the multistage model of cancer development: some...
TRANSCRIPT

The multistage model of cancer development some implications
Gordon Rittera Richard Wilsona Francesco Pompeiab and Dimitriy Burmistrovac
aHarvard University Department of Physics Cambridge USAbExergen Corp Watertown MA USAcMenzie-Cura and Associates Winchester MA USA
The multistage model introduced by Armitage and Doll was very successful at describing manyfeatures of cancer development Doll and Peto noted a significant departure below the prediction of
the model and suggested that this could be due to undercounting of cases at older ages or to the
lsquobiology of extreme old agersquo Moolgavkar pointed out that it could also be due to the approximation
used The recent observation that cancer incidence falls rapidly above age 80 has stimulated new
modelling investigations such as the PompeiWilson beta model (which does reproduce the rapid
fall) In the present paper we argue that Moolgavkarrsquos criticisms while mathematically correct do
not affect the conclusions particularly the constancy of the number of stages across different cancer
registries (Cook Doll and Fellingham 1969 A mathematical model for the age distribution ofcancer in man International Journal of Cancer 4 93112) We discuss several exact solutions
compare them with the most recent data and prove rigorously that the standard ArmitageDoll
multistage model can never reproduce the sharp turnaround in cancer incidence at old age seen in the
data We discuss in detail multistage processes which have a property observed in many laboratory
studies namely that some stages progress much faster than the others We verify mathematically the
intuition that sufficiently fast stages do not appreciably affect the incidence rate of cancer and
discuss implications of this fact for cancer treatment strategies We also show that the simplest
possible modification of the ArmitageDoll model to incorporate cellular senescence just leads tothe PompeiWilson beta model Toxicology and Industrial Health 2003 19 125145
Key words beta model cancer incidence carcinogenesis cellular senescence multistage model old
age
Introduction
The idea that cancer proceeds by a number of stages
was developed by Armitage and Doll (1954) soon
after cancer registries became available in many
countries They noted that cancer incidence was not
very reliable whereas mortality is much more
objective Therefore they plotted cancer mortality
against age rather than incidence However evenmortality was not often well described above age70 since many death certificates had the nebulousentry lsquoold agersquo They therefore only used themultistage model to describe cancer mortality upto age 70 Cancer incidence I(t) is defined byepidemiologists as the rate of diagnosing new casesdivided by the number of persons at risk Similarlynormalized cancer mortality is the rate of cancerdeaths divided by the number of persons at riskArmitage and Doll found that cancer mortalityfitted a function
Address all correspondence to Gordon Ritter Harvard UniversityDepartment of Physics 17 Oxford Street Cambridge MA 02138 USAE-mail ritterfasharvardedu
Toxicology and Industrial Health 2003 19 125145
wwwtihjournalcom
Arnold 2003 1011910748233703th195oa
I a tk1 (1)
where k was interpreted as the number of stages inthe progression of cancer and a is proportional tothe product of the probabilities at each stage Thismodel successfully described several importantknown features of cancer
1 There is usually a latency period between theinsult which appears to cause the cancer and theexpression of the cancer
2 If cancer-causing agents act at different stages ofthe cancer progression and are present at highdoses then a multiplicative synergy is predictedas seen for example between smoking andasbestos exposure
3 Cook et al (1969) found that while k varied fordifferent tumor sites it is constant betweencountries The constant a varied between coun-tries as might be expected from the differentenvironmental situations
These successes led to widespread use of themodel It was soon noted that above age 60 theage-specific mortality rate flattened and fell belowthe curve predicted by Armitage and Doll Twomajor reasons for such a flattening have beensuggested the first is a variation in susceptibility(variation in a) between different members of thesame group being considered and the second is afailure of the mathematical approximations used byDoll and Armitage
Cook Doll and Fellingham considered varia-tions of susceptibility in the population Theyplotted incidence versus age on a logarithmicscale for varying fractions C of sensitive persons(Cook et al 2004 Figure 4 reproduced here asFigure 1) They concluded that if only 1 of thepopulation is susceptible to cancer then at olderages the normalized incidence curves must fallwhen all the 1 have developed cancer
Even if the number of susceptibles is 10 then aflattening occurs Figure 1 reproduced from Cooket al (1969) indicates that by suitable choice of theparameter C one might be able to describe a rapidfall off in incidence a suggestion we discuss laterCook Doll and Fellingham ultimately rejected thispossibility because data showed a constant agefor the peak of cancer incidence while theirparticular assumption of insensitive individuals
indicates that incidence peaks at a younger age
for rarer cancersThe variations in cure rates for cancer in the last
50 years suggest that it is now more important to
discuss incidence rather than mortality and in
addition that incidence may now be much better
determined than 50 years ago In this paper there-
fore we reopen the discussion with an emphasis on
cancer incidence rather than mortalityLet f(t) denote the event rate for the kth change
at time t in a single cell According to our
definition cancer incidence is related to f(t) by
I(t)Nf(t) where N is the number of affected cells
The theoretical model calculates f(t) we will per-
form this calculation exactly for a variety of
interesting situationsIn the case of the ArmitageDoll model
IAD(t)h(t) is an infinitely growing hazard func-
tion and satisfies the criterion that the cumulative
probability of cancer given by
Figure 1 Cook Doll and Fellingham (1969) considered variablesusceptibility in the population This plot shows incidence versus ageon a logarithmic scale for varying fractions C of sensitive personsModified from Cook et al (1969) Figure 4
Multistage model of cancer developmentG Ritter et al
126
F (t)1exp
g
t
0
h(t)dt
(2)
approaches 1 in the limit as t 0 According tothe definition of the hazard function we considerthe differential equation
IAD(t)atk1F (t)
1 F(t) (3)
Solving this equation with the initial conditionF(0)o yields
F (t)1(o1) exp
atk
k
The condition o0 corresponds to fetal cancerwhich while a real condition is ignored in thispaper Hence we are concerned with the limit o00
F(t)1exp(atk=k) (4)
This satisfies limt0F (t)1 which describes thefact that according to this model everyone willdevelop cancer if they do not die of something elseIt is obvious from these considerations thatthe ArmitageDoll approximation (Equation (1))increases without bound and given sufficienttime any susceptible cell eventually becomes ma-lignant
As noted above the ArmitageDoll model(Equation (1)) must be viewed as an approximationThe correct equations were written down byMoolgavkar (1991) and Armitage (1985) and itwas seen that (Equation (1)) is only valid for smallvalues of t It was seen also that (Equation (1))overestimates incidence Moolgavkar (1991) haspublished a simple example supporting the ideathat ArmitageDoll approximation can be inade-quate if the transition rates for cancer stages are notsmall enough His example presupposes a smallernumber of cells N109 in the tissue of concern thanis usually taken In contrast to the ArmitageDollapproximation the hazard function in the exactmodel has a finite asymptote limt0h(t)Nmminwhere mmin is the minimal transition rate Thesuggestion that the failure of the ArmitageDollapproximation may be responsible for the observedflattening of incidence for ages above 60 followsfrom this
Pompei and Wilson (2001) reopened the issue
They noted that since 1955 data on cancer
incidence has improved and data on both incidence
and mortality above age 70 has improved For
discussions of cancer development incidence may
now be more reliable (for many tumor sites) than
mortality due to improvements in cancer treat-
ment Making the assumption that the data and
particularly the SEER data in the United States are
reliable Pompei and Wilson plotted the normalized
cancer incidence data against age finding a rapid
drop in normalized incidence above age 80 which is
not explained by simple parameter adjustments
of the ArmitageDoll model This is illustrated
in Figure 2 for a common cancer (colon) an
Figure 2 SEER data from Pompei et al (2004) and SEER (2002)shown together with the best possible fit of the ArmitageDoll model
Multistage model of cancer developmentG Ritter et al
127
intermediate-incidence cancer (non-Hodgkins lym-phoma) and a rare cancer (brain)
Pompei and Wilson added an extra term toEquation (1) leading to a beta function I
atk1(1bt) which fits the data for most cancersremarkably well and speculated on possible biolo-gical explanations for the extra term PompeiPolkanov and Wilson (Pompei et al 2001) exam-ined the limited data on laboratory mice that havebeen followed for their natural life of 1000 days ormore rather than being sacrificed at 750 days Theage of cancer incidence is not well determined formice but cancer mortality suffers neither fromcomplications of improved treatment nor frommisreporting In the absence of these systematicerrors it was observed that the cancer mortality fellto zero before the end of life This is shown inFigure 3
In the present paper we re-examine the criticismsof Moolgavkar and improve upon the speculationsof Pompei and Wilson This is done byexact calculations in the multistage model In theprocess we derive several closed-form solutionsto the model Following previous studies (Pompeiand Wilson 2001) we continue to make theassumption that the SEER data in the UnitedStates is reliable above age 80 We prove that underthis model as one would expect the probabilitythat a single cell becomes cancerous given infinitetime is 1
Crucial parameters for the multistage model arethe transition rates which we denote mi We give anexact solution for a model with a number ns of slowstages with rate ms and one fast stage with rate mfand show that if mfms the fast stage only slightlyperturbs the final solution and may be neglectedWe show that if the multistage model is assumed tocalculate the probability of cancer for a single cell
then this model can only be consistent with therapid fall-off observed in the data if some unusualbiological assumptions are made
Methods
Assumptions of the multistage model
In this section we repeat starting with the funda-mental assumptions the derivation of the exactmultistage model and suggest various extensionsthereof
In the ArmitageDoll approximation cancerincidence has a simple power law growth This isan approximation to the exact multistage model(Moolgavkar 1978 Moolgavkar et al 1999 Pom-pei and Wilson 2001) described by the Batemanequations (Bateman 1910) The multistage modelmakes several basic assumptions
1 Malignant tumors arise from a series of mod-ifications of a single progenitor cell
2 The process of developing a malignancy isequally likely for all cells in the same tissue
3 The process of developing a malignancy in onecell is independent of the process in any othercell
4 After a malignancy has developed in a cellproliferation to an overt cancer is rapid andinvolves many cells in the same tissue and mayeven involve metastasis to another tissue
Under these assumptions cancer is the last of aseries of k sudden and irreversible changes whichmust take place in a specific order For a cell whichhas experienced (i1) changes the event rate forthe next change is mi We choose the conventionthat a single transition from a state p0 to a state p1
p0 0m1
p1 (5)
corresponds to one lsquostagersquo and has k1 Thisimplies that the number of transition rate constantsm1 mk always equals the number of stages
Cancer in a cell
Cancer is the last of a series of k sudden andirreversible changes which must take place in a cellin a specific order As is standard for Poisson
Figure 3 Mice data from Pompei et al (2001) shown together withthe best possible fit of the ArmitageDoll model
Multistage model of cancer developmentG Ritter et al
128
random processes we neglect the probability of twoor more events taking place in (t tdt) as dt 0 0This means that if a cell is in state pi at time t theprobability of transformation to state pi1 in asmall time interval Dt is given by mi1Dto(Dt)where o(Dt) is very small compared to Dt ando(Dt)=Dt 0 0 as Dt 0 0 Also the probability oftransformation i 0 i j with j1 in time Dt isassumed to be o(Dt) This implies that 1=mi1 is theaverage time required for the cell to go from state ito state i1
The probability to find a cell in the ith stage bythe end of time interval (t tdt) is then given by
pi(tdt) (1mi1dt) pi(t)mi1dt 0
midt pi1(t) (6)
Taking the limit dt 0 0 Equation (6) becomes
p0(t)m1p0(t)
pi(t)mi1pi(t)mipi1(t) (7)
pk(t)mkpk1(t)
The first and the last equations in this system aredifferent because there are no stages before thestage i0 (normal cell) and no stages after the kth
stage which corresponds to cancer The system(Equation (7)) should be completed with appro-priate initial conditions
p0(0)1 pi(0)0 (i1 2 k) (8)
which mean that the cell was normal at time t0The last equation in Equation (7) also gives theprobability to find the cell at its last cancerousstage Following Armitage and Doll we reserve thenotation f(t) for this quantity
f (t)pk(t)mkpk1(t) (9)
Thus f(t) denotes the event rate for the kth
change (corresponding to cancer) at time t in asingle cell
Cancer in a tissue and in humans
The event rate f(t) is a probability distributionfunction (PDF) for appearance of cancerous cellswhich means that f
0f (t)dt1 which means that
given enough time each cell will become cancerousEven though the PDF is convenient for mathema-
tical modelling it is not directly observable Onlydeveloped cancer in a tissue rather than a singlecancer cell could be recognized as a cause of deathor even diagnosed at all In practical studies cancerincidence is commonly observed Following thenotation of Armitage and Doll cancer incidenceI(t) (which is the same quantity as the hazard
function used in biostatistics and denoted there ash(t)) is defined by epidemiologists as the rate ofdiagnosing new cases divided by the number ofpersons at risk
Let us define a cumulative distribution function(CDF) for cancer per cell as F(t)f
t
0f (t)dt that is
the probability for a cell to become a cancer bymoment t Then the probability of at least onecancer cell in a tissue of N cells by the moment t isG(t)1[1F(t)]N This definition assumes theindependence of cells Cancer incidence can bedefined in terms of G(t) as
I(t)G(t)=(1G(t))NF (t)=(1F (t))
Cancer is a rare event even on the tissue leveland therefore F (t)1 and hence the approxi-mation
I(t)Nf (t) (10)
is quite good since the number of cells in a tissue Nis greater than 109 Equation (10) allows fitting ofthe theoretically derived cancer event rate per cellf(t) and the observed incidence of cases in popula-tion I(t) This result was derived by Armitage andby Moolgavkar
In the above the function I(t)Nf (t) is inci-dence of cancer in a tissue or equivalently in aperson When multiplied by the size of a popula-tion this becomes the true number of cancers inthat population at risk or the incidence Howeverthe number of cells N may not be large enough toensure that the ArmitageDoll approximation isaccurate so it is important to discuss this para-meter There are about 1014 cells in a human bodyand in any given tissue they would be proportionalto weight Thus as an order of magnitude approx-imation the colon has roughly 1012 cells Thepostulate that only cells on the surface are im-portant may reduce this to 1011 Therefore in whatfollows we take the number used by Moolgavkar(109 cells) as a lower limit
Multistage model of cancer developmentG Ritter et al
129
The Bateman equations
The exact solution to the multistage model can be
derived by analogy from Batemanrsquos solution of
successive radioactive decays (Bateman 1910)The simplest form of Batemanrsquos solution de-
scribes a linear decay chain leading ultimately to a
stable nucleus More complex forms have also been
used to describe for example the decay of radium
The isotope radium C decays into radium C and
radium C which both decay into lead so the
diagram contains a loop It is highly likely that
these more complex forms are also relevant to
cancer incidence although in the present work we
restrict attention to linear decay chains A k-stage
radioactive decay process depicted schematically
by the diagram
is described by the Bateman equations a set ofcoupled linear first-order differential equations inwhich pi(t) denotes the quantity of objects in state i
at time t
dp0
dtm01p0
dpj
dtmj1 jpj1mj1pj (1B iBk) (11)
dpk
dtmk1kpk1
where mj j1 is the partial rate constant for thetransition from state j to state j1 and mj is thetotal decay rate1 of state j1 The partial decayrates are related to the total decay rates byconstants bj j1 called branching ratios Thisrelationship takes the form
mj j1bj j1mj1
In the case of a linear decay chain the branchingratios equal unity and the partial rate constants aregiven by
mj j1mj1
In this simple case Equation (11) is equivalent tothe equations derived earlier in our discussion ofcancer in a cell Most discussions of the modelmake this assumption However we note that notall lsquoinitiatedrsquo cells proceed to cancer but the vastmajority are either repaired or excreted Somemay undergo clonal expansion and multiply Thiscould be modelled by taking values of thebranching ratio bj j1 less than unity (in the firstcase) or greater than 1 for clonal expansionHowever this is a very simplistic view of clonalexpansion which treats the expansion as a defini-tive process rather than a stochastic one We areattempting to understand this further The reali-zation that the multistage model only takes intoaccount the slow late limiting steps in the cancerprocess allows a theory in which clonal expansionoccurs rapidly but is not noticed as a separatestage
However if these branching ratios are the samefor each cell they may be subsumed in an overallconstant factor in the formula for incidence andthe fitted values of m adjusted accordingly Thenthe algebra is the same as if each and every celleventually becomes cancerous
We will work with linear decay chains in whatfollows In that case the diagram of the processsimplifies to
p0 0m1
p1 0m2 0mk
pk (12)
In the earlier section lsquoCancer in a cellrsquo it wasshown that the same differential equations describethe multistage model for the formation of malig-nant cells Every cell eventually transforms givensufficient time so in a realistic model
pk()g
0
f (t)dt1 (13)
In Lemma 3 we show that Equation (13) is a truemathematical consequence of the Bateman equa-tions with appropriate initial conditions
1In a model describing radioactivity mi1ln(2)=t1=2 where t1=2 is thehalf-life of that species Our notation differs slightly from that ofMoolgavkar who identifies the rate constant with the state being leftrather than with the state to which the transition is being made
Multistage model of cancer developmentG Ritter et al
130
The most general initial conditions for the
system Equation (11) are to allow pi(0) to be arbit-
rary nonzero constants for all i The solution
to the system Equation (11) with general initial
conditions is
for m1 kA simplifying assumption consistent with the
use of Equation (11) to analyse the probability of asingle cell becoming cancerous (or the radioactivedecay of a single nuclide) is that the concentrationsof the later stages (or daughter nuclides) are allinitially zero This assumption is equivalent to theinitial conditions
p0(0)a
pi(0)0 (for all i1) (15)
With these initial conditions the solution (Equa-tion (14)) reduces immediately to the simpler formvalid for m f1 kg
m1(t)cm
Xm
j1
xjm emj t (16)
where cmaYm1
j1
mj1j
Here c1 is an empty product which is 1 byconvention c2m12 etc and we have defined time-independent constants
xjmY
l1m
lj
(mlmj)1 (17)
Equations (16) and (17) were shown for thelinear chain by Moolgavkar (1991) [(A1) p 217] inhis work on the multistage theory of carcinogenesisWhen evaluating the sum in Equation (14) the usershould be warned that the terms tend to be largeand of opposite sign Indeed when the m are equalanother formula has to be used The cancerincidence in a tissue with N cells (which is the
approximation for a person) is Nf(t) to a very highdegree of accuracy and
f (t)pk (t)mkpk1(t)
We state our most important mathematical asser-tions as a series of Lemmas for which we provideproofs in an appendix
Lemma 1 (Solution of the Bateman equations)Assume that mi mj for all i j Then the functionspm(t) defined by Equation (16) and Equation (17)
solve the Bateman equations Equation (11) withinitial conditions Equation (15)
Generally we take the initial condition a1 Itis easy to see that the first nonvanishing term in aTaylor series of pm1(t) is of the ArmitageDollform where m is an index for an intermediate stageIndeed pm1(t) is the constant cm times a linearcombination of exponentials am
j1xjm emj t the nth
term in the Taylor series of this linear combinationis clearly
Xm
j1
xjm
(mjt)n
n (18)
This vanishes by algebraic identity if nBm1 andfor nm1 one may check that am
j1xjmmm1j 1
so to lowest nonvanishing orderpm1(t)cmtm1=(m1)
The next nonvanishing term (nm) also has asimple form Equation (18) with nm becomes
tm
m
Xm
j1
mj tm
(m 1)m
where m1
kak
i1mi denotes the average m We may
then write
pm1(t)cm
tm1
(m 1)(1m t )
Substituting mk for the last stage we find
I(t)atk1(1m t )
where
a
Qk
j1 mj
(k 1) (19)
pm1(t)Xm1
i0
Ym2
j0
mj j1
Xm
j1
pi(0)emj tQl (i 1) m
l j
(ml mj)
26664
37775 (14)
Multistage model of cancer developmentG Ritter et al
131
This is the Taylor expansion discussed by Mool-gavkar It was referred to (Moolgavkar 1978) as theMacLaurin series but that is a special case of themore general Taylor series We use the more generalterm in this paper
As one might expect the solution (Equation(16)) has a simple series expansion which can beexpressed in closed form This is given in thefollowing Lemma
Lemma 2 (Taylor expansion)Let fmi i1 kg be any set of nonzero constants
with mi mj if i j Then
Xm
i1
ximemi tX
j]m1
ajtj
with am11=(m1) and
aj1(1) jm
j
Xi155ijm
mi1mi2
mijm(20)
if jm This implies that
pm1(t)cm
tm1
(m 1)
1
m
Xm
i1
mi
tmO(tm1)
Remark 1 In various applications it is mostconvenient to use the lowest nonvanishing order(the ArmitageDoll approximation) in this seriesexpansion and itrsquos necessary to have conditionswhich guarantee that this truncation is valid Wecompare the lowest nonvanishing order to the(assumably better) approximation obtained byconsidering higher-order terms In particularlet us compare the lowest nonvanishing order(jm1 in the above formulae) to the followingterm In this paper as with the papers ofMoolgavkar we are concerned with the last stageonly and this corresponds to mk in the aboveequations Let Dn denote the nth nonzero term inthe Taylor expansion which has coefficientakn2
By a simple calculation
D2
D1
t m (21)
This formula shows that D2=D1 can be arbi-trarily large by inclusion of a fast stage or
stages each with large m When attempting as
we are doing to find a model to fit the data
from ages 20 to 70 years the constant a in the
first term of Equation (19) is heavily constrained
by the data If k is not varied the constant a
is proportional to the product m1 mk One
can only make m arbitrarily large if this pro-
duct is fixed to the correct value This is
discussed further in the section on determination
of k We will show later that if nf of the stages are
much faster than the remaining nsknf stages
then the exact solution for the k-stage model is
approximately equal to the exact solution for the
remaining ns stages which we call slow stages
Equation (21) indicates that the Taylor series for
k stages converges more slowly than the Taylor
series for ns stages because inclusion of fast
stages increases the average transition constant
In the presence of fast stages a large number of
terms must be included before the Taylor series
becomes accurate and consideration of only the
first and second terms in the expansion is
incorrect However the exact solution for the
model without the fast stages has a different
Taylor series which converges quite rapidly This
realization leads to important practical conclu-
sions that are discussed in more detail later
Specifically if inclusion of a few fast stages set
m103 then Equation (21) shows that D2 is 10
percent of D1 at t100 If the stages are fast
enough to set m102 per year then D2=D1 is
of order one at 100 years It is clear that by
including fast stages D2=D1 can be made arbi-
trarily largeMoolgavkar deliberately chose a set of para-
meters which included some slow and some fast
(but not very fast) stages (see for example
Moolgavkar 1978 Table 1) Although the above
estimate for D2=D1 does hold for Moolgavkarrsquos
example a discussion of his chosen parameters
needs more careful numerical analysis Consider
a tissue of 109 cells affected by a hypothetical
six-stage2 malignant tumor with transition rates
(per year) given by
2Moolgavkar (1991) refers to this as a seven-stage process but onlygives values for six transition constants We assume here that six stagesare meant
Multistage model of cancer developmentG Ritter et al
132
104 2104 34104 7103
8103 9103
In this example
m00046 per year
So our formula Equation (21) predicts D2046D1
at t100 years This is precisely what is observed inFigure 4 which shows the exact solution togetherwith its first five Taylor approximants The numer-ical factor relating the second approximationwhich lies below the exact curve to the firstapproximation lying above the curve at t100years is seen to be about 146
As noted in the introduction 109 cells may be toofew Bearing in mind that the model is used to fitincidence data each value of the six constants m
would have to be reduced by a factor of 2 if N wereincreased to 64109 and the exact formula at aget100 years would then be only 128 times theArmitageDoll approximation
The implications for determination of the valueof k using Moolgavkarrsquos parameters is discussedlater More generally
Dn1
Dn
akn1
akn2
t
t
k n
Pnk
Pn1k
where Pnk is the symmetric degree n polynomial ink variables given by ai155in
mi1 min
These poly-nomials satisfy
Pnk mkPn1k(terms involving m1 mk1)
It follows that for n]2
Dn1
Dn
t
k n
mk
ratio approaching
zero as mk 0
13(22)
The surprising conclusion of Equation (22) isthat if we add one very fast (mm104) stage at theend it may be necessary to consider hundreds orthousands of terms in the Taylor expansion in orderto attain accurate results The rate of convergenceof this expansion is affected strongly by theappearance of fast stages while the exact solutionis not This is a very important paradoxical resultthe solution for which may be found in the earliercomments If there are ns slow stages and nf veryfast stages the exact solution is close to the
ArmitageDoll approximation for the ns slowstages alone the ArmitageDoll approximationfor the full exact model with nsnf stages is notaccurate For equal transition probabilities givenby the single transition constant m the aboveimplies D2=D1mt
Lemma 3 (Normalization)Let p0(t) pk(t) satisfy the k-stage Batemanequations (Equation (11)) with any collection(m1 mk) of transition constants Then
g
0
pk(t)dtYk
i1mi
Xk
j1
xjk
mj
1 (23)
This equation shows that given sufficient timethe cancer process will eventually reach the laststage
General properties of the solutions
The general solution for reasonable values of k has far too many constants to be determined byepidemiological data alone However there arevarious cases in which the solution of the Batemanequations Equations (16) and (17) can be simplifiedenough to depend on only a few independentconstants even if the number of stages is large
Equation (21) implies that the ratio of the secondterm to the first in the Taylor series for I(t)
D2
D1
is proportional to the average m But the product ofthe ms is constrained by the value of the observedincidence With this constraint the average m will
Figure 4 Exact solution for Moolgavkarrsquos example shown with itsfirst five Taylor approximants
Multistage model of cancer developmentG Ritter et al
133
be a minimum when all the values of m are equalFor this important special case D2=D1 is muchsmaller than for example in Moolgavkarrsquos case
Another interesting result is that permuting thestages makes no difference to the final result pk(t)or more generally to mmpm1(t) for any m5k Thisis self-evident physically for radioactive decaywhich is also described by the Bateman equationsThis result used implicitly in the ArmitageDollapproximation is a fundamental property of theexact solution It may be proved as follows Itsuffices to consider a transposition of the ith stageand the jth stage as any permutation maybe represented as a product of transpositionsNow xim is a product of (mlmi)
1 where l iApplying the transposition these terms become(mlmj)
1except for the l j term which be-
comes (mimj)1 The result is thenQ
lj(mlmj)1
xjmA third general result is that if there are say 4
stages which proceed slowly (small equal values ofm) and many stages which act much faster (values ofm that are 10 times larger) then the final resultdepends only on the 4 slow stages This is self-evident physically and was probably implicit in thethinking of earlier users of the multistage modelThis is proven rigorously in Lemma 6 and dis-cussed with a specific example in the section onmeasurement of k
We pay special attention to a simple case (onenot covered by Lemma 1) in which all stages haveequal transition probabilities This simple case isdescribed by Lemma 4
Equal transition ratesLemma 4 (Equal transition rates)Consider the Bateman equations pj (t)m(pj1pj)arising from equal transition rates m1m2
mkm For any natural number j5k1 the solutionis
pj(t)1
jemt(mt) j (24)
In particular for the penultimate stage
pk1(t)1
(k 1)emt(mt)k1
The global maximum of pj(t) occurs at the agetpeak j=m Further as before we have
g
0
pk (t)dtg
0
mpk1(t)dt1
Remark 2 Since f(t) applies to a single cell m isvery small (on the order of 103) and hence for anynonzero number of stages tpeak j=m always occursat a much greater age than those contained in ahuman lifespan This important point is discussedin detail in the earlier section lsquoCan the multistagemodel ever fit the datarsquo
The algebraic simplicity of equation Equation(24) makes it attractive as a starting point for ourdiscussions It has two adjustable constants k andm A useful numerical example is the followingSuppose we would like to know how the exactsolution Equation (24) differs from the first term inits series expansion
pj(t)Xn0
(mt) jn
jn (25)
In general the first term in the series expansion ofexxn (n0 an integer) is xn and jexxnxn j
xn(1ex) In a numerical example with G03 G001 per year and k7 we have mt502for all t in a human lifespan Then frac121emtfrac12502and so for the first Taylor remainder R1 of pj(t) wehave
jR1j51
5k1256106 (26)
Equally separated transition rates
Another case which is amenable to an exactanalytical solution is when the transition constantsmi for the various stages increase by a fixed amountOf course it is not necessary to assume that in theactual cancer process the transition rates occur inincreasing order thanks to invariance of the processunder general permutations as remarked above sothis solution applies to the general situation whenthere exists some arbitrary reordering of thetransition constants so that they increase linearly
Lemma 5 (Equal separation)Suppose that the transition constants mm1m2 mk are uniformly separated so that mi1mi d for all i and for some constant d0 Then for anyn5k the solution is given by
Multistage model of cancer developmentG Ritter et al
134
pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

I a tk1 (1)
where k was interpreted as the number of stages inthe progression of cancer and a is proportional tothe product of the probabilities at each stage Thismodel successfully described several importantknown features of cancer
1 There is usually a latency period between theinsult which appears to cause the cancer and theexpression of the cancer
2 If cancer-causing agents act at different stages ofthe cancer progression and are present at highdoses then a multiplicative synergy is predictedas seen for example between smoking andasbestos exposure
3 Cook et al (1969) found that while k varied fordifferent tumor sites it is constant betweencountries The constant a varied between coun-tries as might be expected from the differentenvironmental situations
These successes led to widespread use of themodel It was soon noted that above age 60 theage-specific mortality rate flattened and fell belowthe curve predicted by Armitage and Doll Twomajor reasons for such a flattening have beensuggested the first is a variation in susceptibility(variation in a) between different members of thesame group being considered and the second is afailure of the mathematical approximations used byDoll and Armitage
Cook Doll and Fellingham considered varia-tions of susceptibility in the population Theyplotted incidence versus age on a logarithmicscale for varying fractions C of sensitive persons(Cook et al 2004 Figure 4 reproduced here asFigure 1) They concluded that if only 1 of thepopulation is susceptible to cancer then at olderages the normalized incidence curves must fallwhen all the 1 have developed cancer
Even if the number of susceptibles is 10 then aflattening occurs Figure 1 reproduced from Cooket al (1969) indicates that by suitable choice of theparameter C one might be able to describe a rapidfall off in incidence a suggestion we discuss laterCook Doll and Fellingham ultimately rejected thispossibility because data showed a constant agefor the peak of cancer incidence while theirparticular assumption of insensitive individuals
indicates that incidence peaks at a younger age
for rarer cancersThe variations in cure rates for cancer in the last
50 years suggest that it is now more important to
discuss incidence rather than mortality and in
addition that incidence may now be much better
determined than 50 years ago In this paper there-
fore we reopen the discussion with an emphasis on
cancer incidence rather than mortalityLet f(t) denote the event rate for the kth change
at time t in a single cell According to our
definition cancer incidence is related to f(t) by
I(t)Nf(t) where N is the number of affected cells
The theoretical model calculates f(t) we will per-
form this calculation exactly for a variety of
interesting situationsIn the case of the ArmitageDoll model
IAD(t)h(t) is an infinitely growing hazard func-
tion and satisfies the criterion that the cumulative
probability of cancer given by
Figure 1 Cook Doll and Fellingham (1969) considered variablesusceptibility in the population This plot shows incidence versus ageon a logarithmic scale for varying fractions C of sensitive personsModified from Cook et al (1969) Figure 4
Multistage model of cancer developmentG Ritter et al
126
F (t)1exp
g
t
0
h(t)dt
(2)
approaches 1 in the limit as t 0 According tothe definition of the hazard function we considerthe differential equation
IAD(t)atk1F (t)
1 F(t) (3)
Solving this equation with the initial conditionF(0)o yields
F (t)1(o1) exp
atk
k
The condition o0 corresponds to fetal cancerwhich while a real condition is ignored in thispaper Hence we are concerned with the limit o00
F(t)1exp(atk=k) (4)
This satisfies limt0F (t)1 which describes thefact that according to this model everyone willdevelop cancer if they do not die of something elseIt is obvious from these considerations thatthe ArmitageDoll approximation (Equation (1))increases without bound and given sufficienttime any susceptible cell eventually becomes ma-lignant
As noted above the ArmitageDoll model(Equation (1)) must be viewed as an approximationThe correct equations were written down byMoolgavkar (1991) and Armitage (1985) and itwas seen that (Equation (1)) is only valid for smallvalues of t It was seen also that (Equation (1))overestimates incidence Moolgavkar (1991) haspublished a simple example supporting the ideathat ArmitageDoll approximation can be inade-quate if the transition rates for cancer stages are notsmall enough His example presupposes a smallernumber of cells N109 in the tissue of concern thanis usually taken In contrast to the ArmitageDollapproximation the hazard function in the exactmodel has a finite asymptote limt0h(t)Nmminwhere mmin is the minimal transition rate Thesuggestion that the failure of the ArmitageDollapproximation may be responsible for the observedflattening of incidence for ages above 60 followsfrom this
Pompei and Wilson (2001) reopened the issue
They noted that since 1955 data on cancer
incidence has improved and data on both incidence
and mortality above age 70 has improved For
discussions of cancer development incidence may
now be more reliable (for many tumor sites) than
mortality due to improvements in cancer treat-
ment Making the assumption that the data and
particularly the SEER data in the United States are
reliable Pompei and Wilson plotted the normalized
cancer incidence data against age finding a rapid
drop in normalized incidence above age 80 which is
not explained by simple parameter adjustments
of the ArmitageDoll model This is illustrated
in Figure 2 for a common cancer (colon) an
Figure 2 SEER data from Pompei et al (2004) and SEER (2002)shown together with the best possible fit of the ArmitageDoll model
Multistage model of cancer developmentG Ritter et al
127
intermediate-incidence cancer (non-Hodgkins lym-phoma) and a rare cancer (brain)
Pompei and Wilson added an extra term toEquation (1) leading to a beta function I
atk1(1bt) which fits the data for most cancersremarkably well and speculated on possible biolo-gical explanations for the extra term PompeiPolkanov and Wilson (Pompei et al 2001) exam-ined the limited data on laboratory mice that havebeen followed for their natural life of 1000 days ormore rather than being sacrificed at 750 days Theage of cancer incidence is not well determined formice but cancer mortality suffers neither fromcomplications of improved treatment nor frommisreporting In the absence of these systematicerrors it was observed that the cancer mortality fellto zero before the end of life This is shown inFigure 3
In the present paper we re-examine the criticismsof Moolgavkar and improve upon the speculationsof Pompei and Wilson This is done byexact calculations in the multistage model In theprocess we derive several closed-form solutionsto the model Following previous studies (Pompeiand Wilson 2001) we continue to make theassumption that the SEER data in the UnitedStates is reliable above age 80 We prove that underthis model as one would expect the probabilitythat a single cell becomes cancerous given infinitetime is 1
Crucial parameters for the multistage model arethe transition rates which we denote mi We give anexact solution for a model with a number ns of slowstages with rate ms and one fast stage with rate mfand show that if mfms the fast stage only slightlyperturbs the final solution and may be neglectedWe show that if the multistage model is assumed tocalculate the probability of cancer for a single cell
then this model can only be consistent with therapid fall-off observed in the data if some unusualbiological assumptions are made
Methods
Assumptions of the multistage model
In this section we repeat starting with the funda-mental assumptions the derivation of the exactmultistage model and suggest various extensionsthereof
In the ArmitageDoll approximation cancerincidence has a simple power law growth This isan approximation to the exact multistage model(Moolgavkar 1978 Moolgavkar et al 1999 Pom-pei and Wilson 2001) described by the Batemanequations (Bateman 1910) The multistage modelmakes several basic assumptions
1 Malignant tumors arise from a series of mod-ifications of a single progenitor cell
2 The process of developing a malignancy isequally likely for all cells in the same tissue
3 The process of developing a malignancy in onecell is independent of the process in any othercell
4 After a malignancy has developed in a cellproliferation to an overt cancer is rapid andinvolves many cells in the same tissue and mayeven involve metastasis to another tissue
Under these assumptions cancer is the last of aseries of k sudden and irreversible changes whichmust take place in a specific order For a cell whichhas experienced (i1) changes the event rate forthe next change is mi We choose the conventionthat a single transition from a state p0 to a state p1
p0 0m1
p1 (5)
corresponds to one lsquostagersquo and has k1 Thisimplies that the number of transition rate constantsm1 mk always equals the number of stages
Cancer in a cell
Cancer is the last of a series of k sudden andirreversible changes which must take place in a cellin a specific order As is standard for Poisson
Figure 3 Mice data from Pompei et al (2001) shown together withthe best possible fit of the ArmitageDoll model
Multistage model of cancer developmentG Ritter et al
128
random processes we neglect the probability of twoor more events taking place in (t tdt) as dt 0 0This means that if a cell is in state pi at time t theprobability of transformation to state pi1 in asmall time interval Dt is given by mi1Dto(Dt)where o(Dt) is very small compared to Dt ando(Dt)=Dt 0 0 as Dt 0 0 Also the probability oftransformation i 0 i j with j1 in time Dt isassumed to be o(Dt) This implies that 1=mi1 is theaverage time required for the cell to go from state ito state i1
The probability to find a cell in the ith stage bythe end of time interval (t tdt) is then given by
pi(tdt) (1mi1dt) pi(t)mi1dt 0
midt pi1(t) (6)
Taking the limit dt 0 0 Equation (6) becomes
p0(t)m1p0(t)
pi(t)mi1pi(t)mipi1(t) (7)
pk(t)mkpk1(t)
The first and the last equations in this system aredifferent because there are no stages before thestage i0 (normal cell) and no stages after the kth
stage which corresponds to cancer The system(Equation (7)) should be completed with appro-priate initial conditions
p0(0)1 pi(0)0 (i1 2 k) (8)
which mean that the cell was normal at time t0The last equation in Equation (7) also gives theprobability to find the cell at its last cancerousstage Following Armitage and Doll we reserve thenotation f(t) for this quantity
f (t)pk(t)mkpk1(t) (9)
Thus f(t) denotes the event rate for the kth
change (corresponding to cancer) at time t in asingle cell
Cancer in a tissue and in humans
The event rate f(t) is a probability distributionfunction (PDF) for appearance of cancerous cellswhich means that f
0f (t)dt1 which means that
given enough time each cell will become cancerousEven though the PDF is convenient for mathema-
tical modelling it is not directly observable Onlydeveloped cancer in a tissue rather than a singlecancer cell could be recognized as a cause of deathor even diagnosed at all In practical studies cancerincidence is commonly observed Following thenotation of Armitage and Doll cancer incidenceI(t) (which is the same quantity as the hazard
function used in biostatistics and denoted there ash(t)) is defined by epidemiologists as the rate ofdiagnosing new cases divided by the number ofpersons at risk
Let us define a cumulative distribution function(CDF) for cancer per cell as F(t)f
t
0f (t)dt that is
the probability for a cell to become a cancer bymoment t Then the probability of at least onecancer cell in a tissue of N cells by the moment t isG(t)1[1F(t)]N This definition assumes theindependence of cells Cancer incidence can bedefined in terms of G(t) as
I(t)G(t)=(1G(t))NF (t)=(1F (t))
Cancer is a rare event even on the tissue leveland therefore F (t)1 and hence the approxi-mation
I(t)Nf (t) (10)
is quite good since the number of cells in a tissue Nis greater than 109 Equation (10) allows fitting ofthe theoretically derived cancer event rate per cellf(t) and the observed incidence of cases in popula-tion I(t) This result was derived by Armitage andby Moolgavkar
In the above the function I(t)Nf (t) is inci-dence of cancer in a tissue or equivalently in aperson When multiplied by the size of a popula-tion this becomes the true number of cancers inthat population at risk or the incidence Howeverthe number of cells N may not be large enough toensure that the ArmitageDoll approximation isaccurate so it is important to discuss this para-meter There are about 1014 cells in a human bodyand in any given tissue they would be proportionalto weight Thus as an order of magnitude approx-imation the colon has roughly 1012 cells Thepostulate that only cells on the surface are im-portant may reduce this to 1011 Therefore in whatfollows we take the number used by Moolgavkar(109 cells) as a lower limit
Multistage model of cancer developmentG Ritter et al
129
The Bateman equations
The exact solution to the multistage model can be
derived by analogy from Batemanrsquos solution of
successive radioactive decays (Bateman 1910)The simplest form of Batemanrsquos solution de-
scribes a linear decay chain leading ultimately to a
stable nucleus More complex forms have also been
used to describe for example the decay of radium
The isotope radium C decays into radium C and
radium C which both decay into lead so the
diagram contains a loop It is highly likely that
these more complex forms are also relevant to
cancer incidence although in the present work we
restrict attention to linear decay chains A k-stage
radioactive decay process depicted schematically
by the diagram
is described by the Bateman equations a set ofcoupled linear first-order differential equations inwhich pi(t) denotes the quantity of objects in state i
at time t
dp0
dtm01p0
dpj
dtmj1 jpj1mj1pj (1B iBk) (11)
dpk
dtmk1kpk1
where mj j1 is the partial rate constant for thetransition from state j to state j1 and mj is thetotal decay rate1 of state j1 The partial decayrates are related to the total decay rates byconstants bj j1 called branching ratios Thisrelationship takes the form
mj j1bj j1mj1
In the case of a linear decay chain the branchingratios equal unity and the partial rate constants aregiven by
mj j1mj1
In this simple case Equation (11) is equivalent tothe equations derived earlier in our discussion ofcancer in a cell Most discussions of the modelmake this assumption However we note that notall lsquoinitiatedrsquo cells proceed to cancer but the vastmajority are either repaired or excreted Somemay undergo clonal expansion and multiply Thiscould be modelled by taking values of thebranching ratio bj j1 less than unity (in the firstcase) or greater than 1 for clonal expansionHowever this is a very simplistic view of clonalexpansion which treats the expansion as a defini-tive process rather than a stochastic one We areattempting to understand this further The reali-zation that the multistage model only takes intoaccount the slow late limiting steps in the cancerprocess allows a theory in which clonal expansionoccurs rapidly but is not noticed as a separatestage
However if these branching ratios are the samefor each cell they may be subsumed in an overallconstant factor in the formula for incidence andthe fitted values of m adjusted accordingly Thenthe algebra is the same as if each and every celleventually becomes cancerous
We will work with linear decay chains in whatfollows In that case the diagram of the processsimplifies to
p0 0m1
p1 0m2 0mk
pk (12)
In the earlier section lsquoCancer in a cellrsquo it wasshown that the same differential equations describethe multistage model for the formation of malig-nant cells Every cell eventually transforms givensufficient time so in a realistic model
pk()g
0
f (t)dt1 (13)
In Lemma 3 we show that Equation (13) is a truemathematical consequence of the Bateman equa-tions with appropriate initial conditions
1In a model describing radioactivity mi1ln(2)=t1=2 where t1=2 is thehalf-life of that species Our notation differs slightly from that ofMoolgavkar who identifies the rate constant with the state being leftrather than with the state to which the transition is being made
Multistage model of cancer developmentG Ritter et al
130
The most general initial conditions for the
system Equation (11) are to allow pi(0) to be arbit-
rary nonzero constants for all i The solution
to the system Equation (11) with general initial
conditions is
for m1 kA simplifying assumption consistent with the
use of Equation (11) to analyse the probability of asingle cell becoming cancerous (or the radioactivedecay of a single nuclide) is that the concentrationsof the later stages (or daughter nuclides) are allinitially zero This assumption is equivalent to theinitial conditions
p0(0)a
pi(0)0 (for all i1) (15)
With these initial conditions the solution (Equa-tion (14)) reduces immediately to the simpler formvalid for m f1 kg
m1(t)cm
Xm
j1
xjm emj t (16)
where cmaYm1
j1
mj1j
Here c1 is an empty product which is 1 byconvention c2m12 etc and we have defined time-independent constants
xjmY
l1m
lj
(mlmj)1 (17)
Equations (16) and (17) were shown for thelinear chain by Moolgavkar (1991) [(A1) p 217] inhis work on the multistage theory of carcinogenesisWhen evaluating the sum in Equation (14) the usershould be warned that the terms tend to be largeand of opposite sign Indeed when the m are equalanother formula has to be used The cancerincidence in a tissue with N cells (which is the
approximation for a person) is Nf(t) to a very highdegree of accuracy and
f (t)pk (t)mkpk1(t)
We state our most important mathematical asser-tions as a series of Lemmas for which we provideproofs in an appendix
Lemma 1 (Solution of the Bateman equations)Assume that mi mj for all i j Then the functionspm(t) defined by Equation (16) and Equation (17)
solve the Bateman equations Equation (11) withinitial conditions Equation (15)
Generally we take the initial condition a1 Itis easy to see that the first nonvanishing term in aTaylor series of pm1(t) is of the ArmitageDollform where m is an index for an intermediate stageIndeed pm1(t) is the constant cm times a linearcombination of exponentials am
j1xjm emj t the nth
term in the Taylor series of this linear combinationis clearly
Xm
j1
xjm
(mjt)n
n (18)
This vanishes by algebraic identity if nBm1 andfor nm1 one may check that am
j1xjmmm1j 1
so to lowest nonvanishing orderpm1(t)cmtm1=(m1)
The next nonvanishing term (nm) also has asimple form Equation (18) with nm becomes
tm
m
Xm
j1
mj tm
(m 1)m
where m1
kak
i1mi denotes the average m We may
then write
pm1(t)cm
tm1
(m 1)(1m t )
Substituting mk for the last stage we find
I(t)atk1(1m t )
where
a
Qk
j1 mj
(k 1) (19)
pm1(t)Xm1
i0
Ym2
j0
mj j1
Xm
j1
pi(0)emj tQl (i 1) m
l j
(ml mj)
26664
37775 (14)
Multistage model of cancer developmentG Ritter et al
131
This is the Taylor expansion discussed by Mool-gavkar It was referred to (Moolgavkar 1978) as theMacLaurin series but that is a special case of themore general Taylor series We use the more generalterm in this paper
As one might expect the solution (Equation(16)) has a simple series expansion which can beexpressed in closed form This is given in thefollowing Lemma
Lemma 2 (Taylor expansion)Let fmi i1 kg be any set of nonzero constants
with mi mj if i j Then
Xm
i1
ximemi tX
j]m1
ajtj
with am11=(m1) and
aj1(1) jm
j
Xi155ijm
mi1mi2
mijm(20)
if jm This implies that
pm1(t)cm
tm1
(m 1)
1
m
Xm
i1
mi
tmO(tm1)
Remark 1 In various applications it is mostconvenient to use the lowest nonvanishing order(the ArmitageDoll approximation) in this seriesexpansion and itrsquos necessary to have conditionswhich guarantee that this truncation is valid Wecompare the lowest nonvanishing order to the(assumably better) approximation obtained byconsidering higher-order terms In particularlet us compare the lowest nonvanishing order(jm1 in the above formulae) to the followingterm In this paper as with the papers ofMoolgavkar we are concerned with the last stageonly and this corresponds to mk in the aboveequations Let Dn denote the nth nonzero term inthe Taylor expansion which has coefficientakn2
By a simple calculation
D2
D1
t m (21)
This formula shows that D2=D1 can be arbi-trarily large by inclusion of a fast stage or
stages each with large m When attempting as
we are doing to find a model to fit the data
from ages 20 to 70 years the constant a in the
first term of Equation (19) is heavily constrained
by the data If k is not varied the constant a
is proportional to the product m1 mk One
can only make m arbitrarily large if this pro-
duct is fixed to the correct value This is
discussed further in the section on determination
of k We will show later that if nf of the stages are
much faster than the remaining nsknf stages
then the exact solution for the k-stage model is
approximately equal to the exact solution for the
remaining ns stages which we call slow stages
Equation (21) indicates that the Taylor series for
k stages converges more slowly than the Taylor
series for ns stages because inclusion of fast
stages increases the average transition constant
In the presence of fast stages a large number of
terms must be included before the Taylor series
becomes accurate and consideration of only the
first and second terms in the expansion is
incorrect However the exact solution for the
model without the fast stages has a different
Taylor series which converges quite rapidly This
realization leads to important practical conclu-
sions that are discussed in more detail later
Specifically if inclusion of a few fast stages set
m103 then Equation (21) shows that D2 is 10
percent of D1 at t100 If the stages are fast
enough to set m102 per year then D2=D1 is
of order one at 100 years It is clear that by
including fast stages D2=D1 can be made arbi-
trarily largeMoolgavkar deliberately chose a set of para-
meters which included some slow and some fast
(but not very fast) stages (see for example
Moolgavkar 1978 Table 1) Although the above
estimate for D2=D1 does hold for Moolgavkarrsquos
example a discussion of his chosen parameters
needs more careful numerical analysis Consider
a tissue of 109 cells affected by a hypothetical
six-stage2 malignant tumor with transition rates
(per year) given by
2Moolgavkar (1991) refers to this as a seven-stage process but onlygives values for six transition constants We assume here that six stagesare meant
Multistage model of cancer developmentG Ritter et al
132
104 2104 34104 7103
8103 9103
In this example
m00046 per year
So our formula Equation (21) predicts D2046D1
at t100 years This is precisely what is observed inFigure 4 which shows the exact solution togetherwith its first five Taylor approximants The numer-ical factor relating the second approximationwhich lies below the exact curve to the firstapproximation lying above the curve at t100years is seen to be about 146
As noted in the introduction 109 cells may be toofew Bearing in mind that the model is used to fitincidence data each value of the six constants m
would have to be reduced by a factor of 2 if N wereincreased to 64109 and the exact formula at aget100 years would then be only 128 times theArmitageDoll approximation
The implications for determination of the valueof k using Moolgavkarrsquos parameters is discussedlater More generally
Dn1
Dn
akn1
akn2
t
t
k n
Pnk
Pn1k
where Pnk is the symmetric degree n polynomial ink variables given by ai155in
mi1 min
These poly-nomials satisfy
Pnk mkPn1k(terms involving m1 mk1)
It follows that for n]2
Dn1
Dn
t
k n
mk
ratio approaching
zero as mk 0
13(22)
The surprising conclusion of Equation (22) isthat if we add one very fast (mm104) stage at theend it may be necessary to consider hundreds orthousands of terms in the Taylor expansion in orderto attain accurate results The rate of convergenceof this expansion is affected strongly by theappearance of fast stages while the exact solutionis not This is a very important paradoxical resultthe solution for which may be found in the earliercomments If there are ns slow stages and nf veryfast stages the exact solution is close to the
ArmitageDoll approximation for the ns slowstages alone the ArmitageDoll approximationfor the full exact model with nsnf stages is notaccurate For equal transition probabilities givenby the single transition constant m the aboveimplies D2=D1mt
Lemma 3 (Normalization)Let p0(t) pk(t) satisfy the k-stage Batemanequations (Equation (11)) with any collection(m1 mk) of transition constants Then
g
0
pk(t)dtYk
i1mi
Xk
j1
xjk
mj
1 (23)
This equation shows that given sufficient timethe cancer process will eventually reach the laststage
General properties of the solutions
The general solution for reasonable values of k has far too many constants to be determined byepidemiological data alone However there arevarious cases in which the solution of the Batemanequations Equations (16) and (17) can be simplifiedenough to depend on only a few independentconstants even if the number of stages is large
Equation (21) implies that the ratio of the secondterm to the first in the Taylor series for I(t)
D2
D1
is proportional to the average m But the product ofthe ms is constrained by the value of the observedincidence With this constraint the average m will
Figure 4 Exact solution for Moolgavkarrsquos example shown with itsfirst five Taylor approximants
Multistage model of cancer developmentG Ritter et al
133
be a minimum when all the values of m are equalFor this important special case D2=D1 is muchsmaller than for example in Moolgavkarrsquos case
Another interesting result is that permuting thestages makes no difference to the final result pk(t)or more generally to mmpm1(t) for any m5k Thisis self-evident physically for radioactive decaywhich is also described by the Bateman equationsThis result used implicitly in the ArmitageDollapproximation is a fundamental property of theexact solution It may be proved as follows Itsuffices to consider a transposition of the ith stageand the jth stage as any permutation maybe represented as a product of transpositionsNow xim is a product of (mlmi)
1 where l iApplying the transposition these terms become(mlmj)
1except for the l j term which be-
comes (mimj)1 The result is thenQ
lj(mlmj)1
xjmA third general result is that if there are say 4
stages which proceed slowly (small equal values ofm) and many stages which act much faster (values ofm that are 10 times larger) then the final resultdepends only on the 4 slow stages This is self-evident physically and was probably implicit in thethinking of earlier users of the multistage modelThis is proven rigorously in Lemma 6 and dis-cussed with a specific example in the section onmeasurement of k
We pay special attention to a simple case (onenot covered by Lemma 1) in which all stages haveequal transition probabilities This simple case isdescribed by Lemma 4
Equal transition ratesLemma 4 (Equal transition rates)Consider the Bateman equations pj (t)m(pj1pj)arising from equal transition rates m1m2
mkm For any natural number j5k1 the solutionis
pj(t)1
jemt(mt) j (24)
In particular for the penultimate stage
pk1(t)1
(k 1)emt(mt)k1
The global maximum of pj(t) occurs at the agetpeak j=m Further as before we have
g
0
pk (t)dtg
0
mpk1(t)dt1
Remark 2 Since f(t) applies to a single cell m isvery small (on the order of 103) and hence for anynonzero number of stages tpeak j=m always occursat a much greater age than those contained in ahuman lifespan This important point is discussedin detail in the earlier section lsquoCan the multistagemodel ever fit the datarsquo
The algebraic simplicity of equation Equation(24) makes it attractive as a starting point for ourdiscussions It has two adjustable constants k andm A useful numerical example is the followingSuppose we would like to know how the exactsolution Equation (24) differs from the first term inits series expansion
pj(t)Xn0
(mt) jn
jn (25)
In general the first term in the series expansion ofexxn (n0 an integer) is xn and jexxnxn j
xn(1ex) In a numerical example with G03 G001 per year and k7 we have mt502for all t in a human lifespan Then frac121emtfrac12502and so for the first Taylor remainder R1 of pj(t) wehave
jR1j51
5k1256106 (26)
Equally separated transition rates
Another case which is amenable to an exactanalytical solution is when the transition constantsmi for the various stages increase by a fixed amountOf course it is not necessary to assume that in theactual cancer process the transition rates occur inincreasing order thanks to invariance of the processunder general permutations as remarked above sothis solution applies to the general situation whenthere exists some arbitrary reordering of thetransition constants so that they increase linearly
Lemma 5 (Equal separation)Suppose that the transition constants mm1m2 mk are uniformly separated so that mi1mi d for all i and for some constant d0 Then for anyn5k the solution is given by
Multistage model of cancer developmentG Ritter et al
134
pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

F (t)1exp
g
t
0
h(t)dt
(2)
approaches 1 in the limit as t 0 According tothe definition of the hazard function we considerthe differential equation
IAD(t)atk1F (t)
1 F(t) (3)
Solving this equation with the initial conditionF(0)o yields
F (t)1(o1) exp
atk
k
The condition o0 corresponds to fetal cancerwhich while a real condition is ignored in thispaper Hence we are concerned with the limit o00
F(t)1exp(atk=k) (4)
This satisfies limt0F (t)1 which describes thefact that according to this model everyone willdevelop cancer if they do not die of something elseIt is obvious from these considerations thatthe ArmitageDoll approximation (Equation (1))increases without bound and given sufficienttime any susceptible cell eventually becomes ma-lignant
As noted above the ArmitageDoll model(Equation (1)) must be viewed as an approximationThe correct equations were written down byMoolgavkar (1991) and Armitage (1985) and itwas seen that (Equation (1)) is only valid for smallvalues of t It was seen also that (Equation (1))overestimates incidence Moolgavkar (1991) haspublished a simple example supporting the ideathat ArmitageDoll approximation can be inade-quate if the transition rates for cancer stages are notsmall enough His example presupposes a smallernumber of cells N109 in the tissue of concern thanis usually taken In contrast to the ArmitageDollapproximation the hazard function in the exactmodel has a finite asymptote limt0h(t)Nmminwhere mmin is the minimal transition rate Thesuggestion that the failure of the ArmitageDollapproximation may be responsible for the observedflattening of incidence for ages above 60 followsfrom this
Pompei and Wilson (2001) reopened the issue
They noted that since 1955 data on cancer
incidence has improved and data on both incidence
and mortality above age 70 has improved For
discussions of cancer development incidence may
now be more reliable (for many tumor sites) than
mortality due to improvements in cancer treat-
ment Making the assumption that the data and
particularly the SEER data in the United States are
reliable Pompei and Wilson plotted the normalized
cancer incidence data against age finding a rapid
drop in normalized incidence above age 80 which is
not explained by simple parameter adjustments
of the ArmitageDoll model This is illustrated
in Figure 2 for a common cancer (colon) an
Figure 2 SEER data from Pompei et al (2004) and SEER (2002)shown together with the best possible fit of the ArmitageDoll model
Multistage model of cancer developmentG Ritter et al
127
intermediate-incidence cancer (non-Hodgkins lym-phoma) and a rare cancer (brain)
Pompei and Wilson added an extra term toEquation (1) leading to a beta function I
atk1(1bt) which fits the data for most cancersremarkably well and speculated on possible biolo-gical explanations for the extra term PompeiPolkanov and Wilson (Pompei et al 2001) exam-ined the limited data on laboratory mice that havebeen followed for their natural life of 1000 days ormore rather than being sacrificed at 750 days Theage of cancer incidence is not well determined formice but cancer mortality suffers neither fromcomplications of improved treatment nor frommisreporting In the absence of these systematicerrors it was observed that the cancer mortality fellto zero before the end of life This is shown inFigure 3
In the present paper we re-examine the criticismsof Moolgavkar and improve upon the speculationsof Pompei and Wilson This is done byexact calculations in the multistage model In theprocess we derive several closed-form solutionsto the model Following previous studies (Pompeiand Wilson 2001) we continue to make theassumption that the SEER data in the UnitedStates is reliable above age 80 We prove that underthis model as one would expect the probabilitythat a single cell becomes cancerous given infinitetime is 1
Crucial parameters for the multistage model arethe transition rates which we denote mi We give anexact solution for a model with a number ns of slowstages with rate ms and one fast stage with rate mfand show that if mfms the fast stage only slightlyperturbs the final solution and may be neglectedWe show that if the multistage model is assumed tocalculate the probability of cancer for a single cell
then this model can only be consistent with therapid fall-off observed in the data if some unusualbiological assumptions are made
Methods
Assumptions of the multistage model
In this section we repeat starting with the funda-mental assumptions the derivation of the exactmultistage model and suggest various extensionsthereof
In the ArmitageDoll approximation cancerincidence has a simple power law growth This isan approximation to the exact multistage model(Moolgavkar 1978 Moolgavkar et al 1999 Pom-pei and Wilson 2001) described by the Batemanequations (Bateman 1910) The multistage modelmakes several basic assumptions
1 Malignant tumors arise from a series of mod-ifications of a single progenitor cell
2 The process of developing a malignancy isequally likely for all cells in the same tissue
3 The process of developing a malignancy in onecell is independent of the process in any othercell
4 After a malignancy has developed in a cellproliferation to an overt cancer is rapid andinvolves many cells in the same tissue and mayeven involve metastasis to another tissue
Under these assumptions cancer is the last of aseries of k sudden and irreversible changes whichmust take place in a specific order For a cell whichhas experienced (i1) changes the event rate forthe next change is mi We choose the conventionthat a single transition from a state p0 to a state p1
p0 0m1
p1 (5)
corresponds to one lsquostagersquo and has k1 Thisimplies that the number of transition rate constantsm1 mk always equals the number of stages
Cancer in a cell
Cancer is the last of a series of k sudden andirreversible changes which must take place in a cellin a specific order As is standard for Poisson
Figure 3 Mice data from Pompei et al (2001) shown together withthe best possible fit of the ArmitageDoll model
Multistage model of cancer developmentG Ritter et al
128
random processes we neglect the probability of twoor more events taking place in (t tdt) as dt 0 0This means that if a cell is in state pi at time t theprobability of transformation to state pi1 in asmall time interval Dt is given by mi1Dto(Dt)where o(Dt) is very small compared to Dt ando(Dt)=Dt 0 0 as Dt 0 0 Also the probability oftransformation i 0 i j with j1 in time Dt isassumed to be o(Dt) This implies that 1=mi1 is theaverage time required for the cell to go from state ito state i1
The probability to find a cell in the ith stage bythe end of time interval (t tdt) is then given by
pi(tdt) (1mi1dt) pi(t)mi1dt 0
midt pi1(t) (6)
Taking the limit dt 0 0 Equation (6) becomes
p0(t)m1p0(t)
pi(t)mi1pi(t)mipi1(t) (7)
pk(t)mkpk1(t)
The first and the last equations in this system aredifferent because there are no stages before thestage i0 (normal cell) and no stages after the kth
stage which corresponds to cancer The system(Equation (7)) should be completed with appro-priate initial conditions
p0(0)1 pi(0)0 (i1 2 k) (8)
which mean that the cell was normal at time t0The last equation in Equation (7) also gives theprobability to find the cell at its last cancerousstage Following Armitage and Doll we reserve thenotation f(t) for this quantity
f (t)pk(t)mkpk1(t) (9)
Thus f(t) denotes the event rate for the kth
change (corresponding to cancer) at time t in asingle cell
Cancer in a tissue and in humans
The event rate f(t) is a probability distributionfunction (PDF) for appearance of cancerous cellswhich means that f
0f (t)dt1 which means that
given enough time each cell will become cancerousEven though the PDF is convenient for mathema-
tical modelling it is not directly observable Onlydeveloped cancer in a tissue rather than a singlecancer cell could be recognized as a cause of deathor even diagnosed at all In practical studies cancerincidence is commonly observed Following thenotation of Armitage and Doll cancer incidenceI(t) (which is the same quantity as the hazard
function used in biostatistics and denoted there ash(t)) is defined by epidemiologists as the rate ofdiagnosing new cases divided by the number ofpersons at risk
Let us define a cumulative distribution function(CDF) for cancer per cell as F(t)f
t
0f (t)dt that is
the probability for a cell to become a cancer bymoment t Then the probability of at least onecancer cell in a tissue of N cells by the moment t isG(t)1[1F(t)]N This definition assumes theindependence of cells Cancer incidence can bedefined in terms of G(t) as
I(t)G(t)=(1G(t))NF (t)=(1F (t))
Cancer is a rare event even on the tissue leveland therefore F (t)1 and hence the approxi-mation
I(t)Nf (t) (10)
is quite good since the number of cells in a tissue Nis greater than 109 Equation (10) allows fitting ofthe theoretically derived cancer event rate per cellf(t) and the observed incidence of cases in popula-tion I(t) This result was derived by Armitage andby Moolgavkar
In the above the function I(t)Nf (t) is inci-dence of cancer in a tissue or equivalently in aperson When multiplied by the size of a popula-tion this becomes the true number of cancers inthat population at risk or the incidence Howeverthe number of cells N may not be large enough toensure that the ArmitageDoll approximation isaccurate so it is important to discuss this para-meter There are about 1014 cells in a human bodyand in any given tissue they would be proportionalto weight Thus as an order of magnitude approx-imation the colon has roughly 1012 cells Thepostulate that only cells on the surface are im-portant may reduce this to 1011 Therefore in whatfollows we take the number used by Moolgavkar(109 cells) as a lower limit
Multistage model of cancer developmentG Ritter et al
129
The Bateman equations
The exact solution to the multistage model can be
derived by analogy from Batemanrsquos solution of
successive radioactive decays (Bateman 1910)The simplest form of Batemanrsquos solution de-
scribes a linear decay chain leading ultimately to a
stable nucleus More complex forms have also been
used to describe for example the decay of radium
The isotope radium C decays into radium C and
radium C which both decay into lead so the
diagram contains a loop It is highly likely that
these more complex forms are also relevant to
cancer incidence although in the present work we
restrict attention to linear decay chains A k-stage
radioactive decay process depicted schematically
by the diagram
is described by the Bateman equations a set ofcoupled linear first-order differential equations inwhich pi(t) denotes the quantity of objects in state i
at time t
dp0
dtm01p0
dpj
dtmj1 jpj1mj1pj (1B iBk) (11)
dpk
dtmk1kpk1
where mj j1 is the partial rate constant for thetransition from state j to state j1 and mj is thetotal decay rate1 of state j1 The partial decayrates are related to the total decay rates byconstants bj j1 called branching ratios Thisrelationship takes the form
mj j1bj j1mj1
In the case of a linear decay chain the branchingratios equal unity and the partial rate constants aregiven by
mj j1mj1
In this simple case Equation (11) is equivalent tothe equations derived earlier in our discussion ofcancer in a cell Most discussions of the modelmake this assumption However we note that notall lsquoinitiatedrsquo cells proceed to cancer but the vastmajority are either repaired or excreted Somemay undergo clonal expansion and multiply Thiscould be modelled by taking values of thebranching ratio bj j1 less than unity (in the firstcase) or greater than 1 for clonal expansionHowever this is a very simplistic view of clonalexpansion which treats the expansion as a defini-tive process rather than a stochastic one We areattempting to understand this further The reali-zation that the multistage model only takes intoaccount the slow late limiting steps in the cancerprocess allows a theory in which clonal expansionoccurs rapidly but is not noticed as a separatestage
However if these branching ratios are the samefor each cell they may be subsumed in an overallconstant factor in the formula for incidence andthe fitted values of m adjusted accordingly Thenthe algebra is the same as if each and every celleventually becomes cancerous
We will work with linear decay chains in whatfollows In that case the diagram of the processsimplifies to
p0 0m1
p1 0m2 0mk
pk (12)
In the earlier section lsquoCancer in a cellrsquo it wasshown that the same differential equations describethe multistage model for the formation of malig-nant cells Every cell eventually transforms givensufficient time so in a realistic model
pk()g
0
f (t)dt1 (13)
In Lemma 3 we show that Equation (13) is a truemathematical consequence of the Bateman equa-tions with appropriate initial conditions
1In a model describing radioactivity mi1ln(2)=t1=2 where t1=2 is thehalf-life of that species Our notation differs slightly from that ofMoolgavkar who identifies the rate constant with the state being leftrather than with the state to which the transition is being made
Multistage model of cancer developmentG Ritter et al
130
The most general initial conditions for the
system Equation (11) are to allow pi(0) to be arbit-
rary nonzero constants for all i The solution
to the system Equation (11) with general initial
conditions is
for m1 kA simplifying assumption consistent with the
use of Equation (11) to analyse the probability of asingle cell becoming cancerous (or the radioactivedecay of a single nuclide) is that the concentrationsof the later stages (or daughter nuclides) are allinitially zero This assumption is equivalent to theinitial conditions
p0(0)a
pi(0)0 (for all i1) (15)
With these initial conditions the solution (Equa-tion (14)) reduces immediately to the simpler formvalid for m f1 kg
m1(t)cm
Xm
j1
xjm emj t (16)
where cmaYm1
j1
mj1j
Here c1 is an empty product which is 1 byconvention c2m12 etc and we have defined time-independent constants
xjmY
l1m
lj
(mlmj)1 (17)
Equations (16) and (17) were shown for thelinear chain by Moolgavkar (1991) [(A1) p 217] inhis work on the multistage theory of carcinogenesisWhen evaluating the sum in Equation (14) the usershould be warned that the terms tend to be largeand of opposite sign Indeed when the m are equalanother formula has to be used The cancerincidence in a tissue with N cells (which is the
approximation for a person) is Nf(t) to a very highdegree of accuracy and
f (t)pk (t)mkpk1(t)
We state our most important mathematical asser-tions as a series of Lemmas for which we provideproofs in an appendix
Lemma 1 (Solution of the Bateman equations)Assume that mi mj for all i j Then the functionspm(t) defined by Equation (16) and Equation (17)
solve the Bateman equations Equation (11) withinitial conditions Equation (15)
Generally we take the initial condition a1 Itis easy to see that the first nonvanishing term in aTaylor series of pm1(t) is of the ArmitageDollform where m is an index for an intermediate stageIndeed pm1(t) is the constant cm times a linearcombination of exponentials am
j1xjm emj t the nth
term in the Taylor series of this linear combinationis clearly
Xm
j1
xjm
(mjt)n
n (18)
This vanishes by algebraic identity if nBm1 andfor nm1 one may check that am
j1xjmmm1j 1
so to lowest nonvanishing orderpm1(t)cmtm1=(m1)
The next nonvanishing term (nm) also has asimple form Equation (18) with nm becomes
tm
m
Xm
j1
mj tm
(m 1)m
where m1
kak
i1mi denotes the average m We may
then write
pm1(t)cm
tm1
(m 1)(1m t )
Substituting mk for the last stage we find
I(t)atk1(1m t )
where
a
Qk
j1 mj
(k 1) (19)
pm1(t)Xm1
i0
Ym2
j0
mj j1
Xm
j1
pi(0)emj tQl (i 1) m
l j
(ml mj)
26664
37775 (14)
Multistage model of cancer developmentG Ritter et al
131
This is the Taylor expansion discussed by Mool-gavkar It was referred to (Moolgavkar 1978) as theMacLaurin series but that is a special case of themore general Taylor series We use the more generalterm in this paper
As one might expect the solution (Equation(16)) has a simple series expansion which can beexpressed in closed form This is given in thefollowing Lemma
Lemma 2 (Taylor expansion)Let fmi i1 kg be any set of nonzero constants
with mi mj if i j Then
Xm
i1
ximemi tX
j]m1
ajtj
with am11=(m1) and
aj1(1) jm
j
Xi155ijm
mi1mi2
mijm(20)
if jm This implies that
pm1(t)cm
tm1
(m 1)
1
m
Xm
i1
mi
tmO(tm1)
Remark 1 In various applications it is mostconvenient to use the lowest nonvanishing order(the ArmitageDoll approximation) in this seriesexpansion and itrsquos necessary to have conditionswhich guarantee that this truncation is valid Wecompare the lowest nonvanishing order to the(assumably better) approximation obtained byconsidering higher-order terms In particularlet us compare the lowest nonvanishing order(jm1 in the above formulae) to the followingterm In this paper as with the papers ofMoolgavkar we are concerned with the last stageonly and this corresponds to mk in the aboveequations Let Dn denote the nth nonzero term inthe Taylor expansion which has coefficientakn2
By a simple calculation
D2
D1
t m (21)
This formula shows that D2=D1 can be arbi-trarily large by inclusion of a fast stage or
stages each with large m When attempting as
we are doing to find a model to fit the data
from ages 20 to 70 years the constant a in the
first term of Equation (19) is heavily constrained
by the data If k is not varied the constant a
is proportional to the product m1 mk One
can only make m arbitrarily large if this pro-
duct is fixed to the correct value This is
discussed further in the section on determination
of k We will show later that if nf of the stages are
much faster than the remaining nsknf stages
then the exact solution for the k-stage model is
approximately equal to the exact solution for the
remaining ns stages which we call slow stages
Equation (21) indicates that the Taylor series for
k stages converges more slowly than the Taylor
series for ns stages because inclusion of fast
stages increases the average transition constant
In the presence of fast stages a large number of
terms must be included before the Taylor series
becomes accurate and consideration of only the
first and second terms in the expansion is
incorrect However the exact solution for the
model without the fast stages has a different
Taylor series which converges quite rapidly This
realization leads to important practical conclu-
sions that are discussed in more detail later
Specifically if inclusion of a few fast stages set
m103 then Equation (21) shows that D2 is 10
percent of D1 at t100 If the stages are fast
enough to set m102 per year then D2=D1 is
of order one at 100 years It is clear that by
including fast stages D2=D1 can be made arbi-
trarily largeMoolgavkar deliberately chose a set of para-
meters which included some slow and some fast
(but not very fast) stages (see for example
Moolgavkar 1978 Table 1) Although the above
estimate for D2=D1 does hold for Moolgavkarrsquos
example a discussion of his chosen parameters
needs more careful numerical analysis Consider
a tissue of 109 cells affected by a hypothetical
six-stage2 malignant tumor with transition rates
(per year) given by
2Moolgavkar (1991) refers to this as a seven-stage process but onlygives values for six transition constants We assume here that six stagesare meant
Multistage model of cancer developmentG Ritter et al
132
104 2104 34104 7103
8103 9103
In this example
m00046 per year
So our formula Equation (21) predicts D2046D1
at t100 years This is precisely what is observed inFigure 4 which shows the exact solution togetherwith its first five Taylor approximants The numer-ical factor relating the second approximationwhich lies below the exact curve to the firstapproximation lying above the curve at t100years is seen to be about 146
As noted in the introduction 109 cells may be toofew Bearing in mind that the model is used to fitincidence data each value of the six constants m
would have to be reduced by a factor of 2 if N wereincreased to 64109 and the exact formula at aget100 years would then be only 128 times theArmitageDoll approximation
The implications for determination of the valueof k using Moolgavkarrsquos parameters is discussedlater More generally
Dn1
Dn
akn1
akn2
t
t
k n
Pnk
Pn1k
where Pnk is the symmetric degree n polynomial ink variables given by ai155in
mi1 min
These poly-nomials satisfy
Pnk mkPn1k(terms involving m1 mk1)
It follows that for n]2
Dn1
Dn
t
k n
mk
ratio approaching
zero as mk 0
13(22)
The surprising conclusion of Equation (22) isthat if we add one very fast (mm104) stage at theend it may be necessary to consider hundreds orthousands of terms in the Taylor expansion in orderto attain accurate results The rate of convergenceof this expansion is affected strongly by theappearance of fast stages while the exact solutionis not This is a very important paradoxical resultthe solution for which may be found in the earliercomments If there are ns slow stages and nf veryfast stages the exact solution is close to the
ArmitageDoll approximation for the ns slowstages alone the ArmitageDoll approximationfor the full exact model with nsnf stages is notaccurate For equal transition probabilities givenby the single transition constant m the aboveimplies D2=D1mt
Lemma 3 (Normalization)Let p0(t) pk(t) satisfy the k-stage Batemanequations (Equation (11)) with any collection(m1 mk) of transition constants Then
g
0
pk(t)dtYk
i1mi
Xk
j1
xjk
mj
1 (23)
This equation shows that given sufficient timethe cancer process will eventually reach the laststage
General properties of the solutions
The general solution for reasonable values of k has far too many constants to be determined byepidemiological data alone However there arevarious cases in which the solution of the Batemanequations Equations (16) and (17) can be simplifiedenough to depend on only a few independentconstants even if the number of stages is large
Equation (21) implies that the ratio of the secondterm to the first in the Taylor series for I(t)
D2
D1
is proportional to the average m But the product ofthe ms is constrained by the value of the observedincidence With this constraint the average m will
Figure 4 Exact solution for Moolgavkarrsquos example shown with itsfirst five Taylor approximants
Multistage model of cancer developmentG Ritter et al
133
be a minimum when all the values of m are equalFor this important special case D2=D1 is muchsmaller than for example in Moolgavkarrsquos case
Another interesting result is that permuting thestages makes no difference to the final result pk(t)or more generally to mmpm1(t) for any m5k Thisis self-evident physically for radioactive decaywhich is also described by the Bateman equationsThis result used implicitly in the ArmitageDollapproximation is a fundamental property of theexact solution It may be proved as follows Itsuffices to consider a transposition of the ith stageand the jth stage as any permutation maybe represented as a product of transpositionsNow xim is a product of (mlmi)
1 where l iApplying the transposition these terms become(mlmj)
1except for the l j term which be-
comes (mimj)1 The result is thenQ
lj(mlmj)1
xjmA third general result is that if there are say 4
stages which proceed slowly (small equal values ofm) and many stages which act much faster (values ofm that are 10 times larger) then the final resultdepends only on the 4 slow stages This is self-evident physically and was probably implicit in thethinking of earlier users of the multistage modelThis is proven rigorously in Lemma 6 and dis-cussed with a specific example in the section onmeasurement of k
We pay special attention to a simple case (onenot covered by Lemma 1) in which all stages haveequal transition probabilities This simple case isdescribed by Lemma 4
Equal transition ratesLemma 4 (Equal transition rates)Consider the Bateman equations pj (t)m(pj1pj)arising from equal transition rates m1m2
mkm For any natural number j5k1 the solutionis
pj(t)1
jemt(mt) j (24)
In particular for the penultimate stage
pk1(t)1
(k 1)emt(mt)k1
The global maximum of pj(t) occurs at the agetpeak j=m Further as before we have
g
0
pk (t)dtg
0
mpk1(t)dt1
Remark 2 Since f(t) applies to a single cell m isvery small (on the order of 103) and hence for anynonzero number of stages tpeak j=m always occursat a much greater age than those contained in ahuman lifespan This important point is discussedin detail in the earlier section lsquoCan the multistagemodel ever fit the datarsquo
The algebraic simplicity of equation Equation(24) makes it attractive as a starting point for ourdiscussions It has two adjustable constants k andm A useful numerical example is the followingSuppose we would like to know how the exactsolution Equation (24) differs from the first term inits series expansion
pj(t)Xn0
(mt) jn
jn (25)
In general the first term in the series expansion ofexxn (n0 an integer) is xn and jexxnxn j
xn(1ex) In a numerical example with G03 G001 per year and k7 we have mt502for all t in a human lifespan Then frac121emtfrac12502and so for the first Taylor remainder R1 of pj(t) wehave
jR1j51
5k1256106 (26)
Equally separated transition rates
Another case which is amenable to an exactanalytical solution is when the transition constantsmi for the various stages increase by a fixed amountOf course it is not necessary to assume that in theactual cancer process the transition rates occur inincreasing order thanks to invariance of the processunder general permutations as remarked above sothis solution applies to the general situation whenthere exists some arbitrary reordering of thetransition constants so that they increase linearly
Lemma 5 (Equal separation)Suppose that the transition constants mm1m2 mk are uniformly separated so that mi1mi d for all i and for some constant d0 Then for anyn5k the solution is given by
Multistage model of cancer developmentG Ritter et al
134
pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

intermediate-incidence cancer (non-Hodgkins lym-phoma) and a rare cancer (brain)
Pompei and Wilson added an extra term toEquation (1) leading to a beta function I
atk1(1bt) which fits the data for most cancersremarkably well and speculated on possible biolo-gical explanations for the extra term PompeiPolkanov and Wilson (Pompei et al 2001) exam-ined the limited data on laboratory mice that havebeen followed for their natural life of 1000 days ormore rather than being sacrificed at 750 days Theage of cancer incidence is not well determined formice but cancer mortality suffers neither fromcomplications of improved treatment nor frommisreporting In the absence of these systematicerrors it was observed that the cancer mortality fellto zero before the end of life This is shown inFigure 3
In the present paper we re-examine the criticismsof Moolgavkar and improve upon the speculationsof Pompei and Wilson This is done byexact calculations in the multistage model In theprocess we derive several closed-form solutionsto the model Following previous studies (Pompeiand Wilson 2001) we continue to make theassumption that the SEER data in the UnitedStates is reliable above age 80 We prove that underthis model as one would expect the probabilitythat a single cell becomes cancerous given infinitetime is 1
Crucial parameters for the multistage model arethe transition rates which we denote mi We give anexact solution for a model with a number ns of slowstages with rate ms and one fast stage with rate mfand show that if mfms the fast stage only slightlyperturbs the final solution and may be neglectedWe show that if the multistage model is assumed tocalculate the probability of cancer for a single cell
then this model can only be consistent with therapid fall-off observed in the data if some unusualbiological assumptions are made
Methods
Assumptions of the multistage model
In this section we repeat starting with the funda-mental assumptions the derivation of the exactmultistage model and suggest various extensionsthereof
In the ArmitageDoll approximation cancerincidence has a simple power law growth This isan approximation to the exact multistage model(Moolgavkar 1978 Moolgavkar et al 1999 Pom-pei and Wilson 2001) described by the Batemanequations (Bateman 1910) The multistage modelmakes several basic assumptions
1 Malignant tumors arise from a series of mod-ifications of a single progenitor cell
2 The process of developing a malignancy isequally likely for all cells in the same tissue
3 The process of developing a malignancy in onecell is independent of the process in any othercell
4 After a malignancy has developed in a cellproliferation to an overt cancer is rapid andinvolves many cells in the same tissue and mayeven involve metastasis to another tissue
Under these assumptions cancer is the last of aseries of k sudden and irreversible changes whichmust take place in a specific order For a cell whichhas experienced (i1) changes the event rate forthe next change is mi We choose the conventionthat a single transition from a state p0 to a state p1
p0 0m1
p1 (5)
corresponds to one lsquostagersquo and has k1 Thisimplies that the number of transition rate constantsm1 mk always equals the number of stages
Cancer in a cell
Cancer is the last of a series of k sudden andirreversible changes which must take place in a cellin a specific order As is standard for Poisson
Figure 3 Mice data from Pompei et al (2001) shown together withthe best possible fit of the ArmitageDoll model
Multistage model of cancer developmentG Ritter et al
128
random processes we neglect the probability of twoor more events taking place in (t tdt) as dt 0 0This means that if a cell is in state pi at time t theprobability of transformation to state pi1 in asmall time interval Dt is given by mi1Dto(Dt)where o(Dt) is very small compared to Dt ando(Dt)=Dt 0 0 as Dt 0 0 Also the probability oftransformation i 0 i j with j1 in time Dt isassumed to be o(Dt) This implies that 1=mi1 is theaverage time required for the cell to go from state ito state i1
The probability to find a cell in the ith stage bythe end of time interval (t tdt) is then given by
pi(tdt) (1mi1dt) pi(t)mi1dt 0
midt pi1(t) (6)
Taking the limit dt 0 0 Equation (6) becomes
p0(t)m1p0(t)
pi(t)mi1pi(t)mipi1(t) (7)
pk(t)mkpk1(t)
The first and the last equations in this system aredifferent because there are no stages before thestage i0 (normal cell) and no stages after the kth
stage which corresponds to cancer The system(Equation (7)) should be completed with appro-priate initial conditions
p0(0)1 pi(0)0 (i1 2 k) (8)
which mean that the cell was normal at time t0The last equation in Equation (7) also gives theprobability to find the cell at its last cancerousstage Following Armitage and Doll we reserve thenotation f(t) for this quantity
f (t)pk(t)mkpk1(t) (9)
Thus f(t) denotes the event rate for the kth
change (corresponding to cancer) at time t in asingle cell
Cancer in a tissue and in humans
The event rate f(t) is a probability distributionfunction (PDF) for appearance of cancerous cellswhich means that f
0f (t)dt1 which means that
given enough time each cell will become cancerousEven though the PDF is convenient for mathema-
tical modelling it is not directly observable Onlydeveloped cancer in a tissue rather than a singlecancer cell could be recognized as a cause of deathor even diagnosed at all In practical studies cancerincidence is commonly observed Following thenotation of Armitage and Doll cancer incidenceI(t) (which is the same quantity as the hazard
function used in biostatistics and denoted there ash(t)) is defined by epidemiologists as the rate ofdiagnosing new cases divided by the number ofpersons at risk
Let us define a cumulative distribution function(CDF) for cancer per cell as F(t)f
t
0f (t)dt that is
the probability for a cell to become a cancer bymoment t Then the probability of at least onecancer cell in a tissue of N cells by the moment t isG(t)1[1F(t)]N This definition assumes theindependence of cells Cancer incidence can bedefined in terms of G(t) as
I(t)G(t)=(1G(t))NF (t)=(1F (t))
Cancer is a rare event even on the tissue leveland therefore F (t)1 and hence the approxi-mation
I(t)Nf (t) (10)
is quite good since the number of cells in a tissue Nis greater than 109 Equation (10) allows fitting ofthe theoretically derived cancer event rate per cellf(t) and the observed incidence of cases in popula-tion I(t) This result was derived by Armitage andby Moolgavkar
In the above the function I(t)Nf (t) is inci-dence of cancer in a tissue or equivalently in aperson When multiplied by the size of a popula-tion this becomes the true number of cancers inthat population at risk or the incidence Howeverthe number of cells N may not be large enough toensure that the ArmitageDoll approximation isaccurate so it is important to discuss this para-meter There are about 1014 cells in a human bodyand in any given tissue they would be proportionalto weight Thus as an order of magnitude approx-imation the colon has roughly 1012 cells Thepostulate that only cells on the surface are im-portant may reduce this to 1011 Therefore in whatfollows we take the number used by Moolgavkar(109 cells) as a lower limit
Multistage model of cancer developmentG Ritter et al
129
The Bateman equations
The exact solution to the multistage model can be
derived by analogy from Batemanrsquos solution of
successive radioactive decays (Bateman 1910)The simplest form of Batemanrsquos solution de-
scribes a linear decay chain leading ultimately to a
stable nucleus More complex forms have also been
used to describe for example the decay of radium
The isotope radium C decays into radium C and
radium C which both decay into lead so the
diagram contains a loop It is highly likely that
these more complex forms are also relevant to
cancer incidence although in the present work we
restrict attention to linear decay chains A k-stage
radioactive decay process depicted schematically
by the diagram
is described by the Bateman equations a set ofcoupled linear first-order differential equations inwhich pi(t) denotes the quantity of objects in state i
at time t
dp0
dtm01p0
dpj
dtmj1 jpj1mj1pj (1B iBk) (11)
dpk
dtmk1kpk1
where mj j1 is the partial rate constant for thetransition from state j to state j1 and mj is thetotal decay rate1 of state j1 The partial decayrates are related to the total decay rates byconstants bj j1 called branching ratios Thisrelationship takes the form
mj j1bj j1mj1
In the case of a linear decay chain the branchingratios equal unity and the partial rate constants aregiven by
mj j1mj1
In this simple case Equation (11) is equivalent tothe equations derived earlier in our discussion ofcancer in a cell Most discussions of the modelmake this assumption However we note that notall lsquoinitiatedrsquo cells proceed to cancer but the vastmajority are either repaired or excreted Somemay undergo clonal expansion and multiply Thiscould be modelled by taking values of thebranching ratio bj j1 less than unity (in the firstcase) or greater than 1 for clonal expansionHowever this is a very simplistic view of clonalexpansion which treats the expansion as a defini-tive process rather than a stochastic one We areattempting to understand this further The reali-zation that the multistage model only takes intoaccount the slow late limiting steps in the cancerprocess allows a theory in which clonal expansionoccurs rapidly but is not noticed as a separatestage
However if these branching ratios are the samefor each cell they may be subsumed in an overallconstant factor in the formula for incidence andthe fitted values of m adjusted accordingly Thenthe algebra is the same as if each and every celleventually becomes cancerous
We will work with linear decay chains in whatfollows In that case the diagram of the processsimplifies to
p0 0m1
p1 0m2 0mk
pk (12)
In the earlier section lsquoCancer in a cellrsquo it wasshown that the same differential equations describethe multistage model for the formation of malig-nant cells Every cell eventually transforms givensufficient time so in a realistic model
pk()g
0
f (t)dt1 (13)
In Lemma 3 we show that Equation (13) is a truemathematical consequence of the Bateman equa-tions with appropriate initial conditions
1In a model describing radioactivity mi1ln(2)=t1=2 where t1=2 is thehalf-life of that species Our notation differs slightly from that ofMoolgavkar who identifies the rate constant with the state being leftrather than with the state to which the transition is being made
Multistage model of cancer developmentG Ritter et al
130
The most general initial conditions for the
system Equation (11) are to allow pi(0) to be arbit-
rary nonzero constants for all i The solution
to the system Equation (11) with general initial
conditions is
for m1 kA simplifying assumption consistent with the
use of Equation (11) to analyse the probability of asingle cell becoming cancerous (or the radioactivedecay of a single nuclide) is that the concentrationsof the later stages (or daughter nuclides) are allinitially zero This assumption is equivalent to theinitial conditions
p0(0)a
pi(0)0 (for all i1) (15)
With these initial conditions the solution (Equa-tion (14)) reduces immediately to the simpler formvalid for m f1 kg
m1(t)cm
Xm
j1
xjm emj t (16)
where cmaYm1
j1
mj1j
Here c1 is an empty product which is 1 byconvention c2m12 etc and we have defined time-independent constants
xjmY
l1m
lj
(mlmj)1 (17)
Equations (16) and (17) were shown for thelinear chain by Moolgavkar (1991) [(A1) p 217] inhis work on the multistage theory of carcinogenesisWhen evaluating the sum in Equation (14) the usershould be warned that the terms tend to be largeand of opposite sign Indeed when the m are equalanother formula has to be used The cancerincidence in a tissue with N cells (which is the
approximation for a person) is Nf(t) to a very highdegree of accuracy and
f (t)pk (t)mkpk1(t)
We state our most important mathematical asser-tions as a series of Lemmas for which we provideproofs in an appendix
Lemma 1 (Solution of the Bateman equations)Assume that mi mj for all i j Then the functionspm(t) defined by Equation (16) and Equation (17)
solve the Bateman equations Equation (11) withinitial conditions Equation (15)
Generally we take the initial condition a1 Itis easy to see that the first nonvanishing term in aTaylor series of pm1(t) is of the ArmitageDollform where m is an index for an intermediate stageIndeed pm1(t) is the constant cm times a linearcombination of exponentials am
j1xjm emj t the nth
term in the Taylor series of this linear combinationis clearly
Xm
j1
xjm
(mjt)n
n (18)
This vanishes by algebraic identity if nBm1 andfor nm1 one may check that am
j1xjmmm1j 1
so to lowest nonvanishing orderpm1(t)cmtm1=(m1)
The next nonvanishing term (nm) also has asimple form Equation (18) with nm becomes
tm
m
Xm
j1
mj tm
(m 1)m
where m1
kak
i1mi denotes the average m We may
then write
pm1(t)cm
tm1
(m 1)(1m t )
Substituting mk for the last stage we find
I(t)atk1(1m t )
where
a
Qk
j1 mj
(k 1) (19)
pm1(t)Xm1
i0
Ym2
j0
mj j1
Xm
j1
pi(0)emj tQl (i 1) m
l j
(ml mj)
26664
37775 (14)
Multistage model of cancer developmentG Ritter et al
131
This is the Taylor expansion discussed by Mool-gavkar It was referred to (Moolgavkar 1978) as theMacLaurin series but that is a special case of themore general Taylor series We use the more generalterm in this paper
As one might expect the solution (Equation(16)) has a simple series expansion which can beexpressed in closed form This is given in thefollowing Lemma
Lemma 2 (Taylor expansion)Let fmi i1 kg be any set of nonzero constants
with mi mj if i j Then
Xm
i1
ximemi tX
j]m1
ajtj
with am11=(m1) and
aj1(1) jm
j
Xi155ijm
mi1mi2
mijm(20)
if jm This implies that
pm1(t)cm
tm1
(m 1)
1
m
Xm
i1
mi
tmO(tm1)
Remark 1 In various applications it is mostconvenient to use the lowest nonvanishing order(the ArmitageDoll approximation) in this seriesexpansion and itrsquos necessary to have conditionswhich guarantee that this truncation is valid Wecompare the lowest nonvanishing order to the(assumably better) approximation obtained byconsidering higher-order terms In particularlet us compare the lowest nonvanishing order(jm1 in the above formulae) to the followingterm In this paper as with the papers ofMoolgavkar we are concerned with the last stageonly and this corresponds to mk in the aboveequations Let Dn denote the nth nonzero term inthe Taylor expansion which has coefficientakn2
By a simple calculation
D2
D1
t m (21)
This formula shows that D2=D1 can be arbi-trarily large by inclusion of a fast stage or
stages each with large m When attempting as
we are doing to find a model to fit the data
from ages 20 to 70 years the constant a in the
first term of Equation (19) is heavily constrained
by the data If k is not varied the constant a
is proportional to the product m1 mk One
can only make m arbitrarily large if this pro-
duct is fixed to the correct value This is
discussed further in the section on determination
of k We will show later that if nf of the stages are
much faster than the remaining nsknf stages
then the exact solution for the k-stage model is
approximately equal to the exact solution for the
remaining ns stages which we call slow stages
Equation (21) indicates that the Taylor series for
k stages converges more slowly than the Taylor
series for ns stages because inclusion of fast
stages increases the average transition constant
In the presence of fast stages a large number of
terms must be included before the Taylor series
becomes accurate and consideration of only the
first and second terms in the expansion is
incorrect However the exact solution for the
model without the fast stages has a different
Taylor series which converges quite rapidly This
realization leads to important practical conclu-
sions that are discussed in more detail later
Specifically if inclusion of a few fast stages set
m103 then Equation (21) shows that D2 is 10
percent of D1 at t100 If the stages are fast
enough to set m102 per year then D2=D1 is
of order one at 100 years It is clear that by
including fast stages D2=D1 can be made arbi-
trarily largeMoolgavkar deliberately chose a set of para-
meters which included some slow and some fast
(but not very fast) stages (see for example
Moolgavkar 1978 Table 1) Although the above
estimate for D2=D1 does hold for Moolgavkarrsquos
example a discussion of his chosen parameters
needs more careful numerical analysis Consider
a tissue of 109 cells affected by a hypothetical
six-stage2 malignant tumor with transition rates
(per year) given by
2Moolgavkar (1991) refers to this as a seven-stage process but onlygives values for six transition constants We assume here that six stagesare meant
Multistage model of cancer developmentG Ritter et al
132
104 2104 34104 7103
8103 9103
In this example
m00046 per year
So our formula Equation (21) predicts D2046D1
at t100 years This is precisely what is observed inFigure 4 which shows the exact solution togetherwith its first five Taylor approximants The numer-ical factor relating the second approximationwhich lies below the exact curve to the firstapproximation lying above the curve at t100years is seen to be about 146
As noted in the introduction 109 cells may be toofew Bearing in mind that the model is used to fitincidence data each value of the six constants m
would have to be reduced by a factor of 2 if N wereincreased to 64109 and the exact formula at aget100 years would then be only 128 times theArmitageDoll approximation
The implications for determination of the valueof k using Moolgavkarrsquos parameters is discussedlater More generally
Dn1
Dn
akn1
akn2
t
t
k n
Pnk
Pn1k
where Pnk is the symmetric degree n polynomial ink variables given by ai155in
mi1 min
These poly-nomials satisfy
Pnk mkPn1k(terms involving m1 mk1)
It follows that for n]2
Dn1
Dn
t
k n
mk
ratio approaching
zero as mk 0
13(22)
The surprising conclusion of Equation (22) isthat if we add one very fast (mm104) stage at theend it may be necessary to consider hundreds orthousands of terms in the Taylor expansion in orderto attain accurate results The rate of convergenceof this expansion is affected strongly by theappearance of fast stages while the exact solutionis not This is a very important paradoxical resultthe solution for which may be found in the earliercomments If there are ns slow stages and nf veryfast stages the exact solution is close to the
ArmitageDoll approximation for the ns slowstages alone the ArmitageDoll approximationfor the full exact model with nsnf stages is notaccurate For equal transition probabilities givenby the single transition constant m the aboveimplies D2=D1mt
Lemma 3 (Normalization)Let p0(t) pk(t) satisfy the k-stage Batemanequations (Equation (11)) with any collection(m1 mk) of transition constants Then
g
0
pk(t)dtYk
i1mi
Xk
j1
xjk
mj
1 (23)
This equation shows that given sufficient timethe cancer process will eventually reach the laststage
General properties of the solutions
The general solution for reasonable values of k has far too many constants to be determined byepidemiological data alone However there arevarious cases in which the solution of the Batemanequations Equations (16) and (17) can be simplifiedenough to depend on only a few independentconstants even if the number of stages is large
Equation (21) implies that the ratio of the secondterm to the first in the Taylor series for I(t)
D2
D1
is proportional to the average m But the product ofthe ms is constrained by the value of the observedincidence With this constraint the average m will
Figure 4 Exact solution for Moolgavkarrsquos example shown with itsfirst five Taylor approximants
Multistage model of cancer developmentG Ritter et al
133
be a minimum when all the values of m are equalFor this important special case D2=D1 is muchsmaller than for example in Moolgavkarrsquos case
Another interesting result is that permuting thestages makes no difference to the final result pk(t)or more generally to mmpm1(t) for any m5k Thisis self-evident physically for radioactive decaywhich is also described by the Bateman equationsThis result used implicitly in the ArmitageDollapproximation is a fundamental property of theexact solution It may be proved as follows Itsuffices to consider a transposition of the ith stageand the jth stage as any permutation maybe represented as a product of transpositionsNow xim is a product of (mlmi)
1 where l iApplying the transposition these terms become(mlmj)
1except for the l j term which be-
comes (mimj)1 The result is thenQ
lj(mlmj)1
xjmA third general result is that if there are say 4
stages which proceed slowly (small equal values ofm) and many stages which act much faster (values ofm that are 10 times larger) then the final resultdepends only on the 4 slow stages This is self-evident physically and was probably implicit in thethinking of earlier users of the multistage modelThis is proven rigorously in Lemma 6 and dis-cussed with a specific example in the section onmeasurement of k
We pay special attention to a simple case (onenot covered by Lemma 1) in which all stages haveequal transition probabilities This simple case isdescribed by Lemma 4
Equal transition ratesLemma 4 (Equal transition rates)Consider the Bateman equations pj (t)m(pj1pj)arising from equal transition rates m1m2
mkm For any natural number j5k1 the solutionis
pj(t)1
jemt(mt) j (24)
In particular for the penultimate stage
pk1(t)1
(k 1)emt(mt)k1
The global maximum of pj(t) occurs at the agetpeak j=m Further as before we have
g
0
pk (t)dtg
0
mpk1(t)dt1
Remark 2 Since f(t) applies to a single cell m isvery small (on the order of 103) and hence for anynonzero number of stages tpeak j=m always occursat a much greater age than those contained in ahuman lifespan This important point is discussedin detail in the earlier section lsquoCan the multistagemodel ever fit the datarsquo
The algebraic simplicity of equation Equation(24) makes it attractive as a starting point for ourdiscussions It has two adjustable constants k andm A useful numerical example is the followingSuppose we would like to know how the exactsolution Equation (24) differs from the first term inits series expansion
pj(t)Xn0
(mt) jn
jn (25)
In general the first term in the series expansion ofexxn (n0 an integer) is xn and jexxnxn j
xn(1ex) In a numerical example with G03 G001 per year and k7 we have mt502for all t in a human lifespan Then frac121emtfrac12502and so for the first Taylor remainder R1 of pj(t) wehave
jR1j51
5k1256106 (26)
Equally separated transition rates
Another case which is amenable to an exactanalytical solution is when the transition constantsmi for the various stages increase by a fixed amountOf course it is not necessary to assume that in theactual cancer process the transition rates occur inincreasing order thanks to invariance of the processunder general permutations as remarked above sothis solution applies to the general situation whenthere exists some arbitrary reordering of thetransition constants so that they increase linearly
Lemma 5 (Equal separation)Suppose that the transition constants mm1m2 mk are uniformly separated so that mi1mi d for all i and for some constant d0 Then for anyn5k the solution is given by
Multistage model of cancer developmentG Ritter et al
134
pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

random processes we neglect the probability of twoor more events taking place in (t tdt) as dt 0 0This means that if a cell is in state pi at time t theprobability of transformation to state pi1 in asmall time interval Dt is given by mi1Dto(Dt)where o(Dt) is very small compared to Dt ando(Dt)=Dt 0 0 as Dt 0 0 Also the probability oftransformation i 0 i j with j1 in time Dt isassumed to be o(Dt) This implies that 1=mi1 is theaverage time required for the cell to go from state ito state i1
The probability to find a cell in the ith stage bythe end of time interval (t tdt) is then given by
pi(tdt) (1mi1dt) pi(t)mi1dt 0
midt pi1(t) (6)
Taking the limit dt 0 0 Equation (6) becomes
p0(t)m1p0(t)
pi(t)mi1pi(t)mipi1(t) (7)
pk(t)mkpk1(t)
The first and the last equations in this system aredifferent because there are no stages before thestage i0 (normal cell) and no stages after the kth
stage which corresponds to cancer The system(Equation (7)) should be completed with appro-priate initial conditions
p0(0)1 pi(0)0 (i1 2 k) (8)
which mean that the cell was normal at time t0The last equation in Equation (7) also gives theprobability to find the cell at its last cancerousstage Following Armitage and Doll we reserve thenotation f(t) for this quantity
f (t)pk(t)mkpk1(t) (9)
Thus f(t) denotes the event rate for the kth
change (corresponding to cancer) at time t in asingle cell
Cancer in a tissue and in humans
The event rate f(t) is a probability distributionfunction (PDF) for appearance of cancerous cellswhich means that f
0f (t)dt1 which means that
given enough time each cell will become cancerousEven though the PDF is convenient for mathema-
tical modelling it is not directly observable Onlydeveloped cancer in a tissue rather than a singlecancer cell could be recognized as a cause of deathor even diagnosed at all In practical studies cancerincidence is commonly observed Following thenotation of Armitage and Doll cancer incidenceI(t) (which is the same quantity as the hazard
function used in biostatistics and denoted there ash(t)) is defined by epidemiologists as the rate ofdiagnosing new cases divided by the number ofpersons at risk
Let us define a cumulative distribution function(CDF) for cancer per cell as F(t)f
t
0f (t)dt that is
the probability for a cell to become a cancer bymoment t Then the probability of at least onecancer cell in a tissue of N cells by the moment t isG(t)1[1F(t)]N This definition assumes theindependence of cells Cancer incidence can bedefined in terms of G(t) as
I(t)G(t)=(1G(t))NF (t)=(1F (t))
Cancer is a rare event even on the tissue leveland therefore F (t)1 and hence the approxi-mation
I(t)Nf (t) (10)
is quite good since the number of cells in a tissue Nis greater than 109 Equation (10) allows fitting ofthe theoretically derived cancer event rate per cellf(t) and the observed incidence of cases in popula-tion I(t) This result was derived by Armitage andby Moolgavkar
In the above the function I(t)Nf (t) is inci-dence of cancer in a tissue or equivalently in aperson When multiplied by the size of a popula-tion this becomes the true number of cancers inthat population at risk or the incidence Howeverthe number of cells N may not be large enough toensure that the ArmitageDoll approximation isaccurate so it is important to discuss this para-meter There are about 1014 cells in a human bodyand in any given tissue they would be proportionalto weight Thus as an order of magnitude approx-imation the colon has roughly 1012 cells Thepostulate that only cells on the surface are im-portant may reduce this to 1011 Therefore in whatfollows we take the number used by Moolgavkar(109 cells) as a lower limit
Multistage model of cancer developmentG Ritter et al
129
The Bateman equations
The exact solution to the multistage model can be
derived by analogy from Batemanrsquos solution of
successive radioactive decays (Bateman 1910)The simplest form of Batemanrsquos solution de-
scribes a linear decay chain leading ultimately to a
stable nucleus More complex forms have also been
used to describe for example the decay of radium
The isotope radium C decays into radium C and
radium C which both decay into lead so the
diagram contains a loop It is highly likely that
these more complex forms are also relevant to
cancer incidence although in the present work we
restrict attention to linear decay chains A k-stage
radioactive decay process depicted schematically
by the diagram
is described by the Bateman equations a set ofcoupled linear first-order differential equations inwhich pi(t) denotes the quantity of objects in state i
at time t
dp0
dtm01p0
dpj
dtmj1 jpj1mj1pj (1B iBk) (11)
dpk
dtmk1kpk1
where mj j1 is the partial rate constant for thetransition from state j to state j1 and mj is thetotal decay rate1 of state j1 The partial decayrates are related to the total decay rates byconstants bj j1 called branching ratios Thisrelationship takes the form
mj j1bj j1mj1
In the case of a linear decay chain the branchingratios equal unity and the partial rate constants aregiven by
mj j1mj1
In this simple case Equation (11) is equivalent tothe equations derived earlier in our discussion ofcancer in a cell Most discussions of the modelmake this assumption However we note that notall lsquoinitiatedrsquo cells proceed to cancer but the vastmajority are either repaired or excreted Somemay undergo clonal expansion and multiply Thiscould be modelled by taking values of thebranching ratio bj j1 less than unity (in the firstcase) or greater than 1 for clonal expansionHowever this is a very simplistic view of clonalexpansion which treats the expansion as a defini-tive process rather than a stochastic one We areattempting to understand this further The reali-zation that the multistage model only takes intoaccount the slow late limiting steps in the cancerprocess allows a theory in which clonal expansionoccurs rapidly but is not noticed as a separatestage
However if these branching ratios are the samefor each cell they may be subsumed in an overallconstant factor in the formula for incidence andthe fitted values of m adjusted accordingly Thenthe algebra is the same as if each and every celleventually becomes cancerous
We will work with linear decay chains in whatfollows In that case the diagram of the processsimplifies to
p0 0m1
p1 0m2 0mk
pk (12)
In the earlier section lsquoCancer in a cellrsquo it wasshown that the same differential equations describethe multistage model for the formation of malig-nant cells Every cell eventually transforms givensufficient time so in a realistic model
pk()g
0
f (t)dt1 (13)
In Lemma 3 we show that Equation (13) is a truemathematical consequence of the Bateman equa-tions with appropriate initial conditions
1In a model describing radioactivity mi1ln(2)=t1=2 where t1=2 is thehalf-life of that species Our notation differs slightly from that ofMoolgavkar who identifies the rate constant with the state being leftrather than with the state to which the transition is being made
Multistage model of cancer developmentG Ritter et al
130
The most general initial conditions for the
system Equation (11) are to allow pi(0) to be arbit-
rary nonzero constants for all i The solution
to the system Equation (11) with general initial
conditions is
for m1 kA simplifying assumption consistent with the
use of Equation (11) to analyse the probability of asingle cell becoming cancerous (or the radioactivedecay of a single nuclide) is that the concentrationsof the later stages (or daughter nuclides) are allinitially zero This assumption is equivalent to theinitial conditions
p0(0)a
pi(0)0 (for all i1) (15)
With these initial conditions the solution (Equa-tion (14)) reduces immediately to the simpler formvalid for m f1 kg
m1(t)cm
Xm
j1
xjm emj t (16)
where cmaYm1
j1
mj1j
Here c1 is an empty product which is 1 byconvention c2m12 etc and we have defined time-independent constants
xjmY
l1m
lj
(mlmj)1 (17)
Equations (16) and (17) were shown for thelinear chain by Moolgavkar (1991) [(A1) p 217] inhis work on the multistage theory of carcinogenesisWhen evaluating the sum in Equation (14) the usershould be warned that the terms tend to be largeand of opposite sign Indeed when the m are equalanother formula has to be used The cancerincidence in a tissue with N cells (which is the
approximation for a person) is Nf(t) to a very highdegree of accuracy and
f (t)pk (t)mkpk1(t)
We state our most important mathematical asser-tions as a series of Lemmas for which we provideproofs in an appendix
Lemma 1 (Solution of the Bateman equations)Assume that mi mj for all i j Then the functionspm(t) defined by Equation (16) and Equation (17)
solve the Bateman equations Equation (11) withinitial conditions Equation (15)
Generally we take the initial condition a1 Itis easy to see that the first nonvanishing term in aTaylor series of pm1(t) is of the ArmitageDollform where m is an index for an intermediate stageIndeed pm1(t) is the constant cm times a linearcombination of exponentials am
j1xjm emj t the nth
term in the Taylor series of this linear combinationis clearly
Xm
j1
xjm
(mjt)n
n (18)
This vanishes by algebraic identity if nBm1 andfor nm1 one may check that am
j1xjmmm1j 1
so to lowest nonvanishing orderpm1(t)cmtm1=(m1)
The next nonvanishing term (nm) also has asimple form Equation (18) with nm becomes
tm
m
Xm
j1
mj tm
(m 1)m
where m1
kak
i1mi denotes the average m We may
then write
pm1(t)cm
tm1
(m 1)(1m t )
Substituting mk for the last stage we find
I(t)atk1(1m t )
where
a
Qk
j1 mj
(k 1) (19)
pm1(t)Xm1
i0
Ym2
j0
mj j1
Xm
j1
pi(0)emj tQl (i 1) m
l j
(ml mj)
26664
37775 (14)
Multistage model of cancer developmentG Ritter et al
131
This is the Taylor expansion discussed by Mool-gavkar It was referred to (Moolgavkar 1978) as theMacLaurin series but that is a special case of themore general Taylor series We use the more generalterm in this paper
As one might expect the solution (Equation(16)) has a simple series expansion which can beexpressed in closed form This is given in thefollowing Lemma
Lemma 2 (Taylor expansion)Let fmi i1 kg be any set of nonzero constants
with mi mj if i j Then
Xm
i1
ximemi tX
j]m1
ajtj
with am11=(m1) and
aj1(1) jm
j
Xi155ijm
mi1mi2
mijm(20)
if jm This implies that
pm1(t)cm
tm1
(m 1)
1
m
Xm
i1
mi
tmO(tm1)
Remark 1 In various applications it is mostconvenient to use the lowest nonvanishing order(the ArmitageDoll approximation) in this seriesexpansion and itrsquos necessary to have conditionswhich guarantee that this truncation is valid Wecompare the lowest nonvanishing order to the(assumably better) approximation obtained byconsidering higher-order terms In particularlet us compare the lowest nonvanishing order(jm1 in the above formulae) to the followingterm In this paper as with the papers ofMoolgavkar we are concerned with the last stageonly and this corresponds to mk in the aboveequations Let Dn denote the nth nonzero term inthe Taylor expansion which has coefficientakn2
By a simple calculation
D2
D1
t m (21)
This formula shows that D2=D1 can be arbi-trarily large by inclusion of a fast stage or
stages each with large m When attempting as
we are doing to find a model to fit the data
from ages 20 to 70 years the constant a in the
first term of Equation (19) is heavily constrained
by the data If k is not varied the constant a
is proportional to the product m1 mk One
can only make m arbitrarily large if this pro-
duct is fixed to the correct value This is
discussed further in the section on determination
of k We will show later that if nf of the stages are
much faster than the remaining nsknf stages
then the exact solution for the k-stage model is
approximately equal to the exact solution for the
remaining ns stages which we call slow stages
Equation (21) indicates that the Taylor series for
k stages converges more slowly than the Taylor
series for ns stages because inclusion of fast
stages increases the average transition constant
In the presence of fast stages a large number of
terms must be included before the Taylor series
becomes accurate and consideration of only the
first and second terms in the expansion is
incorrect However the exact solution for the
model without the fast stages has a different
Taylor series which converges quite rapidly This
realization leads to important practical conclu-
sions that are discussed in more detail later
Specifically if inclusion of a few fast stages set
m103 then Equation (21) shows that D2 is 10
percent of D1 at t100 If the stages are fast
enough to set m102 per year then D2=D1 is
of order one at 100 years It is clear that by
including fast stages D2=D1 can be made arbi-
trarily largeMoolgavkar deliberately chose a set of para-
meters which included some slow and some fast
(but not very fast) stages (see for example
Moolgavkar 1978 Table 1) Although the above
estimate for D2=D1 does hold for Moolgavkarrsquos
example a discussion of his chosen parameters
needs more careful numerical analysis Consider
a tissue of 109 cells affected by a hypothetical
six-stage2 malignant tumor with transition rates
(per year) given by
2Moolgavkar (1991) refers to this as a seven-stage process but onlygives values for six transition constants We assume here that six stagesare meant
Multistage model of cancer developmentG Ritter et al
132
104 2104 34104 7103
8103 9103
In this example
m00046 per year
So our formula Equation (21) predicts D2046D1
at t100 years This is precisely what is observed inFigure 4 which shows the exact solution togetherwith its first five Taylor approximants The numer-ical factor relating the second approximationwhich lies below the exact curve to the firstapproximation lying above the curve at t100years is seen to be about 146
As noted in the introduction 109 cells may be toofew Bearing in mind that the model is used to fitincidence data each value of the six constants m
would have to be reduced by a factor of 2 if N wereincreased to 64109 and the exact formula at aget100 years would then be only 128 times theArmitageDoll approximation
The implications for determination of the valueof k using Moolgavkarrsquos parameters is discussedlater More generally
Dn1
Dn
akn1
akn2
t
t
k n
Pnk
Pn1k
where Pnk is the symmetric degree n polynomial ink variables given by ai155in
mi1 min
These poly-nomials satisfy
Pnk mkPn1k(terms involving m1 mk1)
It follows that for n]2
Dn1
Dn
t
k n
mk
ratio approaching
zero as mk 0
13(22)
The surprising conclusion of Equation (22) isthat if we add one very fast (mm104) stage at theend it may be necessary to consider hundreds orthousands of terms in the Taylor expansion in orderto attain accurate results The rate of convergenceof this expansion is affected strongly by theappearance of fast stages while the exact solutionis not This is a very important paradoxical resultthe solution for which may be found in the earliercomments If there are ns slow stages and nf veryfast stages the exact solution is close to the
ArmitageDoll approximation for the ns slowstages alone the ArmitageDoll approximationfor the full exact model with nsnf stages is notaccurate For equal transition probabilities givenby the single transition constant m the aboveimplies D2=D1mt
Lemma 3 (Normalization)Let p0(t) pk(t) satisfy the k-stage Batemanequations (Equation (11)) with any collection(m1 mk) of transition constants Then
g
0
pk(t)dtYk
i1mi
Xk
j1
xjk
mj
1 (23)
This equation shows that given sufficient timethe cancer process will eventually reach the laststage
General properties of the solutions
The general solution for reasonable values of k has far too many constants to be determined byepidemiological data alone However there arevarious cases in which the solution of the Batemanequations Equations (16) and (17) can be simplifiedenough to depend on only a few independentconstants even if the number of stages is large
Equation (21) implies that the ratio of the secondterm to the first in the Taylor series for I(t)
D2
D1
is proportional to the average m But the product ofthe ms is constrained by the value of the observedincidence With this constraint the average m will
Figure 4 Exact solution for Moolgavkarrsquos example shown with itsfirst five Taylor approximants
Multistage model of cancer developmentG Ritter et al
133
be a minimum when all the values of m are equalFor this important special case D2=D1 is muchsmaller than for example in Moolgavkarrsquos case
Another interesting result is that permuting thestages makes no difference to the final result pk(t)or more generally to mmpm1(t) for any m5k Thisis self-evident physically for radioactive decaywhich is also described by the Bateman equationsThis result used implicitly in the ArmitageDollapproximation is a fundamental property of theexact solution It may be proved as follows Itsuffices to consider a transposition of the ith stageand the jth stage as any permutation maybe represented as a product of transpositionsNow xim is a product of (mlmi)
1 where l iApplying the transposition these terms become(mlmj)
1except for the l j term which be-
comes (mimj)1 The result is thenQ
lj(mlmj)1
xjmA third general result is that if there are say 4
stages which proceed slowly (small equal values ofm) and many stages which act much faster (values ofm that are 10 times larger) then the final resultdepends only on the 4 slow stages This is self-evident physically and was probably implicit in thethinking of earlier users of the multistage modelThis is proven rigorously in Lemma 6 and dis-cussed with a specific example in the section onmeasurement of k
We pay special attention to a simple case (onenot covered by Lemma 1) in which all stages haveequal transition probabilities This simple case isdescribed by Lemma 4
Equal transition ratesLemma 4 (Equal transition rates)Consider the Bateman equations pj (t)m(pj1pj)arising from equal transition rates m1m2
mkm For any natural number j5k1 the solutionis
pj(t)1
jemt(mt) j (24)
In particular for the penultimate stage
pk1(t)1
(k 1)emt(mt)k1
The global maximum of pj(t) occurs at the agetpeak j=m Further as before we have
g
0
pk (t)dtg
0
mpk1(t)dt1
Remark 2 Since f(t) applies to a single cell m isvery small (on the order of 103) and hence for anynonzero number of stages tpeak j=m always occursat a much greater age than those contained in ahuman lifespan This important point is discussedin detail in the earlier section lsquoCan the multistagemodel ever fit the datarsquo
The algebraic simplicity of equation Equation(24) makes it attractive as a starting point for ourdiscussions It has two adjustable constants k andm A useful numerical example is the followingSuppose we would like to know how the exactsolution Equation (24) differs from the first term inits series expansion
pj(t)Xn0
(mt) jn
jn (25)
In general the first term in the series expansion ofexxn (n0 an integer) is xn and jexxnxn j
xn(1ex) In a numerical example with G03 G001 per year and k7 we have mt502for all t in a human lifespan Then frac121emtfrac12502and so for the first Taylor remainder R1 of pj(t) wehave
jR1j51
5k1256106 (26)
Equally separated transition rates
Another case which is amenable to an exactanalytical solution is when the transition constantsmi for the various stages increase by a fixed amountOf course it is not necessary to assume that in theactual cancer process the transition rates occur inincreasing order thanks to invariance of the processunder general permutations as remarked above sothis solution applies to the general situation whenthere exists some arbitrary reordering of thetransition constants so that they increase linearly
Lemma 5 (Equal separation)Suppose that the transition constants mm1m2 mk are uniformly separated so that mi1mi d for all i and for some constant d0 Then for anyn5k the solution is given by
Multistage model of cancer developmentG Ritter et al
134
pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

The Bateman equations
The exact solution to the multistage model can be
derived by analogy from Batemanrsquos solution of
successive radioactive decays (Bateman 1910)The simplest form of Batemanrsquos solution de-
scribes a linear decay chain leading ultimately to a
stable nucleus More complex forms have also been
used to describe for example the decay of radium
The isotope radium C decays into radium C and
radium C which both decay into lead so the
diagram contains a loop It is highly likely that
these more complex forms are also relevant to
cancer incidence although in the present work we
restrict attention to linear decay chains A k-stage
radioactive decay process depicted schematically
by the diagram
is described by the Bateman equations a set ofcoupled linear first-order differential equations inwhich pi(t) denotes the quantity of objects in state i
at time t
dp0
dtm01p0
dpj
dtmj1 jpj1mj1pj (1B iBk) (11)
dpk
dtmk1kpk1
where mj j1 is the partial rate constant for thetransition from state j to state j1 and mj is thetotal decay rate1 of state j1 The partial decayrates are related to the total decay rates byconstants bj j1 called branching ratios Thisrelationship takes the form
mj j1bj j1mj1
In the case of a linear decay chain the branchingratios equal unity and the partial rate constants aregiven by
mj j1mj1
In this simple case Equation (11) is equivalent tothe equations derived earlier in our discussion ofcancer in a cell Most discussions of the modelmake this assumption However we note that notall lsquoinitiatedrsquo cells proceed to cancer but the vastmajority are either repaired or excreted Somemay undergo clonal expansion and multiply Thiscould be modelled by taking values of thebranching ratio bj j1 less than unity (in the firstcase) or greater than 1 for clonal expansionHowever this is a very simplistic view of clonalexpansion which treats the expansion as a defini-tive process rather than a stochastic one We areattempting to understand this further The reali-zation that the multistage model only takes intoaccount the slow late limiting steps in the cancerprocess allows a theory in which clonal expansionoccurs rapidly but is not noticed as a separatestage
However if these branching ratios are the samefor each cell they may be subsumed in an overallconstant factor in the formula for incidence andthe fitted values of m adjusted accordingly Thenthe algebra is the same as if each and every celleventually becomes cancerous
We will work with linear decay chains in whatfollows In that case the diagram of the processsimplifies to
p0 0m1
p1 0m2 0mk
pk (12)
In the earlier section lsquoCancer in a cellrsquo it wasshown that the same differential equations describethe multistage model for the formation of malig-nant cells Every cell eventually transforms givensufficient time so in a realistic model
pk()g
0
f (t)dt1 (13)
In Lemma 3 we show that Equation (13) is a truemathematical consequence of the Bateman equa-tions with appropriate initial conditions
1In a model describing radioactivity mi1ln(2)=t1=2 where t1=2 is thehalf-life of that species Our notation differs slightly from that ofMoolgavkar who identifies the rate constant with the state being leftrather than with the state to which the transition is being made
Multistage model of cancer developmentG Ritter et al
130
The most general initial conditions for the
system Equation (11) are to allow pi(0) to be arbit-
rary nonzero constants for all i The solution
to the system Equation (11) with general initial
conditions is
for m1 kA simplifying assumption consistent with the
use of Equation (11) to analyse the probability of asingle cell becoming cancerous (or the radioactivedecay of a single nuclide) is that the concentrationsof the later stages (or daughter nuclides) are allinitially zero This assumption is equivalent to theinitial conditions
p0(0)a
pi(0)0 (for all i1) (15)
With these initial conditions the solution (Equa-tion (14)) reduces immediately to the simpler formvalid for m f1 kg
m1(t)cm
Xm
j1
xjm emj t (16)
where cmaYm1
j1
mj1j
Here c1 is an empty product which is 1 byconvention c2m12 etc and we have defined time-independent constants
xjmY
l1m
lj
(mlmj)1 (17)
Equations (16) and (17) were shown for thelinear chain by Moolgavkar (1991) [(A1) p 217] inhis work on the multistage theory of carcinogenesisWhen evaluating the sum in Equation (14) the usershould be warned that the terms tend to be largeand of opposite sign Indeed when the m are equalanother formula has to be used The cancerincidence in a tissue with N cells (which is the
approximation for a person) is Nf(t) to a very highdegree of accuracy and
f (t)pk (t)mkpk1(t)
We state our most important mathematical asser-tions as a series of Lemmas for which we provideproofs in an appendix
Lemma 1 (Solution of the Bateman equations)Assume that mi mj for all i j Then the functionspm(t) defined by Equation (16) and Equation (17)
solve the Bateman equations Equation (11) withinitial conditions Equation (15)
Generally we take the initial condition a1 Itis easy to see that the first nonvanishing term in aTaylor series of pm1(t) is of the ArmitageDollform where m is an index for an intermediate stageIndeed pm1(t) is the constant cm times a linearcombination of exponentials am
j1xjm emj t the nth
term in the Taylor series of this linear combinationis clearly
Xm
j1
xjm
(mjt)n
n (18)
This vanishes by algebraic identity if nBm1 andfor nm1 one may check that am
j1xjmmm1j 1
so to lowest nonvanishing orderpm1(t)cmtm1=(m1)
The next nonvanishing term (nm) also has asimple form Equation (18) with nm becomes
tm
m
Xm
j1
mj tm
(m 1)m
where m1
kak
i1mi denotes the average m We may
then write
pm1(t)cm
tm1
(m 1)(1m t )
Substituting mk for the last stage we find
I(t)atk1(1m t )
where
a
Qk
j1 mj
(k 1) (19)
pm1(t)Xm1
i0
Ym2
j0
mj j1
Xm
j1
pi(0)emj tQl (i 1) m
l j
(ml mj)
26664
37775 (14)
Multistage model of cancer developmentG Ritter et al
131
This is the Taylor expansion discussed by Mool-gavkar It was referred to (Moolgavkar 1978) as theMacLaurin series but that is a special case of themore general Taylor series We use the more generalterm in this paper
As one might expect the solution (Equation(16)) has a simple series expansion which can beexpressed in closed form This is given in thefollowing Lemma
Lemma 2 (Taylor expansion)Let fmi i1 kg be any set of nonzero constants
with mi mj if i j Then
Xm
i1
ximemi tX
j]m1
ajtj
with am11=(m1) and
aj1(1) jm
j
Xi155ijm
mi1mi2
mijm(20)
if jm This implies that
pm1(t)cm
tm1
(m 1)
1
m
Xm
i1
mi
tmO(tm1)
Remark 1 In various applications it is mostconvenient to use the lowest nonvanishing order(the ArmitageDoll approximation) in this seriesexpansion and itrsquos necessary to have conditionswhich guarantee that this truncation is valid Wecompare the lowest nonvanishing order to the(assumably better) approximation obtained byconsidering higher-order terms In particularlet us compare the lowest nonvanishing order(jm1 in the above formulae) to the followingterm In this paper as with the papers ofMoolgavkar we are concerned with the last stageonly and this corresponds to mk in the aboveequations Let Dn denote the nth nonzero term inthe Taylor expansion which has coefficientakn2
By a simple calculation
D2
D1
t m (21)
This formula shows that D2=D1 can be arbi-trarily large by inclusion of a fast stage or
stages each with large m When attempting as
we are doing to find a model to fit the data
from ages 20 to 70 years the constant a in the
first term of Equation (19) is heavily constrained
by the data If k is not varied the constant a
is proportional to the product m1 mk One
can only make m arbitrarily large if this pro-
duct is fixed to the correct value This is
discussed further in the section on determination
of k We will show later that if nf of the stages are
much faster than the remaining nsknf stages
then the exact solution for the k-stage model is
approximately equal to the exact solution for the
remaining ns stages which we call slow stages
Equation (21) indicates that the Taylor series for
k stages converges more slowly than the Taylor
series for ns stages because inclusion of fast
stages increases the average transition constant
In the presence of fast stages a large number of
terms must be included before the Taylor series
becomes accurate and consideration of only the
first and second terms in the expansion is
incorrect However the exact solution for the
model without the fast stages has a different
Taylor series which converges quite rapidly This
realization leads to important practical conclu-
sions that are discussed in more detail later
Specifically if inclusion of a few fast stages set
m103 then Equation (21) shows that D2 is 10
percent of D1 at t100 If the stages are fast
enough to set m102 per year then D2=D1 is
of order one at 100 years It is clear that by
including fast stages D2=D1 can be made arbi-
trarily largeMoolgavkar deliberately chose a set of para-
meters which included some slow and some fast
(but not very fast) stages (see for example
Moolgavkar 1978 Table 1) Although the above
estimate for D2=D1 does hold for Moolgavkarrsquos
example a discussion of his chosen parameters
needs more careful numerical analysis Consider
a tissue of 109 cells affected by a hypothetical
six-stage2 malignant tumor with transition rates
(per year) given by
2Moolgavkar (1991) refers to this as a seven-stage process but onlygives values for six transition constants We assume here that six stagesare meant
Multistage model of cancer developmentG Ritter et al
132
104 2104 34104 7103
8103 9103
In this example
m00046 per year
So our formula Equation (21) predicts D2046D1
at t100 years This is precisely what is observed inFigure 4 which shows the exact solution togetherwith its first five Taylor approximants The numer-ical factor relating the second approximationwhich lies below the exact curve to the firstapproximation lying above the curve at t100years is seen to be about 146
As noted in the introduction 109 cells may be toofew Bearing in mind that the model is used to fitincidence data each value of the six constants m
would have to be reduced by a factor of 2 if N wereincreased to 64109 and the exact formula at aget100 years would then be only 128 times theArmitageDoll approximation
The implications for determination of the valueof k using Moolgavkarrsquos parameters is discussedlater More generally
Dn1
Dn
akn1
akn2
t
t
k n
Pnk
Pn1k
where Pnk is the symmetric degree n polynomial ink variables given by ai155in
mi1 min
These poly-nomials satisfy
Pnk mkPn1k(terms involving m1 mk1)
It follows that for n]2
Dn1
Dn
t
k n
mk
ratio approaching
zero as mk 0
13(22)
The surprising conclusion of Equation (22) isthat if we add one very fast (mm104) stage at theend it may be necessary to consider hundreds orthousands of terms in the Taylor expansion in orderto attain accurate results The rate of convergenceof this expansion is affected strongly by theappearance of fast stages while the exact solutionis not This is a very important paradoxical resultthe solution for which may be found in the earliercomments If there are ns slow stages and nf veryfast stages the exact solution is close to the
ArmitageDoll approximation for the ns slowstages alone the ArmitageDoll approximationfor the full exact model with nsnf stages is notaccurate For equal transition probabilities givenby the single transition constant m the aboveimplies D2=D1mt
Lemma 3 (Normalization)Let p0(t) pk(t) satisfy the k-stage Batemanequations (Equation (11)) with any collection(m1 mk) of transition constants Then
g
0
pk(t)dtYk
i1mi
Xk
j1
xjk
mj
1 (23)
This equation shows that given sufficient timethe cancer process will eventually reach the laststage
General properties of the solutions
The general solution for reasonable values of k has far too many constants to be determined byepidemiological data alone However there arevarious cases in which the solution of the Batemanequations Equations (16) and (17) can be simplifiedenough to depend on only a few independentconstants even if the number of stages is large
Equation (21) implies that the ratio of the secondterm to the first in the Taylor series for I(t)
D2
D1
is proportional to the average m But the product ofthe ms is constrained by the value of the observedincidence With this constraint the average m will
Figure 4 Exact solution for Moolgavkarrsquos example shown with itsfirst five Taylor approximants
Multistage model of cancer developmentG Ritter et al
133
be a minimum when all the values of m are equalFor this important special case D2=D1 is muchsmaller than for example in Moolgavkarrsquos case
Another interesting result is that permuting thestages makes no difference to the final result pk(t)or more generally to mmpm1(t) for any m5k Thisis self-evident physically for radioactive decaywhich is also described by the Bateman equationsThis result used implicitly in the ArmitageDollapproximation is a fundamental property of theexact solution It may be proved as follows Itsuffices to consider a transposition of the ith stageand the jth stage as any permutation maybe represented as a product of transpositionsNow xim is a product of (mlmi)
1 where l iApplying the transposition these terms become(mlmj)
1except for the l j term which be-
comes (mimj)1 The result is thenQ
lj(mlmj)1
xjmA third general result is that if there are say 4
stages which proceed slowly (small equal values ofm) and many stages which act much faster (values ofm that are 10 times larger) then the final resultdepends only on the 4 slow stages This is self-evident physically and was probably implicit in thethinking of earlier users of the multistage modelThis is proven rigorously in Lemma 6 and dis-cussed with a specific example in the section onmeasurement of k
We pay special attention to a simple case (onenot covered by Lemma 1) in which all stages haveequal transition probabilities This simple case isdescribed by Lemma 4
Equal transition ratesLemma 4 (Equal transition rates)Consider the Bateman equations pj (t)m(pj1pj)arising from equal transition rates m1m2
mkm For any natural number j5k1 the solutionis
pj(t)1
jemt(mt) j (24)
In particular for the penultimate stage
pk1(t)1
(k 1)emt(mt)k1
The global maximum of pj(t) occurs at the agetpeak j=m Further as before we have
g
0
pk (t)dtg
0
mpk1(t)dt1
Remark 2 Since f(t) applies to a single cell m isvery small (on the order of 103) and hence for anynonzero number of stages tpeak j=m always occursat a much greater age than those contained in ahuman lifespan This important point is discussedin detail in the earlier section lsquoCan the multistagemodel ever fit the datarsquo
The algebraic simplicity of equation Equation(24) makes it attractive as a starting point for ourdiscussions It has two adjustable constants k andm A useful numerical example is the followingSuppose we would like to know how the exactsolution Equation (24) differs from the first term inits series expansion
pj(t)Xn0
(mt) jn
jn (25)
In general the first term in the series expansion ofexxn (n0 an integer) is xn and jexxnxn j
xn(1ex) In a numerical example with G03 G001 per year and k7 we have mt502for all t in a human lifespan Then frac121emtfrac12502and so for the first Taylor remainder R1 of pj(t) wehave
jR1j51
5k1256106 (26)
Equally separated transition rates
Another case which is amenable to an exactanalytical solution is when the transition constantsmi for the various stages increase by a fixed amountOf course it is not necessary to assume that in theactual cancer process the transition rates occur inincreasing order thanks to invariance of the processunder general permutations as remarked above sothis solution applies to the general situation whenthere exists some arbitrary reordering of thetransition constants so that they increase linearly
Lemma 5 (Equal separation)Suppose that the transition constants mm1m2 mk are uniformly separated so that mi1mi d for all i and for some constant d0 Then for anyn5k the solution is given by
Multistage model of cancer developmentG Ritter et al
134
pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

The most general initial conditions for the
system Equation (11) are to allow pi(0) to be arbit-
rary nonzero constants for all i The solution
to the system Equation (11) with general initial
conditions is
for m1 kA simplifying assumption consistent with the
use of Equation (11) to analyse the probability of asingle cell becoming cancerous (or the radioactivedecay of a single nuclide) is that the concentrationsof the later stages (or daughter nuclides) are allinitially zero This assumption is equivalent to theinitial conditions
p0(0)a
pi(0)0 (for all i1) (15)
With these initial conditions the solution (Equa-tion (14)) reduces immediately to the simpler formvalid for m f1 kg
m1(t)cm
Xm
j1
xjm emj t (16)
where cmaYm1
j1
mj1j
Here c1 is an empty product which is 1 byconvention c2m12 etc and we have defined time-independent constants
xjmY
l1m
lj
(mlmj)1 (17)
Equations (16) and (17) were shown for thelinear chain by Moolgavkar (1991) [(A1) p 217] inhis work on the multistage theory of carcinogenesisWhen evaluating the sum in Equation (14) the usershould be warned that the terms tend to be largeand of opposite sign Indeed when the m are equalanother formula has to be used The cancerincidence in a tissue with N cells (which is the
approximation for a person) is Nf(t) to a very highdegree of accuracy and
f (t)pk (t)mkpk1(t)
We state our most important mathematical asser-tions as a series of Lemmas for which we provideproofs in an appendix
Lemma 1 (Solution of the Bateman equations)Assume that mi mj for all i j Then the functionspm(t) defined by Equation (16) and Equation (17)
solve the Bateman equations Equation (11) withinitial conditions Equation (15)
Generally we take the initial condition a1 Itis easy to see that the first nonvanishing term in aTaylor series of pm1(t) is of the ArmitageDollform where m is an index for an intermediate stageIndeed pm1(t) is the constant cm times a linearcombination of exponentials am
j1xjm emj t the nth
term in the Taylor series of this linear combinationis clearly
Xm
j1
xjm
(mjt)n
n (18)
This vanishes by algebraic identity if nBm1 andfor nm1 one may check that am
j1xjmmm1j 1
so to lowest nonvanishing orderpm1(t)cmtm1=(m1)
The next nonvanishing term (nm) also has asimple form Equation (18) with nm becomes
tm
m
Xm
j1
mj tm
(m 1)m
where m1
kak
i1mi denotes the average m We may
then write
pm1(t)cm
tm1
(m 1)(1m t )
Substituting mk for the last stage we find
I(t)atk1(1m t )
where
a
Qk
j1 mj
(k 1) (19)
pm1(t)Xm1
i0
Ym2
j0
mj j1
Xm
j1
pi(0)emj tQl (i 1) m
l j
(ml mj)
26664
37775 (14)
Multistage model of cancer developmentG Ritter et al
131
This is the Taylor expansion discussed by Mool-gavkar It was referred to (Moolgavkar 1978) as theMacLaurin series but that is a special case of themore general Taylor series We use the more generalterm in this paper
As one might expect the solution (Equation(16)) has a simple series expansion which can beexpressed in closed form This is given in thefollowing Lemma
Lemma 2 (Taylor expansion)Let fmi i1 kg be any set of nonzero constants
with mi mj if i j Then
Xm
i1
ximemi tX
j]m1
ajtj
with am11=(m1) and
aj1(1) jm
j
Xi155ijm
mi1mi2
mijm(20)
if jm This implies that
pm1(t)cm
tm1
(m 1)
1
m
Xm
i1
mi
tmO(tm1)
Remark 1 In various applications it is mostconvenient to use the lowest nonvanishing order(the ArmitageDoll approximation) in this seriesexpansion and itrsquos necessary to have conditionswhich guarantee that this truncation is valid Wecompare the lowest nonvanishing order to the(assumably better) approximation obtained byconsidering higher-order terms In particularlet us compare the lowest nonvanishing order(jm1 in the above formulae) to the followingterm In this paper as with the papers ofMoolgavkar we are concerned with the last stageonly and this corresponds to mk in the aboveequations Let Dn denote the nth nonzero term inthe Taylor expansion which has coefficientakn2
By a simple calculation
D2
D1
t m (21)
This formula shows that D2=D1 can be arbi-trarily large by inclusion of a fast stage or
stages each with large m When attempting as
we are doing to find a model to fit the data
from ages 20 to 70 years the constant a in the
first term of Equation (19) is heavily constrained
by the data If k is not varied the constant a
is proportional to the product m1 mk One
can only make m arbitrarily large if this pro-
duct is fixed to the correct value This is
discussed further in the section on determination
of k We will show later that if nf of the stages are
much faster than the remaining nsknf stages
then the exact solution for the k-stage model is
approximately equal to the exact solution for the
remaining ns stages which we call slow stages
Equation (21) indicates that the Taylor series for
k stages converges more slowly than the Taylor
series for ns stages because inclusion of fast
stages increases the average transition constant
In the presence of fast stages a large number of
terms must be included before the Taylor series
becomes accurate and consideration of only the
first and second terms in the expansion is
incorrect However the exact solution for the
model without the fast stages has a different
Taylor series which converges quite rapidly This
realization leads to important practical conclu-
sions that are discussed in more detail later
Specifically if inclusion of a few fast stages set
m103 then Equation (21) shows that D2 is 10
percent of D1 at t100 If the stages are fast
enough to set m102 per year then D2=D1 is
of order one at 100 years It is clear that by
including fast stages D2=D1 can be made arbi-
trarily largeMoolgavkar deliberately chose a set of para-
meters which included some slow and some fast
(but not very fast) stages (see for example
Moolgavkar 1978 Table 1) Although the above
estimate for D2=D1 does hold for Moolgavkarrsquos
example a discussion of his chosen parameters
needs more careful numerical analysis Consider
a tissue of 109 cells affected by a hypothetical
six-stage2 malignant tumor with transition rates
(per year) given by
2Moolgavkar (1991) refers to this as a seven-stage process but onlygives values for six transition constants We assume here that six stagesare meant
Multistage model of cancer developmentG Ritter et al
132
104 2104 34104 7103
8103 9103
In this example
m00046 per year
So our formula Equation (21) predicts D2046D1
at t100 years This is precisely what is observed inFigure 4 which shows the exact solution togetherwith its first five Taylor approximants The numer-ical factor relating the second approximationwhich lies below the exact curve to the firstapproximation lying above the curve at t100years is seen to be about 146
As noted in the introduction 109 cells may be toofew Bearing in mind that the model is used to fitincidence data each value of the six constants m
would have to be reduced by a factor of 2 if N wereincreased to 64109 and the exact formula at aget100 years would then be only 128 times theArmitageDoll approximation
The implications for determination of the valueof k using Moolgavkarrsquos parameters is discussedlater More generally
Dn1
Dn
akn1
akn2
t
t
k n
Pnk
Pn1k
where Pnk is the symmetric degree n polynomial ink variables given by ai155in
mi1 min
These poly-nomials satisfy
Pnk mkPn1k(terms involving m1 mk1)
It follows that for n]2
Dn1
Dn
t
k n
mk
ratio approaching
zero as mk 0
13(22)
The surprising conclusion of Equation (22) isthat if we add one very fast (mm104) stage at theend it may be necessary to consider hundreds orthousands of terms in the Taylor expansion in orderto attain accurate results The rate of convergenceof this expansion is affected strongly by theappearance of fast stages while the exact solutionis not This is a very important paradoxical resultthe solution for which may be found in the earliercomments If there are ns slow stages and nf veryfast stages the exact solution is close to the
ArmitageDoll approximation for the ns slowstages alone the ArmitageDoll approximationfor the full exact model with nsnf stages is notaccurate For equal transition probabilities givenby the single transition constant m the aboveimplies D2=D1mt
Lemma 3 (Normalization)Let p0(t) pk(t) satisfy the k-stage Batemanequations (Equation (11)) with any collection(m1 mk) of transition constants Then
g
0
pk(t)dtYk
i1mi
Xk
j1
xjk
mj
1 (23)
This equation shows that given sufficient timethe cancer process will eventually reach the laststage
General properties of the solutions
The general solution for reasonable values of k has far too many constants to be determined byepidemiological data alone However there arevarious cases in which the solution of the Batemanequations Equations (16) and (17) can be simplifiedenough to depend on only a few independentconstants even if the number of stages is large
Equation (21) implies that the ratio of the secondterm to the first in the Taylor series for I(t)
D2
D1
is proportional to the average m But the product ofthe ms is constrained by the value of the observedincidence With this constraint the average m will
Figure 4 Exact solution for Moolgavkarrsquos example shown with itsfirst five Taylor approximants
Multistage model of cancer developmentG Ritter et al
133
be a minimum when all the values of m are equalFor this important special case D2=D1 is muchsmaller than for example in Moolgavkarrsquos case
Another interesting result is that permuting thestages makes no difference to the final result pk(t)or more generally to mmpm1(t) for any m5k Thisis self-evident physically for radioactive decaywhich is also described by the Bateman equationsThis result used implicitly in the ArmitageDollapproximation is a fundamental property of theexact solution It may be proved as follows Itsuffices to consider a transposition of the ith stageand the jth stage as any permutation maybe represented as a product of transpositionsNow xim is a product of (mlmi)
1 where l iApplying the transposition these terms become(mlmj)
1except for the l j term which be-
comes (mimj)1 The result is thenQ
lj(mlmj)1
xjmA third general result is that if there are say 4
stages which proceed slowly (small equal values ofm) and many stages which act much faster (values ofm that are 10 times larger) then the final resultdepends only on the 4 slow stages This is self-evident physically and was probably implicit in thethinking of earlier users of the multistage modelThis is proven rigorously in Lemma 6 and dis-cussed with a specific example in the section onmeasurement of k
We pay special attention to a simple case (onenot covered by Lemma 1) in which all stages haveequal transition probabilities This simple case isdescribed by Lemma 4
Equal transition ratesLemma 4 (Equal transition rates)Consider the Bateman equations pj (t)m(pj1pj)arising from equal transition rates m1m2
mkm For any natural number j5k1 the solutionis
pj(t)1
jemt(mt) j (24)
In particular for the penultimate stage
pk1(t)1
(k 1)emt(mt)k1
The global maximum of pj(t) occurs at the agetpeak j=m Further as before we have
g
0
pk (t)dtg
0
mpk1(t)dt1
Remark 2 Since f(t) applies to a single cell m isvery small (on the order of 103) and hence for anynonzero number of stages tpeak j=m always occursat a much greater age than those contained in ahuman lifespan This important point is discussedin detail in the earlier section lsquoCan the multistagemodel ever fit the datarsquo
The algebraic simplicity of equation Equation(24) makes it attractive as a starting point for ourdiscussions It has two adjustable constants k andm A useful numerical example is the followingSuppose we would like to know how the exactsolution Equation (24) differs from the first term inits series expansion
pj(t)Xn0
(mt) jn
jn (25)
In general the first term in the series expansion ofexxn (n0 an integer) is xn and jexxnxn j
xn(1ex) In a numerical example with G03 G001 per year and k7 we have mt502for all t in a human lifespan Then frac121emtfrac12502and so for the first Taylor remainder R1 of pj(t) wehave
jR1j51
5k1256106 (26)
Equally separated transition rates
Another case which is amenable to an exactanalytical solution is when the transition constantsmi for the various stages increase by a fixed amountOf course it is not necessary to assume that in theactual cancer process the transition rates occur inincreasing order thanks to invariance of the processunder general permutations as remarked above sothis solution applies to the general situation whenthere exists some arbitrary reordering of thetransition constants so that they increase linearly
Lemma 5 (Equal separation)Suppose that the transition constants mm1m2 mk are uniformly separated so that mi1mi d for all i and for some constant d0 Then for anyn5k the solution is given by
Multistage model of cancer developmentG Ritter et al
134
pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

This is the Taylor expansion discussed by Mool-gavkar It was referred to (Moolgavkar 1978) as theMacLaurin series but that is a special case of themore general Taylor series We use the more generalterm in this paper
As one might expect the solution (Equation(16)) has a simple series expansion which can beexpressed in closed form This is given in thefollowing Lemma
Lemma 2 (Taylor expansion)Let fmi i1 kg be any set of nonzero constants
with mi mj if i j Then
Xm
i1
ximemi tX
j]m1
ajtj
with am11=(m1) and
aj1(1) jm
j
Xi155ijm
mi1mi2
mijm(20)
if jm This implies that
pm1(t)cm
tm1
(m 1)
1
m
Xm
i1
mi
tmO(tm1)
Remark 1 In various applications it is mostconvenient to use the lowest nonvanishing order(the ArmitageDoll approximation) in this seriesexpansion and itrsquos necessary to have conditionswhich guarantee that this truncation is valid Wecompare the lowest nonvanishing order to the(assumably better) approximation obtained byconsidering higher-order terms In particularlet us compare the lowest nonvanishing order(jm1 in the above formulae) to the followingterm In this paper as with the papers ofMoolgavkar we are concerned with the last stageonly and this corresponds to mk in the aboveequations Let Dn denote the nth nonzero term inthe Taylor expansion which has coefficientakn2
By a simple calculation
D2
D1
t m (21)
This formula shows that D2=D1 can be arbi-trarily large by inclusion of a fast stage or
stages each with large m When attempting as
we are doing to find a model to fit the data
from ages 20 to 70 years the constant a in the
first term of Equation (19) is heavily constrained
by the data If k is not varied the constant a
is proportional to the product m1 mk One
can only make m arbitrarily large if this pro-
duct is fixed to the correct value This is
discussed further in the section on determination
of k We will show later that if nf of the stages are
much faster than the remaining nsknf stages
then the exact solution for the k-stage model is
approximately equal to the exact solution for the
remaining ns stages which we call slow stages
Equation (21) indicates that the Taylor series for
k stages converges more slowly than the Taylor
series for ns stages because inclusion of fast
stages increases the average transition constant
In the presence of fast stages a large number of
terms must be included before the Taylor series
becomes accurate and consideration of only the
first and second terms in the expansion is
incorrect However the exact solution for the
model without the fast stages has a different
Taylor series which converges quite rapidly This
realization leads to important practical conclu-
sions that are discussed in more detail later
Specifically if inclusion of a few fast stages set
m103 then Equation (21) shows that D2 is 10
percent of D1 at t100 If the stages are fast
enough to set m102 per year then D2=D1 is
of order one at 100 years It is clear that by
including fast stages D2=D1 can be made arbi-
trarily largeMoolgavkar deliberately chose a set of para-
meters which included some slow and some fast
(but not very fast) stages (see for example
Moolgavkar 1978 Table 1) Although the above
estimate for D2=D1 does hold for Moolgavkarrsquos
example a discussion of his chosen parameters
needs more careful numerical analysis Consider
a tissue of 109 cells affected by a hypothetical
six-stage2 malignant tumor with transition rates
(per year) given by
2Moolgavkar (1991) refers to this as a seven-stage process but onlygives values for six transition constants We assume here that six stagesare meant
Multistage model of cancer developmentG Ritter et al
132
104 2104 34104 7103
8103 9103
In this example
m00046 per year
So our formula Equation (21) predicts D2046D1
at t100 years This is precisely what is observed inFigure 4 which shows the exact solution togetherwith its first five Taylor approximants The numer-ical factor relating the second approximationwhich lies below the exact curve to the firstapproximation lying above the curve at t100years is seen to be about 146
As noted in the introduction 109 cells may be toofew Bearing in mind that the model is used to fitincidence data each value of the six constants m
would have to be reduced by a factor of 2 if N wereincreased to 64109 and the exact formula at aget100 years would then be only 128 times theArmitageDoll approximation
The implications for determination of the valueof k using Moolgavkarrsquos parameters is discussedlater More generally
Dn1
Dn
akn1
akn2
t
t
k n
Pnk
Pn1k
where Pnk is the symmetric degree n polynomial ink variables given by ai155in
mi1 min
These poly-nomials satisfy
Pnk mkPn1k(terms involving m1 mk1)
It follows that for n]2
Dn1
Dn
t
k n
mk
ratio approaching
zero as mk 0
13(22)
The surprising conclusion of Equation (22) isthat if we add one very fast (mm104) stage at theend it may be necessary to consider hundreds orthousands of terms in the Taylor expansion in orderto attain accurate results The rate of convergenceof this expansion is affected strongly by theappearance of fast stages while the exact solutionis not This is a very important paradoxical resultthe solution for which may be found in the earliercomments If there are ns slow stages and nf veryfast stages the exact solution is close to the
ArmitageDoll approximation for the ns slowstages alone the ArmitageDoll approximationfor the full exact model with nsnf stages is notaccurate For equal transition probabilities givenby the single transition constant m the aboveimplies D2=D1mt
Lemma 3 (Normalization)Let p0(t) pk(t) satisfy the k-stage Batemanequations (Equation (11)) with any collection(m1 mk) of transition constants Then
g
0
pk(t)dtYk
i1mi
Xk
j1
xjk
mj
1 (23)
This equation shows that given sufficient timethe cancer process will eventually reach the laststage
General properties of the solutions
The general solution for reasonable values of k has far too many constants to be determined byepidemiological data alone However there arevarious cases in which the solution of the Batemanequations Equations (16) and (17) can be simplifiedenough to depend on only a few independentconstants even if the number of stages is large
Equation (21) implies that the ratio of the secondterm to the first in the Taylor series for I(t)
D2
D1
is proportional to the average m But the product ofthe ms is constrained by the value of the observedincidence With this constraint the average m will
Figure 4 Exact solution for Moolgavkarrsquos example shown with itsfirst five Taylor approximants
Multistage model of cancer developmentG Ritter et al
133
be a minimum when all the values of m are equalFor this important special case D2=D1 is muchsmaller than for example in Moolgavkarrsquos case
Another interesting result is that permuting thestages makes no difference to the final result pk(t)or more generally to mmpm1(t) for any m5k Thisis self-evident physically for radioactive decaywhich is also described by the Bateman equationsThis result used implicitly in the ArmitageDollapproximation is a fundamental property of theexact solution It may be proved as follows Itsuffices to consider a transposition of the ith stageand the jth stage as any permutation maybe represented as a product of transpositionsNow xim is a product of (mlmi)
1 where l iApplying the transposition these terms become(mlmj)
1except for the l j term which be-
comes (mimj)1 The result is thenQ
lj(mlmj)1
xjmA third general result is that if there are say 4
stages which proceed slowly (small equal values ofm) and many stages which act much faster (values ofm that are 10 times larger) then the final resultdepends only on the 4 slow stages This is self-evident physically and was probably implicit in thethinking of earlier users of the multistage modelThis is proven rigorously in Lemma 6 and dis-cussed with a specific example in the section onmeasurement of k
We pay special attention to a simple case (onenot covered by Lemma 1) in which all stages haveequal transition probabilities This simple case isdescribed by Lemma 4
Equal transition ratesLemma 4 (Equal transition rates)Consider the Bateman equations pj (t)m(pj1pj)arising from equal transition rates m1m2
mkm For any natural number j5k1 the solutionis
pj(t)1
jemt(mt) j (24)
In particular for the penultimate stage
pk1(t)1
(k 1)emt(mt)k1
The global maximum of pj(t) occurs at the agetpeak j=m Further as before we have
g
0
pk (t)dtg
0
mpk1(t)dt1
Remark 2 Since f(t) applies to a single cell m isvery small (on the order of 103) and hence for anynonzero number of stages tpeak j=m always occursat a much greater age than those contained in ahuman lifespan This important point is discussedin detail in the earlier section lsquoCan the multistagemodel ever fit the datarsquo
The algebraic simplicity of equation Equation(24) makes it attractive as a starting point for ourdiscussions It has two adjustable constants k andm A useful numerical example is the followingSuppose we would like to know how the exactsolution Equation (24) differs from the first term inits series expansion
pj(t)Xn0
(mt) jn
jn (25)
In general the first term in the series expansion ofexxn (n0 an integer) is xn and jexxnxn j
xn(1ex) In a numerical example with G03 G001 per year and k7 we have mt502for all t in a human lifespan Then frac121emtfrac12502and so for the first Taylor remainder R1 of pj(t) wehave
jR1j51
5k1256106 (26)
Equally separated transition rates
Another case which is amenable to an exactanalytical solution is when the transition constantsmi for the various stages increase by a fixed amountOf course it is not necessary to assume that in theactual cancer process the transition rates occur inincreasing order thanks to invariance of the processunder general permutations as remarked above sothis solution applies to the general situation whenthere exists some arbitrary reordering of thetransition constants so that they increase linearly
Lemma 5 (Equal separation)Suppose that the transition constants mm1m2 mk are uniformly separated so that mi1mi d for all i and for some constant d0 Then for anyn5k the solution is given by
Multistage model of cancer developmentG Ritter et al
134
pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

104 2104 34104 7103
8103 9103
In this example
m00046 per year
So our formula Equation (21) predicts D2046D1
at t100 years This is precisely what is observed inFigure 4 which shows the exact solution togetherwith its first five Taylor approximants The numer-ical factor relating the second approximationwhich lies below the exact curve to the firstapproximation lying above the curve at t100years is seen to be about 146
As noted in the introduction 109 cells may be toofew Bearing in mind that the model is used to fitincidence data each value of the six constants m
would have to be reduced by a factor of 2 if N wereincreased to 64109 and the exact formula at aget100 years would then be only 128 times theArmitageDoll approximation
The implications for determination of the valueof k using Moolgavkarrsquos parameters is discussedlater More generally
Dn1
Dn
akn1
akn2
t
t
k n
Pnk
Pn1k
where Pnk is the symmetric degree n polynomial ink variables given by ai155in
mi1 min
These poly-nomials satisfy
Pnk mkPn1k(terms involving m1 mk1)
It follows that for n]2
Dn1
Dn
t
k n
mk
ratio approaching
zero as mk 0
13(22)
The surprising conclusion of Equation (22) isthat if we add one very fast (mm104) stage at theend it may be necessary to consider hundreds orthousands of terms in the Taylor expansion in orderto attain accurate results The rate of convergenceof this expansion is affected strongly by theappearance of fast stages while the exact solutionis not This is a very important paradoxical resultthe solution for which may be found in the earliercomments If there are ns slow stages and nf veryfast stages the exact solution is close to the
ArmitageDoll approximation for the ns slowstages alone the ArmitageDoll approximationfor the full exact model with nsnf stages is notaccurate For equal transition probabilities givenby the single transition constant m the aboveimplies D2=D1mt
Lemma 3 (Normalization)Let p0(t) pk(t) satisfy the k-stage Batemanequations (Equation (11)) with any collection(m1 mk) of transition constants Then
g
0
pk(t)dtYk
i1mi
Xk
j1
xjk
mj
1 (23)
This equation shows that given sufficient timethe cancer process will eventually reach the laststage
General properties of the solutions
The general solution for reasonable values of k has far too many constants to be determined byepidemiological data alone However there arevarious cases in which the solution of the Batemanequations Equations (16) and (17) can be simplifiedenough to depend on only a few independentconstants even if the number of stages is large
Equation (21) implies that the ratio of the secondterm to the first in the Taylor series for I(t)
D2
D1
is proportional to the average m But the product ofthe ms is constrained by the value of the observedincidence With this constraint the average m will
Figure 4 Exact solution for Moolgavkarrsquos example shown with itsfirst five Taylor approximants
Multistage model of cancer developmentG Ritter et al
133
be a minimum when all the values of m are equalFor this important special case D2=D1 is muchsmaller than for example in Moolgavkarrsquos case
Another interesting result is that permuting thestages makes no difference to the final result pk(t)or more generally to mmpm1(t) for any m5k Thisis self-evident physically for radioactive decaywhich is also described by the Bateman equationsThis result used implicitly in the ArmitageDollapproximation is a fundamental property of theexact solution It may be proved as follows Itsuffices to consider a transposition of the ith stageand the jth stage as any permutation maybe represented as a product of transpositionsNow xim is a product of (mlmi)
1 where l iApplying the transposition these terms become(mlmj)
1except for the l j term which be-
comes (mimj)1 The result is thenQ
lj(mlmj)1
xjmA third general result is that if there are say 4
stages which proceed slowly (small equal values ofm) and many stages which act much faster (values ofm that are 10 times larger) then the final resultdepends only on the 4 slow stages This is self-evident physically and was probably implicit in thethinking of earlier users of the multistage modelThis is proven rigorously in Lemma 6 and dis-cussed with a specific example in the section onmeasurement of k
We pay special attention to a simple case (onenot covered by Lemma 1) in which all stages haveequal transition probabilities This simple case isdescribed by Lemma 4
Equal transition ratesLemma 4 (Equal transition rates)Consider the Bateman equations pj (t)m(pj1pj)arising from equal transition rates m1m2
mkm For any natural number j5k1 the solutionis
pj(t)1
jemt(mt) j (24)
In particular for the penultimate stage
pk1(t)1
(k 1)emt(mt)k1
The global maximum of pj(t) occurs at the agetpeak j=m Further as before we have
g
0
pk (t)dtg
0
mpk1(t)dt1
Remark 2 Since f(t) applies to a single cell m isvery small (on the order of 103) and hence for anynonzero number of stages tpeak j=m always occursat a much greater age than those contained in ahuman lifespan This important point is discussedin detail in the earlier section lsquoCan the multistagemodel ever fit the datarsquo
The algebraic simplicity of equation Equation(24) makes it attractive as a starting point for ourdiscussions It has two adjustable constants k andm A useful numerical example is the followingSuppose we would like to know how the exactsolution Equation (24) differs from the first term inits series expansion
pj(t)Xn0
(mt) jn
jn (25)
In general the first term in the series expansion ofexxn (n0 an integer) is xn and jexxnxn j
xn(1ex) In a numerical example with G03 G001 per year and k7 we have mt502for all t in a human lifespan Then frac121emtfrac12502and so for the first Taylor remainder R1 of pj(t) wehave
jR1j51
5k1256106 (26)
Equally separated transition rates
Another case which is amenable to an exactanalytical solution is when the transition constantsmi for the various stages increase by a fixed amountOf course it is not necessary to assume that in theactual cancer process the transition rates occur inincreasing order thanks to invariance of the processunder general permutations as remarked above sothis solution applies to the general situation whenthere exists some arbitrary reordering of thetransition constants so that they increase linearly
Lemma 5 (Equal separation)Suppose that the transition constants mm1m2 mk are uniformly separated so that mi1mi d for all i and for some constant d0 Then for anyn5k the solution is given by
Multistage model of cancer developmentG Ritter et al
134
pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

be a minimum when all the values of m are equalFor this important special case D2=D1 is muchsmaller than for example in Moolgavkarrsquos case
Another interesting result is that permuting thestages makes no difference to the final result pk(t)or more generally to mmpm1(t) for any m5k Thisis self-evident physically for radioactive decaywhich is also described by the Bateman equationsThis result used implicitly in the ArmitageDollapproximation is a fundamental property of theexact solution It may be proved as follows Itsuffices to consider a transposition of the ith stageand the jth stage as any permutation maybe represented as a product of transpositionsNow xim is a product of (mlmi)
1 where l iApplying the transposition these terms become(mlmj)
1except for the l j term which be-
comes (mimj)1 The result is thenQ
lj(mlmj)1
xjmA third general result is that if there are say 4
stages which proceed slowly (small equal values ofm) and many stages which act much faster (values ofm that are 10 times larger) then the final resultdepends only on the 4 slow stages This is self-evident physically and was probably implicit in thethinking of earlier users of the multistage modelThis is proven rigorously in Lemma 6 and dis-cussed with a specific example in the section onmeasurement of k
We pay special attention to a simple case (onenot covered by Lemma 1) in which all stages haveequal transition probabilities This simple case isdescribed by Lemma 4
Equal transition ratesLemma 4 (Equal transition rates)Consider the Bateman equations pj (t)m(pj1pj)arising from equal transition rates m1m2
mkm For any natural number j5k1 the solutionis
pj(t)1
jemt(mt) j (24)
In particular for the penultimate stage
pk1(t)1
(k 1)emt(mt)k1
The global maximum of pj(t) occurs at the agetpeak j=m Further as before we have
g
0
pk (t)dtg
0
mpk1(t)dt1
Remark 2 Since f(t) applies to a single cell m isvery small (on the order of 103) and hence for anynonzero number of stages tpeak j=m always occursat a much greater age than those contained in ahuman lifespan This important point is discussedin detail in the earlier section lsquoCan the multistagemodel ever fit the datarsquo
The algebraic simplicity of equation Equation(24) makes it attractive as a starting point for ourdiscussions It has two adjustable constants k andm A useful numerical example is the followingSuppose we would like to know how the exactsolution Equation (24) differs from the first term inits series expansion
pj(t)Xn0
(mt) jn
jn (25)
In general the first term in the series expansion ofexxn (n0 an integer) is xn and jexxnxn j
xn(1ex) In a numerical example with G03 G001 per year and k7 we have mt502for all t in a human lifespan Then frac121emtfrac12502and so for the first Taylor remainder R1 of pj(t) wehave
jR1j51
5k1256106 (26)
Equally separated transition rates
Another case which is amenable to an exactanalytical solution is when the transition constantsmi for the various stages increase by a fixed amountOf course it is not necessary to assume that in theactual cancer process the transition rates occur inincreasing order thanks to invariance of the processunder general permutations as remarked above sothis solution applies to the general situation whenthere exists some arbitrary reordering of thetransition constants so that they increase linearly
Lemma 5 (Equal separation)Suppose that the transition constants mm1m2 mk are uniformly separated so that mi1mi d for all i and for some constant d0 Then for anyn5k the solution is given by
Multistage model of cancer developmentG Ritter et al
134
pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

pn1(t)Cdmn emt(1etd)n1 (27)
where CdmnGm
d n 1
G(m=d)G(n)
is a constant
Remark 3 To calculate the penultimate stagepk1(t) simply replace n by k in the abovepk1(t)Cdmk emt(1etd)k1
Remark 4 This model Equation (27) althoughan exact model with unequal transition constantsallows us to find an analytic expression for theposition of the peak Setting p k1(t)0 in Equa-tion (27) leads to
tpeakd1ln
1
d
m(k1)
(28)
This is an exact expression If dm whichcorresponds to the model with equal transitionrates the log can be expanded leading to
tpeakk 1
m (29)
This agrees with what was found in Lemma 4Remark 5 For consistency Lemma 5 should
reduce to the exact solution for equal transitionrates given in Lemma 4 This is true since for anym5k cm clearly approaches mm1 and
pm1
cm
limd00
Xm
j1
xjmemj t
emtlimd00
d1m
Xm
j1
(1)j1
(j 1)(m j)e(j1)dt
emt
G(m)limd00
d1metd (1 etd)m
etd 1
emt tm1
(m 1)
This is consistent with Lemma 4Remark 6 The solution obtained in Lemma 5 is
an analytic expression containing no sums orproducts which would force the number of stagesk to be an integer therefore it is amenable tocomputer simulations which allow k to be acontinuous variable In particular one can
perform nonlinear regression to optimize thevalue of k together with d and m which are theother free parameters in the model given byEquation (27)
Remark 7 The analytic solution for tpeak allowsus to also find the height of the function at its peak
pk1(tpeak)Cdmk
(k 1) d
(k 1) d m
k1
1
(k 1) d
m
m
d
From this one obtains the qualitative observa-tion that large n shifts the peak down To seethis note that the asymptotic expansion for largek is
pk1(tpeak) 0large k
const1
kO
1
k
2
where the constant is em
dd
m
m
d
Gm
d
1
This is
important because it explains why when weattempt to fit real-world data the curve-fittingroutine finds unrealistically high values of k thenumber of stages
Fast and slow stages
In the discussion of Equation (21) we outlinedthe effect of including a stage much faster thanthe others on the Taylor expansion In thissection we analyse this in a more precise way(Figure 5) To illustrate the general effect whichoccurs when one or a few stages are much fasterthan the rest we first consider a simple examplewhich entails the convergence of the two-stagesolution to the single-stage solution as thetransition rate for the second stage becomeslarge We exhibit this convergence in the functionpk(t) which corresponds to the last stage inEquations (30) and (31) If k1 we have (forI(t)pk(t))
I(t)m1em1t (30)
and if k2
Multistage model of cancer developmentG Ritter et al
135
I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

I(t)m1m2
m2 m1
em1tm1m2
m1 m2
em2t (31)
One may now observe directly that Equation (31)converges to Equation (30) in the limit that m2m1In fact this phenomenon is quite general asfollows from Lemma 6
Lemma 6 (Slow and fast stages)Suppose that the first ns stages have transitionconstant ms followed by one stage with transitionconstant mf Then the exact solution is given by
pns1(t)(ms)
ns1
nsemf tg
t
0
e(mfms)xxnx dx
emf t
ms
ms mf
ns11
G(1 ns t(ms mf ))
ns
13(32)
Further we assert that
limmf 0(mf pns1(t))mspns(t) (33)
The statement of Lemma 6 places the fast stageat the end However previously we have shown that
permuting the stages has no effect on the finalsolution ie on the incidence function ThereforeLemma 6 and in particular Equation (33) appliesto a multistage progression in which any one of thestages is very fast compared to the others Byinduction the same is then true of the case withmultiple fast stages
This is an exact solution no approximations havebeen made Our intuition is that if the fast stage isfast enough then it will have no effect on theproperly normalized solution and Equation (32)will reduce to pns
(t) This intuition is rigorouslyexpressed as Equation (33) We note that both sidesof Equation (33) integrate to one This generalizesthe simple observation following Equation (31) Weillustrate this general phenomenon with a specificexample Consider the Bateman equations with ns
stages at rate n and the remaining kns stages atrate m Let g(tm n) denote the solution pk(t)mpk1(t) and let
S(t n)n
nsetn(tn)ns
denote the exact solution for ns slow stages ofrate n To illustrate that the effect of the fast stagesbecomes negligible for mn we plot g(t nn n) andS(t n) on the same graph for increasing values of n with k5 total stages and ns3 slow stages For theslow transition constant we take a reasonable valueof 5103 which is comparable to the valueschosen by Moolgavkar (1991) The relevant Bate-man equations are
p0(t)m p0(t)
p1(t)m p0n p1
p2(t)n p1n p2 p5(t)n p4
We conclude in this numerical example that form50n the effect of the faster stages is in the orderof a few per cent In the limit as m=n 0 weobserve that g(tm n) converges to S(t n) for all t
Results and discussion
On the number of stages
In the introduction we pointed out that a funda-mental success of the multistage model (1) was by
Figure 5 Exact solutions with three slow stages having rates n5103 followed by two lsquofastrsquo stages at rate m Graphs are shown form2n and m50n The solution S(t n) with ns3 slow stages and nofast stages is shown as a dashed line for comparison
Multistage model of cancer developmentG Ritter et al
136
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145
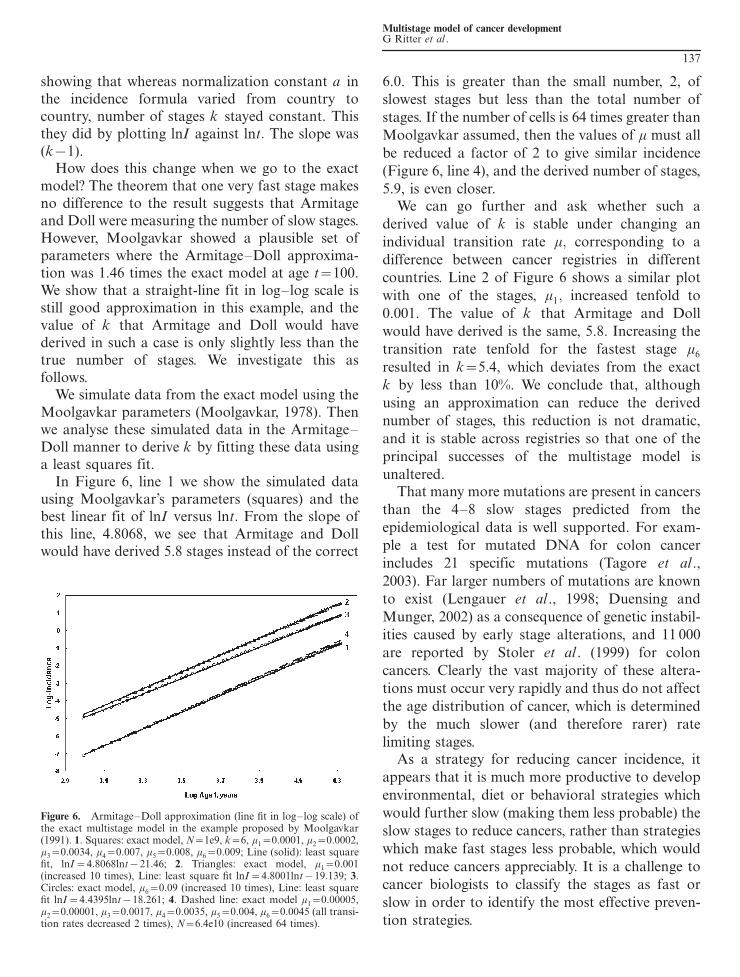
showing that whereas normalization constant a inthe incidence formula varied from country tocountry number of stages k stayed constant Thisthey did by plotting lnI against lnt The slope was(k1)
How does this change when we go to the exact
model The theorem that one very fast stage makesno difference to the result suggests that Armitageand Doll were measuring the number of slow stagesHowever Moolgavkar showed a plausible set ofparameters where the ArmitageDoll approxima-tion was 146 times the exact model at age t100We show that a straight-line fit in loglog scale isstill good approximation in this example and thevalue of k that Armitage and Doll would havederived in such a case is only slightly less than thetrue number of stages We investigate this asfollows
We simulate data from the exact model using theMoolgavkar parameters (Moolgavkar 1978) Thenwe analyse these simulated data in the Armitage
Doll manner to derive k by fitting these data usinga least squares fit
In Figure 6 line 1 we show the simulated datausing Moolgavkarrsquos parameters (squares) and thebest linear fit of lnI versus lnt From the slope ofthis line 48068 we see that Armitage and Dollwould have derived 58 stages instead of the correct
60 This is greater than the small number 2 of
slowest stages but less than the total number of
stages If the number of cells is 64 times greater than
Moolgavkar assumed then the values of m must all
be reduced a factor of 2 to give similar incidence
(Figure 6 line 4) and the derived number of stages
59 is even closerWe can go further and ask whether such a
derived value of k is stable under changing an
individual transition rate m corresponding to a
difference between cancer registries in different
countries Line 2 of Figure 6 shows a similar plot
with one of the stages m1 increased tenfold to
0001 The value of k that Armitage and Doll
would have derived is the same 58 Increasing the
transition rate tenfold for the fastest stage m6
resulted in k54 which deviates from the exact
k by less than 10 We conclude that although
using an approximation can reduce the derived
number of stages this reduction is not dramatic
and it is stable across registries so that one of the
principal successes of the multistage model is
unalteredThat many more mutations are present in cancers
than the 48 slow stages predicted from the
epidemiological data is well supported For exam-
ple a test for mutated DNA for colon cancer
includes 21 specific mutations (Tagore et al
2003) Far larger numbers of mutations are known
to exist (Lengauer et al 1998 Duensing and
Munger 2002) as a consequence of genetic instabil-
ities caused by early stage alterations and 11 000
are reported by Stoler et al (1999) for colon
cancers Clearly the vast majority of these altera-
tions must occur very rapidly and thus do not affect
the age distribution of cancer which is determined
by the much slower (and therefore rarer) rate
limiting stagesAs a strategy for reducing cancer incidence it
appears that it is much more productive to develop
environmental diet or behavioral strategies which
would further slow (making them less probable) the
slow stages to reduce cancers rather than strategies
which make fast stages less probable which would
not reduce cancers appreciably It is a challenge to
cancer biologists to classify the stages as fast or
slow in order to identify the most effective preven-
tion strategies
Figure 6 ArmitageDoll approximation (line fit in loglog scale) ofthe exact multistage model in the example proposed by Moolgavkar(1991) 1 Squares exact model N1e9 k6 m100001 m200002m300034 m40007 m50008 m60009 Line (solid) least squarefit lnI48068lnt2146 2 Triangles exact model m10001(increased 10 times) Line least square fit lnI48001lnt19139 3Circles exact model m6009 (increased 10 times) Line least squarefit lnI44395lnt18261 4 Dashed line exact model m1000005m2000001 m300017 m400035 m50004 m600045 (all transi-tion rates decreased 2 times) N64e10 (increased 64 times)
Multistage model of cancer developmentG Ritter et al
137
There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

There are arguments that numerous stagesare unnecessary for modelling which entail a two-stage clonal expansion (Moolgavkar and Luebeck2003) However it is important to recognize thatthe additional stages are part of the carcino-genesis process and it is clearly of interest topossess models which reflect in as much detail aspossible the correct structure of the carcinogenesisprocess
Can the multistage model ever fit the data
A salient feature of the data is that cancer incidenceappears to fall off fast above age 80 to nearlyzero at age 100 (Pompei et al 2004) Consider anexact model for I(t)Nf(t) with equal transitionrates
Nm (t m)k
et m k
which as discussed above has its peak at tpeakk=m Since k is the number of stages it is at least oforder one and on the other hand if the model isapplied to a single cell then epidemiological datarequires that even for common cancers the transi-tion rate m must be O(103) Therefore tpeak
O(103) and the turnaround can never occur within ahuman lifespan
We now give a second argument that the turn-around is not relevant within a human lifespanNote that pk(t) is a linear combination of exponen-tials so for any set of positive real transitionconstants it increases from zero to a peak turnsover and asymptotically approaches zero as t 0 Qualitatively this has the same feature as thedata and a more careful analysis is necessary to seethe problem
As noted by Heidenreich et al (2002) theincidence function
I(t)Npk(t)
1 pk(t)(34)
increases to a finite asymptote as t 0 Withstages and transition constants based on fittingreal-world data pk(t) is around 109 or smaller for0BtB100 years so in this range Npk(t) is wellapproximated by Equation (34) which is monotoneincreasing However the time scale at which pk(t)
starts to turn over is precisely the scale at whichpk(t) becomes of order one and the approximationpk(t)1 is no longer valid In conclusion we havethe following assertion
Lemma 7 (Turnover)The turnover in pk(t) can never explain the turnoverin the data
It is of course possible to fit an exact solutionwith Ncells1010 to the (monotone increasing) partof the data set between 20 years and 70 yearsWe consider both equal transition constants(Figure 7) and equally separated transition con-stants (Figure 8) In the region shown in Figure 7which corresponds to a human lifespan the lowest-order Taylor approximation to the exact solutionwith equal transition rates is an extremely goodapproximation (see Remark 1 above) and thus theexact curve is indistinguishable to the Armitage
Doll approximation Iatk1Interestingly even though we only fitted the
equal separation model to the data up to age 70one can see in Figure 8 that the equal separationmodel correctly predicted the next few data points
Susceptibility differences
An early attempted explanation of the decrease inincidence after age 80 is that the cancer-susceptiblepeople are being depleted Thus the incidence levelsoff and drops to zero when all the senistive peoplehave developed cancer This was described by Cooket al (1969) who used the ArmitageDoll approx-imation and made the simple assumption thatonly a fixed fraction of population is susceptible(sensitive) eg able to develop cancer We derive a
Figure 7 Fit of exact solution with equal transition constants to malecolon cancer data with Ncells1010
Multistage model of cancer developmentG Ritter et al
138
more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

more general expression for the age-specific inci-dence Let S(t) be the number of sensitive personsat time t Then
d
dtS(t)S(t)I1(t)
where I1(t) is the incidence per sensitive person LetP be the total number of persons at t0 This leadsto
S(t)PCexp
g
t
0
I1(t)dt
where C is the fraction of sensitive persons Theincidence in the population which includes bothsensitive and insensitive persons is
I(t)new cases per unit time
persons at risk at time t
dS(t)=dt
(1 C)P CPexpg
t
0
I1(t)dt
CPeg
t
0I1(t)dtI1(t)
(1 C)P CPegt
0I1(t)dt
I1(t)
1
1 C
C
expg
t
0
I1(t)dt(35)
The choice C1 corresponds to the assumptionthat all people are sensitive and implies I(t)I1(t)With C0 which corresponds to a completelyinsensitive population we have I(t)0 Further forany 0BCB1 we have I(t) 0 0 as t 0 Thus if
there are any immune persons in the populationI(t) will eventually turn over and tend to zerowhich might enable such an equation to fit thedata The challenge is whether with reasonableassumptions the incidence will approach zero forall cancers within a human lifetime as SEER data
suggests If we define G 1 C
C
expf
t
0I1(t)dt then
Equation (35) implies that the equation determiningthe peak I (t)0 takes the form
I 1I2
1
G
G 1
In the general case one would not expect to solvethis except numerically but for the ArmitageDoll
case I1(t)atk1 which implies I 1=(I1)2 k1
atk
and we have
k 1
a
tk(1 C)
Ceatk=k 1 C
Taylor expanding the right hand side gives theapproximate solution
tpeak
k 1
ak(1 C)
1=k
(36)
For typical laboratory numbers k6 a1013C01 the approximation Equation (36) givestpeak65 However this depends sensitively on k
With I1(t) given by the exact model with equaltransition rates (ETR) in a tissue with N cells wefind
IETR(t)
N
G(k)emt(mt)k1
1 1 C
Cexp
N
m
G(k) G(k mt)
G(k)
(37)
The fact that the function IETR(t) presented inEquation (37) is extremely sensitive to its funda-mental parameters presents a problem from amodelling perspective With typical biological para-
meters of N1013 and m103 the factor N=mappearing under the exponential in Equation (37) isof order 1016 To obtain a reasonable function the
Figure 8 Fit of exact equal separation model to male colon cancerdata with Ncells1010 Although the fit was done using data up to age70 the model remains accurate to age 90
Multistage model of cancer developmentG Ritter et al
139
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145
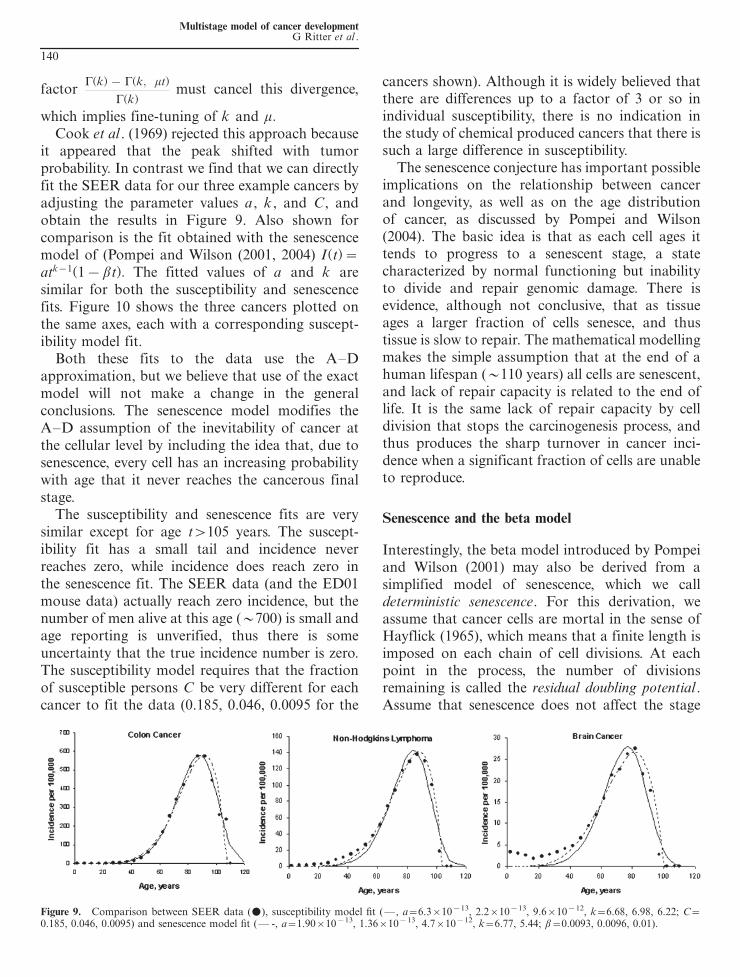
factor
G(k) G(k mt)
G(k)must cancel this divergence
which implies fine-tuning of k and mCook et al (1969) rejected this approach because
it appeared that the peak shifted with tumorprobability In contrast we find that we can directlyfit the SEER data for our three example cancers byadjusting the parameter values a k and C andobtain the results in Figure 9 Also shown forcomparison is the fit obtained with the senescencemodel of (Pompei and Wilson (2001 2004) I(t)atk1(1bt) The fitted values of a and k aresimilar for both the susceptibility and senescencefits Figure 10 shows the three cancers plotted onthe same axes each with a corresponding suscept-ibility model fit
Both these fits to the data use the ADapproximation but we believe that use of the exactmodel will not make a change in the generalconclusions The senescence model modifies theAD assumption of the inevitability of cancer atthe cellular level by including the idea that due tosenescence every cell has an increasing probabilitywith age that it never reaches the cancerous finalstage
The susceptibility and senescence fits are verysimilar except for age t105 years The suscept-ibility fit has a small tail and incidence neverreaches zero while incidence does reach zero inthe senescence fit The SEER data (and the ED01mouse data) actually reach zero incidence but thenumber of men alive at this age (700) is small andage reporting is unverified thus there is someuncertainty that the true incidence number is zeroThe susceptibility model requires that the fractionof susceptible persons C be very different for eachcancer to fit the data (0185 0046 00095 for the
cancers shown) Although it is widely believed thatthere are differences up to a factor of 3 or so inindividual susceptibility there is no indication inthe study of chemical produced cancers that there issuch a large difference in susceptibility
The senescence conjecture has important possibleimplications on the relationship between cancerand longevity as well as on the age distributionof cancer as discussed by Pompei and Wilson(2004) The basic idea is that as each cell ages ittends to progress to a senescent stage a statecharacterized by normal functioning but inabilityto divide and repair genomic damage There isevidence although not conclusive that as tissueages a larger fraction of cells senesce and thustissue is slow to repair The mathematical modellingmakes the simple assumption that at the end of ahuman lifespan (110 years) all cells are senescentand lack of repair capacity is related to the end oflife It is the same lack of repair capacity by celldivision that stops the carcinogenesis process andthus produces the sharp turnover in cancer inci-dence when a significant fraction of cells are unableto reproduce
Senescence and the beta model
Interestingly the beta model introduced by Pompeiand Wilson (2001) may also be derived from asimplified model of senescence which we calldeterministic senescence For this derivation weassume that cancer cells are mortal in the sense ofHayflick (1965) which means that a finite length isimposed on each chain of cell divisions At eachpoint in the process the number of divisionsremaining is called the residual doubling potential Assume that senescence does not affect the stage
Figure 9 Comparison between SEER data (m) susceptibility model fit ( a631013 221013 961012 k668 698 622 C
0185 0046 00095) and senescence model fit ( - a1901013 1361013 471012 k677 544 b00093 00096 001)
Multistage model of cancer developmentG Ritter et al
140
transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

transitions or transition rates in the ArmitageDoll
process Assume a constant (deterministic) cell
division rate then the residual doubling potential
for a cell decreases linearly with ageIf a malignant cell is completely senescent so
that the residual number of doublings is zero then
this cell does not produce observable cancer More
generally if the residual doubling potential is small
then the malignant cell is limited to a small number
of descendants and this produces an unobservable
tumor or one which is effectively destroyed by the
immune system A larger tumor has a proportion-
ally greater chance of survivalArmitageDoll theory assumes that each malig-
nant cell leads to a tumor but naturally this cell
must undergo many divisions to produce an
observable tumor Accordingly the Armitage
Doll model Iatk1 must be modified to reflect
the possibility that the cell as it progresses to
cancer may be simultaneously approaching mor-
tality in the sense of Hayflick The appropriate
modification which takes account of this effect is to
introduce a linear multiplicative factor which must
be of the form (1bt) and which now represents
the conditional probability for an observable tu-
mor given one cell which is malignant (or in the kth
stage of the multistage process) at time t The
incidence function now takes the form of the
PompeiWilson beta model As before the para-
meter b may be determined through curve-fitting
As a caveat to this approach it must be noted that
the existence of in vivo senescence in the human
body has not been established
If the explanation is accepted that cellularsenescence is responsible for the rapid fall off incancers above age 80 there are several conse-quences some of which are discussed in Pompeiand Wilson (2004) For example it is not sufficientfor a drug or environmental agent (or lack thereof)to be shown to reduce cancer If the action of thedrug or agent is to increase senescence to reducecancer then it will be accompanied by the seriousside effect of reduction in longevity Alterations inthe p53 gene have been shown to do this
Melatonin a known antioxidant that reducesDNA damage has been shown in mice experimentsto both increase cancers and increase longevitysuggesting that antioxidants might require morecareful consideration If a drug or environmentalagent could be targeted to one or more specificstages in the multistage cancer interpretation thenperhaps reduction in cancer might be accomplishedwithout reduction in longevity This is seems to bethe case when known strong carcinogens areprevented from acting on cells such as stoppingsmoking or stopping b-naphthylamine inhalation
Another consequence is that drugs such ascortisone which are carcinogenic and dangerousfor use on the young may well be relatively safe foranyone over age 80 for treatment for example ofarthritis Further study of this point is veryimportant If modeling and tests bear this out itwould be welcome news for the aged
At the very least anyone testing or regulating ananticancer drug must be aware of the possibilitythat the drug might be acting as an accelerator ofsenescence which would reduce life expectancy anddemand the appropriate test regimen
Conclusions
This study set out to investigate whether it ispossible to describe cancer incidence above age 80using the exact multistage model but no otherassumptions We prove rigorously that it is notpossible In the process we have proved severaltheorems about the multistage model that may beuseful in further investigations For example weconclude that the multistage model considers onlythe slow stages and that the incidence function isinvariant under permutation of the rate constantsCommon cancers are thought to have 48 slow
Figure 10 Comparison between SEER data for male colon cancer(k) non-Hodgkins lymphoma (^) and brain cancer (I) each withsusceptibility model fit
Multistage model of cancer developmentG Ritter et al
141
stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

stages our conclusion is that there could be manymore fast stages without an appreciable effect onthe age distribution of cancer The possible numberof these lsquofastrsquo stages depends on how much fasterthey are If fast stages are present they maydramatically affect the convergence of the Taylorexpansion
We found that with additional biological as-sumptions it is possible to fit the data and wepresented two possibilities One is the addition ofcellular senescence the other is an assumption thatmost people are not susceptible to cancer and infact for rare cancers very few people are suscep-tible
The incidence data alone cannot decide betweenthe model that there are large differences insusceptibility and the model that cellular senes-cence is important or whether either one is likelyWe merely present the data fits and the problems intheir understanding for others to consider
Acknowledgements
The authors would like to thank Suresh Moolgav-kar for helpful discussions
References
Armitage P 1985 The multistage models of carcinogenesis
Environmental Health Perspectives 53 195201
Armitage P and Doll R 1954 The age distribution of cancer
and a multi-stage theory of carcinogenesis British Journal
of Cancer VIII 112
Bateman H 1910 Solutions of a system of differential
equations occurring in the theory of radioactive transfor-
mation Proceedings of the Cambridge Philosophical
Society 15 42327
Cook PJ Doll R and Fellingham SA 1969 A mathema-
tical model for the age distribution of cancer in man
International Journal of Cancer 4 93112
Duensing S and Munger K 2002 Human papillomaviruses
and centrosome duplication errors modeling the origins
of genomic instability Oncogene 21 624148
Hayflick L 1965 The limited in vitro lifetime of human
diploid cell strains Experimental Cell Research 37 614 36
Heidenreich WF Luebeck EG and Hazelton WD 2002
Multistage models and the incidence of cancer in the
cohort of A-bomb survivors Radiation Research 158
60714
Lengauer C Kinzler KW and Vogelstein B 1998 Genetic
instabilities in human cancers Nature 396 64349
Moolgavkar SH 1978 The multistage theory of carcinogen-
esis and the age distribution of cancer in man Journal of
the National Cancer Institute 61 4952
Moolgavkar SH 1991 Carcinogenesis models an overview
Basic Life Science 58 38796
Moolgavkar SH Krewski D and Schwarz M 1999
Mechanisms of carcinogenesis and biologically-based
models for quantitative estimation and prediction of
cancer risk In Moolgavkar SH Krewski D and Zeise
L et al editors Quantitative Estimation and Prediction of
Cancer Risk IARC Scientific Publications Lyon France
Oxford University Press 131
Moolgavkar SH and Luebeck EG 2003 Multistage carci-
nogenesis and the incidence of human cancer Genes
Chromosomes and Cancer 38 302306
Pompei F and Wilson R 2001 The age distribution of
cancer the turnover at old age Health and Environmental
Risk Assessment 7 161950
Pompei F and Wilson R 2004 A quantitative model of
cellular senescence influence on cancer and longevity
Toxicology and Industrial Health 18 36576
Pompei F Polkanov M and Wilson R 2001 Age distribu-
tion of cancer in mice the incidence turnover at old age
Toxicology and Industrial Health 17 116
Pompei F Lee EE and Wilson R 2004 Cancer turnover
(Letter) httpwwwnaturecomnrc (accessed January
2004)
Pompei F Lee EE and Wilson R Cancer incidence
turnover to age 110+ Gerontological Society of America
Annual Meeting Nov 2004 Washington DC
SEER Public-Use 19951999 when Using SEERStat Sur-
veillance Epidemiology and End Results (SEER) Pro-
gram (wwwseercancergov) SEERStat Database
Incidence-SEER 11 Regs Nov 2001 Sub (19732000)
National Cancer Institute DCCPS Surveillance Research
Program Cancer Statistics Branch released April 2002
based on the November 2001 submission
Stoler DL Chen N and Basik M et al 1999 The onset and
extent of genomic instability in sporadic colorectal tumor
progression Proceedings of the National Academy of
Science 96 1512126
Tagore KS Lawson MJ and Yucaitis JA et al 2003
Sensitivity and specificity of a stool DNA multitarget
assay panel for the detection of advanced colorectal
neoplasia Clinical Colorectal Cancer 3 4753
Multistage model of cancer developmentG Ritter et al
142
Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

Appendix A
Proofs of assertions
Proof of Lemma 1
cm1mm1mcm and xjm1 (mm1mj)1
xjm
for all jm1
d
dtpmcm1
Xm1
j1
xjm1(mj)emj t
cm1
Xm1
j1
xjm1(mm1mj)emj t
cm1
Xm1
j1
mm1xjm1emj t
mm1mcm
Xm1
j1
xjmemj tmm1pm
mm1mpm1mm1pm
as desiredProof of Lemma 2 Equation (20) is proved by
double induction on m and j The series aaj tj
converges absolutely for all t C since Equation(16) is a linear combination of exponentials
Proof of Lemma 3 pk(t)mkpk1(t) so thelemma is equivalent to the statement that for0BiBk
g
0
pi(t)dt1=mi1
Generally speaking pi(t) represents the numberof objects in state i of the process (normalized bythe initial condition) Considering the process forone object forces the initial condition to be p0(0)
1 pi(0)0 i1 and it follows that pi(t)1 if theobject is in state i and zero otherwise It is thenobvious that f
0pi(t)dt does not depend on the rates
to get into state i and is inversely proportional tothe rate to get out of state i Therefore
g
0
pi(t)dt1=mi1
(this can also be proved by a tedious directcomputation) Setting ik1 gives the result that
g
0
mkpk1(t)dt1
as desiredProof of Lemma 4
pj (t)m
1
(j 1)emt(mt)j1
emt(mt)j
j
13
m(pj1pj)
We infer that the global maximum occurs at thesolution of pjpj1 which immediately gives tj=m The value of the integral follows by directcomputation as well as by Lemma 3 in the limit ofequal transition rates
Proof of Lemma 5 mj m(j1)d j]1 sothat mpmj (p j)d This implies that
xjnd1nY
p1n
pj
1
p jd1n
Yj1
i1
1
(i)
Yj1
i1
1
i
d1n(1)j1
(j 1)(n j)
cnYn1
j1
mj Yn2
i0
(m id)dn1
m
d
n1
dn1
Gm
d n 1
G(m=d)
where in the last line we have made use of thePochhammer symbol or lsquoforward factorialrsquo and itsidentity in terms of gamma functions
(a)na(a1) (an1)G(an)=G(a)
We find by direct calculation that
Xn
j1
(1) j1
(j 1)(n j)e(j1)dt
etd(1 etd)n
(etd 1)G(n)
Multistage model of cancer developmentG Ritter et al
143
hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

hence
pn1(t)cn
Xn
j1
xjnemj t
Gm
d n 1
dn1G(m=d)
Xn
j1
(1)j1e(m(j1)d)t
(j 1)(n j)
Gm
d n 1
G(m=d)
emt etd(1 etd)n
(etd 1)G(n)
Gm
d n 1
Gm
d
G(n)
emt(1etd)n1
This completes the proof
Proof of Lemma 6 Let A be any constantIn general the solution of the differential equa-tion
f (t)Af (t)g(t) f (0)0
where g is an arbitrary function and f is to besolved for is given by
f (t)eAt
gt
0
eAxg(x)dx
Suppose that stages zero through ns are describedby the transition constant ms and the stages ns
through k are described by transition constant mf Later we will consider the limit mf 0 whilekeeping ms fixed This means that
d
dtpns1mspns
mf pns1
By the above reasoning with Amf gmspns
this implies that
pns1(t)emf t
gt
0
emf xmspns(x)dx
However from our solution of the model withequal transition rates we know that
pns(x)
1
nsemsx(msx)ns
so we have
pns1(t)(ms)
ns1
nsemf t
gt
0
e(mf ms)xxns dx
This is as far as we can go without the use ofspecial functions To proceed note that
gt
0
eaxxbdxbG(b) G(1 b ta)
ab1
Apply this with amsmf and bns Thisgives
gt
0
e(mfms)xxnsdxnsG(ns) G(1 ns t(ms mf ))
(ms mf )ns1
We infer that
pns1(t)(ms)
ns1(ns G(1 ns tms tmf ))
nsetmf (ms mf )
ns1
emf t
ms
ms mf
ns1
1G(1 ns t(ms mf ))
ns
13
We note that the last line is precisely Equation(32) and completes the proof of that assertion Itremains to examine the limit as mf 0 Ele-mentary methods of asymptotic analysis show thatfor fixed a and frac12zfrac12 0
G(a z)za1ez(1O(z1))
Thus neglecting terms proportional to inversepowers of mf we find
Multistage model of cancer developmentG Ritter et al
144
mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145

mf pns1(t)mf emf t
ms
msmf
ns1
1(ms mf )
nset(msmf )tns
ns
13
emstmf
ms
ms mf
mns
s
tns
ns
emst
ms
ms=mf 1
(mst)
ns
ns
0 ms
1
nsemst(mst)
ns mspns(t)
This completes the proof We remark in passing
that
pns1(t)
ms
ms mf
ns1
etmf g(ns
1 t(msmf ))=ns
where g is the lower incomplete gamma function
Multistage model of cancer developmentG Ritter et al
145