the role of toxoplasma gondii as a possible inflammatory
TRANSCRIPT

Family Medicine and Community Health
SYSTEMATIC REVIEW
Family Medicine and Community Health 2016;4(4):44–62 44www.fmch-journal.org DOI 10.15212/FMCH.2016.0128© 2016 Family Medicine and Community Health
RE
VIE
W
The role of Toxoplasma gondii as a possible inflammatory agent in the pathogenesis of type 2 diabetes mellitus in humans
Aus Molan1, Kazunori Nosaka1, Michael Hunter2,3, Wei Wang1
Abstract
Type 2 diabetes mellitus (T2DM) continues to be a major challenge for public health authorities
worldwide. While potential causes such as obesity, physical inactivity, and dietary patterns have
been proposed to explain the growing epidemic, there may also be unidentified environmental
determinants. An emerging field of research is starting to examine the association of infectious and
environmental pathogens with diabetes. In particular, the potential of these pathogens to cause low-
grade inflammation that facilitates the risk and development of T2DM. An understudied pathogen
of potential interest is the protozoan parasite Toxoplasma gondii (T. gondii). There is limited clinical
evidence supporting the association between chronic T. gondii infection and the development of
many disorders, including T2DM, in both animals and humans. This review (1) addresses the exist-
ing knowledge of the role of T. gondii in the inflammation process leading to T2DM, (2) examines
the current studies describing the relationship between T. gondii and T2DM, and (3) makes recom-
mendations for future studies to determine the role of T. gondii in the pathogenesis of T2DM. We
believe that T. gondii may be an important target for T2DM intervention, and propose a new field
of study, “toxoplasmic type 2 diabetes.”
Keywords: Etiology; inflammation; Toxoplasma gondii infection; toxoplasmosis; diabetes mel-
litus, type 2
1. School of Medical and Health
Sciences, Edith Cowan Univer-
sity, Joondalup, WA, Australia
2. Busselton Population and Med-
ical Research Institute, Busselton,
WA, Australia
3. School of Population Health,
University of Western Australia,
Nedlands, WA, Australia
CORRESPONDING AUTHOR:
Wei Wang, MD, PhD, FFPH
School of Medical and Health
Sciences, Edith Cowan Uni-
versity, 270 Joondalup Drive,
Joondalup WA 6027, Australia
Tel.: +61-8-63043717
E-mail: [email protected]
Received 24 August 2016;
Accepted 18 September 2016
Introduction
Diabetes mellitus is one of the most challeng-
ing public health burdens of the 21st century
[1]. It is a chronic disorder characterized by
persistent hyperglycemia with disturbances
in fat, carbohydrate, and protein metabo-
lism resulting from abnormalities in insulin
secretion, insulin action, or both [1, 2]. Type
2 diabetes mellitus (T2DM; International
Classification of Diseases, Tenth Revision,
Clinical Modification diagnosis code E11.9) is
characterized by reduced insulin production
and an inability of body tissues to fully respond
to insulin (insulin resistance) [1–3], account-
ing for approximately 90% of all diagnosed
diabetes cases [2, 3]. The pathogenesis of
T2DM has been recognized as an ongoing
deterioration of the insulin secretory capacity
of pancreatic β cells that does not allow com-
pensation for an increased peripheral insulin
demand [4, 5]. The prevalence of diabetes is
reaching epidemic levels, with an estimated
global prevalence of 9% among adults aged
18 years or older [6–12]. Additionally, dia-
betes places excessively high human, social,
and economic costs on all countries. In 2013,
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

Molan et al.
45 Family Medicine and Community Health 2016;4(4):44–62
RE
VIE
W
5.1 million deaths were caused by diabetes worldwide (one
person every 6 s) [6, 12]. Looking ahead, it is estimated that
642 million people will be living with diabetes by 2040, which
is a more than 50% increase from 415 million in 2015 [7]. This
is mainly due to the increase in the numbers of people with
T2DM as a result of the increase in life expectancy, genetic
predisposition, physical inactivity, dietary changes, the obe-
sity epidemic, and the decreased mortality rates in diabetic
individuals [1, 3, 6–11]. However, there may also be additional
unidentified novel risk factors, such as subclinical inflamma-
tion caused by infectious agents, that contribute to this rising
prevalence of T2DM [13].
There are epidemiological data that link inflammatory bio-
markers as important risk indicators for future development of
diabetes [14]. In this regard, an emerging field of research is
beginning to investigate the potential of infectious and envi-
ronmental pathogens to cause low-grade inflammation that
may facilitate the risk and development of various metabolic
conditions, including diabetes and obesity. An understud-
ied pathogen of potential interest is the protozoan parasite
Toxoplasma gondii (T. gondii) (International Classification
of Diseases, Tenth Revision, Clinical Modification diagnosis
code B58.9). First described in 1908 by Nicolle and Manceaux
[15], T. gondii infects approximately one third of the world’s
population and is considered one of the most successful human
parasites [16–19]. Humans acquire T. gondii by the ingestion
of food, water, or soil contaminated by oocysts from the defin-
itive hosts, cats (Fig. 1). T. gondii is also transmitted in people
vertically via the maternal placenta and horizontally via blood
transfusion [18, 21–23]. T. gondii is capable of infecting all
warm-blooded animals [24], and toxoplasmosis is a disease
of considerable public health impact – the global prevalence
rates of infection by this parasite are phenomenal, with figures
ranging from 15% to 85% depending on dietary habits, cli-
mate conditions, hygiene standards, and geographical regions
[21, 23, 25–27]. Although T. gondii has a worldwide distribu-
tion and possibly the widest host range of any parasite, there
is only one species (T. gondii) in the genus Toxoplasma [28],
and cats are the only definitive host in which T. gondii sexual
development is known to occur [25, 20].
Toxoplasmosis can present with various nonspecific signs
and symptoms, but most are similar to general flulike indicators
[22, 23, 27]. In immunocompetent individuals, it is thought
that the disease is asymptomatic and that the individual recov-
ers without treatment because of an efficient immune system
which limits the spread of the rapidly multiplying tachyzoites
[22]. Therefore people are not routinely screened for T. gondii
infection unless they are immunocompromised or pregnant
[4, 23–25]. In all cases the parasite remains detectable in the
serum throughout the life of the host, with dormant cysts being
formed in various anatomical sites, including the central nerv-
ous system, often establishing latent toxoplasmosis [18, 22].
T. gondii can infect and replicate in any nucleated host cells,
leading to the production of various inflammatory markers
via the innate acute inflammatory responses and antigen-spe-
cific adaptive immunity [4]. This facilitates a state of chronic
inflammation at various anatomical sites in the host [4].
Chronic T. gondii infection has been linked to several auto-
immune disorders, including thyroid disease, systemic sclero-
sis, rheumatoid arthritis, and inflammatory bowel syndrome
[29]. In addition, several reports have demonstrated a positive
correlation between T. gondii infection and numerous neu-
rological disorders and cancers [4, 22, 23, 28, 30]. However,
T. gondii infection in individuals with T2DM has received
little recognition, and human studies investigating T. gondii
infection and inflammatory biomarkers in T2DM subjects are
not available. This review (1) addresses the existing knowl-
edge of the role of T. gondii in the inflammation process lead-
ing to T2DM, (2) examines the current studies describing
the relationship between T. gondii and T2DM, and (3) makes
recommendations for further studies to determine the role of
T. gondii in the pathogenesis of T2DM using inflammation
as the main outcome. The association between T2DM and
T. gondii, if proven, will have important and significant public
health outcomes. Routine screening for T. gondii infection in
individuals with T2DM would add to the risk prediction for
diabetes to target individuals for early aggressive interven-
tion. Targeting T. gondii and reducing its risk factors could be
used as an Achilles heel to reduce inflammation, leading to
improved glucose tolerance and reduced insulin resistance in
diabetic individuals. We believe that T. gondii latent infection
may have excellent potential as a novel target for T2DM inter-
vention and may pave a path for a new field of study, “toxoplas-
mic type 2 diabetes.”
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

The role of Toxoplasma gondii as a possible inflammatory agent
Family Medicine and Community Health 2016;4(4):44–62 46
RE
VIE
W
Host inflammatory responses to T. gondii infection
The founding study by Frenkel [31] demonstrated that the
transfer of serum from infected to uninfected animals failed
to protect the latter against parasite infection. This knowl-
edge assisted in pointing toward T cells rather than antibod-
ies as the major effectors of resistance to T. gondii infection.
Direct
Indirect
Fig. 1. The complete life cycle of Toxoplasma gondii. Infection consists of an acute phase followed by a chronic phase. The life cycle is divided
into the sexual and asexual stages, where the former occurs in the intestine of the definitive host, while the latter occurs in the intermediate hosts.
After completion of the sexual phase, oocysts released in the feces undergo sporulation to form sporocysts that become a source of infection
for intermediate hosts. When the oocysts are ingested, the internal sporocysts pass through the stomach and excyst in the intestine to release
sporozoites. These sporozoites invade epithelial cells and differentiate into tachyzoites, an actively replicating form of T. gondii. Because of their
active multiplication, tachyzoites cause extensive tissue damage and may disseminate to different tissues of the host. T. gondii infects circulating
cells and can use them as a Trojan horse to gain access to protective tissues such as the brain, where entry of immune cells is restricted. The
tachyzoites eventually undergo stage conversion and become slowly multiplying bradyzoites within tissue cysts. If for some reason the tissue
cyst becomes activated, the bradyzoites can undergo stage conversion and become tachyzoites. The bradyzoite form is the only stage that can
infect feline enterocytes and lead to oocyst production, restarting the life cycle. (Adapted from Hunter and Sibley [20] with permission).
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

Molan et al.
47 Family Medicine and Community Health 2016;4(4):44–62
RE
VIE
W
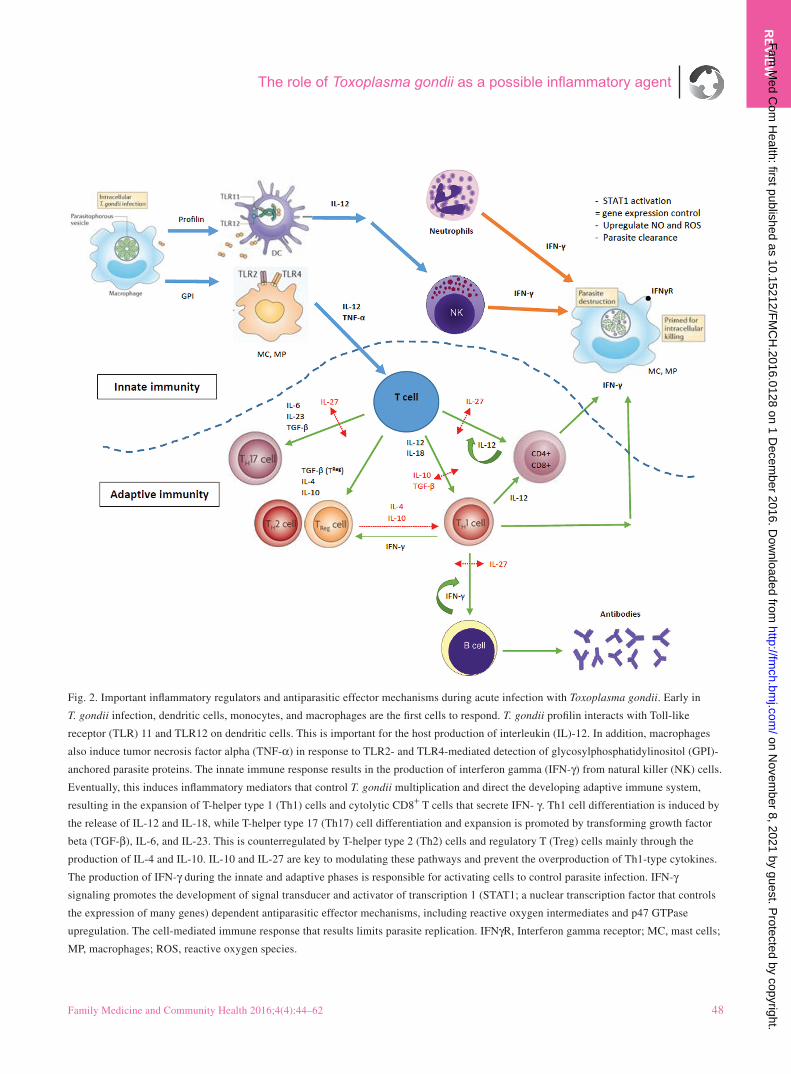
More recently, it has become clear that T. gondii manipulates
and modulates host resistance mechanisms at multiple points
along the proinflammatory pathway which in turn dictate
parasite burden and disease (Fig. 2) [32]. During the acute
phase of this infection, which lasts for less than 10 days, T.
gondii elicits a very strong T-helper type 1 (Th1) cell medi-
ated inflammatory response (Table 1) [41]. The ability of the
host to control T. gondii depends on the subsequent produc-
tion of interleukin (IL)-12 by macrophages and dendritic
cells, resulting from the differentiation and clonal expansion
of host Th1 cells in response to the recognition of conserved
T. gondii microbe molecular structures (glycosylphosphati-
dylinositol anchors and profilin) by Toll-like receptors [32,
92]. In turn, IL-12 stimulates natural killer cells and CD4+
and CD8+ T cells to release interferon gamma (IFN-γ), which
is also central in host resistance to T. gondii infection [32].
The Th1 response, defined by the production of IL-12 and
IFN-γ, is a characteristic of infection with intracellular patho-
gens, including T. gondii [93]. In fact, a significant discovery
that allowed progress toward the identification of essential
recognition and effector mechanisms involved in host resist-
ance to T. gondii infection was the identification of IFN-γ
as the major regulator of cell-mediated immunity to T. gon-
dii in both mouse and human cells [94–97]. Studies show-
ing the importance of T cells in resisting T. gondii infection
are unequivocal. For example, mice which lack functional T
cells are extremely susceptible to T. gondii infection [94, 95].
Likewise, IL-12- or IFN-γ-deficient mice are extremely sus-
ceptible to infection by T. gondii and demonstrate an inabil-
ity to control parasite burden [96, 97]. Furthermore, several
murine models have demonstrated the protection of naïve
mice against T. gondii infection as a result of adoptive trans-
fer of immune T cells [31, 98–100]. With regard to T. gondii
infection in humans, the importance of the adaptive immune
response has been demonstrated by the increase of suscep-
tibility to T. gondii infection in individuals with primary or
acquired T-cell function defects [96].
In addition to the systemic effects, T. gondii also induces
the production of a number of proinflammatory cytokines
(tumor necrosis factor alpha, TNF-α; IL-1; IL-1β; IL-6;
IL-18; IL-12p40; IL-8; IL-17; IL-22; IL-15), anti-inflam-
matory cytokines (transforming growth factor beta, TGF-β;
IL-4; IL-10; IL-27), nitric oxide synthase, and reactive oxy-
gen species that are associated with inflammatory responses
in various sites and cells, including the brain, microglial
cells, astrocytes, and infiltrating CD4+ and CD8+ T cells
[30, 32, 41, 50, 61–67, 92, 101–108]. It is important to note
that excess production of proinflammatory cytokines ends
up causing damage to the host; therefore a delicate balance
between the above mentioned proinflammatory and anti-
inflammatory signals is necessary for the survival of the host
[32]. Maintaining this immune homeostasis during T. gon-
dii infection requires both the capacity to control the host
immune response and the capacity to limit the replication of
the pathogen [93]. To this extent, IL-10, TGF-β, and IL-27
are key downregulators that prevent the overproduction of
Th1-type cytokines, thereby regulating inflammation [32,
92, 103, 104]. More specifically, during T. gondii infection,
the in vivo synthesis of both IL-12 and IFN-γ is modulated
by IL-10 to avoid an excessive immune response that could
cause host tissue damage and widespread inflammation [45,
109]. Paradoxically, while the host immune response lim-
its the tissue spread of the parasite to ensure the survival of
the host, it is also assisting in the survival of the parasite
by converting it into a bradyzoite, an intracellular dormant
resistant form persisting in the muscles, central nervous sys-
tem, and brain tissues [106]. Given that a strong Th1 immune
response is elicited to maintain the dormant bradyzoite stage
[110] and that a Th1 immune response is also maintained to
promote long-lasting immunity [111], hosts infected with T.
gondii remain in an “asymptomatic” state of constant low-
grade inflammation.
T2DM as an inflammatory disease
Inflammation not only participates in host defenses against
environmental and infectious agents such as T. gondii but also
contributes to the pathophysiology of many chronic diseases
[112]. Increased circulatory acute-phase response markers
and proinflammatory cytokines in individuals with T2DM
were first reported in 1997 [113]. Since then, the ideas that
inflammation participates in the pathogenesis of T2DM and
that T2DM is a chronic inflammatory disease have gained
undeniable attention, with many reports describing changes
to immune cell function [4, 43, 53, 55, 114–117]. In addition,
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from
The role of Toxoplasma gondii as a possible inflammatory agent
Family Medicine and Community Health 2016;4(4):44–62 48
RE
VIE
W
Fig. 2. Important inflammatory regulators and antiparasitic effector mechanisms during acute infection with Toxoplasma gondii. Early in
T. gondii infection, dendritic cells, monocytes, and macrophages are the first cells to respond. T. gondii profilin interacts with Toll-like
receptor (TLR) 11 and TLR12 on dendritic cells. This is important for the host production of interleukin (IL)-12. In addition, macrophages
also induce tumor necrosis factor alpha (TNF-α) in response to TLR2- and TLR4-mediated detection of glycosylphosphatidylinositol (GPI)-
anchored parasite proteins. The innate immune response results in the production of interferon gamma (IFN-γ) from natural killer (NK) cells.
Eventually, this induces inflammatory mediators that control T. gondii multiplication and direct the developing adaptive immune system,
resulting in the expansion of T-helper type 1 (Th1) cells and cytolytic CD8+ T cells that secrete IFN- γ. Th1 cell differentiation is induced by
the release of IL-12 and IL-18, while T-helper type 17 (Th17) cell differentiation and expansion is promoted by transforming growth factor
beta (TGF-β), IL-6, and IL-23. This is counterregulated by T-helper type 2 (Th2) cells and regulatory T (Treg) cells mainly through the
production of IL-4 and IL-10. IL-10 and IL-27 are key to modulating these pathways and prevent the overproduction of Th1-type cytokines.
The production of IFN-γ during the innate and adaptive phases is responsible for activating cells to control parasite infection. IFN-γ
signaling promotes the development of signal transducer and activator of transcription 1 (STAT1; a nuclear transcription factor that controls
the expression of many genes) dependent antiparasitic effector mechanisms, including reactive oxygen intermediates and p47 GTPase
upregulation. The cell-mediated immune response that results limits parasite replication. IFNγR, Interferon gamma receptor; MC, mast cells;
MP, macrophages; ROS, reactive oxygen species.
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

Molan et al.
49 Family Medicine and Community Health 2016;4(4):44–62
RE
VIE
W
Tabl
e 1.
Im
mun
e re
spon
se to
Tox
opla
sma
gond
ii a
nd ty
pe 2
dia
bete
s m
ellit
us in
hum
an a
nd a
nim
al m
odel
s
Ana
lyze
d
biom
arke
r
B
iolo
gica
l act
ivity
Sel
ecte
d re
fere
nces
Type
2 d
iabe
tes
mel
litus
res
pons
e
Toxo
plas
ma
gond
ii re
spon
se
IL-1
β
PI. M
edia
tor
of in
flam
mat
ory
resp
onse
. Inv
olve
d in
a v
arie
ty o
f
cellu
lar
activ
ities
(ce
ll pr
olif
erat
ion,
diff
eren
tiatio
n, a
nd a
popt
osis
)
Secr
eted
fro
m m
onoc
ytes
: Mos
ser
and
Edw
ards
[33
], G
iulie
tti e
t al.
[34]
, Hat
anak
a
et a
l. [3
5], P
icku
p [3
6], P
icku
p an
d C
rook
[37
]
Ars
enije
vic
et a
l. [3
8], F
abia
ni e
t al.
[39]
, Tom
asik
et a
l. [4
0]
IL-6
PI
cyt
okin
e an
d A
I m
yoki
ne
(inc
reas
ed d
urin
g ex
erci
se).
Stim
ulat
es im
mun
e re
spon
se
Secr
eted
fro
m m
onoc
ytes
: Mos
ser
and
Edw
ards
[33
], G
iulie
tti e
t al.
[34]
, Hat
anak
a
et a
l. [3
5], P
icku
p [3
6], P
icku
p an
d C
rook
[37
]
Ars
enije
vic
et a
l. [3
8], F
ilise
tti a
nd C
ando
lfi [
41],
Prad
a an
d N
go-T
u [4
2], P
arlo
g et
al.
[21]
, Fab
iani
et a
l. [3
9]
IL-8
PI
. CX
CL
8. I
nduc
es c
hem
otax
is a
nd
phag
ocyt
osis
Secr
eted
fro
m m
onoc
ytes
: Mos
ser
and
Edw
ards
[33
], G
iulie
tti e
t al.
[34]
, Hat
anak
a
et a
l. [3
5], P
icku
p [3
6], P
icku
p an
d C
rook
[37
].
Secr
eted
fro
m β
cel
ls: J
agan
nath
an e
t al.
[43]
Parl
og e
t al.
[21]
IL-1
0
AI.
Dow
nreg
ulat
es e
xpre
ssio
n of
Th1
cyto
kine
s. I
nvol
ved
in r
egul
atio
n of
the
JAK
–STA
T s
igna
ling
path
way
Secr
eted
fro
m β
cel
ls: J
agan
nath
an e
t al.
[43]
.
Not
wel
l stu
died
Ars
enije
vic
et a
l. [3
8, 4
4], F
ilise
tti a
nd C
ando
lfi
[41]
, Gaz
zine
lli e
t al.
[45]
, Fab
iani
et a
l. [3
9]
IL-1
2
PI. I
nvol
ved
in d
iffe
rent
iatio
n of
nai
ve
T c
ells
into
Th1
cel
ls. S
timul
ated
prod
uctio
n of
IFN
-γ a
nd T
NF-
α
Weg
ner
et a
l. [4
6], T
siav
ou e
t al.
[47]
, Win
er
et a
l. [4
8]
Blis
s et
al.
[49]
, Fili
setti
and
Can
dolfi
[41
], G
azzi
nelli
et a
l. [5
0, 4
5], N
guye
n et
al.
[51]
, Par
log
et a
l. [2
1],
Fabi
ani e
t al.
[39]
, Lie
berm
an e
t al.
[60]
IL-1
7
PI. I
nduc
ed a
nd m
edia
tes
PI r
espo
nses
(pro
duct
ion
of I
L-6
, G-C
SF, G
M-C
SF,
IL-1
β, T
GF-
β, T
NF-
α)
Jaga
nnat
han
et a
l. [5
3], W
iner
et a
l. [5
4],
Her
der
et a
l. [5
5], A
ndri
anka
ja e
t al.
[56]
,
Osb
orn
et a
l. [5
7], Y
ang
et a
l. [5
8], A
cost
a-
Rod
rigu
ez e
t al.
[59]
Fabi
ani e
t al.
[39]
, Kel
ly e
t al.
[55]
TG
F-β
A
I. C
ontr
ols
prol
ifer
atio
n, c
ellu
lar
dif f
eren
tiatio
n
Lev
el in
crea
ses
in th
e pr
edia
betic
sta
ge
Fi
liset
ti an
d C
ando
lfi [
41],
Fis
cher
et a
l. [6
1],
Gaz
zine
lli e
t al.
[50]
, Nag
inen
i et a
l. [6
2]
IFN
-γ
PI. A
ctiv
ator
of
mac
roph
ages
and
indu
cer
of M
HC
cla
ss I
I m
olec
ule
expr
essi
on. A
berr
ant I
FN-γ
exp
ress
ion
is a
ssoc
iate
d w
ith a
num
ber
of
auto
infla
mm
ator
y an
d au
toim
mun
e
dise
ases
Jaga
nnat
han
et a
l. [5
3], F
euer
er e
t al.
[63]
,
Win
er e
t al.
[48]
Ara
ujo
and
Slif
er [
64],
Ars
enije
vic
et a
l. [6
5],
Filis
etti
and
Can
dolfi
[41
], G
avri
lesc
u an
d D
enke
rs
[66]
, Gaz
zine
lli e
t al.
[45]
, Her
mes
et a
l. [6
7], K
han
et a
l. [6
8], M
ordu
e et
al.
[69]
, Ngu
yen
et a
l. [5
1],
Peng
et a
l. [1
6], P
arlo
g et
al.
[21]
, Fab
iani
et a
l.
[39]
, Tom
asik
et a
l. [4
0]
TN
F-α
PI
. Inv
olve
d in
sys
tem
ic in
flam
mat
ion
Secr
eted
fro
m m
onoc
ytes
: Mos
ser
and
Edw
ards
[33
], G
iulie
tti e
t al.
[34]
, Hat
anak
a
et a
l. [3
5], P
icku
p [3
6], P
icku
p an
d C
rook
[37
]
Ars
enije
vic
et a
l. [4
4], B
liss
et a
l. [6
1], F
ilise
tti a
nd
Can
dolfi
[41
], G
azzi
nelli
et a
l. [5
0, 4
5], K
han
et a
l.
[68]
, Pra
da a
nd N
go-T
u [4
2], P
arlo
g et
al.
[21]
,
Fabi
ani e
t al.
[39]
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

The role of Toxoplasma gondii as a possible inflammatory agent
Family Medicine and Community Health 2016;4(4):44–62 50
RE
VIE
W
Ana
lyze
d
biom
arke
r
B
iolo
gica
l act
ivity
Sel
ecte
d re
fere
nces
Type
2 d
iabe
tes
mel
litus
res
pons
e
Toxo
plas
ma
gond
ii re
spon
se
IL-1
RA
A
I. M
embe
r of
the
IL-1
fam
ily.
Secr
eted
by
vari
ous
type
s of
cel
ls,
incl
udin
g im
mun
e ce
lls, e
pith
elia
l
cells
, and
adi
pocy
tes,
and
is a
nat
ural
inhi
bito
r of
the
PI e
ffec
t of
IL-1
β
Lev
el in
crea
ses
in th
e pr
edia
betic
sta
ge
IL-2
7
Bel
ongs
to th
e IL
-12
fam
ily.
Reg
ulat
es th
e ac
tivity
of
B a
nd T
lym
phoc
ytes
Lev
el in
crea
ses
in th
e pr
edia
betic
sta
ge
H
all e
t al.
[70]
IL-4
AI.
Ind
uces
dif
fere
ntia
tion
of n
aive
help
er T
cel
ls to
Th2
cel
ls
Ars
enije
vic
et a
l. [3
8], F
ilise
tti a
nd C
ando
lfi [
41],
Parl
og e
t al.
[21]
, Fab
iani
et a
l. [3
9]
Lep
tin
“S
atie
ty h
orm
one.
” M
ade
by a
dipo
se
cells
and
hel
ps to
reg
ulat
e en
ergy
bala
nce
by in
hibi
ting
hung
er
Ver
y hi
gh le
vel i
n pe
ople
with
type
2 d
iabe
tes
mel
litus
and
obe
sity
(hi
gh, >
15 n
g/m
L):
Mitt
endo
rfer
et a
l. [7
2], M
oon
et a
l. [7
3]
Bal
taci
and
Mog
ulko
c [7
1]
Adi
pone
ctin
M
odul
atio
n of
glu
cose
and
lipi
d
met
abol
ism
in in
sulin
-sen
sitiv
e tis
sues
Lev
el d
ecre
ases
in th
e pr
edia
betic
sta
ge
M
ilova
ncov
ic e
t al.
[74]
(un
affe
cted
)
PAI-
1
Key
reg
ulat
or o
f fib
rino
lysi
s,
thus
atte
nuat
es th
e ac
tivity
of
the
fibri
noly
tic s
yste
m
Auw
erx
et a
l. [7
5], V
ague
et a
l. [7
6], J
anss
on
et a
l. [7
7], S
amps
on e
t al.
[78]
, Juh
an-V
ague
et a
l. [7
9], P
ando
lfi e
t al.
[80]
, Gra
nt e
t al.
[81]
,
Nor
dt e
t al.
[82]
, Sch
neid
er a
nd S
obel
[83
],
Sobe
l et a
l. [8
4], A
less
i et a
l. [8
5], C
arm
assi
et a
l. [8
6], C
alle
s-E
scan
don
et a
l. [8
7], M
eigs
et a
l. [8
8]
Tom
asik
et a
l. [4
0]
IL-2
3
Secr
eted
by
activ
ated
den
driti
c ce
lls
and
activ
ated
mac
roph
ages
. IL
-23
func
tions
in in
nate
and
ada
ptiv
e
imm
unity
to r
egul
ate
Th1
7 fu
nctio
n
and
prol
ifer
atio
n
Men
sah-
Bro
wn
et a
l. [8
9], L
ukic
et a
l. [9
0]
(IL
-23
lead
s to
dia
bete
s in
duct
ion)
Muñ
oz e
t al.
[91]
, Lie
berm
an e
t al.
[52]
AI,
Ant
i-in
flam
mat
ory;
G-C
SF, g
ranu
locy
te c
olon
y st
imul
atin
g fa
ctor
; GM
-CSF
, gra
nulo
cyte
–mac
roph
age
colo
ny s
timul
atin
g fa
ctor
; IL
, int
erle
ukin
; IL
-1R
A, i
nter
leuk
in-1
rece
ptor
ant
agon
ist;
IFN
-γ, i
nter
fero
n ga
mm
a; J
AK
, Jan
us k
inas
e; P
AI-
1, p
lasm
inog
en a
ctiv
ator
inhi
bito
r 1;
PI,
pro
infla
mm
ator
y; S
TAT,
sig
nal t
rans
duce
r an
d ac
tivat
or o
f
tran
scri
ptio
n; T
GF-
β tr
ansf
orm
ing
grow
th f
acto
r be
ta; T
h1, T
hel
per
type
1; T
h2, T
hel
per
type
2; T
h17,
T h
elpe
r ty
pe 1
; TN
F-α,
tum
or n
ecro
sis
fact
or a
lpha
.
Tabl
e 1
(con
tinue
d)
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

Molan et al.
51 Family Medicine and Community Health 2016;4(4):44–62
RE
VIE
W
there are also experimental data that provide evidence for a
direct link between inflammation and T2DM, which supports
the notion that this disease is, at least in part, an inflammatory
condition [13, 112].
Serum-based inflammatory biomarkers as a measure-
ment of subclinical inflammation have been implicated in
the development of T2DM, with several cross-sectional and
prospective studies describing elevated circulating levels of
acute-phase proteins as well as chemokines and cytokines
(Table 1). For example, elevated levels of IL-6 (a cytokine
that induces differentiation of Th17 cells), IL-8, IL-17, TNF-
α, TGF-β, and IL-1β secreted from monocytes in T2DM
patients have been reported [33–35, 43, 53–59]. Elevated lev-
els of IL-1β, IL-6, and C-reactive protein are predictive of
T2DM [115, 117, 118], while elevated levels of IL-1 recep-
tor antagonist are observed during obesity and prediabetes
[117]. Moreover, patients with T2DM also have elevated
serum levels of IL-12, a cytokine that promotes Th1 cell dif-
ferentiation and increased INF-γ production [47, 48, 59, 63].
In addition, proinflammatory cytokines are increasingly
thought to contribute to the dysfunction and death of β cells
during the progression of T2DM [119]. More specifically, it
has been established that pancreatic β cells, as well as neural
cells, can be destroyed by several toxic agents and noxious
stimuli, such as reactive oxygen species, NO, and cytokines
(TNF-α, IL-1β, and IFN-γ) [4]. The subsequent proinflam-
matory cytokine balance has been directly linked to T2DM
by several in vivo studies showing that the inhibition of key
inflammatory cytokines protects rats from insulin resistance
[120–124]. These reports suggest that increased cytokine pro-
duction not only precedes but also maintains insulin resist-
ance in animals, hence adding detail to the current model of
T2DM being an inflammatory disease.
Spranger et al. [115] prospectively examined the effects
of IL-1β, IL-6, and TNF-α on the development of T2DM in
more than 27,000 individuals. In addition to observing ele-
vated levels of IL-6 and TNF-α in individuals with T2DM,
they also found that IL-6 was an independent predictor of
T2DM. Their data also showed that patterns of these cytokines
could modify the risk of T2DM by elevating the levels of both
IL-1β and IL-6, rather than elevating the level of IL-6 alone,
which increases the risk of T2DM. This strongly supports the
hypothesis that a subclinical inflammatory reaction has a role
in the pathogenesis of T2DM.
Possible association between obesity, T2DM, and
T. gondii infection
In addition to T2DM, obesity is also thought to be associ-
ated with a chronic systemic inflammatory response that
is characterized by altered cytokine production and activa-
tion of inflammatory signaling pathways [125]. Obesity has
previously been referred to as “a state of chronic inflamma-
tion” [26]. The increase in the production of inflammatory
cytokines, such TNF-α and IL-6, and certain adipokines
during the inflammatory process in obesity has been linked
to the development of insulin resistance by several reports
[126–128]. It is well established that insulin resistance pre-
cedes the onset of T2DM, at which point impaired glucose
tolerance occurs because of insulin deficiency as a result of
β-cell decomposition [129]. While there are several factors
such as genetics, environmental influences, and obesity that
link the development of insulin resistance with T2DM [129],
the possibility that obesity may enhance the release of inflam-
matory markers that trigger the development of insulin resist-
ance has generated interest in the field of diabetes for a number
of reasons. Predominantly, most individuals with T2DM are
overweight or obese, at least in the early phase, and obesity
is an established risk factor for the development of T2DM [2,
130, 131]. The significant increase in the incidence and preva-
lence of diabetes in the last 2 decades can be largely explained
by the global epidemic of obesity [131]. Furthermore, chronic
inflammation is associated with obesity, insulin resistance,
and T2DM, all of which belong to the metabolic diseases
known as “metabolic syndrome” [132].
The first molecular study to establish the link between
obesity, inflammation, and insulin resistance was published
in 1993 by Hotamisligil et al. [133]. They demonstrated that
the inflammatory cytokine TNF-α is constitutively expressed
in adipose tissue and overexpressed in obese rodents [133].
Furthermore, the inhibition of TNF-α in these obese rodents
significantly increased insulin sensitivity by increasing the
peripheral uptake of glucose in response to insulin. Two years
later, Hotamisligil et al. [134] examined the expression patterns
of TNF-α messenger RNA (mRNA) in the adipose tissues of
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

The role of Toxoplasma gondii as a possible inflammatory agent
Family Medicine and Community Health 2016;4(4):44–62 52
RE
VIE
W
19 obese and 18 control premenopausal women using Northern
blot analysis and found that the obese group expressed sig-
nificantly greater (2.5-fold, P<0.001) TNF-α mRNA in their
adipose tissue than the lean, control group. In addition, the
expression levels of TNF-α mRNA in adipose tissue showed
a strong correlation with the level of hyperinsulinemia, which
is an indirect measure of insulin resistance [134]. Lastly, they
also observed an increase in insulin sensitivity and a 45%
decrease in TNF-α mRNA expression (P<0.001) in adipose
tissue when the obese individuals reduced their body weight.
In support of a direct role for inflammation in the develop-
ment of obesity, the above-mentioned results were validated
by a human study that found a significant increase in adipose
TNF-α mRNA levels with increasing adiposity and that a sig-
nificant correlation exists between TNF-α mRNA levels and
percent body fat (P<0.05) [135]. It was also found that TNF-α
levels markedly decreased with weight loss and in some indi-
viduals the decreases were up to 46% of initial levels (P<0.02).
Further studies established TNF-α as a potent insulin receptor
inhibitor, hence further implicating this cytokine in the insulin
resistance of T2DM and obesity [136].
With use of animal models it has also been shown that
diet-induced obese mice have more T cells in adipose tissue
than lean, control mice [137], and diet-controlled weight loss
is associated with reduced adipose tissue inflammation [137].
In addition, the development of insulin resistance and other
metabolic abnormalities, including reduced metabolic rates,
has also been associated with chronic inflammation [138,
139]. Accordingly, it is possible that the chronic proinflamma-
tory state induced by latent T. gondii infection precipitates (in
nonobese individuals), perpetuates, or exacerbates (in obese
individuals) the inflammation-related weight gain. Reeves
et al. [130] reported a positive association between T. gondii
seropositivity and obesity from a sample of 999 human adults.
Their results showed that individuals with positive T. gon-
dii serological findings had twice the odds of being obese
(BMI >30 kg/m2) than seronegative individuals (P=0.01).
Furthermore, it has been proposed that infection with
T. gondii may be associated with obesity because of the abil-
ity of the organism to alter inflammatory fat distribution as
it adjusts and resides in fatty tissues [29]. Certainly, exces-
sive gestational weight gain has been previously reported
from a study comparing pregnant woman infected with T.
gondii with uninfected pregnant woman [120]. In that study,
979 mothers (mean age 30 years, range 19–44 years) were
examined, and 194 (19.2%) tested positive for T. gondii infec-
tion and 758 (80.2%) tested negative for T. gondii infection
during the 16th week of pregnancy. The study authors col-
lected data on maternal weight and other parameters before
pregnancy and at the 16th, 20th, 30th, and 36th weeks of
pregnancy. One of the main findings of the study was that
the mean maternal weight of the T. gondii-infected mothers
before pregnancy was much higher (63.6 kg) than that of the
uninfected mothers (61.5 kg). In addition, the effect of T. gon-
dii infection on weight gain was significant in the 16th week
(P=0.006) and the 20th week (P=0.049) of pregnancy and
nearly significant in the 30th week (P=0.073). Moreover, the
extra weight remained until the end of the pregnancies. In
another study, a murine pregnancy model was used to mimic
toxoplasmosis complications in humans and it was found that
dams infected with high doses of T. gondii tachyzoites had
significant excess body weight gain that led to fetal abortion
or mummified embryos [121]. Consistent with the conclusions
of the study authors, excessive weight gain during pregnancy,
especially in the first trimester, usually indicates a negative
effect of some internal (genetic or epigenetic) or external
(environmental) factor(s) [120].
Since obesity in humans is characterized by hyperleptine-
mia and a body-weight-suppressing response to exogenous
leptin [122], it is very interesting to note that the hormone
leptin has also been implicated in the T. gondii host inflamma-
tory response [29]. Using a rat model, Baltaci and Mogulkoc
[71] found that infection with T. gondii caused a significant
increase of plasma leptin concentrations (P<0.01), with no
change in body weight of the animals. They concluded that
this biomarker, similar in structure to IL-2, exerts a proinflam-
matory action in obese and diabetic individuals.
T. gondii may also influence motivation- and reward-driven
behaviors (e.g., overeating) via altered dopamine pathways.
Animal models have shown that T. gondii influences both
dopamine release and availability of the rate-limiting enzyme
in dopamine synthesis [121]. Dopamine antagonists can block
behavioral changes in T. gondii infected rats, and thus para-
site-induced host behavioral changes that may be driven by
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

Molan et al.
53 Family Medicine and Community Health 2016;4(4):44–62
RE
VIE
W
survival needs may have inadvertent effects on eating patterns
that promote obesity [130].
Accordingly, it can be concluded that the association
between T. gondii infection and obesity is very significant and
could alter the current views on obesity management and other
important public health issues related to obesity, especially
diabetes.
Studies describing the association between
diabetes and T. gondii infection
Only a handful of case–control epidemiological studies that
demonstrate the possible association between T. gondii infec-
tion and T2DM have been published [123, 124, 140, 141].
Most of these studies were conducted in less developed coun-
tries such as Iran, and showed an overall positive association
between T. gondii infection and T2DM (P≤0.001) (Table 2).
Gokce et al. [123] investigated the seropositivity rates
of anti-T. gondii antibodies in 807 individuals with T2DM
(351 men, 456 woman, age range 15–88 years, mean age and
standard deviation 52.8±14.01 years) in comparison with 250
healthy sex- and aged-matched controls (110 men, 140 woman,
age range 18–75 years, mean age and standard deviation
51.9±13.4 years). Using commercial IgG and IgM ELISA kits,
they found IgG antibodies more frequently in the T2DM group
(n=457, 56.6%) than in healthy, control group (n=56, 22.4%,
P<0.001). IgM antibodies were also found in 19 of the patients
with T2DM (2.4%) and only in four controls (1.6%, P=0.3).
Furthermore, a striking correlation was observed between the
duration of diabetes and T. gondii IgG seropositivity (Table 3).
In another serological study, anti-T. gondii IgG antibod-
ies were detected in 60.4% of diabetic patients (n=91) com-
pared with 37.7% of individuals in the healthy, control group
(n=93) [124]. In the same study, the risk of T. gondii infection
in diabetic patients was two-fold higher in comparison with
the healthy controls (odds ratio 2.21, 95% confidence interval
1.6–3.7, P=0.001). However, a similar study also conducted in
2013, by Siyadatpanah et al. [140], did not find a statistically
significant difference in the prevalence of T. gondii in diabetic
(52.6%) and nondiabetic (50.6%) individuals when they meas-
ured the IgG and IgM levels in a cohort of 300 individuals.
Most recently, Saki et al. [141] investigated the prevalence
of anti-T. gondii IgG and IgM antibodies in a cohort of 110 Tabl
e 2.
Des
crip
tion
of d
ata
extr
acte
d fr
om th
e ca
se–c
ontr
ol s
tudi
es d
escr
ibin
g as
soci
atio
n be
twee
n To
xopl
asm
a go
ndii
infe
ctio
n an
d ty
pe 2
dia
bete
s m
ellit
us
Ref
eren
ce
N
Age
M
atch
inga
D
efini
tion
M
easu
re o
f Tox
opla
sma
gond
ii ex
posu
re
Ser
opre
vale
nce
P
T2D
M
posi
tive
(n)
T2D
M
nega
tive
(n)
T2D
M p
ositi
ve
and
Toxo
plas
ma
gond
ii po
sitiv
e (n
) T
2DM
neg
ativ
e
and
Toxo
plas
ma
gond
ii po
sitiv
e (n
)
Saki
et a
l. [1
41]
(201
6, I
ran)
22
0
Adu
lts
No
U
nspe
cifie
d Ig
G/I
gM E
LIS
A (T
rini
ty B
iote
ch,
USA
) and
IFA
kit
(Eur
oim
mun
,
Uni
ted
Kin
gdom
)
11
0 11
0 50
(45
.4%
) 24
(21
.8%
) <0
.001
Siya
datp
anah
et a
l.
[140
] (20
13, I
ran)
30
0
Adu
lts
Uns
peci
fied
Uns
peci
fied
IgG
/IgM
EL
ISA
(V
IRO
,
Ger
man
y)
15
0 15
0 79
(52
.7%
) 76
(50
.6%
) 0.
729
Shir
bazo
u et
al.
[124
] (2
013,
Ira
n)
184
A
dults
Y
es
Uns
peci
fied
IgG
EL
ISA
(Pi
shta
z Te
b Z
aman
Dia
gnos
tics,
Ira
n)
91
93
55
(60
.4%
) 36
(38
.7%
) 0.
003
Gok
ce e
t al.
[123
]
(200
8, T
urke
y)
10
57
15–8
8 ye
ars
Yes
A
DA
Ig
G/I
gM E
LIS
A a
nd I
FAT,
imm
unofl
uore
scen
ce a
ntib
ody
test
(E
uroi
mm
un)
80
7 25
0 45
7 (5
6.6%
) 56
(22
.4%
) <0
.001
Tota
l
1761
–
–
–
–
11
58
603
641
(55.
4%)
192
(31.
8%)
<0.0
01
AD
A, A
mer
ican
Dia
bete
s A
ssoc
iatio
n; I
FA, i
mm
unofl
uore
scen
ce; I
FAT,
; T
2DM
, typ
e 2
diab
etes
mel
litus
.a M
atch
ing
is b
y ag
e an
d se
x al
one.
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

The role of Toxoplasma gondii as a possible inflammatory agent
Family Medicine and Community Health 2016;4(4):44–62 54
RE
VIE
W
diabetic pregnant woman and 110 nondiabetic controls and
found that the prevalence rate of both IgG antibody and IgM
antibody in the former group (42.7% and 2.7% respectively)
was significantly higher than in the latter group (21.8% and
0% respectively). They also examined the prevalence rate of
IgG antibody with the duration of diabetes and reported find-
ings similar to those of Gokce et al. [123] in that the preva-
lence rate of IgG antibody was directly associated with the
duration of diabetes.
An animal study has also shown a positive correlation
between both the T. gondii parasite load and the IgM antibody
titer and blood glucose levels in diabetic rats infected with
T. gondii and monitored at 15-day intervals for 60 days after
induction of diabetes and 105 days after T. gondii infection
[142]. During this period, Hassanain et al. [142] observed a sig-
nificant increase in the number of T. gondii cysts (in the brain)
and blood glucose levels in the infected diabetic group when
compared with the uninfected diabetic group. In addition, the
elevated brain parasite load and the IgM titer corresponded
to the elevated glucose levels in the infected diabetic group,
which led to the view that T. gondii infection can increase the
risk of developing diabetes [142].
Modrek et al. [143] reported a significantly high T. gondii
seroprevalence rate of 70.7% among diabetic patients in Iran
(n=205; age range 13–60 years). In addition, the difference
between the presence of toxoplasma antibodies and fasting
blood glucose levels was statistically significant (P<0.05).
While this study lacked a control group, a meta-analysis of
35 reports to determine the seroprevalence rate of T. gondii
infection in the Iranian general population reported an over-
all seroprevalence rate of 39.3% (95% confidence interval
33.0%–45.7%) [144]. This supports the suggestion of an asso-
ciation between T. gondii infection and diabetes in humans.
In addition, there is also evidence of possible indirect
associations between T. gondii infection and certain forms of
diabetes. For example, in 2015, researchers from the Czech
Republic reported significant findings from a cross-sectional
study looking at latent toxoplasma infection and the levels of
blood glucose in pregnant woman [27]. Serum samples for the
determination of T. gondii seropositivity were collected during
the 9th–12th gestational weeks from two subsets of woman
who were later screened for gestational diabetes mellitus in
the 24th–28th gestational weeks at the General University
Hospital in Prague in 2008. Pregnant women with latent toxo-
plasmosis had significantly higher (P=0.010) blood glucose
levels and a higher prevalence (19.5%) of gestational diabetes
mellitus (n=532, P=0.033) in the 24th–28th gestational weeks
than pregnant women without toxoplasmosis (healthy controls,
12%) (Table 4).
The role of T. gondii in the pathogenesis of T2DM remains
an enigma. One possible explanation could be the inflamma-
tory-mediated destruction of pancreatic β cells which leads to
the reduction in β-cell mass that ultimately contributes to the
failure of the β cell to produce enough insulin. This in turn
would increase the risk of developing acute and chronic pancre-
atitis as well as diabetes [5, 145–147]. Additionally, it is known
that T2DM is associated with an increased risk of developing
acute pancreatitis [148] and that T. gondii infection can cause
pancreatic tissue necrosis [149]. IL-18 and IL-1β are partly
responsible for the development of pancreatic inflammation,
and the neutralization of either of these cytokines decreases
inflammation [93]. Accordingly, individuals infected with T.
gondii may be at greater risk of developing diabetes than unin-
fected individuals. Certainly, insulin has been shown to have a
stimulatory effect on the in vitro replication of T. gondii [131].
Insulin and d-glucose have been shown to have a synergistic
Table 3. Relationship between the seropositivity rates of anti-Toxoplasma gondii antibodies analyzed in 807 individuals with type 2 diabetes
mellitus and duration of the disease (according to Gokce et al. [123])
Duration of T2DM (years) IgG-positive individuals (n) IgG-negative individuals (n)
0–5 39 (16.0%) 193 (83.2%)
6–10 149 (51.6%) 140 (48.4%)
>11 269 (94.1%) 17 (5.9%)
T2DM, type 2 diabetes mellitus.
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

Molan et al.
55 Family Medicine and Community Health 2016;4(4):44–62
RE
VIE
W
dose-responsive stimulating effect on the replication of T. gon-
dii tachyzoites in vitro [150].
Lastly, it has been argued that the simple explanation for
the relationship between T. gondii and T2DM may be that
diabetic patients are more susceptible to parasitic infections
because of the possibility of a suppressed immune system,
decreased arterial perfusion, and neuropathy [151–153].
However, there is evidence to suggest otherwise. A study
looking at parasitic infections in 200 diabetic individuals (16
with type 1 diabetes mellitus and 184 with T2DM) and 1024
nondiabetic individuals found no significant differences with
regard to the incidences of the presence of intestinal parasites
between the T2DM group and the control group (47% and
56% respectively) [154]. In light of above-mentioned reports,
there is a strong clinical and laboratory evidence to support
the notion of a genuine association between chronic latent T.
gondii infection and development of T2DM.
Desirable criteria for good-quality studies
on this topic
If the serological and inflammatory associations between T.
gondii and T2DM are further replicated, this will warrant the
need for further research to clarify whether T. gondii increases
the risk of developing T2DM, and the mechanisms that drive
this relationship, if any. Current reports are limited to associa-
tion studies, meaning that the authors are not able to determine
if there is a causal relationship between T. gondii and T2DM
or obesity, including possible reverse causality (i.e., T2DM
increasing the risk of T. gondii infection) or shared causal-
ity (i.e., common factor causing both T2DM and T. gondii
infection). In addition, studies measuring only T. gondii IgG
and IgM seropositivity cannot shed any light on the possible
inflammatory association between T2DM and T. gondii infec-
tion. Therefore future studies specifically designed to investi-
gate the role of T. gondii in T2DM using inflammation as the
main outcome are urgently needed so as to provide a more
robust link between T. gondii infection and T2DM. To this
extent, studies examining selected inflammatory biomark-
ers that are altered in the serum during and in the absence of
chronic T. gondii infection in individuals with T2DM versus
healthy controls without T2DM will be highly valuable.
Good-quality epidemiological research is warranted to
further help draw conclusions on this important subject. The
design should be a population-based case–control study with
samples representative of all age-groups matched for all pos-
sible confounding parameters. These studies should also use
standard criteria to define various parameters, especially
the diagnosis of T2DM, in addition to setting inclusion and
Table 4. Description of data highlighting the possible indirect association between latent Toxoplasma gondii infection with blood glucose levels
and gestational diabetes mellitus (according to Kankova et al. [27])
Subset 1 (n=191 pregnant women)
OGTT in the 24th–28th GW With Toxoplasma gondii Without Toxoplasma gondii P
Mean baseline BGL (mmol/L) after 8-h fast 5.04 (95% CI 4.92–5.17) 4.88 (95% CI 4.82–4.95) 0.026
Mean BGL (mmol/L) 60 min after administration of
glucose solution
7.73 (95% CI 7.09–8.37) 6.89 (95% CI 6.61–7.16) 0.007
Mean BGL (mmol/L) 120 min after administration of
glucose solution
6.43 (95% CI 6.03–6.83) 5.74 (95% CI 5.55–5.93) 0.001
0.010
Subset 2 (n=532 pregnant women)
GDM in the 24th–28th GW With Toxoplasma gondii (n) Without Toxoplasma gondii (n) P
24/123 (19.5%) 49/409 (12%) 0.033
Blood samples for Toxoplasma gondii analysis were collected in the 9th–12th gestational weeks.
BGL, Blood glucose level; CI, confidence interval; GDM, gestational diabetes mellitus; GW, gestational weeks; OGTT, oral glucose
tolerance test.
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

The role of Toxoplasma gondii as a possible inflammatory agent
Family Medicine and Community Health 2016;4(4):44–62 56
RE
VIE
W
exclusion criteria according to latest definitions of diabetes by
the World Health Organization. Lastly, individuals with psy-
chiatric conditions, including personality disorders, should
be excluded from these studies because of the association
between positive T. gondii serological findings and various
forms of serious mental illness, as previously reported [18,
21, 115].
Concluding remarks
T2DM is characterized by impaired pancreatic β-cell
function resulting in insulin resistance with an underlying
subclinical inflammation. Many reports suggest that obe-
sity and insulin resistance are associated with elevated
circulating levels of inflammatory cytokines such as IL-2,
IL-6, IL-12, IFN-γ, and TNF-α, raising the possibility that
metabolic abnormalities in diabetes may originate from or
be exacerbated by cytokine overproduction. A possible con-
tributor to this prolonged low-grade inflammation that sub-
sequently leads to the clinical expression of T2DM is the
protozoan parasite T. gondii. Cellular immunity has been
established as the key component of the host’s immune
response to infection by T. gondii. A wealth of data points
to the major role of T lymphocytes and type 1 cytokines
during murine T. gondii infection. In the clinical setting,
this has been confirmed by the emergence of T. gondii as
a major opportunistic infectious pathogen during the pro-
gression of many disorders and diseases. Thus T. gondii is
rapidly emerging as a pathogen of potential interest in obe-
sity and diabetes research. Much progress has been made
in recent years in determining how early innate responses,
namely, production of cytokines such as IL-12 and TNF-α,
drive the development of the strong Th1 response to T. gon-
dii. While this response is crucial to the control of infection,
it is becoming increasingly appreciated that an excessively
vigorous response induced by the parasite can lead to patho-
logical changes in the host. Therefore it is important for the
host and parasite to maintain tight control of the inflamma-
tory response, thereby prolonging it to the detriment of the
host. Human studies investigating inflammatory biomarkers
specifically in T2DM individuals with T. gondii infection
are urgently needed to provide a more robust link between
T. gondii infection and T2DM.
Conflict of interest
The authors declare no conflict of interest.
Funding
This work was partly supported by an Australia–China
international joint research grant (NHMRC-APP1112767-
NSFC-81561128020).
References1. Shaw J, Tanamas S. Diabetes: the silent pandemic and its impact
on Australia. Diabetes Australia [Internet]. [accessed 2016
Jul 12]. Available from: www.healthinfonet.ecu.edu.au/key-
resources/bibliography/?lid=22970.
2. American Diabetes Association. Diagnosis and classification of
diabetes mellitus. Diabetes Care 2008;31:S55–60.
3. Diabetes Australia. [Internet] Diabetes in Australia. [accessed
2016 Jul 1]. Available from: www.diabetesaustralia.com.au/dia-
betes-in-australia.
4. Prandota J. T. gondii infection acquired during pregnancy
and/or after birth may be responsible for development of both
type 1 and 2 diabetes mellitus. J Diabetes Metab 2013;4(2):
1000241.
5. Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard
H, Eppler R, et al. Mechanisms of β-cell death in type 2 diabetes.
Diabetes 2005;54(Suppl 2):S108.
6. International Diabetes Federation. [Internet] IDF diabetes atlas
(sixth edition). [accessed 2016 Jul 1]. Available from: www.idf.
org/sites/default/files/EN_6E_Atlas_Full_0.pdf.
7. International Diabetes Federation. [Internet] IDF diabetes atlas
(seventh edition) [Internet]. [accessed 2016 Jul 1]. Available
from: www.diabetesatlas.org/.
8. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence
of diabetes: estimates for the year 2000 and projections for 2030.
Diabetes Care 2004;27(5):1047–53.
9. Lipscombe LL, Hux JE. Trends in diabetes prevalence, incidence,
and mortality in Ontario, Canada 1995–2005: a population-based
study. Lancet 2007;369(9563):750–6.
10. World Health Organization. [Internet] Global status report on
noncommunicable diseases 2014. Geneva: World Health Organi-
zation. [accessed 2016 Jul 12]. Available from: www.who.int/
nmh/publications/ncd-status-report-2014/en/.
11. World Health Organization. [Internet] Diabetes program.
Geneva: World Health Organization. [accessed 2016 Jul 12].
Available from: www.who.int/diabetes/en/.
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

Molan et al.
57 Family Medicine and Community Health 2016;4(4):44–62
RE
VIE
W
12. World Health Organization. [Internet] Global Health Estimates:
Deaths by Cause, Age, Sex and Country, 2000–2012. Geneva,
WHO. [accessed 2016 July 12]. Available from: www.who.int/
healthinfo/global_burden_disease/en/.
13. Pradhan A. Obesity, metabolic syndrome, and type 2 diabetes:
inflammatory basis of glucose metabolic disorders. Nutr Rev
2007;65(12 Pt 2):S152–6.
14. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin
resistance. J Clin Invest 2006;116(7):1793–801.
15. Nicolle C, Manceaux L. Sur une infection à corps de Leishman
(ou organismes voisins) du gondi. C R Acad Sci 1908;147:763–6.
16. Peng HJ, Chen XC, Lindsay DS. A review: competence, compro-
mise, and concomitance-reaction of the host cell to Toxoplasma
gondii infection and development. J Parasitol 2011;94(4):620–8.
17. Centers for Disease Control and Prevention. [Internet] Parasites
– toxoplasmosis (toxoplasma infection). Centers for Disease
Control and Prevention. [accessed 2016 Jul 12]. Available from:
www.cdc.gov/parasites/toxoplasmosis/epi.html.
18. Halonen SK, Weiss LM. Toxoplasmosis. In: Garcia HH, Tanow-
itz HB, Del Brutto OH, editors. Neuroparasitology and tropical
neurology. Handbook of clinical neurology, vol. 114 (3rd series).
Amsterdam: Elsevier; 2013. pp. 125–45.
19. Pappas G, Roussos N, Falagas ME. Toxoplasmosis snapshots:
global status of Toxoplasma gondii seroprevalence and implica-
tions for pregnancy and congenital toxoplasmosis. Int J Parasitol
2009;39(12):1385–94.
20. Hunter CA, Sibley LD. Modulation of innate immunity by Toxo-
plasma gondii virulence effectors. Nat Rev 2012;10(11):766–88.
21. Parlog A, Schluter D, Dunay IR. Toxoplasma gondii-induced
neuronal alterations. Parasite Immunol 2015;37(3):159–70.
22. Cong W, Liu G, Meng Q, Dong W, Qin S, Zhang F, et al. Toxo-
plasma gondii infection in cancer patients: Prevalence, risk fac-
tors, genotypes and association with clinical diagnosis. Cancer
Lett 2015;359(2):307–13.
23. Tenter A, Heckeroth A, Weiss M. Toxoplasma gondii: from ani-
mals to humans. Int J Parasitol 2000;30(12–13):1217–58.
24. Robert-Gangneux F, Dardé ML. Epidemiology of and diag-
nostic strategies for toxoplasmosis. Clin Microbiol Rev
2012;25(2):264–96.
25. Albuquerque MC, Aleixo AL, Benchimol EI, Leandro AC, das
Neves LB, Vicente RT, et al. The IFN-γ +874T/A gene polymor-
phism is associated with retinochoroiditis toxoplasmosis suscep-
tibility. Mem Inst Oswaldo Cruz 2009;104(3):451–5.
26. Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link
between insulin resistance, obesity and diabetes. Trends Immu-
nol 2004;25(1):4–7.
27. Kankova S, Flegr J, Calda P. An elevated blood glucose level and
increased incidence of gestational diabetes mellitus in pregnant
woman with latent toxoplasmosis. Folia Parasitol 2015;62. pii:
2015.056.
28. Dupey JP. The history of Toxoplasma gondii – the first 100 years.
J Eukaryot Microbiol 2008;55(6):467–75.
29. Carter CJ. Toxoplasmosis and polygenic disease susceptibility
genes: extensive Toxoplasma gondii host/pathogen interactome
enrichment in nine psychiatric or neurological disorders. J Pat-
hog 2013;2013:965046.
30. Henriques SA, Brett R, Alexander J, Pratt J, Roberts CW. Neu-
ropsychiatric disease and Toxoplasma gondii infection. Neuro-
immunomodulation 2009;16(2):122–33.
31. Frenkel JK. Adoptive immunity to intracellular infection. J
Immunol 1967;98(6):1309–19.
32. Melo MB, Jensen KD, Saeij J. Toxoplasma gondii effectors are
master regulators of the inflammatory response. Trends Parasitol
2011;27(11):487–95.
33. Mosser DM, Edwards JP. Exploring the full spectrum of mac-
rophage activation. Nat Rev Immunol 2008;8(12):958–69.
34. Giulietti A, van Etten E, Overbergh L, Stoffels K, Bouillon R,
Mathieu C. Monocytes from type 2 diabetic patients have a pro-
inflammatory profile. 1,25-Dihydroxyvitamin D3 works as anti-
inflammatory. Diabetes Res Clin Pract 2007;77(1):47–57.
35. Hatanaka E, Monteagudo PT, Marrocos MS, Campa A. Neutrophils
and monocytes as potentially important sources of proinflamma-
tory cytokines in diabetes. Clin Exp Immunol 2006;146(3):443–7.
36. Pickup JC. Inflammation and activated innate immunity in the
pathogenesis of type 2 diabetes. Diabetes Care 2004;27(3):813–23.
37. Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the
innate immune system? Diabetologia 1998;41(10):1241–8.
38. Arsenijevic D, Girardier L, Seydoux J, Chang HR, Dulloo AG.
Altered energy balance and cytokine gene expression in a murine
model of chronic infection with Toxoplasma gondii. Am J Phys-
iol 1997;272(5 Pt 1):E908–17.
39. Fabiani S, Pinto B, Bonuccelli, Bruschi F. Neurobiological stud-
ies on the relationship between toxoplasmosis and neuropsychi-
atric diseases. J Neurol Sci 2015;351(1-2):3–8.
40. Tomasik J, Schultz TL, Kluge W, Yolken RH, Bahn S, Carruthers
VB. Shared Immune and Repair Markers During Experimental
Toxoplasma Chronic Brain Infection and Schizophrenia. Schizophr
Bull 2016; 42(4):386–95.
41. Filisetti D, Candolfi E. Immune response to Toxoplasma gondii.
Ann Inst Super Sanita 2004;40(1):71–80.
42. Prada J, Ngo-Tu T. Overexpression and protective role of
interleukin6, tumor necrosis factor-alpha and nitric oxides in
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

The role of Toxoplasma gondii as a possible inflammatory agent
Family Medicine and Community Health 2016;4(4):44–62 58
RE
VIE
W
experimental and clinical ocular toxoplasmosis. Int J Infect Dis
2008;12(1):e388.
43. Jagannathan M, McDonnell M, Liang Y, Hasturk H, Hetzel J,
Rubin D, et al. Toll-like receptors regulate B cell cytokine produc-
tion in patients with diabetes. Diabetologia 2010;53(7):1461–71.
44. Arsenijevic D, Girardier L, Seydoux J, Pechere JC, Garcia I,
Lucas R, et al. Metabolic-cytokine responses to a second immu-
nological challenge with LPS in mice with T. gondii infection.
Am J Physiol 1998;274(3 Pt 1):E439–45.
45. Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T,
Cheever A, Kuhn R, et al. In the absence of endogenous IL-10,
mice acutely infected with Toxoplasma gondii succumb to a
lethal immune response dependent upon CD4+ T cells and
accompanied by overproduction of IL-12, IFN-γ, and TNF-α. J
Immunol 1996;157(2):798–805.
46. Wegner M, Winiarska H, Bobkiewicz-Kozłowska T, Dworacka
M. IL-12 serum levels in patients with type 2 diabetes treated
with sulphonylureas. Cytokine 2008;42(3):312–16.
47. Tsiavou A, Degiannis D, Hatziagelaki E, Koniavitou K, Raptis
SA. Intracellular IFN-γ production and IL-12 serum levels in
latent autoimmune diabetes of adults (LADA) and in type 2 dia-
betes. J Interferon Cytokine Res 2004;24(7):381–7.
48. Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al.
Normalization of obesity associated insulin resistance through
immunotherapy. Nat Med 2009;15(8):921–9.
49. Bliss SK, Marshall AJ, Zhang Y, Denkers EY. Human poly-
morphonuclear leukocytes produce IL-12, TNF-alpha and
the chemokines macrophageinflammatory protein-1a and
-1b in response to Toxoplasma gondii antigens. J Immunol
1999;162(12):7369–75.
50. Gazzinelli RT, Amichay D, Scharton-Kersten T, Grunwald E,
Farber JM, Sher A. Role of macrophage-derived cytokines in
the induction and regulation of cell-mediated immunity to Toxo-
plasma gondii. Curr Top Microbiol Immunol 1996;219:127–39.
51. Nguyen TD, Bigaignon G, Markine-Goriaynoff D, Heremans
H, Nguyen TN, Warnier G, et al. Virulent Toxoplasma gondii
strain RH promotes T-cellin dependent overproduction of pro-
inflammatory cytokines IL12 and g-interferon. J Med Microbiol
2003;52(Pt 10):869–76.
52. Lieberman LA, Cardillo F, Owyang AM, Rennick DM, Cua
DJ, Kastelein RA, et al. IL-23 provides a limited mechanism
of resistance to acute toxoplasmosis in the absence of IL-121.
J Immunol 2004;173(3):1887–93.
53. Jagannathan M, McDonnell ME, Shin H, Rehman Q, Hasturk
H, Apovian CM, et al. Elevated proinflammatory cytokine pro-
duction by a skewed T cell compartment requires monocytes
and promotes inflammation in type 2 diabetes. J Immunol
2011;186(2):1162–72.
54. Winer S, Paltser G, Chan Y, Tsui H, Engleman E, Winer D, et al.
Obesity predisposes to Th17 bias. Eur J Immunol 2009;39(9):
2629–35.
55. Herder C, Zierer A, Koenig W, Roden M, Meisinger C, Thorand
B. Transforming growth factor-β1 and incident type 2 diabetes:
results from the MONICA/KORA case-cohort study, 1984–2002.
Diabetes Care 2009;32(10):1921–3.
56. Andriankaja OM, Barros SP, Moss K, Panagakos FS, DeVizio
W, Beck J, et al. Levels of serum interleukin (IL)-6 and gingival
crevicular fluid of IL-1β and prostaglandin E2 among non-smok-
ing subjects with gingivitis and type 2 diabetes. J Periodontol
2009;80(2):307–16.
57. Osborn O, Brownell SE, Sanchez-Alavez M, Salomon D, Gram
H, Bartfai T. Treatment with an interleukin 1 beta antibody
improves glycemic control in diet-induced obesity. Cytokine
2008;44(1):141–8.
58. Yang L, Anderson DE, Baecher-Allan C, Hastings WD,
Bettelli E, Oukka M, et al. IL-21 and TGF-β are
required for differentiation of human TH
17 cells. Nature
2008;454(7202):350–2.
59. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto
F. Interleukins 1β and 6 but not transforming growth factor-β
are essential for the differentiation of interleukin 17–producing
human T helper cells. Nat Immunol 2007;8(9):942–9.
60. Kelly MN, Kolls JK, Happel K, Schwartzman JD, Schwarzen-
berger P, Combe C, et al. Interleukin-17/interleukin-17 receptor-
mediated signaling is important for generation of an optimal
polymorphonuclear response against Toxoplasma gondii infec-
tion. Infect Immun 2005;73(1):617–21.
61. Fischer HG, Nitzgen B, Reichmann G, Hadding U. Cytokine
responses induced by Toxoplasma gondii in astrocytes and
microglial cells. Eur J Immunol 1997;27(6):1539–48.
62. Nagineni CN, Detrick B, Hooks JJ. Transforming growth
factor-beta expression in human retinal pigment epithelial
cells is enhanced by Toxoplasma gondii: a possible role in the
immunopathogenesis of retinochoroiditis. Clin Exp Immunol
2002;128(2):372–8.
63. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A,
et al. Lean, but not obese, fat is enriched for a unique population
of regulatory T cells that affect metabolic parameters. Nat Med
2009;15(8):930–9.
64. Araujo FG, Slifer T. Different strains of Toxoplasma gondii
induce different cytokine responses in CBA/Ca mice. Infect
Immun 2003;71(7):4171–4.
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

Molan et al.
59 Family Medicine and Community Health 2016;4(4):44–62
RE
VIE
W
65. Arsenijevic D, Bilbao FD, Giannakopoulos P, Girardier L,
Samec S, Richard D. A role for interferon-gamma in the hyper-
metabolic response to murine toxoplasmosis. Eur Cytokine Netw
2001;12(3):518–27.
66. Gavrilescu LC, Denkers EY. IFN-γ overproduction and high level
apoptosis are associated with high but not low virulence Toxo-
plasma gondii infection. J Immunol 2001;167(2):902–9.
67. Hermes G, Ajioka JW, Kelly KA, Mui E, Roberts F, Kasza K,
et al. Neurological and behavioral abnormalities, ventricular
dilatation, altered cellular functions, inflammation, and neuronal
injury in brains of mice due to common, persistent, parasitic
infection. J Neuroinflammation 2008;5:48.
68. Khan IA, Schwartzman JD, Matsuura T, Kasper L. A dichotomous
role for nitric oxide during acute Toxoplasma gondii infection in
mice. Proc Natl Acad Sci USA 1997;94(25):13960–95.
69. Mordue DG, Monroy F, La Regina M, Dinarello CA, Sibley
LD. Acute toxoplasmosis leads to lethal overproduction of Th1
cytokines. J Immunol 2001;167(8):4574–84.
70. Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana
CG, et al. The cytokines interleukin 27 and interferon-γ promote
distinct Treg cell populations required to limit infection-induced
pathology. Immunology 2012; 37(3):511–23.
71. Baltaci AK, Mogulkoc R. Plasma leptin levels in rats with induced
Toxoplasma gondii infection. Bratisl Med J 2012;113(2):67–9.
72. Mittendorfer B, Horowitz JF, DePaoli AM, McCamish MA, Pat-
terson BW, Klein S. Recombinant human leptin treatment does
not improve insulin action in obese subjects with type 2 diabetes.
Diabetes 2011;60(5):1474–7.
73. Moon HS, Matarese G, Brennan AM, Chamberland JP, Liu X,
Fiorenza CG, et al. Efficacy of metreleptin in obese patients with
type 2 diabetes: cellular and molecular pathways underlying lep-
tin tolerance. Diabetes 2011;60(6):1647–56.
74. Milovanovi I, Vujani M, Klun I, Bobi B, Nikoli A, Ivovi
V, et al. Toxoplasma gondii infection induces lipid metabolism
alterations in the murine host. Memórias do Instituto Oswaldo
Cruz. 2009;104(2):175–8.
75. Auwerx J, Bouillon R, Collen D, Geboers J. Tissue-type
plasminogen activator antigen and plasminogen activator inhibi-
tor in diabetes mellitus. Arteriosclerosis 1988;8(1):68–72.
76. Vague P, Juhan-Vague I, Aillaud MF, Badier C, Viard R, Alessi
MC, et al. Correlation between blood fibrinolytic activity, plas-
minogen activator inhibitor level, plasma insulin level, and
relative body weight in normal and obese subjects. Metabolism
1986;35(3):250–3.
77. Jansson JH, Johansson B, Boman K, Nilsson TK. Hypo-
fibrinolysis in patients with hypertension and elevated choles-
terol. J Intern Med 1991;229(4):309–16.
78. Sampson M, Kong C, Patel A, Unwin R, Jacobs HS. Ambulatory
blood pressure profiles and plasminogen activator inhibitor (PAI-
1) activity in lean women with and without the polycystic ovary
syndrome. Clin Endocrinol (Oxf) 1996;45(5):623–9.
79. Juhan-Vague I, Vague P, Alessi MC, Badier C, Valadie J, Aillaud
MF, Alan C. Relationships between plasma insulin triglyceride,
body mass index, and plasminogen activator inhibitor 1. Diabetes
Metab 1987;13(3 Pt 2):331–6.
80. Pandolfi A, Cetrullo D, Polishuck R, Alberta MM, Calafiore
A, Pellegrini G, et al Plasminogen activator inhibitor type 1 is
increased in the arterial wall of type II diabetic subjects. Arterio-
scler Thromb Vasc Biol 2001;21(8):1378–82.
81. Grant MB, Ellis EA, Caballero S, Mames RN. Plasminogen
activator inhibitor-1 overexpression in nonproliferative diabetic
retinopathy. Exp Eye Res 1996;63(3):233–44.
82. Nordt TK, Sawa H, Fujii S, Sobel BE. Induction of plasminogen
activator inhibitor type-1 (PAI-1) by proinsulin and insulin in
vivo. Circulation 1995;91(3):764–70.
83. Schneider DJ, Sobel BE. PAI-1 and diabetes: a journey from the
bench to the bedside. Diabetes Care 2012;35(10):1961–7.
84. Sobel BE, Woodcock-Mitchell J, Schneider DJ, Holt RE,
Marutsuka K, Gold H. Increased plasminogen activator inhibi-
tor type 1 in coronary artery atherectomy specimens from type
2 diabetic compared with nondiabetic patients: a potential fac-
tor predisposing to thrombosis and its persistence. Circulation
1998;97(22):2213–21.
85. Alessi MC, Peiretti F, Morange P, Henry M, Nalbone G, Juhan-
Vague I. Production of plasminogen activator inhibitor 1 by
human adipose tissue: possible link between visceral fat accumu-
lation and vascular disease. Diabetes 1997;46(5):860–7.
86. Carmassi F, Morale M, Ferrini L, Dell’Omo G, Ferdeghini M,
Pedrinelli R, et al. Local insulin infusion stimulates expression
of plasminogen activator inhibitor-1 and tissue-type plasmi-
nogen activator in normal subjects. Am J Med 1999;107(4):
344–50.
87. Calles-Escandon J, Mirza SA, Sobel BE, Schneider DJ. Induc-
tion of hyperinsulinemia combined with hyperglycemia and
hypertriglyceridemia increases plasminogen activator inhibitor 1
in blood in normal human subjects. Diabetes 1998;47(2):290–3.
88. Meigs JB, Wilson PW, Fox CS, Vasan RS, Nathan DM,
Sullivan LM, et al. Body mass index, metabolic syndrome, and
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

The role of Toxoplasma gondii as a possible inflammatory agent
Family Medicine and Community Health 2016;4(4):44–62 60
RE
VIE
W
risk of type 2 diabetes or cardiovascular disease. J Clin Endo-
crinol Metab 2006;91(8):2906–12.
89. Mensah-Brown EP, Shahin A, Al-Shamisi M, Wei X, Lukic ML.
IL-23 leads to diabetes induction after subdiabetogenic treat-
ment with multiple low doses of streptozotocin. Eur J Immunol
2006;36(1):216–23.
90. Lukic ML, Shahin A, Mensah-Brown E, Al-Hakim A. Il-23
enhances diabetes induction in the wild type but not in INF-
gamma –/– mice. J Immunol 2007;178:129–36.
91. Muñoz M, Heimesaat MM, Danker K, Struck D, Lohmann
U, Plickert R, et al. Interleukin (IL)-23 mediates Toxoplasma
gondii–induced immunopathology in the gut via matrixmetal-
loproteinase-2 and IL-22 but independent of IL-17. J Exp Med
2009;206(13):3047.
92. Lang C, Grob U, Luder CGK. Subversion of innate and adap-
tive immune responses by Toxoplasma gondii. Parasitol Res
2007;100(2):191–203.
93. Dupont CD, Christian DA, Hunter CA. Immune response and
immunopathology during toxoplasmosis. Semin Immunopathol
2012;34(6):793–813.
94. Gazzinelli RT, Hieny A, Wynn T, Wolf S, Sher A. IL-12 is
required for the T-lymphocyte-independent induction of inter-
feron γ by an intracellular parasite and induces resistance in
T-cell-deficient hosts. Proc Natl Acad Sci USA 1993;90(13):
6115–9.
95. Lindberg RE, Frenkel JK. Toxoplasmosis in nude mice. J Parasi-
tol 1977;63(2):219–21.
96. Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S,
Caspar P, et al. Parasite-induced IL-12 stimulates early IFN-
gamma synthesis and resistance during acute infection with Tox-
oplasma gondii. J Immunol 1994;153(6):2533–43.
97. Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Inter-
feron-gamma: the major mediator of resistance against Toxo-
plasma gondii. Science 1988;240(4851):516–8.
98. Parker SJ, Roberts CW, Alexander J. CD81 T cells are the major
lymphocyte subpopulation involved in the protective immune
response to Toxoplasma gondii in mice. Clin Exp Immunol
1991;84(2):207–12.
99. Suzuki Y, Remington JS. Dual regulation of resistance against
Toxoplasma gondii infection by Lyt-21 and Lyt-11, L3T41 T
cells in mice. J Immunol 1988;140(11):3943–6.
100. Suzuki Y, Remington JS. The effect of anti-IFN-gamma antibody
on the protective effect of lyt-21 immune T cells against toxo-
plasmosis in mice. J Immunol 1990;144(5):1954–6.
101. Montoyoa JG. Laboratory diagnosis of Toxoplasma gondii infec-
tion and toxoplasmosis. J Infect Dis 2002;85(1):S73–82.
102. Walpole IR, Hodgen N, Bower C. Congenital toxoplasmosis: a
large survey in Western Australia. Med J Aust 1991;154(11):720–4.
103. Opsteegh M, Haveman R, Swart AN, Mensink-Beerepoot ME,
Hofhuis A, Langelaar MFM, et al. Seroprevalence and risk fac-
tors for Toxoplasma gondii infection in domestic cats in the
Netherlands. Prev Vet Med 2012;104(3–4):317–26.
104. Kolbekova P, Kourbatova E, Novotna M, Kodym P, Flegr J. New
and old risk-factors for Toxoplasma gondii infection: prospec-
tive cross-sectional study among military personnel in the Czech
Republic. Clin Microbiol Infect 2007;13(10):1012–7.
105. Villena I, Aubert D, Gomis P, Ferté H, Inglard JC, Denis-Bisiaux
H, et al. Evaluation of a strategy for Toxoplasma gondii oocyst
detection in water. Appl Environ Microbiol 2004;70(7):4035–9.
106. VanWormer E, Miller MA, Conrad PA, Grigg ME, Rejmanek
D, Carpenter TE, et al. Using molecular epidemiology to track
Toxoplasma gondii from terrestrial carnivores to marine hosts:
implications for public health and conservation. PLoS Negl Trop
Dis 2014;8(5):e2852.
107. Yap G, Pesin M, Sher A. Cutting edge: IL-12 is required for
the maintenance of IFN-gamma production in T cells mediat-
ing chronic resistance to the intracellular pathogen, Toxoplasma
gondii. J Immunol 2000;165(2):628–31.
108. Gaddi PJ, Yap S. Cytokine regulation of immunopathology in
toxoplasmosis. Immunol and Cell Biol 2007;85(2):155–9.
109. Neyer LE, Grunig G, Fort M, Remington JS, Rennick D, Hunter
CA. Role of interleukin-10 in regulation of T-cell-dependent and
T-cell-independent mechanisms of resistance to Toxoplasma
gondii. Infect Immun 1997;65(5):1675–82.
110. Denkers EY, Gazzinelli RT. Regulation and function of T-cell-
mediated immunity during Toxoplasma gondii infection. Clin
Microbiol Rev 1998;11(4):569–88.
111. Gigley JP, Fox BA, Bzik DJ. Cell-mediated immunity to Toxo-
plasma gondii develops primarily by local Th1 host immune
responses in the absence of parasite replication. J Immunol
2009;182(2):1069–78.
112. Chagas CEA, Borges MC, Martini LA, Rogero MM. Focus
on vitamin D, inflammation and type 2 diabetes. Nutrients
2012;4(1):52–67.
113. Pickup JC, Mattock MB, Chuaney GD, Burt D. NIDDM as a
disease of the innate immune system: association of acute-phase
reactants and interleukin-6 with metabolic syndrome X. Diabeto-
logia 1997;40(11):1286–92.
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

Molan et al.
61 Family Medicine and Community Health 2016;4(4):44–62
RE
VIE
W
114. Tilg H, Moschen AR. Inflammatory mechanisms in the regula-
tion of insulin resistance. Mol Med 2008;14(3–4):222–31.
115. Spranger J, Kroke A, Möhlig M, Hoffmann K, Bergmann MM,
Ristow M, et al. Inflammatory cytokines and the risk to develop
type 2 diabetes: results of the prospective population-based Euro-
pean Prospective Investigation into Cancer and Nutrition (EPIC)-
Potsdam Study. Diabetes 2003;52(3):812–7.
116. Grimble RF. Inflammatory status and insulin resistance. Curr
Opin Clin Nutr Metab Care 2002;5(5):551–9.
117. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory
disease. Nat Rev Immunol 2011;11(2):98–107.
118. Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reac-
tive protein, interleukin 6, and risk of developing type 2 diabetes
mellitus. J Am Med Assoc 2001;286(3):327–34.
119. Ramadan JW, Steiner SR, O’Neill CM, Nunemaker CS. The cen-
tral role of calcium in the effects of cytokines on beta-cell func-
tion: implications for type 1 and type 2 diabetes. Cell Calcium
2011;50(6):481–90.
120. Flegr J. Influence of latent Toxoplasma infection on human per-
sonality, physiology and morphology: pros and cons of the Toxo-
plasma–human model in studying the manipulation hypothesis.
J Exp Biol 2013;216(Pt 1):127–33.
121. Oz HS, Tobin T. Atovaquone ameliorate gastrointestinal toxo-
plasmosis complications in a pregnancy model. Med Sci Monit
2012;18(9):BR337–45.
122. Coppari R, Bjørbæk C. The potential of leptin for treating
diabetes and its mechanism of action. Nat Rev Drug Discov
2012;11(9):692–70.
123. Gokce C, Yazar S, Bayram F, Gundogan K, Yaman O, Sahin I.
Anti-Toxoplasma gondii antibodies in type 2 diabetes. Natl Med
J India 2008;21(1):51.
124. Shirbazou S, Delpisheh A, Mokhetari R, Tavakoli G. Serologic
detection of anti Toxoplasma gondii infection in diabetic patients.
Iran Red Crescent Med J 2013;15(8):701–3.
125. Shih YL, Ho KT, Tsao CH, Chang YH, Shiau MY, Huang CN,
et al. Role of cyotkines in metabolism and type 2 diabetes mel-
litus. Int J Biomed Lab Sci 2013;2(1):1–6.
126. Andersson CX, Gustafson B, Hammarstedt A, Hedjazifar S,
Smith U. Inflamed adipose tissue, insulin resistance and vascular
injury. Diabetes Metab Res Rev 2008;24(8):595–603.
127. Gustafson B, Hammarstedt A, Andersson CX, Smith U.
Inflamed adipose tissue: a culprit underlying the metabolic
syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol
2007;27(11):2276–83.
128. Maury E, Brichard SM. Adipokine dysregulation, adipose tis-
sue inflammation and metabolic syndrome. Mol Cell Endocrinol
2010;314(1):1–16.
129. King GL. The role of inflammatory cytokines in diabetes and its
complications. J Periodontol 2008;79(8 Suppl):1527–34.
130. Reeves GM, Mazaheri S, Snitker S, Langenberg P, Giegling I,
Hartmann A, et al. A positive association between T. gondii sero-
positivity and obesity. Front Public Health 2013;1:73.
131. Oz H. Toxoplasmosis, pancreatitis, obesity, and drug discovery.
Pancreat Disord Ther 2014;4(2):138.
132. Hotamisligil GS. Inflammation and metabolic disorders. Nature
2006;444(7121):860–7.
133. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expres-
sion of tumor necrosis factor-alpha: direct role in obesity-linked
insulin resistance. Science 1993;259(5091):87–91.
134. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman
BM. Increased adipose tissue expression of tumor necrosis fac-
tor-alpha in human obesity and insulin resistance. J Clin Invest
1992;95(5):2409–15.
135. Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo
RB. The expression of tumor necrosis factor in human adipose
tissue. Regulation by obesity, weight loss, and relationship to
lipoprotein lipase. J Clin Invest 1995;95(5):2111–9.
136. Hotamisligil GS, Spiegelman BM. Tumor necrosis fac-
tor a: a key component of the obesity-diabetes link. Diabetes
1994;43(11):1271–8.
137. Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Ver-
non AH. Interferon-gamma, a Th1 cytokine, regulates fat
inflammation: a role for adaptive immunity in obesity. Circ Res
2008;103(5):467–76.
138. Priceman SJ, Kujawski M, Shen S, Cherryholmes GA, Lee H,
Zhang C. Regulation of adipose tissue T cell subsets by Stat3 is
crucial for diet-induced obesity and insulin resistance. Proc Natl
Acad Sci USA 2013;110(32):13079–84.
139. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in
obesity. Annu Rev Immunol 2011;29:415–45.
140. Siyadatpanah A, Tabatabaie F, Oormazdi H, Meamar AH, Razm-
jou E, Hadighi R, et al. Comparison of anti-toxoplasma IgG and
IgM antibodies determined by ELISA method in diabetic and
non-diabetic individuals in west Mazandaran province, Iran,
2011–2012. Ann Biol Res 2013;4(6):281–5.
141. Saki J, Shafieenia S, Foroutan-Rad M. Seroprevalence of toxo-
plasmosis in diabetic pregnant women in southwestern of Iran.
J Parasit Dis 2016;40(4):1586.
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from

The role of Toxoplasma gondii as a possible inflammatory agent
Family Medicine and Community Health 2016;4(4):44–62 62
RE
VIE
W
142. Hassanain MA, El-Fadaly HA, Hassanain NA. Toxoplasma gon-
dii parasite load elevation in diabetic rats as latent opportunistic
character. Ann Trop Med Public Health 2014;7(2):110–5.
143. Modrek M J, Saravani R, Mousavi M, Khorashad AS, Piri M.
Investigation of IgG and IgM antibodies against Toxoplasma
gondii among diabetic patients. Int J Infect 2015;2(3):e27595.
144. Daryani A, Sarvi S, Aarabi M, Mizani A, Ahmadpour E, Shokri
A, et al. Seroprevalence of Toxoplasma gondii in the Iranian
general population: a systematic review and meta-analysis. Acta
Trop 2014;137:185–94.
145. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler
PC. Beta-cell deficit and increased beta-cell apoptosis in humans
with type 2 diabetes. Diabetes 2013;52(1):102–10.
146. Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C,
Yagihashi S. Reduced beta-cell mass and expression of oxida-
tive stress-related DNA damage in the islet of Japanese type II
diabetic patients. Diabetologia 2002;45(1):85–96.
147. Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, et al.
Selective beta-cell loss and alpha-cell expansion in patients
with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab
2003;88(5):2300–8.
148. Gonzalez-Perez A, Schlienger RG, Rodríguez LA. Acute pancre-
atitis in association with type 2 diabetes and antidiabetic drugs.
Diabetes Care 2010;33(12):2580–5.
149. Waree P. Toxoplasmosis: pathogenesis and immune response.
Thammasat Med J 2008;8(4):487–96.
150. Zhu S, Lai D, Li S, Lun Z. Stimulative effects of insulin on
Toxoplasma gondii replication in 3T3-L1 cells. Cell Biol Int
2006;30(2):149–53.
151. Akinbo FO, Olujobi SO, Omoregie R, Egbe E. Intestinal parasitic
infections among diabetes mellitus patients. Biomark Genomic
Med 2013;5(1–2):44–7.
152. Mohtashamipour M, Hoseini SG, Pestehchian N, Yousefi H, Fal-
lah E, Hazratatian T. Intestinal parasitic infections in patients
with diabetes mellitus: a case-control study. J Anal Res Clin Med
2015;3(3):157–63.
153. Bessman AN, Sapico FL. Infections in the diabetic patient: the
role of immune dysfunction and pathogen virulence factors.
J Diabetes Complications 1992;6(4):258–62.
154. Nazligul Y, Sabuncu T, Ozbilge H. Is there a predisposition
to intestinal parasitosis in diabetic patients? Diabetes Care
2001;24(8):1503–4.
Related Information
Family Medicine and Community Health can provide you more papers focusing on diabetes mellitus. Please find them below.
• Correlation between KCNQ1 gene polymorphism and type 2 diabetes mellitus in Huaihai region of China
http://www.ingentaconnect.com/content/cscript/fmch/2014/00000002/00000001/art00003
• Four-year dynamic observation and study on standardized management of elderly patients with type 2 diabetes in Beijing
Yongding Road Community
http://www.ingentaconnect.com/content/cscript/fmch/2014/00000002/00000002/art00005
• Hypomagnesemia is a risk factor for metabolic syndrome and type 2 diabetes mellitus in native Balinese
http://www.ingentaconnect.com/content/cscript/fmch/2013/00000001/00000001/art00003
• Availability and social determinants of community health management service for patients with chronic diseases: An
empirical analysis on elderly hypertensive and diabetic patients in an eastern metropolis of China
http://www.ingentaconnect.com/content/cscript/fmch/2015/00000003/00000001/art00003
• Exploring point-of-care transformation in diabetic care: A quality improvement approach
http://www.ingentaconnect.com/content/cscript/fmch/2015/00000003/00000002/art00004
• Longitudinal study of a community hospital integrated model for diabetes management in the Beijing Jingsong community
http://www.ingentaconnect.com/content/cscript/fmch/2014/00000002/00000001/art00004
on Novem
ber 8, 2021 by guest. Protected by copyright.
http://fmch.bm
j.com/
Fam
Med C
om H
ealth: first published as 10.15212/FM
CH
.2016.0128 on 1 Decem
ber 2016. Dow
nloaded from