the simultaneous speciation of aluminium and iron in a flow-injection system
TRANSCRIPT

ANALYTICA CHIMICA ACTA
ELSEWIER Analytica Chimica Acta 306 (1995) 5-20
The simultaneous speciation of aluminium and iron in a flow-injection system
Nicholas Clarke * , Lars-G&an Danielsson
Dil,ision of Analytical Chemistry, The Royal Institute of Technology, S-100 44 Stockholm, Sweden
Received 18 August 1994; revised 1 December 1994; accepted 1 December 1994
Abstract
It is possible to speciate Al and Fe simultaneously using complexation in a flow-injection system. Oxine is used as the
reagent for Al and Fe(IlI), and l,lO-orthophenanthroline in combination with iodide as the reagent for Fe(H). The reaction products are extracted into chloroform. A diode array detector is used for measurement and a multivariate method, partial
least squares, to separate the contributions to the signal from aluminium, iron(II1) and iron(H). Results show that our system is able to measure the three components correctly in the presence of each other. The sampling frequency is 85 injections/h and the detection limit 0.06 mg/l for all three components. Results obtained with natural water samples were satisfactory. The only cation that might possibly interfere in normal samples is Ca’+. The ‘free’ ions, Al”+, Fe”+ and Fe’+, as well as
some labile complexes, such as AlSO:, are included in the measured parameter. Aluminium-fluoro complexes and large
polymeric hydroxo complexes of Al and Fe(II1) are excluded. Complexes with humic or fulvic acids are normally excluded,
except in the case of Fe(III1, for which they appear to be included to a small extent. The reduction of iron(III1 to iron and the oxidation of iron(R) to iron(III), in both cases mediated by humic substances, could be shown.
Keyword.s: Kinetic methods; Flow injection; Aluminium; Iron; Speciation; Waters; Oxine: l,lO-Orthophenanthroline; Multivariate analysis:
Partial least squares
1. Introduction
The speciation of aluminium and iron is necessary
because the important geochemical and toxicological effects of these elements depend on the species in which they occur. An overview of the geochemistry of aluminium and iron and their biological effects in
natural waters is given by Clarke [l]. Methods for aluminium speciation have been reviewed recently
Corrcspondinp author.
by Clarke et al. [2], and a brief survey of methods for iron speciation is given by Clarke [l]. Some specia- tion methods are not suitable for use on natural water
samples, others require several measurements to be made, which makes them somewhat laborious. The labour involved can often be decreased by using a flow system, such as flow-injection analysis (FIA).
Methods for the simultaneous determination of sev- eral components using FIA have been reviewed by Kuban [3]. For example, FIA methods using diode array detection exist for the simultaneous determina- tion of the total concentrations of iron(W) and alu- minium [4] and of iron(H) and iron(H) [s].
0003.?h70/9S/$O9.50 0 19% Elscvicr Science B.V. All rights reserved
SSDI 0003-2670(94)00674-I

6 N. Clarke, L.-G. Danielwon /Analytica Chimica Acra 306 (1995) 5-20
As far as we know, there exists no method for the
simultaneous speciation of both aluminium and iron. Our intention was to develop such a method based
on the method of Clarke et al. [6,7] for the determi- nation of quickly reacting aluminium (Al,,) using
FIA.
2. Experimental
2.1. Chemicals
Oxine (8_hydroxyquinoline, 8-quinolinol) was ob-
tained from May and Baker, l,lO-orthophenanthro- line from Merck or Riedel-de Haen, sodium acetate
from Merck or Riedel-de Ha&n and NaI from
Mallinckrodt, Merck or J.T. Baker. Nordic Reference
Humic and Fulvic Acids (NRHA and NRFA, respec- tively, characterized by Pettersson et al. [S]), isolated
by the method of Thurman and Malcolm [9], were
provided by the National Swedish Environmental Protection Board. R%dsla Fulvic Acid (RFA, [8]) was provided by the University of Linkoping, Sweden,
where it was isolated by the method of Pettersson et
al. [lo], i.e., adsorption on an anion exchanger (DEAE) without prior pH adjustment, followed by
adsorption on an XAD-8 resin, cation exchange and
lyophilization. Analytical reagent grade FeCl, . 4H,O was obtained from Riedel-de Haen. Carbon
dioxide gas was from DFK, N, and H, gases were
from AGA (PLUS quality). All other chemicals were
from Merck and of analytical reagent grade. For aluminium, iron(II1) and other metal cations, we
have used Titrisol (Merck) or Fixanal (Riedel-de Haen) standard solutions. All chemicals were used
without further purification. All water used had passed through a Modulab Analytical System (Con- tinental Water Systems Co.) or a Milli-Q or Milli-Ro
system (Millipore). Standard solutions at pH 2.5-2.6 were used for
calibration, using hydrochloric acid to obtain the pH. At this pH, it is possible to have both iron(I1) and iron(II1) in solution at the same time, the solution being stable long enough for an experiment to be run. We used equilibrium calculations in order to determine the composition of the standard solutions. The calculations were carried out using a modified version (V 87.02) of HALTAFALL [l 11. All equilib-
rium constants used were, if necessary, corrected for
the right ionic strength using the Giintelberg and Davies approximations [ 121. The constants chosen
were valid for 25” C whenever possible, although in some cases we used constants that were valid for
20” C. The temperature in the laboratory was nor-
mally somewhere between 20 and 25” C, and we
assumed that these constants could be used without
correction for the temperature. We assumed the exis-
tence of the complexes given in the papers used, and that other complexes were not present in significant quantities. The equilibrium constants used for the
aluminium complexes and for the ligands forming complexes with aluminium were the same as those
used previously [7]. Constants for the iron complexes were obtained from Lindsay [13] (Fe hydroxo and
chloro complexes), Bruno et al. [14,15] (Fe carbon-
ato complexes), and a constant for HCl from HIgg [16]. These constants are not given here for reasons
of space; we advise the interested reader to consult
the original references. At a pH of 2.5-2.6, at metal concentrations of up to 2 mg l- ‘, and in the absence of ligands other than OH- and small quantities of
Cll, HCO; and CO:-, nearly all aluminium should
exist as A13+, iron(II1) as Fe3+, FeOH’+ or Fe(OH)i
and iron (II) as Fe’+. Complexes between chloride
ions and aluminium or iron (II) are very weak and can be ignored. FeCI’+ should exist in small
amounts. Carbonato complexes can be ignored.
The standard solutions contained varying quanti-
ties of all 3 components in concentrations from 0.06
to 2.0 mg/l. This was necessary, as a sample may contain all three components, between which there could be an interplay. This interplay might not be taken into account in the model if each standard
contained only one component. All solutions were stored in acid-soaked polyethy-
lene containers. Solutions containing iron(I1) were
always prepared immediately before an experiment, to avoid oxidation of iron(I1). At a pH of ca. 2.5-2.6, the oxidation of iron(B) is sufficiently slow SO that no significant change in the concentrations of iron(I1) and iron(II1) occurs during the few hours necessary to run an experiment. We considered using a redox buffer for the standard solutions, but felt that it would be an advantage to keep the solutions as simple as possible in order to minimize the risk for
unforeseen effects.

N. Clarke. L.-G. Danielsson/Analytica Chimicu Actu 306 (1995) 5-20 7
2.2. Equipment
We used an FIA 08 peristaltic pump (Bifok,
Sweden) with Tygon pump tubes from Technicon. The injector was a Model 7040 valve with a Model 5701 pneumatic actuator and a Model 7163 solenoid
valve, all from Rheodyne. The coils were PTFE tubes from Tecator (HiiganBs, Sweden), with inner
diameters of 0.35, 0.5 or 0.7 mm. For the separation
step, a membrane separator of the type described by
BiickstrSm et al. [ 171 was used. For detection, we used a 996 photodiode array detector (Waters-Milli-
pore), together with an NEC Power Mate 486/66i
computer with Millennium 2010 Chromatography Manager software (Waters-Millipore, version 1.10) and a LaserJet IIIP printer (Hewlett-Packard).
The program Unscrambler v. 3.52 (Camo A/S, Trondheim, Norway) was used for multivariate anal-
ysis of the results. Total aluminium and iron concentrations were
measured using inductively coupled plasma optical
emission spectrometry (ICP-OES), either an ARL 3520 B ICP Analyzer or a Jobin-Yvon 24.
For equilibrium dialysis experiments, Spectra/Par
6 membrane (Spectrum) was used.
2.3. Out&e of the method
The manifold (FIA set-up) is shown in Fig. 1,
together with the flow rates, coil dimensions and
residence times used. The relatively large sample
DB
Fig. 1. Schematic diagram of the flow injection setup. Sa = Sample
stream (flow rate, 1.7 ml/min), C = carrier stream (flow rate, 1.6
ml/min), R = reagent stream (flow rate, 1.2 ml/min), W = water
(flow rate, 0.6 ml/min), DB = displacement bottle containing
chloroform, I = injector (length of injector coil = 1.2 m), RC =
reaction coil (length 0.5 m, residcncc time 1.9-2.1 s), EC =
extraction coil (length 3.0 m, residence time 9.5-10.6 s), S = phase
separator, D = spectrophotometric detector. The tubing in all coils
has an inner diameter of 0.5 mm.
introduction rates (compared to Clarke et al., [7])
gave a larger signal and thus a better detection limit. We injected our samples (normally about 0.24 ml>
into a stream of water. The samples were buffered to pH 5.0 at the same time as the reagent was added.
Buffering is necessary as the reactions of both oxine
and l,lO-orthophenanthroline with metal ions are
pH-dependent. We chose pH 5.0 as many natural
fresh waters have a pH in this region. The use of this
pH will thus minimize the disturbance to the sample
caused by buffering. The reagent solution consists of 6.5 mM oxine, 7
mM l,lO-orthophenanthroline and 0.30 M sodium
iodide in 0.15 M sodium acetate at pH 5.0. Oxine is used as the reagent for Al and Fe(III). Lacroix [18] gave a figure of 3.56 mM for the solubility of
uncharged oxine in water at 18” C. At pH 5.0, un- charged oxine is slightly less than 50% of the total
oxine. This means that the total oxine concentration
used lies near the limit for the solubility of oxine in
water. However, warming the solution to about 60” C caused all oxine to dissolve fairly rapidly. After-
wards, the solution was stable for several weeks, no
precipitation of oxine occurring. Without this warm- ing, complete dissolution of the oxine was hard to
obtain, resulting in an unstable solution and high uncertainty limits for the samples.
For Fe(II), l,lO-orthophenanthroline is used as the reagent. Iodide is added in order to form an
extractable ion-association species with the Fe(II)-
orthophenanthroline complex [19]. A large excess of
both reagents is used. as this should give pseudo-first
order kinetics. Acetate is used as the pH buffer. The
complexes formed are then extracted into chloroform (trichloromethane), which is a powerful extractant
for oxine and metal oxinates (Table A.9 in Ref. [20];
Table 7.18 in Ref. [21]). As the pump tubes are vulnerable to attack by chloroform, a displacement bottle is used to provide the chloroform flow [7]. The displacement bottle has the additional advantage that the pulsations caused by the use of a peristaltic pump are reduced.
Separation of the aqueous and organic phases is necessary before measurement. It is important that none of the aqueous phase passes through the detec- tor; on the other hand, a small loss of organic phase can be permitted. The absorbance in the organic phase is measured at many wavelengths at regular

8 N. Clarke, L.-G. Danirlsson/Analytica Chimica Acta 306 (1995) 5-20
intervals from 350 to 650 nm, at a rate of five
complete spectra/s. 12 or 13 wavelengths at regular intervals from 376 or 353 nm respectively, up to 630
nm are used for evaluation of the results. The noise
level at 353 nm tended to be rather high, due pre-
sumably to absorption by oxine. This meant that a
better result could often be achieved by ignoring this
first wavelength. It might be possible to use fewer than 12 wavelengths, although this would probably
decrease the predictive power of the model.
The signals (absorbances) at each wavelength are integrated, so that the peak areas are obtained. As
each sample is injected four times, the mean peak area for each sample at each wavelength is then
calculated, the contributions to the signal from Al,
Fe(U) and Fe(II1) then being separated using multi- variate analysis. The multivariate method used is
partial least squares (PLS). In this method, a model
is obtained giving the best correlations between two matrices, one (the X matrix) containing the mean
peak areas after blank correction at different wave-
lengths for the standard solutions, the other (the Y matrix) containing the concentrations of the three
components in these solutions. The model obtained is then used to calculate (or predict) the concentra-
tions present in unknown samples, based on the
mean peak areas obtained at the different wave-
lengths (the prediction matrix). Although the matri-
ces are mean centred, they are not scaled, as this is unnecessary when all variables in a block are mea-
sured in the same units [22].
3. Results and discussion
In the method of Clarke et al. [6,7], iron is masked using hydroxylamine and 1, IO-ortho- phenanthroline, thus preventing its reaction with ox-
ine. By leaving out the masking reagent and measur-
ing at more than one wavelength, it should be possi- ble to measure both aluminium and iron [4,23]. However, using oxine alone, it is not possible to measure both iron(I1) and iron(II1). At a pH below 4, no extraction is observed in the case of iron(H), while at higher pH values, iron(I1) is oxidized to iron(II1) (p. 587 in Ref. [21]).
Attempts to use a single-phase system, with 8-hy- droxyquinoline-5-sulphonic acid as reagent for Al,
Fe(II1) and Fe(II), showed that it was possible to separate signals from the three components. How-
ever, the method proved to be impractical for use
with natural waters. The method is described in more detail by Clarke [l].
Acetate was used as the pH buffer. No problems due to the formation of acetato complexes with iron
or aluminium were observed. This is because the
reagents form much stronger complexes with the metals than acetate does.
The oxinates of Al and Fe(II1) are fairly insoluble in water, so it is necessary to extract them into an
organic solvent before measurement. Chloroform was used as the solvent. We found that an extraction time
of about 10 s gave good results. Extraction has the added advantage of enabling the sample to become
more concentrated by extracting from a relatively
large volume of water to a smaller volume of organic solvent, thus improving the detection limit. Also,
extraction minimizes interference due to absorption by humic substances. Humic substances may, how-
ever, irlterfere in another way, as their behaviour as
surfactants may influence the segmentation [24]. This will result in a larger standard deviation for samples containing large quantities of them.
In order to be able to determine both iron(U) and
iron(III), a second reagent was necessary. l,lO-Or- thophenanthroline is an often used reagent for the
determination of iron( the complex formed be-
tween the two being non-extractable. However, the iron(l,lO-orthophenanthroline complex forms
extractable ion-association species with some anions [25]. Among these is iodide [19]. A large excess of
iodide is necessary to obtain a high degree of extrac- tion; however, no improvement was found for iodide concentrations above 0.3 M.
Iodide forms fairly strong complexes with Fe”’ [26]. However, we believe that it is unlikely that these complexes will be formed in significant
amounts in the present system, as oxine forms much
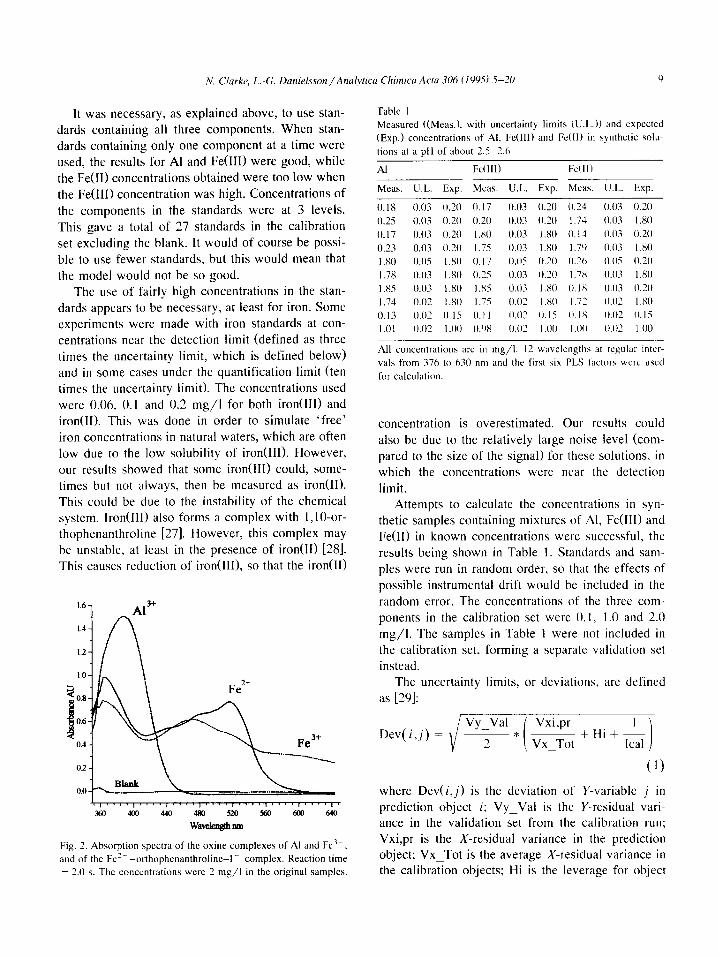
stronger complexes. The spectra obtained with the system are shown
in Fig. 2. The range tested for linearity extended from the detection limit up to 2.0 mg/l for all three components. No deviation from linearity could be seen in the model. The linear range probably extends upward beyond 2.0 mg/l, although this was not tested.
N. Clarke, L.-G. Danielsson/Anal~tica Ckimicu Acta 306 (199.5) S-20 9
It was necessary, as explained above, to use stan- dards containing all three components. When stan- dards containing only one component at a time were
used, the results for Al and Fe(III) were good, while
the Fe(I1) concentrations obtained were too low when the Fe(Il1) concentration was high. Concentrations of
the components in the standards were at 3 levels.
This gave a total of 27 standards in the calibration
set excluding the blank. It would of course be possi-
ble to use fewer standards, but this would mean that
the model would not be so good. The use of fairly high concentrations in the stan-
dards appears to be necessary, at least for iron. Some experiments were made with iron standards at con-
centrations near the detection limit (defined as three
times the uncertainty limit, which is defined below) and in some cases under the quantification limit (ten
times the uncertainty limit). The concentrations used
were 0.06, 0.1 and 0.2 mg/l for both iron(II1) and iron(I1). This was done in order to simulate ‘free’
iron concentrations in natural waters, which are often
low due to the low solubility of iron(II1). However,
our results showed that some iron(II1) could, some- times but not always, then be measured as iron(I1). This could be due to the instability of the chemical system. Iron(II1) also forms a complex with l,lO-or-
thophenanthroline [27]. However, this complex may
be unstable, at least in the presence of iron(I1) [28]. This causes reduction of iron(III), so that the iron(I1)
1.6
0.6 -
0.4 - Fe3+
0.2 -
Fig. 2. Absorption spectra of the wine complexes of Al and Fe3+,
and of the Fc’+ -orthophenanthroline-I- complex. Reaction time
= 2.0 s. The concentrations were 2 mg/l in the original samples.
Table 1 Measured ((Meas.), with uncertainty limits (U.L.)) and expected
(Exp.) concentrations of Al, Fc(II1) and Fe(lI) in synthetic solu-
tions at a pH of about 2.5-2.6
Al Fe(III) Fc(II)
Meas. U.L. Exp. Mcas. U.L. Exp. Meas. U.L. Exp.
0.18 0.03 0.20 0.17 0.03 0.20 0.24 0.03 0.20
0.25 0.03 0.20 0.20 0.03 0.20 I .74 0.03 1.80
0.17 0.03 0.20 I.XO 0.03 1.80 0.11 0.03 0.20
0.23 0.03 0.20 1.75 0.03 I .80 1.7’) 0.03 I .80
1 .x0 0.05 I.XO 0.17 0.05 0.20 0.2h 0.05 0.20
1.7x 0.03 I .x0 0.25 0.03 0.20 1.78 0.03 I .x0
I Xi 0.03 I .x0 I .x5 0.03 I .x0 0.18 0.03 0.20
I .74 0.02 I .x0 1.75 0.02 I .x0 I .72 0.02 I .x0 0.13 0.02 0.15 0. I I 0.03 0.15 0.1s 0.02 0. I5
1.01 0.02 I .OO 0.0X 0.02 I .oo I .oo 0.02 I .oo
All concentrations arc in mg/l. 12 wavclcngths at regular inter-
vals from 376 to 630 nm and the first six PLS tactors wcrc used
for calculation.
concentration is overestimated. Our results could
also be due to the relatively large noise level (com- pared to the size of the signal) for these solutions, in
which the concentrations were near the detection
limit. Attempts to calculate the concentrations in syn-
thetic samples containing mixtures of Al, Fe(III) and
Fe(U) in known concentrations were successful, the results being shown in Table 1. Standards and sam-
ples were run in random order, so that the effects of
possible instrumental drift would be included in the
random error. The concentrations of the three com-
ponents in the calibration set were 0.1, 1.0 and 2.0
mg/l. The samples in Table 1 were not included in the calibration set, forming a separate validation set
instead. The uncertainty limits, or deviations, are defined
as [29]:
Vy_Val Vxi,pr -*-
2 i -
I
Vx Tot +Hi+G
1
(1)
where Dev(i, j) is the deviation of Y-variable j in
prediction object i; Vy_Val is the Y-residual vari- ance in the validation set from the calibration run; Vxi,pr is the X-residual variance in the prediction object; Vx Tot is the average X-residual variance in the calibration objects; Hi is the leverage for object

10 N. Clarke, L.-G. Danielsson /Analytica Chimica Acta 306 (1995) 5-20
i, telling how much influence the object has on the lead to increased uncertainty limits for samples with model; and Ical is the number of calibration objects. large concentrations of organic matter.
This deviation is not a standard formula, but an
empirically found relationship. In Table 1, the uncer-
tainty limits were between 0.02 and 0.05 mg/l for
all samples. All three components in each sample had similar uncertainty limits.
We found that the system could handle samples at a pH as low as 2.5 adequately, without there being
problems with the buffering.
3.1. Sample throughput, repeatability and the detec-
tion limit
The sample throughput was 85 injections/h, us- ing a sample containing 2 mg/l of all three compo-
nents and with a reaction time of 2.1 s and an
extraction time of 10.6 s. The detection limits for Al,
Fe(II1) and Fe(I1) were calculated as three times the uncertainty limits for the calculated concentrations of
the three components in a sample. The sample cho- sen contained 0.15 mg/l of all three components at pH 2.6. We obtained detection limits of 0.06 mg/l
for all three components. This means that the sample
chosen had in fact concentrations around the quan- tification limit.
31 consecutive injections of this sample with the same reaction and extraction times gave a relative
standard deviation of the absorbance values of be-
tween 5 and 12%, depending on wavelength. These relative standard deviations appear at first sight to be
rather high, but in fact the concentrations involved are very low, the concentrations of the three compo-
nents being around the quantification limit. The rela- tive standard deviations are due to the simultaneous variation of all three components. The highest rela- tive standard deviations were obtained at low wave-
lengths, where all three components cause absorp- tion, and where oxine too can absorb. The lowest
relative standard deviations were obtained at 560- 585 nm, where absorption is due mostly to Fe(III).
Many metal cations form extractable complexes
with oxine. Some form complexes with l,lO-ortho-
phenanthroline, although in many cases these are not extractable. We investigated the following ions and
concentrations: Na+ (2.0 mM), Ca*+ (2.0 mM),
Mg*+ (0.8 mM), K+ (0.2 mM), Sr*+ (0.03 mM) and Cr3+, MO”, Mn”, Co*+, Ni2+, CU*+, Zn*+,
Cd”+, Hg2’, Sn4+ and Pb2+ (all 0.02 mM). None of
these ions are likely to interfere with the determina- tion of the iron fractions either as iron(III) or iron(
as the absorbances of the complexes formed were insignificant at wavelengths above 500 nm. Na+,
Mg”+, K+ and Sr*+ do not interfere in the determi-
nation of the aluminium fraction either. All the other
ions interfere or may interfere with the determination
of Al. However, the maximum absorbance of the complexes formed is at around 360 nm instead of 390 nm, as is the case for aluminium trioxinate. The
lowest calculated concentrations of the various cations that would give a signal at 363 nm equivalent
to that of 2.2 PM (0.06 mg/l) Al (also at this wavelength) are shown in Table 2. This aluminium
concentration is the detection limit; approximately three times greater concentrations of interfering ions
would be needed for equivalence with the quantifica-
tion limit. With the exception of Ca, the concentra-
tions needed are far higher than those found nor-
Table 2
Lowest calculated concentrations of various cations that would
give a signal at 363 nm equivalent to that of 2.2 pM (0.06 mg/l)
Al
Cation Concentration equivalent to 2.2 pM
(0.06 mg/l) Al
3.2. Interferences
Humus should not interfere by absorbing at the wavelengths measured, as it is generally not ex- tractable into chloroform [7]. However, it may inter- fere by affecting the segmentation [24]. This might
Ca*+
cr’+
Mob+
Mn”
co*+ Ni?’
cu*+
ZI?+
Cd’+
Hg*+
Sn4+ Pb2+
( /.LM) (mg/l)
696 27.9
7.2 0.37
15.0 1.44
7.2 0.40
1.2 0.071
1.8 0.11
1.8 0.11
1.2 0.078
8.4 0.94
8.4 1.7
9.6 1.1
3.6 0.75

N. Clarke, L-G. Danielsson/Analytica Chimica Acta 306 (199-i) S-20 II
mally in natural fresh waters [30-331. Thus, only Ca, if present in large quantities, is likely to interfere. For calcium, Garrels et al. (Table 5 in Ref. [30]) give
a figure of 15 mg/l in streams, which is below the concentration necessary for interference. In areas in
which acid precipitation is a problem, the concentra- tion of dissolved calcium is often lower than this.
For example, Johnson et al. [34] give figures up to
1.70 mg/l for Falls Brook in New Hampshire. In
areas with calcium-rich rocks (such as limestones),
concentrations may of course be much higher. How- ever, waters in limestone areas have a relatively high pH, and are normally well buffered against acid precipitation, so aluminium speciation is less likely
to be of interest in them. If interference should occur, it might be possible
to correct for it by introducing Ca as a fourth compo-
nent besides Al, Fe(III1 and Fe(B), although this might well cause problems in the determination of
Al. The problem can, however, be avoided easily, simply by not using wavelengths under about 390
nm in the evaluation, although this would mean that fewer wavelengths would be available for the deter- mination of aluminium.
3.3. Characterisation
With a reaction time as short as 1.9-2.1 s, only ‘free’ metal ions (Al’+, Fe”+ and Fe” 1 and certain
labile complexes react. Non-labile complexes (which
may or may not be the same as strong complexes) and colloidal forms of the metals do not have time to be dissociated before extraction begins. That which
is measured is of course an operationally defined parameter. It is thus extremely important that it is known which complexes are included in the mea-
sured fraction. By comparison with the work of Clarke et al. [7], we call our measured fractions Quickly Reacting Aluminium, Iron(III) and Iron(B).
Our Quickly Reacting Aluminium is very similar to, although not identical with, that of Clarke et al.
In order to study which Al and Fe complexes are
included in Quickly Reacting Al, Fe(III) and Fe(B), solutions containing known concentrations of various ligands were run. We compared the results obtained with equilibrium calculations, which were carried out in the same way as those used to determine the composition of the standard solutions (see the Exper-
imental section). Apart from those mentioned above, and those used previously [7], equilibrium constants were obtained from Lindsay [13] (Al-phosphato
complexes), Olson and O’Melia [35] (Fe-silicato complexes), Kotrly and Sucha [26] (Fe-glycinato
complexes), Athavale et al. [36] (Al-malonato com-
plexes) and Cavasino and Di Dio [37] (Fe(IIIl-
malonato complexes and the malonate system). For
the silicate system, constants were obtained from
Sjiiberg et al. [38-411, Bilinski et al. [42] and ehman et al. [43]. In the case of aluminium’s silicato com-
plexes, constants are given both by Browne and Driscoll [44] and by Sjiiberg (personal communica-
tion) for the formation of AlH,SiO,“+. However. these constants differ by about an order of magnitude
after correction for the ionic strength. Browne and
Driscoll give constants for two other complexes as
well. Comparisons were thus made with equilibrium calculations using constants from both sources. There
is also considerable uncertainty concerning the alu- minium-phosphato complexes, the aluminium-
phosphate system being extremely complex (Ghman,
personal communications). In the case of aluminium, we have normally ignored the carbonate system in our calculations. Aluminium’s carbonato complexes
are very weak, and may be neglected in slightly acid
solutions except when the partial pressure of carbon dioxide is high [45]. As acetate and glycine were
used to buffer the pH of the samples, the complexes
formed by these ligands with aluminium or iron
were, where relevant, investigated. For complexes with NRFA, NRHA and RFA, no
constants are known that are valid under all condi- tions, although a conditional constant has been ob-
tained [46]. For Al, we compared our results both with equilibrium calculations using the constants ob-
tained by Lovgren et al. [47] for complexes with organic matter in a concentrated bog water, and with the results obtained by Clarke et al. [6,7,46] with their system for Al speciation. We considered it
valuable to study the behaviour of metal complexes with several different sorts of humus. As humus is rather variable in its composition and characteristics,
humus from different environments, isolated in dif- ferent ways, could behave differently.
It is of course possible that buffering the sample can, by changing the sample’s equilibrium, change the speciation. Howe./er, Clarke et al. [7] found that

12 N. Clarke, L.-G. Danielsson /Analytica Chimica Acta 306 (1995) 5-20
this was not a problem for samples at a pH > 3.5, even in the presence of such a strong ligand as
citrate. Our system is likely to be less troubled by
such problems, as the sample is buffered at the same
time as the reagent is added, rather than before, as
was the case with the system of Clarke et al. [7].
For the characterisation experiments, we consid-
ered it unnecessary to use as many wavelengths as for normal runs, as the number of components to be determined was lower. For solutions containing alu- minium only, a single wavelength at 388 nm was
sufficient. In the case of iron, which could occur
either as iron(III) or iron( 11 wavelengths were
used, at regular intervals from 391 to 627 nm.
Fairly low concentrations were used, in order that
the concentrations of Quickly Reacting metals might be near to those actually found in natural waters.
This is important, as the complexes present can depend on the concentrations. A drawback with the
use of low concentrations was that the spread in the results could be quite large.
The aluminium characterisation experiments were carried out using samples at a pH between 3.3 and
5.5, with the exception of some samples at pH
7.9-10.0 used to study the reaction of AI(O The
pH was buffered using acetate/acetic acid at a con- centration of either 3 mM (for solutions containing
humic substances) or 10 mM for other solutions. No
pH buffer was used for the samples at pH 7.9-10.0. Except in the case of samples containing humic
substances, an ionic strength of 0.1 M, obtained using NaNO,, was used. At this ionic strength, the small variations in ionic strength caused by different
concentrations of the cations and of the ligands should be relatively insignificant, making compari-
son with equilibrium calculations straightforward. The ionic strength of solutions containing humic
substances was not adjusted, so that it should be as near as possible to that of natural fresh waters. Of
course, the presence of the 3 mM pH buffer meant that the ionic strength of these solutions was still higher than that of most natural fresh waters. For the study of aluminium’s carbonato complexes, a high partial pressure of carbon dioxide was needed, which we obtained by passing carbon dioxide gas through the samples. This should give a partial pressure of carbon dioxide equal to 1 atm. The aluminium con- centration used was 37.1 PM (1.0 mg/l). All solu-
Table 3
Degree of inclusion of aluminium bound to various ligands in the
measured parameter, compared to the degree of inclusion of A13+,
with a reaction time of 1.9-2.1 s
AlOH’+
AI(
Large polymeric hydroxo complexes
Fluoro complexes
Also:
AIHPO:
Silicato complexes
A12(OH)2CO:+
A12(OH)200CCH:+
Al(OOC),CH;, AI((OOC)$H,); Complexes with NRFA
Complexes with NRHA
Complexes with RFA
Complexes with natural humus
Complete
75-100%
Nearly none
Nearly none
Complete
O-40%
O-80%
Complete
About 85%
Nearly none
Nearly none
Nearly none
Nearly none
Nearly none/partial
Mixed complexes in which hydroxide is included arc considered
together with the other ligand, e.g. mixed fluoro-hydroxo com-
plexes together with fluoro complexes,
tions were stored in acid-soaked polyethylene con-
tainers. The samples were allowed to stand at least overnight in order to come reasonably close to equi-
librium, so that the species present could be esti- mated using equilibrium calculations.
To correct for loss of metal cations by, for exam-
ple, adsorption on the container walls, total metal concentrations were measured using ICP-OES.
The results are shown in Table 3. The measured
fraction included A13+, AlOH*+, AlSO: and
Al,(OH),CO, . 2-t Al(OH), and the acetato complex
A12(OH)200CCH;+ were included to a large ex- tent. AlHPOl was included either to an insignificant
or to a small extent. Aluminium’s complexes with malonate, a moderate strength organic ligand, were also studied. The complexes formed, A1(00C)2CHl
and A1((OOC),CHZ);, were not included in the measured fraction. Large polymeric hydroxo com-
plexes (such as Al,,(OH),,O~+), the fluoro com- plexes and complexes with NRFA, NRHA or RFA hardly began to react with oxine in our system. The results are in good agreement with those of Clarke et al. [6,7,46,48], although with the present method, AlHPOl reacts to a smaller, and Al(OH), to a larger, extent.
The extent of the reaction of the silicato com- plexes was unclear. If we used Sjoberg’s constant, the results obtained indicated that the (one) silicato

N. Clarke. L-G. Daniel.won /Analytica Chimica Acta 306 (I 993) S-20 I.3
complex hardly began to react. On the other hand, if Browne’s and Driscoll’s constants were used, the results showed that three silicato complexes reacted
jointly to an extent of between 40 and 80%. Clarke
et al. [48] found that, regardless of which constants were used, the silicato complexes were included
completely in the fraction measured in their system.
The reason for the difference is unclear, but it may be due to the presence of different silicato complexes
in the samples. The aluminium-silicate system is complex and not yet well understood, and the forma-
tion of different complexes under slightly different conditions is not at all unlikely.
The extent of the reaction of some aluminium species could not be determined, as they were never
present in sufficiently high concentrations. Of these,
it is likely that AI( and AI(OH), are included
in the measured parameter. Al,(OH)z ‘,
Al &OH), HCO: + , Al(SO,); and AlH,POi’ may
also be included. at least partially. The results of Clarke et al. suggest that weak organic complexes
could be wholly or partially included as well. Characterisation of Fe(II1) was studied in samples
with a pH between 2.5 and 4. The iron concentration
used was 3.6 PM (0.20 mg/l). Initially, glycine was
used as a pH buffer. However, the glycinato complex FeOOCCH,NH:’ is rather strong [26], so that at a
glycine concentration of 10 mM, nearly all iron was
present as this complex in the pH range used (3.2- 3.7). Accordingly, we chose not to use a pH buffer,
but to obtain the right pH using hydrochloric acid instead. As was the case for Al, the ionic strength
was adjusted to 0.1 M with NaNO, for samples not containing humus. The ionic strength of the samples
containing humus was not adjusted in any way. The solutions were stored in acid-soaked polyethylene containers and allowed to stand at least overnight.
Our results are shown in Table 4. The complexes FeOH’+, FeF”, FeHPO,f, FeSO:, the glycinato
complex FeOOCCH2 NH z + , the acetato complexes Fe(OOCCH,)f and Fe(OOCCH,),, and the mal-
onato complex Fe(OOC),CH; were included in the measured parameter. Other complexes such as, for example, Fe(OH)i , were presumably included but were not present in sufficient quantities to be mea- surable. No results could be obtained for the silicato or carbonato complexes, as equilibrium calculations showed them to be present in too low concentrations
Table 4
Degree of inclusion of iron bound to various ligands in the
measured paramctcr. compared to the degree of inclusion of Fe”+,
with a reaction time of 1.0-2.0 s
FeOH’+ Complete
Large polymeric hydroxo complexes/ colloids Nearly none
FcF” Complete
FcSO; Complctc
FeHPO: Complete
FcOOCCH NH’ + 7
Fc(OOCC;, ,?’ .-Fc(OOCCH ?jl
(‘omplete
Cornpick
Fe(OOC),CH;
Complcxca with NRFA
Complcxcs with NRHA
Complexes with RFA
Complexes with natural humus
Complete
Partial
Partial
Partial
Partial
to be measurable under the conditions of our experi-
ments. As the complexes appear to be fairly weak, they may well be included in the measured fraction.
The large polymeric hydroxo complexes and colloids were, on the other hand, not included.
Fe(III) was partially (after standing overnight) reduced to Fe(II) in the presence of NRFA, NRHA
and RFA. This is in agreement with the findings of. for example, Langford and Gutzman [49] and
Ephraim et al. [%)I. However, no reduction in the
combined measured concentrations of Fe(II) and Fe(III) was found. This could mean either that iron’s
humic complexes are included in the measured frac-
tion, or that the pH was too low (3.0-3.4) for measurable quantities of these complexes to be
formed.
Characterisation of the method with respect to Fe(II) complexes presented some problems, due to the necessity of holding the iron in the reduced state
at a suitable pH. Water was held at a suitable p’ value by first passing N, gas through it, followed by
placing platinum foil in the water and passing H1 gas over it. The samples were prepared just before
the experiment. This means, of course, that they had little time to reach equilibrium. However, equilib-
rium calculations showed a preponderance of simple monomeric species, which should be formed quickly.
Therefore, we assumed that we could still use com- parisons with equilibrium calculations. In some cases, the ligands were added from stock solutions made with water that was in equilibrium with oxygen in the air. However. the volumes used of this water

14 N. Clarke, L.-G. Danielson /Analytica Chimica Acta 306 (1995) 5-20
Table 5
Degree of inclusion of iron bound to various ligands in the
measured parameter, compared to the degree of inclusion of Fe*‘,
with a reaction time of 1.9-2.0 s
FeOH+
FeH,PO:
FeCO, (aqj FeOOCCHl
Complexes with NRFA
Complexes with NRHA
Complexes with RFA
Complete
40-70%
Nearly none
Complete
Nearly none
Nearly none
Nearly none
were relatively small, and appeared not to affect the
results. The pH of the samples was between 3.8 and 4.2 for samples containing NRFA, NRHA or RFA.
For these samples, neither the pH nor the ionic strength were adjusted in any way. All other samples were buffered to pH 4.3-5.5 using acetate or to pH
5.1-6.8 using carbonate. Speciation at higher pH values was not studied due to problems with oxida-
tion of the iron. An iron(B) concentration of 9.0-10.6
PM (0.50-0.59 mg/l) was used. The ionic strength
was adjusted to 0.1 M with NaNO, for samples not
containing humus, and the solutions were stored in
acid-soaked polyethylene containers. Fe(I1) does not appear to form strong halides [13], so these com-
plexes were not studied. Iron(I1) sulphides were not investigated either, due to their very low solubility.
The Fe(I1) species that are included in the mea- sured fraction are shown in Table 5. They include
Fe’+, FeOH+ and the acetato complex FeOOCCH_T. The phosphato complex FeH,PO,f is included to an
extent of 40-70%. FeCO,(,,, is excluded. The pres- ence of sulphate caused no reduction in the signal. This could mean that FeSO,r,,, is included in the
measured parameter. However, the possibility of the reduction of sulphate to elemental sulphur, in which case the iron would remain as Fe”+, cannot be ruled
out. There was no evidence of the loss of iron(I1) due to the formation of sulphides.
The presence of NRFA, NRHA and RFA caused a reduction in the signal for Fe(II), probably due to the formation of complexes with this form of iron. However, there was also evidence of oxidation of
Fe(I1) to Fe(III), as Fe(II1) was present in a few samples containing humus despite iron having been added as Fe(I1). Oxidation of Fe(I1) to Fe(II1) in the
presence of natural organic matter has been observed
before. Liang et al. 1511 found that at partial pres-
sures of oxygen of about 0.005 atm, natural organic matter could enhance the rate of oxidation of Fe(I1).
The function of fulvic acids as redox mediators has
been discussed by, for example, Langford et al. [52]. That Fe(II1) complexes with humic matter are
included partially in the measured fraction, while
Fe(I1) complexes with humus are excluded may at first sight seem strange, as Fe(II1) forms stronger
complexes with humus than Fe(I1). The results can
be explained as being due to the rather lower pH
used for the experiments with Fe(II1). At these pH
values, competition between iron(II1) and protons for the binding sites on the humus may make it easier
for the oxine to form complexes with the iron. At a higher pH, nearer to those found in most natural fresh waters, the results might have been different. Also, oxine is a stronger complexing agent for
iron(II1) than 1, lo-orthophenanthroline is for iron(I1). Equilibrium calculations can be used to obtain the
concentration of any desired species or group of
species from the parameter measured by the method,
as long as the concentrations of the relevant ligands and their equilibrium constants are known.
Total dissolved concentrations of Al, Fe(II1) or Fe(I1) could be measured with a suitable pretreat-
ment of the samples.
3.4. Studies of iron-humus interactions using equi-
librium dialysis
In order to further study the interactions of iron and humus in our system, we performed experiments using equilibrium dialysis. The dialysis method was
based on those of LaZerte [53], Berggren 1541 and Clarke et al. [6,46]. The volume of the outer solution
was about 10 1, the experiment being run in a polypropylene bucket.
In the first experiment, the outer solution had an iron(II1) concentration of about 0.1 mg/l (1.8 ,uM) and a pH of 3.0-3.1. This iron concentration is under the quantification limit, but such a low concen- tration was necessary in order to avoid precipitation. Water that had passed through a Milli-Ro system was used. The inner solutions were 15 ml. Six inner solutions were equilibrated with the outer solution simultaneously. One was a control solution, contain-

N. Clarke, L.-G. Danielsson / Analytica Chimica Acta 306 (199.5) 5-20 15
ing only water that had passed through a Modulab system. Others contained 23 mg/l of NRFA, 16
mg/l of NRHA or 21 mg/l of RFA. In addition,
two samples of natural humus-rich water were used. These samples, called GII and Damm A, were from
surface waters in Masbybacken and Svartbticken,
both in Sweden, respectively. The inner solutions were acidified to about the same pH as the outer solution, in order to prevent the precipitation of iron
on the membrane. A cellulose ester membrane, Spec- tra/Por 6, with a nominal molecular weight cut-off
of 1000 was used. The equilibration time was about
142 h. During equilibration, the solutions were
shaken at 60 rpm using a Lab Shaker (Kihner, Basel). The temperature during the experiment was
20.2-20.4” C. After the experiment, the concentra- tions in the inner and outer solutions were measured
both with the FIA system and with ICP-OES. Ten iron standards were used, containing both iron(II1)
and iron in concentrations of 0.06, 0.1 and 0.2 mg/l, together with a blank. Standard solutions in- cluding aluminium at levels of 0.1, 0.5 and 1.0
mg/l, together with iron(II1) and iron( both at 0.1 mg/l, were also used, so as to be able to correct for the presence of aluminium if necessary. This neces-
sity did not arise. The first five PLS factors were
used for calibration.
water samples GII and Damm A (the inner solutions) were 1.94 and 0.84 mg/l, respectively after dialysis.
This iron was probably present throughout the dialy-
sis, as these samples contained total Fe concentra-
tions of 2.48 and 1.40 mg/l, respectively, after acidification but before dialysis. The concentrations measured with the FlA system were 0.42 and 0.2
mg/l Fe(II1) respectively. 0.42 mg/l was outside
the calibrated range in this experiment, but the figure is unlikely to be far wrong as it lies within the normal range for linearity. Figures for iron(I1) were
under the detection limit, although it is possible that
at least some of the iron that was excluded from the
measured fraction may have been present as iron
complexes with humus. It appears, however, that iron(II1) complexes with natural humus may be in-
cluded, at least partially, in the measured fraction. It is interesting to note that iron(II1) was not reduced in
the presence of humus in the natural water samples,
unlike in the case of the isolated humic substances.
We found that the iron concentrations in the outer solution and the control solution were about the same, measured both with the FIA system and with
ICP-OES. For the samples containing NRFA, NRHA and RFA, the total iron concentrations were 0.14, 0.15 and 0.15 mg/l, respectively. Iron concentra-
tions measured with the FIA system were 0.10, 0.08 and 0.11 mg/l, respectively, the iron being present as iron in all three cases. Although these concen-
trations lie under the quantification limit, it is proba- bly safe to say that the concentrations determined using ICP-OES were greater than those determined
using the present method. It seems, therefore, that, at suitable concentrations and at equilibrium, these ref- erence humic substances mediate the complete re-
duction of iron(II1) to iron(I1). The complexes formed by iron and NRFA, NRHA or RFA may be iron complexes or iron(II1) complexes or a mixture of both. In any case, they appear not to be included in the measured fraction.
In the second experiment, reducing conditions were used. The dialysis method used was much the same as that described above. The outer solution,
about 10 1, contained about 0.1 mg/l Fe. A pH of 4.2 was used, the pH being buffered using 3 mM
acetate-acetic acid. Reducing conditions were ob-
tained using the same method as in the characterisa-
tion experiments described earlier. The inner solu- tions were the same as in the first experiment, except
that they were not acidified. Equilibration time was about 114 h, with shaking at 50 rpm. Ten iron
standards were used, containing both iron(II1) and iron in concentrations of 0.1, 0.5 and 1.0 mg/l, together with a blank. Standard solutions including aluminium at levels of 0.1, 0.5 and 1.0 mg/l, to-
gether with iron(II1) and iron(B), both at 0.5 mg/l,
were also used, so as to be able to correct for the presence of aluminium if necessary. In this experi- ment, this turned out to be necessary, as sample GII
contained 0.08 mg/l (3 FM) Quickly Reacting Al after dialysis. This aluminium was presumably or- ganic bound, so its presence means that aluminium’s humic complexes may occasionally be included in Quickly Reacting Al. Another possibility, that alu- minium is released during the dissolution and reduc- tion of Fe(OH),(,,, is less likely, as Fe(III) was not reduced in sample GII (see below).
Total iron concentrations in the natural humus-rich The first six PLS factors were used for calibra-
16 N. Clarke, L.-G. Danielsson /Analytica Chimica Acta 306 (1995) 5-20
tion. As before, the concentrations in the outer solu-
tion and in the control solution were about the same, measured both as Quickly Reacting Fe(R) with the
FIA system and as total Fe with ICP-OES. For the samples containing NRFA, NRHA and RFA, iron
concentrations measured with ICP-OES were 0.30, 0.34 and 0.29 mg/l, respectively. Quickly Reacting
Fe(II1) concentrations were 0.07 mg/l in all three
samples, while Quickly Reacting Fe(I1) concentra-
tions were 0.10, 0.08 and 0.13 mg/l, in the NRFA, NRHA and RFA samples, respectively. The concen-
trations of Quickly Reacting Fe(R) are what would
be expected for uncomplexed Fe(U), supporting our belief that the humic complexes of Fe(R) are not
included in the measured parameter. The appearance
of Quickly Reacting Fe(II1) supports the idea that NRFA, NRHA and RFA can mediate both oxidation and reduction of iron, depending on the conditions.
The iron(III) is probably present as humic com- plexes, providing further evidence that these com- plexes are included partially in Quickly Reacting
Fe(II1). Samples GII and Damm A contained 3.45 and
1.65 mg/l total Fe respectively, after dialysis. No
Quickly Reacting Fe(R) was found. The concentra- tions of Quickly Reacting Fe(III) were 0.69 and 0.48 mg/l in GII and Damm A, respectively. These
results agree with those from the first dialysis experi- ment, the higher concentrations found here being due to the reduced competition for binding sites from
hydrogen ions. It appears certain that some Fe(II1) humic complexes can be included in Quickly React-
ing Fe(III). The results in this case do not prove the
oxidation of Fe(U), as these samples contained 2.72
and 1.44 mg/l total Fe respectively, before dialysis.
3.5. Measurements made on natural waters
Natural water samples taken from the Skjervatjern area, Norway, were analysed for Quickly Reacting
Al, Fe(II1) and Fe(R) using the FIA system, and for
total Al and Fe using ICP-OES (determined by Ann-Catrine Norrstriim, of the Division of Land and
Water Resources, the Royal Institute of Technology).
These samples were taken from different environ- ments, including lake, peat and ground water. The
samples and standards were run alternately in ran-
dom order. The concentrations of the three compo- nents in the standard solutions were 0.1, 0.5 and 1.0
mg/l. 28 standards, including a blank, were used. Water that had passed through a Modulab system was run as a blank for the natural water samples; the same water, acidified to the same pH as the stan- dards, was used as a blank for these. In order to
decide how many wavelengths and PLS factors to
use in the calculations, five of the standards used
were also run as samples. It was assumed that the
model that gave the best results for these, taking both accuracy and precision into account, was the best model to use for the natural water samples as well.
12 wavelengths at regular intervals from 376 to 630 nm and the first five PLS factors were used for calculation of the Quickly Reacting concentrations.
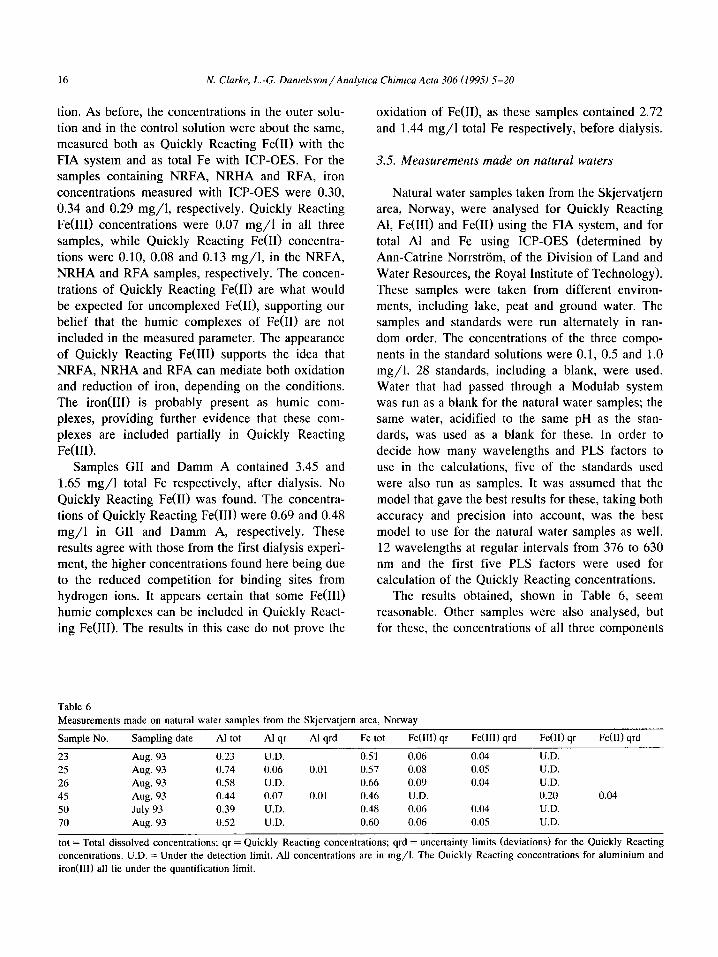
The results obtained, shown in Table 6, seem
reasonable. Other samples were also analysed, but for these, the concentrations of all three components
Table 6
Measurements made on natural water samples from the Skjervatjern area, Norway
Sample No. Sampling date Al tot Al qr Al qrd Fe tot Fe(llI) qr Fe(II1) qrd Fe(H) qr Fe(R) qrd
23 Aug. 93 0.23 U.D. 0.51 0.06 0.04 U.D.
25 Aug. 93 0.74 0.06 0.01 0.57 0.08 0.05 U.D.
26 Aug. 93 0.58 U.D. 0.66 0.09 0.04 U.D.
45 Aug. 93 0.44 0.07 0.01 0.46 U.D. 0.20 0.04
50 July 93 0.39 U.D. 0.48 0.06 0.04 U.D.
70 Aug. 93 0.52 U.D. 0.60 0.06 0.05 U.D.
tot = Total dissolved concentrations; qr = Quickly Reacting concentrations; qrd = uncertainty limits (deviations) for the Quickly Reacting
concentrations. U.D. = Under the detection limit. All concentrations are in mg/l. The Quickly Reacting concentrations for aluminium and
iron(II1) all lie under the quantification limit.

N. Clarke, L.-G. Danielsson/Analytica Chimica Acta 306 (1995) 5-20 17
were under the detection limit. The low values found
may be due to the presence of aluminium and iron in polymeric or colloidal forms.
3.6. Comparison with the method of Clarke et al. 171
A further test made on the method was a compari- son of Quickly Reacting aluminium as determined using the new method with a determination using the
method of Clarke et al. [7]. Surface water samples
from Masbybacken and Svartbacken in Sweden, and from the Lysina catchment in the Czech Republic,
were used. Total dissolved aluminium and total dis-
solved silicon were determined using ICP-OES.
We used the method of Clarke et al. exactly as described, except that the reduction coil was 13 cm
long instead of 14 cm. The buffering, reaction and extraction times were slightly longer than those used by Clarke et al. (1.1, 2.8 and 5.8 s respectively,
compared with 1.0, 2.3 and 5.1 s). However, we do not believe that this should lead to significantly
different results. The experimental procedure was the same as for
the natural water samples from Skjervatjern, except that three of the standards were also used as valida-
tion samples and the first six PLS factors were used
for calculation of the Quickly Reacting concentra- tions of the metals.
The results are shown in Table 7. Good agree- ment was obtained for the Lysina sample, although the new method gave a slightly lower result. For the Swedish samples, lower values were obtained for Quickly Reacting aluminium determined with the
present method. The difference in the results ob-
tamed with the two methods is about 0.1 mg/l for
all samples. A number of explanations are possible: (1) The concentrations are low, for the simultane-
ous aluminium-iron system around or under the
quantification limit. (2) Some of the Swedish samples are rich in
organic matter. Certain organic complexes may be-
have differently in the two systems. (3) In the system of Clarke et al., iron@) is
reduced, this reduction beginning before the reagent stream is merged with the carrier stream. Some
colloidal iron(III) may be being reduced, with release
of bound aluminium.
(4) In the simultaneous aluminium-iron system,
some aluminium could be included in the iron frac-
tions. (5) Although not observed by Clarke et al. [7],
very small amounts of humus could be extracted and
absorb. In the present system, this might be treated as iron, because the humus spectrum resembles the spectra of the iron complexes formed rather than that
of aluminium trioxinate. However, in the system of
Clarke et al. humus would be included as aluminium. (6) The similarity in the differences obtained be-
tween the two sets of results suggests that a back-
ground value of 0.1 mg/l may be being subtracted by the program from the results obtained with the
new system. A higher background value might be
due to absorption by humus that has been extracted into chloroform.
(7) The differences might depend on the presence of silicato complexes. This is unlikely (with the
possible exception of the Lysina sample), because
Table 7
Comparison of Quickly Reacting aluminium determined by the present method and determined by the method of Clarke et al. (1992)
Sample Al,,, (mg 1-l) Al,,, (mg 1-l) U.L. (mg I-‘) Al,, (mg I-‘) SD. (mg I-‘) pH Colour Sir,,, (mg 1-r)
Damm A 0.21 0.06 0.02 0.13 0.02 4.24 Brown 3.84
Damm Es 0.61 0.09 0.03 0.20 0.00 3.78 Brown 2.68
GI 0.16 U.D. 0.02 0.03 0.00 4.88 Yellow 3.92
GII 1.16 0.13 0.02 0.24 0.01 4.11 Brown N.M.
Bo 4 0.44 0.10 0.02 0.17 0.00 3.81 Yellow 4.16
Lysina 1.66 0.61 0.01 0.69 0.03 3.85 Colourless 8.42
Al t<,t = Total dissolved Al; Al,,, = Quickly Reacting Al determined with our present method; U.L. = uncertainty limit for Al,,,; Al,, =
Quickly Reacting Al determined according to Clarke et al. (1992); SD. = standard deviation for Al,,; Si,,,, = total dissolved silicon;
U.D. = under the detection limit, N.M. = not measured.

18 N. Clarke, L.-G. Danielsson /Analytica Chimica Acta 306 (1995) 5-20
both the silicon and aluminium concentrations are
too low. More than one of these factors may be operating.
4. Conclusions
It is possible to measure Quickly Reacting Al,
Fe(R) and Fe(II1) simultaneously using complexation by oxine and l,lO-orthophenanthroline-iodide, fol-
lowed by extraction into chloroform, in a flow-in-
jection system. A reaction time of about 1.9-2.1 s and an extraction time of about 9.5-10.6 s gave good results. This is, as far as we know, the first
time that a method that measures these three compo- nents simultaneously has been presented. The contri-
butions to the signal from the three components can
be separated without difficulty using Partial Least Squares.
The sample throughput is 85 injections/h, and detection limits of 0.06 mg/l for all three compo-
nents were obtained. The relative standard deviation
of the absorbance for 31 consecutive injections of a standard containing 0.15 mg/l of the three compo-
nents varies from 5 to 12%, depending on wave- length. The linear range extends from the detection
limit up to at least 2 mg/l for all components. The only cation that might interfere under normal
circumstances is Ca2+. If present in concentrations of about 28 mg/l, Ca2+ gives a signal equivalent to
that of 60 pg/l Al at 363 nm, the wavelength at
which the absorbance of the calcium complex is
greatest. However, this problem can be avoided by not using wavelengths under 390 nm for evaluation.
In the case of aluminium, the most important species in the measured parameter are Al”+, A10H2+ and AlSOl. Weak organic complexes similar to the
acetato complexes could be included partially. Other weak complexes may also be included. On the other hand, large polymeric hydroxo complexes and fluoro complexes are excluded. AlHPO,f is included to an extent of O-40%. Complexes with humic or fulvic acids are almost always excluded, although excep- tions do, rarely, occur.
The Fe(III) species that are included in the mea- sured parameter include Fe3+, FeOH’+, FeF’+, FeHPOl and FeSO:. Large polymeric hydroxo
complexes are excluded. Complexes with natural and extracted humic substances appear to be included partially.
Among the Fe(R) species that are included are Fe’+ and FeOH+. FeH,POl is included to an
extent of 40-70%. FeCO,(,,, is excluded. Com- plexes with humic and fulvic acids appear also to be excluded.
The reduction of iron(II1) to iron(U) and the
oxidation of iron(R) to iron(III), in both cases medi- ated by humic substances, could be demonstrated. It
appeared that the same humic substances could, un- der different conditions, function as promoters of either oxidation or reduction.
Results obtained with natural water samples were satisfactory.
A comparison made with the system for the deter- mination of Quickly Reacting aluminium developed
by Clarke et al. [7] using natural water samples resulted in slightly lower values obtained with the
present system. Several explanations are possible.
The method has other potential uses as well as the measurement of Quickly Reacting Al, Fe(III) and Fe(R) in natural waters. For example, it could also
be used for the study of the interactions of Fe and Al with humic substances (including their competition for available sites), or for the study of the kinetics of
the reduction of Fe(II1) to Fe(R) and the oxidation of Fe(H) to Fe010 in the presence of humus. It should also be possible to use our method for the determina-
tion of conditional equilibrium constants for humic
complexes of iron(R) and aluminium, in the same way as Clarke et al. [46] did for the humic com-
plexes of aluminium. It might be possible to improve the system, for
example by using a smaller injection volume, as has been proposed by Berggren and Spar& [55] for the system of Clarke et al. This would make it possible to analyze samples containing much higher concen- trations of aluminium and iron. Also, the effect of humic substances on the segmentation would be reduced.
It might also be possible to use fewer wavelengths in the evaluation without sacrificing accuracy. This would have the advantage of simplifying the evalua- tion.
By reducing the reaction time, it might be possi- ble to obtain a different speciation.

N. Clarke, L.-G. Danielsson/Analytica Chimica Acta 306 (1995) S-20 I4
Acknowledgements
We would like to thank Bruce Kowalski, Peter Kaufmann, Steve Banwart, Olle Wahlberg and An-
ders Spar&n and other members of the Department of Analytical Chemistry at the Royal Institute of Tech-
nology for interesting and useful discussions and
suggestions, Ann-Catrine NorrstrGm for providing natural water samples and for making the ICP mea-
surements on these, Lars Lundin, Reiner Giesler and
Jakuh HruSka for providing natural water samples, Folke Ingman for helpful comments on the
manuscript, the National Swedish Environmental Protection Board for providing NRFA and NRHA,
Catharina Pettersson and Bert Allard for providing
RFA, Olle Wahlberg for the loan of platinum foil, Rolf Jansson for technical help and the National Swedish Environmental Protection Board, the Bengt Lundqvist Memorial Foundation, the OK Environ-
mental Foundation and the Futura Foundation for financial support.
References
III
[21
[31 [41
[51
[hl
[71
[Xl
[‘)I
II01
[I II
I121
[I31
N. Clarke, Ph.D. Thesis, The Royal Institute of Technology.
Stockholm, Sweden. 1994.
N. Clarke. L.-G. Danielsson and A. Spar&, Pure Appl.
Chcm.. (1995) in press.
V. Kubln, Crit. Rev. Anal. Chcm., 23 (lYY2) 15.
L.E. L&n, A. Rios, M.D. Luque de Castro and M. Val&rcel,
Lab. Robot. Autom.. I (1989) 295.
M. Blanco, J. Gene, H. Iturriaga. S. Maspoch and J. Riba,
Talanta. 34 (lY87) Y87.
N. Clarke, L.-G. Danielsson and A. Spar&, Finnish Humus
News. 3 ( I YY I ) 253.
N. Clarke, L-G. Danielsson and A. Spa&, Int. J. Environ.
Anal. (‘hem., 4X (1992) 77.
C‘. Pettcrsson, J. Ephraim and B. Allard, in C. Pettersson,
Ph.D. Thesis. University of Linkiiping, Sweden, 1992, Paper
III.
EM. l‘hurman and R.L. Malcolm, Environ. Sci. Technol., IS
( I YX I ) 463.
C. Pettcrshon. I. Arsenic, J. Ephraim, H. BorCn and B.
Allard, Sci. Total Environ.. Xl/82 (1989) 287.
N. Ingri, W. Kakolowicz. L.G. Sill& and B. Warnqvist,
Talanta, I4 (lYh7) 1261.
W. Stumm and J.J. Morgan, Aquatic Chemistry, Wilcy-lnter-
xiencc. New York, 2nd edn.. lY81.
W.L. Lindsay. Chemical Equilibria in Soils, Wiley, New
York. lY7Y.
[14] J. Bruno, W. Stumm, P. Wersin and F. Brandberg, Geochim.
Cosmochim. Acta, 56 (1992) 1139.
[IS] J. Bruno, P. Wersin and W. Stumm, Geochim. Cosmochim.
Acta, 56 (1992) 1139.
[16] G. Higg, AllmIn och oorganisk kemi. Almqvist and Wikscll.
Stockholm. 6th edn., 1963, p. 3 17.
[17] K. Bickstriim, L..-G. Danielsson and L. Nord. Anal. Chim.
Acta. 169 (1985) 43.
[IX] S. Lacroix. Anal. <‘him. Acta, I (1947) 260.
[IY] F. Vydra and R. Pribil, Talanta, 3 (IYS9) 72.
[20] A. Ringbom, Complcxation in Analytical Chemistry, Intcr-
science, New York, 1963.
[21] T. Sekine and Y. Hasegawa, Solvent Extraction Chemistry,
Fundamentals and Applications. Marcel Dekker. New York.
1977.
[22] P. Geladi and B.R. Kowalski, Anal. Chim. Acta. IX5 (19X6)
I.
[23] K. Goto, H. Tamura. M. Onodera and M. Nagatama. Talanta.
21 (IY74) 1x3.
[24] V. KubBn, L.-G. Daniclsson and F. Ingman. Anal. Chcm., 62
( 1990) 2026.
[2S] Z. Marczenko, Separation and Spcctrophotomctric Determi-
1261
[271
[281 l2Yl
[301
[311
[321
[331
[341
[3Sl
1361
I371
[3X1
[391
[401
[411
nation of Elements. Ellis Horwood. Chichester, 1986, p. 332.
S. Kotrlf and L. Sucha, Handbook of Chemical Equilibria in
Analytical Chemistry. Ellis Horwood. Chichester. 19X5. pp.
131, 156-157. 167.
A.E. Harvey Jr., J.A. Smart and ES. Amis. Anal. Chcm. 27
(IYSSI 26.
H. Fadrux and J. Ma@, Analyst, 100 (lY75) 540.
C‘amo A/S. Unscrambler II Version 35 lJscr’\ Guide, Camo
A/S, Trondheim, IYYl, p. 212.
R.M. Garrels. F.T. Mackenzie and C. Hunt, Chemical Cycles
and the Global Environment. Assessing Human Influcnccs.
William Kaufmann. Los Altos. 1975, pp. 33-35.
A.M. Shillcr and I:.A. Boyle. Geochim. Cosmochim. Acta.
51 (lYX7) 3273.
R. Rossmann and J. Barrcs. J. Great Lakes Rcs.. I4 (1988)
1X8.
K.H. Coale and AR. Flegal. Sci. Tot. Environ.. S7/NX
(1089) 2Y7.
N.M. Johnson, C.T. Driscoll, J.S. Eaton. G.E. Likens and
W.H. McDowell, Geochim. Cosmochim. Acta. 45 (lY81)
1421.
L.L. Olson and CR. O’Mclia. J. Inorg. Nucl. Chcm., 35
(lY73) 1977.
V.T. Athavalc, N. Mahadevan, P.K. Mathur and R.M. Sathc.
J. Inorg. Nucl. Chem.. 2Y (1967) 1947.
F.P. Cavasino and E. Di Dio. J. Chcm. Sot. (A). (lY7l)
3176.
S. Sjiiberg, A. Nordin and N. Ingri, Mar. C’hcm.. 10 (1YXl)
521.
S. Sjiibcrg. Y. Hagglund. A. Nordin and N. Ingri. Mar.
Chem.. I3 (1983) 35.
S. Sjiiberg, N. Ingri, A.-M. Nenncr and I..-(). ehman, Inorg.
Biochcm., 24 (1985) 267.
S. Sjiiberg, L.-O. ehman and N. Ingri, Acta Chem. Stand. A.
30 (1085) Y3.

20 N. Clarke, L.-G. Danielsson/Analytica Chimica Acta 306 (1995) 5-20
[42] H. Bilinski, L. Horvath, N. Ingri and S. Sjdberg, .I. Soil Sci., [49] C.H. Langford and D.W. Gutzman, Anal. Chim. Acta, 256
41 (1990) 119. (1992) 183.
1431 L.-O. Ghman, A. Nordin, I. Fattahpour Sedeh and S. Sjoberg,
Acta Chem. Stand., 45 (1991) 335.
[44] B.A. Browne and CT. Driscoll, Science, 256 (1992) 1667.
[45] T. Hedlund, S. Sjiiberg and L.-O. Ghman, Acta Chem.
Stand. A, 41 (1987) 197.
[50] J.H. Ephraim, A.S. Mathuthu and J.A. Marinsky, Complex
Forming Properties of Natural Organic Acids. Part 2. Com-
plexes with Iron and Calcium, Swedish Nuclear Fuel and
Waste Management Company Technical Report 90-28,
Stockholm, 1990.
[46] N. Clarke, L.-G. Danielsson and A. Spar&r, Water Air Soil
Pollut., (1995) in press.
[47] L. Lijvgren, T. Hedlund, L.-O. Ghman and S. Sjoberg, Water
Res., 21 (1987) 1401.
[51] L. Liang, J.A. McNabb, J.M. Paulk, B. Gu and J.F. Mc-
Carthy, Environ. Sci. Technol., 27 (1993) 1864.
[52] C.H. Langford, R. Ray, G.W. Quance and T.R. Khan, Anal.
Lett., 10 (1977) 1249.
[48] N. Clarke, L.-G. Danielsson, A. Hanning, F. Ingman and A.
Spar&r, in L. Maxe (Ed.), Analytical Methodology, Effects
of Acidification on Groundwater in Sweden - Hydrological
and Hydrochemical Processes, Final Report II of the Inte-
grated Groundwater Acidification Project, 1994, pp. 9-14.
[53] B.D. LaZerte, Can. J. Fish. Aquat. Sci., 41 (1984) 766.
[54] D. Berggren, Int. J. Environ. Anal. Chem., 35 (1989) 1.
[55] D. Berggren and A. Spar&r, Int. J. Environ. Anal. Chem.,
(1995) submitted for publication.