alternative to vaccines, researchers have developed drugs that are effective
against most strains of influenza. This is possible because of the invariability of the
active site of one of the surface proteins, neuraminidase (sialidase). In order to
understand how these drugs work, and the reasoning behind the development of new
drugs, it necessary to know how the influenza virus infects and proliferates in its victim.
Mode of Influenza Infection and Drug Targets
After entering its host, usually through the respiratory system, the influenza virus
particles, or virions, must bind to host cells. For this purpose, the virions have
hemagglutinin proteins on their outer coat which bind to the sialic acids displayed on the
host cell's s~rface!'"~ Once bound, the virus enters the host cell by endocytosis. In
order for replication to occur, the virion's ribonucleoprotein must enter the host cell's
nucleus. This is triggered by a signal involving influx of protons through an ion channel,
namely the M2 protein!3 After the ribonucleoprotein enters the nucleus, replication of
the viral RNA ensues. Progeny viral particles are then assembled and bud out of the host
cell into circu~ation!~ Like the host cell, the newly formed virions also contain sialic
acids on their outer coats which can then bind to other viral hemagglutinin and thus forrn
aggregates. To solve this problem the virus employs its sialidase to hydrolyze the sialic
acid residues from its surface. Thus, resulting viral particles are free to infect other
cells.42
It is the three proteins mentioned above, the M2, the hemagglutinin and the
sialidase proteins, that have been targeted for drug design. The two drugs that target the

M2 protein, amantidine and rimantidine, cause adverse side effects and are ineffective as
they are no longer effective against mutated forms of the virus.44 Another problem with
targeting the M2 protein is that only influenza type A contains this protein, therefore
these drugs have no effect on influenza type B virions.
Currently there are no drugs on the market targeting the hemagglutinin protein
responsible for virus-host cell interaction. Wade has suggested that one reason for this
lies in the conformation of the substrate in the binding site of the hemagglutinin enzyme
as well as in the structures of the binding pockets them~elves .~~ It has been shown that
sialic acid binds to the sialidase active site in a boat c~nformation.~~ In contrast, sialic
acid binds to the hemagglutinin binding pocket in a chair conf~rmation.~~ In addition, the
hemagglutinin binding site is shallower and has fewer charged residues than the sialidase
active site. Therefore sialic acid derivatives have a greater number of interactions with
the sialidase compared to the hemagglutinin protein, and drug targets have been designed
to bind in the sialidase active site rather than the hemagglutinin active site.46
Numerous inhibitors of the sialidase protein have been synthesized, a few of
which are discussed presently.
Previous Work: Influenza Sialidase Inhibitors
An effective influenza sialidase inhibitor will either compete with the substrate
for the active site (competitive inhibition) or bind irreversibly to its active site
(irreversible inhibition). In general, good inhibitors mimic the transition state of the
enzyme-substrate reaction. Two ways to emulate the oxacarbenium ion-like transition
state (Figure 6) in these reactions are: (1) conformation; and (2) charge. It has been
shown by X-ray crystallography s t ~ d i e s ~ ~ , ~ ~ that sialic acid is bound in a boat
16

conformation, and several kinetic studies have supported the development of positive
charge in the transition state. 30,34,48
One of the first inhibitors of the influenza virus sialidase discovered is
NeuSAc2en (4), a derivative of sialic acid (Figure 1 1).49 The double bond between
carbons 2 and 3 flattens the ring between C2, C3, C4 and the ring oxygen thus mimicking
an oxacarbenium ion-like transition state conformation. Neu5Ac2en (4) has been
demonstrated to be a fairly good inhibitor of influenza sialidase with an inhibition
constant in the range of 1 0-5 to 1 0-6 M."
A derivative of this first inhibitor is 4-deoxy-4-guanidino-NeuSAc2en, also
known as Zanamivir (5). 51,52,53 Like Neu5Ac2en, it has a double bond between C2 and
C3 thereby mimicking the transition state structure of the sialidase-catalyzed reaction.
Unlike Neu4Ac2en, it contains a guanidino group at C4, which interacts with a
negatively charged pocket on the influenza sialidase active site.54
Figure 11 : Influenza sialidase inhibitors.
H O ~ C o 2 H AcHN Ho* AcHN C02H fDco2R
HO
H N p N H AcHN Neu5Ac2en 4 - - - - H2N NH2
Zanamivir 5 R = CH2CH3 Oseltamivir phosphate
NHAc
(GS 4071) 6 R = H GS4101
- - Peramivir (BCX- 1 8 12) 7 - -
OH
Target Compound 8

The most recent influenza drug on the market is Oseltamivir phosphate (GS 4104)
(6), which is the ethyl ester of the active component, GS 4071 .55956 The major difference
between Zanamivir and GS4 10 1 is that GS4 10 1 has an 0-isopentyl group instead of a
glycerol side chain at C6. This lipophilic side group enables interactions with several
hydrophobic residues that line the binding pocket.55
Another influenza sialidase inhibitor, Peramivir (or BCX-18 12) (7), was in phase
I11 clinical trials when its development was halted.57 Like Zanarnivir, this inhibitor
contains a guanidino group and like Oseltamivir phosphate, it contains an 0-isopentyl
group. The interesting difference between Peramivir and the other drugs on the market is
that it contains a 5-membered carbocyclic ring instead of the usual 6-membered ring.
This suggests that it is the three-dimensional display of the functional groups that interact
with the active site that determines the potency of the inhibit~r.~' Another example of a
five membered ring compound that positions substituents similarly to a six-membered
ring is the compound (+)-mannostatin A (10) (Figure 12), which superimposes well on
the mannosyl cation (9) model. This result was used to explain why (+)-mannostatin A is
a potent inhibitor of the mannosidase
Figure 12: Mannosidase inhibitors.
mannosyl cation 9 (+)-mannostatin A 10

Research Objective
As strains of influenza that have become resistant to treatment with Zanamivir
and Oseltamivir phosphate,60 and Zanamivir displays poor bioavailability and must be
administered as a spray directly to the lungs," new inhibitors which are orally active
must be synthesized. The target compound (8) designed for this research proposal is
pictured in Figure 11. Similar to (34104, a lipophilic O-isopentyl group replaces the
glycerol side chain. In contrast to both Zanamivir and GS4104, the target compound will
have an amino group at C2 rather than a double bond. In this way the positive charge of
the oxacarbenium ion transition state is mimicked. In addition, the positive charge allows
for charge-charge interactions with carboxylate residues in the active site thus
presumably strengthening its bioactivity. Study towards the synthesis of compound 8 is
described in Chapter three.

CHAPTER TWO: ENZYMATIC AND SPONTANEOUS SOLVOLYSIS OF AN a-D-N-ACETYLNEURAMINYL PYRIDINIUM
ZWITTERION
Overview
This chapter presents experiments that probe the mechanism of the spontaneous
solvolysis of 3,4-dihydro-2H-pyrano[3,2-clpyridinium N-acetylneuraminide (DHP
NeuAc) (3), as well as the enzyme-catalyzed hydrolysis of this compound. The enzyrne-
catalyzed reactions are divided into two sections: the first section involves hydrolysis of
a series of pyridinium N-acetylneuraminides catalyzed by several neuraminidases while
the second section focuses on the hydrolysis of DHP NeuAc catalyzed by the
neuraminidase, and two active site mutants, from Micromonospora viridifaciens.
Figure 13: 3,4-Dihydro-2H-pyrano[3,2-clpyridinium N-acetylneuraminide (DHP NeuAc).
Spun tan eous Solvolysis of 3,4-dihydro-2H-pyrano(3,Z-c] pyridinium N- acetylneuraminide
Results
The synthesis of 3,4-dihydro-2H-pyrano[3,2-c] pyridine (DHP) was carried out
according to the procedure outlined by Sliwa and Cordonnier (Scheme I ) . ~ ' A mixture of

morpholine and 1 -benzyl-4-piperidone was heated at reflux to give after distillation
enamine 11 in 89% yield. This material was used without further purification, as the 'H
NMR spectrum agreed with that reported in literat~re.~' The enamine was then
condensed with ethyl acrylate to yield a mixture of two isomeric products 12 and 13 in a
total yield of 94%. Full analysis of the 'H NMR was not attempted, however, there was
an approximate 2: 1 mixture of isomers as seen by the integration of the two separate CH3
ethyl signals. Reduction of the esters 12 and 13, followed by hydrolysis of the enamine
gave the hemiketal 14 which was dehydrated to give 15. Heating this cyclic alkene in the
presence of palladium caused simultaneous aromatization and benzyl protecting group
removal to give the target pyridine 16 in 52% yield. The pKa of its conjugate acid was
determined to be 7.18 * 0.02 by fitting the absorbance (245 nm) versus pH (shown in
Figure 14) to a standard titration equation.
Scheme 1: Synthesis of 3,4-dihydro-2H-pyrano[3,2-c] pyridine.6'
Ph Ph
Ethyl acrylate
Ethanol
89 % Reflux
I 94 %
H 11
4
1. LlAlH,, ether enNa Ph - phnNm X y z f ' d K * 2. H20, H', reflux 2
63% 52% OH 8

Figure 14: Plot of absorbance versus wavelength for pK, determination of DHPH'.
I I I
220 240 260 Wavelength (nm)
Sialyl chloride 18 was synthesized as outlined by Roy and Laferriere and this
route is shown in Scheme 2.62 In detail, the carboxylic acid group was protected as its
methyl ester (17) in 77% yield. Then all of the alcohol groups were protected as acetates,
but under these acid conditions (AcCl, AcOH) the newly formed anomeric acetate is
substituted by a chloride ion to give 18 in 96% yield.

Scheme 2: Synthesis of sialyl chloride 1 8 . ~ ~
AcO C02Me
AcHN AcO 18
Having the pyridine derivative (16) and the sialyl chloride (18) in hand, the
coupling reaction between these compounds was accomplished according to the
procedure developed by Chou et al. (Scheme 3).21 Compound 19 was then deprotected to
yield the target compound N-[(5-acetamido-3,5-dideoxy-~-glyrero-a-~-galacto-non-2-
u l o p y r a n o s y l ) o n a t e ] - 3 ' , 4 ' - d i h y d r o - 2 H - p ~ d i n i u m (DHP NeuAc) (3) in
25% yield.
Scheme 3: Coupling between 18 and 16, and deprotection to yield DHP NeuAc (3).
Y'
AcO +$ Me +
AcHN AcO 18
room temp molecular sieves ACO
44% AcHN
AcO 19
1) NaOMe, MeOH 1

Rate constants for the spontaneous solvolysis of DHP NeuAc in a methanollwater
mixture were then determined and this data is summarized below (Table 1).
Table 1: Observed rate constants for the solvolysis of DHP NeuAc (3) in a methanollwater mixture (vlv), T = 65 OC.'
% MeOH lo5 x k,b, (s-l)
a Mean value of three runs; quoted error = o,-, .
In order to compare the rates of hydrolysis of compound 3 with those of
compound 20 (Figure 1 5),63 the reactions were carried out in the absence of added salts
so that the ionic strength was close to zero. A plot of log(kobs) for compound 3 versus the
log(kobs) for compound 20 is shown in Figure 16. The slope of the best fit linear line is
0.71 rt 0.02, thus indicating that as the polarity of the solvent is reduced (increase in
MeOH content) a smaller rate acceleration occurs for solvolysis of 3 than that reported
for 2 0 . ~ ~

Figure 15: DHP NeuAc (3) and P-D-glucopyranosyl 4'-bromoisoquinolinium tetrafluoroborate salt (20).
HO 4 i r n AcHN / H0 &b HO p\
HO 3
/ 0 20
Figure 16: Plot of 10g(kobr)3 versus log(kobs)20 for the aqueous solvolyses of 3 and 20 at T = 65OC. The kinetic data for compound 20 are taken from reference 62. The line shown is the best linear least-squares fit through the data points.
-
slope = 0.71 + 0.02
The product studies for the solvolysis of 3 were carried out in the presence of the
sterically hindered base, N-methylmorpholine, to ensure stability of the acid-labile
products. Listed in Table 2 are the observed products (see Figure 17 for structures)
formed during the reactions of 3 in 50% and 100% aqueous methanol at 65 OC.

Table 2: Observed products formed during the reactions of compound 3 in aqueous methanol (vlv) at 65
MeOH % Glycal GlyOH a-GlyOMe P-GlyOMe
" All solvents contained 3 mole equivalents of N-methylmorpholine. Percentages do not necessarily add up to 100% because of rounding. Not detected; estimated that less than 5% methyl a-D-N-acetylneuraminide (22) was formed. [H20]/[MeOH] = 2.25 in this solvent mixture.
" Integral for p-D-sialic acid corrected assuming that 7% of the total sialic acid present was in the a-form.
Figure 17: Products of DHP NeuAc solvolysis.
HO
HO C02H AcHN
HO AcHN
HO
GlyOH 22
OH
OH
HO C02H
AcHN AcHN
Discussion
The aim of these experiments was to determine whether solvolysis of zwitterions
such as compound 3 occurs with (pathway a, Scheme 4) or without (pathway b, Scheme
4) intramolecular assistance by the anomeric carboxylate.

Scheme 4: Possible pathways for DHP NeuAc solvolysis.
HO
AcHN
pathway a / pathway b \ HO
AcHN HO
It has been reported that pyridinium glycosides hydrolyze via an DN * AN (SN1)
mechanism in which the transition state involves a significant amount of C-N bond
~ l e a v a ~ e . ~ ~ - ~ ~ Therefore, the expected effect on reaction rate as the solvent polarity is
decreased is different for pathway a and b. In pathway a, the carboxylate forms a
transient a-lactone, displacing the positively charged pyridinium moiety in an
intramolecular ANDN (SN2) reaction. This pathway is analogous to a type 111 SN2 reaction
where an anionic nucleophile attacks a positively-charged molecule and the charge at the
transition state is much less than that of the starting materials. Therefore, a large rate
increase is expected with decrease in the solvent's polarity. In contrast, pathway b
proceeds with the positive charge on the nitrogen being delocalized in the transition state.
As a result, a more modest increase in rate of solvolysis is expected with decreasing
solvent polarity.
The rates of solvolysis of DHP NeuAc are less sensitive to decreasing the solvent
polarity than are those of compound 20, which lacks the anomeric carboxylate group.

Thus, indicating that the carboxylate group is not nucleophilically participating during
departure of the neutral leaving group.
Product studies provided further evidence for the absence of intramolecular
nucleophilic participation of the carboxylate group. If the reaction proceeded via
pathway a, we would expect only the a-anomer of methyl D-N-acetylneuraminide (23) as
a product. However, only P-methyl D-N-acetylneuraminide (24) was observed.
These results are in contrast to the 1 : 1 mixture of a:P methyl D-N-
acetylneuraminides obtained upon solvolysis of compound 2 in 20% v/v MeOH:H20
(Figure 1 8).66367 These two results indicate that compounds 3 and 2 cannot be solvolyzed
via the same intermediate, a solvent-equilibrated oxacarbenium ion 26 (Figure 19).
These results can be resolved, however, when one takes into account that the DHP
leaving group departs as a neutral molecule whereas the cytidine monophosphate leaves
as a negatively charged entity. Previous work on glucopyranosyl fluorides (which also
have negatively charged leaving groups) has shown that they hydrolyze via an ANDN
me~hanism'"'~ whereas a-glucopyranosyl pyridinium salts hydrolyze via a DN * AN
m e ~ h a n i s m . ~ ~ ' ~ ~

Figure 18: CMP-N-acetylneuraminate (2).
The nucleophilic selectivity (kMeOHikHOH) of DHP NeuAc (3) in 50% viv
MeOH:H20 is 1 S8, as calculated by Equation 1.
Equation 1
~ M ~ O H [Gly - OMe] [HOH] -= ~ H O H [Gly - OH] [MeOH]
This value, which is larger than the corresponding nucleophilic selectivity for
compound 20 in the same solvent (kMeOHIkHoH = 1.07) ,~~ is consistent with the cationic
intermediate 25 having a longer lifetime than that of intermediate 26 (Figure 19). The
reported lifetimes for cations 2514 and 2666 in water are 1.0 * lo-" and 2 3 x lo-'' s
respectively.
Figure 19: Glucopyranosyl cation (25) and N-acetylneuraminyl cation (26).

Finally, the product studies also revealed that in 100% MeOH, glycal21 was also
formed. This likely occurs with proton-abstraction by the pyridine leaving group in a
mechanism similar to that suggested by Thibblin and Saeki for the solvolysis of 1-(1-
methyl- 1 -phenylethyl)pyridiniurn cations (Figure 2 0 ) . ~ ~
Figure 20: Glycal formation via proton abstraction by the pyridine leaving group.
Experimental
General Materials and Methods
Methanol was dried by distillation from its magnesium alkoxide salt. Deionized
water was further purified by use of a "Milli-Q ultra pure water" system. NMR spectra
were acquired at operating frequencies of 400 and 100 MHz for 'H and 13c NMR,
respectively, using either CDC13 or DzO as the solvent and internal reference. Coupling
constants are reported in Hertz. The two anomeric methyl N-acetylneuraminides (21 and
22) were purchased from Toronto Research Chemicals Inc. (TRC), and N-
acetylneuraminic acid was purchased from Rose Chemicals. All other chemicals and
reagents were purchased from Sigma-Aldrich.
Morpholine (46.0 g, 0.528 mol) was added to a solution of 1-benzyl-4-piperidone
(50.0 g, 0.264 mol) in dry toluene (160 mL) and the mixture was heated at reflux for 12
3 0

hours in a flask fitted with a Dean-Stark apparatus. After cooling, the solution was
concentrated and the orange syrup distilled, yielding the enamine in 89 % yield which
was used without further purification. b.p. 170-175 OC10.4 ton, lit. b.p. 154-1 57 OC10.2
4-(l-Benzyl-3-carbethoxyethyl-1,2,3,6-tetrahydro-4-pyridyI)-morpholine (12 and 13)
The morpholine enamine 11 (3 1.5 g, 0.122 mol) and ethyl acrylate (1 3.5 g, 0.135
mol) were heated at reflux in absolute ethanol (70 mL) for 20 hours with protection from
moisture. The solvent was then evaporated and the resulting material was distilled to
give the desired ester (41.5 g) in 94 % yield. b.p. 200-210 "C10.3 torr, lit. 189-196 OC10.2
ton"'. 'H NMR (CDCI,): F 1.22-1.26 (6H, two triplets, CH3 of ethyl group, ratio of
isomers approximately 2: 1).
6-Benzyl-8a-hydroxy-3,4,4a,5,6,7,8,8a-octahydro-2H-pyrano-[3,2-c] pyridine (14)
A solution of the enamine ester (43.4 g, 0.12 mol) in ether (1 30 mL) was added
dropwise to a suspension of LiA1H4 (4.6 g, 0.12 mol) in dry ether (1 30 mL) at 0 OC. The
suspension was then heated at reflux for 4 hours, cooled, and absolute ethanol was added
slowly (1 6 mL) followed by 20 % aq H2S04. The solution was heated at 60 OC for 4
hours, then cooled and allowed to stir overnight. The solution was then made basic by
the addition of aq NaOH, and filtered. The resultant white precipitate was placed in a
continuous extraction apparatus and extracted with CHC13. The combined filtrate and
chloroform extract was evaporated to give the crude hemiketal as a yellow solid.
Recrystallization from toluene afforded the product as a white solid in 63 % yield. m.p.
133-134 OC, lit. 135 '(2"'. 'H NMR (CDCl3): 6 1.60-2.33 (8H, m), 2.51-2.55 (lH, m, 3-

CH2,), 2.79-2.84 (IH, m, 3-CH2t,), 3.63-3.67 (1 H, my 2-CH2,), 3.99-4.06 (IH, m, 2-CH2t,),
3.52 (2H, s, CH2-Ph), 7.29-7.32 (5H, m, ArH).
6-Benzyl-3,4,5,6,7,8-hexahydro-2H-pyrano[3,2-] pyridine (15)
p-Toluenesulphonic acid (2.00 g, 11.6 mmol) was added to a solution of the
hemiketal 14 (16.7 g, 67.5 mmol) in toluene (300 mL). The solution was heated at reflux
overnight while water was removed using a Dean-Stark apparatus. The resultant solution
was cooled, and basified with solid potassium carbonate, it was then filtered and the
toluene evaporated under reduced pressure. The resulting yellow liquid was distilled to
produce a clear colorless oil (14.3 g) in 93 % yield. b.p. 135-140 OCI0.2 ton, lit. 123-125
'C/O. 1 torr61 'H NMR (CDC13): F 1.83-1.86 (4H, m, 7-CH2, 5-CH2), 2.12-2.15 (2H, m,
8-CH2), 2.59 (2H, t, J3,2 = 5.8 HZ, 3-CHz), 2.85 (2H, S, 4-CH2), 3.59 (2H, S, CH2-Ph),
3.93-3.96 (2H, m, 2-CH2), 7.27-7.37 (5H, m, Ar).
3,4-Dihydro-2H-pyrano[3,2-clpyridine (16)
To a stirred solution of 15 (1 1.8 g, 52.0 mmol) in dry xylene (100 mL) was added
10% Pd/C (1.3 g). The stirred suspension was heated at reflux for 48 hours before it was
filtered and evaporated to yield a yellow oil. The crude product was then distilled b.p.
50-52 0C10.4 ton, lit 73-74 OCIO.8 ton? to yield a colorless liquid (3.7 g) in 52 % yield.
'H NMR (D20): t j 2.07 (2H, my 2-CH2), 2.88 (2H, t, J4,3 = 6.5 Hz, 4-CHz), 4.48 (2H, t,
J2 ,3 = 5.2 HZ, 2-CH2), 7.21 (lH, d, J8,7 = 6.7 HZ, H-8), 8.25 (IH, d, J7,8 = 6.7 HZ, H-7),
8.32 (1 H, s, H-5). Anal. Calcd for C8HloNO: C, 55.99; H, 5.87; N, 8.16. Found: C,
55.85; H, 5.83; N, 8.12.

Methyl (5-acetamido-3,5-dideoxy-~-glycero-~-~-ga~acto-non-2-ulopyranos~l)0~~t~
(17)
A suspension of sialic acid (3.0 g, 9.7 mmol) and Amberlite (H') resin (2.0 g) in
dry methanol (50 mL) was stirred for 25 hours at room temperature. The mixture was
then filtered and the filtrate concentrated to give a white solid. The solid was then
recrystallized from methanollether to give the desired methyl ester (2.5 g) in 77 % yield.
mp 185-1 87 OC, corrected 190-192 OC, lit 62370171 193.5-194.7 OC 'H NMR (D20): 6 1.90
(lH, t, J3a, 3e + J3a, 4 = 24.7 HZ, H-3a), 2.03 (3H, S, CH3), 2.29 (lH, dd, J3e, 3a= 13.1, J3e, 4 =
4.9Hz, H-3e), 3.53 (lH, dd, J7,8= 13.1, J7,6=0.9Hz, H-7), 3.59 (lH, dd, Jsa,9b= 11.9,
Jg,8=6.4Hz, H-9a), 3.71 (IH, ddd, J8,9a=6.1,J8,9b=2.7, J8,7=9.2 HZ, H-8), 3.80-3.83
(4H, m, H-9b, 0CH3), 3 .go (1 H, t, J5, 4 + J5, = 20.4 Hz, H-5), 4.01 -4.07 (2H, m, H-4, H-
7).
Methyl (5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-~-glycero-~-~-ga1acto-non- 2-ulopyranosy1)-onate chloride (18)
Methanol (7.5 mL, 185 mmol) was added dropwise with stirring to a flask
containing cold (ice-water bath) acetyl chloride (36.8 mL, 5 18 mrnol). Acetic acid (14.7
mL) and compound 17 (2.5 g, 7.6 mmol) were then added and the solution stirred under
nitrogen for 48 hours at room temperature. The solution was concentrated and then co-
evaporated with toluene three times to yield a white foam (3.7 g) which was used
without purification, in 96 % yield. 'H NMR (CDC13): 6 2.04-2.14 (15H, m, CH3), 2.28
(lH, t, J3a, 3e + J3a, 4= 25.0 HZ, H-3a), 2.78 (lH, t, J3e, 3a= 14.0, J3e, 4= 4.9 HZ, H-3e), 3.88
(3H, s, OCH3), 4.06 (lH, dd, J9,9b= 12.5, J9 ,8= 5.5 Hz, H-9a), 4.20 (lH, q, J5,4= J5,6=
J ~ , N H = 10.4 HZ, H-5), 4.34 (lH, dd, J6,5= 11.0, J6,7=2.3 HZ, H-6), 4.41 (lH, dd, Jgb,9a=

12.5, J9b,8=2.7 Hz,H-gb), 5.18 (IH, ddd, J8,ga=5.8, J8,9b=2.7, Jg,7=9.8 HZ, H-8),
5.29-2.48 (2H, my H-4, H-7).
N-[Methyl(5-acetamido-4,7,8,9-tetra-O-acety1-3,5-dideoxy-D-glycero-a-D-galacto- non-2-ulopyranosyl)onate]-3 ',4'-dihydro-2 'H-pyrano[3',2'-clpyridinium tetrafluoroborate (19)
The fully protected N-acetylneuraminyl chloride 18 (1 .OO g, 1.96 mmol) was
added to a suspension of dried 4 A molecular sieves (3.0 g) in anhydrous THF (40 mL).
This mixture was stirred under a nitrogen atmosphere for 10 min then 3,4-dihydro-2H-
pyrano[3,2-clpyridine (6.63 g, 49.0 rnmol) was added and the reaction mixture was
cooled using an icelwater bath in the dark. Subsequently, silver tetrafluoroborate (0.05 g,
2.56 mrnol) was added to the stirred solution which was then allowed to warm to room
temperature, and the reaction was kept in the dark at this temperature for 60 hr. The
resulting solution was concentrated and 40 ml MeOH was added. The mixture was then
sonicated for 10 min, filtered, and the solvent evaporated under reduced pressure. The
crude product was purified by flash chromatograpy (silica gel, MeOH:CH2C12 1 : 10) to
yield a white solid (0.56 g, 44%): Rf0.24 MeOH:CH2C12 1:7 'H NMR (CDC13) 6: 1.89
(4 H, m, CH3, H-3a), 2.01 (3 H, s, CH3), 2.05 (3 H, s, CH3), 2.12 (5 H, m, CH3, H-7'),
2.27 (3 H, S, CH3), 3.00 (2 H, m, H-8'), 3.48 (1 H, dd, J3a,3e = 14.0 HZ, J3e,4 = 7.0 HZ, H-
3e), 4.04 (1 H, dd, J9a,9b = 12.5 HZ, J 9 , 8 = 5.8 HZ, H-9a), 4.21 -4.29 (2 H, m, H-5, H-9b),
4.49 (2 H, t, J6',7* = 5.2 HZ, H-6'), 5.00 (1 H, dd, J6,5 = 11.4 HZ, J6,~ = 3.1 HZ, H-6), 5.42
(1 H, dd, J4,3e = 7.0 HZ, J4,3a = 2.6 HZ, H-4), 5.52 (2 H, m, H-8, H-7), 7.1 1 (1 H, d, J4',3' =
7.4 Hz, H-47, 8.62 (1 H, d, J39,47 = 7.4 HZ, H-37, 8.83 (1 H, s, H-1') 13c NMR (CDC13)
6: 21.5(C-7'), 22.0 (C-8'), 23.1 (CH3), 37.9 (C-3), 48.7 (C-5), 63.1 (C-9), 67.0 + 67.1
(C-7, C-8), 69.4 (C-67, 70.2 (C-4), 73.5 (C-6), 94.1 (C-2), 114.0 (C-47, 121.8 (Ar),
34

139.2 (C-l'), 142.0 (C-3'), 165.7, 167.4, 169.9, 170.0, 170.8, 171.1, 171.6, (C-1, C=O,
Ar).
N-[(5-Acetamido-3,5-dideoxy-D-glycero-a-D-galacto-non-2-ulopyranosyl)onate]- 3',4'-dihydro-2'H-pyrano[3',4'-c)pyridinium. (3)
The protected DHP NeuAc (19) (150 mg, 0.24 mmol) was dissolved in dry
MeOH (8 mL). The solution was stirred in an ice bath and methanok sodium methoxide
was added (3 equivalents, 2.5 mL). AAer stirring for 20 minutes at 0 OC, the solution was
neutralized with Rexyn 101 H' resin, filtered, and washed with MeOH. The product was
concentrated under vacuum without heating and then dissolved in 0.1 M aq. NaOH (5
mL) and stirred for 30 min at 0 OC. The solution was then neutralized with Rexyn 101 H'
resin, filtered, and lyophilized. Purification by reverse-phase HPLC (mobile phase; 5%
aqueous MeOH containing 1 % AcOH (vlv) on a C- 18 column) gave 3 as a white solid in
25% yield. 'H NMR (D2O) 6: 1.89 (1 H, t, J3a,3e + J3a,4 = 22.9 HZ, H-3a), 2.07 (2 H, rn,
H-7'), 2.90 (2 H, t, J7',8' = 6.4 HZ, H-8'), 3.23 (1 H, dd, J3e,3a = 12.2 HZ, J3e,4 = 3.9 HZ, H-
3e) 3.62-3.67 (2 H, m, H-7, H-9a), 3.84-4.04 (5 H, m, H-4, H-5, H-6, H-8, H-9b), 4.5 1 (1
H, t, J67,7~ = 5.1 HZ, H-6'), 7.24 (1 H, d, J3*,49 = 7.4 HZ, H-4'), 8.57 (1 H, dd, J3',1' = 2.0
HZ, J3',4' = 7.4 HZ, H-37, 8.66 (1 H, S, J17,31 = 2.0 Hz, H-1') 13c NMR (D20) 6: 22.2 (C-
7'), 24.2 (C-8'), 24.7 (CH3), 43.1 (C-3), 53.8 (C-5), 65.4 (C-9), 72.6 (C-6'), 70.6 (C-7),
70.7 (C-4), 73.7 (C-8), 77.2 (C-6), 96.5 (C-2), 117.5 (C-4') 126.1 (Ar), 140.7 (C-1 '),
142.4 (C-3'), 171.4 (C-I), 172.3 (Ar), 177.7 (C=O). HRMS (ESI) calcd for Ci9H26N209
[M + H'] 427.1717, found 427.1717.

Kinetics
The solvolysis reactions of compound 3 were conducted at 65 OC, and were
monitored by following the decrease in absorbance at 266 nrn using a Cary 3E UV-Vis
spectrophotometer equipped with the Cary six-cell Peltier constant-temperature
accessory. Reactions were initiated by the injection of a stock solution of compound 3
(1 0 pL, 3 mM) into a waterlmethanol mixture (1 .OO mL) containing N-methylmorpholine
(3 equiv). Rate contants were calculated by non-linear least squares regression of the
absorbance versus time data to a standard first-order rate equation using the computer
program GraFit.
Product Studies
The solvolytic product studies were performed by heating in a sealed vial that
contained a solution of 3 (1.05-1.12 mg) and N-methylmorpholine (3 equiv) in a binary
solvent mixture (1 0 mL) for 7 x tXhyd at 65 OC. After cooling, the reaction vials were
opened, the solvent removed under reduced pressure followed by drying under vacuum
(-0.01 mm Hg) for 48 hrs to afford solid residues that were dissolved in D20 (0.5 mL)
and their 'H NMR spectra were acquired. The products formed were determined by
comparing the newly acquired NMR spectra with those of the two known methyl
sialosides 23 and 24, sialic acid 22, and the glycal21.

Hydrolysis of Pyridinium a-D-N-Acetylneuraminides Catalyzed by Bacterial and Viral Sialidases
Results
The Michaelis-Menten parameters for the enzyme-catalyzed hydrolysis of DHP-
NeuAc by sialidases from Vibrio cholerae (V. cholerae), Clostridium perfringens (C,
perfringens), Salmonella typhimurium (S. typhimurium), and Newcastle disease virus
(NDV) are listed in Table 3. In the standard Michaelis-Menten expression, kcat is the first
order rate constant for the enzyme catalyzed-hydrolysis when [S]>>K,. The term kcatlKm
is the second-order rate constant for the reaction of enzyme and substrate under the
condition [S]<<K,. As the reaction rates for the NDV sialidase-catalyzed hydrolysis of
4-methyl and 3,4-dimethyl pyridinium N-acetylneuraminides were slow, (kcat values of
-1 37 <0.001 s ), kcat values were not measured for the NDV sialidase-catalyzed hydrolysis of
DHP NeuAc.
Table 3: Michaelis-Menten parameters for the sialidase-catalyzed hydrolysis of DHP NeuAc (3) at 37 O C .
V. cholerae 0.150 k 0.014 0.024 1.6 x lo2
C.perfringens 0.224k0.030 5.4 2.4 x lo4
S. typhimurium 6.7 k 2.9 5.8 8.6 x lo2
NDV 0.058 k 0.012 N D ~ N D ~
" Errors are estimated at * 20%. ~ o t determined.

The PI, values on the kinetic parameters k,,, and kc,/Km for the sialidase-catalyzed
hydrolyses of the pyridinium a-D-N-acetylneuraminides are shown in Table 4. The
structures of the pyridinium a-D-N-acetylneuraminides are shown in Figure 2 1. The
kinetic data for compounds 27-30 with % cholerae, C. perfringens and S. typhimurium
sialidases were measured by J.N. Watson and the data for these materials with the NDV
sialidase was measured by J.H. hen.^^
Table 4: Calculated PI, values for kcat and kcatlKm on the siladase-catalyzed hydrolyses of pyridinium a-D-N-acetylneuraminides at 37 O C . ' ~
Sialidase kcat kcat/Km
C. pevfringens -0.32 h 0.19 -0.27 h 0.19
S. typhimuvium -1-0.03 k 0.27 -0.12 k 0.23
V. cholerae -0.89 * 0.25 -0.70 * 0.12
Figure 21: Pyridinium a-D-N-acetylneuraminides.
HO
AcHN
Discussion
The viral neuraminidase from Newcastle disease has two other functions besides
hydrolysis of sialic acid. First, it acts as a hemagglutinin receptor for glycoconjugates,
3 8

binding to sialic acid displayed by host cells. Second, it undergoes a conformational
change that activates a fusion protein, thereby promoting fusion of the virus particle to
host cells.72 In the present study, only the parent pyridinium sialoside 27, was
hydrolyzed by the NDV neuraminidase at a rate significantly higher than background (kcat
of 2.95 s - ' ) . ~ ~ However, DHP NeuAc (3) bound in the active site of this enzyme and
competitively inhibited the enzymatic hydrolysis ofp-nitrophenyl NeuAc with a
dissociation constant (K,) of 58 pM. Recent crystallographic studies by Crennell et al.
have suggested that a "conformational switch" in the NDV neuraminidase active site is
necessary for hydrolysis of substrates to occur.73 It follows then that the unnatural
pyridinium N-acetylneuraminides are binding to the enzyme active site, but perhaps are
which would allow them to be hydrolyzed.
Conformational change has also been proposed to be the rate-limiting step for
influenza type A viral sialidase-catalyzed hydrolysis of the pyridinium N-
a~et~lneuraminides.~~ However, it cannot be inferred that these viral sialidases operate
via a common mechanism. In fact it has been reported that the influenza type A and
NDV viral neuraminidases show different substrate specificities and kinetic
properties.74'75
Corfield et al. have observed that the bacterial neuraminidases also have differing
substrate spe~ificities.~~ Indeed, a similar observation was made in the present study
where DHP NeuAc bound much tighter to the V. cholerae and C. pevfringens enzymes
than it did to the S. typhimurium enzyme.

Taking a closer look at the V. cholerae neurarninidase, the PI, values were -0.89 &
0.25 and -0.70 & 0.12 for kcat and kc,&,, respectively. These large negative values seem
to indicate that breakage of the sialyl-aglycon bond is rate-limiting; however, both plots
appear to curve concave upwards which is normally associated with a change in
mechanism (Figure 22). This would mean that the parent pyridinium NeuAc (27) reacts
via a SN1 mechanism while the 3-methyl (28), the 4-methyl (29), and the 3,4-dimethyl
pyridinium sialoside (30), and DHP NeuAc (3) react via a S N ~ mechanism. If this is the
case, it is expected that the difference between the rates of hydrolysis for compounds 27
and 3 would be less for the enzyme-accelerated reaction that for the spontaneous reaction.
However, the opposite was found where the difference in log (kcat) values for compounds
27 and 3 was greater than the corresponding value for the spontaneous rates of hydrolysis
for these compounds.21 Therefore this curvature in the Brsnsted plot is attributed to a
slowing down of the required conformational change because of steric interactions of the
pyridine substituents with residues in the active site.17 Nevertheless, since the slope of
the Brsnsted plot is negative, conformational change appears to be concerted with C-N
bond cleavage in the rate-determining step. In comparison, Guo and Sinnott also studied
the V. cholerae neuraminidase catalyzed-hydrolysis but withp-nitrophenyl N-
acetylneuraminic acid as a substrate. They concluded that the rate-determining step was
solely cleavage of the C - 0 aryl bondV3'

Figure 22: Plot of log(kcat) versus p ~ , ( ~ - ~ ' ) for the NDV sialidase-catalyzed hydrolysis of compounds 27-30 and 3.37
In contrast to the large negative P1, values for the I? cholerae enzyme, those for S.
typhimurium were indistinguishable from zero. This corresponds to a rate-limiting
conformational change of the enzyme-pyridinium N-acetylneuraminide complex rather
than the cleavage of the C-N bond. Guo et al. concluded from their PI, value of -0.80 that
cleavage of the C-0 bond was rate-limiting in S. typhimurium neuraminidase-catalyzed
hydrolysis of aryl N-acetylneuraminic acids.34 They also postulated that hydrolysis
occurred via a transition state in which the N-acetylneuraminic acid moiety is in a *c5-
chair conformation and that little proton donation to the leaving group had occurred.
Thus it appears that in the case of the less conformationally flexible substrates the
conformational change has been slowed down sufficiently that it is now rate-determining.
The chemical step was also reported to be rate-limiting for hydrolysis of a(2-3)
sialyl-lactose by the C. perfringens neuraminida~e.~~ This also seems to be the case for

the catalyzed hydrolysis of the pyridinium sialosides, as PI, values of -0.32 k 0.19 and
-0.27 h 0.19 were obtained for kcat and kcatlKmrespectively. However these Pig values are
associated with large errors and therefore it is difficult to make any firmer conclusions
from this data.
Experimental
Materials. Buffers and reagents were purchased from Fisher Scientific and Sigma
Chemical Co. The Clostridium perfringens (A99 strain) and Salmonella typhimurium
(LT2 strain) sialidases were purchased from Sigma Chemical Co, while the Vibrio
cholerae (395 strain) and NDV (Hitchner B1 strain) enzymes were purchased from
Calbiochem. Sialic acid was purchased from Rose Scientific andp-nitrophenyl a-D-N-
acetylneuraminide was synthesized by V. Dookhun according to literature
procedures.70~77 The four pyridinium N-acetylneuraminides (27)- (30) were synthesized
by Dr. D.T.H. chou21 and the DHP NeuAc was synthesized as described earlier in this
thesis.
Enzyme kinetics. The kinetic measurements for DHP NeuAc will be described
presently; measurements for the other pyridinium D-N-acetylneuraminides were
performed by Watson and Chen in a similar manner.37278 Sialidase concentrations were
standardized usingp-nitrophenyl a-D-N-acetylneuraminide (pNP NeuAc) in order to
obtain relative kcat values for the DHP NeuAc in the manner reported by Chou et aL2'
The buffers used for the kinetic measurements were as follows: 50 mM sodium acetate,
pH 5.5 for the Newcastle disease virus sialidase; 50 mM sodium acetate, pH 5.1 for the
C. pevfringens sialidase; 50 mM sodium acetate, pH 5.5, 100 mM NaCl for the S.
typhimurium sialidase; and 50 mM sodium acetate, pH 5.5, 150 mM NaC1, 9 mM CaC12
42

for the V. cholevae sialidase. Initial rates were measured at 37 OC by monitoring the
production ofp-nitrophenol at 340 nm for 10 minutes using a Varian Cary 3E UV-vis
spectrophotometer equipped with a Peltier temperature controller. Each 400 pL reaction
was initiated by addition of an enzyme stock solution (50 pL) to a pre-equilibrated
solution of substrate and buffer. Sialidase-catalyzed rates of hydrolysis of DHP NeuAc
were low therefore apparent binding constants (K,) were determined. Five initial rates of
hydrolysis of pNP NeuAc were monitored in the presence of four different concentrations
of DHP NeuAc 3 (0-2 mM) and the kinetic data fit to a competitive binding model using
a nonlinear least-squares fitting algorithm (Grafit). A single point rate determination for
the enzyme-catalyzed hydrolysis of DHP NeuAc, was utilized in order to estimate V,,,
(background hydrolysis was negligible). In these experiments a DHP NeuAc solution (2
mM) was mixed with twenty times the amount of sialidase used in the competitive
binding assays. The so-measured V values were used to estimate V,,, values which were
then substituted into the Michaelis-Menten equation along with the previously
determined Km values and enzyme concentrations to determine kcat values. Extinction
coefficient differences were determined in each buffer by reading the initial absorbance
of DHP NeuAc 3 and the final absorbance of the hydrolysis products.

Hydrolysis of DHP NeuAc Catalyzed by the Micromonospora viridifaciens Neuraminidase
Results
The kinetic parameters for the hydrolysis of two substrates: p-nitrophenyl (pNP)
NeuAc and DHP NeuAc (Figure 23) catalyzed by the wild-type (wt) sialidase from
Micromonospora viridifaciens (M. viridifaciens) and two active-site mutants, the D92G,
and the Y370G mutant are listed below (Table 5). The kcatlKm and catalytic proficiency
values for hydrolysis of DHP NeuAc catalyzed by the Y370G mutant are upper limits as
less than 1 % hydrolysis of a 0.1 mM solution was observed over a period of 1 1 hours.
Figure 23: Substrates for M. viridifaciens neuraminidase-catalyzed hydrolysis.
H O
AcHN AcHN H O HO
p-nitrophenol NeuAc 31 DHP NeuAc 3
Table 5: Kinetic parameters for the wt, D92G, and Y370G M. viridifaciens sialidase- catalyzed hydrolysis of pNP NeuAc and DHP NeuAc 3.
pNP NeuAc DHP NeuAc
Sialidase Catalytic Catalytic a 1 1 proficiency ( ~ - 1 1 ~ kcatIKm (M-~s-') proficiency ( ~ - 1 1 ~
Y370G ( 1 . 1 * 0 . 3 ) ~ 1 0 ~ 2.2 x 10" <5b -4 x lo7
a values provided by ats son.^' 0.1 mM DHP NeuAc 3 showed <1 % hydrolysis over 12 hrs, a similar amount of pNP NeuAc was fully
hydrolyzed in 5 min using the same amount of enzyme Catalytic proficiency is defined as (kcatlK,, , ) /~,at

The extrapolated rate constant for the spontaneous hydrolysis of pNP N ~ U A C ~ ~ at
37 OC is 5.0 x 1 0-6 S-l. The rates for the uncatalyzed hydrolysis of DHP NeuAc were
measured at 65,72, and 80 "C. The measurement at 65 O C is described previously in this
chapter. The rates at 72 and 80 OC were measured in 0.3 M MES buffer at pH 6.2.
Although the rate at 65 OC was measured at a different pH, comparison to the rates at 72
and 80 OC is valid because the rates of hydrolyses of pyridinium N-acetylneuraminides
are independent of pH between 2 and 1 o . ~ ' The rate constant for spontaneous hydrolysis
-8 -1 at 37 OC was determined by extrapolation to be 8.6 x 10 s .
Table 6: Rates of hydrolysis of DHP NeuAc (3).
Temperature (OC) Rate (s-l)
The pKa values for the conjugate acids of the leaving groups (pNP and DHP-H+)
are both 7.18 for pNP NeuAc and DHP NeuAc. Therefore, because the rate of
spontaneous hydrolysis for DHP NeuAc is slower than that for pNP NeuAc, it can be
concluded that DHP is an intrinsically worse leaving group than pNP. Such differences
in rate have been previously reported in the literature where the leaving ability followed
the order ~ - > ~ ~ r - > ~ ( ~ ~ r i d i n e ) , even though the pK,'s of their conjugate acids were
similar.*' Therefore in the present study, catalytic proficiency [($aclKm)lku,,at] is used as

a measure of the degree of rate acceleration caused by the enzyme and this number
should be independent of leaving group abilities.
Dicussion
For the pNP NeuAc substrate there is a slight decrease in enzyme proficiency
when the catalytic residue aspartic acid 92 is mutated in the M. viridifaciens sialidase I (Table 5, data for wt and ~ 9 2 ~ ) . ~ ~ However, the relative drop in kcatlKm is less than a
factor of two. Such a result is consistent with there being little acid-catalysis in the
transition state for hydrolysis of activated aryl sialosides.
For the enzyme-catalyzed hydrolyses of the DHP NeuAc substrate (3), changing
the D92 residue to a glycine results in a slightly higher enzyme proficiency. It was
expected that turnover rate would be the same as the wt since this substrate does not
require acid catalysis by the aspartic acid residue. A possible explanation for the slight
increase in enzyme proficiency is that mutation of this residue creates more room for this
conforrnationally restricted substrate.
When the Y370 residue in M. viridifaciens is mutated to a glycine, a slight
decrease in kcat/& for the pNP NeuAc substrate was observed. Whereas, for the DHP
NeuAc substrate with the tyrosine mutant, kcatlKm decreased by a factor of greater than
3800 with respect to the wt. Indeed, the catalytic proficiency of the Y370G enzyme for
the pNP NeuAc substrate was greater than that for DHP NeuAc by over 4000.
In their study on three Y370 mutants of the M. viridifaciens sialidase, Watson et
al. found that the relative kcatlKm [(kcatlKm for Y370D)/( kcat/& for wt)] for a-(2,3)
sialyl-lactose (a natural substrate) was approximately 2 x 1 o - ~ while that for pNP NeuAc

was approximately 3 x 1 0-2.35 This further highlights the importance of the tyrosine in
enzyme-catalyzed hydrolysis of substrates with poor leaving groups. The reactions
catalyzed by these tyrosine mutants involved inversion of configuration at the substrate's
anomeric centre rather than retention. The authors concluded that in this case, using
activated substrates, a water molecule can efficiently act as the nucleophile to displace
the leaving group.
Kleineidam et al. reported a study that involved the mutagenesis of the proposed
general acid catalytic residue in the Clostridium perfringens (C. perfringens)
neuraminidase whereby kinetic characterization was performed using
4-methylumbelliferyl N-acetylneuraminic acid (MU NeuAc) as substrate. When the MU
NeuAc substrate was tested with two aspartic acid mutants (D100N and D 100E) of C.
perfringens, the reduction in hydrolytic activity (kcat) compared to the wt was 75- and
200-fold respectively.81 Chien et al. also carried out mutagenesis studies with the C.
perfringen sialidase and reported a complete loss of activity for hydrolysis of a-(2,3)
sialyl-lactose with the DlOOV mutant.82 Thus the need for the conserved aspartic acid
residue in catalysis varies depending on the enzyme and substrate used in the reaction.
Previous studies where the conserved tyrosine residue was mutated in the C.
perfringen sialidase were performed by Klieneidam et al. where a molecular activity of
0.005% was observed relative to the wt enzyme for hydrolysis of MU N ~ U A C . ~ '
The results from other studies on active site tyrosine mutants of sialidase
enzymes, including (i) influenza virus s i a l idase (~ /~ee /40)~~ and (ii) human membrane-
associated sialidases4 have been reported to give complete loss of enzyme activity, thus
further highlighting the importance of this residue in catalysis.

Experimental
Materials. Buffers and reagents were purchased from Fisher Scientitfic and
Sigma Chemical Co. The wt sialidase from M. viridifaciens, and theY370G and D92G
mutants were cloned, expressed and purified by Dr. J.N. ats son.^^ Sialic acid was
purchased from Rose Scientific andp-nitrophenyl a-D-N-acetylneuraminide was
synthesized by V. Dookhun according to literature procedures.70377 The DHP NeuAc (3)
was synthesized according to the procedure outlined earlier in Chapter 2 of this report.
Enzyme kinetics. The kinetic measurements were performed at 37 O C in 100 mM
sodium acetate, pH 5.25. The kinetic parameters for the sialoside-catalyzed hydrolysis of
pNP NeuAc by the wild-type and DG mutants were characterized by Dr. J.N. Watson. 78
The sialoside-catalyzed hydrolysis of pNP NeuAc by the Y370G mutant was monitored
by following changes in absorbance versus time at 340 nrn using a Cary-3E UV-vis
spectrophotometer equipped with the Cary Six-Cell Petltier constant-temperature
accessory. Hydrolysis of DHP NeuAc was monitored in a similar fashion, but using a
wavelength of 266 nrn. For all reactions, the substrate was allowed to warm to 37 OC, and
then the reaction was initiated upon addition of the enzyme (50 yL). Binding constants
(Km) and maximal rates were determined by following the rates of hydrolyses of different
concentrations of the substrates with an invariant amount of enzyme, with the exception
of DHP NeuAc (3) with the YG mutant. In the case of the Y370G mutant, less than 1%
hydrolysis of 0.1 mM DHP NeuAc was observed over 11 hours. Therefore rates of
hydrolyses of pNP NeuAc (5.0 x M) were measured in the presence of five different
concentrations of DHP NeuAc. All data was fit to the Michaelis-Menten equation using
a standard nonlinear least-squares program (Grafit) to determine Km and V,,, values; and

in the case of the Y370G mutant, an ICso was determined. From the value and the
concentration of substrate (pNP NeuAc) a binding constant was calculated, based on the
assumption that the inhibition was competitive. Extinction coefficient differences were
determined by measuring the difference in absorbance between the substrate in question
and its hydrolysis products.
Spontaneous hydrolysis kinetics. The hydrolysis reactions of compound 3 were
monitored by following the decrease in absorbance at 266 nrn using a Cary 3E UV-Vis
spectrophotometer equipped with the Cary six-cell Peltier constant-temperature
accessory. The reactions were conducted at 72 and 80 "C in 0.3 M MES buffer, pH 6.20.
Reactions were initiated by the injection of a stock solution of 3 (12.5 pL, 4 mM) into 1
mL of buffer. Rate contants were calculated by non-linear least squares regression of the
absorbance versus time data to a standard first-order rate equation. The rate constant for
the hydrolysis reaction at 37 OC was then extrapolated from the rates at 65 (measured as
described previously in this chapter), 72 and 80 "C.

CHAPTER THREE: PROGRESS TOWARD THE SYNTHESIS OF A NOVEL INFLUENZA INHIBITOR
Overview
This chapter will ouline two proposed syntheses of the target compound 8 as
shown in Figure 24. Both routes start with inexpensive starting materials and involve a
number of steps that have literature precedence. Unfortunately, problems were
encountered with both syntheses and alternative routes will need to be explored.
Figure 24: Target compound.
Proposed Synthesis I
Starting with quinic acid, the alcohol groups (except the one at C2) can be
protected with an isopropylidene and a lactone ring to give compound 32 (Scheme 5).@
The C2 hydroxyl can then be converted to a tosyl group (33).86 1t was proposed that
opening of the lactoneYp7 and displacement with azide would give compound 35.

Scheme 5: Proposed synthesis 1
2,2-dimethoxypropane TsOH $0 P I
reflux in benzene "79 TsCl - pyridine
NaN, * - - - - - - - - - - - - - - - .
DMF
i NaOMe
i MeOHITHF
i O0C t
Once compound 35 was obtained, the stereochemistry at C3 would be inverted by
oxidation then reduction. Subsequently the C3 hydroxyl would be protected with a
benzyl group. The azide group would then be reduced and protected to give 36 shown in
Scheme 6, where Pg stands for a protecting group. The isopropylidene of compound 36
would be replaced with a cyclic sulfate, followed by displacement with azide at C5,
which is trans dia~ial .~ ' Using standard functional group modification, the final product
8 would be synthesized.

Scheme 6: Proposed synthesis 1 continued.
,..' standard functional
Progress
Three of the alcohol groups of quinic acid were protected according to the
procedure outlined by White et al. with the exception that the cyclopentanone was
replaced with 2,2-dimethoxypropane to give the isopropylidene in 52 % yield instead of
the cyclopentylidene.85 The 'H and 13c NMR spectra coincided with the previously
reported chemical shifts and peak multiplicities for this c ~ r n ~ o u n d . ~ ~ ~ ~ ~
The unprotected hydroxyl at C2 was then converted to a tosyl group in 83 % yield
according to procedure outlined by Achiwa and ~ a m a d a . ~ ~ The melting point of this
compound agreed with the reported value, however, there were no previously reported
NMR spectral assignments for comparison. Therefore, the 'H and "C NMR spectra were

assigned using 2D NMR (COSY and HETCOR) as well as comparison to spectra of a
similar compound, the known mesylate deri~ative.~'
The tosylate derivative 33 was subsequently stirred in NaOMe and THF in an
attempt to open the lactone ring (34),87 however, NMR showed that the starting material
had been degraded to give a complex mixture containing at least five compounds (tlc
analysis with ethyl acetate as eluant). Tosylate 31 was then reacted with ammonia gas in
ethanol in an attempt to displace the tosylate to produce an amine group at CI (40),
(Scheme 7).86 However, a complex mixture was obtained and isolation of the desired
product failed. Instead, the tosyl derivative of quinic acid (33) was reacted with sodium
azide in an attempt to synthesize the azide derivative 39. Unfortunately, when this
reaction was tried using hexafluoroisopropanol, a non-nucleophilic polar solvent, at a
temperature of 59 "C no reaction occurred and the starting material was recovered. At
this point it was decided that this synthesis would no longer be pursued and a new
synthetic route was devised.
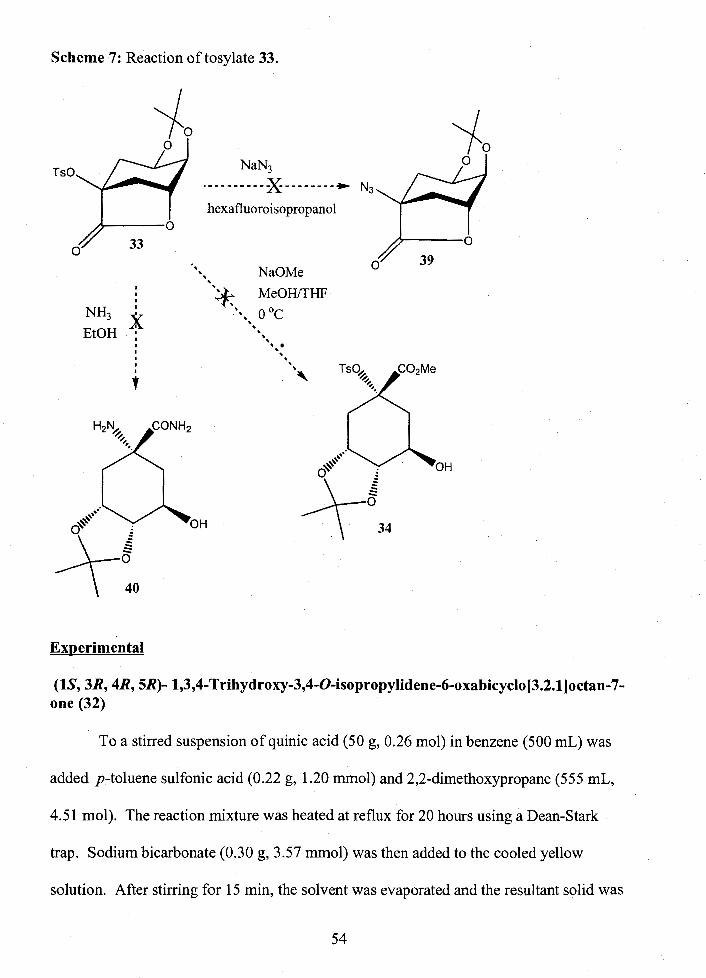
Scheme 7: Reaction of tosylate 33.
I
hexafluoroisopropanol
. NaOMe I
. I I ' . MeomHF
*., 0 "c EtOH : . .
I
. I
. . I I
. i
34
Experimental
(IS, 3R, 4R, 5R)- 1,3,4-Trihydroxy-3,4-O-isopropylidene-6-oxabicyclo[3.2.l]octan-7- one (32)
To a stirred suspension of quinic acid (50 g, 0.26 mol) in benzene (500 mL) was
added p-toluene sulfonic acid (0.22 g, 1.20 rnmol) and 2,2-dimethoxypropane (555 mL,
4.5 1 mol). The reaction mixture was heated at reflux for 20 hours using a Dean-Stark
trap. Sodium bicarbonate (0.30 g, 3.57 mmol) was then added to the cooled yellow
solution. After stirring for 15 min, the solvent was evaporated and the resultant solid was
54

dissolved in dichloromethane (250 mL). Decolorizing carbon was added and the
suspension was heated at reflux for 10 min. The cooled suspension was filtered and
concentrated, and the yellow solid was crystallized from toluene/ethyl acetate to give
white crystals (28.8 g) in 52 % yield. m.p. 138-139 'C litg8 137-138 OC 'H NMR
(CDC13) 6 1.32 (3H9 S, CH3), 1.52 (3H, S, CH3), 2.18 (lH, dd, Jga,S = 3.0 HZ, Jga,ge = 14.7
HZ, H-8a), 2.28-2.39 (2H, m, H-2b, H-Sb), 2.65 (1 H, d, J2a,2e = 12.0 HZ, H-2a), 2.82 (1 H,
S, OH), 4.30 (lH, dt, J4,3 = 2.4 HZ, J4,5 = 6.5 HZ, H-4), 4.49 (lH, ddd, J5,ga = 3.0 HZ, J5,4 =
6.5 Hz,J5,gb=7.3 Hz,H-5),4.72(lH7dd, J 3 , 4 = 2 . 4 H ~ , J 3 , ~ ~ = 6 . 1 Hz,H-3).
(IS, 3R, 4R, 5R)- 3,4-Dihydroxy-3,4-0-isopropylidene-l-(4-methylbenzenesulfonyl)- 6-oxabicyclo[3.2.l]octan-7-one tosylate (33)
To a stirred solution of the lactone 32 (4.59 g, 21.5 mmol) in pyridine (23 mL)
was slowly addedp-tosyl chloride (9.17 g, 48.1 mrnol). The mixture was sonicated until
all of the p-tosyl chloride had dissolved, and this mixture was left standing for a week
until crystals reappeared. The mixture was poured into ice-water, and extracted with
toluene (3 x 50 mL). The organic layer was dried with MgS04, filtered, and concentrated
under reduced pressure. The product was then recrystallized using toluene/hexanes to
afford the desired tosylate (3.59 g) in 78 % yield. corr. m.p. 94-96 OC lits6 96-97 OC. 'H
NMR (CDC13) 6 1.32 (3 H, s, CH3 isopropylidene), 1.53 (3 H, s, CH3 isopropylidene),
2.40 (1 H, dd, JSa,s = 3.5 HZ, J8, sb = 14.4 HZ,, H-8a), 2.45 (3H, s CH3 tosyl), 2.48 (1 H,
ddd, Jgb,2b =2.2 HZ, Jgb,s = 7.7 HZ, H-8b), 2.89 (lH, d,J2a,3 = 11.8 HZ, H-2a), 3.08-3.14
(lH, m, H-2b), 4.30-4.32 (lH, m, H-4), 4.50-4.46 (lH, m, H-5), 4.81 (lH, dd, J3,4 = 2.6
HZ, J3,2b = 6.1 HZ, H-3), 7.35 (2H, d, J = 8.4 HZ, H-3', H-5'), 7.89 (2H, d, J = 8.4 Hz, H-
2', H-6'). 13c NMR (CDC13) 6 21.7 (C-CH3), 24.4 (C-CH3), 27.0 (Ar-CH3), 32.7 (C-2),

36-4 (C-8), 71.1 (C-5), 71.9 (C-4), 75.4 (C-3), 81.1 (C-I), 1 10.3 (C(CH3)*), 128.0 (Ar),
128.2 (Ar), 129.0 (Ar), 129.8 (Ar), 134.3 (Ar), 145.3 (Ar), 172.3 (C-7).
(lR, 3R, 4S, SR)-l-Amino-5-hydroxy-3,4-O-isopropylidene-l-cyclohexy1amide (40)
Ammonia gas was bubbled into a stirred suspension of tosylate 33 (8.4 g, 22.8
mmol) in dry EtOH (100 mL) until the solution clarified. This solution was left standing
for 2 days and then air was passed through the solution for 1 hour. The solution was then
1 concentrated to yield a yellowish-brown syrup. H NMR showed a mixture of several
products which could not be identified.
(lR, 3R, 4S, 5R)-5-hydroxy-3,4-0-isopropylidene-l-(3-toluenesulfony1)-l-cyclohexy1 acetate (34)
Sodium methoxide (0.06 mmol) and THF (2 mL) were stirred in an ice bath and
tosylate 33 (200 mg, 0.54 mmol) was added. The solution was stirred 1 hour at 0 OC, then
neutralized with Arnberlite H+ resin, filtered and evaporated under reduced pressure to
yield a yellow solid. 'H NMR showed degradation of the starting material had occurred
and that none of the desired methyl ester had formed.
(IS, 3R, 4R, 5R)- 1,3,4-trihydroxy-3,4-O-isopropylidene-6-oxabicyclo[3.2.l]octan-7- one azide (39)
Tosylate 33 (50.0 mg, 0.14 mmol) was stirred at 0 OC in a flask containing
hexafluoroisopropanol(3 mL) and dry (3 A) molecular sieves. Sodium azide (1.1
equivalents, 0.15 mmol) was then added and the reaction mixture heated to reflux 5
hours. After cooling, the mixture was filtered and the filtrate diluted with 1 : 1 hexanes:
ethyl acetate, washed with water and saturated sodium hydrogen carbonate, and dried

over MgS04. The resulting mixture was filtered and concentrated. 'H NMR of this
material showed that no reaction had occurred.
Proposed Synthesis 2
The known compound 41 can be prepared via a Diels-Alder reaction between
furan and ethyl acrylate to yield the bicyclic ethyl ester:' which upon hydrolysis92 and
crystallization yields pure endo adduct (41), (Scheme 8). The bicyclic carboxylic acid
could then be protected with a benzyl group (421~' and the double bond converted to an
epoxide (43).93 The epoxide should open with azide to give compound 44. The bicyclic
azide could then be reduced and acetylated, and the alcohol alkylated with various "R" or
alkyl groups, for example with an isopentyl group (46).

Scheme 8: Proposed synthesis 2.
K2C03
1) BF30EtL BnBr + exo product -- 2) 10% aq NaOH DMF
: R-Lg $ base
I 42 C02Bn
1 MCPBA CH2C12
Once alkylated, the bicyclic compound (46) would be ring opened with base to
give the cyclohexene derivative (47) as shown in Scheme 9. Reaction of the cyclohexene
derivative with bromine would afford a bromonium ion which would be reacted with
azide to give 48. This material could then be reduced to the amine. Subsequent radical
induced debromination followed by removal of the benzyl protecting group should yield
the target compound 8.

Scheme 9: Proposed synthesis 2 continued.
i reduction j debromination + debenzylation
Progress
Compound 41 was synthesized in an 11 % yield over two steps. It was planned to
optimize this reaction once it had been shown that the critical reaction, azide opening of
the epoxide, was viable. It is expected that with repeated crystallization of the mother
liquor, as well as optimization of the reaction conditions, the yield would be improved.
Of note, Kotsuki et al. reported a yield of 55% for synthesis of the endo-adduct of methyl
acrylate9' (step 1 of Scheme 8). In addition Tran and Crout reported a 59% yield for the
hydrolysis of a mixture of endo and exo isomers of the methyl acrylate adduct to give the
endo isomer of (step 2, Scheme 8).
The carboxylic acid 41 was then protected with a benzyl in a 76 % yield
and the double bond converted to an epoxide93 in 81 % yield (43). All attempts to open
the epoxide with sodium azide in DMF failed. The reaction was repeated with

ammonium chloride salt as an additive and the solution was heated to reflux but again the
epoxide starting material was recovered unchanged. Different solvents were used,
namely methanol9', and 10% aqueous rne th~x~ethanol~~ but again no reaction occurred.
Degradation of the epoxide occurred when dioxane and DMSO were used. The epoxide
was also dissolved in methanol and ammonia gas bubbled in but once again the starting
material decomposed. Finally the epoxide was stirred with samarium isopropoxide and
azidotrimethylsilane in dichloromethaneg6 but no reaction occurred. In addition, use of
0 97 these reagents in benzene at 80 C produced no discernable reaction. At this point this
synthesis was abandoned. If this present synthesis is to be used, one could make the
aziridine (49), rather than the epoxide (43), which could then be opened by
intramolecular attack of the carboxylate group. However, in this case the stereochemistry
of the free alcohol after ring opening would need to be inverted as shown in Scheme 10.
Scheme 10: Alternative to proposed synthesis 2.
Experimental
(~)-endo-7-Oxabicyclo[2.2.l]hept-5-ene-2-carboxylic acid (41)
Boron trifluoride diethyl etherate (3 mL, 0.024 mol) was added dropwise to a
stirred -78•‹C solution of furan (50 g, 0.73 mol) and ethyl acrylate (24 g, 0.24 mol). The
yellow solution was kept in the freezer for 20 hours and then diluted with CH2C12. The

organic layer was extracted with a saturated sodium hydrogen carbonate, saturated
sodium chloride, dried with MgS04, filtered and concentrated. Ten percent aq NaOH
(170 mL) was added, and the solution stirred overnight at room temperature. The
solution was acidified with concentrated HCl, extracted with CH2C12, dried over MgS04,
filtered and evaporated. The product was then recrystallized using ethyl acetatelhexanes
to give the endo carboxylic acid (3.7 g) in 11 % yield. m.p. 90-91 OC, corrected 95-96 OC
o 98 1 lit 96-98 0c9', 98.0-99.5 C H NMR (CDC13) 6 1.57 (IH, dd, J3,d0,2 = 3.7, J3end0,3exo =
11.7 Hz, H-3 endo), 2.10-2.16 (lH, m, H-3 exo), 3.19-3.15 (lH, m, H-2), 5.04 (lH, d,
J4,3exo=4.6 HZ, H-4), 5.19 (lH, d, J1,2=4.7 HZ, H-1), 6.30 (lH, d, J6,5=5.8 HZ, H-6),
6.47 (lH, d, J5,6 = 5.8 HZ, H-5)
Benzyl (~)-endo-7-oxabicyclo[2.2.1]hept-5-ene-2-carboxylate (42)
To the bicyclic acid 41 (0.5 g, 3.57 mmol) in DMF (5 mL) silver carbonate (1.07
g, 3.88 mmol) was added followed by benzyl bromide (1.5 mL, 12.6 mrnol). The
solution was allowed to stir at room temperature for 1 day, diluted with CH2C12 and
washed with H20 followed by saturated sodium chloride. The organic layer was dried
over MgS04, filtered, and concentrated to yield the desired benzyl ester as a clear
colorless syrup (0.62 g) in 76 %. 'H NMR (CDC13) 6 1.59-1.64 (lH, m, H-3 endo), 2.08-
2.15 (1 H, m, H-3 exo), 3.15-3.19 (1 H, m, H-2), 5.01-5.20 (4H, m, H-1, H-4, CH2-Ph),
6.13(1H,dd,J6,1=1.5,J6,5=5.8H~,H-6),6.42(1H,dd,J5,4=1.8,J5,6=5.8H~,H-5),
7.31-7.40 (5H, m, ArH). 13c NMR (CDC13) 6 28.5 (C-3), 42.9 (C-2), 66.4 (CH2-Ph),
78.8 (C-1), 79.1 (C-4), 128.2 (Ar), 128.3 (Ar), 128.6 (Ar), 135.8 (Ar), 132.5 (C-6), 137.1
(C-5), 172.0 (C=O). Anal. Calcd for C14Hl4O3: C, 73.03; H, 6.13. Found: C, 73.40; H,
6.29.

Benzyl (&)-endo-7-oxabicyclo-5,6-oxirane[2.2,1] he-5-en-2-carboxlate (43)
The benzyl ester 42 (1.03 g, 4.48 mmol) was dissolved in CH2C12 (25 mL) and
this solution was added dropwise to a stirred 0 OC solution of m-chloroperoxybenzoic
acid (1.23 g, 7.13 mrnol) in CH2C12 (75 mL) under nitrogen. The reaction was allowed to
warm to room temperature and stirred for 1 % days. The reaction mixture was then
diluted with CH2C12, washed with saturated sodium hydrogen carbonate, dried with
MgS04, filtered and concentrated. The desired epoxide crystallized from tert-butyl
methyl etherhexanes to yield white needles (1.8 g) in 93 %. 'H NMR (CDC13) 6 1.89
(IH, dd, J3endo~exo = 12.5, J3endo,2= 4.8 HZ, H-3end0), 2.01-2.08 (IH, m, H-3ex0), 3.12-
3.18 (lH, m, H-2), 3.26 (lH, d,J5,6 = 3.4 Hz, H-5), 3.33 (lH, d, J6,5 = 3.4 Hz, H-6), 4.55
(lH, d, J4,3exo = 5.1 HZ, H-4), 4.66 (lH, d, J1 ,2 = 5.1 HZ, H-1), 5.10-5.22 (2H, m, CH2-Ph),
7.35-7.39 (5H, m, aromatics). 13c NMR (CDC13) 8 29.8 (C-3), 46.8 (C-2), 47.9 (C-5),
49.3 (C-6), 66.8 (CH2-Ph), 74.5 (C-1), 74.8 (C-4), 128.3 (Ar), 128.5 (Ar), 128.6 (Ar),
135.3 (Ar), 170.8 (C=O). Anal. Calcd for Cl4HI4o4: C, 68.28; H, 5.73. Found: C,
Benzyl (~)-endo-5-azido-6-hydroxy-7-oxabicyclo-[2.2.l]hept-5-ene-2-carboxylate (44) (Attempted)
Sodium azide (3.0 equivalents, 0.89 mmol), ammonium chloride (26.9 mg, 0.50
mmol), and H20 (20 pL) were added to a stirred solution of compound 43 (72.9 mg, 0.30
mmol) in 2-methoxyethanol(2 mL). The solution was heated to reflux for 3 hours,
cooled and diluted with dichloromethane. The organic layer was washed 3 times with
H20, dried over MgS04, filtered and concentrated. Analysis of the 'H NMR spectrum

showed that no reaction had taken place and the starting material was recovered
unchanged.
The same reaction was repeated using 80 mol equiv of H20 and the solution
heated to reflux overnight, however no reaction took place and the starting material was
recovered.
Benzyl (~)-endo-5-azido-6-hydroxy-7-oxabicyclo-[2.2.l]hept-5-ene-2-carboxy1ate (44) (Attempted)
Sodium azide (1.1 equivalents, 2.72 mmol) was added to a stirred solution of
epoxide 43 (9.3 mg, 0.038 mmol) in dry DMF (0.5 mL). The solution was stirred at room
temperature for 2 $4 days before it was diluted with ethyl acetate, and washed with
saturated sodium hydrogen carbonate and saturated sodium chloride. The organic layer
was then dried over MgS04, filtered and evaporated under reduced pressure to yield the
starting material.
This reaction was repeated with the addition of ammonium chloride (1.2
equivalents) and was heated to reflux overnight. However, 'H NMR showed that no
reaction occurred and the starting material was recovered.
Benzyl (*)-endo-5-azido-6-hydroxy-7-oxabicyclo-[2.2.l]hept-5-ene-2-carboxylate (44) (Attempted)
To a solution of the epoxide 43 (32.0 mg, 0.1 3 mmol) in dioxane (2 mL) was
added sodium azide (29.6 mg, 0.46 mmol) and trimethylsilyl chloride (20 pL, 0.15
rnmol). The solution was heated to reflux overnight, and then it was cooled and
concentrated under reduced pressure. 'H NMR analysis of the crude product showed that
the starting material had decomposed, yet none of the desired product was evident.

Benzyl (i)-endo-5-azido-6-hydroxy-7-oxabicyclo-[2.2.l]hept-5-ene-2-carboxylate (44) (Attempted)
To a stirred solution of epoxide 43 (2 1.8 mg, 0.088 mmol) in DMSO (2 mL) was
added sodium azide (22.0 mg, 0.34 mmol) and ammonium chloride (4.7 mg, 0.088
mmol). The solution was heated at reflux overnight then cooled. The cooled solution
was diluted with CH2C12, washed with saturated sodium hydrogen carbonate, dried with
1 MgS04, filtered and evaporated. H NMR showed that the starting material decomposed.
Benzyl (i)-endo-5-azido-6-hydroxy-7-oxabicyclo-[2.2.1]hept-5-ene-2-carboxylate (44) (Attempted)
Ammonia gas was bubbled into 0 OC MeOH (25 mL) for 3 minutes. The epoxide
43 (29.8 mg, 0.12 mmol) was then added to the MeOH and the solution warmed to room
temperature and stirred overnight. The solution was concentrated under reduced pressure
and the 'H NMR spectrum of the residue showed that the epoxide had decomposed.
Benzyl (~)-endo-5-azido-6-hydroxy-7-oxabicyclo-[2.2.1]hept-5-ene-2-carboxylate (44) (Attempted)
To a stirred solution of epoxide 43 (40.0 mg, 0.16 mmol) in dry MeOH (2 mL)
were added sodium azide (35.2 mg, 0.54 mmol) and ammonium chloride (29.0 mg, 0.59
mmol). The solution was heated to reflux for 8 hours, cooled in the refrigerator
overnight, and then concentrated under reduced pressure. The 'H NMR spectrum of the
residue showed that no reaction had occurred and the starting material was recovered
unchanged.

Benzyl (~)-endo-5-azido-6-hydroxy-7-oxabicycl0-[2.2.1] hept-5-ene-2-carb0~~1~t~ (44)
The epoxide 43 (250 mg, 1.02 mmol) was stirred under N2p) in a flame-dried
flask for 15 minutes without solvent. A suspension of samarium isopropoxide (33.3 mg,
0.102 mmol) in 5 mL of CH2C12 was added to the epoxide and the solution was stirred
another 15 minutes before the addition of trimethylsilyl azide (0.1 5 mL, 0.203 mmol).
The solution was stirred for 3 days and then concentrated under reduced pressure without
heating. The product was then dissolved in 1:4 ethyl acetate:hexanes and filtered through
silica, and concentrated. The product was then stirred with Rexyn 10 1 H' resin (0.13g) in
dry MeOH (5 mL) for 30 minutes, filtered and concentrated. TLC in ethyl acetate:
dichloromethane 1 : 15 showed that no reaction had occurred (Rf 0.48). 'H NMR also
showed that the starting material had not reacted.
Benzyl (~)-endo-5-azido-6-hydroxy-7-oxabicyclo-[2.2.l]hept-5-ene-2-carboxylate (44)
Samarium isopropoxide (1 73.6 mg, 0.53 mmol) in CH2C12 (1 mL) was added to a
stirred solution of trimethylsilylazide (0.15 mL, 1.06 mmol) in dry benzene (5 mL). The
solution was heated to reflux for 5 hours under Nz(,). The epoxide 43 (100 mg, 0.41
mmol) was then dissolved in dry benzene (2 mL) and added to the reagents. After 15
minutes the reaction mixture was allowed to cool and was placed in the refrigerator
overnight. It was then diluted with ether, washed with 3 M HC1 followed by saturated aq
NaHC03, and dried over MgS04. After filtering and concentrating under reduced
1 pressure, H NMR revealed that no reaction had occurred and the starting material was
recovered.

CHAPTER FOUR: CONCLUSIONS
The rates of solvolysis of DHP NeuAc (3) were compared to those for P-D-
glucopyranosyl4'-bromoisoquinolinium tetrafluoroborate (20), which lacks the
carboxylate at the anomeric centre. It was found that the rates of DHP NeuAc (3) were
less sensitive to decreases in the polarity of the solvent, indicating a transition state where
there is no intramolecular nucleophilic participation by the carboxylate. Product studies
confirmed this result in that none of the a-methyl sialoside was observed, as would be
expected if the carboxylate formed a transient a-lactone.
The enzymatic hydrolysis of DHP NeuAc (3) was kinetically characterized with
bacterial sialidases fi-om Vibrio cholerae, Clostridium perfringens, and Salmonella
typhimurium as well as with the sialidase from Newcastle disease virus. Fitting the
kinetic data with that for a series of pyridinium N-acetylneuraminides gave Brsnsted
plots where the slope gave values for PI,. The P1, value for the S. typhimurium sialidase
was essentially zero, indicating that conformational change rather than cleavage of the C-
N bond was rate limiting. Sialylation was found to be rate-determining for the i?
cholerae sialidase, and this step occurs concurrently with a conformational change, while
for the C. perfringens enzyme the chemical step was found to be partially rate-limiting.
The DHP NeuAc compound (3) was found to be a competitive inhibitor of the Newcastle
disease sialidase, with a Ki of 58 yM.
In addition to comparing the mechanisms of different sialidases, the kinetics were
characterized for the Micromonospora viridifaciens sialidase and compared with those of

two active site mutants of the same enzyme. Using DHP NeuAc (3) as a substrate, the
enzyme proficiency was slightly higher with the general acidhase mutant (D92G) than
with the wild-type. It was concluded that this mutation allows for more facile
conformational change with the inflexible substrate. On the other hand, less than 1%
hydrolysis was observed over 11 hours with the Y370G mutant, highlighting the
necessity of this conserved tyrosine to act nucleophilically for hydrolysis of DHP NeuAc.
Knowing these details regarding the catalytic mechanism of sialidases, a new
class of inhibitors was designed. The important feature of this proposed inhibitor (8) is
that the basic nitrogen atom bonded to the anomeric centre should be protonated and thus,
interact with a conserved aspartic acid residue. Two routes toward the synthesis of this
new inhibitor were explored, however, both encountered problems. It is expected that
instead of making the epoxide derivative (43) in the second synthesis, one could instead
make the aziridine and continue with this route.

REFERENCES
Voet, D.; Voet, J.G. Biochemistiy; 2nd ed.; John Wiley & Sons, Inc.: Toronto, 1995.
Sinnott, M.L. 1990. Chem. Rev. 90, 1 171-1 202.
Schauer, R.; Kelm, S.; Reuter, G.; Roggentin, P.; Shaw, L. In Biology of the Sialic Acids; Rosenberg, A., Ed.; Plenum Press: New York, 1995; pp 7-67.
Blix, G. 1936. Hoppe Seyler h Z Physiol Chem 240,43-54.
Corfield, A.P.; Schauer, R. In Sialic Acids: Chemistry, Metabolism and Function; Schauer, R., Ed.; Springer-Verlag: New York, 1982; pp 5-50.
Tsuji, S.; Datta, A.K.; Paulson, J.C. 1996. Glycobioloay 6, V-VII.
Schauer, R. 1982. Adv. Carbohydr. Chem. Biochem. 40,13 1-234.
Sjoberg, E.R.; Manzi, A.E.; Khoo, K.-H.; Dell, A.; Varki, A. 1992. J. Biol. Chem. 267,16200- 1621 1.
Young, P.R.; Jencks, W.P. 1977. J. Am. Chem. Soc. 99,8238-8248.
Craze, G.A.; Kirby, A.J.; Osborne, R. 1978. J. Chem. Soc. Perkin Trans. 2,357- 368.
Knier, B.L.; Jencks, W.P. 1980. J. Am. Chem. Soc. 102,6789-6798.
Sinnott, M.L.; Jencks, W.P. 1980. J. Am. Chem. Soc. 102,2026-2032.
Amyes, T.L.; Jencks, W.P. 1989. J. Am. Chem. Soc. 1 1 1,7888-7900.
Huang, X.; Surry, C.; Hiebert, T.; Bennet, A. J. 1995. J. Am. Chem. Soc. 1 1 7, 10614-10621.
Richard, J.P.; Tsuji, Y. 2000. J. Am. Chem. Soc. 122,3963-3964.
Banait, N.S.; Jencks, W.P. 1991. J. Am. Chem. Soc. 1 13,795 1-7958.
Zhang, Y.; Bommuswamy, J.; Sinnott, M.L. 1994. .I Am. Chem. Soc. 1 16, 7557-7563.

18 Bennet, A.J.; Kitos, T.E. 2002. J. Chem. Soc., Perkin Trans. 2, 1207-1222.
19 Ashwell, M.; Guo, X.; Sinnott, M.L. 1992. J. Am. Chem. Soc. 1 14, 10158- 10166.
20 Horenstein, B.A.; Bruner, M. 1998. J. Am. Chem. Soc. 120, 1357-1362.
21 Chou, D.; Watson, J.; Scholte, A.; Borgford, T.; Bennet, A. 2000. J. Am. Chem. SOC. 122, 8357-8364.
22 Friebolin, H.; Supp, M.; Brossmer, R.; Keilich, G.; Ziegler, D. 1980. Angew. Chem., Int. Ed. Engl. 19, 208-209.
23 McCarter, J.D.; Withers, S.G. 1994. Curr Opin Struct Biol 4, 885-892.
24 Wang, Q.; Graham, R. W.; Trimbur, D.; Warren, R.A. J.; Withers, S.G. 1994. J. Am. Chem. Soc. 116, 11594-11595.
25 Todd, A.E.; Orengo, C.A.; Thornton, J.M. 2001. J. Mol. Biol. 307, 1 1 13-1 143.
26 Saito, M.; Yu, R.K. In Biology of the Sialic Acids; Rosenberg, A., Ed.; Plenum Press: New York, 1995; pp 261 -3 14.
27 Schauer, R. 1985. Trends Biochem Sci 10,357-360.
28 Wilson, J.C.; Angus, D.I.; von Itzstein, M. 1995. J. Am. Chem. Soc. 1 17,42 14- 42 17.
29 Friebolin, H.; Baumann, W.; Brossmer, R.; Keilich, G.; Supp, M.; Ziegler, D.; von Nicolai, H. 1981. Biochem Int 3,321-326.
30 Chong, A.K.J.; Pegg, M.S.; Taylor, N.R.; von Itzstein, M. 1992. Eur JBiochem 207, 335-343.
3 1 Guo, X.; Sinnott, M.L. 1993. Biochem J 296,291-292.
32 Burrneister, W.P.; Ruigrok, R.W.H.; Cusack, S. 1992. EMBOJ. ll,49-56.
33 Varghese, J.N.; McKimm-Breschkin, J.; Caldwell, J.B.; Kortt, A.A.; Colman, P.M. 1992. Proteins: Struct., Funct., Genet. 14, 65-71.
34 Guo, X.; Laver, W.G.; Vimr, E.; Sinnott, M.L. 1994. J. Am. Chem. Soc. 1 16, 5572-5578.
35 Watson, J.N.; Dookhun, V.; Borgford, T.J.; Bennet, A.J. 2003. Biochemistry 42, 12682-1 2690.

Watts, A.G.; Damager, I.; Amaya, M.L.; Buschiazzo, A.; Alzari, P.; Frasch, A.C.; Withers, S.G. 2003. J. Am. Chem. Soc. 125,7532-7533.
Watson, J.; Knoll, T.L.; Chen, J.H.; Chou, D.T.; Borgford, T.J.; Bennet, A.J. 2003. Unpublished work.
Cunven, M.; Dunnell, K.; Ashley, J. 1990. Br. Med. J. 302,425-426.
Kuby, J. In Immunology; Allen, D., Ed.; W. H. Freeman and Company: New York, 1997; p 462.
Laver, W.G.; Bischotberger, N.; Webster, R.G. 1999. ScientiJic American, 56- 65.
Wiley, D.C.; Wilson, I.A.; Skehel, J.J. 1981. Nature 289, 373.
Wilson, I.A.; Skehel, J.J.; Wiley, D.C. 1981. Nature 289, 366.
Hayden, F.G.; Hay, A. 1992. Curr. Top. Microbiol. Immunol. 176, 1 19-1 30.
Woodhead, M.; Lavanchy, D.; Johnston, S.; Colman, P.; Fleming, D. 2000. Int. J. Clin. Pract. 54,604.
Weis, W.; et al. Wiley, D.C. 1988. Nature 333,426-43 1.
Wade, R.C. 1997. Structure 5, 1 139-1 145.
Varghese, J.N.; McKimm-Breschkin, J.; Caldwell, J.B.; Keilich, G.; Colman, P.M. 1992. Proteins: Struct., Funct., Genet. 14, 327-332.
Thomas, A,; Jourand, D.; Bret, C.; Amara, P.; Field, J.J. 1999. J. Am. Chem. Soc. 121,9693-9702.
Meindl, P.; Bodo, G.; Palese, P.; Schulman, J.; Tuppy, H. 1974. Virology 58, 457.
Palese, P.; Schulman, J.L. In Chemoprophylaxis and Viral Infection of the Upper Respiratory Tract; Oxford, J. S., Ed.; CRC Press: FLY U.S.A., 1977; Vol. 1, p 189.
Taylor, N.R.; von Itzstein, M. 1994. J. Med. Chem. 37,616.
von Itzstein, M.; Wu, W.-Y.; Jin, B. 1994. Carbohydr. Res. 259, 301.
von Itzstein, M.; Wu, W.-Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Phan, T.V.; Smythe, M.L.; White, H.F.; Oliver, S.W.; Colman, V.J.; Cameron, J.M.; Penn, C.R. 1993. Nature (London) 37, 1473.

G m , C.U.; Lew, W.; Williams, M.A.; Liu, H.T.; Zhang, S.; Swaminathan, N.; Bischofberger, N,; Chen, M.S.; Mendel, D.B. ; Tai, C .Y.; Laver, W.G.; Stevens, R.C. 1997. J. Am. Chem. Sot. 1 19,68 1-690.
Florio, P.; Thornson, R. J.; Alafaci, A.; Abo, S.; von Itzstein, M. 1999. Bioorg. Med. Chem. Lett, 9,2065.
Anonymous 2002. Antivir. Agents Bull. 15, 162-1 63.
Mineno, T.; Miller, M.J. 2003. J. Org. Chem. 68,6591-6596.
Winkler, D.A. 1996. J. Med. Chem. 39,4332-4334.
Dyason, J.C.; von Itzstein, M. 2001. Aust. J. Chem. 54,663-670.
Sliwa, H.; Cordonnier, G. 1975. J. Heterocyclic Chemistry 12, 809-8 10.
Roy, R.; Laferriere, C.A. 1990. Can. J. Chem. 68,2045.
63 Zhu, J.; Bennet, A.J. 2000. J. Org. Chem. 65,4423-4430.
64 Hosie, L.; Marshall, P.J.; Sinnott, M.L. 1984. J. Chem. Soc., Perkin Trans. 2, 1121-1 131.
Huang, X.; Tanaka, K.S.E.; Bennet, A.J. 1997. J. Am. Chem. Soc. 1 19,11147- 11 154.
Horenstein, B.A.; Bruner, M.J. 1998. J. Am. Chem. Soc. 120, 1357-1362.
Horenstein, B.A.; Bruner, M.J. 1996. J. Am. Chem. Soc. 118, 10371-10379.
Thibblin, A.; Saeki, Y. 1997. J. Org. Chem. 62, 1079-1 082.
Pocar, D.; Bianchetti, G.; Croce, T.D. 1964. Chem. Ber. 97, 1225.
Rothermel, J.; Faillard, H. 1990. Carbohydr. Res. 196, 29-40.
Kuhn, R.; Lutz, P.; MacDonald, D.C. 1966. Chem. Ber. 99, 6 1 1.
Lamb, R.A.; ~olakofsky, D. In Fundamental Virology; 3rd. ed.; Fields, B. N., et al, Eds.; Lippincott Raven, 1996; pp 577-604.

Corfield, A.P.; Higa, H.; Paulson, J.C.; Schauer, R. 1983. Biochem. Biophys, Acta 744, 121-126.
Bouwstra, J.B.; Deyl, C.M.; Vliegenthart, F.G. 1987. Bid. Chem. Hoppe-Seyler 368,269-275.
Eschenfelder, V.; Brossmer, R. 1987. Carbohydr. Res. 162,294-297.
Watson, J.N. In Molecular Biology and Biochemistry; Simon Fraser University: Burnaby, 2003; p 206.
Dookhun, V.; Bennet, A.J. 2003. J. Am. Chem. Soc. in preparation.
Jones, C.C.; Sinnott, M.L. 1977. J. Chem. Soc. Chem. Commun., 767-768.
81 Kleineidam, R.G.; Kruse, S.; Roggentin, P.; Schauer, R. 2001. Biol. Chem. 382, 313-319.
82 Chien, C.-H.; Sham, Y.-J.; Sheu, S.-Y. 1996. Enzyme Microb. Technol. 19, 267-276.
83 Ghate, A.A.; Air, G.M. 1998. Eur JBiochem 258,320-33 1.
84 Wang, Y.; Yamaguchi, K.; Shimada, Y.; Zhao, X.; Miyagi, T. 2001. Eur J Biochem 268,2201 -2208.
85 White, J.D.; Carnrnack, J.H.; Sakuma, K.; Rewcastle, G.W.; Widener, R.K. 1995. J. Org. Chem. 60,3600-361 1.
86 Achiwa, K.; Yarnada, S.-I. 1966. Chem. Pharm. Bull. 14,537-549.
87 Falck, J.R.; Abdali, A.; Wittenberger, S.J. 1990. J. Chem. Soc. Chem. Commun., 953-955.
88 Wang, Z.-X.; Shi, Y. 1997. J. Org. Chem. 62,8622-8623.
89 Flores-Parra, A.; Paredes-Tepox, C.; Joseph-Nathan, P.; Contreras, R. 1990. Tetrahedron 46,4137-4148.

90 Rohloff, J.C.; Kent, K.M.; Postich, M.J.; Becker, M.W.; Chapman, H.H.; Kelly, D.E.; Lew, W .; Louie, M.S .; McGee, L.R.; Prisbe, E.J.; Schultze, L.M.; Yu, R.H.; Zhang, L. 1998. J. Org. Chem. 63,4545-4550.
91 Kotsuki, H.; Asao, K.; Ohnishi, H. 1984. Bull. Chem. Soc. Jpn. 57, 339-3340.
92 Tran, C.H.; Crout, D.H.G. 1998. J. Chem. Soc., Perkin Trans. 1, 1065-1068.
93 Furniss, B.S.; Hannaford, A.J.; Smith, P.W.G.; Tatchell, A.R. In Vogelk Textbook of Practical Organic Chemistry; 5th ed.; Longman Scientific & Technical copublished with John Wiley & Sons, Inc. : New York, 1989; pp 1 134- 1 135.
94 Krajewski, K.; Ciunik, Z.; Siemion, I.Z. 1999. Tetrahedron: Asymmetry 10, 4591-4598.
95 Ogawa, S.; Yato, Y.; Nakamura, K.; Takata, M.; Takagaki, T. 1986. Carbohydr. Res. 148,249-255.
96 Martinez, L.E.; Leighton, J.L.; Carsten, D.H.; Jacobsen, E.N. 1995. J. Am. Chem. Soc. 117,5897-5898.
97 Back, T.; Minksztym, K. 1999. Synlett, 201-203.
98 Ogawa, S.; Uemura, M.; Fujita, T. 1988. Carbohydr. Res. 177,213.