the roles of the voa subunit of the vacuolar - tspace … · · 2014-02-19a subunit of the...
TRANSCRIPT

The roles of the Voa subunit of the vacuolar
H+-ATPase in dense-core vesicle acidification,
transmitter uptake and storage
by
Ner Mu Nar Saw
A thesis submitted in conformity with the requirements for the degree of
Master of Science
Graduate Department of Physiology
University of Toronto
© Copyright by Ner Mu Nar Saw 2011

The roles of the Voa subunit of the vacuolar H+-ATPase in
dense-core vesicle acidification, transmitter uptake and storage
Ner Mu Nar Saw
Master of Science
Graduate Department of Physiology
University of Toronto
2011
Abstract The Vo sector of the vacuolar H+-ATPase is a multi-subunit complex that forms a
proteolipid pore. The largest subunit in this complex is the a subunit which has four
isoforms (a1-a4). The isoform(s) critical for secretory vesicle acidification has yet to be
identified. Using a cell line derived from rat pheochromocytoma in which Voa1 and/or
Voa2 had been down-regulated this study revealed that Voa1, and to a lesser extent,
Voa2 are critical for acidifying dense-core vesicles (DCVs). The acidification defects
resulting from down-regulation of Voa1 and Voa1/ Voa2 were suppressed by the
expression of knockdown-resistant Voa1. Defects in DCV acidification resulted in
reductions in their transmitter uptake and storage. Lastly, Ca2+-dependent peptide
secretion appeared normal in Voa1 and Voa1/ Voa2 knockdown cells. . This study
demonstrated that Voa1 and Voa2 cooperatively regulate dense-core vesicle acidificatio
as well as transmitter uptake/storage, while they may not be critical for dense-core ve
n
sicle
xocytosis.
e
ii

Acknowledgements
First and foremost I would like to express my sincerest gratitude to my supervisor,
Dr. Shuzo Sugita, for all his encouragement and expert guidance during my Master's
study. I also thank Drs. William Trimble and Herbert Gaisano for their helpful insights
and constructive criticisms regarding my project. Next, I would like to thank Nan Chang
(Toronto Western Research Institute) for her instructions on Flow Cytometry. I would
also like to thank Audrey Darabie (University of Toronto) for her instructions on electron
microscopy. Furthermore, I would like to thank Soo-Young Ann Kang for her help with
some of the HPLC measurements and construct making as well as the NPY-hPLAP
secretion assays. Lastly, I would like to thank Gayoung Anna Han and Leon Parsaud for
their help with some of the immunofluorescence confocal microscopy experiments.
iii

Table of Contents Abbreviations.................................................................................................................vi Figure Summary..........................................................................................................vii I. Introduction...............................................................................................................1 i) The Vacuolar H+-ATPase a) Overview of the V-ATPase................................................................3 b) Mechanism and Regulation of the Proton Pump Function of the V-ATPase.........................................................................8 c) Intracellular V-ATPases....................................................................12 d) Plasma Membrane V-ATPases..........................................................14 e) The Vo Domain and Membrane Fusion.............................................16 ii) The Voa Subunit a) Overview of the Voa subunit..............................................................18 b) The Voa Isoforms...............................................................................20 iii) Background of the model used to study Voa function a) The PC12 cell as a model to study Voa function................................23 b) Dense-core vesicles and the regulated secretory pathway.................24 c) Catecholamine synthesis and uptake into dense-core vesicles........................................................................................25 iv) Purpose and significance of this study..........................................................28 II. Hypotheses................................................................................................................29 III. Materials and Methods i) Growth and maintenance of the PC12 cells....................................................30 ii) Construction of fluorescent protein-tagged Voa1, Voa2 and Voa3 constructs and generation of PC12 cell lines expressing these recombinant proteins......................................................................32 iii) Immunofluorescence confocal microscopy...................................................34 iv) Reverse transcription-polymerase chain reaction..........................................37 v) Construction of Voa1 and Voa2 knockdown plasmids and generation of stable Voa1 and/or Voa2 knockdown PC12 cell lines.........................38 vi) Western Blot analyses of the generated PC12 cell lines................................41 vii) Construction of Neuropeptide Y-based reporter constructs.........................43 viii) Transfection of NPY-based reporter constructs into various
iv

PC12 cells and subsequent FACS analyses............................................44 ix) Construction of knockdown-resistant human Voa1 construct and generation of hVoa1-expressing Voa1-knockdown and Voa1/Voa2-double knockdown PC12 cells..............................................46 x) Measurement of [3H]-NA uptake into PC12 cells...........................................47 xi) Measurement of endogenous dopamine in PC12 cells by HPLC..................48 xii) Measurement of NPY-hPLAP secretion from PC12 cells............................50 IV. Results i) Voa1, Voa2, and Voa3 are differentially localized in PC12 cells; Voa4 is not expressed in these cells..........................................................51 ii) Western Blot results for Voa1-, Voa2-knockdown and Voa1/Voa2-double knockdown cells; compensatory up-regulation of V
.....................74
7
normal in Voa1KD and DKD ..................................................................82
... .......... . ...97
vi) Future Directions..........................................................................................101
eferences.....................................................................................................................102
Appendix II.........................................................................................................113
oa2 and Ac45 is seen in Voa1 knockdown cells.....................................................58 iii) Knockdown of Voa1, but not of Voa2, results in a significant reduction in dense-core vesicle acidification............................................61 iv) Double knockdown Voa1 and Voa2 caused dramatic reductions in dense-core vesicle acidification............................................................64 v) The expression of knockdown-resistant human Voa1 suppressed the acidification defects caused by down-regulation of endogenous Voa1 and Voa1/Voa2.............................................................71 vi) Catecholamine uptake is significantly reduced in Voa1/ Voa2 double-knockdown cells...................................................... vii) Endogenous dopamine contents are significantly reduced in Voa1 and/or Voa2 knockdown cells....................................7 viii) Ca2+-dependent regulated secretion of transfected peptide is V. Discussion i) Summary and Conclusion.................................................................................85 ii) Localization of Voa isoforms as determinant for V-ATPase localization........90 iii) Voa1 and Voa2 in DCV acidification; alternate interpretations......................93 iv) The Voa subunit and membrane fusion..................................... .. .. v) The suitability of NPY-epHluorin as a dense-core vesicle pH reporter.....................................................................................................99 R Appendices Appendix I..........................................................................................................112
v

Abbreviations
um cid
FT roblast
aline
ution
tein Receptor
Protein CP Valosin-Containing Protein
[3H]-NA Tritium-labelled noradrenaline (or norepinephrine) DCV Dense-core vesicle DMEM Dulbecco’s Modified Eagle MediEDTA Ethylenediaminetetraacetic aEM Electron Microscopy ER Endoplasmic Reticulum GFP Green Fluorescence Protein GSH Reduced glutathione HEK293- Human Embryonic Kidney-FibMunc18 Mammalian uncoordinated 18NGF Nerve Growth Factor PBS Phosphate-Buffered SPEI Polyethylenimine PFA Paraformaldehyde PI Propidium iodide PSS Physiological Saline SolRNAi RNA interference RT Room Temperature RT-PCR Reverse transcription-Polymerase chain reactionshRNA Short-Hairpin RNA SNAR Soluble NSF Attachment proESNM Silent Nucleotide Mutation SV Synaptic Vesicle TGN Trans Golgi Network Unc Uncoordinated VAMP Vesicle Associated MembraneV
vi

vii
igure Summary
ses
s
protein-fused Voa subunit to be
2 subunits
s
cells
5 in DKD
in Voa2KD
igure 3. Proper localization of NPY-EmGFP into DCVs is seen in both
ct of increasing concentrations of NH4Cl on
ct seen in DKD using the ionophores
results showing the expression of knockdown-resistant human
igure 8. FAC ress the
Voa1 and Voa1/Voa2
re
sfected peptide is not affected by knockdown of Voa1 or
Voa1/ Voa2
F Figure 1. Schematic depiction of the subunit structure of mammalian V-ATPa
Figure 2. Biosynthesis and uptake of catecholamine into dense-core vesicle
Figure 3. Design of shRNA knockdown sequence and predicted structure
Figure 4. Schematic representation of fluorescent
expressed; resulting western blots
Figure 5. Intracellular localization of recombinant Voa1 and Voa
Figure 6. Intracellular localization of recombinant Voa3 subunit
Figure 7. Intracellular localization of endogenous Voa1 and Voa2 subunit
Figure 8. RT-PCR result showing absence of Voa4 mRNA in PC12
Figure 9. Western blot results of Voa1KD, Voa2KD and DKD cells
Figure 10. Upregulation of Voa2 and Ac45 in Voa1KD; upregulation of Ac4
Figure 11. DCV acidification defect is seen in Voa1KD but not
Figure 12. Severe defect in DCV acidification is seen in DKD
F 1
DKD and its control
Figure 14. Lower magnification picture of NPY-EmGFP localization into DCVs
Figure 15. FACS results showing the effe
DKD fluorescence
Figure 16. Calibration of the acidification defe
nigericin and monensin
Figure 17. Western blot
Voa1
F 1 S results showing that knockdown-resistant human Voa1 can supp
acidification defect caused by knockdown of
Figure 19. Uptake of norepinephrine into DCV is ATP-dependent
Figure 20. Uptake of norepinephrine is significantly reduced in DKD
Figure 21. HPLC measurements of whole-cell lysates show that dopamine
content is significantly reduced in Voa1KD, Voa2KD, and DKD
Figure 22. HPLC measurements of partially-purified plasma membrane dense-co
vesicles show that dopamine content is severely reduced in DKD
Figure 23. Secretion of tran

I. Introduction
In eukaryotic cells the generation and maintenance of a low pH in the lumen of
various intracellular organelles is critical for normal functioning of the cells. The pH of
the cytoplasm is about 7.2 while the pH of intracellular organelles such as the Golgi
apparatus (pH 6.0-6.6), endosomes (pH 6.0-6.8), lysosomes (pH 4.5-5.4) and secretory
granules (pH 5.0-5.5) are significantly lower (Saroussi and Nelson, 2009). The
maintenance of a low pH in intracellular organelles is crucial for processes such as post-
translational protein modifications, protein sorting, degradation of proteins and other
macromolecules, and secondary transport of transmitters.
Although knowledge of the molecular players involved in the generation and
maintenance of organellar pH is sparse at this point the concept of how pH regulation can
occur in these organelles is firmly established. The maintenance of a low pH is thought to
be facilitated by at least two factors: the ability of the organelle to pump protons into its
lumen and the ability of the organelle to fine-tune luminal proton concentration via
passive ion transport. The vacuolar H+-ATPase (V-ATPase), which is the focus of this
study, has been prominently established as the primary proton pump in organelles
requiring a low pH. Therefore, theoretically, regulating V-ATPase activity can serve as
one important control point for regulating pH. In fact, there has been much evidence
which describe the different ways in which V-ATPase function can be regulated (more on
this in a later section).
1

Passive ion transport which depends on membrane conductance of the ions in
question serve as a second important control point for pH regulation. In the absence of
passive ion transport, i.e. proton pumping by V-ATPases is the only determinant of
membrane potential, the theoretical maximal H+ concentration can be calculated based
solely on thermodynamic considerations, yielding luminal pH of ~3.0 (Paroutis et al.,
2004). This calculated pH value is about two magnitudes lower than the actual pH values
observed in experimental conditions suggesting that passive ion transport plays a
significant role in determining the steady-state pH of the organelle. In fact, proton 'leak'
through putative H+ channels has been shown to be important for determining steady-
state pH in experiments looking at the profiles of organellar pH increase in the presence
of the V-ATPase inhibitor concanamycin. The importance of proton 'leak' in maintaining
a low pH inside the Golgi has also been inferred from the use of Zn2+ to inhibit Zn2+-
sensitive proton channels (Paroutis et al., 2004).
In addition to proton 'leak', the conductance of other ions may also contribute to
the determination of organellar pH. For example, the influx of Cl- ions to counteract the
build up of membrane potential caused by the proton pump have been shown to be
important for maintaining a low pH in endosomes. Furthermore, it was determined that
chloride channel protein 5 (CLC-5) was the mediator of this chloride conductance as
defects in this chloride channel resulted in defects in endosomal acidification (Gunther et
al., 1998, Igarashi et al., 1998, Luchow et al., 1998). Therefore, it is readily conceivable
that counterion conductance for other ions such as K+ and Na+ which help fine-tune and
maintain organellar pH may exist although the molecular entities required for these
functions have yet to be confirmed.
2

I. i) The Vacuolar H+-ATPase
I. i. a) Overview of the V-ATPase The vacuolar H+-ATPase (V-ATPase) is a multisubunit protein complex that
plays crucial functions in pH regulation in eukaryotic cells and is largely conserved from
yeast to human. The V-ATPase primarily functions as a proton pump, utilizing the energy
from ATP hydrolysis to move protons across membrane barriers such as from the cytosol
into the lumen of intracellular compartments or from the cytosol to the outside of the cell
(Nishi and Forgac, 2002, Forgac, 2007). V-ATPases were first discovered on the
membranes of intracellular organelles of the vacuolar system. For example, these proton
pumps were isolated from chromaffin granules of the adrenal medulla (Cidon and Nelson,
1983), chlathrin-coated vesicles (Stone, 1983) (Forgac and Cantly, 1984) and yeast
vacuoles (Anraku and Wada, 1989). Soon afterwards, the presence of V-ATPases at the
plasma membrane was also confirmed beginning with their discovery on the plasma
membrane of insect epithelial cells (Wieczorek and Klein, 1991).
V-ATPases are structurally and mechanistically related to F-ATPases, ATP
synthases which are exclusively located on the mitochondrial membrane, as well to A-
ATPases, ATP synthases that are found in archaebacteria (Forgac, 2007, Marshansky and
Futai, 2008). Aside from being large multisubunit complexes that are made up of a
peripheral ATP-hydrolytic domain and an integral proton translocation domain the three
3

ATPases use the rotary mechanism (described below) to translocate protons. The major
difference between V-ATPases and the other two ATPases is that V-ATPases normally
function to use the energy from ATP to transport protons against it gradient while the
ATP synthases normally function to produce ATP using the flow of protons down their
concentration gradient (Forgac, 2007).
V-ATPases consist of two domains: a peripheral V1 domain that is 650 kDa and a
membrane integral Vo domain that is 250 kDa (see Figure 1) (Forgac, 2007, Xu et al.,
2007). The V1 domain is composed of eight different subunits [A (70 kDa), B (56 kDa),
C (42 kDa), D (34 kDa), E (31 kDa), F (14 kDa), G (13 kDa), H (50 kDa)] and is
responsible for ATP hydrolysis (Xu et al., 2007). Subunits A and B are arranged such that
three copies of each subunit combine alternately to form a hexamer which contain
binding sites for ATP hydrolysis at the interface of these subunits. In addition to the A-B
hexamer the V1 sector is made up of two copies each of subunit E and G, one or two
copies of subunit H and single copies of the remaining subunits. The mammalian Vo
sector is composed of seven different subunits [a (100-110 kDa), c (16 kDa), c" (21 kDa),
d (38-42 kDa), e (9 kDa), ac45 (45 kDa), and M8-9 (8-9 kDa)] and is responsible for
forming the pathway for proton translocation (Xu et al., 2007). The Vo sector contains a
single copy of the large a subunit, four or five copies of the proteolipid c subunit and
single copies of the remaining subunits (Forgac, 2007).
Since much of the knowledge regarding structure and function of the V-ATPase
that have been gained have come from studies of yeast V-ATPase a brief introduction of
the yeast V-ATPase as this point is appropriate. The subunit composition of the yeast V-
ATPase is very similar to that of the mammalian V-ATPase described above except that
4

the yeast V-ATPase does not contain the accessory proteins ac45 and M8-9. The yeast V-
ATPase also has a gene coding for the proteolipid c' that is not found in mammals. Lastly,
except for the a subunit which has two isoforms (Vph1p and Stv1p) all the other subunits
which make up the yeast V-ATPase are encoded by a single gene (Nishi and Forgac,
2002).
Figure 1: A schematic depiction of the subunit structure of mammalian V-ATPases
Xu et al., Histology and Pathology (2007)
5

In mammalian cells some of the subunits which make up the V-ATPase contain
multiple isoforms. The B, E, and H subunits of V1 each have two isoforms while the C
and G subunits each have three. For the Vo domain the d subunit have two isoforms while
the a subunit have four isoforms, the most of all the V-ATPase subunits. In addition
multiple splice variants have been confirmed for two of the Voa isoforms - with four
splice variants confirmed for Voa1 (Poea-Guyon et al., 2006) and nine confirmed for
Voa3 (Smirnova et al., 2005).
The critical role played by V-ATPases in cellular function is supported by
knockout studies of the V-ATPase subunits. In mice, knockout of the Voc subunit result
in early embryonic lethality (Inoue et al., 1999, Sun-Wada et al., 2000). In particular the
embryos lacking Voc developed to the blastocyst stage and were implanted in the uterine
epithelium but died shortly afterwards. An examination of the cells lacking Voc showed
impaired endocytosis as well as organellar acidification with the Golgi complex
becoming swollen and vacuolated (Sun-Wada et al., 2000). Knockout of the d subunit
resulted in a similar phenotype of early embryonic lethality. The blastocysts from these
knockout mice show impaired acidification, as detected by the membrane permeable
lipophilic weak base acridine orange, after several days of culture (Miura et al., 2003).
The accessory protein Ac45 also appears to be important for early development as
blastocysts lacking the expression of Ac45 suffered from embryonic lethality
(Schoonderwoert and Martens, 2002).
Although there has yet to be any knockout mice data for the Voa subunit to date,
this subunit, particularly Voa1, is also expected to play a critical role in early
development as the loss of the Voa1 homologues Vha100-1 and Unc-32, in Drosophila
6

and C. elegans, respectively, are lethal during development (Oka et al., 2001b, Pujol et al.,
2001, Hiesinger et al., 2005).
The use of pharmacological agents to block V-ATPase proton pump activity has
also been useful for elucidating the importance of organelle acidification. A class of very
potent and specific inhibitors of V-ATPase activity consist of the macrolide antibiotics
bafilomycin and concanamycin which can block proton pump activity at nanomolar
concentrations (Drose and Altendorf, 1997, Forgac, 2007). Bafilomycin A1 has been
used to show the importance of organelle acidification in the sorting of chromogranin A
into dense-core granules (Taupenot et al., 2005), as well as in transmitter uptake into
synaptic vesicles (Moriyama et al., 1992). Incidentally, the use of bafilomycin A1 has
also contributed in revealing the potential role of the Voa subunit in membrane fusion
(discussed later on), a function that is independent of its role in proton pumping (Sun-
Wada et al., 2006, Peri and Nüsslein-Volhard, 2008). Lastly, the use of pharmacological
agents to block V-ATPase activity may prove to be clinically relevant as drugs that are
specific to certain types of V-ATPases are created. For example, FR16735 specifically
inhibits osteoclast V-ATPase and not lysosomal V-ATPase (Niikura et al., 2004) and is
therefore a potential therapeutic agent for osteoporosis.
The importance of V-ATPases in cellular processes and ultimately in organism
survival is seen in the critical importance of pH regulation in the functions of various
intracellular organelles. The next section will describe the mechanism of proton
translocation by V-ATPases as well as the regulation of this proton pumping function.
7

I. i. b) Mechanism and Regulation of the Proton Pump
Function of the V-ATPase
Studies of yeast V-ATPases revealed much of what is known about the
mechanisms of V-ATPase function as a proton pump as well as how this pump activity is
regulated. In addition to the bulbous A-B hexamer which contains three ATP-binding
sites responsible for ATP hydrolysis the V-ATPase has a few other features that are
required for proton translocation. The V-ATPase is structurally organized in such a way
that the A-B hexamer is connected to the Vo domain via a 'central stalk' composed of the
D, F and Vod subunits. The proteolipid subunits c, c', and c" each contain an important
glutamate residue within one of its transmembrane domains and interact to form a ring
which is in contact with this central stalk via the d subunit (Wang et al., 2007). Rotation
of the central stalk, also described as a rotor, as a result of ATP hydrolysis result in
rotation of the proteolipid ring (helical 'swivelling' at the level of each proteolipid subunit)
which facilitates proton translocation (Hirata et al., 2003, Forgac, 2007). Note that
specific inhibitors of V-ATPases such as bafilomycin A1 and concanamycin A are
thought to block proton translocation by interacting with the c subunit and prevent helical
'swivelling' (Forgac, 2007).
In addition to the central stalk the V-ATPase had been shown to contain
'peripheral stalks' which act as stators to prevent the movement of the A-B hexamer
relative to the other V-ATPase subunits that are not part of the rotary system. The
peripheral stalks are thought to be formed by interactions of the N-terminus of the Voa
subunit with the C, E, G, H subunits (Marshansky and Futai, 2008). Additionally the
8

transmembrane domains of the Voa subunit are thought to form two aqueous
hemichannels with one being accessible from the cytosol only and the other being
accessible from the lumen (or extracellular space) only. An arginine residue within a
transmembrane region (TM7) of the a subunit has been shown to be critical for proton
translocation (Kawasaki-Nishi et al., 2001b).
The mechanism of proton translocation thus appears to occur in the following
manner: ATP hydrolysis by the A-B hexamer causes a conformational change of the
hexamer which results in the rotation of the central stalk and the proteolipid ring; the
movement of the proteolipid ring forces protons in the cytosol-accessible hemichannel to
protonate the glutamate residues of the proteolipids as they move into more hydrophobic
regions; finally, further movement of the proteolipids localizes the protonated glutamate
residues to the lumen-accessible hemichannel where the arginine residue of subunit a
facilitate deprotonation and release of the protons into the lumen of the organelle (or
extracellular space).
As mentioned earlier the luminal pH of intracellular organelles is highly regulated
and that one major control point for this regulation appears to be in the regulation of the
proton pump activity of the V-ATPase. Regulation of proton pump activity of the V-
ATPase can be achieved through several means: reversible dissociation of the V1 and Vo
domains, reversible disulphide formation, and differences in coupling efficiencies
between ATP hydrolysis and proton translocation of different Voa isoforms.
In yeast the V-ATPase has been shown to dissociate into the soluble V1 domain
(without the C subunit), the membrane integral Vo domain and the soluble C subunit in
response to glucose depletion (Kawasaki-Nishi et al., 2001c, Forgac, 2007, Jefferies et al.,
9

2008). Once dissociated the V1 complex is no longer capable of ATP hydrolysis while
the Vo complex is no longer capable of proton translocation. Reassembly and reinitiation
of proton transport is restored when glucose is restored. V-ATPase dissociation in
response to glucose depletion is therefore thought to be a mechanism to conserve cellular
ATP in low energy conditions. The dissociation of the V-ATPase complex has als
observed in the mid gut of insect cells during moulting (Forgac, 2007, Jefferies et al.,
2008). Conversely, increased assembly of V-ATPase complexes is observed in dendritic
cells upon activation of antigen processing to decrease lysosomal pH (from 5.4 to 4.5)
and increase lysosomal function (Trombetta et al., 2003). The dissociation and
reassembly of V-ATPases appear to be controlled independently as dissociation (but not
reassembly) requires an intact microtubule network and reassembly (but not dissociation)
requires the RAVE (regulator of the V-ATPase of the vacuolar and endosomal
membranes) complex (Forgac, 2007, Jefferies et al., 2008).
o been
Another way of regulating V-ATPase activity appears to be through the reversible
formation of disulphide bonds between conserved cysteine residues at the nucleotide-
binding site of the catalytic subunit A. The formation of disulphide bonds causes the A
subunit to lose its ATPase activity as seen in V-ATPases purified from bovine chlathrin-
coated vesicles (Feng and Forgac, 1992) as well as in yeast (Oluwatosin and Kane, 1997).
Removal of these disulphide bonds result in the regaining of ATPase activity.
Differences in the coupling efficiencies between of Voa isoforms between ATP
hydrolysis and proton translocation have also been shown to regulate the proton pump
function of V-ATPases. For example, in yeast Vph1p is localized to the central vacuole
while Stv1p is localized to the Golgi/endosome. While the V-ATPases made by each
10

isoform showed similar kinetics of proton translocation as well as similar binding affinity
to ATP and similar sensitivity to pharmacological agents such as concanamycin A,
Stv1p-containing complexes show a lower ratio of proton transport to ATP hydrolysis
than Vph1p-containing complexes (Kawasaki-Nishi et al., 2001c). The carboxyl-terminal
half of the a subunit appear to be important for this difference in coupling efficiency
(Kawasaki-Nishi et al., 2001a). This difference in coupling efficiency can partly explain
the lower pH found in central vacuoles compared to the Golgi.
Control of V-ATPase activity can therefore act as an important regulator of
organelle pH. Combined with differences in the other determinants of pH (e.g. proton
leaks and Cl- conductance) between the various organelles one can imagine how the
internal pH of these different organelles can be fine-tuned for their specific functions.
11

I. i. c) Intracellular V-ATPases
Having introduced the concepts of how the luminal pH of intracellular organelles
may be regulated and the central role played by V-ATPases in this regulation it is now
appropriate to mention some specific functions of organelle acidification. Cytosolic pH is
about 7.2 and organelles such as early endosomes, lysosomes, the Golgi apparatus, and
secretory vesicles all require some degree of acidification for proper function (Saroussi
and Nelson, 2009).
The Golgi apparatus, consisting of the cis-, medial- (also called Golgi stacks), and
trans-Golgi network plays a critical role in protein modification as well as in the sorting
of proteins involved in the constitutive and regulated secretory pathways. The Golgi
display a gradual decrease of pH from the cis- to the trans-Golgi network with the pH of
the cis-Golgi estimated at being ~6.7 and the pH of the trans-Golgi network estimated at
being ~6.0 (Paroutis et al., 2004). Acidification within the Golgi is important for the
function of enzymes that are involved in O-glycosylation, trimming and processing of N-
glycans as well as sulphation of newly made proteins. At the trans-Golgi network (TGN)
the sorting of proteins destined for various places may also require an acidic condition.
For example the pH in the TGN is important for the binding of lysosomal enzymes to the
mannose-6-phosphate receptor destined for the late endosome and eventually to the
lysosome (Schoonderwoert and Martens, 2001).
Early endosomes provide a pathway for endocytosed proteins to be properly
sorted. The low pH of endosomes (pH 6.0-6.8) allow the release of ligands from receptors,
a prerequisite for the sorting of the receptor back to the plasma membrane and the sorting
12

of the ligand to the lysosome for degradation (Saroussi and Nelson, 2009). For example,
low density lipoprotein (LDL) bound to the LDL receptor at the plasma membrane is
endocytosed and transported to the endosome. The low pH of the endosome allow the
LDL to be released from its receptor (Hinton et al., 2009). The LDL is then sorted to the
lysosome for degradation while the LDL receptor is recycled back to the plasma
membrane for further use. Neutralization of endosomal compartments with ionophores,
weak bases or with V-ATPase-specific inhibitors (bafilomycin or concanamycin) can
disrupt this process and prevent proper sorting of the ligand and receptor (Hinton et al.,
2009).
Lysosomes, which are responsible for the degradation of proteins and other
macromolecules, contain proteases, hydrolases and lipases. These enzymes require a low
pH for their function (Saroussi and Nelson, 2009). In fact the pH of lysosomes have been
estimated to be ~5 and can reach as low as 4.5 under the right cues. For example, the
maturation of dendritic cells (antigen presenting cells) is characterized partly by the
lowering of their lysosomal pH which allows them to degrade antigens more rapidly. The
decrease in pH during maturation is made possible by increased assembly of V-ATPases
(recruitment of V1 sectors) on the lysosomal membrane (Trombetta et al., 2003).
The electrochemical gradient generated by the proton pump action of V-ATPases
is also required for the uptake of transmitters into secretory vesicles. This topic will be
more thoroughly discussed in a later section.
13

I. i. d) Plasma Membrane V-ATPases
The proton pump function of V-ATPases has also been shown to be important for
normal physiological processes occurring at the plasma membrane. At the plasma
membrane of specialized cells V-ATPases are known to facilitate physiological processes
such as bone resorption, urine acid-base balance, and sperm maturation.
Bone density is partly regulated by specialized cells called osteoclasts. Osteoclasts
cause bone resorption by first attaching to the surface of calcified bone and forming a
sealed region into which the osteoclast releases digestive enzymes such as cathepsins to
dissolve the bone matrix. Proton pumping into this extracellular space by plasma
membrane V-ATPases facilitate bone resorption by providing a low pH environment
which optimize enzymatic activity (Xu et al., 2007, Jefferies et al., 2008). Failure of the
osteoclast V-ATPases result in osteopetrosis, a disease which is characterized by
thickening of the bone (Xu et al., 2007).
Plasma membrane V-ATPases also play a critical role in urinary acidification and
acid-base balance in the kidney, particularly at the collecting duct of the distal nephron.
The cytoplasm of renal α-intercalated cells become acidified due to the carbonic
anhydrase-catalyzed reaction of CO2 coming from the plasma with H2O which produce
H+ and HCO3-. The apical V-ATPases function to extrude the generated protons into the
renal tubular lumen while Cl-/HCO3- antiporters at the basolateral membrane release
HCO3- into the plasma to prevent the cell from becoming too alkaline (Forgac, 2007,
Jefferies et al., 2008). The failure of V-ATPases to fulfil this role in acid-base balance has
been known to lead to distal renal tubular acidosis (dRTA), a disease characterized by
14

low plasma pH and excessive loss of urinary K+ and Ca2+ (Smith et al., 2000, Stover et al.,
2002).
Another role played by plasma membrane V-ATPases is with regards to sperm
maturation. V-ATPases at the apical membrane of epididymal clear cells maintain a low
pH in the vas deferens and the epididymis through acid secretion. The low pH is required
for sperm maturation and maintenance of sperm in a quiescent state. During sexual
arousal the pH of the vas deferens and epididymis rises due to secretion of HCO3- by
nearby principal cells. The rise in pH stimulates sperm motility (Hinton et al., 2009).
The proton pump activity of V-ATPases, as evident from the given examples, is
critical for many physiological functions. Interestingly, novel functions of the V-ATPase
such as pH sensing and membrane fusion have been proposed in some of the relatively
more recent studies of this protein complex. The role of the V-ATPase in membrane
fusion will be discussed in the next section.
15

I. i. e) The Vo Domain and Membrane Fusion
Recent studies implicate the Vo sector in a novel role that is independent of its
widely accepted role as a critical component of V-ATPase proton pump function -- that
of membrane fusion. The first of these studies came from biochemical analyses in which
the yeast Vo sector was shown to be required for homotypic vacuole fusion (Peters et al.,
2001). Since that landmark study other studies have emerged which show the
involvement of the Vo sector in membrane fusion. In a C. elegans model it was shown
that the Vo sector was critical for the fusion of cuticle-containing vesicles with the
plasma membrane in epidermal cells (Liegeois et al., 2006).
s
-Wada et al., 2006).
In other studies the Voa subunit specifically was shown to play a role in
membrane fusion. Voa1 was shown to be involved in a late step of synaptic vesicle
exocytosis in Drosophila neurons (Hiesinger et al., 2005). In this study loss of vha100-1
(a homologue of Voa1) resulted in the accumulation of synaptic vesicles near the
neuronal plasma membrane. In another study using zebrafish microglia as a model it wa
shown that Voa1 is required for fusion between phagosomes and lysosomes during
phagocytosis (Peri and Nüsslein-Volhard, 2008). Voa3 has also been implicated in
membrane fusion. In a study using pancreatic beta cells Voa3 was found to be localized
on the membranes of insulin-containing secretory granules and to be critical in insulin
secretion from these granules (Sun
16

Mechanistically the Vo sector is proposed to facilitate membrane fusion by first
forming trans-Vo pairs between apposing membranes. Once in close proximity membrane
fusion is thought to take place through the mixing of the hydrophobic c subunits. Pore
opening ultimately results from the radial outward movement of the c subunits (Peters et
al., 2001, Nishi and Forgac, 2002)
17

I. ii) The Voa Subunit
I. ii. a) Overview of the Voa subunit
At ~110 kDa the Voa subunit is the largest subunit that makes up the Vo sector
(Figure 1). In higher eukaryotes such as worm, fly, mouse and humans there exist four
isoforms of Voa. In addition multiple splice variants have been confirmed for some of
these isoforms (Pujol et al., 2001, Smirnova et al., 2005, Poea-Guyon et al., 2006).
The Voa isoforms appear to be well-conserved between different species. For
example, bovine Voa1 exhibit 62-84% amino acid sequence similarity to the C. elegans
Voa isoforms VHA-5, VHA-6, VHA-7 and UNC-32, with UNC-32 sharing the highest
similarity (Oka et al., 2001b). Within species there is also high amino acid sequence
similarity between different isoforms. For example, in mouse the four a isoforms have
about 70% amino acid sequence similarity (Oka et al., 2001a) (Also see Appendix II) .
With respect to topology the Voa subunit consists of a large hydrophilic N-
terminus, followed by 8 or 9 transmembrane domains and either a cytosolic or luminal C-
terminus (Leng et al., 1999, Marshansky and Futai, 2008, Clarke et al., 2010). The N-
terminus is thought to contain the sorting signal of the Voa subunit largely based on a
study of the two yeast Voa isoforms, Vph1p (sorts to the vacuole) and Stv1p (sorts to the
Golgi) (Kawasaki-Nishi et al., 2001a) while the C-terminus is thought to be involved in
the coupling efficiency of ATP hydrolysis to proton translocation (Kawasaki-Nishi et al.,
2001c).
18

Although the exact region is not yet known subunit a, along with subunits B and E
of the V1 sector, have been shown to physically interact with the glycolytic enzyme
aldolase to regulate V-ATPase assembly/disassembly in a glucose-dependent manner (Lu
et al., 2004). The study provided evidence for a direct link between the ATP-generating
glycolytic pathway and the ATP-using V-ATPase activity.
19

I. ii. b) The Voa Isoforms
In mammals Voa1, Voa2, and Voa3 are widely expressed in all tissues with Voa1
more strongly expressed in the brain and Voa3 primarily expressed in osteoclasts (Nishi
and Forgac, 2000, Toyomura et al., 2000). On the other hand, the expression of Voa4
appears to be restricted to some epithelium cells of the kidney (Oka et al., 2001a), the
inner ear (Stover et al., 2002), and the ocular ciliary bodies (Kawamura et al., 2010).
Although Voa1 is expressed ubiquitously it is typically regarded as a neuronal
Voa isoform because of its high expression in neural tissues. For example in worm Unc-
32 (a homologue of Voa1) is predominantly expressed in neurons (Pujol et al., 2001). In
mice Voa1 is also strongly expressed in the brain (Nishi and Forgac, 2000, Toyomura e
al., 2000). There are four known splice variants of V
t
06).
., 2006).
oa1, a1-I to IV, which exhibit
differential tissue expression as well as intracellular sorting (Poea-Guyon et al., 20
Splice variants I and IV are expressed in neurons while splice variants II and III are
ubiquitously expressed in other tissues. a1-I appears to be the splice variant that sorts to
synaptic vesicles and presynaptic plasma membrane in neurons (Poea-Guyon et al
To date there are no known diseases or conditions associated with loss of Voa1
function. The reason for this lack of observable phenotype for Voa1 is very likely due to
the critical importance of this isoform throughout development. For example Unc-32 was
shown to be essential for C. elegans embryogenesis (Oka et al., 2001b).
Similar to Voa1, Voa2 is also widely expressed in various tissues (Nishi and
Forgac, 2000, Toyomura et al., 2000). At the intracellular level this Voa isoform is
strongly associated with functions of the Golgi as it is preferentially localized to the
20

Golgi in most cells types examined. For example Voa2 is localized to the Golgi in
cultured osteoclasts (Toyomura et al., 2003), in neurons (Poea-Guyon et al., 2006), and in
epididymal clear cells (Pietrement et al., 2006). The only exception comes from a study
in which Voa2 was localized to early endosomes of mouse kidney proximal tubule cells
(Hurtado-Lorenzo et al., 2006).
Observations that Voa2 localize to the Golgi agree very well with the association
of certain diseases with the loss or reduction of Voa2. Human diseases in which Voa2
function is lost or reduced are associated with defects of the Golgi, including impaired
glycosylation (Kornak et al., 2008) as well as perturbation in general vesicular trafficking
and tropoelastin secretion (Hucthagowder et al., 2009). The overt effects of these
impaired cell processes include different degrees of wrinkly skin and mental retardation
(Kornak et al., 2008, Hucthagowder et al., 2009)
Although ubiquitously expressed Voa3 is most strongly expressed in osteoclasts,
cells that are specialized for bone resorption (Frattini et al., 2000, Kornak et al., 2000,
Toyomura et al., 2003). Within the osteoclast Voa3 may be found on lysosomes as well
as on the plasma membrane depending on the differentiation state of the cell (Toyomura
et al., 2003). At the plasma membrane Voa3 contributes to osteoclast function through
V-ATPase proton pump activity which acidify the region where bone resorption would
take place to activate the proteases involved. Loss of Voa3 function results in
osteopetrosis in humans (Frattini et al., 2000, Kornak et al., 2000). Voa3 is also suggested
to be involved in secretory granule exocytosis in a process that is independent of its
function as a proton pump (Sun-
Wada et al., 2006).
21

As mentioned earlier the expression of Voa4 is quite restricted compared to the
other Voa isoforms as it is only expressed in some epithelium cells of the kidney (Oka et
al., 2001a, Stehberger et al., 2003), the inner ear (Stover et al., 2002), epididymal clear
cells (Pietrement et al., 2006) and the ocular ciliary bodies (Kawamura et al., 2010). The
importance of Voa4, however, appears to be well established within these cells as can be
seen from the diseases associated with loss of Voa4 function. For example, mutations in
Voa4 cause recessive distal renal tubular acidosis (Smith et al., 2000, Stover et al., 2002,
Stehberger et al., 2003) and in some cases hearing loss in humans (Stover et al., 2002). In
the case of distal renal tubular acidosis, a condition that is characterized by low plasma
pH and excessive urinary loss of K+ and Ca2+, intercalated cells of the distal renal tubule
lose their abilities to expel protons into the collecting duct due to loss of V-ATPase
activity (Stehberger et al., 2003).
22

I. iii) Background of the model used to study Voa
Function
I. iii. a) The PC12 cell as a model to study Voa function
The PC12 cell line was derived from a rat pheochromocytoma, an adrenal
medullary tumor (Greene and Tischler, 1976) and have been used successfully for many
different studies including monoamine biogenesis, protein trafficking and secretory
vesicle exocytosis. There are several reasons why the PC12 cell is a good model to use
for the current study on Voa function.
Firstly, PC12 cells contain many large dense-core vesicles (~1000) which require
a proton gradient for the uptake of catecholamines. Secondly, the PC12 cell is a well-
establish model for neuroendocrine secretion (Arunachalam et al., 2008, Han et al., 2009)
making it useful for testing the potential role of Voa isoforms in exocytotic membrane
fusion. Thirdly, the transfection rate (via electroporation) of PC12 cells is reasonably
high and can reach up to 60% (Martin and Grishanin, 2003). A reasonably high
transfection rate is particularly important for Fluorescence Activated Cell Sorting (FACS)
analysis which is a key technique in this study. Lastly, the technique to infect PC12 cells
in order to isolate stable cell lines that over-express an exogenous protein or have certain
endogenous proteins down-regulated is well-established (Arunachalam et al., 2008, Han
et al., 2009).
23

I. iii. b) Dense-core vesicles and the regulated secretory
pathway
The regulated exocytosis of dense core-vesicles (also called dense-core granules,
DCGs) is one defining function of endocrine and neuroendocrine cells, such as the PC12
cell. In these cells there exists, in addition to a constitutive secretory pathway, a regulated
secretory pathway which ensures the availability of DCGs for stimulated exocytosis.
The biogenesis of DCGs begin at the membrane of the trans-Golgi network (TGN)
with the accumulation/aggregation of 'granulogenic' proteins at defined membrane
regions of the TGN. The granulogenic proteins, which include granins (chromogranins A
and B; secretotogranins II-IV), pro-hormones, pro-neuropeptides and major cargo
proteins accumulate at lipid raft microdomains, membrane regions with high contents in
cholesterol and other lipids (Kim et al., 2006). The aggregation of granulogenic proteins
is thought to provide the driving force for budding of the membrane at the TGN.
Cholesterols and other lipids such as diacylglycerol (DAG) and phosphatidic acids (PAs)
utilize their cone-shaped structures to facilitate negative curvature formation of the Golgi
membrane and eventual budding of the DCG (Kim et al., 2006).
Immediately after budding the DCG is considered to be in an 'immature' state.
Maturation of the DCG begins with increased acidification which gradually lowers the
luminal pH and activate pro-hormone convertases and carboxypeptidases to allow for the
processing of pro-hormones, pro-neuropeptides and other proteins (Kim et al., 2006). The
maturation process also involve the removal of cargoes that were mistakenly packed into
the DCG such as lysosomal enzymes, constitutive secretory proteins and some membrane
24

proteins. These proteins are removed by the budding off of chlathrin-coated constitutive-
like vesicles from the DCG. Finally, the removal of water and condensation of granule
content is required to form the mature DCG.
Several factors are known to regulate dense-core granule biogenesis. Firstly, an
appropriately low pH in the trans-Golgi network is required for the aggregation of
granulogenic proteins. For example, it has been shown that treatment of PC12 cells with
bafilomycin A1 resulted in a significant reduction of sorting of chromogranin A to DCG
as well as a significantly fewer number of secretory granules with dense cores (Taupenot
et al., 2005). The low pH is also required for the negative curvature-inducing lipids such
cholesterol, DAG and PAs to be in their conical forms which facilitate in budding.
Finally the number of DCGs can be regulated by the amount of granulogenic
proteins present at the TGN. In this respect, chromogranin A plays a major role in DCG
biogenesis by protecting granulogenic proteins from degradation through its induction of
the protease inhibitor, protease nexin-1 (Kim and Loh, 2006). Granulogenic proteins
amounts can also be regulated at the post-transcriptional level by polypyrimidine-tract
binding tract protein (PTB) which protects granulogenic protein mRNAs from being
degraded (Knoch et al., 2004).
.
25

I. iii. c) Catecholamine synthesis and uptake into dense-core
vesicles
The synthesis of the catecholamines dopamine, norepinephrine and epinephrine
from tyrosine are catalyzed by several enzymes that work in sequence (Figure 2A)
(Daubner et al., 2011). The amino acid tyrosine is converted to L-Dopa by tyrosine
hydroxylase. L-Dopa is then converted to dopamine (DA) by L-Dopa decarboxylase.
Subsequently, dopamine is converted to norepinephrine (NE) by dopamine-β-
hydroxylase. Lastly NE can be converted to epinephrine (E) by the enzyme
phenylethanolamine-N-methyltransferase. In this pathway the conversion of tyrosine to
L-Dopa by tyrosine hydroxylase is the rate-limiting step (Daubner et al., 2011). In PC12
cells the predominant catecholamine is dopamine due to the lack of dopamine-β-
hydroxylase activity in these cells (Greene and Tischler, 1976).
The uptake of transmitters into secretory vesicles has been shown to require the
proton pump activity of V-ATPases, i.e. the generation of a low luminal pH (Moriyama
et al., 1992). Different transmitters utilize different aspects of this low pH for their uptake
into the vesicle. For example, the uptake of both norepinephrine and glutamate is
disrupted by the inhibition of V-ATPases by bafilomycin A1. However, the uptake of
norepinephrine into secretory vesicles requires the proton (chemical) gradient generated
by the V-ATPases while the uptake of glutamate requires the positive membrane
potential (electrical gradient) generated by the accumulation of H+ inside the vesicles
(Moriyama et al., 1992).
26

Figure 2: Catecholamine synthesis and uptake into dense core vesicles
A) Adapted from Daubner et al., 2011. B) V-ATPase activity is required for the
uptake of catecholamines (CA) into dense-core vesicles.
The uptake of catecholamines, such as norepinephrine is facilitated by vesicular
monoamine transporters (VMATs) located on the vesicular membrane. There exists two
isoforms of this transporter, VMAT1 and VMAT2. VMAT1 is expressed in
neuroendocrine cells while VMAT2 is expressed in neuronal cells. The proton gradient
generated by the vesicle's V-ATPases is used to drive the secondary transport of
catecholamines. Two protons are extruded for every one catecholamine up-taken. In
chromaffin granules the concentration of catecholamines has been found to be as high as
500-1000 mM as a result of this transport system (Camacho et al., 2008).
27

I. iv) Purpose and significance of this study
The importance of V-ATPases in maintaining proper acidification of intracellular
compartments as well as the critical importance of the Voa subunit in this aspect of V-
ATPase function is generally accepted. However, the specific Voa isoforms involved in
the acidification of these compartments have yet to be elucidated. In fact, studies which
involve the removal or down-regulation of specific isoforms of Voa have yet to reveal
acidification defects in the intracellular organelles studied (Sun-Wada et al., 2006, Peri
and Nüsslein-Volhard, 2008). For example, in Voa1 knockdown zebrafish, vesicular
acidification in microglia phagosomes and lysosomes, as measured by the pH-sensitive
dye LysoSensor, appear normal despite the observations of other phenotypes caused by
loss of Voa1 (Peri and Nüsslein-Volhard, 2008). Similarly, despite loss of Voa3, insulin-
containing secretory granules of pancreatic beta cells did not exhibit acidification defect,
as measured by another pH-sensitive dye LysoTracker (Sun-Wada et al., 2006).
The purpose of this study is to determine the Voa isoforms that are required for
dense-core vesicle acidification in a neuroendocrine cell model. If successful the
significance of this study is two fold. Firstly, it will have identified the specific Voa
isoform(s) required for the acidification of a secretory vesicle. Secondly, it will have
established a mammalian neuroendocrine model in which genetic manipulation
techniques can be used to study the acidification of intracellular compartments.
28

II. Hypotheses
Hypothesis #1: The Voa subunit displays isoform-specific intracellular localization
and plays a critical role in targeting the V-ATPase to specific
intracellular organelles.
Specific Aim 1.1: To determine the intracellular localization of Voa1, Voa2,
and Voa3 in PC12 cells
Hypothesis #2: Voa1 and Voa2 cooperatively play critical roles in secretory
vesicle acidification and transmitter uptake/storage.
Specific Aim 2.1: To determine whether knockdown of Voa1, Voa2, and
Voa1/Voa2 in PC12 cells results in acidification defect of
dense-core vesicles.
Specific Aim 2.2: To determine whether defects (if any) in dense-core vesicle
acidification result in reduction of neurotransmitter uptake
and storage.
Hypothesis #3: The Voa subunit may also be critical for exocytotic membrane fusion.
Specific Aim 3.1: To determine whether knockdown of Voa1 and Voa1/Voa2
in PC12 cells results in secretion defects of a transfected
neuropeptide.
29

III. Materials and Methods
III. i) Growth and maintenance of the PC12 cells As described in the introduction the PC12 cell line was derived from a rat adrenal
medullary tumor, or pheochromocytoma (Greene and Tischler, 1976), and has been a
very useful mammalian model for many different studies including neuroendocrine
secretion and monoamine biosynthesis (Martin and Grishanin, 2003). This current study
makes use of PC12 cells as a model for secretory vesicle acidification, neurotransmitter
uptake and storage as well as secretory vesicle exocytosis. Since its original creation
many derivatives of the original PC12 cell line had been produced. The line that is used
as the starting, or wild-type, cell line for all experiments in this current study is that
which was created by Thomas F. Martin. The techniques to grow and maintain this line
of PC12 cells is therefore adapted from those used in Thomas Martin's lab (Klenchin et
al., 1998).
For this study wild-type PC12 cells are maintained in DMEM (Invitrogen or
HyClone, Logan, UT) containing 5% calf serum, 5% equine serum (both from HyClone),
penicillin (100 units/ml)/streptomycin (0.1 mg/ml) (Sigma) (Li et al., 2007, Arunachalam
et al., 2008) and, in some cases, 250 ng/ml Amphotericin B (Sigma) and 1.25 μg/ml
plasmocin (InvivoGen, San Diego, CA). This is the base medium for PC12 cells and will
henceforth be known as 'PC12 medium'. PC12 medium can be supplemented with one or
a combination of selection drugs such as puromycin (2.5 μg/ml), G418/neomycin (0.7
mg/ml) and blasticidin (5 μg/ml) for the purpose isolating populations of cells that had
successfully incorporated a plasmid of interest.
The cells are grown on 10 cm dishes (Sarstedt) kept in 37°C incubator with a 10%
CO2 atmosphere. When the dishes become confluent (90-100%) passaging of the cells is
performed by first aspirating the medium, adding 1 ml of Hank's basal salt solution
containing 1 mM EDTA and then replacing the dish back into the incubator for 3-5
30

minutes to allow gentle detachment of the cells from the dish. After this incubation period
4-8 ml of the appropriate PC12 medium is added to the dish and the loosened cell
aggregates are then triturated about ten times using a 10 ml pipette fitted to a 200 μl
yellow pipetman tip in order to separate the cell aggregates into single cell suspension.
Once thoroughly dispersed the cells are plated onto newly prepared 10 cm dishes
containing the appropriate PC12 medium. Typical ratios for passaging are between 1:6
and 1:8. Each dish is replaced with fresh medium about once every three days and it takes
about one week for the plated cells to reach confluency and be ready for another round of
passaging.
31

III. ii) Construction of fluorescent protein-tagged Voa1, Voa2
and Voa3 constructs and generation of PC12 cell lines
expressing these recombinant proteins
In order to gain information regarding the localization of Voa1, Voa2 and Voa3 in
PC12 cells cell lines which stably express Voa1, Voa2 or Voa3 fused with Emerald green
fluorescent protein (EmGFP) or mCherry were generated. This task was achieved by
using the lentivirus-mediated expression vector pLVX-IRES-blast (purchased from
Clontech, Mountain View, CA). cDNAs of EmGFP and mCherry were obtained by
amplification using PCR from pCMV-EmGFP and pcDNA3.1-myc-HisA-mCherry (a
kind gift from Dr. Herbert Gaisano, University of Toronto, Canada), respectively. The
PCR products were then digested with BamHI and BglII and ligated into the BamHI site
of pLVX-IRES-blast to generate pLVX-EmGFP-IRES-blast and pLVX-mCherry-IRES-
blast. Subsequently, cDNAs of human Voa1 (IMAGE clone ID 5195776), mouse Voa2
(IMAGE clone ID 3670722) and human Voa3 (IMAGE clone IB 5210733) were
amplified by PCR and ultimately ligated to the EcoRI/XbaI site (a1, a2) or XhoI/XbaI site
(a3) of pLVX-EmGFP-IRES-blast and pLVX-mCherry-IRES-blast to make the final
construct pLVX-Voax-EmGFP (or mCherry)-IRES-blast.
To make PC12 cell lines which express these recombinant proteins lentiviral
particles of the constructed expression plasmids were first produced by co-transfecting
each of these plasmids with pCMV8.74 and pMD2G in HEK-293FT cells (FT cells).
Briefly, a transfection mixture containing the desired plasmid (9μg), pCMV8.74 (4.8μg),
pMD2G (3μg), and polyethylenimine (PEI) (40 μl of 1.2 mg/ml, pH 7.2) in 1 ml of 0.15
M sodium chloride (NaCl) solution is thoroughly vortexed within an 1.5 ml Eppendorf
tube and then applied to a confluent dish of FT cells already containing 8 ml of FT
medium. The next day the old FT medium is replaced by fresh medium. Two days later,
the FT medium containing lentiviral particles is harvested by transferring the medium to a
15 ml tube and centrifuging at 800 x g for 3 minutes at 4°C. The supernatant is carefully
transferred to a new 15 ml tube and the tube is kept in a 4°C incubator until use.
32

The lentivirus particles produced by the FT cells are used to infect wild-type
PC12 cells. The lentivirus 'soup' obtained from the supernatant of the FT cells is mixed
with PC12 medium (2.5 ml virus + 0.5 ml PC12 medium) and then applied to the wild-
type PC12 cells in 10 cm dishes that are about 50% confluent. Two days later the
infection medium is replaced by 8 ml of fresh PC12 medium to allow the cells to recover
from the stress of infection. Once the cells become confluent they are eventually selected
with blasticidin-containing (5 μg/ml) PC12 medium to obtain populations of cells that
stably express the recombinant proteins. Selection takes about 2-3 weeks before the
surviving cells have become confluent enough to be passaged, analyzed, or frozen for
later use.
*In this study Ann Kang made the pLVX-hV0a1-mCherry/EmdGFP-IB and pLVX-
mV0a2-mCherry-EmdGFP-IB. She also generated the PC12 cell lines which express
these recombinant proteins.
33

III. iii) Immunofluorescence confocal microscopy
Immunofluorescence confocal microscopy (ICM) was performed for various cell
lines in this study. Wild-type PC12 cells were used in ICM analyses to determine the
localization of endogenous Voa1 and Voa2. Similarly, PC12 cells that stably express
Voa1-EmGFP, Voa2-EmGFP or Voa3-EmGFP were also analyzed using ICM to
indirectly determine Voa subunit localization via the localization of GFP. Lastly, ICM
analyses were performed on V
n a
PY-
oa1/ Voa2 double knockdown cells (DKD; discussed i
later section) and its control which were transfected with Neuropeptide Y-EmGFP (N
EmGFP; discussed in a later section) to confirm the correct localization of NPY-EmGFP
to dense-core vesicles. There were similarities as well as differences in the preparation of
the mentioned cells for ICM:
For all cell lines studied sterilized circular glass cover slips (0.25 mm width, 1.8
cm diameter) were placed in 2.2 cm wells within 12-well cell culture plates. The cover
slips were then coated for 30 minutes with poly-D-lysine (0.1 mg/ml) at room
temperature. Cells were allowed to adhere to the cover slips overnight and then
differentiated on the cover slips for 3-4 days in DMEM containing 100 ng/ml nerve
growth factor (NGF) (Sigma), 1% equine serum, 1% calf serum and P/S. After the
differentiation period cells were washed with Phosphate Buffered Saline (PBS), and fixed
for 15 minutes with PBS containing 4% paraformaldehyde.
For cells stably expressing Voa1-EmGFP, Voa2-EmGFP or Voa3-EmGFP as well
as for DKD cells (and its control) which were transfected with NPY-EmGFP
permeabilization was achieved by incubating for 5 minutes with PBS containing 0.2%
Triton X-100. Subsequently, nonspecific sites were blocked for 1 hour at room
temperature in blocking buffer, PBS containing 0.3% Bovine Serum Albumin (BSA). To
determine the localization of Voa1-EmGFP, Voa2-EmGFP and Voa3-EmGFP a double-
staining procedure was used in which primary antibodies against GFP (rabbit polyclonal,
1:1000 dilution) and synaptotagmin-1 (Cl41.1 mouse monoclonal, 1:1000 dilution),
34

GM130 (mouse monoclonal, 1:500 dilution), EEA1 (goat polyclonal, 1:1000 dilution), or
LAMP1 (mouse monoclonal, 1:1000) in blocking buffer were then applied for 1 hour at
room temperature. Following three washes in blocking buffer, Alexa-488-conjugated goat
anti-rabbit antibody (1:1000 dilution) and rhodamine red-x-conjugated goat anti-mouse
antibody (1:1000 dilution) or Alexa-568-conjugated donkey anti-goat antibody (1:1000
dilution) in blocking buffer were applied to the samples for 1 hour in the dark at room
temperature. Samples were washed again three times in blocking buffer before being
mounted on microscope slides.
Similar steps were taken in preparing control and DKD cells that were transfected
with NPY-EmGFP. In these cells, however, rabbit anti-Secretogranin II polyclonal
antibody (SgII, 1:1000 dilution) and Alexa-568-conjugated goat anti-rabbit antibody
(1:1000 dilution) were used for primary and secondary staining, respectively. The natural
fluorescence of GFP was used to detect the GFP signal in these cells.
For the localization of endogenous Voa1 and Voa2 in wild type PC12 cells similar
steps were taken in preparing these cells up to the cell-fixing stage with 4%
paraformaldehyde. Subsequently, permeabilization of the cells was achieved by
incubating for 15 minutes with 0.1% SDS, 0.4% saponin, 1% normal goat serum (NGS),
and 1% BSA in PBS. Primary antibodies against Voa1 (rabbit polyclonal, 1:1000 dilution)
or Voa2 (rabbit polyclonal, 1:1000 dilution) and synaptotagmin-1 or GM130 in PBS
containing 0.4% saponin, 1% NGS, and 1% BSA were applied overnight to the
permeabilized cells. The next day cells were washed three times (10 minutes each time)
with PSB containing 0.4% saponin, 1% NGS, and 1% BSA. Secondary antibodies against
Voa1 or Voa2 (Alexa-488-conjugated goat anti-rabbit antibody, 1:1000 dilution) and
synaptotagmin-1 or GM130 (rhodamine red-x-conjugated goat anti-mouse antibody,
1:1000 dilution) in PBS containing 0.4% saponin, 1% NGS and 1% BSA were applied
for 1 hr in the dark. Cells were then washed three times (10 minutes each time).
35

All samples were mounted onto microscope slides using Fluoromount-G reagent
(SouthernBiotech, Birmingham, AL). Immunofluorescence staining was recorded with a
Zeiss laser confocal scanning microscope (LSM 510) with an oil immersion objective
lens (63x) and using the appropriate filters.
*Anna Han and Leon Parsaud obtained ICM data for the localization of endogenous Voa1
and Voa2 as well as for the higher magnification pictures of NPY-EmGFP transfected
cells.
36

III. iv) Reverse transcription-polymerase chain reaction
Reverse transcriptase-polymerase chain reaction (RT-PCR) was used in this study
mainly to confirm the (lack of) expression of the Voa4 subunit in PC12 cells. Additional
experiments on Voa1, Voa3 and GAPDH served as positive controls to ensure that RT-
PCR protocol used was working well.
To perform RT-PCR total RNA was first extracted from wild-type PC12 and
normal rat kidney (NRK; CRL-6509, ATCC) cells using RNeasy kit (Qiagen) and by
following its associated protocol on RNA extraction. The QIAGEN One Step RT-PCR
Kit (Qiagen) provided all the necessary reagents for the RT-PCR reaction. The follow
protocol was used: denaturation at 94°C for 30s; annealing at 55°C for 30s; elongation at
72°C for 1 min.; and 30 cycles of reaction.
The following primers were used: Voa1, sense: TCTCCACCCATTCAGAGGAC,
anti-sense CCTTCCATGATCAGCAGGAT, product size: 301 base-pairs (bp); Voa3,
sense: GCTTCCACCTTGGAGAACAG, antisense: CCCAGAGACGCAAGTAGGAG,
product size: 169 bp, Voa4, sense: CATGGGCATCTTCTCCATCT, antisense:
TTGAAGCCAGGTTCCAAATC, product size, 230 bp, GAPDH, sense:
CTCATGACCACAGTCCATGC, antisense: TTCAGCTCTGGGATGACCTT, product
size: 155 bp.
After the reactions the PCR products were electrophoresed on a 1.5% agarose gel
containing ethidium bromide to visualize the bands. Pictures of the gels were taken using
the software FluorS.
37

III. v) Construction of Voa1 and Voa2 knockdown plasmids
a generation of stable Vnd
oa1 and/or Voa2 knockdown PC12
cell lines
In this study we used short hairpin RNA (shRNA) to down-regulate Voa1 and
Voa2 in PC12 cells. shRNAs prevent the expression of a protein of interest by activating
a normal cellular pathway for post-translational gene-silencing (PTGS) known as RNA
interference (RNAi) (Paddison et al., 2002, Manjunath et al., 2009). In this process the
transcribed shRNA is cleaved by a nuclease called DICER to produce ~20 nucleotides
double-stranded RNA known as small interfering RNA (siRNA). The produced siRNA
then associates with a multiprotein complex called the RNA-induced silencing complex
(RISC). Subsequently, the 'guide' strand of this siRNA then target RISC to the desired
mRNA based on sequence homology thereby allowing enzyme Argonaute 2 to cleave the
'passenger' strand as well as the targeted mRNA to prevent the expression of the targeted
gene (Manjunath et al., 2009). There are three main features of a shRNA: a 19-29
nucleotide sequence which is derived from the target gene (passenger strand); a linker
sequence of 4-15 nucleotides which acts as a loop and allow the entire transcribed
nucleotide to have a 'hair pin' shape; and a reverse complement of the targeted 19-29
nucleotide sequence which binds to the homologous sequence of its target mRNA to
cause its degradation by the RISC complex (guide strand).
To knock down the Voa1 gene, we targeted a 21-nucleotide sequence of rat Voa1,
GCTGCTTATTGTTGTGTCAGT (bases 61-81, Voa1KD). Similarly a 21-nucleotide
sequence of rat Voa2 was targeted to knock down Voa2,
GGTGGAGCTCAGAGAAGTCAC (bases 315-335, Voa2KD). For both constructs
CTCGAG was used as a linker sequence. An example of the shRNA design for Voa2 (top)
as well as the predicted short-hair pin structure (bottom) is shown (Figure 3).
38

Figure 3: Design of shRNA knockdown sequence for rat Voa2
A 58 bp oligonucleotide containing the target sequence (passenger strand), the loop, the
reverse complement (guide strand) as well as flanking bases for ligation into the proper
vector (top). Schematic of the predicted structure of the shRNA (bottom).
The designed fifty-eight base-pair oligos containing sense and antisense of the
target sequences were then annealed and subcloned into the AgeI/EcoRI sites of pLKO-
puro (purchased from Sigma, Oakville, ON, Canada) to generate the Voa1 and Voa2
knockdown plasmids, pLKO-puro-Voa1KD and the pLKO-puro-Voa2KD, respectively.
Inserted sequences were verified by sequencing. Additionally, a neomycin resistant
version of the knockdown plasmid was made for Voa2 by replacing the puromycin
resistant gene with the neomycin resistant gene at the SpeI/KpnI sites to generate pLKO-
neo-Voa2KD.
For production of the recombinant lentiviruses for pLKO-puro-Voa1KD and
pLKO-puro-Voa2KD and infection of these viruses into PC12 cells we followed the
procedures for lentiviral production and infection as described earlier (Section III. ii.).
For each recombinant virus, we isolated a pool of heterogeneous cells that had survived
39

puromycin-containing medium over a period of two weeks. The surviving cells were then
subjected to immunoblot analyses using anti-Voa1 and anti-Voa2 antibodies to determine
the efficacy of knockdown for the proteins of interest.
The Voa1 knockdown construct proved to be very efficient at knocking down
Voa1. However, the efficacy of the Voa2 knockdown construct in knocking down Voa2
was not as high. To maximize the knockdown of Voa2 we further infected the
puromycin-resistant cell line with lentiviruses for pLKO-neo-Voa2KD. After selection
with medium containing both puromycin and neomycin we obtained a pool of
heterogeneous cells which are resistant to both puromycin and neomycin. Western blot
analyses confirmed that the knockdown level of Voa2 improved due to this procedure.
The rationale for using two plasmids to target the same sequence of Voa2 was that that
having at least two copies of the knockdown sequences incorporated into the host PC12
cells may have stronger and more stable knockdown effects than having just a single
copy of the knockdown sequence.
D
aining medium. Knock down of both Voa1 and Voa2 was confirmed using
estern blot.
e as the
n
ntil use. We found that the cells maintain their phenotypes for two to three months.
To generate stable Voa1 and Voa2 double knockdown (DKD) cells, we
sequentially infected PC12 cells with lentiviruses generated from pLKO-puro-Voa1K
and pLKO-neo-Voa2KD and isolated a pool of cells which survived puromycin and
neomycin-cont
W
Respective controls for the knockdowns cell were made at the same tim
knockdowns using lentiviruses generated from vectors that did not contain the
knockdown sequence, i.e. pLKO-puro or pLKO-neo only. Once successful knockdown
was confirmed using Western blot cells were grown, frozen and kept in liquid nitroge
u
40

III. vi) Western Blot analyses of the generated PC12 cell lines
ish
omogenate
ing. 20 μg of each prepared samples were loaded and ran in
0% polyacrylamide gel.
ropriate
as then washed 3 times with TBS-T before Luminol was applied and the film developed.
s
a),
Western blot samples were prepared by harvesting a confluent 10 cm culture d
containing the cells of interest. Cells were resuspended in PBS containing a protease
inhibitor and disrupted by passing through a 23 1/2 gauge needle several times to create a
homogenate. After measuring protein concentration an equal volume of the h
was added to sample buffer (10% mercaptoethanol, 10% glycerol, 4% SDS,
Brompohenol Blue in 0.15 M Tris at pH 6.8) and the mixture briefly sonicated. Except
for the samples which would be used to blot for Voa1 and Voa2 every sample was boiled
for 2-3 minutes before load
1
After overnight transfer of the protein onto nitrocellulose membrane each
membrane was prepared by blocking for 1 hr with 0.5% skim milk in TBS-T and then
washed 3 times (15 minutes each time) in TBS-T before the application of the app
primary antibodies (2 hr to overnight). After blotting with primary antibody the
membrane was washed 3 times TBS-T before the application of the appropriated horse
radish peroxidase (HRP)-conjugated secondary antibodies for 45 minutes. The membrane
w
We obtained rabbit polyclonal antibodies against Voa1 from Synaptic System
(Gottingen, Germany) and Santa Cruz Biotechnology (Santa Cruz, CA), GFP from
Invitrogen (Carlsbad, CA) and Calnexin from Sigma (Oakville, ON, Canada); mouse
polyclonal antibodies against Voa2 from Abnova (Taiwan), goat polyclonal antibodies
against EEA1 (Santa Cruz), mouse monoclonal antibodies against Vod1 (clone 34-Z)
from Santa Cruz Biotechnology, syntaxin-1A/1B (clone HPC-1) (Barnstable et al, 1985)
from Sigma, Ac45 (clone 3A2) from Abnova (Taiwan), SNAP-25 (clone SMI 81) from
Covance (Princeton, NJ), GM130 (clone 35) from BD Biosciences (Mississauga, ON,
Canada), LAMP1 (clone LY1C6) from StressMarq Biosciences (Victoria, BC, Canad
GAPDH from Millipore, and DsRed from Clontech. Finally mouse monoclonal anti-
41

synaptotagmin-1 (Cl41.1), rabbit polyclonal anti-V0a2 antibody (Peng et al., 1999) and
anti-VCP/p97 antibody (Sugita and Südhof, 2000) were kind gifts from Drs. Reinh
Jahn (Max Planck Institute for Biophysical Chemistry, Germany), Xiao-Song Xie
(University of Texas Southwestern Medic
ard
al Center at Dallas) and Thomas Südhof
tanford University, CA), respectively. (S
42

III. vii) Construction of Neuropeptide Y-based reporter
constructs
ith
from Dr.
espectively.
and
V-NPY-epHluorin, pCMV-NPY-rpHluorin, and pCMV-NPY-
mGFP, respectively.
In order to detect defects in dense-core vesicle acidification resulting from
knocking down Voa1 and/or Voa2 we used Neuropeptide Y fused with super ecliptic
pHluorin as the reporter construct. The plasmids to express neuropeptide Y fused w
super ecliptic pHluorin, ratiometric pHluorin (rpHluorin) and Emerald GFP were
generated using pCMV5 as the parental plasmid. cDNAs of super ecliptic pHluorin and
ratiometric pHluorin were amplified by PCR from pGM6 and pGM1 (kind gifts
Gero Miesenböck, University of Oxford, UK), respectively. The PCR products
containing epHluorin and rpHluorin were digested with ClaI and XbaI and ligated to the
same sites on pCMV5, generating pCMV-epHluorin and pCMV-rpHluorin, r
pCMV-EmGFP was a kind gift from Dr. Weiping Han (University of Texas
Southwestern Medical Center, Dallas). cDNA of NPY was amplified by PCR on pVenus-
N1-NPY (a kind gift from Atsushi Miyawaki, Riken, Japan) and digested with BglII
ClaI and ligated to the same site of pCMV-epHluorin, pCMV-rpHluorin, or pCMV-
EmGFP generating pCM
E
43

III. viii) Transfection of NPY-based reporter constructs into
PC12 cells and subsequent FACS analyses
ng
nd 25
ghly
electroporated (Capacitance 1 μF, Voltage 330 mV) with a time constant of
3-18 ms.
and
.
ACS analysis the cells were re-plated onto 6-well plates 3 to 4 days
fter transfection.
BS containing 1% calf serum, 1% equine serum
nd 10 μg/ml of propidium iodide (PI).
Transfection of the NPY-based reporter constructs was achieved by
electroporation (Martin and Grishanin, 2003). Cells to be transfected were collected from
10 cm dishes that were 70-90% confluent and resuspended just as they would be duri
passaging. The cells were then transferred to 15 ml tubes and pelleted (800 x g for 3
minutes at 4°C). This cell pellet was washed once with 4-5 ml of Cytomix (120 mM KCl,
0.15 mM CaCl2, 10 mM KH2PO4, 10 mM K2HPO4, 2 mM EGTA, 5 mM MgCl2, a
mM HEPES adjusted to pH 7.5). The cells were again pelleted and then thorou
resuspended in 1-2 ml of Cytomix depending on cell pellet size. 500 μl of this
resuspension was then mixed with 15 μg of plasmid DNA (pCMV5, pCMV-NPY-
epHluorin or pCMV-NPY-EmGFP) and the total content transferred to an electroporation
cuvette and
1
Immediately after transfection the cells were re-plated onto new 10 cm dishes
kept in PC12 medium to allow for recovery. For the purpose of immunofluorescence
confocal microscopy the cells were further re-plated onto circular glass coverslips in 12-
well plates (see Section III.iii for preparation for ICM) three 3 to 4 days after transfection
For the purpose of F
a
One or two days after re-plating to 6-well plates the cells from each well were
harvested and resuspended in 300-500 μl various buffered solutions. Experiments to test
acidification defects were performed in P
a
44

Experiments that tested the effects of different concentrations of NH4Cl on the
NPY-epHluorin were performed in HEPES-buffered saline (pH 7.4) containing 15 mM
HEPES (pH 7.4), 5.6 mM KCl, 140 mM NaCl, 2.2 mM CaCl2, 0.5 mM MgCl, 5.6 mM
Glucose and 10 μg/ml of propidium iodide (PI). NH
al
ue to the rapid influx of
H3 (dissociated from NH4+) into intracellular compartments and the subsequent
ombin
, 0.5
e that facilitates the exchange of H+ and K+. Monensin is an
ther-based ionophore which is capable of crossing lipid membranes of cells and acting
s a Na
,
re
ual
nsity of the cells transfected with
CMV5 was subtracted from the average values of the cells transfected with pCMV-
PY-epHluorin to obtain the effect fluorescence.
4Cl applications were performed with
various (20 mM, 50 mM, 100 mM) concentrations of NH4Cl in substitution of equ
concentrations of NaCl. NH4Cl rapidly alkalinizes luminal pH d
N
c ation of most of these NH3 molecules with luminal H+.
Experiments to calibrate the pH dependence of NPY-epHluorin were performed
in either MES-buffered saline (adjusted to pH 5.5, 6.0, and 6.5) or HEPES-buffered
saline (adjusted to pH 7.0, and 7.5) containing 15 mM HEPES or MES, 140 mM KCl
mM MgCl2, 0.2 mM EGTA, 5 μg/ml nigericin, 5 μM monensin, and 10 μg/ml of PI.
Nigericin is an ionophor
e
a +/H+ antiporter.
In all experiments samples were triturated ten times with a 1 ml pipette and then
passed through a 35 μm nylon mesh strainer (BD Falcon, Cat#352235). For each sample
104 PI negative cells (FL3 channel) were analyzed for GFP intensity (FL1 channel) by
FACS Calibur (BD Biosciences). All samples were analyzed at room temperature. The
FACS machine was calibrated before each session of usage and the same settings we
used for all measurements. FACS allow for the measurement GFP intensity of individ
cells. The average fluorescence values of GFP inte
p
N
45

II. ix) Construction of knockdown-resistant human Voa1
(hVoa1) plasmid and generation of hVoa1-expressing
Voa1-knockdown and Voa1/Voa2-double knockdown
PC12 cells
re
a cell
Voa1 and then knockdown
ndogenous Voa1 or Voa1/ Voa2 from this cell line.
To ensure that the acidification defects observed in Voa1KD and DKD cells we
not due to off-target effects of expressing the Voa1 and Voa2 knockdown sequences it
was necessary to perform a rescue experiment. Our strategy was to first produce
line which express a knockdown-resistant version of
e
To make the knockdown-resistant Voa1-expressing construct we introduced 8
silent nucleotide mutations (SNMs) (GCCTACTGCTGCGTGTCG, underlines indicate
SNM) within the target sequence of the human V
pLVX-
Voa1(SNM)-IRES-blast. pLVX-mCherry-IRES-blast was used a control plasmid.
Ann Kang generated pLVX-hVoa1(SNM)-IRES-blast and pLVX-mCherry-IRES-blast.
a2 as well as retention of the
xpressed human Voa1were confirmed by Western blot.
oa1 cDNA by mutagenesis (compare
with Voa1 knockdown sequence in Section III.v). These SNMs were necessary to protect
the mRNA transcripts transcribed from the Voa1 expression plasmid from being degraded
by the shRNA mediated knockdown of Voa1. The protected human Voa1 cDNA was
then subcloned into the EcoRI/XbaI site of pLVX-IRES-blast to make
h
*
PC12 cell lines stably expressing this human Voa1 gene was generated using the
techniques mentioned previously (Section III. ii ) Once expression of the human Voa1
was confirmed by immunoblot we proceeded to knock down endogenous Voa1 and Voa2
from this cell line using the same techniques used to knockdown wild-type PC12 cells
(Section III. v). Knockdown of endogenous Voa1 and Vo
e
46

III. x) Measurement of [3H]-NA uptake into PC12 cells
od to
all-homogenizer instead of
sing the pharmacological agent streptolysin O.
S, pH
GE
P or 2 mM MgCl2 at 37°C for 60 min. The
olume of each reaction sample was 100 μl.
moved.
was
e protein concentrations of the cells. Each assay was
erformed in quadruplicates.
Uptake of noradrenaline (NA) into secretory vesicles requires a proton gradient
across the vesicular membrane and is ATP-dependent. We used an established meth
measure ATP-dependent uptake of [3H]-NA into dense-core vesicles in PC12 cells
(Ahnert-Hilger et al., 1998, Brunk et al., 2009) with a small modification; the cell’s
plasma membrane was mechanically permeabilized using a b
u
PC12 cells grown on 10 cm dishes until 70-100% confluency were washed with
physiological saline solution (PSS), harvested in K-glutamate buffer (20 mM HEPE
7.1, 140 mM potassium glutamate, 2 mM EGTA, 1 mM MgCl2) and mechanically
permeabilized with a ball-homogenizer (Wang et al., 2000, Wang et al., 2004, Fujita et al.,
2007, Li et al., 2007). The permeabilized PC12 cells were washed once with K-glutamate
buffer and incubated with the same buffer containing 50 nM [3H]-noradrenaline (NA,
Healthcare, Montreal, Quebec, Canada), 450 nM unlabeled NA (Sigma) and 0.5 mM
ascorbic acid in the presence of 2 mM MgAT
v
Reaction was halted by adding 1 ml of ice-cold K-glutamate buffer. Samples
were then centrifuged at 4°C for 3 minutes at 14 000 rpm and the supernatants re
An extra wash with 500 μl of ice-cold K-glutamate buffer was performed. After
centrifugation at 4° C for 3 minutes at 14 000 rpm the pelleted samples were treated with
200 μl of 0.5% Triton X-100 (in deionized water) and subsequently collected for liquid
scintillation counting. The amount of [3H]-NA up-taken into the permeabilized cells
normalized using the respectiv
p
47

III. xi) Measurement of endogenous dopamine in PC12 cells by
HPLC
bile
d to
ards,
these compounds were first established at the amount of 2.5
g/10 μl sample volume.
C12
edium.
ta et
(in the range of 10 – 30 μg/10 μl sample volume) of each sample
f PC12 cells.
Ann Kang and Krzysztof Grzegorczyk performed HPLC on whole-cell lysates.
Concentrations of catecholamines were measured using high performance liquid
chromatography (HPLC), which consists of a delivery pump (Model HP1100, Agilent), a
reversed-phase analytical column (ZORBAX Eclipse XDB-C8, 150 x 4.6 mm i.d., 5 μm,
Agilent), a degasser, and a fluorescence detector. We followed the method developed by
Lakshmana and Raju (1997) that used isocratic assay without derivatization . The mo
phase consisted of sodium acetate (0.02 M), methanol (16%), heptane sulfonic acid
(0.055%), EDTA (0.2 mM), and dibutylamine (0.01% v/v). The solution was adjuste
pH 3.92 with o-phosphoric acid. The flow rate was set to 0.9 ml/min. As stand
noradrenaline, adrenaline and dopamine were dissolved in 0.1 M PCA at the
concentration of 250 ng/ml each, and the volume of injection was set to 10 μl. Good
detection and separation of
n
For whole-cell lysate measurements of the concentrations of catecholamines P
cells grown until 80-100% confluency were harvested from 10 cm dishes in 1 ml of
Hank's buffer containing 1 mM EDTA, and supplemented with 9 ml of PC12 m
The cells were pelleted by centrifugation, washed once with 1 ml of PSS, and
homogenized in 200 μl of 0.1 M PCA. Insoluble materials were removed by
centrifugation (three times) and 10 μl of the cleared solution was applied to HPLC. We
detected the clear peak of dopamine (in the range of 20 – 160 ng/10 μl sample volume).
This result confirms that the primary catecholamine in PC12 cells is dopamine (Fuji
al, 2007). The concentrations of these catecholamines were normalized by the total
protein concentrations
o
*
48

For measuring dopamine concentrations from partially purified dense-core
vesicles we followed the purification protocol established by Martin and Kowalchyk
(1997) (Martin and Kowalchyk, 1997). Briefly, PC12 cells in K-Glutamate buffer were
permeabilized by passing them ten times through a ball-homogenizer (diameter 0.2527”).
The cell homogenate was then centrifuged at 800 x g to pellet the nucleus and other large
cell pieces. The supernatant was then centrifuged at 5000 x g to pellet plasma membrane-
associated dense-core vesicles. After dissolution of this pellet in 0.1M PCA and the
insoluble materials cleared 50 μl of this sample was loaded for HPLC analyses.
In all cases, the total protein concentration was measured using the Bradford
method with bovine serum albumin as a standard.
49

III. xii) Measurement of NPY-hPLAP secretion from PC12
cells
Voa1KD, DKD and Munc18-1/2 double knockdown PC12 cells (Han et al., 2009)
as well as their respective controls were used for NPY-hPLAP secretion measurements.
Cells grown until 70%-80% confluency in 10 cm dishes were transfected with 15 μg of a
reporter plasmid, pCMV-neuropeptide Y(NPY)-hPLAP (Fujita et al., 2007, Li et al., 2007,
Arunachalam et al., 2008) using electroporation (see Section III. viii). After four days, the
cells were harvested and re-plated onto 24-well plates.
Eight or nine days after electroporation, the plated cells were washed once with
PSS, and NPY-hPLAP secretion was stimulated with 200 µl of PSS (containing 145 mM
NaCl, 5.6 mM KCl, 2.2 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, and 15 mM
HEPES, pH 7.4) or High K+-PSS (containing 81 mM NaCl, 70 mM KCl). Secretion was
terminated after a 20-minute incubation period at 37°C by chilling to 0°C. Samples were
then centrifuged at 4°C for 3 minutes and supernatants were removed. The pellets were
solubilized in 200 μl PSS containing 0.1% Triton X-100.
The amounts of NPY-hPLAP secreted into the medium and retained in the cells
were measured by the Phospha-Light Reporter Gene Assay System (Applied Biosystems,
Foster City, CA). The samples were treated at 65°C for 30 minutes to inactivate non-
placental alkaline phosphatases and an aliquot of 10 μl was assayed for placental alkaline
phosphatase activity using the kit. The total volume of the assay was 120 μl. After 5 to 10
minutes of reaction, chemiluminescence was quantified by FB12 luminometer (Berthold
Detection Systems, Zylux Corporation, Oak Ridge, TN).
*NPY-hPLAP secretion assays were performed by Ann Kang. *Munc18-1/2 double knockdown cells were generated by Liping Han.
50

IV. Results
IV. i) Voa1, Voa2, and Voa3 are differentially localized in PC12
cells; Voa4 is not expressed in these cells
To examine the isoform-specific localization of the Voa subunit in
neuroendocrine PC12 cells, we engineered PC12 cells that stably express Voa1, Voa2 or
Voa3 as EmGFP- and/or mCherry-fusion proteins through lentivirus-mediated infec
(Figure 4A) (Section III. ii). Although the expression constructs of the V
tion
analyses (Figure 4B,C).
oa isoforms
clearly produced fluorescence signals in transfected HEK293-FT cells, fluorescent
signals from the stable PC12 cells were very weak suggesting low expression levels of
these fluorescent protein-fused Voa proteins. However, the fusion proteins were detected
using either anti-mCherry or anti-GFP antibodies in Western blot
We therefore used anti-GFP antibody in our immunofluorescence studies to detect
the localizations of the GFP-fused Voa proteins. Specifically, we examined co-
localization of these recombinant proteins with synaptotagmin-1, a marker protein for
secretory vesicles (i.e., both synaptic-like clear microvesicles and dense-core vesicles) as
well as with GM130, a marker protein of cis- and medial-Golgi. We found that Voa1-
EmGFP showed punctate staining both at the soma and at the tip of the neurites (Figure
5). At the tip of the neurites, Voa1-EmGFP co-localized with synaptotagmin-1 (Figure 5).
At the soma the Voa1-EmGFP signals do not coincide with GM130. In contrast, Voa2-
EmGFP showed strong perinuclear staining and co-localized well with GM130 (Figure 5)
while it only weakly co-localized with synaptotagmin-1 (Figure 5).
Unlike Voa1- or Voa2-EmGFP, Voa3-EmGFP did not co-localize with either
synaptotagmin-1 or GM130 (Figure 6). It also did not co-localize with LAMP1, a marker
protein of lysosomes (Figure 6). Voa3-EmGFP, however, strongly co-localized with
EEA1, a marker protein of early endosomes (Figure 6).
51

Figure 4: Fluorescent protein-fused Voa subunit
A) A schematic representation of the fluorescent protein-fused Voa subunit depicts Voa
with a large cytoplasmic N-terminus followed by 8 transmembrane domains and
cytoplasmic C-terminus. The fluorescent protein flanks the Voa subunit's C-terminus. B)
and C) Western blots showing the fluorescent protein-fused Voax with x = 1, 2, or 3. 20
μg of protein was loaded into each well.
52

Figure 5: Stably expressed Voa1 co-localizes with SytI while Voa2 co-localizes with
GM130.
NGF-differentiated PC12 cells that stably express Voa1-EmGFP (A, B) or Voa2-EmGFP
(C, D) were co-stained with anti-GFP rabbit polyclonal antibody (all panels) and either
anti-synaptotagmin-1 mouse monoclonal antibody (A, C) or anti-GM130 mouse
monoclonal antibody (B, D). For secondary staining Alexa 488 conjugated goat anti-
rabbit antibody and rhodamine red-x conjugated goat anti-mouse antibody were used
against the appropriate primary antibodies. Right panels are merged pictures. Scale
bar = 10 μm
53

Figure 6. Stably expressed Voa3 is localized on early endosomes.
NGF-differentiated PC12 cells that stably express Voa3-EmGFP were co-stained with
anti-GFP rabbit polyclonal antibody (all panels) and either anti-synaptotagmin-1 mouse
monoclonal antibody (A), anti-GM130 antibody (B), anti-LAMP1 antibody (D) or anti-
EEA1 goat polyclonal antibody (C). For secondary staining Alexa 488 conjugated goat
anti-rabbit antibody, rhodamine red-x conjugated goat anti-mouse antibody or Alexa 568
conjugated donkey anti-goat antibody were used against the appropriate primary
antibodies. Right panels are merged pictures. Scale bar = 10 μm.
54

The data from fluorescent protein-tagged Voa isoforms suggest that Voa1 is the
major isoform expressed on secretory vesicles. To confirm this observation we examined
whether endogenous Voa1 is also localized on secretory granules using rabbit anti-Voa1
antibody (Santa Cruz Biotechnology). In strong agreement with the Voa1-EmGFP data
(Fig. 5A), we found that endogenous Voa1 displayed a strong enrichment at the tip of the
neurites where it was co-localized with synaptotagmin-1 (Figure 7). Thus, Voa1 with
respect to its intracellular localization, can play a role in secretory vesicle acidification.
Our attempt to confirm the localization of endogenous Voa2 as suggested by
Voa2-EmGFP localization data using rabbit anti-Voa2 antibody was not as conclusive as
the staining by this antibody was less clear. Nonetheless, we found a partial enrichment
of anti-Voa2 staining at the perinuclear region, where Voa2 appeared to partially co-
localize with GM130 (Figure 7), which is in agreement with the Voa2-EmGFP data
(Figure 5).
Taken as a whole, the localization data obtained from immunofluorescence
microscopy suggest that Voa1 and to some extent Voa2 may play a role in secretory
vesicle acidification as they were observed to co-localize with secretory vesicle markers.
55

Figure 7. Endogenous Voa1 co-localizes with syntaptotagmin-1.
NGF-differentiated PC12 cells were co-stained with anti-Voa1 rabbit polyclonal antibody
and antisynaptotagmin-1 mouse monoclonal antibody (A, B) or with anti-Voa2 rabbit
polyclonal antibody and GM130 mouse monoclonal antibody (C). For secondary staining
Alexa 488 conjugated goat anti-rabbit antibody and rhodamine red-x conjugated goat
anti-mouse antibody were used against the appropriate primary antibodies. Right panels
are merged pictures. Scale bar = 10 μm
56

We next performed RT-PCR to determine whether or not Voa4 is expressed in
PC12 cells. We found that Voa4 is present in normal rat kidney (NRK) cells while it is
absent in PC12 cells (Figure 8). In addition we showed by RT-PCR that Voa1, Voa3 as
well as GAPDH, a commonly used control protein, is expressed in both PC12 and NRK
cells. Thus, we excluded any further analysis of Voa4 from this study.
Figure 8: Voa4 is not expressed in PC12 cells.
RT-PCR analyses comparing the expression of Voa1, Voa3, Voa4, and GAPDH between
PC12 and normal rat kidney (NRK) cells
57

IV. ii) Western Blot results for Voa1-, Voa2-knockdown and Voa1/ Voa2-double knockdown cells; compensatory up- regulation of Voa2 and Ac45 is seen in Voa1 knockdown cells
To study the functional significance of Voa1 and Voa2 in neurosecretory cells, we
generated PC12 cell lines in which Voa1, Voa2 or Voa1/Voa2 had been down-regulated
(V
oa1.
oa1KD, Voa2KD and DKD, respectively) together with the controls using lentivirus-
mediated shRNA (Section III. v). A summary of the immunoblot results is shown in
Figure 9.
Western blot results indicate that Voa1 knockdown was highly efficient. Anti-
Voa1 antibody from Synaptic Systems, which is specific to rodent Voa1, and that from
Santa Cruz Biotechnology, which detects both rodent Voa1 and human Voa1 (see Figure
17) showed similar levels of knockdown in Voa1KD and DKD. Knockdown by Voa2 was
less complete as there appeared to be more residual Voa2 as seen in Voa2KD and DKD
using two different anti-Voa2 antibodies - mouse polyclonal anti-Voa2 antibody from
Abnova and rabbit polyclonal anti-Voa2 antibody. Voa1KD exhibited an upregulation of
Voa2 possibly due to a compensatory reaction of the cell to the down-regulation of Voa1.
Another protein that appeared to be upregulated due to the down-regulation of Voa1 is
Ac45, an accessory protein of the Vo sector (Supek et al., 1994), as can be seen in
Voa1KD and DKD cells (Figure 9 and 10).
Examining the expression levels of several other proteins, which include another
subunit of the Vo sector of the V-ATPase (Vod1), t-SNARE proteins (syntaxin-1 and
SNAP-25) (Söllner et al., 1993), a marker protein for cis- and medial-Golgi (GM130)
(Nakamura et al., 1995), a marker protein for the ER (Calnexin) (Wada et al., 1991) and a
general membrane trafficking protein (VCP/p97) (Peters et al., 1990) did not reveal any
significant changes in expression. These observations strengthen the idea that the
upregulation of Voa2 in Voa1KD and upregulation of Ac45 in Voa1KD and DKD cells
are specific and are likely due to a compensatory reaction of the cell in response to the
down-regulation of V
58

Figure 9: Upregulation of Voa2 and Ac45 is seen in Voa1KD.
Western blot summary of A) Voa1KD B) Voa2KD and C) DKD using primary antibodies
indicated on the right. Upregulation of Ac45 is also seen in DKD. 20 μg of sample was
loaded into each well.
59

Figure 10: Significant upregulation of Voa2 and Ac45 in Voa1KD and of Ac45 in
DKD
A) Quantification of the changes in Voa2 and Ac45 expressions between control and
Voa1KD. B) Quantification of the changes in Ac45 expressions between control and
DKD. For both A) and B) Western Blot images of these proteins were quantified using
Image J. The protein expression in the knockdown cells was normalized with that in
control cells. The data are from 4-5 independent blots.
60

IV. iii) Knockdown of Voa1, but not of Voa2, results in a
significant reduction in dense-core vesicle acidification
To examine the effects of Voa knockdown on acidification inside dense-core
vesicles, we transfected NPY-epHluorin into Voa1KD and Voa2KD along with their
respective control and performed fluorescence activated cell sorting (FACS) to quantify
the fluorescence signals (see Section III. viii).
We found a consistent increase in the averaged fluorescence signals of NPY-
epHluorin from Voa1KD cells compared to its control (Figure 11A) which suggests a
disruption of dense-core vesicle acidification. This effect was statistically significant
(Student’s independent t-test, n = 8 each, t14 = 3.20, p < 0.01).
However, from this data alone we cannot rule out the possibility that the Voa1KD
was somehow more transfection-prone and/or accumulate the transfected proteins.
Therefore, we performed immunoblot analyses of the expression levels of transfected
NPY-epHluorin which showed no increases in NPY-epHluorin in Voa1KD compared to
its control (Figure 11C).
Additionally, we transfected both Voa1KD and its control with a construct that
expresses NPY fused with the soluble domains of human placental alkaline phosphatase
(NPY-hPLAP) and quantified its enzymatic activity. We found that the enzymatic
activities of NPY-hPLAP extracted from the transfected cells, and normalized by the total
protein concentrations, were comparable between control and knockdown cells (n = 6
each) (Figure 11D). Taken together the results suggest that the observed increase in the
fluorescence signals of NPY-epHluorin in Voa1 knockdown cells is not due to the
increased expression and/or accumulation of these fluorescent proteins, but rather the
increase in fluorescence is due to an increase in pH inside the dense-core vesicles of the
Voa1 knockdown cells.
61

On the other hand FACS analyses involving Voa2KD and its control showed no
significant differences in fluorescence between them (n= 9 each, t16 = 1.37, p = 0.19)
suggesting that dense-core vesicle acidification is normal in Voa2KD. Combining the
above data it appears that Voa1 is a critical component of dense-core vesicle acidification
while Voa2 is not the major determinant of vesicular pH, at least in the presence of Voa1.
62

Figure 11: Down-regulation of Voa1 but not of Voa2 results in significant reductions
of dense-core vesicles
A) Summary data of FACS analysis of fluorescence signals from control and
Voa1KD that were transfected with NPY-epHluorin (n = 8 each). B) Summary data of
FACS analysis of fluorescence signals form control and Voa2KD there were transfected
with NPY-epHluorin (n = 11 each) A) and B) In each analysis, 104 PI-negative cells were
quantified and their mean value was used for the summary data. (C) Immunoblot analysis
of the transfected NPY-epHluorin in control and Voa1 knockdown cells. Twenty μg of
total homogenates from control and stable Voa1 knockdown PC12 cells that were
electroporated with 15 μg of pCMV-NPY-epHluorin were analyzed by SDS-PAGE and
immunoblotting using anti-GFP antibody. The results of three independent
electroporations are shown. (D) Quantification of the protein expression
of transfected NPY-hPLAP.
63

IV. iv) Double knockdown Voa1 and Voa2 caused dramatic
reductions in dense-core vesicle acidification
Earlier we described an interesting up-regulation of Voa2 in response to the
down-regulation of Voa1 (Figure 10; Section IV. ii). Although Voa2 knockdown alone
did not induce significant changes in acidification of dense-core vesicles (Figure 11),
Voa2 may still play a significant compensatory role in vesicular acidification in the
absence of Voa1. To examine the functional significance of Voa1 and Voa2 as a whole,
we generated Voa1/ Voa2 double knockdown (DKD) cells by infecting PC12 cells with
both Voa1 and Voa2 knockdown constructs (Section III. v) (Figure 9).
When DKD and its control were transfected with NPY-epHluorin and then
quantified with FACS, the distribution of the fluorescence signals in DKD was evidently
shifted to the right when compared with the distribution of the signal of its control
(Figure 12). The averaged signals in DKD was about four times that of its control. This
difference in fluorescence between DKD and its control was highly statistically
significant (n = 13 each, t24 = 5.16, p < 0.0001). To ensure that the observed
fluorescence signals measured were not due to mis-localized NPY-epHluorin we
transfected DKD and its control with NPY-EmGFP in order to view its localization.
Immunofluorescence confocal microscopic analysis of these transfected, nerve growth
factor (NGF)-differentiated cells revealed that NPY-EmGFP strongly co-localized with
Secretogranin II (SgII), a marker for dense-core vesicles, in both DKD and its control
which suggest that sorting of the reporter is not affected by double knockdown of Voa1
and Voa2 (Figure 13 and 14). It therefore appears that dense-core vesicle acidification is
severely defective in DKD. Our results demonstrate that Voa1 and, to a lesser degree,
Voa2 play overlapping roles in the acidification of dense-core vesicles.
We then examined whether the dramatic difference in NPY-epHluorin
fluorescence between control and DKD cells is abolished upon application of NH4Cl
(Section III. viii). If the difference in NPY-epHluorin fluorescence between control and
64

DKD cells is due to the difference in their DCV luminal pH, this difference should be
reduced or abolished when all intracellular organelles become alkalinized. Using HEPES-
buffered saline, we again observed a clear difference in the FACS signal of NPY-
epHluorin between DKD and its control in the absence of NH4Cl (i.e., at 0 mM NH4Cl in
Figure 15, n = 10 each, t18 = 3.81, p < 0.0001). NH4Cl increased the signal of NPY-
epHluorin of both control and DKD cells in a dose-dependent manner. However, the
increase in the NPY-epHluorin signal was steeper in the control cells than in the DKD
(Figure 15). In the presence of 100 mM NH4Cl, there was no statistically significant
difference in NPY-epHluorin signal between DKD and it control and DKD (n = 10 each,
t18 = 1.65, p = 0.12). When normalized by the signal of NPY-epHluorin in the presence
of 100 mM NH4Cl, presumably the maximal possible fluorescence, the signal without
NH4Cl was 13.6% in the control cells whereas the signal without NH4Cl was 41.4% in
DKD (Figure 15). These results confirm that the lumen of dense-core vesicles are
significantly alkalinized in DKD compared to its control.
We further wanted to determine the exact changes in pH inside the dense-core
vesicles caused by knockdown of both Voa1 and Voa2. To accurately measure the
changes in pH, we used a KCl-based solution containing a combination of ionophores,
monensin and nigericin to allow us to dictate organelle luminal pH (Section III. viii)
(Kim et al., 1996, Demaurex et al., 1998) and calibrated the signal of our reporter
construct NPY-pHluorin with respect to pH. Specifically, the FACS signal of NPY-
epHluorin in control and DKD cells that was measured in the normal HEPES-buffered
saline was calibrated with the FACS signal of NPY-epHluorin measured in the KCl
calibration buffer (Figure 16). The results indicate that the average pH inside dense-core
vesicles of the control cells is ~6.0 compared to ~6.6 in DKD. This is a significant shift in
pH of dense-core vesicles by the double knockdown of Voa1 and Voa2, considering that
cytosolic pH is around 7.2.
We also observed that the signal of NPY-epHluorin was higher in DKD than in
the control cells at each respective pH (Figure 16), which suggests that the total amount
of NPY-epHluorin expressed in DKD was higher than its control. A higher amount NPY-
65

epHluorin inside DKD can explain why differences in the signal of NPY-epHluorin
between DKD and controls cells were not completely abolished in the presence of 100
mM NH4Cl (Figure 15). Nevertheless, our calibration experiment clearly indicate a
significant shift in pH in dense-core vesicles resulting from the knockdown of both Voa1
and Voa2.
Figure 12: Down-regulation of both Voa1 and Voa2 results in dramatic reductions in
dense-core vesicle acidification
(A) Examples of FACS analysis of fluorescence signals compare the fluorescence
signal distribution from DKD and its control that are transfected with NPY-epHluorin.
For each sample, 104 PI negative cells were analyzed. (B) Summary data of the FACS
analysis of fluorescence signals from control and Voa1/Voa2 double knockdown cells
that are transfected with NPY-epHluorin (n = 11 each).
66

Figure 13: Sorting of NPY-EmGFP to dense-core vesicle is not affected by knocking
down Voa1 and Voa2.
NGF-differentiated control (upper panels) and DKD (lower panels) cells that were
transfected with NPY-EmGFP and stained with anti-Secretogranin II (SgII) rabbit
polyclonal antibody. For secondary staining rhodamine red-x conjugated goat anti-rabbit
antibody was used. Right panels are merged pictures. Scale bar = 10 μm.
67

Figure 14: Sorting of NPY-EmGFP to dense-core vesicle is not affected by knocking
down Voa1 and Voa2 (lower magnification)
NGF-differentiated control (upper panels) and DKD (lower panels) cells that were
transfected with NPY-EmGFP and stained with anti-Secretogranin II (SgII) rabbit
polyclonal antibody. For secondary staining rhodamine red-x conjugated goat anti-rabbit
antibody was used. Right panels are merged pictures. Scale bar = 10 μm.
68
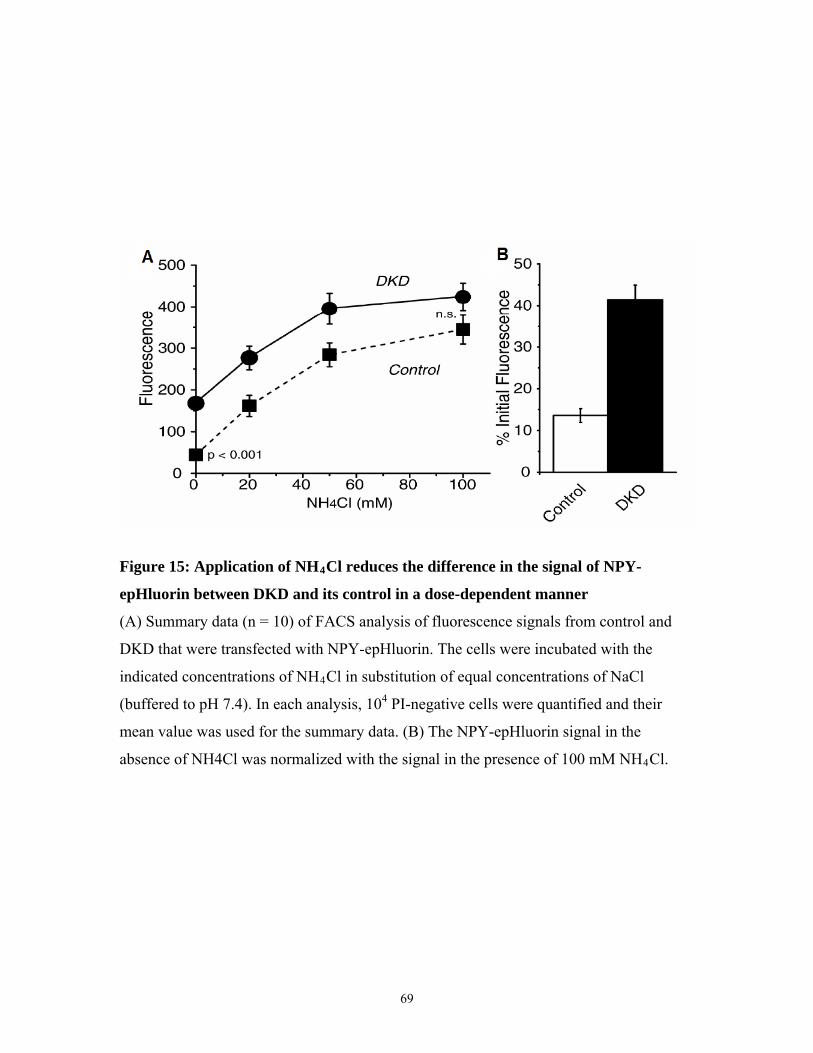
Figure 15: Application of NH4Cl reduces the difference in the signal of NPY-
epHluorin between DKD and its control in a dose-dependent manner
(A) Summary data (n = 10) of FACS analysis of fluorescence signals from control and
DKD that were transfected with NPY-epHluorin. The cells were incubated with the
indicated concentrations of NH4Cl in substitution of equal concentrations of NaCl
(buffered to pH 7.4). In each analysis, 104 PI-negative cells were quantified and their
mean value was used for the summary data. (B) The NPY-epHluorin signal in the
absence of NH4Cl was normalized with the signal in the presence of 100 mM NH4Cl.
69

Figure 16: Calibration of NPY-epHluorin signal with respect to pH reveals a
substantial shift in pH in dense-core vesicles by knockdown of Voa1/Voa2.
Summary data of the FACS analysis of the fluorescence signals from control and DKD
cells that are transfected with NPY-epHluorin (n= 6 each). Cells that were used for the
calibration were incubated with a combination of nigericin (5 μg/ml) and monensin (5
μM) in KCl-based solutions that are either MES-buffered (pH 5.5, 6.0 and 6.5) or
HEPES-buffered (pH 7.0 and 7.5). Cells that were used to measure intact fluorescence
were incubated in an NaCl-based solution that is HEPES-buffered (pH 7.4).
70

IV. v) The expression of knockdown-resistant human Voa1
suppressed the acidification defects caused by down-
regulation of endogenous Voa1and Voa1/ Voa2
To ensure that the acidification defects seen in Voa1KD and DKD is due
specifically to the down-regulation of Voa1 and Voa1/ Voa2, respectively, we tested
whether the expression of knockdown-resistant Voa1 can rescue the acidification defects
observed in Voa1KD and DKD. To this end we first engineered PC12 cells in which
knockdown-resistant human Voa1 is stably expressed (Section III. ix). We then
proceeded to knocking down Voa1 from this cell line to generate a human Voa1-
expressing Voa1KD (hVoa1/Voa1KD) PC12 cell line. We further knocked down Voa2
from hVoa1/Voa1KD to generate hVoa1/DKD.
of
igure
6).
rent
The availability of two anti-Voa1 antibodies which has different species-specific
immunoreactivity was key to confirming the successful generation of our cell lines. Anti-
Voa1 antibody from Synaptic Systems is specific to rodent Voa1 while anti-Voa1
antibody from Santa Cruz detects both rodent and human Voa1. Using these two
antibodies we were able to confirm the expression of human Voa1 in all three groups of
cells (hVoa1-expressing, hVoa1/Voa1KD, hVoa1/DKD) as well as the down-regulation of
endogenous Voa1 in hVoa/Voa1KD and hVoa1/DKD (Figure 17). The knockdown
Voa2 was confirmed using mouse polyclonal anti-Voa2 antibody from Abnova (F
1
Transfection with NPY-epHluorin and subsequent analyses by FACS in diffe
concentrations of NH4Cl revealed that there were no differences in NPY-epHluorin
fluorescence signals from hVoa/Voa1KD and hVoa1/DKD compared to their respective
controls (Figure 18). Knockdown-resistant Voa1 was able to suppress the acidification
defect caused by knockdown of Voa1 and Voa1/Voa2. Therefore, the defect in dense-
core vesicle acidification resulting from knockdown of Voa1 and Voa1/Voa2 was
71

specifically due to the down-regulation of these protein and not due to off-target effects
of the knockdown plasmids. From these results, we also confirmed the critical role of
Voa1 as a regulator of dense-core vesicle acidification as Voa1 expression was able to
suppre
ss even the severe acidification defect that was expected from knocking down both
oa1 and Voa2.
V
Figure 17: Expression of knockdown-resistant human Voa1
(A) Immunoblot analyses of the PC12 cells that stably express mCherry (control) or
human Voa1 (SNM) that is resistant to the Voa1KD shRNA. (B,C) Immunoblot analyses
of the human Voa1(SNM) expressing cells in which endogenous Voa1 or Voa1/Voa2 was
own-regulated. 20 μg of sample was loaded into each well. d
72

Figure 18: Expression of knockdown-resistant Voa1 suppresses the acidification
nd
tions of NH4Cl in
ubstitution of equal concentrations of NaCl (buffered to pH 7.4).
defects caused by knockdown of Voa1 or Voa1/Voa2.
Summary data (n = 6-7) of FACS analysis of fluorescence signals from Voa1KD (A) a
DKD (B) and their respective controls double knockdown cells that expressed human
Voa1(SNM). The cells were incubated with the indicated concentra
s
73

IV. vi) Catecholamine uptake is significantly reduced in Voa1/
Voa2 double-knockdown cells
lar
nse-core vesicles should lead to a
duction in catecholamine uptake into these vesicles.
that the uptake of [3H]-NA was greatly increased in
e presence of MgATP (Figure 19).
n
16
ing
nt across the membrane of dense-core vesicles with Voa1 playing a more important
le.
The uptake of catecholamine into secretory vesicles is mediated by the vesicu
monoamine transporter (VMAT) and requires a proton gradient across the vesicular
membrane that is established by the V-ATPase (Moriyama and Futai, 1990, Amara and
Kuhar, 1993, Schuldiner et al., 1998, Masson et al., 1999, Gasnier, 2000) (Section I.iii.c).
We therefore anticipated that reduced acidification of de
re
We first confirmed the ATP-dependent nature of the uptake of [3H]-NA into
dense-core vesicles by demonstrating
th
We then proceeded to examine the abilities of our knockdown cells - Voa1KD,
Voa2KD and DKD - to uptake [3H]-NA keeping in mind the different severities of (or
lack of) acidification defects they displayed in earlier sections. We found that Voa2KD
did not show any changes in the uptake of [3H]-NA (Figure 20B). While Voa1KD
showed a tendency of reduced [3H]-NA uptake (Figure 20A, control: 9354 ± 1419 dpm,
=9; Voa1KD: 6559 ± 2603 dpm, n = 9) the difference was not statistically significant (t
= 1.68, p = 0.11). However, as expected from its severe phenotype of dense-core vesicle
acidification defect DKD showed a strong reduction (by 60%) in the uptake of [3H]-NA
compared to its control and this effect was highly significant (n = 6 each, t10 = 3.69, p <
0.01) (Figure 20C). Combined, these results suggest that Voa1 and Voa2 play overlapp
roles in the uptake of catecholamines into dense-core vesicles by generating a proton
gradie
ro
74

Figure 19: ATP-dependent [3H]-NA uptake in PC12 cells.
An example of [3H]-NA uptake assays in the presence or absence of 2 mM MgATP using
mechanically perme
abilized wild-type PC12 cells performed in quadruplicates. The error
ars indicate SEM. b
75

Figure 20: Down-regulation of Voa1/Voa2, but not of Voa1 or Voa2, result in
oa2
uble knockdown and its
ontrol (C, n = 6 each) cells. The error bars indicate SEM.
significant decreases in the uptake of [3H]-NA.
[3H]-NA uptake assays in the presence or absence of 2 mM MgATP were performed
using mechanically permeabilized Voa1 knockdown and its control (A, n = 9 each), V
knockdown and its control (B, n = 5 each) or Voa1/Voa2 do
c
76

IV. vii) Endogenous dopamine contents are significantly
reduced in Voa1 and/or Voa2 knockdown cells
s dopamine content correlating to the severity of dense-core
eir
12
opamine
tal protein
lly
s.
tein, n = 15), a difference that was highly significant (t28 = 5.53, p < 0.001)
igure 21D).
We next examined one physiological consequence of reduced catecholamine
uptake in our Voa knockdown cells, namely, dense-core vesicle dopamine content.
Similar to parental adrenal chromaffin cells, PC12 cells store catecholamines inside
dense-core vesicles. Like noradrenaline from the previous section the uptake of dopamine
also requires a proton gradient across the vesicular membrane. We, therefore, expected to
see reductions in endogenou
vesicle acidification defect.
We measured the catecholamine contents of the Voa knockdown cells with th
respective controls using high performance liquid chromatography (HPLC). HPLC
effectively separates adrenaline, noradrenaline and dopamine (Figure 21E,F). In PC
cells dopamine is the major catecholamine whereas noradrenaline is the minor one
(Greene and Tischler, 1976, Fujita et al., 2007) (Figure 21F). The values of d
concentrations obtained were normalized by the corresponding to
concentrations for the Voa knockdown cells and their controls.
We found a reduction of dopamine concentrations inside (0.78 ± 0.06 ng/μg
protein, n = 8) compared with its control (1.48 ± 0.13 ng/μg protein, n = 9), a difference
that was highly significant (t16 = 4.97, p < 0.001) (Figure 21A). Although more modest,
we also observed that the dopamine contents of Voa2KD (0.64 ± 0.08 ng/μg protein, n =
25) were lower than its control (0.95 ± 0.11 ng/μg protein, n = 25), which was statistica
significant (t48 = 2.13, p < 0.05) (Figure 21B). Thus, in addition to Voa1 knockdown,
Voa2 knockdown also result in significant impacts on dopamine storage inside the cell
The most severe reduction in endogenous dopamine content was observed for DKD.
Knocking down both Voa1 and Voa2 resulted in almost a 70% reduction in dopamine
contents in DKD (0.29 ± 0.04 ng/μg protein, n = 15) compared to its control cells (0.91 ±
0.10 ng/μg pro
(F
77

To ensure that the observed differences in dopamine concentrations betwe
control and DKD cells using whole-cell lysates truly reflected the differences in
dopamine concentrations inside dense-core vesicles, we proceeded to measure dopamine
contents from partially purified dense-core vesicles (Martin and Kowalchyk, 1997) of
DKD and its control. We found similar reductions of dopamine concentrations in the
partially purified dense-core vesicles of DKD compared with those of it control (n = 6
each, t10 = 5.20, p < 0.001) (Figure 22B). Our HPLC results indicate that V
en
e
s catecholamine inside the cells,
although Voa2 also appears to play a minor role.
oa1 is th
critical isoform for the maintenance of endogenou
78

79

Figure 21. Down-regulation of Voa1, Voa2 and Voa1/Voa2 causes significant
reductions in the amount of endogenous dopamine stored in PC12 cells.
Endogenous dopamine concentrations were measured using HPLC for Voa1 knockdown
and its control (A, n = 9 each), Voa2 knockdown and its control (B, n = 25 each) or
Voa1/Voa2 double knockdown and its control (D, n = 15 each). Values were normalized
with the total protein concentrations of the samples. The error bars indicate SEM. C)
Sample picture of HPLC data showing differences in dopamine amounts between control
and Voa1/Voa2 double knockdown cells. E) and F) HPLC analysis of catecholamines
from standard samples and PC12 cells. (E) Separation of standard samples (the volume of
the injected sample is 10 μl, the concentration of catecholamines is 0.25 ng/μl each, A:
adrenaline, NA: noradrenaline, DA: dopamine). (F) PC12 cells.
80

Figure 22: Down-regulation of Voa1/Voa2 cause significant reductions in the
amount of endogenous dopamine in purified plasma membrane-associated dense-
core vesicles
Endogenous dopamine concentrations were measured using HPLC for DKD and its
control (B, n = 6 each) from purified plasma membrane-associated DCV. 2.5
μg of this purified sample was immunoblotted with anti-Secretogranin II rabbit
polyclonal antibody (A).
81

IV. viii) Ca2+-dependent regulated secretion of transfected
peptide is normal in Voa1KD and DKD
Up to this point our study had been focused on elucidating the role of the Voa
subunit in dense-core vesicle acidification. To that end we obtained strong evidence that
Voa1 and, to a lesser degree, Voa2 play critical and overlapping roles in the acidification
and transmitter uptake and storage of dense-core vesicles. Recently, however, an
interesting and independent function has been suggested for Voa1 in exocytotic and
phagocytotic membrane fusion (Hiesinger et al., 2005, Peri and Nüsslein-Volhard, 2008,
Williamson et al., 2010).
To examine the potential role of Voa1 in exocytotic membrane fusion we
analyzed Voa1KD as well as DKD for their exocytotic abilities. We assayed the secre
of a transfected peptide, NPY-fused human placental alkaline phosphatase (NPY-hPLA
Section III. xii).
tion
P;
We found that both Voa1KD and its control exhibited robust secretion of
transfected NPY-hPLAP upon high K+ stimulation (70 mM), reaching 25-35% secretion
of the total amount of NPY (Figure 23A). There was no statistically significant difference
in high K+-induced NPY-hPLAP secretion between Voa1KD and its control (n = 8 each,
t14 = 0.28, p = 0.79). There was also no statistical significant difference (n = 6 each, t10
= 1.49, p = 0.17) (Figure 23B) in high K+-induced NPY-hPLAP secretion between DKD
and its control. Thus, peptide secretion appear to be normal in both Voa1KD and DKD.
To ensure that the lack of observed secretion defect was not due to the inability of
the NPY-hPLAP assay to detect such changes we performed this assay on Munc18-1/-2
double-knockdown cells (Han et al., 2009). We observed that the regulated secretion of
NPY-hPLAP was strongly reduced in Munc18-1/-2 (n = 6 each, t10 = 9.80, p < 0.001),
82

indicating that dense-core vesicle secretion is dependent on the Sec1/Munc18 family
proteins (Figure 23C).
Our data, which show that peptide release in Voa1KD as well as DKD is normal
compared to their control, does not support the emerging idea that Voa1 functions
critically for membrane fusion downstream of the SNARE proteins (Hiesinger et al.,
2005), at least for the case of dense-core vesicles.
83

Figure 23. Down-regulation of Voa1 or Voa1/Voa2 does not cause significant defects
in Ca2+-dependent secretion of transfected peptide.
(A) Secretion of transfected NPY-hPLAP was stimulated from control and Voa1KD cells
with or without 70 mM KCl for 20 min. Each experiment was performed in triplicates.
The error bars indicate SEM (n = 8). (B) Secretion of transfected NPY-hPLAP was
stimulated from control and DKD cells with or without 70 mM KCl for 20 min. Each
experiment was performed in triplicates. The error bars indicate SEM (n = 6). (C)
Secretion of transfected NPY-hPLAP was stimulated from control, and Munc18-1/2
double knockdown cells with or without 70 mM KCl for 20 min. Each experiment was
performed in triplicates. The error bars indicate SEM (n = 6).
84

V. Discussion
V. i) Summary and Conclusion
In this study we aimed to elucidate the functions of the Voa subunit of the V-
ATPase in PC12 cells, a well established model of neuroendocrine cell. In particular we
wanted to determine the Voa isoforms involved in dense-core vesicle acidification as well
as examine their potential involvement in dense-core vesicle exocytosis. Using
immunofluorescence confocal microscopy to visualize the localization of GFP-tagged
Voa1-3 subunit as well as endogenous Voa1 and Voa2 we established that Voa1 is
strongly localized to secretory vesicles while Voa2 is strongly localized to the Golgi (at
least to the cis- and medial-Golgi) and to a lesser extent the secretory vesicles (Figures 5
and 7). We also observed that Voa3 is strongly localized to the early endosome and not to
secretory vesicles, the Golgi or lysosomes (Figure 6). Reverse transcription-polymera
chain reaction (RT-PCR) confirmed the lack of V
se
to
the
oa4 expression in PC12 cells which
allowed us to exclude its analyses from this study (Figure 8). Although the presence of
Voa3 in PC12 cells was confirmed by RT-PCR as well as by Western blot we decided
also exclude its analysis with respect to dense-core vesicle acidification to focus on
analyses Voa1 and Voa2.
To elucidate their involvement in dense-core vesicle acidification we knocked
down Voa1 and/or Voa2 using lentivirus-mediated shRNA from PC12 cells. Down
regulation of Voa1 resulted in the upregulation of Voa2 as well as the upregulation of the
85

mammalian V-ATPase accessory protein, Ac45 (Figures 9 and 10). Using NPY-
epHluorin as a pH-sensitive reporter construct for dense-core vesicles in tandem with
FACS analysis we observed that down-regulation of Voa1 alone resulted in a consistent
increase in NPY-epHluorin fluorescence which suggests defects in dense-core vesicle
acidification . This increase in fluorescence was not due to accumulation of the NPY-
epHluorin protein inside the Voa1 knockdown cells as shown by Western blot (Figure 11).
The knockdown of Voa2, on the other hand, did not show an increase in NPY-epHluorin
suggesting that dense-core vesicle acidification is normal in these cells (Figure 11).
Double knockdown of Voa1 and Voa2, i.e. DKD, exhibited a dramatic increase
(~4 fold increase) in NPY-epHluorin fluorescence compared to its control which suggests
that dense-core vesicle acidification is severely impaired in these cells (Figure 12).
Additionally the proper localization of NPY-EmGFP to dense-core vesicles, as evident by
co-localization of this peptide with secretogranin II, in both control and DKD indicate
that sorting of the transfected peptide is not affected by loss of Voa1 and Voa2 (Figures
13 and 14). However, experiments using ammonium chloride to alkalinize all
intracellular compartments of the cell revealed that the maximal NPY-epHluorin
fluorescence of DKD was consistently higher than the maximal NPY-epHluorin
fluorescence of its control (~20-30% higher) (Figure 15). The ammonium chloride data
suggest that the dramatic increase in NPY-epHluorin seen in DKD in normal cond
(in the absence of ammonium chloride) is not all attributable to defects in dense-core
vesicle acidification alone. The accumulation of NPY-epHluorin in DKD compared to
control can explain some of the observed increase in NPY-epHluorin fluorescence
exhibited by DKD. Nevertheless, the major contributor to this large increase in
itions
86

fluorescence appears to be the loss of dense-core vesicle acidification as calibration
experiments using the ionophores nigericin and monensin showed that on average the pH
of dense-core vesicles is ~6.6 in DKD compared to only
~6.0 in its control (Figure 16).
e to
ell
Having obtained strong evidence that knocking down both Voa1 and Voa2
resulted in severe defects in dense-core vesicle acidification it became imperative to show
that the effects observed are specifically due to the loss of Voa1 and Voa2 and not du
off-target effects of using the knockdown strategy. To this end, we generated a PC12 c
line in which knockdown-resistant human Voa1 is expressed and from this cell line we
proceeded to knockdown either Voa1 or both Voa1 and Voa2. The expression of human
Voa1 as well as the down-regulation of endogenous Voa1 and Voa2 in the resulting cell
lines were confirmed by Western blot (Figure 17). Examining these cell lines we
determined that the presence of knockdown-resistant Voa1 prevented the dense-core
vesicle acidification defect caused by loss of endogenous Voa1 and endogenous Voa1/
Voa2 (Figure 18). The phenotype of dense-core vesicle acidification defect seen in
Voa1KD as well as in DKD therefore appears to be specifically caused by the loss of
Voa1 and Voa1/Voa2, respectively.
We further linked the disruption of dense-core vesicle acidification to
physiological processes which include catecholamine uptake and storage. Catecholamine
uptake into secretory vesicle is dependent on the proton gradient generated by V-
ATPases (Moriyama and Futai, 1990). Using a slightly modified version of an established
uptake assay (Ahnert-Hilger et al., 1998, Brunk et al., 2009) we found that the uptake of
[3H]-NA is severely reduced in DKD while it is normal in Voa2KD. For Voa1KD
although a tendency of reduced uptake was observed this reduction was not statistically
87

significant (Figure 20). These results correlated well with earlier observations of the trend
of acidification defects seen in Voa1KD, Voa2KD and DKD. In addition to reduction in
transmitter uptake the endogenous contents of dopamine also appear to be reduced
accordingly with respect to the degree of acidification defects observed in the different
knockdown cells. While both Voa1KD and Voa2KD show significant reductions of
dopamine content as seen in HPLC measurements of whole-cell lysates DKD show the
most dramatic reductions of dopamine content (Figure 21). Additional HPLC
measurements on DKD of dopamine content from partially purified plasma membrane-
associated dense-core vesicles confirmed that it was dense-core vesicle dopamine content
that was dramatically reduced (Figure 22). Therefore, the disruption in dense-core
vesicle acidification appears to have a negative impact on catecholamine uptake and
storage.
Lastly we examined whether Voa1KD and DKD display any defect in dense-core
vesicle exocytosis of a transfected peptide, NPY-hPLAP. We found that the secretion of
NPY-hPLAP was normal in these cells. A previously generated line of Munc18-1/2
double knockdown cells (Han et al., 2009) showed defective secretion of this peptide
which confirmed the critical involvement of the Sec1/Munc18 family proteins in
regulated secretion.
Taken altogether this study provides strong evidence that Voa1 and Voa2 can
(from the localization data) and do (from NPY-epHluorin/FACS data) function critically
in dense-core vesicle acidification and that the loss of these proteins result in predictable
physiological consequences in catecholamine uptake and storage. Voa1 appears to be the
more important player with respect to dense-core vesicle acidification although Voa2
88

appears to play a role as well, especially in the absence of Voa1. To my knowledge, this
study is the first to examine the effect of double knockdown Voa1 and Voa2 on the
acidification of intracellular compartments. It is also the first to successfully show the
disruption of dense-core vesicle acidification resulting from the removal of these two Voa
isoforms. This study, however, does not support the role of the Voa subunit in dense-core
vesicle exocytotic membrane fusion.
89

V. ii) Localization of Voa isoforms as determinant for V-
ATPase localization
The results from this study suggest that in NGF-differentiated PC12 cells Voa1 is
primary localized to secretory vesicles and that Voa2 is primarily localized to the Golgi
with some localization to secretory vesicles (Figure 5). Furthermore, Voa3 is suggested to
be sorted primarily to early endosomes in PC12 cells (Figure 6). The localization data
from this study fully support and complement the ensuing data the Voa1, and to a lesser
extent, Voa2 are critically involved in dense-core vesicle acidification, i.e. the are
localized to dense-core vesicles where they can contribute to V-ATPase function. The
observed differential localization of the Voa isoforms in the studied PC12 cells also
supports the generally accepted idea that Voa isoforms contain important sorting signals
for compartment-specific localization of the V-ATPase.
However, the big picture defining the roles played by Voa isoforms with respect
to determining V-ATPase localization in different cell-types is admittedly more
complicated. While there is strong evidence to suggest that Voa isoforms contain the
localization signals for compartment-specific sorting of V-ATPases the variability of V
isoform localization within different cell types suggest that other factors may also be
involved in the sortin
oa
g process.
A study utilizing chimeras of the two yeast Voa isoforms, Vph1p (normally
localized to the vacuoles) and Stv1p (normally localized to the Golgi), revealed that
domains in the cytosolic N-termini of the isoforms directly determine V-ATPase sorting
to the specific compartments (Kawasaki-Nishi et al., 2001a). Another study utilized the
existence of alternative Voa1 splice variants to study the domains important for
90

compartmental sorting of Voa (Poea-Guyon et al., 2006). In rat, there exist four Voa1
splice variants, a1-I-IV; a1-I contains exon C (an 18-bp insertion between exons 17 and
18), a1-II contains exon N (a 21-bp insertion between exons 4 and 5), a1-III contains
neither while a1-IV contains both exons C and N (Poea-Guyon et al., 2006). The study
revealed that in rat hippocampal neurons a1-I strongly co-localized with the synaptic
vesicle marker, SV2, within axonal varicosities while a1-IV was sorted to the distal
dendrites and axons and only weakly co-localized with SV2. a1-II and a1-III, which are
not normally expressed in neurons, mimicked the localization of a1-IV and a1-I,
respectively (Poea-Guyon et al., 2006). The authors concluded that the alternative
splicing of exon N may modify sorting of a1 to synaptic vesicles, i.e. lack of exon N
result in default sorting to synaptic vesicle. The results also suggest that exon C may
allow for the expression of a1-I and a1-IV in neurons. These two studies provide strong
evidence for the role N-terminus of Voa as a determinant of Voa localization.
A survey of literature, however, reveals that the sorting of Voa isoforms may also
have a cell-specific component. For example, while Voa2 is generally associated with the
Golgi -- in cultured osteoclasts (Toyomura et al., 2003), epididymis clear cells
(Pietrement et al., 2006), hippocampal neurons (Poea-Guyon et al., 2006) -- this isoform
is also detected in early endosomes of kidney proximal tubule cells (Hurtado-Lorenzo et
al., 2006) suggesting that specific cellular processes may also contribute to determining
the sorting of Voa isoforms. Intracellular conditions may also affect sorting of the Voa
isoform. For example, Voa3 which is localized to lysosomes in a mouse macrophage cell
line gets sorted to the plasma membrane after differentiation into osteoclast-like cell
(Toyomura et al., 2003).
91

The existence of multiple isoforms of some of the other V-ATPase subunits and
the findings that some of these isoforms have tissue-specific expression supports the idea
that localization of V-ATPases in a particular membrane in a given cell type may be
driven by a unique isoform signature. Examples include V-ATPases of the kidney and
epididymis (a4, B1, C2b, d2, G3), brain (a1, G2), and osteoclasts (a3, d2) (Forgac, 2007)
Taken altogether, the above studies suggest that while the Voa subunit is likely to
contain a sorting signal for the localization of the V-ATPase due to its high variability
compared to the other V-ATPase subunits there may be other factors that contribute to
intracellular V-ATPase localization.
In this study the criteria for co-localization was the overlap of fluorescence
signals of the Voa isoforms (or GFP-fused versions) with the signals of a specific
intracellular marker. However, localization studies using light microscopy is limited with
respect to the resolution it can achieve. In general, a resolution of 200 nm is considered to
be the limit of light microscopy as signals originating from less that 200 nm apart cannot
be distinguish. Keeping in mind that organelles such as dense-core vesicles (~100 nm
diameter) and synaptic-like microvesicles (~40 nm diameter) which are found in PC12
cells (Martin and Grishanin, 2003) are less than this distance localization data from
fluorescence light microscopy must be interpreted with some reservation. Additional
methods to confirm localization data may include immunogold electron microscopy
(much higher resolution than light microscopy) or the biochemical purification of the
organelle of interest (such as dense-core vesicles) (Wegrzyn et al., 2010) followed by
Western blot analyses.
92

V. iii) Voa1 and Voa2 in dense-core vesicle acidification;
alternate interpretations of our current data
Examining the results from this study I believe that the conclusion which I
reached, that Voa1, and to a lesser extent, Voa2 are critical for dense-core vesicle
acidification is a reasonable one. However, like almost every study critical analyses of the
results as well as of the techniques used which lead to alternative interpretations of the
data is entirely possible.
My interpretation of the results from this study depended on two key assumptions.
Firstly, the sorting of the transfected pH-sensitive reporter NPY-epHluorin in
undifferentiated PC12 cells is specific to dense-core vesicles and is not affected by down-
regulation of Voa1 and/or Voa2. The use of FACS to examination dense-core vesicle
acidification is absolutely dependent on this assumption because although FACS analysis
allow the quantification of fluorescence from many cells it cannot distinguish between
different sources of the fluorescence signals within each cell. Secondly, the knockdown
of Voa1 and/or Voa2 did not significantly affect the formation and maintenance of dense-
core vesicles. Changes is dense-core vesicle numbers will affect interpretation of the
results obtained from catecholamine loading assays as well as total dopamine content.
With respect to NPY-epHluorin sorting we found that in NGF-differentiated
control as well as DKD cells NPY-EmGFP appears to have been sorted correctly as this
protein strongly co-localized with secretogranin II, a dense-core vesicle marker. However,
all the evidence to support the roles of Voa1 and Voa2 in dense-core vesicle acidification
93

were obtained from undifferentiated PC12 cells, which have a different morphology from
NGF-differentiated PC12 cells (Greene and Tischler, 1976). Also it is not unreasonable
to expect that the sorting of NPY-epHluorin would be affected to some degree by the loss
of the Voa subunit, particularly Voa2, as this isoform has been shown to be localized to
the Golgi (Pietrement et al., 2006, Poea-Guyon et al., 2006) and to be important for
Golgi-related functions (Kornak et al., 2008, Hucthagowder et al., 2009). Additionally,
the ammonium chloride experiments suggest that there is some accumulation of NPY-
epHluorin inside DKD compared to its control (Figure 15). Taken together these
criticisms may lead to the interpretation that the dramatic increase in fluorescence seen by
knocking down both Voa1 and Voa2 is caused by loss of Voa2 via mislocalization of
NPY-epHluorin to a compartment of higher pH, such as the Golgi.
However, my original conclusion that Voa1 is critical for dense-core vesicle
appears to still be solid despite these criticisms. The fact that endogenous Voa1 as well as
overexpressed Voa1-EmGFP strongly localize to secretory vesicles (Figures 5 and 7),
combined with the observations that knockdown of Voa1 alone caused significant
increase in NPY-epHluorin fluorescence in the absence of NPY-epHluorin accumulation
(Figure 11) as well as the observation that knockdown-resistant Voa1 prevented the
increase of NPY-epHluorin fluorescence seen in Voa1KD and DKD (Figures 17 and 18)
strengthen my conclusion that Voa1 is critical for dense-core vesicle acidification.
To alleviate the concerns that increased in fluorescence of NPY-epHluorin is due
mis-localized or accumulated NPY-epHluorin an additional experiment that is
complementary to the FACS analysis can be performed. In this experiment a modified
version of NPY-epHluorin which contains an additional fluorescent protein that is
94

relatively pH-insensitive (such as NPY-mCherry-epHluorin) can be used as the reporter
construct. PC12 cells transfected with this construct and then differentiated with NGF to
promote growth of the neurite tips can be examined live under a fluorescent microscope.
Quantification of the fluorescent signals can be performed exclusively at the tips of the
neurite where dense-core vesicles are located. The epHluorin fluorescence can then be
normalized by the mCherry signal to obtain a pH-dependent signal. This experiment is
complementary to FACS analysis in that it is capable of obtaining fluorescence signals
from a region of interest albeit at the expense of examining a significantly fewer number
of cells.
The second major assumption of this study is relevant to data obtained from the
functional experiments, i.e. those dealing with the physiological effects caused by loss of
dense-core vesicle acidification. In this study, we found that catecholamine uptake
(Figure 20) as well as storage (Figures 21 and 22) is perturbed to varying degrees with
the down-regulation of Voa1 and/or Voa2 and that this perturbation is roughly correlated
with the degree of dense-core vesicle acidification defects observed. However, these data
would be interpreted quite differently if the number of dense-core vesicles themselves
were affected by the knockdown of Voa1 and/or Voa2 as the electron microscopy
analysis of control and DKD would suggest (Appendix Figure 1). If there are fewer
dense-core vesicles as a result of loss of Voa1 and Voa2 then the dramatic decrea
in catecholamine uptake (Figure 20) and storage (Figures 21 and 22) in DKD compared
to its control can be directly attributed to there being fewer dense-core vesicles in DKD
ses seen
.
The interpretation of the electron micrographs themselves with respect of dense-
core vesicle numbers can be misleading, however. The loss of dopamine, the major
95

catecholamine in PC12 cells (Klenchin et al., 1998, Martin and Grishanin, 2003), from
dense-core vesicles can significantly impair its detection by electron microscopy (Martin
and Kowalchyk, 1997) as the vesicles are no longer as 'dense'. I therefore felt it
reasonable to ignore this data when interpreting the catecholamine uptake and storage
data. In fact Western blot analysis of secretogranin II, a marker for dense-core vesicles as
well as a regulator of DCV biosynthesis (Courel et al., 2010), from plasma-membrane
associated dense-core vesicles suggests that DCV numbers remain unchanged (Figure 22).
The use of amperometric measurements of dopamine oxidation can verify the loss of
quantal dopamine content within the dense-core vesicles of DKD and would greatly
complement the HPLC experiments.
96

V. iv) The Voa subunit and membrane fusion
As mentioned in an introductory section recent studies have suggested that the Vo
sector, and in some cases the Voa subunit specifically, is involved in the process of
membrane fusion (Peters et al., 2001, Bayer et al., 2003, Hiesinger et al., 2005, Peri and
Nüsslein-Volhard, 2008, Williamson et al., 2010). To test this interesting yet still
controversial idea that the Voa subunit may be involved in membrane fusion we
performed an experiment which measured the secretion of a transfected peptide, NPY-
hPLAP. We found no significant changes in NPY-hPLAP secretion from Voa1KD as
well as DKD which suggest that the mechanisms of exocytotic are intact in these cells
contrast we found that Munc18-1/2 double knockdown cells exhibit impaired secretion of
NPY-hPLAP confirming SNAREs-dependent nature of exocytotic membrane fusion
(Söllner et al., 1993, Jahn et al., 2003, Jahn, 2004, Sudhof, 2004).
. In
The lone experiment of NPY-hPLAP secretion which we performed in this study
is admittedly not sufficient to fully address the potential role of the Voa subunit in
exocytotic membrane fusion. Firstly, the time course of the peptide secretion assay of 20
minutes, although sufficient for detecting gross defects in the exocytotic mechanism (i.e.
in Munc18-1/2 double knockdown cells), may not be ideal for detecting more subtle
changes (if any) caused by the loss of Voa1 and Voa2. For example, the time course for
dense-core vesicle exocytosis is about 20-30 seconds (Martin and Grishanin, 2003)
meaning that changes in the kinetics of dense-core vesicle exocytosis will likely escape
detection by this assay.
97

In addition to the inherent limits of the peptide secretion assay the lack of
observed secretory defects may be due to residual Voa. Although the knockdown of Voa1
can be considered extremely efficient there is residual Voa1 which may be sufficient to
allow it to perform other functions (if any). Residual Voa2 and Voa3 in DKD may also
play a role in exocytotic membrane fusion.
Lastly, the results from the peptide secretion assay may only be relevant for
dense-core vesicle exocytosis and not for synaptic vesicle exocytosis. Although the
exocytosis of both types of vesicles is known to be mediated by SNARE proteins each
type of vesicle differ in their morphology, membrane protein complements, biogenesis,
trafficking/recycling pathways and release kinetics. For example, exocytosis from
synaptic vesicles occurs in the order of less than a millisecond while exocytosis from
dense-core vesicles takes place in the order of seconds to minutes (Martin and Grishanin,
2003). Therefore, the Voa subunit, as suggested by some recent studies may still play a
critical role in membrane fusion of some organelles.
98

V. v) The suitability of NPY-epHluorin as a dense-core vesicle
pH reporter
In this study the rationale for using NPY-epHluorin as an indicator of dense-core
vesicle pH include the following: 1) Neuropeptide Y is normally sorted to dense-core
vesicles as a part of the regulated secretory pathway; 2) This peptide is secreted during
exocytosis.
The first point takes advantage of normal cellular activity to sort NPY to dense-
core vesicles giving this reporter some specificity. The use of a biological molecule is in
contrast to the use of pH-sensitive dyes such as LysoSensor and LysoTracker which tend
to accumulate in all acidic organelles. As a result while FACs analyses can be performed
on thousands of cells transfected with NPY-epHluorin in a very short time using pH-
sensitive dyes such as LysoSensor and LysoTracker require careful examination of each
cells, e.g. staining with organelle markers to confirm correct localization of the dye with
every single experiment.
This time consuming process results in fewer cells being studied and potentially
higher variability of the results. The fact that this peptide is secreted also allows for
studies using FACs as no extraneous signals originating from the plasma membrane
caused by exocytosis would be retained which can offset fluorescence measurements.
However, these same characteristics of NPY may also be a disadvantage to using
this molecule as a pH reporter. Firstly, disruption of acidification at the Golgi (potentially
due to knocking down Voa1 and Voa2) may cause mislocalization of this peptide as pH is
99

known to be an important regulator DCV biogenesis. Secondly, potential changes in
exocytosis due to loss of Voa1 and Voa2 may cause differential accumulation of DCV
and therefore lead to differential accumulation of NPY-epHluorin in the cells.
100

V. vi) Future Directions
In this study using PC12 cells we have established a mammalian neuroendocrine
cell model in which Voa1 and/or Voa2 are stably down-regulated. Using the pH-sensitive
reporter NPY-epHluorin we successfully showed the dense-core vesicle acidification
defect resulting from loss of Voa1 and Voa1/Voa2. Furthermore, using wild-type PC12
cells as well as PC12 overexpressing Voa subunit fused with GFP we determined the
localization of Voa1-3 in these cells.
With the above tools as a starting point it would be interesting to see whether the
loss of one or more Voa isoforms affects the localization of the remaining isoforms as this
will suggest potential compensatory functions of the Voa isoforms. For example, down-
regulation of Voa1 leads to the upregulation of Voa2. Does this upregulation of Voa2
result in greater trafficking of this isoform to secretory vesicles? It would also be
interesting to know the localization of Voa3-GFP in the absence of Voa1 and Voa2.
Using the cell lines already established we can also examine how the loss of one
or more of the Voa isoforms affect the expression and localization of the other subunits of
the V-ATPase. So far we have looked at the expression of Vod and Ac45. Ac45 was
upregulated in Voa1KD as well as DKD. These types of study should lead to a better
understanding of the relationship of the Voa subunit to the other subunits of the V-
ATPase.
Lastly, using the established Voa knockdown cells the question of Voa's
involvement in exocytotic membrane fusion can be more fully addressed using more
time-sensitive methods. For example, dense-core vesicle exocytosis from the available
PC12 cells can be examined using capacitance measurements.
101

References
Ahnert-Hilger G, Nürnberg B, Exner T, Schäfer T, Jahn R (1998) The heterotrimeric G
protein Go2 regulates catecholamine uptake by secretory vesicles. EMBO J
17:406-413.
Amara S, Kuhar M (1993) Neurotransmitter transporters: recent progress. Annu Rev
Neurosci 16:73-93.
Arunachalam L, Han L, Tassew N, He Y, Wang L, Xie L, Fujita Y, Kwan E, Davletov B,
Monnier P, Gaisano H, Sugita S (2008) Munc18-1 is critical for plasma
membrane localization of syntaxin1 but not of SNAP-25 in PC12 cells. Mol Biol
Cell 19:722-734.
Bayer M, Reese C, Buhler S, Peters C, Mayer A (2003) Vacuole membrane fusion: V0
functions after trans-SNARE pairing and is coupled to the Ca2+-releasing channel.
J Cell Biol 162:211-222.
Brunk I, Blex C, Speidel D, Brose N, Ahnert-Hilger G (2009) Ca2+-dependent activator
proteins of secretion promote vesicular monoamine uptake. J Biol Chem
284:1050-1056.
Camacho M, Machado JD, Alvarez J, Borges R (2008) Intravesicular calcium release
mediates the motion and exocytosis of secretory organelles - A study with adrenal
chromaffin cells. Journal of Biological Chemistry 283:22383-22389.
Clarke M, Maddera L, Engel U, Gerisch G (2010) Retrieval of the Vacuolar H(+)-
ATPase from Phagosomes Revealed by Live Cell Imaging. Plos One 5.
Courel M, Soler-Jover A, Rodriguez-Flores JL, Mahata SK, Elias S, Montero-Hadjadje
M, Anouar Y, Giuly RJ, O'Connor DT, Taupenot L (2010) Pro-hormone
Secretogranin II Regulates Dense Core Secretory Granule Biogenesis in
Catecholaminergic Cells. Journal of Biological Chemistry 285:10030-10043.
Daubner SC, Le T, Wang S (2011) Tyrosine hydroxylase and regulation of dopamine
synthesis. Archives of Biochemistry and Biophysics 508:1-12.
Demaurex N, Furuya W, D'Souza S, Bonifacino JS, Grinstein S (1998) Mechanism of
acidification of the trans-Golgi network (TGN). In situ measurements of pH using
retrieval of TGN38 and furin from the cell surface. J Biol Chem 273:2044-2051.
102

Drose S, Altendorf K (1997) Bafilomycins and concanamycins as inhibitors of V-
ATPases and P-ATPases. Journal of Experimental Biology 200:1-8.
Feng Y, Forgac M (1992) CYSTEINE 254 OF THE 73-KDA A SUBUNIT IS
RESPONSIBLE FOR INHIBITION OF THE COATED VESICLE (H+)-
ATPASE UPON MODIFICATION BY SULFHYDRYL-REAGENTS. Journal of
Biological Chemistry 267:5817-5822.
Forgac M (2007) Vacuolar ATPases: rotary proton pumps in physiology and
pathophysiology. Nat Rev Mol Cell Biol 8:917-929.
Frattini A, Orchard P, Sobacchi C, Giliani S, Abinun M, Mattsson J, Keeling D,
Andersson A, Wallbrandt P, Zecca L, Notarangelo L, Vezzoni P, Villa A (2000)
Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a
subset of human autosomal recessive osteopetrosis. Nat Genet 25:343-346.
Fujita Y, Xu A, Xie L, Arunachalam L, Chou T, Jiang T, Chiew S, Kourtesis J, Wang L,
Gaisano H, Sugita S (2007) Ca2+-dependent activator protein for secretion 1 is
critical for constitutive and regulated exocytosis but not for loading of transmitters
into dense core vesicles. J Biol Chem 282:21392-21403.
Gasnier B (2000) The loading of neurotransmitters into synaptic vesicles. Biochimie
82:327-337.
Greene L, Tischler A (1976) Establishment of a noradrenergic clonal line of rat adrenal
pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad
Sci USA 73:2424-2428.
Gunther W, Luchow A, Cluzeaud F, Vandewalle A, Jentsch TJ (1998) ClC-5, the
chloride channel mutated in Dent's disease, colocalizes with the proton pump in
endocytotically active kidney cells. Proceedings of the National Academy of
Sciences of the United States of America 95:8075-8080.
Han L, Jiang T, Han G, Malintan N, Xie L, Wang L, Tse F, Gaisano H, Collins B,
Meunier F, Sugita S (2009) Rescue of Munc18-1 and -2 double knockdown
reveals the essential functions of interaction between Munc18 and closed syntaxin
in PC12 cells. Mol Biol Cell 20:4962-4975.
103

Hiesinger P, Fayyazuddin A, Mehta S, Rosenmund T, Schulze K, Zhai R, Verstreken P,
Cao Y, Zhou Y, Kunz J, Bellen H (2005) The v-ATPase V0 subunit a1 is required
for a late step in synaptic vesicle exocytosis in Drosophila. Cell 121:607-620.
Hinton A, Bond S, Forgac M (2009) V-ATPase functions in normal and disease processes.
Pflugers Arch 457:589-598.
Hirata T, Iwamoto-Kihara A, Sun-Wada G, Okajima T, Wada Y, Futai M (2003) Subunit
rotation of vacuolar-type proton pumping ATPase: relative rotation of the G and
C subunits. J Biol Chem 278:23714-23719.
Hucthagowder V, Morava E, Kornak U, Lefeber D, Fischer B, Dimopoulou A, Aldinger
A, Choi J, Davis E, Abuelo D, Adamowicz M, Al-Aama J, Basel-Vanagaite L,
Fernandez B, Greally M, Gillessen-Kaesbach G, Kayserili H, Lemyre E, Tekin M,
Türkmen S, Tuysuz B, Yüksel-Konuk B, Mundlos S, Van Maldergem L, Wevers
R, Urban Z (2009) Loss-of-function mutations in ATP6V0A2 impair vesicular
trafficking, tropoelastin secretion and cell survival. Hum Mol Genet 18:2149-
2165.
Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada GH, Bourgoin S,
Casanova J, Wildeman A, Ausiello D, Brown D, Marshansky V (2006) V-ATPase
interacts with ARNO and Arf6 in early endosomes and regulates the protein
degradative pathway. Faseb Journal 20:A1225-A1225.
Igarashi T, Gunther W, Sekine T, Inatomi J, Shiraga H, Takahashi S, Suzuki J, Tsuru N,
Yanagihara T, Shimazu M, Jentsch TJ, Thakker RV (1998) Functional
characterization of renal chloride channel, CLCN5, mutations associated with
Dent's(Japan) disease. Kidney International 54:1850-1856.
Inoue H, Noumi T, Nagata M, Murakami H, Kanazawa H (1999) Targeted disruption of
the gene encoding the proteolipid subunit of mouse vacuolar H+-ATPase leads to
early embryonic lethality. Biochimica Et Biophysica Acta-Bioenergetics
1413:130-138.
Jahn R (2004) Principles of exocytosis and membrane fusion. Ann NY Acad Sci
1014:170-178.
Jahn R, Lang T, Südhof T (2003) Membrane fusion. Cell 112:519-533.
104

Jefferies K, Cipriano D, Forgac M (2008) Function, structure and regulation of the
vacuolar (H+)-ATPases. Arch Biochem Biophys 476:33-42.
Kawamura N, Tabata H, Sun-Wada G-H, Wada Y (2010) Optic Nerve Compression and
Retinal Degeneration in Tcirg1 Mutant Mice Lacking the Vacuolar-Type H(+)-
ATPase a3 Subunit. Plos One 5.
Kawasaki-Nishi S, Bowers K, Nishi T, Forgac M, Stevens TH (2001a) The amino-
terminal domain of the vacuolar proton-translocating ATPase a subunit controls
targeting and in vivo dissociation, and the carboxyl-terminal domain affects
coupling of proton transport and ATP hydrolysis. Journal of Biological Chemistry
276:47411-47420.
Kawasaki-Nishi S, Nish T, Forgac M (2001b) Arg-735 of the 100-kDa subunit a of the
yeast V-ATPase is essential for proton translocation. Proceedings of the National
Academy of Sciences of the United States of America 98:12397-12402.
Kawasaki-Nishi S, Nishi T, Forgac M (2001c) Yeast V-ATPase complexes containing
different isoforms of the 100-kDa a-subunit differ in coupling efficiency and in
vivo dissociation. Journal of Biological Chemistry 276:17941-17948.
Kim JH, Lingwood CA, Williams DB, Furuya W, Manolson MF, Grinstein S (1996)
Dynamic measurement of the pH of the Golgi complex in living cells using
retrograde transport of the verotoxin receptor. J Cell Biol 134:1387-1399.
Kim T, Gondre-Lewis MC, Arnaoutova I, Loh YP (2006) Dense-core secretory granule
biogenesis. Physiology 21:124-133.
Kim T, Loh YP (2006) Protease nexin-1 promotes secretory granule biogenesis by
preventing granule protein degradation. Molecular Biology of the Cell 17:789-798.
Klenchin VA, Kowalchyk JA, Martin TFJ (1998) Large dense-core vesicle exocytosis in
PC12 cells. Methods-a Companion to Methods in Enzymology 16:204-208.
Knoch KP, Bergert H, Borgonovo B, Saeger HD, Altkruger A, Verkade P, Solimena M
(2004) Polypyrimidine tract-binding protein promotes insulin secretory granule
biogenesis. Nature Cell Biology 6:207-214.
Kornak U, Reynders E, Dimopoulou A, van Reeuwijk J, Fischer B, Rajab A, Budde B,
Nürnberg P, Foulquier F, Lefeber D, Urban Z, Gruenewald S, Annaert W,
Brunner H, van Bokhoven H, Wevers R, Morava E, Matthijs G, Van Maldergem
105

L, Mundlos S, Group AD-tS (2008) Impaired glycosylation and cutis laxa caused
by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet 40:32-
34.
Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, Hasan C, Bode U,
Jentsch T, Kubisch C (2000) Mutations in the a3 subunit of the vacuolar H(+)-
ATPase cause infantile malignant osteopetrosis. Hum Mol Genet 9:2059-2063.
Leng XH, Nishi T, Forgac M (1999) Transmembrane topography of the 100-kDa a
subunit (Vph1p) of the yeast vacuolar proton translocating ATPase. Journal of
Biological Chemistry 274:14655-14661.
Li G, Han L, Chou T, Fujita Y, Arunachalam L, Xu A, Wong A, Chiew S, Wan Q, Wang
L, Sugita S (2007) RalA and RalB function as the critical GTP sensors for GTP-
dependent exocytosis. J Neurosci 27:190-202.
Liegeois S, Benedetto A, Garnier JM, Schwab Y, Labouesse M (2006) The V0-ATPase
mediates apical secretion of exosomes containing Hedgehog-related proteins in
Caenorhabditis elegans. Journal of Cell Biology 173:949-961.
Lu M, Sautin YY, Holliday LS, Gluck SL (2004) The glycolytic enzyme aldolase
mediates assembly, expression, and activity of vacuolar H+-ATPase. Journal of
Biological Chemistry 279:8732-8739.
Luchow A, Gunther W, Cluzeaud F, Vandewalle A, Jentsch TJ (1998) A possible role of
the chloride channel ClC-5 in endocytosis in the kidney. Molecular Biology of the
Cell 9:342A-342A.
Manjunath N, Wu H, Subramanya S, Shankar P (2009) Lentiviral delivery of short
hairpin RNAs. Advanced Drug Delivery Reviews 61:732-745.
Marshansky V, Futai M (2008) The V-type H+-ATPase in vesicular trafficking: targeting,
regulation and function. Curr Opin Cell Biol 20:415-426.
Martin TF, Kowalchyk JA (1997) Docked secretory vesicles undergo Ca2+-activated
exocytosis in a cell-free system. J Biol Chem 272:14447-14453.
Martin TFJ, Grishanin RN (2003) PC12 cells as a model for studies of regulated secretion
in neuronal and endocrine cells. Neurons: Methods and Applications for the Cell
Biologist 71:267-286.
106

Masson J, Sagné C, Hamon M, El Mestikawy S (1999) Neurotransmitter transporters in
the central nervous system. Pharmacol Rev 51:439-464.
Miura GI, Froelick GJ, Marsh DJ, Stark KL, Palmiter RD (2003) The d subunit of the
vacuolar ATPase (Atp6d) is essential for embryonic development. Transgenic
Research 12:131-133.
Moriyama Y, Futai M (1990) H+-ATPASE, A PRIMARY PUMP FOR
ACCUMULATION OF NEUROTRANSMITTERS, IS A MAJOR
CONSTITUENT OF BRAIN SYNAPTIC VESICLES. Biochemical and
Biophysical Research Communications 173:443-448.
Moriyama Y, Maeda M, Futai M (1992) THE ROLE OF V-ATPASE IN NEURONAL
AND ENDOCRINE SYSTEMS. Journal of Experimental Biology 172:171-178.
Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis T, Warren
G (1995) Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol
131:1715-1726.
Niikura K, Takano M, Sawada M (2004) A novel inhibitor of vacuolar ATPase,
FR167356, which can discriminate between osteoclast vacuolu ATPase and
lysosomal vacuolar ATPase. British Journal of Pharmacology 142:558-566.
Nishi T, Forgac M (2000) Molecular cloning and expression of three isoforms of the 100-
kDa a subunit of the mouse vacuolar proton-translocating ATPase. J Biol Chem
275:6824-6830.
Nishi T, Forgac M (2002) The vacuolar (H+)-atpases - Nature's most versatile proton
pumps. Nature Reviews Molecular Cell Biology 3:94-103.
Oka T, Murata Y, Namba M, Yoshimizu T, Toyomura T, Yamamoto A, Sun-Wada G,
Hamasaki N, Wada Y, Futai M (2001a) a4, a unique kidney-specific isoform of
mouse vacuolar H+-ATPase subunit a. J Biol Chem 276:40050-40054.
Oka T, Toyomura T, Honjo K, Wada Y, Futai M (2001b) Four subunit a isoforms of
Caenorhabditis elegans vacuolar H+-ATPase. Cell-specific expression during
development. J Biol Chem 276:33079-33085.
Oluwatosin YE, Kane PM (1997) Mutations in the CYS4 gene provide evidence for
regulation of the yeast vacuolar H+-ATPase by oxidation and reduction in vivo.
Journal of Biological Chemistry 272:28149-28157.
107

Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS (2002) Short hairpin
RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes
& Development 16:948-958.
Paroutis P, Touret N, Grinstein S (2004) The pH of the secretory pathway: Measurement,
determinants, and regulation. Physiology 19:207-215.
Peri F, Nüsslein-Volhard C (2008) Live imaging of neuronal degradation by microglia
reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133:916-927.
Peters C, Bayer M, Bühler S, Andersen J, Mann M, Mayer A (2001) Trans-complex
formation by proteolipid channels in the terminal phase of membrane fusion.
Nature 409:581-588.
Peters J, Walsh M, Franke W (1990) An abundant and ubiquitous homo-oligomeric ring-
shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and
NSF. EMBO J 9:1757-1767.
Pietrement C, Sun-Wada GH, Da Silva N, McKee M, Marshansky V, Brown D, Futai M,
Breton S (2006) Distinct expression patterns of different subunit Isoforms of the
V-ATPase in the rat epididymis. Biology of Reproduction 74:185-194.
Poea-Guyon S, Amar M, Fossier P, Morel N (2006) Alternative splicing controls
neuronal expression of v-ATPase subunit a1 and sorting to nerve terminals.
Journal of Biological Chemistry 281:17164-17172.
Pujol N, Bonnerot C, Ewbank J, Kohara Y, Thierry-Mieg D (2001) The Caenorhabditis
elegans unc-32 gene encodes alternative forms of a vacuolar ATPase a subunit. J
Biol Chem 276:11913-11921.
Saroussi S, Nelson N (2009) Vacuolar H(+)-ATPase-an enzyme for all seasons. Pflugers
Arch 457:581-587.
Schoonderwoert VTG, Martens GJM (2001) Proton pumping in the secretory pathway.
Journal of Membrane Biology 182:159-169.
Schoonderwoert VTG, Martens GJM (2002) Targeted disruption of the mouse gene
encoding the V-ATPase accessory subunit Ac45. Molecular Membrane Biology
19:67-71.
Schuldiner S, Steiner-Mordoch S, Yelin R (1998) Molecular and biochemical studies of
rat vesicular monoamine transporter. Adv Pharmacol 42:223-227.
108

Smirnova AS, Morgun A, Shulzhenko N, Silva I, Gerbase-DeLima M (2005)
Identification of new alternative splice events in the TCIRG1 gene in different
human tissues. Biochemical and Biophysical Research Communications 330:943-
949.
Smith A, Skaug J, Choate K, Nayir A, Bakkaloglu A, Ozen S, Hulton S, Sanjad S, Al-
Sabban E, Lifton R, Scherer S, Karet F (2000) Mutations in ATP6N1B, encoding
a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal
tubular acidosis with preserved hearing. Nat Genet 26:71-75.
Stehberger PA, Schulz N, Finberg KE, Karet FE, Giebisch G, Lifton RP, Geibel JP,
Wagner CA (2003) Localization and regulation of the ATP6V0A4 (a4) vacuolar
H+-ATPase subunit defective in an inherited form of distal renal tubular acidosis.
Journal of the American Society of Nephrology 14:3027-3038.
Stover E, Borthwick K, Bavalia C, Eady N, Fritz D, Rungroj N, Giersch A, Morton C,
Axon P, Akil I, Al-Sabban E, Baguley D, Bianca S, Bakkaloglu A, Bircan Z,
Chauveau D, Clermont M, Guala A, Hulton S, Kroes H, Li Volti G, Mir S, Mocan
H, Nayir A, Ozen S, Rodriguez Soriano J, Sanjad S, Tasic V, Taylor C, Topaloglu
R, Smith A, Karet F (2002) Novel ATP6V1B1 and ATP6V0A4 mutations in
autosomal recessive distal renal tubular acidosis with new evidence for hearing
loss. J Med Genet 39:796-803.
Sudhof T (2004) The synaptic vesicle cycle. Annu Rev Neurosci 27:509-547.
Sun-Wada G, Toyomura T, Murata Y, Yamamoto A, Futai M, Wada Y (2006) The a3
isoform of V-ATPase regulates insulin secretion from pancreatic beta-cells. J Cell
Sci 119:4531-4540.
Sun-Wada GH, Murata Y, Yamamoto A, Kanazawa H, Wada Y, Futai M (2000) Acidic
endomembrane organelles are required for mouse postimplantation development.
Developmental Biology 228:315-325.
Supek F, Supekova L, Mandiyan S, Pan Y, Nelson H, Nelson N (1994) A novel
accessory subunit for vacuolar H(+)-ATPase from chromaffin granules. J Biol
Chem 269:24102-24106.
109

Söllner T, Whiteheart S, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P,
Rothman J (1993) SNAP receptors implicated in vesicle targeting and fusion.
Nature 362:318-324.
Taupenot L, Harper KL, O'Connor DT (2005) Role of H+-ATPase-mediated acidification
in sorting and release of the regulated secretory protein chromogranin A -
Evidence for a vesiculogenic function. Journal of Biological Chemistry 280:3885-
3897.
Toyomura T, Murata Y, Yamamoto A, Oka T, Sun-Wada GH, Wada Y, Futai M (2003)
From lysosomes to the plasma membrane - Localization of vacuolar type H+-
ATPase with the a3 isoform during osteoclast differentiation. Journal of
Biological Chemistry 278:22023-22030.
Toyomura T, Oka T, Yamaguchi C, Wada Y, Futai M (2000) Three subunit a isoforms of
mouse vacuolar H(+)-ATPase. Preferential expression of the a3 isoform during
osteoclast differentiation. J Biol Chem 275:8760-8765.
Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I (2003) Activation of
lysosomal function during dendritic cell maturation. Science 299:1400-1403.
Wada I, Rindress D, Cameron P, Ou W, Doherty Jn, Louvard D, Bell A, Dignard D,
Thomas D, Bergeron J (1991) SSR alpha and associated calnexin are major
calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem
266:19599-19610.
Wang L, Li G, Sugita S (2004) RalA-exocyst interaction mediates GTP-dependent
exocytosis. J Biol Chem 279:19875-19881.
Wang Y, Cipriano D, Forgac M (2007) Arrangement of subunits in the proteolipid ring of
the V-ATPase. J Biol Chem 282:34058-34065.
Wang Y, Sugita S, Sudhof T (2000) The RIM/NIM family of neuronal C2 domain
proteins. Interactions with Rab3 and a new class of Src homology 3 domain
proteins. J Biol Chem 275:20033-20044.
Wegrzyn JL, Bark SJ, Funkelstein L, Mosier C, Yap A, Kazemi-Esfarjani P, La Spada
AR, Sigurdson C, O'Connor DT, Hook V (2010) Proteomics of Dense Core
Secretory Vesicles Reveal Distinct Protein Categories for Secretion of
110

Neuroeffectors for Cell-Cell Communication. Journal of Proteome Research
9:5002-5024.
Williamson W, Wang D, Haberman A, Hiesinger P (2010) A dual function of V0-
ATPase a1 provides an endolysosomal degradation mechanism in Drosophila
melanogaster photoreceptors. J Cell Biol 189:885-899.
Xu J, Cheng T, Feng HT, Pavlos NJ, Zheng MH (2007) Structure and function of V-
ATPases in osteoclasts: potential therapeutic targets for the treatment of osteolysis.
Histology and Histopathology 22:443-454.
111

Appendix I: Examination of Voa1/Voa2 double knockdown cells using Electron Microscopy
Appendix Figure 1: Analysis of dense-core vesicle of control vs DKD using electron
microscopy. (A) An example of a typical electron micrograph from each cell line; control
(left); DKD. (right) (B) Summary of the number of DCVs/electron micrograph. (C)
Distribution of DCVs.
112

Appendix II: Sequence alignments of human Voa isoforms
Appendix Figure 2: Alignment of protein sequences of Voa isoforms highlighting
conserved residues.
113