the mechanism of transamination - jbc.org · that there is concomitant loss of activity (5)....
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMIBTRY Vol. 245, No. 4, Issue of February 25, pp. 806-813, 1970
Printed in U.S.A.
The Mechanism of Transamination
FUNCTION OF THE HISTIDYL RESIDUE AT THE ACTIVE SITE OF SUPERNATANT ASPARTATE TRANSAMINASE*
(Received for publication, September 8, 1969)
D. L. PETERSONI AND M. MARTINEZ-CARRION
From the Department of Chemistry, Biochemistry and Biophysics Program, University of Notre Dame, Notre Dame, Indiana 46556
SUMMARY
Controlled photooxidation inactivates supernatant glu- tamate aspartate transaminase through the destruction of histidyl residues only. Since previous studies have failed to show alterations in the over-all enzyme structure, the mechanism of transamination has been examined before and after photooxidation, taking advantage of the spectral changes associated with the enzyme prosthetic group, pyri- doxal phosphate, after substrate or inhibitor binding. By spectrophotometric titration of the active site, photooxidized enzyme retains its ability to bind the dicarboxylic acids, glutarate, oxaloacetate, and a-ketoglutarate, to form non- covalent enzyme-substrate complexes. Binding of (Y- methyl aspartate was also measured spectrophotometrically. Because the interaction of the enzyme with radioactive sub- strate can be trapped by reduction of the enzyme-substrate complex with sodium borohydride, it is shown that aspartate in the forward reaction and a-ketoglutarate in the back reaction form the initial aldimine and ketimine complexes, respectively. The affinity for each substrate or substrate analogue is greater in the case of photooxidized enzyme under normal buffer conditions. Native enzyme exhibits relatively large changes in the dissociation constant with changes in the buffer anion concentration, while photo- oxidized enzyme fails to show these competitive anion effects. Photooxidized enzyme forms all of the visible enzyme substrate intermediates characteristic of the half- transamination reaction between an amino acid and its keto acid analogue, except for the semiquinoid intermediate which absorbs at 490 mp. This inability correlates with the failure to catalyze the exchange of the a-hydrogen of the amino acid with tritium from tritiated water. From these data and from studies on the rate of the forward and back reaction with cysteine suEnate and Lu-ketoglutarate, respec- tively, it is concluded that the primary defect after photo- oxidation of the active site histidine of supernatant glu- tamate aspartate transaminase occurs in the removal of the a-hydrogen from the aldimine Schiff’s base in the forward reaction. A role of proton acceptor is proposed for the histidine residue at the active site.
The identification of amino acids which comprise the active site of an enzyme and the assignment of specific roles to them are essential for an understanding of the mechansim of enzymatic catalysis. The assignment of roles to amino acid residues at the active site has generally been based on studies of the pH dependence of kinetic parameters or on experiments involving chemical modification of these amino acids. The former method is presumptive, in that assignment of a pK to a specific amino acid in a protein involves considerable suppositions. Although chemical modifications more clearly define an amino acid as essential, much difficulty is encountered in assigning a role, since the product of such modification may be totally inactive enzyme. This difficulty has been circumvented in some cases by the use of minimally altered enzymes, whose activity, although decreased, is not destroyed. This is exemplified by the works on carboxy- peptidase (1) and chymotrypsin (2). Another approach is pos- sible when an enzyme is characterized by visible changes upon reaction with substrate. For example, Miller and Schwert (3) have shown that destruction of presumed histidine residues re- sults in the inability of lactic dehydrogenase to form the highly fluorescent enzyme-NADH-oxaloacetate complex.
Supernatant glutamate aspartate transaminase (EC 2.6.1 .‘I) should be particularly useful for modification studies. Consider- able detail of its mechanism of catalysis is known (4). The over- all reaction is separable into half-reactions, as shown in Scheme I, where the over-all reaction between aspartate, a-ketoglutarate, glutamate, and oxaloacetate is represented as the sum of two half-reactions, A and B, each of which may be readily studied individually. The unique spectral properties of pyridoxal phos- phate enzymes, and of aspartate transaminase, in particular, allow several of the catalytic steps of each half-reaction to be observed separately and directly. In this manner, pyridoxal phosphate acts as a natural reporter of events occurring at the active site. The detection of these events is independent of the ability of the enzyme to carry out the over-all catalysis. The
* This work was supported by Grants AM-12227 and HE-11448 from the National Institutes of Health, United States Public Health Service. Preliminary reports on parts of this work have anneared (10.15). This work is based on a dissertation submitted (by D. L. ‘P.j in partial fulfillment of the requirement for the de- gree of Doctor of Philosophy at the University of Notre Dame.
$ Predoctoral Fellow of the National Science Foundation.
by guest on June 9, 2019http://w
ww
.jbc.org/D
ownloaded from

Issue of February 25, 1970 D. L. Peterson and M. Martinez-Carrion 807
effect of virtually any modification of the enzyme on this series of observable steps can be determined.
We have previously shown that photooxidation of the super- natant glutamate aspartate transaminase in the presence of meth- ylene blue results in the destruction of histidine residues only, and that there is concomitant loss of activity (5). Because of the correlation of the first order rate constants for the photoxidation of the histidines and the loss of catalytic activity, only 1 of the histidyl residues was assigned as responsible for the loss of en- zymatic activity (5). We saw no gross protein conformational changes and both photoxidized and native apoenzyme bound 2 moles of either pyridoxal phosphate or pyridoxamine phosphate (6). The photoxidized holoenzyme has also been shown to be identical with the native pyridoxal enzyme in absorption spectral and circular dichroism properties (6).
Since the over-all structure of the enzyme appears to be un- affected, but activity is lost by the destruction of a histidine resi- due, we have investigated the possible role of this residue in the mechanism of transamination. In this paper we report the effect of photooxidation on the enzyme binding of substrates to form the various distinguishable enzyme-substrate intermediates and on the enzyme’s ability to interconvert these complexes.
EXPERIMENTAL PROCEDURE
Materials
Cysteine sulfinic acid, pyridoxal phosphate, hydroxyfumarate, pyridoxamine phosphate, and oc-methyl-nn-aspartic acid were purchased from Sigma; erythro-fi-hydroxy-nn-aspartic acid, oxalo- acetic acid, aspartic acid, uniformly labeled a-ketoglutaric acid- r4C, and uniformly labeled aspartic acid-14C were purchased from Calbiochem. Supernatant glutamate aspartate transaminase was prepared as previously described (7).
METHODS
Spectrophotometric Studies-All spectra were recorded with a Cary model 15 recording spectrophotometer equipped with a 0- to O.l-absorbance unit expanded scale slide wire. Circular dichro- ism spectra were recorded with a Cary model 60 recording spec- tropolarimeter equipped with a circular dichroism attachment.
Photooxidation of Supernatant Glutamate Aspartate Transam- inase-Photooxidation was performed in 0.05 M potassium phos- phate, pH 7.5, as previously described (5). Enzyme activity was determined according to the method of Lis (8). The activity of native and photooxidized apoenzyme was determined after re- combination in lop4 M pyridoxal phosphate.
Binding of Substrates-The dissociation constants of the en- zyme substrate complexes with methyl aspartate, oxaloacetate, oc-ketoglutarate, and glutarate were determined from the changes in optical density at 430 rnp, when microliter aliquots of concen- trated substrate solution were added to the pyridoxal form of the enzyme.
The dissociation constant for erythro-P-hydroxy-nn-aspartate may be measured in the native enzyme by following the appear- ance of the semiquinoid complex characterized by absorption at 490 rnp upon addition of aliquots of substrate. This is not possi- ble for the photooxidized enzyme, however, since this complex is is not formed (see “Results”), nor can a decrease in the optical density at 360 rnp be followed, because the initial aldimine en- zyme substrate complex absorbs at this wave length. However, we have shown that the enzyme substrate aldimine complex with
Aspartate -l- Epyridoxal * Epyridoxamine -/- oxaloacetate (A)
Epyridoxmine i- a-ketoglutaratee Epyridorar + glutamate Aspartate + cu-ketoglutarate+glutamate + oxaloacetate 09
SCHEME I
hydroxyaspartate exhibits no dichroicity at 360 rnp, and, there- fore, is readily distinguishable from the free enzyme which ex- hibits a dichroic maximum at this wave length. The dissociation constant may be determined spectropolarimetrically by following the disappearance of the 360 rnp dichroic band of either native or photooxidized pyridoxal enzyme upon addition of microliter ali- quots of substrate solution.
Binding of ar-ketoglutarate to the pyridoxamine form of the enzyme and aspartate to the pyridoxal form of the enzyme was shown using radioactive substrate and reducing the enzyme sub- strate Schiff’s base complex with sodium borohydride. After re- moval of the unbound substrate by passage through a Sephadex G-25 column, 2.5 x 75 cm, in 0.05 M potassium phosphate, pH 7.5, enzyme concentration was determined spectrophotometri- tally at 280 rnp and radioactivity was determined by counting in a Packard Tri-Carb liquid scintillation counter in Bray’s solution.
Tritium Exchange with Hydroxyaspartate-The ability of native and photooxidized enzyme to catalyze the exchange of the hydro- gen of the a-carbon of hydroxyaspartate with tritium from triti- ated water was determined by removal of O.l-ml aliquots from a reaction mixture containing 2.6 X lop5 M enzyme, 0.05 M
erythro-hydroxy-nn-aspartate, TzO, 100 @i, and 0.1 M sodium tetraborate, pH 9.1, in a total volume of 0.95 ml. The reaction was stopped with an equal volume of 10% trichloracetic acid. The amino acid was repeatedly recrystallized from water with acetone, and the precipitate was collected by centrifugation. The final precipitate was dissolved in 1.0 ml of water. Amino acid concentration was determined by the quantitative ninhydrin method of Rosen (9). Radioactivity was determined in Bray’s solution. Counting efficiency was determined by the addition of a known volume of tritium standard.
THEORY AND RESULTS
The inactivation of an enzyme by modification of an amino acid may result from (a) gross conformational changes, (b) ina- bility to bind coenzyme, (c) inability to bind substrate, or (d) failure to carry out the conversion of bound substrate to product. In the case of photooxidized glutamate aspartate transaminase, the first two possibilities have been eliminated on the basis of previous work (6). It is important, therefore, to determine which of the latter two possibilities is correct. I f substrate is not bound, then the critical histidine may function directly or indi- rectly in the binding process or the maintenance of the proper local conformation which allows this binding. However, if sub- strate does bind, the histidine must be involved either directly or indirectly in some later function, such as the conversion to prod- ucts.
Steady state kinetic data show no change in the K,, but a de- crease in the Vmax after photooxidation (10). The decrease in the V,,, is proportional to the degree of photooxidation and has been interpreted as showing that the photooxidized enzyme is completely inactive (10). This type of approach, however, cannot be used to distinguish between the possible means of in- activation. Because of the unique spectral properties of pyri- doxal phosphate enzymes, which allow the direct observation of
by guest on June 9, 2019http://w
ww
.jbc.org/D
ownloaded from
808 Function of Histidine Residue in Aspartate Transaminase Vol. 245, No. 4
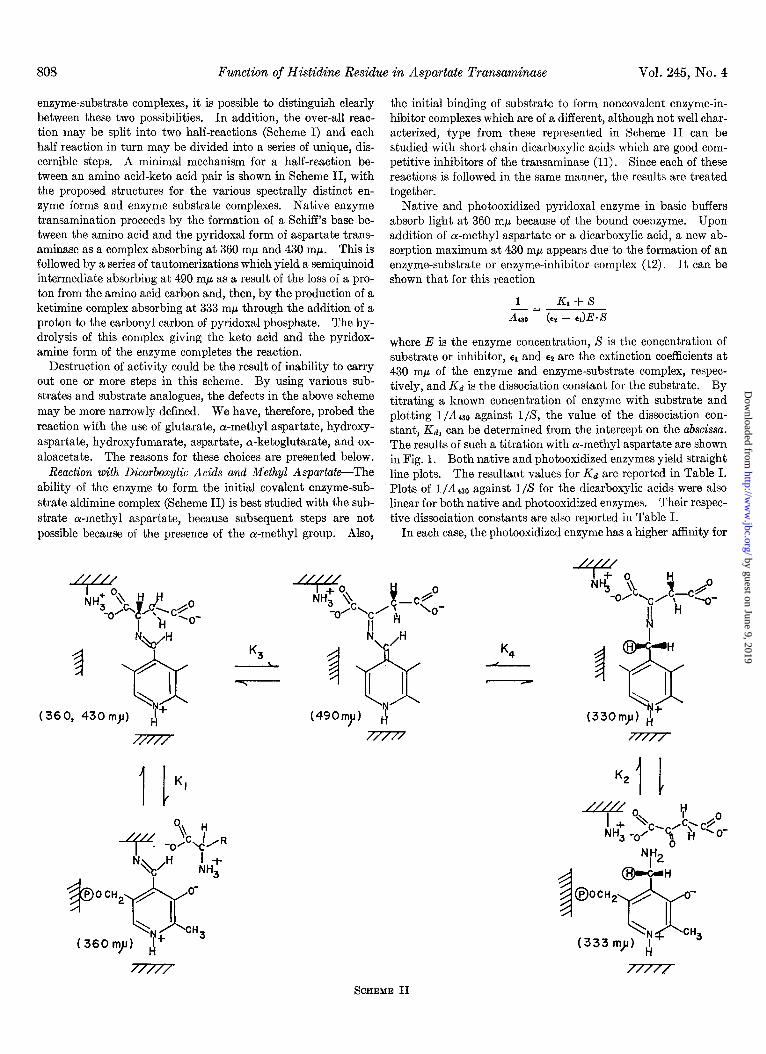
enzyme-substrate complexes, it is possible to distinguish clearly between these two possibilities. In addition, the over-all reac- tion may be split into two half-reactions (Scheme I) and each half reaction in turn may be divided into a series of unique, dis- cernible steps. A minimal mechanism for a half-reaction be- tween an amino acid-keto acid pair is shown in Scheme II, with the proposed structures for the various spectrally distinct en- zyme forms and enzyme substrate complexes. Native enzyme transamination proceeds by the formation of a Schiff’s base be- tween the amino acid and the pyridoxal form of aspartate trans- aminase as a complex absorbing at 360 rnp and 430 mp. This is followed by a series of tautomerizations which yield a semiquinoid intermediate absorbing at 490 rnp as a result of the loss of a pro- ton from the amino acid carbon and, then, by the production of a ketimine complex absorbing at 333 mn through the addition of a proton to the carbonyl carbon of pyridoxal phosphate. The hy- drolysis of this complex giving the keto acid and the pyridox- amine form of the enzyme completes the reaction.
Destruction of activity could be the result of inability to carry out one or more steps in this scheme. By using various sub- strates and substrate analogues, the defects in the above scheme may be more narrowly defined. We have, therefore, probed the reaction with the use of glutarate, a-methyl aspartate, hydroxy- aspartate, hydroxyfumarate, aspartate, a-ketoglutarate, and ox- aloacetate. The reasons for these choices are presented below.
Reaction with Dicarboxylic Acids and Methyl Aqartate-The ability of the enzyme to form the initial covalent enzyme-sub- strate aldimine complex (Scheme II) is best studied with the sub- strate a-methyl aspartate, because subsequent steps are not possible because of the presence of the a-methyl group. Also,
(36
1 tQH K3
/ -cx I’ \ I
0, 430my) N+ 4
/..///
the initial binding of substrate to form noncovalent enzyme-in- hibitor complexes which are of a different, although not well char- acterized, type from these represented in Scheme II can be studied with short chain dicarboxylic acids which are good com- petitive inhibitors of the transaminase (11). Since each of these reactions is followed in the same manner, the results are treated together.
Native and photooxidized pyridoxal enzyme in basic buffers absorb light at 360 rnp because of the bound coenzyme. Upon addition of a-methyl aspartate or a dicarboxylic acid, a new ab- sorption maximum at 430 rnp appears due to the formation of an enzyme-substrate or enzyme-inhibitor complex (12). It can be shown that for this reaction
1 KI + S -= Aso k* - E,)E.X
where E is the enzyme concentration, S is the concentration of substrate or inhibitor, cl and ~2 are the extinction coefficients at 430 rnp of the enzyme and enzyme-substrate complex, respec- tively, and Kd is the dissociation constant for the substrate. By titrating a known concentration of enzyme with substrate and plotting l/A430 against l/S, the value of the dissociation con- stant, Kd, can be determined from the intercept on the abscissa. The results of such a titration with a-methyl aspartate are shown in Fig. 1. Both native and photooxidized enzymes yield straight line plots. The resultant values for Kd are reported in Table I. Plots of l/Adz0 against l/S for the dicarboxylic acids were also linear for both native and photooxidized enzymes. Their respec- tive dissociation constants are also reported in Table I.
In each case, the photooxidized enzyme has a higher affinity for
SCHEME II
by guest on June 9, 2019http://w
ww
.jbc.org/D
ownloaded from

Issue of February 25, 1970 D. L. Peterson and M. Martinez-Carrion 809
0.1 0.2
1, mM-’ oiMeAsp
FIG. 1. Spectrophotometric titration of native (Curve 1) and photooxidized (Curve 2) aspartate transaminase with a-methyl- nn-aspartate. Titrations were performed at 430 rnp with 8.9 X 10-S M native and 2.9 X 10-s M photooxidized enzyme in 0.05 M sodium pyrophosphate buffer, pH 8.8. a-Methyl aspartate was added with a precision microburette. The photooxidized enzyme had lost 80% of its original activity.
TABLE I
Dissociation constants of inhibitors with native and photooxidized supernatant glutamate aspartate transaminase
The binding of oxalacetate was studied with 0.5-ml samples of native, 9.8 X 10-z M, or photooxidized, 9.1 X 10e6 M, enzyme con- taining 0.05 M sodium pyrophosphate, pH 8.8, by titrating with 5-J aliquots of 1.0 M oxalacetate, neutralized to pH 8.8. Changes in optical density at 430 rnp were followed spectrophotometrically. Reported values were determined graphically as described in the text. The dissociation constants foror-ketoglutarate were deter- mined in the same manner, except that 1.09 X 10m4 M native and 3.98 X 10-b M photooxidized enzyme were used. The binding of glutarate was studied with 4.3 X lo+ M native or 3.1 X 10m6 M photooxidized enzyme containing 0.08 M Tris-HCl, pH 8.2, by titrating with 5-~1 aliquots of 100 mM glutarate, pH 8.2.
I EWApX
Substrate Native Photooxidized
Kd Kd (deionized) Ka (deifkxd)
llzY tn.%4
MethylaspartateG. . . 3.1 1.3 1.6 1.6 Oxalacetate. . . 190 1.7 70 Ketoglutarate. . . 65 6.1 10 Glutarate. . . . . 4 1 1.6 1.6
0 Details in Fig. 1.
the substrate than does the native enzyme. These, however, represent apparent dissociation constants only, since for native enzyme Jenkins has shown them to depend on the concentration of buffer anions (13). Fig. 2 shows the results of titrating native enzyme with a-methyl aspartate in the presence of different con- centrations of buffer anions. When the values of Kd are plotted against buffer anion concentration, a straight line is obtained whose intercept on the ordinate is the dissociation constant in the absence of anions. Titrations of photooxidized enzyme under the same conditions yield values for Kd which also give a straight line on the secondary plots. It is apparent that the value of the
2 16C
I I
/ SO
/
k 40
20
_il_j____
IO
1 - I I I 2 4 6
&,,M-’ ’ a- MeAsp
FIG. 2. Effect of anion concentration on the dissociation con- stant of a-methyl-nn-aspartate. Right, spectrophotometric titra- tions of native transaminase in different concentrations of Tris- HCl, pH 8.2. Samples, 0.4 ml, of 1.99 X 10-S M enzyme were titrated by addition of 5-~1 aliquots of 0.5 M a-methyl-nn-aspar- tate. Numbers refer to sodium chloride millimolar concentra- tions. Left, plots of the observed dissociation constants obtained from the spectrophotometric titration against the buffer anion concentration for native (1) and photooxidized (2) enzymes.
I I 40 80
[Cl-] mM
FIG. 3. Effect of anion concentration on the dissociation con- stant of glutarate. Kd values were obtained at the anion concen- tration indicated by titration with glutarate as described in Table I.
dissociation constant for photooxidized enzyme does not change significantly with changes in buffer anion concentration, whereas the native enzyme exhibits relatively large changes.
Similar results are obtained when the dicarboxylic acids are used as substrate as shown for glutarate in Fig. 3. Again, the photooxidized enzyme exhibits little variation with change in anion concentration, while the native enzyme exhibits relatively large changes. The values of the dissociation constants in the absence of anions for a-methylaspartate, oxaloacetate, cr-keto- glutarate, and glutarate are reported in Table I. In the absence of anions, the native enzyme has a higher affinity for each of the substrates than does the photooxidized enzyme.
Reaction with erythro-&Hydroxy-L-aspart&--Since, as shown above, photooxidized enzyme can bind substrates to form the initial complexes, it is of interest to determine whether it is also capable of forming the subsequent visible intermediates. In an
by guest on June 9, 2019http://w
ww
.jbc.org/D
ownloaded from

Function of Histidine Residue in Aspartate Transaminase Vol. 245, No. 4
equilibrium mixture of aspartate and native enzyme, an absorp- tion maximum at 490 rnp has been observed (13). This inter- mediate was assigned the structure shown in Scheme II and re- sults from the tautomerization of the initial aldimine Schiff’s base to the semiquinoid-like intermediate. In the case of aspartate, however, the amount of this intermediate is too small to study readily. This problem has been circumvented by using the sub- strate analogue, erythro-fl-hydroxyaspartate. In an equilibrium mixture with the transaminase, the same intermediates are formed with erythro-P-hydroxyaspartate, except that a greatly increased amount of the 490 rnp species is seen (14). The ability
I I 1 I I
300 350 400 450 500
“P
FIG. 4. Reaction of native (Curse 1) and photooxidized (Curve 2) (40% residual activity) asp&ate transaminase with erythro-fi- hydroxy-nn-aspartate, by difference spectroscopy. Each cuvette contained 1.0 ml of 4.8 X 10-e M enzyme in 0.1 M sodium tetra- borate, pH 9.1. HsO, 25,~1, was added to the reference side, while 25 ~1 of 0.1 M hydroxyaspartate were added to the sample side.
16
12
8
e
4
IO
- 2 ae
~~
5
2 IO 20
3 I/S (mM-‘1 A
1 A / 3 ‘\A’
FIG. 5. Reaction of native and photooxidized enzyme with er@ro-p-hydroxy-nn-aspartate. The reaction was followed spectropolarimetrically. Curve 1, native or photooxidized en- zyme in 0.1 M sodium tetraborate buffer, pH 9.1. Curve 8, native enzyme after the addition of hydroxy-nn-aspartate. Curve 8, photooxidized enzyme (20% residual activity) after the addition of a saturating concentration of erythro-p-hydroxy-nn-aspartate. Inset, titration of native (1) and photooxidized (2) enzyme with hydroxyaspartate. The samples containing 4.7 X 10-b M enzyme and 0.1 M sodium tetraborate, pH 9.1, were titrated with lo-p1 aliquots of 9.5 mM hydroxyaspartate.
of photooxidized enzyme to carry out this tautomerization has, therefore, been examined.
Native enzyme reacts with erythro-P-hydroxyaspartate to yield an enzyme substrate complex which absorbs at 490 rnp. On the other hand, photooxidized enzyme produces less of this interme- diate, the amount proportional to the residual activity (Fig. 4). Experiments with stopped flow techniques show that the rate of production of the smaller amounts of this intermediate is identical with that of the native enzyme (15). Apparently, only those enzyme molecules which remain unaltered are able to form this complex. It is also evident from Fig. 4 that photooxidized en- zyme cannot bypass this intermediate since the increase at 330 rnp and the decrease at 360 rnp are also proportional to the re- sidual activity.
Photooxidized enzyme does bind hydroxyaspartate, however, as judged by circular dichroism, to form an enzyme substrate intermediate which absorbs at 360 mp (Fig. 5). At high pH, both native and photooxidized enzyme possess a dichroic max- imum of the same amplitude at 360 rnp, which disappears com- pletely when an excess of erythro-P-hydroxyaspartate is added. By titrating a known concentration of enzyme with this amino acid, a dissociation constant may be determined graphically from a plot 1/0360 against l/X, where 13~~~ is the amplitude at 360 mp and S is the concentration of hydroxyaspartate (Fig. 5). The plots are linear for both native and photooxidized enzyme and the photooxidized enzyme has a slightly higher affinity for the substrate than does native enzyme.
Reaction with Hydroxyjumarate-Since photoxidized enzyme cannot form the semiquinoid intermediate in the forward reac- tion, we were interested in determining whether it is able to pro- duce this intermediate in the back reaction. Jenkins has shown that, in the presence of magnesium ions, hydroxyfumarate de- composes to form oxaloylglycolate (IS), the unstable transamina- tion product of hydroxyaspartate, and that this readily reacts with native pyridoxamine enzyme to yield a mixture of pyridoxal enzyme and the same intermediate which also absorbs at 490 rnh. As seen in Fig. 6, photooxidized pyridoxamine enzyme produces less of this intermediate, the amount being proportional to the residual activity. It is also apparent that the reaction does not bypass this intermediate nor is the equilibrium shifted, since the increase at 360 rnp is also proportional to the residual activity.
Tritium Exchange with erythro-/3-Hydroxyaspartate-As shown
Native
A 0.2
Photooxidized
FIG. 6. The reaction of pyridoxamine enzyme with hydroxy- fumarate by difference spectroscopy. Each cuvette contained 5 X 10-6 M enzyme, 0.05 M Tris-HCl (pH 7.5), and 2 mM MgCl. To the sample side, 5 ~1 of a saturated solution of hydroxyfumarate neutralized to pH 8 were added. An equal volume of water was added to the reference side. Solid line, native enzyme; dashed line, photooxidized enzyme (30% residual activity).
by guest on June 9, 2019http://w
ww
.jbc.org/D
ownloaded from

Issue of February 25, 1970 D. L. Peterson and M. Martinez-Carrion 811
in Scheme II, the formation of the semiquinoid complex which absorbs at 490 rnE.1 requires the loss of a proton from the a-carbon of the amino acid. Since, with hydroxyaspartate, the photo- oxidized enzyme cannot form the semiquinoid complex, one might expect the rate of exchange of the a-hydrogen of this substrate with tritium from tritiated water to be greatly reduced.
Fig. 7 shows the variation in the ability to catalyze this ex- change as a function of the degree of photooxidation, where the rate of exchange is proportional to the residual activity. Photo- oxidized enzyme is, apparently, unable to catalyze this reaction or does so only at a greatly reduced rate.
Binding of Aspartate and a-Ketoglutarate-The ability of native and photooxidized pyridoxal and pyridoxamine forms of the en- zyme to bind aspartate and cw-ketoglutarate, respectively, is shown in Table II. Both photooxidized and native pyridoxal enzyme readily bind aspartate; photooxidized enzyme binds a greater amount, presumably reflecting a higher affinity for the substrate or a larger half-life of the enzyme substrate complex(es) . Photooxidized pyridoxamine enzyme binds significant amounts of
MINUTES
FIQ. 7. Exchange of erythro-hydroxyaspartate and tritium- labeled water. Filled circles, native enzyme; open circles, photo- oxidized enzyme (60% residual activity) ; squares, photooxidized enzyme (40% residual activity). Details in text.
TABLE II Binding of aspartate and cy-ketoglutarate to pyridoxal or
pyridoxamine forms of native and photooxidized supernatant glutamate aspartate transaminase
Native, lo-’ M, or photooxidized, 1.2 X lo-’ M, enzyme in 0.05 M Tris-HCl, pH 7.5, was reacted with aspartate 0.05 mrvr, with 4.4 X 107 dpm per rmole. NaBHe, 0.1 ml, containing 1 mg per ml was added. Unbound substrate was removed by passage through a Sephadex G-25 column, 2.5 X 75 cm. The protein fraction was collected and the concentration was determined spectrophoto- metrically. Radioactivity was determined in Bray’s solution. In the binding of a-ketoglutarate, the reaction mixture and pro- cedurewere the same at 2.78 mM a-ketoglutarate with 7 X 106 dpm per pmole.
Native
ElUyItIe Aspartate Ketoglutarate
@VZ/p?iWlL? cpmjwtm1e
Pyridoxal. . . . . . . . 3.3 x 104 Pyridoxamine. . . . . . . . . . 5 x 102
Photooxidized Pyridoxal. . . 1.4 x 106 Pyridoxamine. . . . . 1.4 x 106
cr-ketoglutarate, while native enzyme binds little. This is con- sistent with the known nature of the equilibrium between the pyridoxal and pyridoxamine forms of the native enzyme. In the presence of cY-ketoglutarate, pyridoxamine enzyme is essentially, instantaneously, and completely converted to the pyridoxal form. If photooxidized pyridoxamine enzyme is produced by recon- stitution of photooxidized apoenzyme with pyridoxamine phos- phate, the back reaction is restricted, as shown below. If the substrate is bound, it should be readily trapped. We did, in fact observe this.
Reaction with Cysteine SulJinate-Native pyridoxal enzyme reacts with excess cysteine sulfinate to produce pyridoxamine enzyme and an unstable transamination product which decom- poses to yield pyruvate (17). Since 1 mole of pyruvate is formed for each mole of bound pyridoxal phosphate of both native and photooxidized enzyme (Table III), as judged by coupling the re- action with lactic dehydrogenase and DPNH, we concluded that the enzyme is completely converted to the pyridoxamine form for both enzymes. That pyridoxamine enzyme is produced from the pyridoxal enzyme is seen also by fluorescence and circular dichroism measurements. Up on addition of sodium hydroxide to pH 13, native and photooxidized enzymes produced after transamination with cysteine sulfinic acid exhibit essentially iden- tical fluorescence enhancement at 410 mp. For native enzyme, this is due to the release of bound pyridoxamine phosphate (18). The photooxidized enzyme exhibits the same increase indicating the same amount of bound pyridoxamine phosphate. The cir- cular dichroism spectra of both native and photooxidized enzyme produced in this reaction exhibit a single maximum at 333 mp (Fig. 8). Although photooxidized enzyme possesses only 75% of the amplitude of the native protein, it has only 30% residual catalytic activity. As both contain the same amount of pyri- doxamine phosphate, this difference must arise from slight varia- tions in the environment of the coenzyme.
The rate of the reaction with cysteine sulfinate has been ex- amined (Fig. 9). Native enzyme reacts essentially instantane- ously but photooxidized enzyme gives a biphasic reaction with an
TABLE III Conversion of pyridoxal enzyme to pyridoxamine enzyme by-cysteine
sulfinate The pyridoxal phosphate content of native and photooxidized
enzyme (30yo residual activity) was determined spectrophoto- metrically at pH 5.3 in acetate buffer as described in the text. The amount of pyruvate formed was determined by coupling the reaction with lactic dehydrogenase. The reaction mixture con- tained: 0.1 mg of lactic dehydrogenase; 0.1 M sodium pyrophos- phate, pH 8.8; 10-d M DPNH; and native enzyme, 8.2 X 10-s M, or photooxidized transaminase, 4.04 X 10-6 M. The change in opti- cal density at 340 m upon the addition of excess cysteine sulfinate was recorded; 6.22 X 108 ~-1 cm-l was the extinction coefficient used to determine the moles of DPNH consumed. The release of pyridoxamine phosphate from the enzyme in 0.05 M potassium phosphate, pH 7.5, after treatment with NaOH to pH 13 was measured as the enhancement in fluorescence of the coenzyme and is expressed in arbitrary units.
I Enzyme Pyridoxal
phosphate Pyruvate
Native.. . . . . Photooxidized. . . . . . .
/.mmles pmoles
80 82 28.5 40.3 40 29
by guest on June 9, 2019http://w
ww
.jbc.org/D
ownloaded from

812 Function of Hi&dine Residue in Aspartate Transaminase Vol. 245, No. 4
2.4
e:/jpl& , 1 300 350 400 450 500
FIG. 8. Circular dichroism spectra of native and photooxidized pyridoxamine enzyme before and after addition of cr-ketoglu- tarate. Curve 1, native pyridoxamine enzyme prepared either by reaction with a lo-fold molar excess of cvsteine sulfinate in 0.04 M
sodium pyrophosphate, pH 8.8, or by reconstitution of apoenzyme with excess pyridoxamine phosphate in the same buffer. Excess cysteine sulfinate was removed by dialysis against pyrophosphate buffer for 4 hours. Pyridoxamine phosphate was removed bv passage through a Sephadex G-25 column. Curve 2, same as i, after the addition of 8 X 10mp M a-ketodutarate neutralized to DH 8.0. Curve 3, photooxidized pyridoxal enzyme (35y0 residual ac- tivity) with a lo-fold molar excess of cysteine sulfinate as de- scribed for native enzyme. Curve 4, same as S, after addition of 8 X lo-4 M oc-ketoglutarate. Curve 5, photooxidized pyridoxamine enzyme prepared by recombination of apoenzyme (20% residual activity) with excess pyridoxamine phosphate, as described for native enzyme. Curve 6, same as 6, after addition of 8 X 10-J M
a-ketoglutarate. All spectra were normalized to an enzyme con- centration of 5.3 X 10-S M and with l.O-cm path length cuvettes.
initial phase which is instantaneous and proportional to the residual activity and a final slow phase. This second phase ap- parently represents the reaction of those molecules which have been modified by photooxidation. From a plot of log (A t - A,) against time, we obtained a straight line and a 3.3 x 10V3 see-1 value for the first order rate constant of the slow phase.
Reaction of Pyricloxamine Enzyme with oL-Ketoglutarate-Na- tive pyridoxamine enzyme produced either by reaction with cysteine sulfinate or by reconstitution of apoenzyme with pyri- doxamine phosphate readily reacts with ketoglutarate to form the pyridoxal form of the enzyme (Fig. 8). Photooxidized pyridox- amine enzyme produced by reaction of the pyridoxal form with cystine sulfinate can also be completely reconverted to the pyridoxal enzyme by transamination with a-ketoglutarate (Fig. 8). However, photooxidized pyridoxamine enzyme produced by recombination of photooxidized apoenzyme with pyridoxamine phosphate does not carry out this back transamination. Also, as is evident in Fig. 8, the ellipticity at 333 rnp is very low even though photooxidized apoenzyme binds pyridoxamine phos- phate (6).
DISCUSSION
Photooxidized enzyme is not inhibited in its ability to form the initial aldimine or ketimine complexes, either with the nat- ural substrates, aspartate and Lu-ketoglutarate, or with the sub- strate analogues, a-methylaspartate or hydroxyaspartate. Since the photooxidized enzyme is actually a mixture of photo- oxidized and native enzyme, it is necessary to be certain that the observations are not made on only the residual native enzyme. If only this residual enzyme were binding substrate, the dissocia- tion constant should remain unchanged upon photooxidation, and the extinction coefficient for the enzyme substrate complex should decrease to a value proportional to the residual activity.
0.40
I
A360 !
b
Olrl;ii=__;_ --o---- -o- ____ Q-----0-.
0.20 I I 1 I I I I
2o min 40 60
FIG. 9. Reaction of native (solid line) and photooxidized, 30% residual activity, (dash line) enzymes with cysteine sulfinate. Samples of enzyme, 0.5 ml, containing 0.05 M Tris-HCI, pH 8.2, were allowed to equilibrate for 15 min at 25”. The reaction was initiated by the addition of 25 ~1 of 10 PM per ml of cysteine sul- finate and the change in optical density at 360 rnp was recorded. Inset, first order plot of the reaction of photooxidized enzyme with cysteine sulfinate.
In fact, the dissociation constant decreases very significantly after photooxidation, while, as seen for methylaspartate (ordinate intercept in Fig. l), the extinction coefficients remain essentially unchanged. In the case of erythro-/3-hydroxyaspartate, the com- plete disappearance of the 360 rnp dichroic maximum for both native and photooxidized enzyme clearly shows that both species bind substrate.
The dissociation constants obtained in the presence of buffer represent apparent affinities only, since they depend upon the buffer anion concentration. Jenkins has shown that there are two sites which bind the carboxyl groups of the substrate and that, at high pH, one of these sites binds an anion as a competitive inhibitor which must be displaced by the substrate (11). The photooxidized enzyme does not exhibit this competitive ion ef- fect, and, as a result, has a higher apparent affinity for the sub- strate under normal buffer conditions. The true dissociation constant, in the absence of any buffer anions, is smaller for native enzyme than for photooxidized enzyme. Since only histidine residues have been destroyed, the anion-binding site might be such a residue. However, this seems unlikely because the anion- binding site appears to have a pK greater than 11 (17, 19). On the other hand, we have observed local conformational changes at the active site after photooxidation (6). These local altera- tions could result in a separation of the anion and substrate bind- ing sites, thereby abolishing the competitive anion effect. Other possible explanations could be proposed, however, since under normal assay conditions the photooxidized enzyme is catalytically inactive, and under these same conditions the affinity for sub- strate is greater than that of the native enzyme, it is apparent that effects on K1, Kz (Scheme II), or K, cannot explain the in- action.
Inactivation is normally followed by assay of the total reaction shown in Scheme I. Inability to carry out the forward reaction of each half-reaction, back reaction, or both would result in loss of activity. The reaction with cysteine sulfinate clearly shows that the forward reaction can occur, but only at a greatly re- duced rate. The back reaction has also been shown. However,
by guest on June 9, 2019http://w
ww
.jbc.org/D
ownloaded from

Issue of February 25, 1970 D. L. Peterson and Al. Martinez-Carrion 813
the rate of the back reaction was too great to measure with the methods described. Inactivation, therefore, seems to be primar- ily due to reduction of the rate for the forward reaction.
Since substrates bind to the photooxidized enzyme, this reduc- tion in rate must reflect an inability to convert the initial com- plex to products. Spectral studies indicate that the photo- oxidized enzyme is capable of making all of the enzyme-substrate intermediates except the semiquinoid complex absorbing at 490 rnp. The failure to form this intermediate has been related to the inability to remove the a-hydrogen of the amino acid. An analogous effect has been observed in rabbit muscle aldolase. Hoffee et al. (18, 20) have shown that destruction of histidine residues causes a great decrease in the ability of aldolase to catalyze the exchange between hydroxyacetone phosphate and tritium labeled water. This is particularly interesting in view of the similarity in mechanisms of the two enzymes, each pro- ceeding through a Schiff’s base intermediate.
The equilibrium between the various enzyme substrate com- plexes is pH-independent (21). On this basis, we concluded that a group within the protein is involved in the tautomerization steps. In model systems, imidazole has been shown to be an effective catalyst (22)) and Ayling, Dunathan, and Snell(23) have suggested that either a lysine or histidine may serve a similar role in the enzymatic reaction of pyridoxamine-pyruvate trans- aminase. Since photooxidation destroys activity by altering only histidine residues (6), and because the only observable ef- fects on the mechanism which can readily account for this inac- tivation are effects on those steps known to involve the removal of protons from the enzyme substrate complexes, it seems that the histidyl residue functions as a general base catalyst for inter- conversion of these complexes, most probably, in the removal of the a-proton of the substrate amino acid.
REFERENCES 1. VALLEE, B. L., Fed. Proc., 23. 8 (1964). 2. KNOWLES, J. R., Biochem. J., 96, 180 (1965).
3.
4.
5.
6.
7.
8. 9.
10.
11.
12.
13.
14. 15.
16. JENKINS, W. T., J. Biol. Chem., 236, 1121 (1961). 17. JENKINS, W. T. AND D’ARI, L., J. Biol. Chem., 241,2845 (1966). 18. FARRELLY, J. G., AND CHURCHICH, J. E., Biochim. Biophys.
19.
20.
21. 22.
23.
MILLBR, D. B. S., AND SCHWERT, G. W., J. Biol. Chem., 238, 3249 (1963).
SNELL, E. E., BRAUNSTEIN, A. E., SEVERIN, E. S., AND TORCHINSKY, Yu. M. (Editors), Pyridoxal catalysis: enzymes and model sustems. Interscience Publishers. New York. 1968.
MARTINEZ-CARRION, M., Tu~aNo, C., RIVA,‘F., AND FA~ELLA, P., J. Biol. Chem., 242; 1426 (1967) . .
MARTINEZ-CARRION. M.. KUCZENSKI. R.. TIEMEIER. D. C.. AND PETERSON, D: L., 3. Biol. Chek., 246, 799 (197Oj. ’
M.~RTINEZ-C.\RRION, M., TUR~NO, C., CHIANCONE, E., Boss.%, F., GIARTOSIO, A., RIVA, F., END FASLLLA, P., J. Biol. Chem., 242, 2397 (1967).
LIS, H., Biochim. Biophys. Acta, 28, 191 (1958). ROSEN, H., Arch. Biochem. Biophys., 67,10 (1957) MARTINEZ-CARRION, M., in K. YAMADA, N. KATUNUMA, AND
H. WAD.I (Editors), Symposium on pyridoxal enzymes, Maruzen Company, Ltd., Tokyo, 1968, p. 19.
JENKINS, W. T., AND D’ARI, L., J. Biol. Chem., 241, 5667 (1966).
FASELLI, P., GIARTOSIO, A., AND HAMMES, G., Biochemistry, 6, 197 (1966).
JENKINS, W. T., AND TAYLOR, R. T., J. Biol. Chem., 240, 2907 (1965).
JENKINS, W. T., J. Biol. Chem., 236, 1121 (1961). MARTINEZ-C.LRRION, M., AND PETERSON, D. L., Symposium
on mechanism of enzyme action, vol. 19, Federation of European Biochemical Societies, Academic Press, London, P. 229.
Acta, 168, 280 (1968). JENKINS. W. T.. in E. E. SNELL. A. E. BRAUNSTEIN. E. S.
SEVER~N, AND Y. M. TORCI&KY (Editors), Py&doxaE catalysis: enzymes and model systems, Interscience Pub- lishers, New York, 1968, p. 324.
HOFFEE, P., LAI, C. Y., PUGH, E. L., .~ND HORECKER, B. L., Proc. Nat. Acad. Sci. U. S. A., 67, 107 (1967).
JINKINS, W. T., J. Biol. Chem., 239, 1742 (1964). THANASSI, J. W., BUTLER, A. R., AND BRUICE, T. C., Bio-
chemistry, 4, 1463 (1965). AYLING, J. E., DUNATHAN, H. C., AND SNELL, E. E., Biochemis -
try, 7, 4537 (1968).
by guest on June 9, 2019http://w
ww
.jbc.org/D
ownloaded from

D. L. Peterson and M. Martinez-CarrionTRANSAMINASE
RESIDUE AT THE ACTIVE SITE OF SUPERNATANT ASPARTATE The Mechanism of Transamination: FUNCTION OF THE HISTIDYL
1970, 245:806-813.J. Biol. Chem.
http://www.jbc.org/content/245/4/806Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/245/4/806.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on June 9, 2019http://w
ww
.jbc.org/D
ownloaded from