the permeability transition pore complex: another … · the permeability transition pore complex:...
TRANSCRIPT

The permeability transition pore complex: another viewAndrew P. Halestrap *, Gavin P. McStay, Samantha J. Clarke
Department of Biochemistry, University of Bristol, Bristol BS8 1TD, UK
Received 14 August 2001; accepted 20 December 2001
Abstract
Mitochondria play a critical role in initiating both apoptotic and necrotic cell death. A major player in this process is the mitochondrialpermeability transition pore (MPTP), a non-specific pore, permeant to any molecule of < 1.5 kDa, that opens in the inner mitochondrialmembrane under conditions of elevated matrix [Ca2+], especially when this is accompanied by oxidative stress and depleted adeninenucleotides. Opening of the MPTP causes massive swelling of mitochondria, rupture of the outer membrane and release of intermembranecomponents that induce apoptosis. In addition mitochondria become depolarised causing inhibition of oxidative phosphorylation andstimulation of ATP hydrolysis. Pore opening is inhibited by cyclosporin A analogues with the same affinity as they inhibit the peptidyl–prolylcis-trans isomerase activity of mitochondrial cyclophilin (CyP-D). These data and the observation that different ligands of the adeninenucleotide translocase (ANT) can either stimulate or inhibit pore opening led to the proposal that the MPTP is formed by a Ca-triggeredconformational change of the ANT that is facilitated by the binding of CyP-D. Our model is able to explain the mode of action of a widerange of known modulators of the MPTP that exert their effects by changing the binding affinity of the ANT for CyP-D, Ca2+ or adeninenucleotides. The extensive evidence for this model from our own and other laboratories is presented, including reconstitution studies thatdemonstrate the minimum configuration of the MPTP to require neither the voltage activated anion channel (VDAC or porin) nor any otherouter membrane protein. However, other proteins including Bcl-2, BAX and virus-derived proteins may interact with the ANT to regulatethe MPTP. Recent data suggest that oxidative cross-linking of two matrix facing cysteine residues on the ANT (Cys56 and Cys159) playsa key role in regulating the MPTP. Adenine nucleotide binding to the ANT is inhibited by Cys159 modification whilst oxidation of Cys56
increases CyP-D binding to the ANT, probably at Pro61. © 2002 Société française de biochimie et biologie moléculaire / Éditionsscientifiques et médicales Elsevier SAS. All rights reserved.
Keywords: Adenine nucleotide translocase; Cyclophilin-D; Calcium; Oxidative stress; Mitochondrial swelling; Reconstitution; MPTP mechanism; Voltageactivated anion channel
1. Introduction
The primary role of mitochondria in healthy cells is theprovision of ATP to support normal cell function and, untilrecently, this had been the major focus of research intomitochondrial function. However, in recent years the centralrole of mitochondria in apoptotic and necrotic cell death hasbecome apparent, and a major player in this arena is the
mitochondrial permeability transition (MPT). The MPTrefers to the massive swelling and depolarisation of mito-chondria that occurs under some conditions, most notably asa result of calcium overload and oxidative stress. The causeof the MPT is the opening of a non-specific pore in the innermitochondrial membrane, known as the mitochondrial per-meability transition pore (MPTP). A major consequence ofthe MPT is the uncoupling of oxidative phosphorylation.This will prevent mitochondria making ATP by oxidativephosphorylation and also, by reversal of the mitochondrialproton-translocating ATPase, actively stimulate the hydroly-sis of ATP produced by glycolysis. Unrestrained, this willinevitably lead the cell towards necrotic death. Indeed poreopening has been shown to be a critical event in the necroticcell death that occurs when tissues are subjected to insultssuch as reperfusion injury and chemical toxins (see [1,2]).
Abbreviations: ANT, adenine nucleotide translocase; CAT, carboxy-atractyloside; BKA, bongkrekic acid; CsA, cyclosporin A; CyP, cyclophi-lin; IMM, inner mitochondrial membrane; MPT, mitochondrial permeabi-lity transition; MPTP, mitochondrial permeability transition pore; NEM,N-ethylmaleimide; CuP, copper phenanthroline; PAO, phenylarsine oxide;VDAC, voltage activated anion channel
* Corresponding author. Tel.: +44 117 928 8592; fax: +44 117 928 8274.E-mail address: [email protected] (A.P. Halestrap).
Biochimie 84 (2002) 153–166
© 2002 Société française de biochimie et biologie moléculaire / Éditions scientifiques et médicales Elsevier SAS. All rights reserved.PII: S 0 3 0 0 - 9 0 8 4 ( 0 2 ) 0 1 3 7 5 - 5

In addition to its role in necrosis, transient MPTP openingmay be involved in apoptosis through the release of cyto-chrome c and other pro-apoptotic molecules. These thenactivate the caspase cascade that sets apoptosis in motion[1–3]. However, it seems probable that only if subsequentMPTP closure occurs will ATP levels be maintained, ensur-ing that cell death continues down an apoptotic rather thana necrotic pathway [4]. Nevertheless, mitochondria play animportant role in apoptosis even in the absence of the MPTsince release of pro-apoptotic factors such as cytochrome c,apoptosis inducing factor (AIF) and Smac/Diablo from theinter-membrane space of mitochondria may occur throughchanges in the outer membrane permeability. These areinduced by pro-apoptotic proteins such as BAX and Bid[3,5].
The main emphasis of the work performed in thislaboratory over the past 12 years has been to elucidate themolecular mechanism of the MPTP and its role in reperfu-sion injury of the ischaemic heart. In this review we willrestrict our attention to the former and the reader is referredelsewhere for a review of the extensive work from this andother laboratories on the role of the MPTP in reperfusioninjury in the heart and brain, and the development ofprotective strategies that may be applicable to clinicalsituations [1,6,7].
2. The MPT in historical context
The phenomenon of the MPT was first describedseveral decades ago as a massive swelling of mito-chondria that accompanies calcium overload. It wasinitially thought to be a result of non-specific damageto the inner mitochondrial membrane (IMM) byphospholipases [8]. However, pioneering studies,initiated by Hunter and Haworth in the late seventies[9,10] and confirmed 7 years later by Martin Cromp-ton and colleagues [11], implied that the pore is aunique molecular entity that allows the passage ofany molecule of < 1500 daltons across the IMM, andcan be rapidly closed by chelation of calcium. Be-cause the MPTP allows rapid passage of protons, itsopening is accompanied by depolarisation of themitochondria and uncoupling of oxidative phospho-rylation. In addition, the equilibration of all smallsolutes across the IMM leaves behind high concen-trations of proteins in the matrix and these exert acolloidal osmotic pressure that is responsible for theextensive swelling of mitochondria associated withthe MPT [1,12]. Our own interest in the MPTP arosefrom an investigation into the regulation of ATPproduction by hormones since these studies led us toinvestigate the physiological role of changes in mito-chondrial volume. A brief summary of this workfollows because it provided us with important cluesabout the mechanism of the MPTP.
3. Mitochondrial swelling — MPTP-dependentand independent mechanisms
Studies on the hormonal regulation of hepatic metabo-lism in this and other laboratories during the seventies andearly eighties had demonstrated that stimulation of respira-tion and oxidative phosphorylation are critical events [13].It became apparent that these effects involve an increase inmitochondrial [Ca2+] and activation of the three calcium-sensitive mitochondrial dehydrogenases, pyruvate dehydro-genase, isocitrate dehydrogenase and 2-oxoglutarate dehy-drogenase. The resulting increase in matrix NADH/NAD+
stimulates respiration leading to an increase in protonmotive force (pmf) and ATP production [14]. However, therise in NADH is only transient and is followed by a declineto basal levels even though rates of respiration, ATPproduction and the pmf remain elevated. This re-oxidationof NADH is associated with a calcium-mediated increase inmitochondrial matrix volume of 20–40% that could bedetected by a small decrease in light scattering as well as bydirect measurement of volume with 3H2O and [14C]-sucrose. Such small increases in matrix volume cause astimulation of NADH oxidation by increasing the rate ofelectron flow from Complex 1 into ubiquinone, an effectthat can be mimicked by 1–5 nM valinomycin, a potassiumionophore. This provides an explanation of why the hor-monally induced increase in mitochondrial NADH/NAD+ isonly transient whilst the pmf and ATP production ratesremain stimulated (see [13,15]). These results have recentlybeen confirmed at the single cell level (see [16]).
The calcium-dependent increase in matrix volume iscaused by entry of K+ ions through a potassium channel,driven by the membrane potential, with proton-drivenco-transport of the phosphate anion providing charge com-pensation. Osmotically obliged water follows, causing therelatively modest (20–40%) swelling of the matrix (c.f. themassive swelling that accompanies the permeability transi-tion). Extensive studies on this calcium activated potassiumchannel led to the conclusion that the protein responsible isthe adenine nucleotide translocase (ANT) an integral innermembrane protein whose normal role is the translocation ofATP and ADP across the IMM. This is converted into achannel when the ATP/ADP binding sites are not occupiedby nucleotides, an event which happens rarely under restingconditions. However, when matrix [Ca2+] is elevated, pyro-phosphate (PPi) accumulates in the matrix and may tran-siently displace adenine nucleotides from some ANT mol-ecules. The increase in [PPi] is caused by the powerfulinhibition of pyrophosphatase by micromolar [Ca2+], butelevating matrix [PPi] independently of Ca2+, for exampleby provision of butyrate, can also cause a modest increase inmitochondrial matrix volume [15].
It was during these studies that Martin Crompton re-ported that the permeability transition could be specificallyinhibited by cyclosporin A (CsA) [17]. This key observationled us to consider whether the physiological changes in
154 A.P. Halestrap et al. / Biochimie 84 (2002) 153–166

mitochondrial volume we had been measuring in responseto hormones might be related to the massive swellingassociated with the permeability transition. The lack of aneffect of CsA on the physiological response clearly demon-strates that this is not the case [18] but introduced thelaboratory to the permeability transition pore and thepossibility that under some conditions, the ANT can act asa channel as well as a transporter [19].
4. Experimental protocols used to studythe permeability transition in isolated mitochondria
The MPT can be studied in isolated mitochondria by avariety of methods (see [1,2]). These include:
• Measuring the swelling of mitochondria by monitoringthe associated decrease in light scattering. As mito-chondria swell their refractive index changes and theyscatter less light. This can be detected as a decrease inlight absorbance measured with a spectrophotometer.To avoid any complications that changes in the redoxstate of respiratory chain components might cause, thewavelength of the incident light should be at theisosbestic point for the cytochromes (520 nm).
• Following the loss of mitochondrial membrane poten-tial using fluorescent dyes or a TPMP+ electrode. Adisadvantage of this technique is that uncoupling doesnot necessarily mean that the MPTP has opened, and itis important to confirm this by showing inhibition ofthe response by CsA.
• Measurement of the release of accumulated calciumwith a calcium-sensitive dye or electrode. The samecriticisms apply to this technique as to measurements ofthe membrane potential.
• Measurement of [14C] sucrose permeation into mito-chondria. This is a very reliable technique that has beenused primarily by Crompton’s laboratory (see [2]), butrequires specialised apparatus.
In this laboratory we routinely employ light scattering toinvestigate the mechanism of the MPTP and prefer to usede-energised mitochondria (no respiratory substratespresent and the electron transport chain inhibited withrotenone and antimycin A). A calcium ionophore is alsoadded to ensure that calcium equilibrates across the innermitochondrial membrane. Although such conditions are farremoved from the in vivo situation, they minimise thepossibility of indirect effects that added reagents may haveon the permeability transition. For example, any moleculethat depolarises the mitochondria following calcium accu-mulation will cause opening of the MPTP, whilst anyinhibitor of calcium entry into mitochondria will prevent theMPTP. Yet in neither case is the effect of the reagent directlyon the MPTP mechanism. We have also found that moreconsistent results can be obtained using mitochondria thatare left on ice for 18–24 h before use. The reason for this isthat, following their preparation, mitochondria lose an
increasing proportion of their adenine nucleotides duringthe first few hours of storage on ice. This causes thesensitivity of the MPTP towards calcium to change substan-tially over the time period of an experiment, whereasleaving mitochondria for 18 h before use minimises thiseffect. Another ‘trick’ that ensures reproducible results is toperform the light scattering experiment in an iso-osmoticKSCN buffer. The reason behind this choice of buffer is thatit is often used for determining mitochondrial potassiumfluxes, but it so happens that its mild chaotropic actionsensitises the MPT to calcium. The mechanism underlyingthis effect will be discussed below.
A variation of the light scattering technique, first intro-duced by Hunter and Haworth [9,10], is to pre-swell themitochondria by opening the MPTP with excess calciumand then induce shrinkage of the mitochondria in thepresence of polyethylene glycol (PEG) of about 4 kDa. Thismolecule is too large to permeate the MPTP, and thus exertsan osmotic pressure on the matrix even when the MPTP isopen. The advantage of this technique is that the composi-tion of the matrix can be varied at will once the MPTP isopen in the pre-swollen state, and thus matrix effectors suchas [ADP] can be studied very readily. The extent of poreopening is reflected in the rate at which the light scatteringdecreases upon PEG addition.
5. Characteristics of the MPTP
The primary trigger for opening of the MPTP is a rise inmatrix [Ca2+], but the concentration required is highlydependent on the prevailing conditions (see [1,8,20]). Sev-eral factors are known to greatly enhance the sensitivity ofthe pore to [Ca2+], of which the most potent and relevant tothe cellular setting are oxidative stress, adenine nucleotidedepletion, increased inorganic phosphate concentrations andmitochondrial depolarisation. Under energised conditions(but not de-energised conditions), the pore is also sensitiveto ligand-induced conformational changes of the ANT beingactivated by carboxyatractyloside (CAT) and inhibited bybongkrekic acid (BKA). Low pH (< 7.0) is a potent inhibitorof the MPTP probably as a result of protons competing withCa2+ for its binding at the trigger site [9,21,22]. Thespecificity of this site for calcium appears to be absolute andother divalent cations such as Sr2+, Mn2+, Ba2+ and Mg2+
act as inhibitors [9,22]. In addition to the calcium-specifictrigger site on the matrix surface, there is another inhibitorydivalent cation binding site on the external surface of theinner membrane with a Ki for Mg2+ of about 0.3 mM [23].An important factor that must be considered in relation tothe effects of Mg2+ on the MPTP is that ATP bindsmagnesium tightly (Kd ∼ 10 –4 at pH 7.0) and the Mg–ATPcomplex does not bind to the ANT. Thus, energisation ofmitochondria, which in its own right protects against MPTPopening, will also decrease free [ATP] and [Mg2+] thatinhibit pore opening and the overall effect may be hard to
A.P. Halestrap et al. / Biochimie 84 (2002) 153–166 155

predict. More recently, it has been shown that ubiquinoneanalogues can act either as activators or inhibitors of theMPTP [24]. A much fuller list of known effectors of theMPTP may be found elsewhere [8].
A well-documented feature of the permeability transitionis that for any individual mitochondria it is an all or nonephenomenon. That is, the mitochondria are either ‘normal’or massively swollen with no intermediate state beingapparent (see [20]). However, within a population, indi-vidual mitochondria possess different sensitivities to induc-ers of the MPTP. Thus, the magnitude of the decrease inlight scattering observed in a suspension of mitochondriareflects the number of mitochondria in the population thathave undergone the permeability transition rather than aprogressive and concerted increase in volume of all themitochondria. The stochastic nature of the MPT can readilybe explained since once a single pore opens, rapid protonentry and adenine nucleotide loss will induce more pores toopen, leading to a full transition. Other mitochondria withinthe population will then take up the calcium that is releasedby those that have undergone the transition and some ofthese will now undergo the transition themselves. This willlead to a progressive opening of all the mitochondria that isreflected in the progressive decrease in light scattering [20].
6. The molecular mechanism of the MPTP
6.1. The role of cyclophilin D
The discovery that opening of the MPTP could beinhibited specifically by sub-micromolar concentrations ofthe immunosuppressive drug, cyclosporin A (CsA) [17]provided the impetus for our own research into the molecu-lar mechanism of the MPTP. The immunosuppressive effectof CsA involves its binding to a cytosolic protein calledcyclophilin-A (CyP-A) [25] and this led us to investigate therole of a matrix CsA binding protein (cyclophilin). Cyclo-philins exhibit peptidyl–prolyl cis-trans isomerase (PPIase)activity, and catalyse the interconversion between cis andtrans conformations of peptide bonds adjacent to prolineresidues [25]. As such they are ideally suited for enablingthe conformational change in a membrane protein thatwould be required to induce formation of a pore. We wereable to demonstrate that the mitochondrial matrix containssuch a CsA-sensitive PPIase [19] and that the K0.5 values ofseveral CsA analogues for the inhibition of this PPIaseactivity correlated with their potency as inhibitors of poreopening [19,26,27]. This led us to propose in 1990 that theMPTP was formed from an interaction between the adeninenucleotide translocase (ANT) and mitochondrial CyP [19]as discussed further below. Subsequently we were able topurify, clone and sequence the CsA-sensitive mitochondrialPPIase [28,29] and hence confirm that it was a uniquecyclophilin, the rat equivalent of the human CyP-3 geneproduct [30] and distinct from cytosolic cyclophylin A. Now
known as cyclophilin D (CyP-D), this nuclear encodedprotein has a mitochondrial targeting presequence that iscleaved after translocation of the protein into the matrix.Cleavage occurs at one of two points leading to matureproteins of about 17.6 kDa (minor product) and 18.6 kDa(major product) [28]. Subsequently, Crompton and col-leagues also purified and sequenced mitochondrial CyP-D[31] and confirmed that the protein can be imported into themitochondria before cleavage of its presequence [32].Northern blots demonstrate that mRNA for CyP-D ispresent in rat muscle, heart, liver, kidney and brain and is ofidentical size (1.5 kb) in all tissues [29]. This makes itunlikely that there are differently spliced tissue-specificisoforms that might account for the different sensitivities ofmitochondria from various tissues.
For a soluble protein such as CyP-D to play a role in theopening of the MPTP it must interact with a membraneprotein and induce a conformational change when triggeredby Ca2+. Indeed, it could be envisaged that factors known tosensitize mitochondria to pore opening, such as oxidativestress, might do so by enhancing binding of CyP-D to itstarget membrane protein. In support of this we demonstratedthat oxidative stress induced by t-butyl hydroperoxide(TBH) or diamide treatment increase CyP-D binding to theinner mitochondrial membrane in parallel with increasingthe sensitivity of pore opening to [Ca2+] [33,34]. Interest-ingly, in order to show this effect consistently, the mito-chondrial membranes had to be prepared in iso-osmoticKSCN medium to stabilise the complex between CyP-D andits membrane target protein. Stabilisation could also beachieved by the addition of low concentrations of guani-dinium hydrochloride, implying that it is the chaotropicproperty of KSCN that is responsible for its stabilisingeffects [34]. This suggests that the CyP-D forms a complexwith the target protein, inducing a conformational changethat exposes more of the protein surface to the aqueousmedium. Such an effect might be predicted for the formationof a channel. Another factor that was shown to enhance bothCyP-D binding and MPTP opening in response to [Ca2+] isan increase in matrix volume [34]. The cause of this isunknown although it may reflect the accessibility of CyP-Dto its target membrane binding site. Whatever the mecha-nism, one significant consequence is that once the perme-ability transition has occurred and mitochondria have swol-len, more CyP-D is likely to become bound making reversalof the MPT more difficult. In our experiments, no matterwhat conditions were used to stimulate binding of CyP tothe IMM, the process was almost totally prevented by CsA.However, in our hands other known modulators of theMPTP such as matrix [Ca2+], [ADP], pH or membranepotential were without effect [33–35]. In contrast , Bernardiand colleagues have demonstrated an inhibitory effect oflow pH on CyP-D binding to sub-mitochondrial particles.The effect was blocked by the histidine reagent diethylpy-rocarbonate which also blocks the inhibitory effect of pH onthe MPTP [36]. These experiments were performed in low
156 A.P. Halestrap et al. / Biochimie 84 (2002) 153–166

ionic strength media where a large number of other matrixproteins also remained bound to the membrane at low pH,and thus it is possible that the effect of pH was onnon-specific binding of CyP-D to charged groups on thephospholipids or membrane proteins. Crompton and col-leagues used similar conditions when labelling the mem-brane bound CyP-D with photoactivatable CsA derivatives[37]. Their data suggested that Ca2+ might enhance andADP diminish CyP binding under such conditions, but ourown experiments did not confirm this [34,38].
Although it appears to be well established that CsAblocks pore opening by binding to CyP-D, there are data toindicate that CyP binding may not be required for the MPTPto open at very high matrix [Ca2+]. Thus, Novgorodov et al[39] and Crompton and Andreeva [40], using differenttechniques, have shown that at high matrix [Ca2+] inhibitionof pore opening by CsA is overcome. We have confirmedthis in both heart mitochondria [26] and liver mitochondria[33–35]. Yet under the same conditions, CsA is able toprevent almost totally the binding of CyP-D to the innermitochondrial membrane [33,35]. In addition, studies on themegachannel of patched clamped mitochondria have shownthat an inhibitory effect of CsA is overcome at higher [Ca2+][41]. Although it is dangerous to assume that the megachan-nel studied electrophysiologically is necessarily a manifes-tation of the same molecular entity as the MPTP [42], itdoes appear that CyP-D binding is not absolutely essentialfor the MPTP to open, but may rather sensitise the processto [Ca2+]. This is not unreasonable if it is assumed that poreopening involves a cis-trans isomerisation around a prolineresidue that causes a conformational change of the protein.This process could occur independently of CyP-D binding,but be greatly enhanced by the bound CyP-D.
6.2. The role of the adenine nucleotide translocase
6.2.1. Indirect evidenceAn involvement of the ANT in pore opening was first
proposed by Hunter and Haworth [10] and more convincingevidence provided by LeQuoc and LeQuoc [43] and our-selves [19,35]. The early evidence was based largely uponthe observation that in energised mitochondria, any reagentsuch as CAT that stabilised the ‘c’ conformation of the ANT,sensitised the MPT to [Ca2+], whilst any reagent such asBKA that stabilised the ‘m’ conformation of the ANT, madethe MPT less sensitive to [Ca2+]. In passing it should benoted that many workers have used CAT and BKA as‘specific’ activators and inhibitors of the MPTP in culturedcell models of apoptosis. In doing so they fail to recognisethat the primary effect of these reagents will be to preventATP/ADP exchange across the mitochondrial inner mem-brane, with profound effects on cellular metabolism irre-spective of any effect on the MPT. It should also be notedthat CAT and its less potent analogue atractyloside bind tothe ANT with very high affinity (Kd < 1 µM), and thus theiruse at 100–50 000 µM in some published experiments may
well induce non-specific effects quite unrelated to theirbinding to the ANT.
Matrix ADP is another important modulator of poreopening that acts by decreasing the sensitivity of thecalcium trigger site to [Ca2+]. There are two ADP bindingsites with Ki values of about 1 and 25 µM. The high affinitysite is blocked by the inhibitor CAT and is therefore thoughtto be associated with the ANT [9,10,35,44,45]. The identityof the second site is less clear but may well be anextramitochondrial binding site for adenine nucleotides onthe ANT. We tested the ability of a range of nucleotides toinhibit the MPT, and found that apart from ADP, only ATPand deoxy-ADP inhibit with K0.5 values 500 and 20 timesgreater than ADP respectively. This correlates with theiraffinity for the matrix binding site of the ANT [35]. Bernardiand colleagues have provided strong evidence that theMPTP is voltage-regulated, being activated as the mem-brane potential becomes less negative (see [20,46]). Wehave suggested that the membrane potential is sensed by theANT itself through an effect on adenine nucleotide binding.This is possible because the ANT catalyses an electrogenicexchange of ATP4– for ADP3– with a mechanism that maywell involve a potential driven conformational changealtering the affinity of the ANT for adenine nucleotides oneither side of the membrane [47,48]. In support of thishypothesis, we have demonstrated that in mitochondriadepleted of adenine nucleotides by pyrophosphate treat-ment, not only is the MPT much more sensitive to [Ca2+],but it is also no longer voltage sensitive [19,27].
Adenine nucleotide binding is antagonised by oxidativestress induced by reagents such as t-butylhydroperoxide(TBH) or diamide and also by thiol reagents such asphenylarsine oxide (PAO), a powerful activator of the MPT[35,49]. PAO has the greatest effect of the reagents tested,raising the K0.5 for ADP inhibition of the MPT to > 500 µM[35]. We have shown that this effect is accompanied bycovalent modification of the ANT [35] which may explainwhy PAO is a more potent stimulus of the MPT thandiamide or TBH, and yet has a smaller effect on CyP-Dbinding [34]. Oxidative stress and PAO also shift thevoltage dependence of the MPT, allowing the pore to openat more negative potentials. This is exactly what would bepredicted if thiol modification inhibits adenine nucleotidebinding to the matrix surface of the ANT whilst membranepotential enhances this binding.
6.2.2. CyP-D binds specifically to the ANTThe data described above strongly supports a critical role
for the ANT in the formation of the MPTP, as we originallyproposed in 1990 [19], but falls short of proof. In order toconfirm that CyP-D does bind specifically to the ANT underconditions favouring opening of the MPTP we over-expressed CyP-D as a fusion protein with glutathione-S-transferase (GST) for use as a CyP-D affinity column. Whensolubilised IMMs were passed over this column and weaklybinding proteins washed off, only one protein remained
A.P. Halestrap et al. / Biochimie 84 (2002) 153–166 157
bound and this was the ANT. Binding was inhibited bypre-treatment of the GST–CyP-D with CsA and enhancedwhen the inner membranes were prepared from mitochon-dria subjected to oxidative stress with diamide [38]. Insimilar studies, Crompton and colleagues found that boththe voltage activated anion channel (VDAC, also known asporin) and the ANT bound tightly to the GST–CyP column,but in contrast to our own studies they were unable toprevent binding with CsA [50]. These apparent discrepan-cies may be the result of major differences in the experi-mental protocol used in the two studies. Thus, Crompton etal. used unfractionated heart mitochondria whereas our ownstudies employed purified inner membranes from livermitochondria. Since heart and liver mitochondria possessdifferent isoforms of the ANT (mainly ANT1 and ANT2,respectively), it is possible that the relative affinities of theseisoforms for CyP-D and VDAC are different. Anotherdifference is that Crompton solubilised the mitochondriawith the zwitterionic detergent CHAPS whilst in our ownstudies the IMMs were solubilised in the non-ionic deter-gent Triton-X100.
6.2.3. Thiol groups on the ANT play a critical role in poreopening
Bernardi and colleagues have provided data that suggesttwo distinct thiol groups are implicated in modulatingMPTP activity [51,52]. One is sensitive to oxidation ofglutathione, for example by TBH or diamide, and isprotected by both monobromobimane and N-ethyl-maleimide. The other responds to the redox state of matrixNAD(P), and is protected by N-ethylmaleimide but notmonobromobimane. It is the latter site that accounts for thewell documented stimulatory effect of oxidation of matrixNADH on the MPT, perhaps through the mediation ofthioredoxin or lipoamide [53]. The ANT is known to havethree cysteine residues that show differential reactivitytowards various thiol reagents and oxidising agents in aconformation dependent manner [54–56]. These cysteinesmay well represent the thiol groups that regulate bothCyP-D binding and the inhibitory effects of ADP andmembrane potential on the MPT [35]. In support of this,eosine maleimide, that specifically attacks cysteine 159 ofthe ANT within the adenine nucleotide binding site [54,57],also abolishes the ability of ADP to inhibit the opening ofthe MPTP [35]. Furthermore, the ANT binds specifically toa phenylarsine oxide column [35] and this binding isprevented by pre-treatment with eosine maleimide or dia-mide (unpublished data of Gavin McStay and AndrewHalestrap).
These data suggest that PAO and diamide treatment arelikely to cross-link either Cys159 with Cys56 or Cys256 withCys56. The latter has particular attractions because CyP-D islikely to bind to a proline residue in view of its peptidyl-–prolyl cis-trans isomerase activity, and there is a prolineresidue, Pro61, close to Cys56 that we suggested in ouroriginal model might be the binding site for CyP-D [19].
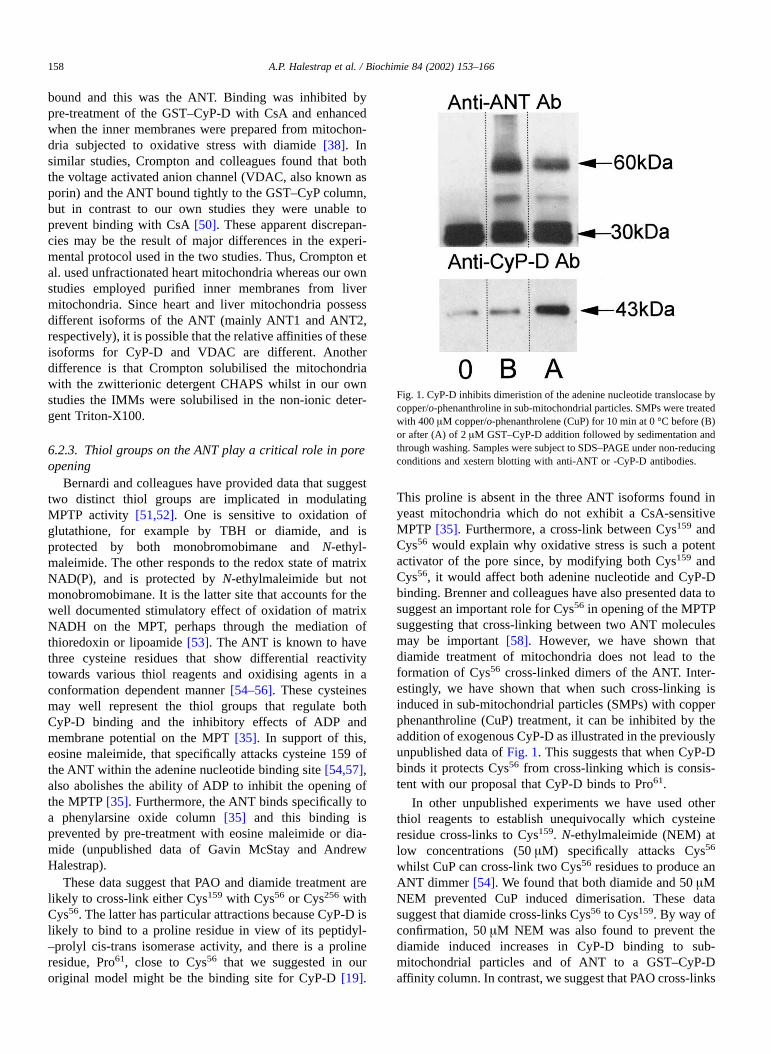
This proline is absent in the three ANT isoforms found inyeast mitochondria which do not exhibit a CsA-sensitiveMPTP [35]. Furthermore, a cross-link between Cys159 andCys56 would explain why oxidative stress is such a potentactivator of the pore since, by modifying both Cys159 andCys56, it would affect both adenine nucleotide and CyP-Dbinding. Brenner and colleagues have also presented data tosuggest an important role for Cys56 in opening of the MPTPsuggesting that cross-linking between two ANT moleculesmay be important [58]. However, we have shown thatdiamide treatment of mitochondria does not lead to theformation of Cys56 cross-linked dimers of the ANT. Inter-estingly, we have shown that when such cross-linking isinduced in sub-mitochondrial particles (SMPs) with copperphenanthroline (CuP) treatment, it can be inhibited by theaddition of exogenous CyP-D as illustrated in the previouslyunpublished data of Fig. 1. This suggests that when CyP-Dbinds it protects Cys56 from cross-linking which is consis-tent with our proposal that CyP-D binds to Pro61.
In other unpublished experiments we have used otherthiol reagents to establish unequivocally which cysteineresidue cross-links to Cys159. N-ethylmaleimide (NEM) atlow concentrations (50 µM) specifically attacks Cys56
whilst CuP can cross-link two Cys56 residues to produce anANT dimmer [54]. We found that both diamide and 50 µMNEM prevented CuP induced dimerisation. These datasuggest that diamide cross-links Cys56 to Cys159. By way ofconfirmation, 50 µM NEM was also found to prevent thediamide induced increases in CyP-D binding to sub-mitochondrial particles and of ANT to a GST–CyP-Daffinity column. In contrast, we suggest that PAO cross-links
Fig. 1. CyP-D inhibits dimeristion of the adenine nucleotide translocase bycopper/o-phenanthroline in sub-mitochondrial particles. SMPs were treatedwith 400 µM copper/o-phenanthrolene (CuP) for 10 min at 0 °C before (B)or after (A) of 2 µM GST–CyP-D addition followed by sedimentation andthrough washing. Samples were subject to SDS–PAGE under non-reducingconditions and xestern blotting with anti-ANT or -CyP-D antibodies.
158 A.P. Halestrap et al. / Biochimie 84 (2002) 153–166
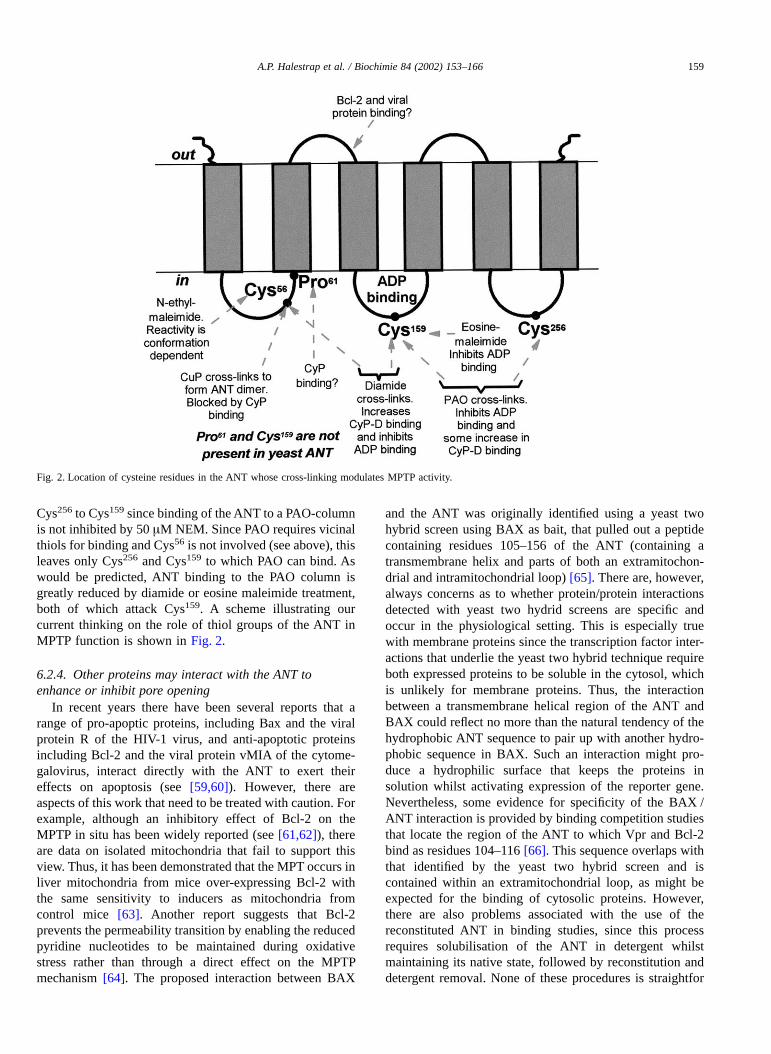
Cys256 to Cys159 since binding of the ANT to a PAO-columnis not inhibited by 50 µM NEM. Since PAO requires vicinalthiols for binding and Cys56 is not involved (see above), thisleaves only Cys256 and Cys159 to which PAO can bind. Aswould be predicted, ANT binding to the PAO column isgreatly reduced by diamide or eosine maleimide treatment,both of which attack Cys159. A scheme illustrating ourcurrent thinking on the role of thiol groups of the ANT inMPTP function is shown in Fig. 2.
6.2.4. Other proteins may interact with the ANT toenhance or inhibit pore opening
In recent years there have been several reports that arange of pro-apoptic proteins, including Bax and the viralprotein R of the HIV-1 virus, and anti-apoptotic proteinsincluding Bcl-2 and the viral protein vMIA of the cytome-galovirus, interact directly with the ANT to exert theireffects on apoptosis (see [59,60]). However, there areaspects of this work that need to be treated with caution. Forexample, although an inhibitory effect of Bcl-2 on theMPTP in situ has been widely reported (see [61,62]), thereare data on isolated mitochondria that fail to support thisview. Thus, it has been demonstrated that the MPT occurs inliver mitochondria from mice over-expressing Bcl-2 withthe same sensitivity to inducers as mitochondria fromcontrol mice [63]. Another report suggests that Bcl-2prevents the permeability transition by enabling the reducedpyridine nucleotides to be maintained during oxidativestress rather than through a direct effect on the MPTPmechanism [64]. The proposed interaction between BAX
and the ANT was originally identified using a yeast twohybrid screen using BAX as bait, that pulled out a peptidecontaining residues 105–156 of the ANT (containing atransmembrane helix and parts of both an extramitochon-drial and intramitochondrial loop) [65]. There are, however,always concerns as to whether protein/protein interactionsdetected with yeast two hydrid screens are specific andoccur in the physiological setting. This is especially truewith membrane proteins since the transcription factor inter-actions that underlie the yeast two hybrid technique requireboth expressed proteins to be soluble in the cytosol, whichis unlikely for membrane proteins. Thus, the interactionbetween a transmembrane helical region of the ANT andBAX could reflect no more than the natural tendency of thehydrophobic ANT sequence to pair up with another hydro-phobic sequence in BAX. Such an interaction might pro-duce a hydrophilic surface that keeps the proteins insolution whilst activating expression of the reporter gene.Nevertheless, some evidence for specificity of the BAX /ANT interaction is provided by binding competition studiesthat locate the region of the ANT to which Vpr and Bcl-2bind as residues 104–116 [66]. This sequence overlaps withthat identified by the yeast two hybrid screen and iscontained within an extramitochondrial loop, as might beexpected for the binding of cytosolic proteins. However,there are also problems associated with the use of thereconstituted ANT in binding studies, since this processrequires solubilisation of the ANT in detergent whilstmaintaining its native state, followed by reconstitution anddetergent removal. None of these procedures is straightfor
Fig. 2. Location of cysteine residues in the ANT whose cross-linking modulates MPTP activity.
A.P. Halestrap et al. / Biochimie 84 (2002) 153–166 159

ward as will be considered below. Despite these misgivings,it remains an attractive hypothesis that other proteins maytarget the ANT, the most abundant protein of the innermitochondrial protein, and exert their effects on apoptosis ornecrosis by modulating the MPT.
6.3. Other possible components of the MPTP
It is important to recognise that the permeability transi-tion is fundamentally an inner membrane phenomenonassociated with uncoupling of the mitochondria, swelling ofthe matrix and unfolding of the inner membrane cristae.Indeed, the MPT can be observed in mitoplasts, mitochon-dria from which the outer membrane is removed, withidentical properties to that observed with normal mitochon-dria [35]. This is not to deny that proteins of the outermembrane may be involved in the MPT, and it is certainlytrue that the MPT leads to outer membrane rupture. How-ever, at the heart of the MPTP must be a channel across theinner membrane. Our data strongly suggest that the onlyinner membrane protein to which CyP-D binds is the ANTwhereas the data of Crompton et al. [50] suggest that VDACmay also bind, perhaps through a secondary interaction withthe ANT. This highlights an ongoing controversy in theliterature as to the minimum configuration of the MPTP.Does the open pore consist only of the ANT and CyP-D aswe originally proposed or are other components requiredsuch as VDAC and the peripheral benzodiazepine receptor(see [1–3]). These proteins were shown to co-purify with theANT as a complex under some conditions [67] and are alsothought to interact at contact sites, points of intimate contactbetween the inner and outer membranes [68].
It was these observations that led Zoratti and Szabo intheir seminal review [41] to propose that VDAC and theperipheral benzodiazepine receptor might be integral com-ponents of the MPTP. Although this was only a hypothesis,and not based on experimental evidence, it was perceived asestablished fact by many of those working on apoptosis whowere becoming aware of the critical role of mitochondria inthis process. Since that time the groups of Kroemer andBrdizka have suggested that other proteins associated withthe contact sites, such hexokinase and creatine kinase mayalso be involved in the MPTP [61]. Indeed fractions ofdetergent solubilised proteins containing these components,along with CyP-D, could be reconstituted into proteolipo-somes to form a calcium activated, CsA-sensitive pore[58,69]. However, which of the components are essential topore formation could not be established by such techniques.The evidence is perhaps most compelling for VDAC sinceCrompton and colleagues were able to reconstitute theANT–VDAC complex eluted from a GST–CyP-D affinitycolumn glutathione into proteoliposomes to form a calciumactivated pore that was inhibited by CsA [50]. However, asrecognised by Crompton et al., this does not demonstratethat VDAC is essential for MPTP formation, and thequestion remains as to the minimum configuration of the
MPTP? Is VDAC an essential component or does it play aregulatory role? Are there other proteins that are essentialfor MPTP function or regulation? Can both the major ANTisoforms form the MPTP and with similar affinities forCyP-D and other regulatory proteins such as VDAC? Themost powerful approach to answering these questions wouldbe to reconstitute the active MPTP complex from purifiedcomponents, but this has proved extremely difficult to dowith any consistency.
6.4. Reconstitution of the MPTP
Support for an involvement of the ANT in the formationof the MPTP had come from earlier studies in which it hadbeen shown that thiol reagents were able to convert thereconstituted ANT from a specific antiporter to a non-specific channel [70]. Subsequently it was shown that thereconstituted ANT can also form non-specific pores whenexposed to high (millimolar) concentrations of calcium[71,72]. These pores differ from the MPTP in that they arenot sensitive to CsA, but their permeability properties aresimilar. Initial reports of reconstitution of the MPTP fromthe laboratories of Brdiczka and Kroemer used crudefractions from whole homogenised brain rather than purifiedcomponents and are thus hard to interpret (see [61]).Subsequently, Kroemer’s group went on to use the purified,reconstituted ANT in studies on the MPTP (see [59], whilstCrompton’s group reported reconstitution of CsA-sensitivepore activity using the CyP-D/ANT/VDAC complex iso-lated by glutathione elution from GST–CyP-D affinitycolumn [50]. However, in our view these experiments fail totake into account three major problems associated with anyattempt to reconstitute the MPTP from its componentproteins and thus make interpretation of the results obtaineddifficult.
• The first problem is that of solubilising the membraneproteins in a suitable detergent without denaturing themand maintaining their activity through purification. Forexample, it is well documented that unless stabilised bybinding of a high affinity ligand such as BKA oratractyloside (ATR), the ANT rapidly denatures whensolubilised in detergent [73]. It is significant that inlaboratories specialising in measuring the activity ofthe reconstituted ANT, studies are performed at lowtemperature and over a millisecond time scale [48,73].In contrast, recent studies using the reconstituted ANTfor investigating the MPTP have confirmed ANT activ-ity by measuring transport over 1 h at room tempera-ture (see e.g. [74]).
• The second problem is the need to obtain sealed,detergent-free proteoliposomes, with the ANT in adefined orientation, preferably with the matrix surfacefacing out to allow binding of exogenous cyclophilin. Aparticular problem in many reported studies is the useof an inappropriate step for the removal of Triton-X100following reconstitution of the solubilised proteins. The
160 A.P. Halestrap et al. / Biochimie 84 (2002) 153–166

low critical micellar concentration and large micellarsize of this detergent means that it cannot be properlyremoved from proteoliposomes by dilution and gelfiltration [75]. Since even very low concentrations ofTriton-X100 can exert profound effects on both theinhibitor sensitivity of VDAC [76] and on the generalpermeability of lipososomes [77], results obtained inthis way must be treated with caution.
• The third problem is that a method must be found toassay pore opening in small amounts of reconstitutedmaterial. The published studies from other laboratorieshave used the release of a radioactive, fluorescent orchromogenic marker molecule entrapped within theproteoliposome. In many cases the release was mea-sured after 30–60 min at room temperature (e.g.[62,66] yet total release of the marker from a proteoli-posome would be expected to occur almost instanta-neously once the MPTP had opened. Furthermore, thisapproach does not allow continuous recording of poreactivity to determine kinetics of activation and regula-tion since the release of the marker is an all or nonephenomenon.
Aware of the problems listed above, we have performedour own studies using rapidly purified ANT reconstitutedinto proteoliposomes in the presence and absence of CyP-D[78]. The most reliable published purifications of the ANTutilise high affinity ligands, such as BKA or ATR to stabilisethe ANT during detergent solubilisation and subsequentpurification [73]. Unfortunately, in our hands, CyP-D doesnot bind to the ATR–ANT and BKA–ANT complexes [38],and thus we cannot use this approach to maintain ANTstability. As an alternative, we have endeavoured to use avery rapid purification procedure for the ANT, using Triton-X100 solubilised heart mitochondria with hydroxyapatitechromatography followed by ion exchange FPLC [72] andimmediate reconstitution of the pure protein fraction inproteoliposomes, using Biobeads to remove detergent. Toassay the reconstituted MPTP we have developed a continu-ous spectrophotometric technique in which malate dehydro-genase is entrapped within the proteoliposomes and is onlyaccessible to its substrates (NADH and oxaloacetate) whenthe pore opens. This is monitored by the decrease in A340 asNADH is oxidised, and thus it is possible to determine therate at which pores open as well as the fraction of pores thatare open under any particular condition. We have used thistechnique to demonstrate that in the presence of recombi-nant CyP-D, the purified, reconstituted ANT does formCsA-sensitive pores at micromolar [Ca2+] (Fig. 3). Thesedata are important because they confirm that the minimumconfiguration of the MPTP includes only the ANT andCyP-D, and any role for other proteins such as VDAC islikely to be regulatory rather than essential. However, wehave not been able to use the reconstituted system toconfirm this or to study detailed kinetics and regulation ofthe pore because the success rate for reconstitution of activeMPTP has been < 10%. We have concentrated our efforts on
establishing the reasons for this pore success rate and howit can be overcome. Recent data suggest that our standardpurification procedure for the ANT causes substantial –S-S-cross-linking of Cys56 to form ANT dimers that we know tobe inactive in pore formation. The problem has been thatwhen reagents such as dithiothreitol are used to maintain thereduced state of the ANT the purification procedure nolonger works.
7. Genetic approaches to studying the MPTP
It does not take much imagination to recognise that apowerful approach to elucidating the mechanisms of theMPTP would be a genetic approach in which proposedcomponents of the MPTP are either knocked out totally orsubjected to site-directed mutagenesis. Yeast would seemthe organism of choice for such studies especially since itappears that their mitochondria do not display a conven
Fig. 3. Reconstitution of the MPTP from purified ANT and CyP-D. Thepurified ANT was reconstituted into proteoliposomes containing entrappedmalate dehydrogenease (MDH). Conversion of NADH to NAD+ by theentrapped malate dehydrogenase is only possible when the MPTP opens.Ca2+ and recombinant CyP-D were added externally and pore openingfollowed by the oxidation of NADH measured spectrophotometrically at340 nm. Data are taken from [78] where further details may be found.
A.P. Halestrap et al. / Biochimie 84 (2002) 153–166 161

tional CsA-sensitive MPT [35,79] and yeast ANT knockoutsare available. Unfortunately, expressing active mammalianANT isoforms in yeast mitochondria has proved extremelydifficult but recent reports suggest that it can be done if theN-terminus of the yeast ANT is spliced onto the mammalianisoform [80]. In conjunction with the expression mamma-lian CyP-D this may provide an alternative approach toreconstitution in studying the molecular mechanism of theMPTP and has the advantage of being accessible to site-directed mutagenesis. However, it must always be borne inmind that over-expression of an IMM protein may disruptnormal mitochondrial function and lead to cell death irre-spective of whether or not it induces the formation of theMPTP. This may be the real basis of the report thatoverexpression of ANT-1 in mammalian cells can domi-nantly induce apoptosis [81]
8. A model of the MPTP that explains the modeof action of different modulators
In Fig. 4 we summarise our current understanding of themechanism of the MPTP, which represents a developmentof the model originally presented in 1990 [19], and whichforms the basis of most models in the literature. We proposethat CyP-D binds to the ANT on Pro61 and that this bindingis greatly enhanced when Cys56 is cross-linked (e.g. byoxidative stress) to Cys159. Yeast mitochondria lack boththese residues and do not possess a CsA-sensitive MPTP[35,79]. We suggest that Ca2+ binds to a site on the ANTitself, perhaps involving some of the many aspartate andglutamate residues on intramitochondrial loops of the ANT,but to date we have no information on the exact location.Binding of adenine nucleotides to the substrate binding siteof the ANT greatly reduces the sensitivity of the pore to[Ca2+], probably by masking the calcium binding site.Binding of calcium is thought to trigger the conformationalchange required to induce pore formation. This is likely toinvolve a cis-trans isomerisation of the peptide bond adja-cent to Pro61 that is facilitated by CyP-D binding but nottotally dependent on it. This is consistent with the ability ofhigh (mM) [Ca2+] to convert the reconstituted to a pore inthe absence of CyP-D [71]. Furthermore, at high calciumconcentrations or upon adenine nucleotide depletion, thepore becomes insensitive to CsA [27,35].
Our model is able to provide a plausible explanation ofthe effects of most known regulators of the pore that may actto modulate binding of adenine nucleotides, calcium orCyP-D to their respective sites as summarised in Table 1.Thus, any intervention that reduces adenine nucleotidebinding enhances pore opening. Factors acting this wayinclude adenine nucleotide depletion, increased matrixphosphate or pyrophosphate concentrations (competes forthe nucleotide binding site), and the conformational state ofthe ANT. The latter can be influenced by depolarisation(decreases the affinity of the matrix binding site for ADP)
and specific ligands of the ANT such as carboxyatractylo-side (decrease matrix ADP binding affinity) and BKA(increases matrix ADP binding affinity). Furthermore, modi-fication of a specific thiol group on the ANT (Cys159) eitherby oxidative stress or thiol reagents such as eosine maleim-ide or phenylarsine oxide, also decreases adenine nucleotidebinding and can account for the ability of these agents toactivate the pore [35]. Factors that enhance cyclophilinbinding and hence increase the sensitivity of the MPTP to[Ca2+] include oxidative stress (Cys56–Cys159 cross-linking), chaotropic agents and increased matrix volume. Incontrast, CsA has the opposite effect by preventing CyP-Dbinding. Bernardi and colleagues have presented data toshow that the effect of low pH involves a specific histidineresidue and that this may modulate CyP binding to the innermitochondrial membrane [36]. However, in our experimentswe did not detect such an effect of low pH on CyP bindingto either inner membranes [34] or to the purified ANT [38].Rather, we suggest that protons (low pH), Mg2+ and otherdivalent cations compete directly with Ca2+ at the triggersite.
The modes of action of two other potent inhibitors of theMPTP are not so clear. Trifluoperazine is a potent inhibitorof the MPT under energised but not de-energised conditions[35]. It was originally thought to act indirectly throughinhibition of phospholipase A2, preventing the accumulationof free fatty acids that are known to stimulate the MPTP,probably through interaction with the ANT. However, inhi-bition occurs even without changes in free fatty acidaccumulation and is now thought to be mediated by aneffect on surface membrane charge that changes the voltagesensitivity of the MPTP [82]. The mechanism of action ofubiquinone analogues as either as activators or inhibitors ofthe MPTP is also unclear [24,83]. Earlier work had shownthat the probability of pore opening varied according to therate of electron transfer through Complex 1 of the respira-tory chain respiratory [83]. These data led Fontaine andcolleagues to suggest that components of Complex 1 may beinvolved in the formation and/or regulation of the MPTP[24, 84]. However, the recent observation that the uncou-pling proteins UCP1, UC2 and UCP3 require oxidisedubiquinone to function suggest an alternative mechanism[85, 86]. Since the UCPs are close relatives of ANT it wouldseem quite likely that a ubiquinone binding site may alsoexist on the ANT.
9. Conclusions
Although falling short of definitive proof, the availableevidence strongly supports a model for the MPTP in whichthe ANT and CyP-D are the key components, with otherproteins in the outer mitochondrial membrane and cytosol(such as VDAC and Bcl-2) playing regulatory roles. How-ever, urgently required are reproducible protocols for thereconstitution of the active MPTP from its pure components
162 A.P. Halestrap et al. / Biochimie 84 (2002) 153–166
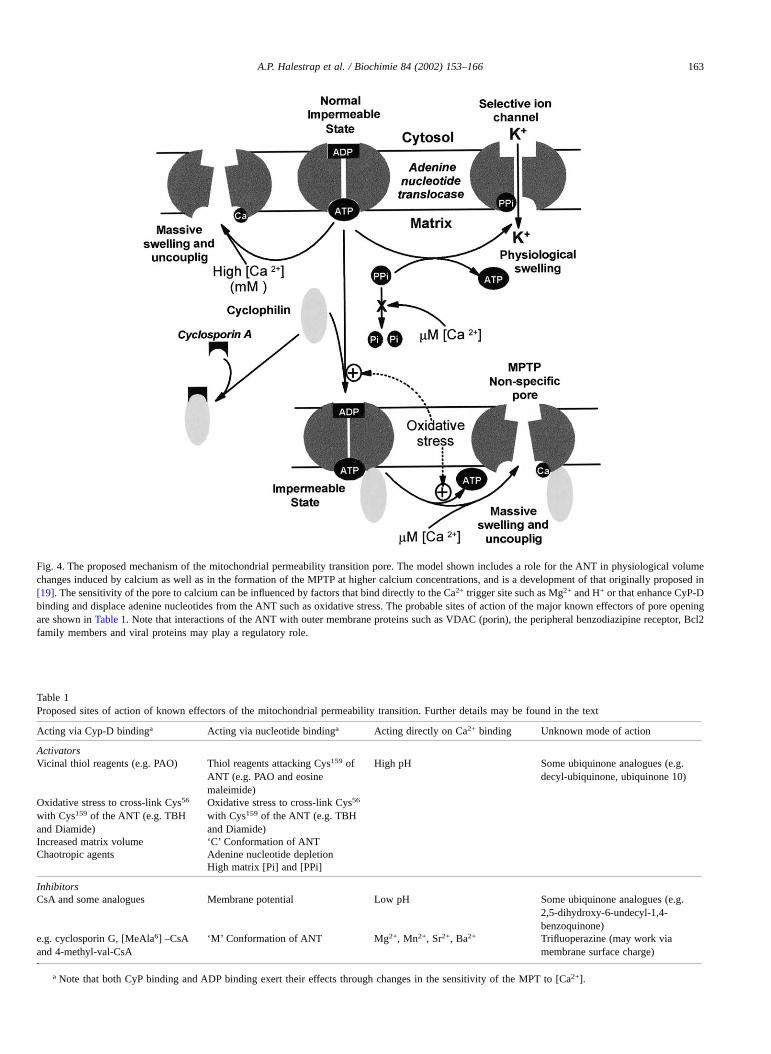
Table 1Proposed sites of action of known effectors of the mitochondrial permeability transition. Further details may be found in the text
Acting via Cyp-D bindinga Acting via nucleotide bindinga Acting directly on Ca2+ binding Unknown mode of action
ActivatorsVicinal thiol reagents (e.g. PAO) Thiol reagents attacking Cys159 of
ANT (e.g. PAO and eosinemaleimide)
High pH Some ubiquinone analogues (e.g.decyl-ubiquinone, ubiquinone 10)
Oxidative stress to cross-link Cys56
with Cys159 of the ANT (e.g. TBHand Diamide)
Oxidative stress to cross-link Cys56
with Cys159 of the ANT (e.g. TBHand Diamide)
Increased matrix volume ‘C’ Conformation of ANTChaotropic agents Adenine nucleotide depletion
High matrix [Pi] and [PPi]
InhibitorsCsA and some analogues Membrane potential Low pH Some ubiquinone analogues (e.g.
2,5-dihydroxy-6-undecyl-1,4-benzoquinone)
e.g. cyclosporin G, [MeAla6] –CsAand 4-methyl-val-CsA
‘M’ Conformation of ANT Mg2+, Mn2+, Sr2+, Ba2+ Trifluoperazine (may work viamembrane surface charge)
.a Note that both CyP binding and ADP binding exert their effects through changes in the sensitivity of the MPT to [Ca2+].
Fig. 4. The proposed mechanism of the mitochondrial permeability transition pore. The model shown includes a role for the ANT in physiological volumechanges induced by calcium as well as in the formation of the MPTP at higher calcium concentrations, and is a development of that originally proposed in[19]. The sensitivity of the pore to calcium can be influenced by factors that bind directly to the Ca2+ trigger site such as Mg2+ and H+ or that enhance CyP-Dbinding and displace adenine nucleotides from the ANT such as oxidative stress. The probable sites of action of the major known effectors of pore openingare shown in Table 1. Note that interactions of the ANT with outer membrane proteins such as VDAC (porin), the peripheral benzodiazipine receptor, Bcl2family members and viral proteins may play a regulatory role.
A.P. Halestrap et al. / Biochimie 84 (2002) 153–166 163

and a suitable assay to study its activity and regulation inreal time. Progress is being made in this direction, but themajor hurdle remains the maintenance of ANT activityduring detergent solubilisation, purification and reconstitution. Once this hurdle is overcome, it is likely that many ofthe present controversies will be resolved which may aid inthe design of drugs to protect tissues from cell death causedby a range of insults such as ischaemia/reperfusion andsome chemical toxins. Conversely, triggering the opening ofthe MPTP might provide another target for chemotherapy.
Acknowledgements
This work was supported by project grants from theMedical Research Council and the British Heart Foundationand a CASE Studentship to GPM from the BBSRC inassociation with MitoKor.
References
[1] A.P. Halestrap, The Mitochondrial Permeability Transition: its mo-lecular mechanism and role in reperfusion injury, in: G.C. Brown,D.G. Nicholls, C.E. Cooper (Eds.), Biochemical Society Symposiumno 66 - Mitochondria and Cell Death, Portland Press, London, 1999,pp. 181–203.
[2] M. Crompton, The mitochondrial permeability transition pore and itsrole in cell death, Biochem. J. 341 (1999) 233–249.
[3] G. Kroemer, J.C. Reed, Mitochondrial control of cell death, Nat. Med.6 (2000) 513–519.
[4] P. Nicotera, M. Leist, Energy supply and the shape of death in neuronsand lymphoid cells, Cell Death Differ. 4 (1997) 435–442.
[5] J.C. Martinou, D.R. Green, Breaking the mitochondrial barrier, Nat.Rev. Mol. Cell Biol. 2 (2001) 63–67.
[6] A.P. Halestrap, P.M. Kerr, S. Javadov, K.Y. Woodfield, Elucidatingthe molecular mechanism of the permeability transition pore and itsrole in reperfusion injury of the heart, Biochim. Biophys. Acta 1366(1998) 79–94.
[7] M.S. Suleiman, A.P. Halestrap, E.J. Griffiths, Mitochondria andmyocardial protection, Pharmacol. Ther. 89 (2001) 29–46.
[8] T.E. Gunter, D.R. Pfeiffer, Mechanisms by which mitochondriatransport calcium, Am. J. Physiol. 258 (1990) C755–C786.
[9] R.A. Haworth, D.R. Hunter, The Ca2+-induced membrane transitionin mitochondria. II. Nature of the Ca2+ trigger site, Arch. Biochem.Biophys. 195 (1979) 460–467.
[10] D.R. Hunter, R.A. Haworth, The Ca2+ -induced membrane transitionin mitochondria. I. The protective mechanisms, Arch. Biochem.Biophys. 195 (1979) 453–459.
[11] M. Crompton, A. Costi, L. Hayat, Evidence for the presence of areversible Ca2+-dependent pore activated by oxidative stress in heartmitochondria, Biochem. J. 245 (1987) 915–918.
[12] M. Crompton, The mitochondrial permeability transition pore and itsrole in cell death, Biochem. J. 341 (1999) 233–249.
[13] A.P. Halestrap, Regulation of mitochondrial metabolism throughchanges in matrix volume, Biochem. Soc. Trans. 22 (1994) 522–529.
[14] J.G. McCormack, A.P. Halestrap, R.M. Denton, The role of calciumions in the regulation of mammalian intramitochondrial metabolism,Physiol. Rev. 70 (1990) 391–425.
[15] A.P. Halestrap, The regulation of the matrix volume of mammalianmitochondria in vivo and in vitro, and its role in the control ofmitochondrial metabolism, Biochim. Biophys. Acta. 973 (1989)355–382.
[16] G.A. Rutter, R. Rizzuto, Regulation of mitochondrial metabolism byER Ca2+ release: an intimate connection, Trends Biochem. Sci. 25(2000) 215–221.
[17] M. Crompton, H. Ellinger, A. Costi, Inhibition by cyclosporin A of aCa2+-dependent pore in heart mitochondria activated by inorganicphosphate and oxidative stress, Biochem. J. 255 (1988) 357–360.
[18] A.M. Davidson, A.P. Halestrap, Partial Inhibition by Cyclosporin A ofthe swelling of liver mitochondria in vivo and in vitro induced bysub-micromolar [Ca2+] but not by butyrate. Evidence for two distinctswelling mechanisms, Biochem. J. 268 (1990) 147–152.
[19] A.P. Halestrap, A.M. Davidson, Inhibition of Ca2+-induced largeamplitude swelling of liver and heart mitochondria by Cyclosporin Ais probably caused by the inhibitor binding to mitochondrial matrixpeptidyl–prolyl cis-trans isomerase and preventing it interacting withthe adenine nucleotide translocase, Biochem. J. 268 (1990) 153–160.
[20] P. Bernardi, Mitochondrial transport of cations: channels, exchangers,and permeability transition, Physiol. Rev. 79 (1999) 1127–1155.
[21] A.P. Halestrap, Calcium-dependent opening of a non-specific pore inthe mitochondrial inner membrane is inhibited at pH values below 7- implications for the protective effect of low pH against chemical andhypoxic cell damage, Biochem. J. 278 (1991) 715–719.
[22] P. Bernardi, S. Vassanelli, P. Veronese, R. Colonna, I. Szabo,M. Zoratti, Modulation of the mitochondrial permeability transitionpore - effect of protons and divalent cations, J. Biol. Chem. 267(1992) 2934–2939.
[23] P. Bernardi, P. Veronese, V. Petronilli, Modulation of the mitochon-drial cyclosporin a-sensitive permeability transition pore. 1 evidencefor 2 separate Me2+ binding sites with opposing effects on the poreopen probability, J. Biol. Chem. 268 (1993) 1005–1010.
[24] L. Walter, V. Nogueira, X. Leverve, M.P. Heitz, P. Bernardi, E. Fon-taine, Three classes of ubiquinone analogs regulate the mitochondrialpermeability transition pore through a common site, J. Biol. Chem.275 (2000) 29521–29527.
[25] A. Galat, Peptidylproline cis-trans-isomerases–immunophilins, Eur.J. Biochem. 216 (1993) 689–707.
[26] E.J. Griffiths, A.P. Halestrap, Further evidence that cyclosporin-Aprotects mitochondria from calcium overload by inhibiting a matrixpeptidyl–prolyl cis-trans isomerase - implications for the immuno-suppressive and toxic effects of cyclosporin, Biochem. J. 274 (1991)611–614.
[27] E.J. Griffiths, A.P. Halestrap, Mitochondrial non-specific pores remainclosed during cardiac ischaemia, but open upon reperfusion, Bio-chem. J. 307 (1995) 93–98.
[28] C.P. Connern, A.P. Halestrap, Purification and N-terminal sequencingof peptidyl–prolyl cis-trans-isomerase from rat liver mitochondrialmatrix reveals the existence of a distinct mitochondrial cyclophilin,Biochem. J. 284 (1992) 381–385.
[29] K.Y. Woodfield, N.T. Price, A.P. Halestrap, cDNA cloning of ratmitochondrial cyclophilin, Biochim. Biophys. Acta 1351 (1997)27–30.
[30] D.J. Bergsma, C. Eder, M. Gross, H. Kersten, D. Sylvester, E. Appel-baum, D. Cusimano, G.P. Livi, M.M. Mclaughlin, K. Kasyan,T.G. Porter, C. Silverman, D. Dunnington, A. Hand, W.P. Prichett,M.J. Bossard, M. Brandt, M.A. Levy, The cyclophilin multigenefamily of peptidyl–prolyl isomerases - characterization of threeseparate human isoforms, J. Biol. Chem. 266 (1991) 23204–23214.
[31] A. Tanveer, S. Virji, L. Andreeva, N.F. Totty, J.J. Hsuan, J.M. Ward,M. Crompton, Involvement of cyclophilin D in the activation of amitochondrial pore by Ca2+ and oxidant stress, Eur. J. Biochem. 238(1996) 166–172.
[32] N. Johnson, A. Khan, S. Virji, J.M. Ward, M. Crompton, Import andprocessing of heart mitochondrial cyclophilin D, Eur. J. Biochem. 263(1999) 353–359.
[33] C.P. Connern, A.P. Halestrap, Recruitment of mitochondrial cyclophi-lin to the mitochondrial inner membrane under conditions of oxida-tive stress that enhance the opening of a calcium-sensitive non-specific channel, Biochem. J. 302 (1994) 321–324.
164 A.P. Halestrap et al. / Biochimie 84 (2002) 153–166

[34] C.P. Connern, A.P. Halestrap, Chaotropic agents and increased matrixvolume enhance binding of mitochondrial cyclophilin to the innermitochondrial membrane and sensitize the mitochondrial permeabil-ity transition to [Ca2+], Biochemistry 35 (1996) 8172–8180.
[35] A.P. Halestrap, K.Y. Woodfield, C.P. Connern, Oxidative stress, thiolreagents, and membrane potential modulate the mitochondrial perme-ability transition by affecting nucleotide binding to the adeninenucleotide translocase, J. Biol. Chem. 272 (1997) 3346–3354.
[36] A. Nicolli, E. Basso, V. Petronilli, R.M. Wenger, P. Bernardi,Interactions of cyclophilin with the mitochondrial inner membraneand regulation of the permeability transition pore, a cyclosporinA-sensitive channel, J. Biol. Chem. 271 (1996) 2185–2192.
[37] L. Andreeva, A. Tanveer, M. Crompton, Evidence for the involvementof a membrane-associated cyclosporin-A-binding protein in the Ca2+-activated inner membrane pore of heart mitochondria, Eur. J. Bio-chem. 230 (1995) 1125–1132.
[38] K. Woodfield, A. Ruck, D. Brdiczka, A.P. Halestrap, Direct demon-stration of a specific interaction between cyclophilin-D and theadenine nucleotide translocase confirms their role in the mitochon-drial permeability transition, Biochem. J. 336 (1998) 287–290.
[39] S.A. Novgorodov, T.I. Gudz, Y.M. Milgrom, G.P. Brierley, Thepermeability transition in heart mitochondria is regulated synergisti-cally by ADP and cyclosporin-A, J. Biol. Chem. 267 (1992)16274–16282.
[40] M. Crompton, L. Andreeva, On the interactions of Ca2+ and cy-closporin A with a mitochondrial inner membrane pore: A study usingcobaltammine complex inhibitors of the Ca2+ uniporter, Biochem. J.302 (1994) 181–185.
[41] M. Zoratti, I. Szabo, Electrophysiology of the inner mitochondrialmembrane, J. Bioenerg. Biomembr. 26 (1994) 543–553.
[42] T.A. Lohret, R.C. Murphy, T. Drgon, K.W. Kinnally, Activity of themitochondrial multiple conductance channel is independent of theadenine nucleotide translocator, J. Biol. Chem. 271 (1996)4846–4849.
[43] K. LeQuoc, D. LeQuoc, Involvement of the ADP/ATP carrier incalcium-induced perturbations of the mitochondrial inner membranepermeability: importance of the orientation of the nucleotide bindingsite, Arch. Biochem. Biophys. 265 (1988) 249–257.
[44] S.A. Novgorodov, T.I. Gudz, Y.E. Kushnareva, D.B. Zorov,Y.B. Kudrjashov, Effect of ADP/ATP antiporter conformational stateon the suppression of the nonspecific permeability of the innermitochondrial membrane by cyclosporine-A, FEBS Lett. 277 (1990)123–126.
[45] R.A. Haworth, D.R. Hunter, Control of the mitochondrial permeabil-ity transition pore by high-affinity ADP binding at the ADP/ATPtranslocase in permeabilized mitochondria, J. Bioenerg. Biomembr.32 (2000) 91–96.
[46] P. Bernardi, Modulation of the mitochondrial cyclosporin-a-sensitivepermeability transition pore by the proton electrochemical gradient -evidence that the pore can be opened by membrane depolarization,J. Biol. Chem. 267 (1992) 8834–8839.
[47] J.H.M. Soverijn, L.A. Huisman, J. Rosing, N. Kemp, Comparison ofADP and ATP as substrates for the adenine nucleotide translocator inrat liver mitochondria, Biochem. Biophys. Acta 305 (1973) 185–198.
[48] T. Gropp, N. Brustovetsky, M. Klingenberg, V. Muller, K. Fendler,E. Bamberg, Kinetics of electrogenic transport by the ADP/ATPcarrier, Biophys. J. 77 (1999) 714–726.
[49] E. Lenartowicz, P. Bernardi, G.F. Azzone, Phenylarsine oxide inducesthe cyclosporin-A-sensitive membrane permeability transition in ratliver mitochondria, J. Bioenerg. Biomembr. 23 (1991) 679–688.
[50] M. Crompton, S. Virji, J.M. Ward, Cyclophilin-D binds strongly tocomplexes of the voltage-dependent anion channel and the adeninenucleotide translocase to form the permeability transition pore, Eur.J. Biochem. 258 (1998) 729–735.
[51] P. Costantini, B.V. Chernyak, V. Petronilli, P. Bernardi, Modulation ofthe mitochondrial permeability transition pore by pyridine nucleotidesand dithiol oxidation at two separate sites, J. Biol. Chem. 271 (1996)6746–6751.
[52] B.V. Chernyak, P. Bernardi, The mitochondrial permeability transitionpore is modulated by oxidative agents through both pyridine nucle-otides and glutathione at two separate sites, Eur. J. Biochem. 238(1996) 623–630.
[53] A. Bindoli, M.T. Callegaro, E. Barzon, M. Benetti, M.P. Rigobello,Influence of the redox state of pyridine nucleotides on mitochondrialsulfhydryl groups and permeability transition, Arch. Biochem. Bio-phys. 342 (1997) 22–28.
[54] E. Majima, H. Koike, Y.M. Hong, Y. Shinohara, H. Terada, Charac-terization of Cysteine Residues of Mitochondrial ADP/ATP Carrierwith the SH-Reagents Eosin 5-maleimide and N-ethylmaleimide,J. Biol. Chem. 268 (1993) 22181–22187.
[55] E. Majima, Y. Shinohara, N. Yamaguchi, Y.M. Hong, H. Terada,Importance of loops of mitochondrial ADP/ATP carrier for itstransport activity deduced from reactivities of its cysteine residueswith the sulfhydryl reagent eosin-5-maleimide, Biochemistry 33(1994) 9530–9536.
[56] E. Majima, K. Ikawa, M. Takeda, M. Hashimoto, Y. Shinohara,H. Terada, Translocation of loops regulates transport activity ofmitochondrial ADP–ATP carrier deduced from formation of a specificintermolecular disulfide bridge catalyzed by copper-o-phenanthroline,J. Biol. Chem. 270 (1995) 29548–29554.
[57] E. Majima, N. Yamaguchi, H. Chuman, Y. Shinohara, M. Ishida,S. Goto, H. Terada, Binding of the fluorescein derivative eosin Y tothe mitochondrial ADP/ATP carrier: characterization of the adeninenucleotide binding site, Biochemistry 37 (1998) 424–432.
[58] P. Costantini, A.S. Belzacq, H. LaVieira, N. Larochette, M.A. de-Pablo, N. Zamzami, S.A. Susin, C. Brenner, G. Kroemer, Oxidationof a critical thiol residue of the adenine nucleotide translocatorenforces Bcl-2-independent permeability transition pore opening andapoptosis, Oncogene 19 (2000) 307–314.
[59] H.L.A. Vieira, D. Haouzi, C. ElHamel, E. Jacotot, A.S. Belzacq,C. Brenner, G. Kroemer, Permeabilization of the mitochondrial innermembrane during apoptosis: impact of the adenine nucleotide trans-locator, Cell Death Differ. 7 (2000) 1146–1154.
[60] P. Boya, B. Roques, G. Kroemer, Viral and bacterial proteinsregulating apoptosis at the mitochondrial level, EMBO J. 20 (2001)4325–4331.
[61] I. Marzo, C. Brenner, N. Zamzami, S.A. Susin, G. Beutner, D. Brdic-zka, R. Remy, Z.H. Xie, J.C. Reed, G. Kroemer, The permeabilitytransition pore complex: a target for apoptosis regulation by caspasesand Bcl-2-related proteins, J. Exp. Med. 187 (1998) 1261–1271.
[62] C. Brenner, H. Cadiou, H.L.A. Vieira, N. Zamzami, I. Marzo,Z.H. Xie, B. Leber, D. Andrews, H. Duclohier, J.C. Reed, G. Kro-emer, Bcl-2 and Bax regulate the channel activity of the mitochon-drial adenine nucleotide translocator, Oncogene 19 (2000) 329–336.
[63] J.C. Yang, A. Kahn, G. Cortopassi, Bcl-2 does not inhibit thepermeability transition pore in mouse liver mitochondria, Toxicology151 (2000) 65–72.
[64] A. Kowaltowski, A.E. Vercesi, G. Fiskum, Bcl-2 prevents mitochon-drial permeability transition and cytochrome c release via mainte-nance of reduced pyridine nucleotides, Cell Death Differ. 7 (2000)903–910.
[65] I. Marzo, C. Brenner, N. Zamzami, J.M. Jurgensmeier, S.A. Susin,H.L.A. Vieira, M.C. Prevost, Z.H. Xie, S. Matsuyama, J.C. Reed,G. Kroemer, Bax and adenine nucleotide translocator cooperate in themitochondrial control of apoptosis, Science 281 (1998) 2027–2031.
A.P. Halestrap et al. / Biochimie 84 (2002) 153–166 165

[66] E. Jacotot, K.F. Ferri, C. ElHamel, C. Brenner, S. Druillennec,J. Hoebeke, P. Rustin, D. Metivier, C. Lenoir, M. Geuskens,H.L.A. Vieira, M. Loeffler, A.S. Belzacq, J.P. Briand, N. Zamzami,L. Edelman, Z.H. Xie, J.C. Reed, B.P. Roques, G. Kroemer, Controlof mitochondrial membrane permeabilization by adenine nucleotidetranslocator interacting with HIV-1 viral protein R and Bcl-2, J. Exp.Med. 193 (2001) 509–519.
[67] M.W. Mcenery, A.M. Snowman, R.R. Trifiletti, S.H. Snyder, Isolationof the Mitochondrial Benzodiazepine Receptor - Association with theVoltage-Dependent Anion Channel and the Adenine Nucleotide Car-rier, Proc. Natl. Acad. Sci. USA 89 (1992) 3170–3174.
[68] D. Brdiczka, Contact sites between mitochondrial envelope mem-branes - structure and function in energy-transfer and protein-transfer,Biochim. Biophys. Acta 1071 (1991) 291–312.
[69] G. Beutner, A. Ruck, B. Riede, D. Brdiczka, Complexes betweenporin, hexokinase, mitochondrial creatine kinase and adenylate trans-locator display properties of the permeability transition pore. Impli-cation for regulation of permeability transition by the kinases,Biochim. Biophys. Acta 1368 (1998) 7–18.
[70] T. Dierks, A. Salentin, C. Heberger, R. Kramer, The mitochondrialaspartate/glutamate and ADP/ATP carrier switch from obligate coun-terexchange to unidirectional transport after modification by SH-reagents, Biochim. Biophys. Acta 1028 (1990a) 268–280.
[71] N. Brustovetsky, M. Klingenberg, Mitochondrial ADP/ATP carriercan be reversibly converted into a large channel by Ca2+, Biochem-istry 35 (1996) 8483–8488.
[72] A. Ruck, M. Dolder, T. Wallimann, D. Brdiczka, Reconstitutedadenine nucleotide translocase forms a channel for small moleculescomparable to the mitochondrial permeability transition pore, FEBSLett. 426 (1998) 97–101.
[73] M. Klingenberg, E. Winkler, S.G. Huang, ADP/ATP carrier anduncoupling protein, Methods Enzymol. 260 (1995) 369–389.
[74] A.S. Belzacq, E. Jacotot, H.L. Vieira, D. Mistro, D.J. Granville,Z. Xie, J.C. Reed, G. Kroemer, C. Brenner, Apoptosis induction bythe photosensitizer verteporfin: identification of mitochondrial ad-enine nucleotide translocator as a critical target, Cancer Res. 61(2001) 1260–1264.
[75] J.L. Rigaud, D. Levy, G. Mosser, O. Lambert, Detergent removal bynon-polar polystyrene beads - Applications to membrane proteinreconstitution and two-dimensional crystallization, Eur. Biophys.J. Biophys. Lett. 27 (1998) 305–319.
[76] G. Bathori, A. Fonyo, E. Ligeti, Trace amounts of Triton X-100modify the inhibitor sensitivity of the mitochondrial porin, Biochim.Biophys. Acta 1234 (1995) 249–254.
[77] G.M. Alder, W.M. Arnold, C.L. Bashford, A.F. Drake, C.A. Pasternak,U. Zimmermann, Divalent cation-sensitive pores formed by naturaland synthetic melittin and Triton X-100, Biochim. Biophys. Acta1061 (1991) 111–120.
[78] A.P. Halestrap, E. Doran, J.P. Gillespie, A. O’Toole, Mitochondriaand cell death, Biochem, Soc. Trans. 28 (2000) 170–177.
[79] S. Manon, X. Roucou, M. Guerin, M. Rigoulet, B. Guerin, Minire-view: Characterization of the yeast mitochondria unselective channel:a counterpart to the mammalian permeability transition pore?J. Bioenerg. Biomembr. 30 (1998) 419–429.
[80] M. Hashimoto, Y. Shinohara, E. Majima, T. Hatanaka, N. Yamazaki,H. Terada, Expression of the bovine heart mitochondrial ADP/ATPcarrier in yeast mitochondria: significantly enhanced expression byreplacement of the N-terminal region of the bovine carrier by thecorresponding regions of the yeast carriers, Biochim. Biophys. Acta1409 (1999) 113–124.
[81] M.K.A. Bauer, A. Schubert, O. Rocks, S. Grimm, Adenine nucleotidetranslocase-1, a component of the permeability transition pore, candominantly induce apoptosis, J. Cell Biol. 147 (1999) 1493–1501.
[82] K.M. Broekemeier, D.R. Pfeiffer, Inhibition of the mitochondrialpermeability transition by cyclosporin a during long time frameexperiments: relationship between pore opening and the activity ofmitochondrial phospholipases, Biochemistry 34 (1995) 16440–16449.
[83] E. Fontaine, F. Ichas, P. Bernardi, A ubiquinone-binding site regulatesthe mitochondrial permeability transition pore, J. Biol. Chem. 273(1998) 25734–25740.
[84] E. Fontaine, O. Eriksson, F. Ichas, P. Bernardi, Regulation of thepermeability transition pore in skeletal muscle mitochondria - Modu-lation by electron flow through the respiratory chain complex, J. Biol.Chem. 273 (1998) 12662–12668.
[85] K.S. Echtay, E. Winkler, M. Klingenberg, Coenzyme, Q is anobligatory cofactor for uncoupling protein function, Nature 408(2000) 609–613.
[86] K.S. Echtay, E. Winkler, K. Frischmuth, M. Klingenberg, Uncouplingproteins 2 and 3 are highly active H+ transporters and highlynucleotide sensitive when activated by coenzyme Q (Ubiquinone),Proc. Natl. Acad. Sci. USA 98 (2001) 1416–1421.
166 A.P. Halestrap et al. / Biochimie 84 (2002) 153–166