the neurology · of neurology and professor of clinical neurology, paracelsus medical university,...
TRANSCRIPT

v o l u m e 5 n u m b e r 3 | W I n T e r 2 0 1 3
Selected reports from the
66th Annual meeting of the American epilepsy Society
Alison m. Pack, mDGuest editor
CONTINUING EDUCATION FOR PHYSICIANS: 2.5 CREDITS AVAILABLE
NeurologyThe
REPORT
This activity is supported by an educational grant from Eisai Inc.

Guest Editor: Alison M. Pack, MD
The opinions or views expressed in this publication are those of the authors and do not necessarily reflect the opinions or recommendations of Eisai Inc., the University of Cincinnati, or the publisher, Direct One Communications, Inc. Please consult the full prescribing information before using any medication mentioned in this publication.
This publication was made possible through an educational grant from Eisai Inc.
Copyright © 2013 by Direct One Communications, Inc. All rights reserved. Printed in the USA.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 1
V O L U M E 5 N U M B E R 3 | W I N T E R 2 0 1 3
Selected Reports from the 66th Annual Meeting of the American Epilepsy Society
Alison M. Pack, MD, Guest Editor
NeurologyThe
REPORT
4 IntroductionAlison M. Pack, MDThe Neurological Institute of New York, Columbia University Medical Center, New York, New York
6 FromMoleculestoCells,Networks,andSeizures:HowDoesaGeneCauseEpilepsy?Belinda Oyinkan Marquis, MDState University of New York Downstate Medical Center, Brooklyn, New York
14 AnticonvulsantTreatmentofEpilepsyandRefractoryStatusEpilepticus:RecentClinicalTrialsSarah Aminoff Kelley, MDJohns Hopkins Hospital, Baltimore, Maryland
19 PharmacotherapyofMedicallyRefractoryPartial-OnsetEpilepsyCynthia M. Correll, MDColumbia Comprehensive Epilepsy Center, Columbia University Medical Center, New York, New York
24 OptimalUseoftheNewestAntiepilepticDrugsandGenericsPeter Pressman, MDUniversity of California, San Francisco, School of Medicine, San Francisco, California
35 UpdateonRecentGlobalStudiesofPerampanel,aNewSelectiveAMPAAntagonistLisa Aenlle-Matusz, MD, MPHEmory University School of Medicine, Atlanta, Georgia
40 CMEPostTestandEvaluation

2 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
RATIONALE AND PURPOSERecent years have brought many new insights into the origins of epilepsy and novel medications that can reduce seizure frequency. However, too many patients continue to experience seizures that are refractory to current treatment. The reports in this edition of The Neurology Report take a close look at genetic predisposition to seizure activity, antiepileptic drugs (AEDs) recently approved by the US Food and Drug Administration (FDA), combination therapies, and emerging uses of AEDs approved for other purposes. The authors discuss how a genetic predisposition to epilepsy may be identified, evaluated, and managed; the results of recent clinical trials of AEDs with novel mechanisms of action that show significant promise in treating patients with epilepsy; optimal combinations of AEDs based on knowledge of their pharmacokinetics, mechanisms of action, and clinical trial evidence; factors related to treatment adherence; recent developments in treating refractory status epilepticus; and an analysis of results from recent global trials of perampanel. The articles within are based upon selected presentations delivered at the 66th Annual Meeting of the American Epilepsy Society, held November 30 to December 4, 2012, in San Diego, California.
The articles in this issue, written from the academic perspective of physicians-in-training at leading medical institutions, summarize the import of these new findings and place them into clinical context. This activity has been developed and approved by a planning committee of nationally recognized thought leaders to meet a perceived educational need to provide neurologists,
About This CME Activityneurosurgeons, and other physicians with diagnostic and therapeutic strategies to help them perform their medical roles.
LEARNING OBJECTIVESAfter studying this issue of The Neurology Report, participants in this educational activity should be able to:
• Interpret data on novel AEDs and use of these drugs in patients of different ages and with different comorbid conditions.
• Outline the discovery of genes associated with epilepsy, their variability, and their impact on clinical practice.
• Review the results of recent clinical trials of novel AEDs in adults and children with different seizure disorders and comorbidities.
• Summarize recent studies of newer AEDs, alone and in combination, in patients with medically refractory seizures.
• Discuss the influence of drug interactions and adverse effects of AEDs and the risks/benefits of using generic AEDs.
TARGET AUDIENCENeurologists, neurosurgeons, and other physicians significantly involved in the management of patients with epilepsy should find participation in this educational activity valuable.
ACCREDITATION AND CREDIT DESIGNATIONThis activity has been planned and implemented in
accordance with the Essential Areas and Policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of the University of Cincinnati and Direct One
Communications, Inc. The University of Cincinnati is accredited by the ACCME to provide continuing medical education for physicians.
The University of Cincinnati designates this Enduring Material Activity for a maximum of 2.5 AMA PRA Category 1 Credits ™. Physicians should only claim credit commensurate with the extent of their participation in the activity.
CREDIT AVAILABILITYActivity release date: February 19, 2013 Expiration date: February 20, 2014
METHOD OF PARTICIPATIONThis Enduring Material Activity is available in print and online at www.NeurologyReport.com and consists of an introduction, five articles, a postactivity assessment, and an evaluation. Estimated time to complete the activity is 2.5 hours.
To receive credit, participants must read the CME information on these two pages, including the learning objectives and disclosure statements, as well as the full content of this monograph, and then complete the post test and evaluation form online at www.NeurologyReport.com. Upon successful completion of the post test (80% correct) and evaluation form, a CME certificate of participation will be awarded automatically. The certificate may be printed directly from the Web site or e-mailed and printed later.
There are no fees for participating in or receiving credit for this activity.
CME REVIEWERRick Ricer, MD Adjunct Professor of Family Medicine University of Cincinnati Cincinnati, Ohio

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 3
CME ACCREDITATIONSusan P. Tyler, MEd, CMP, CCMEP Director, Continuing Medical Education University of Cincinnati Cincinnati, Ohio
FACULTY DISCLOSURESAll faculty members (or anyone else in a position to control content, such as activity planners) are required to complete a Disclosure of Commercial Interest and Resolution form and to cooperate with identified methods for resolving conflict of interest prior to participating in the activity. The University of Cincinnati requires disclosure to the learners of all relevant financial relationships and adheres strictly to the ACCME Standards for Commercial Support.
Alison M. Pack, MD, Associate Professor of Clinical Neurology, The Neurological Institute of New York, Columbia University Medical Center, New York, New York, has nothing to disclose.
Belinda Oyinkan Marquis, MD, a Clinical Neurophysiology Fellow (Epilepsy) at the State University of New York Downstate Medical Center, Brooklyn, New York, has nothing to disclose.
Sarah Aminoff Kelley, MD, a Pediatric Neurophysiology Fellow at Johns Hopkins Hospital, Baltimore, Maryland, has nothing to disclose.
Cynthia M. Correll, MD, a Neurophysiology Fellow at the Columbia Comprehensive Epilepsy Center, Columbia University Medical Center, New York, New York, has nothing to disclose.
Peter Pressman, MD, a Behavioral Neurology Fellow at the University of California, San Francisco, School of Medicine, San Francisco, California, has nothing to disclose.
Lisa Aenlle-Matusz, MD, MPH, a
Clinical Neurophysiology/Epilepsy Fellow at Emory University School of Medicine, Atlanta, Georgia, has nothing to disclose.
Rick Ricer, MD, has nothing to disclose.
Susan P. Tyler, MEd, CMP, CCMEP, has nothing to disclose.
Jacqueline Keenan and Edwin Geffner of Direct One Communications, Inc., have nothing to disclose.
DISCLAIMERThis activity is an independent educational activity under the direction of the University of Cincinnati. The activity was planned and implemented in accordance with the Essential Areas and Policies of the ACCME, the Ethical Opinions/Guidelines of the American Medical Association, the US Food and Drug Administration (FDA), the Office of Inspector General of the US Department of Health and Human Services, and the Pharmaceutical Research and Manufacturers of America Code on Interactions With Healthcare Professionals, thus assuring the highest degree of independence, fair balance, scientific rigor, and objectivity.
However, the planning committee, faculty, University of Cincinnati, Eisai Inc., and Direct One Communications, Inc. shall in no way be liable for the currency of information or for any errors, omissions, or inaccuracies in this activity. The opinions and recommendations presented herein are those of the faculty and do not necessarily reflect the views of the provider, producer, or grantors.
Participants in this activity are encouraged to refer to primary references or full prescribing information resources.
DISCLOSURE OF UNAPPROVED/OFF-LABEL USEDiscussions concerning drugs, dosages, devices, and procedures
may reflect the clinical experience of the planning committee or faculty, may be derived from the professional literature or other sources, or may suggest uses that are investigational and not approved labeling or indications.
In this issue of The Neurology Report, Dr. Kelley describes the investigational use of lacosamide for treating refractory status asthmaticus and acute repetitive seizures and the unapproved combination of valproate and lamotrigine for managing drop seizures in children. Dr. Correll discusses the unapproved use of eslicarbazepine acetate for carbamazepine-refractory seizures and the off-label use of pregabalin monotherapy for partial-onset seizures. Dr. Pressman describes the investigational use of lacosamide in adults with generalized seizures, children with focal and mixed focal-generalized seizures, and patients with status asthmaticus; summarizes emerging, unapproved uses for rufinamide and clobazam in both adults and children; and reviews the mechanisms of action, indications, major side effects, and drug interactions of two AEDs that have been approved for marketing in Europe but not in the US (stiripentol and eslicarbazepine acetate).
CONTACT INFORMATIONWe would like to hear your comments regarding this or other educational activities produced by Direct One Communications, Inc. In addition, suggestions for future activities are welcome. Contact us at:
Direct One Communications, Inc. 1424 Ridge Road Syosset, NY 11791 Phone: 516-364-1020 Fax: 516-364-4217 Website: www.CMEdirect.net
AboutThisCMEActivity

4 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
Introduction Selected Reports from the 66th Annual Meeting of the American Epilepsy SocietyAlison M. Pack, MD, Guest Editor
The Neurological Institute of New York, Columbia University Medical Center, New York, New York
Dr. Pack is Associate Professor of Clinical Neurology, The Neurological Institute of New York, Columbia University Medical Center, New York, New York.
Advances in antiepileptic drug (AED) therapy and dramatic surgical procedures have saved the lives of countless indi-
viduals who have been diagnosed with epilepsy. Despite this progress, however, too many adults and children continue to live with disabling seizures that disrupt their lives and make education, work, and leisure activities impossible or much more difficult than they ordinarily would be.
During the 66th Annual Meeting of the American Epilepsy Society, held November 30 to December 4, 2012, in San Diego, California, over 4,000 neu-rologists and medical professionals from around the globe participated in sym-posia, scientific exhibitions, lectures, poster and platform presentations, and course offerings that provided cutting-edge information on the prevention and treatment of epilepsy. During this meeting, five fellows in neurology at-tended scientific sessions and symposia to create the articles in this issue of The Neurology Report. These papers touch on many facets of epilepsy research, from the possible genetic basis of the disease to the advantages and disad-vantages of conventional therapies to the promise of newer-generation AEDs and drug combinations.
n FROM MOLECULES TO CELLS, NETWORKS, AND SEIZURES: HOW DOES A GENE CAUSE EPILEPSY?
Genetic predisposition is at the root of hundreds of medical conditions, and laboratory researchers have made great progress in discovering the mysteries
of the human genome and how they affect our health. Belinda Oyinkan Marquis, MD, of the State University of New York Downstate Medical Center, discusses classes of copy number vari-ants and their relationship to epilepsy development. In addition, she describes new techniques to analyze genetic varia-tions, specific mutations that have been linked to particular types of epilepsy, and methods to test genetic theories in vitro. Dr. Marquis details different types of ongoing genomic research, including projects being conducted by the Epi4K Consortium, a group that is focusing upon the genetics of epilepsy. Finally, this paper delves into ways that new information on genetics and the genome as they relate to epilepsy may influence treatment of the disease.
n ANTICONVULSANT TREATMENT OF EPILEPSY AND REFRACTORY STATUS EPILEPTICUS: RECENT CLINICAL TRIALS
Because multiple agents often are prescribed to relieve patients of epileptic symptoms, physicians try to select the most effective AEDs available that cause the fewest adverse reactions and drug interactions. Sarah Aminoff Kelley, MD, of Johns Hopkins Hospital, recounts the results of multiple clinical studies evaluat-ing older and newer AEDs in patients with seizures that were not easily controlled, intractable, or refractory. Dr. Kelley covers research on add-on lacosamide therapy for refractory status epilepticus and pro-vides an overview of common side effects of AEDs. Likewise, she describes drug interactions between AEDs and other
types of medications. Finally, Dr. Kelley addresses controversies related to the use of single or multiple AEDs, including the economic advantages of combination therapy, the efficacy of one combination in children with drop seizures, and the possibility of treating specific symptoms in patients with intractable partial-onset seizures.
n PHARMACOTHERAPY OF MEDICALLY REFRACTORY PARTIAL-ONSET EPILEPSY
Cynthia M. Correll, MD, of the Co-lumbia Comprehensive Epilepsy Center at Columbia University Medical Center, confronts options and recommendations for AED use in patients with refractory seizures. In her article, she describes sev-eral research presentations of studies on the safety and efficacy of the novel AEDs eslicarbazepine acetate, perampanel, lacosamide, pregabalin, rufinamide, and extended-release lamotrigine. The studies presented included patients with seizure disorders and mental depression, long-term outcomes of AEDs, the switching of immediate-release forms to extended-release drugs, polytherapy based on mechanism of action, and patient compli-ance to AED regimens.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 5
AlisonM.Pack,MD Introduction
n OPTIMAL USE OF THE NEWEST ANTIEPILEPTIC DRUGS AND GENERICS
The eight new AEDs approved by the US Food and Drug Administration (FDA) over the past 8 years feature dy-namic mechanisms that control seizures via different pathways. Peter Pressman, MD, of the University of California, San Francisco, School of Medicine, provides an overview of stiripentol, lacosamide, rufinamide, eslicarbazepine acetate, vigabatrin, ezogabine (retigabine), clo-bazam, and perampanel in terms of their mechanisms of action, pharmacokinetics, approved indications, off-label uses, use with other AEDs, drug interactions, and adverse effects. In addition, Dr. Pressman reflects on data from studies compar-
ing the bioequivalence and therapeutic equivalence of brand-name drugs with their generic versions.
n UPDATE ON RECENT GLOBAL STUDIES OF PERAMPANEL, A NEW SELECTIVE AMPA ANTAGONIST
Lisa Aenlle-Matusz, MD, MPH, pro-vides details on perampanel, a non-competitive, selective AMPA antagonist recently approved by the FDA as add-on therapy of partial-onset seizures with or without secondary generalized seizures in patients ≥ 12 years of age. Four research groups performed separate analyses on data from three placebo-controlled phase III studies of patients with partial-onset seizures with or without secondary generalized seizures to learn more about
responder rates and freedom from sei-zures in perampanel-treated and placebo groups, the effect of perampanel therapy on seizure frequency, the efficacy and safety of perampanel among North American patients, and the effect of the drug on quality of life. One other study using computer simulation and peram-panel pharmacokinetic data investigated how delayed or missed perampanel doses affect plasma drug levels.
The authors of this edition of The Neu-rology Report have contributed articles reflecting the breadth and intensity of current research on various issues related to the pharmacologic management of epilepsy. The future certainly holds great promise that these discoveries, if con-firmed, will improve the lives of countless patients with epilepsy.

6 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
The genetic spectrum of epilepsy is evolving at a breakneck pace, with scientists and clinicians continually reporting new find-
ings that may affect the everyday practice of neurologists who diagnose and treat patients with seizures. At a symposium held during the 66th Annual Meeting of the American Epilepsy Society, speakers addressed the process of gene discovery (ie, how mutations and chromosome deletions and duplications are identified in populations and single individuals), genetic variability (ie, how the same mutation can be responsible for differ-ent phenotypes), the management of patients with a genetically related seizure disorder, and the provision of specific information and counseling for patients diagnosed with certain mutations or chromosome defects.
n Gene Discovery in epilepsyBased on a presentation by Heather C. Mefford, MD, PhD, Assistant Professor of Pediatrics, Division of Genetic Medicine, University of Washington, and Attending Physician, Medical Genetics Clinic, Seattle Children’s Hospital, Seattle, Washington
New methods of genomic analysis have broadened our understanding of
the human genome. The emergence of genome-scanning technologies has un-covered an unexpectedly large portion of structural variations in the genome. These microscopic and submicroscopic variants include deletions, duplications, insertions, inversions, and transloca-tions, which are collectively known as copy number variants (CNVs).1 These CNVs are microdeletions or microdu-plications of segments of the genome that range from a few hundred base pairs to several hundred megabases (Mb). A change in copy number is revealed by comparing two or more genomes.1 CNVs play an important role in genetically complex epilepsies that can either be inherited or occur de novo.2
There are two classes of CNVs: recur-rent and nonrecurrent. Recurrent CNVs often are de novo mutations, individually more frequent, and the cause of some known microdeletions. Three large, recur-rent microdeletions at 15q13.3, 16p13.11, and 15q11.2 are each present in 0.5%–1% of patients with epilepsy.3 Microdeletions of 15q13.3 are associated with idiopathic generalized epilepsies (odds ratio, 68).4 These microdeletions are also established risk factors for related disorders, which
include intellectual disability, autism, and schizophrenia.5 Known medical conditions related to microdeletion dis-orders are Angelman, Prader-Willi, and Williams-Beuren syndromes.
However, rare, nonrecurrent CNVs are also important. Nonrecurrent CNVs occur throughout the genome and are not sequence-dependent. Large (> 2 Mb), rare CNVs, including the deletion at 16p13.11, are enriched in patients with diverse epilepsy syndromes.6 Rare CNVs also are found in 10% of patients with various types of epilepsy3 and 8% of patients with epileptic encephalopathies.7
LaboratoryMethodsThe development of new experimen-
tal and computational strategies, such as comparative genome hybridization (CGH) and single-nucleotide polymor-phism (SNP) arrays, has allowed human structural genetic variations to be ana-lyzed at a higher resolution.1
CGH, the most robust method for performing genome-wide scans to find novel CNVs, uses labeled fragments from a genome of interest (proband). These fragments are hybridized with a second differentially labeled genome to arrays
From Molecules to Cells, Networks, and Seizures: How Does a Gene Cause Epilepsy?Belinda Oyinkan Marquis, MDState University of New York Downstate Medical Center, Brooklyn, New York
Abstract The genetics of epilepsy are evolving rapidly, with each discovery potentially having therapeutic implications. The field has developed from the miracle of gene discovery to an understanding of the functional impact of genetic changes and variability to a new therapeutic, scientific, and clinical perspective of epilepsy. At a symposium held during the 66th Annual Meeting of the American Epilepsy Society, experts on the genetic road map of epilepsy and related medical conditions discussed the discovery of genes associated with different types of epilepsy, their variability, and their impact on clinical practice.
Dr. Marquis is a Clinical Neurophysiology Fellow (Epilepsy) at the State University of New York Downstate Medical Center, Brooklyn, New York.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 7
BelindaOyinkanMarquis,MD From Molecules to Cells, Networks, and Seizures: How Does a Gene Cause Epilepsy?
that are spotted with cloned DNA frag-ments to reveal copy number differences between the two genomes.1 CGH has revealed many more CNVs in humans than previously recognized.8
SNParraysare DNA arrays that de-tect polymorphisms within a population. An array contains the target nucleic acid sequence. One or more labeled allele-specific oligonucleotide (ASO) probes are applied, and a detection system that records and interprets the hybridization signal is used.9 SNP arrays are useful for detecting unilateral disomy and consan-guinity.
ModesofAnalysisCNV analysis provides new oppor-
tunities to examine the range of genetic variations associated with disease. They point to novel candidate genes that may be used for targeted sequencing in large epilepsy cohorts. Further testing with array CGH studies is warranted for pa-tients who have epileptic encephalopathy and “epilepsy plus” syndromes: epilepsy in addition to brain malformations, intellectual disability, autism spectrum disorder, or congenital anomalies.
Gene-sequencinganalysishas im-proved by significant leaps over the past 30 years. The classic Sanger sequencing method involves a DNA primer, DNA polymerase, and deoxynucleopeptides. Sample DNA is denatured and copied by polymerase chain reaction, sequenced, and then analyzed. However, this method is time-consuming and expensive.
The Sanger sequencing method has been supplanted by next-generation sequencing, which can analyze millions of fragments of DNA simultaneously at significantly less expense. This method captures exomes, which are the part of the genome formed by exons; these cod-ing portions of expressed genes provide the genetic blueprint used in the syn-thesis of proteins and other functional gene products. This information-rich extraction is then run through automated next-generation sequencing methods.
Gene discovery is accomplished using three clinical strategies: (1) trio analysis, (2) family analysis, and (3)
analysis of multiple, unrelated affected individuals (Figure 1). Trio analysis involves sequencing of the mother and father of an affected child to investigate a de novo mutation in the child. Family analysis involves sequencing selected individuals within a family to establish a pattern of inheritance (ie, recessive, dominant, X-linked). Analysis of mul-tiple unrelated affected individuals looks for mutations in the same gene.
Exome sequencing for diagno-sis is moving into the clinical setting rapidly. As the technology advances, its cost will continue to fall, and the technique will become more accessible. Next-generation gene panels currently
are available commercially to sequence many genes, although they are not all-inclusive. Therefore, clinicians who use these tools must focus on a known disease or set of genes. If a specific di-agnosis is suspected, it is recommended that clinicians investigate a particular gene (eg, SCN1A for Dravet syndrome, MECP2 for Rett syndrome). In cases of a nonspecific epilepsy-plus syndrome or multiple causative genes, CNV test-ing or gene-panel analysis should be considered. If an interesting phenotype and a supportive family history are involved, whole-exome sequencing can be considered if it is financially feasible (Figure 2).
Sequence multiple individuals
Look for mutations in the same gene across multiple
a�ected individuals
Sequence selected individuals
Use inheritance pattern
Recessive, dominant, X-linked pattern
Sequence mother + father + child
Look for de novo changein child
Severe, de novo disorders
Trio analysis Family analysis Multiple, unrelated,a�ected individuals
FiGUre 1 Three strategies for gene discovery. Adapted from a slide presented by Heather Mefford, MD, PhD.
Suspect a speci�c diagnosis?
Search for copynumber variants
Order a gene-panel analysis
Test for a speci�c geneDravet syndrome: SCN1A
Rett syndrome: MECP2
Sequence whole exome
Nonspeci�c, “epilepsy-plus” syndrome*
Many possiblecausative genes
No answersInteresting phenotype
Interesting familyGood insurance
* Epilepsy in addition to brain malformations, intellectual disability, autism spectrum disorder, or congenital anomalies
FiGUre 2 Pathways to a diagnosis using exome sequencing. Adapted from a slide presented by Heather Mefford, MD, PhD.

8 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
BelindaOyinkanMarquis,MD From Molecules to Cells, Networks, and Seizures: How Does a Gene Cause Epilepsy?
n How Do we Determine tHe FUnctional impact oF Genetic cHanGes?
Based on a presentation by Jack Parent, MD, Associate Professor of Neurology and Co-Director, Comprehensive Epilepsy Program, University of Michigan Medical School, Ann Arbor, Michigan
The growing advances in genetic sequencing broaden the opportunity to discover possible disease-causing muta-tions. The key elements to consider when evaluating suspected mutations and vari-ants include changes in an amino-acid se-quence made by the mutation, evolution-ary conservation of the altered sequence, and the effects on protein function or structure resulting from the variation.
After a gene is determined to be patho-genic, the next step is to determine how a particular mutation functions and leads to epilepsy. The effects of gene mutations on proteins are manifested in various ways. Function may be lost when a protein either does not engage in its native function or is degraded. This commonly occurs with autosomal-recessive or autosomal-dominant haploinsufficiency, in which quantitative re-duction of gene expression to 50% of normal levels results in an abnormality.10 Conversely, function may be gained when a normal novel protein has a de novo or “toxic” new func-tion. Finally, there are dominant-negative mutations, in which the mutant protein suppresses some other normal protein.
Dravet syndrome, or severe myoclonic encephalopathy of infancy (SMEI), is a prime example of mutations leading to epilepsy. This epileptic encephalopathy is related to early-onset seizures and is associated with cognitive impairments and 15% mortality by adolescence.11 Most often it is caused by a de novo loss of func-tion mutation in the neuronal sodium-channel gene SCN1A, which leads to haploinsufficiency of NaV1.1 channels.12 Voltage-gated sodium channels have criti-cal roles in the initiation and propagation of action potentials and are crucial regula-tors of neuronal excitability. Mutations in the NaV1.1 channel gene SCN1A cause genetically distinct epilepsy syndromes.
EvaluatingCauseandEffectThe functional effects of a mutation
must be tested by creating cell-culture models.
Heterologousexpressionsystemscan be created with the use of Xenopus oo-cytes, human embryonic kidney cells, or other progenitor cells. These systems are used to express mutant human genes and are easily studied using electrophysiologic methods. Further, they may be used for protein expression and targeting, enzyme assays, and defined protein interactions. However, heterologous expression does not mimic the effects in neurons or in vivo.13
Primary neuron cultures are ad-vantageous, because they allow affected neuron cells to be evaluated as they func-tion. However, they are technically more difficult to cultivate; their expression cannot be well controlled; and, without being in a native environment, the cells may change in culture.
Transgenicknockoutmousemod-els showed that mice with a heterozy-gous functioning Scn1a+/– gene (Dravet syndrome model) exhibited a reduced sodium current in interneurons.14 These mouse models were unique in that the phenotype of the Scn1a+/– mice pheno-copied human SMEI. The finding that haploinsufficiency of a sodium channel causes epilepsy was unsuspected, because reduced sodium current could lead to in-excitability rather than hyperexcitability. The theory of an alternate effect underly-ing the causation of epilepsy is based upon loss of sodium current in hippocampal inhibitory interneurons able to transmit or secrete γ-aminobutyric acid (GABA). Intractable epilepsy could result from failure in the excitability of hippocampal GABAergic inhibitory neurons.14
There are, however, limitations to the value of transgenic mouse models. Murine models do not harbor a patient’s other genetic variations, and there can be vari-ability in the breeding of transgenic mice. In addition, gene expression may vary among different lines of transgenic mice.
Induced pluripotent stem cell(iPSC)technologyoffers enormous po-tential for understanding disease states by creating models of the genetic milieu on a microscopic level. The technique involves
direct reprogramming of somatic cells to a stem cell (pluripotent state). The iPSCs can then be modified genetically and differentiated into relevant tissue to be examined for disease mechanisms or to screen for new therapies.
Dravet syndrome can be better under-stood with iPSCs. As an example, imagine two subjects with Dravet syndrome—one harbored an SCN1A splice site muta-tion and the other, an SCN1A nonsense mutation. After iPSCs derived from Dravet syndrome patient fibroblasts were compared with those of controls, Dravet iPSC neurons showed increased sodium current and increased excitability, firing repetitively and bursting spontaneously.
iPSCs also have been created to study sudden unexplained death of epileptic patients (SUDEP). Ion channelopathies likely predispose patients to SUDEP.11 Individuals with Dravet syndrome and generalized epilepsy with fever symptoms plus another SCN1A channelopathy have a high incidence of SUDEP. The Nav1.1 subunit is expressed in cardiac myocytes. In work not yet published, Dravet syndrome patient fibroblasts were reprogrammed to iPSCs and then modi-fied and differentiated to become cardiac myocytes; the same was performed with controls. Cardiac myocytes derived from the iPSCs of controls displayed orga-nized and uniform contractility. Cardiac myocytes derived from Dravet syndrome iPSCs displayed faster and irregular myo-cyte contraction. Electrophysiologic stud-ies showed increased sodium currents in cardiac myocytes from Dravet syndrome patients and mutant mice, and the altered sodium currents affected cardiomyocyte potential, leading to an arrhythmogenic state.
However, iPSCs have limitations. Although the cells may be genetically manipulated for reprogramming to avoid genetic integration, learning how to make regional and subtype-specific neurons is still a challenge, since incomplete matura-tion and abnormal growth may result. As-sessing iPSC-derived neurons in vivo by transplanting iPSC neurons from Dravet syndrome patients and controls into a rat hippocampus and then taking brain slices

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 9
BelindaOyinkanMarquis,MD From Molecules to Cells, Networks, and Seizures: How Does a Gene Cause Epilepsy?
for hippocampal recording in a natural environment may overcome this problem.
With cell culture and animal models, the key is to identify and confirm potential epilepsy-causing mutations. The patient-specific models of epilepsy mutations provide abundant knowledge and allow future researchers to set a new goal of creating personalized therapy. With mul-tiple models of genetic epilepsies, it may be possible to perform drug screening, to test existing therapies, and to analyze new therapies and toxicities in the future.
n wHat can we learn aboUt epilepsy From Genome seqUences?
Based on a presentation by David Goldstein, PhD, Richard and Pat Johnson Distinguished University Professor, Director of the Center for Human Genome Variation, and Professor of Molecular Genetics and Microbiology and Biology, Duke University School of Medicine, Durham, North Carolina
Genetic mechanisms that influence the development of epilepsy in the vast major-ity of patients remain unknown.
GeneIdentificationinComplexEpilepsies
Historically, gene-identification stud-ies have been performed using linkage studies and association approaches.
Linkagestudiesdraw upon genetic and phenotypic information from mul-tiple generations to map regions of the genome that are co-inherited with familial disease. Regions of interest from linkage studies tend to be relatively large due to the few opportunities for recombination within the families investigated.
Associationstudiesinvestigate vari-ants at individual loci that occur more commonly in unrelated patients than controls. Different unrelated individuals with a unique recombination history are evaluated; therefore, the size of the as-sociated region theoretically should be much smaller, and specific susceptibility genes may be more easily identified than possible with a linkage study. To identify specific susceptibility genes with a par-ticular linkage, linkage studies are often followed by association mapping.15
Candidate-geneassociationstudiesare the next step in gene identification.
These studies focus on genes likely to be involved in disease pathophysiology. Candidate-gene studies in patients with epilepsy have been less than successful, partially because of inadequate knowledge of disease pathophysiology needed to select candidate genes.
As a result, genome-wide asso-ciation studies (GWAS) have been employed to understand variants. GWAS use genotyping chips to “tag” variants that may be common in a population.16 GWAS have been useful for identifying secure risk factors for common diseases.
However, GWAS have failed to identify many variants of large effect in complex diseases such as epilepsy. Consequently, researchers have a blossoming interest in the possibility that variants too rare to
be represented well on GWAS chips may be important contributors to common diseases, including the epilepsies.
This has led to considerable interest in the use of next-generation sequencing, which allows for the near-comprehensive characterization of genetic variants across the genome to identify rare risk factors for disease.17 The rare-variant hypothesis is supported by the recent observation that rare CNVs can have large effects on neu-ropsychiatric disease risk.18 This observa-tion, along with the fact that only 9% of variants reported as disease-associated in the Human Gene Mutation Database are CNVs,19 strongly suggests that other types of rare variants, such as single-nucleotide, insertion, and/or deletion variants, may be important contributors to complex diseases. Next-generation sequencing has
great potential to identify susceptibility genes for complex diseases; however, large sample sizes will be required, because lo-cus and allelic heterogeneity are predicted to be high, and all genomes carry a large number of rare functional variants.20
TheEpi4KConsortiumThe National Institute of Neurologi-
cal Disorders and Stroke (NINDS) has recognized the importance of accelerating progress in this area of epilepsy research. In 2010, the NINDS issued a Funding Opportunity Announcement for the cre-ation of the Epi4K Consortium,21 a “center without walls” to focus on the genetics of human epilepsy. This collaborative study aims to sequence at least 4,000 subjects with epilepsy. The geographically bound-less structure consists of three cores and four scientific projects, as well as a steering committee made up of primary investiga-tors and representatives from NINDS. Three projects will analyze specific sets of epilepsy cohorts (namely, epileptic encephalopathies, multiplex families and pairs, and prognosis) drawn from seven large-scale genetic studies conducted around the world, and one will apply cutting-edge analytic techniques related to the detection of CNVs.
Project1of Epi4K addresses the ge-netics of epileptic encephalopathies that are refractory to medication and associ-ated with comorbid cognitive dysfunction and behavioral disturbances. The two more common types are infantile spasm and Lennox-Gastaut syndrome.
Infantile spasm is the most common of the epileptic encephalopathies, occurring in 1 in 3,000 live births and becoming noticeable at 4–12 months of age.22 This syndrome is associated with the character-istic electroencephalographic pattern of hypsarrhythmia, which is the sine qua non of the syndrome. Spasms are associated with an electrodecremental response. Ap-proximately 50%–60% of infantile spasm cases are associated with developmental brain malformations, tuberous sclerosis complex, chromosomal syndromes, or metabolic conditions. Many patients with infantile spasm or other early-onset epileptic encephalopathies may experi-
Rare variants, such as single-nucleotide, insertion, and/or deletion variants, may be important contributors to complex diseases such as epilepsy.

10 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
BelindaOyinkanMarquis,MD From Molecules to Cells, Networks, and Seizures: How Does a Gene Cause Epilepsy?
ence an evolution to Lennox-Gastaut syndrome, suggesting a likely shared etiology.23
Lennox-Gastaut syndrome typically is noticed at 1–8 years of age and is char-acterized by mixed seizure types (tonic, atonic, myoclonic, atypical absence, focal, and generalized) and intellectual disabilities.24 Its cause is unknown in about 25%–35% of cases; the others are symptomatic of structural or metabolic abnormalities.
Participants in Project 1 are being contributed by the Epilepsy Phenome/Genome Project (EPGP),25 an actively en-rolling, 5-year, NINDS-funded study that will involve 5,250 members of families with epilepsy. Participants will undergo detailed phenotyping and analysis of medical records, electroencephalography, and magnetic resonance imaging. All Project 1 samples are from the infantile spasm/Lennox-Gastaut syndrome arm of the EPGP.
In preliminary results obtained from whole exome sequencing of 165 trios (75 infantile spasm and 90 Lennox-Gastaut syndrome probands and parents) to iden-tify new genes and pathways conferring risk of epileptic encephalopathies, there were 180 de novo mutations enriched in specific gene sets. These mutations in-cluded 14 causal de novo mutations in six known genes for epileptic encephalopa-thy (SCN1A, SCN8A, STXBP1, SCN2A, CDKL5, and KCNQ2). Of these 14 muta-tions, 13 were newly identified as causal.
Projects 2–4.The objective of Proj-ect 2 is to identify genomic variations that influence risk for common forms of idiopathic generalized epilepsies and non-lesional focal epilepsies found in families with two or more affected members. The objective of Project 3 is to explore the re-lationship between genetic variation and prognosis. The Project 4 objective is to apply novel computational algorithms to next-generation sequencing data to detect epilepsy-associated CNVs.
The goal of the Epi4K Consortium is to emphasize careful phenotyping of subjects and to increase accessibility to next-gener-ation sequencing, state of-the-art methods for genomic analysis, and the advantages
of large-scale collaborations. If successful, this collaborative process will advance the identification of the causes of many forms of so-called idiopathic and cryptogenic epilepsy, which will drive the development of drugs used to treat epilepsy and, pos-sibly, disease-modifying or antiepilepto-genic agents. This study also opens doors for understanding the biologic bases of related genetic risks and assessing their translational implications.15
n How can Genetic inFormation impact manaGement?
Based on a presentation by Samuel Berkovic, MD, FRS, Laureate Professor of Medicine, University of Melbourne, Melbourne, Victoria, Australia
The genetic understanding of epi-lepsy causation continues to expand with continued advances in technology and research. Clinicians must now learn to apply the information and decide how it will influence the management of patients with epilepsy.
Traditionally, neurologists have be-lieved that after congenital cortical mal-formations, trauma, and stroke, idiopathic forces are the cause of approximately 75% of epilepsy cases.26 Until recently, familial aggregation studies, twin studies, and multiplex family studies have been the sources of gene discovery related to epilepsy.
Familialaggregationstudiescom-pare the frequency of a disease in par-ticular relatives to controls and determine the recurrence risk ratio. The overall recurrence risk ratio for epilepsy is 2.5 in first-degree relatives, 4–9 for generalized epilepsy, 2–3 for partial epilepsy, and 3–5 for febrile seizures, suggesting that there is a large degree of genetic inheritance in particular for generalized epilepsies.
Twin studies support this premise. Vadlamudi et al27 evaluated a cohort of twins to examine the genetics of epilepsy syndromes. In all, 418 twin pairs were studied, with one or both of each pair reporting seizures. More monozygous pairs were concordant for seizures than were dizygous pairs. When analyzed according to epilepsy syndrome, the case-wise concordances for generalized
epilepsies (99 patients; monozygous, 0.73; dizygous, 0.33) were greater than were those for partial epilepsies (103 patients; monozygous, 0.34; dizygous, 0.04), with intermediate values for febrile seizures (180 patients; monozygous, 0.60; dizygous, 0.14) and unclassified epilepsies (36 patients; monozygous, 0.43; dizygous, 0.13). The investigators concluded that genetic factors are particularly important in the generalized epilepsies, but they also play a role in the partial epilepsies. In ad-dition, the high frequency of concordant monozygous pairs having the same major syndrome strongly suggests there are syndrome-specific genetic determinants, rather than a broad genetic predisposition, to seizures.28
Multiplex family studiesselect for familial units having two or more af-fected members. Many gene discoveries, such as autosomal-dominant nocturnal frontal-lobe epilepsy, have resulted from these studies.
PracticalUseofTheseDiscoveriesThe growing precision in gene identi-
fication has advanced the understanding of epilepsy causation. Epilepsies formerly broadly classified as being idiopathic in nature now are being identified as genetic in origin.
Monogenic epilepsies often are related to dominant inheritance and can display genetic heterogeneity with pleiotropic expression. In other words, one gene can influence many phenotypic traits. Such genes can cause ion-channel subunit ab-normalities, such as sodium or potassium voltage-gated channels or ligand-gated channels (eg, nicotinic or GABA-asso-ciated channels). In addition, causative monogenic non–ion-channel genes have been identified. Some examples are the LGI1 gene associated with temporal lobe epilepsy, the GLUT1 gene associated with early-onset absence seizures, and the PRRT2 gene associated with a spectrum of neurologic disorders, including benign familial infantile epilepsy, febrile seizures, childhood absence seizures, paroxysmal dyskinesias, migraines, and hemiplegic migraines.29 Complex epilepsies have been associated with common variants

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 11
BelindaOyinkanMarquis,MD From Molecules to Cells, Networks, and Seizures: How Does a Gene Cause Epilepsy?
and rare variants associated with CNVs.De novo mutagenesis has unexpectedly
shown significant importance in various epilepsies, particularly Dravet syndrome. On the basis of a study of monozygotic twins showing discordant inheritance of SCN1A,30,31 the timing of de novo muta-genesis is linked to parental germ-line mutations, postzygotic mutations, and mosaicism. In this study, most cases of the de novo mutation occurred in the parental germ line, giving rise to a mutated sperm or egg. Mutations in SCN1A occur more frequently in the male germ line. In one set of monozygotic twins, the proband displayed the SCN1A mutation in multiple cell lines, and the twin was unaffected; this suggested that mutagenesis occurred
early in postzygotic development at the two cell–premorula stage.
Mosaicism refers to the presence of two different cell lines after fertilization, which indicates the timing of mutagen-esis. Somatic mosaicism occurs in non–sex cells, whereas germline mosaicism occurs in gamete-forming cells. In Dravet syndrome, somatic mosaicism is found when mildly affected parents have one or more affected offspring. Germ-line mosaicism is inferred in cases of unaf-fected parents having multiple affected offspring.27 These are important identifi-cations for genetic counseling. Gametal mutations are associated with negligible recurrence risk, whereas a germ-line mosaicism is associated with a high risk
of recurrence (Figure 3).27,32–38
The expanding role of genetics in the daily practice of neurology and manage-ment of epilepsy patients exemplifies how important it is for neurologists to improve genetic literacy. The initial step is to obtain a family history and to make a precise diagnosis of a clinical syndrome. This information will serve as a foundation for choosing appropriate testing. Clinicians should consider performing high-yield tests in the appropriate setting when faced with a particular clinical presentation. For example, a patient presenting with epi-lepsy plus multiple comorbid syndromic features, such as intellectual disability, autistic spectrum disorder, congenital anomalies, or brain malformations, may
FiGUre 3 Timing of mutations in Dravet syndrome. Gray boxes show the timing of mutations, and green boxes describe evidence from twin and family studies. Most cases of this syndrome are believed to be due to de novo mutations in gametes (usually paternal31), although in non-twin patients with the syndrome, postzygotic mutation cannot be ruled out. Somatic mosaicism has been described in mildly affected parents (father or mother) with a child or children with Dravet syndrome.32–34 Germ-line mosaicism in fathers occurs in the adult testis, whereas germ-line mosaicism in mothers (not shown) occurs in early development, because oocytes are mature by 5 months of fetal life. Germ-line mosaicism is inferred when a healthy parent has multiple affected children.32,34–37 Reproduced, with permission, from Vadlamudi et al.27
Premorula mutation
Frequency in Dravet syndromeunknown; discordant
monozygous twins
Germ-line(gametal) mutation
Most cases of Dravet syndrome;concordant monozygous twins
Germ-line mosaicism(male)
Una�ected parent;multiple a�ected children
Somatic mosaicism
Mildly a�ected parent;child with Dravet syndrome

12 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
BelindaOyinkanMarquis,MD From Molecules to Cells, Networks, and Seizures: How Does a Gene Cause Epilepsy?
be tested with CGH and SNP arrays for CNVs. In patients with a clinical presen-tation in adolescence or adulthood that suggests GLUT1 encephalopathy, early-onset absence epilepsy, or epilepsy and paroxysmal exercise-induced dyskinesias, SLC2A1 gene sequencing should be con-sidered. A confirmed diagnosis can be treated with a ketogenic diet. In children presenting with signs and symptoms con-sistent with Dravet syndrome, sodium-channel blockers should be avoided to prevent exacerbation of seizures.
Dravet syndrome should also be con-sidered in adults with a suggestive history; an abnormal gait can be a helpful diagnos-tic clue in an undiagnosed patient. A study by Rodda et al38 showed that children with Dravet syndrome show progressive gait deterioration in the second decade of life; they tend to have a crouch gait and skeletal malalignment.39 In families har-boring a female inheritance of syndromes suggestive of Dravet syndrome that do not feature an SCN1A mutation, testing for a PCDH19 mutation may be in order. Mutations in PCDH19 (which encodes protocadherin 19 on the X chromosome) were identified as epilepsy in females with mental retardation (EFMR) or a Dravet-like phenotype. In this case, heterozygous females are affected, whereas hemizygous males are spared.40
In daily practice, genetic diagnoses aid in determining causation and thereby avoid unnecessary testing. The results, however, may alter treatment, as in indi-viduals having GLUT1 or SCN1A muta-tions. Diagnosis is essential for counseling and for discussing the risk of recurrent de novo mutations or other unusual inheri-tance patterns.
n tHe promise oF epilepsy Genetics: a personal anD scientiFic perspective
Based on a presentation by Tracy Dixon-Salazar, PhD, Postdoctoral Fellow, University of California at San Diego, San Diego, California
Gene identification using exome se-quencing may be studied by evaluating consanguineous families or small fami-lies with many linkage peaks. Via such analysis, genomic variants are filtered
and prioritized to show gene mutations relevant to disease causation (Figure 4).
Next-generation exome sequencing has moved from the realm of research to the clinic. It will have a significant impact on diagnosis and management, because greater knowledge of genetic disease may reduce costs, advance accuracy, and point to unsuspected, yet treatable, conditions. This hypothesis was studied by Dixon-Salazar et al,41 who used whole-exome sequencing in 118 patients diagnosed with pediatric-onset neurodevelop-mental disease; most known causes had been excluded. The authors identified 22 previously unidentified disease-causing genes (19% of the cohort) and further established exome sequencing as a useful tool for gene discovery. Exome sequenc-ing uncovered 10 probands (8% of the cohort) having mutations in genes that were associated with a disease that was dif-ferent from that initially diagnosed. Thus, exome sequencing was shown to yield a correct diagnosis. Upon further medical evaluation, these mutations accounted for each proband’s disease, leading to a change in diagnosis and some associated changes in patient management. These results provided evidence that genomic strategies may clarify the diagnosis of certain patients with neurodevelopmental disorders.
An example of a potentially treatable neurodevelopmental disorder that may be treated following gene identification is the BCKDK mutation, which is as-sociated with autism. Novarino et al42 have identified inactivating mutations in BCKDK in consanguineous families with autism, epilepsy, and intellectual disabil-ity. The encoded protein is responsible for
phosphorylation-mediated inactivation of the E1α subunit of BCKDK. Patients with homozygous BCKDK mutations display reductions in BCKDK messenger RNA and protein, E1α phosphorylation, and plasma branched-chain amino acids. Bckdk knockout mice show abnormal brain amino acid profiles and neurobe-havioral deficits that respond to dietary supplementation. These findings suggest that autism presenting with intellectual disability and epilepsy caused by BCKDK mutations may represent a potentially treatable syndrome.
An unpublished case study of a 19-year-old female with Lennox-Gastaut syndrome of unknown cause is a fitting example of another treatable case. She was diagnosed at 32 months of age, had been experiencing 100–250 seizures per month, and was unresponsive to over 26 trials of antiepileptic therapy. Exome-sequencing analysis of this patient identified numerous high-impact vari-ants in calcium-channel subunits. The most common genetic variant of L-type calcium channels is gain of function; therefore, the receptor may have allowed for increased calcium influx that led to increased calcium at the synapse and increased neurotransmitter release. This theory was supported by increased seizure frequency with calcium supplementation. After weighing the risk and benefits, in-vestigators evaluated the use of off-label verapamil, a calcium-channel blocker, in this patient. Administration of verapamil reduced seizure frequency by 75%–80%. Interestingly, this patient was the daughter of the presenter and the leading example of the implications of exome sequencing on epilepsy management.
FiGUre 4 Exome sequencing to evaluate consanguineous families or small families with many linkage peaks. Adapted from a slide presented by Tracy Dixon-Salazar, PhD.
Prio
ritiz
eFi
lter Variants
identi�ed
Indels, splicecoding, non-synonymous
Homozygousvariants
Genotype notin controls
Variantsin linkageintervals
Type ofmutation
Conservationacross species
Proteindamageprediction
Relevanceto disease
Sequenceexome
MappingVariant calling
Variant�ltering
Variantprioritization

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 13
BelindaOyinkanMarquis,MD From Molecules to Cells, Networks, and Seizures: How Does a Gene Cause Epilepsy?
Exome sequencing has great potential to allow greater understanding of novel disease-causing genes in genetically en-riched families, identification of known causes of disease, correction of diagnosis and prognosis, and direction of treatment. Exome sequencing will change the con-ceptualization of epilepsy. Soon, analysis of unique de novo mutations and genes in patients with epilepsy is expected to be the rule rather than the exception. With relevant disease-causing genes, the pos-sibility of repurposing a drug or designing a novel drug to treat a specific gene defect is plausible. This new paradigm for diag-nosis and management opens the doors for the personalized treatment of patients with epilepsy.
RefeRences
1. Feuk L, Carson A, Scherer S. Structural varia-tion in the human genome. Nature. 2006;7:85–97.
2. Scheffer IE, Berkovic SF. Copy number vari-ants—an unexpected risk factor for the idiopathic generalized epilepsies. Brain. 2010:133:7–8.
3. Mefford HC, Muhle H, Ostertag P, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962.
4. Dibbens LM, Mullen S, Helbig I, et al. Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for dis-orders with complex inheritance. Hum Mol Genet. 2009;18:3626–3631.
5. Mefford HC, Mulley JC. Genetically complex epilepsies, copy number variants and syndrome constellations. Genome Med. 2010;2:71.
6. Heinzen EL, Radtke RA, Urban TJ, et al. Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet. 2010;86:707–718.
7. Mefford HC, Yendle SC, Hsu C, et al. Rare copy number variants are an important cause of epileptic encephalopathies. Ann Neurol. 2011;70:974–985.
8. Mills RE, Walter K, Stewart C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65.
9. Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311.
10. Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disor-ders. J Clin Invest. 2005;115:2010–2017.
11. Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia. 2011;52:44–49.
12. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mu-tations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–1332.
13. Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia. 2010;51:1650–1658.
14. Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneu-rons in a mouse model of severe myoclonic epilepsy in infancy. Nature. 2006;9:1142–1149.
15. Cardon LR, Bell JI. Association study designs for complex diseases. Nat Rev Genet. 2001;2:91–99.
16. Barrett JC, Cardon LR. Evaluating cover-age of genome-wide association studies. Nat Genet. 2006;38:659–662.
17. Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425.
18. Need AC, Goldstein DB. Whole genome association studies in complex diseases: where do we stand? Dialogues Clin Neurosci. 2010;12:37–46.
19. Stenson PD, Mort M, Ball EV, et al. The Hu-man Gene Mutation Database: 2008 update. Genome Med. 2009;1:13.
20. Pelak K, Shianna KV, Ge D, et al. The char-acterization of twenty sequenced human genomes. PLoS Genet. 2010;6:e1001111.
21. The Epi4K Consortium. Epi4K: gene discov-ery in 4,000 genomes. Epilepsia. 2012;53:1457–1467.
22. Frost FD, Hrachovy RA. Infantile Spasms. Boston, MA; Kluwer Academic Publishers; 2003.
23. Osborne JP, Lux AL, Edwards SW, et al. The underlying etiology of infantile spasms (West syndrome): information from the United King-dom Infantile Spasms Study (UKISS) on contem-porary causes and their classification. Epilepsia. 2010;51:2168–2174.
24. Camfield PR. Definition and natural history of Lennox-Gastaut syndrome. Epilepsia. 2011:52:3–9.
25. Epilepsy Phenome/Genome Project. 2007. http://www.epgp.org. Accessed December 11, 2012.
26. Hauser WA, Kurland LT. The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967. Epilepsia. 1975;16:1–66.
27. Vadlamudi L, Dibbens LM, Lawrence KM, et al. Timing of de novo mutagenesis—a twin study of sodium-channel mutations. N Engl J Med. 2010;363:1335–1340.
28. Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy
syndromes. Ann Neurol. 1998;43:435-445.29. Marini C, Conti V, Mei D, et al. PRRT2
mutations in familial infantile seizures, paroxysmal dyskinesia, and hemiplegic migraine. Neurology. 2012;79:2109–2114.
30. Arnheim N, Calabrese P. Understand-ing what determines the frequency and pattern of human germline mutations. Nat Rev Genet. 2009;10:478–488.
31. Heron SE, Scheffer IE, Iona X, et al. De novo SCN1A mutations in Dravet syndrome and related epileptic encephalopathies are largely of paternal origin. J Med Genet. 2010;47:137–141.
32. Gennaro E, Santorelli FM, Bertini E, et al. Somatic and germline mosaicisms in severe myoclonic epilepsy of infancy. Biochem Biophys Res Commun. 2006;341:489–493
33. Marini C, Mei D, Cross JH, Guerrini R. Mo-saic SCN1A mutation in familial severe myoclonic epilepsy of infancy. Epilepsia. 2006;47:1737–1740.
34. Marini C, Scheffer IE, Nabbout R, et al. SCN1A duplications and deletions detected in Dra-vet syndrome: implications for molecular diagnosis. Epilepsia. 2009;50:1670–1678.
35. Depienne C, Arzimanoglou A, Trouillard O, et al. Parental mosaicism can cause recurrent transmission of SCN1A mutations associated with severe myoclonic epilepsy of infancy. Hum Mutat. 2006;27:389–398.
36. Morimoto M, Mazaki E, Nishimura A, et al. SCN1A mutation mosaicism in a family with severe myoclonic epilepsy in infancy. Epilepsia. 2006;47:1732–1736.
37. Selmer KK, Eriksson AS, Brandal K, Egeland T, Tallaksen C, Undlien DE. Parental SCN1A muta-tion mosaicism in familial Dravet syndrome. Clin Genet. 2009;76:398–403.
38. Rodda JM, Scheffer IE, McMahon JM, Berkovic SF, Graham HK. Progressive gait deterio-ration in adolescents with Dravet syndrome. Arch Neurol. 2012;69:873–878.
39. Jansen FE, Sadleir LG, Harkin LA, et al. Severe myoclonic epilepsy of infancy (Dravet syndrome): recognition and diagnosis in adults. Neurology. 2006;67:2224–2226.
40. Genes in infantile epileptic encephalopa-thies. In: Depienne C, Gourfinkel-An I, Baulac S, et al, eds. Jasper’s Basic Mechanisms of the Epilepsies. 4th ed. Bethesda, MD: National Center for Biotech-nology Information; 2012.
41. Dixon-Salazar TJ, Silhavy JL, Udpa N, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012;4:138ra78.
42. Novarino G, El-Fishawy P, Kayserili H, et al. Mutations in BCKD-kinase lead to a poten-tially treatable form of autism with epilepsy. Science. 2012;338:394–397.

14 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
Currently, there are over 20 an-ticonvulsants available to treat seizures and epilepsy.1 Despite the fact that newer medications
are being developed with potentially higher efficacy, better safety profiles, and even novel mechanisms of action, up to one third of patients are still considered to have intractable epilepsy.2 These patients incur tremendous healthcare expenses, suffer lost wages, and carry many comor-bidities, such as depression and anxiety, that affect their day-to-day activities. Furthermore, they suffer both the effects of their disease and the side effects of their medication, which include acute adverse reactions, such as sedation, dizziness, and irritability, and potentially long-term reactions that may lead to decreased bone mineralization or liver injury.
Additionally, there are as many as 200,000 people in the United States alone who suffer from status epilepticus each year.3 Status epilepticus leads to mortality in up to 20% of cases,4 and up to 40% of af-fected patients are refractory to a standard initial treatment protocol that includes use of benzodiazepines and phenytoin
or fosphenytoin.5 These patients suffer from many iatrogenic complications of therapy, such as hypotension and infec-tion, and experience great physical costs.6 Those who survive often have significant neurologic sequelae, which greatly impact their ability to work, care for themselves, and function as active members of society.
During the 66th Annual Meeting of the American Epilepsy Society held in San Diego, California, investigators presented studies that explored numerous aspects of anticonvulsant use among individuals with easily controlled epilepsy, intractable epilepsy, and refractory status epilepticus (RSE). They addressed hypotheses regard-ing how both old and new anticonvulsants can be used more effectively to treat sei-zures and RSE, as well as safety concerns related to the use of these medications.
n LACOSAMIDE FOR THE TREATMENT OF REFRACTORY STATUS EPILEPTICUS
Because RSE causes significant mor-bidity and mortality, aggressive manage-ment is warranted. When the standard protocol of a benzodiazepine given with
one or two additional anticonvulsants fails or the seizures continue for a prolonged period, they are considered refractory.5
A patient with refractory seizures has no clear next best choice for treatment. Some older agents, such as valproic acid and phenobarbital, are used frequently, and newly available anticonvulsants, such as levetiracetam7 and even enteral (via nasogastric tube) topiramate,8 often are tried. Alternatively, anesthetics or higher doses of barbiturates and benzodiazepines may be used, although they have many associated iatrogenic side effects.
Lacosamide is a relatively new anticon-vulsant that was approved for marketing by the European Commission and the US Food and Drug Administration for adjunctive treatment of partial epilepsy in 2008.9 Lacosamide has a novel mechanism of action that enhances slow inactivation of the sodium channel without affecting the fast activity.9 It has shown similar ef-ficacy in both parenteral and oral forms without causing many of the sedating and cardiovascular effects of other parenteral options,10 and it has few drug-drug in-teractions.11 These characteristics make lacosamide an ideal medication for treat-ment of the critically ill patient.
Anticonvulsant Treatment of Epilepsy and Refractory Status Epilepticus: Recent Clinical TrialsSarah Aminoff Kelley, MDJohns Hopkins Hospital, Baltimore, Maryland
Abstract Choosing the most effective anticonvulsant that causes the fewest adverse effects for a patient with epilepsy or status epilepticus is an ongoing challenge. In addition, even with the regulatory approval of many new anticon-vulsants, the goal of seizure freedom for all patients has not been achieved. At the 66th Annual Meeting of the American Epilepsy Society, clinical researchers presented the results of numerous clinical trials of older and new antiepileptic agents. Specifically, they addressed the use of lacosamide for treating refrac-tory status epilepticus, drug-drug interactions and side effects related to the use of antiepileptic drugs, and comparisons of monotherapy versus the use of combination regimens.
Dr. Kelley is a Pediatric Neurophysiology Fellow at Johns Hopkins Hospital, Baltimore, Maryland.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 15
SarahAminoffKelley,MD Anticonvulsant Treatment of Epilepsy and Refractory Status Epilepticus
ClinicalTrialsofLacosamide
Prior studies evaluating the use of lacosamide in patients with RSE demon-strate a lack of data to support its efficacy and safety in this clinical setting.12 A case series in a small sample of patients (n = 9) found no evidence that lacosamide was effective for the treatment of RSE. Using the authors’ predefined criteria, no subject responded to lacosamide treat-ment.13 In contrast, data from a study of 34 RSE patients suggested that lacosamide may be effective and safe.14 The authors demonstrated seizure cessation in about 65% of patients, with half experiencing improvement < 12 hours after taking an average dose of approximately 350 mg. No lacosamide-treated subject had an adverse event. Similarly, other previously published small retrospective studies demonstrated the efficacy and safety of lacosamide given in the intensive care set-ting for acute repetitive seizures,15 as well as convulsive and nonconvulsive status epilepticus.16–18
Several studies presented at the 2012 Annual Meeting of the American Epilepsy Society looked at larger numbers of RSE patients treated with lacosamide. They presented their data on efficacy and safety.
Add-onparenterallacosamide.Sut-ter and colleagues19 evaluated the safety and efficacy of parenteral lacosamide given as an add-on treatment for RSE. This study is one of the largest reported to date; it also is the only controlled clinical trial using this drug. The authors compared results from consecutive lacosamide-treated patients observed from January 2005 through December 2011 with those of historical controls who were treated prior to 2005.
In all, 111 patients were studied; 59 were given lacosamide. These patients were similar to controls in all aspects ex-cept that controls were slightly older and more likely to have suffered nonconvulsive status epilepticus or stroke. Patients given lacosamide were more likely to experience earlier termination of their RSE and less likely to die. In fact, approximately 40% experienced immediate cessation of their seizures after lacosamide treatment; this
also led to decreased use of anesthetic medications and a potential decrease in overall morbidity and mortality.
No serious adverse outcomes were as-sociated with using lacosamide. The only significant differences in care between the two groups of subjects were the addition of the drug being studied, increased use of topiramate, and more frequent continu-ous electroencephalographic (EEG) mon-itoring. When patients given topiramate were removed from the analysis, however, no difference was found. Whether or not continuous EEG monitoring played a role in decreased mortality in this study is unknown. The role of continuous EEG monitoring in the critically ill patient continues to be investigated.
Use in children and adults. Alam and colleagues20 examined the use of lacosamide in 178 patients between 6 and 90 years of age who received lacosamide for treatment of status epilepticus or acute repetitive seizures. The majority of the patients were treated for nonconvulsive status epilepticus. All patients were moni-tored with continuous video EEG. All but 42 patients were excluded; some patients were excluded for prior lacosamide use, and 7 patients had post-anoxic brain inju-ry and were later separated out due to their complete lack of response to lacosamide. Of the remaining 35 patients, 57% experi-enced seizure termination after receiving a median initial 150-mg dose of lacosamide. Some individuals required subsequent ti-tration. In this study, lacosamide was most often the third- or fourth-line medication used. Other anticonvulsants most com-monly tried > 50% of the time included levetiracetam, phenytoin, lorazepam, and midazolam. No patient experienced a seri-ous adverse event.
Newey and Hantus21 retrospectively looked at 84 adult patients who began seizing after being placed on continu-ous EEG monitoring and subsequently progressed to status epilepticus and lacosamide treatment. More than half of these patients suffered from noncon-vulsive status epilepticus. Wide-ranging etiologies, including stroke, tumor, and epilepsy, were studied. The patients were naïve to lacosamide therapy. After treat-
ment with two other anticonvulsants (most often levetiracetam and phenytoin) failed to improve their symptoms, 15.7% of patients responded within 4 hours of using lacosamide, and 82% experienced relief by 48 hours after lacosamide was given. No adverse events were noted in relation to blood pressure, liver or kidney function, or PR interval.
These retrospective studies all dem-onstrated the efficacy and safety of lacos-amide for treatment of convulsive and nonconvulsive RSE and acute repetitive seizures. Varying doses of lacosamide re-sulted in similar efficacy. Only one of the studies included children; the data did not include a specific analysis of the drug’s ef-fects in younger patients. Prospective and randomized studies are needed to validate these findings, as well as to determine the lowest effective dose in order to minimize adverse events. Future studies also should evaluate the efficacy and safety of lacos-amide therapy in children.
n DRUG INTERACTIONS AND SIDE EFFECTS
Lacosamide may be a valuable adjunc-tive treatment for RSE for many reasons, including its limited drug-drug interac-tions.11 Anticonvulsant medications often affect the metabolism of other anticonvul-sants and drug classes. This is more often the case for older antiepileptic drugs, such as phenytoin, and not for newer genera-tion agents, such as levetiracetam.
PerampanelPlusEnzyme-InducingDrugs
Interactions with anticonvulsants usu-ally occur via the cytochrome P450 sys-tem, which may be induced or inhibited by various medications.22
Perampanel is a selective, noncompeti-tive antagonist of α-amino-3-hydroxy-5-methyl-4-isox azolepropionic acid (AMPA)-type glutamate receptors.23 It was recently approved in both the United States and Europe for adjunctive treat-ment of partial seizures. Perampanel is mainly metabolized by the cytochrome P3A4 system. Use of carbamazepine, oxcarbazepine, and phenytoin increases the clearance of perampanel.24

16 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
SarahAminoffKelley,MD Anticonvulsant Treatment of Epilepsy and Refractory Status Epilepticus
Laurenza and colleagues25 investigated whether the use of perampanel with these enzyme-inducing medications was safe and efficacious. The investigators used data collected during three large, phase III, double-blind pivotal trials of peram-panel.23,26,27 Patients were studied for 19 weeks.
Plasma concentrations of perampanel were lower in patients using an enzyme-inducing medication, although the levels continued to increase proportionally with greater doses of medication. Perampanel administration effectively lowered the number of seizures experienced by both groups, but patients having a higher plas-ma concentration of the drug experienced greater efficacy. This finding indicates that dosing should differ for patients taking enzyme-inducing medications; it may be altered by either starting patients at higher doses or titrating the perampanel dosage more frequently.
Safety and tolerability were comparable at the same dose whether a patient was using enzyme-inducing medications or not. However, neither group experienced a significantly greater number of adverse effects at the same dose, which raises questions as to whether the side effects of perampanel are dose-independent and whether concomitant use of this drug with certain anticonvulsants (specifically, enzyme inducers) leads to an increase in adverse effects. These questions were not addressed in this study.
StatinsPlusEnzyme-InducingDrugs
Physicians have been concerned that enzyme-inducing anticonvulsants could lower statin levels, decrease the clinical effectiveness of these drugs, and increase the risk of cardiovascular disease. These effects have been shown in healthy vol-unteers28 and patients29,30 taking these medications concomitantly. A prior pharmacokinetic study in healthy volun-teers showed decreased atorvastatin levels with coadministration of phenytoin but not lamotrigine.28 The results of another study demonstrated an increase in low-density lipoprotein levels in patients using enzyme-inducing anticonvulsants.30
Karve and colleagues31 evaluated the use of both cytochrome P450-inducing drugs (eg, phenytoin, carbamazepine, phenobarbital, primidone) and noninduc-ing agents (eg, topiramate, oxcarbazepine, lamotrigine, gabapentin, pregabalin, leve-tiracetam, zonisamide, tiagabine, valproic acid) with hydroxymethylglutaryl-coen-zyme A reductase inhibitors (statins) that are metabolized by the cytochrome P450 system. The authors looked retrospec-tively at a very large cohort and examined the combination of enzyme-inducing or nonenzyme-inducing anticonvulsants used with hepatically metabolized (cyto-chrome P450 system) or nonhepatically metabolized anticonvulsants. They in-vestigated whether cardiovascular events occurred more frequently with certain
combinations of these medications and compared the results with those of in-dividuals who used only anticonvulsant medication. Patients were followed for a minimum of 60 days. The authors did not find an increased risk of myocardial infarction, stroke, transient ischemic at-tack, congestive heart failure, or angina in patients taking an anticonvulsant and a statin regardless of whether or not they used an enzyme-inducing anticonvulsant or a hepatically metabolized statin.
These data, however, were collected from a chart review of primary care clinic notes. Drug levels were not included.
NewerAnticonvulsantsandPotentialVascularRisks
Use of older anticonvulsants such as valproic acid may cause metabolic changes
and long-term consequences (eg, weight gain, abnormal bone metabolism). The issue of metabolic changes was addressed recently when Chuang and others32 dis-cussed the use of older enzyme-inducing and enzyme-inhibiting anticonvulsants as well as lamotrigine. Use of the older medications led to increased levels of markers of vascular risk (ie, cholesterol, homocysteine, other markers) that did not occur with administration of lamotrigine. However, the risk of a negative vascular outcome has not been well studied in patients using newer anticonvulsants.
Kim and colleagues33 investigated whether monotherapy with levetiracetam, oxcarbazepine, or topiramate in newly diagnosed epilepsy patients caused altered levels of blood markers that would indi-cate increased vascular risk. They specifi-cally examined lipid profile, homocysteine level, and apolipoprotein B (ApoB):ApoB/A1 ratio after 6 months of treatment. They found a significant increase in homocys-teine levels and ApoB:ApoB/A1 ratio in patients starting anticonvulsants, but they detected no clear difference between the various anticonvulsants used. Thus, newer anticonvulsants may cause changes similar to those caused by older medica-tions and may lead to an increased risk of atherosclerosis in the future. This study, however, did not address whether lifestyle changes after a new epilepsy diagnosis could contribute to these findings. The results suggested that cardiovascular risk factors should be monitored in all patients taking anticonvulsant medications.
n ANTICONVULSANT MONOTHERAPY AND COMBINATION THERAPY
Up to one third of patients with epi-lepsy do not respond to initial or subse-quent anticonvulsant choices,34 even with the introduction of new anticonvulsants35 and new drug combinations.36 Which monotherapy or combination regimen is most effective for specific types of sei-zures and which anticonvulsants may be more useful than others in patients with certain genetic abnormalities remain unanswered. A number of investigators examined the question of monotherapy
Newer anticonvulsants may cause changes similar to those caused by older medications, leading to an increased risk of atherosclerosis.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 17
SarahAminoffKelley,MD Anticonvulsant Treatment of Epilepsy and Refractory Status Epilepticus
versus combination therapy for particular seizure diagnoses and symptoms.
EconomicAdvantagesofCombinationTherapy
Cavazos and colleagues37 hypoth-esized that patients may have better seizure control and subsequently lower healthcare costs if they tried combina-tion therapy when monotherapy failed. Over 34,000 patients with a first-time diagnosis of partial-onset seizure in 2009 were identified from an insurance data-base. The authors excluded patients who experienced a number of disorders often treated with anticonvulsants (eg, neuro-pathic pain, migraine, bipolar disorder) and then analyzed data from individuals on sequential monotherapy, combination therapy from the onset of treatment, or monotherapy followed by combination therapy. In all, 58.4% of patients who had never used more than one anticonvulsant were excluded; they were assumed to have achieved control of their seizures with a single agent. This percentage compared favorably with previous data on response to first-line monotherapy.34
The costs incurred from inpatient and outpatient (including pharmacy) services were then examined. In comparison with individuals in the other two groups, patients treated with combination ther-apy were hospitalized less and had lower medical costs. These findings suggest that improved seizure control may be obtained with combination therapy in an insured, mostly working-class population.
ValproatePlusLamotrigineinChildrenWithDropSeizures
Thome-Souza and Valente38 inves-tigated the efficacy of valproate and lamotrigine in 60 children with drop seizures. Previous results from small stud-ies39 suggested that this drug choice could be effective for such young patients.
The majority of children were given valproate, lamotrigine, and a benzodi-azepine for their seizures. About 25% achieved complete control of their drop seizures, and a little over half had 50%–75% control of their drop seizures. There was no control group, and the use of other
anticonvulsants was not examined. These results suggest that this drug combination may be effective for the treatment of drop attacks in children.
TreatingSpecificSymptomsofIntractablePartialSeizures
One additional study looked at anti-convulsant efficacy in patients with in-tractable partial seizures who had specific seizure symptoms, as opposed to a specific seizure syndrome or seizure classifica-tion. The authors suggested that treating patients based on the old International League Against Epilepsy classification40 (ie, simple or complex partial seizures versus generalized seizures) may not be the most effective way to choose the ap-propriate anticonvulsant.
Sugai and colleagues41 used clinical experience with certain anticonvulsants in patients with various intractable sei-zure symptoms to analyze the efficacy of anticonvulsants within their cohort. They looked at almost 300 children and young adults who had one to three seizure symptoms. The majority of patients had frontal-lobe epilepsy. A responder rate of > 75% was noted with use of potassium bromide, zonisamide, or lamotrigine in patients with tonic seizures, use of zonisamide in those with secondarily generalized tonic-clonic seizures, use of carbamazepine in those with clonic seizures, and use of phenytoin in those with hypermotor seizures. The authors also noted a responder rate of < 25% with use of valproate for partial seizures and of 25%–50% with carbamazepine therapy for secondarily generalized tonic-clonic seizures. Based on these data, the authors surmised that carbamazepine may not be as useful for partial seizures and valproic acid may not be as useful for intractable epilepsy as commonly believed.
REFERENCES
1. Perucca E, Tomson T. The pharmacologi-cal treatment of epilepsy in adults. Lancet Neurol. 2011;10:446–456.
2. Perucca E, French J, Bialer M. Development of new antiepileptic drugs: challenges, incentives, and recent advances. Lancet Neurol. 2007;6:793–804.
3. Lowenstein DH. Treatment options for status epilepticus. Curr Opin Pharmacol. 2005;5:334–339.
4. Proceedings and Abstracts of the First
London Colloquium on Status Epilepticus, Univer-sity College London, April 12–15, 2007. Epilepsia. 2007;48(suppl 8):1–109.
5. Lowenstein DH. The management of re-fractory status epilepticus: an update. Epilepsia. 2006;47(suppl 1):35–40.
6. Gilbert DL, Glauser TA. Complications and costs of treatment of refractory generalized convul-sive status epilepticus in children. J Child Neurol. 1999;14:597–601.
7. McTague A, Kneen R, Kumar R, Spinty S, Appleton R. Intravenous levetiracetam in acute repetitive seizures and status epilepticus in chil-dren: experience from a children’s hospital. Seizure. 2012;21:529–534.
8. Hottinger A, Sutter R, Marsch S, Ruegg S. Topiramate as an adjunctive treatment in patients with refractory status epilepticus: an observational cohort study. CNS Drugs. 2012;26:761–772.
9. Chung SS. New treatment option for partial-onset seizures: efficacy and safety of lacosamide. Ther Adv Neurol Disord. 2010;3:77–83.
10. Biton V, Rosenfeld WE, Whitesides J, Fountain NB, Vaiciene N, Rudd GD. Intravenous lacosamide as replacement for oral lacosamide in patients with partial-onset seizures. Epilepsia. 2008;49:418–424.
11. Beyreuther BK, Freitag J, Heers C, Krebsfanger N, Scharfenecker U, Stohr T. Lacosamide: a review of preclinical properties. CNS Drug Rev. 2007;13:21–42.
12. Fernandez EM, Franck AJ. Lacosamide for the treatment of refractory status epilepticus. Ann Pharmacother. 2011;45:1445–1449.
13. Goodwin H, Hinson HE, Shermock KM, Karanjia N, Lewin JJ 3rd. The use of lacosamide in refractory status epilepticus. Neurocrit Care. 2011;14:348–353.
14. Miró J, Toledo M, Santamarina E, et al. Ef-ficacy of intravenous lacosamide as an add-on treat-ment in refractory status epilepticus: a multicentric prospective study. Seizure. 2012;22:77–79.
15. Parkerson KA, Reinsberger C, Chou SH, Dworetzky BA, Lee JW. Lacosamide in the treatment of acute recurrent seizures and periodic epileptiform patterns in critically ill patients. Epilepsy Behav. 2011;20:48–51.
16. Cherry S, Judd L, Muniz JC, Elzawahry H, LaRoche S. Safety and efficacy of lacosamide in the intensive care unit. Neurocrit Care. 2012;16:294–298.
17. Mnatsakanyan L, Chung JM, Tsimerinov EI, Eliashiv DS. Intravenous lacosamide in refrac-tory nonconvulsive status epilepticus. Seizure. 2012;21:198–201.
18. Koubeissi MZ, Mayor CL, Estephan B, Rashid S, Azar NJ. Efficacy and safety of intrave-nous lacosamide in refractory nonconvulsive status epilepticus. Acta Neurol Scand. 2011;123:142–146.
19. Sutter R, Marsch S, Ruegg S. Safety and efficacy of intravenous lacosamide for adjunctive treatment of refractory status epilepticus: a large comparative cohort study. Presented at the Joint An-nual Meeting of the Swiss SNG Neurological Society, the Swiss Society of Biological Psychiatry SGBP, and the Swiss Society for Behavioral Neurology; Novem-ber 8–10, 2012; Basel, Switzerland.
20. Alam K, Mullin PD, Park S, Berger K, Rosen-

18 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
SarahAminoffKelley,MD Anticonvulsant Treatment of Epilepsy and Refractory Status Epilepticus
gart AJ. Lacosamide in the treatment of refractory status epilepticus and repetitive seizures. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Di-ego, CA. Poster P2 237.
21. Newey CR, Hantus S. Intravenous lacos-amide is safe and effective in treating refractory status epilepticus in a critically-ill population: a large retrospective case series. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster P2 227.
22. Hukkanen J. Induction of cytochrome P450 enzymes: a view on human in vivo findings. Expert Rev Clin Pharmacol. 2012;5:569–585.
23. French JA, Krauss GL, Biton V, et al. Ad-junctive perampanel for refractory partial-onset seizures: randomized phase III study 304. Neurology. 2012;79:589–596.
24. Fycompa [package insert]. Woodcliff Lake, NJ: Eisai, Inc; October 2012.
25. Laurenza A, Gidal B, Hussein Z, et al. Evaluation of efficacy and safety of perampanel in the presence of concomitant CYP3A4-inducing AEDS: analyses from the perampanel phase 3 clinical trials. Presented at the 66th Annual Meeting of the Ameri-can Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster P2 211.
26. French JA, Krauss GL, Steinhoff BJ, et al. Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305. Epilepsia. 2013;54:117–125.
27. Krauss GL, Serratosa JM, Villanueva V, et al. Randomized phase III study 306: adjunctive
perampanel for refractory partial-onset seizures. Neurology. 2012;78:1408–1415.
28. Bullman J, Nicholls A, Van Landingham K, et al. Effects of lamotrigine and phenytoin on the pharmacokinetics of atorvastatin in healthy volun-teers. Epilepsia. 2011;52:1351–1358.
29. Gedde-Dahl A, Devold HM, Molden E. Statin medication in patients treated with antiepilep-tic drugs in Norway. Pharmacoepidemiol Drug Saf. 2012;21:881–885.
30. Candrilli SD, Manjunath R, Davis KL, Gidal BE. The association between antiepileptic drug and HMG-CoA reductase inhibitor co-medication and cholesterol management in patients with epilepsy. Epilepsy Res. 2010;91:260–266.
31. Karve S, Mitra D, Rajagopalan K, Blum D, Grinnell T, Bollu V. Impact of concomitant use of an-tiepileptic drugs and statins on risk of cardiovascular events. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–Decem-ber 4, 2012; San Diego, CA. Poster P2 205.
32. Chuang YC, Chuang HY, Lin TK, et al. Ef-fects of long-term antiepileptic drug monotherapy on vascular risk factors and atherosclerosis. Epilep-sia. 2012;53:120–128.
33. Kim DW, Shen Y, Lee S, Kim JH. Effects of new generation antiepileptic drugs on vascular risk factors in newly diagnosed epilepsy patients. Pre-sented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster P2 214.
34. Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–319.
35. French JA. Refractory epilepsy: clinical overview. Epilepsia. 2007;48(suppl 1):3–7.
36. Nicholas JM, Ridsdale L, Richardson MP, Ashworth M, Gulliford MC. Trends in antiepileptic drug utilisation in UK primary care 1993–2008: cohort study using the general practice research database. Seizure. 2012;21:466–470.
37. Cavazos R, Simons R, Fain R, Powers A, Wang Z. Health outcomes associated with sequential monotherapy and combination therapy with antiepi-leptic drugs in patients with partial onset seizures. Presented at the 66th Annual Meeting of the Ameri-can Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster P2 202.
38. Thome-Souza M, Valente K. Maintenance of valproate and lamotrigine efficacy during one year in a large series of patients with drop attacks. Pre-sented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster P2.212.
39. Machado VH, Palmini A, Bastos FA, Rotert R. Long-term control of epileptic drop attacks with the combination of valproate, lamotrigine, and a benzodiazepine: a “proof of concept,” open label study. Epilepsia. 2011;52:1303–1310.
40. Berg AT, Berkovic SF, Brodie MJ, et al. Re-vised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commis-sion on classification and terminology, 2005–2009. Epilepsia. 2010;51:676–685.
41. Sugai K, Nakagawa E, Komaki H, et al. Effective antiepileptic drugs for intractable partial epilepsies in children and young adults are different among actual seizure symptoms. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster P2 128.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 19
Pharmacotherapy of Medically Refractory Partial-Onset EpilepsyCynthia M. Correll, MDColumbia Comprehensive Epilepsy Center, Columbia University Medical Center, New York, New York
Abstract Anticonvulsant medication options and recommendations continue to expand as we search for the best methods to treat patients with medically refractory epilepsy. At a poster session held during the 66th Annual Meeting of the American Epilepsy Society, presenters discussed recent studies examining the newer antiepileptic drugs eslicarbazepine acetate, perampanel, lacosamide, pregabalin, rufinamide, and extended-release lamotrigine and provided informa-tion on rational polytherapy and medication adherence.
Dr. Correll is a Neurophysiology Fellow at the Columbia Comprehensive Epilepsy Center, Columbia University Medical Center, New York, New York.
Some 20%–40% of patients with epilepsy continue to have un-controlled seizures despite the availability of over 20 drugs,
vagal nerve stimulation, and surgical resection.1 To combat this problem, the field of epilepsy treatment is constantly expanding with new medications and therapeutic modalities. In addition, clini-cal researchers continue to study the best use of our current medication options.
At a poster presentation held during the 66th Annual Meeting of the American Epilepsy Society, experts addressed the latest research involving newer medica-tions and issues regarding treatment ef-ficacy and safety. Presenters focused on the use of eslicarbazepine acetate with concomitant carbamazepine therapy and in patients with mental depression; the long-term outcomes of adjunctive lacos-amide treatment, adjunctive perampanel therapy, and pregabalin monotherapy; the conversion from immediate-release to
extended-release lamotrigine use; the re-sults of polytherapy based on mechanism of action; and adherence to antiepileptic regimens.
n ESLICARBAZEPINE ACETATE THERAPY IN SPECIFIC PATIENT POPULATIONS
PatientsRefractorytoCarbamazepineBased on a presentation by Eugen Trinka MD, MSc, Chairman of Neurology and Professor of Clinical Neurology, Paracelsus Medical University, Salzberg, Austria
Eslicarbazepine acetate is a once-daily antiepileptic drug (AED) approved by the European Medical Agency in 2009 as adjunctive therapy in adults in partial-onset seizures.2 This medication is chemically related to carbamazepine, but it has a different mechanism of action on voltage-gated sodium channels.3 Given its chemical similarities to carbamazepine, eslicarbazepine acetate may be effective and tolerable in patients for whom carba-mazepine failed to accomplish complete seizure control.
Gil-Nagel et al4 studied both efficacy (specifically, median relative reduction in seizure frequency) and tolerability (incidence of treatment-emergent adverse events [TEAEs]) in patients using esli-carbazepine acetate. Study subjects were pooled from two phase III, multicenter,
double-blind, randomized, placebo-con-trolled studies. Prior to randomization, participants must have had four or more documented partial-onset seizures over a 4-week period while using one to three AEDs. The patients were then random-ized to one of four daily dosing treatment groups: 400, 800, or 1,200 mg of eslicar-bazepine acetate or placebo. Patients were followed for a 12-week maintenance pe-riod and 4-week tapering period. Efficacy and tolerability were compared among three carbamazepine groups: those tak-ing no carbamazepine, those taking ≤ 800 mg/d of carbamazepine, and those taking > 800 mg/d of carbamazepine.
When compared with the placebo group (< 10% reduction in seizure fre-quency), the groups using 800 mg/d of eslicarbazepine acetate (> 20% reduction) and 1,200 mg of the drug (> 30% reduc-tion) experienced a significantly reduced seizure frequency in all three carbamaze-pine groups. An eslicarbazepine acetate dose-dependent increase in TEAEs and TEAEs leading to discontinuation of eslicarbazepine acetate was noted; this increase was further elevated in patients on carbamazepine, especially those using doses exceeding 800 mg/d. The most fre-quent TEAEs reported were dizziness, dip-lopia, headache, somnolence, and nausea.
The authors concluded that adjunc-tive eslicarbazepine acetate was effective independent of carbamazepine use. This was exemplified by a significant reduc-tion in seizure frequency among subjects who had previously been on more than 800 mg/d of carbamazepine who were randomized to receive 800 or 1,200 mg/d of eslicarbazepine acetate. The incidence of TEAEs among patients us-ing eslicarbazepine acetate increased in a

20 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
CynthiaM.Correll,MD Pharmacotherapy of Medically Refractory Partial-Onset Epilepsy
dose-dependent manner and was further increased in subjects also treated with carbamazepine, especially if the dose was above 800 mg/d.
PatientswithDepressiveSymptomsBased on a presentation by Mar Carreno, MD, Department of Neurology, Hospital Clinic, Barcelona, Spain
Depression and depressive symptoms, frequent comorbidities associated with epilepsy, have been correlated with poor seizure control and increased AED side effects.5–7 Use of eslicarbazepine acetate previously was associated with improved depressive symptoms during an open-label extension of phase III multicenter trials of eslicarbazepine acetate given as adjunctive treatment for partial-onset seizures.8,9
Carreno and others10 studied the efficacy and tolerability of eslicarbazepine acetate given to subjects with partial-onset seizures and depressive symptoms. Subjects pooled from two phase III multicenter studies had to have had at least four documented partial-onset seizures over a 4-week period. They were randomized to receive 400, 800, or 1,200 mg/d of eslicarbazepine acetate or placebo. Subjects were analyzed over a 12-week maintenance period based on the presence or absence of depressive symp-toms, defined as scores ≥ 10 or < 10 on the Montgomery-Asberg depression rating scale (MADRS), respectively.
Median relative reduction in seizure frequency was significantly reduced in the 800- and 1,200-mg dosing groups having MADRS scores ≥ 10 and < 10. Seizure fre-quency reduction was greater in subjects having a MADRS score < 10 as compared with those having a MADRS scores ≥ 10; the statistical significance of this dif-ference was not discussed. Incidence of TEAEs was again dose-dependent and not significantly different between subjects with MADRS scores ≥ 10 or < 10.
The authors concluded that adjunc-tive therapy with 800 or 1,200 mg/d of eslicarbazepine acetate was effective in decreasing seizure frequency when com-pared with placebo in patients both with and without depressive symptoms. They also concluded that subjects with depres-sive symptoms displayed less effective seizure control in all three groups using
eslicarbazepine acetate and the placebo group. The incidence of TEAEs during eslicarbazepine acetate therapy also in-creased in a dose-dependent manner in this study and was similar among patients with and without depressive symptoms.
n LONG-TERM EFFICACY AND SAFETY OF NEWER ANTICONVULSANT MEDICATIONS
PerampanelasAdjunctiveTherapyBased on a presentation by Georgia Montouris, MD, Assistant Professor of Neurology, Boston University School of Medicine, Boston, Massachusetts
Perampanel recently was approved by both the US Food and Drug Administra-tion (FDA) and the European Medicines Agency as adjunctive therapy for partial-onset seizures following publication of three phase III multicenter, double-blind, randomized, placebo-controlled trials.11–15 Montouris and colleages16 studied the efficacy, measured as median percent re-duction in seizure frequency, and safety, measured as TEAEs and serious adverse events (SAEs), of this drug.
The researchers studied 1,218 patients who participated in an extension trial following their involvement in a 23-week, double-blind, randomized, controlled trial. Patients in the double-blind study were assigned to 2, 4, 8, or 12 mg/d of perampanel or placebo. During the exten-sion trial, all patients completed a 16-week blinded conversion period during which the perampanel dosage was titrated from an initial dose of 2 mg/d up to a maximal tolerated dose (up to 12 mg/d); patients were maintained on this dose as toler-ated. The mean dose during the extension period was 10.2 ± 2.3 mg, and 71.6% of remained in the study up to the cutoff date (week 52).
Prior to perampanel titration in the extension trial, the median percent seizure reduction was 18.6% for the double-blind placebo group and 31.7% for the double-blind perampanel group. After the conver-sion/titration period, the initial double-blind placebo group (49.3% median percent seizure reduction) had achieved a median percent seizure frequency reduc-
tion similar to that of the double-blind perampanel group (46.5% median percent seizure reduction); these reductions in seizure frequency were maintained over the 52-week study period.
The TEAE and serious adverse event incidence during the extension period for the double-blind placebo and perampanel groups were not significantly different. Withdrawal rates due to adverse events during the extension period also were not significantly different at 10.1% and 9.8%, respectively. The most common TEAEs reported were dizziness, somnolence, fatigue, and headache.
The authors concluded that patients on perampanel during the extension period of this study reported seizure reduction rates and TEAEs similar to those seen in the perampanel group participating in the initial double-bind, randomized, controlled studies.
PregabalinasMonotherapyBased on a presentation by Lorraine Yurkewicz, MD, Director of Clinical Services, Pfizer Inc, New York, New York
Yurkewicz and others17 evaluated the safety and efficacy of pregabalin monotherapy in an extension study that followed a 20-week, randomized, double-blind, controlled study. In all, 73 patients were transitioned from doses of 150–600 mg/d to 300 mg/d over 7 days. The subjects continued on pregabalin monotherapy for 24 weeks, allowing dose adjustments of 150–600 mg/d.
A total of 58 patients (79.5%) com-pleted 24 weeks of therapy (mean dura-tion, 174 days; median average dose, 473.7 mg/d). Some 31 subjects (42.5%) had a TEAE, most commonly convulsion, head-ache, nausea, arthralgia, and weight gain. Four of these patients discontinued the medication due to these adverse events, and five patients discontinued the medica-tion due to insufficient clinical response. Ten subjects (13.7%) were seizure-free throughout the open-label study.
Overall, the side-effect profile was similar to those of previous trials evalu-ating this medication. A total of 80% of subjects completed 6 months of pregaba-lin monotherapy, with 13.7% of subjects remaining seizure-free.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 21
CynthiaM.Correll,MD Pharmacotherapy of Medically Refractory Partial-Onset Epilepsy
LacosamideasAdjunctiveTherapyintheElderlyBased on a presentation by William Rosenfeld, MD, The Comprehensive Epilepsy Care Center of Children and Adults, St. Louis, Missouri
Rosenfeld and others18 studied the long-term safety and efficacy outcomes of treatment with lacosamide, a novel drug approved for adjunctive treatment of adults with partial-onset seizures.19,20 Participants in the study were a subgroup of elderly patients (age by end of the trials, ≥ 65 years) who were enrolled in three open-label extension trials lasting up to 8 years.21–24 Safety outcomes were defined as TEAEs, serious adverse events, and patient withdrawal due to TEAEs. Ef-ficacy outcome was defined as the percent change from baseline seizures frequency.
Of the 1,054 patients enrolled in the open-label extension trials, 33 were ≥ 65 years of age by the end of the trial; 21 (63.6%) of the patients completed the open-label extension trial, and 18 (85.7%) continued on lacosamide after the trial ended. In all, 90.9%, 75.8%, and 42.4% of the 33 patients continued taking lacosamide for more than 1, 3, and 5 years, respectively, during the open-label extension trial. A to-tal of 60.1% reached the median modal dose of 400 mg/d for this subgroup. The median percent reduction from baseline seizure frequency for the subgroup was 63%, 58%, and 67% for patients who completed 1, 3, and 5 years, respectively, of lacosamide therapy, which was not significantly differ-ent from that of the general subjects in the extension trial. Dizziness, falls, contusion, sinusitis, cognitive disorder, tremor, head-aches, depression, and cough were the most frequent TEAEs noted. No new types of TEAEs were reported among this subgroup over the prolonged treatment period. Four patients (12.1%) withdrew from the study due to TEAEs.
The authors concluded that despite the limitations of this small subgroup analysis, which extended up to 8 years, elderly patients with partial-onset seizures in the lacosamide open-label extension trials had efficacy and safety outcomes that were similar to those of the entire lacosamide-treated patient population in the extension trial.
ConversionfromImmediate-ReleasetoExtended-ReleaseLamotrigineBased on a presentation by Melissa R. Osborn, BSN, RN, and Patsy Ramey, MSN, RN, Department of Neurology, Vanderbilt University, Nashville, Tennessee
Use of immediate-release lamotrigine may lead to both peak dose toxicity and reduced seizure threshold due to low trough levels. To determine whether extended-release lamotrigine can reduce these effects, Osborn and colleagues 25 analyzed data on patients who underwent conversion from the immediate-release form to the extended-release form over a 2-year period from 2009–2011 as they related to seizure control (namely, me-dian reduction in seizure frequency) and change in adverse experience profile.
Overall, 55 patients were included in the analysis. Nineteen patients converted to the same dose of extended-release la-motrigine, 21 converted to a higher dose, and 7 converted to a lower dose. Analyz-ing all patients, there was a significant reduction in seizure frequency (46% re-duction) after conversion to the extended-release form. The percent reduction in seizure frequency also was significantly reduced after conversion to extended-release lamotrigine among the group that converted to the same lamotrigine dose. Seven patients reported improvement in adverse effects.
The authors concluded that a switch from immediate-release lamotrigine to an extended-release form of the drug resulted in both an improvement in seizure control and adverse effects.
n RUFINAMIDE USAGE PATTERNSBased on a presentation by Elif Silva MD, Eisai, Inc, Woodcliff Lake, New Jersey
Rufinamide, a drug approved by the FDA for adjunctive therapy of seizures associated with Lennox-Gastaut syn-drome,26 has been on the market since 2008. Silva and colleagues27 performed a retrospective longitudinal cohort study to evaluate use and dosing patterns of this drug among commercially insured pa-tients. Data were collected from medical and pharmacy claims data in the Truven Health MarketScan Commercial Database over 2.5 years, from 2008 until 2011. Data
was gathered on patients initiating rufin-amide therapy who had ≥ 6 months of pre- and post-enrollment date information.
The authors specifically examined demographics (age and gender), clini-cal characteristics (comorbid illnesses, concomitant AEDs used), persistence (time to discontinuation of rufinamide), and dose ratio of rufinamide (observed modal dose/calculated ideal dose). A total of 495 patients met the final inclu-sion criteria (mean age,16.9 ± 12.7 years); 40.8% of patients were children 4–12 years of age. Comorbid neurologic conditions included developmental delay (19.7%), growth delay (18%). and mental retarda-tion (14.1%). Most common concomitant AEDs used included lamotrigine (33.1%), levetiracetam (32.7%), divalproex so-dium (28.6%), diazepam (23.9%), topi-ramate (21.6%), clonazepam (20.3%), and zonisamide (18.9%). Over half of the patients were on three or more AEDs before starting rufinamide. Dose ratios of 0.25–0.75 of the ideal dose were noted in 51% of patients, whereas dose ratios of 0.75–1.25 were found in 34%. The aver-age time to discontinuation of rufinamide was 237 ± 204 days (median, 184 days). Patients who took longer than 30 days to reach their modal dose were less likely to discontinue the medication over the study.
The authors concluded that clini-cal use of rufinamide is most common among patients already on combination AED regimens; this most often was noted among those using at least three AEDs. Many patients (62%) did not reach 0.75 of the recommended dose of 45 mg/kg/d or the maximum dose of 3,200 mg/d of rufinamide. The fact that patients who took longer to reach their modal dose were less likely to discontinue the medica-tion suggested that a slower titration may be more tolerable.
n COMBINATION THERAPY BASED ON MECHANISM OF ACTION
Based on a presentation by Jose E. Cavazos, MD, PhD, University of Texas Health Science Center at San Antonio, San Antonio, Texas
Seizure control may be improved through rational polypharmacy by com-bining drugs with synergistic or different

22 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
CynthiaM.Correll,MD Pharmacotherapy of Medically Refractory Partial-Onset Epilepsy
mechanisms of action.28–30 Wang and others31 analyzed persistence (defined as duration of treatment) and healthcare use (number of hospital or emergency room visits) in patients using various AED combination regimens in this cross-sectional, retrospective analysis of medi-cal and pharmacy claims from 2004 to 2011. Patients > 18 years of age who had a second AED added on at the index date and who had at least 90 days of pre-index documentation were included in the study and followed for at least 12 months.
Four categories of mechanism of action were defined. Sodium-channel block-ers included carbamazepine, ethotoin, fosphenytoin, lacosamide, lamotrigine, oxcarbazepine, and phenytoin. The gam-ma-aminobutyric acid (GABA) analogs included clonazepam, diazepam, gabap-entin, phenobarbital, pregabalin, primi-done, tiagabine, and vigabatrin. Leveti-racetam was defined as a synaptic vesicle protein 2A binding (SV2) agent. The fol-lowing drugs were included in a multiple-mechanisms category: divalproex sodium, felbamate, topiramate, valproate sodium, valproic acid, and zonisamide. Based on these categories, a total of 8,615 patients were then assigned to one of seven groups receiving drugs with these combinations: sodium-channel blocker/sodium-channel blocker, sodium-channel blocker/SV2 agent, sodium-channel blocker/drug with multiple mechanisms, a sodium-channel blocker/GABA analog, GABA analog/GABA analog, GABA analog/SV2 agent, and GABA analog/drug with multiple mechanisms.
There were significant differences in baseline gender characteristics among the groups, with males accounting for 48% of the group using sodium-channel blocker/SV2 agent and only 28.4% of the group using GABA analog/GABA analog. Patients using sodium-channel blocker/SV2 agent had the longest mean treat-ment duration (507 ± 506 days), whereas patients using GABA analog/GABA ana-log had the short mean treatment dura-tion (344 ± 345 days). The likelihood of treatment discontinuation for the various combination groups also was compared to discontinuation rates seen among patients
using a sodium-channel blocker/sodium-channel blocker. Those patients using a sodium-channel blocker/SV2 agent were significantly less likely to discontinue therapy (hazard ratio [HR], 0.817; P < 0.001), and those using a GABA analog/GABA analog or GABA analog/drug with multiple mechanisms were more likely to discontinue therapy (HR, 1.252 and 1.172, respectively; P < 0.002).
The group using a sodium-channel blocker/SV2 agent had the lowest percent-age of patients requiring emergency de-partment or hospital visits (61% and 42%, respectively); the group using a GABA analog/GABA analog had the highest (81% and 68%, respectively). Patients on a combination of a GABA analog with any other AED had a significantly lower likelihood of hospitalization (odds ratio [OR], 0.716; 95% confidence interval [CI], 0.539–0.952; P = 0.021) when compared with patients using a GABA analog/GABA analog. Patients using a sodium-channel blocker/any other AED had a significantly lower likelihood of emergency depart-ment visitation when compared with those using a sodium-channel blocker/sodium-channel blocker (OR, 0.853; 95% Cl, 0.742–0.980; P = 0.025).
Limitations to this study included dif-fering opinions on mechanisms of action for the various agents, efficacy and safety differences among drugs within the same categories, and the inclusion of only com-mercially insured patients. The authors concluded that patients on combinations of AEDs having different mechanisms of action exhibited both increased persis-tence and decreased healthcare use. Fur-ther, variations in baseline characteriza-tions such as gender may have contributed to these differences.
n A REVIEW OF ANTIEPILEPTIC MEDICATION ADHERENCE
Based on a presentation by Alexis Economos, MD, Department of Neurology, University of Miami, Miami, Florida
Poor adherence to a recommended medication regimen is a significant im-pediment to the goal of seizure freedom. Economos and others32 performed a criti-cal review of adherence to antiepileptic regimens using a literature review of the
following databases: Cochrane Library, MEDLINE, EMBASE, and Cumulative Index to Nursing and Allied Health Litera-ture (CINAHL). They reviewed 24 studies citing nonadherence rates of 25%–79%. Factors affecting nonadherence included population demographics, medication side effects, medication dosing schedules, and number of medications used.
Adherence was worse among the elderly and adolescents, with children showing better adherence if they came from a higher socioeconomic state or their parents were married.33–38 Use of newer AEDs was related to better com-pliance, presumably due to their better side-effect profiles.39 Less frequent dosing was also associated with increased adher-ence.40 There were conflicting reports on monotherapy versus polytherapy, with one article citing better adherence on monotherapy since patients believed that polytherapy was associated with increased side effects.40,41 However, another article cited better adherence on polytherapy, because patients using more than one drug believed it was more important to take their medications.42
To improve adherence, it has been sug-gested that a more tailored approach be taken for each individual or specific popu-lation of patients.43 These may include the use of lower-cost medications, simpler dosing regimens, and self-management websites; the culturally relevant promo-tion of disease awareness and understand-ing; and the association of medication administration to a specific time, place, or activity.44,45
REFERENCES
1. Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000; 342:314–319.
2. European Medicines Agency. Zebenix (esli-carbazepine acetate): summary of product charac-teristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000988/WC500047225.pdf. Accessed De-cember 28, 2012.
3. Soares-da-Silva P, Hebeisen S. Slow and fast inactivation of voltage-gated sodium channels by eslicarbazepine and carbamazepine. Epilepsia. 2012; 53(suppl 5):54.
4. Gil-Nagel A, Trinka E, Chaves J, et al. A post- hoc exploratory analysis of the effect of esli-carbazepine acetate as adjunctive treatment in adult patients with partial-onset seizures refractory to

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 23
CynthiaM.Correll,MD Pharmacotherapy of Medically Refractory Partial-Onset Epilepsy
carbamazepine. Presented at the 66th Annual Meet-ing of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster 3.228.
5. Hitiris N, Mohanraj R, Norrie J, Sills GJ, Brodie MJ. Predictors of pharmacoresistant epilepsy. Epilepsy Res. 2007;75:192–196.
6. Petrovski S, Szoecke CEI, Jones NC, et al. Neuropsychiatric symptomatology predicts seizure recurrence in newly treated patients. Neurology. 2010; 75:1015–1021.
7. Kanner AM, Barry JJ, Gilliam F, Hermann B, Meador KF. Depressive and anxiety disorders in epilepsy: do they differ in their potential to worsen common AED-related adverse events? Epilepsia. 2012;53:1104–1108.
8. Halasz P, Cramer JAM, Hodoba D, et al. Longer term efficacy and safety of eslicarbazepine acetate: results of a 1-year open-label extension study in partial-onset seizures in adults with epilepsy. Epilepsia. 2010; 51:1963–1969.
9. Hufnagel A, Ben-Menachem E, Gabbai AA, et al. Long term safety and efficacy of eslicarbazepine acetate as adjunctive therapy in the treatment of partial-onset seizures in adults with epilepsy: results of a 1-year open-label extension study. Epilepsy Res. 2012. Epub ahead of print.
10. Carreno M, Ben-Menachem E, O’Brien TJ, et al. A post-hoc exploratory analysis of the effect of eslicarbazepine acetate as adjunctive treatment in adult patients with partial-onset seizures and comorbid clinically relevant depressive symptoms. Presented at the 66th Annual Meeting of the Ameri-can Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster 3.229.
11. Fycompa [package insert]. Woodcliff Lake, NJ: Eisai, Inc; 2012.
12. Fycompa [package insert]. Hatfield, Eng-land: Eisai Europe Ltd; 2012.
13. French J. Global phase III trial of peram-panel, a selective, non-competitive AMPA receptor antagonist, as adjunctive therapy in patients with refractory partial-onset seizures. Presented at the 63rd Annual Meeting of the American Academy of Neurology; April 9–16, 2011; Honolulu, HI. Abstract LBS 002.
14. Loring DW, Lowenstein DH, Barbaro NM, et al. Common data elements in epilepsy research: development and implementation of the NINDS epilepsy CDE project. Epilepsia. 2011;52:1186–1191.
15. Krauss GL, Serratosa JM, Villanueva V, et al. Randomized phase III study 306: adjunctive perampanel for refractory partial-onset seizures. Neurology. 2012;78:1408–1415.
16. Montouris G, Yang H, Williams B, Zhou S, Laurenza A, Fain R. Efficacy and safety of peram-panel in patients with treatment-resistant partial-onset seizures after conversion from double-blind placebo to open-label perampanel. Presented at the 66th Annual Meeting of the American Epilepsy So-ciety; November 30–December 4, 2012; San Diego, CA. Poster 3.241.
17. Yurkewicz L, Kwan P, Fakhoury R, Pitman V, Knapp L. Long-term safety and efficacy of pregabalin monotherapy in patients with partial onset seizures:
an open-label, extension study. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster 3.237.
18. Rosenfeld W, McShea C, Doty P. Evaluation of long-term treatment with lacosamide for partial-onset seizures in the elderly. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster 3.234.
19. European Medicines Agency. Vimpat (lacosamide): summary of product characteristics for lacosamide. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Infor-mation/human/000863/WC500050338.pdf. May 22, 2012. Accessed December 28, 2012.
20. Vimpat [package insert]. Smyrna, GA:UCB, Inc; 2008.
21. Rosenfeld W, Fountain N, Kaubrys G, et al. Lacosamide: long-term safety and efficacy in partial-onset seizures. Presented at the 29th International Epilepsy Congress, August 28–September 1, 2011; Rome, Italy.
22. Husain A, Chung S, Faught E, et al. Long-term safety and efficacy in patients with uncontrolled partial-onset seizures treated with adjunctive lacos-amide: results from a phase III open-label extension trial. Epilepsia. 2012; 53:521–528.
23. Rosenow F, Kelemen A, Ben-Menachem E, et al. Long-term adjunctive lacosamide in patients with uncontrolled partial-onset seizures: results from the SP774 phase III open-label extension trial. Presented at the 29th International Epilepsy Congress, August 28–September 1, 2011; Rome, Italy.
24. Rosenfeld W, Husain A, Rosenow F, McShea C, Isojarvi J, Doty P. Evaluation of long-term treat-ment with lacosamide for partial-onset seizures: a pooled analysis of open-label extension trials, Pre-sented at the 65th Annual Meeting of the American Epilepsy Society; December 2–6, 2011; Baltimore, MD. Abstract 3.233.
25. Osborn MR, Ramey P, Abou-Khalil B. Benefits of conversion from immediate release lamotrigine to extended release lamotrigine in individuals with drug-resistant epilepsy or adverse effects. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–Decem-ber 4, 2012; San Diego, CA. Poster 3.255.
26. Banzel (rufinamide) [package insert]. Woodcliff Lake, NJ; Eisai Co, Ltd.; 2011.
27. Silva E, Margolis JM, Wang Z, Copher R, Labiner D. Rufinamide dosing patterns in commer-cially-insured pediatric and adult patients. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Di-ego, CA. Poster 3.242.
28. Brodie MJ, French JA. Management of epilepsy in adolescents and adults. Lancet. 2000;356:323–329.
29. St. Louis EK. Truly “rational” polytherapy: maximizing efficacy and minimizing drug interac-tions, drug load, and adverse effects. Curr Neuro-pharmacol. 2009;7:96–105.
30. Brodie MJ, Sills GJ. Combing AEDs–rational polytherapy? Seizure. 2011;20:369–375.
31. Wang Z, Margolis JM, Copher R, Cavazos JE. Comparison of treatment duration of AED combination therapies base on mechanism of ac-tion in partial onset seizures. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster 3.248.
32. Economos A, Cheng J, Carrazana E. AEDs and adherence: a critical review. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster 3.262.
33. Sander JW. The use of AEDs—principles and practice. Epilepsia. 2004(suppl 6);45:28–34.
34. Ettinger AB, Bater GA. Best clinical and research practice in epilepsy of old people: focus on AED adherence. Epilepsy Behav. 2009;15:S60–S63.
35. Zeber JE, Copeland LA, Pugh MJV. Varia-tion in AED adherence among older patients with new-onset epilepsy. Ann Pharmacother. 2010;44:1896–1904.
36. Modi AC, Rausch JR, Glauser RA. Pat-terns of nonadherence to AED therapy in children with newly diagnosed epilepsy. JAMA. 2011; 305:1669–1676.
37. Kyngas H. Compliance with health regimens of adolescents with epilepsy. Seizure. 2000;9:598–604.
38. Michaud PA, Suris JC, Viner R. The Ado-lescent With a Chronic Condition: Epidemiology, Developmental Issues, and Health Care Provision. Geneva: World Health Organization, Department of Child and Adolescent Health and Develop-ment; 2007. http://whqlibdoc.who.int/publica-tions/2007/9789241595704_eng.pdf. Accessed December 28, 2012.
39. Beghi, E, Beghi M, Cornaggia DM. The use of recently approved AEDs: use with caution, use in refractory patients or use as first-line indications? Expert Rev Neurother. 2011;11:1759–1767.
40. Reyna MD, Medina MT, Nicolas O et al. Adherence and complementary and alternative medicine use among Honduran people with epilepsy. Epilepsy Behav. 2009;14:645–650.
41. Carpay JA, Aldenkamp AP, Von Donselaar CA. Complaints associated with the use of AEDs: results from a community-based study. Seizure. 2005;14:198–206.
42. Buck D, Jacoby A, Baker GA, Chadwick DW. Factors influencing compliance with AED regimes. Seizure. 1997;6:87–93.
43. Al-Ageel S, Al-Sabhan J. Strategies for improving adherence to AED treatment in pa-tients with epilepsy. Cochrane Database Syst Rev. 2011;(1):CD008312.
44. Diliorio G, Escoffery C, McCarly F. Evalu-ation of WebEase: an epilepsy self- management website. Health Educ Res. 2009;24:185–197.
45. Helde G, Brodtkorb E, Brathen G, Bovim G. An easily performed group education programme for patients with uncontrolled epilepsy: a pilot study. Seizure. 2003;12:497–501.

24 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
Epilepsy is one of the most com-mon and serious neurologic disorders. According to an Insti-tute of Medicine report,1 1 in 26
people will have epilepsy over his or her lifetime. Approximately 30% of people with epilepsy do not attain complete control of their seizures, even after trying several medications.2 Neurosurgery may be a therapeutic option for some patients, but several contraindications to surgery can prevent a good outcome.3
For patients with refractory epilepsy who are not good neurosurgical candi-dates, new antiepileptic drugs (AEDs) may offer some hope, especially when such medications have novel structures or new molecular targets. In addition to new brand-name agents, generic versions of existing medications are becoming increasingly prevalent. Controversies about the true equivalence of original brand-name agents and their generic counterparts have been debated for years, and physicians must judge use of these generic medications according to the best available evidence.
At the Annual Fundamentals Sym-
posium offered during the 66th Annual Meeting of the American Epilepsy Society, a series of presentations focused on the best use of the newest AEDs. Speakers also addressed controversies in manag-ing generic medications in patients with epilepsy. Experts in the field discussed use of newer AEDs in patients with refractory epilepsy, matching novel agents to specific epileptic syndromes, the adverse effects related to use of these drugs, and the ad-ministration of generic drugs based on a firm understanding of the best scientific data available.
The session was chaired by Michael Privitera, MD, Professor of Neurology, University of Cincinnati College of Medi-cine, and Director, Epilepsy Center, UC Neuroscience Institute, Cincinnati, Ohio.
n MECHANISM OF ACTION OF THE NEW AEDs
Based on a presentation by Misty D. Smith, PhD, Research Assistant Professor of Pharmacology and Toxicology , Investigator in the Anticonvulsant Drug Development Program, University of Utah, Salt Lake City, Utah
Since 2007, eight AEDs have been ap-proved by the US Food and Drug Adminis-
tration (FDA) and/or the European Union (EU). In chronologic order of introduction, those drugs are stiripentol, lacosamide, rufinamide, eslicarbazepine acetate, viga-batrin, ezogabine (retigabine), clobazam, and perampanel (Tables 1 and 2).
When considering the mechanism of action of an AED, it is important to remember that we do not have full knowl-edge of all potential sites of action within in vivo systems. These drugs likely have multiple sites of action, and no one action of any given AED completely accounts for its observed clinical effects (ie, efficacy, toxicity, tolerability).
Most established AEDs affect either re-duction of excitatory neurotransmission; enhancement of inhibitory neurotrans-mission of γ-aminobutyric acid (GABA); or modification of sodium, potassium, or calcium ion conductance. To increase the likelihood of improved seizure control, the new AEDs are structurally novel, able to engage new molecular targets, or next-generation compounds.
StiripentolStiripentol is an aromatic alcohol that
is structurally unrelated to other AEDs. In 2007, the European Medicine Agency authorized marketing of the drug for adjunctive therapy with clobazam and val-proic acid to treat refractory generalized
Optimal Use of the Newest Antiepileptic Drugs and Generics Peter Pressman, MDUniversity of California, San Francisco, School of Medicine, San Francisco, California
Abstract At least 60% of people with epilepsy can have their seizures completely controlled by medications, yet seizure freedom remains elusive for the remaining 40%. Within the past 5 years, eight new antiepileptic drugs (AEDs) have been approved to prevent seizures in patients with epilepsy. Many of these medica-tions have exciting new mechanisms that offer new routes to seizure control. In addition to FDA-approved indications, several emerging therapeutic possibilities are being explored for these novel AEDs. The astute clinician must remain aware of the unique pharmacokinetic properties and potential drug interactions and side effects associated with these new drugs. At the 66th Annual Meeting of the American Epilepsy Society, experts discussed the mechanisms of action, phar-macokinetics, approved indications, emerging uses, drug interactions, and side effects of new AEDs. They also reviewed the results of past and ongoing studies concerned with therapeutic equivalence of AED use in patients with epilepsy.
Dr. Pressman is a Behavioral Neurology Fellow at the University of California, San Francisco, School of Medicine, San Francisco, California.
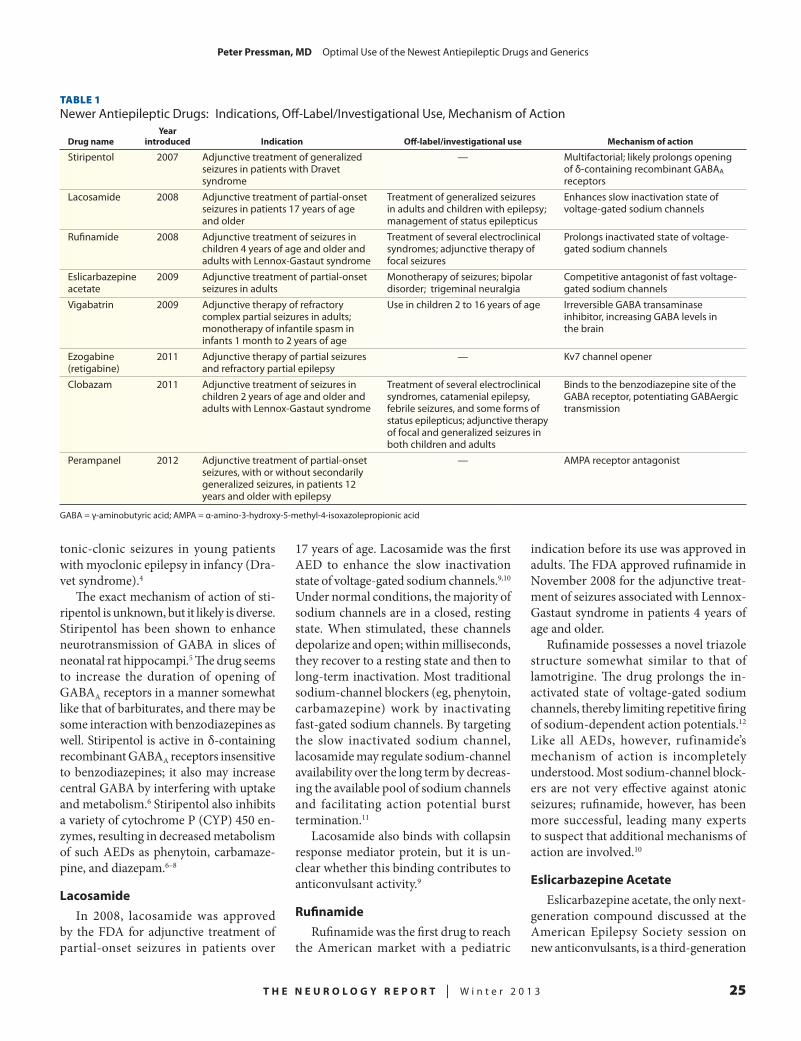
T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 25
PeterPressman,MD Optimal Use of the Newest Antiepileptic Drugs and Generics
tonic-clonic seizures in young patients with myoclonic epilepsy in infancy (Dra-vet syndrome).4
The exact mechanism of action of sti-ripentol is unknown, but it likely is diverse. Stiripentol has been shown to enhance neurotransmission of GABA in slices of neonatal rat hippocampi.5 The drug seems to increase the duration of opening of GABAA receptors in a manner somewhat like that of barbiturates, and there may be some interaction with benzodiazepines as well. Stiripentol is active in δ-containing recombinant GABAA receptors insensitive to benzodiazepines; it also may increase central GABA by interfering with uptake and metabolism.6 Stiripentol also inhibits a variety of cytochrome P (CYP) 450 en-zymes, resulting in decreased metabolism of such AEDs as phenytoin, carbamaze-pine, and diazepam.6–8
LacosamideIn 2008, lacosamide was approved
by the FDA for adjunctive treatment of partial-onset seizures in patients over
17 years of age. Lacosamide was the first AED to enhance the slow inactivation state of voltage-gated sodium channels.9,10 Under normal conditions, the majority of sodium channels are in a closed, resting state. When stimulated, these channels depolarize and open; within milliseconds, they recover to a resting state and then to long-term inactivation. Most traditional sodium-channel blockers (eg, phenytoin, carbamazepine) work by inactivating fast-gated sodium channels. By targeting the slow inactivated sodium channel, lacosamide may regulate sodium-channel availability over the long term by decreas-ing the available pool of sodium channels and facilitating action potential burst termination.11
Lacosamide also binds with collapsin response mediator protein, but it is un-clear whether this binding contributes to anticonvulsant activity.9
RufinamideRufinamide was the first drug to reach
the American market with a pediatric
indication before its use was approved in adults. The FDA approved rufinamide in November 2008 for the adjunctive treat-ment of seizures associated with Lennox-Gastaut syndrome in patients 4 years of age and older.
Rufinamide possesses a novel triazole structure somewhat similar to that of lamotrigine. The drug prolongs the in-activated state of voltage-gated sodium channels, thereby limiting repetitive firing of sodium-dependent action potentials.12 Like all AEDs, however, rufinamide’s mechanism of action is incompletely understood. Most sodium-channel block-ers are not very effective against atonic seizures; rufinamide, however, has been more successful, leading many experts to suspect that additional mechanisms of action are involved.10
EslicarbazepineAcetateEslicarbazepine acetate, the only next-
generation compound discussed at the American Epilepsy Society session on new anticonvulsants, is a third-generation
TABLE 1Newer Antiepileptic Drugs: Indications, Off-Label/Investigational Use, Mechanism of Action
YearDrugname introduced Indication Off-label/investigationaluse Mechanismofaction
Stiripentol 2007 Adjunctive treatment of generalized — Multifactorial; likely prolongs opening seizures in patients with Dravet of δ-containing recombinant GABAA
syndrome receptorsLacosamide 2008 Adjunctive treatment of partial-onset Treatment of generalized seizures Enhances slow inactivation state of seizures in patients 17 years of age in adults and children with epilepsy; voltage-gated sodium channels and older management of status epilepticusRufinamide 2008 Adjunctive treatment of seizures in Treatment of several electroclinical Prolongs inactivated state of voltage- children 4 years of age and older and syndromes; adjunctive therapy of gated sodium channels adults with Lennox-Gastaut syndrome focal seizures Eslicarbazepine 2009 Adjunctive treatment of partial-onset Monotherapy of seizures; bipolar Competitive antagonist of fast voltage- acetate seizures in adults disorder; trigeminal neuralgia gated sodium channelsVigabatrin 2009 Adjunctive therapy of refractory Use in children 2 to 16 years of age Irreversible GABA transaminase complex partial seizures in adults; inhibitor, increasing GABA levels in monotherapy of infantile spasm in the brain infants 1 month to 2 years of age Ezogabine 2011 Adjunctive therapy of partial seizures — Kv7 channel opener (retigabine) and refractory partial epilepsy Clobazam 2011 Adjunctive treatment of seizures in Treatment of several electroclinical Binds to the benzodiazepine site of the children 2 years of age and older and syndromes, catamenial epilepsy, GABA receptor, potentiating GABAergic adults with Lennox-Gastaut syndrome febrile seizures, and some forms of transmission status epilepticus; adjunctive therapy of focal and generalized seizures in both children and adultsPerampanel 2012 Adjunctive treatment of partial-onset — AMPA receptor antagonist seizures, with or without secondarily generalized seizures, in patients 12 years and older with epilepsy
GABA = γ-aminobutyric acid; AMPA = α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid

26 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
PeterPressman,MD Optimal Use of the Newest Antiepileptic Drugs and Generics
TABLE 2Newer Antiepileptic Drugs: Pharmacokinetics, Dosage, Major Side Effects, Drug Interactions
ProteinDrugname Tmax,h t½,h bound,% Dosagea Majorsideeffects Druginteractions Comments
Stiripentol 1.5 4.5 –13 99 50–100 mg/kg Ataxia, drowsiness, CYP450 inhibitor; prolongs Not FDA approved daily; maximum weight loss metabolism of other AEDs dose, 4 g (eg, phenytoin, diazepam, carbamazepine)Lacosamide 0.5–4 13 15 50–100 mg/d in at Dizziness Other CYP inducers affect Large fluctuations in least two divided lacosamide’s metabolism serum concentrations doses; maintenance dose, 200–400 mg/dRufinamide 4–6 6–10 26–35 400–800 mg/d in QT-interval Inducers affect rufinamide’s Nonlinear dose versus two equally divided shortening, fatigue, metabolism; valproic acid serum concentration doses, followed by headache, nausea may increase rufinamide’s relationship; food increases of 400– serum concentration by up intake increases 800 mg/d every to 50% bioavailability other day, up to a maximum of 3,200 mg/d in two equally divided dosesEslicarbazepine 2–3 20–40 40 400 mg every other Dizziness, headache, CYP inducer, resulting in a Prodrug, with 100% acetate day for first 2 weeks, diplopia 12%–16% increase in of dose converted to ten 400 mg daily clearance of carbamazepine, its main metabolite, lamotrigine, and topiramate eslicarbazepine; not FDA approvedVigabatrin 1 7.5 0 Adults: 500 mg twice Peripheral vision May enhance effects of Half-life of biologic daily, followed by loss central nervous system activity exceeds weekly increases of depressants elimination half-life; 500 mg, up to 3 g/d; biologic half-life for infantile spasms, depends upon 50 mg/kg in two GABA-T resynthesis divided daily doses to start, followed by 25–50 mg/kg per day every 3 days up to 150 mg/kg dailyEzogabine 0.5 8 80 100 mg three times Urinary retention, Unusual two-way interaction Limited experience in (retigabine) daily, followed by dizziness, with lamotrigine clinical practice weekly increases of somnolence not more than 150 mg/d, up to 200–400 mg three times dailyClobazam 1–3 10–30 85 Patients up to 30 kg: Similar to Other inducers affect the Extensive experience 5 mg once daily for benzodiazepine metabolism of clobazam; in clinical practice in 1 week, followed by toxicity polymorphisms exist, Europe; may be less 5 mg twice daily for with slow metabolizers sedating and slower 1 week and then 10 having increased adverse to develop tolerance mg twice daily; effects than other patients over 30 kg: benzodiazepines 10 mg once daily for 1 week, followed by 10 mg twice daily for 1 week and then 20 mg twice daily Perampanel 0.5–1.5 70–100 96 2 mg once daily at Dizziness, headache, CYP enzyme inducers Long half-life means bedtime, followed by somnolence, dose- (carbamazepine, phenytoin, that it can take 14 days increases of 2 mg/d related neuropsych- oxcarbazepine) may decrease to reach steady state; at not less than weekly iatric disturbances plasma perampanel levels by absorption delayed intervals, up to 4–8 mg 50%–67%; levonorgestrel- 2 hours when taken once daily at bedtime containing contraceptives with food; limited may be less effective clinical experience
AEDs = antiepileptic drugs; FDA = US Food and Drug Administration; GABA = γ-aminobutyric acida Oral dosage; lacosamide also may be given intravenously

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 27
PeterPressman,MD Optimal Use of the Newest Antiepileptic Drugs and Generics
drug to carbamazepine and a second-generation agent to oxcarbazepine. It was approved in the EU in 2009 for adjunctive treatment of partial seizures in adults.13 In the United States, the drug is currently being tested in phase III, double-blind, randomized, controlled clinical trials in adults with partial-onset seizures and in phase II trials for possible use in the treat-ment of bipolar disorder.14
Like its forebears, eslicarbazepine acetate is a competitive antagonist of fast voltage-gated potassium channels that stabilizes the inactivated neuronal state.13 The drug also potentiates GABA currents. Unlike oxcarbazepine, esli-carbazepine acetate may be given once daily.15 A greater proportion (95%) of eslicarbazepine is converted into an active metabolite than is oxcarbazepine (80%).10 Eslicarbazepine acetate also may enhance slow inactivation of voltage-dependent sodium channels in a manner similar to that of lacosamide.
VigabatrinVigabatrin, a GABA analog, was ap-
proved in 2009 for adjunctive treatment of adults with refractory complex partial seizure; it was also approved as mono-therapy for pediatric patients 1 month to 2 years of age who have infantile spasm. The benefits of therapy must outweigh the risks of potential vision loss, which occurs in up to one third of patients treated with vigabatrin.
The mechanism of action of vigabatrin is the irreversible inhibition of GABA transaminase, the enzyme responsible for catabolism of GABA in presynaptic terminals and glial cells.16 Vigabatrin also inhibits the vesicular GABA transporter and increases extracellular concentrations of GABA in the central nervous system, which, in turn, increases tonic inhibition in extrasynaptic GABA receptors with prolonged activation.10
Ezogabine(Retigabine)Ezogabine (known as retigabine in
Europe) is the first AED to target and open a voltage-gated potassium channel. By targeting the low-threshold KCNQ (Kv7) channel, ezogabine has a hyperpolarizing
effect on neurons and reduces neuronal hyperexcitability.17 At supratherapeutic concentrations, it also enhances GABAA-activated currents.18
The drug was approved in 2011 as adjunctive therapy for patients over 18 years of age who have been diagnosed with partial seizures and refractory partial epilepsy. A notable side effect related to ezogabine therapy is urinary retention due to the presence of voltage-gated po-tassium channel subunits Kv7.2–Kv7.5 in the bladder urothelium.19,20
ClobazamClobazam is a structurally unique
1,5-benzodiazepine, meaning the nitro-gen atoms in a heterocyclic ring are in the 1 and 5 positions rather than the 1 and 4 positions of older benzodiazepines.21, 22 It was first approved in Australia in 1970; it has been used for years in Europe. The drug was approved by the FDA in 2011 for adjunctive therapy of Lennox-Gastaut syndrome in patients > 2 years of age. Use of this medication has resulted in up to a 70% reduction in drop seizures in these patients.23
Like other benzodiazepines, clobazam potentiates GABAergic neurotransmis-sion by binding to the benzodiazepine site of the GABAA receptor. In comparison with 1,4-benzodiazepines, clobazam is less lipophilic and acidic, better tolerated, and less sedating. In addition, patients using clobazam develop tolerance to the drug more slowly than do those using other AEDs.24
PerampanelPerampanel is the first inotropic
α-amino-3-hydroxy-5-methyl-4-isox-azolepropionic acid (AMPA) glutamate receptor antagonist. As such, it provides researchers with a tool for better under-standing the role of the AMPA receptor in refractory seizure disorders and rep-resents a new therapy for epilepsy. As a selective noncompetitive antagonist of neuronal AMPA receptors, perampanel reduces fast excitatory signaling in the brain critical to generation and spread of epileptic activity.10,25 The drug was first approved in the EU; in October 2012,
perampanel was approved by the FDA for treatment of partial-onset seizures.
Understanding the mechanisms of action of AEDs can assist neurologists in making logical selections of an AED for either monotherapy or polytherapy, and it may prevent drug selections that worsen patient outcomes. Established AEDs have diverse targets; however, about a third of epilepsy patients remain refractory to drug treatment, and failure of one medi-cation predicts the failure of medications in the future.26
We can only hope that as we continue to find new medications, we will see con-tinued benefits for our individual patients and for patients with epilepsy as a whole.
n CLINICAL PHARMACOKINETICS AND DRUG INTERACTIONS
Based on a presentation by Cecilie Johannessen Landmark, PhD, Associate Professor of Pharmacy and Biomedical Science, Faculty of Health Sciences, Oslo University College and Akerhus University College of Applied Sciences, Oslo, Norway
The term pharmacokinetics refers to all processes of a drug after administra-tion, including absorption, distribution, metabolism, and excretion. Each pharma-cokinetic process has distinct parameters to consider, such as the area under the curve (AUC), steady-state concentration, half-life, and volume of distribution (Vd).
When considering the pharmaco-kinetic properties of any medication, especially anticonvulsants, one must keep in mind that pharmacokinetics vary by age, gender, physiologic changes (eg, pregnancy), ethnicity, and environ-mental factors. There is often a 10-fold difference in pharmacokinetic variability among patients given the same dose of an AED.
Much of this variability is determined by pharmacokinetic interactions. Such interactions between AEDs may cause no significant change or may lead to toxicity and adverse effects. The efficacy of one or more drugs may be increased or lost. For example, giving valproic acid and lamotrigine together may result in a synergistic effect. On the other hand, giving two sodium-channel blockers may potentiate their adverse effects. Because

28 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
PeterPressman,MD Optimal Use of the Newest Antiepileptic Drugs and Generics
up to 50% of patients with epilepsy must consider using two or more AEDs at a time to control their seizures, drug inter-actions are an important concern.27 The wide range of possible interactions and consequences reinforces the importance of therapeutic drug monitoring (Table 3).
In addition to interactions with other AEDs, physicians must be cognizant of interactions between these classes of drugs and other medications. For example, interactions with oral contraceptives are important in young women. Patients us-ing oral contraceptives are susceptible to CYP3A4-mediated induction by AEDs such as carbamazepine, felbamate, ox-carbazepine, topiramate, rufinamide, eslicarbazepine acetate, and perampanel.28 Conversely, there is UDP-glucuronosyl-transferase (UGT)-mediated induction of lamotrigine and valproic acid by con-traceptives. Older patients on warfarin should be wary of CYP2C9-mediated induction of warfarin by carbamazepine and eslicarbazepine acetate and inhibition by felbamate and stiripentel.29
Each of the newer AEDs has a distinct pharmacokinetic profile.
LacosamideLacosamide is well absorbed, but its
maximum concentration in the blood
(Tmax) is reached 0.5–4 hours following administration. Lacosamide serum con-centrations have shown high fluctuations during the day, with a steep increase during the first 3 hours after administra-tion.30 Fluctuations during the day may be reduced by taking the drug three times daily instead of two. Such dosing also may improve tolerability in patients who expe-rience adverse reactions from lacosamide.
Lacosamide is available in both oral and intravenous (IV) forms and is pre-dominantly excreted renally.31
RufinamideThere is a nonlinear relationship be-
tween dose and serum concentrations of rufinamide, and its absorption after oral administration is dose-dependent. The bioavailability of the drug depends upon food intake, with less absorption occur-ring in the absence of food. Rufinamide is not highly protein-bound (26%–35%).
The metabolism of rufinamide is non–CYP-dependent hydrolysis with a short half-life; however, the drug has other enzyme-inducing properties, and various inducers affect its metabolism. For example, valproic acid inhibits the metabolism of rufinamide, leading to up to a 50% increase in rufinamide serum concentrations.32 The concentration-dose relationship is nonlinear, and children have about 19% lower serum levels than do adults due to increased clearance.33
Therapeutic drug monitoring is es-pecially recommended when this drug is used, as serum concentrations differ markedly between patients.
EslicarbazepineAcetateEslicarbazepine acetate is a prodrug;
100% of a dose is converted into its active metabolite, eslicarbazepine. Its Tmax is 2–3 hours. Protein binding is about 40%, with a Vd of 2.7 L/kg. There is a linear relation-ship between dose and serum concentra-tion. This medication can induce other CYP isoenzymes, leading to a 12%–16% increase in clearance of carbamazepine, lamotrigine, and topiramate. It also can induce the metabolism of oral contra-ceptives. Excretion of eslicarbazepine is
lower in patients with renal or hepatic impairment.13,14,34,35
VigabatrinThe pharmacokinetics of this irrevers-
ible GABA-transaminase inhibitor are easily learned, because it has a low po-tential for pharmacokinetic interactions. Vigabatrin is 100% bioavailable, with no protein binding and a Tmax of 1 hour. The drug has been used in Europe for many years. Although only more recently did it become available in the United States, vigabatrin has been used to a limited extent, because it has caused irreversible peripheral vision loss in many patients.36
Because vigabatrin is a suicide inhibi-tor of GABA-transaminase, the half-life of biological activity exceeds the half-life of the drug concentration in the serum.37 The half-life of biologic activity likely de-pends most on the regeneration of GABA transaminase, which may take up to 6 days from drug administration.38
EzogabineEzogabine has a higher Vd (6.2 L/kg)
than do many other medications. It is me-tabolized by UGT and N-acetylation. This medication can cause an unusual two-way interaction with lamotrigine, leading to a 20% increase in clearance.39,40
ClobazamClobazam is not a new drug outside of
the United States—it has long been used in Europe. The bioavailability of clobazam is close to 100%, with moderately high protein binding at 85% and a Vd of 1 L/kg. Clobazam is metabolized not only by CYP3A4 but also by CYP2C19. This is important, since there are polymorphisms of this enzyme that could lead to pharma-cogenetic variability. In patients with slow CYP metabolism, use of this drug may lead to increased adverse effects akin to benzodiazepine toxicity. Lower doses are called for in patients with renal or hepatic impairment.36,41
PerampanelPerampanel is generally well absorbed,
but that absorption can be delayed by 2 hours when it is taken with food. It is also
TABLE 3 Effects of Antiepileptic Drug Combinations
Enzyme-inducing Decreaseserumdrugs: concentrationsof:Carbamazepine Other enzyme inducers Phenobarbital ValproateEthosuximide LamotriginePhenytoin TiagabineFelbamate ZonisamideOxcarbazepineTopiramate Rufinamide Enzyme-inhibiting Increaseserumdrugs: concentrationsof:Valproate Other enzyme inhibitors Felbamate CarbamazepineStiripentol LacosamideOxcarbazepine RufinamideTopiramate Ethosuximide Phenobarbital Phenytoin Lamotrigine Rufinamide
Source: Cecilie Johannessen Landmark, PhD; used with permission

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 29
PeterPressman,MD Optimal Use of the Newest Antiepileptic Drugs and Generics
96% protein-bound, in a manner similar to that of valproic acid. This means there are possible displacement interactions between perampanel and other highly bound AEDs.
Perampanel also has a high Vd at 77 L/kg. Perampanel has a long half-life, reaching steady-state serum levels after 14 days. Clearance of perampanel can be in-creased two- to threefold by concomitant use of carbamazepine, oxcarbazepine, or phenytoin. Perampanel is also an inducer of oral contraceptives in women. There is limited experience with this drug in clinical practice.18,42
ImpactonClinicalCareandPracticeThe impact of drug interactions vary
due to considerable differences in phar-macokinetics between patients. In clinical practice, therapeutic drug monitoring is helpful for adjusting dosage if interac-tions occur. Wherever possible, avoiding potentially harmful drug interactions is the best course, along with a discussion with the patient of all potential interac-tions (Table 4).36,43
n EFFICACY AND ADVERSE EFFECTS OF NEWER AEDs IN APPROVED INDICATIONS
Based on a presentation by R. Edward Faught, Jr, MD, Professor of Neurology, Emory University, and Chief of Service, Neurology, Emory University Hospital Midtown, Atlanta, Georgia
Well-known reports have suggested the increasing futility of attempting more antiepileptic therapy after a patient has failed two or three AEDs. The results of lesser-known studies, however, have sug-gested that it may be worthwhile to keep trying new medications in patients with refractory epilepsy. Rates of response (defined as at least a 50% reduction in sei-zures) have demonstrated that even after administration of many ineffective AEDs, at least 26.5% of patients have responded favorably to the use of a new AED.44,45
What most people with epilepsy truly want, however, is to be free of seizures. Whereas < 20% of patients will attain this goal after trying a third or fourth drug, others experience improvement, show-ing that there is always some hope of a
meaningful benefit, even in people with refractory epilepsy.
LacosamideA meta-analysis of clinical trials of
lacosamide demonstrated a leveling of dose response after about 400 mg/d was given.46 There seemed to be a linear rela-tionship between dose and side effects, as discerned by the number of patients leaving the clinical trial. Almost 25% of patients suffered dizziness, the most com-mon side effect, when taking 400 mg/d of lacosamide. This side effect seemed to worsen when lacosamide was combined with another sodium-channel blocker. Other side effects included vertigo, ataxia, balance disorders, coordination abnor-malities, and diplopia.
The recommended dosage of lacos-amide is 50 mg given twice daily for the first week; thereafter, the dosage is in-creased in weekly intervals by 100 mg/d in two divided doses, as tolerated, until a goal of 200–400 mg/d in two divided doses is reached. A more conservative method involves simply cutting these doses in half and taking twice as long to titrate the dose upward.
Advantages of lacosamide include the patient’s ability to take the drug just twice a day, although it may be better tolerated if taken three times daily. Use of the drug has been related to few, if any, drug interac-tions. Lacosamide adds particularly well to levetiracetam, topiramate, or pregaba-lin. A relatively low rate of somnolence,
rash, or cognitive side effects has been noted with its use.
Disadvantages of lacosamide use in-clude its modest effectiveness—treatment at 400 mg/d has led to seizure reduction just 20% greater than that observed with placebo (however, the placebo response was high at > 20%). Dizziness caused by the drug limits the physician’s ability to use it with phenytoin, carbamazepine, oxcarbazepine, or lamotrigine, because these combinations tend to worsen diz-ziness. Monotherapy with lacosamide is not proven, however, and dosage levels for use of this drug as a single agent are not established.
EzogabineEzogabine has the unique method
of action of facilitating and prolonging potassium-channel opening, thereby in-hibiting repetitive neuronal firing.18 Use of 1,200 mg/d resulted in a linear decrease in seizure frequency of up to 35.2%.47
Side effects are generally dose-related and include somnolence, dizziness, and confusion.
Ezogabine has an unusual side effect for an AED, urinary retention, which can be severe. In one study, 8% of treated patients had some complaint of voiding difficulty, and 2% had experienced urinary retention.48 Caution is advised for use of the drug in patients with an enlarged prostate or other problems with urinary voiding. The recommended starting dose is 100 mg three times a day, which should then be titrated upward to a total daily dose of 600–1,200 mg in three divided doses.
Benefits of ezogabine therapy include a novel mechanism of action, no significant drug interactions, renal excretion, a low rate of rash, and a low incidence of cogni-tive complaints. Drawbacks include only modest efficacy, the need to dose three times daily, and urinary retention.
RufinamideRufinamide therapy is particularly suc-
cessful for treating atonic seizures or “drop attacks.” Previously, the most commonly used drugs for this indication included felbamate, lamotrigine, and topiramate.
TABLE 4Propensity of Selected Antiepileptic Drugs to Interact with Other Therapeutic Agents
Drug Low Intermediate High
Clobazam •Eslicarbazepine •acetatea
Ezogabine •(retigabine)Lacosamide •Perampanel •Rufinamidea •Vigabatrin •
a Antiepileptic drugs that may cause drug interactions and have enzyme-inducing propertiesSource: Johannessen and Patsalos36; Bialer et al43

30 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
PeterPressman,MD Optimal Use of the Newest Antiepileptic Drugs and Generics
Felbamate is the most effective of these three drugs; however, it reduces just over 40% of such seizures.49
Rufinamide has a similar success rate in the treatment of atonic seizures in pa-tients with Lennox-Gastaut syndrome but offers a better side-effect profile. Dosing for children starts at 10 mg/kg per day given in two divided doses (target dose, 45 mg/kg per day). In adults, the starting dose is 200 mg twice daily (maximum dose, 3,200 mg/d). Rufinamide is prone to interact with other AEDs.
ClobazamClobazam was designed to produce
less somnolence and tolerance than other benzodiazepines. In patients weighing > 30 kg, dosing ranges from 10 to 40 mg/d given in two divided doses. A conserva-tive starting dose would be 5 mg/d given for 1 week, followed by 5-mg/d increases at weekly intervals to reach a goal of 20 mg/d. At a dose of 1 mg/kg, the reduc-tion in weekly drop seizures was relatively impressive (68%).23 Clobazam often is added to regimens including valproic acid, felbamate, lamotrigine, or topiramate.
n EMERGING USES OF THE NEWER AEDs IN STATUS EPILEPTICUS AND EPILEPSY
Based on a presentation by Howard P. Goodkin, MD, PhD, Shure Professor of Neurology and Pediatrics, Division Director of Pediatric Neurology, and Co-Vice Chair for Research in the Department of Neurology, University of Virginia, Charlottesville, Virginia
Off-label uses of drugs are sometimes regarded with suspicion, especially by those outside of the medical profession. Prescribing medications for an indica-tion not specified by the drug company, however, is sanctioned by the FDA. Regu-lations on drug labeling, as delineated in the Kefauver-Harris Amendment to the Federal Food, Drug, and Cosmetic (FD&C) Act of 1962,50 restrict only the marketing of that drug, stating that “an FDA-approved drug may be labeled, pro-moted, and advertised only for those uses for which the drug’s safety and effective-ness have been established.” The FD&C Act does not limit how a physician may use an approved drug, stating that “‘unap-
proved’ or more precisely ‘unlabeled’ uses may be appropriate and rational in certain circumstances, and may, in fact, reflect approaches to drug therapy that have been extensively reported in the medical literature.”50
Off-label use of medication is both an accepted medical practice and quite com-mon, with 21% of prescriptions written for off-label use.51 Off-label prescription relies on medical judgment and should be performed in “good faith, in the best inter-est of the patient, and without fraudulent intent.”52 Physicians should consider the existence of an equally effective on-label alternative and the rationale for off-label use, including published scientific evi-dence and the standard of care regarding the patient’s condition.52
Within the field of epilepsy, common off-label uses include extension to other syndromes or seizure types, use in status epilepticus, and use of a medication in a pediatric or adult population.
LacosamideEmerging uses of lacosamide include
prescribing for generalized seizures in adults, extending the use of lacosamide to children, and using the drug in patients with status epilepticus.
The use of lacosamide in generalized seizures is supported by case reports and series, as well as evidence of a > 50% decrease in the frequency of epileptic inci-dents in 18 of 24 patients with generalized tonic-clonic seizures.53
At least four studies have investigated the use of lacosamide in children, with two studies examining use of the drug for
focal epilepsy and two investigating its use for a mix of focal and general epilepsy.54–57 Investigators noted 30%–50% reductions in seizure frequency with lacosamide use, but patients frequently dropped out of studies because of side effects.
Results on the use of lacosamide in status epilepticus are conflicting. In one study,58 all seven cases of status epilepticus improved within 24 hours of IV lacos-amide administration. In another,59 none of the treated patients experienced reso-lution of signs and symptoms within the study criteria of 4 hours of IV lacosamide use, and only two experienced a decrease in seizure frequency in the days following. The authors noted one, and possibly two, cases of angioedema related to lacosamide administration. The results of a retro-spective study showed cessation of status epilepticus in 17 of 38 patients (response rate, 45%) given lacosamide, with no ad-verse events.60 In another study, 25 of 31 patients (81%) who received lacosamide had cessation of status epilepticus.61
RufinamideTherapeutic trends now include ad-
junctive treatment of focal seizures in children and adults. Multiple small case reports and case series describe electro-clinical syndromes such as malignant migrating partial epilepsy, epilepsy with myoclonic absence, Dravet syndrome, myoclonic astatic epilepsy, West syn-drome, multifocal encephalopathy with bifrontal spike-wave discharges, and other unspecified symptomatic or cryptogenic generalized epilepsy.62–69 In almost all cases, responses ranged and varied, and no clear picture emerged from the data.
Various studies have investigated the use of rufinamide for adjunctive treatment of focal seizures in children and adults. The largest was a 24-week, multicenter, phase II clinical study of 647 patients 15–65 years of age that featured a 12-week prospective baseline phase before randomization into a double-blind, paral-lel group, five-arm treatment phase.70 A large proportion of subjects completed the study. The primary endpoint of a linear trend for dose response was established, with a similar rate of adverse events ob-
Off-label use of medication is both an accepted medical practice and quite common, with 21% of prescriptions written for off-label use.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 31
PeterPressman,MD Optimal Use of the Newest Antiepileptic Drugs and Generics
served between treatment and placebo arms; headache, fatigue, and dizziness were among the most common adverse events.
A subsequent randomized, double-blind, placebo-controlled, parallel-group, multicenter study was performed, in which rufinamide doses were titrated up to a dose of 3,200 mg/d and then main-tained for an 84-day treatment phase. A significant difference between the treat-ment and placebo groups was found (P = 0.007). Side effects were also greater in the treatment group, with dizziness, fatigue, nausea, somnolence, and diplopia being the most frequent adverse events.71
ClobazamAdditional uses for clobazam currently
being investigated include treatment of other electroclinical syndromes, mono-therapy for focal or generalized seizure in adults, and adjunctive therapy for fo-cal or generalized seizure in adults and children. Other possible indications for the future include status epilepticus and febrile seizures.
Case series have described the use of clobazam in West syndrome, Dravet syndrome, myoclonic astatic epilepsy, Landau-Klefner syndrome, Jeavons syndrome, and unspecified epileptic en-cephalopathies.72–75
Because clobazam has existed since 1970, more primary studies have been done on this drug than on previously dis-cussed AEDs. The largest trial to address the potential use of clobazam in adults and children with focal or generalized seizures was a double-blind, crossover study involving 129 patients in 1987.76 In all, 20 patients became seizure-free. Adverse reactions included drowsiness, dizziness, depressive mood, and aggres-siveness. Another double-blind crossover study done in 1990 that focused on the treatment of children found a > 50% reduction in seizure frequency among 11 of 21 patients.77
SummaryCase series suggest an emerging role
for lacosamide in treating adults with generalized seizures, children with
epilepsy, and patients with status epilep-ticus. Case series suggest an emerging role for rufinamide in treating several electroclinical syndromes, and the re-sults of double-blind, placebo-controlled studies support a role for rufinamide in the adjunctive treatment of focal sei-zures. Results from case series suggest a possible role for clobazam in treating several electroclinical syndromes, febrile seizures, and some forms of status epi-lepticus. Outcomes from double-blind, placebo-controlled trials support a role for clobazam in the adjunctive treatment of focal and generalized seizures in both children and adults.
n GENERIC AEDs: FACTS AND FICTION
Based on a presentation by Michael D. Privitera, MD, Professor of Neurology, University of Cincinnati College of Medicine, and Director, Epilepsy Center, UC Neuroscience Institute, Cincinnati, Ohio
Several million doses of generic AEDs are taken every day by people with epilepsy. Not only do these medications benefit individual patients due to their lower costs, but the FDA estimates that $56.7 billion were saved in 2002 alone due to generic substitution, signifying the power of generic drugs in combating healthcare costs.
For years, neurologists and patient advocates have expressed concern that FDA rules on generic medication allow too much variability across formula-tions of anticonvulsants and, in turn, that these variations cause health prob-lems. According to the FDA, there is no reliable documentation of generic drugs causing problems, and the agency maintains that formulations are safely interchangeable.
In 2007, the American Academy of Neurology (AAN) reiterated the con-cerns of many of its constituents, stating that small variations in concentrations between name brands and their generic equivalents could cause toxic effects or sei-zures when taken by people with epilepsy; however, the AAN did not cite evidence for this claim.78 In 2009, the US Senate Appropriations Committee insisted that the FDA report how it was funding stud-
ies to resolve questions of AED generic equivalence.79
DefinitionsofEquivalenceWhen discussing the controversy
of equivalence between generics and brand-name medications, it is important to be clear about certain terminology. The term bioequivalence means that the pharmacokinetic parameters of the AUC and the maximum serum concentration (Cmax) fall within a certain range. The term bioequivalence usually is used to compare a single generic with a brand-name drug. Therapeutic equivalence means that two products have an equal clinical benefit for a patient. In the case of an AED, this means that two drugs would have equal tolerability and seizure control. Finally, the term switchability means that there is no change in thera-peutic effect when one drug is exchanged for another.
These terms have meanings that are similar, but, in fact, the differences may be quite significant. For example, the FDA requires rigorous testing of bioequiva-lence, but it does not demand proof of therapeutic equivalence. It is assumed that if the plasma concentration-time curve and Cmax fall within specified limits, thera-peutic equivalence will follow. Typically, bioequivalence studies are done using one dose in healthy adults—not in people with epilepsy or those using concomitant medications or with comorbid conditions. As long as both the AUC and Cmax fall within 80%–125% of those of the brand-name drug, the generic drug passes FDA standards. Most generic drugs do very well in meeting those standards.
TheCruxoftheProblemThe argument of many neurologists
essentially is that the FDA standards of bioequivalence do not reliably lead to actual therapeutic equivalence or switch-ability. The research literature is full of retrospective studies supporting this position. In Canada, switching from a brand-name AED to its generic equivalent has been associated with a higher rate of switching back to the original medication, when compared with statins for treating

32 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
PeterPressman,MD Optimal Use of the Newest Antiepileptic Drugs and Generics
hyperlipidemia or selective serotonin reuptake inhibitors for treating depres-sion, presumably due to lack of tolerability or diminished efficacy when switching AEDs.80 Furthermore, use of generic AEDs has been associated with a greater need for emergency services.81 In another study, switching to generics was found to have no effect on epilepsy-related events.82
An assessment of five generic carbam-azepine products was performed on data obtained via the Freedom of Information Act.83 A model using these data demon-strated AUC variations of up to 21% and variations in Cmax reaching 40%.
On the other hand, there are sev-eral caveats to these retrospective studies. Switchbacks may have resulted from in-correct attribution to the generic medica-tion by physicians or patients. There was no control in these studies for adherence, stress, or sleep deprivation. There is also a possible placebo effect, in which the state-ment by physicians that a generic might not perform as well as a brand-name drug helped to bring about those results. There has been no rigorous assessment of seizure frequency or blood levels of AEDs.
ImprovingEquivalenceResearchGiven the current state of evidential
equipoise regarding the true therapeutic equivalence and switchability of generic and brand-name AEDs, an opportunity exists for better controlled studies. Three such study protocols have been designed and are currently being enacted. All of these studies are investigating the generic forms of lamotrigine.
The FDA funded the first two research projects, which are combined to form the Equivalence Among Antiepileptic Drug Generic and Brand Products in People with Epilepsy (EQUIGEN) studies. The most disparate generic products were recommended using the abbreviated new drug application data given to the FDA for generic approval plus dissolution charac-teristics. The first study looked at chronic dosing in people with epilepsy, comparing a high-range generic with a low-range generic. The study of chronic medication dosing is more like real life, but there was a concern that such a chronic study may
introduce more variables that enhance or minimize pharmacokinetic differences. Mixing one dose of a drug could alter the bioequivalence results. The second study, then, was a single-dose study in patients with epilepsy to allow for the potential effects of concomitant medications.
The third study is being managed by the University of Maryland. Called BEEP, it is comparing the brand-name drug Lamictal with the most commonly dispensed generic version of lamotrigine, marketed by Teva.
Results of these studies are expected early in 2013. This research is focusing on bioequivalence rather than therapeu-tic equivalence. Even if bioequivalence is solidly established, the question of therapeutic equivalence will remain. A series of studies still needs to be done on therapeutic equivalence as well.
Although more data are pending, it is recommended that physicians research cost differences and recognize that pa-tients who are pregnant, have a history of status epilepticus, or are seizure-free and driving a vehicle are at higher risk than others. Patients should be counseled about unauthorized formulation substitution and the need to call their physician if the pills in a newly refilled prescription look different from those obtained previously. This opportunity can also be used to coun-sel patients on medication adherence and avoidance of such triggers as alcohol and sleep deprivation.
n CONCLUSION
Over the past few years, new medi-cations have been approved to treat epilepsy. Many of these new drugs have novel mechanisms of action, such as tar-geting AMPA receptors or voltage-gated potassium channels. In addition to their indications for medication-resistant par-tial seizures or Lennox-Gastaut syndrome, many of these drugs have additional emerging uses. Such off-label uses include status epilepticus as well as idiopathic generalized seizures.
In addition to understanding new brand-name therapeutics, physicians also must learn the best way to integrate generic medications into their practices.
Potential risks of switching from a brand-name drug to a generic medication or between different generics and potential cost savings related to such switches must be discussed with patients.
REFERENCES
1. England MJ, Liverman CT, Schultz AM, Strawbridge LM. Epilepsy across the spectrum: Promoting health and understanding: a summary of the Institute of Medicine report. Epilepsy Behav. 2012;25:266–276.
2. Kwan P, Brodie MJ. Refractory epilepsy: a progressive, intractable but preventable condition? Seizure. 2002;11:77–84.
3. Ryvlin P, Rheims S. Epilepsy surgery: eligibil-ity criteria and presurgical evaluation. Dialogues Clin Neurosci. 2008;10:91–103.
4. Chiron C, Dulac O. The pharmacologic treat-ment of Dravet syndrome. Epilepsia. 2011;52(suppl 2):72–75.
5. Plosker G. Stiripentol: in severe myoclonic epilepsy of infancy (Dravet syndrome). CNS Drugs. 2012;26:993–1001.
6. Nabbout R, Chiron C. Stiripentol: an example of antiepileptic drug development in childhood epilepsies. Eur J Paediatr Neur. 2012;16(suppl 1):S13–S17.
7. Chiron C, Marchand MC, Tran A, et al. Sti-ripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet. 2000;356:1638–1642.
8. Fisher J. Interactions between modulators of the GABA(A) receptor: stiripentol and benzodiaz-epines. Eur J Pharmacol. 2011;654:160–165.
9. Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS. Progress report on new antiepileptic drugs: a summary of the Ninth EILAT Conference (EILAT IX). Epilepsy Res. 2009;83:1–43.
10. Bialer M, White HS. Key factors in the dis-covery and development of new antiepileptic drugs. Nat Rev Drug Discov. 2010;9:68–82.
11. Fleidervish IA, Friedman A, Gutnick MJ. Slow inactivation of Na+ current and slow cumulative spike adaptation in mouse and guinea-pig neocorti-cal neurones in slices. J Physiol. 1996;493:83–97.
12. White HS, Franklin MR, Kupferberg HJ, Schmutz M, Stables JP, Wolf HH. The anticonvulsant profile of rufinamide (CGP 33101) in rodent seizure models. Epilepsia. 2008;49:1213–1220.
13. Bialer M, Soares-da-Silva P. Pharmacokinet-ics and drug interactions of eslicarbazepine acetate. Epilepsia. 2012;53:935–946.
14. Almeida L, Falcao A, Maia J, Mazur D, Gellert M, Soares-da-Silva P. Single-dose and steady-state pharmacokinetics of eslicarbazepine acetate (BIA 2-093) in healthy elderly and young subjects. J Clin Pharmacol. 2005;45:1062–1066.
15. Rauchenzauner M, Luef G. Eslicarbazepine acetate for partial-onset seizures. Expert Rev Neu-rother. 2011;11:1673–1681.
16. Rogawski MA, Loscher W. The neuro-biology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–564.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 33
PeterPressman,MD Optimal Use of the Newest Antiepileptic Drugs and Generics
17. Brodie MJ, Lerche H, Gil-Nagel A, et al. Efficacy and safety of adjunctive ezogabine (reti-gabine) in refractory partial epilepsy. Neurology. 2010;75:1817–1824.
18. Stephen LJ, Brodie MJ. Pharmacotherapy of epilepsy: newly approved and developmental agents. CNS Drugs. 2011;25:89–107.
19. Gil-Nagel A, Brodie MJ, Leroy R, et al. Safety and efficacy of ezogabine (retigabine) in adults with refractory partial-onset seizures: interim results from two ongoing open-label studies. Epilepsy Res. 2012;102:117–121.
20. Porter RJ, Burdette DE, Gil-Nagel A, et al. Retigabine as adjunctive therapy in adults with par-tial-onset seizures: integrated analysis of three pivot-al controlled trials. Epilepsy Res. 2012;101:103–112.
21. Sankar R. GABA(A) receptor physiology and its relationship to the mechanism of action of the 1,5-benzodiazepine clobazam. CNS Drugs. 2012;26:229–244.
22. Seif-Eddeine H, Ng Y-T. Clobazam for pa-tients with Lennox-Gastaut syndrome and epilepsy. Expert Rev Neurother. 2012;12:385–393.
23. Ng YT, Conry JA, Drummond R, Stolle J, Weinberg MA. Randomized, phase III study results of clobazam in Lennox-Gastaut syndrome. Neurol-ogy. 2011;77:1473–1481.
24. Buck ML. Clobazam: a new option for children with refractory seizures. Pediatric Phar-macotherapy. December 2011. Available at: http://www.medicine.virginia.edu/clinical/departments/pediatrics/education/pharm-news/current/201112.pdf. Accessed January 8, 2013.
25. Krauss GL, Serratosa JM, Villanueva V, et al. Randomized phase III study 306: adjunctive perampanel for refractory partial-onset seizures. Neurology. 2012;78:1408–1415.
26. Brodie MJ, Barry SJE, Bamagous GA, Norrie JD, Kwan P. Patterns of treatment response in newly diagnosed epilepsy. Neurology. 2012;78:1548–1554.
27. Stephen LJ, Brodie MJ. Antiepileptic drug monotherapy versus polytherapy: pursuing seizure freedom and tolerability in adults. Curr Opin Neurol. 2012;25:164–172.
28. Dutton C, Foldvary-Schaefer N. Contracep-tion in women with epilepsy: pharmacokinetic in-teractions, contraceptive options, and management. Int Rev Neurobiol. 2008;83:113–134.
29. Chaudhry AS, Urban TJ, Lamba JK, et al. CYP2C9*1B promoter polymorphisms, in linkage with CYP2C19*2, affect phenytoin autoinduction of clearance and maintenance dose. J Pharmacol Exp Ther. 2010;332:599–611.
30. Sattler A, Schaefer M, May TW, Rambeck B, Brandt C. Fluctuation of lacosamide serum concen-trations during the day and occurrence of adverse drug reactions—first clinical experience. Epilepsy Res. 2011;95:207–212.
31. Ben-Menachem E, Biton V, Jatuzis D, Abou-Khalil B, Doty P, Rudd GD. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48:1308–1317.
32. Perucca E, Cloyd J, Critchley D, Fuseau E. Rufinamide: clinical pharmacokinetics and concentration-response relationships in patients with epilepsy. Epilepsia. 2008;49:1123–1141.
33. May TW, Boor R, Rambeck B, Jurgens U, Korn-Merker E, Brandt C. Serum concentrations of rufinamide in children and adults with epilepsy: the influence of dose, age, and comedication. Ther Drug Monit. 2011;33:214–221.
34. Falcao A, Fuseau E, Nunes T, Almeida L, Soares-da-Silva P. Pharmacokinetics, drug interactions and exposure-response relationship of eslicarbazepine acetate in adult patients with partial-onset seizures: population pharmacokinetic and pharmacokinetic/pharmacodynamic analyses. CNS Drugs. 2012;26:79–91.
35. Fontes-Ribeiro C, Nunes T, Falcao A, et al. Eslicarbazepine acetate (BIA 2-093): relative bio-availability and bioequivalence of 50 mg/mL oral suspension and 200mg and 800mg tablet formula-tions. Drugs RD. 2005;6:253–260.
36. Johannessen Landmark C, Patsalos PN. Drug interactions involving the new second- and third-generation antiepileptic drugs. Expert Rev Neurother. 2010;10:119–140.
37. Lindberger M, Luhr O, Johannessen SI, Lars-son S, Tomson T. Serum concentrations and effects of gabapentin and vigabatrin: observations from a dose titration study. Ther Drug Monit. 2003;25:457–462.
38. Sheean G, Schramm T, Anderson DS, Eadie MJ. Vigabatrin--plasma enantiomer con-centrations and clinical effects. Clin Exp Neurol. 1992;29:107–116.
39. Hermann R, Knebel NG, Niebch G, Rich-ards L, Borlak J, Locher M. Pharmacokinetic interac-tion between retigabine and lamotrigine in healthy subjects. Eur J Clin Pharmacol. 2003;58:795–802.
40. Yamada M, Welty TE. Ezogabine: an evalua-tion of its efficacy and safety as adjunctive therapy for partial-onset seizures in adults. Ann Pharmacother. 2012;46:1358–1367.
41. Johannessen Landmark C, Johannessen SI, Tomson T. Host factors affecting antiepileptic drug delivery-pharmacokinetic variability. Adv Drug Deliv Rev. 2012;64:896–910.
42. Hibi S, Ueno K, Nagato S, et al. Discovery of 2-(2-oxo-1-phenyl-5-pyridin-2-yl-1,2-dihydro-pyridin-3-yl)benzonitrile (perampanel): a novel, noncompetitive alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropanoic acid (AMPA) receptor antago-nist. J Med Chem. 2012;55:10584–10600.
43. Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS. Progress report on new anti-epileptic drugs: a summary of the Tenth EILAT Con-ference (EILAT X). Epilepsy Res. 2010;92:89–124.
44. Schiller Y, Najjar Y. Quantifying the re-sponse to antiepileptic drugs: effect of past treatment history. Neurology. 2008;70:54–65.
45. Callaghan BC, Anand K, Hesdorffer D, Hauser WA, French JA. Likelihood of seizure remis-sion in an adult population with refractory epilepsy. Ann Neurol. 2007;62:382–389.
46. Zaccara G, Perucca P, Loiacono G, Giovan-nelli F, Verrotti A. The adverse event profile of lacos-amide: a systematic review and meta-analysis of ran-domized controlled trials. Epilepsia. 2013;54:66–74.
47. Porter RJ, Partiot A, Sachdeo R, Nohria V, Alves WM. Randomized, multicenter, dose-ranging trial of retigabine for partial-onset seizures. Neurol-ogy. 2007;68:1197–1204.
48. US Food and Drug Administration. NDA 22-345 Potiga (ezogabine) tablets: appendix A, REMS. March 9, 2021. http://www.fda.gov/down-loads/Drugs/DrugSafety/PostmarketDrugSafety-InformationforPatientsandProviders/UCM261933.pdf. Accessed January 8, 2013.
49. Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70:1950–1958.
50. Use of unapproved drugs for approved indications. FDA Drug Bull. 1982;12:5–6.
51. Radley D, Finkelstein S, Stafford R. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166:1021–1026.
52. American Academy of Pediatrics Com-mittee on Drugs. Uses of drugs not described in the package insert (off-label uses). Pediatrics. 2002;110:181–183.
53. Harden CL, Cohn A, Lowe M, Serrano E. Initial post marketing experience with lacos-amide in adult patients with epilepsy. Epilepsy Res. 2012;98:260–263.
54. Gavatha M, Ioannou I, Papavasiliou AS. Effi-cacy and tolerability of oral lacosamide as adjunctive therapy in pediatric patients with pharmacoresistant focal epilepsy. Epilepsy Behav. 2011;20:691–693.
55. Guilhoto LM, Loddenkemper T, Gooty VD, et al. Experience with lacosamide in a series of children with drug-resistant focal epilepsy. Pediatr Neurol. 2011;44:414–419.
56. Heyman E, Lahat E, Levin N, Berkovitch M, Gandelman-Marton R. Preliminary efficacy and safety of lacosamide in children with refractory epi-lepsy. Eur J Paediatr Neurol. 2012;16:15–19.
57. Rastogi RG, Ng YT. Lacosamide in refrac-tory mixed pediatric epilepsy: a prospective add-on study. J Child Neurol. 2012;27:492–495.
58. Albers JM, Möddel G, Dittrich R, et al. Intravenous lacosamide—an effective add-on treatment of refractory status epilepticus. Seizure. 2011;20:428–430.
59. Goodwin H, Hinson HE, Shermock KM, Karanjia N, Lewin JJ 3rd. The use of lacosamide in refractory status epilepticus. Neurocrit Care. 2011;14:348–353.
60. Kellinghaus C, Berning S, Immisch I, et al. Intravenous lacosamide for treatment of status epilepticus. Acta Neurol Scand. 2011;123:137–141.
61. Hofler J, Unterberger I, Dobesberger J, Kuchukhidze G, Walser G, Trinka E. Intravenous lacosamide in status epilepticus and seizure clusters. Epilepsia. 2011;52:e148–e152.
62. Vendrame M, Poduri A, Loddenkemper T, Kluger G, Coppola G, Kothare SV. Treatment of malignant migrating partial epilepsy of infancy with rufinamide: report of five cases. Epileptic Disord. 2011;13:18–21.
63. Hausler M, Kluger G, Nikanorova M. Epilepsy with myoclonic absences—favourable response to add-on rufinamide treatment in 3 cases. Neuropediatrics. 2011;42:28–29.
64. Coppola G. Update on rufinamide in childhood epilepsy. Neuropsychiatr Dis Treat. 2011;7:399–407.
65. Joseph JR, Schultz RJ, Wilfong AA. Ru-

34 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
PeterPressman,MD Optimal Use of the Newest Antiepileptic Drugs and Generics
finamide for refractory epilepsy in a pediatric and young adult population. Epilepsy Res. 2011;93:87–89.
66. Kluger G, Kurlemann G, Haberlandt E, et al. Effectiveness and tolerability of rufinamide in children and adults with refractory epilepsy: first Eu-ropean experience. Epilepsy Behav. 2009;14:491–495.
67. Mueller A, Boor R, Coppola G, et al. Low long-term efficacy and tolerability of add-on rufin-amide in patients with Dravet syndrome. Epilepsy Behav. 2011;21:282–284.
68. Olson HE, Loddenkemper T, Vendrame M, et al. Rufinamide for the treatment of epileptic spasms. Epilepsy Behav. 2011;20:344–348.
69. von Stulpnagel C, Coppola G, Striano P, Muller A, Staudt M, Kluger G. First long-term expe-rience with the orphan drug rufinamide in children with myoclonic-astatic epilepsy (Doose syndrome). Eur J Paediatr Neurol. 2012;16:459–463.
70. Elger CE, Stefan H, Mann A, Narurkar M, Sun Y, Perdomo C. A 24-week multicenter, random-ized, double-blind, parallel-group, dose-ranging study of rufinamide in adults and adolescents with inadequately controlled partial seizures. Epilepsy Res. 2010;88:255–263.
71. Biton V, Krauss G, Vasquez-Santana B, et al. A randomized, double-blind, placebo-controlled,
parallel-group study of rufinamide as adjunctive therapy for refractory partial-onset seizures. Epi-lepsia. 2011;52:234–242.
72. Sheth RD, Ronen GM, Goulden KJ, Pen-ney S, Bodensteiner JB. Clobazam for intractable pediatric epilepsy. J Child Neurol. 1995;10:205–208.
73. Schmidt D. Clobazam for treatment of intractable epilepsy: a critical assessment. Epilepsia. 1994;35(suppl 5):S92–S95.
74. Perry M, Bailey L, Malik S, Gilson C, Kote-cha A, Hernandez A. Clobazam for the treatment of intractable epilepsy in children. J Child Neurol. 2013;28:34–39.
75. Silva RC, Montenegro MA, Guerreiro CAM, Guerreiro MM. Clobazam as add-on therapy in children with epileptic encephalopathy. Can J Neurol Sci. 2006;33:209–213.
76. Koeppen D, Baruzzi A, Capozza M, et al. Clobazam in therapy-resistant patients with partial epilepsy: a double-blind placebo-controlled cross-over study. Epilepsia. 1987;28:495–506.
77. Keene DL, Whiting S, Humphreys P. Clobazam as an add-on drug in the treatment of refractory epilepsy of childhood. Can J Neurol Sci. 1990;17:317–319.
78. Liow K, Barkley GL, Pollard JR, Harden CL,
Bazil CW. Position statement on the coverage of anticonvulsant drugs for the treatment of epilepsy. Neurology. 2007;68:1249–1250.
79. Eleventh US Congress. Agriculture, Rural Development, Food and Drug Administration, and Related Agencies Appropriations Bill, 2010. Public Law 111-80. October 21, 2009. Available at: http://www.gpo.gov/fdsys/pkg/PLAW-111publ80/pdf/PLAW-111publ80.pdf. Accessed January 8, 2013.
80. Duh MS, Andermann F, Paradis PE, Weiner J, Manjunath R, Cremieux PY. The economic con-sequences of generic substitution for antiepileptic drugs in a public payer setting: the case of lamotrig-ine. Dis Manag. 2007;10:216–225.
81. Zachry WM, 3rd, Doan QD, Clewell JD, Smith BJ. Case-control analysis of ambulance, emergency room, or inpatient hospital events for epilepsy and antiepileptic drug formulation changes. Epilepsia. 2009;50:493–500.
82. Devine ST, Weisbart E, Barron J, Behm A. Acute epilepsy exacerbations in patients switched between A-rated anti-epileptic drugs. Curr Med Res Opin. 2010;26:455–463.
83. Krauss GL, Caffo B, Chang YT, Hendrix CW, Chuang K. Assessing bioequivalence of generic antiepilepsy drugs. Ann Neurol. 2011;70:221–228.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 35
Recently, the US Food and Drug Administration and European Medicines Agency granted marketing approval for the use
of perampanel as adjunctive therapy for partial-onset seizures with or without secondary generalized seizures in patients ≥ 12 years of age. This noncompeti-tive, selective antagonist of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) is mainly metabolized by the cytochrome P 3A4 (CYP3A4) system. Clearance of the drug is increased in pa-tients who are also using carbamazepine, oxcarbazepine, and/or phenytoin.1
At the 66th Annual Meeting of the American Epilepsy Society held recently in San Diego, California, experts in the management of epilepsy reviewed sev-eral studies that analyzed data from three
important research projects to investigate different facets of adjunctive perampanel therapy in patients with partial-onset seizures with or without secondary gener-alized seizures. The presenters addressed responder rates, seizure freedom, seizure frequency, efficacy and safety among patients residing in North America, and the effect of perampanel therapy on patient quality of life (QOL). One other study investigated the impact of delayed or missed perampanel doses on plasma drug levels using computer simulation involving pharmacokinetic data.
n INVESTIGATING THE EFFECTS OF PERAMPANEL
Investigators involved in three placebo-controlled, phase III trials—studies 304 [ClinicalTrials.gov ID # NCT00699972],2 305 [ID # NCT00699582],3 and 306 [ID # NCT00700310]4—reported primary data on the adjunctive use of perampanel in patients with refractory partial seizures. In these studies, patients ≥ 12 years of age who were diagnosed with uncontrolled partial seizures despite treatment with up to three antiepileptic drugs (AEDs) were randomized to receive 8 or 12 mg of perampanel or placebo (studies: 304
[North America, Central America, South America] and 305 [Europe, Asia, South Africa, North America, Australia]) or 2, 4, or 8 mg of perampanel or placebo (study 306 [Australia, Europe, and Asia]).
These studies included three periods: 6 weeks for baseline therapy, 6 weeks for dosage titration, and 13 weeks for mainte-nance therapy. Patients used daily diaries to keep track of seizure activity.
ResponderRatesandFreedomfromSeizures
Ben-Menachem and others5 reported pooled analyses of responder rates and seizure freedom as found in these clinical trials. In addition, the researchers assessed the impact of concomitant AED therapy on responder rates in patients treated with adjunctive perampanel.
In the intent-to-treat (ITT) data-set, which included all randomized and treated patients having seizure data, the investigators assessed responder rates (ie, percentages of patients having ≥ 50% or ≥ 75% reductions in the frequency of all partial seizures, complex partial plus sec-ondary generalized seizures, or secondary generalized seizures only) as compared with baseline. They also classified seizure freedom during maintenance therapy for all individuals with partial seizures and secondary generalized seizures in the maintenance completer dataset.
In all, 1,480 patients were randomized and treated. Among the 1,478 patients in the pooled ITT analysis dataset, adminis-tration of 4–12 mg of perampanel resulted in greater 50% and 75% responder rates and seizure freedom rates than did use
Update on Recent Global Studies of Perampanel, a New Selective AMPA AntagonistLisa Aenlle-Matusz, MD, MPHEmory University School of Medicine, Atlanta, Georgia
Abstract Several posters at the 66th Annual Meeting of the American Epilepsy Society presented analyses of three important placebo-controlled studies in-vestigating the adjunctive use of perampanel in patients with refractory partial seizures. The investigators reviewed data on responder rates, seizure freedom, seizure frequency, efficacy and safety among patients living in North America, and quality of life. In addition, the results of a computer simulation using perampanel pharmacokinetic data to determine the impact of delayed or missed doses on plasma drug levels were discussed.
Dr. Aenlle-Matusz is a Clinical Neurophysiology/Epilepsy Fellow at Emory University School of Medicine, Atlanta, Georgia.

36 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
LisaAenlle-Matusz,MD,MPH Update on Recent Global Studies of Perampanel, a New Selective AMPA Antagonist
TABLE 1Response Rates for All Partial-onset, Complex Partial, and Secondary Generalized Seizures (Baseline vs Maintenance Period)
75%responserate,n/N(%)
Perampanel Perampanel Perampanel Perampanel Placebo 2mg/d 4mg/d 8mg/d 12mg/d
All partial-onset 27/441 (6.1) 18/180 (10.0) 2/172 (12.2)a 75/431 (17.4)b 43/254 (16.9)b
seizuresCP + SG seizures 44/405 (10.9) 25/167 (15.0) 28/157 (17.8)a 82/393 (20.9)b 48/233 (20.6)b
SG seizures 42/173 (24.3) 22/68 (32.4) 22/71 (31.0) 73/157 (46.5)b 37/95 (39.9)a
CP + SG = complex partial with or without secondary generalization; SG = complex partial with secondary generalizationa P < 0.06 vs placebob P < 0.001 vs placeboSource: Ben-Menachem et al5
of placebo (Table 1).5 Response rates for all patients with partial-onset seizures improved at higher perampanel doses, irrespective of the concomitant AEDs they used.
Previous analyses showed a benefit of 8 mg of perampanel over 12 mg of the drug.6 Furthermore, pharmacokinetic and pharmacodynamic data showed a linear exposure-efficacy relationship across doses of 2–12 mg no matter what AEDs were used concomitantly.7
Patients used a mean of 2.2 concomi-tant AEDs at baseline, most commonly carbamazepine, valproic acid, lamotrig-ine, and levetiracetam. The improve-ments in 50% responder rates observed with perampanel 8 mg or 12 mg versus placebo were roughly the same regardless of which of the four most common AEDs were used. There were, however, greater improvements with concomitant valproic acid, lamotrigine, or levetiracetam use than with concomitant carbamazepine therapy. Carbamazepine increases per-ampanel clearance but does not affect the relationship between perampanel plasma levels and therapeutic response.8 In comparison with the placebo group, patients given 4 mg of perampanel showed improvement in all partial-onset seizures and complex partial with secondary gen-eralized seizures when valproic acid or lamotrigine also was given.
SeizureFrequencyUsing these same three placebo-con-
trolled phase III studies 304,2 305,3 and 306,4 Kwan et al9 pooled data to investigate changes in seizure frequency by seizure
type and concomitant AEDs used.The mean age of the patients enrolled
in these three studies was 35 years, and the mean time since diagnosis of epilepsy was 21 years. The median seizure frequency over 28 days was 11.6 for partial-onset seizures, 8.2 for complex partial and sec-ondary generalized seizures, and 3.4 for secondary generalized seizures.
The ITT analysis included 1,478 pa-tients. At baseline, most of these patients were receiving at least two AEDs con-comitantly, most commonly carbamaze-pine, valproic acid, lamotrigine, and/or levetiracetam. The investigators analyzed the median percentage change in seizure frequency over a 28-day period by ran-domized dose.
When compared with placebo, ad-ministration of 4–12 mg of adjunctive perampanel resulted in a decreased frequency of all types of partial-onset seizures and secondary generalized sei-zures; these reductions were found in the general population and in patient subgroups receiving any of the four AEDs most commonly given. The great-est differences were seen among patients using 8 mg of perampanel as compared with a placebo group for all partial-onset seizures (–28.8% vs –12.8%, respectively), complex partial seizures with secondary generalized seizures (–35.6% vs –13.9%), and secondary generalized seizures only (–62.9% vs –19.4%). There did not appear to be a significant difference in outcome among patients using 8 mg of perampanel and those using 12 mg of the drug; how-ever, other analyses involving both actual dose and within-patient responses have
shown a 12-mg dose of perampanel to be more beneficial than an 8-mg dose.6
EfficacyandSafetyinNorthAmericanPatients
Vasquez et al10 presented data show-ing once-daily adjunctive perampanel therapy to be effective in patients with treatment-resistant partial-onset seizures. Information on North American patients (United States, 294 patients; Canada, 25 patients) was derived from the two phase III North American studies (studies 3042 and 3053). The majority of patients had complex partial seizures, both with and without secondary generalization.
The studies were conducted in three phases that included prerandomization (baseline), double-blind treatment, and follow-up. The study participants were ≥ 12 years of age and had refractory partial-onset seizures despite treatment with up to three concomitant AEDs (unpublished data, December 2011). The primary efficacy endpoint was median percent change in seizure frequency per 28 days during the double-blind treatment phase versus baseline. The secondary endpoints were safety and 50% responder rate, defined as the proportion of patients experiencing a ≥ 50% reduction in seizure frequency per 28 days in the maintenance period when compared with baseline (unpublished data, December 2011). Safety analyses included incidence rates of treatment-emergent adverse events and reasons for discontinuation (unpublished data). The average age, gender, race, and seizure types were comparable across all perampanel dosage subgroups, including the placebo group.
The overall phase III ITT population (n = 318) and North American cohort (n = 319) had long-standing disease (mean duration, 21.1 years and 23.9 years, respectively), often with no known etiology (48.4% and 53.8%, respectively; unpublished data, March 2011). Most of the participants had complex partial seizures (85.5% and 89.6%) and a history of seizures with secondary generalization (69.5% and 72%; unpublished data, March 2011). The majority of participants were using two enzyme-inducing or noninduc-
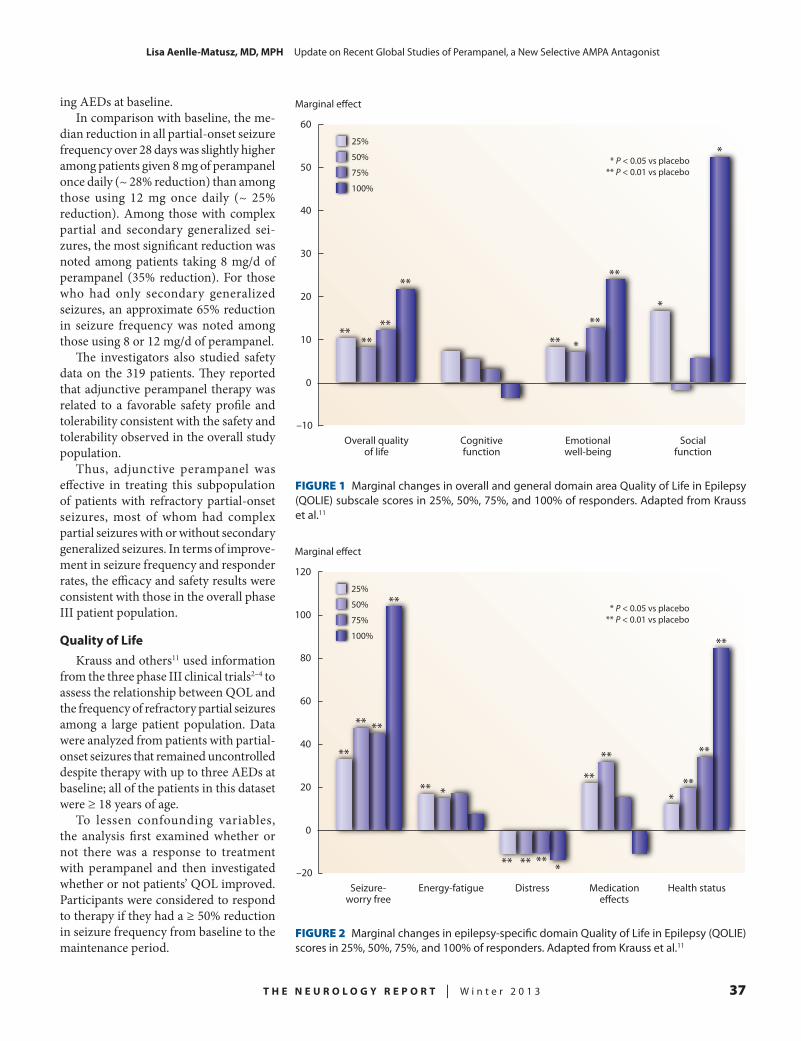
T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 37
LisaAenlle-Matusz,MD,MPH Update on Recent Global Studies of Perampanel, a New Selective AMPA Antagonist
FIGURE 1 Marginal changes in overall and general domain area Quality of Life in Epilepsy (QOLIE) subscale scores in 25%, 50%, 75%, and 100% of responders. Adapted from Krauss et al.11
FIGURE 2 Marginal changes in epilepsy-specific domain Quality of Life in Epilepsy (QOLIE) scores in 25%, 50%, 75%, and 100% of responders. Adapted from Krauss et al.11
Marginal e�ect
30
50
40
60
20
10
0
–10Overall quality
of lifeCognitivefunction
Emotionalwell-being
Socialfunction
25%
50%
75%
100%
* P < 0.05 vs placebo** P < 0.01 vs placebo
****
**
**
** *
**
**
*
*
Marginal e�ect
60
100
80
120
40
20
0
–20Seizure-
worry freeEnergy-fatigue Distress Medication
e�ectsHealth status
25%
50%
75%
100%
* P < 0.05 vs placebo** P < 0.01 vs placebo
**
** **
**
** *
*** ** **
**
**
**
*
**
**
ing AEDs at baseline.In comparison with baseline, the me-
dian reduction in all partial-onset seizure frequency over 28 days was slightly higher among patients given 8 mg of perampanel once daily (~ 28% reduction) than among those using 12 mg once daily (~ 25% reduction). Among those with complex partial and secondary generalized sei-zures, the most significant reduction was noted among patients taking 8 mg/d of perampanel (35% reduction). For those who had only secondary generalized seizures, an approximate 65% reduction in seizure frequency was noted among those using 8 or 12 mg/d of perampanel.
The investigators also studied safety data on the 319 patients. They reported that adjunctive perampanel therapy was related to a favorable safety profile and tolerability consistent with the safety and tolerability observed in the overall study population.
Thus, adjunctive perampanel was effective in treating this subpopulation of patients with refractory partial-onset seizures, most of whom had complex partial seizures with or without secondary generalized seizures. In terms of improve-ment in seizure frequency and responder rates, the efficacy and safety results were consistent with those in the overall phase III patient population.
QualityofLifeKrauss and others11 used information
from the three phase III clinical trials2–4 to assess the relationship between QOL and the frequency of refractory partial seizures among a large patient population. Data were analyzed from patients with partial-onset seizures that remained uncontrolled despite therapy with up to three AEDs at baseline; all of the patients in this dataset were ≥ 18 years of age.
To lessen confounding variables, the analysis first examined whether or not there was a response to treatment with perampanel and then investigated whether or not patients’ QOL improved. Participants were considered to respond to therapy if they had a ≥ 50% reduction in seizure frequency from baseline to the maintenance period.

38 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
LisaAenlle-Matusz,MD,MPH Update on Recent Global Studies of Perampanel, a New Selective AMPA Antagonist
The investigators administered the Quality of Life in Epilepsy (QOLIE)-31P (Santa Monica, CA; RAND Health Communications) questionnaire at baseline and at the end of treatment. This questionnaire contained 30 ques-tions across the seven subscales of seizure-worry, overall QOL, emotional well-being, energy-fatigue, cognitive function, medication effects, and social function and one item on overall health status. The investigators noted significant improvements in QOL (≥ 50% reduction of seizure frequency) both in patients receiving placebo and those given per-ampanel. Further, 33.8% of 903 patients given perampanel were responders. To be considered clinically meaningful in terms of QOLIE-31P scores, the investigators used the previously defined minimally important difference (MID) for QOLIE-31P of 11.8% change in overall score.12
Of the 1,478 patients in the pooled ITT analysis set, 959 perampanel-treated pa-tients from the three studies had QOLIE-31P scores. Complete data for inclusion in the multivariate analysis were available for 742 patients. Overall, 62.4% of patients had baseline QOLIE-31P scores < 50, and the mean improvement at end of treat-ment was 12.5% (P = 0.01).
The mean time since epilepsy diagnosis in this population was 20 years, and the mean age varied from 34–38 years of age by dosage of perampanel. Most patient demographics and clinical characteristics were relatively equally distributed among the different treatment groups. However, the baseline mean seizure frequency var-ied greatly among treatment groups (placebo group, 27 seizures; 4 mg of perampanel, 70 seizures). This difference may contribute to the finding that patients given 4 mg of perampanel did not show as much of a reduction in seizure frequency as did those given 8 or 12 mg of the drug.
Marginal changes in overall QOLIE-31P scores and the domains of emotional well-being, seizure worry, energy-fatigue, distress, medication effects, and overall health status were significantly improved in 50% of responders when compared with nonresponders (Figures 1 and 2).11 In all responders, as response rates increased
from 20% to 100%, there was a greater increase in marginal overall QOLIE-31P score changes and in the general domain areas of emotional well-being and social function (Figure 1).11 In epilepsy-specific domains (seizure-worry and health sta-tus), there also was a greater increase in QOLIE-31P scores as the response level increased (Figure 2).11 When compared with nonresponders, responders had a greater mean improvement in overall QOLIE-31P scores (an additional 21.1% beyond that achieved in nonresponders) and the subscales for seizure-worry (55.3%), cognitive function (35.1%), and emotional well-being (38.1%) that were statistically significant. These mean im-provements were greater than the MID.
The authors concluded that a ≥ 50% reduction in seizure frequency was as-sociated with significant improvements in QOL, both with use of perampanel and placebo. In treated patients, increasing response rates were related to marginal overall changes in QOLIE-31P results and changes in the emotional well-being, social function, seizure-worry, and health status subscales. Clinical responses ob-served with perampanel treatment trans-lated into improved QOLIE-31P scores. A 48% reduction in seizure frequency is a valid treatment goal, even if complete seizure freedom is not achieved.
Finally, refractory partial seizures have a major impact on well-being. Most patients in this analysis had overall QOLIE-31P scores < 50 at baseline. Re-
sponse to perampanel corresponded with significantly improved QOL according to the QOLIE-31P MID.
n IMPACT OF DELAYED/MISSED DOSES ON PERAMPANEL PLASMA LEVELS
According to the results of population pharmacokinetic analyses,13 inducers of CYP3A4 cause increases in perampanel clearance. For example, oral clearance of perampanel is increased by about threefold with concomitant use of car-bamazepine and approximately twofold with coadministration of oxcarbazepine or phenytoin. Many neurologists believe that administration of AEDs that have a long half-life may facilitate medica-tion adherence by reducing the needed frequency of dosing; in addition, it may minimize peak-to-trough fluctuations, particularly in patients who have dif-ficulty adhering to their medication regimens. Perampanel has an elimination half-life of 105 hours in people who have not used enzyme inducers, so it can be administered once daily.
Gidal et al14 studied the impact of delayed or missed doses on perampanel plasma concentrations. They developed pharmacokinetic population parameters using validated data from 19 phase I stud-ies (unpublished data). These data likely may be generalized to other populations, since the 606 volunteers taking part in the pharmacokinetic studies were ≥ 18 years of age and included healthy males and females, people with hepatic impair-ment, and substance abusers. Volunteers received either one dose of perampanel (range, 0.2–36 mg) or repeated once-daily doses of perampanel (range, 1–12 mg/d).
Using NONMEM version 7.2 (ICON plc; Dublin, Ireland) with PDx-pop ver-sion 5 (ICON plc), the researchers per-formed simulations for a typical patient taking 8 mg of perampanel once daily. The investigators were interested in the effect on plasma perampanel concentrations if a dose was missed and replacement of the missing dose was delayed, if a dose was missed and then scheduled therapy was resumed, or a dose was missed in the presence or absence of carbamazepine.
Many neurologists believe that administration of AEDs that have a long half-life may facilitate medication adherence by reducing the needed dosing frequency.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 39
LisaAenlle-Matusz,MD,MPH Update on Recent Global Studies of Perampanel, a New Selective AMPA Antagonist
In patients who missed a dose and did not use any other enzyme-inducing AEDs, perampanel trough concentrations imme-diately preceding the next scheduled dose declined mildly (18.4%). In the presence of enzyme-inducing AEDs, the predicted reduction in trough concentration was moderate (43.9%; unpublished data). Replacing a missed dose within 12 hours resulted in a similar mild decline in the trough concentration in the absence (5%) and presence (13.9%) of carbamazepine (unpublished data). Likewise, replacing a missed dose after 12 hours resulted in a mild decline in trough concentration in the absence of carbamazepine (9.7%) and a moderate decline in the presence of carbamazepine (25.2%). Increases in peak perampanel concentrations also were minor following dose replacement after a delay of 6 hours (no carbamazepine, 1.7%; carbamazepine, 2.5%) and 12 hours (no carbamazepine, 2.5%; carbamazepine, 5.3%). Changes in peak concentrations also were minor following replacement of a missed dose.
The investigators concluded that given the pharmacokinetic characteristics of perampanel, plasma concentrations of the drug fluctuated less than might be expected for a drug having a short half-life, which supports once-daily dosing of perampanel. The fluctuation index of perampanel increases with use of enzyme-inducing AEDs. However, because the efficacy of perampanel appears to be related to drug exposure (ie, plasma con-centration), the data support the idea that declines in trough concentrations are not likely to be as abrupt if a dose is missed.
These data are consistent with the recom-mendation that, in the event of a missed dose, dosing should be resumed the fol-lowing day at the usual daily dose.15 Fur-ther, supplementing a missed dose 6–12 hours later can lessen predicted declines without resulting in excessive spikes in plasma perampanel concentrations.
REFERENCES
1. French JA, Krauss GL, Biton V, et al. Ad-junctive perampanel for refractory partial-onset seizures: randomized phase III study 304. Neurology. 2012;79:589–596.
2. French J. Global phase III trial of peram-panel, a selective, non-competitive AMPA receptor antagonist, as adjunctive therapy in patients with refractory partial-onset seizures. Presented at the 63rd Annual Meeting of the American Academy of Neurology; April 9–16, 2011; Honolulu, HI. Abstract LBS.002.
3. French J, Elger C, Goldberg-Stern H, et al. Use of perampanel, a selective, noncompetitive AMPA receptor antagonist, as adjunctive therapy in patients with refractory partial-onset seizures: results of a global phase III study. Epilepsia. 2011;52(suppl 6):10. Abstract 020.
4. Krauss GL, Serratosa JM, Villanueva V, et al. Randomized phase III study 306: adjunctive perampanel for refractory partial-onset seizures. Neurology. 2012;78:1408–1415.
5. Ben-Menachem E, Perucca E, Squillacote D, Yang H, Zhu J, Laurenza A. Perampanel improves responder rates, irrespective of concomitant anti-epileptic drugs, and increases in seizure freedom: a pooled analysis of three phase III trials. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Di-ego, CA. Poster 1.234.
6. Kramer L, Perucca E, Ben-Menachem E, et al. Perampanel, a selective, non-competitive AMPA receptor antagonist as adjunctive therapy in patients with refractory partial-onset seizures: a dose response analysis from phase III studies. Neurology. 2012;78: abstract P06.117.
7. Hussein Z, Ferry J, Kraus G, Squillacote D, Laurenza A. Demographic factors and concomitant
antiepileptic drugs have no effect on the pharmaco-dynamics of perampanel. Neurology. 2012;78(suppl 1): abstract P06.127.
8. Laurenza A, Ferry J, Hussein Z. Popula-tion pharmacokinetics and pharmacodynamics of perampanel: a pooled analysis from three phase III trials. Epilepsy Curr. 2012;12(suppl 1): abstract 2.231.
9. Kwan P, Brodie M, Squillacote D, Yang H, Zhu J, Laurenza A. Adjunctive perampanel is effective against partial seizures, irrespective of concomitant antiepileptic drugs (AEDs): a pooled analysis of three phase III trials. Presented at the 66th Annual Meet-ing of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster 1.239.
10. Vasquez B, Yang H, Williams B, Zhous S, Laurenza A, Fain R. Efficacy and safety of once-daily adjunctive perampanel, a selective AMPA antagonist in patients with treatment-resistant partial-onset seizures: the North American experience. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Di-ego, CA. Poster 1.241.
11. Krauss G, Faught E, Simons W. Relation-ship between quality of life and the frequency of refractory partial seizures: a pooled analysis of three phase III trials of perampanel. Presented at the 66th Annual Meeting of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster P1.240.
12. Wiebe S, Matijevic S, Eliasziw M, Derry PA. Clinically important change in quality of life in epilepsy. J Neurol Neurosurg Psychiatry. 2002;73:116–120.
13. Hussein Z, Critchley D, Ferry J, Laurenza A. Population pharmacokinetics of perampanel, a selective, non-competitive AMPA receptor antagonist, in patients with refractory partial-onset seizures participating in a randomized, double-blind, placebo-controlled phase III study. Presented at the 29th International Epilepsy Con-gress; August 28–September 1, 2011; Rome, Italy. Abstract p821.
14. Gidal B, Majid O, Ferry J, et al. Impact of delayed dose or missed dose on perampanel plasma concentrations. Presented at the 66th Annual Meet-ing of the American Epilepsy Society; November 30–December 4, 2012; San Diego, CA. Poster P2.211.
15. Fycompa [package insert]. Woodcliff Lake, NJ.; Eisai Inc; October 2012.

40 T H E N E U R O L O G Y R E P O R T | V o l u m e 5 N u m b e r 3
CME Post TestUsing this page as a worksheet, select the best answer to each question based on your reading of the articles in this issue of The Neurology Report, then complete the evaluation on the facing page and see the instructions below it to obtain CME credit.
1. A large, recurrent microdeletion of 15q13.3 is specifically associated with:
a. Lennox-Gastaut syndromeb. West syndromec. Prader-Willi syndromed. Idiopathic generalized epilepsy
2. A severe myoclonic encephalopathy of infancy most often caused by the de novo loss of function mutation in the gene SCN1A is:
a. Rett syndromeb. Dravet syndromec. Angelman syndromed. Benign familial infantile epilepsy
3. A novel antiepileptic drug (AED) that enhances slow inactivation of the sodium channel without affecting fast activity is:
a. Lacosamideb. Clobazamc. Rufinamided. Eslicarbazepine acetate
4. In testing AEDs in young patients with various intractable seizure symptoms, Sugai et al noted a responder rate > 75% with use of:
a. Valproate for partial seizuresb. Carbamazepine for secondarily generalized tonic-
clonic seizuresc. Phenytoin for focal seizuresd. Potassium bromide, zonisamide, or lamotrigine for
tonic seizures
5. In evaluating the efficacy and tolerability of eslicarbazepine acetate in patients with partial-onset seizures, Carreno et al found that patients with depressive symptoms:
a. Had a significantly higher incidence of treatment-emergent adverse effects than did patients without depressive symptoms
b. Showed poorer seizure control than did patients without depressive symptoms
c. Experienced greater seizure control than did patients without depressive symptoms
d. Were more likely to discontinue treatment with eslicarbazepine acetate than did patients without depressive symptoms
6. In a retrospective study of adjunctive rufinamide therapy in patients with Lennox-Gastaut syndrome, Silva et al observed that:
a. Rufinamide therapy was most commonly started in patients already using at least four other AEDs.
b. The most commonly used concomitant AED was divalproex sodium.
c. One in five of the patients was mentally retarded.d. Patients who took longer to reach their modal dose
of rufinamide were less likely to discontinue the medication.
7. The first FDA-approved AED to target and open a voltage-gated potassium channel was:
a. Vigabatrinb. Ezogabinec. Perampaneld. Zonisamide
8. A disadvantage of lacosamide therapy is:a. Significant drug interactionsb. Urinary retentionc. Cognitive complaintsd. Dizziness
9. In an analysis of the results of three global clinical trials of perampanel in patients with partial-onset seizures, Ben-Menachem et al found that higher doses of the drug improved response rates:
a. When perampanel was used aloneb. Only when carbamazepine also was usedc. Only when levetiracetam also was usedd. Irrespective of the concomitant AED used
10. Given the pharmacokinetics of perampanel, a missed dose of the drug should be taken:
a. Immediately, at the full recommended dose, when the patient remembers to take it
b. Immediately, at half the recommended dose, when the patient remembers to take it
c. Six to 12 hours later at the full recommended daily dose
d. The following day at the full recommended daily dose
T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 3 41
Your candid and thorough completion of this evaluation will help the University of Cincinnati improve the quality of its CME activities. Thank you for your participation. Strongly agree Agree Disagree1. As a result of this activity, I am more knowledgeable about …
a. Newer antiepileptic drugs (AEDs) and their use in patients of different ❑ ❑ ❑ages and with different comorbid conditions.
b. The discovery of genes associated with epilepsy, their variability, and their ❑ ❑ ❑impact on clinical practice.
c. The results of recent clinical trials of novel AEDs in adults and children ❑ ❑ ❑with different seizure disorders and comorbidities.
d. Recent studies of newer AEDs, alone and in combination, in patients with ❑ ❑ ❑medically refractory seizure disorders.
e. The influence of drug interactions and adverse effects of AEDs and the ❑ ❑ ❑risks/benefits of using generic AEDs.
Strongly agree Agree Disagree2. I found the content of this educational activity …
a. Clearly written and well organized. ❑ ❑ ❑
b. Accurate and timely. ❑ ❑ ❑
c. Related to its overall objectives. ❑ ❑ ❑
d. Free from commercial bias. ❑ ❑ ❑
e. Relevant to my own clinical practice. ❑ ❑ ❑
Yes No Don’t know3. Did the information you received from this CME activity:
a. Confirm the way you currently manage your patients? ❑ ❑ ❑
b. Suggest new options for managing your patients that you might apply ❑ ❑ ❑in the future?
Patient Board CME management review credit
4. I used the information in this issue for … (check all that apply) ❑ ❑ ❑
5. Approximately how long (in minutes) did it take you to complete this activity, minutes including this evaluation?
Evaluation
Instructions for Obtaining CME CreditTo receive CME credit for this free educational activity and a certificate of participation from the University of Cincinnati:
• Study the educational material presented in this issue of The Neurology Report.• Using page 40 as a worksheet, answer all of the post-test questions based on the content of the articles.• Visit www.NeurologyReport.com on the Web by February 20, 2014, select this issue of The Neurology Report, and
click “CME Credit” to apply for credit online and complete the post test and evaluation.• Complete the registration form, enter your post-test answers from the worksheet on page 40, and respond to all of the
questions on the evaluation form, then click the button to submit your answers. The full text of each article may be accessed at www.NeurologyReport.com, should you need to refer to it again.
• If you answer correctly at least 8 (80%) of the 10 post-test questions, you will immediately receive credit for completing this educational activity and can access your CME certificate online by clicking the “Certificate” button at the bottom of the evaluation form. Follow the on-screen instructions to print or e-mail your certificate.
