the role of skeletal muscle in glucose transport, glucose...
TRANSCRIPT

The Role of Skeletal Muscle in Glucose Transport, Glucose Homeostasis, and Insulin Resistance: Implications for Physical Therapy
Skeletal muscle has a fundamentally important role in the maintenance of nor- mal glucose homeostasis and in regulating whole-body carbohydrate metabolism. In this review, we discuss the regulation of skeletal muscle glucose transport by muscular activity and inactivity. A l a ~ e number of patients routinely seen by pbysical therapists exhibit some form of skeletal muscle insulin resistance. There- fore, we? discuss how skeletal muscle insulin resistance can be localized to a rela- tively snzall muscle mass, or in other circumstances can affect a l a ~ e proportion of the muscle mass leading to disturbances in whole-body glucose homeostasis. We review the mechanisms and regulation of skeletal muscle glucose transport as backgmund for understanding how dejects in this pmess may contribute to the underlying pathogenesis of insulin resistance. Research into the events regulating glucose entry into skeletal muscles has consi'derable impact on how pbysical ther- apy exercise prescriptions may beneJit patients with disturbances in carbohydrate metabolism. With an understanding of the principles of proper exercise prescrip- tion, pbysical therapists can use exercise training as a primary therapeutic inter- vention to improve local muscle and whole-body glucose utilization, and thereby minimize insulin resistance. ISinacore DR, Gulve EA. The role of skeletal muscle in glucose transport, glucose homeostasis, and insulin resistance: implications for physical therapy. Phys Ther. 1993;73:878-891.]
Key Words: Carbohydrate metabolism, Glucose, Insulin resistance, Skeletal muscle,
Physical therapists often focus on deficits in muscular functions while attempting to correct muscle weak- ness, improve endurance, or control inappropriate muscular movements. Unfortunately, physical therapists have largely ignored the role of skeletal muscle in the maintenance of glucose homeostasis and the deficits in skele-
tal muscle glucose metabolism that result from muscular inactivity.
The primary sources of energy for skeletal muscle are carbohydrates and fat. Uptake of glucose is important during exercise in order to supply fuel to the working muscles and after exercise for the replenishment of
DR Sinacore, PhD, PT, is Assistant Professor, Program in Physical Therapy, Washington University School of Medicine, Box 8083, 660 S Euclid Ave, St Louis, MO 63110 (USA). Address all correspon- dence to Dr Sinacore.
EA Gulve, PhD, is Research Assistant Professor, Section of Applied Physiology, Department of Medi- cine, Washington University School of Medicine.
Research in which Dr Sinacore participated was partially supported by a Doctoral Scholarship Award from the Foundation for Physical Therapy Inc. Research in which Dr Gulve participated was supported by grants DK18986 and AGO0425 and by Individual National Research Service Award AGO5404 from the National Institutes of Health.
Davld R Slnacore Erlc A Gulve
muscle glycogen stores. An over- whelming body of evidence now exists that acute muscle contractile activity and chronic exercise improve skeletal muscle glucose transport and whole-body glucose homeostasis.
The need for physical therapists to recognize, understand, and exploit the role of skeletal muscle in glucose homeostasis is underscored by the high prevalence of patients with un- derlying skeletal muscle insulin resis- tance. Resistance to the actions of insulin in these patients can be con- fined to isolated muscle groups (eg, following immobilization of muscle groups by casting), o r it can take on more severe proportions, such as that found in patients with impaired glu-
Physical Therapy /Volume 73, Number 12December 1993 878 / 59

- Table. Major Factors InJuencing Glucose Homeostasik
Intestinal glucose absorption
Pancreatic insulin secretion
Hepatic glucose balance
Glucose extraction (glycolysis and glycogen synthesis stimulated by insulin)
Glucose output (inhibited by insulin)
Peripheral glucose uptake
Insulin-insensitive tissues (eg, brain, red blood cells)
Insulin-sensitive tissues (skeletal and cardiac muscle, adipose tissue)
Insulin counter-regulatory hormones (hormones that increase blood glucose)
Catecholamines, glucagon, glucocorticoids, growth hormone
Renal glucose reabsorption
cose tolerance (IGT) or non-insulin- dependent diabetes mellitus (NIDDM, or type 11 diabetes mellitus).
The purpose of this review is to ex- amine the role of skeletal muscle in maintaining normal glucose homeo- stasis and in enhancing insulin's ac- tion to promote the uptake and dis- posal of glucose. We discuss how skeletal muscle resistance to insulin- stimulated glucose transport is in- duced in a number of clinical condi- tions commonly encountered by physical therapists. Lastly, we summa- rize and review selected aspects of an appropriate exercise prescription, mainly for patients with IGT or NIDDM who are insulin resistant, and other factors that may influence glu- cose utilization by individual mus- cle(~) or the whole body.
Much of the discussion in this review is based on animal research. Our understanding of insulin action in normal and insulin-resistant states has been advanced tremendously by ani- mal research. Although results from animal research can often be applied
on adipocyte insulin action, liver metabolism, and pancreatic insulin secretion are beyond the scope of this review. *
The Role of Skeletal Muscle In Glucose Horneostasls
Before addressing the importance of physical activity in maintaining glu- cose homeostasis, we will discuss the role of skeletal muscle in glucose metabolism and define some essential terminology. First, we will review the regulation of blood glucose. Next, we will describe the pathways for the transport of glucose into skeletal muscle. Finally, we will discuss the pathophysiology of insulin action in skeletal muscle and its contribution to disturbances of whole-body glucose metabolism.
Regulatlon of Blood Glucose Levels
Blood glucose levels are ordinarily controlled within a very narrow range over a 24-hour period.1 In any nondi- abetic population in the fasted state,
qualitatively to humans, quantitative blood glucose values generally vary differences (eg, in precise magnitudes between 3.4 and 6.2 mM (60-100 or in the time course of events) often mg/dL). Although there may be some exist between animal studies and variations, several studies suggest that, situations in humans. Exercise effects within any given individual, normal
'For a discussion of these effects, refer to Sat0 Y, Poortmans J, Hashimoto I, Oshida Y. Integration of medical and sport sciences. In: Hebbelinck M, Shephard RJ, eds. Medicine and Spotl Science. Basel, Swiuerland: S Karger AG, Medical and Scientific Publishers; 1992:37.
pU/mL=6 pM of insulin
blood glucose levels in the postab- sorptive state (ie, the period between meals in which energy must be sup- plied by the body's endogenous fuel stores) are strictly maintained within 20.3 mM (5 mg/dL).2,3 Even postpran- dially (ie, after a meal), blood glucose concentrations increase only 1 to 2 mM, and rarely, if ever, exceed 7.8 to 8.4 mM (140-150 mg/dL).* This strict maintenance of glycemia is normally achieved by a complex set of interac- tions between hormones that act in a coordinated fashion primarily on the liver, pancreatic P-cells, and skeletal muscles.* Insulin-mediated glucose uptake and utilization by peripheral tissues (primarily skeletal muscle) play a pivotal role in these coordi- nated efforts to maintain normal glu- cose homeostasis (Table).
The maintenance of glycemia is de- pendent on the sensitivity of skeletal muscles to the actions of the polypep- tide hormone, insulin. In the basal state, the pancreas normally secretes about 25 ng . kg-'. min-' of insulin.5 In the non-insulin-resistant state, fasting insulin levels may vary from 5 to 20 pLJ/rn~.6+ Postprandial levels may increase severalfold, with peak values reaching 100 pU/mL or high- er.5~6 Insulin acts to promote the transport and disposal of glucose in insulin-sensitive peripheral tissues, specifically striated muscle and adi- pose tissue. Quantitatively, skeletal muscle is responsible for the vast majority of insulin-mediated glucose disposal.3~7-9 Insulin's effects on the liver (eg, inhibition of hepatic glucose production from smaller precursors, o r gluconeogenesis, and inhibition of glycogen breakdown, or glycogenoly- sis) also act to maintain euglycemia (normal blood glucose levels).
Under most conditions, the uptake and utilization of glucose by periph- eral tissues is tightly balanced by hepatic glucose production and out- put, so as to strictly regulate glycemia while meeting fluctuating metabolic requirements. The rapid sensing of elevated glucose levels, subsequent increase in insulin secretion by P-cells, and insulin-mediated glucose transport and disposal into peripheral
60 / 879 Physical Therapy/Volume 73, Number 12December 1 9 3

tissues are all critical steps for lower- ing blood glucose levels after a meal and establishing glucose homeostasis. It is not surprising that defects in all three sites (pancreas, liver, and skele- tal muscle), singularly or collectively, have been implicated in conditions characterized by abnormal glucose homeostasis (eg, in NIDDM).4
The recognition that skeletal muscle can develop resistance to the actions of insulin has important conse- quences for physical therapists. Though rarely considered and even less often assessed, skeletal muscle insulin resistance is very prevalent in patients seen by physical therapists. Insulin resistance occurs after short periods of muscular inactivity, in denemation, or after inactivity due to bed rest. Most commonly, the insulin resistance is localized to individual muscles or small muscle groups, resulting in negligible effects on whole-body glucose metabolism. Disturbances in whole-body glucose homeostasis, however, may occur if insulin resistance develops in a large proportion of the muscle mass.
In conditions such as insulin- dependent diabetes mellitus (IDDM, or type I diabetes mellitus), NIDDM, and IGT,* skeletal muscle insulin resistance contributes significantly to the overall disturbances in glucose homeostasis.3~4J0 Insulin resistance develops in obesity11 and is currently considered a prominent feature of chronic diseases such as hyperten- sion, dyslipidemia, and atherosclerotic cardiovascular disease.12J3 For this review, however, we will focus on the insulin resistance that may occur in specific muscle groups and in patients with NIDDM and IGT, because we believe these individuals represent the majority of patients with insulin resistance seen by physical therapists.
*For definition and clinical criteria for classification, diagnosis of diabetes mellitus and other categories I
1979;28:1039-1057.
Glucose Transport Into Skeletal Muscle
Insulln-stimulated glucose transport. The transport of glucose into skeletal muscles is the initial, and under many physiological conditions the rate-limiting, event in glucose metabolism (ie, the slowest step in the pathway that limits the overall rate of muscle glucose metabolism).14 Therefore, glucose transport is an event of major regulatory importance, as well as of potential disturbance, in glucose homeostasis. Despite the fundamental importance of skeletal muscle glucose transport, only re- cently have we begun to appreciate the complex molecular events leading to glucose entry into mammalian muscle cells. It has been recognized for decades that glucose entry, utiliza- tion, and disposal in resting skeletal muscles are greatly enhanced by insulin. The sequence of molecular events leading to insulin-mediated glucose transport is initiated at the muscle cell membrane by the binding of insulin to its plasma membrane receptor.ls Immediately upon bind- ing, the insulin-receptor complexes internalize by receptor-mediated en- docytosis to initiate several intracellu- lar sequences of events, one of which is to facilitate glucose transport into skeletal and cardiac muscle and adi- pose tissue.
In the early 1 9 8 0 ~ ~ it was discovered that insulin rapidly mobilized carrier proteins from an intracellular pool and shuttled them to the plasma mernbrane.16.17 These carrier proteins allow glucose to be transported across the cell membrane, where it is rapidly phosphorylated by intracellu- lar enzymes and used according to the cells' metabolic requirements. Soon after, a family of glucose trans- porter proteins was identified.lsJ9 The members of this family, which are specifically expressed in different tissues, are referred to as isofom (ie, proteins that are similar, but not iden-
see National Data Group. Classification and sf glucose intolerance. Diabetes.
tical, in their amino acid sequences). The subtle differences in chemical composition allow for differences in functions between family members. Glucose transporter isoforms differ in properties such as their maximal rates of glucose transport, their ability to bind glucose, and the regulation of their activity by various hormones.l9
Skeletal muscles contain at least three different isoforms of the glucose transporter. The GLUTl isoform is a minor isoform that is located in per- ineural tissue and within muscle ~ells.20~2~ The GLUTl isoform's func- tion is currently under debate, but it may mediate basal (ie, insulin- and contraction-independent) glucose transport.l9 The presence of a GLUT5 isoform in skeletal muscle has re- cently been demonstrated.22 The GLUT5 isoform is present in low concentrations, and its function is unknown; it may actually be the major transport protein for fructose.22
The primary isoform, located almost exclusively in striated muscle and adipose tissue, is GLUT4.23-25 Insulin stimulates the rapid "translocation" of GLUT4, that is, the movement of GLUT4 proteins from intracellular membrane pools (inactive storage sites) to their sites of insertion into the plasma membrane.26127 Once in- serted into the plasma membrane, the transporters mediate glucose entry into the cell's interior. Many unan- swered questions remain regarding the specific molecular events that occur between insulin binding and insertion of transporter proteins into the plasma membrane.
Contractlon-stimulated glucose transport. Glucose transport in skel- etal muscles can be activated by fac- tors other than insulin. Muscle con- tractile activity itself is capable of stimulating glucose transport indepen- dently of i n ~ u l i n . ~ ~ 3 ~ Evidence for the existence of a contraction-activated pathway has been provided by a vari- ety of experimental approaches, in- cluding the hind-limb perfusion tech- nique and the incubation of isolated skeletal muscles. Increases in glucose transport can be induced by a bout of
Physical Therapy/Volume 73, Number 12December 1993

exercise,31>2 by stimulation of mus- cles in situ,33 and by stimulation of isolated muscles in vi t r0.~~,3~ In addi- tion to contractile activity, this path- way can be activated by hypoxia.35
Support for the existence of separate pathways comes from numerous stud- ies demonstrating additivity of the maximal effects of insulin and exer- ~ise.2~,3132.3~,36 That is, after glucose transport activity has been stimulated by a maximally effective concentration of insulin, increasing the insulin con- centration further results in no greater increase in glucose transport activity. If an exercise stimulus is superimposed on the insulin stimulus, however, glucose transport can be further increased. Similarly, maximal effects of insulin and hypoxia are additive.35 In contrast, maximal hy- poxic stimuli are not additive with effects of exercise or contractile activi- ty.35 These findings suggest that mus- cle contractions and hypoxia activate a common pathway for glucose trans- port that is distinct from that stimu- lated by insulin.
The existence of a separate pathway from insulin for activating glucose transport has obvious functional con- sequences. Insulin released following a meal will stimulate glucose uptake into all skeletal muscles (albeit with magnitudes that differ among mus- cles). This glucose uptake is in accor- dance with insulin's anabolic role for promoting energy storage. The pres- ence of a contraction-mediated path- way allows for a selective increase in glucose uptake during exercise into working muscles.
The sequence of events by which muscle contractions and hypoxia stimulate glucose transport is incom- pletely understood. A great deal of indirect evidence, however, suggests that this pathway is activated by an increase in intracellular calcium (Ca++). Agents that increase intracel- lular ~ a + + concentration stimulate glucose tran~port37~O and inhibitors of sarcoplasmic reticulum Ca++ re- lease prevent the activation of glucose tran~port.35~39.40 In contrast, insulin- stimulated glucose transport does not
appear to be mediated by increases in intracellular Ca++.39r41
Although the events that initiate the activation of glucose transport through the insulin and contraction/ hypoxia pathways are different, late steps in these pathways may be the same. Like insulin, contractile activity and hypoxia are known to stimulate the translocation of GLUT4 proteins from intracellular compartments to the plasma rnembra11e.~~*~3 Whether insulin and contractionshypoxia act on the same or different intracellular pools of transporters is not known.
Studies44s45 have demonstrated that maximal glucose transport varies between different muscles in propor- tion to the total concentration of GLUT4 protein. One recent study, examining skeletal muscles of differ- ent fiber type compositions, demon- strated that the relative contributions of the contractionhypoxia and insulin pathways to total glucose transport activity differ between rnu~cles.~4 The significance of this finding is not fully apparent, but may indicate that mus- cles can regulate the activities of these two pathways in accordance with differing functional requirements.
Skeletal Muscle as a Major Slte of lnsulln Resistance
Historically, the contribution of skele- tal muscle to glucose homeostasis in the post absorptive and postprandial states has been underestimated. In large part, this has been due to the widespread belief that the mainte- nance of glycemia was the exclusive function of the l i ~ e r . ~ 6 Also, until just over a decade ago, insulin deficiency resulting from pancreatic P-cell de- fects was still considered the primary pathogenetic factor responsible for the insulin resistance of NIDDM.3
Nearly 90% of insulin-mediated glu- cose uptake occurs in peripheral tissues rather than in the splanchnic bed (gut and liveQ.4 Skeletal muscle comprises 40% to 50% of the total body mass; therefore, it is quantita- tively the major tissue responsible for insulin-dependent glucose utiliza-
tion.4sR.9 Estimates of the total glucose uptake in adipose tissue are uniformly low (typically less than 5%).7*R247 Fur- thermore, the overwhelming majority of glucose entering muscle cells in the postabsorptive state is stored as gly~ogen.48~49 Both the rate and extent of in vivo glycogen synthesis are de- pendent on several factors, including the sensitivity of skeletal muscles to ambient insulin levels.48
Although the site of primary defect in NIDDM is still under debate, insulin resistance in skeletal muscle is now recognized as a prominent feature of NIDDM and perhaps several other chronic diseases.1Or'2 This belief is based on various observations. First, abnormally high circulating levels of insulin in the blood (ie, hyperinsulin- emia) fail to maintain blood glucose levels, resulting in hyperglycemia. Second, studies with animal models of diabetes and humans with NIDDM or IGT have directly demonstrated the existence of skeletal muscle insulin resistance.l3
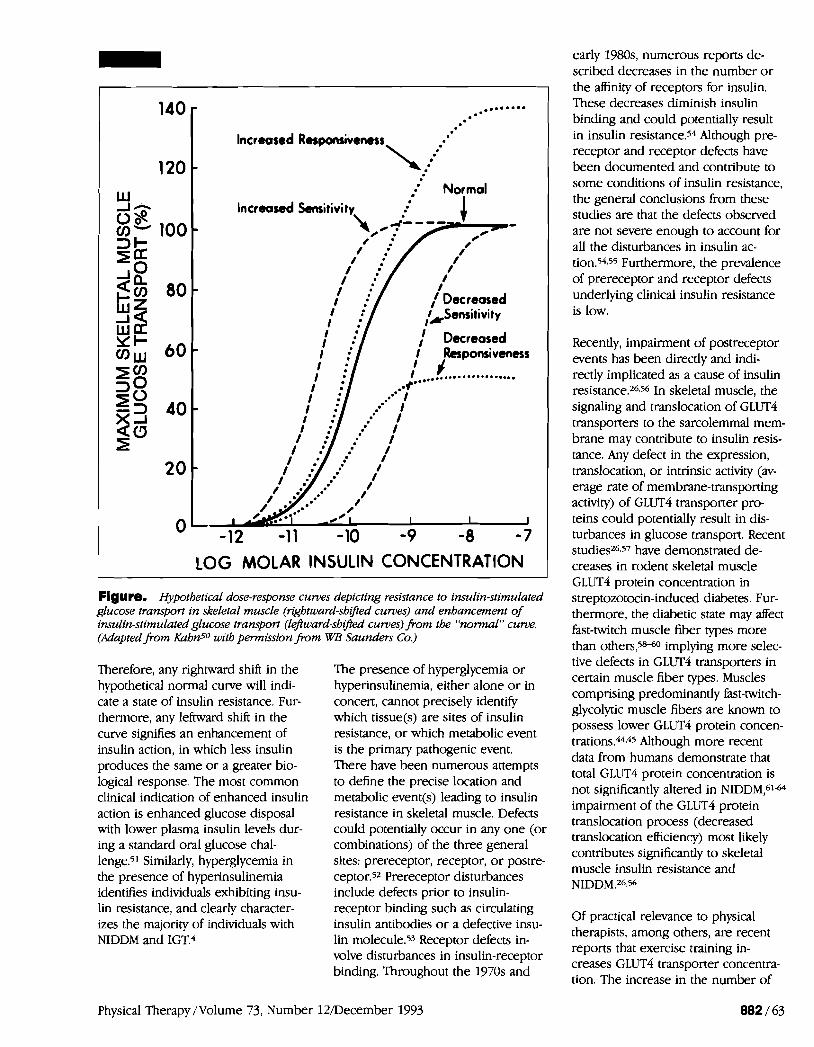
Insulin resistance has been defined by the concentrations of insulin that are required to produce a desired biolog- ical response, such as the uptake of glucose into insulin-sensitive tissues.50 Studies have shown that insulin resis- tance can be present in the liver, adipose tissue, and skeletal muscle, though adipose tissue appears quanti- tatively less important than the liver or skeletal muscles.4
The concept of insulin resistance can be visualized by examining hypotheti- cal dose-response curves for insulin action (Figure) .so The Figure shows that higher concentrations of insulin are needed to produce the same level of glucose transport when muscles are insulin "resistant." When slightly higher physiological concentrations of insulin are needed to promote the same level of glucose transport, but maximal responses are still obtained, the muscle has decreased sensitivity to insulin action. When maximally effec- tive concentrations of insulin result in diminished rates of glucose transport, however, the muscle has decreased responsiveness to insulin action.
Physical Therapy /Volume 73, Number 12December 1993
Increased Respmsiveness
\; Normal
Increased h i t i v i ty. h i t i v i ty
/
I : I . '
i / :/ l ~ e c r e a s e d I : 1 Sensitivity I . '
LOG MOLAR INSULIN CONCENTRATION 1 Figure. Hypothetical dose-response curves depicting resistance to insulin-stimulated glucose transport in skeletal muscle (rightward-shifted curves) and enhancement of insulin-stimulated glucose transport (leftward-shifted curves) from the "normul" c u m . (Adapted from Kahn5O with permission from WB Saunders Co.)
Therefore, any rightward shift in the hypothetical normal curve will indi- cate a state of insulin resistance. Fur- thermore, any leftward shift in the curve signifies an enhancement of insulin action, in which less insulin produces the same or a greater bio- logical response. The most common clinical indication of enhanced insulin action is enhanced glucose disposal with lower plasma insulin levels dur- ing a standard oral glucose chal- lenge.51 Similarly, hyperglycemia in the presence of hyperinsulinemia identifies individuals exhibiting insu- lin resistance, and clearly character- izes the majority of individuals with NIDDM and IGT.4
The presence of hyperglycemia or hyperinsulinemia, either alone or in concert, cannot precisely identify which tissue(s) are sites of insulin resistance, or which metabolic event is the primary pathogenic event. There have been numerous attempts to define the precise location and metabolic event@) leading to insulin resistance in skeletal muscle. Defects could potentially occur in any one (or combinations) of the three general sites: prereceptor, receptor, or postre- ceptor.52 Prereceptor disturbances include defects prior to insulin- receptor binding such as circulating insulin antibodies or a defective insu- lin molecule.53 Receptor defects in- volve disturbances in insulin-receptor binding. Throughout the 1970s and
early 1980s, numerous reports de- scribed decreases in the number o r the affinity of receptors for insulin. These decreases diminish insulin binding and could potentially result in insulin re~istance.5~ Although pre- receptor and receptor defects have been documented and contribute to some conditions of insulin resistance, the general conclusions from these studies are that the defects observed are not severe enough to account for all the disturbances in insulin ac- ti0n.5~~55 Furthermore, the prevalence of prereceptor and receptor defects underlying clinical insulin resistance is low.
Recently, impairment of postreceptor events has been directly and indi- rectly implicated as a cause of insulin resistance.26~56 In skeletal muscle, the signaling and translocation of GLUT4 transporters to the sarcolemmal mem- brane may contribute to insulin resis- tance. Any defect in the expression, translocation, or intrinsic activity (av- erage rate of membrane-transporting activity) of GLUT4 transporter pro- teins could potentially result in dis- turbances in glucose transport. Recent studies26,57 have demonstrated de- creases in rodent skeletal muscle GLUT4 protein concentration in streptozotocin-induced diabetes. Fur- thermore, the diabetic state may affect fast-twitch muscle fiber types more than others,5&0 implying more selec- tive defects in GLUT4 transporters in certain muscle fiber types. Muscles comprising predominantly fast-twitch- glycolytic muscle fibers are known to possess lower GLUT4 protein concen- trations.44s45 Although more recent data from humans demonstrate that total GLUT4 protein concentration is not significantly altered in NIDDM,61-64 impairment of the GLUT4 protein translocation process (decreased translocation efficiency) most likely contributes significantly to skeletal muscle insulin resistance and NIDDM.26856
Of practical relevance to physical therapists, among others, are recent reports that exercise training in- creases GLUT4 transporter concentra- tion. The increase in the number of
Physical Therapy/Volume 73, Number 12December 1993

proteins available for translocation, assuming a fixed percentage of GLUT4 proteins are available for translocation, may be the major mech- anism underlying the observed im- provements in glucose metabolism after exercise training. This possibility needs to be tested in future studies. An additional possibility is that exer- cise training of persons with diabetes could reverse the impairment in translocation efficiency.
Regulation of Skeletal Muscle Glucose Transport by ActMty and InactMty
Glucose transport into skeletal muscle is not a static process proceeding continuously at the same rate. Trans- port activity can be modulated acutely (ie, minutes to hours) by changes in the hormonal environment (eg, secre- tion of insulin after a meal) or by sudden changes in the level of physi- cal activity. The glucose transport process also adapts to chronic changes in physical activity, for exam- ple, during prolonged bed rest o r following a regular program of exer- cise (ie, training). In this section, we will discuss responses to specific alterations in physical activity.
lnactlvlty
Physical inactivity leads to a reduction in glucose t01erance.~- Bed rest is the primary model used for examin- ing effects of inactivity of large muscle masses in humans. Bed rest or whole- body inactivity results in decreased whole-body insulin-mediated glucose disposal,@-72 which is due in large measure to impaired insulin-mediated skeletal muscle glucose disposal.71-73 The effects of inactivity are rapidly manifested. Peripheral insulin resis- tance is evident within just 3 days of absolute bed r e ~ t . 7 ~ In obese individu- als, who were already insulin-resistant with compensatory hyperinsulinemia, 4 days of bed rest led to a further reduction in glucose tolerance.75 Insulin resistance appears to progress further with increasing duration of bed rest.67
The mechanism for the reduction in skeletal muscle insulin action with physical inactivity has been partially explained. The development of insu- lin resistance following bed rest ap- pears to be independent of dimin- ished gravitational stress, as evidenced by the finding that immobilization in the upright position also results in peripheral re~istance.7~ Furthermore, the effect of inactivity on skeletal muscle insulin action is not due to increases in hormones o r metabolites that antagonize insulin action.69272273 Evidence suggests that the reduction in insulin-mediated glucose disposal following inactivity is due to a postin- sulin receptor defect.
The reduction in glucose tolerance during bed rest is not associated with changes in insulin binding, at least in circulating m o n o ~ y t e s . ~ ~ ~ ~ 5 This find- ing is in accord with the results from immobilization studies. Inactivity results in decreased skeletal muscle insulin responsi~eness,7~~~~ a condi- tion associated with a postbinding defect.54.55 Studies in rats have shown that the decrease in skeletal muscle glucose uptake is due, at least in part, to a reduction in glucose transporter c~ncentrat ion.~~ Future research should attempt to identlfy the specific transporter isoforms that are de- creased following physical inactivity (cf, the effects of denervation).
Ambulation following a period of bed rest has long been known to improve glucose t ~ l e r a n c e . ~ M ~ The time re- quired to normalize glucose tolerance depends on the amount of activity undertaken during the reambulation phase, as well as the duration of inac- tivity. In one study using 2 weeks of bed rest followed only by ambulation, without additional exercise, insulin action remained greatly diminished after 1 week of recovery and only partly restored by 2 weeks of recov- ery.73 TWO studies using bed rest of 3 weeks or less followed by walking programs during the reambulation phase demonstrated a normalization of glucose tolerance to pre-bed-rest levels within 1 week of beginning ambulati0n.~5,~~ Thus, the addition of the exercise program during the
reambulation phase accelerates the recovery of insulin action. When indi- viduals are active during the bed-rest period, deterioration in glucose toler- ance can be significantly diminished but not completely pre~ented.74~76 This effect may be independent of the type of exercise performed (eg, iso- tonic versus isometric) but is propor- tional to the total caloric expendi- ture.76 TO completely prevent the hyperinsulinemia that develops dur- ing 2 weeks of bed rest, Dolkas and GreenleaP6 have calculated that at least 1,000 kcal of supplemental exer- cise must be expended per day. This amount of energy expenditure may be impractical in most clinical situa- tions; however, lesser amounts of exercise could be encouraged.
lmmobilizatlon
In v i t r ~ n * ~ ~ and in vivo79~~~ studies have shown that limb immobilization results in skeletal muscle insulin resis- tance for glucose uptake and glycogen synthesis. The impairment in glycogen synthesis is due to at least two factors: (1) impairment of insulin's ability to stimulate glycogen synthase activity and (2) reductions in the supply of glucose secondary to a decrease in transport activity.78 In humans, 1 week of lower-extremity casting combined with the use of crutches during non-weight-bearing ambulation re- sulted in a reduction in insulin- stimulated glucose uptake in the im- mobilized limb.80 Studies with animal models have demonstrated that insu- lin resistance for glucose transport can occur within the first day or two of immobilization.78~79 In mice that have had their hindlimbs casted, re- ductions in glucose transport have been noted within as early as 6 hours following immobilization; however, it is not clear whether the impairment in the early phase of this immobiliza- tion model is partly due to a stress response, with its accompanying in- crease in glucocorticoid concentra- t i o n ~ . ~ ~ The insulin resistance ob- served within 1 day of immobilization was not due to a decrease in insulin binding or activity of the enzyme hexokinase (which phosphorylates glucose immediately after entry into
Physical Therapy / lVolume 73, Number 12December 1993

skeletal muscle); instead, a decrease in insulin responsiveness suggested a postbinding defect.78 Thus, inactivity leads to insulin resistance regardless of whether a large proportion of the body's musculature is inactive or whether the inactivity is localized to a small muscle mass.
In situations in which a small muscle mass is immobilized, glucose toler- ance would not be expected to be impaired. Clinicians should recognize, however, that in addition to recover- ing strength, flexibility, and endur- ance in previously immobilized mus- cles, there needs to be a restoration of insulin-stimulated glucose transport and glycogen synthesis. The ability to replenish glycogen stores will in turn enhance muscle endurance perfor- mance in recovering muscles. Studies with animals suggest that exercise protocols that develop endurance would be sufficient to restore glucose transport activity following immobili- zation. Future research should be directed toward determining whether such exercise protocols will restore glucose transport activity following inactivity in humans.
Denervation
Denemtion is known to induce se- vere insulin resistance for glucose uptake in skeletal muscles.8~-84 In animal models, the onset of insulin resistance can be very rapid, with a reduction in glucose transport evident as early as 3 to 6 hours after denerva- tion.82.w The resistance continues to progress over the next several days.82~84~85 Insulin resistance can develop in fast-twitch and in slow- twitch muscle fibers82 and also in- volves impairment of glycogen synthesis.82*83
The mechanism by which denemtion decreases glucose transpon appears to be complex. Analogous to the effect of immobilization, the initial defect induced by denervation is not mediated by a decrease in insulin binding,82r86 although this may occur following a prolonged period of de- nervation,60 nor is it uue to an inabil- ity to phosphorylate gl~cose.82~83 A
reduction in insulin responsiveness strongly implicates a postreceptor defect, as suggested by Burant et al.B2 Furthermore, although most investiga- tors have focused on the insulin path- way, denemtion also results in re- duced activity of the contraction- stimulated pathway for glucose tran~port.~5 Initially, resistance to stimulated glucose transport occurs in the absence of changes in the concen- tration of GLUT4 protein,85~~~ suggest- ing that the initial defect involves some aspect of the translocation pro- cess or more proximal steps in the signaling pathways.82 Within 3 days after denervation, however, GLUT4 concentration decreases; this reduc- tion in GLUT4 protein may be respon- sible for the further deterioration in insulin action measured several days after denemtion.85*87
Single Bout of Exercise
Exercise has different effects on skele- tal muscle glucose transport activity depending on whether one is examin- ing (1) the effects of a single bout of exercise in the untrained state or (2) the effects of repeated bouts of exer- cise (ie, exercise training). These two circumstances will be discussed sepa- rately. Unless stated otherwise, results from experiments using nondiabetic animals or individuals are presented.
A single bout of exercise has several effects on the transport of glucose into skeletal muscle. One effect is to directly stimulate glucose transport via the muscle contractionfiypoxia- activated pathway (ie, an enhance- ment of glucose transport activity measurable in the absence of insulin). A second effect is to enhance the insulin-stimulated pathway for glucose transpon. These two effects of exer- cise have different time courses. The increase in transport activity mea- sured in the absence of insulin gener- ally reverses within a few hours after cessation of exercise.32~88 The increase in insulin action becomes apparent after the increase in glucose transport, measured in the absence of insulin, has partially or completely worn off.32.33,89,90
Individual bouts of endurance exer- cise of moderate intensity and 0.5 to 2 hours' duration increase insulin ac- tion in animals and in humans. Using a variety of different model systems (eg, in isolated muscles,32,91 in the perfused rat hindquarter?2 and in vivo933%), a single bout of exercise increases insulin sensitivity and can also enhance insulin responsiveness for glucose transport. The exercise- induced increase in insulin respon- siveness, however, persists for much shorter time periods than does the increased insulin sensitivity.90
When muscles are removed from the body and stimulated to contract in a defined incubation medium consisting of salts, glucose, and an appropriate buffer to maintain pH, glucose trans- port activity measured in the absence of insulin is enhanced to the same extent as that measured when mus- cles are removed after a bout of exer- cise.34~95 In contrast, when muscles are stimulated to contract in the same medium, insulin-stimulated glucose transport is not enhanced95 These findings suggest that some factor(s) normally present in vivo is (are) re- quired for the increase in insulin action that is induced by a single bout of exercise. This factor is not required for activation of the contractile activi- tyhypoxia pathway.28.34,35*95 Taken together, the experimental evidence reinforces the notion that a bout of exercise has two distinct effects on skeletal muscle glucose transport: (1) It activates the muscle contraction pathway, and (2) after a delay, it en- hances the insulin-activated pathway.
The factors required for the exercise- induced increase in insulin action may be humoral factors or substances released from nerve terminals, such as sympathomimetic agents. When muscles from sedentary animals are stimulated to contract in situ, how- ever, insulin sensitivity is increased by contractions.33 This finding suggests that changes in humoral factors (eg, changes in hormone concentrations) that normally occur during exercise are not necessary for the enhance- ment of insulin sensitivity. An impor- tant area of future research is to iden-
Physical Therapy /Volume 73, Number

tify the essential factor(s). Insulin itself does not appear to be the factor, because one study96 has suggested that a permissive amount of insulin (ie, a concentration of insulin that is not responsible for the effect by itself, but that must be present for exercise to exert its effects) does not need to be present during muscle contrac- tions for the subsequent enhancement of insulin action.
Whatever the factor(s) required for the exercise-induced increase in insu- lin action, once this process has been activated, it can persist even in the absence of humoral or other factors present in vivo. When muscles are removed from animals immediately after exercise and then incubated in vitro for 3 hours before the measure- ment of glucose transport activity, insulin sensitivity is still greatly en- hanced compared with that measured in muscles from sedentary r a t ~ . 9 ~
Enhanced glucose uptake after exer- cise is not restricted to the insulin- stimulated pathway for glucose trans- port. A single bout of exercise also augments the effect of a subsequent submaximal hypoxic stimulus.95 This finding suggests that a prior bout of exercise also enhances the ability of muscle to subsequently activate glu- cose transport via the contractile activ- ity pathway.
Factors regulating reversal of enhanced lnsulln sensltlvlty after a ~lngle bout of exercise. Mainte- nance of enhanced insulin action after a single bout of exercise is inversely related to the carbohydrate composi- tion of the diet. When animals are fasted or fed a carbohydrate-free diet after a bout of exercise, insulin sensi- tivity remains markedly enhanced for at least 2 days.% In contrast, with a carbohydrate-rich diet, the increase in insulin sensitivity reverses to values characteristic of the sedentary state between 3 and 18 hours after cessa- tion of exercise.m
The relationship between dietary carbohydrate content and reversal of enhanced insulin sensitivity can be understood by considering the effect
of diet on muscle glycogen levels. Studies performed more than 25 years ago demonstrated that a single bout of glycogen-depleting exercise results in replenishment of glycogen stores to levels greater than those measured in the sedentary state (gly- cogen s~percompensation).9~ Carbo- hydrate loading to "supercompensate" muscle glycogen stores in individuals competing in long-distance events is a technique widely used.w.lO'J
More recent studies examining glu- cose transport have provided an ex- planation for this phenomenon. In muscles from animals deprived of carbohydrate after exercise, in which insulin action remains augmented, muscle glycogen levels remain far below the supercompensated level.% Insulin action returns completely to values seen in sedentary rats only under conditions in which muscle glycogen stores are supercompen- sated. These findings suggest that the increase in insulin sensitivity after exercise, coupled with an adequate source of carbohydrate, is an impor- tant factor contributing to muscle glycogen s~percompensat ion.3~~~
Studies examining the effects of di- etary manipulation on insulin action after exercise cannot discriminate between a direct effect of carbohy- drate uptake into muscle or indirect effects of carbohydrate feeding. When muscles are incubated in the absence of carbohydrates after exercise, insu- lin action remains greatly enhanced relative to that measured in muscles from sedentary rats." In contrast, incubation of muscles with glucose in the presence of insulin results in reversal of the enhanced insulin sen- sitivity. This finding suggests that glucose in combination with low concentrations of insulin can directly regulate the reversal of enhanced insulin sensitivity. Furthermore, the signal by which glucose and insulin down-regulate the elevated insulin sensitivity after exercise! appears to be generated during intracellular metabo- lism of glucose rather than during the actual transport of glucose across the cell membrane.97 Thus, these studies provide an explanation for the mecha-
nisms underlying the utility of the carbohydrate loading technique.99JO'J
Exerclse Tralnlng
Studies using animal models have demonstrated that endurance exercise training increases glucose transport into skeletal Exercise training increases insulin responsive- ness and maximal glucose transport elicited by the combined stimuli of insulin and muscle contra~tions.l~F-~~7 StudieslOF-lo7 have shown little or no effect of training on basal glucose transport activity. In humans, both cross-sectional studies"J3J0'+1"J and longitudinal studies1l1Jl2 have shown increased insulin action for whole- body glucose disposal in endurance exercise-trained individuals. The training-induced increase in insulin action in humans also occurs in skele- tal muscle.l12.
As previously noted, GLUT4 makes up the vast majority of skeletal muscle glucose tran~porters.23-~5 The GLUT4 concentration is closely correlated with maximal glucose transport capac- ity in different skeletal mu~cles,4~,~5 suggesting that GLUT4 concentration is an important determinant of a mus- cle's capacity for glucose transport. This relationship led investigators to examine whether the increase in glucose transport activity induced by training might be mediated by an increase in the concentration of mus- cle GLUT4 proteins. In animals, skele- tal muscle GLUT4 protein concentra- tion is increased by several different modes of exercise (ie, by treadmill training,1071113 s~irnming,l05~107 and wheel runninglo6J14). In contrast, GLUT1 protein concentration is not altered by exercise training.lo6.114 The increase in GLUT4 concentration is specific for muscles utilized during training activity106 and does not occur in response to a single bout of exer- cise.1°7J15 Studies utilizing isolated muscles have demonstrated that the relative training-induced enhancement of maximal glucose transport activity is directly proportional to the increase in GLUT4 protein c~ncentration.l~J"
Physical Therapy ./Volume 73, Number 12December 1993

Thus, endurance exercise training, which increases the concentrations of enzymes involved in carbohydrate metabolism,ll6 also increases maximal insulin-stimulated glucose transport, total glucose transport capacity, and GLUT4 protein concentration. Further- more, these increases can occur in the absence of any changes in muscle fiber type c ~ m p o s i t i o n . ~ ~ Recent cross-sectional studies in humans have shown increased skeletal muscle GLUT4 protein concentrations in the trained state.115
In most studies of the effects of train- ing on glucose transport and GLUT4 protein concentrations, muscles con- taining predominately fast-twitch fi- bers were examined. A recent study107 demonstrated that treadmill and swimming exercise increase glucose transport and GLUT4 protein concen- tration in the soleus muscle, which is composed of predominately slow- twitch fibers. In this regard, it is inter- esting that a previous study utilizing wheel running found no increase in soleus muscle glucose transport activ- ity or GLUT4 protein concentration when normalized to muscle mass.1o6 Wheel running is unique in that it induces hypertrophy of the soleus muscle, perhaps as a result of the stretch on this muscle during activity periods.117 Wheel running induced an increase in total glucose transport and total GLUT4 content in the soleus muscle, suggesting that glucose trans- port increases only in proportion to the increase in muscle mass.106 These results suggest that the induction of an increase in GLUT4 protein concentra- tion by endurance exercise is inhibited by the presence of an accompanying hypertrophic stimulus. Muscles that do not undergo hypertrophy in response to wheel running show increases in glucose transport activity and GLUT4 protein concentration.lW
Further investigation is required to determine whether this relationship holds true with more conventional stimuli for muscle hypertrophy such as weight training. Few studies to date have examined the effects of weight training on glucose homeostasis. One recent however, suggested
that repeated participation in activities involving a relatively high number of weight-lifting repetitions may improve glucose tolerance. The mechanism for the improvement in glucose tolerance is not known, but may be related to the increase in total muscle mass available for glucose disposal.119
An apparent contradiction in the liter- ature is that not all training studies in animals and humans have shown increases in all indexes of glucose homeostasis.12%'22 One reason for many of these negative findings is probably that, in the absence of a significant reduction in body fat con- tent, the effects of exercise training on glucose transport activity persist for only a few days. Initially, investigators assumed that training effects on glu- cose transport would persist for sev- eral weeks or even months, analogous to the effects of training on oxidative enzymes. 116~123 The glucose trans- porter proteins, however, may have much shorter half-lives than the oxi- dative enzymes, resulting in a much faster loss of activity following cessa- tion of training. Indirect evidence for this comes from detraining studies that have shown a deterioration of insulin action within 1 week after cessation of exercise (see "Detrain- ing" section). The implication of these findings is that exercise must be per- formed on a regular basis in order to sustain beneficial effects on glucose disposal.
Utlllzatlon of blood glucose In the trained state. The effects of endur- ance training to increase maximally stimulated glucose transport activity as described might seem to be in contra- diction with the well-known ability of exercise training to induce a shift toward greater fat utilization during exercise."6 Endurance training is known to reduce reliance on carbohy- drates as an energy source during submaximal exercise.lZ4-126 Training decreases the turnover of not only endogenous glycogen stores but also the turnover and oxidation of blood glu~ose.126~127
This apparent contradiction can be understood by distinguishing between
effects of training manifested at rest versus those manifested during exer- cise. During the intervening periods between exercise bouts, the training- induced increase in insulin-mediated glucose transport activity results in an enhanced ability of previously exer- cised muscles to replenish glycogen stores. When measured during exer- cise of the same absolute exercise intensity, however, the utilization of blood glucose and muscle glycogen is lower in the trained state, which in turn spares liver as well as muscle glycogen stores from excessive deple- tion. Thus, the training-induced in- crease in glucose transport activity evident at rest either is not manifested during exercise or is overridden by factors that shift the rate-limiting step for glucose uptake to some subse- quent step in glucose metabolism.127 Both effects of training act to mini- mize net reductions in muscle glyco- gen, improving the ability of individu- als to tolerate repeated bouts of exercise.
Detraining
In well-trained individuals, 10 days of inactivity has been shown to result in a deterioration of glucose tolerance, with a concomitant increase in the plasma insulin response to oral glu- cose.lZ8 Studies have also demon- strated that relatively short-term de- training (5-14 days) results in increased insulin secretion in re- sponse to a standardized glucose loadI29 and to a reduction in whole- body insulin-mediated glucose dispos- a1.108,1293130 In one study,lO8 insulin action was greatly diminished just 5 days after the last bout of exercise. The reduction in whole-body glucose disposal after detraining is due in large measure to impaired insulin- mediated glucose disposal in skeletal muscle.ll2 Exercise training has sev- eral effects on glucose tolerance and insulin action. The first of these effects are mediated by the relatively rapid effects of acute exercise and exercise training on insulin sensitivity and responsiveness, as previously dis- cussed. In addition, long-term exer- cise results in a reduction in body fat, particularly in central adiposity, which
Physical Therapy/Volume 73, Number 12December 1993

most likely leads to an additional improvement in insulin action.'31 This btter effect of exercise training is very important in the management of glu- cose intolerance in obese individuals, particularly those with NIDDM. Short- term detraining, which does not alter body fat, would be expected to result in loss of the rapid effects of exercise on glucose metabolism, but not in loss of longer-term adaptations resulting from reductions in central adiposity.
Muscle Damage and lnsulln Reslstance
Certain types of exercise, particularly those that involve eccentric contrac- tions, can lead to muscle dam- age.'32-135 Indexes of damage include muscle soreness, decreased range of motion, inflammation, and elevations in serum creatine kinase activity. Other studies have shown that exer- cise that induces muscle damage is also characterized by impaired glyco- gen resynthesis,'3Fl37 which can oc- cur in both slow-twitch and fast-twitch fibers.137 O'Reilly et all36 showed that following exercise consisting of ec- centric contractions, glycogen resyn- thesis can be impaired for prolonged periods (at least 10 days with their experimental protocol).
Costill et a11s7 have demonstrated that changes in glycogen synthase activity cannot account for the entire impair- ment in glycogen resynthesis follow- ing eccentric exercise. Recent data indicate that insulin resistance of glucose transport may account for the impaired glycogen resynthesis after exercise that induces muscle dam- age.105s107,138 Ploug et all05 showed that a single prolonged bout of exer- cise in untrained rats reduced insulin- stimulated glucose uptake in muscles containing high proportions of slow- twitch or fast-twitch muscle fibers. Another study using untrained rats showed a tendency for insulin resis- tance to develop in soleus muscles after a single 90-minute bout of tread- mill exercise.107 The implication of these findings is that when untrained individuals begin to exercise, they should gradually increase exercise duration and intensity to avoid induc-
ing muscle damage and the accompa- nying insulin resistance.
Kirwan et a1138 recently examined the effects of downhill running in un- trained individuals. Downhill running, which resulted in muscle soreness and elevated creatine kinase activity, also reduced the glucose disposal rate by about 40%. Neither hepatic glu- cose production nor basal glucose turnover was altered by the downhill run. In contrast, cycle ergometer exercise, an activity that did not in- duce muscle damage, did not induce insulin resistance. This study indicates that insulin-mediated glucose dis- posal, most likely into skeletal muscle, is impaired by eccentric exercise that results in muscle damage.
Thus, previous reports that exercise with eccentric contractions impairs glycogen resynthesis can be explained by the findings that muscle damage also impairs glucose uptake and dis- posal. Whether the impairment in glucose uptake can account entirely for the impaired glycogen resynthesis remains to be determined. The impli- cation of these studies is that although most forms of exercise enhance insu- lin action in skeletal muscle, certain forms of exercise, particularly those that can lead to muscle damage, can actually induce insulin resistance in skeletal muscle. This insulin resistance in turn leads to impaired glycogen resynthesis, which can persist for prolonged periods. Thus, if eccentric contractions are at levels that may induce injury, they should probably comprise a minimal proportion of an individual's total activity pattern. The impairment of glycogen resynthesis that occurs with muscle damage can be partially overcome by ingesting a diet high in carbohydrates, although carbohydrate feeding does not com- pletely offset the effects of muscle damage.137 Although less well studied, high-intensity, concentric weight train- ing could, in some circumstances, result in muscle damage, especially if the exercise is novel. Therefore, insu- lin resistance may potentially increase following this form of exercise. The susceptibility for muscle damage may be increased in atrophied muscles,
but this possibility needs to be examined.
Exerclse Prescrlptlon In Insulln=Reslstant IndMduals
We believe there are direct implica- tions regarding glucose uptake and disposal for practicing physical thera- pists. As a specific example of exercise prescription for the insulin-resistant state, we will discuss conditions of whole-body insulin resistance. Exercise prescriptions for conditions character- ized by resistance in isolated muscle groups have not been adequately defined.
The exercise prescription should be thoughtfully constructed to maximize glucose utilization and enhance insu- lin action, yet minimize any potential risks to the patient. Among other factors, the exercise prescription must consider the severity of the diabetic state. In individuals with poorly con- trolled diabetes, exercise may actually worsen the diabetic state.'39 An indi- vidual's diabetes, therefore, should be well controlled prior to the initiation of any exercise program. In addition, because persons with NIDDM, IDDM, or IGT have a much higher incidence of cardiovascular disease,l2 it is im- perative that these individuals be thoroughly evaluated prior to initiat- ing an exercise program. Individual exercise prescriptions should be based on graded exercise test results along with the referring physicians' evaluation of the potential risks asso- ciated with exercise. Several impor- tant considerations in an exercise prescription for the insulin-resistant individual will be discussed including frequency, intensity, and duration of exercise.
As discussed previously, a growing body of evidence suggests that im- provements in glucose homeostasis and insulin sensitivity are diminished rapidly (within a few days) after the last bout of aerobic exercise. If im- provements are quickly lost and re- versed after as little as 48 to 72 hours without exercise, then the maximum interval between exercise sessions should not exceed 48 to 72 hours.
Physical Therapy/Volume 73, Number 12December 1993

Although it has not been adequately investigated, daily exercise training could conceivably be of greater bene- fit; in practice, however, many individ- uals may find it difficult to comply with a daily exercise regimen. Exercise prescribed on alternate days can be beneficial, assuming that the intensity and duration of exercise are adequate.
Exercise intensity is a critical compo- nent of the exercise prescription. The intensity of aerobic exercise must be optimized to induce positive changes, yet minimize potential complications such as hypoglycemia, hyperglycemia, or musc:uloskeletal injury. Exercise intensities ranging between 60% and 90% of maximal oxygen consumption @02max) have generally resulted in improved glycemic control in patients with IGT or mild to moderate NIDDM.14s146 The results of these studies suggest that increasing the exercise intensity improves the likeli- hood of inducing a positive adapta- tion in glucose homeostasis. Few studies, however, have examined the dose responses for exercise training and adaptations in glucose homeosta- sis. This is a very important area that needs to be examined in greater detail.
As is common in clinical practice, when prescribing exercise intensity on the basis of age-predicted maximum heart rate or heart rate reserve, note that 60% of ~o ,max corresponds to approximately 70% of age-predicted maximum heart rate.l@3@435) The thera- pist, therefore, should prescribe an exercise intensity based on the per- centage of maximum heart rate that is high enough to induce positive adapta- tions in glucose homeostasis.
To enhance insulin's action on glu- cose transport and glycogen resynthe- sis, the prescribed exercise should result in a reduction of muscle glyco- gen stores. The utilization of glycogen stores in an exercising muscle is di- rectly linked to the intensity and dura- tion of the exercise. In one study,147 in which bicycle ergometer exercise was performed at an intensity of 83% of h m a x , more than half of the
stored glycogen was utilized within 1 hour.
As part of exercise prescription, the duration must be considered. Admit- tedly, less is known about how long exercise must be performed to derive optimal benefits. Because 80% to 90% of all patients with NIDDM have ac- companying obesity, the duration of exercise should be long enough to allow a considerable caloric expendi- ture, which will in turn lead to a reduction in adiposity. A considerable body of evidence suggests that fat loss, independent of exercise, im- proves insulin a ~ t i 0 n . l ~ ~ Recent evi- dence indicates that reductions in abdominal fat may account for the observed improvements in insulin action following weight loss.131 Exer- cise programs that are combined with dieting for fat loss should therefore result in greater improvements in insulin sensitivity than either exercise or weight loss alone. The prescription for the duration of aerobic exercise should consider maximizing the total energy expenditure, because this is the most important factor influencing weight loss and indirectly insulin action.
Durations of as little as 20 to 30 min- utes of continuous aerobic exercise have proven beneficial in improving glucose homeostasis,l42 although 40 to 60-minute durations would expend twice as much energy and still be feasible to perform for many individu- als on a daily or alternateday program.
Skeletal muscles have a vital role in maintaining normal glucose homeo- stasis and in regulating whole-body glucose metabolism. Although rarely considered, skeletal muscle insulin resistance is a common condition in patients seen by physical therapists. Acute muscle contractile activity (exer- cise) and repetitive endurance exer- cise (training) appear to improve glucose metabolism through indepen- dent pathways for skeletal muscle glucose transport. Given the proper exercise prescription, individuals who are insulin resistant can enhance their
sensitivity to insulin action within small groups of skeletal muscles or potentially improve whole-body glu- cose homeostasis.
More information is required in order to define specific protocols to amelio- rate insulin resistance in small muscle groups. For example, it is very impor- tant for clinicians to know whether current protocols for improving mus- cle strength and endurance after inac- tivity are sufficient to recover glucose metabolism and to define the time course for such changes. Determina- tion of whether protocols for electri- cal stimulation provide suitable means to ameliorate insulin resistance is also needed. Excessively forceful muscle contractions during the early phase of electrical stimulation could actually exacerbate insulin resistance due to transient muscle damage. In addition, more information is needed regard- ing the acute and chronic effects of resistance training. Thus, although our understanding of skeletal muscle glucose metabolism has advanced greatly over the past decade, more research is essential for the treatment of specific clinical conditions routinely encountered by physical therapists.
Acknowledgments
Dr Sinacore acknowledges the Ameri- can Physical Therapy Association's Section on Research and the Retreat Program Committee, particularly Lynn Snyder-Mackler, ScD, PT, and Stuart Binder-Macleod, PhD, PT, for provid- ing support in the form of a graduate student stipend (to DRS) to attend the research retreat on "Muscle Function in Normal and Pathological States" held August 18-23, 1991, in New Hampton, NH.
References
1 Malherbe C, de Gasparo M, de Hettogh R, Hoet JJ. Circadian variations of blood sugar and plasma insulin levels in man. Diabetolo- gia. 1969;5:397-404. 2 Kopf 4 Tchobroutsky G, Eschwege E. Serial postprandial blood glucose levels in 309 sub- jects with and without diabetes. Diabetes. 1973; 22:834-846. 3 DeFronzo RA, Ferrannini E, Koivisto V. New concepts in the pathogenesis of and treatment
Physical Therapy /Volume 73, Number 12December 1993

of non-insulin-dependent diabetes mellitus Am J Med 1983;75:52-81. 4 DeFronzo RA. The triumvirate: P-cell, mus- cle, liver-A collusion responsible for NIDDM. Diabetes. 1988;37:667487. 5 Escalante DA, Davidson J, Garber AJ. Maxi- mizing glycemic control. Clinical Diabetes. 1993;11:34. 6 Tchobroutsky G, Kopf A, Eschwege E, Assan R. Serial postprandial plasma insulin levels in 117 subjects with and without diabetes. Diabe- tes. 1973;22:825-833. 7 Katz LD, Glickman MS, Rappopon S, et al. Splanchnic and peripheral disposal of oral glu- cose in man. Diabetes. 1983;32:675479. 8 Kraegen EW, James DE, Jenkens AB, Chis- holm DJ. Dose response curves for in vivo insulin sensitivity in individual tissues of rats. Am J Pbysiol. 1985;248:E353-E362. 9 Baron AD, Brechtel G, Wallace P, Edelman SV. Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. Am J Pbysiol. 1988;255:E769-E774. 10 DeFronzo RA, Simonson D, Ferrannini E. Hepatic and peripheral insulin resistance: a common feature in non-insulin-dependent and insulin-dependent diabetes. Diabetologia. 1982;23:313-319. 11 Bjorntorp P, Berchtold P, Holm J, et al. The glucose uptake of human adipose tissue in obesity. Eur J Clin Invest. 1971;1:480485. 12 DeFronzo RA, Ferrannini E. Insulin resis- tance: a multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Di- abetes Care. 1%1;14:173-194. 13 Reaven GM. Insulin resistance, hyperinsu- linemia, hypertriglyceridemia, and hyperten- sion: parallels between human disease and rodent models. Diabetes Care. 1991;14:195- 203. 14 Ziel FH, Venkatesan N, Davidson MB. Glu- cose transport is rate-limiting for skeletal mus- cle glucose metabolism in normal and STZ- induced diabetic rats. Diabetes. 1988;37:885- 890. 15 Kahn CR. Membrane receptors for hor- mones and neurotransmitters. J Cell Biol. 1976;70:161-186. 16 Suzuki K, Kono T. Evidence that insulin causes translocation of the glucose transport activity to the plasma membrane from an intra- cellular storage site. Proc Natl Acad Sci USA. 1980;77:2542-2545. 17 Cushman SW, Wardzala LJ. Potential mech- anism of insulin action on glucose transport in the isolated rat adipose cell: apparent translo- cation of intracellular transport systems to the plasma membrane. J Biol Cbem. 1980;255: 47584762. 18 Bell GI, Kayano T, Buse JB, et al. Molecular biology of mammalian glucose transporters. Diabetes Care. 1990;13:19%208. 19 Mueckler M. Family of glucose-transporter genes: implications for glucose homeostasis and diabetes. Diabetes. 1990;39:611. 20 Froehner SC, Davies A, Baldwin SA, Lien- hard GE. The blood-nerve barrier is rich in glucose transporter. J Neurocytol. 1988;17:173- 178. 21 Handberg A, Kayser L, Hoyer PE, Vinten J. A substantial part of GLUT-1 in crude mem- branes from muscle originates from perineural sheaths. Am J Pbysiol. 1992;262:E721-E727.
22 Hundal HS, Ahmed A, Guma A, et al. Bio- chemical and immunocytochemical localiza- tion of the "GLUT5 glucose transporter" in human skeletal muscle. Biocbem J. 1992;286: 339-343. 23 James DE, Strube M, Mueckler M. Molecu- lar cloning and characterization of an insulin- regulatable glucose transporter. Nature. 1989; 338:83-87. 24 Klip A, Paquet MR. Glucose transport and glucose transporters in muscle and their meta- bolic regulation. Diabetes Care. 1990;13:22% 243. 25 Calderhead DM, Kitagawa K, Lienhard GE, Gould GW. Translocation of the brain-type glucose transporter largely accounts for insulin stimulation of glucose transport in BC3H-1 myocytes. BiocbemJ 1990;269:597401. 26 Klip A, Ramlal T, Bilan PJ, et al. Recruit- ment of GLUT-4 transporters by insulin in dia- betic rat skeletal muscle. Biocbem Biopbys Res Commun. 1990;172:72%736. 27 Hirshman MF, Goodyear LJ, Wardzala LJ, et a]. Identification of an intracellular pool of glucose transporters from basal and insulin- stimulated rat skeletal muscle. J Biol Cbem. 1990;265:987-991, 28 Nesher R, Karl I, Kipnis DM. Dissociation of effects of insulin and contraction on glucose transport in rat epitrochlearis muscle. Am J Pbysiol. 1985;249:02264232. 29 Wallberg-Henriksson H, Holloszy JO. Acti- vation of glucose transport in diabetic muscle: responses to contraction and insulin. Am J Physiol. 1985;249:C2334237, 30 Ploug T, Galbo H, Richter EA. Increased muscle glucose uptake during contractions: no need for insulin. Am J Pbysiol. 1984;247:E726 E73 1. 31 Garetto LP, Richter !LA, Goodman MN, Ru- derman NB. Enhanced muscle glucose metab- olism after exercise in the rat: the two phases. Am J Pbysiol. 1984;246:E471-E475. 32 Wallberg-Henriksson H, Constable SH, Young DA, Holloszy JO. Glucose transport into rat skeletal muscle: interaction between exer- cise and insulin. J Appl Pbysiol. 1988;65:909- 913. 33 Richter EA, Garetto LP, Goodman MN, Ru- derman NB. Enhanced muscle glucose metab- olism after exercise: modulation by local fac- tors. Am J Pbysiol. 1984;246:8476E482. 34 Constable SH, Favier RJ, Cartee GD, et al. Muscle glucose transpon: interactions of in vitro contractions, insulin, and exercise. J Appl Pbysiol. 1988;64:2329-2332. 35 Cartee GD, Douen AG, Ramlal T, et al. Stimulation of glucose transport in skeletal muscle by hypoxia. J Appl Pbysiol. 1991;70: 1593-1600. 36 Zorzano A, Balon TW, Goodman MN, Ru- derman NB. Additive effects of prior exercise and insulin on glucose and AIB uptake by muscle. Am JPbysiol. 1986;251:E21-E26. 37 Holloszy JO, Narahara HT. Enhanced per- meability to sugar associated with muscle con- traction: studies of the role of Ca++. J Gen Pbysiol. 1967;50:551-562. 38 Clausen T, Elbrink J, Dahl-Hansen AB. The relationship between the transport of glucose and cations across cell membranes in isolated tissues: the role of cellular calcium in the acti- vation of the glucose transport system in rat
soleus muscle. Biocbim Biopbys Acta. 1975; 375:292-308. 39 Valant P, Erlij D. K+-stimulated sugar up- take in skeletal muscle: role of cytoplasmic Ca2+. Am J Physiol. 1983;245:C1254132. 40 Youn JH, Gulve FA, Holloszy JO. Calcium stimulates glucose transpon in skeletal muscle by a pathway independent of contraction. Am J Physol. 1991;260:C5554561, 41 Klip A, Li G, Logan WJ. Role of calcium ions in insulin action on hexose transport in L, muscle cells. Am J Pbysiol 1984;247:E297- E304. 42 Douen AG, Ramlal T, Rastogi S, et al. Exer- cise induces recruitment of the insulin- responsive glucose transporter: evidence for distinct intracellular insulin- and exercise- recruitable transporter pools in skeletal mus- cle. J Biol Cbem. 1990;265:13427-13430. 43 Goodyear LJ, Hirshman MF, Honon ES. Exercise-induced translocation of skeletal mus- cle glucose transporters. Am J Pbysiol. 1991; 261:E795-E799. 44 Henriksen EJ, Bourey RE, Rodnick KJ, et al. Glucose transporter protein content and glu- cose transport capacity in rat skeletal muscles. Am J Pbysiol. 1990;259:E593-E598, 45 Kern M, Wells JA, Stephens JM, et a]. Insu- lin responsiveness in skeletal muscle is deter- mined by glucose transporter (GLUT4) protein level. BiocbemJ 1990;270:397400. 46 Beam A, Billing B, Sherlock S. Hepatic glu- cose output and hepatic insulin sensitivity in diabetes mellitus. Lancet. 1951;1:69%701. 47 Ferrannini E, Simonson DC, Katz ID, et al. The disposal of an oral glucose load in pa- tients with non-insulin-dependent diabetes. Metabolism. 1988;37:79-85. 48 Bogardus C, Thuillez P, Ravussin E, et al. Effect of muscle glycogen depletion on in vivo insulin action in man. J Clin Invest. 1983;72: 1605-1610. 49 Lillioja S, Mott DM, Zawadzki JK, et a]. Glu- cose storage is a major determinant of in vivo insulin resistance in subjects with normal glu- cose tolerance. J Clin Endocrinol Metab. 1986; 62:922-927. 50 Kahn CR. Insulin resistance, insulin insen- sitivity, and insulin unresponsiveness: a neces- sary distinction. Metabolism. 1978;27(suppl 2):1893-1902. 51 Bergman RN, Finegood DT, Ader M. As- sessment of insulin sensitivity in vivo. Endocr Rev. 1985;6:45-86. 52 Rizza RA, Mandarino LJ, Gerich JE. Mecha- nisms of insulin resistance: assessment using the insulin dose-response curve in conjunction with insulin-receptor binding. Am J Med. 1981; 70~169-176. 53 Tager H, Given B, Baldwin D, et al. A structurally abnormal insulin causing human diabetes. Nature. 1979;281:122-125. 54 Kahn CR. Role of insulin receptors in insulin-resistant states. Metabolism. 1980;29: 45-66, 55 Kolterman OG, Gray RS, Griffin J, et al. Receptor and postreceptor defects contribute to the insulin resistance in noninsulin- dependent diabetes mellitus. J Clin Invest. 1981;68:957-969. 56 King PA. Horton ED, Hirshman MF, Horton ES, Insulin resistance in the Zucker rat (fa/fa) skeletal muscle is associated with a failure of
Physical Therapy I 'Volume 73, Number 12December 1993

glucose transporter translocation. J Clin Invest. 1992;90: 1568-1575. 57 Garvey W, Huecksteadt TP, Birnbaum MJ. Pretranslational suppression of an insulin- responsive glucose transporter in rats with diabetes mellitus. Science. 1989;245:6@63. 58 Armstrong RB, I a n u m CD. Decay of succi- nate dehydrogenase activity in rat skeletal muscle following streptozotocin injection. Horn Metab Res. 1976;8:392-394. 59 Chen V, I anum CD. Metabolic alterations in skeletal muscle of chronically streptozotocin- diabetic rats. Arch Biochem Biophys. 1982;217: 131-138. 60 Sinacore DR. In Vitro Autoradiographic Localization of '25~-Insulin Binding Sites in Rat Skeletal Muscle. Morgantown, WVa: West Virginia IJniversity; 1992. Dissertation. 61 Dohm GL, Elton CW, Friedman JE, et al. Decreased expression of glucose transporter in muscle from insulin-resistant patients. Am J Pbysiol. 1991;260:E459-E463. 62 Eriksson J, Koranyi L, Bourey R, et al. Insulin resistance: in Type 2 (non-insulindependent) diabetic patients and their relatives is not associ- ated with a defect in the expression of the insulin-responsive glucose transporter (GLUT-4) gene in human skeletal muscle. Diabetologia. 1992;35:14>147. 63 Pedersen 0 , Bak JF, Andersen PH, et a]. Evidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDM. Diabetes. 1990;39:865-870. 64 Handberg A, Vaag A, Damsbo P, et al. Expression of insulin-regulatable glucose transporters in skeletal muscle from Type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1990;33:625427. 65 Blotner H. Effect of prolonged physical inactivity on tolerance of sugar. Arch Intern Med 1945;75:3944. 66 Lutwak L, Whedon GD. The effect of physi- cal cond~tioning on glucose tolerance. Clinical Research 1959;7:143-144. 67 Biihr BA. ~ b e r den EinfluB langer dauem- der korperlicher Inaktivitit auf die Blutzuck- erkurve nach order Glukosebelastung. Helv Med Acta. 1963;30:156-175. 68 Altman DF, Baker SD, McCally M, Piemme TE. Carbohydrate and lipid metabolism in man during prolonged bed rest. Clin Res. 1969;17: 543. Abstract. 69 Jennings G, Nelson L, Nestel P, et al. The effects of changes in physical activity on major cardiovascular risk factors, hemodynamics, sympathetic function, and glucose utilization in man: a controlled study of four levels of activ- ity. Circulation. 1986;73:30-40. 70 Stuart CA, Shangraw RE, Prince MJ, et al. Bed-rest-induced insulin resistance occurs primarily in muscle. Metabolism. 1988;37:802- 806. 71 Fukishi T, Kano T, Inque K, Sugimoto E. Decrease in muscle glucose transporter num- ber in chronic physical inactivity in rats. Am J Pbysiol. 1991;26O:E403-E410. 72 Mikines KJ, Richter EA, Dela F, Galbo H. Seven days of bed rest decrease insulin action on glucose uptake in leg and whole body. J Appl Pbysiol. 1991;70:1245-1254. 73 Lipnian RL, Schnure JJ, Bradley EM, Lecocq FR. Impairment of peripheral glucose utiliza- tion in normal subjects by prolonged bed rest. J Lab CIin Med. 1970;76:221-230.
74 Lipman RL, Raskin P, Love T, et al. Glucose intolerance during decreased physical activity in man. Diabetes. 1972;21:101-107. 75 Misbin RI, Moffa AM, Kappy MS. lnsulin binding to monocytes in obese patients treated with carbohydrate restriction and changes in physical activity. J Cltn Endocrinol Metab. 1983;56:273-278. 76 Dolkas CB, Greenleaf JE. Insulin and glu- cose responses during bed rest with isotonic and isometric exercise. JAppl Pbysiol. 1977;43: 1033-1038. 77 Seider MJ, Nicholson WF, Booth FW. Insu- lin resistance for glucose metabolism in dis- used soleus muscle of mice. Am J Pbysiol. 1982;242:E12-E18. 78 Nicholson WF, Watson PA, Booth FW. Glu- cose uptake and glycogen synthesis in muscles from immobilized limbs. JAppl Pbysiol. 1984; 56:431-435. 79 Vissing J, Ohkuwa T, Ploug T, Galbo H. Effect of prior immobilization on muscular glucose clearance in resting and running rats. Am J Physiol. 1988;255:E456-E462. 80 Richter EA, Kiens B, Mizuno M, Strange S. Insulin action in human thighs after one- legged immobilization. J Appl Pbysiol. 1989; 67:19-23. 81 Forsayeth JR, Gould MK. Inhibition of insulin-stimulated xylose uptake in denervated rat soleus muscle: a post-receptor effect. Dia- betologia. 1982;23:511-516. 82 Burant CF, Lemmon SK, Treutelaar MK, Buse MG. Insulin resistance of denemted rat muscle: a model for impaired receptor- function coupling. Am J Physiol. 1984;247: E657-E666. 83 Smith RL, Lawrence JC. Insulin action in denervated rat hemidiaphragms, J Biol Chem. 1984;259:2201-2207. 84 Tudnsky J. Dynamics of insulin resistance in denervated slow and fast muscles in vivo. Am J Pbysiol. 1987;252:R531-R537. 85 Henriksen EJ, Rodnick KJ, Mondon CE, et al. Effect of denervation or unweighting on GLUT-4 protein in rat soleus muscle. J Appl Pbysiol. 1991;70:2322-2327. 86 Smith RL, Lawrence JC. Insulin action in denervated rat skeletal muscle. J Biol Chem. 1985;260:273-278. 87 Block NE, Menick DR, Robinson KA, Buse MG. Effect of denervation on the expression of two glucose transporter isoforms in rat hind- limb muscle, J Clin Invest. 1991;88:1546-1552. 88 Young DA, Wallberg-Henriksson H, Sleeper MD, Holloszy JO. Reversal of the exercise- induced increase in muscle permeability to glucose. Am J Pbysiol. 1987;253:E331-E335. 89 Zorzano A, Balon TW, Goodman MN, Ru- derman NB. Glycogen depletion and increased insulin sensitivity and responsiveness in mus- cle after exercise. Am J Pbysiol. 1986;251:E664- E669. 90 Cartee GD, Young DA, Sleeper MD, et al. Prolonged increase in insulin-stimulated glu- cose transport in muscle after exercise. Am J PLysiol. 1989;256:E494-E499. 91 Davis TA, Klahr S, Tegtmeyer ED, et al. Glucose metabolism in epitrochlearis muscle of acutely exercised and trained rats. Am J Pbysiol. 1986;250:E137-E143. 92 Richter EA, Garetto LP, Goodman MN, Ru- derman NB. Muscle glucose metabolism fol-
lowing exercise in the rat: increased sensitivity to insulin, J Clin Invest. 1982;69:785-793. 93 Richter EA, Mikines KJ, Galbo H, Kiens B. Effect of exercise on insulin action in human skeletal muscle. J Appl Physiol. 1989;66:876- 885. 94 Mikines KJ, Sonne B, Farrell PA, et al. Effect of physical exercise on sensitivity and respon- siveness to insulin in humans. Am J Pbysiol. 1988;254:E248-E259, 95 Cartee GD, Holloszy JO. Exercise increases susceptibility of muscle glucose transport to activation by various stimuli. Am J Pbysiol. 1990;258:E390-E393. 96 Richter EA, Ploug T, Galbo H. Increased muscle glucose uptake after exercise: no need for insulin during exercise. Diabetes. 1985;34: 1041-1048. 97 Gulve EA, Cartee GD, Zierath JR, et al. Re- versal of enhanced muscle glucose transport aher exercise: roles of insulin and glucose. Am JPbysiol. 1990;259:E685-E691. 9 8 Bergstrom J, Hultman E. Muscle glycogen synthesis after exercise: an enhancing factor localized to the muscle cells in man. Nature. 1966;210(5033):309-310. 99 Bergstrom J, Hermansen L, Hultman E, Sal- tin B. Diet, muscle glycogen and physical per- formance. Acta Physiol S c a d 1967;71:140- 150. 100 McArdle WD, Katch FI, Katch VL. Erercke Physiology: Energy, Nutrlrion and Human Per- formance. 3rd ed. Philadelphia, Pa: Lea & Fe- biger; 1991. 101 Berger M, Kemmer FW, Becker K, et al. Effect of physical training on glucose tolerance and on glucose metabolism of skeletal muscle in anaesthetized normal rats. Diabetologia. 1979;16:179-184. 102 James DE, Kraegen EW, Chisholm DJ. Ef- fects of exercise training on in vivo insulin action in individual tissues of the rat. J Clin Invest. 1985;76:657-666. 103 Mikines w, Sonne 8 , Farrell PA, et al. Ef- fect of training on the dose-response relation- ship for insulin action in men. JAppl Pbysiol. 1989;66:695-703. 104 Rodnick KJ, Reaven GM, Azhar S, et al. Effects of insulin on carbohydrate and protein metabolism in voluntaly running rats. Am J Pbysiol. 1990;259:E706-E714. 105 Ploug T, Stallknecht BM, Pedersen 0, et al. Effect of endurance training on glucose transport capacity and glucose transporter ex- pression in rat skeletal muscle. Am J Pbysiol. 1990;259:E77%E786. 106 Rodnick KJ, Henriksen EJ, James DE, Hol- loszy 10. Exercise-training, glucose transport- ers, and glucose transpon in rat skeletal mus- cles. Am J Pbysiol. 1992;262:C9414. 107 Slentz CA, Gulve EA, Rodnick KJ, et al. Glucose transporter number and maximal transpon are increased in endurance-trained rat soleus. J Appl Physiol 1992;73:486492. 108 Burstein R, Polychronakos C, Toews CJ, et al. Acute reversal of the enhanced insulin action in trained athletes: association with in- sulin receptor changes. Diabetes. 1985;34:756- 760. 109 King DS, Dalsky GP, Staten Mq et al. In- sulin action and secretion in endurance- trained and untrained humans, JAppl Pbysiol. 1987;63:2247-2252.
Physical Therapy/Volume 73, Number 12December 1993

110 Rodnick KJ, Haskell WI, Swislocki AUI, et al. Improved insulin action in muscle, liver, and adipose tissue in physically trained human subjects. Am J Physiol. 1987;253:E489-E495. 111 Oshida Y, Yamanouchi K, Hayamizu S, Sato Y. Long-term mild jogging increases insu- lin action despite no influence on body mass index or Vo,max. JAppl Physiol. 1989;66:220& 2210. 112 Dela F, Mikines KJ, Von Linstow M, et al. Effect of training on insulin-mediated glucose uptake in human muscle. Am. J PhysioC 1992; 263:E1134E1143. 113 Friedrnan JE, Sherman WM, Reed MJ, et al. Exercise training increases glucose trans- porter protein GLUT4 in skeletal muscle of obese Zucker (falfa) rats. FEBS Lett. 1990;268: 13-16. 114 Goodyear IJ, Hirshman MF, Valyou PM, Horton ES. Glucose transporter number, func- tion, and subcellular distribution in rat skeletal muscle after exercise training. Diabetes. 1992; 41:1091-1099. 115 Hournard JA, Egan PC, Neufer PD, et al. Elevated skeletal muscle glucose transporter levels in exercise-trained middle-aged men. Am J Physiol. 1991;261:E437-E443. 116 Holloszy JO, Booth FW. Biochemical ad- aptations to endurance exercise in muscle. Ann Rev Physiol. 1976;38:273-291. 117 Rodnick KJ, Reaven GM, Haskell WL, et al. Variations in running activity and enzymatic adaptations in voluntary running rats. JAppl Physiol. 1989,66:1250-1257. 118 Smutok MA, Reece C, Kokkinos PF, et al. Aerobic versus strength training for risk factor intervention in middle-aged men at high risk for coronary heart disease. Metabolism. 1993; 42:177-184. 119 Hurley BF, Kokkinos PF. Effects of weight training on risk factors for coronary artery dis- ease. Sports Medicine. 1987;4:231-238. 120 Ruderman NB, Ganda OP, Johansen K. E5ect.s of physical training on glucose toler- ance and plasma lipids in maturity onset dia- betes mellitus. Diabetes. 1979;28(suppl 1):89- 92. 121 Schneider SH, Amorosa LF, Khachadurain AK, Ruderman NB. Studies on the mechanism of improved glucose control during regular exercise in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1984;26:355-360. 122 Trovati M, Cana Q, Cavalot F, et al. Influ- ence of physical training on blood glucose
control, glucose tolerance, insulin secretion, and insulin action in non-insulin-dependent diabetic patients. Diabetes Care. 1984;7:416- 420. 123 Coyle EF, Martin WH, Sinacore DR, et al. Time course of loss of adaptations after stop- ping prolonged intense endurance training. J Appl Physiol. 1984;57: 1857-1864. 124 Karlsson J, Nordesjo GO, Saltin B. Muscle glycogen utilization during exercise after train- ing. Acta Pbysiol S c a d 1974;90:210-217. 125 Baldwin KM, Fitts RH, Booth FW, et al. Depletion of muscle and liver glycogen during exercise: protective effect of training. PJZuegus Arch. 1975;354:203-212. 126 Brooks GA, Donovan CM. Effect of endur- ance training on glucose kinetics during exer- cise. Am J Physiol. 1983;244:E505-E512. 127 Coggan AR, Kohrt WM, Spina RJ, et al. Endurance training decreases plasma glucose turnover and oxidation during moderate- intensity exercise in man. JAppl Physiol. 1990; 68:%0-996. 128 Heath GW, Gavin JR 111, Hinderliter JM, et al. Effects of exercise and lack of exercise on glucose tolerance and insulin sensitivity. J Appl Phys'ol. 1983;55:512-517. 129 King DS, Dalsky GP, Clutter WE, et al. Effects of exercise and lack of exercise on in- sulin sensitivity and responsiveness. J Appl Physiol. 1988;64:1942-1946. 130 Mikines KJ, Sonne B, Tronier B, Galbo H. Effects of acute exercise and detraining on in- sulin action in trained men. JAppl Physiol. 1989;66:704-711, 131 Bjdrntorp P. Abdominal obesity and the development of non-insulin-dependent diabe- tes mellitus. Diab Metab Rev. 1988;4:615-622. 132 Friden J, Sjostrom M, Ekblom B. A mor- phological study of delayed muscle soreness. Expen'mtia. 198 1;37:506-507. 133 Newham D, Mills K, Quigley B, Edwards R. Pain and fatigue after concentric and eccen- tric contractions. Clin Sci Lond. 1983;64:55-62. 134 Hikida R, Staron R, Hagerman F, et al. Muscle fiber necrosis associated with human marathon runners. J Neurol Sci. 1983;59:185- 203. 135 Evans WJ. Muscle damage: nutritional considerations. Intl J Sport Nutr. 1991; 1:214- 224. 136 O'Reilly KP, Warhol MJ, Fielding RA, et al. Eccentric exercise-induced muscle damage
impairs muscle glycogen repletion. J Appl Physiol. 1987;63:252-256. 137 Costill DL, Pascoe DD, Fink WJ, et al. Im- paired muscle glycogen resynthesis after ec- centric exercise. J Appl Physiol. 1990;69:4650. 138 Kirwan JP, Hickner RC, Yarasheski KE, et al. Eccentric exercise causes transient insu- lin resistance in young subjects. JAppl Physiol. 1992;72:2197-2202. 139 Wallberg-Henriksson H. Acute exercise: fuel homeostasis and glucose transport in insulin-dependent diabetes mellitus. Med Sci Sports Exerc. 1989;2 1:356-361. 140 Saltin B, Lindgarde F, Houston M, et al. Physical training and glucose tolerance in mid- dle aged men with chemical diabetes. Diabe- tes. 1979;28(suppl 1):30-32. 141 Reitman JS, Vasquez B, Klimes I, Nagulesparan M. Improvement of glucose homeostasis after exercise training in non- insulin-dependent diabetes. Diabetes Care. 1984;7:434-441. 142 Bogardus C, Ravussin E, Robbins DC, et al. Effects of physical training and diet ther- apy on carbohydrate metabolism in patients with glucose intolerance and non- insulin-dependent diabetes mellitus. Diabetes. 1984;33:311-318. 143 Krotkiewski M, Ltjnnroth P, Mandroukas K, et al. The effects of physical training on insulin secretion and effectiveness and on glu- cose metabolism in obesity and Type 2 (non- insulin-dependent) diabetes mellitus. Diabeto- logia. 1985;28:881-890. 144 Devlin JT, Horton ES. E5ects of prior high-intensity exercise on glucose metabolism in normal and insulin-resistant men. Diabetes. 1985;34:973-979. 145 Holloszy JO, Schultz J, Kusnierkiewicz J, et al. Effects of exercise on glucose tolerance and insulin resistance. Acta Med Scand Suppl. 1986;711:55-65. 146 Rogers MA, Yamamoto C, King DS, et al. Improvement in glucose tolerance after 1 week of exercise in patients with mild NIDDM. Diabetes Care. 1988;11:613-618. 147 Gollnick PD. Selective glycogen depletion pattern in human muscle fibers after exercise of varying intensity and at varying pedalling rates. J Physiol (Lond). 1974;241:45-57. 148 National Institutes of Health. Consensus development conference on diet and exercise in non-insulin-dependent diabetes mellitus. Diabetes Care. 1987;10:639-644.
Physical Therapy/Volume 73, Number 12December 1993