the role of the ras superfamily small g-proteins in the
TRANSCRIPT

Lawrence UniversityLux
Lawrence University Honors Projects
Spring 2010
The role of the Ras superfamily small G-proteins inthe proinflammatory environment of rhinovirus-exposed monocytic-lineage cellsMichael SchreiberLawrence University
Follow this and additional works at: https://lux.lawrence.edu/luhp
Part of the Respiratory Tract Diseases Commons, and the Virus Diseases Commons© Copyright is owned by the author of this document.
This Honors Project is brought to you for free and open access by Lux. It has been accepted for inclusion in Lawrence University Honors Projects by anauthorized administrator of Lux. For more information, please contact [email protected].
Recommended CitationSchreiber, Michael, "The role of the Ras superfamily small G-proteins in the proinflammatory environment of rhinovirus-exposedmonocytic-lineage cells" (2010). Lawrence University Honors Projects. 51.https://lux.lawrence.edu/luhp/51

The role of the Ras superfamily small G-proteins in the proinflammatory environment of
rhinovirus-exposed monocytic-lineage cells
Michael SchreiberFaculty Advisor: David J. HallDepartment of ChemistryLawrence UniversityAppleton, WISpring 2010

This work is prepared for Honors at Graduation in
Independent Study at Lawrence University,
Appleton, Wisconsin.
I hereby reaffirm the Lawrence University Honor Code.
__________________________________
Michael Thomas Schreiber
1

Table of Contents
...................................................................................Acknowledgements ! 4
.......................................................................................................Abstract! 5
................................................................................................Introduction! 6...............................................................................Asthma exacerbations and the common cold! 6
......................................................................................................Biology of human rhinoviruses! 8
............................Epithelial cells and alveolar macrophages in airway and immune physiology! 13
..............Epithelial cells and alveolar macrophages in HRV-induced infection and inflammation! 17
...........Signal transduction: mechanism of the HRV receptor-mediated inflammatory response! 20
...........................................Biological endpoints of HRV receptor-mediated signal transduction! 25
..........An alternative route: the interferons and toll-like receptors in the inflammatory response! 27
..............Role of the small G-proteins in HRV receptor-mediated signal transduction cascades! 28
............................................................................................Investigative goals and hypotheses! 31
.............................................................................................................Summary of key findings! 32
.............................................................................Materials and Methods! 33.....................................................................................................................................Reagents! 33
............................Isolation and purification/maturation of human blood monocyte-lineage cells! 34
..................................................................................................................................Cell culture! 36
.............................................................................Preparation of human rhinovirus (HRV) stock! 36
...................................................................................Preparation of pharmacological inhibitors! 36
........................................Preparation of recombinant GST-fusion protein for pull-down assays! 36
...............................................Pull-down assay for small molecular-weight G-protein activation! 38
...................................................................................Inhibitor time course for MAPK activation! 40
............................................................................................Immunoblot (Western blot) analysis! 40
.................................Inhibitor time course for cytokine, toll-like receptor, and interferon assays! 41
2

...........................................Enzyme-linked immunosorbant assay (ELISA) for cytokine release! 42
................................................................................................Statistical analysis of ELISA data! 43
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays for Toll-like .....................................................................receptor-3 expression and interferon response! 44
.............................................................................................MTS assay for inhibitor cytotoxicity! 45
......................................................................................................Results ! 46The small G-proteins Rac, Cdc42, and Ras are not activated in the cell lines HeLa and A549
.............................................................................following exposure to HRV 1A or HRV 16! 46
The small G-protein Rac is differentially activated in human and bovine serum-cultured ....................................................macrophages following exposure to HRV 1A and HRV 16! 46
Pharmacological inhibition of Rac prevents activation of the stress-activated p38 MAPK ........................................................following macrophage exposure to HRV 1A or HRV 16! 48
Pharmacological inhibition of Rac downregulates macrophage release of the proinflammatory cytokine MCP-1, but not the release of RANTES following exposure to HRV 1A or HRV 16!49
Pharmacological inhibition of Rac upregulates macrophage expression of the viral innate .....................................immune receptor TLR3 following exposure to HRV 1A and HRV 16! 50
Pharmacological inhibition of Rac upregulates macrophage expression of interferon-α following ..............................................................................................................exposure to HRV 1A! 51
......................................................Pharmacological inhibition of Rac is not inherently cytotoxic! 52
................................................................................................Discussion! 54
...............................................................................................Future Work! 59
................................................................................................References ! 61
.........................................................Appendix I: Supplemental Figures ! 68
..............................Appendix II: Manuscript Submitted for Publication! 70
3

Acknowledgements
I would like to thank Dr. David J. Hall for providing scope and framework to my research and Dr. David Thompson, Dr. Beth De Stasio, and Dr. Judith Humphries for providing helpful criticism along the way.
I would also like to thank Lawrence University alumni Dana Raugi, Megan Wilson, and Bryce Schuler for their contributions to the larger data sets presented here.
I am grateful to the Paul Bertics laboratory, Jim Gern, and Wai-Ming Lee at the University of Wisconson-Madison for their contribution of viral stocks and necessary reagents.
This work was financially supported by the Ronald E. McNair Postbaccalaureate Achievement Program, the Lawrence University Henry Merritt Wriston Scholarship, NIH R15 AI065505-01A1 and NSF 0521112.
4

AbstractRhinoviral infections are a major cause of asthma exacerbations, and though productive
rhinovirus infection occurs predominantly in the bronchial epithelial cells of the upper airway,
monocytic-lineage cells are implicated in establishing the inflammatory microenvironment
observed during rhinoviral asthma exacerbation. It has remained unclear whether small G-protein
activation plays a role in establishing this inflammatory microenvironment. The small molecular-
weight G-proteins are known to be activated in a variety of cell types upon exposure to a range
of viruses. However, it is unclear if small G-protein activation during viral exposure is a
byproduct of receptor attachment, is important for viral replication, or is playing a role in
mediating the inflammatory response. Human rhinovirus (HRV) is unique in that nearly
genetically identical viruses bind the intercellular adhesion molecule (ICAM)-1 or low-density
lipoprotein (LDL) receptors. Furthermore, HRV is capable of eliciting a signaling response in
both epithelial cells, where productive HRV infection occurs, and in macrophages, where there is
an immune response without productive infection. In this study, Rac, cell division cycle (cdc)42,
and Ras were neither activated in epithelial cell lines nor capable of hindering viral replication.
We demonstrated that the small G-protein Rac is differentially activated when major- and minor-
group HRV bind to macrophage ICAM-1 or LDL receptors. Inhibition of Rac in macrophages
attenuated the activation of the stress kinase p38 and the release of the proinflammatory cytokine
MCP-1, but inhibition of Rac did not affect the release of the proinflammatory cytokine
RANTES. Rac inhibition also upregulated the toll-like receptor TLR3 and interferon-α. These
findings suggest that Rac is important in establishing the inflammatory microenvironment that is
initiated in the human airway upon exposure to rhinovirus.
5

IntroductionAsthma exacerbations and the common cold
Asthma is a human airway disease that reduces the quality of life of millions —
particularly children — and that generates a large socioeconomic impact (Gern and Busse 1999).
Although living with asthma is difficult on a day-to-day basis, asthma sufferers face greater, and
potentially life-threatening, challenges during an asthma exacerbation. Asthma exacerbations
may be defined as an acute onset of asthma symptoms (such as cough, wheezing, shortness of
breath, and reduced lung function) much worse than the baseline symptoms experienced by a
particular patient (Johnston 2007). Developing new treatment options for asthma exacerbations is
of vital importance, as exacerbations are responsible for the majority of the health costs and the
majority of the deaths associated with asthma (Johnson 2007).
Asthma exacerbations are most often triggered by upper respiratory tract viral infections.
Among these upper respiratory tract infections, the most frequent is the common cold. The
common cold, in turn, is caused most often by infection with human rhinovirus (HRV) (Johnston
and others 1995). Because HRV is responsible for the majority of common cold infections, it is
also causes the majority of asthma exacerbations (Gern and Busse 1999). Indeed, a 1995 study
showed that HRV infections were present in 80 percent of nine- to eleven-year-olds presenting
with asthma exacerbations during emergency room visits (Johnston and others 1995). In 2003,
another study found similar results in adults (Greenberg 2003). Although these studies are only
correlational, they provide strong evidence for the role of HRV in asthma exacerbations.
The asthma exacerbations produced by HRV infection are characterized by a number of
physiological changes, including airway inflammation, airway obstruction and remodeling, and
enhancement of airway hyperresponsiveness, each of which is a hallmark of clinical asthma
6

(Gern and Busse 1999). The
term “inflammation,” when
applied to the airway of a
person with HRV, describes
the process of recruiting
immune cells to the lung,
with the aim of mounting a
more effective immune
response to the HRV present
there (Holtzman and others 2002). In order to bring immune cells to the lung, the inflammatory
response induces vasodilation, which heightens blood flow and increases the number of immune
cells available to enter the airway (Fixman, Stewart, Martin 2007). For these immune cells to
enter the airway from the blood, it is necessary to increase vascular permeability, or the
“leakiness” of the endothelial barrier between the bloodstream and the interior of the lung
(Holtzman and others 2002). The inflammatory response increases vascular permeability by
activating endothelial cells and altering their cytoskeletal structure, causing them to contract
(Fixman, Stewart, Martin 2007). Over time, chronic airway inflammation gives way to airway
remodeling, which is defined as an increase in airway wall thickness, airway fibrosis, and airway
vascularity (Homer and Elias 2005) (Figure 1). The airway hyperresponsiveness that HRV
infection induces is marked by a sensitivity to agonists of bronchoconstriction and
bronchospasm. In other words, hyperresponsiveness increases the susceptibility of the airway to
tightening, leading to wheeze and cough. If left untreated, the combination of these HRV-induced
7
Figure 1. Airway inflammation and remodeling in asthma.This cutaway diagram compares the normal airway (left) with the inflamed, remodeled airway of an asthmatic (right). Notable characterisitcs of the asthmatic lung include constriction of airway smooth muscle, a thickening of the airway wall, and an accumulation of mucus and intravasate in the airway cavity (http://adam.about.com/).

symptoms may become life threatening.
Importantly, although the association of HRV with
asthma exacerbation is well documented at the
macroscopic level of airway remodeling, etc., the
molecular basis for HRV-induced asthma
exacerbation is not well understood.
Biology of human rhinoviruses
In order to understand the molecular basis
for HRV-induced asthma exacerbation, it is
important to understand the core biology of the rhinoviruses themselves. Human rhinoviruses are
members of the virus family picornaviridae, a family of positive-sense RNA viruses. Although
rhinovirus infections are generally mild, other salient members of the picornaviridae family are
capable of producing severe pathology in otherwise healthy individuals. These other
picornaviruses include the enteroviruses, such as poliovirus and the Coxsackie viruses (Stanway
1990). The picornaviridae possess purely proteinaceous and nearly spherical outer shells
measuring approximately 20-30 nm in diameter. These outer shells are more properly described
as icosahedral capsids (Figure 2). The HRV capsid, made up of a repeating pattern of four
structural proteins termed VP1-VP4, surrounds a viral RNA genome approximately eight
kilobases long that encodes 12 functional viral proteins (Stanway 1990). Taken together, a capsid
and RNA genome make up a single particle of HRV, called a virion. Because the HRV capsid is
composed solely of protein, it lacks the outer, host cell-like phospholipid membrane that
8
Figure 2. Diagram of an icosahedral viral capsid.This diagram illustrates the nearly spherical geometry of an icosahedron and draws attention to the "canyon" present at the five-fold axis of symmetry that is important in receptor recognition. Adopted from Rossmann (1989).

enveloped viruses possess.
Outside of its human
host, the HRV virion is
relatively inert. However,
once HRV successfully
enters its host, it is able to
begin its viral life cycle.
Generally, virologists divide
viral life cycles into a
number of phases. In order
to go on to produce a
second round of infection,
HRV must attach to its host cell receptor, gain entry into the host cell, liberate its RNA genome
from its protective capsid, replicate its genome, and create new capsids (Figure 3).
Although HRV has so far been described as a single, monolithic virus, there are actually
many, extremely similar viruses that collectively are called HRV; these individual variants are
called serotypes (Palmenberg and others 2009). Named for their ability to specifically bind
distinct sets of antibodies, the serotypes of HRV are analogous to strains of bacteria or breeds of
dog, in that the HRV serotypes possess subtle phenotypic differences while sharing nearly
identical genomes. The large number of HRV serotypes, in excess of 100, are usually
phylogenetically divided into three groups, HRVA, HRVB, and HRVC, based on sequence
comparison (Palmenberg and others 2009). Notably, despite their extreme similarity, various
9
Figure 3. Picornaviral life cycle.This diagram illustrates a typical picornaviral life cycle, such as that of HRV. The virus binds to a host cell receptor, is internalized by endocytosis, and uncoats within an endosome. Following uncoating, the virus positive-sense RNA escapes into the cytoplasm where host-cell ribosomes synthesize the viral polyprotein. Autoproteolytic cleavage produces functional viral proteins. Once viral protein is available, genome replication takes place using the viral RNA-dependent RNA polymerase enzyme. Finally, the newly produced viral RNA and protein are packaged into newly assembled virions. Not shown is cellular lysis and the initiation of a second round of infection. Adapted from the Polio Information Center Online (Dove 2002).

HRV serotypes do not all bind the same host cell receptor (Greenberg 2003). Thus, HRV can also
be subdivided into groups based on which of two cellular receptors a particular HRV serotype
will bind and use to gain entry into a host cell (Greenberg 2003). Human rhinoviruses belonging
to the major group, such as the HRV serotype 16, bind the intercellular adhesion molecule-1
(ICAM-1) receptor (Greve and others 1989), whereas human rhinoviruses belonging to the minor
group, such as the HRV serotype 1A, bind the low-density lipoprotein receptor (LDL-R) (Hofer
and others 1994). A later section is devoted to describing the interactions of the HRV serotypes
with their host receptors.
When HRV binds with one of its host cell receptors, it may be taken into the cell through
a number of mechanisms. The most important mechanism of entry appears to be clathrin-
dependent receptor-mediated endocytosis. Clathrin lines pits in the extracellular membrane that
colocalize with membrane-bound receptors, such as the HRV host cell receptors. Interestingly,
ICAM-1 is not known to localize to clathrin-coated pits, as the ICAM-1 protein contains no
localization tag that has been associated with clathrin (DeTulleo and Kirchhausen 1998).
However, HRV 14, a major-group virus that binds to ICAM-1, requires an active clathrin-
dependent endocytic pathway for infection in HeLa cells (DeTulleo and Kirchhausen 1998),
suggesting that ICAM-1 may in fact still associate with clathrin-coated pits. Notably, the minor-
group HRV receptor LDL-R is most densely expressed in clathrin-coated pits, and this receptor is
known to be internalized via the clathrin-dependent pathway (DeTulleo and Kirchhausen 1998).
During the process of internalization, the clathrin-coated pits bud inward, or invaginate, once the
receptor is occupied by its ligand. The uptake of a receptor-ligand complex through the clathrin-
dependent mechanism results in both the receptor and its ligand entering a membrane-bound
10

compartment known as the endosome. The endosome eventually merges with a digestive
compartment called a lysosome, and the conditions within the newly formed endolysosome are
much more acidic. The change in pH may induce the HRV capsid to fall apart in a process
known as viral uncoating, thereby releasing the viral genome and making it available to proceed
with the rest of the HRV life cycle (Brabec and others 2003).
The “positive-sense” RNA of the HRV virion is mostly host mRNA like and is
immediately ready for translation into protein upon entering into the cytoplasm of the host cell.
However, the HRV positive-sense RNA differs from host mRNA in a few key ways that allow it
to be preferentially translated in host cells. Most host mRNA contains a methylated cap at its 5-
prime end that facilitates the host mRNA’s entry into the host translational apparatus; the 5’
methyl cap is recognized by the small subunit of the ribosome, and the binding of this cap
recruits the large ribosomal subunit during the initial steps of translation. In the HRV mRNA-like
genome, there is no 5’ methyl cap. However, the HRV RNA does contain an alternative structure
to the 5’ methyl cap that allows for the HRV RNA to initiate translation within the ribosome. The
alternative structure used by HRV is described as an internal ribosome entry site (IRES). The
IRES represents a portion of the HRV genome with extensive secondary structure that is able to
mimic the function of the host mRNA 5’ methyl cap. In order to preferentially translate its own
genetic information, HRV has evolved a protease activity involved in cleaving host cellular
machinery that plays a role in translating 5’ methyl-capped mRNA, namely the host eIF4G
(Haghighat and others 1996). Notably, IRES-mediated translation is not affected by this protease
activity, so the virus is able to downregulate host protein synthesis while sustaining production of
its own proteins. In other words, the translated rhinoviral protein products upregulate production
11

of the proteins coded for by the viral RNA at the expense of the processes of the host cell.
Although IRES-mediated translation produces a large amount of viral protein, the viral
protein produced by preferential translation of the HRV RNA genome is not immediately
available in a functional form. Because the HRV genome is made up of only one continuous
RNA molecule and only one open reading frame, only one continuous protein is translated. The
translation product is polycistronic, made up of the protein products of a number of rhinovirus
genes. In order for the products of these individual genes to be functional, they must be cleaved
out of the polyprotein. Cleavage from the polyprotein is accomplished by protease activity
inherent to the polyprotein itself, which is called autoproteolytic; the polyprotein contains
proteases that are able to cleave themselves out and seperate the rest of the polyprotein into its 12
functional protein units (Figure 4) (Sarkany and Polgar 2003). The primary viral proteases
responsible for autoproteolytic cleavage in HRV are 2A and 3C (Ryan and Flint 1997).
The rhinoviral RNA replicase proteins, also known as RNA-dependent RNA-
polymerases, left unchecked, eventually induce the production of a large quantity of viral RNA
and viral protein. Following the assembly of viral RNA and protein into new virions, the host cell
may lyse, or burst, sending forth a host of newly crafted virus particles, or virions, that can carry
out further infection. However, host defense mechanisms, such as innate immunity, are often able
to abort the rhinovirus life cycle
before it is complete (Johnston 1995).
Also, it is important to note
that, while the core biology of
rhinovirus infection is important to
12
Figure 4. Picornavirus genome and polyprotein structure.Shown here is the generalized structure of the picornavirus genome. The polyprotein produced during translation of the picornavirus positive-sense genome is necessarily organized in the same fashion. The proteins corresponding to each segment are labelled above the diagram. The capsid proteins are designated by "CP." Adapted from Stanway (1990).

understanding the pathology of respiratory infections and HRV-induced asthma exacerbations,
the present study focuses on the role of HRV as an agonist of ICAM-1 and LDL-R-mediated
signaling cascades. In particular, we focus on the HRV receptor-mediated signaling that occurs in
the cells predominantly present in the airway, namely epithelial cells and immune cells such as
the alveolar macrophages.
Epithelial cells and alveolar macrophages in airway and immune physiology
The mucosal airway lining, composed of epithelial cells, provides a surface barrier that
guards against the entry of airborne pathogens (Velden and Versnel 1998). The epithelial cells are
also responsible for secreting mucus and clearing mucus-trapped foreign material from the
airway via cilliary action (Velden and Versnel 1998). Thus, the epithelial barrier serves as a first
line of defense as part of the non-specific immune system, before even innate immunity comes
into play. However, epithelial cells also have important functions in innate immunity. When
exposed to pathogens, epithelial cells upregulate expression of the antigen-presenting major
histocompatibility (MHC) class I molecules and provide costimulatory signals to macrophages
and other innate immune cells (Message and Johnston 2004). Epithelial cells are also capable of
synthesizing and releasing both pro- and anti-inflammatory cytokines with a wide range of
immunomodulatory activities that may include recruiting, activating, inhibiting, or inducing
differentiation of innate immune cells (Mills, Davies, Devalia 1999).
Notably for the pathogenesis of asthma, epithelial cells also influence the Th1/Th2 skew
of the helper T-cell adaptive response (Message and Johnston 2004). Whereas the Th1 response
is important in antiviral immune responses, the Th2 response is associated with overproduction
13

of immunoglobulin E (IgE) and concomitant allergy, asthma, and hypersensitivity (Magnan and
others 2000). While mast cells and eosinophils were initially implicated as the primary immune
cells responsible for producing airway inflammation, focus has shifted to the T helper type 2
(Th2) cells, leading to the “Th2 hypothesis for asthma.” By releasing cytokines such as IL-4, -5,
-9, and -13, Th2 cells encourage immune responses important in clearing parasitic infections,
including production of certain antibody classes such as IgE, and they are also associated with
eosinophilia, mast cell and granulocyte degranulation, and other hallmarks of the IgE response
correlated with allergy and asthma (Levine and Wenzel 2010). Within the context of the lung, the
Th2 response is also associated with macrophage enrichment and macrophage-mediated
inflammation (Homer and Elias 2005). In contrast, Th1 cells encourage cell-mediated immune
responses important in clearing viral and bacterial infections through their release of cytokines
such as IFN-ɣ and IL-2. The cytokine profiles expressed by Th1 and Th2 cells are reciprocally
inhibitory, i.e. Th1 cytokines suppress Th2 responses and vice versa (Salvi, Suresh Babu,
Holgate 2001). Classically, asthma has been associated with high Th2 cytokine levels and IFN-ɣ
depletion (Salvi, Suresh Babu, Holgate 2001). However, the Th2 hypothesis has increasingly
been criticized for being overly simplistic and reductionist (Salvi, Suresh Babu, Holgate 2001). It
is clear that neither the Th1 nor Th2 response fully dominates in HRV-induced asthma, and both
arms of the T-cell response clearly are important to this condition (Salvi, Suresh Babu, Holgate
2001).
Though the epithelial lining itself contributes significantly to airway immunity, the cells
of the immune system that associate with the epithelium play at least as significant a role. The
immune cells most often associated with the epithelial lining of the airway are macrophages
14

(Lohmann-Matthes, Steinmuller, Franke-Ullmann 1994). Macrophages are cells of the innate
immune system, and, as such, they are responsible for producing an initial, general response to
pathogens invading the airway. This response is often called nonspecific, as the macrophages
produce the same types of responses in reply to a broad variety of pathogens that express
conserved motifs, rather than tailoring the response to any specific pathogen. The primary
response characteristic of macrophages is known as phagocytosis, a process through which
macrophages engulf and destroy pathogens and foreign debris (Martinez and others 2008). When
macrophages perform phagocytosis, they extend projections from the cell body called
pseudopodia that surround a pathogen or piece of debris. When fully surrounded, the foreign
particle or pathogen is taken into the macrophage within a compartment called a phagosome. The
phagosome later merges with a digestive cellular compartment within the macrophage called a
lysosome, and the combined compartment, called a phagolysosome, degrades the foreign particle
or pathogen. Following degradation, the destroyed components of the foreign particle or
pathogen are exported to the macrophage cell surface, where they are bound by cell surface
proteins known as the class II major histocompatibility complex (MHC). The MHC-bound
degradation products of phagocytosis are often antigenic, and they may serve to stimulate cells
of the adaptive immune system through a process known as antigen presentation. Thus, while
macrophages are predominantly associated with the nonspecific, innate immune system, they
also play a role in the adaptive immune system as professional antigen-presenting cells (APCs).
In addition to phagocytosis and antigen-presentation, macrophages also play a vital role
in initiating and regulating the extracellular signaling that takes place between immune cells and
other cells, such as epithelial cells. The extracellular signaling capacity of the macrophages is
15

largely a product of the macrophages’ ability to synthesize and release pro- and anti-
inflammatory signaling molecules called cytokines. In releasing cytokines, macrophages play an
essential role in mediating immunity by relaying signals that recruit, activate, and induce the
differentiation of immune cells such as other macrophages, neutrophils, natural killer cells, and
T-cells (Hu, Chakravarty, Ivashkiv 2008).
Although macrophages exert most of their effector functions in exposed tissues such as
the airway epithelium, they do not originate there. In fact, macrophages arrive in specific tissues
after a long process of growth and development (Geissmann and others 2010). Macrophages are
derived from hematopoetic stem cells that reside in the bone marrow. These stem cells give rise
to myeloid hematopoetic progenitor cells, which mature into progressively more differentiated
cell types such as monoblasts and monocytes. The monocytes circulate in the blood, and they
differentiate into macrophages upon leaving the blood stream, or extravasating. The monocytes
extravasate into specific tissues in response to a chemokine concentration gradient, and they
mature into macrophages in response to a variety of stimuli such as cell-cell contact, exposure to
pathogens, and stimulation by cytokines such as IL-6 (Chomarat and others 2003). Some
macrophages are recruited in the absence of any infection or injury and remain in tissues as
resident, or sentinel macrophages (Lohmann-Matthes, Steinmuller, Franke-Ullmann 1994).
Although macrophages perform similar functions in various tissue types, the macrophages
recruited to each tissue type possess distinct phenotypes, and they are known by diverse names.
For example, macrophages local to the brain are known as microglial cells, whereas the
macrophages of the skin are known as Langerhans cells, and those of the liver are Kupffer cells.
16

The macrophages most important to the present study are those found in the airway, where they
are often called pulmonary or alveolar macrophages.
Epithelial cells and alveolar macrophages in HRV-induced infection and inflammation
HRV may infect a new host when an infected individual coughs or sneezes droplets of
mucus or saliva that are subsequently inhaled. These sputum droplets tend to settle on the
mucosal surface of the upper respiratory tract, where the HRV encounters epithelial cells. Once
HRV has pervaded a mucosal epithelial surface, it produces a characteristic pathology. The
pathology of rhinovirus infection in the upper respiratory tract is well understood, as the
symptoms of infection are generally those of the common cold. However, the symptoms do not
develop immediately as HRV enters the host. Although various researchers have described the
exact timing differently, it is common for HRV to be described as having an incubation period of
12 hours to 3 days before symptoms develop (Rotbart and Hayden 2000). Thus, the hallmarks of
acute infection develop within days from initial exposure. The median duration of HRV infection
has been reported as one week (Arruda and others 1997).
Notably, the symptoms that appear are not directly a consequence of the actions of HRV
itself. Rather, the symptoms are a result of the immune response to the infection. Irritation and
edema result from the recruitment and subsequent invasion of immune cells to the site of
infection at the mucosal epithelium. The response is almost entirely a function of innate
immunity, primarily involving the effector functions of macrophages and neutrophils. Late in the
infection, the cytotoxic T lymphocytes of the adaptive arm of the immune system play a larger
role, as these cells begin to be activated and recruited to kill infected cells (Message and
17

Johnston 2004).
Epithelial cells located in the upper respiratory tract are extremely similar in phenotype to
the epithelial cells along the lower respiratory tract, but HRV preferentially infects cells of the
upper respiratory (Arruda and others 1995). The explanation for this interesting finding remains
somewhat controversial. Many researchers have noted that the preferential infection of the upper
respiratory may be a function of the fact that the epithelial cells of the upper respiratory tract are
located nearer to HRV entry sites, such as the nose, eyes, and mouth. Others have taken a more
mechanistic route to explaining the discrepancy, describing infection as happening
predominantly in the upper respiratory because of the low temperature present there. The
temperature in the upper respiratory is approximately 33 °C, while the higher temperature in the
lower respiratory has been experimentally determined to be less conducive to the rhinoviral life
cycle and subsequent rounds of infection (Message and Johnston 2004). For a time, HRV
researchers dogmatically accepted that rhinovirus was not infectious in the lower respiratory
because of its higher temperature; however, this view has recently been challenged. Critics of the
temperature-gradient hypothesis have noted that HRV is also capable of filtering into the lower
airway and triggering a variety of receptor-mediated signal transduction pathways (Papadopoulos
and others 2000).
Within the lower airway, HRV contacts epithelial cells and alveolar macrophages, the
predominant immune cells present in the lung (Lohmann-Matthes, Steinmuller, Franke-Ullmann
1994; Peters-Golden 2004). Both of these cell types possess receptors for HRV (Hall and others
2005; Subauste and others 1995), and both are capable of proinflammatory signaling (Barnes,
Chung, Page 1998; Levine 1995). As described previously, HRV productively infects epithelial
18

cells (Bardin and others 1994), though macrophages are considered by most researchers not to be
a site of productive HRV replication (Gern and others 1996; Laza-Stanca and others 2006).
However, the proper pathology of rhinovirus in the lower respiratory has only recently been
described, and many gaps in understanding remain to be filled. Though it is widely understood
that rhinovirus infection is capable of producing some inflammation in the lungs, it has been
unclear whether the HRV or cytokines produced in response to it in the upper respiratory merely
trigger an inflammatory response or whether rhinovirus actually infects the lower respiratory
epithelium of the lung. This question has recently been addressed using the technique of in situ
hybridization. In an in situ hybridization study, a fluorescently labeled strand of RNA
complementary to a target strand is used to locate the target strand in a given tissue within a
living organism. Such studies have revealed the production of active rhinovirus RNA in the
tissue of the lung (Papadopoulos and others 2000). The active replication of rhinoviral RNA in
the lung certainly provides evidence for the hypothesis that HRV is infectious in the lung,
although production of viral RNA does not necessarily mean that rhinovirus virions successfully
formed around the RNA in the lower respiratory, escaped from host cells, and proceeded to
produce a second round of infection. In other words, though the in situ hybridization studies
demonstrate that HRV is able to complete the early steps in its life cycle, namely receptor
attachment, host cell entry, uncoating, and RNA replication, these studies do not demonstrate that
HRV is able to complete the later stages of its life cycle in the lower respiratory, namely
production of viral protein, encapsulation of novel positive-sense viral RNAs, host cell lysis, and
initiation of a second round of infection.
19

Other studies have shown that rhinovirus is endocytosed by lung epithelial cells and that
lung immune cells mobilize a response against rhinovirus (Papadopoulos and others 2000).
Immune cells that have been collected from the lung mobilize a response against RV in vitro,
with particularly strong responses derived from alveolar macrophages and cytotoxic T-
lymphocytes. Experimentally infected individuals experience upregulation of proinflammatory
mediators in the lung, as well. In any case, macrophages are known to be important in
establishing a proinflammatory microenvironment in the lung (Barnes, Chung, Page 1998).
Signal transduction: mechanism of the HRV receptor-mediated inflammatory response
The production of biological endpoints such as inflammation by cells responding to HRV
is accomplished by means of an intracellular signal transduction cascade. As they are broadly
understood, signal transduction cascades allow a cell to respond to changes in its environment.
Signal transduction is generally initiated by the extracellular domain of a transmembrane cell-
surface protein (a receptor) binding to a ligand, such as an HRV virion, which causes a
conformational change in the transmembrane receptor protein. Signal transduction pathways are
referred to as cascades because they are composed of many intermediate steps that occur in a
sequential, bifurcating sequence. Theses steps often involve protein conformational change,
phosphorylation events, proteolytic cleavage, etc. (Figure 5).
In any case, the signaling cascade is initiated when the ligand (HRV virion) binds to its
receptor (ICAM-1 or LDL-R). Ligands bind to their cell-surface receptors through a specific
complementarity of size, shape and charge between the ligand and the receptor. HRV is not the
intended ligand for either the ICAM-1 receptor, which normally functions in adhesion, or the
20
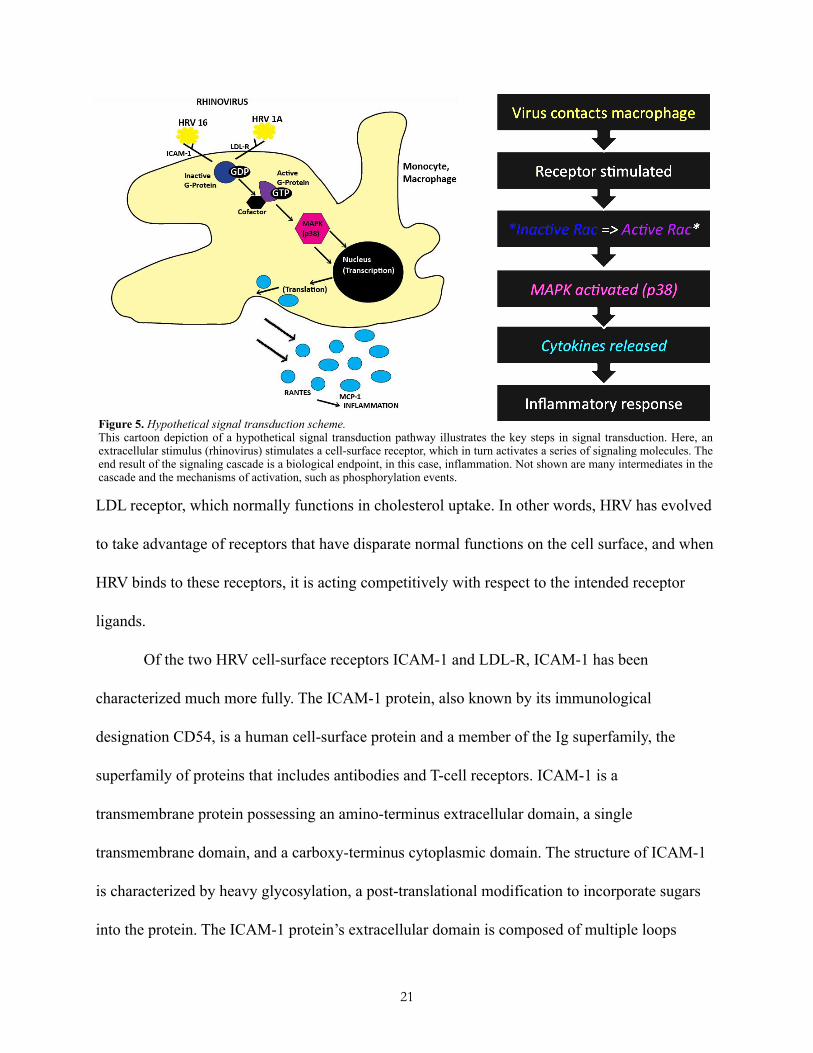
LDL receptor, which normally functions in cholesterol uptake. In other words, HRV has evolved
to take advantage of receptors that have disparate normal functions on the cell surface, and when
HRV binds to these receptors, it is acting competitively with respect to the intended receptor
ligands.
Of the two HRV cell-surface receptors ICAM-1 and LDL-R, ICAM-1 has been
characterized much more fully. The ICAM-1 protein, also known by its immunological
designation CD54, is a human cell-surface protein and a member of the Ig superfamily, the
superfamily of proteins that includes antibodies and T-cell receptors. ICAM-1 is a
transmembrane protein possessing an amino-terminus extracellular domain, a single
transmembrane domain, and a carboxy-terminus cytoplasmic domain. The structure of ICAM-1
is characterized by heavy glycosylation, a post-translational modification to incorporate sugars
into the protein. The ICAM-1 protein’s extracellular domain is composed of multiple loops
21
Figure 5. Hypothetical signal transduction scheme.This cartoon depiction of a hypothetical signal transduction pathway illustrates the key steps in signal transduction. Here, an extracellular stimulus (rhinovirus) stimulates a cell-surface receptor, which in turn activates a series of signaling molecules. The end result of the signaling cascade is a biological endpoint, in this case, inflammation. Not shown are many intermediates in the cascade and the mechanisms of activation, such as phosphorylation events.

created by disulfide bridges
within the protein’s
polypeptide structure (Bella
and others 1998). The
presence of heavy
glycosylation and other
structural characteristics of
ICAM-1 lend the protein binding sites for a number of immune-associated ligands. The immune-
associated ligands to which ICAM-1 binds include macrophage adhesion ligand-1 (Mac-1),
leukocyte function associated antigen-1 (LFA-1), and fibrinogen (Bella and others 1998). These
three proteins are generally expressed on endothelial cells and leukocytes, and they bind to
ICAM-1 to facilitate transmigration of leukocytes across vascular endothelia in processes such as
extravasation and the inflammatory response (Figure 6). As a result of these binding
characteristics, ICAM-1 has classically been assigned the function of intercellular adhesion,
particularly adhesion involving cells of the immune system. Because HRV binding competes
with the normal functioning of the ICAM-1 receptor, HRV may facilitate its own evasion of the
immune system in part by preventing immune cells from adhering to the epithelial surface
through ICAM-1. Notably, HRV has another unique adaptation involving its receptor ligand that
also involves evading the immune response; rhinovirus virions form a cleft or “canyon”
immediately around the ligand for the ICAM-1 receptor (Stanway 1990), and this canyon makes
it difficult for antibodies to bind in the area surrounding the ligand. In the adaptive immune
response, antibodies of the classes IgM and IgA react to rhinovirus by attempting to neutralize
22
Figure 6. Structural schematic of ICAM-1 receptor.This schematic of ICAM-1 highlights the shortness of the receptor's c-terminal cytoplasmic domain and comparatively long extracellular domain. The lollypop-like structures represent sites of heavy glycosylation. The binding sites for HRV and the integrin cellular adhesion molecules macrophage antigen-1 (Mac-1) and lymphocyte function-associated antigen-1 (LFA-1) are also labelled. Adapted from Blaber et al.

the HRV receptor binding site. Therefore, the canyon adaptation allows rhinovirus to escape
neutralization and successfully evade the adaptive immune response (Rossmann 1989).
However, the role these canyons play on the surface of minor-group rhinoviruses, which bind the
LDL receptor, has not yet been fully described. Interestingly, ICAM-1 is constitutively expressed
only at low levels on the surface of respiratory epithelial cells; however, rhinovirus infection is
capable of upregulating ICAM-1 expression, thereby increasing the success of subsequent rounds
of infection (Winther and others 2002).
Importantly, signal-transducing molecules must either contain or associate with proteins
that can activate other signaling intermediates via events such as phosphorylation. ICAM-1 has
been evaluated for the kinase activity that would allow ICAM-1 or a protein associated with it to
phosphorylate another signaling protein, inducing a conformational change and perpetuating a
signal transduction cascade. This evaluation demonstrated that ICAM-1 does not contain
intrinsic kinase activity in its relatively short cytoplasmic domain, meaning that, in order to be a
signaling molecule, ICAM-1 would have to associate with a kinase protein (Holland and Owens
1997) The association of ICAM-1 with kinase proteins is highly probable, as ICAM-1 ligation
triggers phosphorylation in associated Src-related protein tyrosine kinases, most notably the
kinase p56lyn (Holland and Owens 1997). ICAM-1 ligation coupled to p56lyn activity also triggers
signaling function in Raf-1 and MAP kinases such as p38, two proteins with known signaling
properties in immune responses, primarily in immune responses contributing to the inflammatory
response (Holland and Owens 1997). However, signaling molecules in the ICAM-1 signaling
cascade between p56lyn and Raf-1 have not been identified, and p56lyn is not known to directly
interact with Raf-1. In fact, Raf-1 is known to be an effector molecule for the small G-proteins,
23

implicating these signaling molecules, the focus of the present study, in the ICAM-1 cascade
(Takai, Sasaki, Matozaki 2001).
Although LDL-R is the more poorly understood of the receptors implicated in HRV
infectivity, recent work has succeeded in characterizing many aspects of the LDL receptor’s
structure and function. As previously mentioned, the LDL receptor normally functions in the
cellular uptake of cholesterol, and LDL-R is the receptor associated with minor-group HRV such
as HRV 1A. The LDL receptor is a member of a larger LDLR-related family of cell-surface
receptors, all of which are endocytic receptors, responsible for uptake of extracellular materials
(May, Bock, Herz 2003). The LDL receptors present on macrophages recognize such cholesterol-
carrying compounds as apolipoprotein B-100 and apolipoprotein E (Fogelman and others 1988;
Lestavel and Fruchart 1994; Pifat-Mrzljak 1989). The expression of LDL-R is normally tightly
regulated, and LDL-R is quickly upregulated or downregulated in response to fluctuating levels
of intracellular cholesterol (Fogelman and others 1988). Like ICAM-1, the majority of the LDL-
R protein is extracellular (Daniels and others 2009).
Although the LDL receptor has not traditionally been associated with signaling activity,
recent evidence implicates LDL-R in signal transduction cascades involving modulation of ion
gradients, response to extracellular signaling molecules, and activation of a variety of
intracellular protein kinases (May, Bock, Herz 2003). Though HRV-induced signaling pathways
involving LDL-R are not well understood, infection with minor-group HRV correlates with
upregulation of LDL-R (Suzuki and others 2001), analogous to the ICAM-1 upregulation
experienced with major-group viruses. Minor-group virus infection is also associated with
cytokine release and an increase in the activity of the transcription factor nuclear factor κB (NF-
24

κB), a transcription factor with known immunomodulatory properties (Suzuki and others 2001).
Importantly, LDL-R localizes to clathrin-coated pits (Daniels and others 2009), a fact that
supports the clathrin-dependent mechanism of HRV endocytosis.
Biological endpoints of HRV receptor-mediated signal transduction
Previous studies link HRV receptor-mediated signal transduction to a number of
biological endpoints associated with inflammation. Although epithelial cells are able to produce
some of these biological endpoints, many of them are produced predominately by innate immune
cells such as the alveolar macrophages (Alam and others 1996). These biological endpoints
include activation of inflammation-associated transcription factors such as the nuclear factor κB
(NF-κB) (Mogensen and Paludan 2001) and release of inflammatory cytokines such as
interleukin-6 (IL-6), IL-8 (Papi and Johnston 1999), IL-1β, tumor necrosis factor-α (TNF-α)
(Wark and others 2007), MCP-1 (Hall and others 2005), and RANTES (Folkard, Westwick,
Millar 1997). Among these biological endpoints, we selected MCP-1 (monocyte chemotactic
protein-1) and RANTES (regulated upon activation, normal T-cell expressed and secreted) for
further study because of their association with differential levels of asthma severity.
Both MCP-1 and RANTES belong to the C-C class of the β-chemokine supergene family,
a family of chemokines with known proinflammatory properties (Conti and DiGioacchino May-
June 2001). Chemokines, or chemotactic cytokines, are secreted proteins capable of attracting
immune cells to the site of their release, and they are also capable of acting as extracellular
signaling molecules, allowing immune cells to communicate with one another (Olszewska-
Pazdrak and others 1998). Both MCP-1 and RANTES attract immune cells along a concentration
25

gradient, meaning that they recruit immune cells to migrate from regions of low chemokine
concentration to regions of high chemokine concentration (Olszewska-Pazdrak and others 1998).
The chemokine MCP-1 has many functions related to airway inflammation and asthma
severity. Notably, MCP-1 participates in the recruitment of white blood cells with regulatory and
effector functions, including alveolar macrophages, as well as in polarization toward the Th2
immune phenotype that has been associated with asthma (described above) (Conti and
DiGioacchino May-June 2001; Rose, Sung, Fu 2003). Further evidence for the role of MCP-1 in
asthma comes from the observation that single nucleotide polymorphisms in the MCP-1 gene are
associated with distinct levels of asthma severity (Szalai and others 2001). Importantly, MCP-1
has been linked specifically to the inflammation associated with exposing alveolar macrophages
to HRV; indeed, previous work demonstrates that MCP-1 is released by macrophages responding
to HRV (Hall and others 2005).
Like MCP-1, the chemokine RANTES is closely associated with asthma exacerbations.
Strikingly, there is evidence for the importance of RANTES in asthma at the DNA, mRNA and
protein levels. At the DNA level, polymorphisms in the RANTES gene promoter are associated
with various ages of asthma onset (Hizawa and others 2002). At the mRNA level, RANTES has
been found to be constitutively expressed in the normal airway, but upregulated in patients with
mild asthma (Berkman and others 1996). At the protein level, RANTES concentrations are above
normal in the bronchoalveolar lavage fluid (collected after rinsing the lung) of nine out of ten
asthmatics (Folkard, Westwick, Millar 1997). RANTES is known to attract eosinophils,
basophils, and T-cells during inflammatory immune responses, which may contribute to asthma
exacerbation (Berkman and others 1996; Folkard, Westwick, Millar 1997).
26

Additionally, RANTES can selectively enhance IgE production by B-cells, which may lead to
greater airway hypersensitivity during an asthma exacerbation (Folkard, Westwick, Millar 1997).
An alternative route: the interferons and toll-like receptors in the inflammatory response
Although much of the HRV-induced airway inflammation is attributable to cell surface
receptor-mediated signaling, some proinflammatory molecules may be produced by other means.
Specifically, the production of the proinflammatory molecules known as the interferons may be
attributable to HRV-induced signaling that initiates within cells. The interferons are cytokines
produced in response to the presence of many viruses. Interferons function by inducing an
antiviral state in nearby cells, which is accomplished by shutting down these cells’ translational
apparatus (Sen 2001). Previous work has implicated dysregulation of the interferon response as
one of the key contributors to the pathogenesis of HRV-induced asthma exacerbations (Johnston
2007).
One route to interferon production begins with stimulation of the toll-like receptors, a
class of innate immune receptors. These receptors are highly expressed in the airway alveolar
macrophages and epithelial cells relevant to HRV-induced asthma exacerbation (Beutler 2004;
Message and Johnston 2004). Toll-like receptors fall into the category of pattern recognition
receptors (PRRs), receptors that are stimulated by pathogen-associated molecular patterns
(PAMPs) such the dsRNA produced during viral replication. In the case of HRV, the dsRNA
recognized is produced by the HRV RNA-dependent RNA polymerase enzyme during
replication. The HRV RNA polymerase is responsible for transcribing the positive-sense HRV
genome into a negative-sense RNA antigenome, which the polymerase then uses as a template
27

for new postive-sense RNA genomes. The HRV dsRNA produced during this process stimulates
PRRs such as MDA-5 and Toll-like receptor 3 (TLR3). Whereas MDA-5 is a cytosolic receptor,
TLR3 is a chiefly endosomal receptor. TLR3 responds to the presence of dsRNA in the
endosome by signaling through the adaptor protein MyD88, activating the transcription factor
NF-κB, and inducing the synthesis of type I interferons (IFN-α/ß) (Alexopoulou and others 2001;
Takeda and Akira 2005). Interestingly, TLR3 has also been shown to mediate HRV-induced
mucus overproduction during asthma exacerbation (Zhu and others 2009), and TLR3 stimulation
in macrophages may lead to expression of IL-10, a potent signaling molecule with
immunosuppressive properties (Saraiva and O'Garra 2010). Furthermore, TLR3 is upregulated in
bronchial epithelial cells following HRV exposure, and TLR3 inhibition has been linked to
downregulation of the inflammatory cytokine RANTES (Hewson and others 2005). Biological
endpoints following from TLR3 stimulation by HRV in airway epithelial cells include release of
inflammatory cytokines, chemokines, and antimicrobial peptides (Bals and Hiemstra 2004).
Role of the small G-proteins in HRV receptor-mediated signal transduction cascades
The small G-proteins, also known as the Ras small guanosine triphosphatase (GTPase)
superfamily, are proteins that possess signaling activity relatively upstream in a number of
important signal transduction pathways (Wennerberg, Rossman, Der 2005). Notably, many of the
small G-proteins and their associated downstream signaling cascades are implicated in viral life
cycles, particularly within epithelial cells. Many of the G-proteins are anchored to the inner
surface of the cellular membrane by lipid tails, so they tend to be among the first signaling
molecules to take place in transmembrane receptor-mediated signal transduction cascades. The
28

pathways that involve small G-
protein activity have biological
endpoints that include cytoskeletal
rearrangement, cytokine release,
and virus uptake and replication
(Kolokoltsova and others 2008).
The small G-proteins function as
molecular switches, alternating
between an inactive, guanosine
diphosphate (GDP)-bound state and
an active, guanosine triphosphate
(GTP)-bound state. A class of enzymes known as guanosine nucleotide exchange factors (GEFs)
catalyze the conversion of small G-proteins to their active state, while another class of enzymes
known as the GTPase-activating proteins (GAPs) encourage the small G-proteins to activate their
inherent GTPase activity, cleaving GTP to GDP and returning the G-protein to its inactive state
(Figure 7). When the G-proteins are in the active, GTP-bound state, they are able to recruit
effector molecules such as kinases that then can elaborate the signaling cascade.
One of the most prominent members of the small GTPase superfamily encoded in
humans is the 21-kilodalton protein Ras. In addition to its pro-growth role as a proto-oncogene,
Ras has been shown to be important in a number of immune cell signaling pathways. In
particular, Ras has been shown to mediate control of IL-2 gene promoters during the activation
of T-cells (Rayter and others 1992). Ras has also been assigned roles in viral infection and
29
Figure 7. G-protein activation cycle.Small G-proteins such as Ras activate when an upstream event (such as a receptor binding its ligand) stimulates a guanine nucleotide exchange factor (GEF). To activate the G-protein, the GEF triggers a conformational change in the G-protein that facilitates GDP/GTP exchange. Now active, the G-protein may bind cofactors and turn on downstream signaling events. The G-protein reverts to its inactive state and ceases to signal when a GTPase-activating protein (GAP) stimulates the hydrolytic activity inherent to the G-protein, converting GTP to GDP. Adapted from Weinberg, The Biology of Cancer (2007).

replication. For example, Ras activation is known to be modulated by vesicular stomatitis virus
in tumor cells (Balachandran, Porosnicu, Barber 2001), and Ras activation has been described as
important to the replication and pathogenesis of the enteroviruses (Huber and others 1999). As
mentioned earlier, the Ras effector molecule Raf-1 has been associated with ICAM-1 mediated
inflammatory signal transduction (Blaber and others 2003).
Other small G-proteins, such as those of the Rho family, have also been assigned
important roles in cell signaling (Etienne-Manneville and Hall 2002). The Rho GTPases Rac and
Cdc42 are particularly important regulators of signaling related to cytoskeletal rearrangement
through actin reorganization (Burridge 2004) and of signaling related to endocytosis and
exocytosis (Ridley 2001). These proteins are also important regulators of toll-like receptor-
mediated proinflammatory signaling (Bokoch 2005), and their activity has been implicated in
viral pathogenesis. The Rho-family G-protein RhoA, for example, is activated in HeLa cells
responding to HRV (Dumitru, Dreschers, Gulbins 2006). Notably, Rac and Cdc42 activity has
been shown to contribute to the cytopathogenicity of hepatitis C virus (Brazzoli and others
2008). The Rho family G-proteins Rac, Cdc42, and RhoA are also capable of acting as upstream
mediators of mitogen-activated protein kinase (MAPK) activation, including the activation of
JNK, ERK1/2, and p38 (Brown and others 1996; Hall 1998; Bagrodia and others 1995; Dumitru,
Dreschers, Gulbins 2006). Importantly, the stress kinase p38 has been implicated in the ICAM-1-
mediated inflammatory signal transduction cascade (Blaber and others 2003).
The specific G-proteins involved in HRV-induced inflammatory signaling mediated by
the ICAM-1 and LDL receptors or toll-like receptors such as TLR3 have not been previously
described. Additionally, the extent to which inflammatory signaling cascades initiated at the
30

ICAM-1 receptor and inflammatory signaling cascades initiated at the LDL receptor might differ
at the G-protein level has not been well understood.
Investigative goals and hypotheses
As alluded to earlier, the diversity of small G-protein function within the immune system
makes these proteins likely to have interesting roles in elaborating the HRV-induced
inflammatory response. Because many of the small G-proteins are associated with viral life
cycles and proinflammatory signaling pathways, notably that involving ICAM-1, we sought to
explore the role of these G-proteins in the proinflammatory environment of rhinovirus-induced
asthma exacerbation. Many previous explorations have focused on describing signaling much
later in the HRV-induced signaling cascade, at the levels of MAPK phosphorylation (Hall and
others 2005), transcription factor activation (Laza-Stanca and others 2006), or inflammatory
cytokine release (Alam and others 1996). Thus, early signaling events associated with HRV
receptor-mediated signal transduction and viral replication have not yet been well characterized,
either in epithelial cells or macrophages. Because these early signaling events remain poorly
understood, we have chosen to investigate a role in HRV signaling pathways for the small G-
proteins, focusing on the macrophage cell population because of its well-demonstrated
inflammatory capacity. Upon identifying a G-protein as important in the inflammatory HRV-
induced signaling cascade, we sought to inhibit the G-protein in order to determine the effect on
later components in the cascade, and, ultimately, the biological endpoints relevant to the
exacerbation of asthma, namely the release of proinflammatory cytokines. If activation of a
particular G-protein is necessary for the HRV-induced signaling cascade to take place, inhibiting
31

that G-protein should prevent the occurrence of the proinflammatory biological endpoints. As
such, our investigations may provide targets for novel therapeutic strategies against HRV-
induced asthma exacerbation.
As well, though differences between signaling pathways induced by major- and minor-
group HRV binding to ICAM-1 or LDL-R may exist, these differences have not been thoroughly
explored. We pursued exploring these differences at the level of small G-protein activation, as
investigations with the phylogenetically close and nearly genetically identical major-group HRV
16 and minor-group 1A serotypes, which are both members of the HRVA classification
(Palmenberg and others 2009), may provide fresh insight into signaling differences dependent on
virus receptor specificity alone; the viral life cycle for both of these HRV serotypes once they
enter the cell is virtually identical, so later events are unlikely to contribute to differences
between the inflammatory signaling cascades induced by the two HRV serotypes.
Summary of key findings
In the present study, we determined that the small G-proteins Rac, Cdc42, and Ras are not
activated in epithelial cells responding to HRV exposure. However, we also determined that Rac
is activated in macrophages responding to HRV. We further demonstrated that Rac activation in
macrophages is part of a proinflammatory signaling cascade leading to mitogen-activated protein
kinase (MAPK) p38 activation and MCP-1 release. Additionally, we observed an attenuation of
the TLR3-mediated interferon response following HRV-induced Rac activation in macrophages.
Taken together, these results provide a fuller understanding of the pro- and anti-inflammatory
signaling milieu initiated during HRV-induced asthma exacerbation and reinforce the importance
32

of certain G-proteins in initiating the proinflammatory environment. As such, further
investigating the roles played by the G-proteins may provide targets novel for therapeutic
strategies against HRV-induced asthma.
Materials and MethodsReagents
For monocyte preparation, Lymphocyte Separation Media and HBSS were purchased
from CellGro (Manassas, VA), and ACK lysing buffer from BioWhittaker (Walkersville, MD).
Sigma Chemical Company (St. Louis, MO) was the source for protease inhibitor cocktail. Sterile-
filtered, heat-inactivated human (type AB, male only) serum was obtained from BioWhittaker
(#14-498E, Walkersville, MD). Sterile-filtered bovine growth serum was obtained from HyClone
(#ANH19293, Logan, UT). The Rac inhibitor NSC32766 was obtained from Tocris (Ellisville,
MO). Immunoblotting reagents were purchased from a variety of suppliers including Santa Cruz
Biotechnology of Santa Cruz, CA (horseradish peroxidase-conjugated goat anti-rabbit IgG, anti-
Rac antisera, Grb2 antisera, anti-Cdc42 antisera and anti-Ras antisera), Pierce of Springfield, IL
(SupersignalTM chemiluminescence substrate reagents and immobilized glutathione agarose
beads), R&D systems of Minneapolis, MN (anti-MCP1 and anti-RANTES antibody pairs), and
Cell Signaling of Beverly, MA (anti-phospho-p38 MAPK antisera). Anti-CD14 and anti-CD86
was purchased from Becton Dickinson (San Jose, CA). For the MTS assay, CellTiter 96 AQueous
One Solution was purchased from Promega (Fitchberg, WI). For RNA extraction, TRIZOL was
obtained from Invitrogen (Carlsbad, CA). For cDNA preparation, the Omniscript Reverse
Transcriptase Kit was obtained from Qiagen (Germantown, MD) and oligo(dT)15 primer was
obtained from Integrated DNA Technologies (Coralville, IA). For qRT-PCR assays, Sybr Green
33

Universal PCR Master Mix with No AmpErase UNG was obtained from Applied Biosystems
(Foster City, CA). Paired qPCR primers for analysis of TLR3 and beta-actin expression levels
were obtained from Qiagen (Germantown, MD). The Human Interferon α, β Response RT²
Profiler™ PCR Array System was obtained from SABiosciences (Frederick, MD).
Isolation and purification/maturation of human blood monocyte-lineage cells
The protocol used for collecting blood samples was approved by the Lawrence University
Institutional Review Board. Human blood samples were collected from male and female college
students after the students had provided informed consent to the blood collection procedure
(Figure 8). Whole blood in volumes up to 120 mL was collected through a venous catheter into
two 60-mL sterile syringes containing the anticoagulant EDTA at a final concentration of 1 mM.
The whole blood samples were transferred to a sterile glass bottle and were diluted 1:2 with
HBSS. The concentration of EDTA was brought to 2 mM. The diluted blood was transferred to
six sterile 50-mL conical tubes containing 10 mL of Lymphocyte Separation Medium using a
sterile serological pipettor. Blood cells were separated using a density gradient by centrifugation
at 2,000 RPM for 30 minutes at room temperature, without brake, using a Beckman TJ-6
34
Figure 8. Isolation and purification/maturation of human blood monocyte-lineage cells.A) Whole blood from male and female college students who report being in normal health is collected through a venus catheter into a 60-mL syringe. B) The whole blood is separated by centrifugation. C) Monocytes are collected from the "buffy coat" PBMC layer, introduced to human or bovine serum, and allowed to adhere to a plastic tissue culture plate, where they mature for approximately one to two weeks. Nonadherent cells are removed by washing. Adapted from Bryce Schuler, Angelique Van't Wout, and Dana Raugi.

centrifuge and a TH-4 rotor. Leukocytes were collected from the buffy coat interface between the
plasma and erythrocyte layers using a 3.5-mL sterile disposable Pasteur pipet. The collected
leukocytes were transferred to a fresh sterile 50-mL conical tube and were brought to a volume
of 50 mL with sterile HBSS. The leukocytes were then centrifuged for 10 minutes at 1,700 RPM.
The HBSS was removed with an aspirator and the cell pellet was resuspended in 2 mL of ACK
Lysing Buffer. The resuspension was incubated for 2 minutes to lyse any remaining erythrocytes.
The lysing buffer was inactivated with 45 mL of HBSS and the cell suspension was centrifuged
for 10 minutes at 1,200 RPM. The HBSS was removed with an aspirator and the cells were
resuspended in RPMI 1640 containing 2% penicillin and streptomycin and either 5% sterile-
filtered, heat-inactivated human (type AB) serum or 10% sterile-filtered bovine growth serum at
approximately 1 million cells/mL. The cell solution was transferred to 12-well tissue culture
plates at 1 mL/well. The cells were maintained in a 37 °C / 5% carbon dioxide humidified
incubator. The cells were checked to ensure they had adhered to the tissue culture plate
approximately 2 hours after plating. After 24 hours had elapsed, the medium (containing any
non-adherent cells) was removed and replaced with fresh medium, yielding a culture almost
exclusively composed of monocytes. From 10 to 14 days were allowed to elapse for the
monocytes to differentiate into macrophages, dendritic cells, and other cells of the monocyte
lineage. Differentiation was ensured through Fluorescence-Activated Cell Sorting (FACS)
analysis for the presence of the Cluster of Differentiation 14 (CD-14), a cell surface molecule
expressed on macrophages. The analysis was conducted with a BD FACSCaliber analytical flow
cytometer and CellQuest software. Cell populations were approximately 90% CD-14 positive.
35

Cell culture
HeLa cells, A549 cells, and human peripheral blood monocyte-derived macrophages
were cultured in RPMI 1640 (Cellgro) with 5% human AB serum (Cellgro, Manassa, VA) and
1% penicillin/streptomycin (Gibco) at 37˚C in a humidified incubator with 5% CO2.
Preparation of human rhinovirus (HRV) stock
Human rhinovirus (HRV) serotypes 16 and 1A were gifts from the Jim Gern laboratory
and Wai-Ming Lee at the University of Wisconsin - Madison. HRV was grown in HeLa cells and
subsequently sedimented through a sucrose step gradient to remove exogenous protein and other
contaminants. The HRV was then titered to 109 infectious particles/mL and stored at -80 ℃ as
previously described (Hall and others 2005). RPMI 1640 enriched with human serum was used
to prepare all necessary dilutions of both virus serotypes before virus was applied.
Preparation of pharmacological inhibitors
Pharmacological inhibitors were used to determine the involvement of signaling
molecules in the pathways leading to inflammatory cytokine release. NSC23766 was used at
various concentrations to inhibit ras-related C3 botulinum toxin substrate (Rac). Serial dilutions
of NSC23766 were prepared in RPMI 1640 enriched with human or bovine serum as required by
assay protocols.
Preparation of recombinant GST-fusion protein for pull-down assays
BL21pLysS bacteria were transformed with 10 ng of the appropriate GST-fusion protein
36

expressing construct and plated onto luria broth (LB) agar plates containing ampicillin (50 µg/
mL). These bacteria plates were incubated overnight at 37 ºC in a non-CO2 incubator. After
incubation, single colonies were selected from plates and were grown overnight in 50 mL
aliquots of terrific broth (4% w/v) containing ampicillin (50 µg/mL). The overnight growth was
performed in an incubator shaker at 37 ºC. After overnight growth, the 50 mL quantities of
bacterial broth were diluted by placing them in 300 mL of fresh terrific broth containing
ampicillin (50 µg/mL). The freshly diluted culture was allowed to grow for one hour in the 37ºC
incubator shaker. The production of recombinant GST-fusion protein was induced by adding 1 M
isopropylthiogalactaside (IPTG) to the bacterial culture to a final concentration of 0.1 mM. After
a further 3 hours at 37 ºC in the incubator shaker, the culture was centrifuged for 5 minutes at
5,000 RPM (SS34, Sorval) and 4 ºC. The resultant pellet was resuspended in 10 mL bacterial
lysis buffer containing freshly added protease inhibitors (2 mM DTT, PMSF, aprotinin,
benzamidine, leupeptin). The bacterial lysate was sonicated four times while on ice. Each
sonication was for 1 minute, and repetitions were separated by 1-minute rests. The sonicated
lysate was centrifuged for 20 minutes at 10,000 RPM and 4 ºC (SS34, Sorval). Following this
centrifugation, supernatant was removed to 20 1.8-mL microcentrifuge tubes in 500-µL aliquots
and stored at -80 ºC for up to 3 months.
Before use in experiments, the newly produced fusion protein was assayed for quality and
quantity by SDS-PAGE. To this end, a 500-µL aliquot of protein-containing supernatant was
incubated with 50 µL of immobilized glutathione beads (agarose, 50% slurry, washed 5x in PBS)
for 30 minutes. The coupled beads were washed three times in 1x GST-lysis buffer with protease
inhibitors (2 mM DTT, PMSF, aprotinin, benzamidine, leupeptin). After the final wash, GST
37

buffer was removed with an insulin syringe, and beads were resuspended in 40 µL of 1x
Laemmli sample buffer. The resuspended bead samples were denatured at 95 ºC for 5 minutes
and subsequently analyzed by SDS-PAGE with Coomassie staining.
Pull-down assay for small molecular-weight G-protein activation
The pull-down assay protocol was developed from one previously published (Chapman
and Hall 2005) (Figure 9). Immobilized
glutathione agarose beads were washed
5x with PBS and were resuspended in
PBS as a 50% slurry. All reagents were
kept on ice. The appropriate GST-fusion
protein was coupled to the glutathione
beads by mixing 200 µL of the beads
with 500 µL of the frozen GST-fusion
protein in a 1.8-mL microcentrifuge
tube. For Rac and cdc42 pull-down
assays, GST-Pak was used. For Ras
pull-down assays, GST-Raf was used.
To the GST-fusion protein and
glutathione bead mixture,
approximately 1 mL of 3x GST buffer
and 5 µL of protease inhibitor was
38
Figure 9. The Pull-down technique.Shown here is a cartoon depiction of the steps involved in executing the pull-down assay for G-protein activation. Cells are treated with HRV in a time course. After various time points, cells are broken open (lysed), thereby causing the G-proteins of the cell to spill out. In order to quantify only the G-proteins that have been activated in response to HRV treatment, the cofactor known to bind to the G-protein is conjugated to a heavy bead, and the GTP-bound, active G-protein is "pulled down" by centrifugation while the inactive, GDP-bound G-protein is washed away. The active G-protein is then quantified by SDS-PAGE and Western blot.

added. The GST-fusion protein and beads were allowed to couple by rotating the microcentrifuge
tubes at 4 ℃ for 30 minutes. Following this incubation period, the GST-fusion protein coupled
beads were washed 5x with 500 µL of 1x GST buffer prepared with protease inhibitor (1 µL/
mL). The GST buffer was removed using an insulin syringe, and the coupled beads were
resuspended in 300 µL of 1x GST buffer. The coupled bead solution was transferred to six 1.8-
mL microcentrifuge tubes at 50 µL per tube, and tubes were left on ice until the time course
experiment was complete. Blood monocyte-derived macrophages (1 million cells/well in a 12-
well tissue culture plate) were exposed to HRV 1A or HRV 16 for durations of 2, 5, 10, 15, and
30 minutes. When the time course was complete, the tissue culture medium was removed from
the wells with an aspirator and 500 µL of 1x GST buffer was added to each well. In order to
prepare a whole cell extract positive control, 100 µL of Laemelli electrophoresis sample buffer
was added to an additional well. The positive control sample was transferred to a 1.8-mL
microcentrifuge tube and placed on ice until the electrophoresis was performed. The other wells
were scraped to detach the adherent cultured macrophages, and the resulting cell suspensions
were agitated using a vortex mixer and placed on ice for 2 minutes. The cell suspensions were
then centrifuged at 4 ℃ for 5 minutes. The supernatants were transferred to the tubes containing
the GST-fusion protein coupled bead solutions, and the tubes were rotated for 30 minutes at 4 ℃.
After the rotation was complete, the coupled beads were washed three times with 500 µL 1x
GST buffer. After the final wash, the GST buffer was removed using an insulin syringe and 40
µL of 1x Laemelli electrophoresis sample buffer was added to each tube. The samples were then
denatured at 95 ℃ for 5 minutes in preparation for sodium dodecyl sulphate polyacrylamide gel
electrophoresis (SDS-PAGE).
39

Inhibitor time course for MAPK activation
Blood monocyte-derived
macrophages (1 million cells/well in a
12-well tissue culture plate) were
exposed to HRV 1A or HRV 16 for
durations of 15, 30, 60, and 90
minutes (Figure 10). The inhibitor
NSC23766 was used to pretreat some
wells exposed to HRV at the 90-
minute time point. Inhibitor
pretreatments occurred 30 minutes before virus exposure began. When the time course was
complete, the tissue culture medium was removed from the wells with an aspirator and 200 µL of
1X Laemelli electrophoresis sample buffer was added. The wells were scraped to detach the
adherent cultured macrophages, and the sample buffer was transferred to 1.8 mL microcentrifuge
tubes. The samples were sonicated at an output of 4V to shear cellular DNA, and the samples
were denatured at 95 ℃ for 5 minutes in preparation for sodium dodecyl sulphate
polyacrylamide gel electrophoresis (SDS-PAGE).
Immunoblot (Western blot) analysis
The samples and pre-stained broad-range SDS-PAGE standards were loaded into 12%
polyacrylamide slab gels. Samples were added to each gel lane in amounts that ensured equal
protein loading. Electrophoresis was performed at 180 volts for 1 hour or until loading dye
40
Figure 10. Sample time-course plate diagram.This plate diagram depicts a sample plate diagram used in performing a time-course experiment. Cells are exposed to HRV for various amounts of time. Wells marked "+NSC" are pretreated with the Rac inhibitor NSC23766. Following the times marked, cells are lysed, and the cellular lysates proceed to be analyzed by SDS-PAGE and Western blot techniques.

cleared the gel. The proteins contained in the gel were transferred to polyvinylidene difluoride
(PVDF) membranes by applying a 350 mA current over 2 hours, and the resulting blots were
blocked overnight at 4 ℃ in 5% milk (w/v) prepared in 1X tris-buffered saline with Tween 20
(TBS-T). Rabbit anti-phospho primary antibodies (Rac1, cdc42, Ras, p38, GRB-2 [internal
control, the growth factor receptor-bound protein 2]) were diluted in 5% milk/TBS-T (w/v) and
applied to blots at a 1:1000 concentration. The primary antibodies were allowed to bind to the
blots for 1 hour at 37 ℃ with agitation. Following an infinite dilution in TBS-T, anti-rabbit
secondary horseradish peroxidase-linked antibody was applied to blots for 1 hour according to
the manufacturer's instructions. Following this application and a second infinite dilution in TBS-
T, blots were rinsed in TBS-T for 30 minutes and visualized using 500 µL SuperSignal® West
Pico Luminol/Enhancer Solution and 500 µL West Pico Stable Peroxide Solution, 20 µL of
SuperSignal® West Femto Maximum Sensitivity Substrate and a KODAK Image Station 4000
MM (Kodak) and Kodak MI Imaging Software (version 4.0.3). Image capture times were
determined automatically using the software.
Inhibitor time course for cytokine, toll-like receptor, and interferon assays
Inhibitor time courses took place over 24 hours. Experiments were conducted with 12-
well tissue culture plates of blood monocyte-derived macrophages (1 million cells per well).
Before beginning the 24-hour time course, growth medium was replaced to ensure that proteins
present in the supernatant were released following experimental treatment rather than during
growth and differentiation. Following media replacement, cell plates were returned to the 37 ℃
incubator for one hour. The Rac inhibitor NSC23766 was applied to a final concentration of 10
41

µM 15 minutes before HRV was applied. Both HRV 16 and HRV 1A were applied at a
multiplicity of infection (MOI) of 10, meaning that approximately 10 HRV particles were present
per cell. Following the 24-hour time course, the media supernatants were removed to cluster
tubes and stored at -20 ℃ until enzyme-linked immunosorbant assay (ELISA) could be
performed. Remaining adherent monocyte derived macrophages were lysed in TRIZOL and
RNA was extracted according to the manufacturer protocol.
Enzyme-linked immunosorbant assay (ELISA) for cytokine release
Following the inhibitor time course for cytokine release, the sandwich enzyme-linked
immunosorbant assay (ELISA) technique was used to probe for MCP-1 and RANTES release
(Figure 11). Half-size 96 well ELISA/Immuno/Assay (EIA) plates were coated overnight at 4°C
with coating buffer containing the concentrations of monoclonal capture antibody recommended
by the manufacturer (R&D Systems). The plates were washed three times with 1x phosphate
buffered saline with Tween 20 (PBS-T) to remove excess antibody, and a standard curve of
successive 1:2
dilutions of protein
was made. The
standards and the
various samples were
added to the 96-well
plate in triplicate at
25 µL per well, and
42
Figure 11. Enzyme-linked immunosorbant assay proceedure.A) A plastic plate is coated with antibodies that recognize the target protein of interest. After the target protein is bound by these capture antibodies, a second antibody that binds the target protein is added, which is then recognized by another antibody linked to an enzyme that will produce a color change when its substrate is added. B) Darker wells in the ELISA plate are associated with the presence of more target protein. Quantification of target protein is accomplished by comparison to a standard curve of color readings from a set of wells of known target protein concentration. Adapted from Bryce Schuler and New England Biolabs.

the plate was incubated at 4°C overnight. Monoclonal detection antibody was added to the plate
at concentrations recommended by the manufacturer (R&D Systems, Sanquin) after washing
three times in 1x PBS-T and incubating the plate at room temperature for 1 hour. The plate was
washed three times in 1x PBS-T. A 1:10,000 dilution of streptavidin-horseradish peroxidase
(HRP)-40 was added to the wells, and the plate was incubated for 20 minutes. The plate was then
washed three times in 1x PBS-T, and 50 µL of TMB 1 component HRP microwell substrate
solution was added to each well. When a blue color developed such that a gradation between
standards could be visually detected, the reaction was stopped with 1 M hydrochloric acid.
Optical density (absorbance) was read at 450 nm using a Molecular Devices ThermoMAX
microplate reader with Spectrosoft software (version 6.2). Protein concentrations were calculated
by averaging the triplicate values and interpolating from the standard curve.
Statistical analysis of ELISA data
After the ELISAs had been performed, the data were pooled for comparison. To
determine whether differences between control and treated groups were significant, SPSS
(originally the Statistical Package for the Social Sciences; version 16.0) was used to perform an
analysis of variance (ANOVA) (αreject = 0.10). Following detection of significant differences
between groups by ANOVA, post-hoc Bonferroni-adjusted Student’s t-tests (αreject = 0.10) were
used to resolve individual differences between groups.
43

Quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays for Toll-like
receptor-3 expression and interferon response
To perform cDNA synthesis in preparation for qPCR analysis, total isolated RNA
collected from 24-hour time course experiments was used with the Omniscript Reverse
Transcriptase Kit (Qiagen) and oligo(dT)15 primer (IDT) according to the protocols provided by
Qiagen. The samples were denatured (65°C for 5 min), chilled (4°C for 5 min) and amplified
(37°C for 1 h) using a MJ research thermocycler.
Toll-like receptor-3 quantitative PCR assays were performed using an ABI 7500 (Applied
Biosystems, Foster City, CA) and Sybr Green Universal PCR Master Mix with No AmpErase
UNG. The paired qPCR primers used in the assay and cDNA samples were added to this master
mix. The thermal cycler was set to perform an initial set-up (95°, 10 min) and 40 cycles of
denaturation (95°, 15 sec) followed by annealing/extension (60°, 1 min). The threshold cycle (Ct)
was calculated using the 7500 Sequence Detection Software (version 1.3.1) and the baseline and
threshold values were manually set by using the log view of the amplification plots. The
threshold was placed above the background signal but within the lower one-third of the linear
phase of the amplification plot. The relative amount of TLR3 mRNA was determined by
subtracting the threshold cycle values for the gene from the threshold cycle values for the
internal control gene beta-actin (∆Ct).
Interferon response quantitative PCR assays were performed using an ABI 7500
thermocycler in combination with the Human Interferon α, β Response RT² Profiler™ PCR
Array System according to the protocol provided by the manufacturer (SABiosciences). The
44

threshold cycle (Ct) was calculated using the 7500 Sequence Detection Software (version 1.3.1)
and the baseline and threshold values were manually set by using the log view of the
amplification plots. The threshold was placed above the background signal but within the lower
one-third of the linear phase of the amplification plot. Analysis of the data was carried out using
the RT2 Profiler PCR Array Data Analysis application suite available at the manufacturer's Web
site (http://www.sabiosciences.com/pcr/arrayanalysis.php).
MTS assay for inhibitor cytotoxicity
A 96-well tissue culture plate was plated with HeLa cells cultured in RPMI 1640
containing 2% penicillin and streptomycin and 10% sterile-filtered bovine growth serum at 5,000
cells per well in a volume of 50 µL. For the virus treatments, no virus, HRV 1A, or HRV 16 was
added to wells at multiplicities of infection (MOI) of 0.1, 1, and 10. The Rac inhibitor
NSC23766 either was not applied or was applied at 0.1 µM, 1 µM, and 10 µM concentrations.
All experimental treatments were conducted in triplicate. Each well was brought to a final
volume of 100 µL using the bovine growth media, and the cell plate was incubated overnight in a
37 ℃ / 5% carbon dioxide incubator. CellTiter 96 AQueous One Solution (Promega) was used
according to the manufacturer’s instructions. After adding 20 µL of the CellTiter 96 AQueous One
Solution to each well of the 96-well plate, the plate was incubated for 1 hour in the 37 ℃
incubator. After the incubation was complete, optical density (absorbance) was read at 490 nm
using a Molecular Devices ThermoMAX microplate reader with Spectrosoft software (version
6.2).
45

ResultsThe small G-proteins Rac, Cdc42, and Ras are not activated in the cell lines HeLa and A549
following exposure to HRV 1A or HRV 16
Because the small G-proteins have been associated with the replication and infectivity of
several viruses, we sought to explore the roles played by some of the most prominent G-proteins
during the infection of classic epithelial cell lines known to be productively infected by HRV. We
were especially interested in the Rho-family G-proteins Rac and Cdc42 in light of the fact that
the related G-protein RhoA was shown to be activated in HeLa cells responding to HRV
(Dumitru, Dreschers, Gulbins 2006). We selected HRV 16 and HRV 1A for use because they are
genetically closely related, in the HRVA group, but bind to different receptors (Palmenberg and
others 2009). Interestingly, pull-down assays in HeLa and A549 cells for the small G-proteins
Rac and Ras following HRV 1A and HRV 16 exposure revealed little to no G-protein activation
(see Appendix I).
The small G-protein Rac is differentially activated in human and bovine serum-cultured
macrophages following exposure to HRV 1A and HRV 16
Though the G-proteins Rac, Cdc42, and Ras were not activated in HeLa or A549 cells
responding to HRV, we suspected that these proteins might be involved in the macrophage
response to HRV. Previous work has identified the macrophage as a key mediator of the
proinflammatory environment initiated during HRV infection (Gern and others 1996; Hall and
others 2005). In addition, previous work had demonstrated a differential macrophage
inflammatory response to HRV that was serum-dependent (Schuler and Lawrence University.
2009), so we sought to determine whether culturing macrophages in human or bovine serum
46

would alter the
kinetics of G-protein
activation. While we
found that the G-
proteins Cdc42 and
Ras were not activated
in macrophages
(Appendix I), pull-
down assays for the
G-protein Rac
demonstrated serum-
independent Rac
activation in
macrophages
following exposure to
HRV 16 or HRV 1A (Figure 12, 13). HRV was applied at a MOI of 10 in accord with previous
results (Hall and others 2005). The activation of Rac occurred with differential kinetics that
depended upon which HRV serotype provided the stimulus. In response to HRV 1A exposure
(LDL-R tropic), macrophages experienced escalating Rac activation from 2 minutes through 10
minutes followed by declining activation through the 30-minute time point (Figure 12A, 13A). In
response to HRV 16 exposure (ICAM-1 tropic), Rac activation escalated throughout the 30-
minute time course (Figure 12B, 13B).
47
Figure 12. Rac activation following human serum-cultured macrophage exposure to HRV.Activated Rac was pulled down and assayed by SDS-PAGE and immunoblot following exposure of blood monocyte-derived macrophages cultured in human serum to HRV 1A (Panel A) and HRV 16 (Panel B) at an MOI of 10. Each blot is representative of five independent experiments.
Figure 13. Rac activation following bovine serum-cultured macrophage exposure to HRV.Activated Rac was pulled down and assayed by SDS-PAGE and immunoblot following exposure of blood monocyte-derived macrophages cultured in bovine serum to HRV 1A (Panel A) and HRV 16 (Panel B) at an MOI of 10. Each blot is representative of five independent experiments.
HRV 1A
2 5 10 15 30
WCE
2 5 10 15 30
WCE
HRV 16

Pharmacological inhibition of Rac prevents activation of the stress-activated p38 MAPK
following macrophage exposure to HRV 1A or HRV 16
Previous studies have described the important role played by p38 MAPK in
proinflammatory signaling cascades leading to activation of the transcription factor ATF-2 and
the release of the proinflammatory cytokine monocyte chemotactic protein-1 (MCP-1) (Hall and
others 2005). Having established that Rac activation occurs in macrophages responding to HRV,
we sought to determine whether Rac activation is a necessary upstream event for p38 activation.
We chose to block Rac activation using the pharmacological Rac inhibitor NSC23766, selected
based on its previous effective use at similar concentrations in alveolar macrophages (Haberzettl
and others 2008) and other immune cells (Mitchell and others 2008). This small molecule
inhibitor was rationally designed to specifically block the interactions of Rac with the guanine
nucleotide exchange factors (GEFs) responsible for facilitating Rac activation (Akbar and others
2006). We demonstrated that blocking Rac activation using the NSC23766 inhibitor decreased
macrophage p38 activation in response to either HRV 16 or HRV 1A exposure; the activation of
p38 escalated over a 90-minute time course when macrophages were exposed to either HRV 1A
or HRV 16, but applying NSC23766 30 minutes before applying the virus greatly attenuated p38
activation at the 90-minute time point (Figure 14).
48
Figure 14. Activation of MAPK p38 in macrophages exposed to HRV.Selected 90-minute time points were treated with the Rac inhibitor NSC23766 30 minutes before HRV 1A and 16 treatment at an MOI of 10. Activation of p38 MAPK was assayed by SDS-PAGE and immunoblot using an anti-phospho-p38 antibody. Equal protein loading was ensured by using GRB-2 as an internal control and probing with an anti-GRB-2 antibody (not shown). The blot is representative of five independent experiments. Macrophages were cultured in human serum.
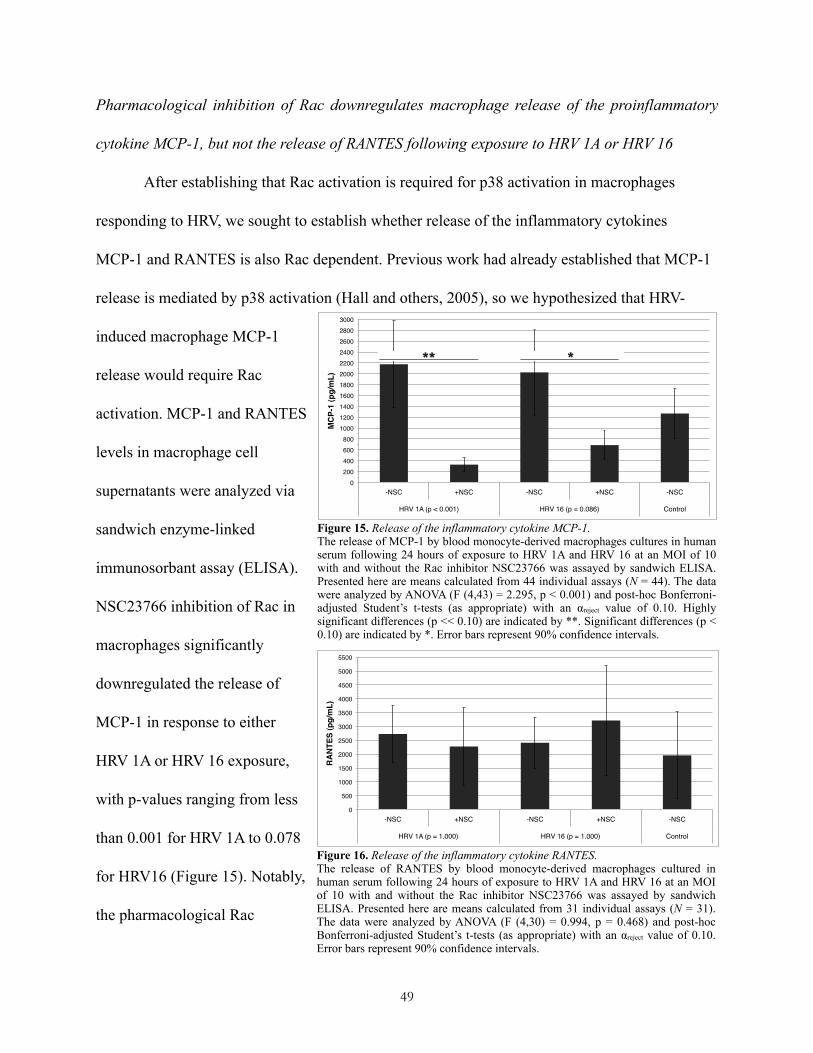
Pharmacological inhibition of Rac downregulates macrophage release of the proinflammatory
cytokine MCP-1, but not the release of RANTES following exposure to HRV 1A or HRV 16
After establishing that Rac activation is required for p38 activation in macrophages
responding to HRV, we sought to establish whether release of the inflammatory cytokines
MCP-1 and RANTES is also Rac dependent. Previous work had already established that MCP-1
release is mediated by p38 activation (Hall and others, 2005), so we hypothesized that HRV-
induced macrophage MCP-1
release would require Rac
activation. MCP-1 and RANTES
levels in macrophage cell
supernatants were analyzed via
sandwich enzyme-linked
immunosorbant assay (ELISA).
NSC23766 inhibition of Rac in
macrophages significantly
downregulated the release of
MCP-1 in response to either
HRV 1A or HRV 16 exposure,
with p-values ranging from less
than 0.001 for HRV 1A to 0.078
for HRV16 (Figure 15). Notably,
the pharmacological Rac
49
Figure 15. Release of the inflammatory cytokine MCP-1.The release of MCP-1 by blood monocyte-derived macrophages cultures in human serum following 24 hours of exposure to HRV 1A and HRV 16 at an MOI of 10 with and without the Rac inhibitor NSC23766 was assayed by sandwich ELISA. Presented here are means calculated from 44 individual assays (N = 44). The data were analyzed by ANOVA (F (4,43) = 2.295, p < 0.001) and post-hoc Bonferroni-adjusted Student’s t-tests (as appropriate) with an αreject value of 0.10. Highly significant differences (p << 0.10) are indicated by **. Significant differences (p < 0.10) are indicated by *. Error bars represent 90% confidence intervals.
0!200!400!600!800!
1000!1200!1400!1600!1800!2000!2200!2400!2600!2800!3000!
-NSC! +NSC! -NSC! +NSC! -NSC!
HRV 1A (p < 0.001)! HRV 16 (p = 0.086)! Control!
MC
P-1
(pg/
mL)!
0!
500!
1000!
1500!
2000!
2500!
3000!
3500!
4000!
4500!
5000!
5500!
-NSC! +NSC! -NSC! +NSC! -NSC!
HRV 1A (p = 1.000)! HRV 16 (p = 1.000)! Control!
RA
NTE
S (p
g/m
L)!
Figure 16. Release of the inflammatory cytokine RANTES.The release of RANTES by blood monocyte-derived macrophages cultured in human serum following 24 hours of exposure to HRV 1A and HRV 16 at an MOI of 10 with and without the Rac inhibitor NSC23766 was assayed by sandwich ELISA. Presented here are means calculated from 31 individual assays (N = 31). The data were analyzed by ANOVA (F (4,30) = 0.994, p = 0.468) and post-hoc Bonferroni-adjusted Student’s t-tests (as appropriate) with an αreject value of 0.10. Error bars represent 90% confidence intervals.

inhibitor did not significantly downregulate the release of RANTES (Figure 16). Since Rac
inhibition did not affect macrophage secretion of RANTES in response to HRV, we explored
other G-proteins for a signaling capacity that might function in RANTES production. However,
pull-down assays for the related G-proteins Cdc42 and Ras did not detect activation of these G-
proteins following macrophage exposure to HRV 1A or HRV 16 (see Appendix I).
Pharmacological inhibition of Rac upregulates macrophage expression of the viral innate
immune receptor TLR3 following exposure to HRV 1A and HRV 16
Because Rac-mediated signaling in blood monocyte-derived macrophages seemed
important to the elaboration of an HRV-induced inflammatory response by means of activation of
p38 and release of MCP-1, we sought to determine whether Rac-mediated signaling would also
be important to the interferon response, as mediated by the toll-like receptors, particularly TLR3.
As described above, TLR3 is a chiefly endosomal receptor that responds to the presence of
dsRNA (such as might be produced during the HRV life cycle) by signaling through the adaptor
protein MyD88 and inducing the synthesis of type I interferons (IFN-α/ß) by activating the
transcription factor NF-kappaB (Alexopoulou and others 2001; Takeda and Akira 2005). We
explored the effect of pharmacological Rac inhibition and HRV 1A and HRV 16 exposure on
blood monocyte-derived macrophage TLR3 expression by performing 24-hour time courses with
these stimuli followed by RNA extraction, reverse transcription, and quantitative polymerase
chain reaction (qPCR). Interestingly, treatment with HRV 1A alone produced an appreciable
upregulation of TLR3, whereas treatment with HRV 16 alone did not.
50

However, when either HRV 1A treatment or HRV 16 treatment was combined with Rac inhibitor
NSC23766 treatment, a substantial upregulation of TLR3 expression was observed (Figure 17).
Pharmacological inhibition of Rac upregulates macrophage expression of interferon-α following
exposure to HRV 1A
The TLR3 signaling pathway is closely associated with the interferon response, so we
sought to explore the effects of Rac inhibition on the regulation of the interferons in HRV-treated
macrophages. Blood monocyte-derived macrophages were treated in a 24-hour time course with
HRV and either the Rac inhibitor NSC23766 or no inhibitor, total RNA was extracted, and
cDNA was synthesized in preparation for assaying the interferon response by qPCR. Despite the
51
Fold
Cha
nge
from
Con
trol
NSC HRV 16 HRV 16 NSC
HRV 1A HRV 1A NSC
Figure 17. Regulation of TLR3 by HRV and Rac inhibitor NSC23766.Expression of toll-like receptor-3 (TLR3) was assayed in blood monocyte-derived macrophages cultured in human serum by qRT-PCR following 24 hours of treatment with HRV 1A or HRV 16 at an MOI of 10 with and without the Rac inhibitor NSC23766. The data are normalized to expression of the housekeeping gene beta-actin and are expressed as gene expression fold change from the untreated control. Error bars represent standard error (N = 4).

high variability in gene
expression between the NSC
treated and uninhibited
experimental conditions
(Figure 18), application of the
Rac inhibitor to HRV 1A-
treated macrophages did seem
to upregulate expression of
interferon-alpha (IFNA1)
(Table 1). Rac inhibition did
not affect the regulation of the
beta or gamma interferons
(Table 1).
Pharmacological inhibition
of Rac is not inherently
cytotoxic
After we established
that Rac was activated in
macrophages responding to
HRV and characterizing the
response of downstream inflammatory mediators to pharmacological Rac inhibition, we sought
52
Figure 18. Regulation of interferons and related genes by the Rac inhibitor NSC23766 in conjunction with HRV 1A treatment.Expression of the interferons and related genes was assayed in blood monocyte-derived macrophages cultured in human serum by qRT-PCR following 24 hours of treatment with HRV 1A at an MOI of 10 with and without the Rac inhibitor NSC23766. The data are normalized to the average expression of a suite of housekeeping genes and are expressed as log2-transformed gene expression fold change for NSC treatment relative to gene expression for HRV 1A treatment alone, i.e. darker shades of red indicate greater expression for HRV 1A +NSC treatment relative to HRV 1A treatment alone.
Table 1. Regulation of the alpha- and beta-interferons by the Rac inhibitor NSC23766 in conjunction with HRV 1A treatment.Expression of the alpha-interferons (IFNA-1,-2,-4) and beta-interferon (IFNB1) was assayed in blood monocyte-derived macrophages cultured in human serum by qRT-PCR following 24 hours of treatment with HRV 1A at an MOI of 10 with and without the Rac inhibitor NSC23766. The data are normalized to the average expression of a suite of housekeeping genes and are ultimately expressed as log2-transformed gene expression fold change for NSC treatment relative to gene expression for HRV 1A treatment alone, i.e. larger positive values indicate an increase in expression for HRV 1A +NSC treatment relative to HRV 1A treatment alone.

to determine whether Rac inhibition exerted any cytotoxic effects. Without determining that the
pharmacological Rac inhibitor NSC23766 is not inherently cytotoxic, it would be difficult to
interpret attenuation of signaling events downstream of Rac as not merely resulting from the
confounding variable of cell death. In order to determine inhibitor cytotoxicity, HeLa cells were
treated with the Rac inhibitor NSC23766 alone or with the inhibitor in addition to HRV, and
treated HeLa cells were then incubated for 24 hours. Although HRV 1A and HRV 16 at the
highest MOI of 10 greatly reduced the viability of treated HeLa cells as determined by MTS cell
viability assay, the application of the NSC23766 inhibitor neither enhanced cell death in response
to virus nor possessed any inherent cytotoxicity (Figure 19). We confirmed that Rac inhibitor did
53
!"
!#$"
!#%"
!#&"
!#'"
!#("
!#)"
!#*"
!#+"
!#,"
$"
$#$"
-./01.2"
34-"!#$"56"
34-"$"56
"
34-"$!"56"
789"$:"6
;<"!#$"
789"$:"6
;<"!#$"34-"!#$"56"
789"$:"6
;<"!#$"34-"$"56"
789"$:"6
;<"!#$"34-"$!"56
"
789"$:"6
;<"$"
789"$:"6
;<"$"34-"!#$"56"
789"$:"6
;<"$"34-"$"56
"
789"$:"6
;<"$"34-"$!"56"
789"$:"6
;<"$!"
789"$:"6
;<"$!"34-"!#$"56"
789"$:"6
;<"$!"34-"$"56"
789"$:"6
;<"$!"34-"$!"56
"
789"$)"6
;<"!#$"
789"$)"6
;<"!#$"34-"!#$"56"
789"$)"6
;<"!#$"34-"$"56"
789"$)"6
;<"!#$"34-"$!"56
"
789"$)"6
;<"$"
789"$)"6
;<"$"34-"!#$"56"
789"$)"6
;<"$"34-"$"56
"
789"$)"6
;<"$"34-"$!"56"
789"$)"6
;<"$!"
789"$)"6
;<"$!"34-"!#$"56"
789"$)"6
;<"$!"34-"$"56"
789"$)"6
;<"$!"34-"$!"56
"
Figure 19. MTS assay for cytotoxicity of Rac.Viability of cells exposed to various amounts of HRV 1A, HRV 16, and Rac inhibitor NSC23766 was assessed by MTS assay. HeLa cells were exposed to no virus, HRV 1A, or HRV 16. For the virus treatments, multiplicities of infection of 0.1, 1, and 10 were used. The Rac inhibitor NSC23766 either was not applied or was applied at 0.1 µM, 1 µM, and 10 µM concentrations. Absorbance at 490 nm following application of MTS reagent was used as a measure of cell viability. Error bars represent standard error (N = 3).

not affect cell viability by repeating this experiment using the A549 cell line and obtaining
similar results (data not shown). In addition, we directly observed (via microscopy) that Rac
inhibition did not affect the viability of macrophages exposed to HRV (data not shown).
DiscussionRecent studies have shown that the Ras superfamily small G-proteins may play a role in
viral replication (Brazzoli and others 2008) and in the inflammatory response (Bagrodia and
others 1995), and studies specific to HRV demonstrate that the small G-protein RhoA is
important to HRV-induced cell signaling in epithelial cells (Dumitru, Dreschers, Gulbins 2006).
We hypothesized that the RhoA-related small G-proteins Rac and Cdc42 as well as the G-protein
Ras would be involved in HRV replication or proinflammatory signaling within epithelial cells.
Interestingly, Rac, Cdc42, and Ras were not activated in epithelial cells exposed to HRV.
However, this result is consistent with previous findings, which indicate that Rac is not important
to p38 MAPK activation in HeLa cells (Dumitru, Dreschers, Gulbins 2006).
Although epithelial cells are the most studied cells that may be involved in HRV-induced
secretion of cytokines, a number of studies indicate that the macrophage is also important in
HRV infection and proinflammatory receptor-mediated signal transduction (Barnes, Chung, Page
1998; Sen 2001). While human macrophages are not productively infected with HRV (Gern and
others 1996), they are known to express the HRV receptors ICAM-1 and LDL-R (Hall and others
2005) and to release inflammatory cytokines including MCP-1 (Hall and others 2005) and IP-10
(Korpi-Steiner and others 2006) in response to HRV exposure. We observed Rac activation in
human macrophages responding to HRV (Figure 12, 13), whereas none of the G-proteins Rac,
Cdc42, and Ras was activated during the response of epithelial cells to HRV exposure (see
54

Appendix I). This result suggests an important and specific role for macrophages in the
inflammatory rhinoviral response. Interestingly, this finding is not consistent with a previous
study, which attributed the inflammatory signal transduction capacity of the HRV receptor
ICAM-1 to the Ras effector molecule Raf-1 (Blaber and others 2003). However, in this previous
study, ICAM-1 was stimulated with antibody rather than HRV, so the results may not be directly
comparable. Nonetheless, our finding that Ras is not activated in response to HRV ICAM-1
ligation in either epithelial cells or macrophages suggests a Ras-independent inflammatory
signaling mechanism within these cells.
HRV is often classified according to which of two cellular receptors a particular serotype
of HRV will bind (Greenberg 2003). According to this classification scheme, the major group of
HRV binds to the intercellular adhesion molecule 1 (ICAM-1) receptor (Greve and others 1989),
whereas the minor group of HRV binds to the low density lipoprotein (LDL) receptor (Hofer and
others 1994). Previous researchers have often chosen to generally examine only signaling
responses to major-group rhinovirus, neglecting the minor-group viruses (Griego and others
2000; Hall and others 2005; Spurrell and others 2005). Although one might assume that a
“general response” to major- and minor-group HRV would occur, our work with the small G-
protein Rac suggests otherwise. When we examined Rac activation kinetics in macrophages
exposed to either HRV 16, a major-group virus, or HRV 1A, a minor-group virus, we observed
that Rac activation occurred with different kinetics depending on which of the HRV types
provided the stimulus (Figure 12, 13). This observation of differential kinetics supports the
notion that signaling pathways leading from the ICAM-1 receptor binding major-group HRV and
the LDL receptor binding minor-group HRV may differ.
55

Additionally, pharmacological inhibition of the G-protein Rac suppressed
proinflammatory signaling involving phosphorylation of the MAPK p38 (Figure 14). The
activation of p38, a mitogen-activated protein kinase (MAPK), has previously been associated
with the production of inflammatory cytokines such as MCP-1 in macrophages exposed to HRV
(Hall and others 2005). Consistent with this previous work, we determined that pharmacological
inhibition of the small G-protein Rac attenuated the release of MCP-1 (Figure 15). These results
implicate Rac in the proinflammatory signaling pathways initiated in macrophages upon
exposure to HRV.
Previous work has also noted that activation of the p38/MCP-1 pathway in blood
monocyte-derived macrophages exposed to HRV is dependent on the serum type used to culture
the macrophages. Indeed, only macrophages cultured in human serum, not bovine serum, were
observed to phosphorylate p38 and release MCP-1 in response to HRV (Schuler and Lawrence
University. 2009). Notably, culturing macrophages in human serum provides a better model of
alveolar macrophages in the asthmatic lung, as the increased endothelial permeability that occurs
in the asthmatic lung results in the increased presence of human serum there (Velden and Versnel
1998). Because human serum contains human antibodies, or immunoglobulin (Ig), whereas
bovine serum does not, a likely hypothesis to explain the dependence of MCP-1 release on serum
type is that activation of the p38/MCP-1 cascade is dependent on stimulation of macrophage Ig
receptors by the Ig present in human serum. Support for this hypothesis has come from the result
that culturing macrophages in bovine serum enriched with human IgG (and thus capable of
stimulating the IgG receptor on macrophages, FcγR) is able to recover the p38/MCP-1 activation
normally observed in human serum upon exposure to HRV (Schuler and Lawrence University.
56

2009). However, although Rac is known to be an upstream regulator of the p38/MCP-1 cascade,
it has remained unclear whether Rac activation in response to HRV exposure would differ
between bovine serum-cultured and human-serum cultured macrophages. When we assayed Rac
activation in bovine and human serum-cultured macrophages exposed to HRV, we observed no
difference in Rac activation kinetics between bovine and human serum-cultured macrophages
(Figure 12, 13). Thus, any regulation of the p38/MCP-1 cascade that is serum dependent does not
occur at the level of the G-proteins or above, and any regulation of the p38/MCP-1 cascade that
is controlled by human IgG binding to FcγR either occurs through a pathway unrelated to Rac or
occurs through a downstream interaction.
Although pharmacological inhibition of the small G-protein Rac successfully blocked the
phosphorylation of the MAPK p38 (Figure 14) and the release of the proinflammatory cytokine
MCP-1 (Figure 15) following HRV exposure to macrophages, Rac inhibition was not successful
in blocking the release of the proinflammatory cytokine RANTES (Figure 16). RANTES seems
to be important to the inflammatory airway environment observed during asthma exacerbations,
as RANTES is chemotactic for eosinophils, which have been implicated in the airway
remodeling that takes place during asthma exacerbation (Nissim Ben Efraim and Levi-Schaffer
2008). MCP-1 and RANTES levels in the bronchoalveolar lavage fluid of asthmatics are
elevated well above the baseline levels observed in normal individuals (Alam and others 1996).
Furthermore, the release of MCP-1 and RANTES has been associated with bronchial
hyperresponsiveness and with leukocyte recruitment to the airway (Conti and DiGioacchino
2001). That Rac inhibition has no noticeable effect on RANTES release makes sense given that
RANTES-induced chemotaxis is known to be Rac dependent in macrophages (Weiss-Haljiti and
57

others 2004). In other words, although Rac inhibition does not prevent RANTES release, Rac
inhibition likely does make alveolar macrophages RANTES insensitive, thereby attenuating the
runaway proinflammatory signaling associated with asthma severity, to the extent that this
proinflammatory intercellular signaling is attributable to RANTES.
The insensitivity of RANTES regulation to Rac inhibition may also be related to the
effects of Rac inhibition on TLR3 regulation and the interferon response. HRV exposure
following Rac inhibition led to a substantial upregulation of TLR3 (Figure 17) and a noticeable
upregulation of IFN-α (Table 1). Thus, while Rac inhibition has apparent anti-inflammatory
properties in the context of the p38/MCP-1 cascade, Rac inhibition may also have some
paradoxic proinflammatory activity, enhancing the response of the TLR3/IFN-α cascade.
Assuming that there is some cross-talk between these two signaling pathways, Rac inhibition
may simultaneously induce both upregulation and downregulation of the release of RANTES,
leading to a net observation of RANTES regulation being insensitive to Rac inhibition. Such an
observation is consistent with the results here described (Figure 16). In order to explore this
hypothesis further, it will be necessary to replicate the results of the interferon and TLR3 qPCR
assays with a broader sample base and to further characterize the dependence of RANTES on
p38- and TLR3-mediated signaling.
Epithelial cells remain the primary focus of many researchers studying HRV-induced
exacerbation of asthma (Peters-Golden 2004), and it is widely understood that epithelial cells are
capable of contributing to the proinflammatory environment initiated in the airway of an
asthmatic during HRV infection (Levine 1995). Based on our results, the signaling events leading
to the release of cytokines from these cells would appear to be mechanistically different from the
58

signaling events leading to their release from macrophages. Notwithstanding that the role of Rac
in HRV-induced macrophage signaling presents one significant mechanistic difference between
inflammatory signaling cascades in these two cell types, further study is needed to clarify the
differences between epithelial cell and macrophage HRV-induced signaling cascades.
Future WorkThere is still much to be learned about the role of the Ras-superfamily G-proteins in the
inflammatory environment of the HRV-exposed airway.
Because the Ras superfamily is so large, there are still many G-proteins that have not
been examined in this context, such as the G-protein Rho, a protein that is closely related to the
Rac protein implicated here.
In addition, the extent to which Rac is truly proinflammatory remains somewhat unclear.
Although Rac is clearly involved in the proinflammatory p38/MCP-1 signaling cascade, Rac
activation apparently exerts a suppressive effect on the TLR3/IFN-α pathway. Thus, more
experiments are needed to determine the net effect of Rac inhibition on the proinflammatory
microenvironment found in the HRV-exposed lung. A logical next step in characterizing Rac’s
role might be to perform co-culture experiments, in which macrophages are cultured in
combination with epithelial cells and similar combinations of HRV and Rac inhibitor are applied.
These experiments may more accurately model the airway, where epithelial cells and
macrophages are able to signal to each other, and may provide clearer evidence for Rac’s
proinflammatory and anti-inflammatory roles.
Because many of the manipulations described here involve the use of pharmacological
inhibition, which may have some nonspecific effects, it will be necessary to confirm these results
59

using other techniques of Rac inhibition. In particular, performing experiments in which Rac is
inhibited using a dominant-negative or RNAi approach and obtaining similar results to those
demonstrated here would provide clear evidence that the Rac inhibitor NSC23766 has acted
specifically to inhibit Rac and no other signaling molecules.
Should Rac inhibition continue to prove useful in attenuating the macrophage
inflammatory response to HRV, developing a Rac inhibitor into a drug that may be prescribed as
a treatment for HRV-induced exacerbations of asthma may be warranted. Although delivering
Rac inhibitor systemically would likely have catastrophic side effects due to Rac’s ubiquitous
and important roles in cell signaling cascades, an inhaled form of the drug would likely have
only localized effects (Lipworth 1996), acting on the lung epithelium and airway immune cells,
such as the alveolar macrophages. Similar localized effectiveness has been demonstrated for
many of the asthma medications currently available in inhaled form (Lipworth 1996). Because
asthma is a disease that is localized to the airway, delivering Rac inhibitor via an inhaled vehicle
should prove very effective; however, it will still be necessary to demonstrate drug safety and
efficacy. Preliminary drug trials for safety might be performed in a mouse model, while
preliminary drug trials for efficacy would likely still need to be performed in human volunteers,
under highly regulated conditions, as there is currently no good mouse model of HRV-induced
asthma (Tuthill and others 2003).
60

ReferencesAkbar H, Cancelas J, Williams DA, Zheng J, Zheng Y. 2006. Rational design and applications of a rac
GTPase–Specific small molecule inhibitor. In: Methods in enzymology. William E. Balch, Channing J. Der,and Alan Hall, editor. Academic Press. 554 p.
Alam R, York J, Boyars M, Stafford S, Grant JA, Lee J, Forsythe P, Sim T, Ida N. 1996. Increased MCP-1, RANTES, and MIP-1alpha in bronchoalveolar lavage fluid of allergic asthmatic patients Am J Respir Crit Care Med 153(4 Pt 1):1398-404.
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-[kappa]B by toll-like receptor 3. Nature 413(6857):732-8.
Arruda E, Pitkaranta A, Witek T,Jr, Doyle C, Hayden F. 1997. Frequency and natural history of rhinovirus infections in adults during autumn. J Clin Microbiol 35(11):2864-8.
Arruda E, Boyle TR, Winther B, Pevear DC, Gwaltney JM,Jr, Hayden FG. 1995. Localization of human rhinovirus replication in the upper respiratory tract by in situ hybridization J Infect Dis 171(5):1329-33.
Bagrodia S, Dérijard B, Davis RJ, Cerione RA. 1995. Cdc42 and PAK-mediated signaling leads to jun kinase and p38 mitogen-activated protein kinase activation. J Biol Chem 270(47):27995-8.
Balachandran S, Porosnicu M, Barber GN. 2001. Oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, ras, or myc function and involves the induction of apoptosis. J Virol 75(7):3474-9.
Bals R and Hiemstra PS. 2004. Innate immunity in the lung: How epithelial cells fight against respiratory pathogens. Eur Respir J 23(2):327-33.
Bardin P, Johnston S, Sanderson G, Robinson B, Pickett M, Fraenkel D, Holgate S. 1994. Detection of rhinovirus infection of the nasal mucosa by oligonucleotide in situ hybridization. Am J Respir Cell Mol Biol 10(2):207-13.
Barnes PJ, Chung KF, Page CP. 1998. Inflammatory mediators of asthma: An update. Pharmacol Rev 50(4):515-96.
Bella J, Kolatkar PR, Marlor CW, Greve JM, Rossmann MG. 1998. The structure of the two amino-terminal domains of human ICAM-1 suggests how it functions as a rhinovirus receptor and as an LFA-1 integrin ligand. Proceedings of the National Academy of Sciences of the United States of America 95(8):4140-5.
Berkman N, Krishnan V, Gilbey T, Newton R, O'Connor B, Barnes P, Chung K. 1996. Expression of RANTES mRNA and protein in airways of patients with mild asthma. Am J Respir Crit Care Med 154(6):1804-11.
Beutler B. 2004. Inferences, questions and possibilities in toll-like receptor signalling. Nature 430(6996):257-63.
Blaber R, Stylianou E, Clayton A, Steadman R. 2003. Selective regulation of ICAM-1 and RANTES gene expression after ICAM-1 ligation on human renal fibroblasts. J Am Soc Nephrol 14(1):116-27.
Bokoch GM. 2005. Regulation of innate immunity by rho GTPases. Trends Cell Biol 15(3):163-71.
61

Brabec M, Baravalle G, Blaas D, Fuchs R. 2003. Conformational changes, plasma membrane penetration, and infection by human rhinovirus type 2: Role of receptors and low pH. J Virol 77(9):5370-7.
Brazzoli M, Bianchi A, Filippini S, Weiner A, Zhu Q, Pizza M, Crotta S. 2008. CD81 is a central regulator of cellular events required for HCV infection of human hepatocytes. J Virol .
Brown JL, Stowers L, Baer M, Trejo J, Coughlin S, Chant J. 1996. Human Ste20 homologue hPAK1 links GTPases to the JNK MAP kinase pathway. 6(5):598-605.
Burridge K. 2004. Rho and rac take center stage. Cell 116(2):167.
Chapman K and Hall DJ. 2005. Quantifying the activation of the small molecular weight G-protein ras. Biochemistry and Molecular Biology Education 33(1):22-7.
Chomarat P, Dantin C, Bennett L, Banchereau J, Palucka AK. 2003. TNF skews monocyte differentiation from macrophages to dendritic cells. J Immunol 171(5):2262-9.
Conti P and DiGioacchino M. 2001. MCP-1 and RANTES are mediators of acute and chronic inflammation Allergy Asthma Proc 22(3):133-7.
Daniels TF, Killinger KM, Michal JJ, Wright RW,Jr, Jiang Z. 2009. Lipoproteins, cholesterol homeostasis and cardiac health. Int J Biol Sci 5(5):474-88.
DeTulleo L and Kirchhausen T. 1998. The clathrin endocytic pathway in viral infection EMBO J 17(16):4585-93.
Dove A. PICO - Polio Life Cycle [Internet]. New York: Columbia University Department of Microbiology; c2002 [cited 2010 5/10/2010]. Available from: http://microbiology.columbia.edu/pico/Chapters/Cellular.html .
Dumitru CA, Dreschers S, Gulbins E. 2006. Rhinoviral infections activate p38MAP-kinases via membrane rafts and RhoA. Cell Physiol Biochem 17(3-4):159-66.
Etienne-Manneville S and Hall A. 2002. Rho GTPases in cell biology Nature 420(6916):629-35.
Fixman ED, Stewart A, Martin JG. 2007. Basic mechanisms of development of airway structural changes in asthma. Eur Respir J 29(2):379-89.
Folkard S, Westwick J, Millar A. 1997. Production of interleukin-8, RANTES and MCP-1 in intrinsic and extrinsic asthmatics. Eur Respir J 10(9):2097-104.
Fogelman AM, Van Lenten BJ, Warden C, Haberland ME, Edwards PA. 1988. Macrophage lipoprotein receptors. Journal of Cell Science.Supplement 9:135-49.
Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. 2010. Development of monocytes, macrophages, and dendritic cells. Science 327(5966):656-61.
Gern JE, Dick EC, Lee WM, Murray S, Meyer K, Handzel ZT, Busse WW. 1996. Rhinovirus enters but does not replicate inside monocytes and airway macrophages. J Immunol 156(2):621-7.
Gern JE and Busse WW. 1999. Association of rhinovirus infections with asthma. Clin Microbiol Rev 12(1):9-18.
62

Gern J, Vrtis R, Kelly E, Dick E, Busse W. 1996. Rhinovirus produces nonspecific activation of lymphocytes through a monocyte-dependent mechanism. J Immunol 157(4):1605-12.
Greenberg SB. 2003. Respiratory consequences of rhinovirus infection Arch Intern Med 163(3):278.
Greve JM, Davis G, Meyer AM, Forte CP, Yost SC, Marlor CW, Kamarck ME, McClelland A. 1989. The major human rhinovirus receptor is ICAM-1. Cell 56(5):839-47.
Griego SD, Weston CB, Adams JL, Tal-Singer R, Dillon SB. 2000. Role of p38 mitogen-activated protein kinase in rhinovirus-induced cytokine production by bronchial epithelial cells. J Immunol 165(9):5211-20.
Haberzettl P, Schins RPF, Hohr D, Wilhelmi V, Borm PJA, Albrecht C. 2008. Impact of the fc{gamma}II-receptor on quartz uptake and inflammatory response by alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 294(6):L1137-1148.
Haghighat A, Svitkin Y, Novoa I, Kuechler E, Skern T, Sonenberg N. 1996. The eIF4G-eIF4E complex is the target for direct cleavage by the rhinovirus 2A proteinase. J Virol 70(12):8444-50.
Hall A. 1998. Rho GTPases and the actin cytoskeleton. Science 279(5350):509-14.
Hall DJ, Bates ME, Guar L, Cronan M, Korpi N, Bertics PJ. 2005. The role of p38 MAPK in rhinovirus-induced monocyte chemoattractant protein-1 production by monocytic-lineage cells. J Immunol 174(12):8056-63.
Hewson CA, Jardine A, Edwards MR, Laza-Stanca V, Johnston SL. 2005. Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J Virol 79(19):12273-9.
Hizawa N, Yamaguchi E, Konno S, Tanino Y, Jinushi E, Nishimura M. 2002. A functional polymorphism in the RANTES gene promoter is associated with the development of late-onset asthma. Am J Respir Crit Care Med 166(5):686-90.
Hofer F, Gruenberger M, Kowalski H, Machat H, Huettinger M, Kuechler E, Blaas D. 1994. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc Natl Acad Sci USA 91:1839-42.
Holland J and Owens T. 1997. Signaling through intercellular adhesion molecule 1 (ICAM-1) in a B cell lymphoma line. Journal of Biological Chemistry 272(14):9108-12.
Holtzman MJ, Morton JD, Shornick LP, Tyner JW, O'Sullivan MP, Antao A, Lo M, Castro M, Walter MJ. 2002. Immunity, inflammation, and remodeling in the airway epithelial barrier: Epithelial-viral-allergic paradigm. Physiol Rev 82(1):19-46.
Homer RJ and Elias JA. 2005. Airway remodeling in asthma: Therapeutic implications of mechanisms. Physiology 20(1):28-35.
Hu X, Chakravarty SD, Ivashkiv LB. 2008. Regulation of interferon and toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms Immunol Rev 226:41-56.
Huber M, Watson KA, Selinka H, Carthy CM, Klingel K, McManus BM, Kandolf R. 1999. Cleavage of RasGAP and phosphorylation of mitogen-activated protein kinase in the course of coxsackievirus B3
63

replication. J Virol 73(5):3587-94.
Johnston SL. 1995. Natural and experimental rhinovirus infections of the lower respiratory tract. American Journal of Respiratory and Critical Care Medicine 152(4 Pt 2):S46-52.
Johnston SL. 2007. Innate immunity in the pathogenesis of virus-induced asthma exacerbations. Proc Am Thorac Soc 4(3):267-70.
Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, Symington P, O'Toole S, Myint SH, Tyrrell DAJ, et al. 1995. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ 310(6989):1225-9.
Kolokoltsova OA, Domina AM, Kolokoltsov AA, Davey RA, Weaver SC, Watowich SJ. 2008. Alphavirus production is inhibited in neurofibromin 1-deficient cells through activated RAS signalling. Virology 377(1):133-42.
Korpi-Steiner NL, Bates ME, Lee W, Hall DJ, Bertics PJ. 2006. Human rhinovirus induces robust IP-10 release by monocytic cells, which is independent of viral replication but linked to type I interferon receptor ligation and STAT1 activation. J Leukoc Biol 80(6):1364-74.
Laza-Stanca V, Stanciu LA, Message SD, Edwards MR, Gern JE, Johnston SL. 2006. Rhinovirus replication in human macrophages induces NF-{kappa}B-dependent tumor necrosis factor alpha production. J Virol 80(16):8248-58.
Lestavel S and Fruchart JC. 1994. Lipoprotein receptors. Cell Mol Biol 40(4):461-81.
Levine SJ. 1995. Bronchial epithelial cell-cytokine interactions in airway inflammation J Investig Med 43(3):241-9.
Levine SJ and Wenzel SE. 2010. Narrative review: The role of Th2 immune pathway modulation in the treatment of severe asthma and its phenotypes. Ann Intern Med 152(4):232-7.
Lipworth BJ. 1996. Pharmacokinetics of inhaled drugs Br J Clin Pharmacol 42(6):697-705.
Lohmann-Matthes M, Steinmuller C, Franke-Ullmann G. 1994. Pulmonary macrophages. Eur Respir J 7(9):1678-89.
Magnan AO, Mely LG, Camilla CA, Badier MM, Montero-Julian FA, Guillot CM, Casano BB, Prato SJ, Fert V, Bongrand P, et al. 2000. Assessment of the Th1/Th2 paradigm in whole blood in atopy and asthma . increased IFN-gamma -producing CD8+ T cells in asthma. Am J Respir Crit Care Med 161(6):1790-6.
Martinez FO, Sica A, Mantovani A, Locati M. 2008. Macrophage activation and polarization. Front Biosci 13:453-61.
May P, Bock HH, Herz J. 2003. Integration of endocytosis and signal transduction by lipoprotein receptors. Sci STKE 2003(176):pe12.
Message SD and Johnston SL. 2004. Host defense function of the airway epithelium in health and disease: Clinical background. J Leukoc Biol 75(1):5-17.
Mills PR, Davies RJ, Devalia JL. 1999. Airway epithelial cells, cytokines, and pollutants. Am J Respir Crit Care Med 160(5 Pt 2):S38-43.
64

Mitchell T, Lo A, Logan MR, Lacy P, Eitzen G. 2008. Primary granule exocytosis in human neutrophils is regulated by rac-dependent actin remodeling. Am J Physiol Cell Physiol 295(5):C1354-1365.
Mogensen TH and Paludan SR. 2001. Molecular pathways in virus-induced cytokine production. Microbiol Mol Biol Rev 65(1):131-50.
Nissim Ben Efraim AH and Levi-Schaffer F. 2008. Tissue remodeling and angiogenesis in asthma: The role of the eosinophil Ther Adv Respir Dis 2(3):163-71.
Normal versus asthmatic bronchiole [Internet] [cited 2010 5/10/2010]. Available from: http://adam.about.com/encyclopedia/Normal-versus-asthmatic-bronchiole.htm
Olszewska-Pazdrak B, Casola A, Saito T, Alam R, Crowe SE, Mei F, Ogra PL, Garofalo RP. 1998. Cell-specific expression of RANTES, MCP-1, and MIP-1alpha by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J Virol 72(6):4756-64.
Palmenberg AC, Spiro D, Kuzmickas R, Wang S, Djikeng A, Rathe JA, Fraser-Liggett CM, Liggett SB. 2009. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 324(5923):55-9.
Papadopoulos N, Bates P, Bardin P, Papi A, Leir S, Fraenkel D, Meyer J, Lackie P, Sanderson G, Holgate S, et al. 2000. Rhinoviruses infect the lower airways. J Infect Dis 181(6):1875-84.
Papi A and Johnston SL. 1999. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-kappa B-mediated transcription. J Biol Chem 274(14):9707-20.
Peter A.B. Wark, Bucchieri F, Johnston SL, Gibson PG, Hamilton L, Mimica J, Zummo G, Holgate ST, Attia J, Thakkinstian A, and others. 2007. IFN-gamma-induced protein 10 is a novel biomarker of rhinovirus-induced asthma exacerbations. J Allergy Clin Immunol 120(3):586-93.
Peters-Golden M. 2004. The alveolar macrophage: The forgotten cell in asthma. Am J Respir Cell Mol Biol 31(1):3-7.
Pifat-Mrzljak G. 1989. [Macrophage lipoprotein metabolism]. Liječnički Vjesnik 111(6-7):224-7.
Rayter SI, Woodrow M, Lucas SC, Cantrell DA, Downward J. 1992. p21ras mediates conrol of IL-2 gene promoter function in T cell activation. EMBO 11(12):4549-56.
Ridley AJ. 2001. Rho family proteins: Coordinating cell responses Trends Cell Biol 11(12):471.
Rose CE,Jr, Sung SS, Fu SM. 2003. Significant involvement of CCL2 (MCP-1) in inflammatory disorders of the lung Microcirculation 10(3-4):273-88.
Rossmann M. 1989. The canyon hypothesis. hiding the host cell receptor attachment site on a viral surface from immune surveillance. J Biol Chem 264(25):14587-90.
Rotbart HA and Hayden FG. 2000. Picornavirus infections: A primer for the practitioner. Arch Fam Med 9(9):913-20.
Ryan M and Flint M. 1997. Virus-encoded proteinases of the picornavirus super-group. J Gen Virol 78(4):699-723.
65

Salvi SS, Suresh Babu K, Holgate ST. 2001. Is asthma really due to a polarized T cell response toward a helper T cell type 2 phenotype? Am J Respir Crit Care Med 164(8):1343-6.
Saraiva M and O'Garra A. 2010. The regulation of IL-10 production by immune cells. Nature Reviews Immunology 10(3):170-81.
Sarkany Z and Polgar L. 2003. The unusual catalytic triad of poliovirus protease 3C. Biochemistry 42(2):516-22.
Schuler B and Lawrence University. 2009. Differential MAPK and cytokine response to human rhinovirus by human macrophages is a function of receptor-mediated signal transduction / Bryce Schuler.
Sen GC. 2001. Viruses and interferons Annu Rev Microbiol 55:255-81.
Spurrell JCL, Wiehler S, Zaheer RS, Sanders SP, Proud D. 2005. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol 289(1):L85-95.
Stanway G. 1990. Structure, function and evolution of picornaviruses J Gen Virol 71 ( Pt 11)(Pt 11):2483-501.
Subauste MC, Jacoby DB, Richards SM, Proud D. 1995. Infection of a human respiratory epithelial cell line with rhinovirus. induction of cytokine release and modulation of susceptibility to infection by cytokine exposure J Clin Invest 96(1):549-57.
Suzuki T, Yamaya M, Kamanaka M, Jia YX, Nakayama K, Hosoda M, Yamada N, Nishimura H, Sekizawa K, Sasaki H. 2001. Type 2 rhinovirus infection of cultured human tracheal epithelial cells: Role of LDL receptor. Am J Physiol Lung Cell Mol Physiol 280(3):L409-420.
Szalai C, Kozma GT, Nagy A, Bojszko A, Krikovszky D, Szabo T, Falus A. 2001. Polymorphism in the gene regulatory region of MCP-1 is associated with asthma susceptibility and severity J Allergy Clin Immunol 108(3):375-81.
Takai Y, Sasaki T, Matozaki T. 2001. Small GTP-binding proteins. Physiol Rev 81(1):153-208.
Takeda K and Akira S. 2005. Toll-like receptors in innate immunity. Int Immunol 17(1):1-14.
Tuthill TJ, Papadopoulos NG, Jourdan P, Challinor LJ, Sharp NA, Plumpton C, Shah K, Barnard S, Dash L, Burnet J, et al. 2003. Mouse respiratory epithelial cells support efficient replication of human rhinovirus. J Gen Virol 84(10):2829-36.
Velden VH and Versnel HF. 1998. Bronchial epithelium: Morphology, function and pathophysiology in asthma. Eur Cytokine Netw 9(4):585-97.
Weinberg RA. 2007. The biology of cancer. New York: Garland Science.
Weiss-Haljiti C, Pasquali C, Ji H, Gillieron C, Chabert C, Curchod M, Hirsch E, Ridley AJ, van Huijsduijnen RH, Camps M, et al. 2004. Involvement of phosphoinositide 3-kinase γ, rac, and PAK signaling in chemokine-induced macrophage migration. Journal of Biological Chemistry 279(41):43273-84.
Wennerberg K, Rossman KL, Der CJ. 2005. The ras superfamily at a glance. J Cell Sci 118(5):843-6.
66

Winther B, Arruda E, Witek TJ, Marlin SD, Tsianco MM, Innes DJ, Hayden FG. 2002. Expression of ICAM-1 in nasal epithelium and levels of soluble ICAM-1 in nasal lavage fluid during human experimental rhinovirus infection. Arch Otolaryngol Head Neck Surg 128(2):131-6.
Zhu L, Lee P, Lee W, Zhao Y, Yu D, Chen Y. 2009. Rhinovirus-induced major airway mucin production involves a novel TLR3-EGFR-dependent pathway. Am J Respir Cell Mol Biol 40(5):610-9.
67

Appendix I: Supplemental Figures
68
HRV 1A
2 5 10 15 30 WCE
2 5 10 15 30 WCE
HRV 16
Figure A1. Rac activation following HeLa epithelial cell exposure to HRV.Activated Rac was pulled down and assayed by SDS-PAGE and immunoblot following exposure of HeLa cells to HRV 1A (Panel A) and HRV 16 (Panel B) at an MOI of 10. Each blot is representative of five independent experiments. “WCE” designates whole cell extract, which serves as a positive control.
HRV 1A
2 5 10 15 30 WCE
2 5 10 15 30 WCE
HRV 16
Ras
Ras
Figure A2. Ras activation following HeLa epithelial cell exposure to HRV.Activated Ras was pulled down and assayed by SDS-PAGE and immunoblot following exposure of HeLa cells to HRV 1A (Panel A) and HRV 16 (Panel B) at an MOI of 10. Each blot is representative of five independent experiments. “WCE” designates whole cell extract, which serves as a positive control.
HRV 1A
2 5 10 15 30 WCE
2 5 10 15 30 WCE
HRV 16
Figure A3. Rac activation following A549 epithelial cell exposure to HRV.Activated Rac was pulled down and assayed by SDS-PAGE and immunoblot following exposure of A549 cells to HRV 1A (Panel A) and HRV 16 (Panel B) at an MOI of 10. Each blot is representative of five independent experiments. “WCE” designates whole cell extract, which serves as a positive control.

69
Figure A5. Cdc42 activation following macrophage exposure to HRV.Activated Cdc42 was pulled down and assayed by SDS-PAGE and immunoblot following exposure of blood monocyte-derived macrophages to HRV 1A (Panel A) and HRV 16 (Panel B) at an MOI of 10. Each blot is representative of five independent experiments. “WCE” designates whole cell extract, which serves as a positive control.
Figure A4. Ras activation following A549 epithelial cell exposure to HRV.Activated Ras was pulled down and assayed by SDS-PAGE and immunoblot following exposure of A549 cells to HRV 1A (Panel A) and HRV 16 (Panel B) at an MOI of 10. Each blot is representative of five independent experiments. “WCE” designates whole cell extract, which serves as a positive control.
Ras
HRV 1A
2 5 10 15 30 WCE
2 5 10 15 30 WCE
HRV 16
Ras
HRV 1A
2 5 10 15 30 WCE
2 5 10 15 30 WCE
HRV 16
Cdc42
Cdc42
HRV 1A
2 5 10 15 30 WCE
2 5 10 15 30 WCE
HRV 16
Ras
Ras
Figure A6. Ras activation following macrophage exposure to HRV.Activated Ras was pulled down and assayed by SDS-PAGE and immunoblot following exposure of blood monocyte-derived macrophages to HRV 1A (Panel A) and HRV 16 (Panel B) at an MOI of 10. Each blot is representative of five independent experiments. “WCE” designates whole cell extract, which serves as a positive control.

Appendix II: Manuscript Submitted for Publication
(Submitted for publication to the journal Innate Immunity 31 March 2010)
The role of the Ras superfamily small G-proteins in the proinflammatory environment of rhinovirus-exposed monocytic-lineage cells
Michael Schreiber*, Bryce Schuler* and David J. Hall*
*Department of Chemistry, Lawrence University, Appleton, WI 54911
Running title: Rac activation by rhinovirus in monocyte-lineage cells
Keywords: CCL2, CCL5, MCP-1, p38, Rac
Corresponding Author:
David J. Hall, Department of Chemistry, Lawrence University, 711 East Boldt Way, Appleton,
WI 54911
Phone: 920-993-6173
Fax: 920-832-6962
Email: [email protected]
70

AbstractRhinoviral infections are a major cause of asthma exacerbations, and though productive
rhinovirus infection occurs predominantly in the bronchial epithelial cells of the upper airway,
monocytic-lineage cells are implicated in establishing the inflammatory microenvironment
observed during rhinoviral asthma exacerbation. It has remained unclear whether small G-protein
activation plays a role in establishing this inflammatory microenvironment. The small molecular-
weight G-proteins are known to be activated in a variety of cell types upon exposure to a range
of viruses. However, it is unclear if small G-protein activation during viral exposure is a
byproduct of receptor attachment, is important for viral replication, or is playing a role in
mediating the inflammatory response. Human rhinovirus (HRV) is unique in that nearly
genetically identical viruses bind the intercellular adhesion molecule (ICAM)-1 or low-density
lipoprotein (LDL) receptors. Furthermore, HRV is capable of eliciting a signaling response in
both epithelial cells, where productive HRV infection occurs, and in macrophages, where there is
an immune response without productive infection. In this study, Rac, cell division cycle (cdc)42,
and Ras were neither activated in epithelial cell lines nor capable of hindering viral replication.
We demonstrated that the small G-protein Rac is differentially activated when major- and minor-
group HRV bind to macrophage ICAM-1 or LDL receptors. Inhibition of Rac in macrophages
attenuated the activation of the stress kinase p38 and the release of the proinflammatory cytokine
CCL2, but inhibition of Rac did not affect the release of the proinflammatory cytokine CCL5.
These findings suggest that Rac is important in establishing the inflammatory microenvironment
that is initiated in the human airway upon exposure to rhinovirus.
71

IntroductionAsthma exacerbations are most often triggered by upper respiratory tract viral infections,
infections for which the primary etiologic agent is human rhinovirus (HRV).1 Thus, HRV is
responsible for both the majority of common cold infections and the majority of asthma
exacerbations.2 Although the association of HRV with asthma exacerbation is well documented,
the molecular basis for HRV-induced asthma exacerbation is not well understood.
HRV is a member of the virus family picornaviridae, a family of positive-sense RNA
viruses with naked icosahedral capsids measuring approximately 20-30 nm wide. Other salient
members of the picornaviridae family include the enteroviruses, such as poliovirus and the
Coxsackie viruses. HRV serotypes are phylogenetically divided into three groups, HRVA,
HRVB, and HRVC, based on sequence comparison.3 However, HRV can also be subdivided into
groups based on which of two cellular receptors a particular HRV serotype will bind.4 Major-
group HRV, such as the HRV serotype 16, binds the intercellular adhesion molecule 1 (ICAM-1)
receptor,5 whereas minor-group HRV, such as the HRV serotype 1A, binds the low-density
lipoprotein receptor (LDL-R).6
HRV is capable of filtering into the lower airway and triggering a variety of receptor-
mediated signal transduction pathways. Within the lower airway, HRV contacts epithelial cells
and alveolar macrophages, the predominant immune cells present in the lung.7,8 Both of these
cell types possess receptors for HRV,9,10 and both are capable of proinflammatory signaling.11,12
HRV productively infects epithelial cells,13 while macrophages are not a site of productive HRV
replication.14,15 However, macrophages are known to be important in establishing a
proinflammatory microenvironment in the lung.12 Previous studies link HRV receptor-mediated
signal transduction to a number of biological endpoints associated with inflammation. Although
72

epithelial cells are able to produce some of these biological endpoints, many of them are
produced predominately by innate immune cells such as the alveolar macrophages.16 These
biological endpoints include activation of inflammation-associated transcription factors such as
the nuclear factor κB (NF-κB) and release of inflammatory cytokines such as interleukin-6
(IL-6), interleukin-8 (IL-8), CCL2, IL-1β and TNF-α.17,18,10,19
Though differences between signaling pathways induced by major- and minor-group
HRV may exist, these differences have not been thoroughly explored, particularly in
macrophages. As well, many previous explorations have focused on describing signaling at the
levels of MAPK phosphorylation,10 transcription factor activation,15 or inflammatory cytokine
release.16 Thus, early signaling events associated with HRV receptor-mediated signal
transduction and viral replication have not yet been well characterized, either in epithelial cells
or macrophages. Because these early signaling events remain poorly understood, we have chosen
to investigate a role in HRV signaling pathways for the small molecular weight G-proteins,
focusing on the macrophage cell population because of its well-demonstrated inflammatory
capacity.
Many of the small G-proteins and their associated downstream signaling cascades are
implicated in viral life cycles, particularly within epithelial cells. The Rho GTPases Rac and
Cdc42, for example, are implicated in the cytopathogenicity of hepatitis C virus.20 Furthermore,
Ras activation is known to be modulated by vesicular stomatitis virus in tumor cells and is
important to the replication and pathogenesis of the enteroviruses.21,22 The Ras-superfamily G-
protein Arf is important in the infectivity of coxsackievirus type B3 in HeLa cells and is required
for poliovirus RNA replication.23,24,25 The Rho-family G-protein RhoA is activated in HeLa cells
73

responding to HRV.26 In addition, Major group B coxsackievirus internalization is regulated by
an occludin-dependent macropinocytosis mechanism that requires the activity of the G-proteins
Rab5, Rab34, and Ras.27 Similarly, induction of dominant-negative Rab5 is known to attenuate
foot-and-mouth-disease virus infectivity.28
Because the small molecular-weight G-proteins are associated with viral life cycles and
proinflammatory signaling pathways, we sought to explore the role of these G-proteins in the
proinflammatory environment of rhinovirus-induced asthma exacerbation. Within this
environment, we were particularly interested in macrophages, since the role of the G-proteins in
macrophage signaling cascades is poorly understood relative to epithelial cells. Such
investigations may provide new targets for novel therapeutic strategies against HRV-induced
asthma exacerbation. Additionally, investigations with the phylogenetically close major-group
HRV 16 and minor-group 1A serotypes, which are both members of the HRVA classification,
may provide fresh insight into signaling differences dependent on virus receptor tropism.29
In the present study, we determined that the small G-proteins Rac, Cdc42, and Ras were
not activated in epithelial cells responding to HRV exposure. However, we also determined that
Rac is activated in macrophages responding to HRV. We further demonstrated that Rac activation
in macrophages is part of a proinflammatory signaling cascade leading to mitogen-activated
protein kinase (MAPK) p38 activation and CCL2 release. These results provide a fuller
understanding of the proinflammatory signaling milieu initiated during HRV-induced asthma
exacerbation and reinforce the importance of certain G-proteins in initiating this
proinflammatory environment.
74

Materials and Methods
Reagents
For monocyte preparation, Lymphocyte Separation Media and HBSS were purchased
from CellGro, (Manassas, VA) and ACK lysing buffer from BioWhittaker, (Walkersville, MD).
Sigma Chemical Company (St. Louis, MO) was the source for protease inhibitor cocktail. The
Rac inhibitor NSC32766 was obtained from Tocris (Ellisville, MO). Immunoblotting reagents
were purchased from a variety of suppliers including Santa Cruz Biotechnology of Santa Cruz,
CA (horseradish peroxidase-conjugated goat anti-rabbit IgG, anti-Rac antisera, Grb2 antisera,
anti-Cdc42 antisera and anti-Ras antisera), Pierce of Springfield, IL (SupersignalTM
chemiluminescence substrate reagents and immobilized glutathione agarose beads), R&D
systems of Minneapolis, MN (anti-MCP1 and anti-CCL5 antibody pairs), and Cell Signaling of
Beverly, MA (anti-phospho-p38 MAPK antisera). Anti-CD14 and anti-CD86 was purchased
from Becton Dickinson (San Jose, CA). The ROS visualization reagent 2'-7'-
Dichlorodihydrofluorescein diacetate (DCFDA) and propidium iodide for cell staining were
obtained from Invitrogen of Carlsbad, CA.
Isolation and purification/maturation of human blood monocyte-lineage cells
The protocol used for collecting blood samples was approved by the Lawrence University
Institutional Review Board. Human blood samples were collected as described previously.10
Whole blood was separated by density gradient, and leukocytes were collected from the buffy
coat interface between the plasma and erythrocyte layers as described previously.10 Collected
cells were distributed to 12-well tissue culture plates and cultured in RPMI 1640 (CellGro,
75

Manassas, VA) containing 2% penicillin and streptomycin (Gibco, Grandisland, NY) and 5%
sterile-filtered, heat-inactivated human (type AB) serum (BioWhittaker, Walkersville, MD).
Adherent monocytes were matured for 7-10 days until a macrophage phenotype was determined
by flow cytometry. Purified human macrophages were lifted off the plate with Cell Dissolution
Solution (Sigma, St. Louis, MO) and the cell population was evaluated for CD14, CD86 positive
cells and viability (annexin V) by flow cytometry.32 Cell populations were typically 95% viable
and 90% CD14-positive.
Cell culture
HeLa cells, A549 cells, and human peripheral blood monocyte-derived macrophages were
cultured in RPMI 1640 (Cellgro) with 5% human AB serum (Cellgro, Manassa, VA) and 1%
penicillin/streptomycin (Gibco) at 37˚C in a humidified incubator with 5% CO2.
Preparation of human rhinovirus (HRV) stock
Human rhinovirus (HRV) serotypes 16 and 1A were gifts from the Jim Gern laboratory
and Wai-Ming Lee at the University of Wisconsin at Madison. Both serotypes are from the
HRVA group.29 HRV was grown in HeLa cells and subsequently sedimented through a sucrose
step gradient to remove exogenous protein and other contaminants. The HRV was then titered at
109 infectious particles/mL and stored at -80 ℃ as previously described.10 RPMI 1640 enriched
with human serum was used to prepare all necessary dilutions of both virus serotypes before
virus was applied. Virus preparations were tested for endotoxin as previously described and
found to be endotoxin-free.10
76

Pull-down assay for small molecular-weight G-protein activation
BL21pLysS bacteria were transformed to express the appropriate GST-fusion proteins,
and the proteins were harvested and stored as described previously.30 The pull-down assay
protocol was developed from one previously published.30 Macrophages (1 million cells/well in a
12-well tissue culture plate) were exposed to HRV 1A or HRV 16 at a multiplicity of infection
(MOI) of 10 in a time course. Cells were lysed in GST buffer, and cell lysates were added to
glutathione agarose beads bound to GST-fusion protein. For Rac and Cdc42 pull-down assays,
GST-Pak was used. For Ras pull-down assays, GST-Raf was used. Beads were cleared by
centrifugation, and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)
followed by immunoblot was performed.
SDS-PAGE and immunoblot analysis
Macrophages (1 million cells/well in a 12-well tissue culture plate) were exposed to
HRV 1A or HRV 16 at an MOI of 10 in a time course. Selected wells were pretreated with 10
μM inhibitor NSC23766 30 minutes before virus treatment. Cell lysates were analyzed for
MAPK activation using SDS-PAGE and immunoblot as previously described.10 Rabbit primary
antibodies were used to probe for the presence of Rac, Cdc42, Ras, phospho-p38, and Grb-2.
Blots were visualized using horseradish peroxidase-coupled secondary antibodies on a KODAK
Image Station 4000 MM (Kodak) and Kodak MI Imaging Software (version 4.0.3).
77

Inhibitor time course for cytokine release
Inhibitor time courses for examining cytokine release took place over 24 hours. The Rac
inhibitor NSC23766 was applied to a final concentration of 10 µM 30 minutes before HRV was
applied. Both HRV 16 and HRV 1A were applied at a MOI of 10. Following the 24-hour time
course, the media supernatants were removed to cluster tubes and stored at -20 ℃ until enzyme-
linked immunosorbant assay (ELISA) could be performed.
Enzyme-linked immunosorbant assay
Following the inhibitor time course for cytokine release, the sandwich enzyme-linked
immunosorbant assay (ELISA) technique was used to probe for CCL2 and CCL5 release. Half-
size 96 well ELISA/Immuno/Assay (EIA) plates were coated overnight at 4 °C with coating
buffer containing the concentrations of monoclonal capture antibody recommended by the
manufacturer (R&D Systems). The plates were washed three times with 1x phosphate buffered
saline with Tween 20 (PBS-T) to remove excess antibody, and a standard curve of successive 1:2
dilutions of protein was prepared. The standards and the various samples were added to the 96-
well plate in triplicate at 25 µL per well, and the plate was incubated at 4°C overnight.
Monoclonal detection antibody was added to the plate at concentrations recommended by the
manufacturer (R&D Systems) after washing three times in 1x PBS-T and incubating the plate at
room temperature for 1 hour. The plate was again washed three times in 1x PBS-T. A 1:10,000
dilution of streptavidin-HRP was added to the wells, and the plate was incubated for 20 minutes.
The plate was then washed three times in 1x PBS-T, and 50 µL of TMB component HRP
78

microwell substrate solution (BioFX, Owing Mills, MD) was added to each well. When a blue
color developed such that a gradation between standards could be visually detected, the reaction
was stopped with 1 M hydrochloric acid. Optical density (absorbance) was read at 450 nm using
a Molecular Devices ThermoMAX microplate reader with Spectrosoft software (version 6.2).
Protein concentrations were calculated by averaging the triplicate values and interpolating from
the standard curve.
Statistical analysis of ELISA data
After the ELISAs had been performed, the data were pooled for comparison. To
determine whether differences between control and treated groups were significant, SPSS
(originally the Statistical Package for the Social Sciences; version 16.0) was used to perform an
analysis of variance (ANOVA) (αreject = 0.10). Following detection of significant differences
between groups by ANOVA, post-hoc Bonferroni-adjusted Student’s t-tests (αreject = 0.10) were
used to resolve individual differences between groups.
ROS measurements
Cells were disassociated by trypsin and centrifuged for two minutes at 4000 rpm. The
supernatant was removed, and the pellet resuspended in medium by gentle agitation at a
concentration of approximately 1x106 cells/ml and kept on ice until the experiment was
performed. The cells were loaded with 10 µM DCFDA for 30 min at 37°C, transferred to flow
cytometry tubes (5·105 cells/tube), and treated at room temperature as described in the figure
legends. Flow cytometry was performed by measuring 10,000 cells on a BD Biosciences
79

FACaliber flow cytometer. Propidium iodide-stained cells were not included in the analysis.
Histogram statistics were analyzed by the program CellQuest (Becton Dickinson, San Jose, CA).
ResultsThe small G-proteins Rac, Cdc42, and Ras are not activated in the cell lines HeLa and A549
following HRV 1A or HRV 16 exposure
Because the small G-proteins have been associated with the replication and infectivity of
several viruses, we sought to explore the roles played by some of the most prominent G-proteins
during the infection of classic epithelial cell lines known to be productively infected by HRV. We
were especially interested in the Rho-family G-proteins Rac and Cdc42 in light of the fact that
the related G-protein RhoA was shown to be activated in HeLa cells responding to HRV.26 We
selected HRV 16 and HRV 1A for use because they are genetically closely related, in the HRVA
group, but bind to different receptors.29 Interestingly, pull-down assays in HeLa and A549 cells
for the small G-proteins Rac, Cdc42, and Ras following HRV 1A and HRV 16 exposure revealed
little to no G-protein activation (data not shown).
The small G-protein Rac is differentially activated in macrophages following HRV 1A and HRV
16 exposure
Though the G-proteins Rac, Cdc42, and Ras were not activated in HeLa or A549 cells
responding to HRV, we suspected that these proteins might be involved in the macrophage
response to HRV. Previous work has identified the macrophage as a key mediator of the
proinflammatory environment initiated during HRV infection.10,31 While we found that the G-
proteins Cdc42 and Ras were not activated in macrophages (data not shown), pull-down assays
80

for the G-protein Rac demonstrated Rac activation in macrophages following exposure to HRV
16 or HRV 1A (Figure 1). HRV was applied at a MOI of 10 in accord with previous results.10 The
activation of Rac occurred with differential kinetics that depended upon which HRV serotype
provided the stimulus. In response to HRV 1A exposure (LDL-R tropic), macrophages
experienced escalating Rac activation from 2 minutes through 10 minutes followed by declining
activation through the 30-minute time point (Figure 1A). In response to HRV 16 exposure
(ICAM-1 tropic), Rac activation escalated throughout the 30-minute time course (Figure 1B).
Production of reactive oxygen species is not induced in HRV-exposed macrophages
Because the production of ROS is important for various macrophage inflammatory functions
including bacterial killing, we first sought to confirm that P2X7 agonists can stimulate the
production of ROS in monocyte derived macrophages as previously described.32 In this regard,
treatment of these cells with the selective P2X7 agonist BzATP (250 µM) for 5–30 min induced
ROS production as measured by DCFDA fluorescence (Figure 2A). Within 5 minutes of BzATP
treatment, a shift in DCFDA fluorescence was observed versus control-treated cells (gray-shaded
histogram versus open histogram). This increase reached a plateau of approximately 6-fold
following 30 minutes of BzATP treatment and did not increase further with longer treatment
(data not shown). Under the present conditions, the addition of HRV 16 or HRV 1A at a MOI of
10 did not stimulate a detectable increase in ROS generation over a 30-minute time course
(Figure 2B).
81

Pharmacological inhibition of Rac prevents activation of the stress-activated p38 MAPK
following exposure of macrophages to HRV 1A or HRV 16
Previous studies have described the important role played by p38 MAPK in
proinflammatory signaling cascades leading to activation of the transcription factor ATF-2 and
the release of the proinflammatory cytokine CCL2.10 We demonstrated that the Rac inhibitor
NSC23766 decreased macrophage p38 activation in response to either HRV 16 or HRV 1A
exposure (Figure 3). We selected the NSC23766 inhibitor based on its previous effective use at
similar concentrations in alveolar macrophages and other immune cells.33,34 Although p38
activation escalated over a 90-minute time course when macrophages were exposed to either
HRV 1A or HRV 16, applying NSC23766 30 minutes before applying the virus decreased p38
activation throughout the 90-minute time course.
Pharmacological inhibition of Rac downregulates the release of the proinflammatory cytokine
CCL2, but not the release of CCL5
CCL2 and CCL5 levels in cell supernatants were analyzed via sandwich enzyme-linked
immunosorbant assay (ELISA). NSC23766 inhibition of Rac in macrophages significantly
downregulated the release of CCL2 in response to either HRV 1A or HRV 16 exposure, with p-
values ranging from less than 0.001 for HRV 1A to 0.086 for HRV16 (Figure 4). Notably, the
Rac inhibitor did not significantly downregulate the release of CCL5 (Figure 5).
Because Rac inhibition did not affect macrophage secretion of CCL5 in response to HRV,
we suspected that another of the G-proteins might function in CCL5 production. However, pull-
82

down assays for the related G-proteins Cdc42 and Ras did not detect activation of these G-
proteins following macrophage exposure to HRV 1A or HRV 16 (data not shown).
Discussion
Recent studies have shown that the Ras superfamily small G-proteins may play a role in
viral replication and in the inflammatory response,20,35 and studies specific to HRV demonstrate
that the small G-protein RhoA is important to HRV-induced cell signaling in epithelial cells.26
We hypothesized that the RhoA-related small G-proteins Rac and Cdc42 as well as the G-protein
Ras would be involved in HRV replication within epithelial cells. Interestingly, Rac, Cdc42, and
Ras were not activated in epithelial cells exposed to HRV. This result is consistent with previous
findings, which indicate that Rac is not important to p38 MAPK activation in HeLa cells.26
Although epithelial cells are the most studied cells involved in HRV-induced cytokines, a
number of studies indicate that the macrophage is also important in HRV infection.12,36 Human
macrophages are not productively infected with HRV,14 but they are known to express the HRV
receptors ICAM-1 and LDL-R and release inflammatory cytokines including CCL2 and IP-10 in
response to HRV exposure.10,37 We observed Rac activation in human macrophages responding
to HRV (Figure 1), whereas none of the G-proteins Rac, Cdc42, and Ras was activated during the
response of epithelial cells to HRV exposure, suggesting an important and specific role for
macrophages in the inflammatory rhinoviral response.
HRV is often classified according to which of two cellular receptors a particular serotype
of HRV will bind.4 According to this classification scheme, the major group of HRV binds to the
intercellular adhesion molecule 1 (ICAM-1) receptor,5 whereas the minor group of HRV binds to
83

the low density lipoprotein (LDL) receptor.6 Previous researchers have often chosen to generally
examine only signaling responses to major-group rhinovirus, neglecting the minor-group viruses.
10,38,39 Although one might assume that a “general response” to major- and minor-group HRV
would occur, our work with the small G-protein Rac suggests otherwise. When we examined Rac
activation kinetics in macrophages exposed to either HRV 16, a major-group virus, or HRV 1A, a
minor-group virus, we observed that Rac activation occurred with different kinetics depending
on which of the HRV types provided the stimulus (Figure 1). This observation of differential
kinetics supports the notion that signaling pathways leading from ICAM-1 binding major-group
HRV and the LDL receptor binding minor-group HRV may differ.
Pharmacological inhibition of the G-protein Rac suppressed proinflammatory signaling
involving phosphorylation of the MAPK p38 (Figure 3). The activation of p38, a mitogen-
activated protein kinase (MAPK), has previously been associated with the production of
inflammatory cytokines such as CCL2 in macrophages exposed to HRV.10 Consistent with this
previous work, we determined that pharmacological inhibition of the small G-protein Rac
attenuated the release of CCL2 (Figure 4). These results implicate Rac in the proinflammatory
signaling pathways initiated in macrophages upon exposure to HRV.
Although pharmacological inhibition of the small G-protein Rac successfully blocked the
phosphorylation of the MAPK p38 (Figure 3) and the release of the proinflammatory cytokine
CCL2 (Figure 4) following HRV exposure to macrophages, Rac inhibition was not successful in
blocking the release of the proinflammatory cytokine CCL5 (Figure 5). CCL5 seems to be
important to the inflammatory airway environment observed during asthma exacerbations, as
CCL5 is chemotactic for eosinophils, which have been implicated in the airway remodeling that
84

takes place during asthma exacerbation.40 CCL2 and CCL5 levels in the bronchoalveolar lavage
fluid of asthmatics are elevated well above the baseline levels observed in normal individuals.16
Furthermore, the release of CCL2 and CCL5 has been associated with bronchial
hyperresponsiveness and with leukocyte recruitment to the airway.41
Epithelial cells remain the primary focus of many researchers studying HRV-induced
exacerbation of asthma,7 and it is widely understood that epithelial cells are capable of
contributing to the proinflammatory environment initiated in the airway of an asthmatic during
HRV infection.11 The signaling events leading to the release of cytokines from these cells would
appear to be mechanistically different from the signaling events leading to their release from
macrophages. Notwithstanding that the role of Rac in HRV-induced macrophage signaling
presents one significant mechanistic difference between inflammatory signaling cascades in
these two cell types, further study is needed to clarify the differences between epithelial cell and
macrophage HRV-induced signaling cascades.
Interestingly, the HRV-induced and Rac-mediated proinflammatory signaling observed in
macrophages does not appear to induce ROS production, a hallmark of classically activated
macrophages.42 Since macrophages treated with HRV do not produce ROS (Figure 3), we do not
believe HRV exposure induces classical activation of macrophages.
85

AcknowledgementsWe are grateful to Dana Raugi and Megan Wilson for their contributions to the data
presented here. We would also like to thank the Paul Bertics laboratory, Jim Gern and Wai-Ming
Lee at the University of Wisconsin-Madison for their contribution of viral stocks and reagents.
This work was supported by the Ronald E. McNair Postbaccalaureate Achievement Program, the
Henry Merritt Wriston Scholarship, NIH R15 AI065505-01A1 and NSF 0521112.
86

References1. Johnston SL, Pattemore PK, Sanderson G et al. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ 1995;310:1225-1229.2. Gern JE and Busse WW. Association of Rhinovirus Infections with Asthma. Clin Microbiol Rev 1999;12:9-18.3. Palmenberg AC, Spiro D, Kuzmickas R et al. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 2009;324:55-59.4. Greenberg SB. Respiratory Consequences of Rhinovirus Infection Arch Intern Med 2003;163:278.5. Greve JM, Davis G, Meyer AM et al. The major human rhinovirus receptor is ICAM-1. 1989;56:839-847.6. Hofer F, Gruenberger M, Kowalski H et al. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc Natl Acad Sci USA 1994;91:1839-1842.7. Peters-Golden M. The Alveolar Macrophage: The Forgotten Cell in Asthma. Am J Respir Cell Mol Biol 2004;31:3-7.8. Lohmann-Matthes M, Steinmuller C and Franke-Ullmann G. Pulmonary macrophages. Eur Respir J 1994;7:1678-1689.9. Subauste MC, Jacoby DB, Richards SM and Proud D. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure J Clin Invest 1995;96:549-557.10. Hall DJ, Bates ME, Guar L et al. The Role of p38 MAPK in Rhinovirus-Induced Monocyte Chemoattractant Protein-1 Production by Monocytic-Lineage Cells. J Immunol 2005;174:8056-8063.11. Levine SJ. Bronchial epithelial cell-cytokine interactions in airway inflammation J Investig Med 1995;43:241-249.12. Barnes PJ, Chung KF and Page CP. Inflammatory Mediators of Asthma: An Update. Pharmacol Rev 1998;50:515-596.13. Bardin P, Johnston S, Sanderson G et al. Detection of rhinovirus infection of the nasal mucosa by oligonucleotide in situ hybridization. Am J Respir Cell Mol Biol 1994;10:207-213.14. Gern JE, Dick EC, Lee WM et al. Rhinovirus enters but does not replicate inside monocytes and airway macrophages. J Immunol 1996;156:621-627.15. Laza-Stanca V, Stanciu LA, Message SD et al. Rhinovirus Replication in Human Macrophages Induces NF-{kappa}B-Dependent Tumor Necrosis Factor Alpha Production. J Virol 2006;80:8248-8258.16. Alam R, York J, Boyars M et al. Increased MCP-1, RANTES, and MIP-1alpha in bronchoalveolar lavage fluid of allergic asthmatic patients Am J Respir Crit Care Med 1996;153:1398-1404.17. Mogensen TH and Paludan SR. Molecular pathways in virus-induced cytokine production. Microbiol Mol Biol Rev 2001;65:131-150.18. Papi A and Johnston SL. Rhinovirus Infection Induces Expression of Its Own Receptor Intercellular Adhesion Molecule 1 (ICAM-1) via Increased NF-kappa B-mediated Transcription. J Biol Chem 1999;274:9707-9720.
87

19. Peter A.B. Wark, Bucchieri F, Johnston SL et al. IFN-gamma-induced protein 10 is a novel biomarker of rhinovirus-induced asthma exacerbations. J Allergy Clin Immunol 2007;120:586-593.20. Brazzoli M, Bianchi A, Filippini S et al. CD81 is a central regulator of cellular events required for HCV infection of human hepatocytes. J Virol 2008.21. Balachandran S, Porosnicu M and Barber GN. Oncolytic Activity of Vesicular Stomatitis Virus Is Effective against Tumors Exhibiting Aberrant p53, Ras, or Myc Function and Involves the Induction of Apoptosis. J Virol 2001;75:3474-3479.22. Huber M, Watson KA, Selinka H et al. Cleavage of RasGAP and Phosphorylation of Mitogen-Activated Protein Kinase in the Course of Coxsackievirus B3 Replication. J Virol 1999;73:3587-3594.23. Marchant D, Sall A, Si X et al. ERK MAP kinase-activated Arf6 trafficking directs coxsackievirus type B3 into an unproductive compartment during virus host-cell entry. J Gen Virol 2009;90:854-862.24. Belov GA, Feng Q, Nikovics K et al. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog 2008;4:e1000216.25. Belov GA, Habbersett C, Franco D and Ehrenfeld E. Activation of cellular Arf GTPases by poliovirus protein 3CD correlates with virus replication. J Virol 2007;81:9259-9267.26. Dumitru CA, Dreschers S and Gulbins E. Rhinoviral infections activate p38MAP-kinases via membrane rafts and RhoA. Cell Physiol Biochem 2006;17:159-166.27. Coyne CB, Shen L, Turner JR and Bergelson JM. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2007;2:181-192.28. Johns HL, Berryman S, Monaghan P et al. A dominant-negative mutant of rab5 inhibits infection of cells by foot-and-mouth disease virus: implications for virus entry. J Virol 2009;83:6247-6256.29. Palmenberg AC, Spiro D, Kuzmickas R et al. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution Science 2009;324:55-59.30. Chapman K and Hall DJ. Quantifying the activation of the small molecular weight G-protein ras. 2005;33:22-27.31. Gern J, Vrtis R, Kelly E et al. Rhinovirus produces nonspecific activation of lymphocytes through a monocyte-dependent mechanism. J Immunol 1996;157:1605-1612.32. Lenertz LY, Gavala ML, Hill LM and Bertics PJ. Cell signaling via the P2X(7) nucleotide receptor: linkage to ROS production, gene transcription, and receptor trafficking Purinergic Signal 2009;5:175-187.33. Haberzettl P, Schins RPF, Hohr D et al. Impact of the Fc{gamma}II-receptor on quartz uptake and inflammatory response by alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2008;294:L1137-1148.34. Mitchell T, Lo A, Logan MR et al. Primary granule exocytosis in human neutrophils is regulated by Rac-dependent actin remodeling. Am J Physiol Cell Physiol 2008;295:C1354-1365.35. Bagrodia S, Dérijard B, Davis RJ and Cerione RA. Cdc42 and PAK-mediated Signaling Leads to Jun Kinase and p38 Mitogen-activated Protein Kinase Activation. J Biol Chem 1995;270:27995-27998.
88

36. Sen GC. Viruses and interferons Annu Rev Microbiol 2001;55:255-281.37. Korpi-Steiner NL, Bates ME, Lee W et al. Human rhinovirus induces robust IP-10 release by monocytic cells, which is independent of viral replication but linked to type I interferon receptor ligation and STAT1 activation. J Leukoc Biol 2006;80:1364-1374.38. Spurrell JCL, Wiehler S, Zaheer RS et al. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol 2005;289:L85-95.39. Griego SD, Weston CB, Adams JL et al. Role of p38 Mitogen-Activated Protein Kinase in Rhinovirus-Induced Cytokine Production by Bronchial Epithelial Cells. J Immunol 2000;165:5211-5220.40. Nissim Ben Efraim AH and Levi-Schaffer F. Tissue remodeling and angiogenesis in asthma: the role of the eosinophil Ther Adv Respir Dis 2008;2:163-171.41. Conti P and DiGioacchino M. MCP-1 and RANTES are mediators of acute and chronic inflammation Allergy Asthma Proc 2001;22:133-137.42. Rickard AJ and Young MJ. Corticosteroid receptors, macrophages and cardiovascular disease. J Mol Endocrinol 2009;42:449-459.
89

Figure LegendsFigure 1.
Rac activation following macrophage exposure to HRV 1A (Panel A) and HRV 16 (Panel B) at
an MOI of 10. Activated Rac was pulled down and assayed by SDS-PAGE and immunoblot
according to procedure in Materials and Methods. Each blot is representative of five independent
experiments.
Figure 2.
Measurement of ROS production by monocyte derived macrophages treated with human
rhinovirus. A) Monocyte-derived macrophages (1·105 cells/mL) were loaded with 10 µM
DCFDA for 30 min at 37°C followed by treatment of the cells with either vehicle (Control) or
250 µM BzATP at room temperature. The samples were then analyzed at the indicated times as
stated under Materials and Methods. The fluorescence of 10,000 cells was monitored by flow
cytometry and is presented as DCFDA fluorescence intensity versus the percent of maximum
events. The data are representative of three independent experiments. B) Monocyte-derived
macrophages were incubated with 10 µM DCFDA as stated above followed by treatment with
either vehicle (Control) , HRV 1A at an MOI of 10, or HRV 16 at an MOI of 10 at room
90
temperature and analyzed after 30 minutes by flow cytometry. The data are representative of
three independent experiments.
Figure 3.
Activation of the MAPK p38 in macrophages exposed to HRV 1A and HRV 16 at an MOI of 10
in a 90-minute time course. Selected 90-minute time points were treated with the Rac inhibitor
NSC23766 30 minutes before HRV treatment. Activation of p38 MAPK was assayed by SDS-
PAGE and immunoblot using an anti-phospho-p38 antibody. Equal protein loading was ensured
by using GRB-2 as an internal control and probing with an anti-GRB-2 antibody (not shown).
The blot is representative of five independent experiments.
A B
91
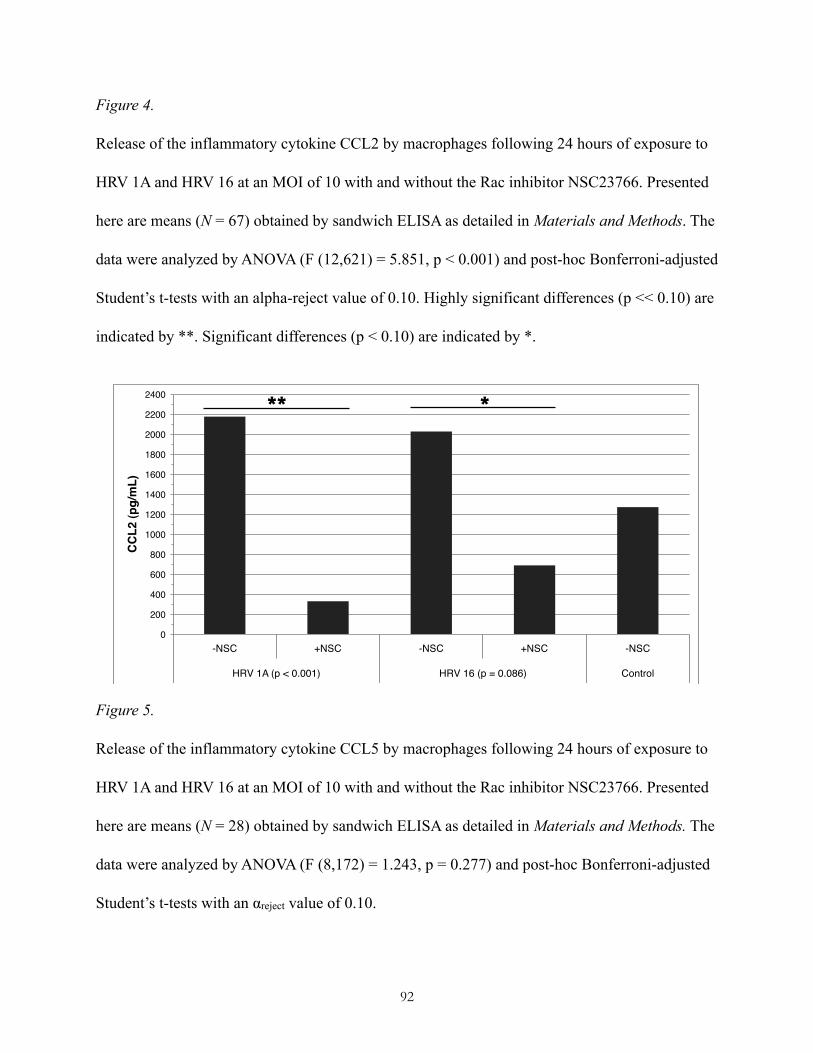
Figure 4.
Release of the inflammatory cytokine CCL2 by macrophages following 24 hours of exposure to
HRV 1A and HRV 16 at an MOI of 10 with and without the Rac inhibitor NSC23766. Presented
here are means (N = 67) obtained by sandwich ELISA as detailed in Materials and Methods. The
data were analyzed by ANOVA (F (12,621) = 5.851, p < 0.001) and post-hoc Bonferroni-adjusted
Student’s t-tests with an alpha-reject value of 0.10. Highly significant differences (p << 0.10) are
indicated by **. Significant differences (p < 0.10) are indicated by *.
Figure 5.
Release of the inflammatory cytokine CCL5 by macrophages following 24 hours of exposure to
HRV 1A and HRV 16 at an MOI of 10 with and without the Rac inhibitor NSC23766. Presented
here are means (N = 28) obtained by sandwich ELISA as detailed in Materials and Methods. The
data were analyzed by ANOVA (F (8,172) = 1.243, p = 0.277) and post-hoc Bonferroni-adjusted
Student’s t-tests with an αreject value of 0.10.
0!
200!
400!
600!
800!
1000!
1200!
1400!
1600!
1800!
2000!
2200!
2400!
-NSC! +NSC! -NSC! +NSC! -NSC!
HRV 1A (p < 0.001)! HRV 16 (p = 0.086)! Control!
CCL2
(pg/
mL)
!
92
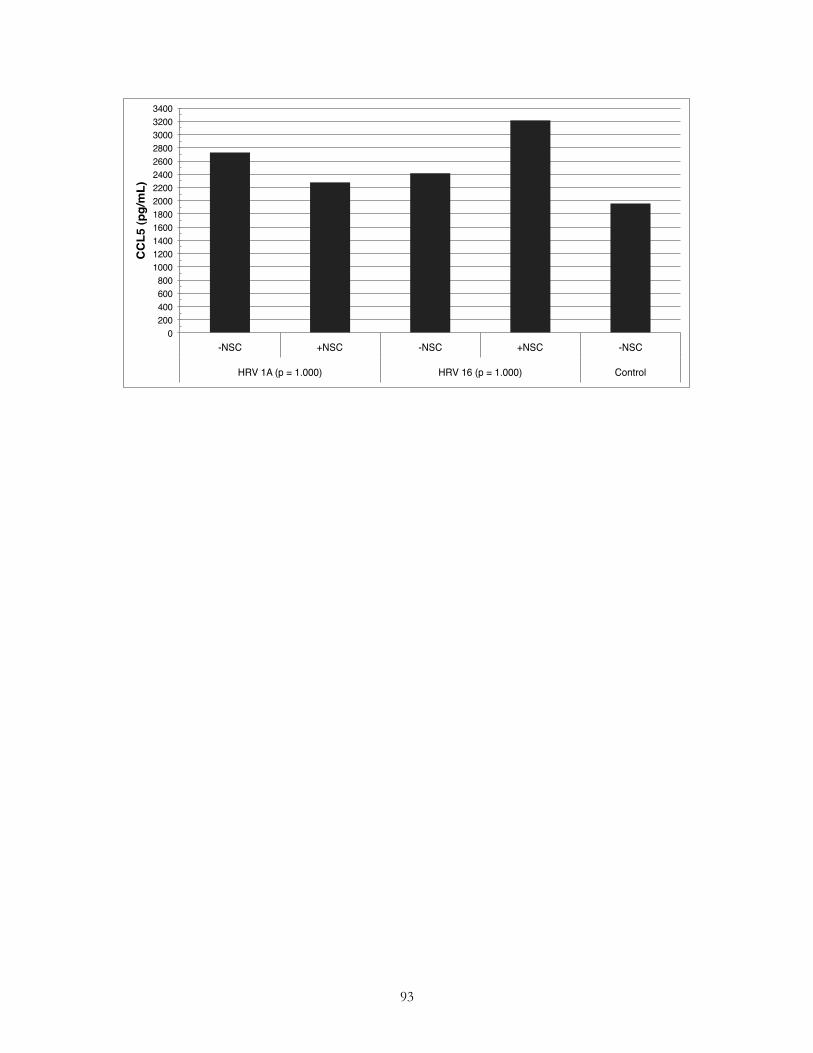
0!200!400!600!800!
1000!1200!1400!1600!1800!2000!2200!2400!2600!2800!3000!3200!3400!
-NSC! +NSC! -NSC! +NSC! -NSC!
HRV 1A (p = 1.000)! HRV 16 (p = 1.000)! Control!
CCL5
(pg/
mL)
!
93