the effects of adenosine a 2b receptor inhibition on vegf and nitric...
TRANSCRIPT

http://informahealthcare.com/rnfISSN: 0886-022X (print), 1525-6049 (electronic)
Ren Fail, Early Online: 1–9! 2014 Informa Healthcare USA, Inc. DOI: 10.3109/0886022X.2014.900404
LABORATORY STUDY
The effects of adenosine A2B receptor inhibition on VEGF and nitricoxide axis-mediated renal function in diabetic nephropathy
Leena Patel1 and Aswin Thaker2
1Department of Pharmacology, Ramanbhai Patel College of Pharmacy, Charotar University of Science and Technology, Anand, Gujarat, India and2Department of Pharmacology & Toxicology, College of Veterinary Science and Animal Husbandry, Anand Agricultural University, Anand, Gujarat,
India
Abstract
Diabetic nephropathy (DN) is the most common cause of end-stage renal disease worldwide.The pathophysiologic mechanisms of diabetic nephropathy are incompletely understoodbut include overproduction of various growth factors and cytokines. Upregulation ofvascular endothelial growth factor (VEGF) is a pathogenic event occurring in most forms ofpodocytopathy; however, the mechanisms that regulate this growth factor induction are notclearly identified. A2B receptors have been found to regulate VEGF expression under hypoxicenvironment in different tissues. One proposed hypothesis in mediating diabetic nephropathyis the modulation of VEGF-NO balance in renal tissue. We determined the role of adenosine A2B
receptor in mediating VEGF overproduction and nitrite in diabetic nephropathy. The renalcontent of A2B receptors and VEGF was increased after 8 weeks of diabetes induction. The renaland plasma nitrite levels were also reduced in these animals. In vivo administration of A2B
adenosine receptor antagonist (MRS1754) inhibited the renal over expression of VEGF andadverse renal function parameters. The antagonist administration also improved the kidneytissue nitrite levels. In conclusion, we demonstrated that VEGF induction via adenosinesignaling might be the critical event in regulating VEGF-NO axis in diabetic nephropathy.
Keywords
A2B Adenosine receptor, diabeticnephropathy, nitric oxide, streptozotocin,VEGF
History
Received 9 November 2013Revised 28 January 2014Accepted 19 February 2014Published online 28 March 2014
Introduction
Diabetic nephropathy (DN) is the most common cause of end-
stage renal disease. About one-third of the patients who
develop diabetes eventually suffer from DN. The costs of DN
are significantly higher than those from other diabetic
complications because the patients are subjected to hemodi-
alysis programs and renal transplant when failure occurs.
Thus, the burden of DN on public health is enormous.1 The
pathomechanisms leading to these changes are not yet clearly
understood and therefore, therapeutic approaches for relief
of this disease are scarce or do not permit a favorable
pharmacological intervention.
Vascular endothelial growth factor (VEGF-A) is one of the
critical mediators in DN. It is constitutively expressed in
podocytes, proximal tubular cells and medullary thick
ascending limb cells in the juxtamedullary region of the
normal kidney. Evidence is emerging that VEGF plays a
critical role in maintaining renal homeostasis.2,3 An alteration
in VEGF-A expression has been shown in a variety of renal
diseases. Altered (increased or decreased) expression of
VEGF leads to glomerular dysfunction and proteinuria.
Both circulating and local VEGF-A levels are high in diabetes
and the excessive VEGF-A has been shown to have a key role
in mediating glomerular hypertrophy, proteinuria and retin-
opathy.4–6 VEGF-A induced excessive angiogenesis has been
extensively found in retinopathy, the same has been also seen
in nephropathy.7 The precise mechanism is unclear for
contradictory status of VEGF-A in diabetic and non-diabetic
kidney disease. Hypoxia has been found to stimulate VEGF-A
expression. Adenosine is a critical mediator in physiological
adaptation to hypoxia and contribute to diseases as diverse as
inflammation and carcinogenesis.8 Once liberated in the
extracellular space, adenosine is either recycled or interacts
with cell surface adenosine receptors. Presently, four subtypes
of G protein-coupled receptors exist, designated A1, A2A, A2B
or A3. A2B receptors are the lower affinity receptors and they
have been found to induce angiogenesis.9
The role of adenosine receptors in cellular dysfunction
mediating progression of DN has been studied recently.
Ex vivo exposure of rat kidney glomeruli to adenosine leads to
an increase in VEGF-A content. Activation of A2B receptor
subtype augments expression, and releases VEGF-A beyond
basal levels in rat glomeruli.10 Uncoupling of VEGF-nitric
oxide (VEGF–NO) axis has been studied as the factor
mediating DN.11 Hypoxia has been found to play a critical
role in pathogenesis of DN.12 We thus hypothesize that
Address correspondence to Leena Patel, Department of Pharmacology,Ramanbhai Patel College of Pharmacy, Charotar University of Scienceand Technology, CHARUSAT Campus, Changa – 388 421, Ta. Petlad,Dist. Anand, Gujarat, India. Tel: +91- 9913523315; Fax: 02697-265021;E-mail: [email protected]
Ren
Fai
l Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Ond
okuz
May
is U
niv.
on
05/0
6/14
For
pers
onal
use
onl
y.

hypoxic condition produced in hyperglycemic environment
of DN may change the expression of adenosine receptors
to angiogenic phenotype increasing levels of VEGF-A. The
accelerated state of VEGF-A together with reduced endothe-
lial NO bioavailability, in diabetic kidney; results in
uncoupling of the VEGF–NO axis. The event consequently
causes VEGF-A to produce diverse biologic effects that could
contribute to DN. Herein, we examined the effects of A2B
antagonist on VEGF–NO axis-mediated renal function in
mice model of DN.
Materials and methods
Chemicals and reagents
Streptozotocin and nitrate reductase were purchased from
Sigma Chemical Co., Milwaukee, WI. An enzyme linked
immunosorbent assay (ELISA) kit for the estimation of
VEGF-A was purchased from RayBiotech Inc., Norcross, GA.
MRS1754 were purchased from Abcam plc.UK. DAN (2,3-
diaminonaphthalene), sodium nitrite and nicotinamide aden-
ine dinucleotide phosphate (NADP) were purchased from
Himedia Laboratories, Mumbai, India. Kits for the estimation
of plasma creatinine and blood urea nitrogen (BUN) were
purchased from Crest Biosystem, Goa, India.
Animal model and experimental protocol
All animal experiments were conducted in accordance with
the guidelines of CPCSEA, India. Male C57BL/6 mice were a
kind gift from Zydus Research Centre, Ahmedabad (body
weight: 22 ± 2 g). Animals were maintained at a room
temperature in a light (12 h light/12 h dark)-controlled
environment with access to food and water ad libitum. One
week after the acclimatization animals were randomly
assigned to different treatment as depicted in Table 1.
Induction of diabetes
The low dose streptozotocin (STZ) protocol described by
Animal Models of Diabetic Complications Consortium
(AMDCC) was followed for induction of diabetes.13 In
brief, mice were fasted prior to injection for 4 h. A single
intraperitoneal STZ (50 mg/kg) injection was administered to
each mouse for 5 days consecutively (n¼ 12). Mice were
supplied with 10% sucrose water to avoid sudden hypogly-
cemia post-injection. A normal control group of mice were
injected with 400mL vehicle (sodium citrate buffer). Body
weight and blood glucose (SD CHECK� GOLD Blood
Glucose Meter, SD Biosensor, Korea) were monitored 1 week
after STZ injection and every week thereafter. Mice with
blood glucose levels of4300 mg/dL were considered diabetic.
Following 8 weeks of diabetes induction (n¼ 6), the mice
were treated with the antagonist of A2B adenosine receptor,
MRS1754 (1 mg/kg, i.p.) for two weeks. The animals in the
antagonist control group were administered MRS1754
(1 mg/mL) for two weeks (n¼ 6).
Collection of urine, blood and tissue samples
Mice were housed in metabolic cages for collection of 24 h
urine samples. Blood was drawn from retro orbital tract. The
setting up of cages and collecting samples were carried out
between 15:00 and 16:00 to avoid food derived creatinine
interference. At the end of the treatment all the animals were
sacrificed and whole kidneys were removed. The kidneys
were weighed and frozen in liquid nitrogen in RNA later� for
isolation of total RNA. The samples were stored at �80 �Cuntil used for biochemical analysis.
Renal function parameters
Plasma and urine creatinine (modified Jaffe’s kinetic method),
BUN (GLDH kinetic method) were measured by commer-
cially available kits following manufacturers’ instructions.
Urine albumin (Bromocresol green method using mouse
albumin as standard) was measured to calculate urinary
albumin excretion (UAE) from 24 h urine samples. Creatinine
clearance was calculated according to the U/P�V principle
for the matching plasma and urine samples.14
Quantitative VEGF-A determinations
Kidney tissues were washed with cold phosphate buffered
saline (PBS) and homogenized (10%w/v) in ice bath. The
homogenate was then centrifuged at 20,000 rpm at 4 �C. The
supernatant was stored at �80 �C till further analysis. VEGF-
A protein was measured in plasma and homogenate by ELISA
following the manufacturer’s protocols (RayBiotech Inc.,
Norcross, GA).
Quantitative NO determinations
As an indicator of nitric oxide bioavailability, nitrite and
nitrate were estimated in urine and kidney homogenate by
spectrofluorometric analysis. The method was adopted from
the literature and modified slightly.15 Briefly, 10 mL of nitrate
reductase (0.5 U/mL) was added to 100 mL of sample, after
4 h; 20 mL of DAN (0.05 mg/mL), 130 mL HCl (1.5 N) were
added. After 10 min, the reaction was stopped by 130 mL of
NaOH (2 N). The resultant solution was diluted to 2 mL
and the emission scan was recorded by a spectrofluorometer
(LS 55 Fluorescence spectrometer, Perkin Elmer) exciting at
360 nm and reading at 415 nm. Sodium nitrite was used as
reference standard.
Real time quantitative PCR for VEGF-A and A2B mRNA
RNeasy Mini Kit (Qiagen, Valencia, CA) was used for the
RNA extraction according to the manufacturer’s instructions.
Reverse transcription of total RNA to cDNA was performed
Table 1. Allocation of treatment to animals.
Groups Group I (n¼ 6) Group II (n¼ 6) Group III (n¼ 6) Group IV (n¼ 6)
Treatment Vehicle(Sodium citrate buffer)
Streptozotocin +Vehicle of MRS1754(for two weeks corresponding to group III)
Streptozotocin+ MRS1754 MRS1754
2 L. Patel and A. Thaker Ren Fail, Early Online: 1–9
Ren
Fai
l Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Ond
okuz
May
is U
niv.
on
05/0
6/14
For
pers
onal
use
onl
y.

with the Verso cDNA Synthesis Kit (Thermo Scientific,
ABgene and Surrey, UK) in a DNA Thermal cycler (Perkin-
Elmer Applied Biosystems, Foster City, CA) with random
hexamers as primers. The quality of DNA and total RNA was
checked by bioanalyzer (2100 Bioanalyzer Instrument,
Agilent Technologies, Santa Clara, CA). The real-time
PCR was performed (7500 Fast, Applied Biosystems, Grand
Island, NY) using the Quanti Tect SYBR Green PCR Kit
(Qiagen, Valencia, CA), with the cDNA synthesized above as
template in a reaction; following manufacturer‘s instructions.
Specific primers (Table 2) used for the mouse adenosine A2B
receptor (gene bank accession number GI:145966718) and
VEGF-A were adopted from the earlier literature.16,17 The
gene for glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) was used as reference. Fold change in gene
expression was calculated for each gene. Melt curves were
performed upon completion of the cycles to ensure that the
non-specific products were absent.
Histochemistry and assessment of glomerulosclerosis
Kidney tissue was fixed in formalin, embedded in paraffin,
sectioned and stained with Masson trichrome reagent. One
hundred glomeruli were randomly selected for determination
of glomerulosclerosis. Glomeruli that exhibited adhesion of
the capillary tuft to Bowman’s capsule, capillary obliteration,
mesangial expansion or segmental tuft sclerosis were defined
as glomerulosclerotic. The extent of glomerular damage was
expressed as the percentage of glomeruli that exhibited
sclerosis. The degree of glomerulosclerosis was graded from
1 to 4 points according to the percentage of effected glomeruli
(1� 10%, 2� 10–20%, 3� 20–50%, and 4� 50%). Blind
analysis was done on all sections by one observer.
Statistical analysis
Data are presented as the mean ± standard error of mean
(SEM). Statistical analysis was performed using Systat13
(syatat Inc., San Jose, CA). For comparisons of continuous
variables, a test of normality was performed (Shapiro–Wilk
test) prior to assessing statistical significance using either a t-
test (parametric) or a Fligner-Wolfe test (nonparametric)
when comparing two groups. Association between the
expressions of both genes and, VEGF-A and NO levels,
were analyzed by Pearson’s correlation coefficient.
Results
General parameters
Following 10 weeks of diabetes induction, plasma glucose
concentration (Figure 1a), kidney hypertrophy index
(Figure 1b), and food intake (Figure 1c) were significantly
increased compared with non-diabetic control animals
(p50.05). Similarly a significant decrease in body weight
(p50.05, Figure 1d) was observed in animals treated with
streptozotocin. The treatment with A2B adenosine receptor
antagonist did not alter the increased levels of plasma
glucose, food intake and body weight significantly. The
treatment with antagonist significantly recovered kidney
hypertrophy index in diabetic animals (p50.05). However,
antagonist administration had no effect on all above param-
eters when administered to vehicle treated animals (MRS
control group).
Renal function parameters
Serum creatinine (Figure 2a), blood urea nitrogen (Figure 2b)
and urinary protein excretion (Figure 2d) were increased in
animals after 10 weeks of diabetes (p50.05). Similarly,
creatinine clearance was lower in diabetic animals compared
with non-diabetic control animals (Figure 2c, p50.05). Urine
albumin levels in control and control MRS animals were
below the detection limit of the method employed for
analysis. The treatment with A2B adenosine receptor antag-
onist recovered blood urea nitrogen, serum creatinine,
creatinine clearance and urinary protein excretion in diabetic
mice (p50.05). Moreover, no change in renal functions was
found in vehicle treated animals (MRS control group) after
two weeks of antagonist treatment.
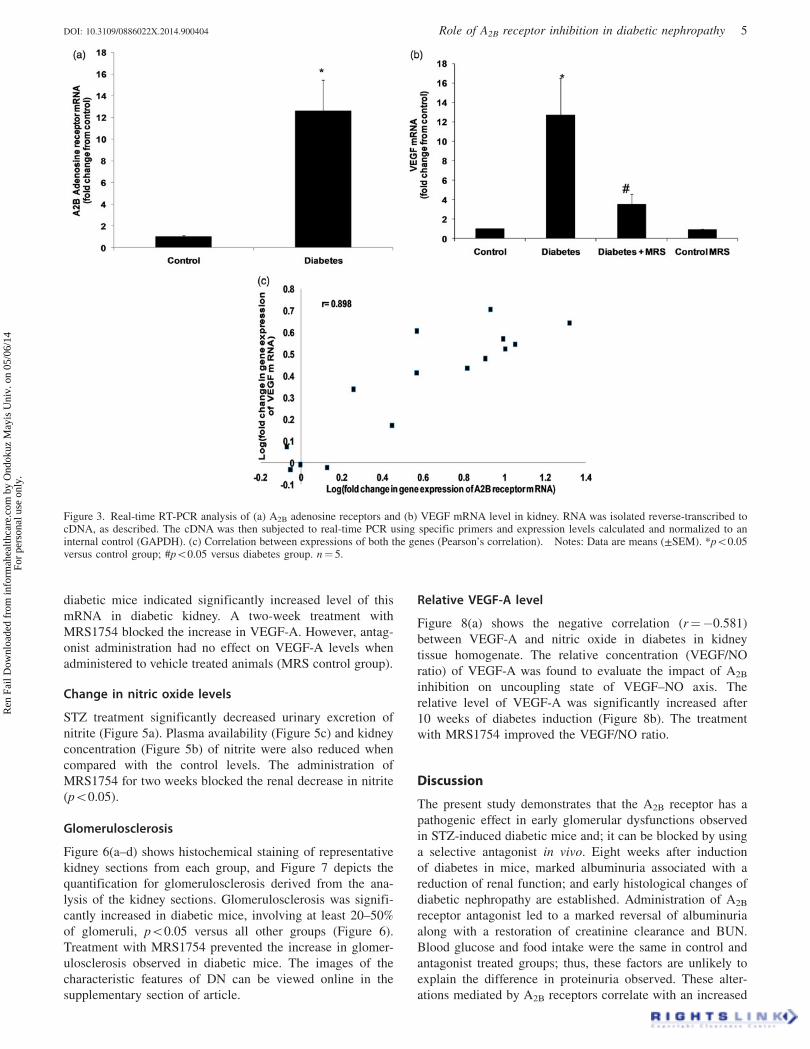
Change in VEGF-A and A2B adenosine receptor geneexpression
To evaluate the impact of diabetes on expression level of
VEGF-A and A2B receptors in mice kidney, we examined
mRNA levels in kidneys of normal and STZ-induced diabetic
animals. The changes in mRNA level were evaluated based on
results from real-time PCR performed on cDNA transcribed
from RNA isolated from whole kidney. There was a
significant increase in the mRNA level of receptor in diabetic
kidney, 10 weeks after diabetes induction (Figure 3, p50.05).
In diabetic animals, the level of A2B receptor and VEGF-A
mRNA were raised 11- and 12–fold, respectively.
Administration of MRS1754 for two weeks blocked the
renal increase in VEGF-A expression. Additionally, when
antagonist was administered to vehicle treated animals (MRS
control group); a partial but not significant decrease in
VEGF-A expression was observed. The expression of A2B
receptor and VEGF-A genes was positively correlated
(Figure 3c, r¼ 0.575).
VEGF-A protein levels
VEGF-A protein measurement by ELISA supported results
obtained by gene expression study (p50.05, Figure 4b).
Comparison of VEGF-A protein level in kidneys of normal and
Table 2. Sequences of primer used in real time PCR.
Primers
Entity Sense sequence Antisense sequence
VEGF GTTCACTGTGAGCCTTGTTCAG GTCACATCTGCAAGTACGTTCGGAPDH TTCACCACCATGGAGAAGGC GGCATGGACTGTGGTCATGAA2B receptor TTGGCATTGGATTGACTC TATGAGCAGTGGAGGAAG
DOI: 10.3109/0886022X.2014.900404 Role of A2B receptor inhibition in diabetic nephropathy 3
Ren
Fai
l Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Ond
okuz
May
is U
niv.
on
05/0
6/14
For
pers
onal
use
onl
y.
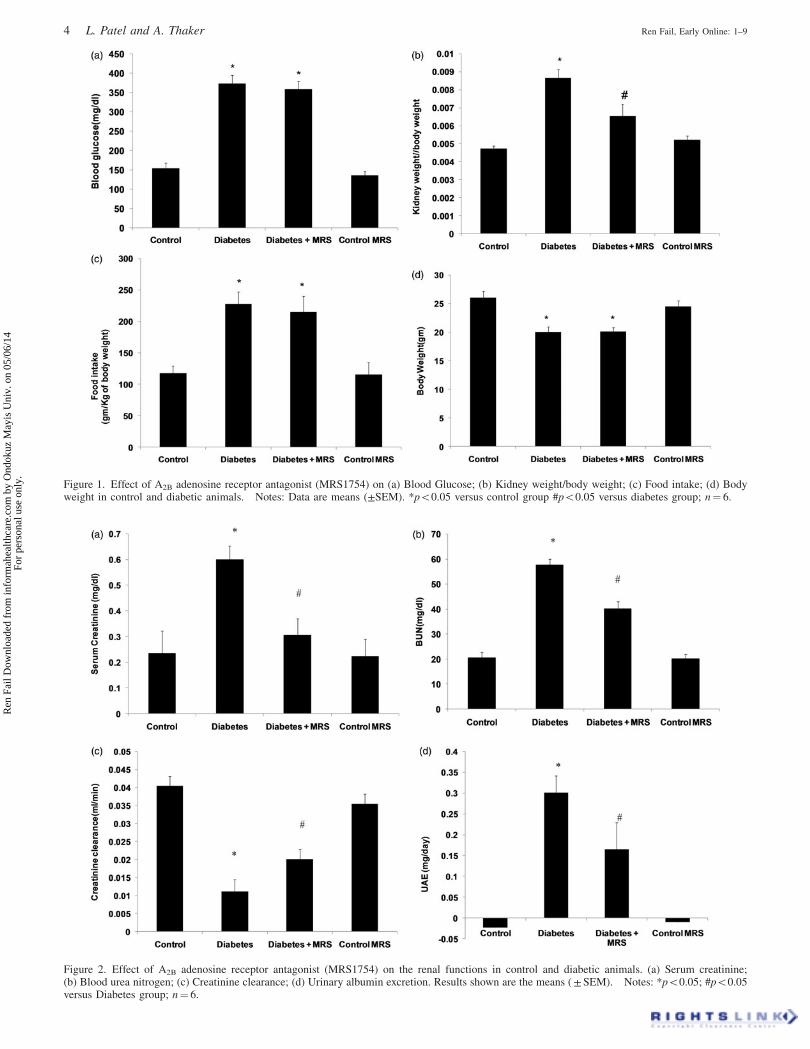
Figure 2. Effect of A2B adenosine receptor antagonist (MRS1754) on the renal functions in control and diabetic animals. (a) Serum creatinine;(b) Blood urea nitrogen; (c) Creatinine clearance; (d) Urinary albumin excretion. Results shown are the means ( ± SEM). Notes: *p50.05; #p50.05versus Diabetes group; n¼ 6.
Figure 1. Effect of A2B adenosine receptor antagonist (MRS1754) on (a) Blood Glucose; (b) Kidney weight/body weight; (c) Food intake; (d) Bodyweight in control and diabetic animals. Notes: Data are means (±SEM). *p50.05 versus control group #p50.05 versus diabetes group; n¼ 6.
4 L. Patel and A. Thaker Ren Fail, Early Online: 1–9
Ren
Fai
l Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Ond
okuz
May
is U
niv.
on
05/0
6/14
For
pers
onal
use
onl
y.
diabetic mice indicated significantly increased level of this
mRNA in diabetic kidney. A two-week treatment with
MRS1754 blocked the increase in VEGF-A. However, antag-
onist administration had no effect on VEGF-A levels when
administered to vehicle treated animals (MRS control group).
Change in nitric oxide levels
STZ treatment significantly decreased urinary excretion of
nitrite (Figure 5a). Plasma availability (Figure 5c) and kidney
concentration (Figure 5b) of nitrite were also reduced when
compared with the control levels. The administration of
MRS1754 for two weeks blocked the renal decrease in nitrite
(p50.05).
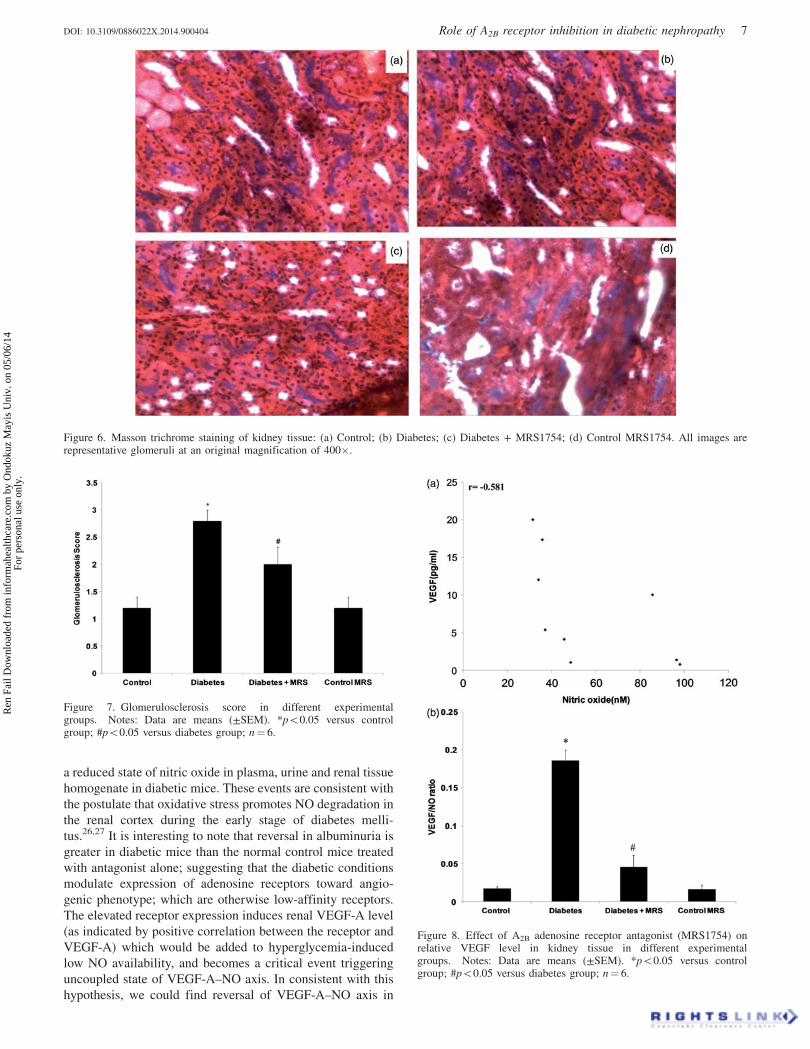
Glomerulosclerosis
Figure 6(a–d) shows histochemical staining of representative
kidney sections from each group, and Figure 7 depicts the
quantification for glomerulosclerosis derived from the ana-
lysis of the kidney sections. Glomerulosclerosis was signifi-
cantly increased in diabetic mice, involving at least 20–50%
of glomeruli, p50.05 versus all other groups (Figure 6).
Treatment with MRS1754 prevented the increase in glomer-
ulosclerosis observed in diabetic mice. The images of the
characteristic features of DN can be viewed online in the
supplementary section of article.
Relative VEGF-A level
Figure 8(a) shows the negative correlation (r¼�0.581)
between VEGF-A and nitric oxide in diabetes in kidney
tissue homogenate. The relative concentration (VEGF/NO
ratio) of VEGF-A was found to evaluate the impact of A2B
inhibition on uncoupling state of VEGF–NO axis. The
relative level of VEGF-A was significantly increased after
10 weeks of diabetes induction (Figure 8b). The treatment
with MRS1754 improved the VEGF/NO ratio.
Discussion
The present study demonstrates that the A2B receptor has a
pathogenic effect in early glomerular dysfunctions observed
in STZ-induced diabetic mice and; it can be blocked by using
a selective antagonist in vivo. Eight weeks after induction
of diabetes in mice, marked albuminuria associated with a
reduction of renal function; and early histological changes of
diabetic nephropathy are established. Administration of A2B
receptor antagonist led to a marked reversal of albuminuria
along with a restoration of creatinine clearance and BUN.
Blood glucose and food intake were the same in control and
antagonist treated groups; thus, these factors are unlikely to
explain the difference in proteinuria observed. These alter-
ations mediated by A2B receptors correlate with an increased
Figure 3. Real-time RT-PCR analysis of (a) A2B adenosine receptors and (b) VEGF mRNA level in kidney. RNA was isolated reverse-transcribed tocDNA, as described. The cDNA was then subjected to real-time PCR using specific primers and expression levels calculated and normalized to aninternal control (GAPDH). (c) Correlation between expressions of both the genes (Pearson’s correlation). Notes: Data are means (±SEM). *p50.05versus control group; #p50.05 versus diabetes group. n¼ 5.
DOI: 10.3109/0886022X.2014.900404 Role of A2B receptor inhibition in diabetic nephropathy 5
Ren
Fai
l Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Ond
okuz
May
is U
niv.
on
05/0
6/14
For
pers
onal
use
onl
y.

expression of this receptor in mice diabetic kidney. Results
obtained are comparable to the earlier in-vivo and in-vitro
studies.18,19 The differential expression of adenosine receptor
was first described by Pawelczyk et al.20 The alteration turns
off the inhibition on VEGF-A release accompanied by an
increase in the production of this growth factor release in
diabetic state.
The mechanism by which A2B receptor antagonist mediate
protection in STZ-induced diabetic nephropathy is not known.
The pathomechanism leading to diabetic nephropathy
involves release of diverse growth factors, increased VEGF-
A being the most often involved. VEGF-A has been found to
act in autocrine manner to induce proteinuria caused by
podocytopathy. A2B receptors have been investigated for
attenuating kidney injury in diabetes directly through effects
on signaling pathways inducing VEGF-A. A2B receptor
antagonist was demonstrated to block the increased release
of TGF-b from diabetic glomeruli in vitro and the
myofibroblast transdifferentiation of mesangial cells
in vivo.21 In these studies, intervention of VEGF-A induction
was a remarkable event. Therefore, interception of both the
TGF-b and VEGF-A signaling via A2B receptors may give
new insights into drug discovery. We also correlate the
increase in low affinity A2B receptor with an increase in
VEGF-A expression.
Several studies have shown that the VEGF-A expression in
renal podocytes increased upon exposure to high glucose
concentration and it is intracellular mediated by PKC-alpha
and ERK1/2 signalling molecules.22 Our results also support
increased expression of VEGF-A in renal tissue in diabetic
state. The in vitro studies also showed reversal of increased
VEGF-A in the presence of MRS1754.10,23 In agreement with
this evidence, our study in vivo implies A2B receptors could
be a transducer of hyperglycemic conditions to mediate
VEGF-A increase in diabetic mielu.
A low renal VEGF-A was observed in many types of
experimental kidney diseases, and administration of VEGF-A
was found to be protective.24 A contradiction in terms occurs
in diabetes, wherein renal VEGF-A levels are high and a
detrimental effect of VEGF-A on kidney disease has been
shown.4,5 Normally, VEGF-A stimulates endothelial nitric
oxide release; thus, elevated NO and VEGF-A act in
coordination with each other as a trophic factor for vascular
endothelium. The surplus NO, derived from the endothelial
cell; acts as an inhibitory factor that prevents excess
endothelial cell proliferation, vascular smooth muscle cell
proliferation and macrophage infiltration. In diabetic state,
where NO bioavailability is reduced; high levels of VEGF-A
lead to excessive endothelial cell proliferation, stimulation of
macrophage chemotaxis and vascular smooth muscle cell
activation.25 Consistent with this, in our study, we could find
Figure 4. Effect of A2B adenosine receptor antagonist (MRS1754) on (a)plasma; (b) homogenate concentration of VEGF protein, measured byELISA in different experimental groups. Notes: Data are means(±SEM). *p50.05 versus control group; #p50.05 versus diabetesgroup, n¼ 6.
Figure 5. A2B adenosine receptor antagonist (MRS1754) on (a) kidneytissue (b) urine nitrite levels in control and diabetic mice. Notes: Dataare means (±SEM). *p50.05 versus control group; #p50.05 versusdiabetes group; n¼ 6.
6 L. Patel and A. Thaker Ren Fail, Early Online: 1–9
Ren
Fai
l Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Ond
okuz
May
is U
niv.
on
05/0
6/14
For
pers
onal
use
onl
y.
a reduced state of nitric oxide in plasma, urine and renal tissue
homogenate in diabetic mice. These events are consistent with
the postulate that oxidative stress promotes NO degradation in
the renal cortex during the early stage of diabetes melli-
tus.26,27 It is interesting to note that reversal in albuminuria is
greater in diabetic mice than the normal control mice treated
with antagonist alone; suggesting that the diabetic conditions
modulate expression of adenosine receptors toward angio-
genic phenotype; which are otherwise low-affinity receptors.
The elevated receptor expression induces renal VEGF-A level
(as indicated by positive correlation between the receptor and
VEGF-A) which would be added to hyperglycemia-induced
low NO availability, and becomes a critical event triggering
uncoupled state of VEGF-A–NO axis. In consistent with this
hypothesis, we could find reversal of VEGF-A–NO axis in
Figure 6. Masson trichrome staining of kidney tissue: (a) Control; (b) Diabetes; (c) Diabetes + MRS1754; (d) Control MRS1754. All images arerepresentative glomeruli at an original magnification of 400�.
Figure 8. Effect of A2B adenosine receptor antagonist (MRS1754) onrelative VEGF level in kidney tissue in different experimentalgroups. Notes: Data are means (±SEM). *p50.05 versus controlgroup; #p50.05 versus diabetes group; n¼ 6.
Figure 7. Glomerulosclerosis score in different experimentalgroups. Notes: Data are means (±SEM). *p50.05 versus controlgroup; #p50.05 versus diabetes group; n¼ 6.
DOI: 10.3109/0886022X.2014.900404 Role of A2B receptor inhibition in diabetic nephropathy 7
Ren
Fai
l Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Ond
okuz
May
is U
niv.
on
05/0
6/14
For
pers
onal
use
onl
y.

diabetic animals after 2 weeks of antagonist administration.
A2B antagonist may also directly modulate NO which is
indicated by increase in NO levels; 2 weeks after MRS1754
treatment.
The current literature evidence indicates Angiotensin II
(Ang II) as contributing factor to induce VEGF-A expression
in diabetic glomerular tissue. Intra-renal renin and angioten-
sinogen levels are increased in diabetic animals. High glucose
has been demonstrated to induce renin and Ang II production
in glomerular cells.28,29 A2BAR antagonist blocked the
progression of renal fibrosis generated in the ADA�/�animals; in mice infused with Ang II or subjected to unilateral
ureteral obstruction. These studies suggest a common patho-
genic pathway involving adenosine signaling in chronic
kidney disease.30 Recently, notch I signaling have also been
proved to increase VEGF-A and podocytopathy.31 Therefore,
it remained to be revealed if A2B receptors also interrelate
activation of the notch I signaling. Our major contribution
is to demonstrate that this pathogenic pathway occurs
early in the progression of the diabetic renal disease, and
supports the option of A2BAR as a pharmacological target
intervention in DN.
In patients with cancer treated with bevacizumab, a
humanized monoclonal anti-VEGF-A antibody, thrombotic
microangiopathy developed as a complication.32
Administration of A2B antagonist may inhibit pathological
change in VEGF, mediated by hypoxic diabetic environment
instead of the widespread non-specific inhibition which
might have resulted into adverse effects. The correlation
between the activities of these low-affinity receptors with
increased ligand availability have already been observed in
diabetic kidney in experimental animals.30 In accordance
with these findings, elevated adenosine levels in plasma of
clinically manifested DN patients were also found.33 Hence,
the measurement of adenosine levels in plasma and urine
or expression of these receptors in biopsy specimen could
represent a novel specific marker for early diagnosis or
patient at risk for development of DN. It has been described
that A2B receptor blockade reverses insulin resistance in
type II diabetes animal models and points toward another
therapeutic potential.34 A2B receptor blockers useful in
humans such as CVT-6883 are being developed in phase 1
trial,35 may be a novel alternative for the management of
DN patients.
Our studies do not exclude the possibility that A2B receptor
antagonist induce a favorable intraglomerular hemodynamic
effect to reduce proteinuria. It is possible that A2B receptor
antagonist mitigate proteinuria through direct effects by
blocking vasoactive inflammatory mediators. Additional
studies are necessary to address this issue.
In summary, our study demonstrates that administration
of selective A2B receptor antagonist attenuates renal dys-
function in DN. We believe that the renal tissue-protective
effect of A2B receptor antagonist is mediated primarily by
inhibiting induction of VEGF-A; thereby, preventing dis-
traction of VEGF–NO axis in diabetic kidney. We conclude
that A2B receptor antagonist represent a novel therapeutic
option for the treatment of diabetic kidney disease and for
potentially other diabetic complications; where VEGF-A is
the culprit.
Acknowledgments
The authors thank Dr. C. J. Joshi (Department of Animal
Biotechnology, Anand Agricultural University, Anand,
Gujarat, India) for providing facility to carry out gene
expression studies and ELISA experiments. They also thank
Ms. Pankti Desai (Department of Pharmaceutical Chemistry
and Analysis, Ramanbhai Patel College of Pharmacy) for
assistance in spectrofluorometric experiments. Authors are
also thankful to Dr D. J. Godasara (Department of Veterinary
Pathology, Anand Agricultural University, Anand, Gujarat,
India) for assistance in histopathological studies.
Declaration of interest
The authors state no conflict of interest.
References
1. Peter Rossing MDD. Diabetic nephropathy: worldwide epidemicand effects of current treatment on natural history. Curr DiabetesReports. 2006;6(6):479–483.
2. Futrakul N, Butthep P, Laohareungpanya N, Chaisuriya P,Ratanabanangkoon K. A defective angiogenesis in chronic kidneydisease. Renal Fail. 2008;30(2):215–217.
3. Schrijvers BF, Flyvbjerg A, De Vriese AS. The role of vascularendothelial growth factor (VEGF) in renal pathophysiology. KidneyInt. 2004;65(6):2003–2017.
4. Tsuchida K, Makita Z, Yamagishi S, et al. Suppression oftransforming growth factor beta and vascular endothelial growthfactor in diabetic nephropathy in rats by a novel advanced glycationend product inhibitor, OPB-9195. Diabetologia. 1999;42(5):579–588.
5. De Vriese AS, Tilton RG, Elger M, Stephan CC, Kriz W, LameireNH. Antibodies against vascular endothelial growth factor improveearly renal dysfunction in experimental diabetes. J Am Soc Nephrol.2001;12(5):993–1000.
6. Ho C, Hsu Y-C, Tseng C-C, Wang F-S, Lin CL, Wang JY.Simvastatin alleviates diabetes-induced VEGF-mediated nephro-pathy via the modulation of Ras signaling pathway. Renal Fail.2008;30(5):557–565.
7. Xu L, Kanasaki K, Kitada M, Koya D. Diabetic angiopathy andangiogenic defects. Fibrogen Tissue Repair. 2012;5:13. doi:10.1186/1755-1536-5-13.
8. Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEBJ. 2006;20(13):2242–2250.
9. Feoktistov I, Goldstein AE, Ryzhov S, et al. Differential expressionof adenosine receptors in human endothelial cells role of A2Breceptors in angiogenic factor regulation. Circ Res. 2002;90(5):531–538.
10. Valladares D, Quezada C, Montecinos P, et al. Adenosine A2Breceptor mediates an increase on VEGF-A production in rat kidneyglomeruli. Biochem Biophys Res Commun. 2008;366(1):180–185.
11. Nakagawa T. Uncoupling of the VEGF-endothelial nitric oxide axisin diabetic nephropathy: an explanation for the paradoxical effectsof VEGF in renal disease. Am J Physiol-Renal Physiol. 2007;292(6):F1665–F1672.
12. Palm F, Nordquist L, Wilcox CS, Hansell P. Oxidative Stress andHypoxia in the Pathogenesis of Diabetic Nephropathy. Studies onRenal Disorders. Springer; 2011:559–586.
13. Wu KK, Huan Y. Streptozotocin-induced Diabetic Models in Miceand Rats. Current Protocols in Pharmacology. John Wiley andSons; 2008:5.47.1–5.47.14.
14. Eisner C, Faulhaber-Walter R, Wang Y, et al. Major contribution oftubular secretion to creatinine clearance in mice. Kidney Int. 2009;77(6):519–526.
15. Rao AM, Dogan A, Hatcher J, Dempsey R. Fluorometric assay ofnitrite and nitrate in brain tissue after traumatic brain injury andcerebral ischemia. Brain Res. 1998;793(1):265–270.
8 L. Patel and A. Thaker Ren Fail, Early Online: 1–9
Ren
Fai
l Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Ond
okuz
May
is U
niv.
on
05/0
6/14
For
pers
onal
use
onl
y.

16. Kreckler LM, Wan TC, Ge Z-D, Auchampach JA. Adenosineinhibits tumor necrosis factor-a release from mouse peritonealmacrophages via A2A and A2B but not the A3 adenosine receptor.J Pharmacol Exp Therapeut. 2006;317(1):172–180.
17. Liu J, Jha P, Lyzogubov VV, Tytarenko RG, Bora NS, Bora PS.Relationship between complement membrane attack complex,chemokine (CC motif) ligand 2 (CCL2) and vascular endothelialgrowth factor in mouse model of laser-induced choroidalneovascularization. J Biol Chem. 2011;286(23):20991–21001.
18. Cardenas A, Toledo C, Oyarzun C, et al. Adenosine A2B receptor-mediated VEGF induction promotes diabetic glomerulopathy.Laboratory Invest. 2013;93:135–144.
19. Valladares D, Quezada C, Montecinos P, et al. Adenosine A2B
receptor mediates an increase on VEGF-A production in rat kidneyglomeruli. Biochem Biophys Res Commun. 2008;366(1):180–185.
20. Pawelczyk T, Grden M, Rzepko R, Sakowicz M, Szutowicz A.Region-specific alterations of adenosine receptors expression levelin kidney of diabetic rat. Am J Pathol. 2005;167(2):315–325.
21. Roa H, Gajardo C, Troncoso E, et al. Adenosine mediatestransforming growth factor-beta 1 release in kidney glomeruli ofdiabetic rats. FEBS Lett. 2009;583(19):3192–3198.
22. Hoshi S, Nomoto K-I, Kuromitsu J, Tomari S, Nagata M.High glucose induced VEGF expression via PKC and ERK inglomerular podocytes. Biochem Biophys Res Commun. 2002;290(1):177–184.
23. Karczewska J, Piwkowska A, Rogacka D, Stepinski J, Angielski S,Jankowski M. Purinergic modulation of glucose uptake intocultured rat podocytes: Effect of diabetic milieu. BiochemBiophys Res Commun. 2011;404(2):723–727.
24. Kang D-H, Kim Y-G, Andoh TF, et al. Post-cyclosporine-mediatedhypertension and nephropathy: amelioration by vascular endothe-lial growth factor. Am J Physiol-Renal Physiol. 2001;280(4):F727–F736.
25. Nakagawa T, Sato W, Sautin YY, et al. Uncoupling ofvascular endothelial growth factor with nitric oxide as a
mechanism for diabetic vasculopathy. J Am Soc Nephrol. 2006;17(3):736–745.
26. Baynes JW, Thorpe SR. Role of oxidative stress in diabeticcomplications: a new perspective on an old paradigm. Diabetes.1999;48(1):1–9.
27. Xia Z, Nagareddy PR, Guo Z, Zhang W, McNeill JH. AntioxidantN-acetylcysteine restores systemic nitric oxide availability andcorrects depressions in arterial blood pressure and heart rate indiabetic rats. Free Radical Res. 2006;40(2):175–184.
28. Singh R, Singh AK, Alavi N, Leehey DJ. Mechanism of increasedangiotensin II levels in glomerular mesangial cells cultured in highglucose. J Am Soc Nephrol. 2003;14(4):873–880.
29. Vidotti D, Casarini D, Cristovam P, Leite C, Schor N, Boim M.High glucose concentration stimulates intracellular renin activityand angiotensin II generation in rat mesangial cells. Am J Physiol-Renal Physiol. 2004;286(6):F1039–F1045.
30. Dai Y, Zhang W, Wen J, Zhang Y, Kellems RE, Xia Y. A2Badenosine receptor-mediated induction of IL-6 promotes CKD.J Am Soc Nephrol. 2011;22(5):890–901.
31. Lin C-L, Wang F-S, Hsu Y-C, et al. Modulation of notch-1signaling alleviates vascular endothelial growth factor-mediateddiabetic nephropathy. Diabetes. 2010;59(8):1915–1925.
32. Schraermeyer U, Julien S. Formation of immune complexes andthrombotic microangiopathy after intravitreal injection of bevaci-zumab in the primate eye. Graefes Arch Clin Exp Ophthalmol.2012;250(9):1303–1313.
33. Xia J-F, Liang Q-L, Hu P, Wang Y-M, Li P, Luo G-A. Correlationsof six related purine metabolites and diabetic nephropathyin Chinese type 2 diabetic patients. Clin Biochem. 2009;42(3):215–220.
34. Figler RA, Wang G, Srinivasan S, et al. Links between insulinresistance, adenosine A2B receptors, and inflammatory markers inmice and humans. Diabetes. 2011;60(2):669–679.
35. Kalla RV, Zablocki J. Progress in the discovery of selective, highaffinity A2B adenosine receptor antagonists as clinical candidates.Purinergic Signal. 2009;5(1):21–29.
Supplementary material available online
Supplementary Figures I–IV
DOI: 10.3109/0886022X.2014.900404 Role of A2B receptor inhibition in diabetic nephropathy 9
Ren
Fai
l Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Ond
okuz
May
is U
niv.
on
05/0
6/14
For
pers
onal
use
onl
y.