the estrogen receptor cofactor spen functions as a tumor ... · the nuclear hormone receptor family...
TRANSCRIPT

Molecular and Cellular Pathobiology
The Estrogen Receptor Cofactor SPEN Functionsas a Tumor Suppressor and Candidate Biomarkerof Drug Responsiveness in Hormone-DependentBreast CancersSt�ephanie L�egar�e1,2, Luca Cavallone2, Aline Mamo2, Catherine Chabot2,Isabelle Sirois1,2, Anthony Magliocco3, Alexander Klimowicz3, Patricia N. Tonin4,5,Marguerite Buchanan2, Dana Keilty1,2, Saima Hassan1,2, David Laperri�ere6,Sylvie Mader6,7, Olga Aleynikova8, and Mark Basik1,2
Abstract
The treatment of breast cancer has benefitted tremendouslyfrom the generation of estrogen receptor-a (ERa)–targetedtherapies, but disease relapse continues to pose a challengedue to intrinsic or acquired drug resistance. In an effort todelineate potential predictive biomarkers of therapy respon-siveness, multiple groups have identified several uncharacter-ized cofactors and interacting partners of ERa, including SplitEnds (SPEN), a transcriptional corepressor. Here, we demon-strate a role for SPEN in ERa-expressing breast cancers. SPENnonsense mutations were detectable in the ERa-expressingbreast cancer cell line T47D and corresponded to undetectableprotein levels. Further analysis of 101 primary breast tumorsrevealed that 23% displayed loss of heterozygosity at the SPENlocus and that 3% to 4% harbored somatically acquired muta-tions. A combination of in vitro and in vivo functional assays
with microarray-based pathway analyses showed that SPENfunctions as a tumor suppressor to regulate cell proliferation,tumor growth, and survival. We also found that SPEN bindsERa in a ligand-independent manner and negatively regulatesthe transcription of ERa targets. Moreover, we demonstrate thatSPEN overexpression sensitizes hormone receptor–positivebreast cancer cells to the apoptotic effects of tamoxifen, buthas no effect on responsiveness to fulvestrant. Consistent withthese findings, two independent datasets revealed that highSPEN protein and RNA expression in ERa-positive breasttumors predicted favorable outcome in patients treated withtamoxifen alone. Together, our data suggest that SPEN isa novel tumor-suppressor gene that may be clinically usefulas a predictive biomarker of tamoxifen response in ERa-pos-itive breast cancers. Cancer Res; 75(20); 4351–63. �2015 AACR.
IntroductionApproximately 70% of breast cancers express the estrogen
receptor-a (ERa) and/or its transcriptional target, the progester-one receptor (PgR), and are dependent on hormones for their
growth, proliferation, and survival (1). The ERa is a member ofthe nuclear hormone receptor family of ligand-dependent tran-scription factors that regulates gene transcription in the presenceof its ligand by binding cis-regulatory motifs, also known asestrogen response elements, lying upstream of target genes or viatethering to DNA by other transcription factors, including AP1and SP1. DNA-bound ERa recruits the basal transcriptionalmachinery and induces the expression of genes implicated innumerous cancer signaling pathways, a process tightly regulatedby complex dynamic interactions with coactivators and corepres-sors (2, 3). Although the ERa has been shown to be one of themost important mitogenic drivers in breast cancer in a multitudeof preclinical and clinical studies, genomic events affecting theESR1 gene are only observed in metastatic breast cancers (4, 5).Interestingly, mutations and chromosomal aberrations appear tooccur at higher rates in coactivators (e.g., AIB1) and corepressors(e.g., GATA3, NCOR1, and NCOR2) of the ERa, suggesting thattheir regulation of the receptor's genomic actions may be inti-mately linked to the development of hormone-dependenttumors.
With protumorigenic functions affecting proliferation,growth, and survival, the ERa is the main oncogenic driver inbreast cancer and represents an important target for the treat-ment of hormone-responsive tumors. Therapies targeting the
1Department of Surgery and Oncology, McGill University, Montr�eal,Qu�ebec, Canada. 2Department of Oncology and Surgery, Lady DavisInstitute for Medical Research, Montr�eal, Qu�ebec, Canada. 3Depart-ment of Pathology, University of Calgary, Calgary, Alberta, Canada.4Department of Human Genetics, McGill University and The ResearchInstitute of the McGill University Health Centre, Montr�eal, Qu�ebec,Canada. 5Department ofMedicine,McGill UniversityandTheResearchInstitute of the McGill University Health Centre, Montr�eal, Qu�ebec,Canada. 6Institut de recherche en immunologie et canc�erologie, IRIC,Montr�eal, Qu�ebec, Canada. 7Department de Biochimie, Universit�e deMontr�eal, Montr�eal,Qu�ebec,Canada. 8Department of Pathology, Jew-ish General Hospital, Montr�eal, Quebec, Canada.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author: Mark Basik, 3755 Chemin de la Cote Ste-Catherine,RoomE421, Montr�eal, QC H3T 1E2, Canada. Phone: 514-340-8222, ext. 4210; Fax:514-340-8716; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-14-3475
�2015 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 4351
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

ERa in breast cancer include antiestrogens, such as tamoxifenand fulvestrant, and inhibitors of estrogen biosynthesis (aro-matase inhibitors). Tamoxifen is a selective ER modulator thatcompetes with estrogen for the ligand-binding domain of theER and induces an alternative conformation that preventscoactivator binding to the receptor but favors recruitment andinteraction with corepressors (6). Although tamoxifen hasbeen successfully used to treat both early and late hormonereceptor–positive breast cancers, more than 50% of patientswith advanced hormone-responsive tumors will progress orexperience disease relapse due to intrinsic and acquired resis-tance to tamoxifen (7). Although many mechanisms of resis-tance to tamoxifen have been characterized in vitro, none haveshown clinical utility as biomarkers guiding treatment withtamoxifen in patients with ERa- and/or PgR-positive tumors.
Over the last decades, a number of previously uncharacterizedERa partners and cofactors were identified, including Split Ends(SPEN), a protein with essential regulatory roles in transcriptio-nal repression (8–12). SPEN, which is also known as SMRT/HDAC1-associated repressor protein (SHARP), is a large nuclearprotein of 402 kDa characterized by four N-terminal RNA-recog-nition motifs and a highly conserved C-terminal SPOC (SpenParalog and Ortholog C-terminal) domain. In various modelorganisms, SPEN has been shown to be critical during embryo-genesis and throughout development, in part, due to its regula-tion of the Notch, TCF/LEF, and EGFR signaling pathways(8, 9, 13). SPEN has also been identified as an estrogen-induciblecofactor able to integrate nuclear hormone receptor activationand repression (10). Despite some evidence for SPEN beingimplicated in endocrine regulation and development, SPEN func-tions have not been investigated in breast cancer.
Using an innovative unbiased integrative genomic approach,we identified mutations in the SPEN gene in a breast cancer cellline and four primary breast tumors (14). We also found thatSPEN inhibits tumor growth and modulates the transcriptionof ERa-target genes, including PGR and BCL2. Moreover, wedemonstrate that SPEN expression predicts response to tamox-ifen in vitro and in clinical samples. Together, our findings showthat SPEN is a novel tumor-suppressor gene and a candidatepredictive biomarker of tamoxifen response in ERa-positivebreast cancers.
Materials and MethodsCell lines
MCF-7 and T47D cells were obtained from the ATCC and werecultured in DMEM supplemented with 10% fetal bovine serum(FBS; Wisent). BT20 and MDA-MB-436 were obtained from theATCC and cultured in EMEM and RPMI-1640 from ATCC sup-plemented with 10% FBS, respectively.
Cell viability assayCells were plated at a density of 2,500 cells per well in 96-well
plate for experiments performed in DMEM supplemented with10%FBS and5,000 cells perwell in 96-well plate for assays carriedunder 1% FBS conditions. At 1, 2, 4, and 7 days, cell viability wasassessed by replacing the medium with a 10% Alamar Bluesolution (Invitrogen) prepared in DMEM. Cells were incubatedwith the solution at 37�C for 4 hours and fluorescence measuredwith a plate reader, FLUOstar Optima, using 560 nm (Excitation)and 590 nm (Emission) filter settings.
Colony formation assayCells were seeded at a density of 5,000 cells per well in 6-well
plates in 1 mL of soft agar (0.3% soft agar in DMEM with 10%FBS), plated onto 2mLof solidified agar (0.7% soft agar inDMEMwith 10% FBS) and incubated at 37�C. Medium (250 mL) wassupplemented to each well twice weekly for 4 weeks. Colonieswere scored electronically using an automated cell colony counter(GelCount; Oxford Optronix).
PlasmidsThe full-length human SPEN cDNA cloned into the
pDream2.1/MCS expression vector was purchased from Gen-Script. SPEN-expressing vector and the empty control vector weretransfected in T47D cells using Attractene (Qiagen) accordingto the manufacturer's protocol. After 72 hours, cells were seededin 6-well plates in DMEM with 10% FBS supplemented withneomycin (Life Technologies) at a concentration of 250 or500 mg/mL Clones were isolated and maintained in selectionmedia throughout culturing period.
SPEN knockdown by RNA interferenceTwo SPEN MISSIONshRNA plasmids with the follow-
ing sequences: SPEN shRNA 1, 50-CCGGCCTGTGGTAAAGGT-GGTGTTTCTCGAGAAACACCACCTTTAC-CACAGGTTTTTG-30
(TRCN0000075165) and SPEN shRNA 2, 50-CCGGCGGCTC-CATCATCAATGACATCTCGAGAT-GTCATTGATGAT-GGAGCC-GTTTTTG-30 (TRCN0000075166) were purchased from Sigma-Aldrich. pLKO.1-puro Non-Target Control and SPEN-specificshRNA plasmids were transfected in MCF-7 cells using Attrac-tene (Qiagen). After 72 hours, cells were seeded in 6-well platesin DMEM plus 10% FBS supplemented with puromycin (LifeTechnologies) at a concentration of 0.5, 1.0, or 2.0 mg/m.Clones were isolated and maintained in selection mediathroughout the culturing period.
RNA isolation and qRT-PCRTotal RNA was isolated from subconfluent cultures using the
RNeasy Mini Kit (Qiagen Sciences) and reversed transcribed intocDNA with the iScript cDNA Synthesis Kit (Bio-Rad). qRT-PCRwere performed with TaqMan Gene Expression Assays (AppliedBiosystems) and with the following probes: SPEN (Invitrogen,Hs00209232_m1), PGR (Invitrogen, Hs01556702_m1), and18S(Invitrogen, Hs99999901_s1).
DNA microarray expression profiling and analysisExpression profiling was conducted according to the manu-
facturer's instructions and following the One-Color Microarray-Based Gene Expression Analysis Protocol from Agilent Tech-nologies. Briefly, integrity and concentration of the input RNAwas evaluated with the Agilent 2011 Bioanalyzer. One hundrednanograms of total RNA was reverse transcribed into cRNA,amplified and labeled with Cy3 dye. The resulting labeled cRNAwas purified using the RNeasy Mini Kit (Qiagen Sciences)according to the instructions of the manufacturer and hybrid-ized to a Sure Print G3 Human GE 8�60 K Microarray (AgilentTechnologies) for 17 hours at 65�C. The array was then washedand scanned on the Agilent DNA Microarray scanner at aresolution of 3 mmol/L. Images were extracted and normalizedwith Feature Extraction software version 9.5. Expressionvalues of three biologic RNA replicates for each probe in the
L�egar�e et al.
Cancer Res; 75(20) October 15, 2015 Cancer Research4352
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

expression array were analyzed using Gene Spring GX (AgilentTechnologies).
Statistical analysisMicroarray expression data were processed with GeneSpring
GX software. Data were normalized to the 75th percentile of allvalues on the microarrays and to the median expression levels ofall samples. The normalized gene expression data were filtered onflags and only those classified as detectedwere allowed to pass thefilter and included in the analysis. The expression profiles of genesdifferentially expressed by more than 1.5-fold based on threebiologic replicates were compared using two-tailed unpairedt tests.
Ingenuity pathway analysisMicroarray data were analyzed using the Ingenuity Pathways
Analysis (IPA) software available at: http://www.ingenuity.com. Genes up- or downregulated by at least 1.50-fold (P <0.05) in Spen 1, SPEN sh1, and SPEN sh3 compared with theirrespective controls were considered for further analyses. Func-tions (P < 0.05) predicted to be increased (z-score > 0.75) inSPEN sh1 and SPEN sh3 relative to the nontarget shRNAtransfected MCF-7 clone (NT2) and anticipated to be decreased(z-score < �0.75) in Spen 1 compared with its empty vectortransfected T47D clone (CTL1) were analyzed. Functions con-sistently modulated were considered as regulated by SPEN.Similarly, an analysis of the upstream regulators predicted tobe responsible for the observed gene expression changes wasconducted. All molecule types were included in the analysisexcept for chemicals and miRNAs. Upstream regulators (P <0.05) predicted to be activated (z-score > 0.75) in SPEN sh1 andSPEN sh3 relative to the nontarget shRNA-transfected MCF-7clone (NT2) and anticipated to be inhibited (z-score < �0.75)in Spen 1 compared with its empty vector T47D control (CTL1)were analyzed. Those common to all pairs of cell lines wereconsidered as modulated by SPEN. The expression data oftumors from The Cancer Genome Atlas (TCGA) database werealso analyzed with IPA. For this analysis, a log2 ratio of 2 wasused to obtain a list of approximately 3,000 differentiallyexpressed genes between the tumor and normal-matched mam-mary sample. In each case, the predicted score of activation(z-score) was computed for the ER and plotted against the log2ratio of SPEN RNA expression in tumors compared with nor-mal-matched breast tissue samples. A log2 ratio of �0.5 wasused to stratify patients into high and low SPEN-expressinggroups.
Fluorescence microscopy to quantify cells with chromatincondensation and membrane permeabilization
Cells were seeded at a density of 2.5 � 105 cells per well in 6-well plates and incubated at 37�C. Seventy-two hours after plat-ing, 20 mL of Hoechst 33342 (HO, 100 mg/mL; Sigma) was addedto the culture medium and plates incubated at 37�C for 10minutes. Cells were then stained with 2 mL of propidium iodide(PI; 5 mg/mL; Sigma) and analyzed by fluorescence microscopyon an Axiovert 40 CFL (Zeiss) microscope. The percentage ofnormal, apoptotic, and necrotic cells were estimated in six ran-dom fields per condition. Apoptotic cells showed highly pyknoticnuclei stained with either HO or PI in the early and late phases ofapoptosis, respectively.
Survival assaysFor experiments with tamoxifen, cells were seeded at a density
of 2,500 cells per well in 96-well plates in complete medium andincubated at 37�C. Twenty-four hours after plating, the mediumwas replaced with 100 mL of hormone-depleted medium [DMEMwithout phenol red (Gibco) supplemented with 10% charcoal-stripped FBS (Gibco)]. Another 24 hours later, 4-hydroxy-tamox-ifen (Sigma-Aldrich Inc.) or its vehicle was added to each well in100 mL of hormone-depleted medium. Cell viability was mea-sured 5 days later with a 10% Alamar Blue solution.
ImmunoprecipitationCells were rinsed with ice-cold PBS, harvested, and lysed in
lysis buffer (250 mmol/L NaCl, 0.5% NP-40, 5 mmol/L EDTA,50 mmol/L Tris) freshly supplemented with protease inhibitors[5 mmol/L sodium fluoride, 1 mmol/L sodium orthovanadate,1 mmol/L phenylmethylsulfonylfluoride, 10 mg/mL aprotinin,and 10 mg/mL leupeptin]. After Micro-BCA quantification(Thermo Scientific), at least 250 mg of proteins were incubatedovernight with 1 mg of anti-SPEN antibody (Sigma; HPA015825)or IgG control antibody (Abcam; ab46540-1). Protein A Dyna-beads beads (Life Technologies) were added for 1.5 hours andwashed three times with 400 mL of lysis buffer. Precipitated beadswere then incubated at 95�C for 7 minutes in 60 mL of Laemmlisample buffer followed by a centrifugation at 13,000 rpm for 1minute. Proteinswere ran in a precast 4% to20%gel (Bio-Rad) for4 hours at 120 V and transferred to a nitrocellulose membraneovernight at 33 V. Membranes were blocked with 5% BSA in TBSTand immunoblotted overnight with an anti-SPEN antibody (Sig-ma; HPA015825, 1:500).
Western blottingSubconfluent cells were collected by trypsinization, washed in
ice-cold PBS, and lysed in lysis buffer freshly supplemented withprotease inhibitors. Lysates were then placed on a rocker machinefor 30minutes and centrifuged for 5minutes at 4�C. Supernatantswere subjected to Bradford quantification and 50 mg of proteinswere loaded and ran by SDS-PAGE in an 8% gel for 1 hour.Proteins were transferred to nitrocellulose membranes and incu-bated with PgR (1:1,000; Cell Signaling Technology) and a-tubu-lin (1:10,000; Abcam) antibodies at 4�Covernight. Protein bandswere detected using the Amersham ECL Western Blotting Detec-tion Reagent.
Clonogenic assaysCells were seeded at a density of 2.5 � 104 cells per cm2 and
grown in DMEM supplemented with 10% FBS for 24 hours.Media was then replaced with 2 mL of hormone-depleted medi-um [DMEMwithout phenol red (Gibco) supplementedwith 10%charcoal-stripped FBS (Gibco)]. Another 24 hours later, 5 or10 mmol/L of tamoxifen or its vehicle was added to the cells inhormone-depleted medium. Cells were stained with a fixingsolution containing crystal violet 5 days after the addition of thedrug.
Stimulation with 17b-estradiolCells were seeded at a density of 5,000 cells per well in
96-well plates in complete medium and incubated at 37�C.Twenty-four hours after plating, the medium was replaced with200 mL of hormone-depleted medium [DMEM without phenol
SPEN Is a Tumor Suppressor in ERa-Positive Breast Cancers
www.aacrjournals.org Cancer Res; 75(20) October 15, 2015 4353
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

red (Gibco) supplemented with 10% charcoal-stripped FBS(Gibco)]. Another 24 hours later, media was replaced with1 nmol/L 17b-estradiol in hormone-depleted DMEM plus 10%charcoal-stripped FBS for 24 hours. Cell viability was measuredat day 0, 2, 5, and 7 with a 10% Alamar Blue solution and foldinduction in proliferation by estradiol compared with 100%control was determined by dividing fluorescence valuesobtained in the presence of estrogen over that obtained withthe vehicle.
Patient cohortThe tamoxifen 50/50 tissuemicroarray (TMA) cohort is a power
series, derived from the Calgary Tamoxifen Breast Cancer Cohort(Cal-TBCC), which includes 50 patients alive 5 years after diag-nosis and 50 patients who died of breast cancer within 5 years ofdiagnosis. The Cal-TBCC is a retrospective database and containsdemographic data, clinical data, and pathologic data from aparent cohort of 819 breast cancer patients diagnosed between1985 and 2000 at the Tom Baker Cancer Center in Calgary,Alberta, Canada. Inclusion criteria: confirmed diagnosis of inva-sive breast carcinoma and treatment with primary surgical inter-vention with or without postoperative local radiotherapy, fol-lowed by adjuvant tamoxifen endocrine therapy (20 mg orally/day) for 5 years, regardless of ER or PgR status. HER2 status wasnot systematically performed at the time of diagnosis for thepatients in this cohort. Exclusion criteria: if diagnostic biopsy orprimary surgical tissue specimens were unavailable, if patientsreceived prior or adjuvant chemotherapy. A total of 534 cases metthe criteria with a median follow-up time of 82.1 months. Theclinical and initial pathology data were retrieved, data for pro-gression-free survival at 5 years were retrieved, and formalin-fixedparaffin-embedded tissue blocks were acquired and replicate0.6-mm cores were built into TMAs. All tissues were fixed in10% neutral buffered formalin and embedded in paraffin accord-ing to standard procedures for the time period. Ethical approvalfor the use of human tissue samples was obtained from theConjoint Health Research Ethics Board.
Breast tumorsOne hundred consecutive breast tumors were collected as
part of a government-funded (FRSQ) tumor bank at the CentreHospitalier de l'Universit�e de Montr�eal (Montr�eal, QC, Canada)from 2000 to 2003. Patients had signed informed consent forbreast tumor banking. Tissues sections for each tumor showed>70% tumor cells as determined by an H&E staining for eachsample. DNA was extracted using the Qiagen DNAmp extractionkit according to the manufacturer's instructions.
aCGH of breast tumorsDNA quality was assessed using a 2100 bioanalyzer using a
DNA 12000 Lab ChIP Kit (Agilent Technologies). Array compar-ative genomic hybridization (aCGH) was performed as previous-ly reported (15).
Loss-of-heterozygosity assessment using polymorphicmicrosatellite repeat markers
Loss-of-heterozygosity (LOH) analysis for the region sur-rounding 1p36.1 was performed using the AFM217zc3a poly-morphic marker (Supplementary Table S4). PCR was per-formed in 12.5-mL volume containing 100 ng of genomic DNA,
1mCi of 35S[dATP], 1� PCR buffer, 200 nm each of dCTP,dGTP, and dTTP, 50 pmol of each primer, and 0.5 U of Taqpolymerase. The PCR conditions were as follow: 94�C for5 minutes (1 cycle), 94�C for 30 seconds, 60�C for 30 seconds,and 72�C for 30 seconds (29 cycles) followed by an incubationat 72�C for 10 minutes (1 cycle). Products were electrophoresedon denaturing gels and autoradiographed at room temperaturefor 5 days. LOH was scored on the basis of the difference in therelative intensity of signals representing two alleles in tumorDNA samples. All samples positive for LOH or allelic imbal-ance at individual loci were analyzed twice in independentassays and Sanger sequenced.
Mutation analysis of SPENMutation analyses were performed as previously described
(14). Briefly, the genomic sequence of the protein-encodingregion of SPENwas obtained from the UCSC Santa Cruz GenomeBioinformatics Site available at: http://genome.ucsc.edu. Primerpairs for PCR amplification and sequencing of SPEN were gen-erated using the Primer3 software available at: http://bioinfo.ut.ee/primer3-0.4.0/ (Supplementary Table S5). PCR productsderived from breast tumors were purified as per the manufac-turer's recommendations using theQIAquick PCRPurification Kit(Qiagen Sciences) and sequenced at the McGill University (Mon-tr�eal, QC, Canada) and Genome Quebec Innovation Center andthe TGEN DNA sequencing facility. Sequence chromatogramswere aligned and analyzed with the Staden Package (http://staden.sourceforge.net/) and the Mutation surveyor software ver-sion 3.24 by using the full-length SPEN reference sequenceNM_015001.
Fluorescence-activated cell sortingFor cell-cycle analyses, cells were detached with trypsin,
washed in PBS supplemented with 5 mmol/L EDTA, suspendedin a fixing solution (1 mL of PBS, 5 mmol/L EDTA for 3 mL of100% ethanol), and incubated at �20�C for at least 24 hours.Then, cells were washed with PBS/EDTA and resuspended in 2mL of staining solution (PBS, 50 mg/mL PI, and 20 mg/mLRNAse A). For Annexin V/PI staining experiments, cells weredetached with trypsin and washed once with PBS. Then, theywere resuspended in 200 mL of Annexin V binding solution(1�) and stained with 4 mL of PI and 4 mL of Annexin V. Forboth assays, cell fluorescence signals were determined imme-diately after staining using a FACScalibur flow cytometer (Bec-ton Dickinson). The analysis was performed using the BDCellQuest (Becton Dickinson), ModFit (Becton Dickinson),and FlowJo software.
Intersection probabilitiesTo determine the statistical significance of intersection between
two lists of genes, we assessed the probability of this intersectionto occur by performing 10,000 independent simulations withrandomly selected lists of genes of the same size. P values werecalculated using a hypergeometric test.
Chromatin immunoprecipitationCells were plated (1.2 � 106 cells in 150-mm culture dishes)
and grown for 72 hours inDMEMcontaining 10%FBS. Cells werefixed at room temperature for 30 minutes in 2 mmol/L Ethyleneglycol bis(succinimidyl succinate) (16) followedwith 10minutes
L�egar�e et al.
Cancer Res; 75(20) October 15, 2015 Cancer Research4354
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

in 1% formaldehyde. Glycine was added (125 mmol/L finalconcentration) and left at room temperature for 5 minutes toquench formaldehyde. Cells were rinsed twice and scraped in ice-cold PBS. Collected cells were then centrifuged at 1,400 rpm for 5minutes and lysed into lysis buffer (50 mmol/L HEPES-KOH pH7.5, 140 mmol/L NaCl, 1 mmol/L EDTA, 1% Triton X-100, 0.1%sodium deoxycholate, 0.1% SDS) freshly supplemented withprotease inhibitors. Sampleswere incubated on ice for 10minutesand sonicated to obtain fragments of 300 to 1,000 bp in size(setting: 25%, 10 times 15 seconds). After centrifugation (15minutes 13 000 rpm, 4�C), the sheared chromatin was dilutedin dilution buffer (1% Triton X-100, 2 mmol/L EDTA pH 8, 20mmol/L Tris–HCl pH 8, 150mmol/L) freshly supplemented withprotease inhibitors. Samples were then incubated with 1.5 mg ofantibody (SPEN: HPA015825, Sigma ERa: sc-543, Santa CruzBiotechnology, IgG: ab46540-1, Abcam) at 4�C with rotationovernight in a final volume of 1 mL. DNA–protein interactionswere washed three times for 10 minutes with wash buffer (0.1%,1% Triton X-100, 2mmol/L EDTA pH 8, 20mmol/L Tris–HCl pH8, 150 mmol/L NaCl) and once for 10 minutes with final washbuffer (0.1%, 1%TritonX-100, 2mmol/L EDTApH8, 20mmol/LTris–HCl pH 8, 500 mmol/L NaCl). Chromatin was eluted with350 mL of elution buffer (1% SDS, 100 mmol/L NaHCO3) byrotation for 15 minutes and reverse cross-linked with 4 mL ofproteinase K solution at 55�C for 90 minutes followed by anovernight incubation at 65�C. DNA was purified using theDNeasy Mini Spin columns (Qiagen) and following the manu-facturer's instructions. PCR amplification of a DNA fragment, 311kb upstream of the PGR transcription start site was performedusing the following primers: forward 50-CCA CTT TGC CAC ATGACA TC REV-30 REV 50-AAC TCC CAA GGG ACC ATT TC-30.
ResultsSPEN is mutated and underexpressed in breast tumors
Using an integrative genomic approach, we previously identi-fied a number of genes containing nonsense mutations andmapping to regions exhibiting LOH in breast cancer cell lines(14, 17). Briefly, this approach involved the profiling of cell linestreated with emetine, an inhibitor of the nonsense-mediated RNAdecay (NMD) pathway (17). Genes whose transcript levels wereincreased by emetine treatment potentially contained a nonsensemutation andwere prioritized on the basis of those that aligned toregions of LOH or deletion as assayed by genotyping with single-nucleotide polymorphisms (SNP) arrays. Using this strategy, weidentified and further established the AT-rich interactive domain1a (ARID1A) as a candidate tumor-suppressor gene in breastcancer, a finding that has since been confirmed by the discoveryofARID1Amutations in breast tumors by several groups (16, 18).Similarly, we identified an insertion/truncation mutation atnucleotide 1184 in the SPEN gene and LOH at the SPEN locus(chromosome 1p36), resulting in undetectable SPEN proteinlevels in the ERa-positive, T47D breast cancer cell line (Supple-mentary Fig. S1A–S1C).
To assess the prevalence of somatically acquired genomicaberrations and mutations affecting SPEN in breast cancer, weperformed aCGH or microsatellite PCR at the AFM217zc3a poly-morphicmarker on a cohort of 101primary breast tumors (Fig. 1Aand Supplementary Table S1). We found that 22.8% (23 of 101)of tumors had LOH or a copy number loss at the SPEN locus.Sequencing of the protein-encoding exons and splice-site junc-
tions of SPEN revealed that 17.4% (4 of 23) of tumors with LOHharbored a somatic mutation within the gene. Two missensemutations (P2158A, A3327T) were detected in two differentsamples, whereas the same nonsense mutation (Q3141X) wasfound in two other tumors (Fig. 1B and C).
To corroborate the prevalence of genomic and genetic altera-tions affecting SPEN, we then surveyed data from the TCGA andthe catalogue of somatic mutations in cancer (COSMIC). Wefound that SPEN RNA expression is downregulated in invasivebreast carcinoma compared with normal breast tissue (P ¼1.43E�07) and that SPEN is homo- or heterozygously deletedin 27.2% of breast cancer samples (Supplementary Fig. S2A andS2B; refs. 19–22), similar to the prevalence of LOH in ourown tumor set. Data analysis also revealed that tumors har-boring nonsense mutations in SPEN express low to very lowSPEN mRNA levels, even in the absence of copy number lossand that breast cancer samples with LOH at the 1p36 locusexpress significantly lower SPEN transcript levels comparedwith tumors without copy number alterations (SupplementaryFig. S2B; refs. 20, 22). In both databases, somatic mutations inSPEN were reported in 2% to 3% of breast cancers as well as inmany other cancer types, with a prevalence reaching 14.3% and11% in cervical and endometrial cancers, respectively (Supple-mentary Fig. S2C; ref. 23).
SPEN acts as a tumor-suppressor gene in ER-positive breastcancer cells
Given the suggested role of SPEN in endocrine regulationand our identification of an insertion–truncation mutation inSPEN in the ERa-positive T47D breast cancer cell line, weattempted to investigate the functions of SPEN in the contextof ERa-positive breast cancers. We first transfected a SPEN-expressing vector in T47D cells and isolated two stable clones(T47D-Spen 1–2; Fig. 2A and B), with varying degrees ofrestored SPEN expression. T47D-SPEN cells displayed a markedreduction of proliferation in both normal and low serumconditions (Fig. 2C and Supplementary Fig. S3A) and exhibitedincreased sensitivity to apoptosis under serum-starved growthconditions (Supplementary Fig. S3B). Reintroduction of SPENinto T47D cells did not lead to their accumulation in G1 or G2
either in 10% or 1% FBS conditions, suggesting that theobserved effects are not due to cell-cycle arrest (SupplementaryFig. S3C and S3D). A soft agar assay revealed that restoration ofSPEN levels in T47D cells abrogates anchorage-independentgrowth (Fig. 2D). To further establish SPEN as a tumor-sup-pressor gene in breast cancer, in vivo xenografts studies wereperformed with BALB-c nude mice implanted with 60 days slowrelease 17b-estradiol pellets and injected with T47D controlcells in one mammary fat pad and T47D-SPEN cells injectedcontralaterally. Of a total of 8 mice that developed palpabletumors, 7 developed tumors from control cells without tumorsarising from contralateral SPEN-overexpressing clones (Fig. 2Eand Supplementary Fig. S3E and S3F). Notably, only one tumorderived from T47D-SPEN cells was collected after completionof the study and its volume was much smaller than controltumors. Together, these in vitro and in vivo results support thetumor-suppressive functions of SPEN in breast cancer.
Then, to further characterize SPEN functions in ERa-positivebreast cancer cells, we silenced its expression in another hor-mone-dependent breast cancer cell line, MCF-7, which expresshigh endogenous levels of the protein (Supplementary Fig. S1B
SPEN Is a Tumor Suppressor in ERa-Positive Breast Cancers
www.aacrjournals.org Cancer Res; 75(20) October 15, 2015 4355
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

and S1C). Using two different short-hairpin RNAs targetingSPEN, four MCF-7-shRNA-SPEN clones showing decreasedSPEN expression were generated (Fig. 3A and B and Supple-mentary Fig. S3G–S3I). shRNA-mediated knockdown ofSPEN slightly increased proliferation (Supplementary Fig.S3J) but markedly increased colony formation in soft agarassays (Fig. 3C and Supplementary Fig. S3K) and reduced therate at which MCF-7 cells undergo apoptosis under low serumconditions (Supplementary Fig. S3L). As in T47D-SPEN clones,no significant difference in cell-cycle distribution was observed
between MCF-7-shRNA-SPEN clones and their control (datanot shown). The results of the functional assays performed withMCF-7 cells are complementary to those obtained with T47Dcells and demonstrate that SPEN regulates proliferation, tumorgrowth, and survival in ERa-positive breast cancer cells. Inter-estingly, using a cohort of 1,784 breast cancer patients withluminal A tumors, we assessed the clinical significance of SPENexpression and found that high SPEN RNA levels were signi-ficantly associated with good survival (HR, 0.78; P ¼ 0.005)over 20 years (Fig. 3D; ref. 24).
Figure 1.SPEN genomic alterations and mutations in breast cancer. A, representative image depicting copy number anomalies of chromosome 1 in a breast tumorsample measured using the 244 K aCGH microarray. Individual probe values are represented as normalized log2 ratios. Red dots represent probeswith increased copy number values (log2 ratio > 1) and green dots represent probes with decreased copy number values (log2 ratio < �1). DNA copynumber losses are apparent at the telomeric end of the 1p arm, including the genomic locus containing SPEN. B, sequencing chromatograms of SPEN atthe site of mutation in the four primary tumor samples (bottom) and their corresponding normal tissues (top). Arrows, mutation sites in tumor samples.C, schematic diagram of SPEN protein domains (RRM, RNA recognition motif; NLS, nuclear localization sequence; RID, receptor interaction domain; SPOC,SPEN paralog and ortholog C-terminal domain), and locations of somatically acquired mutations resulting in the amino acid alterations, P2158A, Q3141X, andA3327T found in the sequence analysis of breast tumors.
L�egar�e et al.
Cancer Res; 75(20) October 15, 2015 Cancer Research4356
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

Then, to evaluate whether the tumor-suppressive function ofSPEN extends to ER-negative breast cancer cells, we silenced itsexpression using siRNAs in BT20 and MDA-MB-436, two triple-negative breast cancer cell lines expressing high RNA and proteinlevels of SPEN (Supplementary Fig. S4A and S4B), and measuredSPEN effects on proliferation. Contrary to our expectations, wefound that SPEN knockdown limited the proliferation of both celllines in proliferation assays, suggesting that SPEN may haveproproliferative functions in ERa-negative breast cancer cells(Supplementary Fig. S4C–S4F). Consistent with these findingsis the observation that SPEN RNA expression levels are predictiveof poor prognosis (HR, 1.49; P ¼ 0.016) in a cohort of 581patients with basal breast tumors (Supplementary Fig. S4G;ref. 24). Although further experiments are required to define theroles of SPEN in basal breast cancers, our results suggest that SPENhas opposing functions in ERa-positive and ERa-negative cancercells while its expression may serve as a useful marker for patientprognostication and stratification in both subtypes.
SPEN regulates the expression of genes related to cell deathTo delineate the transcriptional program regulated by SPEN
in ERa-positive breast cancers, gene expression profiling usingDNA microarrays was conducted on RNA from one of the twoclones generated with the SPEN expression vector (Spen 1, thehighest expressor), as well as one of the two clones generatedwith each of the shRNA hairpin vectors, SPEN shRNA1 (SPENsh1) and SPEN shRNA2 (SPEN sh3), along with their respec-tive controls. Microarray results confirmed the knockdown oroverexpression of SPEN in each stably transfected clone (Sup-plementary Table S2). IPA performed with genes significantly(P < 0.05) altered (�1.5-fold) in each clone compared withits control revealed that the largest proportion of genes(32%–37%) in our dataset are associated with "cell death andsurvival" (Supplementary Fig. S5A). Using the DeathBasedatabase, we found that 31% of genes reported to be involvedin cell death (27 of 86) are differentially expressed in SPENclones compared with their controls (Supplementary Fig. S5B–
Figure 2.SPEN regulates cell growth and survivalin T47D cells. A, representative blotshowing immunoprecipitated SPENprotein levels in SPEN-overexpressingclones (Spen 1 and 2) and the controlclone (CTL1). Immunoprecipitation withnonspecific rabbit IgG in Spen 1 cellswas done as a negative control. B, qRT-PCR of SPEN expression in SPEN-overexpressing T47D cells (Spen 1-2)relative to control clones (CTL1-2).Represented is the mean (�SEM)expression value of SPEN in fourbiologic replicates, normalized to thecontrol clone (CTL1; ��� , P < 0.005;�� , P < 0.01; � , P < 0.05). C, growthcurves of SPEN-overexpressing T47Dclones (Spen 1-2) and control clones(CTL1-2) grown in DMEM plus 1% FBS.Data points represent meanfluorescence values (�SEM) of fourexperiments performed inquadruplicates. D, representativeimages of soft agar colony assaysperformedwith T47D Spen 1 and Spen 2clones and the control clones. Bargraphs represent the mean (�SEM)number of colonies formed per well infour experiments performed intriplicates. E, tumor volume in fat padsof nude mice injected bilaterally withCTL1 and SPEN-overexpressing T47Dclones. Data points represent the meantumor volume (�SEM) of 7 CTL1 and 1Spen 1 primary tumors.
SPEN Is a Tumor Suppressor in ERa-Positive Breast Cancers
www.aacrjournals.org Cancer Res; 75(20) October 15, 2015 4357
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

S5D; ref. 25). Notably, most of these differentially expressedgenes implicated in cell death and survival (e.g., BCL2, BMF,and BIK) are localized at the mitochondrial membrane andparticipate in apoptosis induced by intracellular signals. Wealso found that the expression of BCL2, an estrogen-target genethat has strong antiapoptotic functions, is significantly down-regulated in both SPEN-overexpressing T47D clones (Supple-mentary Fig. S5F), a finding that may explain, at least in part,the increased susceptibility of T47D-Spen 1 and T47D-Spen 2to apoptosis. Then, to better define the tumor-suppressiveactivity of SPEN in breast cancer, we focused on functionspredicted to be increased in MCF-7-SPEN-sh1 and MCF-7-SPEN-sh3 silenced cells and decreased in T47D-Spen 1. Withthese criteria, only the biologic function of "cell survival" wasidentified as consistently repressed by SPEN expression (Sup-plementary Fig. S5E). The data obtained from transcriptomeanalyses thus point to a role for SPEN in the regulation of cellviability and these results are consistent with our findings ofcells grown under low serum and anchorage-independentconditions.
Transcriptional regulation of the ERa targets by SPENFurther analyses with IPA revealed that most upstream reg-
ulators affected by the modulation of SPEN expression areinvolved in the regulation of transcription (Supplementary Fig.S6A), providing additional evidence that SPEN participates in
the organization of transcriptional programs. In addition, wefound that the ER is the only upstream regulator whose pre-dicted activation is inversely correlated with SPEN expression(Fig. 4A). To strengthen this relationship between SPEN and theER, we then cross-referenced our microarray data with twopublicly available lists of 7,095 and 5,342 genes bound byERa and ERb, respectively, within 25 kb of their transcriptionstart site as assayed by ChIP-seq experiments in MCF-7 cellstreated with 17b-estradiol (26, 27). In both cases, almost halfthe number of genes significantly upregulated in MCF-7-shRNA-SPEN cells compared with their control were includedin these lists (P ¼ 0.00003; Supplementary Fig. S6B–S6E;ref. 28). Interestingly, no such enrichment was observed withthe lists of downregulated genes in MCF-7-shRNA-SPEN cells.Then, because ERa and ERb often dimerize with one another,we next examined whether the observed enrichments werespecific for genes bound by both receptors and/or uniquelybound by ERa or ERb. Interestingly, we found a significantoverlap between our microarray data and genes bound by bothreceptors or reported to be uniquely bound by ERa uponstimulation with estrogen. No significant overlap with genessolely bound by ERb was observed, suggesting that our data aremainly enriched for ERa but not ERb target genes. In linewith these results, we also observed that restoration of SPENlevels significantly dampens estradiol-induced proliferationin T47D cells (Fig. 4B; P ¼ 0.002). Next, to evaluate whether
Figure 3.SPEN regulates cell growth and survivalin MCF-7 cells. A, representative blotshowing immunoprecipitated SPENprotein levels in MCF-7 (SPEN sh1-sh4)clones and their control (NT, Non-Target shRNA-transfected control).Immunoprecipitation of SPEN in T47Dcells was done as a negative control. B,qRT-PCR of SPEN expression in SPEN-silenced MCF-7 clones (SPEN sh1-sh4)relative to their control. Represented isthe mean (�SEM) expression value ofSPEN in four biologic replicates,normalized to the control (NT2).C, representative images of soft agarcolony assays performed with MCF-7clones and their control (NT1). Bargraphs represent the mean (�SEM)number of colonies formed per well infive experiments performed intriplicates (��� , P < 0.005; ��, P < 0.01;� , P < 0.05). D, Kaplan–Meier graphsdepicting progression-free survival of1,784 patients with luminal A breasttumors, stratified according to high andmoderate/weak (black) expression ofSPEN.
L�egar�e et al.
Cancer Res; 75(20) October 15, 2015 Cancer Research4358
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475
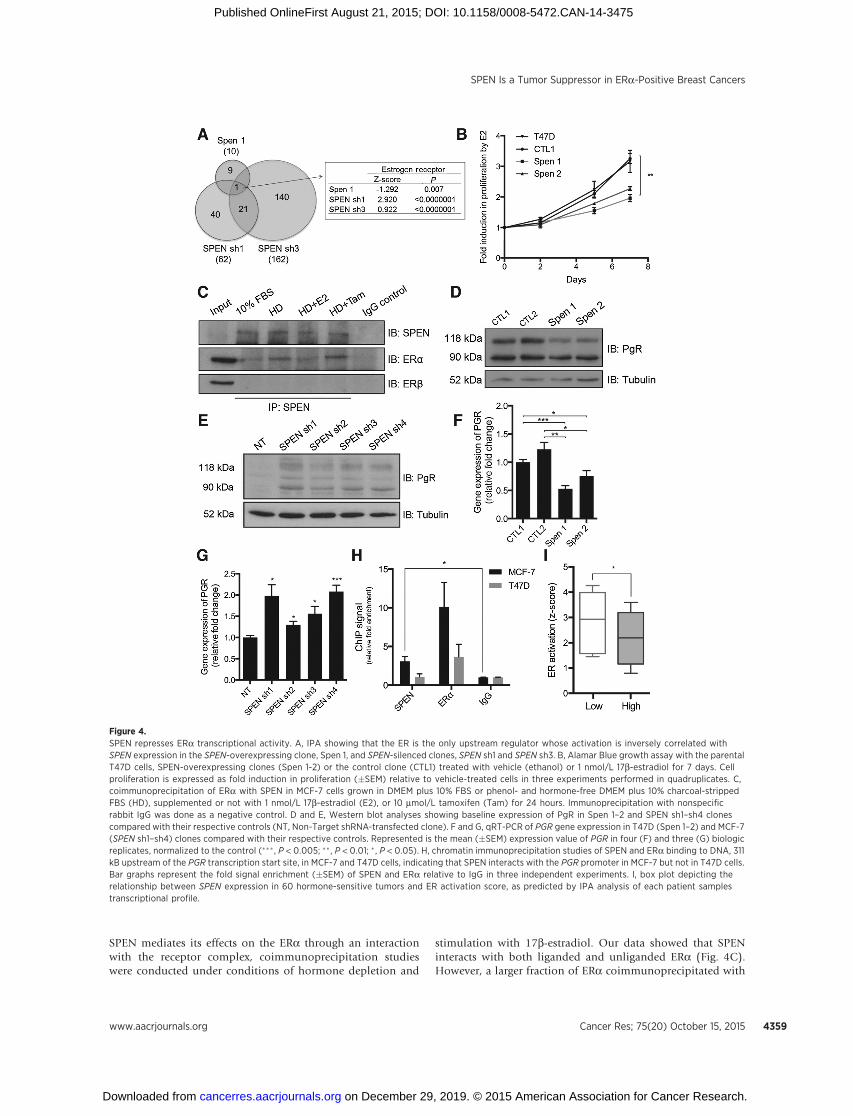
SPEN mediates its effects on the ERa through an interactionwith the receptor complex, coimmunoprecipitation studieswere conducted under conditions of hormone depletion and
stimulation with 17b-estradiol. Our data showed that SPENinteracts with both liganded and unliganded ERa (Fig. 4C).However, a larger fraction of ERa coimmunoprecipitated with
Figure 4.SPEN represses ERa transcriptional activity. A, IPA showing that the ER is the only upstream regulator whose activation is inversely correlated withSPEN expression in the SPEN-overexpressing clone, Spen 1, and SPEN-silenced clones, SPEN sh1 and SPEN sh3. B, Alamar Blue growth assay with the parentalT47D cells, SPEN-overexpressing clones (Spen 1-2) or the control clone (CTL1) treated with vehicle (ethanol) or 1 nmol/L 17b-estradiol for 7 days. Cellproliferation is expressed as fold induction in proliferation (�SEM) relative to vehicle-treated cells in three experiments performed in quadruplicates. C,coimmunoprecipitation of ERa with SPEN in MCF-7 cells grown in DMEM plus 10% FBS or phenol- and hormone-free DMEM plus 10% charcoal-strippedFBS (HD), supplemented or not with 1 nmol/L 17b-estradiol (E2), or 10 mmol/L tamoxifen (Tam) for 24 hours. Immunoprecipitation with nonspecificrabbit IgG was done as a negative control. D and E, Western blot analyses showing baseline expression of PgR in Spen 1–2 and SPEN sh1–sh4 clonescompared with their respective controls (NT, Non-Target shRNA-transfected clone). F and G, qRT-PCR of PGR gene expression in T47D (Spen 1–2) and MCF-7(SPEN sh1–sh4) clones compared with their respective controls. Represented is the mean (�SEM) expression value of PGR in four (F) and three (G) biologicreplicates, normalized to the control (��� , P < 0.005; �� , P < 0.01; � , P < 0.05). H, chromatin immunoprecipitation studies of SPEN and ERa binding to DNA, 311kB upstream of the PGR transcription start site, in MCF-7 and T47D cells, indicating that SPEN interacts with the PGR promoter in MCF-7 but not in T47D cells.Bar graphs represent the fold signal enrichment (�SEM) of SPEN and ERa relative to IgG in three independent experiments. I, box plot depicting therelationship between SPEN expression in 60 hormone-sensitive tumors and ER activation score, as predicted by IPA analysis of each patient samplestranscriptional profile.
SPEN Is a Tumor Suppressor in ERa-Positive Breast Cancers
www.aacrjournals.org Cancer Res; 75(20) October 15, 2015 4359
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

SPEN under hormone-depleted conditions, a finding that isconsistent with corepressors interacting more strongly with un-bound nuclear receptors. Interestingly, no interaction betweenSPEN and ERb was detected under these conditions, suggestingthat SPEN preferentially interacts with ERa over ERb, consistentwith our aforementioned transcriptomic analysis. Then, tofurther support a role for SPEN in the repression of ERa-depen-dent transcription, the expression of the PgR, a primary ERatarget gene, was measured in our SPEN transfectants (29). Weobserved an inverse relationship between SPEN and PgRexpression, at both mRNA and protein levels (Fig. 4D–G andSupplementary Table S2). In addition, chromatin immunopre-cipitation experiments performed with MCF-7 and T47D cellsrevealed that SPEN modestly but consistently interacts withDNA upstream of the PgR gene at a site also bound by the ERain MCF-7 cells but not in T47D cells (Fig. 4H and Supplemen-tary Fig. S6G). Interestingly, SPEN interaction with DNAat this genomic site was restored by its reexpression in T47Dcells (Supplementary Fig. S6H). Together, these results provideevidence for SPEN as a negative regulator of the in ERa-depen-dent transcriptional program in ERa-positive breast cancercells.
SPEN genomic content and expression levels predict ERactivation
To begin to evaluate the clinical significance of our findingsthat suggest a tumor-suppressive role for SPEN in ERa-positivebreast cancers due to its regulation of ERa-dependent tran-scription, the expression profiles of 60 luminal A breast tumorsand their normal-matched counterpart were extracted from theTCGA database and subjected to pathway analysis using IPA.A pairwise comparison of SPEN RNA expression in this subsetof 60 tumors showed that low SPEN expression predicts higherER activation (Fig. 4I). As anticipated, no such correlation wasobserved in a subset of 40 basal breast tumors (data notshown). Taken together, our data suggest that SPEN repressesER-driven transcription in ERa positive but not hormonereceptor–negative breast tumors and that ERa activity is strong-ly enhanced in breast cancer cells expressing low SPEN mRNAlevels.
SPEN sensitizes ERa-positive breast cancer cells to tamoxifenHaving shown that SPEN represses the transcription of genes
downstream of the ERa, we hypothesized that SPEN may affectcellular responses to the antiestrogen, tamoxifen. Using phar-macologically relevant tamoxifen concentrations, we foundthat SPEN silencing confers resistance to the drug in cellviability and clonogenic assays while its overexpression sub-stantially increases sensitivity to tamoxifen (Fig. 5A and B;Supplementary Table S3; Supplementary Fig. S6A–S6E). Inter-estingly, induction of apoptosis following tamoxifen treatmentwas considerably reduced in MCF-7-shRNA clones and 2- to3-fold higher in T47D-SPEN cells than in their respectivecontrols (Fig. 5C and D; Supplementary Fig. S6F–S6H), indi-cating that SPEN sensitizes cells to tamoxifen-induced apo-ptosis. Notably, modulation of SPEN expression did not affectcells sensitivity to ICI 182780 (fulvestrant; SupplementaryFig. S8A–S8D), a pure ERa antagonist that induces ERa deg-radation, suggesting that SPEN's interaction with an intactERamay be critical to predispose cells to apoptosis in responseto tamoxifen treatment.
The clinical significance of SPEN expression levels on tamox-ifen response was determined by assessing its nuclear stainingin TMAs containing triplicate core biopsies from 100 early-stagebreast cancer patients treated exclusively with adjuvant tamox-ifen. This cohort was composed of 50 patients who had diseaserecurrence and died from breast cancer and 50 patients withoutrecurrence or death from the disease. Tissues were scoredaccording to SPEN nuclear staining intensity and percentageof SPEN-positive tumor cells. Kaplan–Meier survival analysesof the 65 hormone-responsive (ERaþPgRþHER2�) breasttumors revealed that high SPEN protein expression was signif-icantly predictive of favorable clinical outcome (P ¼ 0.029;Fig. 5E). Moreover, high SPEN RNA expression was stronglycorrelated with good relapse-free survival (HR, 0.55; P ¼0.0055) in an independent cohort of 424 luminal A ER-positivepatients treated with tamoxifen alone (Fig. 5F; refs. 24, 30).This prognostic effect was not observed in patients with ERa-negative tumors (Supplementary Fig. S7I), suggesting thatSPEN expression levels may serve as a predictive biomarker oftamoxifen response in hormone-sensitive breast cancers.
DiscussionOne of the hallmarks of cancer is the accumulation of
genetic mutations in tumor cell DNA, resulting in the activa-tion of oncogenes and the loss of tumor-suppressor genes(31). Although their expression is usually lost in many tumorscompared with normal tissues due to genetic and epigeneticevents, the limited expression of tumor-suppressor genes maynevertheless be of clinical relevance. Our identification ofSPEN as a tumor-suppressor gene and candidate predictivemarker of tamoxifen response, due to its repressive activity ofERa-dependent transcription in breast cancer, demonstratesthat the study of tumor-suppressor genes might uncover novelmechanisms of drug resistance and biomarkers of drugresponse.
Using a genomic approach, we discovered LOH at the SPENlocus in 23% of breast cancers. Sequencing of SPEN in tumorswith LOH at the 1p36 locus identified four nonsynonymousmutations, including a nonsense mutation recurrent in twoindependent tumors, adding to the 29 mutations in breasttumors reported in the COSMIC database (23). This finding,in addition to our observation that samples from TCGA withnonsense mutations in SPEN express low SPEN mRNA levels,suggest that SPEN gene expression might be regulated by theNMD pathway in breast cancer cells, a mechanism that we haveshown to be responsible for undetectable SPEN protein levelsin T47D cells.
To establish SPEN as a tumor-suppressor gene and define itsfunctions in breast cancer, we used an in vitromodel with MCF-7and T47D cells, two ERa-positive breast cancer cell lines expres-sing high and very low SPEN protein levels, respectively. Theforced expression of SPEN in T47D cells and its knockdown inMCF-7 cells unmasked an inhibitory effect on cell proliferation,growth, and survival. In addition, gene expression analyses ofMCF-7 and T47D clones identified "cell survival" as the majorbiologic function affected by the modulation of SPEN expres-sion. The enrichment of our SPEN-regulated gene expressionprofiles for pro- and antiapoptotic genes provides a molecularmechanism for the altered response of SPEN-silenced MCF-7and SPEN-overexpressing T47D cells to apoptotic stimuli, such
L�egar�e et al.
Cancer Res; 75(20) October 15, 2015 Cancer Research4360
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

as suspension in soft agar and growth under serum-deprivedconditions.
Further analysis of our microarray data revealed that SPENregulates the expression of a number of genes downstream ofthe ER, confirming its role as a repressor of the ERa-dependentsignaling pathway in breast cancer cells. These results extendthose of Shi and colleagues (10), who demonstrated that SPENinhibits estradiol-induced ERa-dependent transcription in aluciferase assay. Although not derived from experiments con-ducted with breast cancer cells, their results were highly sug-gestive of SPEN having repressive effects on ERa transcription.In a yeast-two hybrid assay, they also showed that SPENinteracts with other ERa cofactor, including SRA, an RNAmolecule with coactivating functions, and SMRT and NCoR,
two ERa corepressors. Our observation that SPEN represses theexpression of ERa target genes, including the transcription ofthe PGR, and that it coimmunoprecipitates with the ERacomplex in the presence of estradiol, demonstrates that endog-enous SPEN can bind and repress ligand-bound ERa. Althoughmost corepressors bind unliganded receptors, we have demon-strated that SPEN acts on both estradiol-bound and unboundreceptors, a property unique to few ERa coregulators. Thisfunction may be particularly important in limiting the genomiceffects of 17b-estradiol and preventing uncontrolled cell pro-liferation and survival between successive hormonal cycles inthe normal breast. Indeed, our finding that SPEN knockdownpotentiates the proliferation of the normal-like epithelial cellline, MCF10A, exposes a possible a role for SPEN in preventing
Figure 5.SPEN regulates ERa-positive breastcancer cells response to tamoxifen. Aand B, Alamar Blue survival assayperformed with T47D (Spen 1–2) andMCF-7 (SPEN sh1–sh4) clones treatedwith tamoxifen (T47D: 8 mmol/L, MCF-7:6 mmol/L) or its vehicle for 5 days. Bargraphs represent the mean percentageof survival (�SEM) of tamoxifen-treatedcells relative to vehicle-treated cells inthree (T47D) and at least four (MCF-7)experiments performed inquadruplicates. C and D, Annexin-V/PIflow cytometric analyses performedwith T47D (Spen 1–2) and MCF-7 (SPENsh1–sh4) clones treated with tamoxifenor its vehicle for 5 days. Bar graphsrepresent the percentage of PI-positive(PIþ) cells (�SEM) collected aftertreatment with tamoxifen or its vehiclein five experiments (��� , P < 0.005;�� , P <0.01; �, P <0.05). E and F, Kaplan–Meier graphs depictingprogression-freesurvival of ERaþ/PgRþ/HER2� patientstreated with tamoxifen alone, stratifiedaccording to high and moderate/weak(blue–black) expression of SPEN.
SPEN Is a Tumor Suppressor in ERa-Positive Breast Cancers
www.aacrjournals.org Cancer Res; 75(20) October 15, 2015 4361
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

cellular transformation. Genetic aberrations affecting SPEN innormal and premalignant breast epithelial cells could thereforepotentiate ERa transcriptional activity, especially during thefollicular and estrogen-driven phase of the menstrual cycle, andlead to the expression of estrogen-responsive genes, includingthe PgR as well as antiapoptosis-related genes, all of which maycontribute to cell survival and cancer development.
Given our results showing that SPEN expression establishes aproclivity to apoptosis in ERa-positive breast cancer cells, andthat it represses the transcriptional activity of the ERa, weevaluated the sensitivity of SPEN-silenced MCF-7 and SPEN-overexpressing T47D cells to the antiestrogen, tamoxifen, adrug that antagonizes the ERa. Whereas several mechanismsof resistance to tamoxifen are known, such as ERa phosphor-ylation at S167, HER2 overexpression, and hyperactivation ofthe PI3K pathway, no predictive biomarker for tamoxifenresponse is in current clinical use other than the ERa and thePgR (21, 32–35). Using an in vitro breast cancer model withMCF-7 and T47D cells, we showed that SPEN affects tamoxifenbut not fulvestrant sensitivity in ERa-positive breast cancercells. SPEN protein and RNA expression in two patient cohortsthat had received tamoxifen therapy alone in the adjuvantsetting support our findings, with significantly better outcomesfor patients with breast tumors showing high SPEN expressionthan for those with low/moderate SPEN expression. Althoughfurther clinical validation is needed, our findings suggest that asubpopulation of ERa-positive early-stage breast cancerpatients may not benefit from adjuvant tamoxifen. In addition,our observation that SPEN expression does not affect cellssensitivity to ICI182780, a drug that induces ERa degradation,suggests that SPEN interaction with the ERa may be critical toestablish a proclivity for cell death in response to tamoxifentreatment. It will thus be important in future studies to alsoevaluate the effect of SPEN expression on sensitivity to otherendocrine therapies, such as aromatase inhibitors, which donot alter intracellular ERa levels. In addition to having poten-tial clinical applications, such studies may provide new insightsinto SPEN functions in the context of antiestrogen drugresponse. Taken together, our data indicate that breast cancersexpressing SPEN may be more sensitive to tamoxifen-inducedapoptosis and that SPEN expression could serve as a marker oftamoxifen response in ERa-positive breast cancers.
It is noteworthy that modulation of SPEN expression con-sistently affected serum deprivation and tamoxifen-inducedapoptosis in both T47D and MCF-7 breast cancer cell lines,given that they have different defects in apoptotic mechanisms:T47D cells contain a p53 mutation and MCF-7 cells do notexpress the apoptotic mediator caspase-3 (36, 37). With geno-mic and nongenomic actions affecting proliferation, migration,and apoptosis, the ERa has a central role in the biology ofbreast cancer. Indeed, prior studies have demonstrated that theERa can protect breast cancer cells from program-induced celldeath, in part, by modulating the expression of apoptosis-related genes, such as BCL2, BIK, and BMF (16, 38, 39). Where-as very few of the well-established ERa-target genes (i.e.,GREB1, TFF1, CCND1, etc.) were differentially expressed inour microarray data besides PGR, the modulation of SPENexpression in MCF-7 and T47D cells affected the expressionof a number of apoptosis-related genes, including BCL2, BIK,and BMF. Our finding that approximately 35% of genesinvolved in "cell death and survival" and regulated by SPEN
in each of our SPEN clones are also downstream of the ER (P <0.0001; Supplementary Fig. S6F) suggests that the antiapopto-tic transcriptional program regulated by the ERa is likelycontrolled by SPEN (28).
Generally, our findings suggest that the inactivation of SPENby deletion and/or intragenic mutation may contribute tobreast tumor formation and progression. As SPEN mutationswere reported in other cancer types, we can speculate that SPENtumor-suppressive functions extend to many other tissues.Future studies should therefore seek to evaluate the functionsof SPEN in other cancers, such as cervical and endometrialcancer, in which the occurrence of SPEN mutations is very high(14% and 11%, respectively). In conclusion, our results estab-lish SPEN as a regulator of ERa-dependent transcriptionof apoptosis-related genes in breast cancer and provide func-tional and clinical evidence for SPEN as a tumor-suppressorgene and a candidate predictive biomarker of tamoxifenresponse in ERa-positive breast cancers.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: S. L�egar�e, L. Cavallone, P.N. Tonin, M. BasikDevelopment of methodology: S. L�egar�e, A. Mamo, C. Chabot, I. Sirois,A. Magliocco, A. Klimowicz, P.N. Tonin, M. Buchanan, D. Keilty, M. BasikAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): S. L�egar�e, A. Magliocco, A. Klimowicz, P.N. Tonin,M. Buchanan, D. Keilty, M. BasikAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): S. L�egar�e, L. Cavallone, I. Sirois, A. Magliocco,A. Klimowicz, S. Hassan, D. Laperri�ere, S. Mader, O. Aleynikova, M. BasikWriting, review, and/or revision of the manuscript: S. L�egar�e, C. Chabot,I. Sirois, A. Magliocco, P.N. Tonin, S. Mader, M. BasikAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): S. L�egar�e, I. Sirois, A. Magliocco, A. Klimowicz,M. Buchanan, M. BasikStudy supervision: C. Chabot, P.N. Tonin, M. Basik
AcknowledgmentsThe authors thank past and current members of the M. Basik laboratory,
especially Banujan Balachandran and Elaheh Ahmadzadeh, for technical assis-tance, as well as Isabelle Royal, Vincent Gigu�ere, and Volker Blank for assistanceand discussion. The authors also acknowledge the help of Kathy Ann Forner andChristian Young from the animal care and flow cytometry facility, respectively,of the Lady Davis Institute for Medical Research.
Grant SupportThis study was supported by a McGill Integrated Cancer Research
Training Program studentship (FRN53888) and a doctoral award from theFonds de recherche du Qu�ebec—Sant�e (S. L�egar�e); the Eileen Iwanickipostdoctoral fellowship in Breast Cancer Research from the CanadianInstitutes of Health Research in partnership with the Breast Cancer Societyof Canada (I. Sirois); and by grants from the Cancer Research Society andthe FRQS R�eseau de Cancer Axe Cancer du Sein et Ovaire as well as a grantfrom the Quebec Breast Cancer Foundation (M. Basik, P.N. Tonin, andS. Mader).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received November 25, 2014; revised May 21, 2015; accepted July 10, 2015;published OnlineFirst August 21, 2015.
L�egar�e et al.
Cancer Res; 75(20) October 15, 2015 Cancer Research4362
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

References1. Osborne CK, Hobbs K, Clark GM. Effect of estrogens and antiestrogens on
growth of human breast cancer cells in athymic nude mice. Cancer Res1985;45:584–90.
2. Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics andsufficiency in estrogen receptor-regulated transcription. Cell 2000;103:843–52.
3. Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, et al. Estrogenreceptor-alpha directs ordered, cyclical, and combinatorial recruitmentof cofactors on a natural target promoter. Cell 2003;115:751–63.
4. RobinsonDR,WuYM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1mutations in hormone-resistant metastatic breast cancer. Nat Genet2013;45:1446–51.
5. ToyW, Shen Y,WonH,Green B, Sakr RA,WillM, et al. ESR1 ligand-bindingdomainmutations in hormone-resistant breast cancer. Nat Genet 2013;45:1439–45.
6. Fleming FJ, Hill AD, McDermott EW, O'Higgins NJ, Young LS. Differentialrecruitment of coregulator proteins steroid receptor coactivator-1 andsilencing mediator for retinoid and thyroid receptors to the estrogenreceptor-estrogen response element by beta-estradiol and 4-hydroxyta-moxifen in human breast cancer. J Clin Endocrinol Metab 2004;89:375–83.
7. Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr RelatCancer 2004;11:643–58.
8. Doroquez DB, Orr-Weaver TL, Rebay I. Split ends antagonizes the Notchand potentiates the EGFR signaling pathways during Drosophila eye devel-opment. Mech Dev 2007;124:792–806.
9. Oswald F,Winkler M, Cao Y, Astrahantseff K, Bourteele S, Kn€ochelW, et al.RBP-Jkappa/SHARP recruits CtIP/CtBP corepressors to silence Notch targetgenes. Mol Cell Biol 2005;25:10379–90.
10. Shi Y, Downes M, Xie W, Kao HY, Ordentlich P, Tsai CC, et al. Sharp, aninducible cofactor that integrates nuclear receptor repression and activa-tion. Genes Dev 2001;15:1140–51.
11. Ariyoshi M, Schwabe JW. A conserved structural motif reveals the essentialtranscriptional repression function of Spen proteins and their role indevelopmental signaling. Genes Dev 2003;17:1909–20.
12. Sanchez-Pulido L, Rojas AM, vanWely KH,Martinez AC, Valencia A. SPOC:a widely distributed domain associated with cancer, apoptosis and tran-scription. BMC Bioinformatics 2004;5:91.
13. Feng Y, Bommer GT, Zhai Y, Akyol A, Hinoi T,Winer I, et al.Drosophila splitends homologue SHARP functions as a positive regulator of Wnt/beta-catenin/T-cell factor signaling in neoplastic transformation. Cancer Res2007;67:482–91.
14. Mamo A, Cavallone L, Tuzmen S, Chabot C, Ferrario C, Hassan S, et al. Anintegrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene 2012;31:2090–100.
15. Hosein AN, Wu M, Arcand SL, Lavall�ee S, H�ebert J, Tonin PN, et al. Breastcarcinoma-associated fibroblasts rarely contain p53 mutations or chro-mosomal aberrations. Cancer Res 2010;70:5770–7.
16. Tolhurst RS, Thomas RS, Kyle FJ, Patel H, Periyasamy M, Photiou A, et al.Transient over-expression of estrogen receptor-alpha in breast cancer cellspromotes cell survival and estrogen-independent growth. Breast CancerRes Treat 2011;128:357–68.
17. Mercola D, Welsh J. From mRNA to tumor suppressor. Nat Genet 2004;36:937–8.
18. Cornen S, Adelaide J, Bertucci F, Finetti P, Guille A, Birnbaum DJ, et al.Mutations and deletions of ARID1A in breast tumors. Oncogene 2012;31:4255–6.
19. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al.ONCOMINE: a cancer microarray database and integrated data-miningplatform. Neoplasia 2004;6:1–6.
20. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al.Integrative analysis of complex cancer genomics and clinical profiles usingthe cBioPortal. Sci Signal 2013;6:pl1.
21. LeeKY, Lee JW,NamHJ, Shim JH, SongY, KangKW.PI3-kinase/p38 kinase-dependent E2F1 activation is critical for Pin1 induction in tamoxifen-resistant breast cancer cells. Mol Cells 2011;32:107–11.
22. Cerami E,Gao J,DogrusozU,Gross BE, Sumer SO, Aksoy BA, et al. The cBiocancer genomics portal: an open platform for exploring multidimensionalcancer genomics data. Cancer Discov 2012;2:401–4.
23. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al.COSMIC: mining complete cancer genomes in the Catalogue of SomaticMutations in Cancer. Nucleic Acids Res 2011;39(Database issue):D945–50.
24. Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. Anonline survival analysis tool to rapidly assess the effect of 22,277 genes onbreast cancer prognosis using microarray data of 1,809 patients. BreastCancer Res Treat 2010;123:725–31.
25. Diez J, Walter D, Munoz-Pinedo C, Gabaldon T. DeathBase: a data-base on structure, evolution and function of proteins involved inapoptosis and other forms of cell death. Cell Death Differ 2010;17:735–6.
26. Joseph R,Orlov YL,HussM, SunW,Kong SL,Ukil L, et al. Integrativemodelof genomic factors for determining binding site selection by estrogenreceptor-alpha. Mol Systems Biol 2010;6:456.
27. Grober OM, Mutarelli M, Giurato G, Ravo M, Cicatiello L, De Filippo MR,et al. Global analysis of estrogen receptor beta binding to breast cancercell genome reveals an extensive interplay with estrogen receptor alphafor target gene regulation. BMC Genomics 2011;12:36.
28. Fury W, Batliwalla F, Gregersen PK, Li W. Overlapping probabilitiesof top ranking gene lists, hypergeometric distribution, and stringencyof gene selection criterion. Conf Proc IEEE Eng Med Biol Soc 2006;1:5531–4.
29. Bourdeau V, Deschenes J, Laperriere D, Aid M, White JH, Mader S.Mechanisms of primary and secondary estrogen target gene regulationin breast cancer cells. Nucleic Acids Res 2008;36:76–93.
30. Mihaly Z, Kormos M, Lanczky A, Dank M, Budczies J, Sz�asz MA, et al.A meta-analysis of gene expression-based biomarkers predicting outcomeafter tamoxifen treatment in breast cancer. Breast Cancer Res Treat 2013;140:219–32.
31. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell2011;144:646–74.
32. Arpino G, Green SJ, AllredDC, LewD,Martino S, Osborne CK, et al. HER-2amplification, HER-1 expression, and tamoxifen response in estrogenreceptor-positive metastatic breast cancer: a southwest oncology groupstudy. Clin Cancer Res 2004;10:5670–6.
33. Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S,Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation ofestrogen receptor alpha: a new model for anti-estrogen resistance. J BiolChem 2001;276:9817–24.
34. Larsen MS, Bjerre K, Lykkesfeldt AE, Giobbie-Hurder A, Laenkholm AV,Henriksen KL, et al. Activated HER-receptors in predicting outcome of ER-positive breast cancer patients treated with adjuvant endocrine therapy.Breast 2012;21:662–8.
35. Yamashita H, Nishio M, Kobayashi S, Ando Y, Sugiura H, Zhang Z, et al.Phosphorylation of estrogen receptor alpha serine 167 is predictive ofresponse to endocrine therapy and increases postrelapse survival in met-astatic breast cancer. Breast Cancer Res 2005;7:R753–64.
36. O'Connor PM, Jackman J, Bae I, Myers TG, Fan S, Mutoh M, et al.Characterization of the p53 tumor suppressor pathway in cell lines ofthe National Cancer Institute anticancer drug screen and correlationswith the growth-inhibitory potency of 123 anticancer agents. Cancer Res1997;57:4285–300.
37. Janicke RU. MCF-7 breast carcinoma cells do not express caspase-3. BreastCancer Res Treat 2009;117:219–21.
38. Hur J, Chesnes J, Coser KR, LeeRS,GeckP, Isselbacher KJ, et al. The BikBH3-only protein is induced in estrogen-starved and antiestrogen-exposedbreast cancer cells and provokes apoptosis. Proc Natl Acad Sci U S A2004;101:2351–6.
39. Perillo B, Sasso A, Abbondanza C, Palumbo G. 17beta-estradiol inhibitsapoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol Cell Biol2000;20:2890–901.
www.aacrjournals.org Cancer Res; 75(20) October 15, 2015 4363
SPEN Is a Tumor Suppressor in ERa-Positive Breast Cancers
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475

2015;75:4351-4363. Published OnlineFirst August 21, 2015.Cancer Res Stéphanie Légaré, Luca Cavallone, Aline Mamo, et al. Hormone-Dependent Breast CancersSuppressor and Candidate Biomarker of Drug Responsiveness in The Estrogen Receptor Cofactor SPEN Functions as a Tumor
Updated version
10.1158/0008-5472.CAN-14-3475doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2017/10/17/0008-5472.CAN-14-3475.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/75/20/4351.full#ref-list-1
This article cites 39 articles, 14 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/75/20/4351.full#related-urls
This article has been cited by 5 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/75/20/4351To request permission to re-use all or part of this article, use this link
on December 29, 2019. © 2015 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 21, 2015; DOI: 10.1158/0008-5472.CAN-14-3475