theoretical study of co adsorption on yttrium-doped gold clusters auny (n = 1–9)
TRANSCRIPT

Chemical Physics Letters 498 (2010) 296–301
Contents lists available at ScienceDirect
Chemical Physics Letters
journal homepage: www.elsevier .com/locate /cplet t
Theoretical study of CO adsorption on yttrium-doped gold clusters AunY (n = 1–9)
Ling Lin a, Peter Lievens b, Minh Tho Nguyen a,c,⇑a Department of Chemistry, Katholieke Universiteit Leuven, B-3001 Leuven, Belgiumb Laboratory of Solid State Physics and Magnetism, Katholieke Universiteit Leuven, B-3001 Leuven, Belgiumc Institute for Computational Science and Technology of HoChiMinh City, ThuDuc, HoChiMinh City, Viet Nam
a r t i c l e i n f o a b s t r a c t
Article history:Received 25 June 2010In final form 26 August 2010Available online 31 August 2010
0009-2614/$ - see front matter � 2010 Elsevier B.V. Adoi:10.1016/j.cplett.2010.08.068
⇑ Corresponding author at: Department of ChemLeuven, B-3001 Leuven, Belgium. Fax: +32 16 32 79 9
E-mail address: [email protected] (
CO adsorption on AunY (n = 1–9) is investigated using density functional theory, and compared with puregold clusters Aun+1. CO is confirmed to prefer on-top binding to the least coordinated Au atom for thepure gold clusters, whereas for Y-doped gold clusters, the most favorable sites are dependent on the clus-ter size; the Au on-top sites are favored from n = 6. CO behaves as an electron acceptor, and the C–Ovibrational frequency of the metal–CO complexes correlates with the degree of charge transfer to CO.Reactivity with CO is decreased after one gold atom is replaced by one yttrium for most sizes.
� 2010 Elsevier B.V. All rights reserved.
1. Introduction
Gold is a noble metal but when its size is reduced to the nano-meter range, it loses the nobleness and becomes highly reactive[1,2]. There have been in recent years a large number of studieson the catalytic properties of gold clusters towards different organ-ic reactions, especially the carbon monoxide combustion [1]. As afundamental step in the CO combustion on gold clusters, a COadsorption on Aun is essential for understanding the catalyticmechanism, and thus has attracted much attention. The bondingbetween Aun and CO is generally described using the Blyholdermodel, which considers the bonding in terms of donor–acceptorinteractions [3].
CO adsorption on the neutral, anionic, and cationic gold clustershave been studied experimentally [4–9], and it was found that forneutral gold clusters Aun (n = 9–68), the reactivity towards CO wasstrongly temperature-dependent and remarkably size-dependent.The latter property was discussed qualitatively in terms of elec-tronic shell structure [4]. For gold anions Au�n (n = 2–19), highlysize-dependent reactivity towards CO adsorption was also ob-served, with the small Au�n (n = 1–3) clusters showing no reactivityat room temperature [5–8]. The Au2CO� and Au3CO� were exper-imentally detected at lower temperatures, and the binding ener-gies were predicted to be <0.5 eV using RRKM theory [8]. Forgold cations Auþn (n = 1–65), size-dependent reactivity has alsobeen found, and there is an overall decrease of the CO binding en-ergy with increasing cluster size [9].
Theoretical studies on the CO adsorption on gold clustersmainly focused on the smaller sizes. A recent study of CO reactingwith Au0;�1
n (n = 2–10) [10] indicated that the adsorption strength
ll rights reserved.
istry, Katholieke Universiteit2.M.T. Nguyen).
of CO generally followed the order: cationic > neutral > anionic,which agrees well with a previous study [11], and this may be ex-plained with the electron donating nature of CO during adsorption[12]. Irrespective of the charge state, CO was found to prefer theon-top sites [11], and this configuration was also dominant forthe larger gold clusters Aun (n > 6) [13]. There was no odd–evenalternation observed for the CO binding energy, which is differentfrom the behavior of H adsorption [13]. However, an odd–evenoscillation has been found for the C–O vibrational frequency ofneutral complexes AunCO (n = 1–6) [11,13]. In the series of Aun
(n = 5–10), CO was found to preferentially absorb on top of theleast coordinated gold atom, except for Au5 and Au7, where thebridge sites are more favorable. This was attributed to an enhance-ment of p-back donation in bridge isomers. The C–O vibrationalfrequency also shows an odd–even oscillation [14]. A combinedexperimental and theoretical study on Au2ðCOÞ�1;0
n (n = 1–3) wascarried out recently, and the neutral Au2(CO)n (n = 1–2) clusterswere found to be linear. However, the Au2(CO)� anion is bentdue to the Renner–Teller effect, whereas the Au2ðCOÞ�2 anion re-mains linear, since the Renner–Teller effect is quenched by strongspin–orbit effects. The Au2ðCOÞ�3 anion is a physisorbed complex, inwhich the third CO is weakly interacting with a linear Au2ðCOÞ�2[15].
CO was predicted to behave as an electron acceptor in the reac-tions with Aun in some studies, since the CO moiety in the AunCOcomplexes is negatively charged [16,17]. A vibrational spectromet-ric study of the CO adsorbed gold anions and cations showed thatthere was a red-shift for the CO frequency with increasing CO cov-erage of the clusters, and this was considered as an indication thatthe p-back donation only makes a minor contribution to the Aun–CO bonding, especially for the cations [18,19].
CO adsorption on doped gold clusters has also been investigatedboth experimentally and theoretically. Introduction of an impurityatom in the gold clusters not only changes their geometrical and

L. Lin et al. / Chemical Physics Letters 498 (2010) 296–301 297
electronic property, but also their chemical reactivity. In a studyabout the reaction of CO with hydrogen- and alkali-metal dopedgold clusters, it was predicted that the interaction energy ofAun�1H with CO is significantly enhanced as compared with pureAun (n = 2–8) clusters. There is a certain enhancement for Aun�1Liin a few cases, while the sodium as a dopant exerts no significanteffect on the interaction energy [20]. The reactivity of gold clusterswith CO was found to be enhanced after introduction of Pt or Pd,and CO favored interaction with the dopant instead of the Auatoms [21–24].
A different behavior has been predicted for the silver and cop-per doped gold clusters MmAun (M = Ag, Cu, m + n = 2, 3) [24], Cu-mAun (m + n 6 6) [25] and AgmAuþn (m + n = 4–6) [26,27] for whichthe dopant atoms tend to decrease the reactivity of the pure goldclusters with CO. For the AgmAuþn cluster, CO consistently bindsto a gold atom and the binding energy ranges from 0.65 to1.09 eV, which decreases with increasing silver content [26,27].For the smaller anions AgmAu�n (m + n = 2–3), experimental studiesshowed that they are rather inactive toward CO adsorption [9], andthis is previously attributed to the large s–d valence level splitting,which is unfavorable in the framework of the Blyholder model [28–30].
The adsorption of a single CO group and four other isoelectronicgroups (NO+, BF, CN�, and BO�) to Au12W has been investigatedtheoretically, and the on-top sites are favored for all five complexesconsidered, among which the Au12WCO has the largest HOMO–LUMO gap, making it the most stable in terms of kinetic stability[31]. Another theoretical study shows that Au12W can be coveredby 12 CO molecules, and remains Ih symmetry. The binding energyper carbonyl was predicted to be �100 kJ mol�1 up to the last one[32].
In this context, it is important to explore the behavior of differ-ent dopants on the reactivity of gold clusters. In this work, we setout to perform a systematic investigation about CO adsorption onthe yttrium-doped gold clusters AunY (n = 1–9), the structures ofwhich have been investigated with a combined study of far-infra-red multiple photon dissociation (FIR-MPD) spectroscopy andquantum chemical calculations [33,34]. In contrast to pure goldclusters, which exhibit exclusively planar lowest-energy structuresfor small sizes, several of the studied AunY species are three-dimensional. This is particularly the case for Au4Y and Au9Y. Sev-eral of the lowest-energy structures are quasi-2D, that is, slightlydistorted from planar shapes.
The main objective of this study is to probe on how the interac-tion between CO and AunY depends on the size, structure, and elec-tronic properties of the clusters, and how the impurity atom Yinfluences the reactivity. For this purpose, the interaction of COwith the pure Aun+1 system is also determined using the same the-oretical methods.
2. Computational methods
Geometry optimizations and subsequent vibrational frequencycalculations are carried out using density functional theory (DFT)with the BP86 functional [35,36] with the aid of the GAUSSIAN03 pro-gram packages [37]. The correlation consistent cc-pVDZ-PP basisset [38,39] in which PP stands for an effective core potential replac-ing the cores, is employed for both gold and yttrium atoms, whilefor carbon and oxygen, the all electron cc-pVDZ basis set [40] isused. Different binding sites are considered for CO adsorption onthe lowest-lying cluster isomers. The interaction is evaluated onthe basis of the calculated binding energy, which is simply definedas Eb = E(CO) + E(M) � E(MCO) (E(X) stands for the total energy ofsystem X), and corrected for the zero-point energies. The naturalbond orbital (NBO) charges taken for evaluating the charge trans-
fers in the interactions are computed using the NBO 5.G code[41] integrated into the GAUSSIAN program [37].
Single-point-energy calculations with a larger basis set, namelycc-pVTZ-PP [38,39] for Au, Y, and cc-pVTZ [40] for C and O, wereperformed using the optimized structures for the small sizes. Com-paring the binding energies obtained from two levels (cf. Table S1in Supplementary material), it is seen that there is no large change(by at most 0.08 eV) when single-point energy calculations withthe larger basis set is used. Therefore only the results obtainedwith the cc-pVDZ-PP for Au and cc-pVDZ for C and O basis setsare reported in the present study.
3. Results and discussion
3.1. Geometries and energies of AunCO
The structures of small pure gold clusters have been studiedextensively theoretically [42,43], and we thus took the results fromRef. [39], in which the structure of Au7 predicted agrees well withthe result from a combined experimental and theoretical study[44]. Geometries of Aun are reoptimized with the BP86 functionaland cc-pVDZ-PP basis set. The shapes of the optimal Aun+1CO(n = 1–9) complexes are shown in Figure 1.
For all sizes of the pure gold clusters studied, the on-top atomsconstitute the most favorable adsorption site. For Au5 and Au7, theAu/Au bridge site is similarly favorable (Fig. 1). Our results aboutthe optimal AunCO complexes agree well with a previous studyfor the smaller size n = 2–6 [13], but the binding energies predictedat the present level are a little higher. CO adsorption on the largersizes (n = 7–10, 13) of gold clusters were also studied in that arti-cle, but 3D structures of Aun were employed [13]. For the largergold clusters Aun (n = 5–10), our results agree well with a previousstudy except for Au5, for which a Au/Au bridge site was predictedto be favored by 0.14 eV with respect to the on-top site of Au [14].
The experimental data about the CO binding energy with neu-tral gold clusters have not been reported so far, while for the smallAu�n (n = 2, 3) anions, it has been found that the theoretical valuesare substantially larger than the RRKM data derived from experi-ment [9]. However, for the gold cations Auþn (n = 1–8, 20), the cal-culated CO binding energies have been predicted to be inreasonable agreement with the experimental results [8].
3.2. Geometries and energies of AunYCO
As mentioned above, geometrical parameters of AunY (n = 1–9)have recently been reported [33,34], and the isomers listed inFigure S1 in the Supplementary material are taken into accountfor the CO adsorption in the present study. Shapes of the resultingAunYCO complexes are shown in Figure 2, along with the calcu-lated relative energies. The CO binding energies for the most stableAunYCO structure are also given.
For the diatomic AuY, CO prefers to bind to the Y/Au bridgeforming the AuYCO complex 1a (Cs, Fig. 2), with a binding energyof 0.69 eV, while complex 1b formed by CO binding on-top to theAu atom is 0.51 eV higher in energy. In the AuYCO complex 1a,the C–Y distance of 2.426 Å is slightly shorter than that of C–Au(2.540 Å). After CO adsorption, the Au–Y bond length is elongatedby 0.045 Å.
CO can bind to the Y atom of Au2Y and form 2a, with a bindingenergy of 1.19 eV. Complex 2b formed by CO on-top binding to Auatom is 0.99 eV less stable than 2a, therefore, CO adsorption on thedopant Y is more favorable. Upon CO attachment, the Au2YCO com-plex 2a still keeps the C2v symmetry of Au2Y. The larger binding en-ergy implies stronger interaction between CO and Au2Y, however,

Figure 1. Shape of the most stable Aun+1CO (n = 1–9) complexes and the CO binding energies (eV).
298 L. Lin et al. / Chemical Physics Letters 498 (2010) 296–301
the Au–Y bond length is only slightly elongated (from 2.653 to2.672 Å).
CO adsorbs preferentially on a Y/Au bridge site of Au3Y (D3h),and it tends to break the high symmetry and leads to a low sym-metry structure, with a binding energy of 0.64 eV (3a, Cs). ThisAu3YCO complex with an Au–Y–C triangle is more favored thanthe Au–C complex 3b, with a similar ratio as compared to thoseof AuY (Fig. 2). The Au–Y distance within the Au–Y–C triangle of3a is 2.735 Å, which is elongated by 0.079 Å upon CO adsorption,whereas the distances between Y and the other two Au atoms re-main unchanged.
The lowest Au4YCO complex (4a) is formed by CO attacking thebridge site of the Cs Au4Y cluster, and the CO binding energy is0.76 eV. The other three complexes (4b–d) are slightly higher inenergy than 4a. The AuY distance in the Au–Y–C triangle of themost optimal 4a is elongated by 0.084 Å upon CO attachment(from 2.750 Å).
Au5Y appears different from all the smaller clusters, since COfavors to bind to the bridge site of two gold atoms of the Cs Au5Yisomer, with a binding energy of 1.03 eV (5a, Fig. 2). Complexes(5b–d) with CO binding to other sites of the two lowest-lyingAu5Y isomers are much higher in energy (>0.2 eV). The Au–Cdistance in the most stable Au5YCO 5a is predicted to be 2.076 Å.
The Au6Y cluster has a C2v point group [33,34] and CO can bindon-top to one Au atom (6a), or a Au/Y bridge site (6b), with thebinding energies being both 0.73 eV (Fig. 2), which is �0.14 eVlarger than that of CO connecting the Au–Au bond (6c). In thecomplex 6a, the Au–C distance is predicted to be 1.978 Å. In theAu–Y–C triangle of complex 6b, the C–Y distance of 2.518 Å isshorter than that of C–Au (2.593 Å).
For Au7Y, the complex 7a (Cs) formed from a CO on-top bindingto one Au of the C2v symmetric Au7Y isomer has a binding energy of0.96 eV, and the Y atom is slightly distorted from the plane. Opti-mization of CO bonds to one gold atom of the Cs isomer of Au7Y
also converges to 7a, which is �0.2 eV more stable than the bridgecomplex 7b. The Au–C distance of 1.959 Å in 7a is shorter than thatof 1.978 Å in 6a, as the interaction of CO with Au7Y is stronger thanthat with Au6Y; there is thus a certain correlation between inter-molecular bond distances and binding energies.
The four isomers whose relative energies range within 0.08 eV(Fig. S1) [34] are all considered for the CO adsorption for Au8Y,and the shape of three optimal Au8YCO complexes are shown inFigure 2. Again the most favorable complex (8a) is with CO on-top binding to a gold atom, and the binding energy is 1.02 eV.The other two complexes (8b–c) are slightly higher in energy. Inthe optimal complex 8a, the Au–C distance is predicted to be1.952 Å, which is similar as that in 7a (1.959 Å).
For Au9Y, our calculations suggest that CO favors to attack onegold atom and form complex 9a with a binding energy of0.74 eV, while 9b formed by CO binding to the Y atom is energet-ically unfavorable by 0.10 eV. Upon CO adsorption, the structure ofAu9Y is slightly distorted, with the attacked gold atom deviatingfrom the central Y atom in 9a. The Au–C distance is 1.962 Å, whichis slightly longer than those in 7a and 8a, but shorter than that in6a, and this combines very well with the binding energies.
3.3. Comparison of Aun+1CO with AunYCO
Figure 3 illustrates the variations of the binding energies of COadsorption on both systems AunY and Aun+1 (n = 1–9). For the puregold cluster, the CO binding energies with Aun (n = 2–4) are calcu-lated to be�1.70 eV, while there is a drop for the larger sizes, and itreaches the valley at n = 6, with a smallest binding energy of1.02 eV. It is notable that the CO binding energies to Au5, Au7,and Au8 are all 1.21 eV. As the cluster grows, it seems there is atrend to decrease in the CO binding energies with some exceptions.For the Y-doped gold clusters, the largest binding energy is pre-dicted for Au2Y (1.19 eV). The CO binding energies with AunY are

Figure 2. Shape and relative energies (eV) of the different low-lying isomers of AunYCO (n = 1–9) complexes. The CO binding energies (eV) in the most stable AunYCO aregiven in angle brackets.
Figure 3. The binding energies of CO with AunY and Aun+1 (n = 1–9) as a function ofcluster size.
Figure 4. CO stretching frequencies of the optimal AunYCO and Aun+1CO (n = 1–9)complexes as a function of cluster size.
L. Lin et al. / Chemical Physics Letters 498 (2010) 296–301 299
smaller than those with the Aun+1 (except Au5Y), implying that thereplacement of a gold atom with yttrium will generally decreasethe reactivity of gold clusters with CO.
Figure 4 illustrates the changes of the C–O vibrational frequencyof AunYCO and Aun+1CO (n = 1–9) complexes with the cluster size.
300 L. Lin et al. / Chemical Physics Letters 498 (2010) 296–301
Within the Aun+1CO adducts, there is a very slight odd–even oscil-lation, which agrees well with what was reported in previous stud-ies [11,13,14]. On the other hand, after adsorption on Y-doped goldclusters, the C–O vibrational frequency shows more intense stag-gering. Figure 4 points out that the CO frequencies of AunYCO areall smaller than those of Aun+1CO. As the C–O stretching of the iso-lated CO molecule in the gas phase amounts to 2107 cm�1 (calcu-lated value), a substantial red-shift can thus be observed when COis adsorbed on gold clusters, and even larger red-shift takes placewhen it is adsorbed on the Y-doped gold clusters.
After the interaction with pure gold clusters Aun+1 (n = 1–9), theC–O distance is only marginally elongated in the range of 0.005–0.01 Å with respect to that in the free CO molecule, suggesting thatCO undergoes only very weak distortion upon adsorption on Aun.In contrast, for the Y-doped gold clusters AunY (n = 1–9), the elon-gation of C–O distance is much more significant (0.008–0.05 Å) fol-lowing complexation. Comparing the trend of changes in COfrequencies with that in C–O bond lengths (Fig. S2) and bindingenergies (Fig. 3), it can be concluded that the CO frequency de-creases nearly linearly with the increasing C–O bond length in bothpure and Y-doped gold clusters, but the frequency shifts are not di-rectly correlated with the binding energies.
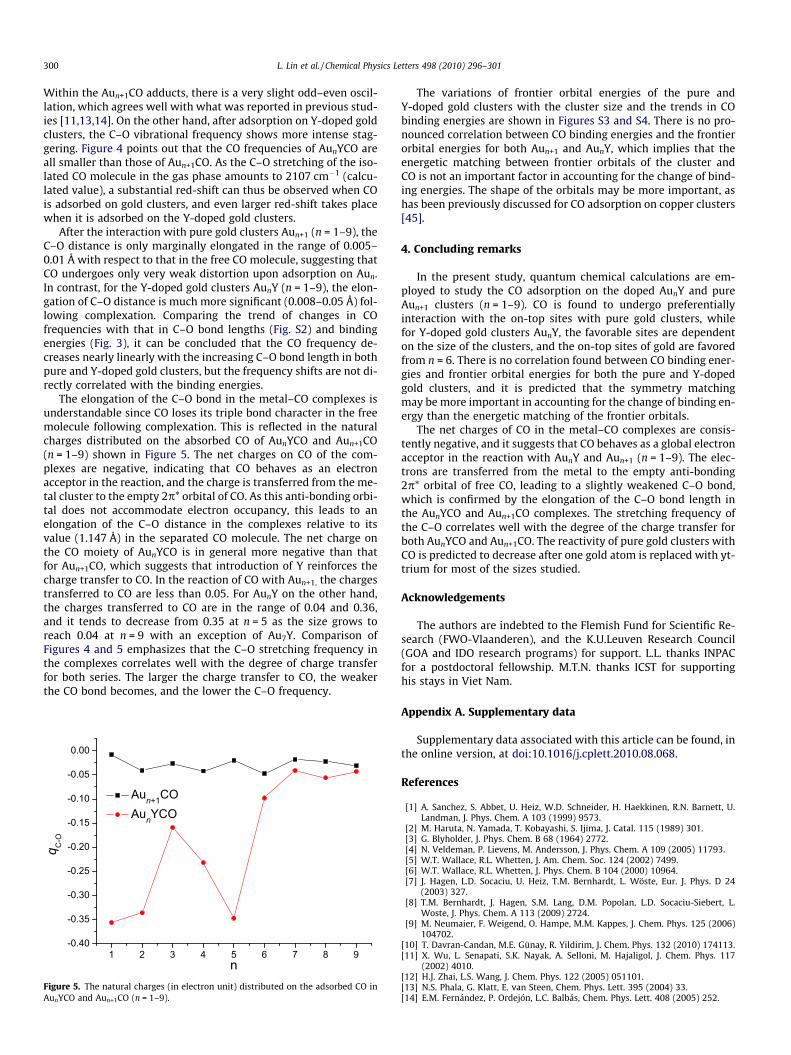
The elongation of the C–O bond in the metal–CO complexes isunderstandable since CO loses its triple bond character in the freemolecule following complexation. This is reflected in the naturalcharges distributed on the absorbed CO of AunYCO and Aun+1CO(n = 1–9) shown in Figure 5. The net charges on CO of the com-plexes are negative, indicating that CO behaves as an electronacceptor in the reaction, and the charge is transferred from the me-tal cluster to the empty 2p* orbital of CO. As this anti-bonding orbi-tal does not accommodate electron occupancy, this leads to anelongation of the C–O distance in the complexes relative to itsvalue (1.147 Å) in the separated CO molecule. The net charge onthe CO moiety of AunYCO is in general more negative than thatfor Aun+1CO, which suggests that introduction of Y reinforces thecharge transfer to CO. In the reaction of CO with Aun+1, the chargestransferred to CO are less than 0.05. For AunY on the other hand,the charges transferred to CO are in the range of 0.04 and 0.36,and it tends to decrease from 0.35 at n = 5 as the size grows toreach 0.04 at n = 9 with an exception of Au7Y. Comparison ofFigures 4 and 5 emphasizes that the C–O stretching frequency inthe complexes correlates well with the degree of charge transferfor both series. The larger the charge transfer to CO, the weakerthe CO bond becomes, and the lower the C–O frequency.
Figure 5. The natural charges (in electron unit) distributed on the adsorbed CO inAunYCO and Aun+1CO (n = 1–9).
The variations of frontier orbital energies of the pure andY-doped gold clusters with the cluster size and the trends in CObinding energies are shown in Figures S3 and S4. There is no pro-nounced correlation between CO binding energies and the frontierorbital energies for both Aun+1 and AunY, which implies that theenergetic matching between frontier orbitals of the cluster andCO is not an important factor in accounting for the change of bind-ing energies. The shape of the orbitals may be more important, ashas been previously discussed for CO adsorption on copper clusters[45].
4. Concluding remarks
In the present study, quantum chemical calculations are em-ployed to study the CO adsorption on the doped AunY and pureAun+1 clusters (n = 1–9). CO is found to undergo preferentiallyinteraction with the on-top sites with pure gold clusters, whilefor Y-doped gold clusters AunY, the favorable sites are dependenton the size of the clusters, and the on-top sites of gold are favoredfrom n = 6. There is no correlation found between CO binding ener-gies and frontier orbital energies for both the pure and Y-dopedgold clusters, and it is predicted that the symmetry matchingmay be more important in accounting for the change of binding en-ergy than the energetic matching of the frontier orbitals.
The net charges of CO in the metal–CO complexes are consis-tently negative, and it suggests that CO behaves as a global electronacceptor in the reaction with AunY and Aun+1 (n = 1–9). The elec-trons are transferred from the metal to the empty anti-bonding2p* orbital of free CO, leading to a slightly weakened C–O bond,which is confirmed by the elongation of the C–O bond length inthe AunYCO and Aun+1CO complexes. The stretching frequency ofthe C–O correlates well with the degree of the charge transfer forboth AunYCO and Aun+1CO. The reactivity of pure gold clusters withCO is predicted to decrease after one gold atom is replaced with yt-trium for most of the sizes studied.
Acknowledgements
The authors are indebted to the Flemish Fund for Scientific Re-search (FWO-Vlaanderen), and the K.U.Leuven Research Council(GOA and IDO research programs) for support. L.L. thanks INPACfor a postdoctoral fellowship. M.T.N. thanks ICST for supportinghis stays in Viet Nam.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.cplett.2010.08.068.
References
[1] A. Sanchez, S. Abbet, U. Heiz, W.D. Schneider, H. Haekkinen, R.N. Barnett, U.Landman, J. Phys. Chem. A 103 (1999) 9573.
[2] M. Haruta, N. Yamada, T. Kobayashi, S. Ijima, J. Catal. 115 (1989) 301.[3] G. Blyholder, J. Phys. Chem. B 68 (1964) 2772.[4] N. Veldeman, P. Lievens, M. Andersson, J. Phys. Chem. A 109 (2005) 11793.[5] W.T. Wallace, R.L. Whetten, J. Am. Chem. Soc. 124 (2002) 7499.[6] W.T. Wallace, R.L. Whetten, J. Phys. Chem. B 104 (2000) 10964.[7] J. Hagen, L.D. Socaciu, U. Heiz, T.M. Bernhardt, L. Wöste, Eur. J. Phys. D 24
(2003) 327.[8] T.M. Bernhardt, J. Hagen, S.M. Lang, D.M. Popolan, L.D. Socaciu-Siebert, L.
Woste, J. Phys. Chem. A 113 (2009) 2724.[9] M. Neumaier, F. Weigend, O. Hampe, M.M. Kappes, J. Chem. Phys. 125 (2006)
104702.[10] T. Davran-Candan, M.E. Günay, R. Yildirim, J. Chem. Phys. 132 (2010) 174113.[11] X. Wu, L. Senapati, S.K. Nayak, A. Selloni, M. Hajaligol, J. Chem. Phys. 117
(2002) 4010.[12] H.J. Zhai, L.S. Wang, J. Chem. Phys. 122 (2005) 051101.[13] N.S. Phala, G. Klatt, E. van Steen, Chem. Phys. Lett. 395 (2004) 33.[14] E.M. Fernández, P. Ordejón, L.C. Balbás, Chem. Phys. Lett. 408 (2005) 252.

L. Lin et al. / Chemical Physics Letters 498 (2010) 296–301 301
[15] Y.-L. Wang, H.-J. Zhai, L. Xu, J. Li, L.-S. Wang, J. Phys. Chem. A 114 (2010) 1247.[16] L. Jiang, Q. Xu, J. Phys. Chem. A 109 (2005) 1026.[17] D.W. Yuan, Z. Zeng, J. Chem. Phys. 120 (2004) 6574.[18] A. Fielicke, G. von Helden, G. Meijer, B. Simard, D.M. Rayner, J. Phys. Chem. B
109 (2005) 23935.[19] A. Fielicke, G. von Helden, G. Meijer, D.B. Pedersen, B. Simard, D.M. Rayner, J.
Am. Chem. Soc. 127 (2005) 8416.[20] N.K. Jena, K.R.S. Chandrakumar, S.K. Ghosh, J. Phys. Chem. C 113 (2009) 17885.[21] C. Song, Q. Ge, L. Wang, J. Phys. Chem. B 109 (2005) 22341.[22] M.M. Sadek, L. Wang, J. Phys. Chem. A 110 (2006) 14036.[23] W.Q. Tian, M. Ge, F. Gu, T. Yamada, Y. Aoki, J. Phys. Chem. A 110 (2006) 6285.[24] A.M. Joshi, M.H. Tucker, W.N. Delgass, K.T. Thomson, J. Chem. Phys. 125 (2006)
194707.[25] Y. Zhao, Z. Li, J. Yang, Phys. Chem. Chem. Phys. 11 (2009) 2329.[26] M. Neumaier, F. Weigend, O. Hampe, M.M. Kappes, J. Chem. Phys. 125 (2006)
104308.[27] M. Neumaier, F. Weigend, O. Hampe, M.M. Kappes, Faraday Discuss. 138
(2008) 393.[28] C. Elschenbroich, A. Salzer, Organometallics, VCH, Weinheim, Germany, 1989.[29] L. Lian, P.A. Hackett, D.M. Rayner, J. Chem. Phys. 99 (1993) 2583.[30] H.M. Lee, M. Ge, B.R. Sahu, P. Tarakeshwar, K.S. Kim, J. Phys. Chem. B 107
(2003) 9994.[31] Y. Fu, J. Li, S.-G. Wang, J. Mol. Model. 16 (2010) 9.
[32] M.P. Johansson, P. Pyykkö, Chem. Commun. 46 (2010) 3762.[33] L. Lin, P. Gruene, P. Claes, G. Meijer, A. Fielicke, P. Lievens, M.T. Nguyen,
ChemPhysChem 9 (2008) 2431.[34] L. Lin, P. Claes, P. Gruene, G. Meijer, A. Fielicke, M.T. Nguyen, P. Lievens,
ChemPhysChem 11 (2010) 1932.[35] A.D. Becke, Phys. Rev. A 38 (1988) 3098.[36] J.P. Perdew, Phys. Rev. B 33 (1986) 8822.[37] M.J. Frisch et al., GAUSSIAN03, Revision C.02, Gaussian Inc., Wallingford, CT, 2004.[38] K.A. Peterson, C. Puzzarini, Theor. Chem. Acc. 114 (2005) 283.[39] K.A. Peterson, D. Figgen, M. Dolg, H. Stoll, J. Chem. Phys. 126 (2007) 124101.[40] T.H. Dunning Jr., J. Chem. Phys. 90 (1989) 1007.[41] E.D. Glendening, J.K. Badenhoop, A.E. Reed, J.E. Carpenter, J.A. Bohmann, C.M.
Morales, F. Weinhold, NBO 5.G, Theoretical Chemistry Institute, University ofWisconsin, Madison, WI, 2001.
[42] X.B. Li, H.Y. Wang, X.D. Yang, Z.H. Zhu, Y.J. Tang, J. Chem. Phys. 126 (2007)084505.
[43] E.M. Fernández, J.M. Soler, I.L. Garzón, L.C. Balbás, Phys. Rev. B 70 (2004)165403.
[44] P. Gruene, D.M. Rayner, B. Redlich, A.F.G. van der Meer, J.T. Lyon, G. Meijer, A.Fielicke, Science 321 (2008) 674.
[45] L. Holmgren, H. Grönbeck, M. Andersson, A. Rosén, Phys. Rev. B 53 (1996)16644.