therapeutic nanoreactors - structural biology brussels
TRANSCRIPT

Vrije Universiteit Brussel Faculteit Wetenschappen
Vakgroep Bio-Ingenieurswetenschappen Onderzoeksgroep Ultrastructuur
Academiejaar 2007-2008
Therapeutic Nanoreactors: Combining Chemistry and Biology in a novel Triblock
Coploymer Drug Delivery System
Proefschrift voorgedragen tot het bekomen van de graad van doctor in de toegepaste biologische wetenschappen
Thesis submitted in fulfillment of the requirements for the degree of doctor
(PhD) in applied biological sciences
Ir. An Ranquin
Promotoren: Prof. Dr. ir. Jan Steyaert Dr. Patrick Van Gelder Dit werk kwam tot stand in het kader van een specialisatiebeurs van het IWT-Vlaanderen (Instituut voor de aanmoediging van Innovatie door Wetenschap en Technologie in Vlaanderen) en een BOF mandaat van de Vrije Universiteit Brussel

Published by the research Group of Ultrastructure. ULTR, Vrije Universiteit Brussel, Pleinlaan 2, 1050 Brussels, Belgium Departement of Molecular and Cellular Interactions, VIB, Belgium. Appart from any fair dealing for the purpose of research or private study or criticism or review, this publication may not be reproduced, stored in a retrieval system, or transmitted in any form or any means, electronic, mechanical, photocopying, recording, scanning, or otherwise, without the prior permission in writing of the publisher. Ranquin – Therapeutic Nanoreactors: Combining Chemistry and Biology in a Novel Triblock Copolymer Drug Delivery System – 2008. PhD Thesis Vrije Universiteit Brussel, Brussels, Belgium Keywords: ADEPT, nanoreactors, triblock copolymers, enzyme-prodrug therapy, drug delivery. ISBN: 9789081294829

I
Acknowledgements
This research was funded by the Instituut voor de aanmoediging van Innovatie door
wetenschap en technologie in Vlaanderen (IWT) and the Vrije Universiteit Brussel
(VUB) in collaboration with Prof. W. Meier from the University of Basel (UB) who
kindely provided us with PMOXA-PDMS-PMOXA polymers.
The past six years have been filled with periods of joy and sorrow, happiness and
despair so it goes without saying that I have a ‘few’ people to thank for support
during these periods. First I would like to thank my promoter Prof. Dr. J. Steyaert
for giving me the opportunity to conduct this research at the Ultrastructure lab. A
special thanks goes to my co-promotor Dr. P. Van Gelder who stood by me during
the last six years. Thanks a lot Patrick for the meaningful discussions, great ideas,
guidance and for revising this manuscript. I would also like to thank the members or
former members of the membrane group: Kris, Kim and Gerard and Anneke and
Wim V. for introducing me into the world of enzymology. A special thanks goes to
Caroline who I could always depend on for experiments, meaningful discussions and
her organization skills which makes Caroline a great colleague and a good friend. I
would also like to thank Ronny for the help with AFM experiments, Els for the
nanobody constructs and info as well as her knowledge on yeast expression and
Lieven in times of computer crisis. And off course I have to thank all of my
colleagues at the ULTR lab for the amazing atmosphere: Elke, Karo, Adinda, Celine,
Sarah, Khadija, Inge, Maia DK, Nathalie, Mike, Klaas, Wim C., Kathy, Nadine,
Marieke, Bruno,…
There are also several people from other labs that deserve my gratitude. The people
from CMIM: Nick, Lea, Jo and Benoit thanks a lot for the good advice. The people
from SWITCH: Maia DB and Hannah, thanks for supplying me week after week with
fresh neuroblastoma cell cultures. Also a special thanks to the people from Basel:
Alexandra, Samantha, Caroline, Per, Fabian, Olivier, Sandrine and off course
Wolfgang for your opinions and knowledge and for entertaining me on my trips to
Basel.

II
There are also some people from outside work that I would like to thank. First and
foremost my partner Jeroen who sometimes had to put up with my bad moods
when things didn’t go well and for always believing in me and supporting me. Also
thanks a lot baby for making yummy dinner each night for the last two months, I
really appreciate it. Special thanks to my baby boy Stanneke who was always there
(for the last 9,5 months anyway) to put a smile on my face, even after only 5 hours
of sleep. Also a thanks to my parents for supporting me and believing in me. And
thanks mum for the beautiful cover.
I can only hope that I didn’t forget anyone at the end of this long list. If I did, I
sincerely apologise.

III
Table of contents Chapter 1: Introduction 1
1.1 Chemotherapy: current status 3 1.2 Enzyme-prodrug therapy 6
1.2.1 ADEPT 6 1.2.2 GDEPT 11
1.3 Liposomes as drug delivery vehicles in cancer therapy 16 1.3.1 Long-circulating liposomes 17
1.3.2 Targeting liposomes to tumor tissue 18 1.3.3 Stimuli-sensitive liposomes 21 1.3.4 Conclusion 27
1.4 Polymer vesicles 28 1.5 References 30
Chapter 2: State of the art: a nanoreactor coming to life 39
2.1 Aim of the work: a novel directed enzyme-prodrug strategy 41
2.2 PMOXA-PDMS-PMOXA triblock copolymers 42 2.3 Bacterial outer membrane proteins 44
2.3.1 Aspecific porines OmpF and PhoE 45 2.3.2 Specific porin Tsx 47
2.4 Nucleoside hydrolase of Trypanosoma vivax 49 2.5 Purine nucleoside analogs as prodrugs 54
2.5.1 Deoxyadenosine analogs 54 2.5.2 6-Thioguanosine 57
2.6 Targeting of nanoreactors 58 2.6.1 Passive targeting 58 2.6.2 Active targeting with single domain
antibodies 59 2.7 References 65
Chapter 3: Encapsulation of therapeutic nucleoside hydrolase in functionalised nanocapsuls 71
3.1 Introduction 73 3.2 Materials and methods 75
3.2.1 Purification of T. vivax nucleoside hydrolase 75 3.2.2 Purification of E. coli porins OmpF and PhoE 76 3.2.3 Electrophysiology 76

IV
3.2.4 Preparation of proteoliposomes and swelling assays 77
3.2.5 Preparation of enzyme encapsulating proteoliposomes 78
3.2.6 Fluorescence measurements 78 3.3 Results 78
3.3.1 Purification and functional characterisation of TvNH and porins 78
3.3.2 Solute transport through OmpF and PhoE 80 3.3.3 Activity of nucleoside hydrolase
encapsulated in proteoliposomes 81 3.3.4 Interactions between TvNH and liposomes 83
3.4 Discussion 86 3.5 References 88
Chapter 4: Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles 91
4.1 Introduction 93 4.2 Materials and methods 94
4.2.1 Purification of T. vivax nucleoside hydrolase 94 4.2.2 Cloning, expression and purification of Tsx 94 4.2.3 Preparation of nanoreactors 96 4.2.4 DLS measurements 97 4.2.5 Reducing sugar assay 97 4.2.6 Atomic Force Microscopy (AFM) 97 4.2.7 Transmission Electron Microscopy (TEM) 98 4.2.8 Fluorescent labelling of TvNH 98 4.2.9 In vitro nanoreactor-macrophage
interactions 99 4.3 Results 100
4.3.1 Polymer batch variability 100 4.3.2 AFM and TEM images of nanoreactors 104 4.3.3 Fluorescently labelled Tsx-TvNH
nanoreactors 106 4.3.4 In vitro interaction of fluorescent
nanoreactors with macrophages 107 4.4 Discussion 109 4.5 References 111
Chapter 5: Therapeutic nanoreactors: combining chemistry and biology in a novel triblock copolymer drug delivery system 113
5.1 Introduction 115 5.2 Materials and methods 119

V
5.2.1 Purification of TvNH 119 5.2.2 Purification of E. coli porins OmpF and Tsx 119 5.2.3 Preparation of nanoreactors 120 5.2.4 Trypsin digestion 120
5.3 Results 121 5.3.1 Kinetic parameters of TvNH 121 5.3.2 Preparation of nanoreactors 122 5.3.3 Encapsulation efficiency of TvNH 123 5.3.4 Activity of nanoreactors 124
5.4 Discussion 127 5.5 References 128
Chapter 6: Nanoreactor mediated prodrug activation and killing of neuroblastoma cells 131
6.1 Introduction 133 6.2 Materials and methods 135
6.2.1 Nucleoside analogs and nucleobase analogs 135 6.2.2 Cytotoxicity assay 136 6.2.3 EC50 determination 136 6.2.4 Swelling assay with Tsx proteoliposomes 137 6.2.5 Preparation of TvNH nanoreactors 137 6.2.6 Enzymatic activity assay 138 6.2.7 Limulus Amebocyte Lysate (LAL) assay 138
6.3 Results 139 6.3.1 Cytotoxicity screening of prodrugs and drugs 139 6.3.2 EC50 determination of 6-thioguanosine and
6-thioguanine 141 6.3.3 6-Thioguanosine activation by human purine
nucleoside phosphorylase (hPNP) 143 6.3.4 Transport of 6-thioguanosine and 6-
thioguanine by Tsx 144 6.3.5 Kinetics of Tsx-TvNH nanoreactors 145 6.3.6 Cytotoxicity of 6-thioguanosine activated by
TvNH encapsulating nanoreactors 146 6.4 Discussion 148 6.5 References 153
Chapter 7: Production of cAbLys-3 mutants for selective coupling to nanoreactors 155
7.1 Introduction 157 7.2 Materials and methods 159
7.2.1 Site directed mutagenesis of cAbLys-3 159

VI
7.2.2 Bacterial expression and purification of cAbLys-3 mutants 160
7.2.3 Expression of cAbLys-3 S17C in Pichia pastoris 161
7.2.4 Enzyme-Linked Immuno Sorbent Assay (ELISA) 163
7.3 Results 164 7.3.1 Production of cAbLys-3 mutants in E. coli 164 7.3.2 Functionality of cAbLys-3 mutants 167 7.3.3 Production of cAbLys-3 S17C in P. pastoris 167
7.4 Discussion 168 7.5 References 170 7.6 Appendix 171
Chapter 8: General discussion 175 Summary 183 Samenvatting 187 Publications 191

Chapter 1 Introduction
1
Chapter 1:
Introduction

Chapter 1
2

Introduction
3
1.1 Chemotherapy: current status
The first drug used for cancer chemotherapy, Mustard gas, was not originally
intended for that purpose, but used as a chemical warfare agent. Scientist
discovered that it had a toxic effect on rapidly growing tumor cells. That
experience led researchers to look for other substances that might have similar
effects. As a result, many other drugs have been developed to treat cancer, and
drug development since then has exploded into a multi-billion dollar industry.
Most chemotherapeutic drugs work by impairing mitosis (cell division),
therefore specifically targeting fast-dividing cells and inducing apoptosis.
Unfortunately this means that non-malignant fast dividing cells such as those
responsible for hair growth and for replacement of the intestinal epithelium
(lining) are also often affected. This causes severe side effects to patients
receiving chemotherapy. Since most chemotherapeutics affect rapidly growing
tumors, chemotherapy is mostly used to treat tumors with high growth rates
such as acute myelogenous leukaemia and the aggressive lymphomas, including
Hodgkin's disease. Young tumors are treated more effectively because
mechanisms regulating cell growth are usually still preserved. With succeeding
generations of tumor cells, differentiation is typically lost, growth becomes less
regulated, and tumors become less responsive to most chemotherapeutic agents.
Near the centre of some solid tumors, cell division has effectively ceased, making
them insensitive to chemotherapy. Therefore, solid tumors are usually treated by
radiation therapy and surgery due to the fact that the chemotherapeutic agent
often does not reach the core of the tumor. Another problem occurs when cancer
cells become more resistant to chemotherapy treatments as a result of reduced
drug accumulation in the tumor cells. In 1976, Juliano and Ling discovered the
first surface glycoprotein responsible for altering drug permeation in drug
resistant tumor cells, p-glycoprotein (Juliano and Ling, 1976). It is responsible
for the active efflux of drugs from cancer cells. Since then, other multidrug
resistance proteins have been identified, including the multidrug resistance-
associated protein (MRP1, ABCC1) (Abe et al., 1995), identified in small cell lung
carcinoma and the breast cancer resistance protein (mitoxantrone resistance

Chapter 1
4
protein, ABCG2) (Doyle et al., 1998). Medications to inhibit the function of these
multidrug resistance proteins can enhance the efficacy of chemotherapy.
The majority of chemotherapeutic drugs can be divided into: alkylating agents
(cisplatin, carboplatin, oxaliplatin,…) (Alderden et al., 2006; Canetta et al., 1985;
Graham et al., 2004), antimetabolites (fludarabine, 2-chlorodeoxyadenosine, 2'-
deoxycoformycin...) (Cheson, 1992), anthracyclines (doxorubicin,
mitoxantrone,...) (Lown, 1993), alkaloids (camptothecin, paclitaxel,...) (Wall and
Wani, 1995), monoclonal antibodies (trastuzumab, cetuximab, rituximab,...)
(Albanell et al., 2003) and other antitumor agents. All of these drugs affect cell
division or DNA synthesis and function in some way. Some newer agents don't
directly interfere with DNA. These include the new tyrosine kinase inhibitor
imatinib mesylate (Gleevec® or Glivec®) (Droogendijk et al., 2006), which
directly targets a molecular abnormality in certain types of cancer (chronic
myelogenous leukaemia, gastrointestinal stromal tumors).
In addition, some drugs may be used which modulate tumor cell behaviour
without directly attacking those cells. Hormone treatments fall into this category
of adjuvant therapies.
One of the major difficulties in chemotherapy is the dosage: if the dose is too
low, it will be ineffective against the tumor, while at excessive doses the toxicity
(side-effects) will be intolerable to the patient. This has led to the formation of
detailed "dosing schemes" in most hospitals, which give guidance on the correct
dose and adjustment in case of toxicity. Harmful and lethal toxicity from
chemotherapy limits the dosage of drugs that can be given. Some tumors can be
destroyed by sufficiently high doses of chemotherapeutic agents. Unfortunately,
these high doses cannot be given because they would be fatal to the patient.
Several efforts have been undertaken to increase the tolerated dose of
chemotherapeutics. These include Haematopoietic stem cell transplant
approaches, isolated infusion approaches, targeted delivery mechanisms and
prodrug therapies. Since chemotherapeutic toxicity mainly affects haematopoietic
cells, cell transplants are used in combination with chemotherapeutics to
decrease damage to haematopoietic tissue. However, years of research has
yielded little proof of efficacy. Therefore haematological malignancies such as
myeloma, lymphoma, and leukaemia remain the main indications for stem cell
transplants.

Introduction
5
Isolated limb perfusion (often used in melanoma), or isolated infusion of
chemotherapy into the liver or the lung have been used to treat some tumors.
This way, a very high dose of chemotherapy can be delivered to tumor sites
without causing overwhelming systemic damage. Unfortunately, while these
approaches can be useful against solitary or limited metastases, they are - by
definition - not systemic and therefore do not treat distributed metastases or
micro metastases. Another way of increasing the dosage of therapeutics is the
use of specifically targeted delivery vehicles to increase effective levels of
cytotoxins for tumor cells while reducing effective levels for other cells. This
should result in an increased tumor kill and/or reduced toxicity. Specifically
targeted delivery vehicles have a differentially higher affinity for tumor cells by
interacting with tumor specific or tumor associated antigens. In addition to their
targeting component, they also carry a payload which is the chemotherapeutic
agent. Specifically targeted delivery vehicles vary in their stability, selectivity and
choice of target, but in essence they all aim to increase the maximum effective
dose that can be delivered to the tumor cells. Reduced systemic toxicity means
that they can also be used in sicker patients, and that they can carry new
chemotherapeutic agents that would have been far too toxic to deliver via
traditional systemic approaches.
Finally another way to increase dosage is the use of prodrugs. Prodrugs are
non toxic precursors that are designed to be transformed after administration to
form a pharmacologically active species. Such prodrugs are divided into two
categories: prodrugs designed to increase bioavailability and prodrugs designed
to deliver anticancer agents locally at the site of the tumor to increase specificity
and decrease systemic toxicity. The first category of prodrugs is used in case of
poorly soluble anti cancer agents. They are usually metabolically transformed to
their active compound. The latter category however is specifically activated at
the tumor site. Activation of the prodrugs can be achieved by the tumor
environment, enzymes specifically up-regulated in tumor tissue, enzymes
excreted by tumor cells or exogenous enzymes directed to tumor tissue.

Chapter 1
6
1.2 Enzyme-prodrug therapy
Enzyme-prodrug therapies were developed to deliver exogenous enzymes to
tumor tissue to selectively convert a relatively non-toxic prodrug to an active
drug. This leads to a higher local drug concentration in the tumor, improving the
anti tumor effect. Additionally, this lowers the systemic drug concentration
hereby reducing unwanted side effects that accompany conventional cancer
chemotherapy. The enzyme can either be delivered by an antibody-enzyme
fusion protein (antibody-directed enzyme-prodrug therapy, ADEPT) or by a
vector carrying the gene encoding for the exogenous enzyme (gene-directed
enzyme-prodrug therapy, GDEPT). When a viral vector is used for gene delivery,
the latter is also referred to as viral-directed enzyme-prodrug therapy (VDEPT)
(Figure 1).
Figure 1: Schematic overview of enzyme-prodrug therapy
1.2.1. ADEPT
Antibody directed enzyme-prodrug therapy is a two step approach where
selectivity for the target is achieved by an antibody in an antibody-enzyme fusion
complex. The antibody binds to antigens that are preferentially expressed on the
surface of tumor cells, or in the tumor interstitium. In the first step, the
active enzyme
prodrug
active drugantibody
antigen
active drug
prodrug
prodrugactiveenzyme
enzyme cDNA
enzyme mRNAtranscription
enzymetranslation
posttranslationalmodification
??
enzyme cDNA
viral transduction
physical transduction
VDEPT
GDEPT
ADEPT
CYTOPLASM
active enzyme
prodrug
active drug
prodrug
active drugantibody
antigen
active drug
prodrug
prodrugactiveenzyme
enzyme cDNA
enzyme mRNAtranscription
enzyme cDNA
enzyme mRNAtranscription
enzyme
posttranslationalmodification
??
enzyme cDNA
viral transduction
physical transduction
VDEPT
GDEPT
ADEPT
CYTOPLASM
active enzyme
prodrug
active drug
prodrug
active drugantibody
antigen
active drug
prodrug
prodrugactiveenzyme
enzyme cDNA
enzyme mRNAtranscription
enzyme cDNA
enzyme mRNAtranscription
enzymetranslation
posttranslationalmodification
??
enzyme cDNA
viral transduction
physical transduction
VDEPT
GDEPT
ADEPT
CYTOPLASM
active enzyme
prodrug
active drug
prodrug
active drugantibody
antigen
active drug
prodrug
prodrugactiveenzyme
enzyme cDNA
enzyme mRNAtranscription
enzyme cDNA
enzyme mRNAtranscription
enzyme
posttranslationalmodification
??
enzyme cDNA
viral transduction
physical transduction
VDEPT
GDEPT
ADEPT
CYTOPLASM

Introduction
7
antibody-enzyme conjugate is administered and accumulates in tumor tissue.
After clearance of non bound antibody-enzyme conjugates, a non toxic prodrug is
injected in the second step. This non-toxic prodrug can then be converted to a
cytotoxic drug by the antibody-enzyme complex which is located in the tumor.
There are two important features of this system. First, one molecule of antibody-
enzyme conjugate is able to catalyse the conversion of many molecules of
prodrug which enables higher drug concentrations at the tumor compared to
direct injection of the drug. Secondly, tumor cells which do not express the
antigen that is targeted by the antibody-enzyme complex are killed due to the
bystander effect.
There are some specific requirements for enzymes that are used in ADEPT. It
is important that they exert catalytic properties that are different from any
endogenous enzyme to prevent activation of prodrugs at other sites in the body.
They should also catalyse a scission reaction and be active and stable under
physiological conditions. Finally, they should affect high catalytic turnover.
The enzymes used in ADEPT can be characterised in three categories: (i)
enzymes of non-mammalian origin and with no mammalian homologue. These
enzymes have great potential to be used in ADEPT since activation of prodrugs
by endogenous enzymes in the blood and healthy tissue is avoided. Enzymes of
bacterial origin in particular are of interest since they are readily available on a
large scale due to their lack of posttranslational modifications. Their main
disadvantage is their potential to elicit an immune response. This is very
problematic since circulating host anti-conjugate antibodies may interfere with
successive treatment. Enzymes in this category include carboxypeptidase G2
(CPG2), cytosine deaminase (CD), β-lactamase (β-L), penicillin G amidase (PGA),
penicillin V amidase (PVA), etc. (ii) Enzymes of non-mammalian origin with a
mammalian homologue. It is very important that the mammalian homologue of
the enzyme used is only present at low concentrations in the blood or healthy
tissue. Furthermore the mammalian homologue should have different catalytic
properties than the enzyme used to activate the prodrug. This includes different
turnover rates, different optimal conditions such as pH or different structural
requirements for substrates. Enzymes in this category include E. coli β-
glucuronidase (β-G) which has an optimal pH of 6.8 compared to 5.3 for the
mammalian homologue and a higher turnover rate and E. coli nitroreductase that

Chapter 1
8
exhibits different structural requirements for substrates than the human
homologue DT-diaphorase. (iii) Enzymes of mammalian origin. Their main
advantage is the reduction to elicit a host immune response. Unfortunately use in
ADEPT is limited due to the fact that their presence in humans is likely to elicit
prodrug activation in the blood and healthy tissue. Examples include alkaline
phosphatase (AP), α–galactosidase (α-G) and carboxypeptidase A (CPA). In case
of CPA, the risk of conversion of the prodrug by the human enzyme was
circumvented by site specific mutation of the enzyme. This mutant T268G CPA is
able to convert bulky analogues such as 3-cyclopentyltyrosine methotrexate
whereas the wild-type enzyme is not (Smith et al., 1997). Table 1 gives an
overview of selected examples of ADEPT in enzyme-prodrug cancer therapy.
The antibodies (Abs) that bind to tumor-associated antigens are a key
component in ADEPT since they ensure the specific activation of prodrugs in
tumor tissue. There are some specific requirements for Abs used in ADEPT. For
instance, they should bind to tumor cells with high affinity but exert minimal
binding to normal tissue. Furthermore, covalent binding to the enzyme must not
impair the ability of the Abs to bind the associated antigen, nor should it alter the
enzyme activity. Ideally, the non-bound Ab-enzyme conjugate should be rapidly
cleared from the blood. This can be achieved by using a secondary antibody that
binds the Ab-enzyme conjugate. This was demonstrated by Sharma et al
(Sharma et al., 1994). Their study was performed with a monoclonal anti-
carcinoembryonic antigen antibody fragment A5B7-F(ab’)2 conjugated to the
bacterial enzyme, carboxypeptidase G2 (CPG2), in LS174T xenografted mice. A
monoclonal antibody (SB43), directed at CPG2, was used, which inactivates
CPG2 in vitro and in vivo. SB43 was galactosylated so that it had sufficient time
to form a complex with plasma CPG2, resulting in the inactivation and clearance
of the complex from plasma via the carbohydrate-specific receptors in the liver.
Injection of SB43gal 19 hours after administration of the conjugate significantly
reduced the amount of conjugate present in the blood without affecting
conjugate levels in the tumor. They also used a different approach in which the
conjugate was galactosylated so that it is rapidly cleared from the blood by the
asialoglycoprotein receptors in the liver. Localization of the conjugate was
achieved by blocking this receptor for about 8 hours with a single injection of an
inhibitor that binds competitively to the receptor. This allowed tumor localization

Introduction
9
of the conjugate followed by a rapid clearance of the galactosylated conjugate
from the blood as the inhibitor was consumed. The conjugate had a tumor to
blood ratio of 45:1 after 24 hours, which increased to 100:1 at 72 hours after the
conjugate injection.
Table 1: Selected examples of ADEPT in enzyme-prodrug therapy
Enzyme Antibody Prodrug Ref
β-glucosidase HMFG1 Amygdalin (Syrigos et al., 1998)
human β-glucosidase
humanised CEA specific binding region
Prodrugs of anthracyclines
(Florent et al., 1998)
human β-glucosidase
anti-CD20 1H4 Doxorubicin (Haisma et al., 1998)
human β-glucosidase
anti-CEA BW431 Doxorubicin (Bosslet et al., 1992)
carboxypeptidase G2
anti-CEA A5B7 CMDA (Stribbling et al., 1997)
carboxypeptidase G2
W14
p-N-bis (2-Chloroethyl)benzoyl glutamic acid
(Bagshawe et al., 1988; Springer et al., 1991)
alkaline phosphatase
anti-tumor associated carbohydrate L6
Etoposide phosphate (Senter et al., 1989)
Penicillin amidase anti-tumor associated carbohydrate L6
Doxorubicin
(Kerr et al., 1990; Vrudhula et al., 1993)
β-lactamase anti-CEA CEM2314 Desacetylvinblastine hydrazide
(Meyer et al., 1993)
β-lactamase cab-CEA5 nanobody 7-(4-carboxybutanamido) cephalosporin
(Cortez-Retamozo et al., 2004)
Another important factor in ADEPT is the ability of the Ab-enzyme conjugate to
penetrate the tumor. It is well known that tumor vasculature is leaky which
enables macromolecules to extravasate to the tumor interstitium. Furthermore,
the lymphatic drainage system is impaired so that macromolecules are retained
in the interstitium for a prolonged time. This is called the enhanced permeability
and retention effect (EPR) (Figure 4) (Jain, 1987). Due to this EPR, it is relatively

Chapter 1
10
easy for Ab-enzyme conjugates to enter the tumor interstitium. However, further
penetration of the tumor is much more difficult for macromolecules. This leads to
an inadequate distribution of Ab-enzyme conjugates in the tumor tissue (Jain,
1990). To overcome this limitation, Ab fragments, which include F(ab’)2, F(ab’),
scFv and nanobodies, rather than intact antibodies have been used (Figure 2).
These Ab fragments show an increased interstitial rate of transport and
additionally a more rapid clearance from the blood. Using shorter Ab fragments
also decreases the chance of eliciting a host immune response towards the Ab-
enzyme conjugate. A further decreased immunogenicity can be achieved by
combining the regions of the murine antibody that are responsible for the
antigen recognition with human antibody fragments. Such antibodies can be
“chimerized” (murine variable region and human constant region; antibodies
called –iximab) or “humanized” (additional replacement of the murine framework
regions within the complementarity determining regions by human residues,
antibodies called –umab) (Vaughan et al., 1998). Initial comparisons between
murine and chimeric monoclonal antibodies were very promising. For example
use of the chimeric MAb 17-1A in the treatment of colorectal adenocarcinoma
showed improved pharmacokinetics together with a significant reduction in
immunogenicity (LoBuglio et al., 1989). However, not all chimeric MAb’s are
successful in reducing immunogenicity. Nevertheless, many of them as well as
humanised MAb’s have progressed through clinical trials (Reichert et al., 2005).
Figure 2: schematic representation of different antibody fragments used in ADEPT
VL
CH3
CH1
CH2
VH
CL
CH1CL VH
VL
CH1VH
CL
VL
F(ab’) 50 kDa F(ab’)2 100kDa
Monoclonal antibody
CH3
VHH
VHH
CH2
Single chain camel antibody
Nanobody 15 kDa
VLVH
scFv 25 kDa
VL
CH3
CH1
CH2
VH
CL
CH3
CH1
CH2
VH
CL
CH1CL VH
VL
CH1CL VH
VL
CH1VH
CL
VL
F(ab’) 50 kDa F(ab’)2 100kDa
Monoclonal antibody
CH3
VHH
VHH
CH2
Single chain camel antibody
Nanobody 15 kDa
VLVH
scFv 25 kDa

Introduction
11
Since many ADEPT strategies make use of a bacterial enzyme, there is a high
risk that a host immune response towards this bacterial enzyme is elicited. A way
of overcoming this is to use so called abzymes. Abzymes are antibodies raised
against the transition intermediate of an enzyme substrate. Given the strong
binding affinity towards this transition state, they are able to catalyse the
conversion from substrate to product. Since it is an antibody, it can be
chimerized or humanised to minimize immunogenicity. In antibody-directed
abzyme prodrug therapy (ADAPT) a bispecific antibody is used were one arm is
the catalytic antibody and the other arm is the targeting antibody (Kakinuma et
al., 2002).
In conclusion, although ADEPT offers much advantages compared to
conventional therapy, there are many clinical limitations. In poorly vascularised
tumors, delivery of large conjugates is restricted and it is impossible to deliver
conjugates to all tumor cells. Because the enzyme level is low, it is difficult to
generate adequate levels of active drug to reach lethal doses. Furthermore, the
binding of conjugates is limited by antigen heterogeneity. The biggest problem
with ADEPT however, remains the immunogenicity of the antibody-enzyme
conjugate. Although there are many ways of overcoming this immunogenicity,
development is very costly and difficult.
1.2.2. GDEPT
GDEPT, or suicide gene therapy, is also a two step approach. In the first step
the gene encoding a prodrug activating enzyme is directed to tumor tissue. In a
second step the prodrug is administered and subsequently activated by the
prodrug activating enzymes present in tumor cells. Success or failure of GDEPT
strategies not only rely on choosing the right enzyme-prodrug combination but
also on the efficient gene transfection and sustained expression of the genes in
tumor tissue. Therefore the design of appropriate delivery vectors remains the
biggest challenge in suicide gene therapy. In the past two decades, numerous
viral and non-viral methods for transduction were developed.
Since viral vectors naturally evolved to efficiently transfect host cells and
posses the appropriate molecular mechanisms for gene delivery, they are widely
used for suicide gene therapy. In these vectors, the genes necessary for the
replication phase of the virus are replaced by the gene encoding the prodrug

Chapter 1
12
activating enzyme. In this manner, non replicative viruses are made that can
infect target cells and introduce genes either by integrating them into the
genomic DNA of the target cell or by residing on a plasmid. Since viruses have
evolved as parasites, they elicit an immune response that reduces their clinical
use. Despite this, several viral vectors have been engineered for suicide gene
therapy. Interest has centred on four types: adenoviruses, retroviruses
(including lentiviruses), adeno-associated viruses and herpes simplex virus type
1. One major challenge of viral vectors is the efficient targeting of the vectors to
the cells of interest: to achieve successful gene therapy, the appropriate genes
must be delivered to and expressed in target cells, without harming non-target
cells.
One way to obtain tumor specific expression of the prodrug activating enzyme
is by transcriptional targeting. In this approach tumor-specific enhancer-
promoters are used thus allowing transcription of the gene in tumor cells only.
Many tumor-specific enhancer-promoter sequences were discovered and used to
target adenoviruses to various tissues, for a summery see Table 2. These
adenoviral vectors carrying suicide genes controlled by tumor specific regulatory
elements demonstrated both targeting and efficacy. However, transcriptional
targeting does not prevent transfection of healthy tissue and toxic effects related
to this dislocation of viral vectors.
Another way to achieve tumor specific expression of the suicide gene is by
direct targeting of the viral vectors to the surface of tumor cells by using the
native tropism (host range) of the viral vector. However, this native tropism
often does not meet the therapeutic need. Native tropism may not be able to
specifically transfect tumor tissue and therefore needs to be diminished to avoid
toxic side effects. Therefore many different mechanisms were developed to
approve the targeting of viral vectors to specific tissues, including pseudotyping,
adaptor systems and genetic approaches. For a complete overview see Table 3.

Introduction
13
Table 2: selected examples of transcriptional targeting to various tumors
Enhancer-promotor Target tissue Ref
hAFP (human α-fetoprotein) Hepatocellular carcinoma (Kaneko et al., 1995; Li et al., 2001)
hALA (human α-lactalbumin) Mammary cells (breast cancer)
(Anderson et al., 2000)
hCEA (human carcinoembryonicantigen)
Colorectal carcinoma (Zhang et al., 2003)
Cox-2 (cyclooxygenase-2) Gastrointestinal cancer (Yamamoto et al., 2001)
GRP (gastrin releasing peptide) Lung cancer (Morimoto et al., 2001)
L-plastin Ovarian and bladder cancer (Peng et al., 2001)
rPB (rat probasin) Prostate cancer (Andriani et al., 2001; Lu and Steiner, 2000)
PSA (human prostate-specific antigen)
Prostate cancer (Gotoh et al., 1998)
SLPI (secretory leukoprotease inhibitor)
Cervical and ovarian cancer (Barker et al., 2003)
Survivin Suvivin-positive tumor cells (Zhu et al., 2004)
hTERT (human telomerase reverse transcriptase)
Telomerase-positive tumors (Bilsland et al., 2003; Gu et al., 2000; Huang et al., 2004)
Tg (rat thyroglobulin) Thyroid carcinoma (Shimura et al., 2001; Zhang et al., 2001)
Tyrosinase Melanoma (Nettelbeck et al., 2002; Siders et al., 1996)
BLG (ovine β-lactoglobulin) Mammary cells (breast cancer)
(Anderson et al., 2000)
ErbB2 ErbB2-positive tumor cells (Vassaux et al., 1999)

Chapter 1
14
Table 3: Tropism changing strategies for viral targeting
Approach Principle Ref
Adaptor systems
Receptor-ligand A tumor specific ligand is fused with the viral
receptor
(Pereboev et al., 2004; Snitkovsky and Young, 2002)
Bispecific antibodies
Two antibodies that bind the viral vector and the target cell are coupled
(Bartlett et al., 1999; Wurdinger et
al., 2005)
Chemical linkage The targeting molecule is chemically coupled
to the viral particle (Eto et al., 2005)
Avidin-Biotin Coupling biotin to the vector and avidin to
the targeting molecule (Arnold et al., 2006)
Antibody A tumor specific antibody that binds to a
genetically incorporated Ig-binding domain of the vector
(Tai et al., 2003)
Genetic systems
Serotype switching Using a different serotype of the same family (Wu et al., 2006)
Pseudotyping Using a viral attachment protein from a
different strain or family (Cronin et al., 2005)
Targeting motifs Small targeting peptides that are inserted into the capsid or viral attachment protein
(Gollan and Green, 2002; Stachler and
Bartlett, 2006)
Single-chain antibody
A single chain antibody is inserted in the viral attachment protein
(Chowdhury et al., 2004; Hedley et al., 2006; Yang et al.,
1998)
Mosaic viral attachment proteins
Two viral attachment proteins with different properties are combined
(Pereboeva et al., 2004)
In 2007, 70 % of gene therapy clinical trials used viral vectors for gene
delivery. However, there are many drawbacks in using viral vectors for gene
delivery such as their immunogenicity, cytotoxicity as well as the risk of
insertional mutagenesis. Therefore non-viral vectors have important safety
advantages over viral vectors.

Introduction
15
Conventional non-viral vectors include diverse liposomal formulations (DNA
lipoplexes) (Karmali and Chaudhuri, 2007; Liu et al., 2003), cationic peptides
(Fabre and Collins, 2006) and polymers (DNA polyplexes) such as
polyetylenimine (Boussif et al., 1995; Lungwitz et al., 2005). They interact with
DNA to facilitate cell entry by binding or enveloping DNA through a charge
interaction. These vectors however have poor in vivo transfection efficiency and
only confer transient gene expression. This is partially due to the ability of the
non-viral vector–DNA complex to interact with blood plasma proteins,
undesirable cells and the extracellular matrix. Once inside the target cell,
additional requirements for transfection include the need for the DNA to escape
the liposome or endosome, resistance to cytoplasmic degradative enzymes such
as nucleases, and passage through the double-membrane structure of the
nuclear envelope. Finally, but importantly, plasmid DNA that is delivered to the
nucleus is generally not replicated and is lost when the nuclear envelope is
degraded during mitosis.
To overcome these problems, novel non-viral vectors with increased in vivo
stability have been developed, which have a reduced affinity for extracellular
proteins and cell surfaces, enabling them to reach target cells (Kichler, 2004;
Knorr et al., 2007). Furthermore, the inclusion of ligands for receptor-mediated
endocytosis (Kircheis et al., 2001; Kursa et al., 2003), endosomal disruption
sequences (Cho et al., 2003; Funhoff et al., 2005) and nuclear-import signals
(Bremner et al., 2004; Zanta et al., 1999) have improved the passage of non-
viral vectors into the cell, and into its nucleus. In combination, these modular
non-viral vectors mimic the ability of viruses to overcome the cellular barriers to
DNA delivery through mechanisms that are analogous to those of viral vectors
(Uherek et al., 1998).
The enzyme-prodrug combinations used in GDEPT are similar to those used in
ADEPT. The most frequently used and best characterised prodrug activating
enzymes include thymidine kinase from herpes simplex virus type 1, cytosine
deaminase, cytochrome P450 reductase, nitroreductase, carboxypeptidase G2
and purine nucleoside phosphorylase. For an overview see Table 4.
Taken together, suicide gene therapy shows great potential to improve
chemotherapy by decreasing unwanted cytotoxic side effects. However, both
viral and non-viral approaches suffer from several drawbacks such as inefficient

Chapter 1
16
gene transfection and prolonged gene expression, pathogenicity and
immunogenicity.
Table 4: Selected examples of suicide gene therapy systems
Enzyme Prodrug Vector Ref
herpes simplex virus 1 Thymidine kinase
ganciclovir Adenovirus Ad-CMV-
UTk, Polyethylenimine
(Mathis et al., 2006) (Iwai et
al., 2002)
yeast Cytosine deaminase
5-fluorocytosine Adenovirus Ad-CMV-CD (Zeng et al., 2007)
Cytochrome P 2B1 Cyclophosphamide Adenovirus Ad-CYP2B1 (Huch et al., 2006)
E. coli Nitroreductase
Nitrocompound CB1954
Adenovirus Ad-hTR-NTR (Bilsland et al., 2003; Plumb et
al., 2001) E. coli Purine nucleoside phosphorylase
6-methylpurine 2’-deoxyriboside
Plasmid phTERT-ePNP, Adenovirus Ad2-Tyr2-
PNP
(Zhou et al., 2007) (McCart et al., 2002)
Carboxypeptidase G2
Mustard compound ZD2767P
Adenovirus AdV-hTERT-CPG2
(Schepelmann et al., 2007)
Human deoxycytidine kinase
Cytosine arabinoside Adenovirus Ad-hdCk, Retrovirus Rv-hdCK
(Manome et al., 1996)
D. melanogaster Deoxyribonucleoside kinase
Cytosine arabinoside, 5-fluorocytosine
lipoplexes (Kamiya et al., 2006)
β-glucuronidase glucuronide prodrug of
doxorubicin (DOX-GA3)
poly(2-(dimethylamino)ethyl
methacrylate) polyplexes
(Fonseca et al., 1999)
1.3 Liposomes as drug delivery vehicles in cancer therapy
Liposomes were suggested as drug carriers in cancer chemotherapy by
Gregoriadis et al. in 1974 (Gregoriadis et al., 1974). Since then, the interest in
liposomes has increased and liposome systems are now being extensively
studied as drug carriers. Moreover, liposomes are the most advanced of the
particulate drug carriers and are now considered to be a mainstream drug
delivery technology with breakthrough developments resulting in the approval of

Introduction
17
several liposomal drugs, such as Doxyl® (Ortho Biotech), DaunoXome® (Gilead
Sciences, Inc.), caelyx® (Schering-Plough, Inc.), ...
In general, hydrophilic drugs can be entrapped in the aqueous interior and
hydrophobic drugs can be incorporated in the bilayer (Figure 3). Amphiphilic
drugs that are weak bases or weak acids can also be loaded into the liposome
interior using remote loading methods. Three basic requirements need to be
fulfilled if liposomes are to be successful in delivering drugs specifically to
cancerous tissue: (i) prolonged blood circulation, (ii) sufficient tumor
accumulation, (iii) controlled drug release and uptake by tumor cells with a
release profile matching the pharmacodynamics of the drug.
Initially, the research in liposome drug delivery systems suffered from the
very fast blood clearance, due to adsorption of plasma proteins (opsonins) to the
‘naked’ phospholipid membrane and complement activation. Subsequently
triggering recognition and uptake of the liposomes predominantly by Kuppfer
cells of the reticuloendothelial system (RES) (Ishida et al., 2002). Liposomes
typically have a half-life of approximately 0,6 hours resulting in complete
removal from the bloodstream within several hours (Blume and Cevc, 1993). A
major advance in the field of liposomes came with the development of long-
circulating liposomes or Stealth® liposomes.
1.3.1. Long-circulating liposomes
Different methods have been suggested to achieve long circulation of
liposomes in vivo, including coating the liposome surface with inert,
biocompatible polymers, such as poly(ethylene glycol) (PEG). These PEG chains
form a protective layer over the liposome surface and prevent liposome
interactions with opsonins and subsequent clearance of liposomes (Klibanov et
al., 1990) (Figure 3). An important feature of protective polymers is their
flexibility, which allows a relatively small number of surface-grafted polymer
molecules to create an impermeable layer over the liposome surface (Torchilin et
al., 1994). Long-circulating liposomes demonstrate dose-independent, non-
saturable, log-linear kinetics and increased bioavailability (Allen and Hansen,
1991). Although, PEG remains the standard for the steric protection of
liposomes, other polymers also posses stealth-like properties such as methyl and
ethyl polyoxazolines (Woodle et al., 1994; Zalipsky et al., 1996), poly-N-

Chapter 1
18
vinylpyrrolidones (Torchilin et al., 2001), and polyvinyl alcohols (Takeuchi et al.,
2001). Long-circulating liposomes, or stealth liposomes, are now being
investigated in detail and are widely used in biomedical in vitro and in vivo
studies and have found their way into clinical practice (Gabizon, 2001).
More recently, research has focussed on attaching PEG in a removable fashion
to facilitate liposome capture by cells. For instance, in suicide gene therapy
uptake of the liposomal vector is required for therapeutic activity. After PEG-
liposomes accumulate at the target site, through the enhanced permeability and
retention (EPR) effect (Jain, 1987), the PEG coating is detached locally by
proteolysis (Hatakeyama et al., 2007) or mild thiolysis (Zalipsky et al., 1999) at
the tumorsite allowing facilitated endocytosis of the lipoplexes
Figure 3: Schematic representation of a long circulating liposome
1.3.2. Targeting liposomes to tumor tissue
Both conventional and stealth liposomes are able to accumulate in tumor
tissue due to the enhanced permeation and retention effect (EPR). Leaky tumor
vasculature in combination with an impaired lymphatic drainage, allows
macromolecules ranging from 10 to 500nm to infiltrate solid tumor tissue (Figure
4) (Jain, 1987; Matsumura and Maeda, 1986). This passive targeting results in

Introduction
19
several-fold increased drug concentrations in solid tumors relative to those
obtained with non-macromolecular-complexed free drugs (Northfelt et al., 1996).
Figure 4: Schematic overview of the enhanced permeation and retention effect (EPR). Vascular endothelial cells are depicted in light blue, macromolecules in dark blue.
In attempts to further increase the bioavailability of liposomal drugs at the
target site, recent efforts in the liposome field have focused on the active
targeting of liposomes to specific tissues by coupling ligands to their surface.
Targeting moieties can essentially be any molecule that selectively recognizes
and binds to target antigens or receptors over-expressed or selectively expressed
on cancer cells and include antibody molecules, or fragments thereof, peptides,
carbohydrates, glycoproteins or receptor ligands. To date antibodies or antibody
fragments that bind tumor associated antigens, folate -that binds to the folate
receptor induced on the surface of actively growing cancer cells- or transferrin –
that binds to the integrin receptor expressed on the surface of endothelial cells of
the neo-vasculature of growing tumors - are the most extensively researched
ligands for targeting liposomes to tumor tissue.
Liposomes can be coated with antibodies or antibody fragments either by
directly attaching the antibody to the liposome phospholipid head group or to the
distal end of the PEG polymer. The latter approach has proven most successful,
due to better accessibility of the antibody towards its target (Maruyama et al.,
1999; Sapra and Allen, 2003). However, the attachment of antibodies directly to
the liposome surface is also a viable method (Maruyama et al., 1995) since
shielding of the bound antibody by the PEG chains decreases the antibody-

Chapter 1
20
mediated clearance and immunogenicity of the liposomes. The coating of
liposomes with antibodies directed against tumor-associated targets consists of a
fine balance between coating with a sufficient amount of antibodies to achieve
target binding and tumor retention on one side and enhanced RES clearance with
an increased number of antibodies per liposome on the other (Maruyama et al.,
1999). An optimal coating ratio of 10–30 antibody molecules per liposome was
shown to give the most efficient delivery of drugs to tumors with limited increase
in RES uptake (Maruyama et al., 1995; Sapra and Allen, 2003).
Immunoliposomes can be targeted to surface molecules expressed either in the
vascular system or in the extravascular system on tumor cell membranes. The
most readily accessible target sites for immunoliposomes are the vascular
endothelial surface of growing tumors and circulating cells related to the immune
system.
1.3.2.1. Coupling mechanisms
In general, covalently linking ligands either to the surface of liposomes or the
distal end of PEG chains is based on three main reaction methods, which are
quite efficient and selective: reaction between activated carboxyl groups and
amino groups, which yields an amide bond; reaction between pyridyldithiols and
thiols, which yields disulphide bonds; and reaction between maleimide
derivatives and thiols, which yields thioether bonds.
Coupling of ligands directly to the surface of liposomes can be achieved via
linker lipids such as N-glutaryl-phosphatidylethanolamine (NGPE), N-(4'-(4"-
maleimidophenyl)butyroyl)dioleoylphosphatidylethanolamine (MPB-DOPE) or N-
(3'-(pyridyldithio)propionoyldioleoylphosphatidylethanolamine (PDP-DOPE) that
are incorporated in the liposome membrane. NGPE is activated by 1-ethyl-3(3-
dimethylaminopropyl)carbodiimide (EDC) and N-hydroxy-sulphosuccinimide
(sulpho-NHS) to interact with an amino group of the ligand to yield an amide
bond. MPB-DOPE and PDP-DOPE can be linked to thiol-containing ligands forming
a thioether bond or disulphide bond, respectively. In addition PDP-DOPE can be
linked to ligands that are activated by succinimidyl-4-(p-
maleimidophenyl)butyrate (SMPB) by means of a thioether bond.
For coupling ligands to the distal end of the PEG chain, several commercially
available PEG derivatives can be used such as maleimide-PEG (Mal-PEG), PDP-

Introduction
21
PEG, hydrazide-PEG (Hz-PEG) and p-nitrophenylcarbonyl-PEG (pNP-PEG). Mal-
PEG and PDP-PEG coupling is identical to MPB-DOPE and PDP-DOPE. Hz-PEG
reacts with oxidized carbohydrates on the ligand to form a hydrazon bond
whereas pNP-PEG binds aminogroups via a carbamate bond.
Non covalent linking of ligands to the liposome surface or distal end of the PEG
chain is also possible via the biotin-avidin coupling method. Avidin that has four
binding sites for biotin, serves as a crosslinker between biotinylated ligand and
biotinylated lipids or biotinylated PEG chains.
1.3.3. Stimuli-sensitive liposomes
Triggered release of liposome encapsulated drugs at the site of the tumor can
enhance tumor specific drug delivery. In conventional liposomes, the release of
encapsulated drug occurs gradually due to leaking of the substance or
degradation of the liposome. To date many stimuli-sensitive liposomes are being
developed that release their substance in one single burst as a result of
destabilization of the liposome membrane caused by a change in the
environment.
1.3.3.1. pH-sensitive liposomes
It is well known that the extracellular environment of solid tumors is acidic
with a pH ranging from 6.5 to 7.2 compared to the pH of the blood and normal
tissue (7.5) (Stubbs et al., 2000; Tannock and Rotin, 1989). However, the most
acidic regions in solid tumors are located far from the tumor vasculature thus
making it difficult for liposomes to reach these acidic regions (Helmlinger et al.,
1997). In addition, it is technically very difficult to design pH responsive
liposomes that are stable in the blood (pH 7.5) and instable at pH 6.5 due to the
small change in acidity. Therefore using the acidic tumor microenvironment for
triggered release has not been very successful. A more viable strategy is the
exploitation of the very acidic environment in endosomes and lysosomes where
the pH is lower than 5.0. When it is desired to deliver the drugs to the
cytoplasm, liposomes can be non-specifically or specifically (receptor mediated)
internalized to endosomes by endocytosis and subsequently delivered to
lysosomes. In the lysosome, both liposomes and encapsulated drug can be

Chapter 1
22
degraded by metabolic enzymes. Therefore it is important that liposomes can
escape the endosome after internalization. Intensive research has focussed on
acid triggered drug delivery using fusogenic liposomes. After cell internalization,
the drop in pH in the endosome triggers a phase transition of the lipid bilayer,
resulting in the fusion of the liposome membrane with the endosomal membrane
(Hafez and Cullis, 2001) (Figure 5).
Figure 5: Destabilization of the endosomal membrane: After being endocytosed by the cell and taken inside the endosome, the liposome containing stimuli (pH)-sensitive components, such as lipids (a) in the membrane can undergo pH-dependent membrane destabilization and initiate the destabilization of the endosomal membrane allowing drug (b) efflux into the cell cytoplasm.
A main strategy has been to stabilize the non-bilayer forming lipid
diacylphosphatidylethanolamine (DOPE) with micellar forming lipids such as PEG-
coated lipids. Mixtures of these lipids can form stable liposomes. After acid
catalyzed cleavage of the PEG chain from the PEG-lipids in the liposomes, the
liposomes become fusogenic and fuse with the endosome membrane leading to
drug release into the cytoplasm of the cell (Gerasimov et al., 1999; Guo and
Szoka, 2001; Ishida et al., 2001). Mildly acidic amphiphiles such as
diacylsuccinylglycerols, cholesterol hemisuccinate and oleic acids can also be
used to destabilize the liposome membrane and promote fusion by protonation at
acidic pH (Chu et al., 1990; Collins et al., 1990; Duzgunes et al., 1985)
1.3.3.2. Thermosensitive liposomes
Since it is possible to induce local hyperthermia (LHT) by external heating via
high frequency waves or internal heating, research has also focussed on
producing heat sensitive liposomes. The idea originated from the understanding
Nature Reviews Drug Discovery 4, 145-160 (2005)

Introduction
23
that liposomes become highly leaky to water soluble contents near the gel-to-
liquid crystalline phase transition temperature of its membrane (Blok et al.,
1975). Yatvin and co-workers introduced this concept in 1978. They used
dipalmitoylphosphatidylcholine (DPPC) liposomes with a phase transition
temperature of 41°C and added small amounts of distearoylphosphatidylcholine
(DSPC) as a co-lipid to adjust its transition temperature to 42°C. They showed
that these liposomes delivered more than four times as much methotrexate to
murine tumors heated to 42°C as to unheated control tumors (Weinstein et al.,
1979; Yatvin et al., 1978). Several other formulations based on this composition
have been designed including sterically stabilized liposomes (Maruyama et al.,
1993; Merlin, 1991). For instance Gaber et al. designed long circulating
thermosensitive liposomes made of a mixture of DPPC/hydrogenated soy
phosphatidylcholine (HSPC)/cholesterol (CHOL)/DSPC-PEG lipids that released
more than 60% of their contents when heated at 42 °C for 0.5 h in vitro (Gaber
et al., 1995). More recently, thermosensitive polymers have been used to
produce heat-sensitive liposomes. These polymers become water insoluble above
critical temperature called the lower critical solution temperature (LCST). At the
molecular level this means that chains of these polymers undergo a coil-to-
globule transition above the LCST (Figure 6). When such thermosensitive
polymers are fixed on liposome membranes, the temperature-dependent change
of their solubility can destabilize liposomes resulting in the release of their
content (Kono, 2001). Among thermosensitive polymers, poly(N-
isopropylacrylamide) (poly(NIPAM)) is most frequently used. This polymer is
known to exhibit a drastic change in water-solubility in a very narrow
temperature region near 32°C (Schild, 1992). In addition, its LCST can be
adjusted to a desired temperature by copolymerization with co-monomers with
varying hydrophilicity or hydrophobicity. In general, incorporation of hydrophobic
co-monomers decreases the LCST whereas hydrophilic co-monomers lead to an
increase in LCST (Feil et al., 1993). Han et al. were able to produce
thermosensitive liposomes by modification of the surface of
DPPC/HSPC/CHOL/DSPC-PEG liposomes with polyNIPAM/acrylamide copolymers.
By varying the acrylamide content they obtained copolymers with LCST’s ranging
from 33°C to 47°C. These copolymer modified liposomes showed increased burst
release of encapsulated doxorubicin around the LCST of the
polyNIPAM/acrylamide copolymer (Han et al., 2006).

Chapter 1
24
The use of thermosensitive liposomes has a significant advantage over other
triggering concepts because hyperthermia increases the tumor blood flow and
microvascular permeability (Kong and Dewhirst, 1999; Song et al., 1984). This
results in higher liposome tumor accumulation. Unfortunately, thermosensitive
liposomes can not be used to treat metastatic tumors since the location of the
tumor must be known and the tumor site must be accessible to local
hyperthermia.
Figure 6: Effect of temperature on the solubility of thermosensitive polymers grafted on the liposomal surface
1.3.3.3. photosensitive liposomes
Liposomes can be made photosensitive by using lipids that undergo various
transformations such as isomerisation, fragmentation or polymerization upon
photoexcitation, hereby destabilizing the liposomal membrane.
Firstly, photoisomerizable lipids were exploited by Bisby et al. to allow UV-
triggered release from liposomes (Bisby et al., 2000a). For this purpose they
used the lipid azobenzene-glycero-phosphocholine. Trans-cis isomerisation upon
UV irradiation resulted in destabilization of the liposome membrane and fast
release of encapsulated doxorubicin (Figure 7). However, the use of UV-light is
not very suitable for biological applications due to the potential damage to
healthy tissue. Bisby and coworkers also discovered that incorporation of
cholesterol (up to 25 mol%) in the liposomal membrane induces fast release of
encapsulated drug upon illumination at 470 nm (Bisby et al., 2000b). These
cholesterol containing liposomes therefore have greater therapeutic potential.
Adv Drug Deliv Rev 53 (2001) 307– 319

Introduction
25
Figure 7: Trans-cis isomerization upon UV irradiation of Bis-Azo PC
Secondly, sensitized photo-oxidation has proven to be another viable method
for light triggered release. Thompson et al. used plasmenylcholine that is cleaved
into single chain surfactants via sensitized photo-oxidation of the plasmalogen
vinyl ether linkage (Thompson et al., 1996) (Figure 8). This results in
destabilization of the liposome membrane and an increased permeability.
Zinc phthalocyanine, tin octabutoxy phthalocyanine and bacteriochlorophyll-a
were investigated as sensitizing agents. Bacteriochlorophyll-a produced the
fastest release and absorbs at a wavelength of 820 nm which allows tissue
penetration of ≥ 0,8 cm.
Biochem Biophys Res Commun 276(1), 169–173 (2000)

Chapter 1
26
Figure 8: Singlet oxygen-mediated photo-oxidation of plasmalogen vinyl ether linkage.
Finally, drug release from liposomes by photopolymerization was addressed by
Bondurant et al. who reported a PEG-liposome formulation containing 1,2-bis[10-
(2′,4′-hexadienoyloxy)-decanoyl]-sn-glycero-3-phosphocholine (bis-SorbPC), a
photosensitive lipid that forms a cross-linked lipid network upon exposure to UV-
light (Bondurant et al., 2001). Leaking of encapsulated drugs occurs during the
polymerization process due to the formation of defects in the bilayer (Spratt et
al., 2003) (Figure 9). Again, the use of UV-light is not very suitable for biological
applications. The incorporation of a cyanine dye into the PEG-liposomes made
them also sensitive to visible light, thus increasing the therapeutic potential
(Mueller et al., 2000).
Biochim Biophys Acta 1279 25-34(1996)

Introduction
27
Figure 9: Photopolymerization triggered release via Bis-SorPC. The photopolymerization-induced reduction in the surface area of the polymerizable domains during UV irradiation is shown on the right.
1.3.4. Conclusion
Since the 1970s, when it was first suggested that liposomes could be used as
drug carriers in the treatment of cancer, a significant amount of research has
been performed to optimize and utilize the liposomal carriers successfully in the
treatment of various diseases. Cancer has especially been a disease where
considerable efforts have been made to use liposomal drug delivery systems to
gain increased efficacy and limited toxicity of various chemotherapeutics. A few
drug carriers have appeared on the market, however, the clinical success with
respect to efficacy in cancer therapy, when compared with the free drug, has
been limited even though improved toxicity profiles are found and many
promising preclinical experiments have been reported. Although targeting
liposomal formulations to tumor tissue increases the accumulation in tumor
tissue, this thus not necessarily result in improvement of therapeutic efficacy
(Andresen et al., 2005). This is mainly caused by the obstruction of liposome
extravasation after the first liposomes have bound to target cells lining the blood
vessels. Another obstacle relates to the fact that immunoliposomes in general
Biochim Biophys Acta 1611, 35–43 (2003)

Chapter 1
28
show enhanced liposome clearance (Andresen et al., 2005). Furthermore, it is
important to notice that doxorubicin is often the drug of choice when developing
active release strategies. This drug is able to diffuse over an intact liposome
membrane, caused by the destruction of the pH gradient used to load the
liposomes with doxorubicin. Therefore it is not the best drug to prove an active
release concept.
It is clear that the use of liposomal formulations in cancer therapy holds great
promise but many hurdles still have to be overcome before resulting in clinical
success.
1.4 Polymer vesicles
Nowadays, many different delivery vehicles are being investigated but the best
known examples are lipid vesicles or liposomes that are made of closed lipid
bilayers. Although liposomes were originally used to study biological membranes,
they were introduced in the 1970’s as drug delivery vehicles. Owing to their low
molecular weight (MW < 1 kDa), aggregation of lipids results in molecularly thin
membranes that posses a dynamical, physical softness. As a consequence, many
lipid vesicle properties such as encapsulant retention, membrane stability, and
degradation are not particularly well controlled. In order to obtain more robust
membranes with controllable properties, extensive efforts were made within the
last decade to design polymeric vesicles. This has led to a wide range of
container systems made of diblock copolymers, triblock copolymers or highly
branched polymeric dendrimers. Block copolymers have a similar basic structure
as lipids but consist of distinct polymer chains covalently linked in a series of two
or more segments. Amphiphilic block copolymers are composed of at least one
hydrophilic block and one hydrophobic block, causing self-assembly in aqueous
solutions to nanometer-sized suprastructures. In the absence of solvent, block
copolymers can assume a wide variety of ordered morphologies such as the
lamellar phase. The transition from an isotropically disordered state to an
ordered state such as the lamellar phase is controlled by three parameters: (i)
the molecular weight of the copolymer, (ii) the mass or volume fraction ƒ of each
block and (iii) the effective interaction energy ε between monomers in the blocks
(Discher and Eisenberg, 2002). Upon addition of solvent, the lamellar phase can

Introduction
29
swell and rearrange to form rod-like or spherical structures. Typical bilayer
particles are formed, consisting of a core comprised of their insoluble part
surrounded by a corona of their soluble part (Nardin et al., 2001). The driving
forces for the self-organization are the difference in solubility of the blocks and
the constraint imposed by the chemical linkage between the blocks. Depending
on their concentration, molecular shape, hydrophobic-to-hydrophilic balance and
block-length, micelles, vesicles, cylinders or rod-like structures are formed
(Stoenescu et al., 2004). Compared to the self-assembled structures formed by
lower molecular weight amphiphilic molecules such as lipids or surfactants, block
copolymers self-assemble into significantly more stable particles. This higher
stability is due to the larger size of the hydrophobic part and the slower
dynamics of the underlying copolymer molecules caused by a higher
entanglement (Battaglia and Ryan, 2005; Meier, 2000). It is this increased
stability, along with their self-assembled nanometer-sized structures that make
block copolymers so attractive for biomedical applications such as drug delivery
(Nardin et al., 2004).
Numerous polymers have already been used for the hydrophobic block and
include inert PEE (polyethylethylene), PS (polystyrene), PDMS
(polydimethylsiloxane), PBD (polybutadiene) and the degradable PLA (polylactic
acid) and PCL (polycaprolactone). Hydrophilic blocks have been synthesized from
PEG, the negatively charged PAA (polyacrylic acid), crosslinkable PMOXA
(polymethyloxazoline) and the most common PEO (polyethylene oxide) (Discher,
2007).

Chapter 1
30
1.5 References
Abe, T., Koike, K., Ohga, T., Kubo, T., Wada, M., Kohno, K., Mori, T., Hidaka, K. and Kuwano, M. (1995) Chemosensitisation of spontaneous multidrug resistance by a 1,4-dihydropyridine analogue and verapamil in human glioma cell lines overexpressing MRP or MDR1. Br J Cancer, 72, 418-423.
Albanell, J., Codony, J., Rovira, A., Mellado, B. and Gascon, P. (2003) Mechanism of action of anti-HER2 monoclonal antibodies: scientific update on trastuzumab and 2C4. Adv Exp Med Biol, 532, 253-268.
Alderden, R., Hall, M. and Hambley, T. (2006) The Discovery and Development of Cisplatin. journal of chemical education, 83, 673-816.
Allen, T.M. and Hansen, C. (1991) Pharmacokinetics of stealth versus conventional liposomes: effect of dose. Biochim Biophys Acta, 1068, 133-141.
Anderson, L.M., Krotz, S., Weitzman, S.A. and Thimmapaya, B. (2000) Breast cancer-specific expression of the Candida albicans cytosine deaminase gene using a transcriptional targeting approach. Cancer Gene Ther, 7, 845-852.
Andresen, T.L., Jensen, S.S. and Jorgensen, K. (2005) Advanced strategies in liposomal cancer therapy: problems and prospects of active and tumor specific drug release. Prog Lipid Res, 44, 68-97.
Andriani, F., Nan, B., Yu, J., Li, X., Weigel, N.L., McPhaul, M.J., Kasper, S., Kagawa, S., Fang, B., Matusik, R.J., Denner, L. and Marcelli, M. (2001) Use of the probasin promoter ARR2PB to express Bax in androgen receptor-positive prostate cancer cells. J Natl Cancer Inst, 93, 1314-1324.
Arnold, G.S., Sasser, A.K., Stachler, M.D. and Bartlett, J.S. (2006) Metabolic biotinylation provides a unique platform for the purification and targeting of multiple AAV vector serotypes. Mol Ther, 14, 97-106.
Bagshawe, K.D., Springer, C.J., Searle, F., Antoniw, P., Sharma, S.K., Melton, R.G. and Sherwood, R.F. (1988) A cytotoxic agent can be generated selectively at cancer sites. Br J Cancer, 58, 700-703.
Barker, S.D., Coolidge, C.J., Kanerva, A., Hakkarainen, T., Yamamoto, M., Liu, B., Rivera, A.A., Bhoola, S.M., Barnes, M.N., Alvarez, R.D., Curiel, D.T. and Hemminki, A. (2003) The secretory leukoprotease inhibitor (SLPI) promoter for ovarian cancer gene therapy. J Gene Med, 5, 300-310.
Bartlett, J.S., Kleinschmidt, J., Boucher, R.C. and Samulski, R.J. (1999) Targeted adeno-associated virus vector transduction of nonpermissive cells mediated by a bispecific F(ab'gamma)2 antibody. Nat Biotechnol, 17, 181-186.
Battaglia, G. and Ryan, A.J. (2005) Bilayers and interdigitation in block copolymer vesicles. J Am Chem Soc., 127, 8757-8764.
Bilsland, A.E., Anderson, C.J., Fletcher-Monaghan, A.J., McGregor, F., Evans, T.R., Ganly, I., Knox, R.J., Plumb, J.A. and Keith, W.N. (2003) Selective ablation of human cancer cells by telomerase-specific adenoviral suicide gene therapy vectors expressing bacterial nitroreductase. Oncogene, 22, 370-380.
Bisby, R.H., Mead, C. and Morgan, C.G. (2000a) Active uptake of drugs into photosensitive liposomes and rapid release on UV photolysis. Photochem Photobiol, 72, 57-61.
Bisby, R.H., Mead, C. and Morgan, C.G. (2000b) Wavelength-programmed solute release from photosensitive liposomes. Biochem Biophys Res Commun, 276, 169-173.
Blok, M.C., van der Neut-Kok, E.C., van Deenen, L.L. and de Gier, J. (1975) The effect of chain length and lipid phase transitions on the selective permeability properties of liposomes. Biochim Biophys Acta, 406, 187-196.
Blume, G. and Cevc, G. (1993) Molecular mechanism of the lipid vesicle longevity in vivo. Biochim Biophys Acta, 1146, 157-168.
Bondurant, B., Mueller, A. and O'Brian, D. (2001) Photoinitiated destabilization of sterically stabilized liposomes. Biochimica et Biophysica acta, 1511, 113-122.

Introduction
31
Bosslet, K., Czech, J., Lorenz, P., Sedlacek, H.H., Schuermann, M. and Seemann, G. (1992) Molecular and functional characterisation of a fusion protein suited for tumour specific prodrug activation. Br J Cancer, 65, 234-238.
Boussif, O., Lezoualc'h, F., Zanta, M.A., Mergny, M.D., Scherman, D., Demeneix, B. and Behr, J.P. (1995) A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A, 92, 7297-7301.
Bremner, K.H., Seymour, L.W., Logan, A. and Read, M.L. (2004) Factors influencing the ability of nuclear localization sequence peptides to enhance nonviral gene delivery. Bioconjug Chem, 15, 152-161.
Canetta, R., Rozencweig, M. and Carter, S.K. (1985) Carboplatin: the clinical spectrum to date. Cancer Treat Rev, 12 Suppl A, 125-136.
Cheson, B.D. (1992) New antimetabolites in the treatment of human malignancies. Semin Oncol, 19, 695-706.
Cho, Y.W., Kim, J.D. and Park, K. (2003) Polycation gene delivery systems: escape from endosomes to cytosol. J Pharm Pharmacol, 55, 721-734.
Chowdhury, S., Chester, K.A., Bridgewater, J., Collins, M.K. and Martin, F. (2004) Efficient retroviral vector targeting of carcinoembryonic antigen-positive tumors. Mol Ther, 9, 85-92.
Chu, C.J., Dijkstra, J., Lai, M.Z., Hong, K. and Szoka, F.C. (1990) Efficiency of cytoplasmic delivery by pH-sensitive liposomes to cells in culture. Pharm Res, 7, 824-834.
Collins, D., Litzinger, D.C. and Huang, L. (1990) Structural and functional comparisons of pH-sensitive liposomes composed of phosphatidylethanolamine and three different diacylsuccinylglycerols. Biochim Biophys Acta, 1025, 234-242.
Cortez-Retamozo, V., Backmann, N., Senter, P.D., Wernery, U., De Baetselier, P., Muyldermans, S. and Revets, H. (2004) Efficient cancer therapy with a nanobody-based conjugate. Cancer Res, 64, 2853-2857.
Cronin, J., Zhang, X.Y. and Reiser, J. (2005) Altering the tropism of lentiviral vectors through pseudotyping. Curr Gene Ther, 5, 387-398.
Discher, D.E. (2007) Emerging applications of polymersomes in delivery: From molecular dynamics to shrinkage of tumors. Progress in Polymer Science, 32, 838-857.
Discher, D.E. and Eisenberg, A. (2002) Polymer vesicles. Science., 297, 967-973. Doyle, L.A., Yang, W., Abruzzo, L.V., Krogmann, T., Gao, Y., Rishi, A.K. and Ross, D.D.
(1998) A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A, 95, 15665-15670.
Droogendijk, H.J., Kluin-Nelemans, H.J., van Doormaal, J.J., Oranje, A.P., van de Loosdrecht, A.A. and van Daele, P.L. (2006) Imatinib mesylate in the treatment of systemic mastocytosis: a phase II trial. Cancer, 107, 345-351.
Duzgunes, N., Straubinger, R.M., Baldwin, P.A., Friend, D.S. and Papahadjopoulos, D. (1985) Proton-induced fusion of oleic acid-phosphatidylethanolamine liposomes. Biochemistry, 24, 3091-3098.
Eto, Y., Gao, J.Q., Sekiguchi, F., Kurachi, S., Katayama, K., Maeda, M., Kawasaki, K., Mizuguchi, H., Hayakawa, T., Tsutsumi, Y., Mayumi, T. and Nakagawa, S. (2005) PEGylated adenovirus vectors containing RGD peptides on the tip of PEG show high transduction efficiency and antibody evasion ability. J Gene Med, 7, 604-612.
Fabre, J.W. and Collins, L. (2006) Synthetic peptides as non-viral DNA vectors. Curr Gene Ther, 6, 459-480.
Feil, H., Bae, Y., Feijen, J. and Kim, S. (1993) Effect of Comonomer Hydrophilicity and Ionization on the Lower Critical Solution Temperature of N-isopropylacrylamide Copolymers. Macromolecules, 26, 2496-2500.
Florent, J.C., Dong, X., Gaudel, G., Mitaku, S., Monneret, C., Gesson, J.P., Jacquesy, J.C., Mondon, M., Renoux, B., Andrianomenjanahary, S., Michel, S., Koch, M., Tillequin, F., Gerken, M., Czech, J., Straub, R. and Bosslet, K. (1998) Prodrugs of anthracyclines for use in antibody-directed enzyme prodrug therapy. J Med Chem, 41, 3572-3581.

Chapter 1
32
Fonseca, M.J., Storm, G., Hennink, W.E., Gerritsen, W.R. and Haisma, H.J. (1999) Cationic polymeric gene delivery of beta-glucuronidase for doxorubicin prodrug therapy. J Gene Med, 1, 407-414.
Funhoff, A.M., van Nostrum, C.F., Lok, M.C., Kruijtzer, J.A., Crommelin, D.J. and Hennink, W.E. (2005) Cationic polymethacrylates with covalently linked membrane destabilizing peptides as gene delivery vectors. J Control Release, 101, 233-246.
Gaber, M.H., Hong, K., Huang, S.K. and Papahadjopoulos, D. (1995) Thermosensitive sterically stabilized liposomes: formulation and in vitro studies on mechanism of doxorubicin release by bovine serum and human plasma. Pharm Res, 12, 1407-1416.
Gabizon, A.A. (2001) Pegylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest, 19, 424-436.
Gerasimov, O.V., Boomer, J.A., Qualls, M.M. and Thompson, D.H. (1999) Cytosolic drug delivery using pH- and light-sensitive liposomes. Adv Drug Deliv Rev, 38, 317-338.
Gollan, T.J. and Green, M.R. (2002) Redirecting retroviral tropism by insertion of short, nondisruptive peptide ligands into envelope. J Virol, 76, 3558-3563.
Gotoh, A., Ko, S.C., Shirakawa, T., Cheon, J., Kao, C., Miyamoto, T., Gardner, T.A., Ho, L.J., Cleutjens, C.B., Trapman, J., Graham, F.L. and Chung, L.W. (1998) Development of prostate-specific antigen promoter-based gene therapy for androgen-independent human prostate cancer. J Urol, 160, 220-229.
Graham, J., Mushin, M. and Kirkpatrick, P. (2004) Oxaliplatin. Nat Rev Drug Discov, 3, 11-12.
Gregoriadis, G., Wills, E.J., Swain, C.P. and Tavill, A.S. (1974) Drug-carrier potential of liposomes in cancer chemotherapy. Lancet, 1, 1313-1316.
Gu, J., Kagawa, S., Takakura, M., Kyo, S., Inoue, M., Roth, J.A. and Fang, B. (2000) Tumor-specific transgene expression from the human telomerase reverse transcriptase promoter enables targeting of the therapeutic effects of the Bax gene to cancers. Cancer Res, 60, 5359-5364.
Guo, X. and Szoka, F.C., Jr. (2001) Steric stabilization of fusogenic liposomes by a low-pH sensitive PEG--diortho ester--lipid conjugate. Bioconjug Chem, 12, 291-300.
Hafez, I.M. and Cullis, P.R. (2001) Roles of lipid polymorphism in intracellular delivery. Adv Drug Deliv Rev, 47, 139-148.
Haisma, H.J., Sernee, M.F., Hooijberg, E., Brakenhoff, R.H., vd Meulen-Muileman, I.H., Pinedo, H.M. and Boven, E. (1998) Construction and characterization of a fusion protein of single-chain anti-CD20 antibody and human beta-glucuronidase for antibody-directed enzyme prodrug therapy. Blood, 92, 184-190.
Han, H.D., Shin, B.C. and Choi, H.S. (2006) Doxorubicin-encapsulated thermosensitive liposomes modified with poly(N-isopropylacrylamide-co-acrylamide): drug release behavior and stability in the presence of serum. Eur J Pharm Biopharm, 62, 110-116.
Hatakeyama, H., Akita, H., Kogure, K., Oishi, M., Nagasaki, Y., Kihira, Y., Ueno, M., Kobayashi, H., Kikuchi, H. and Harashima, H. (2007) Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther, 14, 68-77.
Hedley, S.J., Auf der Maur, A., Hohn, S., Escher, D., Barberis, A., Glasgow, J.N., Douglas, J.T., Korokhov, N. and Curiel, D.T. (2006) An adenovirus vector with a chimeric fiber incorporating stabilized single chain antibody achieves targeted gene delivery. Gene Ther, 13, 88-94.
Helmlinger, G., Yuan, F., Dellian, M. and Jain, R.K. (1997) Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med, 3, 177-182.
Huang, Q., Zhang, X., Wang, H., Yan, B., Kirkpatrick, J., Dewhrist, M.W. and Li, C.Y. (2004) A novel conditionally replicative adenovirus vector targeting telomerase-positive tumor cells. Clin Cancer Res, 10, 1439-1445.

Introduction
33
Huch, M., Abate-Daga, D., Roig, J.M., Gonzalez, J.R., Fabregat, J., Sosnowski, B., Mazo, A. and Fillat, C. (2006) Targeting the CYP2B 1/cyclophosphamide suicide system to fibroblast growth factor receptors results in a potent antitumoral response in pancreatic cancer models. Hum Gene Ther, 17, 1187-1200.
Ishida, T., Harashima, H. and Kiwada, H. (2002) Liposome clearance. Biosci Rep, 22, 197-224.
Ishida, T., Kirchmeier, M.J., Moase, E.H., Zalipsky, S. and Allen, T.M. (2001) Targeted delivery and triggered release of liposomal doxorubicin enhances cytotoxicity against human B lymphoma cells. Biochim Biophys Acta, 1515, 144-158.
Iwai, M., Harada, Y., Tanaka, S., Muramatsu, A., Mori, T., Kashima, K., Imanishi, J. and Mazda, O. (2002) Polyethylenimine-mediated suicide gene transfer induces a therapeutic effect for hepatocellular carcinoma in vivo by using an Epstein-Barr virus-based plasmid vector. Biochem Biophys Res Commun, 291, 48-54.
Jain, R.K. (1987) Transport of molecules in the tumor interstitium: a review. Cancer Res, 47, 3039-3051.
Jain, R.K. (1990) Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res, 50, 814s-819s.
Juliano, R.L. and Ling, V. (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta, 455, 152-162.
Kakinuma, H., Fujii, I. and Nishi, Y. (2002) Selective chemotherapeutic strategies using catalytic antibodies: a common pro-moiety for antibody-directed abzyme prodrug therapy. Journal of immunological methods, 269, 269-281.
Kamiya, H., Ochiai, H., Harashima, H., Ito, M. and Matsuda, A. (2006) Transient expression of Drosophila melanogaster deoxynucleoside kinase gene enhances cytotoxicity of nucleoside analogs. Nucleosides Nucleotides Nucleic Acids, 25, 553-560.
Kaneko, S., Hallenbeck, P., Kotani, T., Nakabayashi, H., McGarrity, G., Tamaoki, T., Anderson, W.F. and Chiang, Y.L. (1995) Adenovirus-mediated gene therapy of hepatocellular carcinoma using cancer-specific gene expression. Cancer Res, 55, 5283-5287.
Karmali, P.P. and Chaudhuri, A. (2007) Cationic liposomes as non-viral carriers of gene medicines: resolved issues, open questions, and future promises. Med Res Rev, 27, 696-722.
Kerr, D.E., Senter, P.D., Burnett, W.V., Hirschberg, D.L., Hellstrom, I. and Hellstrom, K.E. (1990) Antibody-penicillin-V-amidase conjugates kill antigen-positive tumor cells when combined with doxorubicin phenoxyacetamide. Cancer Immunol Immunother, 31, 202-206.
Kichler, A. (2004) Gene transfer with modified polyethylenimines. J Gene Med, 6 Suppl 1, S3-10.
Kircheis, R., Wightman, L., Schreiber, A., Robitza, B., Rossler, V., Kursa, M. and Wagner, E. (2001) Polyethylenimine/DNA complexes shielded by transferrin target gene expression to tumors after systemic application. Gene Ther, 8, 28-40.
Klibanov, A.L., Maruyama, K., Torchilin, V.P. and Huang, L. (1990) Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett., 268, 235-237.
Knorr, V., Allmendinger, L., Walker, G.F., Paintner, F.F. and Wagner, E. (2007) An acetal-based PEGylation reagent for pH-sensitive shielding of DNA polyplexes. Bioconjug Chem, 18, 1218-1225.
Kong, G. and Dewhirst, M.W. (1999) Hyperthermia and liposomes. Int J Hyperthermia, 15, 345-370.
Kono, K. (2001) Thermosensitive polymer-modified liposomes. Adv Drug Deliv Rev, 53, 307-319.
Kursa, M., Walker, G.F., Roessler, V., Ogris, M., Roedl, W., Kircheis, R. and Wagner, E. (2003) Novel shielded transferrin-polyethylene glycol-polyethylenimine/DNA complexes for systemic tumor-targeted gene transfer. Bioconjug Chem, 14, 222-231.

Chapter 1
34
Li, Y., Yu, D.C., Chen, Y., Amin, P., Zhang, H., Nguyen, N. and Henderson, D.R. (2001) A hepatocellular carcinoma-specific adenovirus variant, CV890, eliminates distant human liver tumors in combination with doxorubicin. Cancer Res, 61, 6428-6436.
Liu, D., Ren, T. and Gao, X. (2003) Cationic transfection lipids. Curr Med Chem, 10, 1307-1315.
LoBuglio, A.F., Wheeler, R.H., Trang, J., Haynes, A., Rogers, K., Harvey, E.B., Sun, L., Ghrayeb, J. and Khazaeli, M.B. (1989) Mouse/human chimeric monoclonal antibody in man: kinetics and immune response. Proc Natl Acad Sci U S A, 86, 4220-4224.
Lown, J.W. (1993) Anthracycline and anthraquinone anticancer agents: current status and recent developments. Pharmacol Ther, 60, 185-214.
Lu, Y. and Steiner, M.S. (2000) Transcriptionally regulated adenoviruses for prostate-specific gene therapy. World J Urol, 18, 93-101.
Lungwitz, U., Breunig, M., Blunk, T. and Gopferich, A. (2005) Polyethylenimine-based non-viral gene delivery systems. Eur J Pharm Biopharm, 60, 247-266.
Manome, Y., Wen, P.Y., Dong, Y., Tanaka, T., Mitchell, B.S., Kufe, D.W. and Fine, H.A. (1996) Viral vector transduction of the human deoxycytidine kinase cDNA sensitizes glioma cells to the cytotoxic effects of cytosine arabinoside in vitro and in vivo. Nat Med, 2, 567-573.
Maruyama, K., Ishida, O., Takizawa, T. and Moribe, K. (1999) Possibility of active targeting to tumor tissues with liposomes. Adv Drug Deliv Rev, 40, 89-102.
Maruyama, K., Takizawa, T., Yuda, T., Kennel, S.J., Huang, L. and Iwatsuru, M. (1995) Targetability of novel immunoliposomes modified with amphipathic poly(ethylene glycol)s conjugated at their distal terminals to monoclonal antibodies. Biochim Biophys Acta, 1234, 74-80.
Maruyama, K., Unezaki, S., Takahashi, N. and Iwatsuru, M. (1993) Enhanced delivery of doxorubicin to tumor by long-circulating thermosensitive liposomes and local hyperthermia. Biochim Biophys Acta, 1149, 209-216.
Mathis, J.M., Williams, B.J., Sibley, D.A., Carroll, J.L., Li, J., Odaka, Y., Barlow, S., Nathan, C.O., Li, B.D. and DeBenedetti, A. (2006) Cancer-specific targeting of an adenovirus-delivered herpes simplex virus thymidine kinase suicide gene using translational control. J Gene Med, 8, 1105-1120.
Matsumura, Y. and Maeda, H. (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res, 46, 6387-6392.
McCart, J.A., Wang, Z.H., Xu, H., Hu, Y., Park, B., Alexander, H.R. and Bartlett, D.L. (2002) Development of a melanoma-specific adenovirus. Mol Ther, 6, 471-480.
Meier, W. (2000) Polymer nanocapsules. Chem. Soc. Rev., 29, 295-303. Merlin, J.L. (1991) In vitro evaluation of the association of thermosensitive liposome-
encapsulated doxorubicin with hyperthermia. Eur J Cancer, 27, 1031-1034. Meyer, D.L., Jungheim, L.N., Law, K.L., Mikolajczyk, S.D., Shepherd, T.A., Mackensen,
D.G., Briggs, S.L. and Starling, J.J. (1993) Site-specific prodrug activation by antibody-beta-lactamase conjugates: regression and long-term growth inhibition of human colon carcinoma xenograft models. Cancer Res, 53, 3956-3963.
Morimoto, E., Inase, N., Mlyake, S. and Yoshizawa, Y. (2001) Adenovirus-mediated suicide gene transfer to small cell lung carcinoma using a tumor-specific promoter. Anticancer Res, 21, 329-331.
Mueller, A., Bondurant, B. and O'Brien, D.F. (2000) Visible-Light-Stimulated Destabilization of PEG-liposomes. Macromolecules, 33, 4799-4804.
Nardin, C., Bolikal, D. and Kohn, J. (2004) Nontoxic block copolymer nanospheres: design and characterization. Langmuir., 20, 11721-11725.
Nardin, C., Widmer, J., Winterhalter, M. and Meier, W. (2001) Amphiphilic block copolymer nanocontainers as bioreactors. Eur. Phys. J. E, 4, 403-410.
Nettelbeck, D.M., Rivera, A.A., Balague, C., Alemany, R. and Curiel, D.T. (2002) Novel oncolytic adenoviruses targeted to melanoma: specific viral replication and cytolysis by expression of E1A mutants from the tyrosinase enhancer/promoter. Cancer Res, 62, 4663-4670.

Introduction
35
Northfelt, D.W., Martin, F.J., Working, P., Volberding, P.A., Russell, J., Newman, M., Amantea, M.A. and Kaplan, L.D. (1996) Doxorubicin encapsulated in liposomes containing surface-bound polyethylene glycol: pharmacokinetics, tumor localization, and safety in patients with AIDS-related Kaposi's sarcoma. J Clin Pharmacol, 36, 55-63.
Peng, X.Y., Won, J.H., Rutherford, T., Fujii, T., Zelterman, D., Pizzorno, G., Sapi, E., Leavitt, J., Kacinski, B., Crystal, R., Schwartz, P. and Deisseroth, A. (2001) The use of the L-plastin promoter for adenoviral-mediated, tumor-specific gene expression in ovarian and bladder cancer cell lines. Cancer Res, 61, 4405-4413.
Pereboev, A.V., Nagle, J.M., Shakhmatov, M.A., Triozzi, P.L., Matthews, Q.L., Kawakami, Y., Curiel, D.T. and Blackwell, J.L. (2004) Enhanced gene transfer to mouse dendritic cells using adenoviral vectors coated with a novel adapter molecule. Mol Ther, 9, 712-720.
Pereboeva, L., Komarova, S., Mahasreshti, P.J. and Curiel, D.T. (2004) Fiber-mosaic adenovirus as a novel approach to design genetically modified adenoviral vectors. Virus Res, 105, 35-46.
Plumb, J.A., Bilsland, A., Kakani, R., Zhao, J., Glasspool, R.M., Knox, R.J., Evans, T.R. and Keith, W.N. (2001) Telomerase-specific suicide gene therapy vectors expressing bacterial nitroreductase sensitize human cancer cells to the pro-drug CB1954. Oncogene, 20, 7797-7803.
Reichert, J.M., Rosensweig, C.J., Faden, L.B. and Dewitz, M.C. (2005) Monoclonal antibody successes in the clinic. Nat Biotechnol, 23, 1073-1078.
Sapra, P. and Allen, T.M. (2003) Ligand-targeted liposomal anticancer drugs. Prog Lipid Res, 42, 439-462.
Schepelmann, S., Ogilvie, L.M., Hedley, D., Friedlos, F., Martin, J., Scanlon, I., Chen, P., Marais, R. and Springer, C.J. (2007) Suicide gene therapy of human colon carcinoma xenografts using an armed oncolytic adenovirus expressing carboxypeptidase G2. Cancer Res, 67, 4949-4955.
Schild, H. (1992) Poly(N-isopropylacrylamide): experiment, theory and application. Progress in Polymer Science, 17, 163-249.
Senter, P.D., Schreiber, G.J., Hirschberg, D.L., Ashe, S.A., Hellstrom, K.E. and Hellstrom, I. (1989) Enhancement of the in vitro and in vivo antitumor activities of phosphorylated mitomycin C and etoposide derivatives by monoclonal antibody-alkaline phosphatase conjugates. Cancer Res, 49, 5789-5792.
Sharma, S.K., Bagshawe, K.D., Burke, P.J., Boden, J.A., Rogers, G.T., Springer, C.J., Melton, R.G. and Sherwood, R.F. (1994) Galactosylated antibodies and antibody-enzyme conjugates in antibody-directed enzyme prodrug therapy. Cancer, 73, 1114-1120.
Shimura, H., Suzuki, H., Miyazaki, A., Furuya, F., Ohta, K., Haraguchi, K., Endo, T. and Onaya, T. (2001) Transcriptional activation of the thyroglobulin promoter directing suicide gene expression by thyroid transcription factor-1 in thyroid cancer cells. Cancer Res, 61, 3640-3646.
Siders, W.M., Halloran, P.J. and Fenton, R.G. (1996) Transcriptional targeting of recombinant adenoviruses to human and murine melanoma cells. Cancer Res, 56, 5638-5646.
Smith, G.K., Banks, S., Blumenkopf, T.A., Cory, M., Humphreys, J., Laethem, R.M., Miller, J., Moxham, C.P., Mullin, R., Ray, P.H., Walton, L.M. and Wolfe, L.A., 3rd. (1997) Toward antibody-directed enzyme prodrug therapy with the T268G mutant of human carboxypeptidase A1 and novel in vivo stable prodrugs of methotrexate. J Biol Chem, 272, 15804-15816.
Snitkovsky, S. and Young, J.A. (2002) Targeting retroviral vector infection to cells that express heregulin receptors using a TVA-heregulin bridge protein. Virology, 292, 150-155.
Song, C.W., Lokshina, A., Rhee, J.G., Patten, M. and Levitt, S.H. (1984) Implication of blood flow in hyperthermic treatment of tumors. IEEE Trans Biomed Eng, 31, 9-16.

Chapter 1
36
Spratt, T., Bondurant, B. and O'Brien, D.F. (2003) Rapid release of liposomal contents upon photoinitiated destabilization with UV exposure. Biochim Biophys Acta, 1611, 35-43.
Springer, C.J., Bagshawe, K.D., Sharma, S.K., Searle, F., Boden, J.A., Antoniw, P., Burke, P.J., Rogers, G.T., Sherwood, R.F. and Melton, R.G. (1991) Ablation of human choriocarcinoma xenografts in nude mice by antibody-directed enzyme prodrug therapy (ADEPT) with three novel compounds. Eur J Cancer, 27, 1361-1366.
Stachler, M.D. and Bartlett, J.S. (2006) Mosaic vectors comprised of modified AAV1 capsid proteins for efficient vector purification and targeting to vascular endothelial cells. Gene Ther, 13, 926-931.
Stoenescu, R., Graff, A. and Meier, W. (2004) Asymmetric ABC-Triblock Copolymer Membranes Induce a Directed Insertion of Membrane Proteins. Macromol Biosci, 4, 930-935.
Stribbling, S.M., Martin, J., Pedley, R.B., Boden, J.A., Sharma, S.K. and Springer, C.J. (1997) Biodistribution of an antibody-enzyme conjugate for antibody-directed enzyme prodrug therapy in nude mice bearing a human colon adenocarcinoma xenograft. Cancer Chemother Pharmacol, 40, 277-284.
Stubbs, M., McSheehy, P.M., Griffiths, J.R. and Bashford, C.L. (2000) Causes and consequences of tumour acidity and implications for treatment. Mol Med Today, 6, 15-19.
Syrigos, K.N., Rowlinson-Busza, G. and Epenetos, A.A. (1998) In vitro cytotoxicity following specific activation of amygdalin by beta-glucosidase conjugated to a bladder cancer-associated monoclonal antibody. Int J Cancer, 78, 712-719.
Tai, C.K., Logg, C.R., Park, J.M., Anderson, W.F., Press, M.F. and Kasahara, N. (2003) Antibody-mediated targeting of replication-competent retroviral vectors. Hum Gene Ther, 14, 789-802.
Takeuchi, H., Kojima, H., Yamamoto, H. and Kawashima, Y. (2001) Evaluation of circulation profiles of liposomes coated with hydrophilic polymers having different molecular weights in rats. J Control Release, 75, 83-91.
Tannock, I.F. and Rotin, D. (1989) Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res, 49, 4373-4384.
Torchilin, V.P., Levchenko, T.S., Whiteman, K.R., Yaroslavov, A.A., Tsatsakis, A.M., Rizos, A.K., Michailova, E.V. and Shtilman, M.I. (2001) Amphiphilic poly-N-vinylpyrrolidones: synthesis, properties and liposome surface modification. Biomaterials, 22, 3035-3044.
Torchilin, V.P., Omelyanenko, V.G., Papisov, M.I., Bogdanov, A.A., Jr., Trubetskoy, V.S., Herron, J.N. and Gentry, C.A. (1994) Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta, 1195, 11-20.
Uherek, C., Fominaya, J. and Wels, W. (1998) A modular DNA carrier protein based on the structure of diphtheria toxin mediates target cell-specific gene delivery. J Biol Chem, 273, 8835-8841.
Vassaux, G., Hurst, H.C. and Lemoine, N.R. (1999) Insulation of a conditionally expressed transgene in an adenoviral vector. Gene Ther, 6, 1192-1197.
Vaughan, T.J., Osbourn, J.K. and Tempest, P.R. (1998) Human antibodies by design. Nat Biotechnol, 16, 535-539.
Vrudhula, V.M., Senter, P.D., Fischer, K.J. and Wallace, P.M. (1993) Prodrugs of doxorubicin and melphalan and their activation by a monoclonal antibody-penicillin-G amidase conjugate. J Med Chem, 36, 919-923.
Wall, M.E. and Wani, M.C. (1995) Camptothecin and taxol: discovery to clinic--thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Res, 55, 753-760.
Weinstein, J.N., Magin, R.L., Yatvin, M.B. and Zaharko, D.S. (1979) Liposomes and local hyperthermia: selective delivery of methotrexate to heated tumors. Science, 204, 188-191.

Introduction
37
Woodle, M.C., Engbers, C.M. and Zalipsky, S. (1994) New amphipatic polymer-lipid conjugates forming long-circulating reticuloendothelial system-evading liposomes. Bioconjug Chem, 5, 493-496.
Wu, Z., Asokan, A. and Samulski, R.J. (2006) Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther, 14, 316-327.
Wurdinger, T., Verheije, M.H., Raaben, M., Bosch, B.J., de Haan, C.A., van Beusechem, V.W., Rottier, P.J. and Gerritsen, W.R. (2005) Targeting non-human coronaviruses to human cancer cells using a bispecific single-chain antibody. Gene Ther, 12, 1394-1404.
Yamamoto, M., Alemany, R., Adachi, Y., Grizzle, W.E. and Curiel, D.T. (2001) Characterization of the cyclooxygenase-2 promoter in an adenoviral vector and its application for the mitigation of toxicity in suicide gene therapy of gastrointestinal cancers. Mol Ther, 3, 385-394.
Yang, Q., Mamounas, M., Yu, G., Kennedy, S., Leaker, B., Merson, J., Wong-Staal, F., Yu, M. and Barber, J.R. (1998) Development of novel cell surface CD34-targeted recombinant adenoassociated virus vectors for gene therapy. Hum Gene Ther, 9, 1929-1937.
Yatvin, M.B., Weinstein, J.N., Dennis, W.H. and Blumenthal, R. (1978) Design of liposomes for enhanced local release of drugs by hyperthermia. Science, 202, 1290-1293.
Zalipsky, S., Hansen, C.B., Oaks, J.M. and Allen, T.M. (1996) Evaluation of blood clearance rates and biodistribution of poly(2-oxazoline)-grafted liposomes. J Pharm Sci, 85, 133-137.
Zalipsky, S., Qazen, M., Walker, J.A., 2nd, Mullah, N., Quinn, Y.P. and Huang, S.K. (1999) New detachable poly(ethylene glycol) conjugates: cysteine-cleavable lipopolymers regenerating natural phospholipid, diacyl phosphatidylethanolamine. Bioconjug Chem, 10, 703-707.
Zanta, M.A., Belguise-Valladier, P. and Behr, J.P. (1999) Gene delivery: a single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc Natl Acad Sci U S A, 96, 91-96.
Zeng, H., Wei, Q., Huang, R., Chen, N., Dong, Q., Yang, Y. and Zhou, Q. (2007) Recombinant adenovirus mediated prostate-specific enzyme pro-drug gene therapy regulated by prostate-specific membrane antigen (PSMA) enhancer/promoter. J Androl, 28, 827-835.
Zhang, M., Li, S., Nyati, M.K., DeRemer, S., Parsels, J., Rehemtulla, A., Ensminger, W.D. and Lawrence, T.S. (2003) Regional delivery and selective expression of a high-activity yeast cytosine deaminase in an intrahepatic colon cancer model. Cancer Res, 63, 658-663.
Zhang, R., Straus, F.H. and DeGroot, L.J. (2001) Adenoviral-mediated gene therapy for thyroid carcinoma using thymidine kinase controlled by thyroglobulin promoter demonstrates high specificity and low toxicity. Thyroid, 11, 115-123.
Zhou, J.H., Tang, B., Liu, X.L., He, D.W. and Yang, D.T. (2007) hTERT-targeted E. coli purine nucleoside phosphorylase gene/6-methylpurine deoxyribose therapy for pancreatic cancer. Chin Med J (Engl), 120, 1348-1352.
Zhu, Z.B., Makhija, S.K., Lu, B., Wang, M., Kaliberova, L., Liu, B., Rivera, A.A., Nettelbeck, D.M., Mahasreshti, P.J., Leath, C.A., Barker, S., Yamaoto, M., Li, F., Alvarez, R.D. and Curiel, D.T. (2004) Transcriptional targeting of tumors with a novel tumor-specific survivin promoter. Cancer Gene Ther, 11, 256-262.


Chapter 2 State of the art: a nanoreactor coming to life
39
Chapter 2:
State of the art: a nanoreactor
coming to life

Chapter 2
40

State of the art: a nanoreactor coming to life
41
2.1 Aim of the work: a novel directed enzyme-prodrug strategy
Since both antibody-directed enzyme-prodrug therapy (ADEPT) and gene-
directed enzyme-prodrug therapy (GDEPT) (Chapter 1) suffer from several
drawbacks, such as immunogenicity of the antibody-enzyme conjugate,
inefficient transfection of gene delivery vectors, unsustained gene expression,
pathogenesis of viral vectors and the risk of insertional mutagenesis, we want to
introduce a novel enzyme–prodrug system based on triblock copolymeric
vesicles.
Patrick Van Gelder, my co-promotor was working in collaboration with Prof. W.
Meier at the university of Basel where they inserted a bacterial outer membrane
protein maltoporin (LamB) (Ranquin and Van Gelder, 2004) into the triblock
copolymeric membrane of nanometer-sized vesicles composed of poly(2-
methyloxazoline)-β-poly(dimethyl siloxane)- β-poly(2-methyloxazoline) (PMOXA-
PDMS-PMOXA). They showed that maltoporin is still functional after insertion in
the membrane. Moreover, lambda phage is still able to bind to its receptor and
inject its DNA through the porin into the polymeric vesicles (Graff et al., 2002).
This clearly showed that otherwise impermeable polymeric vesicles can be
permeabilized for various solutes by incorporation of bacterial porins. Nardin et
al. also demonstrated that enzymes can be trapped inside these polymeric
vesicles without loss of function by encapsulating β-lactamase and inserting
OmpF in the membrane (Nardin et al., 2000b).
In the lab of Prof. J. Steyaert, my promoter, nucleoside hydrolases from
various origins were extensively researched for several years. These enzymes
catalyse the hydrolysis of nucleosides into the respective nucleobase and ribose
(Versees et al., 2001). They in particular studied the nucleoside hydrolase of
Trypanosoma vivax (TvNH) that is capable of hydrolysing several nucleoside
analogs into the respective nucleobase analog. Since many chemotherapeutics
used to date are nucleobases they saw the potential of TvNH to function as a
prodrug activating enzyme in cancer therapy.
Patrick Van Gelder brought these two visions together and thus the idea was
born to use such PMOXA-PDMS-PMOXA vesicles that are permeabilized by

Chapter 2
42
bacterial porins, to encapsulate prodrug-activating enzymes and use such
nanoreactors as a novel prodrug activating system. This system is likely to have
many advantages over ADEPT since the enzyme is shielded from the
environment and thus protected from proteolytic degradation and binding to
components of the immune system. In addition, each therapeutic unit holds a
high concentration of enzymes as compared to ADEPT where only one enzyme is
present per therapeutic unit.
In this project the two main goals are: validating the TvNH as a prodrug
activating enzyme and ultimately deliver a proof of principle that nanoreactors
that are permeabilized with bacterial porins and encapsulate a prodrug activating
enzyme can function as prodrug activating systems.
In this chapter the individual building blocks of these nanoreactors are
described in more detail.
2.2 PMOXA-PDMS-PMOXA triblock copolymers
Amphiphilic block copolymers are composed of at least one hydrophilic block
and one hydrophobic block, covalently linked. They are similar to low molar-mass
amphiphiles such as surfactants or lipids, and can self-assemble into a plethora
of lyotropic mesophases (Alexandridis et al., 1998; Forster and Plantenberg,
2002). At low concentrations, they self assemble into micelles of various shapes
and at higher concentrations into lyotropic liquid crystalline phases. Typical
bilayer particles are formed upon self-assembly in aqueous solutions, consisting
of a core comprised of their insoluble part surrounded by a corona of their
soluble part (Nardin et al., 2001). The driving forces for the self-organization are
the difference in solubility of the blocks and the constraint imposed by the
chemical linkage between the blocks. Depending on their concentration,
molecular shape, hydrophobic-to-hydrophilic balance and block-length, micelles,
vesicles, cylinders or rod-like structures are formed.
In this work the ABA triblock copolymer poly(2-methyloxazoline)-b-
poly(dimethyl siloxane)-b-poly(2-methyloxazoline) (PMOXA-PDMS-PMOXA) is
used which was synthesized by the group of Wolfgang Meier (Figure 1) or
purchased from Polymer Source Inc.. For a given composition of PMOXA-PDMS-

State of the art: a nanoreactor coming to life
43
PMOXA, a homogeneous liquid crystalline phase is observed in water at
concentrations above 48 wt. %. Below 48 wt. % the system shows a broad
miscibility gap where the lamellar phase coexists with excess water. The phase
behaviour of this triblock copolymer is similar to that of typical bilayer forming
lipids like lecithin and it self assembles into vesicular structures in dilute aqueous
solutions which consist of a spherically closed triblock copolymer membrane as
shown by Nardin et al (Nardin et al., 2000a).. These vesicles have a size range of
50 to 500 nm as shown by cryo-TEM. Furthermore, cryo-TEM images also
revealed a membrane thickness of 10 nm. The mechanical and visco-elastical
properties of this PMOXA-PDMS-PMOXA membrane were characterized by
applying short electric field pulses to giant free-standing membranes (Nardin,
2000). Electrical field pulses are used to charge the membrane resulting in
electric stress. Above a critical voltage the membrane will rupture and a fast
discharge across the rupture occurs. Analysis of the discharge kinetics gives
information about the strength and stability of the membrane. This experiment
indicated that PMOXA-PDMS-PMOXA membranes were considerably more stable
than conventional black lipid membranes and posses a high flexibility provided by
the PDMS middle block. Crosslinking the PMOXA-PDMS-PMOXA polymers via
methacrylate endgroups further increased the stability. The reason for this higher
stability compared to conventional lipid membranes, is the larger size of the
hydrophobic part and the slower dynamics of the underlying copolymer
molecules caused by a higher entanglement (Battaglia and Ryan, 2005). Since
the formation of membrane-like structures of the PMOXA-PDMS-PMOXA triblock
copolymers in aqueous solutions, closely resembles the behaviour of lipid
molecules, the polymer membranes can be regarded as a mimetic of biological
membranes. To test this hypothesis, Nardin et al. successfully reconstituted the
outer membrane protein OmpF in the membrane of PMOXA-PDMS-PMOXA
triblock copolymeric vesicles (Nardin et al., 2001). Although the polymer
membrane (10 nm) is two- to threefold thicker than a conventional lipid
membrane, the functionality of the membrane protein was fully preserved upon
reconstitution in the polymer membrane. This is possible due to the high
flexibility of the hydrophobic block and the conformational freedom of the
polymer molecules, which allows a block copolymer membrane to adapt to the
specific geometric and dynamic requirements of membrane proteins without
considerable loss of free energy (Pata and Dan, 2003). Currently, other

Chapter 2
44
membrane proteins have been reconstituted in the PMOXA-PDMS-PMOXA
membrane, including LamB (Graff et al., 2002), aquaporin (Stoenescu et al.,
2004) and FhuA (Nallani et al., 2006).
Finally, the PMOXA outer blocks posses similar stealth properties as PEG which
makes PMOXA-PDMS-PMOXA based vesicles very interesting for use in
biomedical applications (Woodle et al., 1994).
Figure 1: composition of PMOXA-PDMS-PMOXA polymers. In this representation the polymers were functionalised with methacrylate endgroups for polymer crosslinking via UV irradiation. X en y represent the amount of dimethyl siloxane and methyloxazoline repeats respectively. For the polymers used in this thesis, x and y are 72 and 18 respectively or 54 and 21 respectively.
2.3 Bacterial outer membrane proteins
The outer membrane protects Gram-negative bacteria against a harsh
environment. At the same time, the embedded proteins fulfil a number of tasks
that are crucial to the bacterial cell, such as solute and protein translocation, as
well as signal transduction. Unlike membrane proteins from all other sources,
integral outer membrane proteins do not consist of transmembrane α-helices,
but instead fold into antiparallel β-barrels.
The outer membrane (OM) is an asymmetrical membrane consisting of
phospholipids and mostly lipopolysaccharides (LPS) in the inner and outer
monolayer, respectively. Transport across the outer membrane is mediated by
channel forming proteins. General porins like OmpF, OmpC and PhoE allow
passive diffusion of small hydrophilic solutes with molecular weights up to 600
Da. The flux of solutes through these porins is proportional to the permeability
and the concentration gradient between the periplasmic space and the outside

State of the art: a nanoreactor coming to life
45
medium. If this concentration gradient becomes too small, the flux can only be
maintained by increasing the permeability. One possible option to do this is to
increase the number of channels. However, only small increases in flux can be
gained using this option: the theory of diffusion (Berg, 1993) tells us that only
1% covering of the cell surface with channels is sufficient to reach 50% of the
flux of a cell that is completely covered with channels. Alternatively, higher
fluxes could potentially be obtained by increasing the radius (r) of the channel
with the consequence that the channel surface increases with r2. Unfortunately,
toxins and bile salts would then easily enter the cell and the protective function
of the OM would be lost. Therefore, other porins have evolved that facilitate the
uptake of certain solutes by the presence of a specific binding site. Examples are
LamB, ScrY and Tsx, which are specific for maltodextrins, sucrose and
nucleosides respectively.
In this thesis two aspecific porins, OmpF and PhoE and one specific porin, Tsx
were used.
2.3.1. Aspecific porins OmpF and PhoE
OmpF, the receptor for bacteriophage T2, is one of the general porins and is
abundantly present in the outer membrane. PhoE on the other hand is a
phosphate-limitation induced porin that is only expressed under phosphate
starvation. Both porins allow transport of solutes up to 600 Da. OmpF is a
slightly cation selective porin with a molecular mass of 37 kDa whereas PhoE is
an anion selective porin with a molecular mass of 36.8 kDa. The transport of
solutes through both porins is osmotically regulated and therefore solely
governed by the concentration gradient across the outer membrane. They both
fold into 16 stranded β-barrels and insert in the outer membrane as homotrimers
(Cowan et al., 1992) (Figure 2). Loop L3 plays an important factor in pore
permeability because it folds back into the barrel thus forming a constriction
zone at half the height of the channel, giving it an hourglass-like shape.
Residues at this constriction zone on L3 and the opposing barrel wall contribute
to the ion-selectivity filter by creating a transverse electrostatic field. Site
directed mutagenesis revealed that lysine and arginine residues determine the
anion selectivity of PhoE together with additional charges at the mouth and
inside the barrel (Bauer et al., 1989; Benz et al., 1989). In OmpF, the cation

Chapter 2
46
selectivity is determined by three arginine residues and two aspartate residues at
the constriction zone (Phale et al., 2001). OmpF and PhoE, like all other porins
are extremely stable proteins that can resist denaturation in the presence of 5M
guanidinium chloride or 2% SDS at 70 °C. It was shown by Phale et al. (Phale et
al., 1998) that the latching loop L2 contributes strongly to this extreme stability
because it bends over the wall of an adjacent monomer. Additionally, the trimer
hydrophobic interface adds to the robustness of the trimeric porin (Phale et al.,
1998; Van Gelder et al., 1996).
Figure 2: Bacterial β-barrel porins OmpF and PhoE as seen from the plane of the membrane (OmpF) and from the top of the membrane (PhoE). Loop L3 is bend back into the barrel.
Conductance measurements revealed that both porins can exist in an open or
closed state, depending on the transmembrane voltage (Delcour, 1997). Charged
residues in the channel are most probably responsible for this voltage gating
since replacement of these residues result in a changed voltage sensitivity (Saint
et al., 1996; Van Gelder et al., 1997). Interestingly, OmpF and PhoE show an
opposite voltage dependency. Moreover, the voltage dependency of PhoE can be
changed to match that of OmpF by substituting the charged residues at the
constriction zone by those of OmpF (Samartzidou and Delcour, 1998). Therefore
it is believed that charged residues at the constriction zone are responsible for
voltage gating.
Since both OmpF and PhoE are aspecific porins that show only a slight ion
selectivity and allow transport of solutes up to 600 Da, they are suited for
L3
OmpF PhoE

State of the art: a nanoreactor coming to life
47
transport of nucleoside(analogues) and nucleobase(analogues) through the
nanoreactor membrane.
2.3.2. Specific porin Tsx
Tsx was discovered as the receptor for bacteriophage T6, hence its name Tsx.
Tsx is a substrate-specific transporter with a molecular mass of 31.5 kDa that
has low affinity (µM to mM) binding sites for its substrates which are nucleosides
and deoxynucleosides. These binding sites are saturable and allow efficient
diffusion at low concentration gradients (Benz et al., 1988; Maier et al., 1988).
Although Tsx is substrate specific, it allows diffusion of small solutes such as
serine (Heuzenroeder and Reeves, 1981; Luckey and Nikaido, 1980) and also
transports albicidin, a relatively high molecular weight (850 Da) antibiotic (Birch
et al., 1990).The importance of the Tsx protein for nucleoside uptake becomes
apparent only at low substrate concentrations (<1µM). At higher concentrations,
Tsx becomes dispensable and transport of nucleosides occurs through general
porins. In tsx mutants, the rate of uptake of adenosine and thymidine is strongly
reduced but interestingly the rate of uptake of cytidine is unaffected.
Furthermore, conductance measurements showed that the saturation constant Ks
for cytidine, 2x10-2 M is very high compared to adenosine (6x10-4 M), meaning
that the affinity of Tsx for cytidine is very low (Maier et al., 1988). This is
probably related to the fact that the expression of the tsx gene is under a
negative control of cytR and cytidine is the effector molecule of the cytR
repressor. The apparent Tsx-independent permeation of cytidine across the outer
membrane at very low concentrations may allow cytidine to alert the cell to the
presence of other exogenous nucleosides. Comparison of the in vivo Tsx
dependent transport of adenosine and adenine arabinoside indicated that Tsx
does not strongly differentiate between nucleosides with different pentose
moieties (Krieger-Brauer and Braun, 1980). Tsx does not seem to play a role in
the transport of free nucleobases or monophosphate nucleosides (Benz et al.,
1988; McKeown et al., 1976; van Alphen et al., 1978). Reconstituted Tsx forms a
very low-conductance channel of 10 pS in 1M KCl compared to 1.9 nS for OmpF
(Buehler et al., 1991), which is indicative for a narrow pore (Benz et al., 1988;
Maier et al., 1988).

Chapter 2
48
The crystal structure of Tsx in complex with nucleosides was solved in 2004 by
Ye and Van den Berg (Ye and Van Den Berg, 2004).
Figure 3: Crystal structure of the E. coli Tsx channel. Surface representation viewed from the extracellular side (A). Cut away side view showing the nucleoside binding sites Nuc0, Nuc1 and Nuc2 in thymidine soaked crystals. The aromatic residues that line the channel and that are involved in nucleoside binding are indicated in cyan.
Tsx is a monomeric protein that folds into a 12 stranded β-barrel. There are no
extracellular loops that bend back into the barrel to form an hourglass shaped
constriction. Instead the channel has a much longer constriction zone, spanning
almost the entire membrane. The cross section of the channel in the plane of the
membrane shows a keyhole-like shape (Figure 3a). Transport of the nucleosides
through the channel is achieved via a greasy slide mechanism (Van Gelder et al.,
2002): the substrate binds through hydrophobic contacts with pairs of aromatic
residues located on opposite sides of the channel and through hydrogen-bonding
interactions with ionisable residues. Binding and release of the substrate by
these weak binding sites results in the movement of the substrates through the
pore (Figure 3b). The net direction of transport is governed by the concentration
gradient of the substrate across the outer membrane. Both the nucleobase
moiety and the riboside moiety contribute to the binding.

State of the art: a nanoreactor coming to life
49
Since Tsx is able to efficiently transport nucleosides at submicromolar
concentrations, it is very suitable to permeabilise nanoreactors for nucleosides
and nucleoside analogs.
2.4 Nucleoside Hydrolase of Trypanosoma vivax
Nucleoside hydrolases (or nucleoside N-ribohydrolases, NHs) comprise a
superfamily of structurally related metalloproteins with a unique β–sheet
topology. They are glycosidases that catalyze the hydrolysis of the N-glycosidic
bond between the anomeric carbon atom of ribose and purine or pyrimidine base
to form the free nucleobase and ribose (Figure 4). They all exert a stringent
specificity towards the ribose moiety but their preference towards the nature of
the nucleobase moiety is more variable. All members of this superfamily have
the characteristic N-terminal motif DXDXXXDD. Scanning genomes for the
occurrence of this fingerprint showed that NHs are widely distributed in nature.
They have been found in bacteria (Ogawa et al., 2001; Petersen and Moller,
2001), protozoa (Cui et al., 2001; Pelle et al., 1998), yeasts (Kurtz et al., 2002),
insects (Ribeiro and Valenzuela, 2003), mesozoa (Versees et al., 2003), plants,
amphibians and fish. Interestingly they were not found in mammals. Their
metabolic role is best understood in parasitic protozoa were they are part of the
salvage pathway for the scavenging of purines from their environment. Parasitic
protozoa rely on this purine salvage pathway for their survival since they are
unable to synthesize purines de novo. These protozoan NHs have been
extensively studied since they are potential targets for treatment. Protozoan NHs
are classified into three categories based on their substrate specificity: the base-
aspecific inosine-uridine preferring NHs,(IU-NH) the purine-specific inosine-
adenosine-guanosine NHs (IAG-NH) and the 6-oxopurine specific inosine-
guanosine NHs (IG-NH).

Chapter 2
50
Figure 4: Catalytic reaction of NHs. The NH-catalysed hydrolysis of a ribonucleoside (examplified by inosine).
The NH of Trypanosoma vivax (TvNH) used in this project belongs to the IAG-
NH class of nucleoside hydrolases with a molecular mass of 37 kDa.
Trypanosoma vivax causes trypanosomiasis, or sleeping sickness, in domestic
animals and wildlife. Therefore it was well studied as a possible target in our lab.
The substrate specificity was first determined by measuring the activity for
various substrates (Table 1). A faster turnover (kcat ↑) and higher substrate
affinity (KM ↓) resulted in a higher specificity for purines as compared to
pyrimidines (Versees et al., 2001). In 2001 Versées et al. solved the crystal
structure of TvNH which gave much insight in the mechanism of IAG-NHs since it
was the first structure of an IAG-NH that was solved. They solved the structure
of TvNH and TvNH in complex with 3-deaza-adenosine, an adenosine analogue
that is an inhibitor of the TvNH and binds to the active site with high affinity
(Figure 5A). TvNH is a homodimer, with each monomer being a single domain,
globular protein. Each subunit consists of 10 β-strands, 12 α-helices and three
short 310 helices (Figure 5B). Eight of the ten β-strands form a central mixed
sheet with 7 parallel strands (β1-β6 and β10) and one antiparallel strand (β7).
The parallel strands β1-β6 and α-helices 1-7 form a motif that resembles a
Rossman fold. The first βαβαβ motif of the Rossman fold is known to be involved
in nucleotide binding. Since TvNH is specific for nucleosides instead of
nucleotides, the consensus sequences GXGXXG involved in phosphate- binding,
is replaced in TvNH by an aspartate-rich region that binds Ca2+.
The active sites are located at the C-terminal ends of the core 8-stranded β-
sheet of both subunits and is formed by strands β1, β2, β4 and β5 and helices
α1, α3, α10 and α11. A bound calcium ion is also situated in the active site. 3-

State of the art: a nanoreactor coming to life
51
deaza-adenosine is bound in the active site with its ribose interacting with the
Ca2+ and the purine base pointed towards the exit of the active-site. The bound
ribose adopts an envelope conformation whereas the purine base adopts a syn
conformation towards the ribose. When 3-deaza-adenosine is bound, the Ca2+-
ion interacts with two hydroxyl groups of the ribose. Furthermore, the enzyme
forms hydrogen bonds with all three hydroxyl groups of the ribose, explaining
the high specificity toward the ribose moiety of the substrates. The 3-deaza-
adenine leaving group is involved in extensive aromatic stacking interactions with
two tryptophan residues (Trp 83 and 260). It is this aromatic stacking that
contributes to the purine selectivity of TvNH.
Figure 5: Structure (A) and topology (B) of a TvNH subunit
The enzymatic reaction occurs via a SN1mechanism with an oxocarbenium ion
transition state. The nucleophile in this reaction is probably a Ca-bound water
molecule that is activated by Asp10 by abstraction of a proton. In many other
enzymes such as IU-NHs, phosphorylases and AMP-nucleosidases,
hydrolysis/phosphorolysis of the N-glycosidic bond in nucleosides and nucleotides
commonly involves the protonation of the leaving nucleobase concomitant with
nucleophilic attack. In TvNH however no general acid or hydrogen bond donor
could be identified and leaving group activation occurs by another mechanism:
aromatic stacking of the purine base by tryptophans 83 and 260 lowers the pka
of the leaving group by several units, allowing protonation of N-7 by a solvent
molecule (Versees et al., 2004) (Figure 6)
The aromatic stacking of the heterocyclic nucleobase is an important feature of
this enzyme, which makes TvNH promiscuous towards the nature of the
BA BA

Chapter 2
52
nucleobase moiety. In our lab several nucleoside analogues were tested as
substrate for TvNH (Table 1). These data clearly show that when the ribose
moiety is altered like in 2’-deoxy nucleoside analogues, the activity of TvNH is
greatly diminished. Alternatively, when the nucleobase moiety is altered like in
purine riboside, 6-thioguanosine..., the activity of TvNH is retained. One
exception is p-nitrophenyl riboside. This is due to the fact that p-nitrophenyl
riboside cannot be protonated, however it is susceptible to decomposition when
the ribosyl group is converted to the oxocarbenium ion, resulting in reduced but
not diminished activity of TvNH.
The nucleobase analogues of several compounds tested are known cytotoxic
agents such as 2-fluoroadenine, 2-chloroadenine, 6-methylpurine and 6-
thioguanine. Therefore TvNH is a good candidate for prodrug activation.
Figure 6: Schematic representation of the interactions in the active site of nucleoside hydrolase of T. vivax and a nucleoside.

State of the art: a nanoreactor coming to life
53
Table 1: kinetic parameters for nucleosides and nucleoside analogues of TvNH
Compound kcat (S-1) Km (µM) Kcat/Km
(s-1mM-1) Kd (µM)
Adenosine 2.58 8 322
Guanosine 2.31 2.33 991
Inosine 5.19 5.37 966
Cytidine 0.338 925 0.36
Uridine 0.022 586 0.037
Para-nitrophenylriboside 0.206 257 0.8
Xanthosine 2.35 1760 1.3
Purine riboside 3.92 3.79 1034
2’-deoxyadenosine 0.0043 23 0.19
3’-deoxyadenosine 0.00031 223 0.0014
5’-deoxyadenosine 0.030 656 0.046
7-methylguanosine 3.45 2.13 1610
3-methylcytidine 0.98 1.97 497
3-deaza-adenosine 0.2
7-deaza-adenosine 356
5’-deoxyguanosine 0.079 504 0.16 5’-deoxy-7-methylguanosine
4.33 126.9 34.1
2-fluoroadenosine 1.86 39.05 47.6
6-iodopurineriboside 2.17 <2 >1085
8-bromoadenosine
6-thioguanosine 1.77 3.56 497
2-chloroadenosine 1.97 2.70 729
Nicotinamide riboside 1.59 4.82 330
6-methylpurine riboside 4.3 < 10 > 430
2.5 Purine nucleoside analogs as prodrugs
Nucleoside analogs are one of the most common classes of drugs used to treat
cancer. Cytotoxic nucleoside analogs are antimetabolites that can be
incorporated into DNA or RNA macromolecules or inhibit enzymes involved in
nucleoside synthesis, hereby inducing apoptosis. When using nucleoside analogs
as prodrugs, it is important that the nucleoside analog prodrug is non toxic and
cannot be metabolised by endogenous enzymes to a toxic form. Furthermore,
the nucleobase analog drug should be toxic and cannot be metabolised by
endogenous enzymes to an inactive form.

Chapter 2
54
2.5.1. Deoxyadenosine analogs
The development of 2-chloro-2’ deoxyadenosine (CdA) and 2-fluoro-2’-
deoxyadenosine (FdA) derives from the understanding of the pathogenesis of
adenosine deaminase (ADA) deficiency. In this metabolic disease, 2’-
deoxyadenosine is accumulated due to the enzyme defect. 2’-deoxyadenosine is
then further metabolised by deoxycytidine kinase (dCK), nucleoside
monophospate kinase and nucleoside diphospohate kinase to form the metabolite
2’-deoxyadenosine triphosphate (dATP) which is toxic in higher concentrations
(Figure 7). Since dCK is highly upregulated in lymphocytes this results in the
immunodeficiency related to ADA-deficiency. This understanding led Carson and
co-workers to synthesize 2’-deoxyadenosine analogs that also exert a
lymphocytic activity but are no substrates of ADA and can therefore not be
neutralised by deamination (Carson et al., 1980). Of these analogs, CdA had the
most favourable therapeutic ratio with an ID50 (% inhibition of growth) of 0,003
µM, determined on the T-lymphoblastoid cell line CCRF-CEM. FdA had an ID50 of
0,15 µM (Carson et al., 1980; Parker et al., 1998). From experiments with dCK-
deficient lymphoblasts, it was clear that activation by dCK is the main route for
cytotoxicity. When dCK-deficient lymphoblasts were used, the ID50 increased a
1000 fold and a 100 fold for CdA and FdA respectively. Whereas deficiency of
deoxyadenosine kinase led to a 10 fold increase of the ID50 of CdA and had no
effect on the ID50 of FdA. Furthermore, cytotoxicity of CdA and FdA was
completely blocked by addition of deoxycytidine.
The cytotoxicity involves several mechanisms. Like dATP, they inhibit
ribonucleotide reductase, hereby reducing the pool of deoxynucleotide
triphosphates for DNA synthesis and enhancing their own cytotoxicity by self-
potentiation. They also inhibit DNA- and RNA polymerase after incorporation into
DNA and RNA respectively, hereby terminating chain elongation (Parker et al.,
1988). However, neither mechanism can account for the remarkable cytotoxicity
in resting cells where both ribonucleotide reductase activity and DNA synthesis
are extremely low (Carson et al., 1983). The most likely explanation is the
interference of CdA and FdA with DNA repair (Pettitt et al., 2000) and activation
of the caspase 9/caspase 3 death pathway by interaction with the pro-apoptotic
factor Apaf1 (Genini et al., 2000).

State of the art: a nanoreactor coming to life
55
Figure 7: metabolic pathways of 2-fluoro-2’-deoxyadenosine and 2-fluoroadenine
CdA, also known as Cladribine, is used as a therapeutic agent to treat chronic
lymphocytic leukaemia and hairy-cell leukaemia (Carrera et al., 1991). It was
discovered by Bontemps et al. that one of the major catabolites in plasma and
urine of CdA, 2-chloroadenine (CAde), is also cytotoxic (Bontemps et al., 2000).
Breakdown of CdA to CAde is probably due to acid hydrolysis in the stomach,
degradation by gut bacterial phosphorylases and by phosphorolytic cleavage in
the liver. The initial phosphorylation of CAde is catalysed by the adenine
phosphoribosyltransferase (APRT) since it is inhibited in the presence of adenine.
Further phosphorylation to 2-chloro-ATP is catalysed by adenylate kinase and
nucleoside diphosphate kinase (Bontemps et al., 2000). An identical metabolic
pathway was found for FdA (Parker et al., 1998) (Figure 7).
Although 2-chloro-ATP and 2-fluoro-ATP, like 2-chloro-dATP induce apoptosis,
this is done by a different pathway (Barbieri et al., 1998). Parker and co-workers
showed that F-ATP inhibits protein, DNA and RNA synthesis. The effect on DNA
synthesis is probably the result of a shortage of enzymes involved in DNA
synthesis. This means that 2-fluoroadenine is also cytotoxic to non proliferating
cells which was confirmed by Parker et al. using non proliferating CCRF-CEM
cells, MRC-5 cells (human lung fibroblasts) and Balb-3T3 cells (mouse embryonic
cells) (Parker et al., 1998).
In conclusion, the nucleoside analogs CdA and FdA are only toxic to
lymphocytes due to the necessary activation by deoxycitidine kinase. In contrast,
2-fluoro-2’-deoxyadenosine
2-fluoro-dAMPATP
ADPdCK
2-fluoro-dADPATP
ADPN-MPK
2-fluoro-dATPNTP
NDPN-DPK
2-fluoroadenine
PRibose-1P
E.coli PNP
2-fluoro-AMPPRPP
PPiAPRT
2-fluoro-ADPATP
ADPAdK
2-fluoro-ATPNTP
NDPN-DPK
APRT = adenine phosphoribosyltransferase
dCK = deoxycytidine kinase
N-MPK = nucleosidemonophosphate kinase
N-DPK = nucleoside diphosphatekinase
AdK = adenylate kinase
PNP = purine nucleosidephosphorylase
2-fluoro-2’-deoxyadenosine
2-fluoro-dAMPATP
ADPdCK
2-fluoro-dADPATP
ADPN-MPK
2-fluoro-dATPNTP
NDPN-DPK
2-fluoroadenine
PRibose-1P
E.coli PNP
2-fluoro-AMPPRPP
PPiAPRT
2-fluoro-ADPATP
ADPAdK
2-fluoro-ATPNTP
NDPN-DPK
APRT = adenine phosphoribosyltransferase
dCK = deoxycytidine kinase
N-MPK = nucleosidemonophosphate kinase
N-DPK = nucleoside diphosphatekinase
AdK = adenylate kinase
PNP = purine nucleosidephosphorylase

Chapter 2
56
the nucleobase analogs 2-chloroadenine and 2-fluoroadenine are toxic to other
cell types due to a different activation pathway. Another advantage is the
cytotoxicity of both 2-chloroadenine and 2-fluoroadenine on resting cells. Taken
together, this makes 2-chloro-2’-deoxyadenosine and 2-fluoro-2’-
deoxyadenosine good prodrug candidates to treat non-lymphocytic tumors.
For instance, as shown by Parker et al., 2-fluoro-2’-deoxyadenosine and
another deoxyadenosine analog 6-methylpurine deoxyribose (6-MPdR) are good
substrates for E. coli purine nucleoside phosphorylase (PNP) that cleaves them
into 2-fluoroadenine or 6-methylpurine and ribose-1-phosphate (Parker et al.,
2003) (Figure 7). In contrast, they are not cleaved by human PNP because it
cannot cleave adenosine and adenosine analogs. Parker et al. successfully
transfected D54 gliomas with the gene encoding E.coli PNP. These transfected
tumor cells were subsequently injected into the flanks of nude mice (NCr-nu) and
then treated intraperitoneally with FdA and 6-MPdR. This resulted in excellent
antitumor activity for both nucleoside analogs. 6-MPdR is a very good prodrug
candidate to be used in suicide gene therapy with E. coli PNP since its nucleobase
6-methylpurine has high bystander activity. This means that 6-methylpurine can
diffuse to neighbouring cells that do not express E. coli PNP. Gadi et al. showed
that less than 1% of PNP expressing cells is sufficient to kill an entire cell
population in vitro owing to the membrane permeability of 6-methylpurine (Gadi
et al., 2000). Parker et al. found that also in vivo tumors in which only 20 % of
the cells express E.coli PNP are destroyed by FdA and 6-MPdR due to this
bystander activity (Parker et al., 2003).
Although CdA was not tested as a prodrug in combination with E. coli PNP,
Bzowska and Kazimierczuk, showed that 2-chloro-2’-deoxyadenosine can also be
efficiently cleaved by E.coli PNP into 2-chloroadenine and ribose monophosphate
(Bzowska and Kazimierczuk, 1995).

State of the art: a nanoreactor coming to life
57
2.5.2. 6-Thioguanosine
Although little is known about the cytotoxic effects of 6-thioguanosine, the
nucleobase 6-thioguanine is a well known cytotoxic agent. It has been used to
treat acute lymphoblastic leukaemia (ALL) over the last 45 years. It is also used
to treat inflammatory bowel disease and autoimmune conditions and to reduce
organ rejection after transplantation (Al Hadithy et al., 2005). Before exerting its
cytotoxicity, 6-thioguanine needs to be activated by hypoxanthine-guanine
phosphoribosyl transferase (HGPRT) to form 6-thioguanosine monophosphate
(TGMP). TGMP can then further be metabolised to form 6-thioguanosine
triphosphate (TGTP) by guanylate kinase and nucleoside diphosphate kinase or it
can be metabolised to form 6-thio-2’-deoxyguanosine triphosphate (TdGTP) by
nucleoside diphosphate reductase and nucleoside diphosphate kinase (Figure 8).
Although TGMP is a substrate of guanylate kinase, the reaction velocity for this
substrate is only 0.04% of the velocity of the natural substrate GMP. This leads
to an accumulation of TGMP which inhibits de novo purine synthesis through
negative feedback inhibition of the enzyme phosphoribosyl amine synthetase
(McCollister et al., 1964) and progressive irreversible inhibition of inosine 5’-
phosphate dehydrogenase (Hampton, 1963). Since the binding affinity to
guanylate kinase of TGMP is comparable to that of GMP, TGMP furthermore acts
as a competitive inhibitor of guanylate kinase with a Ki of 60µM (Miech et al.,
1969). This inhibition leads to a drastic decrease in GDP and GTP concentrations.
Since GDP and GTP are required for nucleic acid synthesis and function as
coenzymes of vital enzymatic reactions, this inhibition also contributes to the
cytotoxic action of 6-thioguanine.
Another key event in the toxicity of 6-thioguanine is the incorporation of
TdGTP into DNA. Interestingly, highly lethal concentrations of 6-thioguanine do
not immediately interfere with cell growth or cell cycle progression but only after
completion of the cell cycle in which TdGTP was incorporated into the DNA. The
cells treated with 6-thioguanine are predominantly arrested in the G2 phase of
the cell cycle (Wotring and Roti Roti, 1980). Maybaum and Mandel confirmed
that this was the result of chromatid disruption (Maybaum and Mandel, 1983).

Chapter 2
58
Figure 8: Metabolic pathways of 6-thioguanine
2.6 Targeting of nanoreactors
2.6.1. Passive targeting
Targeting of drugs can be either active or passive. In 1986, Maeda and
Matsumura introduced the concept of passive targeting to solid tumors by means
of the enhanced permeability and retention effect (EPR) (Matsumura and Maeda,
1986). It is well know that the vascular permeability of tumors is enhanced due
to the action of secreted factors such as kinin. This allows macromolecules to
diffuse from the bloodstream into the tumor interstitium. Furthermore, the
lymphatic drainage system is impaired so that macromolecules are retained in
the interstitium for a prolonged time (Jain, 1987). Particles ranging from 10 to
500 nm in size are able to extravasate through these leaky blood vessels (Figure
9) and remain there for a longer period. A perfect example of this passive
targeting is pegylated lyposomal doxorubicine (Doxil®) which is a long term
circulating carrier (t1/2 ~ 3 days) that is accumulated in tumor tissue (Green and
Rose, 2006). Doxil is used to treat relapsing ovarian cancer and AIDS-related
Kaposi’s sarcoma. Compared to conventional doxorubicin, plasma levels of Doxil
are higher since doxorubicin is encapsulated in liposomes and remains in the
blood for a prolonged time. Furthermore, clearance from the bloodstream is 9
6-thioguanine
6-thioguanosine monophosphate
6-thioguanosine diphosphate
6-thioguanosine triphosphate
6-thio-deoxyguanosine diphosphate
6-thio-deoxyguanosine triphosphate
PRPP
PPi
HGPRT
ATP
ADPGK
N-DPKNTP
NDP
Thioredoxin 2SH
Thioredoxin S-S
N-DPR
NTP
NDPN-DPK
HGPRT = hypoxanthine-guanosinephosphoribosyltransferase
GK = Guanylate kinase
N-DPK = nucleosidediphosphatekinase
N-DPR = nucleosidediphosphatereductase6-thioguanine
6-thioguanosine monophosphate
6-thioguanosine diphosphate
6-thioguanosine triphosphate
6-thio-deoxyguanosine diphosphate
6-thio-deoxyguanosine triphosphate
PRPP
PPi
HGPRT
ATP
ADPGK
N-DPKNTP
NDP
Thioredoxin 2SH
Thioredoxin S-S
N-DPR
NTP
NDPN-DPK
6-thioguanine
6-thioguanosine monophosphate
6-thioguanosine diphosphate
6-thioguanosine triphosphate
6-thio-deoxyguanosine diphosphate
6-thio-deoxyguanosine triphosphate
PRPP
PPi
HGPRT
ATP
ADPGK
N-DPKNTP
NDP
Thioredoxin 2SH
Thioredoxin S-S
N-DPR
NTP
NDPN-DPK
HGPRT = hypoxanthine-guanosinephosphoribosyltransferase
GK = Guanylate kinase
N-DPK = nucleosidediphosphatekinase
N-DPR = nucleosidediphosphatereductase

State of the art: a nanoreactor coming to life
59
times less than compared to doxorubicin alone. This is the effect of pegylation of
the liposomes which decreases the uptake of the liposomes by macrophages,
predominantly kuppfer cells. Finally, tissue distribution of Doxil is 25 times less
than compared to doxorubicin alone. This means that doxil is predominantly
accumulated in tumor tissue and not in healthy tissue due to the EPR effect.
Doxorubicin is known to exhibit cardiotoxicity as an effect of the accumulation in
the heart. This cardiotoxicity is remarkably reduced when doxil formulations are
used (Safra et al., 2000).
Figure 9: Schematic overview of the enhanced permeation and retention effect (EPR). Drug molecules are represented as dark blue dots.
2.6.2. Active targeting with single domain antibodies
The first idea of active drug targeting called the ‘magic bullet’ approach was
proposed by Ehrlich in the nineteenth century. The magic bullet is composed of
two units, the drug unit and the targeting unit which is responsible for delivering
the drug to the designated site. The following substances can be used as
targeting moieties: antibodies and their fragments, lectins, lipoproteins,
hormones, saccharides, peptides, polynucleotides, folate… Monoclonal antibodies
are the most frequently used. Research shows that several body compartments
and pathologies can be targeted including components of the cardiovascular
system, the reticulo-endothelial system, lymphatic system, tumors, infarcts,
inflammations, infections and transplants. In the early years of targeted drug
therapy, the drug was directly linked to the antibody (= immunotoxin). Later on

Chapter 2
60
soluble or insoluble carriers where loaded with multiple active molecules and
conjugated to the targeting unit, thus delivering a high amount of drug per
targeting unit.
In this project, the antigen binding domain of single chain antibodies derived
from camelids is used for targeting purposes. These naturally occurring single
chain antibodies devoid of a light chain were discovered by Hamers et al. in 1993
(Hamers-Casterman et al., 1993) (Figure 10). These so called heavy-chain
antibodies (HCAb) are present in the serum of camels, dromedaries and llamas.
It should be emphasized that they posses both the conventional heterotetrameric
antibodies common to all vertebrates as well as the homodimeric heavy chain
antibodies. The relative proportion seems to vary with an average of about 50 %
of each type. Whether a conventional antibody or HCAb is raised, depends on the
antigen.
Figure 10: shematic representation of a conventional monoclonal antibody and a heavy chain antibody
HCAbs have a lower molecular weight of 100 kDa compared to conventional
antibodies because they lack the CH1 domain (Hamers-Casterman et al., 1993).
To structurally compensate for the absence of CH1, HCAbs have a longer hinge
between the variable domain called VHH and CH2. Up until now, 3 different types
of hinges have been found: a short hinge (11 amino acids) and a long hinge (35
amino acids) in dromedary and a long hinge (29 amino acids) in llama (Hamers-
Casterman et al., 1993; Vu et al., 1997). Both longer hinges contain a repeated
Pro-Xaa motif that is assumed to adopt a helical conformation that functions as a
rigid rod-like spacer (Evans et al., 1986; Roditi et al., 1989).
VL
CH3
CH1
CH2
VH
CL
CH1CL VH
VL
CH1VH
CL
VL
F(ab’) 50 kDa F(ab’)2 100kDa
Monoclonal antibody
CH3
VHH
VHH
CH2
Single chain camel antibody
Nanobody 15 kDa
VLVH
scFv 25 kDa
VL
CH3
CH1
CH2
VH
CL
CH3
CH1
CH2
VH
CL
CH1CL VH
VL
CH1CL VH
VL
CH1VH
CL
VL
F(ab’) 50 kDa F(ab’)2 100kDa
Monoclonal antibody
CH3
VHH
VHH
CH2
Single chain camel antibody
Nanobody 15 kDa
VLVH
scFv 25 kDa

State of the art: a nanoreactor coming to life
61
Because there is no light chain in HCAbs, their antigen binding site is
composed of a single domain, VHH, compared to the VH-VL paired antigen
binding site of conventional antibodies. Sequence analysis of various llama VHH
domains revealed that they all belong to the VHIII family (Muyldermans et al.,
1994; Vu et al., 1997). Although they belong to the same family, their sequences
differ more from each other than expected for members of the same family.
Conventionally, members of the same family share a nucleotide sequence
identity above 80 % whereas VHH sequence identities range from 58 to 90%.
This lower identity probably reflects a high rate of somatic mutation which can
lead to increased diversity.
To compensate for the absence of the light chain, adaptations in the VHH
domain are necessary. For instance, there are four important hydrophobic to
hydrophilic amino acid substitutions in the framework 2 region of the VHH
domain: Leu11Ser, Val37Phe, Gly44Glu, Leu45Arg/Cys and Trp47Gly (Figure
11). These residues normally interact with the VL domain. Consequently,
substituting them to more hydrophilic residues will impair the ability of the VHH
to form heterodimers with a VL domain and increase the solubility and stability of
the HCAbs (Dumoulin et al., 2002). This was confirmed by Davies and
Riechmann who analysed the effect of three of these mutations (G44E, L45R and
W47G) on a human VHIII. Non mutated VH domains aggregated at a protein
concentration above 1 mg/ml whereas the aggregation of the mutated VH was
significantly reduced (Davies and Riechmann, 1994).
Besides these VH/VHH hallmark substitutions in the framework regions, also
the hypervariable regions differ from each other. First, the average CDR3 region
of the VHH domain is about twice as long as in the VH domain. This results in a
higher antigen binding surface, hereby increasing the repertoire. Secondly, the
CDR1 region is elongated with 4 residues (27-30). In conventional antibodies
these 4 residues form the loop connecting the two β-sheets of the
immunoglobulin fold. Their hypervariability in VHHs shows their involvement in
antigen binding. Finally, the long CDR3 region is connected to the CDR1 region
via a disulfide bridge. This disulphide bond restricts the conformational flexibility
of the long CDR3 loop in the absence of a bound antigen. Immobilising the loop
in this manner will minimise the entropic penalty upon binding of the antigen.

Chapter 2
62
This long CDR3 loop has another interesting advantage. Lauwereys et al.
found that a large amount of HCAbs that were isolated after immunisation with
enzymes, are competitive enzyme inhibitors that bind to the active site of the
enzymes (Lauwereys et al., 1998). This was further investigated for HCAbs that
bind to the active site of lysozyme. This enzyme inhibition is due to the convex
shape of the VHH, predominantly formed by the CDR3 loop that protrudes in the
convex substrate binding pocket of lysozyme (De Genst et al., 2006). In
contrast, conventional antibodies that are competitive enzyme inhibitors are rare
because the antigen-binding surface is either flat or concave (Padlan, 1996).
Therefore it is clear that HCAbs recognize epitopes that are currently out of reach
for conventional antibodies.
Figure 11: schematic representation of VH and VHH sequences. Hallmark amino acid substitutions in the framework regions are shown.
Since VHHs are single domain antigen binding molecules, their isolation and
expression is more straight forward and easier as compared to the smallest
antigen binding fragment, ScFv, of conventional antibodies. In case of the ScFv
fragments which are a combination of a VH and VL domain, first a library of
cloned VH and VL domains is generated after immunisation. Since there are
several VH and VL gene families, several sets of primers are necessary to clone
the whole repertoire. Subsequently binders to a specific antigen are selected
from random combinations of these VH and VL domains. This means that large
libraries need to be screened in order to isolate the native combinations and that
FR1 FR2 FR3
CDR1 CDR3CDR2
S11 F37E44R45G47
VHH
FR1 FR3FR2
CDR1 CDR2 CDR3
L11 V37G44L45W47
VH
FR1 FR2 FR3
CDR1 CDR3CDR2
S11 F37E44R45G47
VHH
FR1 FR2 FR3
CDR1 CDR3CDR2
FR1 FR2 FR3
CDR1 CDR3CDR2
S11 F37E44R45G47
VHH
FR1 FR3FR2
CDR1 CDR2 CDR3
L11 V37G44L45W47
VH
FR1 FR3FR2
CDR1 CDR2 CDR3
FR1 FR3FR2
CDR1 CDR2 CDR3
L11 V37G44L45W47
VH

State of the art: a nanoreactor coming to life
63
many binders with lower affinity and stability will be obtained. In case of the
VHH, only one library of VHH domains needs to be generated from which binders
are selected via phage display vectors. This significantly reduces the cloning and
screening labour to isolate the native binder. Furthermore, only one set of
primers is needed to clone the whole repertoire because all VHH domains belong
to the same family. Finally, expression of the VHHs in E.coli generally results in
5-10 mg/l of VHH which is about 10 times higher than the production of ScFvs
(Arbabi Ghahroudi et al., 1997).
VHHs isolated from immunised libraries are highly specific for the target
antigen with Kd constants in the nanomolar range (Muyldermans and Lauwereys,
1999), which is similar to the affinity of most scFv, and do not cross-react with
other non-related antigens. This was demonstrated by cAb-Lys3 that was
isolated after immunisation with hen egg-white lysozyme. cAb-Lys3 binds henn
lysozyme as well as turkey lysozyme that only differs by 6 amino acid residues
but not to human lysozyme which is more distantly related (Arbabi Ghahroudi et
al., 1997).
In conclusion, VHH are distinguished from the scFv by some unique
properties: size, stability, solubility, ease of cloning a library and selecting highly
specific antigen binders, in vivo maturation and high expression levels. Therefore
it is expected that they will perform better than other antibody fragments in
numerous applications such as diagnostics, in vivo imaging, targeting and as
crystallisation aids. Since their discovery in 1993, numerous VHH have been
isolated against different antigens, for multiple purposes as shown in Table 2.

Chapter 2
64
Table 2: selected examples of VHHs and their applications.
Antigen VHH Application Ref
Variant surface glycoprotein of Trypanosomes
NbAn33 Trypanosome
targeting of Tr-Apol-I (Baral et al., 2006)
Prostate specific antigen cAbPSA-N7 and
cAbPSA-N50 Prostate cancer
diagnostics (Saerens et al., 2005)
Carcinoembryogenic antigen
cAbCEA-5 Tumor targeting
(Cortez-Retamozo
et al., 2004)
Human lysozyme cAbHUL-6 Inhibition of amyloid
fibril formation
(Dumoulin et al., 2003)
MazE cAbmaz1 Crystallisation aid (Loris et al.,
2003)
Neisseria meningitidis LPS VHH5G Inhibition of LPS mediated sepsis
(El Khattabi et al., 2006)
Epidermal growth factor receptor VIII
ORBI-83 VHH Tumor targeting (Omidfar et al., 2007)
caffeine VSA2 Caffeine detection (Ladenson
et al., 2006)

State of the art: a nanoreactor coming to life
65
2.7 References
Al Hadithy, A.F., de Boer, N.K., Derijks, L.J., Escher, J.C., Mulder, C.J. and Brouwers, J.R. (2005) Thiopurines in inflammatory bowel disease: pharmacogenetics, therapeutic drug monitoring and clinical recommendations. Dig Liver Dis, 37, 282-297.
Alexandridis, P., Olsson, U. and Lindman, B. (1998) Arecord nine different phases (four cubic, two hexagonal, and one lamellar liquid crystalline and two micellar solutions) in a ternary isothermal system of an amphiphilic block copolymer and selective solvents (water and oil). langmuir, 14, 2627-2638.
Arbabi Ghahroudi, M., Desmyter, A., Wyns, L., Hamers, R. and Muyldermans, S. (1997) Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett, 414, 521-526.
Baral, T.N., Magez, S., Stijlemans, B., Conrath, K., Vanhollebeke, B., Pays, E., Muyldermans, S. and De Baetselier, P. (2006) Experimental therapy of African trypanosomiasis with a nanobody-conjugated human trypanolytic factor. Nat Med, 12, 580-584.
Barbieri, D., Abbracchio, M.P., Salvioli, S., Monti, D., Cossarizza, A., Ceruti, S., Brambilla, R., Cattabeni, F., Jacobson, K.A. and Franceschi, C. (1998) Apoptosis by 2-chloro-2'-deoxy-adenosine and 2-chloro-adenosine in human peripheral blood mononuclear cells. Neurochem Int, 32, 493-504.
Battaglia, G. and Ryan, A.J. (2005) Bilayers and interdigitation in block copolymer vesicles. J Am Chem Soc., 127, 8757-8764.
Bauer, K., Struyve, M., Bosch, D., Benz, R. and Tommassen, J. (1989) One single lysine residue is responsible for the special interaction between polyphosphate and the outer membrane porin PhoE of Escherichia coli. J Biol Chem, 264, 16393-16398.
Benz, R., Schmid, A., Maier, C. and Bremer, E. (1988) Characterization of the nucleoside-binding site inside the Tsx channel of Escherichia coli outer membrane. Eur J Biochem, 176, 699-705.
Benz, R., Schmid, A., Van der Ley, P. and Tommassen, J. (1989) Molecular basis of porin selectivity: membrane experiments with OmpC- PhoE and OmpF-PhoE hybrid proteins of Escherichia coli K-12. Biochim Biophys Acta, 981, 8-14.
Berg, H. (1993) Random walks in biology. Princeton university press. Birch, R.G., Pemberton, J.M. and Basnayake, W.V. (1990) Stable albicidin resistance in
Escherichia coli involves an altered outer-membrane nucleoside uptake system. J Gen Microbiol, 136, 51-58.
Bontemps, F., Delacauw, A., Cardoen, S., Van Den Neste, E. and Van Den Berghe, G. (2000) Metabolism and cytotoxic effects of 2-chloroadenine, the major catabolite of 2-chloro-2'-deoxyadenosine. Biochem Pharmacol, 59, 1237-1243.
Buehler, L.K., Kusumoto, S., Zhang, H. and Rosenbusch, J.P. (1991) Plasticity of Escherichia coli porin channels. Dependence of their conductance on strain and lipid environment. J Biol Chem, 266, 24446-24450.
Bzowska, A. and Kazimierczuk, Z. (1995) 2-Chloro-2'-deoxyadenosine (cladribine) and its analogues are good substrates and potent selective inhibitors of Escherichia coli purine-nucleoside phosphorylase. Eur J Biochem, 233, 886-890.
Carrera, C.J., Piro, L.D., Saven, A., Beutler, E., Terai, C. and Carson, D.A. (1991) 2-Chlorodeoxyadenosine chemotherapy triggers programmed cell death in normal and malignant lymphocytes. Adv Exp Med Biol, 309A, 15-18.
Carson, D.A., Wasson, D.B., Kaye, J., Ullman, B., Martin, D.W., Jr., Robins, R.K. and Montgomery, J.A. (1980) Deoxycytidine kinase-mediated toxicity of deoxyadenosine analogs toward malignant human lymphoblasts in vitro and toward murine L1210 leukemia in vivo. Proc Natl Acad Sci U S A, 77, 6865-6869.

Chapter 2
66
Carson, D.A., Wasson, D.B., Taetle, R. and Yu, A. (1983) Specific toxicity of 2-chlorodeoxyadenosine toward resting and proliferating human lymphocytes. Blood, 62, 737-743.
Cortez-Retamozo, V., Backmann, N., Senter, P.D., Wernery, U., De Baetselier, P., Muyldermans, S. and Revets, H. (2004) Efficient cancer therapy with a nanobody-based conjugate. Cancer Res, 64, 2853-2857.
Cowan, S.W., Schirmer, T., Rummel, G., Steiert, M., Ghosh, R., Pauptit, R.A., Jansonius, J.N. and Rosenbusch, J.P. (1992) Crystal structures explain functional properties of two E. coli porins. Nature, 358, 727-733.
Cui, L., Rajasekariah, G.R. and Martin, S.K. (2001) A nonspecific nucleoside hydrolase from Leishmania donovani: implications for purine salvage by the parasite. Gene, 280, 153-162.
Davies, J. and Riechmann, L. (1994) 'Camelising' human antibody fragments: NMR studies on VH domains. FEBS Lett, 339, 285-290.
De Genst, E., Silence, K., Decanniere, K., Conrath, K., Loris, R., Kinne, J., Muyldermans, S. and Wyns, L. (2006) Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc Natl Acad Sci U S A, 103, 4586-4591.
Delcour, A.H. (1997) Function and modulation of bacterial porins: insights from electrophysiology. FEMS Microbiol Lett, 151, 115-123.
Dumoulin, M., Conrath, K., Van Meirhaeghe, A., Meersman, F., Heremans, K., Frenken, L.G., Muyldermans, S., Wyns, L. and Matagne, A. (2002) Single-domain antibody fragments with high conformational stability. Protein Sci, 11, 500-515.
Dumoulin, M., Last, A.M., Desmyter, A., Decanniere, K., Canet, D., Larsson, G., Spencer, A., Archer, D.B., Sasse, J., Muyldermans, S., Wyns, L., Redfield, C., Matagne, A., Robinson, C.V. and Dobson, C.M. (2003) A camelid antibody fragment inhibits the formation of amyloid fibrils by human lysozyme. Nature, 424, 783-788.
El Khattabi, M., Adams, H., Heezius, E., Hermans, P., Detmers, F., Maassen, B., van der Ley, P., Tommassen, J., Verrips, T. and Stam, J. (2006) Llama single-chain antibody that blocks lipopolysaccharide binding and signaling: prospects for therapeutic applications. Clin Vaccine Immunol., 13, 1079-1086. Epub 2006 Aug 1023.
Evans, J.S., Levine, B.A., Trayer, I.P., Dorman, C.J. and Higgins, C.F. (1986) Sequence-imposed structural constraints in the TonB protein of E. coli. FEBS Lett, 208, 211-216.
Forster, S. and Plantenberg, T. (2002) From self-organizing polymers to nanohybrid and biomaterials. Angew Chem Int Ed Engl, 41, 689-714.
Gadi, V.K., Alexander, S.D., Kudlow, J.E., Allan, P., Parker, W.B. and Sorscher, E.J. (2000) In vivo sensitization of ovarian tumors to chemotherapy by expression of E. coli purine nucleoside phosphorylase in a small fraction of cells. Gene Ther, 7, 1738-1743.
Genini, D., Adachi, S., Chao, Q., Rose, D.W., Carrera, C.J., Cottam, H.B., Carson, D.A. and Leoni, L.M. (2000) Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood, 96, 3537-3543.
Graff, A., Sauer, M., Van Gelder, P. and Meier, W. (2002) Virus-assisted loading of polymer nanocontainer. Proc Natl Acad Sci U S A, 99, 5064-5068. Epub 2002 Mar 5026.
Green, A.E. and Rose, P.G. (2006) Pegylated liposomal doxorubicin in ovarian cancer. Int J Nanomedicine, 1, 229-239.
Hamers-Casterman, C., Atarhouch, T., Muyldermans, S., Robinson, G., Hamers, C., Songa, E.B., Bendahman, N. and Hamers, R. (1993) Naturally occurring antibodies devoid of light chains. Nature, 363, 446-448.
Hampton, A. (1963) Reactions of Ribonucleotide Derivatives of Purine Analogues at the Catalytic Site of Inosine 5'-Phosphate Dehydrogenase. J Biol Chem, 238, 3068-3074.
Heuzenroeder, M.W. and Reeves, P. (1981) The tsx protein of Escherichia coli can act as a pore for amino acids. J Bacteriol, 147, 1113-1116.

State of the art: a nanoreactor coming to life
67
Jain, R.K. (1987) Transport of molecules in the tumor interstitium: a review. Cancer Res, 47, 3039-3051.
Krieger-Brauer, H.J. and Braun, V. (1980) Functions related to the receptor protein specified by the tsx gene of Escherichia coli. Arch Microbiol, 124, 233-242.
Kurtz, J.E., Exinger, F., Erbs, P. and Jund, R. (2002) The URH1 uridine ribohydrolase of Saccharomyces cerevisiae. Curr Genet, 41, 132-141.
Ladenson, R.C., Crimmins, D.L., Landt, Y. and Ladenson, J.H. (2006) Isolation and characterization of a thermally stable recombinant anti-caffeine heavy-chain antibody fragment. Anal Chem, 78, 4501-4508.
Lauwereys, M., Arbabi Ghahroudi, M., Desmyter, A., Kinne, J., Holzer, W., De Genst, E., Wyns, L. and Muyldermans, S. (1998) Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. Embo J, 17, 3512-3520.
Loris, R., Marianovsky, I., Lah, J., Laeremans, T., Engelberg-Kulka, H., Glaser, G., Muyldermans, S. and Wyns, L. (2003) Crystal structure of the intrinsically flexible addiction antidote MazE. J Biol Chem, 278, 28252-28257.
Luckey, M. and Nikaido, H. (1980) Specificity of diffusion channels produced by lambda phage receptor protein of Escherichia coli. Proc Natl Acad Sci U S A, 77, 167-171.
Maier, C., Bremer, E., Schmid, A. and Benz, R. (1988) Pore-forming activity of the Tsx protein from the outer membrane of Escherichia coli. Demonstration of a nucleoside-specific binding site. J Biol Chem, 263, 2493-2499.
Matsumura, Y. and Maeda, H. (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res, 46, 6387-6392.
Maybaum, J. and Mandel, H.G. (1983) Unilateral chromatid damage: a new basis for 6-thioguanine cytotoxicity. Cancer Res, 43, 3852-3856.
McCollister, R.J., Gilbert, W.R., Jr., Ashton, D.M. and Wyngaarden, J.B. (1964) Pseudofeedback Inhibition of Purine Synthesis by 6-Mercaptopurine Ribonucleotide and Other Purine Analogues. J Biol Chem, 239, 1560-1563.
McKeown, M., Kahn, M. and Hanawalt, P. (1976) Thymidine uptake and utilization in Escherichia coli: a new gene controlling nucleoside transport. J Bacteriol, 126, 814-822.
Miech, R.P., York, R. and Parks, R.E., Jr. (1969) Adenosine triphosphate-guanosine 5'-phosphate phosphotransferase. II. Inhibition by 6-thioguanosine 5'-phosphate of the enzyme isolated from hog brain and sarcoma 180 ascites cells. Mol Pharmacol, 5, 30-37.
Muyldermans, S., Atarhouch, T., Saldanha, J., Barbosa, J.A. and Hamers, R. (1994) Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng, 7, 1129-1135.
Muyldermans, S. and Lauwereys, M. (1999) Unique single-domain antigen binding fragments derived from naturally occurring camel heavy-chain antibodies. J Mol Recognit, 12, 131-140.
Nallani, M., Benito, S., Onaca, O., Graff, A., Lindemann, M., Winterhalter, M., Meier, W. and Schwaneberg, U. (2006) A nanocompartment system (Synthosome) designed for biotechnological applications. J Biotechnol, 123, 50-59. Epub 2005 Dec 2020.
Nardin, C., Hirt, T., Leukel, J. and Meier, W. (2000a) polymerized ABA triblock copolymer vesicles. langmuir, 16, 1035-1041.
Nardin, C., Thoeni, S., Widmer, J., Winterhalter, M. and Meier, W. (2000b) Nanoreactors based on (polymerized) ABA-triblock copolymer vesicles. Chem Comm, 1433-1434.
Nardin, C., Widmer, J., Winterhalter, M. and Meier, W. (2001) Amphiphilic block copolymer nanocontainers as bioreactors. Eur. Phys. J. E, 4, 403-410.
Nardin, C., Winterhalter, M., Meier, W. (2000) Giant Free-Standing ABA Triblock Copolymer Membranes. Langmuir, 16, 7708-7712.
Ogawa, J., Takeda, S., Xie, S.X., Hatanaka, H., Ashikari, T., Amachi, T. and Shimizu, S. (2001) Purification, characterization, and gene cloning of purine nucleosidase from Ochrobactrum anthropi. Appl Environ Microbiol, 67, 1783-1787.

Chapter 2
68
Omidfar, K., Rasaee, M.J., Kashanian, S., Paknejad, M. and Bathaie, Z. (2007) Studies of thermostability in Camelus bactrianus (Bactrian camel) single-domain antibody specific for the mutant epidermal-growth-factor receptor expressed by Pichia. Biotechnol Appl Biochem, 46, 41-49.
Padlan, E.A. (1996) X-ray crystallography of antibodies. Adv Protein Chem, 49, 57-133. Parker, W.B., Allan, P.W., Hassan, A.E., Secrist, J.A., 3rd, Sorscher, E.J. and Waud, W.R.
(2003) Antitumor activity of 2-fluoro-2'-deoxyadenosine against tumors that express Escherichia coli purine nucleoside phosphorylase. Cancer Gene Ther, 10, 23-29.
Parker, W.B., Allan, P.W., Shaddix, S.C., Rose, L.M., Speegle, H.F., Gillespie, G.Y. and Bennett, L.L., Jr. (1998) Metabolism and metabolic actions of 6-methylpurine and 2-fluoroadenine in human cells. Biochem Pharmacol, 55, 1673-1681.
Parker, W.B., Bapat, A.R., Shen, J.X., Townsend, A.J. and Cheng, Y.C. (1988) Interaction of 2-halogenated dATP analogs (F, Cl, and Br) with human DNA polymerases, DNA primase, and ribonucleotide reductase. Mol Pharmacol, 34, 485-491.
Pata, V. and Dan, N. (2003) The effect of chain length on protein solubilization in polymer-based vesicles (polymersomes). Biophys J, 85, 2111-2118.
Pelle, R., Schramm, V.L. and Parkin, D.W. (1998) Molecular cloning and expression of a purine-specific N-ribohydrolase from Trypanosoma brucei brucei. Sequence, expression, and molecular analysis. J Biol Chem, 273, 2118-2126.
Petersen, C. and Moller, L.B. (2001) The RihA, RihB, and RihC ribonucleoside hydrolases of Escherichia coli. Substrate specificity, gene expression, and regulation. J Biol Chem, 276, 884-894.
Pettitt, A.R., Sherrington, P.D. and Cawley, J.C. (2000) Role of poly(ADP-ribosyl)ation in the killing of chronic lymphocytic leukemia cells by purine analogues. Cancer Res, 60, 4187-4193.
Phale, P.S., Philippsen, A., Kiefhaber, T., Koebnik, R., Phale, V.P., Schirmer, T. and Rosenbusch, J.P. (1998) Stability of trimeric OmpF porin: the contributions of the latching loop L2. Biochemistry, 37, 15663-15670.
Phale, P.S., Philippsen, A., Widmer, C., Phale, V.P., Rosenbusch, J.P. and Schirmer, T. (2001) Role of charged residues at the OmpF porin channel constriction probed by mutagenesis and simulation. Biochemistry, 40, 6319-6325.
Ranquin, A. and Van Gelder, P. (2004) Maltoporin: sugar for physics and biology. Res Microbiol, 155, 611-616.
Ribeiro, J.M. and Valenzuela, J.G. (2003) The salivary purine nucleosidase of the mosquito, Aedes aegypti. Insect Biochem Mol Biol, 33, 13-22.
Roditi, I., Schwarz, H., Pearson, T.W., Beecroft, R.P., Liu, M.K., Richardson, J.P., Buhring, H.J., Pleiss, J., Bulow, R., Williams, R.O. and et al. (1989) Procyclin gene expression and loss of the variant surface glycoprotein during differentiation of Trypanosoma brucei. J Cell Biol, 108, 737-746.
Saerens, D., Frederix, F., Reekmans, G., Conrath, K., Jans, K., Brys, L., Huang, L., Bosmans, E., Maes, G., Borghs, G. and Muyldermans, S. (2005) Engineering camel single-domain antibodies and immobilization chemistry for human prostate-specific antigen sensing. Anal Chem, 77, 7547-7555.
Safra, T., Muggia, F., Jeffers, S., Tsao-Wei, D.D., Groshen, S., Lyass, O., Henderson, R., Berry, G. and Gabizon, A. (2000) Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol, 11, 1029-1033.
Saint, N., Lou, K.L., Widmer, C., Luckey, M., Schirmer, T. and Rosenbusch, J.P. (1996) Structural and functional characterization of OmpF porin mutants selected for larger pore size. II. Functional characterization. J Biol Chem, 271, 20676-20680.
Samartzidou, H. and Delcour, A.H. (1998) E.coli PhoE porin has an opposite voltage-dependence to the homologous OmpF. Embo J, 17, 93-100.
Stoenescu, R., Graff, A. and Meier, W. (2004) Asymmetric ABC-Triblock Copolymer Membranes Induce a Directed Insertion of Membrane Proteins. Macromol Biosci, 4, 930-935.

State of the art: a nanoreactor coming to life
69
van Alphen, L., Verkleij, A., Leunissen-Bijvelt, J. and Lugtenberg, B. (1978) Architecture of the outer membrane of Escherichia coli. III. Protein-lipopolysaccharide complexes in intramembraneous particles. J Bacteriol, 134, 1089-1098.
Van Gelder, P., de Cock, H. and Tommassen, J. (1996) Assembly of bacterial outer membrane proteins. In von Heijne, G. (ed.), Membrane protein assembly. R G Landes Company, Georgetown,TX, pp. 63-82.
Van Gelder, P., Dumas, F., Bartoldus, I., Saint, N., Prilipov, A., Winterhalter, M., Wang, Y., Philippsen, A., Rosenbusch, J.P. and Schirmer, T. (2002) Sugar transport through maltoporin of Escherichia coli: role of the greasy slide. J Bacteriol, 184, 2994-2999.
Van Gelder, P., Saint, N., Phale, P., Eppens, E.F., Prilipov, A., van Boxtel, R., Rosenbusch, J.P. and Tommassen, J. (1997) Voltage sensing in the PhoE and OmpF outer membrane porins of Escherichia coli: role of charged residues. J Mol Biol, 269, 468-472.
Versees, W., Decanniere, K., Pelle, R., Depoorter, J., Brosens, E., Parkin, D.W. and Steyaert, J. (2001) Structure and function of a novel purine specific nucleoside hydrolase from Trypanosoma vivax. J Mol Biol, 307, 1363-1379.
Versees, W., Loverix, S., Vandemeulebroucke, A., Geerlings, P. and Steyaert, J. (2004) Leaving group activation by aromatic stacking: an alternative to general acid catalysis. J Mol Biol, 338, 1-6.
Versees, W., Van Holsbeke, E., De Vos, S., Decanniere, K., Zegers, I. and Steyaert, J. (2003) Cloning, preliminary characterization and crystallization of nucleoside hydrolases from Caenorhabditis elegans and Campylobacter jejuni. Acta Crystallogr D Biol Crystallogr, 59, 1087-1089.
Vu, K.B., Ghahroudi, M.A., Wyns, L. and Muyldermans, S. (1997) Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol Immunol, 34, 1121-1131.
Woodle, M.C., Engbers, C.M. and Zalipsky, S. (1994) New amphipatic polymer-lipid conjugates forming long-circulating reticuloendothelial system-evading liposomes. Bioconjug Chem, 5, 493-496.
Wotring, L.L. and Roti Roti, J.L. (1980) Thioguanine-induced S and G2 blocks and their significance to the mechanism of cytotoxicity. Cancer Res, 40, 1458-1462.
Ye, J. and Van Den Berg, B. (2004) Crystal structure of the bacterial nucleoside transporter Tsx. EMBO J, 23, 3187-3195. Epub 2004 Jul 3122.


Chapter 3 Encapsulation of therapeutic NH in functionalised nanocapsules
71
Chapter 3: Encapsulation of
therapeutic nucleoside hydrolase
in functionalised nanocapsules
Gerard Huysmans, An Ranquin, Lode Wyns, Jan Steyaert, Patrick Van Gelder
Abstract
Liposomes are introduced as encapsulating carrier for prodrug activating enzymes. Inosine- adenosine- guanosine preferring nucleoside hydrolase of Trypanosoma vivax, a potential prodrug activating enzyme, was encapsulated in functionalized dioleyl-phosphatidylglycerol/egg-phosphatidylglycerol (DOPC/EPG) liposomes. First, transport of nucleosides through general diffusion porins OmpF and PhoE was measured in swelling assays, after which fully functional nanoreactors were developed. Enzyme catalysis of p-nitrophenylriboside, a substrate analogue for nucleoside hydrolases, was significantly higher in permeabilized vesicles than in control vesicles without porins. Residual activity of control vesicles possibly resides in an interaction between the enzyme and the liposomes. This interaction was not of electrostatic nature, since it remained unaffected after the addition of high salt or after perturbation of liposome surface charge and charge density. With these vesicles, we have introduced a new strategy for prodrug therapy, combining the benefits of ADEPT and liposome targeting strategies. Part of this work was published in Journal of Controlled Release 102 (2005) 171–179

Chapter 3
72

Encapsulation of therapeutic NH in functionalised nanocapsules
73
3.1 Introduction
Cancer is a major death cause throughout the world. Current strategies as
radiotherapy and surgery have only gained limited success in the early stages of
the disease and mostly chemotherapy is necessary, because the tumor is only
detected in a later stage (Aghi et al., 2000). Most anticancer drugs are anti-
proliferative agents that display little selectivity, also damaging normal
proliferating cells and thus have a limited therapeutic index. As a consequence,
anticancer drugs are administered in suboptimal conditions, promoting therapy
failure as well as the development of drug resistant cancer cells. Recent
developments in oncogenesis have introduced more selective chemotherapeutics
or lead to direct targeting of anticancer drugs or prodrugs to the tumor site
(Allen, 2002; Torchilin, 2000).
Liposomes can carry a large load of drugs to the tumor site without the need
for covalent linkage between the carrier and the drug. Consequently, antibody
directed liposomes have been proposed as carriers to deliver drugs or prodrugs
locally to the tumor (Gregoriadis, 1995). Subsequent destabilization of the
liposome in the micro-environment of the tumor allows the release of the
therapeutics. Prodrugs can then be metabolized through the physical properties
of the micro-environment or through endogenous enzymes (Denny, 2001).
In combination with endogenous enzymes the use of prodrugs is rather
limited. Therefore intensive research is made into two promising paths to
broaden the range of usable prodrugs (Greco and Dachs, 2001; Senter and
Springer, 2001; Xu and McLeod, 2001): antibody directed enzyme prodrug
therapy or ADEPT and gene-directed enzyme prodrug therapy or GDEPT. Both
strategies are two-step processes, in which firstly an exogenous enzyme is
delivered to the tumor site and in which during the second step this enzyme is
used to locally convert a systematically administered prodrug into a toxic agent.
In ADEPT strategies, the enzyme is targeted to the tumor site in a fusion with a
monoclonal antibody; in GDEPT, the so called “suicide gene” for the enzyme is
brought directly into the cancer cells, mostly through a viral vector. As for
ADEPT, the major obstacle might be the immune response of the host to the
exogenous enzyme (Allen, 2002), hurdles in GDEPT are the membrane

Chapter 3
74
permeability for prodrugs and the lacking of an efficient gene transfer (Springer
and Niculescu-Duvaz, 2000).
With our research objectives, we introduce a complementary strategy, being
therapeutic nanoreactors. By combining the benefits of the above described
therapies, we wish to overcome current problems with ADEPT and GDEPT,
through encapsulating an exogenous enzyme in a non-immunogenic and
permeabilized carrier. As in ADEPT, the reactor will be targeted to the tumor
through a cancer specific monoclonal antibody. Encapsulation of the exogenous
enzyme makes evasion of the host immune system possible and permeabilization
will ensure that prodrugs and hydrolysed metabolites can pass through the
vesicle wall.
Initially, liposome formulations will be developed to encapsulate the enzyme.
Already a plethora of liposome encapsulated enzymes have found applications in
biomedical industry, reviewed by (Walde and Ichikawa, 2001), but
proteoliposomes in the described formulations only function as a stabilizing and
protective carrier rather than being a functional reactor shell. Permeabilization
will be obtained with prokaryotic outer membrane proteins in the nanovesicle
shell. Reconstitution of outer membrane proteins in liposomes is widely used for
functional characterization of these proteins (Nikaido, 2003) and has recently
been explored in terms of nanoreactors with acetylcholinesterase (Colletier et al.,
2002; Nasseau et al., 2001) and β-lactamase (Graff et al., 2001). In both
reactors, OmpF was functionally capable of transporting the enzymatic substrate
(acetylcholine and ampicillin, respectively) inside the reactor and of releasing the
metabolized reaction products back into the environment.
Here, we report our first findings based upon the encapsulation of the inosine–
adenosine–guanosine preferring nucleoside hydrolase (TVNH; EC 3.2.2.1) of
Trypanosoma vivax (Versees et al., 2001) in porin permeabilized liposomes
(Figure 1) and show that these reactors are significantly more active than control
reactors without porins. Nucleoside hydrolases catalyse the hydrolysis of the N-
glycosidic bond in ribonucleosides. Given that no homologues of this class of
enzymes are found in mammalian cells (Hammond and Gutteridge, 1984), these
enzymes are attractive for use in cancer prodrug therapy since endogenous
hydrolysation of the prodrug will be limited.

Encapsulation of therapeutic NH in functionalised nanocapsules
75
Figure 1: schematic representation of a therapeutic nanoreactor with a lipid shell encapsulating TVNH and functionalised with bacterial outer membrane protein OmpF. Substrate analogue 6-methyl-purine riboside, a potential prodrug, diffuses into the reactor through OmpF and is converted into toxic 6-methylpurine, which is released back into the environment through diffusion.
3.2 Materials and methods
3.2.1. Purification of T. vivax nucleoside hydrolase
Purification of the wild type TVNH was performed as described by Versées et
al. (Versees et al., 2001). Shortly, TVNH was purified in a two step purification
scheme from Escherichia coli strain WK6 (Zell and Fritz, 1987) containing the
gene coding for the enzyme in a pQE-30 expression vector (Qiagen). In a two-
step purification protocol, the presence of an N-terminal His6-tag allowed
recovery of the protein on a Ni-NTA affinity column (Qiagen). pH gradient step
elution from the affinity column was followed by gel filtration on a superdex-200
column (Amersham Bioscience). SDS polyacrylamide gel electrophoresis
confirmed the purity of the enzyme.

Chapter 3
76
3.2.2. Purification of E. coli porins OmpF and PhoE
BL21(DE3) ∆lamB ∆ompC ∆ompA ompF::Tn5 and BL21(DE3) ∆lamB ompR
∆ompA strains were used for overproduction of OmpF and PhoE, respectively, as
described by Prilipov et al. (Prilipov et al., 1998). Overproduced porins were
subsequently purified by combining detergent extraction methods according to
Agterberg et al. (Agterberg et al., 1990) and Garavito and Rosenbusch (Garavito
and Rosenbusch, 1986). After cell harvesting by centrifugation, the cell pellets
were resuspended in 2% SDS 20 mM Tris–HCl pH 8 and shaken for 1 h at 60 °C
to obtain cell lysis. Lysates were pelleted by ultracentrifugation (20,000 rpm, 30
min, 4 °C) and resuspended in 0.125% octylpolyoxyethylene (oPOE) 20 mM
NaH2PO4 pH 7.3 for pre-extraction at 37 °C for 45 min. After a second cycle of
ultracentrifugation, pellets were resuspended in the same phosphate buffer with
3% oPOE for overnight extraction at 4°C and an additional 45 min at 37°C.
Finally, 10 mM EDTA was added to the supernatant after ultracentrifugation, to
strip of lipopolysaccharides. Purified proteins were concentrated (amiconR MWCO
5000) and dialyzed (Spectra/Por MWCO 6500-8000) against 20 mM NaH2PO4 150
mM NaCl 1% oPOE pH 7.3. SDS-polyacrylamide gel electrophoresis ascertained
the purity of both porins.
3.2.3. Electrophysiology
Black lipid membrane experiments were carried out as described previously by
Van Gelder et al. (Van Gelder et al., 2000) Conductance properties of OmpF and
PhoE channels were characterized in symmetrical solutions of 1 M KCl 1 mM
CaCl2 10 mM Tris pH 7.4, using soy bean phosphatidylcholine (Sigma) bilayers
over the circular aperture in the Teflon septum. Porins inserted spontaneously
after injection. Steady-state (I/V) curves were recorded with a BLM-120 Bilayer
Membrane Amplifier (Bio-Logic) and digitalized through a Powerlab/4SP
(ADInstruments). Conductance histograms result from at least 250 channel
events.

Encapsulation of therapeutic NH in functionalised nanocapsules
77
3.2.4. Preparation of proteoliposomes and swelling assays
Production of proteoliposomes was initially described by Nikaido and co-
workers (Luckey and Nikaido, 1980; Nikaido et al., 1991; Nikaido and
Rosenberg, 1983). Briefly, dried lipids (DOPC/EPG 4:1) were dissolved in diethyl
ether and a smooth film was formed under a gentle stream of nitrogen. After
drying, vesicles were spontaneously formed upon the addition of buffer (20 mM
Hepes pH 7.0). Porins were reconstituted at a concentration of approximately 20
ng µl-1 in buffer. Subsequently, all liposomes were sonicated and dried. For
swelling assays, liposomes were resuspended in 17% dextrane (MW 40,000,
Fluka) and shaken vigorously after 1 h. Swelling assays were performed as
reported previously by Nikaido (Nikaido and Rosenberg, 1983). Liposome
swelling upon the addition of substrate was followed 1 min
spectrophotometrically at 500 nm and transport flux was determined as follows:
dtdA
Ai⎟⎠⎞
⎜⎝⎛=Φ
1
Where Φ is the transport flux, Ai the initial absorption and dA/dt the
absorption difference during the measured time. Transport of substrates was
measured under isotoneous concentration as determined with raffinose, a higher
molecular weight sugar that is unable to diffuse through the porins. Under this
concentration, the scattering signal of the liposomes remains constant
throughout the measuring time within the sensitivity of the measurement,
indicating no swelling nor shrinking of the liposomes and making visualization of
solute transport unambiguously possible. Accordingly, arabinose, ribose, glucose,
inosine and cytidine added at the isotoneous concentration were tested for
transport through porins.
3.2.5. Preparation of enzyme encapsulating proteoliposomes
For the development of enzyme encapsulating proteoliposomes, sonicated and
dried liposomes as described above were resuspended in buffer containing 0.5
mg ml-1 TVNH. Subsequently, eight freeze–thaw cycles and extrusion through

Chapter 3
78
200 nm polycarbonate filters provided uniform and unilamellar proteoliposomes.
Uniform size distribution was visualized through dynamic light scattering at 532
nm with a scattering angle of 90° (Laser-Spectroscatter 201, RiNA netzwerk RNA
technologien). Free enzyme was removed through metal affinity chromatography
(QIAexpress Ni-NTA Protein purification system, QIAGEN). Activity of the
reactors was followed spectrophotometrically at 400 nm by enzymatic hydrolysis
of 1 mM p-nitrophenylriboside in buffer.
3.2.6. Fluorescence measurements
Fluorescence of free enzyme and of free enzyme in the presence of liposomes
(20–80 µl ml-1) was followed between 300 and 400 nm with a BowmaR Series 2
Luminescence Spectrometer after excitation at 280 nm (slit width of 4 nm and
scan speed of 1nm s-1).
3.3 Results
3.3.1. Purification and functional characterization of TVNH and porins
TVNH purification resulted in a single band on Coomassie stained SDS-PAGE
(Figure 2). Although kinetic parameters were slightly different from previously
reported values (Versees et al., 2001), the enzymatic characteristics of the
protein showed that it was fully functional (Table 1) and measured differences in
kinetic parameters are most likely due to the different buffer system.
SDS extraction of porins from the cell-envelope fraction yielded highly pure
material (Figure 2). The trimeric form, which runs on SDS-PAGE at 95 kDa when
non-boiled, is converted to a 36-kDa band when heated at 100 °C for 10 min.
The double band observed in the trimer lane for both porins is due to residual
lipopolysaccharides. Full stripping of lipopolysaccharides, however, is not
necessary for our purposes.

Encapsulation of therapeutic NH in functionalised nanocapsules
79
Figure 2: SDS-page gels after Coomassie staining of (a) TVNH, (b) OmpF and (c) PhoE. Lanes 2 and 3 in (b) and (c) represent non-boiled and boiled porins, respectively. Arrows indicate monomers.
Table 1: Kinetic constants of TVNH in 20 mM Hepes pH 7
TVNH TVNH + liposomes
inosine ρ-nitrophenylriboside ρ-nitrophenylriboside
kcat (s-1) 1.226 ± 0.043 0.081 ± 0.003 0.018 ± 0.002
KM (µM) 2.63 ± 0.63 1325 ± 101 2487 ± 571
Reconstitution of the purified porins in planar bilayers or black lipid
membranes (BLM) allows to visualize (and thus ascertain) their functionality
through determination of their conductance. The channel transitions from current
traces of OmpF reconstituted in planar membranes clearly showed one
population with a unit step value of 1.66 ± 0.44 nS which is in fair agreement
with Buehler et al. (Buehler et al., 1991) (Figure 3). Closure of the channels was
observed above the critical voltage of 155 mV as reported by Saint et al. (Saint
et al., 1996). Similarly, the measurement was repeated for PhoE, resulting in a
channel conductance of 1.22 ± 11 nS and closure above 135 mV.
1 2 1 2 3 1 2 3

Chapter 3
80
Figure 3: Conductance measurements of OmpF (A) and PhoE (B). Conductance histograms result from at least 250 channel events
3.3.2. Solute transport through OmpF and PhoE
OmpF and PhoE are general diffusion porins with a molecular exclusion limit of
roughly 600 Da. There is no selectivity for the transported molecules, although
OmpF shows slight cation selectivity while PhoE favours transport of anionic
molecules (Koebnik et al., 2000). Transport efficiency through porins was
ascertained for low molecular weight sugars (arabinose, ribose and glucose) and
nucleosides (cytidine and inosine) by reconstitution of purified porins in
proteoliposomes (Figure 4). Results for both porins were compared after
normalization to diffusion of arabinose. All tested solutes were transported
efficiently, with the exception of inosine: as inosine was still moderately
transported through OmpF, practically no diffusion was observed through PhoE.
Most likely, the voluminous hypoxantine residue lies at the basis of this transport
inefficiency. Interestingly, although only different in the orientation of its
hydroxyl groups in regard to arabinose, ribose diffused more efficiently through
either OmpF or PhoE and was even transported in the absence of porins (results
not shown). As ribose is most likely the slow release product in the reaction
scheme of nucleoside hydrolases (Vandemeulebroucke et al., 2003), this efficient
diffusion comes as an unexpected advantage

Encapsulation of therapeutic NH in functionalised nanocapsules
81
Figure 4: Transport of molecular substrates through porins OmpF (■) and PhoE (□). Transport fluxes were normalized to and recalculated as a percentage arabinose transport, for which transport efficiency was stated 100%.
3.3.3. Activity of nucleoside hydrolase encapsulated in proteoliposomes
Size distributions of proteoliposomes containing 0.5 mg ml-1 TVNH with and
without reconstituted porins were determined by dynamic light scattering. No
significant difference in radius was found between the different reactors: the
control reactors without porins had a mean hydrodynamic radius of 57.28 ±
10.11 nm, while OmpF and PhoE containing reactors displayed radii of 56.90 ±
6.85 and 52.92 ± 11.55 nm, respectively.
The enzymatic activity of encapsulated TVNH was determined using p-
nitrophenyl riboside as a substrate. Due to the low solubility of p-
nitrophenylriboside, it was impossible to perform swelling assays with this
substrate, but based upon the structural analogy with cytidine (Figure 5) and its
comparable molecular weight (271 and 243.2 for p-nitrophenylriboside and
cytidine, respectively), no transport problems through either one of the porins
was expected. Comparison of the hydrolytic activity of OmpF and PhoE
permeabilized reactors showed that both proteoliposomes were equally active.
Since there was no significant difference in hydrodynamic radius of the porins
OmpF and PhoE, it is obvious that diffusion of the enzyme substrate across both
porins occurred at comparable rates.

Chapter 3
82
Figure 5: Molecular structures of cytidine (left) and p-nitrophenylriboside (right).
Controls clearly were less active than either OmpF or PhoE permeabilized
liposomes (Figure 6). The residual enzymatic activity of the control vesicles
might stem from free TVNH after the affinity chromatography separation step,
but can also occur from adsorbed enzyme at the liposome surface. However, the
higher activity of permeabilized liposomes cannot be a result of more adsorbed
enzyme. The hydrodynamic radii of control and functionalized vesicles were equal
within experimental errors and furthermore there is no scientific ground to
assume that more protein will adsorb to a porin containing liposome.

Encapsulation of therapeutic NH in functionalised nanocapsules
83
Figure 6: Activity of TVNH encapsulated in OmpF (▲) and PhoE (o) permeabilized liposomes and in non permeabilized control vesicles (■). Liposomes consisting of DOPC and EPG in a 4:1 ratio were developed. Activity was measured with 1 mM final concentration p-nitrophenolriboside in 20 mM Hepes pH 7.0 at 400nm. Error bars are too small to show.
Another plausible explanation to the hydrolyzing activity of controls is that the
liposome lipid bilayer is permeable to the more hydrophobic p-
nitrophenylriboside. This seems unlikely with the rather high molecular weight of
p-nitrophenylriboside. Nevertheless, p-nitrophenylriboside might locally
destabilize the lipid bilayer and thus initiate its transport across the bilayer. In
this respect, Walde and Marzetta (Walde and Marzetta, 1998) already reported
membrane permeability for higher molecular weight hydrophobic molecules as
substrates to chymotrypsin encapsulating liposomes. Furthermore, Chakrabarti
et al. (Chakrabarti et al., 1994) reported membrane permeability for ADP with
polynucleotide phosphorylase encapsulated in POPC-liposomes. With the
surprising transport efficiency of ribose in mind, we could suggest a favourable
interaction of the phenyl part of p-nitrophenylriboside with the lipid bilayer.
3.3.4. Interactions between TVNH and liposomes
Adsorption of encapsulated enzymes on lipid surfaces is reported numerously
in literature and was reviewed recently (Walde and Ichikawa, 2001). To
determine whether or not the TVNH interacts with the liposomes, additional
experiments were performed with empty and non functionalized control
liposomes as described for swelling assays, to which TVNH was added externally

Chapter 3
84
and co-incubated for 90 min prior to activity measurements with p-
nitrophenylriboside. Thus, inhibition of TvNH activity was observed dependent on
liposome concentration and incubation time (results not shown). Determination
of the kinetic parameters resulted in a decrease in kcat as well as an increase in
KM in comparison to earlier published results from our group (Versees et al.,
2001) (Table 1). However, it still remains unclear whether these data reveal a
real shift in kinetic parameters or whether a (partial) inactivation of part of the
enzyme results in an apparent change of the parameters.
Although we could not unambiguously determine the cause of this inhibition,
fluorescence experiments do indicate a direct or indirect interaction between the
TvNH and the liposomes. TVNH shows a characteristic strong fluorescence signal
which peaks around 340 nm due to two nearly surface exposed tryptophan
residues, responsible for aromatic stacking in the catalytic cleft. Upon the
addition of liposomes, the fluorescence signal of these tryptophans is quenched
in a strongly concentration dependent manner, suggesting shielding of these
residues from the aqueous environment (Figure 7). In contrast, fluorescence of
rhodamine in control experiments with liposomes was not quenched.
Furthermore, this quenching implicates that these interacting TVNH molecules
will be inactivated or at least severely hampered in catalytic activity, explaining
the altered kinetic parameters discussed above. Indeed, as less enzyme
molecules are catalytically active, the conversion rate will decrease, leading to a
lower kcat. An additional increase in KM could point to the above suggested
interaction between the substrate and the liposomes, facilitating substrate
transport inside the vesicles. This leads to an increase in intravesicular substrate
concentration in such a manner that the external substrate concentration is
lower, resulting in an increased apparent KM.

Encapsulation of therapeutic NH in functionalised nanocapsules
85
Figure 7: Fluorescence scan of TVNH between 300 and 400 nm in absence (a) and presence of different amounts of liposome preparations (20 (b), 40 (c) and 80 µl ml-1 (d) after 90 min incubation at 30 °C. Liposomes (20 µl ml-1) alone showed no inherent fluorescence (e). Emission spectrum was measured after excitation at 280 nm.
In an attempt to prevent this enzyme–liposome interaction, a series of
liposomes with different net charge and charge density were constructed. This
can be established in a controllable manner through varying the DOPC/EPG ratio.
Since the enzyme charge around the catalytic cleft is predominantly negative
(Versees et al., 2001), a series of more negative liposomes was developed by
increasing the EPG content. No significant changes in catalysis as compared to
previous liposome measurements were observed upon the addition of the TVNH
(Figure 8). Furthermore, the addition of salt up to 1 M is unable to abolish the
interaction (results not shown). We therefore conclude that the enzyme–
liposome interaction cannot be of electrostatic nature. Instead, a strong
hydrophobic force is found more likely to be responsible for the interaction.
To confirm this hypothesis we looked at the effect of various detergents on the
activity of TVNH. The activities were measured in the presence of detergent with
the substrate inosine in 20mM Hepes pH7.0. The enzymatic reaction was
followed spectrophotometrically at 280 nm and compared to the activity of TvNH
in absence of detergent (Table 2). Since al detergents have a severe negative
effect on the activity of TvNH, it is plausible to say that the interaction between
TvNH and liposomes is of a hydrophobic nature.

Chapter 3
86
Figure 8: Activity of TvNH in presence of DOPC:EPG liposomes after 90 min co-incubation. The enzyme/liposome ratio is in agreement as described for the development of the nanoreactors. Activities were normalized to the liposome density
Table 2: Effects of detergents on the activity of TvNH. Detergents were used at a final concentration of 2x the critical micelle concentration.
detergent % TvNH activity
remaining
Anzergent 3-10 6% Anzergent 3-12 9% SB12 20% n-nonyl-β-D-glucoside 9% n-decyl- β -D-glucoside 39% n-dodecyl- β -D-glucoside 24% n-octyl- β -D-maltoside 4% n-nonyl- β -D-maltoside 11% n-decyl- β -D-maltoside 2% n-undecyl- β -D-maltoside 7%

Encapsulation of therapeutic NH in functionalised nanocapsules
87
3.4 Discussion
This report is a pioneering step to introduce an alternative approach to
liposome based cancer treatment and a new principle for prodrug strategies.
Nucleoside hydrolase of T. vivax, a potential attractive enzyme for cancer
therapy, was encapsulated in liposomes, which were functionalized with specific
porins of E. coli. By liposome swelling assays, we could show the successful
diffusion of the substrates for these enzymes. The enzymatic activity showed
that functionalized reactors were more effective in substrate hydrolysation than
control reactors.
At this point, we were unable to establish whether the shielding of the
tryptophan residues of TvNH was a result from permanent adsorption on the
liposome surface or from the formation of liposome induced enzyme aggregates.
The latter, however, would lead to a less clear liposome concentration dependent
enzyme inactivation since at low liposome concentration the vesicle surface is
still large enough to initiate aggregation of more enzyme molecules. This might
pose problems in terms of immunogenicity.
The techniquely less demanding liposome approach was chosen here to
demonstrate this concept of functionalized nanoreactors. Further experiments
will concentrate on a novel kind of polymer based vesicles instead of natural
lipids. We recently showed that functional Tsx, a nucleoside/nucleotide specific
outer membrane protein of E. coli, could be incorporated in these polymer
vesicles. These polymer vesicles might shield the porins from the immune
system since the polymeric membrane is thicker than a lipid bilayer, and at the
same time prevent non-specific adsorption of enzymes.
This strategy may prove to be an attractive alternative to ADEPT and GDEPT,
overcoming distinct hurdles of these therapies.

Chapter 3
88
3.5 References
Aghi, M., Hochberg, F. and Breakefield, X.O. (2000) Prodrug activation enzymes in cancer gene therapy. J Gene Med, 2, 148-164.
Agterberg, M., Adriaanse, H., Lankhof, H., Meloen, R. and Tommassen, J. (1990) Outer membrane PhoE protein of Escherichia coli as a carrier for foreign antigenic determinants: immunogenicity of epitopes of foot-and- mouth disease virus. Vaccine, 8, 85-91.
Allen, T. (2002) Ligand-targeted therapeutics in anticancer therapy. nature reviews cancer, 2, 750-763.
Buehler, L.K., Kusumoto, S., Zhang, H. and Rosenbusch, J.P. (1991) Plasticity of Escherichia coli porin channels. Dependence of their conductance on strain and lipid environment. J Biol Chem, 266, 24446-24450.
Chakrabarti, A.C., Breaker, R.R., Joyce, G.F. and Deamer, D.W. (1994) Production of RNA by a polymerase protein encapsulated within phospholipid vesicles. J Mol Evol, 39, 555-559.
Colletier, J.P., Chaize, B., Winterhalter, M. and Fournier, D. (2002) Protein encapsulation in liposomes: efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotechnol, 2, 9.
Denny, W. (2001) Prodrug strategies in cancer therapy. European Journal of medicinal chemistry, 36, 577-595.
Garavito, R.M. and Rosenbusch, J.P. (1986) Isolation and crystallization of bacterial porin. Methods Enzymol, 125, 309-328.
Graff, A., Winterhalter, M. and Meier, W. (2001) Nanoreactors from polymer-stabilized liposomes. Langmuir, 17, 919-923.
Greco, O. and Dachs, G.U. (2001) Gene directed enzyme/prodrug therapy of cancer: historical appraisal and future prospectives. J Cell Physiol, 187, 22-36.
Gregoriadis, G. (1995) Engineering liposomes for drug delivery: progress and problems. Trends Biotechnol, 13, 527-537.
Hammond, D.J. and Gutteridge, W.E. (1984) Purine and pyrimidine metabolism in the Trypanosomatidae. Mol Biochem Parasitol, 13, 243-261.
Koebnik, R., Locher, K.P. and Van Gelder, P. (2000) Structure and function of bacterial outer membrane proteins: barrels in a nutshell [In Process Citation]. Mol Microbiol, 37, 239-253.
Luckey, M. and Nikaido, H. (1980) Specificity of diffusion channels produced by lambda phage receptor protein of Escherichia coli. Proc Natl Acad Sci U S A, 77, 167-171.
Nasseau, M., Boublik, Y., Meier, W., Winterhalter, M. and Fournier, D. (2001) Substrate-permeable encapsulation of enzymes maintains effective activity, stabilizes against denaturation, and protects against proteolytic degradation. Biotechnol Bioeng, 75, 615-618.
Nikaido, H. (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev, 67, 593-656.
Nikaido, H., Nikaido, K. and Harayama, S. (1991) Identification and characterization of porins in Pseudomonas aeruginosa. J Biol Chem, 266, 770-779.
Nikaido, H. and Rosenberg, E.Y. (1983) Porin channels in Escherichia coli: studies with liposomes reconstituted from purified proteins. J Bacteriol, 153, 241-252.
Prilipov, A., Phale, P.S., Van Gelder, P., Rosenbusch, J.P. and Koebnik, R. (1998) Coupling site-directed mutagenesis with high-level expression: large scale production of mutant porins from E. coli. FEMS Microbiol Lett, 163, 65-72.

Encapsulation of therapeutic NH in functionalised nanocapsules
89
Saint, N., Lou, K.L., Widmer, C., Luckey, M., Schirmer, T. and Rosenbusch, J.P. (1996) Structural and functional characterization of OmpF porin mutants selected for larger pore size. II. Functional characterization. J Biol Chem, 271, 20676-20680.
Senter, P.D. and Springer, C.J. (2001) Selective activation of anticancer prodrugs by monoclonal antibody-enzyme conjugates. Adv Drug Deliv Rev, 53, 247-264.
Springer, C.J. and Niculescu-Duvaz, I. (2000) Prodrug-activating systems in suicide gene therapy. J Clin Invest, 105, 1161-1167.
Torchilin, V. (2000) drug targeting. European Journal of Pharmaceutical Sciences, 11, 81-91.
Van Gelder, P., Dumas, F. and Winterhalter, M. (2000) Understanding the function of bacterial outer membrane channels by reconstitution into black lipid membranes [In Process Citation]. Biophys Chem, 85, 153-167.
Vandemeulebroucke, A., Versees, W., De Vos, S., Van Holsbeke, E. and Steyaert, J. (2003) Pre-steady-state analysis of the nucleoside hydrolase of Trypanosoma vivax. Evidence for half-of-the-sites reactivity and rate-limiting product release. Biochemistry, 42, 12902-12908.
Versees, W., Decanniere, K., Pelle, R., Depoorter, J., Brosens, E., Parkin, D.W. and Steyaert, J. (2001) Structure and function of a novel purine specific nucleoside hydrolase from Trypanosoma vivax. J Mol Biol, 307, 1363-1379.
Walde, P. and Ichikawa, S. (2001) Enzymes inside lipid vesicles: preparation, reactivity and applications. Biomol Eng, 18, 143-177.
Walde, P. and Marzetta, B. (1998) Bilayer permeability-based substrate selectivity of an enzyme in liposomes. Biotechnol Bioeng, 57, 216-219.
Xu, G. and McLeod, H.L. (2001) Strategies for enzyme/prodrug cancer therapy. Clin Cancer Res, 7, 3314-3324.
Zell, R. and Fritz, H.J. (1987) DNA mismatch-repair in Escherichia coli counteracting the hydrolytic deamination of 5-methyl-cytosine residues. Embo J, 6, 1809-1815.


Chapter 4 Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
91
Chapter 4: Comparison and
characterisation of PMOXA-PDMS-PMOXA
vesicles Abstract The phase behaviour of triblock copolymers in aqueous solutions is controlled by their chemical composition, the length and structure of the individual blocks and the molecular architecture of the whole polymer. In addition the molecular weight distribution of the individual blocks has a profound effect on the phase behaviour of such systems. Small changes in these properties can lead to different phase behaviour in aqueous solutions. Therefore we screened several PMOXA-PDMS-PMOXA polymers to see which polymer batches are able to spontaneously self-assemble in to nanometer-sized vesicles. Two polymer batches that show such phase behaviour were identified. In addition we were able to produce enzymatically active nanoreactors with these batches by encapsulating TvNH in the polymeric vesicles and incorporating the porin Tsx in the polymeric membrane. The nanoreactors were analysed by DLS, AFM and TEM. We also performed a preliminary experiment to determine the interaction of such nanoreactors with macrophages by fluorescently labelling the encapsulated TvNH with Alexa®555.

Chapter 4
92

Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
93
4.1 Introduction
Block copolymers consist of hydrophilic and hydrophobic blocks and behave
similar to conventional surfactants in aqueous solutions. They self assemble into
micelles and vesicles of different size and shape and at high concentrations they
form lyotropic liquid crystalline phases. Their phase behaviour is predominantely
controlled by their chemical composition, the length and structure of the
individual blocks and the molecular architecture of the whole polymer. In
addition the molecular weight distribution of the individual blocks has a profound
effect on the phase behaviour of such systems (Nardin and Meier, 2001). The
ABA triblock copolymer poly(2-methyloxazoline)-b-poly(dimethyl siloxane)-
bpoly(2-methyloxazoline) (PMOXA-PDMS-PMOXA) can self assemble into various
structures in aqueous solutions, including nanotubes, free standing films and
vesicular structures, depending on the experimental conditions and constitution
of the polymer (Sauer and Meier, 2004). This implies that even small changes in
the block copolymer composition can lead to different phase behaviour. The
presence of impurities can also have an effect on the phase behaviour of triblock
copolymers. Thus, there is a high risk for batch variability, since it is very difficult
to precisely control the synthesis and length of both hydrophilic and hydrophobic
blocks. This is something we indeed observed with the PMOXA-PDMS-PMOXA
polymers.
In this study we show the different behaviour of different batches of
PMOXA18-PDMS72-PMOXA18 and PMOXA21-PDMS54-PMOXA21 and the ability of
these triblock copolymers to form self-assembled enzymatically active
nanoreactors that encapsulated nucleoside hydrolase of Trypanosoma vivax
(TvNH) and are permeabilized by the bacterial outer membrane channel Tsx. In a
second part we characterised nanoreactors formed by PMOXA21-PDMS54-PMOXA21
via AFM and TEM and performed a preliminary test to see whether such
nanoreactors can be taken up by macrophages, since uptake by macrophages or
other antigen presenting cells is the first step in the activation of the immune
system.

Chapter 4
94
4.2 Materials and methods
4.2.1. Purification of T. vivax nucleoside hydrolase
Purification of the wild type TVNH was performed as described by Versées
et al. (Versees et al., 2001). Shortly, TVNH was purified in a two step purification
scheme from Escherichia coli strain WK6 (Zell and Fritz, 1987) containing the
gene coding for the enzyme in a pQE-30 expression vector (Qiagen). In a two
step purification protocol, the presence of an N-terminal His6-tag allowed
recovery of the protein on a Ni-NTA affinity column (Qiagen). pH gradient step
elution from the affinity column was followed by gel filtration on a superdex-200
column (Amersham Bioscience). SDS polyacrylamide gel electrophoresis
confirmed the purity of the enzyme (chapter 3).
4.2.2. Cloning, expression and purification of Tsx
For cloning of Tsx, a pGtsx plasmid was constructed (Figure 1). The pGompf
plasmid as described by Prilipov et al. served as a template. Firstly, the gene
encoding OmpF was cut from the vector with restriction enzymes PstI and ClaI.
Secondly, the sequence 5’-
GAACACCACCACCACCACCACCTTGTTCCGCGTGGTAGTAT-3’ encoding a His6-tag
followed by a trombine cleaving sequence was ligated in the vector. Finally the
gene encoding tsx was cloned in this new vector

Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
95
pGnhistromtsx3733 bps
500
1000
15002000
2500
3000
3500
XbaIEcoRIPacI
BsiHKAIBsp1286I
BsrGIPstI
ClaIXhoIISmlIBsmI
BtgIBpu10I
NruIBsiHKAI++EcoRV
Bpu1102IBmrIXcmIVan91IOliI
BamHIXhoII
Bpu1102IEcoO109ISmlI
AccIIIXhoII
NaeINgoMIV
BanIIBsp1286I
BsaAIDraIII
PsiI
BsiHKAIBsp1286I
XhoIIXhoIISmlI
XmnIBsiHKAI
Bsp1286IBsaHI
ScaI
BpmIBmrI
AhdI
XhoIIXhoII
XhoIISmlI
XhoII
SmlIBsiHKAI
Bsp1286I
SmlI
SapIXhoII
nhistrom
Tsx insert
Figure 1: schematic representation of the pGtsx vector. The gene encoding Tsx is cloned between restriction sites ClaI and BamHI (depicted in red) and the sequence encoding the His6-tag and trombine cleaving site is cloned between PstI and ClaI (depicted in blue).
BL21(DE3) ∆lamB ompR ∆ompA strains were used for overproduction of Tsx
as described by Prilipov et al. (Prilipov et al., 1998). For this purpose, the gene
encoding Tsx was cloned in the pGompf plasmid. For purification purposes an
Nterminal 6His-tag was added. Overproduced porins were subsequently purified
by combining detergent extraction methods of Agterberg et al. (Agterberg et al.,
1990) and Garavito and Rosenbusch (Garavito and Rosenbusch, 1986). After cell
harvesting by centrifugation, the cell pellets were resuspended in 2% SDS 20
mM Tris pH 8.0 and shaken for 1 h at 60 °C to obtain cell lysis. Lysates were
pelleted by ultracentrifugation (20,000xg, 30 min, 4 °C) and resuspended in
0.125% oPOE 20 mM NaH2PO4 pH 7.3 for pre-extraction at 37 °C for 45 min.
After a second cycle of ultracentrifugation, pellets were resuspended in the same
phosphate buffer with 3% oPOE for overnight extraction at 4°C and an additional
45 min at 37°C. Finally, 10 mM EDTA was added to the supernatant after
ultracentrifugation, obtaining stripping of lipopolysaccharides. Since Tsx was
expressed with an N-terminal 6His-tag, it was further purified on a Ni-NTA
affinity column (Qiagen) and gel filtration on a superdex-200 column (Amersham
Bioscience) to remove contaminating proteins.

Chapter 4
96
4.2.3. Preparation of nanoreactors
4.2.3.1. Lamellar film rehydration method
Block copolymers and porins are mixed in ethanol to a molar ratio of 10 to 1.
This solution is then dried at the bottom of a glass tube under vacuum to remove
all the remaining solvent. Subsequently, the lamellar film is rehydrated for 24 h
at 4°C by adding PBS pH 7.4 containing 54 µM TvNH to a final polymer
concentration of 1%. This results in swelling of the lamellar film and formation of
vesicles. Finally, extrusion with a polycarbonate filter of 0.2 µm gives
monodisperse vesicles. Non encapsulated TvNH is removed by Ni-NTA affinity
chromatography due to the presence of an N-terminal His6-tag.
4.2.3.2. Solvent evaporation method
A solution of purified porin is mixed with copolymer in ethanol to the desired
molar ratio of protein to polymer (1/10). To encapsulate the enzyme in the
interior of the vesicles, the homogeneous polymer-porin solution is added drop
wise to PBS, pH 7.4 buffer containing TvNH (54 µM) to a final polymer
concentration of 1% and stirred for two hours at room temperature. During this
incubation period the nanoreactors are formed by self-assembly and ethanol is
evaporated. The solution is stirred for an additional 48 h at 4°C. Then the
resulted dispersion is repeatedly extruded through a 0.2 µM polycarbonate filter
to obtain monodisperse vesicles. The non encapsulated TvNH is removed by Ni-
NTA affinity chromatography.
4.2.3.3. Direct dispersion method
In the direct suspension method the polymer is suspended in PBS pH 7.4
containing 54 µM TvNH at a final concentration of 1% and gently stirred at 4°C
overnight to form self-assembled vesicles. To destabilize the polymer membrane
of the vesicles, O.5 % Triton X-100 is subsequently added and the suspension is
sonicated twice in a water bath sonicator for 10s. Porins are added shortly after
the sonication step and will incorporate inside the polymeric wall of the
nanoreactors. Biobeads (5g/ml) (BioRad) are added to remove the detergent

Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
97
Vesicles were repeatedly extruded through a polycarbonate filter and non
encapsulated TvNH is removed by Ni-NTA affinity chromatography..
4.2.4. DLS measurements
The size and polydispersity of the nanoreactors was determined via DLS
(laser-spectroscatter 201 by RiNA GmbH, Berlin, Germany) at 532 nm with a
scattering angle of 90°. Ten data sets were recorded and the size distribution
was analyzed using the software CONTIN.
4.2.5. Reducing sugar assay
To determine the enzymatic activity of encapsulated TvNH for guanosine in the
nanoreactors we used a reducing sugar assay as described by Parkin (Parkin,
1996). Briefly, the enzymatic reaction was stopped by adding a CuSO4–solution.
The Cu2+ is reduced to Cu+
by the reaction product ribose. This reduced Cu reacts
with neocuproin to form a complex. Color (yellow) development of this complex
was achieved by heating the solution 8 min at 95°C and the OD at 450nm was
measured. A standard curve with known ribose concentration was used to
determine the extinction coefficient under the assay conditions.
4.2.6. Atomic Force Microscopy (AFM)
Freshly cleaved mica was functionalized with 3-aminopropyltriethoxysilane
(APES) to allow adsorption of the nanoreactors to the mica chip. This was
done by placing two cups with 30 µl APES and 10 µl N,N-diisopropylethylamine,
respectively, at the bottem of a desiccator, the mica chips were placed at the
upper part of the desiccator and this was incubated for 2 h. The mica chips were
left to cure for an additional 48 h. Subsequently, 20 µl of a 10Ox diluted sample
of nanoreactors prepared with PMOXA21-PDMS54-PMOXA21 via the solvent
evaporation method was placed on the APES-mica chip. After 30 min of
incubation at room temperature, the chip was rinsed 4X with PBS to wash of non
adsorbed nanoreactors. The chip was finally wetted with PBS for scanning in
liquid.

Chapter 4
98
A Nanoscope IIIa atomic force microscope (Digital Instruments/Veeco) in
tapping mode at room temperature was used to acquire 512 x 512 pixel images.
We used an NP tip (Veeco), a 125-µm cantilever with a nominal spring constant
of 50 N m-1 and resonant frequencies in a range of 279 to 362 kHz. The scan
rate was 1.5 Hz and the scan size was 1.5 x 1.5 µm. Images were flattened prior
to analysis using the NanoScope 6.11r1 software (Digital Instruments/Veeco).
4.2.7. Transmission Electron Microscopy (TEM)
To obtain a support film for TEM measurements, carbon was coated onto
copper/formvar (polyvinyl formal) grids by evaporating a uniform sheet of carbon
on the grids using a vacuum evaporator. Samples were prepared by depositing
10 µl of undiluted nanoreactor solution (stock of 10mg polymer/ml) on the
carbon-coated side of the grids. After 30-60 seconds adsorption, the excess of
liquid was removed by touching the grid surface with a filter paper. The grid was
then washed 3 times with a drop of distilled water.
The specimen was stained by lowering the grid twice on a drop of negative
stain solution (1-2 % uranyl acetate) for 20 seconds.
Grids were analysed using a Phillips Tecnai 10 electron microscope operating
at 80 kV. Micrographs were recorded with a SIS camera.
4.2.8. Fluorescent labelling of TvNH
To fluorescently label TvNH we used the amine reactive Alexa®555 dye
(Molecular probes). This fluorescent dye has spectra that are identical to those
of the Cy3 dye with an absorption maximum at 555 nm and a fluorescence
emission maximum at 565 nm. This amine reactive succinimidyl ester forms a
stable amide bond with amine groups on the protein. Coupling of the Alexa®555
dye was performed as follows. TvNH was dialysed to 0.1 M sodium bicarbonate
buffer pH 8.3 to a final concentration of 20 mg/ml. Alexa®555 was dissolved in
DMSO to a final concentration of 10mg/ml. 1ml of the TvNH sample was mixed with
100 µl Alexa®555 and incubated for 1 h at room temperature under continuous
stirring. The reaction was stopped by adding 1.5 M of hydroxylamine pH 8.5. After

Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
99
one hour of incubation at room temperature, non bonded Alexa®555 was removed
via gelfiltration on a superdex 75 High Resolution column. The running buffer used
for the gelfiltration was Phosphate Buffered Saline (PBS) pH 7.4. The degree of
labelling (DOL) was determined by:
[ ] dyeproteinMWA
DOLε
max=
Where Amax is the absorption of the protein-dye complex at 555 nm, MW is
the molecular weight of the protein and εdye is the extinction coefficient of the
dye at 555 nm (150.000 cm-1 M-1). The protein concentration in the protein-dye
complex is determined by measuring the absorption at 280 nm and using the
Lambert-Beer law. However, a correction needs to be made for the absorbance
of the dye at 280 nm by using the following equation:
( )CFAAAprotein max280 −=
Where A280 is the absorption of the protein-dye complex at 280 nm and CF is
the correction factor of the dye at 280 nm (0.08).
4.2.9. In vitro nanoreactors-macrophages interactions
Macrophages were recruited to the peritoneum by injection of thioglycolate.
After 24 h 5.107 peritoneal exudate cells (PEC’s) were harvested. The cells
were cultured in 36 well plates at 37°C in RPMI-1640 medium (Invitrogen)
supplemented with 10 % heat-inactivated fetal calf serum, 0.03% L-glutamine,
100 mg/ml streptomycin, 100 mg/ml penicillin, 1mM nonessential amino acids
and 1 mM sodium pyruvate (Invitrogen). After 24 h of incubation the medium
was removed and 50 µl of undiluted fluorescent Alexa®555 nanoreactors was
added. As a negative control, 50 µl of labelled nanoreactors were added to Wells
without macrophages. Pictures were taken after 15 min, 45 min and 17h with an
eclipse TE2000-S microscope (Nikon) fitted with a Chroma filter for excitation
540-580nm and emission 600nm.

Chapter 4
100
4.3 Results
4.3.1. Polymer batch variability
Initial experiments were conducted using the PMOXA18-PDMS72-PMOXA18
triblock copolymer, kindly provided by Prof. W. Meier from the department of
chemistry at the university of Basel (Ranquin et al., 2005). Preparation of
nanoreactors was very reproducible until a new polymer batch was provided.
This new polymer had an identical composition to the first polymer batch.
However, results previously obtained could not be reproduced. With our first
batch of polymer, we used the lamellar film method to produce nanoreactors.
After rehydration of the polymer film, the size of vesicles ranges from 50 nm
to 1µM with a mean diameter of ± 160 nm (Figure 2 A). After extrusion a more
monodisperse vesicle population was obtained with a mean radius of ± 70nm
(Figure 2 B).
With the new polymer batch, large aggregates are formed after film
rehydration with a mean radius of ± 2.2 µm (Figure 3A). These aggregates can
not be extruded since they are too large. Sonication of these aggregates results
in smaller aggregates with a radius ranging from 160 nm to 1,2 µM (Figure 3 B).
After successive extrusion through a 0.45 µM and 0.2 µm filter, a more
monodisperse population was obtained with a mean radius of ± 240 nm (Figure
3 C). Moreover, these nanoreactors were not active, although sonication had no
effect on the activity of the TvNH enzyme (data not shown).

Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
101
A B
Figure 2: DLS measurements of Tsx permeabilized nanoreactors composed of PMOXA18-PDMS72-PMOXA18 (Basel), prepared via the lamellar film rehydration method before extrusion (A) and after extrusion (B).
Preparation via the solvent exchange method resulted in smaller aggregates
but again no activity was found for such aggregates. Finally, preparation via the
direct method resulted in the formation of relatively small aggregates with a
radius of 238 nm (Figure 4 A). Incorporation of Tsx was achieved by destabilizing
the polymer membrane with 0.05 % TritonX-100. After incorporation of Tsx,
TritonX-100 was removed via biobeads. After removal of TritonX-100 we again
obtained very large aggregates with a mean diameter of 1 µm (Figure 4 C).

Chapter 4
102
A B
C
Figure 3: DLS measurements of lamellar film preparation method after film rehydration (A), after sonication (B) and after extrusion (C).
We tested all three preparation methods for several polymer batches without
obtaining small (< 200 nm) active nanoreactors until finally we purchased
PMOXA21-PDMS54-PMOXA21 from Polymer Source Inc. This polymer has the
same hydrophilic and hydrophobic building blocks as the polymer received from
Basel but there is a small difference in the block lengths. The Basel polymer has
a hydrophilic block length of 18 and a hydrophobic block length of 72. Although
the hydrophilic blocks of the Polymer Source polymer are similar in length as the
Basel polymer, the hydrophobic block length is significantly shorter. This might

Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
103
be an advantage for incorporating porins since the polymer membrane thickness
will be more in accordance to the natural environment of porins, i.e. a
phospholipid membrane.
A B
C D
Figure 4: DLS measurements of the direct preparation method after vesicle formation(A), after incorporation of Tsx via TritonX-100 (B) and after removal of TritonX-100 via Biobeads (C). Tsx permeabilized nanoreactors composed of PMOXA21-PDMS54-PMOXA21 (Polymer Source Inc.) prepared via the solvent evaporation method (D).

Chapter 4
104
Nanoreactors with the Polymer Source polymer were prepared via the solvent
exchange method. After formation and removal of non encapsulated TvNH via Ni-
NTA, we obtained vesicles with a mean radius of ± 100 nm (Figure 4D). Since
the sample is already monodisperse, extrusion is not necessary. The activity of
the nanoreactors was measured for guanosine using a reducing sugar assay.
Guanosine concentrations ranging from 50 to 1000 µM were tested and the
obtained data was fitted to a Michaelis-Menten equation (Figure 5). With this
polymer we were able to make small (100 nm radius) and active nanoreactors.
Furthermore, these results were very reproducible. Therefore we chose to use
this polymer for our future experiments.
Figure 5: Activity of Tsx permeabilized nanoreactors that encapsulate TvNH, composed of PMOXA21-PDMS54-PMOXA21 (purchased from Polymer Source Inc.). Reaction rates were determined using a reducing sugar assay. The data were fitted to a Michaelis-Menten equation.
4.3.2. AFM and TEM images of nanoreactors
AFM images clearly show nanoreactors with low polydispersity (Figure 6 A and
B). Analysis of the cross section of the nanoreactors shows a radius of
approximately 75 nm and a height of 10-20 nm (Figure 6C). This is not in
agreement with the results of DLS measurements which showed a mean radius
of 104 nm. Similar observations were made by Meier and co-workers (personal
communication) and this discrepancy between AFM and DLS results was
attributed to the collapse of the vesicles on the mica chip.

Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
105
A B
C
Figure 6: Atomic force microscopy images. Height image (A), amplitude image (B) and nanoreactor cross sections (C). Images were analysed using the Nanoscope software.

Chapter 4
106
Figure 7: TEM images of nanoreactors. Samples were analysed using a Phillips Tecnai 10 electron microscope operating at 80 kV. Micrographs were recorded on with a SIS camera.
Nanoreactors of approximately 170 nm in diameter can be detected on TEM
images (Figure 7). This size is in good agreement with the size measured in DLS
(200 nm) (data not shown). Although the image is of low resolution, the sample
forms uniformly sized vesicles of spherical shape.
4.3.3. Fluorescently labelled Tsx-TvNH nanoreactors
The amine reactive dye Alexa®555 was used to fluorescently label the
encapsulated enzyme TvNH. After removal of the non bonded dye via
gelfiltration, the approximate number of dye molecules per protein molecule
(degree of labelling) was calculated to be 0.5. This fluorescently labelled TvNH
(5µM) was used to prepare fluorescent nanoreactors via the solvent evaporation
method. To perform in vitro macrophage uptake experiment, the fluorescent
vesicles were additionally purified on a superdex 75 HR gelfiltration column with
with PBS, pH 7.4 as running buffer (results not shown). Fluorescent vesicles with
a mean radius of ± 70 nm (Figure 8) were obtained.

Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
107
Figure 8: Analysis of the size-distributions of fluorescent Tsx-TvNH nanoreactors by DLS.
4.3.4. In vitro interaction of fluorescent nanoreactors with macrophages
By producing fluorescent nanoreactors the uptake by or binding to
macrophages can be visualised by fluorescent microscopy. As a negative control
fluorescent nanoreactors were added to wells that do not contain macrophages.
Fluorescent microscopy images of this negative control show a slight increase in
fluorescence over time at the bottom of the well (Figure 9, left panel). In
contrast, when fluorescent nanoreactors are added to cultured macrophages, the
fluorescence increases rapidly near the macrophages (Figure 9, right panel).
After 17 h of incubation, there is a strong fluorescence present on the
macrophages, indicating that nanoreactors are concentrated at the cell surface.
However no clear uptake of vesicles is seen. This concentration of vesicles at the
surface of macrophages, can be the result of nonspecific binding, specific binding
to cell surface receptors of the macrophages or entrapment of the vesicles in the
extra cellular matrix of the macrophage cell culture.

Chapter 4
108
After 15 min
After 45 min
After 17 hours
Figure 9: Fluorescent images of the interaction between macrophages and fluorescent nanoreactors. Left panel: negative control where no macrophages were cultured before adding the fluorescent nanoreactors, Right panel: macrophages were cultured and after removal of culture medium, fluorescent nanoreactors were added. Pictures were taken on an eclipse TE2000-S (Nikon) fluorescent microscope with a Chroma filter for excitation 540-580nm and emission 600nm.

Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
109
4.4 Discussion
Nanoreactors based on triblock copolymers are a completely new and
promising field. Therefore if pharmacological applications on an industrial scale
are to be envisioned it is of uttermost importance that the synthesis of the
polymers can be precisely controlled in order that the different polymer batches
have identical phase behaviour.
Given that the phase behaviour of triblock copolymers in aqueous solutions
depends on the composition, length and structure of the individual blocks as well
as on the molecular architecture of the whole polymer and the molecular weight
distribution of the individual blocks, it is very important to precisely control these
properties to assure the reproducibility of the experiments performed with them.
However, due to the nature of the synthesis of the polymer, it is very difficult to
precisely control the length and molecular weight distribution. From our
experience it is clear that small changes can have profound effect on the
behavior of the polymer in aqueous solutions. Polymers with identical
composition but from a different batch, behave very differently in aqueous
solution where one polymer batch self assembles into nanometer sized vesicles
while another results in the formation of micrometer sized aggregates. This
different behavior was observed for different production methods. The larger
aggregates could be nanotubes or free standing films since no enzymatic activity
could be detected. Several PMOXA-PDMS-PMOXA triblock copolymers with
different compositions were screened for their ability to form enzymatically active
vesicles in aqueous solutions (data not shown). Finally, a commercially available
PMOXA21-PDMS54-PMOXA21, that self-assembles into nanoreactors with a mean
diameter of 150 - 200 nm via the solvent evaporation method was found. By fine
tuning the production method, monodisperse vesicles were obtained without
extrusion. Furthermore, the production of such nanoreactors is highly
reproducible even with different batches.
The size and shape of the nanoreactors was analysed via DLS, AFM and TEM.
AFM images (height and amplitude) showed a monodisperse population.
However, the size determined via AFM does not concur with the size determined
by DLS. With AFM, a mean diameter of 150 nm and a height of 15 nm were
found whereas the mean diameter in DLS was 200 nm. This discrepancy can be

Chapter 4
110
explained by a fundamental difference in both techniques. DLS measurements
are performed in solution whereas in AFM measurements the vesicles are
adsorbed to a mica chip. Adsorption of the vesicles to the chip can lead to
deformation of the vesicles. This was also observed by the group of W. Meier and
is explained as the collapse of the vesicles on the chip which gives the vesicles a
shape comparable with a deflated ball. Also force effects of the sampling AFM tip
might contribute to deformation of the vesicles.
Although the resolution of the TEM image is very low, it also shows a spherical
shaped monodisperse population with a mean diameter of 170 nm. Since
macrophages play an important role in both innate and adaptive immunity we
investigated whether macrophages are able to engulf nanoreactors. Uptake of
nanoreactors can result in the activation of macrophages leading to the
production and secretion of effector molecules such as IL-1β, IL-6, IL-10, IL-12,
IL-18, INF-α/β and TNF-α (Martinez et al., 2008). In addition, breakdown of the
nanoreactors in the lysosome and antigen presentation at the cell surface can
activate CD4+ T-cells leading to a humane or cellular immune response.
Therefore we performed a preliminary experiment to evaluate the in vitro uptake
of fluorescent nanoreactors by macrophages. In this experiment no clear uptake
of nanoreactors was observed during a 17 h period. Since the culture medium
was removed before adding the nanoreactors, the results after 17 h have to be
interpreted with caution. It is well known that deprivation from culture medium
for more than several hours will lead to cell death. Due to the absence of a
positive control, we can not be sure that the lack of a fluorescence signal inside
the macrophages means that the reactors are not taken up by the macrophages.
However, these observations are in good agreement with the findings of Broz et
al. (Broz et al., 2007). They also used PMOXA-PDMS-PMOXA vesicles to target
macrophages. Their goal was to promote macrophage uptake of the vesicles by
targeting the scavenger receptor A1 from macrophages with polyA. Unliganded
vesicles were not taken up. Nevertheless, the uptake of nanoreactors by
macrophages demands further investigation.

Comparison and characterisation of PMOXA-PDMS-PMOXA vesicles
111
4.5 references
Agterberg, M., Adriaanse, H., Lankhof, H., Meloen, R. and Tommassen, J. (1990) Outer membrane PhoE protein of Escherichia coli as a carrier for foreign antigenic determinants: immunogenicity of epitopes of foot-and- mouth disease virus. Vaccine, 8, 85-91.
Broz, P., Marsch, S. and Hunziker, P. (2007) Targeting of vulnerable plaque macrophages with polymer-based nanostructures. Trends Cardiovasc Med., 17, 190-196.
Garavito, R.M. and Rosenbusch, J.P. (1986) Isolation and crystallization of bacterial porin. Methods Enzymol, 125, 309-328.
Martinez, F., Sica, A., Mantovani, A. and Locati, M. (2008) Macrophage activation and polarization. Front Biosci. , 13, 453-461.
Nardin, C. and Meier, W. (2001) Polymerizable amphiphilic block copolymers: From nanostructured hydrogels to nanoreactors and ultrathin films. Chimia, 55, 142- 146.
Parkin, D.W. (1996) Purine-specific nucleoside N-ribohydrolase from Trypanosoma brucei brucei. Purification, specificity, and kinetic mechanism. J Biol Chem, 271, 21713- 21719.
Prilipov, A., Phale, P.S., Van Gelder, P., Rosenbusch, J.P. and Koebnik, R. (1998) Coupling site-directed mutagenesis with high-level expression: large scale production of mutant porins from E. coli. FEMS Microbiol Lett, 163, 65-72.
Ranquin, A., Versees, W., Meier, W., Steyaert, J. and Van Gelder, P. (2005) Therapeutic Nanoreactors: Combining Chemistry and Biology in a Novel Triblock Copolymer Drug Delivery System. Nano Lett, 5, 2220-2224.
Sauer, M. and Meier, W. (2004) Polymer nanocontainers for drug delivery. In, pp. 224- 237.
Versees, W., Decanniere, K., Pelle, R., Depoorter, J., Brosens, E., Parkin, D.W. and Steyaert, J. (2001) Structure and function of a novel purine specific nucleoside hydrolase from Trypanosoma vivax. J Mol Biol, 307, 1363-1379.
Zell, R. and Fritz, H.J. (1987) DNA mismatch-repair in Escherichia coli counteracting the hydrolytic deamination of 5-methyl-cytosine residues. Embo J, 6, 1809-1815.


Therapeutic nanoreactors: combining chemistry and biology in a novel triblock copolymer drug delivery system
113
Chapter 5:
Therapeutic nanoreactors:
combining chemistry and biology in a novel
triblock copolymer drug delivery system
Ranquin An, Versées Wim, Meier Wolfgang, Steyaert Jan and Patrick Van Gelder
Abstract Triblock copolymeric nanoreactors are introduced as an alternative for liposomes as encapsulating carrier for prodrug activating enzymes. Inosine–adenosine–guanosine preferring nucleoside hydrolase of Trypanosoma vivax, a potential prodrug activating enzyme, was encapsulated in nanometer-sized vesicles constructed of PMOXA-PDMS-PMOXA triblock copolymers. The nanoreactor is functionalized by incorporation of bacterial porins, OmpF or Tsx, in the reactor wall. Efficient cleavage of three natural substrates and one prodrug, 2-fluoroadenosine by the nanoreactors, was demonstrated. Part of this work was published in Nanoletters 5(11) (2005) 2220-2224

Chapter 5
114

Therapeutic nanoreactors: combining chemistry and biology in a novel triblock copolymer drug delivery system
115
5.1 Introduction
Currently chemotherapy is one of the major strategies to treat cancer
patients. Despite its success, it is limited by several drawbacks such as low
bioavailability of the chemotoxin, low drug concentrations at the tumor site,
systemic toxicity, lack of specificity and the appearance of drug-resistant tumors.
To overcome these problems many different strategies have been developed
including improved drug formulations (e.g. liposomes) (Vail et al., 2004),
resistance modulators (Bauer et al., 2005) and antidote/toxicity modifiers
(Anderson, 2005). Selective local enzymatic activation of prodrugs is also a
possibility to increase drug concentrations in the tumor and decrease systemic
toxicity (Denny, 2001), thus improving the therapeutic index. Unfortunately
human tumors rarely express useful activating enzymes at high concentrations.
Therefore exogenous enzymes need to be used and directed to the tumor.
Directed enzyme-prodrug therapies involve two stages. In the first step the
activating enzyme is directed to the tumor. In the second step the non-toxic
prodrug is systemically administered and subsequently converted in high local
concentrations of an anticancer drug by the enzyme at the tumor site. The
targeting of the enzyme can either be mediated by antibodies, termed antibody-
directed enzyme-prodrug therapy (ADEPT) or by a gene-vector, termed gene-
directed enzyme-prodrug therapy (GDEPT). Both ADEPT and GDEPT suffer from
the same shortcomings. First, most activating enzymes are immunogenic.
Second, the efficient targeting remains an obstacle and finally, most prodrugs
are also activated by endogenous enzymes.
Several strategies have been proposed to decrease the immunogenicity of the
activating enzyme such as antibody-directed abzyme prodrug therapy (ADAPT)
(Kakinuma et al., 2002). Abzymes are catalytic antibodies that are raised against
a stable transition state analog and can be humanized to reduce immunogenicity.
Their catalytic activity however is usually 1000 fold lower compared to enzymes
that catalyze the same reaction (Avalle et al., 2000). Another way to avoid
immunogenicity is polymer-directed enzyme-prodrug therapy (PDEPT) (Satchi et
al., 2001). In this approach a polymer-drug conjugate is injected first and

Chapter 5
116
accumulates in the tumor tissue by a mechanism called enhanced permeability
and retention effect (EPR) (Matsumura and Maeda, 1986). Afterwards an
enzyme-polymer conjugate is injected that activates the prodrug at the tumor
site. The polymer shields the enzyme from its environment, thereby reducing
immunogenicity. Unfortunately, synthesis of polymer-enzyme conjugates
typically have a low yield and reduced activity of the enzyme (Ashihara et al.,
1978).
We recently proposed a novel strategy in the form of therapeutic nanoreactors
to avoid immunogenicity without loss of enzyme activity (Huysmans et al.,
2005). In this scheme the enzyme is shielded from the immune system by
encapsulation in liposomes. To make these liposomes functional, they were
permeabilized by channel forming proteins to allow diffusion of the substrate and
product but not the enzyme through the reactor wall. Unfortunately, these
reactors showed some uncontrollable characteristics, potentially leading to
immunogenicity of these liposomes. Furthermore, liposome based carriers are
unstable in blood serum and need to be grafted with PEG or other polymers to
increase the average circulation time in the body (Cattel et al., 2004).
In this study we introduce a more promising kind of nanoreactor in which the
reactor wall is composed of the amphiphilic ABA triblock copolymer, poly(2-
methyloxazoline)-poly(dimethylsiloxane)-poly(2-methyloxazoline) (PMOXA-
PDMS-PMOXA) (Figure 1). This copolymer has an average molecular weight of
8660 g/mol and self assembles to form stable vesicular structures in aqueous
solutions (Graff et al., 2002). Containers with controlled mean diameters of 200
nm can be obtained by successive extrusion. Vesicles made of this triblock
copolymer are more stable and less permeable, especially in dilute solutions,
compared to liposomes (Nardin and Meier, 2001). This is due to the higher
length of the hydrophobic block of the polymer, slower dynamics and
intermolecular steric stabilization. Furthermore, these vesicles are completely
covered with PMOXA that has similar stealth properties as PEG. Stealth
properties allow vesicles to escape clearance by the immune system because of
low adsorption of plasma proteins and low hepatosplenic uptake (Woodle et al.,
1994). This results in longer blood circulation times. Also, non specific uptake of

Therapeutic nanoreactors: combining chemistry and biology in a novel triblock copolymer drug delivery system
117
non permeabilized PMOXA-PDMS-PMOXA nanocontainers by COS-7 cells and
THP-1-derived macrophages is completely absent in vitro (Broz et al., 2005),
which makes these nanocontainers biocompatible and good candidates for in vivo
use.
Figure 1: Schematic representation of a functionalized nanoreactor build up of PMOXA-PDMS-PMOXA, permeabilized by the bacterial outer membrane protein Tsx and encapsulated with Trypanosoma vivax Nucleoside Hydrolase (TvNH).
Despite a polymeric membrane thickness of 10 nm, three times wider than a
biological lipid membrane, channel forming proteins have previously been
incorporated in PMOXA-PDMS-PMOXA vesicles without loss of function (Meier et
al., 2000). This is probably due to the high flexibility of the hydrophobic PDMS
block and the presence of shorter polymers which segregate around the protein.
A recent mean field analysis also indicated that protein incorporation only causes
a minor energy penalty in the polymeric membrane (Pata and Dan, 2003). We
permeabilized our nanoreactors by incorporation of two different bacterial outer
membrane, channel forming proteins, called porins (OmpF and Tsx) in the vesicle
wall. OmpF forms a 16-stranded transmembrane β-barrel that functions as a
molecular sieve, allowing concentration driven diffusion of solutes < 600 Da
(Koebnik et al., 2000). Tsx on the other hand forms a 12-stranded β-barrel and
allows specific transport of nucleosides and nucleotides (Ye and Van Den Berg,

Chapter 5
118
2004). Since it has a binding site for nucleosides in the interior of the channel,
rapid transport of nucleosides at very low concentrations is possible compared to
slow diffusion through the nonspecific porin OmpF. By permeabilizing the
nanoreactors with OmpF or Tsx, small substrates can be transported across the
vesicle membrane to reach the interior where they are activated by the enzyme.
Subsequently products can diffuse out of the vesicle to the exterior.
As a proof of principle for our system we chose the purine specific nucleoside
hydrolase of Trypanosoma vivax (TvNH) as prodrug activating enzyme. This
enzyme is a member of the nucleoside hydrolase superfamily that catalyses the
hydrolysis of the N-glycosidic bond of β-ribonucleoside forming the free nucleic
base and ribose (Versees and Steyaert, 2003). These enzymes are widely
distributed in nature but they are not present in mammals. Since TvNH is purine
specific, its natural substrates are inosine, adenosine and guanosine.
Crystallographic data on the T. vivax enzyme showed that the ribose is tightly
bound to the enzyme with all its hydroxyl groups involved in multiple
stereoselective H-bonds (Versees et al., 2001). This makes the enzyme highly
specific towards the ribose moiety. The nucleic base in contrast is stacked
between two tryptophans residues and forms very few specific interactions with
the enzyme. Consequently TvNH is less specific towards the nucleic base moiety.
This feature makes TvNH a promising candidate for enzyme-prodrug strategies
since many known chemotoxins are nucleobase analogs (Klopfer et al., 2004;
Parker et al., 1998).
To explore the possibility of using TvNH encapsulating nanoreactors as
prodrug activating moieties, we measured activities of such nanoreactors for
three natural substrates and one prodrug 2-fluoroadenosine.

Therapeutic nanoreactors: combining chemistry and biology in a novel triblock copolymer drug delivery system
119
5.2 Materials and methods
5.2.1. Purification of TvNH
Purification of the wild type TVNH of T. vivax was performed as described by
Versées et al. (Versees et al., 2001). Shortly, TVNH was purified in a two step
purification scheme from E. coli strain WK6 (Zell and Fritz, 1987) containing the
gene for the enzyme in a pQE-30 expression vector (Qiagen). In a two-step
purification protocol, the presence of an N-terminal His6-tag allowed recovery of
the protein on a Ni-NTA affinity column (Qiagen). pH gradient step elution from
the affinity column was followed by gel filtration on a superdex-200 column
(Amersham Bioscience). SDS polyacrylamide gel electrophoresis confirmed the
purity of the enzyme.
5.2.2. Purification of E. coli porins OmpF and Tsx
BL21(DE3) ∆lamB ompR ∆ompA strains were used for overproduction of OmpF
and Tsx as described by Prilipov et al. (Prilipov et al., 1998). For this purpose,
the gene encoding Tsx was cloned in the pGompf plasmid. For purification
purposes an N-terminal 6His-tag was added. Overproduced porins were
subsequently purified by combining detergent extraction methods of Agterberg et
al. (Agterberg et al., 1990) and Garavito and Rosenbusch (Garavito and
Rosenbusch, 1986). After cell harvesting by centrifugation, the cell pellets were
resuspended in 2% SDS 20 mM Tris pH 8.0 and shaken for 1 h at 60 °C to obtain
cell lysis. Lysates were pelleted by ultracentrifugation (20,000xg, 30 min, 4 °C)
and resuspended in 0.125% oPOE 20 mM NaH2PO4 pH 7.3 for pre-extraction at
37 °C for 45 min. After a second cycle of ultracentrifugation, pellets were
resuspended in the same phosphate buffer with 3% oPOE for overnight
extraction at 4°C and an additional 45 min at 37°C. Finally, 10 mM EDTA was
added to the supernatant after ultracentrifugation, obtaining stripping of
lipopolysaccharides. Purified proteins were concentrated (amiconR MWCO 5000)
and dialyzed (Spectra/Por MWCO 6500-8000) against 20 mM NaH2PO4 150 mM
NaCl 1% oPOE pH 7.3. Since Tsx was expressed with an N-terminal 6His-tag, it
was further purified on a Ni-NTA affinity column (Qiagen) and gel filtration on a

Chapter 5
120
superdex-200 column (Amersham Bioscience) to remove contaminating proteins.
SDS-polyacrylamide gel electrophoresis ascertained the purity of both porins.
5.2.3. Preparation of nanoreactors
To produce permeabilized nanoreactors, a 1 % (w/v) polymer (PMOXA18-
PDMS72-PMOXA18)-ethanol solution was mixed with water solubilised porins,
purificated according to Prilipov et al. (Prilipov et al., 1998). Typically 250 µl of
polymer-ethanol solution was mixed with porin solution to give a final porin
concentration of 0.01 µg/µl or 0.1 µg/µl. This results in a molecular ratio of porin
to polymer of 1:100 and 1:10 respectively. Since a vesicle with a diameter of
about 200 nm contains about 13000 triblock copolymer molecules, this results in
130 to 1300 porin molecules per vesicle (Nardin, 2000). The solution was then
dried to produce a lamellar polymer/porin film. This film was subsequently
rehydrated for several hours under continues stirring in 20 mM Hepes, pH 7.0
containing ± 50 µM of the prodrug activating enzyme TvNH. To obtain a
monodisperse nanoreactor sample, the solution was extruded several times
through a polycarbonate filter with a pore diameter of 200 nm. Since TvNH has
a 6His-tag, non encapsulated prodrug activating TvNH was removed on a Ni-NTA
affinity column (Amersham Biosciences). After each step the samples were
analyzed by DLS to determine the size of the nanoreactors and the polydispersity
of the sample (laser-spectroscatter 201 by RiNA GmbH, Berlin, Germany) at 532
nm with a scattering angle of 90°. Ten data sets were recorded and the size
distribution was analyzed using the software CONTIN.
5.2.4. Trypsin digestion
To make sure that no free TvNH was present in the nanoreactor samples, a
Trypsin digestion was performed. The activity of samples of the nanoreactors
permeabilized with Tsx before Ni-NTA affinity chromatography, the flow through
and the elution was followed spectrophotometrically at 400nm using 1mM p-
nitrophenyl riboside as substrate. This activity was measured again after
incubation at 37°C for 1hour in the presence of Trypsin (16 units).

Therapeutic nanoreactors: combining chemistry and biology in a novel triblock copolymer drug delivery system
121
5.2.5. Activity measurements of nanoreactors
The enzymatic activity of the nanoreactors for three natural substrates
inosine, adenosine and guanosine- and one prodrug, 2-fluoroadenosine was
determined using a reducing sugar assay as described by Parkin (Parkin, 1996).
Briefly, the enzymatic reaction was stopped by adding a CuSO4–solution. The
Cu2+ is reduced to Cu+ by the reaction product ribose. This reduced Cu reacts
with neocuproin to form a complex. Color development (Yellow) of this complex
was achieved by heating the solution 8 min at 95°C and the OD at 450nm was
measured. A standard curve with known ribose concentration was used to
determine the extinction coefficient under the assay conditions.
5.3 Results
5.3.1. Kinetic parameters of TvNH
To ensure the possibility to use nanoreactors encapsulating TvNH as prodrug
activators, the kinetic parameters of TvNH for four prodrugs, 2-fluoroadenosine,
2-chloroadenosine 6-methylpurine riboside and 6-thioguanosine were determined
(Table 1). We found that these prodrugs are efficiently hydrolyzed to their
cytotoxic base with the same efficiency as the natural substrates. Thus, TvNH
can be used as a prodrug activating enzyme in combination with these prodrugs.
Table 1: Kinetic parameters of TvNH for natural substrates and prodrugs
kcat (s -1) KM (µM)
Inosine 5.19 ± 0.08 5.37 ± 0.42
Adenosine 2.6 ± 0.2 8 ± 1.8
Guanosine 2.31 ± 0.11 2.33 ± 0.47
2-fluoroadenosine 1.86 ± 0.11 39.05 ± 7.99
2-chloroadenosine 1.41 ± 0.05 4.56 ± 0.91
6-methylpurine riboside
4.34 ± 0.13 < 10

Chapter 5
122
5.3.2. Preparation of nanoreactors
Size distributions of nanoreactors containing 50 µM TVNH and reconstituted
porins OmpF and Tsx were determined with dynamic light scattering before and
after extrusion through a polycarbonate filter whit a pore diameter of 200 nm.
Before extrusion, reactors with a mean diameter of 362 nm were obtained
whereas the mean diameter decreases to 200nm after the extrusion. No
significant differences were found between OmpF (Figure 2) and Tsx (results not
shown) permeabilized reactors.
Figure 2: Analysis of the size-distributions of OmpF-functionalized nanoreactors by DLS before extrusion (A) and after extrusion (B). Measurements were carried out at 90° and 532 nm on a laser-spectroscatter 201 by RiNa GmbH, Berlin, Germany. Size distributions were determined by CONTIN analysis.
Nanoreactors were subsequently purified on a Ni-NTA column to remove non-
encapsulated TvNH. DLS measurements confirmed that this purification step has
no effect on the size distribution of the nanoreactors (results not shown).
To ensure that this purification step completely removes all non-encapsulated
TvNH, a Trypsin digestion was performed. Enzymatic activity of the sample
before Ni-NTA affinity chromatography, the flow through and the elution (eluted
with 500 µM imidazol) was measured before incubation with Trypsin. This activity
was stated as 100 % activity for each sample. After incubation for 1 hour with
Trypsin, the activity of the different samples was measured again (Figure 3).

Therapeutic nanoreactors: combining chemistry and biology in a novel triblock copolymer drug delivery system
123
Figure 3: Activity measurements of nanoreactors before and after Trypsin digestion. The activity of each sample before Trypsin digestion was stated as 100 % activity. The activity was measured spectrophotometrically at 400 nm with 500 µM of p-nitrophenyl riboside.
For the sample before Ni-NTA and the elution sample, the activity is greatly
decreased by Trypsin digestion which shows that the digestion was successful.
However it was not complete since there is still 55% and 28% of the activity
present, respectively. For the purified nanoreactors (sample flow through)
digestion with Trypsin has no effect on the activity which means that the enzyme
is protected by encapsulation in the polymeric vesicles and that no non-
encapsulated TvNH was present in the sample.
5.3.3. Encapsulation efficiency of TvNH
Since TvNH is deactivated by detergents (see chapter 3), it was impossible to
determine the encapsulated enzyme concentration by dissolving the nanoreactor
and measuring the enzyme activity. Therefore the efficiency of encapsulation was
determined by comparative SDS-page analysis. Known quantities of free TvNH
(0.1-3 µg) were run on an SDS-PAGE together with an aliquot of the nanoreactor
sample (Figure 4). To assure complete disruption of the nanoreactors, 0.5 %
TritonX-100 was added to the sample. The intensity of each coomassie blue
stained TvNH band was measured with Intelligent Quantifier software (Bio Image
Systems Inc., Michigan, USA) to determine the quantity of TvNH in the
nanoreactor sample. A standard curve of band intensity versus known amount of
TvNH was calculated and used to determine the quantity of TvNH in the
nanoreactor sample. This leads to an estimated encapsulation efficiency of 15 %.
0
20
40
60
80
100
120
Before NiNta Flow through Ni-NTA Elution Ni-NTA
% a
ctiv
ity

Chapter 5
124
Two protein bands are visible in the nanoreactor sample corresponding to TvNH
and porin. The porin band however is running at a higher molecular weight than
the purified porin, probably due to strong interaction between polymer and porin.
Therefore we couldn’t use comparative SDS-page analysis to determine the
amount of incorporated porin.
Figure 4: SDS -PAGE analysis for determination of the encapsulation efficiency of TvNH. Free TvNH (0.1-3 µg) was run together with an aliquot of nanoreactor sample (X).
5.3.4. Activity of nanoreactors
To determine the activity of the nanoreactors and compare it to the free
enzyme, we used three natural substrates of TvNH: inosine, guanosine and
adenosine. Additionally we measured the activity of 2-fluoroadenosine to
evaluate the prodrug activating properties of the nanoreactors. 2-
fluoroadenosine is similar to the known prodrug 2-fluoro-2’-deoxyadenosine
which is used in combination with E.coli purine nucleoside phosphorylase (PNP)
in ADEPT and GDEPT therapies (Parker et al., 2003). Activities for all substrates
were measured using a reducing sugar assay described by Parkin et al. (Parkin,
1996)
First of all the activity of non permeabilized nanoreactors with encapsulated
TvNH was measured (data not shown). These nanoreactors showed no activity at
all which is an improvement compared to the liposomal nanoreactors (Huysmans
et al., 2005). For the permeabilized nanoreactors, the rate of product formation
V was determined for various substrate concentrations (0-1000 µM) and for porin
ratios 1:100 and 1:10 OmpF and 1:100 Tsx. These rates were fitted to a
hyperbolic curve to determine apparent kinetic constants (Figure 5) (Table 2).
For fitting purposes, at least 10 individual points were measured per experiment.
NH (µg) 3 2.5 1.5 1 0.5 0.3 0.2 0.1 X
OmpF
TvNH
NH (µg) 3 2.5 1.5 1 0.5 0.3 0.2 0.1 X
OmpF
TvNH
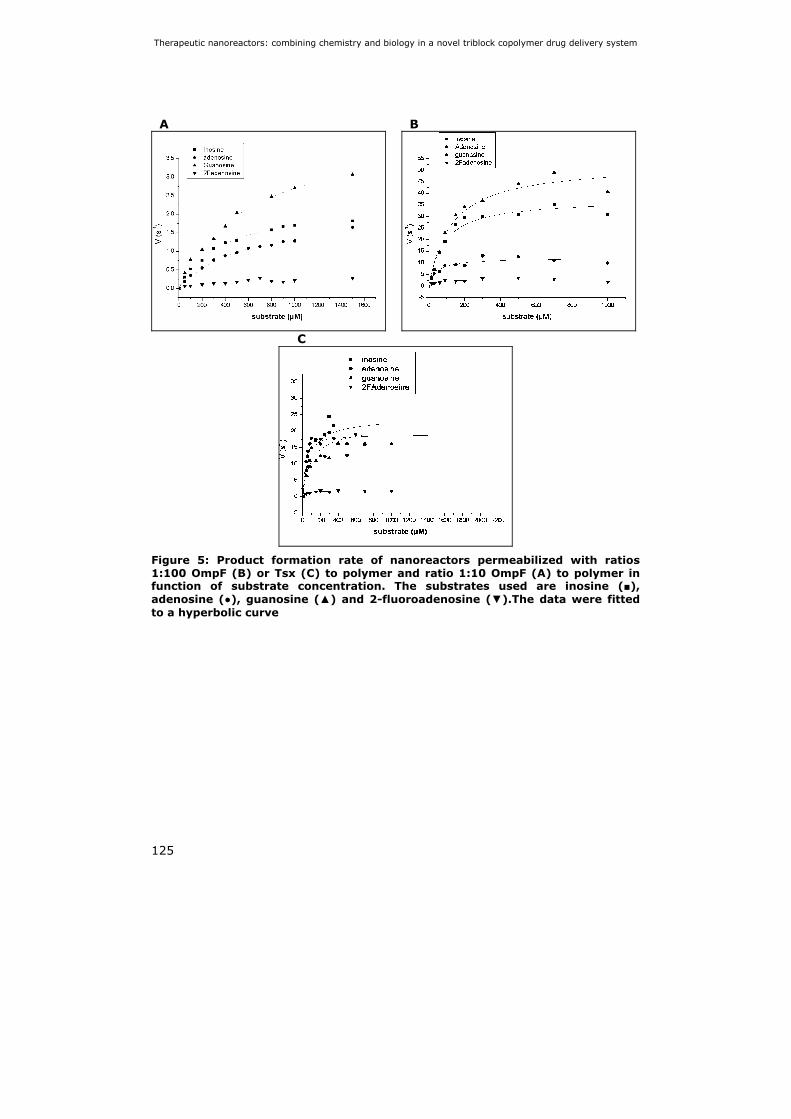
Therapeutic nanoreactors: combining chemistry and biology in a novel triblock copolymer drug delivery system
125
A B
C
Figure 5: Product formation rate of nanoreactors permeabilized with ratios 1:100 OmpF (B) or Tsx (C) to polymer and ratio 1:10 OmpF (A) to polymer in function of substrate concentration. The substrates used are inosine (■), adenosine (●), guanosine (▲) and 2-fluoroadenosine (▼).The data were fitted to a hyperbolic curve

Chapter 5
126
Table 2: Kinetic properties of nanoreactors permeabilized with OmpF or Tsx
The (KM)app value corresponds to the exterior substrate concentration at which
the rate of product formation (V) is half its maximal value (Vmax). Vmax divided by
the total enzyme concentration equals (kcat)app.
In all cases we saw that (KM)app > KM,enzyme. This observation indicates that the
substrate concentration is higher outside the nanoreactor as compared to inside.
Furthermore, the activity of the nanoreactors is a function of the porin to
polymer ratio in the reactor wall. When this ratio increases 10 times (from 1:100
to 1:10) for OmpF permeabilized nanoreactors, the (KM)app and (kcat)app also
increase approximately 10 times. This observation is also seen when Tsx, a
nucleoside specific transporter, is used. All data indicate that the (kcat)app value is
correlated to the amount and the nature of the porin used. It thus appears that
the activity of the nanoreactors is limited by transport at low porin
concentrations. At higher porin concentrations or when the specific transporter
Tsx is used, the (kcat)app value of the nanoreactors is higher than that of the free
enzyme. This might indicate that under these conditions, the activity of the
nanoreactors is no longer limited by transport. The exact reason for this
exceptionally high activity of the nanoreactors is as yet not understood.
Inosine Adenosine Guanosine 2-Fadenosine
kcat (s -1) 5.2 ± 0.1 2.6 ± 0.2 2.3 ± 0.1 1.9 ± 0.1 (TvNH)free
KM (µM) 5.4 ± 0.4 8.0 ± 1.8 2.3 ± 0.5 39.1 ± 8.0 (kcat)app (s -1) 2.3 ± 0.1 2.2 ± 0.1 4.3 ± 0.2 0.3 ± 0.1 1/100 OmpF
nanoreactors (KM)app (µM) 356 ± 23 602 ± 71 582 ± 57 396 ± 169 (kcat)app (s -1) 37.5 ± 1.9 12.1 ± 0.8 52.8 ± 3.1 2.9 ± 0.3 1/10 OmpF
nanoreactors (KM)app (µM) 84 ± 15 41 ± 12 125 ± 23 35 ± 19 (kcat)app (s -1) 24.1 ± 1.5 19.0 ± 1.5 21.5 ± 1.1 1.9 ± 0.1 1/100 Tsx
nanoreactors (KM)app (µM) 80 ± 18 24 ± 17 100 ± 18 38 ± 10

Therapeutic nanoreactors: combining chemistry and biology in a novel triblock copolymer drug delivery system
127
5.4 Discussion
In this study we propose a new therapeutic tool based on nanoreactors that
are composed of PMOXA18-PDMS72-PMOXA18 triblock copolymers. These
nanoreactors were functionalized by encapsulating the prodrug activating
enzyme TvNH and permeabilizing the reactor wall with bacterial membrane
porins OmpF and Tsx.
We were able to produce a monodisperse sample of nanoreactors with a mean
size of 200nm. Since particles with a size of 10-500 nm are able to extravasate
through the leaky blood vasculature of tumor tissue, this is a good size for
passive targeting to tumors via the enhanced permeability and retention effect
(Jain, 1987).
We furthermore proved that the encapsulated enzyme is completely protected
from degradation by proteases which is an enormous advantage compared to
ADEPT strategies were the prodrug activating enzyme is exposed to the
environment.
It was demonstrated that these nanoreactors can efficiently hydrolyze
different substrates including the prodrug 2-fluoroadenosine, resulting in the
release of the cytotoxic molecule, 2-fluoroadenine. Furthermore, nanoreactors
that were not permeabilized by porins showed no activity at all which is a great
improvement compared to liposomal nanoreactors (Huysmans et al., 2005). For
nanoreactors with a high porin content or when the nucleoside specific
transporter Tsx is used we found an (kcat)app that is higher than the kcat of the
free enzyme. It seems unlikely that encapsulation of the enzyme increases the
turn over rate. Therefore this high (kcat)app is probably caused by an
overestimation of the amount of encapsulated enzyme. This means that the
encapsulation efficiency is greater than calculated via the comparative SDS-PAGE
analysis (Knight and Chambers, 2003).
Evidently, these nanoreactors are flexible systems that can be used with
different enzyme/substrate combinations and targeted to different tumor tissues
or organs. Depending on the functionalities of the nanoreactors, they could be
applied in other fields than cancer therapy such as gene-, RNAi- or drug delivery,
diagnostics and in vivo imaging.

Chapter 5
128
5.5 References
Agterberg, M., Adriaanse, H., Lankhof, H., Meloen, R. and Tommassen, J. (1990) Outer membrane PhoE protein of Escherichia coli as a carrier for foreign antigenic determinants: immunogenicity of epitopes of foot-and- mouth disease virus. Vaccine, 8, 85-91.
Anderson, B. (2005) Dexrazoxane for the prevention of cardiomyopathy in anthracycline treated pediatric cancer patients. Pediatr Blood Cancer, 44, 584-588.
Ashihara, Y., Kono, T., Yamazaki, S. and Inada, Y. (1978) Modification of E. coli L-asparaginase with polyethylene glycol: disappearance of binding ability to anti-asparaginase serum. Biochem Biophys Res Commun, 83, 385-391.
Avalle, B., Friboulet, A. and Thomas, D. (2000) Catalysis by anti-idiotypic antibodies. Chem Immunol, 77, 80-88.
Bauer, K.S., Karp, J.E., Garimella, T.S., Wu, S., Tan, M. and Ross, D.D. (2005) A phase I and pharmacologic study of idarubicin, cytarabine, etoposide, and the multidrug resistance protein (MDR1/Pgp) inhibitor PSC-833 in patients with refractory leukemia. Leuk Res, 29, 263-271.
Broz, P., Benito, S.M., Saw, C., Burger, P., Heider, H., Pfisterer, M., Marsch, S., Meier, W. and Hunziker, P. (2005) Cell targeting by a generic receptor-targeted polymer nanocontainer platform. J Control Release, 102, 475-488.
Cattel, L., Ceruti, M. and Dosio, F. (2004) From conventional to stealth liposomes: a new Frontier in cancer chemotherapy. J Chemother, 16 Suppl 4, 94-97.
Denny, W. (2001) Prodrug strategies in cancer therapy. European Journal of medicinal chemistry, 36, 577-595.
Garavito, R.M. and Rosenbusch, J.P. (1986) Isolation and crystallization of bacterial porin. Methods Enzymol, 125, 309-328.
Graff, A., Sauer, M., Van Gelder, P. and Meier, W. (2002) Virus-assisted loading of polymer nanocontainer. Proc Natl Acad Sci U S A, 99, 5064-5068. Epub 2002 Mar 5026.
Huysmans, G., Ranquin, A., Wyns, L., Steyaert, J. and Van Gelder, P. (2005) Encapsulation of therapeutic nucleoside hydrolase in functionalised nanocapsules. J Control Release, 102, 171-179.
Jain, R.K. (1987) Transport of molecules in the tumor interstitium: a review. Cancer Res, 47, 3039-3051.
Kakinuma, H., Fujii, I. and Nishi, Y. (2002) Selective chemotherapeutic strategies using catalytic antibodies: a common pro-moiety for antibody-directed abzyme prodrug therapy. journal of immunological methods, 269, 269-281.
Klopfer, A., Hasenjager, A., Belka, C., Schulze-Osthoff, K., Dorken, B. and Daniel, P.T. (2004) Adenine deoxynucleotides fludarabine and cladribine induce apoptosis in a CD95/Fas receptor, FADD and caspase-8-independent manner by activation of the mitochondrial cell death pathway. Oncogene, 23, 9408-9418.
Knight, M.I. and Chambers, P.J. (2003) Problems associated with determining protein concentration: a comparison of techniques for protein estimations. Mol Biotechnol, 23, 19-28.
Koebnik, R., Locher, K.P. and Van Gelder, P. (2000) Structure and function of bacterial outer membrane proteins: barrels in a nutshell [In Process Citation]. Mol Microbiol, 37, 239-253.
Matsumura, Y. and Maeda, H. (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res, 46, 6387-6392.
Meier, W., Nardin, C. and Winterhalter, M. (2000) Reconstitution of Channel Proteins in (Polymerized) ABA Triblock Copolymer Membranes Angew Chem Int Ed Engl., 39, 4599-4602.
Nardin, C., Hirt, T., Leukel, J., Meier, W. (2000) Polymerized ABA Triblock Copolymer Vesicles. Langmuir, 16, 1035-1041.

Therapeutic nanoreactors: combining chemistry and biology in a novel triblock copolymer drug delivery system
129
Nardin, C. and Meier, W. (2001) Polymerizable amphiphilic block copolymers: From nanostructured hydrogels to nanoreactors and ultrathin films. Chimia, 55, 142-146.
Parker, W.B., Allan, P.W., Hassan, A.E., Secrist, J.A., 3rd, Sorscher, E.J. and Waud, W.R. (2003) Antitumor activity of 2-fluoro-2'-deoxyadenosine against tumors that express Escherichia coli purine nucleoside phosphorylase. Cancer Gene Ther, 10, 23-29.
Parker, W.B., Allan, P.W., Shaddix, S.C., Rose, L.M., Speegle, H.F., Gillespie, G.Y. and Bennett, L.L., Jr. (1998) Metabolism and metabolic actions of 6-methylpurine and 2-fluoroadenine in human cells. Biochem Pharmacol, 55, 1673-1681.
Parkin, D.W. (1996) Purine-specific nucleoside N-ribohydrolase from Trypanosoma brucei brucei. Purification, specificity, and kinetic mechanism. J Biol Chem, 271, 21713-21719.
Pata, V. and Dan, N. (2003) The effect of chain length on protein solubilization in polymer-based vesicles (polymersomes). Biophys J, 85, 2111-2118.
Prilipov, A., Phale, P.S., Van Gelder, P., Rosenbusch, J.P. and Koebnik, R. (1998) Coupling site-directed mutagenesis with high-level expression: large scale production of mutant porins from E. coli. FEMS Microbiol Lett, 163, 65-72.
Satchi, R., Connors, T.A. and Duncan, R. (2001) PDEPT: polymer-directed enzyme prodrug therapy. I. HPMA copolymer-cathepsin B and PK1 as a model combination. Br J Cancer, 85, 1070-1076.
Vail, D.M., Amantea, M.A., Colbern, G.T., Martin, F.J., Hilger, R.A. and Working, P.K. (2004) Pegylated liposomal doxorubicin: proof of principle using preclinical animal models and pharmacokinetic studies. Semin Oncol, 31, 16-35.
Versees, W., Decanniere, K., Pelle, R., Depoorter, J., Brosens, E., Parkin, D.W. and Steyaert, J. (2001) Structure and function of a novel purine specific nucleoside hydrolase from Trypanosoma vivax. J Mol Biol, 307, 1363-1379.
Versees, W. and Steyaert, J. (2003) Catalysis by nucleoside hydrolases. Curr Opin Struct Biol, 13, 731-738.
Woodle, M.C., Engbers, C.M. and Zalipsky, S. (1994) New amphipatic polymer-lipid conjugates forming long-circulating reticuloendothelial system-evading liposomes. Bioconjug Chem, 5, 493-496.
Ye, J. and Van Den Berg, B. (2004) Crystal structure of the bacterial nucleoside transporter Tsx. EMBO J, 23, 3187-3195. Epub 2004 Jul 3122.
Zell, R. and Fritz, H.J. (1987) DNA mismatch-repair in Escherichia coli counteracting the hydrolytic deamination of 5-methyl-cytosine residues. Embo J, 6, 1809-1815.


Chapter Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
131
Chapter 6: Nanoreactor
mediated prodrug activation and killing
of neuroblastoma cells
Abstract
In this study we introduce a novel enzyme-prodrug system as an alternative to antibody-directed enzyme-prodrug therapy (ADEPT), based on a nanoreactor concept. The nanoreactor is a triblock copolymeric vesicle composed of poly(2-methyloxazoline)-poly(dimethylsiloxane)-poly(2-methyloxazoline) (PMOXA-PDMS-PMOXA) that encapsulates the prodrug activating enzyme nucleoside hydrolase of Trypanosoma vivax (TvNH). To permeabilize this vesicle for substrates and products the bacterial nucleoside specific porin Tsx, is incorporated in the polymer membrane. After screening of three compounds: 2-fluoroadenosine, 2-chloroadenosine and 6-thioguanosine we found that 6-thioguanosine is significantly less toxic than the nucleobase 6-thioguanine. The EC50’s are 9,2 and 4,8 µM for 6-thioguanosine after 48 h and 72h of incubation respectively and 3,3 and 1,4 µM for 6-thioguanine after 48 h and 72 h of incubation respectively. We further evaluated the cytotoxic effect of the nanoreactors in combination with the prodrug 6-thioguanosine on neuroblastoma cell cultures and found that 0,33 mg/ml of nanoreactors and 5 µM of 6-thioguanosine is sufficient to kill 80 % of the cell population after 72 h of incubation.
Manuscript in preparation

Chapter 6
132

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
133
6.1 Introduction
Enzyme-prodrug therapies were developed to selectively convert a relatively
non-toxic prodrug to an active drug at the tumor site. This leads to a higher local
drug concentration in the tumor thus improving the anti tumor effect.
Additionally, this lowers the systemic drug concentration hereby reducing
unwanted side effects that accompany conventional cancer chemotherapy. The
enzyme can either be delivered by an antibody-enzyme fusion protein
(antibodydirected enzyme-prodrug therapy, ADEPT) (Bagshawe et al., 2004) or
by a vector carrying the gene encoding for the exogenous enzyme (gene-
directed enzyme-prodrug therapy, GDEPT) (Dachs et al., 2005). Both therapeutic
strategies suffer from difficulties such as immunogenicity of the antibody-enzyme
conjugate in ADEPT systems or inefficient gene transfection, prolonged gene
expression, pathogenicity and immunogenicity in GDEPT strategies.
One way to reduce the immunogenicity of exogenous enzymes, predominantly
from bacterial origin (Senter and Springer, 2001) (Syrigos et al., 1998)
(Stribbling et al., 1997) that are used in enzyme-prodrug therapy is to
encapsulate the enzyme in liposomes. Thus, the enzyme is shielded from the
environment and protected from proteolysis and interaction with components of
the immune system. Such liposomes have to be grafted with polyethyleneglycol
(PEG) molecules to improve their stability in blood plasma and to increase their
circulation time. An alternative for liposome based systems is to make use of
block copolymers that are able to self-organize into nanometersized polymeric
vesicles. The polymeric vesicles are composed of the amphiphilic triblock
copolymer poly(2-methyloxazoline)-poly(dimethylsiloxane)-poly(2-
methyloxazoline) (PMOXA-PDMS-PMOXA) (chapter 2 Figure 1). It was
demonstrated by Nardin et al. that PMOXA-PDMS-PMOXA membranes are
considerably more stable than conventional phospholipid membranes and posses
a high flexibility provided by the PDMS middle block (Nardin, 2000).
Furthermore, the PMOXA outer blocks posses similar stealth properties as PEG
which makes PMOXA-PDMS-PMOXA based vesicles promising for use in
biomedical applications (Woodle et al., 1994). Previously, Meier and co-workers

Chapter 6
134
were able to reconstitute bacterial channelforming proteins such as OmpF
(Nardin et al., 2001) and LamB (Graff et al., 2002) in the triblock copolymer
shell. This way, molecules are able to enter and leave the vesicle’s interior. This
finding opened the door to the development of nanoreactors which can be used
in cancer therapy. Many different enzymes have been used in enzyme-prodrug
strategies including Escherichia cloaca β-lactamase (Cortez-Retamozo et al.,
2004), herpes simplex thymidine kinase (Iwai et al.2002; Mathis et al., 2006), E.
coli nitroreductase (Bilsland et al., 2003; Plumb eal., 2001), yeast cytosine
deaminase (Zeng et al., 2007) and E. coli purinnucleoside phosphorylase (Parker
et al., 2003; Zhou et al., 2007)
We have recently introduced a new strategy for enzyme-prodrug therapy
(Ranquin et al., 2005) (Figure 1) based on nanoreactors. The nucleoside
hydrolase of Trypanosoma vivax (TvNH), was encapsulated inside the polymeric
vesicle (Versees et al., 2001). Although Trypanosoma vivax nucleoside hydrolase
(TvNH) catalyses the hydrolysis of nucleosides and several nucleoside analogues
(Ranquin et al., 2005), it has never been tested as a prodrug activating enzyme.
To make sure substrates and products can readily diffuse in and out of
thepolymeric vesicles, Tsx, a nucleoside specific porin, was incorporated into
thepolymeric membrane. We previously showed that such vesicles were able
tohydrolyse Inosine, adenosine, guanosine and the nucleoside analog 2-
fluoroadenosine (Ranquin et al., 2005).
In this study we validate such a nanoreactor system as an alternative toADEPT
and GDEPT. We have tested the cytotoxic capacity of these nanoreactorson N2A
neuroblastoma cells. To that purpose we have screened severalnucleoside
analogs that are hydrolysed by TvNH into cytotoxic nucleobaseanalogs as
potential prodrugs. 2-fluoroadenine and 2-chloroadenine are knowncytotoxic
agents that are phosporylated by cells and exert their cytotoxic actionat the
nucleoside triphosphate level (Parker et al., 1988). Moreover, 2-fluoro-2’-
deoxyadenosine is already used as a prodrug in combination with E. coli
purinenucleoside phosphorylase in GDEPT systems (Parker et al., 2003). Since
TvNH isunable to hydrolyse 2’-deoxyadenosine and 2’-deoxyadenosine analogs,
we explored whether 2-fluoroadenosine and 2-chloroadenosine can also serve as
prodrugs in combination with TvNH. 6-thioguanine is another known toxic
compound that has been used to treat acute lymphoblastic leukaemia (ALL) over

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
135
the last 45 years. It has also been used to treat inflammatory bowel disease
andorgan rejection after transplantation. Although little is known about the
cytotoxic effects of the nucleoside 6-thioguanosine, it is also a possible prodrug
candidate in our screening.
Figure 1: Schematic representation of a Tsx permeabilized nanoreactor that encapsulates TvNH and is composed of PMOXA-PDMS-PMOXA.
6.2 Materials and Methods
6.2.1. Nucleoside analogs and nucleobase analogs
2-Fluoroadenosine and 2-chloroadenosine were purchased from Sigma and 6-
thioguanosine was purchased from Acor. To obtain the respective nucleobases, a
complete hydrolysis was performed with TvNH and followed
spectrophotometrically at 280 nm for 2-fluoroadenosine, 262 nm for 2-
chloroadenosine and 355nm for 6-thioguanosine. . The reaction was carried out
with 1µM of TvNH at 37°C in PBS pH 7.4. After complete hydrolysis, TvNH was

Chapter 6
136
removed from the solutions by filtration on a Microcon centrifugal filter device
(Millipore) with a cutt-off of 30 kDa.
6.2.2. Cytotoxicity Assay
Neuroblastoma cells (N2A) were plated at a cell density of 104 cells per well in
100 µl Dulbecco’s Modified Eagle’s Medium / Nutrient Mixture F-12 Ham’s Liquid
Medium (DMEM/F12) (GIBCO)). The cells were allowed to adhere to the plates
and were grown at 37°C. After 24 h solutes were added and the cells were grown
at 37°C for an additional 24 h-72 h. The cell viability was determined using an in
vitro toxicity test based on neutral red uptake (purchased from Sigma). The dye
neutral red (Toluylene Red) is taken up by viable cells via active transport and
stored in lysosomes. In contrast non viable cells are unable to take up this dye.
In our cytotoxicity assays, 2µl of neutral red solution was added to each well and
uptake was allowed for 45 min. at 37°C. Subsequently the medium was removed
and the wells were washed with 100 µl of Hanks Balanced Salt Solution (HBSS)
to remove residual dye. The neutral red that was taken up by viable cells was
then solubilised by adding 100 µl of solubilisation solution (50 % ethanol, 1%
Acetic acid). After 10 min of solubilisation at room temperature, the absorbance
at 540 nm of the dye was measured spectrophotometrically. Each condition was
measured fivefold. The Neutral Red uptake for untreated control cells that were
grown in medium for 48 h and 72 h was stated as 100 % cell survival. 0,5 %
sodium azide was used as a positive control and incubated for 4 h. The dye
uptake for this condition was stated as 0 % cell survival.
6.2.3. EC50 determination
To determine the EC50 values of 6-thioguanosine and 6-thioguanine, cells
were cultured as described in 6.2.2. After 24 h of growth, concentrations ranging
from 0 to 200 µM of both compounds were added to the cells. Each concentration
was measured fivefold. After 48 h and 72 h cell survival was determined using
the neutral red in vitro toxicity assay as described in 6.2.2. The survival of
untreated control cells was stated as 100 %. The obtained data were fitted with
the logistic dose response equation from the programme Origin:

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
137
( )
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛+
−+=
p
xx
AAAy
0
212
1
Where A1 and A2 are the maximum and minimum survival percentages
respectively, x0 is the concentration of solute at the midpoint of the sigmoid
curve, i.e. the concentration were half of the toxic effect is observed (EC50) and p
is the power number of the sigmoid curve.
6.2.4. Swelling assays with Tsx proteoliposomes
Proteoliposomes were produced as described by Nikaido and co-workers
(Luckey and Nikaido, 1980; Nikaido et al., 1991; Nikaido and Rosenberg, 1983).
Briefly, 1,2-Dioleoyl-sn-Glycero-3-phosphocholine (DOPC) and egg phosphatidyl
glycerol (EPG) were mixed in a 4:1 ratio and dissolved in diethyl ether.
Subsequently, a smooth film was formed under a gentle stream of nitrogen. After
drying, liposomes were spontaneously formed upon the addition of Phosphate
Buffered Saline (PBS). Proteoliposomes with reconstituted Tsx, were made by
rehydration in PBS that contained purified Tsx (20 µg ml-1). Subsequently,
liposomes were sonicated for 5 seconds in a water bath sonicator. Swelling
assays were performed as described previously by Nikaido (Nikaido and
Rosenberg, 1983). Liposome swelling upon the addition of substrate was
followed for 5 min spectrophotometrically at 500 nm and the transport flux was
determined as follows:
dtdA
Ai⎟⎠⎞
⎜⎝⎛=Φ
1
Where Φ is the transport flux, Ai the initial absorption and dA/dt the
absorption difference during the measured time. Adenosine, adenine, ribose, 6-
thioguanosine and 6-thioguanine were added to the proteoliposome solution at a
final concentration of 0.8 mM. Because of the low solubility of the tested solutes,
the swelling assay was performed under non-isotoneous concentrations.

Chapter 6
138
6.2.5. Preparation of TvNH encapsulated nanoreactors
Nanoreactors permeabilized by Tsx were prepared as follows: PMOXA21-
PDMS54-PMOXA21 (Polymer Source Inc.) was dissolved in ethanol at a
concentration of 8.3 % for 1h at room temperature. Subsequently, Tsx, cloned
and purified as described in chapter 4, was added at a final concentration of
0.1%. This ethanol solution was added drop-wise to a PBS solution containing
150 µM of TvNH (expressed and purified as described in chapter 3) under
continuous stirring. The solution was stirred for an additional 72 h at 4°C. To
remove non-encapsulated TvNH, 1ml of PBS-buffered Ni-NTA resin (GE
Healthcare) was added and allowed to bind his-tagged TvNH for 24 h. The
nanoreactors were separated from the Ni-NTA resin on a PD10 column. The size
and polydispersity of the nanoreactors was determined via DLS
(laserspectroscatter 201 by RiNA GmbH, Berlin, Germany) at 532 nm with a
scattering angle of 90°. Ten data sets were recorded and the size distribution
was analyzed using the software CONTIN.
6.2.6. Enzymatic activity Assay
To determine the activity of Tsx permeabilized nanoreactors and humane
purine nucleoside phosphorylase (hPNP) the rate of hydrolysis of 6-thioguanosine
was measured spectrophotometrically at 355nm. The rate of hydrolysis of
guanosine was measured spectrophotometrically at 300 nm. The reaction was
carried out in PBS, pH 6.6 at 37°C to measure the activity of nanoreactors. To
measure the activity of hPNP, the reaction was carried out in 50 mM Potassium
phosphate buffer pH 7.0. Different concentrations of 6-thioguanosine and
guanosine ranging from 0-1 mM were tested. The obtained reaction rates were
fitted to a Michaelis-Menten equation to determine Vmax and KM :
SKSV
VM +
×= max

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
139
6.2.7. Limulus Amebocyte Lysate (LAL) assay
To determine the endotoxin concentration in the fluorescent nanoreactor
sample, a LAL assay was performed (Cambrex). This test is based on the
activation of a protoenzyme present in the Limulus Amebocyte lysate by
endotoxin. This activated protoenzyme cleaves the colourless substrate Ac-Ile-
Glu-Ala-Arg-pNA to form p-nitroaniline (pNA). The pNA release is continually
measured spectrophotometrically at 405 nm throughout the incubation period.
The concentration of endotoxin in a given sample is calculated from its reaction
time by comparison to the reaction time of solutions containing known amounts
of endotoxin standard.
A sample of 1000X diluted nanoreactors was used to determine the endotoxin
concentration. To obtain a standard curve, E. coli O55:B5 derived endotoxin
(Cambrex) was used in the following concentrations: 0, 0.005, 0.05, 0.5, 5 en 50
endotoxin units (EU)/ml. As a monitor for product inhibition or enhancement, a
positive product control (PPC) was performed by spiking the nanoreactor samples
with 50 EU. Prior to adding the LAL, the samples were heated at 37 °C for 10
min. The calculations of the EU in the nanoreactor samples and the recovery of
EU in the PPC were done by WinKQCL software (Cambrex).
6.3 Results
6.3.1. Cytotoxicity screening of prodrugs and drugs
First we compared the cytotoxicity of prodrugs versus drugs on neuroblastoma
cell lines (N2A cells). The toxic effects on this cell line of 2-fluoroadenosine, 2-
chloroadenosine, 6-thioguanosine, 2-fluoroadenine, 2-chloroadenine and 6-
thioguanine were measured at two concentrations (25 µM and 2.5 µM). A
viability assay was performed after 24 h and 48 h incubation (Figure 2). We
found that 2-fluoroadenosine is very toxic for N2A cells with dye uptake
percentages of approximately 30 and 10 % at 2.5 and 25 µM respectively after
24 h incubation and a dye uptake percentage of 5 % after 48 h incubation
(Figure 2A). Furthermore, the cytotoxicity of the nucleoside is higher than the
cytotoxicity of the nucleobase which disqualifies this molecule as a good prodrug
candidate. 2-Chloroadenosine and 2-chloroadenine show only moderate cytotoxicity

Chapter 6
140
even at the highest concentration and longer incubation periods. As for 2-
fluoroadenosine, the cytotoxicity of 2-chloroadenosine is slightly higher than the
cytotoxicity of its nucleobase. As a result, 2-cloroadenosine is not a good prodrug
candidate (Figure 2B).
In case of 6-thioguanosine, both the nucleoside and nucleobase show no
significant toxicity after 24 h of incubation. After 48 h however, the cytotoxicity of
the nucleobase 6-thioguanine is significantly increased leading to cell survival
percentages of 30 % (Figure 2C). This delayed cytotoxicity is in good agreement with
the findings of Wotring and Roti Roti (Wotring and Roti Roti, 1980). They found that
incorporation of the active metabolite 6-thioGTP into newly synthesized DNA leads to
cell cycle arrest in the G2 phase. Therefore the cytotoxic action of 6-thioguanine only
becomes apparent after completion of the cell cycle in which the 6-thioGTP was
incorporated into the DNA. 6- Thioguanosine however, does not show a significant
increase in cytotoxicity after 48 h. This means that thioguanosine is a good prodrug
candidate for enzymeprodrug therapy.

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
141
A B
05
1015202530354045
25 µM/24 h 2.5 µM/24 h 25 µM/48 h 2.5 µM/48 h
% c
ell s
urvi
val
2-fluoroadenosine2-fluoroadenine
0102030405060708090
100
25 µM/24h 2.5 µM/24h 25 µM/48h 2.5 µM/48h
% c
ell s
urvi
val
2Cl-adenosine2Cl-adenine
C
0
20
40
60
80
100
120
25 µM/24 h 2.5 µM/24 h 25 µM/48 h 2.5 µM/48 h
% c
ell s
urvi
val
6-thioguanosine6-thioguanine
Figure 2: In vitro cytotoxicity screening of candidate prodrugs. (A) compares the cytotoxicity of the prodrug 2-fluoroadenosine to the drug 2-fluoroadenine, (B) compares the prodrug 2-chloroadenosine to the drug 2-chloroadenine and (C) compares the prodrug 6-thioguanosine to the drug 6-thioguanine (C) on N2A cell cultures. Cell survival was determined after 24h and 48h via a neutral red based toxicity assay.
6.3.2. EC50 determination of 6-thioguanosine and 6-thioguanine
Given that 6-thioguanosine is a promising prodrug candidate, we determined
the EC50 for the nucleoside analog, 6-thioguanosine, and the nucleobase analog,
6-thioguanine. To this end we measured the cell viability for concentrations of
both compounds, ranging from 0-200 µM. Since the cytotoxic action of 6-
thioguanine only becomes apparent after 24h, cell survival was measured after

Chapter 6
142
48 h and 72 h of incubation. Cell survival was determined using a Neutral Red
based in vitro toxicity assay (Figure 3, Table 1).
A B
Figure 3: EC50 determination of 6-thioguanosine (A) and 6-thioguanine (B) for N2A cells. Cell survival for several concentrations ranging from 0-200 µM after 48h (■) and 72h (▲) incubation was measured via a Neutral Red based toxicity assay. The data were fitted with the logistic dose response equation in Origin, to determine the EC50 values.
Table 1: EC50 values of 6-thioguanosine and 6-thioguanine.
6-thioguanosine 6-thioguanine
48 h 72 h 48 h 72 h
EC50 9,2 ± 1,3 4,8 ± 0,5 µM 3,3 ± 0,7 µM 1,4 ± 0,2 µM
For 6-thioguanosine cell survival showed an initial plateau phase with a start
of cell death after approximately 2 and 3 µM after 48 and 72 h, respectively. In
contrast 6-thioguanine showed an immediate decrease in cell viability. The EC50
of 6-thioguanine is approximately 3 times lower than the EC50 of 6-thioguanosine
(Table 1), indicating that 6-thioguanine is more toxic to N2A cells than 6-
thioguanosine. Especially at low concentrations (0-10 µM) 6-thioguanine exerts a
high cytotoxicity in comparison to 6-thioguanosine. When used in this
concentration range, 6-thioguanosine can be used as a prodrug.

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
143
6.3.3. 6-Thioguanosine activation by human purine nucleoside phosphorylase (hPNP)
To validate 6-thioguanosine as a prodrug for enzyme-prodrug therapy, it is
important to know that it can not be activated by an endogenous enzyme. This
would lead to toxic side effects and loss of the tumor specificity of the therapy.
Since 6-thioguanosine is also toxic for neuroblastoma cells at high concentrations
we believe that it is activated by the endogenous enzyme purine nucleoside
phosphorylase (PNP). PNP catalyses the cleavage of the glycosidic bond of ribo
and deoxyribonucleosides in the presence of inorganic orthophosphate (Pi) as a
second substrate to generate the purine base and ribo(deoxyribo)-1-phosphate.
Although PNP is a ubiquitous enzyme that is present in many organisms, the
substrate specificity may differ from species to species. Human PNP (hPNP) only
hydrolyses 6-oxopurine nucleosides such as guanosine and inosine or nucleoside
analogs that have an atom of similar electron density at the 6 position (Bzowska
et al., 2000). Therefore 6-thioguanosine may be hydrolyzed by hPNP to form 6-
thioguanine. This is particularly important since hPNP is highly expressed in
lymphocytes. This is well illustrated by the PNP deficiency syndrome in which
patients suffer from severe T-cell immunodeficiency or decreased B-cell function
(Giblett et al., 1975; Markert, 1991) In our experiments we only observed
toxicity of 6-thioguanosine at concentrations > 5 or 10 µM after 48 h and 72 h of
incubation, respectively. To determine whether this cytotoxicity is caused by
hydrolysis of 6-thioguanosine by hPNP, we determined the activity of this
enzyme for 6-thioguanosine and compared it to the activity for the natural
substrate guanosine. The rate of hydrolysis was measured
spectrophotometrically at 300 nm for guanosine and 355 nm for 6-thioguanosine
in 50 mM potassium phosphate buffer pH 7.0. Substrate concentrations ranging
from 0 to 1mM were tested (Figure 4).

Chapter 6
144
A B
Figure 4: Enzymatic activity of human PNP for guanosine (A) and 6-thioguanosine (B). Reactions were carried out in 50 mM potassium phosphate buffer pH 7.0 at 37°C. The data were fitted to a Michaelis–Menten equation.
The affinity of hPNP for 6-thioguanosine (451 µM) is much lower as compared
to guanosine (72 µM), illustrated by a higher KM value for 6- thioguanosine. But
because hPNP has a high turnover rate for 6-thioguanosine, hydrolysis of 6-
thioguanosine by hPNP might lead to lymphocyte cytotoxicity.
6.3.4. Transport of 6-thioguanosine and 6-thioguanine by Tsx
To validate 6-thioguanosine as a prodrug in combination with Tsx
permeabilized nanoreactors, it is important to test if 6-thioguanosine can reach
the encapsulated TvNH allowing activation of 6-thioguanosine to 6-thioguanine.
In addition the produced 6-thioguanine has to be transported to the exterior of
the nanoreactor after formation. Therefore the Tsx mediated transport of 6-
thioguanosine and 6-thioguanine was analysed. To this end we performed
swelling assays with proteoliposomes that incorporate Tsx. Swelling was
recorded spectrophotometrically at 500 nm for 5 min to calculate the flux of
different substrates. Adenosine, which is well transported by Tsx (Benz et al.,
1988), was used as a positive control. In addition, transport of adenine and

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
145
ribose by Tsx proteoliposomes was also measured (Figure 5). Our results indicate
that
Figure 5: Transport through Tsx proteoliposomes. The transport was determined via a swelling assay under non- isotoneous concentrations. Each solute was added to the proteoliposome solution at a final concentration of 0,8 mM. Swelling was measured spectrophotometrically at 500 nm for 5 min.
6.3.5. Kinetics of Tsx-TvNH nanoreactors
Nanoreactors with a mean radius of 103 nm (as determined by DLS, data not
shown) that are permeabilized by Tsx and that encapsulate TvNH were prepared
as described above (6.2.5).
The rate of hydrolysis of 6-thioguanosine by these nanoreactors was
determined spectrophotometrically at 355 nm in a PBS buffer pH 6.6 at 37°C. 6-
Thioguanosine concentrations ranging from 0-300 µM were used in initial rate
experiments and the data were fitted to a Michaelis-Menten equation to
determine the apparent Vmax and apparent KM (Figure 6). Vmax is 1.44 (mg/ml)-1
min-1 and KM is 38 µM. The KM value for these nanoreactors is approximately 10
times higher as compared to the KM of free TvNH for 6-thioguanosine (3.56 µM).
These results are comparable to results obtained previously for inosine,
guanosine, adenosine and 2- fluoroadenosine (Ranquin et al., 2005). This higher
apparent KM value of the nanoreactors can be explained by a difference in
exterior and interior substrate concentrations. If the rate of transport is lower
than the rate of hydrolysis, the interior substrate concentration will be lower than
0
0,005
0,01
0,015
0,02
0,025
0,03Φ
(min
)-1Adenosine
Adenine
ribose
6-thioguanosine
6-thioguanine

Chapter 6
146
the exterior concentration. The interior substrate concentration however is not
known and thus the known exterior concentration is used to determine the
apparent KM value. If we assume that encapsulation of TvNH in the polymeric
nanoreactors does not alter the kinetic behaviour of the enzyme we can conclude
that the interior substrate concentration is 10 times lower than the exterior
concentration. From SDS page analysis the encapsulated enzyme concentration
used in our enzymatic activity was estimated to be 7.5 10-9 M (data not shown).
From this we estimated the kcat for the encapsulated enzyme to be 1.05 s-1 which
is comparable to the kcat value of the free TvNH for 6-thioguanosine ( 1.77 s-1 ).
Figure 6: Enzymatic activity of Tsx permeabilzed nanoreactors that encapsulate TvNH. The reaction rates were measured spectrophotometrically at 355 nm in PBS pH 6.6 at 37 °C. The data were fitted to the Michaelis-Menten equation.
6.3.6. Cytotoxicity of 6-thioguanosine activated by TvNH encapsulating nanoreactors
To explore the therapeutic potential of Tsx permeabilized nanoreactors that
encapsulate TvNH in combination with the prodrug 6-thioguanosine, we
measured the cytotoxicity of the nanoreactors in the presence and absence of
the prodrug. A LAL assay was performed to determine the endotoxin content of
the sample. De endotoxin content was < 50 EU/ml. Since we used only 5 and 10
µl of nanoreactors sample in the assay, interference of endotoxin is not

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
147
expected. Two concentrations of nanoreactors, 0.33 mg/ml and 0.66 mg/ml were
tested in combination with 5, 10, 15 and 20 µM of the prodrug 6-thioguanosine.
The cell survival was measured after 72 h of incubation. As a positive control 0.5
% sodium azide was added and incubated for 4 h. To evaluate the effect of 6-
thioguanosine in combination with the nanoreactors, the dye uptake was stated
as 100% for cells were 0.33 mg/ml or 0.66 mg/ml nanoreactors was added
without prodrug. Comparison of these conditions to the untreated control cells
shows that adding nanoreactors to the culture medium has a minor effect on cell
growth (Figure 7). From the EC50 experiments we know that 4.8 µM of 6-
thioguanosine results in 50 % cell kill after 72 h. However we calculated the time
necessary to hydrolyse 5 µM of 6-thioguanosine with 0.33 mg/ml and 0.66
mg/ml nanoreactors to be 5.4 h and 2.7 h respectively. To completely hydrolyse
20 µM of 6-thioguanosine, 7.7 h and 3.9 h are needed respectively. This means
that the cells are only exposed to 6-thioguanosine for a short term. For both
nanoreactor concentrations used, the survival percentage is already close to the
minimal value and does not decrease much by increasing the 6-thioguanosine
concentration. Furthermore, addition of 0.5 % sodium azide to untreated cells
result in similar dye uptake percentages, suggesting that even at low
concentrations of 6-thioguanosine, all cells are killed. This is to be expected
when the nanoreactor activity is similar in culture medium as in PBS since
approximately 1 µM of 6-thioguanine is sufficient to kill 50% of the cell
population. At 5 µM 6-thioguanosine, this concentration is reached by 0.33
mg/ml and 0.66 mg/ml nanoreactors after 20 min and 10 min respectively. At 20
µM 6-thioguanosine, this is reached after 5 min and 2.5 min respectively. For the
same reason, there is no difference in cytotoxicity between 0.33 mg/ml and 0.66
mg/ml nanoreactors.

Chapter 6
148
Figure 7: Cytotoxicity of 6-thioguanosine activated by Tsx permeabilized nanoreactors that encapsulate TvNH. 5, 10, 15 and 20 µM of 6-thioguanosine (6TG) was added and the survival percentage was determined after 72 h of incubation via a neutral red based toxicity assay. Cell survival was stated as 100% when only nanoreactors were added but no 6-thioguanosine.
6.4 Discussion
Enzyme-prodrug therapy systems generally involve two steps. In the first step
the prodrug- activating enzyme is directed towards tumor tissue. In the second
step, the prodrug is administered systemically. Since the prodrug-activating
enzyme is only present in tumor tissue, the cytotoxic compound will only be
formed at the site of the tumor. A good prodrug needs to have the following
characteristics: (i) it should be relatively non toxic to avoid damage to healthy
tissue accompanied by severe side effects for the patient, which is the case for
many chemotherapeutics used to date. (ii) In addition, it is very important that
the prodrug is not activated by endogenous enzymes. This will also lead to
damage to healthy tissue and the tumor specificity of the therapy will be lost.
(iii) Finally, the prodrug needs to be efficiently activated by the prodrug
activating enzyme at the site of the tumor.
Many chemotherapeutic drugs are nucleoside analogs or nucleobase analogs
that interfere with DNA synthesis or cell division by incorporating into newly
synthesized DNA or by inhibition of enzymes involved in DNA synthesis or
nucleoside metabolism.

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
149
In a first part of this study, we want to validate TvNH as a prodrug activating
enzyme for cancer therapy. TvNH, which catalysis the hydrolysis of nucleosides
into the corresponding nucleobase and ribose, can be compared to the known
prodrug activating E. coli purine nucleoside phosphorylase (EcPNP). This enzyme
catalyses a similar enzymatic reaction by cleaving the glycosidic bond of ribo-and
deoxyribonucleosides in the presence of inorganic orthophosphate (Pi) as a
second substrate to generate the purine base and ribo(deoxyribo)-1-phosphate.
EcPNP is used in combination with the 6-aminodeoxynucleoside analog 2-fluoro-
2’-deoxyadenosine.
Although TvNH is able to cleave several nucleosides and nucleoside analogs
(chapter 2 table 1) it displays little activity towards 2’-deoxyadenosine with a
kcat value that is 1000X smaller and a KM value 4 times larger as compared to
adenosine. Therefore it can not be used to hydrolyse 2-fluoro-2’-
deoxyadenosine. TvNH however can efficiently cleave 2-fluoroadenosine and 2-
chloroadenosine with a similar kcat value and a similar KM value for 2-
chloroadenosine and with a KM value 5 times larger for 2-fluoroadenosine as
compared to adenosine. Another compound, 6-thioguanosine that is efficiently
cleaved by TvNH with similar kcat and KM values as compared to guanosine,
shows great potential as a prodrug. Although little is known about the toxicity of
6-thioguanosine, the nucleobase 6-thioguanine has been used to treat acute
lymphoblastic leukaemia, inflammatory bowel disease and to reduce organ
rejection after transplantation (Al Hadithy et al., 2005).
In an initial screening, the cytotoxicity of the prodrug candidates 2-
fluoroadenosine, 2-chloroadenosine and 6-thioguanosine on neuroblastoma cell
cultures (N2A cells) was compared to the cytotoxicity of the respective
nucleobase analogs.
Our experiment showed that 2-fluoroadenosine can not be used as a prodrug
since its cytotoxicity is higher than that of the nucleobase 2-fluoroadenine
(Figure 2A). This high cytotoxicity can be explained by the action of adenosine
kinase that metabolises 2-fluoroadenosine to form 2-fluoroadenosine
monophosphate. This can be further metabolised to 2-fluoroATP by the
subsequent action of adenylate kinase and nucleoside-diphosphate kinase or to
2-fluorodATP by the subsequent action of adenylate kinase, ribonucleotide
reductase and nucleoside-diphosphate kinase. In contrast 2-Fluoroadenine is

Chapter 6
150
activated to 2-fluoroadenosine monophosphate by hypoxanthine
phosphoribosyltransferase. A difference in expression level or activity of
adenosine kinase and hypoxanthine phosphorybosyltransferase towards 2-
fluoroadenosine and 2-fluoroadenine respectively, can explain the difference in
cytotoxicity of both compounds.
On the other hand 2-chloroadenosine shows only moderate cytotoxicity
towards neuroblastoma cells (Figure 2B), suggesting that 2-chloroadenosine is
not or slowly metabolised by one of the enzymes necessary to convert it to the
toxic triphosphate metabolites; adenosine kinase, nucleoside diphosphate kinase
or ribonucleotide reductase. Given the lack of high cytotoxicity of 2-
chloroadenine, 2-chloroadenosine is not suitable as a prodrug.
6-thioguanosine showed significantly lower toxicity than 6-thioguanine (Figure
2C). The cytotoxic effect of 6-thioguanine only becomes apparent after 24h
which is in agreement with the cytotoxic mechanism of 6-thioguanine. Cells are
capable of finishing the cell cycle in which 6-thiodGTP was incorporated because
this does not interfere with DNA elongation. Incorporation of 6-thiodGTP however
introduces severe chromatid disruption which results in cell cycle arrest
predominantly in the G2 phase (Maybaum and Mandel, 1983; Wotring and Roti
Roti, 1980). Determination of the EC50 values of 6-thioguanosine and 6-
thioguanine confirms the difference in cytotoxicity between both compounds
(Figure 3). However 6-thioguanosine also display toxicity at higher
concentrations which we believe is caused by hydrolyses of 6-thioguanosine by
PNP. Humane PNP can hydrolyse 6-oxonucleosides such as inosine and
guanosine. Although we only found cytotoxicity at higher concentrations it is
important to determine whether hPNP can efficiently hydrolyze 6-thioguanosine
since hPNP is highly expressed in lymphocytes, in particular T-cells. This is
illustrated by PNP deficiency which causes severe T-cell immunodeficiency
(Giblett et al., 1975; Markert, 1991). We therefore determined the activity of
hPNP for 6-thioguanosine and compared it to the activity for guanosine (Figure
4). Although hPNP has a lower affinity for 6-thioguanosine than for guanosine,
the turnover rate for 6-thioguanosine is higher than for guanosine. This result
suggests that 6-thioguanosine could be toxic towards lymphocytes, resulting in
immunodeficiency.

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
151
In a second part of this study we want to validate the use of polymeric
nanoreactors as an alternative for ADEPT and GDEPT strategies. We introduced
the concept of nanoreactors in 2005 to confine an enzymatic reaction to the
inside of polymeric vesicles composed of the amphiphilic triblock copolymer
copolymer poly(2-methyloxazoline)-poly(dimethylsiloxane)-poly(2-
methyloxazoline) (PMOXA-PDMS-PMOXA) (chapter 2 Figure 1). This system can
be used as an alternative to ADEPT. The advantages of using such nanoreactors
are the protection of the encapsulated enzyme from proteolysis in the blood,
avoiding immunogenicity of the enzyme by shielding it from components of the
immune system and introducing a large amount of enzymes per antibody-reactor
conjugate as compared to only one enzyme molecule in an antibody-enzyme
conjugate in ADEPT. To this end we encapsulated TvNH inside the PMOXA-
PDMSPMOXA vesicles (Figure 1). The triblock copolymeric membrane is
permeabilized by incorporation of the bacterial porin Tsx. This incorporation will
allow transport of TvNH substrates and products across the polymeric membrane
since Tsx is a nucleoside specific porin.
Activity measurements of Tsx permeabilized nanoreactors for 6-thioguanosine
showed that it is efficiently hydrolyzed with a similar estimated kcat value and an
apparent KM value 10 times higher as compared to free enzyme (Figure 6). This
10 times higher KM is in agreement with earlier results obtained with inosine,
adenosine, guanosine and 2-fluoroadenosine (Ranquin et al., 2005) and can be
explained by a difference in exterior and interior substrate concentrations. If the
rate of transport is lower than the rate of hydrolysis, the interior substrate
concentration will be lower than the exterior concentration. The apparent KM
value was determined based on the exterior concentration. If we assume that
encapsulation of TvNH in the polymeric nanoreactors does not alter the kinetic
behaviour of the enzyme we can conclude from our determined KM value that
the interior substrate concentration is 10 times lower than the exterior
concentration. Thus the rate of transport is 10 times lower than the rate of
enzymatic conversion.
Finally, we used Tsx permeabilized and TvNH encapsulated nanoreactors in
combination with 6-thioguanosine in a cytotoxicity assay to determine whether
such nanoreactors can be used as a novel enzyme-prodrug system. In our
cytotoxicity experiment we stated the cell survival of cells that were only treated

Chapter 6
152
with nanoreactors as 100 % survival. Although nanoreactors have a negative
effect on cell growth, this is only a minor effect and it is independent of the
nanoreactor concentration. As a positive control we used the cytotoxin sodium
azide at a final concentration of 0.5 %. This concentration is sufficient to
completely kill a cell population within 4 h. The cell survival after treatment with
Sodium azide was therefore stated as 0%. We demonstrated that 5 µM of 6-
thioguanosine and 0.33 mg/ml nanoreactors are sufficient to almost completely
kill the neuroblastoma cell culture within 72 h (Figure 7). Although 5µM of 6-
thioguanosine is also toxic and leads to 50 % cell kill after 72 h (Figure 3), the
exposure to this high concentration of 6-thioguanosine is very short. We
calculated the time to completely hydrolyze 6-thioguanosine to 6-thioguanine to
be 20 min for 0.33 mg/ml nanoreactors. Therefore we believe that the
cytotoxicity is completely caused by the action of the produced 6-thioguanine.
These results show that Tsx-TvNH nanoreactors indeed have great potential as
a novel enzyme-prodrug system.

Nanoreactor mediated prodrug activation and killing of neuroblastoma cells
153
6.5 References
Al Hadithy, A.F., de Boer, N.K., Derijks, L.J., Escher, J.C., Mulder, C.J. and Brouwers, J.R. (2005) Thiopurines in inflammatory bowel disease: pharmacogenetics, therapeutic drug monitoring and clinical recommendations. Dig Liver Dis, 37, 282-297.
Bagshawe, K., Sharma, S. and Begent, R. (2004) Antibody-directed enzyme prodrug therapy (ADEPT) for cancer. Expert Opin Biol Ther, 4, 1777-1789.
Benz, R., Schmid, A., Maier, C. and Bremer, E. (1988) Characterization of the nucleosidebinding site inside the Tsx channel of Escherichia coli outer membrane. Eur J Biochem, 176, 699-705.
Bilsland, A.E., Anderson, C.J., Fletcher-Monaghan, A.J., McGregor, F., Evans, T.R., Ganly, I., Knox, R.J., Plumb, J.A. and Keith, W.N. (2003) Selective ablation of human cancer cells by telomerase-specific adenoviral suicide gene therapy vectors expressing bacterial nitroreductase. Oncogene, 22, 370-380.
Bzowska, A., Kulikowska, E. and Shugar, D. (2000) Purine nucleoside phosphorylases: properties, functions, and clinical aspects. Pharmacol Ther, 88, 349-425.
Cortez-Retamozo, V., Backmann, N., Senter, P.D., Wernery, U., De Baetselier, P., Muyldermans, S. and Revets, H. (2004) Efficient cancer therapy with a nanobodybased conjugate. Cancer Res, 64, 2853-2857.
Dachs, G., Tupper, J. and Tozer, G. (2005) From bench to bedside for gene-directed enzyme prodrug therapy of cancer. Anticancer drugs, 16, 349-359.
Giblett, E.R., Ammann, A.J., Wara, D.W., Sandman, R. and Diamond, L.K. (1975) Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet, 1, 1010-1013.
Graff, A., Sauer, M., Van Gelder, P. and Meier, W. (2002) Virus-assisted loading of polymer nanocontainer. Proc Natl Acad Sci U S A, 99, 5064-5068. Epub 2002 Mar 5026.
Iwai, M., Harada, Y., Tanaka, S., Muramatsu, A., Mori, T., Kashima, K., Imanishi, J. and Mazda, O. (2002) Polyethylenimine-mediated suicide gene transfer induces a therapeutic effect for hepatocellular carcinoma in vivo by using an Epstein-Barr virus-based plasmid vector. Biochem Biophys Res Commun, 291, 48-54.
Luckey, M. and Nikaido, H. (1980) Specificity of diffusion channels produced by lambda phage receptor protein of Escherichia coli. Proc Natl Acad Sci U S A, 77, 167-171. Markert, M.L. (1991) Purine nucleoside phosphorylase deficiency. Immunodefic Rev, 3, 45-81.
Mathis, J.M., Williams, B.J., Sibley, D.A., Carroll, J.L., Li, J., Odaka, Y., Barlow, S., Nathan, C.O., Li, B.D. and DeBenedetti, A. (2006) Cancer-specific targeting of an adenovirus-delivered herpes simplex virus thymidine kinase suicide gene using translational control. J Gene Med, 8, 1105-1120.
Nardin, C., Widmer, J., Winterhalter, M. and Meier, W. (2001) Amphiphilic block copolymer nanocontainers as bioreactors. Eur. Phys. J. E, 4, 403-410.
Nardin, C., Winterhalter, M., Meier, W. (2000) Giant Free-Standing ABA Triblock Copolymer Membranes. Langmuir, 16, 7708-7712.
Nikaido, H., Nikaido, K. and Harayama, S. (1991) Identification and characterization of porins in Pseudomonas aeruginosa. J Biol Chem, 266, 770-779.
Nikaido, H. and Rosenberg, E.Y. (1983) Porin channels in Escherichia coli: studies with liposomes reconstituted from purified proteins. J Bacteriol, 153, 241-252.
Parker, W.B., Allan, P.W., Hassan, A.E., Secrist, J.A., 3rd, Sorscher, E.J. and Waud, W.R. (2003) Antitumor activity of 2-fluoro-2'-deoxyadenosine against tumors that express Escherichia coli purine nucleoside phosphorylase. Cancer Gene Ther, 10, 23-29.

Chapter 6
154
Parker, W.B., Bapat, A.R., Shen, J.X., Townsend, A.J. and Cheng, Y.C. (1988) Interactio of 2-halogenated dATP analogs (F, Cl, and Br) with human DNA polymerases, DN primase, and ribonucleotide reductase. Mol Pharmacol, 34, 485-491.
Plumb, J.A., Bilsland, A., Kakani, R., Zhao, J., Glasspool, R.M., Knox, R.J., Evans, T.R and Keith, W.N. (2001) Telomerase-specific suicide gene therapy vector expressing bacteria nitroreductase sensitize human cancer cells to the pro-dru CB1954. Oncogene, 20, 779 7803.
Ranquin, A., Versees, W., Meier, W., Steyaert, J. and Van Gelder, P. (2005) Therapeutic Nanoreactors: Combining Chemistry and Biology in a Novel Triblock Copolymer Drug Delivery System. Nano Lett, 5, 2220-2224.
Senter, P.D. and Springer, C.J. (2001) Selective activation of anticancer prodrugs by monoclonal antibody-enzyme conjugates. Adv Drug Deliv Rev, 53, 247-264.
Stribbling, S.M., Martin, J., Pedley, R.B., Boden, J.A., Sharma, S.K. and Springer, C.J. (1997) Biodistribution of an antibody-enzyme conjugate for antibody-directed enzyme prodrug therapy in nude mice bearing a human colon adenocarcinoma xenograft. Cancer Chemother Pharmacol, 40, 277-284.
Syrigos, K.N., Rowlinson-Busza, G. and Epenetos, A.A. (1998) In vitro cytotoxicity following specific activation of amygdalin by beta-glucosidase conjugated to a bladder cancer-associated monoclonal antibody. Int J Cancer, 78, 712-719.
Versees, W., Decanniere, K., Pelle, R., Depoorter, J., Brosens, E., Parkin, D.W. and Steyaert, J. (2001) Structure and function of a novel purine specific nucleoside hydrolase from Trypanosoma vivax. J Mol Biol, 307, 1363-1379.
Woodle, M.C., Engbers, C.M. and Zalipsky, S. (1994) New amphipatic polymer-lipid conjugates forming long-circulating reticuloendothelial system-evading liposomes. Bioconjug Chem, 5, 493-496.
Wotring, L.L. and Roti Roti, J.L. (1980) Thioguanine-induced S and G2 blocks and their significance to the mechanism of cytotoxicity. Cancer Res, 40, 1458-1462.
Zeng, H., Wei, Q., Huang, R., Chen, N., Dong, Q., Yang, Y. and Zhou, Q. (2007) Recombinant adenovirus mediated prostate-specific enzyme pro-drug gene therapy regulated by prostate-specific membrane antigen (PSMA) enhancer/promoter. J Androl, 28, 827-835.
Zhou, J.H., Tang, B., Liu, X.L., He, D.W. and Yang, D.T. (2007) hTERT-targeted E. coli purine nucleoside phosphorylase gene/6-methylpurine deoxyribose therapy for pancreatic cancer. Chin Med J (Engl), 120, 1348-1352.

Chapter 7 Production of cAbLys-3 mutants for selective coupling to nanoreactors
155
Chapter 7: Production of cAbLys-
3 mutants for selective coupling to
nanoreactors Abstract To specifically crosslink cAbLys-3 to the surface of PMOXA-PDMS-PMOXA polymeric nanoreactors using the heterobifunctional crosslinker N-(p-maleimidophenyl)isocyanate, cAbLys-3 S17C, S134C and S141C mutants were prepared and expressed in E.coli. These cAbLys-3 mutants are still able to bind lysozyme with similar affinity as the wild-type cAbLys-3. Since the expression yield in E. coli is very low, an alternative expression system using Pichia Pastoris was introduced which improved the yield 60 fold.

Production of cAbLys-3 mutants for selective coupling to nanoreactors
156

Chapter 7
157
7.1 Introduction
It is well know that the vascular permeability of tumors is enhanced due to the
action of secreted factors such as kinin. This allows macromolecules to diffuse
from the bloodstream into the tumor interstitium. Furthermore, the lymphatic
drainage system is impaired so that macromolecules are retained in the
interstitium for a prolonged time (Jain, 1987; Matsumura and Maeda, 1986).
Therefore particles ranging from 10-500 nm, such as liposomes, can be passively
targeted to tumor tissue due to this enhanced permeation and retention effect
(EPR) (Bornmann et al., 2008; Green and Rose, 2006; Koo et al., 2005). In
addition the attachment of ligands can further improve the accumulation of
nanoparticles in tumor tissue. Monoclonal antibodies are the most frequently
used ligands for targeting to tumor tissue.
In this project we want to use the antigen binding domain of single chain
antibodies derived from camelids for targeting purposes. These naturally
occurring single chain antibodies devoid of a light chain were discovered by
Hamers et al. in 1993 (Hamers-Casterman et al., 1993). These so called heavy-
chain antibodies (HCAb) are present in the serum of camels, dromedaries and
llamas. Due to the missing light chain in HCAbs, their antigen binding site is
composed of a single domain, VHH, compared to the VH-VL paired antigen
binding site of conventional antibodies. As a result VHHs are distinguished from
the smallest antigen binding domain, scFv, of conventional antibodies by some
unique properties: size, stability, solubility, ease of cloning a library and selecting
highly specific antigen binders, in vivo maturation and high expression levels
(Arbabi Ghahroudi et al., 1997; Muyldermans and Lauwereys, 1999). Since their
discovery VHHs, now called nanobodies, have been used for multiple purposes
such as diagnostics (Saerens et al., 2005), tumor targeting (Cortez-Retamozo et
al., 2004) and crystallisation aids (Loris et al., 2003).
We have recently introduced a new strategy for enzyme-prodrug therapy
(Ranquin et al., 2005) based on nanoreactors. The nucleoside hydrolase of
Trypanosoma vivax (TvNH), that cleaves nucleoside(analogs) in
nucleobase(analogs), was encapsulated inside the polymeric vesicle to activate
nucleoside analog prodrugs. To make sure substrates and products can readily

Production of cAbLys-3 mutants for selective coupling to nanoreactors
158
diffuse in and out of the polymeric vesicles, Tsx, a nucleoside specific porin (Benz
et al., 1988) was incorporated into the polymeric membrane. Since these
nanoreactors showed promising in vitro anti-tumor activity in combination with
the prodrug 6-thioguanosine, we want to target these nanoreactors to tumor
tissue by linking a nanobody to the surface of the reactors.
The nanoreactors are composed of the triblock copolymer poly(2-
methyloxazoline)-b-poly(dimethyl siloxane)-b-poly(2-methyloxazoline) (PMOXA-
PDMS-PMOXA). The endgroup of the hydrophilic block PMOXA that is available for
nanobody coupling is a hydroxyl group. Although there is a plethora of linkers
available for the chemical crosslinking of proteins, only one commercially
available linker was found that is reactive towards hydroxyl groups: the
heterobifunctional linker N-(p-maleimidophenyl)isocyanate (PMPI) (Pierce). The
isocyanate end of PMPI can react with hydroxyl groups to form carbamate
linkages whereas the maleimide end reacts with sulfhydryls to form a thioether
bond.
As a proof of principle we want to couple cAbLys-3 to the surface of
nanoreactors. This nanobody binds to lysozyme with nanomolar range affinity
(Desmyter et al., 1996) and is well characterised. Although nanobodies posses
four highly conserved cysteine residues (which form two disulphide bridges),
these cysteine residues are buried in the three dimensional structure and
therefore unavailable for linkage to the PMPI linker. Therefore we introduced
additional accessible cysteine residues at position 17, 134 and 141 by
substitution of serine residues.
Since expression of the cAbLys-3 serine to cysteine mutants in Escherichia coli
resulted in very low yields, we additionally used expression in Pichia pastoris to
obtain high quantities of mutant cAbLys-3. P. pastoris reaches high cell densities
and the strong methanol inducible alcohol oxidase promotor can be used for
expression. Furthermore by including the α-factor signal sequence, the protein is
secreted into the medium allowing easy purification.

Chapter 7
159
7.2 Materials and Methods
7.2.1. Site directed mutagenesis of cAbLys-3
S17C, S134C and S141C point mutations were introduced to cAbLys-3 via
site-directed mutagenesis using the QuickChangeTM Site-directed Mutagenesis
KIT from Stratagene. As template for PCR reactions we used the plasmid
pHen6c-cAbLys-3 (Figure 1) (Conrath et al., 2001). The primers used for
introducing the serine to cysteine mutations are given in Table 1. After
amplification of the plasmid using these primers and PfuTurboTM DNA
polymerase, methylated, non-mutated parental DNA was digested with DpnI.
Subsequently the mutated DNA was transformed to XL1-Blue supercompetent
cells (Invitrogen) that are capable of repairing the nicks in the mutated DNA.
This results in the formation of circular plasmid DNA that contains the mutated
cAbLys-3 gene. The faithful introduction of the mutations was confirmed by DNA
sequencing (ABI prism 3100 genetic analyzer, Applied Biosystems). Finally, the
plasmid was transformed to E. coli WK6 cells for expression.
Plac operatorPelB
CAbLys-3
His-tagpHenCAbLys-3
3638 bps
AmpR
Plac operatorPelB
CAbLys-3
His-tagpHenCAbLys-3
3638 bps
AmpR
Figure 1: Schematic representation of the pHen6c-cAbLys-3 plasmid. The plasmid contains a plac operator, the gene encoding cAbLys-3 with a pelB signal sequence and a C-terminal His6-tag, an Ampicillin Resistance gene (AmpR) and Ori (not shown on the figure).

Production of cAbLys-3 mutants for selective coupling to nanoreactors
160
Table 1: Primers used for the site-directed mutagenesis of cAbLys-3 and pPicZα colony PCR. Base substitutions for serine to cysteine mutations are depicted in bold
Reverse primer 5’-ACAGGAGAGTCTCAGACACCCTCCAGCCTGCACC-3’ S17C
Forward primer 5’-GGTGCAGGCTGGAGGGTGTCTGAGACTCTCCTGT-3’
Reverse primer 5’-GATGGTGATGGTGGTGGCAGGAGACGGTGACCTG-3’ S134C
Forward primer 5’-CAGGTCACCGTCTCCTGCCACCACCATCACCATC-3’
Reverse primer 5’-CGACGGCCAGTGAATTCTAGCAGTGATGGTGATGGTGGTG-3’ S141C
Forward primer 5’-CACCACCATCACCATCACTGCTAGAATTCACTGGCCGTCG-3’
Reverse primer 5’-CCTACAGTCTTACGGTAAACG-3’ pPicZα
Forward primer 5’-GACTGGTTCCAATTGACAAGC-3’
7.2.2. Bacterial expression and purification of cAbLys-3 mutants
Large scale production of cAbLys-3 mutants, S17C, S134C and S141C was
performed in shaker flasks by growing the E. coli WK6 cells in Terrific Broth (TB)
(Table 2) supplemented with 0.1% (w/v) glucose and 100 µg/ml ampicillin until
an OD600 of 0.6–0.9 was obtained. Expression was then induced with 1 mM IPTG
for 16 hours at 37°C. After pelleting, the cells were resuspended in 0.2 M Tris-
HCL pH 8.0, 0.5 mM EDTA, 0.5 M sucrose (TES). The periplasmic proteins were
extracted by an osmotic shock by diluting the resuspended cells 3x in 50 mM
Tris-HCL pH 8.0, 0.12 mM EDTA, 0.12 M sucrose. This periplasmic extract was
subsequently loaded onto pre-equilibrated (50 mM Hepes pH 7.0, 1M NaCl) Ni-
NTA Sepharose (GE healthcare), and after washing with 50 mM Hepes pH 7.0,
1M NaCl, 10mM imidazol, the bound proteins were eluted with 50 mM Hepes pH
7.0, 1M NaCl, 1M imidazol. Finally the protein was concentrated on Vivaspin
(Vivascience) concentrators with a cut-off of 5 kDa and loaded onto a Superdex-
75 16/60 (Pharmacia) gelfiltration chromatography column, equilibrated with
phosphate buffered saline (PBS). The purity of the cAbLys-3 mutants was
evaluated by Coomassie brilliant blue stained SDS-PAGE.

Chapter 7
161
7.2.3. Expression of cAbLys-3 S17C in Pichia Pastoris
7.2.3.1. Cloning
pHen6c-cAbLys-3 S17C was digested with PstI and EcoRI. The restriction
fragment containing cAbLys-3 S17C was ligated into a PstI, EcoRI digested
pPiCZα vector (Figure 2) (Invitrogen). The ligation mixture was subsequently
transformed to electro-competent Top10 cells and subsequently grown at 37°C
for 24 h on Luria Broth (LB) plates (Table 2). To identify clones that posses
pPICZα-cAbLys-3 S17C, a colony PCR was performed with pPICZα forward and
reverse primers (Table 1). The sequence of the positive clones was confirmed by
DNA sequencing (ABI prism 3100 genetic analyzer, Applied Biosystems). Two
positive pPICZα-CAbLys-3 S17C plasmids were isolated via a Qiagen midiprep
and linearised with BstXI for transformation to P. Pastoris X-33 cells.
7.2.3.2. Transformation
Electro-competent Pichia Pastoris X-33 cells (Invitrogen) were prepared as
follows. The cells were grown on a YPD plate (Table 2) at 30 °C for 48 h to obtain
single colonies. A single colony was picked and grown in 5ml YPD for 24 h at
30°C. This overnight cell culture was used to inoculate 250 ml YPD medium.
After 18 h at 30 °C, the cells (OD600 = 1.3) were collected and washed twice with
sterile cold water. The cells were made electro-competent by adding 800 µl 1M
sorbitol.
Linearised pPICZα-cAbLys-3 S17C plasmid (10 µg) was added to 80 µl electro-
competent cells and electroporated with an E.coli pulser (BioRad) at 1.5kV. 1 ml
1 M sorbitol was added and the cells were incubated for 1 h at 30 °C.
Subsequently 200 µl 5x YPD medium was added and after 1 h incubation at
30°C, the cells were plated on YPDS plates containing 100 µg/ml, 250 µg/ml and
500 µg/ml Zeocine. Cells were grown on these plates for 2 days at 30 °C.

Production of cAbLys-3 mutants for selective coupling to nanoreactors
162
Figure 2: Schematic representation of the PicZα-cAbLys-3 S17C plasmid. This plasmid contains, a 5’AOX1 promotor region, the gene coding for cAbLys-3S17C with an α-factor signal sequence and a C-terminal His6-tag, followed by an AOX1 transcription termination region, a Zeocine Resistance gene followed by a CYC1 transcription termination region and an Ori.
7.2.3.3. Expression and purification
Single colonies from the 250 µg/ml and 500 µg/ml Zeocine plates were used
to inoculate 10 ml YPG medium (Table 2). After 48 h at 30 °C this culture was
used to inoculate three flasks of 250 ml BGY (Table 2) medium at an OD600 of
0.04. After 24 h at 30 °C, the cells were pelleted and resuspended in three flasks
of 250 ml BMY (Table 2) medium to induce expression of cAbLys-3 S17C. During
the following 36 h the cultures were re-induced 4 times with 0,5 % methanol.
After a total of 48 h induction at 30 °C, the cells were removed and NaCl and
imidazol were added to the supernatant at a final concentration of 1 M and 20
mM, respectively, and purificated as described under 7.2.2.

Chapter 7
163
Table 2: Composition of culture media used.
TB LB YPD∗ YPG YPDS∗ BGY BMY Trypton 12 g/l 10 g/l 20 g/l 20 g/l 20 g/l 20 g/l 20 g/l Yeast extract 24 g/l 5 g/l 10 g/l 10 g/l 10 g/l 10 g/l 10 g/l NaCl 5 g/l 10 g/l Glucose 20 g/l 20 g/l Glycerol 30 g/l 30 g/l Sorbitol 182.2 g/l K-phosphate pH 6.0 200 mM 200 mM
Methanol 0.5 % Biotin 0.4 mg/l 0.4 mg/l ∗ To make culture plates, 20 g/l agar was added
7.2.4. Enzyme-Linked Immuno Sorbent Assay (ELISA)
F96 Maxisorb Nunc-immunoplates were coated overnight at 4 °C with 1 µg/ml
lysozyme in PBS. To avoid nonspecific adsorption of antibodies to the
immunoplates, they were blocked for two hours at room temperature with 1%
(w/v) caseïne in PBS. Three concentrations, 0.1 µM, 1 µM and 10 µM of cAbLys-3
S17C, S134C and S141C were added to the lysozyme coated plates and
incubated for 2 hours at room temperature. To detect bound cAbLys-3, firstly,
mouse α-His6 antibody was added, followed by a goat α-mouse antibody-
peroxidase conjugate. Each antibody was incubated at room temperature for two
hours. After incubation with each antibody, the plate was washed with PBS, 0.01
% (v/v) Tween to remove unbound antibody. Signal development was achieved
by adding 0.004 % peroxidase substrate tetramethyl benzidine (TMB) and 3 %
(v/v) H2O2 in 0.1 M phosphate buffer pH 6.0, 2% (w/v) citric acid. After 30 min
the enzymatic reaction was stopped by adding 1N H2SO4. Finally colour
development was measured at 45O nm on a SpectraMax multiwell plate reader
(Molecular Devices). To determine the nonspecific binding of α-mouse antibody-
peroxidase conjugate, this antibody was added to lysozyme coated wells without
cAbLys-3 mutant antibody and to wells without mouse α-His6 antibody. As a
positive control, 10 µM cAbLys-3 was used.

Production of cAbLys-3 mutants for selective coupling to nanoreactors
164
7.3 Results
7.3.1. Production of cAbLys-3 mutants in E. coli
To target nanoreactors to tumor sites we like to couple camelid antibodies, so
called nanobodies®, to the polymers of the nanoreactor. Since the reactive group
of PMOXA is a hydroxyl group we will use the hetero-bifunctional linker PMPI
where the isocyanate end can react with hydroxyl groups and the maleimide end
is able to reacts with sulfhydryls.
Although nanobodies posses four highly conserved cysteine residues (which form
two disulphide bridges), these cysteine residues are buried in the three
dimensional structure and therefore unavailable for linkage to the PMPI linker
(Figure 3). Therefore cAbLys-3 mutants were made to introduce additional
cysteine residues for coupling to the nanoreactors. This method has an important
advantage compared to the more conventional coupling methods that use amine-
or carboxyl- reactivity. By introducing additional cysteines, the coupling is highly
specific and can be controlled whereas amine- or carboxyl- coupling is highly
unspecific due to the abundance of these groups in proteins.
Since serine to cysteine substitutions introduce only a minor structural
change, we identified two serine residues, S17 and S134 that are located far
from the cAbLys-3 antigen binding site (Figure 3). Additionally we introduced an
extra cysteine residue at position 141, at the end of the C-terminal His6-tag. This
residue is not visible in the X-ray structure.
Mutants were originally cloned and expressed in E. coli WK6 cells. Purification
of these three mutants resulted in a single band on Coomassie stained SDS-
PAGE (Figure 4).

Chapter 7
165
Figure 3: Three dimensional structure of cAbLys-3 (blue) and Lysozyme (green). Cysteine residues in cAbLys-3 are depicted in pink, ser17 and ser134 are depicted in red.
A B C D
Figure 4: SDS-PAGE analysis of purified cAbLys-3 S17C (A), cAbLys-3 S134C (B) and cAbLys-3 S141C (C), expressed in E. coli and cAbLys-3 S17C expressed in P. pastoris
We obtained 0.23 mg/l culture, 0.20 mg/l and 0.15 mg/l of cAbLys-3 S134C,
S141 and S17C respectively. This low yield is probably the result of different
codon usage in E. coli as compared to camelidae. Especially eukaryotic glycine,
arginine, lysine and leucine codons are rarely used by E.coli (Figure 5).

Production of cAbLys-3 mutants for selective coupling to nanoreactors
166
Figure 5: Codon usage of the cAbLys-3 sequence in E. coli. The Y-axis represents the relative adaptiveness of E. coli to use the codon. A relative adaptiveness smaller than 50 % is depicted in grey. The analysis was performed using Graphical Codon Usage Analyser software (www.gcua.de).

Chapter 7
167
7.3.2. Functionality of cAbLys-3 mutants
To determine whether these mutants are unaffected in antigen binding, we
performed an Enzyme-Linked Immuno Sorbent Assay (ELISA) (Figure 6). The
results clearly show that cAbLys-3 S17C and cAbLys-3 S134C are able to bind
lysozyme with similar affinities as wild type cAbLys-3. Moreover, the binding
shows a concentration dependent signal. CAbLys-3 S141C on the other hand
shows only a low binding signal as compared to the wild type nanobody.
Lysozyme + + + + + + + α-His6 + + + + + α-mouse∼peroxidase + + + + + +
cAbLys-3 S17C + cAbLys-3 S134C + cAbLys-3 S141C + cAbLys-3 +
Figure 6: ELISA to determine antigen binding capacity of cAbLys-3 mutants.
7.3.3. Production of cAbLys-3 S17C in P. Pastoris
Although we were able to produce pure cAbLys-3 S17C, S134C and S141C
that are still able to bind lysozyme in E. coli, the yield was very low. Therefore,
an alternative expression method in P. Pastoris was used to obtain larger
quantities of purified nanobody. Since cAbLys-3 S17C has the best lysozyme
binding properties this mutant was preferred for high yield expression in P.
00,1
0,20,30,4
0,50,6
0,70,8
OD450
100 nM1 µM10 µMnegative controls

Production of cAbLys-3 mutants for selective coupling to nanoreactors
168
Pastoris. By using a PicZα plasmid, the expression of cAbLys-3 S17C can be
induced by methanol since it contains an AOX1 promotor region. Due to the
presence of an α-factor signal sequence, the expressed protein is secreted into
the culture medium. Expression in P. Pastoris resulted in pure cAbLys-3 S17C
with a yield of 8.7 mg/l culture. This is approximately 60 times higher than the
yield obtained with E. coli (Figure 4).
7.4 Discussion
Targeting of enzymes and/or lipidic or polymeric vehicles to tumor sites is
mostly accomplished by chemical coupling of antibodies. In this study we want to
couple camelid antibodies to PMOXA-PDMS-PMOXA triblock copolymers. As a
model antibody we used cAbLys-3, an antibody directed against lysozyme. In
future experiments, the nanoreactor-antibody complex can than conveniently be
used to target Lewis Lung carcinoma cells (3LL cells) which express lysozyme on
their plasma membrane (personal communication)
The presence of an hydroxyl group on the polymers forced us to use the
hetero bifunctional linker PMPI which can be coupled to the polymer via its
isocyanate end and to the antibody with its maleimide group to form a thioether
bond with sulfhydryl residues.
Therefore, the solved X-ray structure of cAbLys-3 was scrutinized for possible
site-directed cysteine mutants that wouldn’t interfere with antigen binding. We
were able to construct three cAbLys-3 serine to cysteine mutants, S17C, S134C
and S141C that still can bind lysozyme (Figure 6). CAbLys-3 S17C and cAbLys-3
S134C bind lysozyme with similar affinity as the wild type cAbLys-3. CAbLys-3
S141C, however, has a lower binding capacity as compared to the wild type. This
is unexpected since residue 141 is located equally far from the antigen binding
site as residue 134. Since residue 141 is not part of the visible folded structure
but situated just behind the C-terminal His6-tag, we believe the lower signal in
the ELISA assay is caused by interference of binding of the mouse α-His antibody
to the His6-tag of cAbLys-3 S141C.

Chapter 7
169
Expression of the cAbLys-3 mutants in E. coli yields only a low amount of
protein (0.15 – 0.23 mg/l culture medium) as compared to WT cAbLys-3 (4
mg/l) which can be explained by misfolding of the nanobody. Introduction of an
additional cysteine can result in malformation of disulphide bridges and
misfolding of the protein. Formation of correct disulphide bridges is highly
controlled in the periplasmic space and involves an intricate cascade of redox
proteins. The oxidation of sulfhydryl residues is mainly catalysed by the
periplasmic chaperone DsbA which is recycled through an oxidation reaction with
the membrane protein DsbD (Kadokura and Beckwith, 2002). Failure of this
system by over expression of the target protein or by non-prokaryotic
heterologous targets might result in the misfolding and subsequent aggregated
of the latter. That part of the antibodies are indeed misfolded and aggregation is
confirmed by the presence of cAbLys-3 mutant protein in the insoluble fraction
after the periplasmic extraction (data not shown). However the yield could not be
improved by performing the periplasmic extraction in the presence of
Dithiothreitol (DTT) (data not shown). The reason for the low yield of cAbLys-3 in
general can be explained by the difference in codon usage between E. coli and
higher eukaryotes such as camelidae (Figure 5). Especially eukaryotic glycine,
arginine, lysine and leucine codons are rarely used by E.coli. Since a complete
change of these codons to the ones preferred by E. coli would be time consuming
and very costely, we used an eukaryotic expression host, P. pastoris. Although
codon usage of the cAbLys-3 sequences are far from ideal for expression in P.
pastoris, larger expression yields can be obtained through high cell densities and
strong promoters. The expression yield for cAbLys-3 S17C was improved 60 fold
by using P. pastoris.
Although preliminary experiments for the coupling of TvNH-Tsx nanoreactors
to cAbLys-3 have been performed, they were unsuccessful (data not shown).
Much effort still has to be put into fine tuning the chemical reaction and finding
an efficient assay for the detection of nanobody-coupled nanoreactors.

Production of cAbLys-3 mutants for selective coupling to nanoreactors
170
7.5 References
Arbabi Ghahroudi, M., Desmyter, A., Wyns, L., Hamers, R. and Muyldermans, S. (1997) Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett, 414, 521-526.
Benz, R., Schmid, A., Maier, C. and Bremer, E. (1988) Characterization of the nucleoside-binding site inside the Tsx channel of Escherichia coli outer membrane. Eur J Biochem, 176, 699-705.
Bornmann, C., Graeser, R., Esser, N., Ziroli, V., Jantscheff, P., Keck, T., Unger, C., Hopt, U.T., Adam, U., Schaechtele, C., Massing, U. and von Dobschuetz, E. (2008) A new liposomal formulation of Gemcitabine is active in an orthotopic mouse model of pancreatic cancer accessible to bioluminescence imaging. Cancer Chemother Pharmacol, 61, 395-405.
Conrath, K.E., Lauwereys, M., Galleni, M., Matagne, A., Frere, J.M., Kinne, J., Wyns, L. and Muyldermans, S. (2001) Beta-lactamase inhibitors derived from single-domain antibody fragments elicited in the camelidae. Antimicrob Agents Chemother, 45, 2807-2812.
Cortez-Retamozo, V., Backmann, N., Senter, P.D., Wernery, U., De Baetselier, P., Muyldermans, S. and Revets, H. (2004) Efficient cancer therapy with a nanobody-based conjugate. Cancer Res, 64, 2853-2857.
Desmyter, A., Transue, T.R., Ghahroudi, M.A., Thi, M.H., Poortmans, F., Hamers, R., Muyldermans, S. and Wyns, L. (1996) Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat Struct Biol, 3, 803-811.
Green, A.E. and Rose, P.G. (2006) Pegylated liposomal doxorubicin in ovarian cancer. Int J Nanomedicine, 1, 229-239.
Hamers-Casterman, C., Atarhouch, T., Muyldermans, S., Robinson, G., Hamers, C., Songa, E.B., Bendahman, N. and Hamers, R. (1993) Naturally occurring antibodies devoid of light chains. Nature, 363, 446-448.
Jain, R.K. (1987) Transport of molecules in the tumor interstitium: a review. Cancer Res, 47, 3039-3051.
Kadokura, H. and Beckwith, J. (2002) Four cysteines of the membrane protein DsbB act in concert to oxidize its substrate DsbA. Embo J, 21, 2354-2363.
Koo, O.M., Rubinstein, I. and Onyuksel, H. (2005) Role of nanotechnology in targeted drug delivery and imaging: a concise review. Nanomedicine, 1, 193-212.
Loris, R., Marianovsky, I., Lah, J., Laeremans, T., Engelberg-Kulka, H., Glaser, G., Muyldermans, S. and Wyns, L. (2003) Crystal structure of the intrinsically flexible addiction antidote MazE. J Biol Chem, 278, 28252-28257.
Matsumura, Y. and Maeda, H. (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res, 46, 6387-6392.
Muyldermans, S. and Lauwereys, M. (1999) Unique single-domain antigen binding fragments derived from naturally occurring camel heavy-chain antibodies. J Mol Recognit, 12, 131-140.
Ranquin, A., Versees, W., Meier, W., Steyaert, J. and Van Gelder, P. (2005) Therapeutic Nanoreactors: Combining Chemistry and Biology in a Novel Triblock Copolymer Drug Delivery System. Nano Lett, 5, 2220-2224.
Saerens, D., Frederix, F., Reekmans, G., Conrath, K., Jans, K., Brys, L., Huang, L., Bosmans, E., Maes, G., Borghs, G. and Muyldermans, S. (2005) Engineering camel single-domain antibodies and immobilization chemistry for human prostate-specific antigen sensing. Anal Chem, 77, 7547-7555.

Chapter 7
171
7.6 Appendix
Another way of targeting nanoreactors to specific cell types can be
accomplished by adhesins that specifically recognize sugars present on the target
cells. These sugars can be part of glycosylated proteins or of lipids such as
ceramides.
Ed Conway, from the Centrum voor Transgene Technologie en Gentherapie
(KULeuven), is studying the acute renal failure syndrome (Kindt et al., 2008). He
and his co-workers are able to relief the symptoms in a mouse model by
injection of naked plasmid DNA that encodes for the anti-apoptotic protein
survivin. However, non-targeted DNA injection results in systemic expression of
survivin, a protein that is also upregulated in tumor tissue. Therefore there is
great interest in using polymeric vesicles for directing the plasmid construct to
kidney cells.
For targeting of plasmid encapsulated vesicles, a bacterial adhesion, PapG II,
was cloned, expressed and purified. PapG II is the adhesion at the tip of the P
pilus that mediates attachment of uropathogenic Escherichia coli to the
uroepithelium of the human kidney via binding to globotriaosylceramides
(Dodson et al., 2001).
For high expression, the gene was cloned with the gateway® technology
(invitrogen). This technology is a universal cloning method that takes advantage
of the site-specific recombination of bacteriophage lambda to provide a rapid and
highly efficient way to move DNA sequences into multiple vector systems. We
isolated the PapG II encoding gene from the genomic DNA of E. coli APEC 54. For
coupling purposes a C-terminal Lys3-tag was introduced via PCR. Firstly, a gene
construct with flanking attB recombination sequences was made via PCR with
attB forward and attB reverse primers (Table 1). This construct was then
inserted into a gateway® donor vector (pDONR) via recombination, catalyzed by
BP clonase™ II. The plasmid was subsequently transformed to Ca-competent E.
coli DH5α cells and cultured on kanamycine plates. Since the pDONR vector has
a kanamycine resistance gene, cells that were not transformed with pDONR, can
not grow. Additionally, the pDONR vector contains the gene sequence of the
toxin ccdB, a toxin, between the att recombination sequences. Therefore, the

Production of cAbLys-3 mutants for selective coupling to nanoreactors
172
cells transformed with plasmids that do not contain the PapG II gene are killed
and only cells with the PapG II vector can grow. Via an identical recombination
method, the gene coding for PapGII was cloned into a gateway® destination
vector (pDEST) that contains an ampicillin resistance gene. Subsequently the
pDEST was transformed to E. coli BL21 (DE3) C43 cells and cultured on ampicillin
plates. After each transformation step a colony PCR was performed with secL A1
and secB primers (Table 1) to select positive clones and the sequence was
confirmed by DNA sequencing (ABI prism 3100 genetic analyzer, Applied
Biosystems).
Table 1: primers used for the cloning of PapG II via the gateway® technology. Recombination sequences are depicted in bold, the sequence encoding the Lys3-tag is depicted in gray
Primer Sequence attB forward primer
5’- GGGGACAAGTTTGTACAAAAAAGCAGGCTTAAGAAGGAGATATACCATG
AAAAAATGGTTCCCAGCTTTGTTATTTTC-3’ attB reverse primer
5’-GGGGACCACTTTGTACAAGAAAGCTGGGTATTACTTCTTCTTGCCGATATT
CTTAAATAAGAATAACAT -3’
secL A1 5’-CTCTCGCGT TAACGCTAGC ATGGAT-3’ secL B 5’-GTAACATCAGAGATTTTGAGACAC-3’
Large scale production of PapG II was performed in shaker flasks by growing the
bacteria in Luria Broth (LB) and 100 µg/ml ampicillin, at 28 °C, until an OD600 of
0.6–0.9 was obtained. Expression was then induced with 0,2 mM IPTG for 16
hours at 28°C. After pelleting the cells, they were resuspended in 50 mM Hepes
pH 7.0, 2.5 mM EDTA, 30 % (w/v) sucrose. The periplasmic proteins were
extracted by an osmotic shock by diluting the resuspended cells 3x in 50 mM
Hepes pH 7.0. PapG II was purified from this periplasmic extract via cation
exchange chromatography on a 30S source column (Pharmacia). The column
was equilibrated, loaded and washed with 50 mM Hepes pH 7.0. PapG II was
eluted with 50 mM Hepes pH 7.0, 1 M NaCl by applying a gradient. Finally the
protein was concentrated on Vivaspin (Vivascience) concentrators with a cut-off
of 5 kDa and loaded onto a Superdex-75 16/60 (Pharmacia) gelfiltration
chromatography column, which was equilibrated with phosphate buffered saline
(PBS). The purity of PapG II was evaluated by Coomassie brilliant blue stained
SDS-PAGE.

Chapter 7
173
Figure 1: SDS-PAGE analysis of E. coli PapG II
References Dodson, K.W., Pinkner, J.S., Rose, T., Magnusson, G., Hultgren, S.J. and Waksman, G.
(2001) Structural basis of the interaction of the pyelonephritic E. coli adhesin to its human kidney receptor. Cell, 105, 733-743.
Kindt, N., Menzebach, A., Van de Wouwer, M., Betz, I., De Vriese, A. and Conway, E.M. (2008) Protective role of the inhibitor of apoptosis protein, survivin, in toxin-induced acute renal failure. Faseb J, 22, 510-521.


Chapter 8 General discussion
175
Chapter 8:
General discussion

Chapter 8
176
8.1 General discussion
We succeeded in showing the potential of polymeric nanoreactors as a novel
prodrug activating system, and thus delivered a first proof of principal (chapter
6). This system can have a great advantage compared to conventional
antibodydirected enzyme-prodrug therapy (ADEPT). One of the biggest concerns
in ADEPT is the immunogenicity of the antibody-enzyme conjugate. This can be
diminished by altering T cell epitopes (Harding et al., 2005), but this is a very
labour intensive process. By encapsulating the enzyme in nanoreactors, it is
shielded from the environment thus protecting it from proteolytic degradation
and detection by the immune system. Moreover when using nanoreactors a high
concentration of enzyme can be targeted to the tumor per therapeutic unit
whereas only one enzyme molecule is targeted to the tumor per therapeutic unit
in ADEPT. We showed that when the enzyme is encapsulated in polymeric
vesicles that are permeabilized by porins, the enzymatic activity still remains
high (Ranquin et al., 2005).
Although we were not yet successful in attaching nanobodies to the surface of
the nanoreactors for targeting purposes, passive targeting of polymeric vesicles
is also a possibility due to the enhanced permeation and retention effect (EPR) of
leaky tumor vasculature (Matsumura and Maeda, 1986) (Jain, 1987). We did
construct the necessary cAbLys-3 cysteine mutants for attachment to the
hydroxyl endgroup of PMOXA via the heterobifunctional PMPI linker (chapter 7).
The biggest issue in the coupling of cAbLys-3 cysteine mutants to nanoreactors is
finding an accurate way for the detection of targeted nanoreactors and
separation of targeted and non-targeted reactors. There are some alternative
coupling strategies that could also be used. For instance, the streptavidin-biotin
coupling method as described by Broz et al. (2005) is a possibility. For this
purpose biotinylated PMOXA21-PDMS54-PMOXA21 polymers were already made.
However, the production method of biotinylated nanoreactors is not yet
optimized. Since biotin is a bulky group compared to the nanobodies, random
attachment might interfere with antigen binding and thus it will be important to
specifically biotinylate the nanobodies. Another option for targeting of
nanoreactors by nanobodies is to attach an α-helical membrane anchor to the C
terminus of the nanobody. This way the nanobody can be inserted into the

General discussion
177
polymeric membrane in a similar way as the porin. However approximately 50 %
of the inserted nanobodies will be pointed to the interior of the vesicles instead of
the exterior. Moreover, since the polymeric membrane (10 nm) is much thicker
than a biological membrane (4 nm) a very large linker region between anchor
and nanobody will be needed.
To evaluate the true therapeutic potential of the nanoreactors, a lot of
questions need to be answered concerning the in vivo behaviour of the
nanoreactors.
Firstly, it is important to determine whether these nanoreactors are able to
elicit an immune response. Uptake by macrophages or other antigen presenting
cells is an important first step in eliciting an immune response. Therefore we
already performed a preliminary macrophage uptake experiment with fluorescent
nanoreactors (chapter 5). Although this experiment is far from conclusive, the
lack of macrophage uptake seen in this experiment is supported by earlier work
on the same type of vesicles by Broz et al (Broz et al., 2005). Here, a poly A
ligand had to be attached to the surface of PMOXA-PDMS-PMOXA vesicles to
target the A1 scavenger receptor of macrophages and to promote macrophage
uptake. A similar experiment with fluorescent macrophages was done by De
Vocht et al. (manuscript in preparation). They used FACS analysis to determine
whether fluorescent nanoreactors are taken up by macrophages. They found that
only a small percentage (7%) of nanoreactors is associated with macrophages.
Additionally, they analysed the secretion of several cytokines to determine if
macrophages are activated in the presence of nanoreactors. Cytokine secretion
was only observed for samples that were contaminated with endotoxin, as
determined via a LAL assay (personal communication). This result also indicates
that the reactors are not taken up by macrophages and do not activate
macrophages. A second experiment with endotoxin free samples in vitro and in
vivo is on the way, to confirm previous results and determine the uptake and
activation in vivo.
Secondly, it is important to determine the biodistribution and toxicity of the
reactors that are injected intravenously or intraperitonealy since accumulation in
organs such as the hart, liver and kidney can lead to severe toxicity. Although we
6). The in vivo toxicity towards various cell types however can not be predicted.

Chapter 8
178
Since polymeric vesicles are comparable with liposomes, accumulation of
nanoreactors in the liver is expected. This accumulation might lead to liver
toxicity. For this reason, De Vocht et al. evaluated the toxicity of nanoreactors
towards hepatocytes in vitro (manuscript in preparation). They observed no
significant toxicity for nanoreactor concentrations ranging from 50-1000 µg/ml
as compared to the positive control. Although these results are promising and
polymers made from PMOXA-PDMS-PMOXA blocks are approved by the FDA for
use in contact-lens material, the in vivo toxicity towards various cell types
remains unknown as is the case for many block copolymer particles (micelles,
nanotubes …). The available published data are scattered bits and pieces mostly
concerned with in vitro tests on cell cultures. For instance, silicium containing
compounds such as siloranes were shown to have a very low genotoxic potential
on cultured mouse fibroblast cells (Kostoryz et al., 2007). Possibly similar results
will be obtained with PDMS blocks but a definite proof is still missing. It is clear
that an extensive study on the biodistribution and toxicity is needed to provide
the necessary answers.
Thirdly, it is important that nanocontainers can be removed from the body by
degradation and secretion since the PDMS middle block is not degradable.
Although we validated the nanoreactor prodrug activation system in
combination with nucleoside hydrolase of Trypanosoma vivax and 6-
thioguanosine, it can easily be used with other known prodrug activating
enzymes such as carboxipeptidase G2 (Stribbling et al., 1997), β-lactamase
(Meyer et al., 1993) (Cortez-Retamozo et al., 2004), alkaline phosphatase
(Senter et al., 1989), E. coli purine nucleoside phosphorylase (Parker et al.,
2003), etc.. Furthermore nanoreactors can also be used for purposes other than
prodrug activation. For instance, they have potential to be used in enzyme
replacement therapy. Nowadays, the enzymes used in enzyme replacement
therapy are pegylated to prolong the blood circulation (Brewerton et al., 2003).
By incorporating the enzyme in PMOXA-PDMS-PMOXA polymers blood circulation
times might be further increased resulting in less frequent injections of the
enzyme.
Nanoreactors can also be useful in diagnostics where screening of a certain
blood compound is performed with an antibody-enzyme conjugate. However, the
saw no reactor dependent toxicity towards neuroblastoma cells in vitro (chapter

General discussion
179
antibody to enzyme ratio (1:1) in these conventional methods is rather poor.
Using antibody linked nanoreactors can improve the detection signal since a high
concentration of enzymes is linked to the detecting antibody. As a result, the
detection limit of the conventional technique can be improved allowing for
disease detection in an early stage.
Finally, nanoreactors can potentionally be used for in vivo biosensing of blood
glucose levels. For the monitoring of glucose levels in diabetes patients, a
glucose sensor is used. Practically all of the reported sensors that continuously
measure glucose levels are electrochemically based and take advantage of the
reaction of glucose with oxygen. This reaction is catalyzed by glucose oxidase
and forms gluconic acid and peroxide (Wilson et al., 1992). The sensors are
multilayered devices where the enzyme is trapped between two semipermeable
membranes. The inner membrane is connected to an electrode to detect
produced radicals. Instead of using such multilayered devices, a simpler devices
based on glucose oxidase encapsulated nanoreactors, can be envisioned.

Chapter 8
180
8.2 References
Brewerton, L., Fung, E. and Snyder, F. (2003) Polyethyleenglycol-conjugated adenosine phosphorylase: development of alternative enzyme therapy for adenosine deaminase deficiency. Biochim biophys acta, 1637, 171-177.
Broz, P., Benito, S.M., Saw, C., Burger, P., Heider, H., Pfisterer, M., Marsch, S., Meier, W. and Hunziker, P. (2005) Cell targeting by a generic receptor-targeted polymer nanocontainer platform. J Control Release, 102, 475-488.
Cortez-Retamozo, V., Backmann, N., Senter, P.D., Wernery, U., De Baetselier, P., Muyldermans, S. and Revets, H. (2004) Efficient cancer therapy with a nanobodybased conjugate. Cancer Res, 64, 2853-2857.
Harding, F., Liu, A., Stickler, M., Razo, Chin, R., Faravashi, N., Viola, W., Graycar, T., Yeung, V., Aehle, W., Meijer, D., Wong, S., Rashid, M., Valdes, A. and Schellenberger, V. (2005) A beta-lactamase with reduced immunogenicity for the targeted delivery of chemotherapeutics using antibody-directed enzyme prodrug therapy. Mol Cancer Ther, 4, 1791-1800.
Jain, R.K. (1987) Transport of molecules in the tumor interstitium: a review. Cancer Res, 47, 3039-3051.
Kostoryz, E., Zhu, Q., Zhao, H., Glaros, A. and Eick, J. (2007) Assessment of cytotoxicity and DNA damage exhibited by siloranes and oxiranes in cultured mammalian cells. Mutat Res, 634, 156-162.
Matsumura, Y. and Maeda, H. (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res, 46, 6387-6392.
Meyer, D.L., Jungheim, L.N., Law, K.L., Mikolajczyk, S.D., Shepherd, T.A., Mackensen, D.G., Briggs, S.L. and Starling, J.J. (1993) Site-specific prodrug activation by antibody-beta-lactamase conjugates: regression and long-term growth inhibition of human colon carcinoma xenograft models. Cancer Res, 53, 3956-3963.
Parker, W.B., Allan, P.W., Hassan, A.E., Secrist, J.A., 3rd, Sorscher, E.J. and Waud, W.R. (2003) Antitumor activity of 2-fluoro-2'-deoxyadenosine against tumors that express Escherichia coli purine nucleoside phosphorylase. Cancer Gene Ther, 10, 23-29.
Ranquin, A., Versees, W., Meier, W., Steyaert, J. and Van Gelder, P. (2005) Therapeutic Nanoreactors: Combining Chemistry and Biology in a Novel Triblock Copolymer Drug Delivery System. Nano Lett, 5, 2220-2224.
Senter, P.D., Schreiber, G.J., Hirschberg, D.L., Ashe, S.A., Hellstrom, K.E. and Hellstrom, I. (1989) Enhancement of the in vitro and in vivo antitumor activities of phosphorylated mitomycin C and etoposide derivatives by monoclonal antibodyalkaline phosphatase conjugates. Cancer Res, 49, 5789-5792.
Stribbling, S.M., Martin, J., Pedley, R.B., Boden, J.A., Sharma, S.K. and Springer, C.J. (1997) Biodistribution of an antibody-enzyme conjugate for antibody-directed enzyme prodrug therapy in nude mice bearing a human colon adenocarcinoma xenograft. Cancer Chemother Pharmacol, 40, 277-284.

General discussion
181
Wilson, G., Zhang, Y., Reach, G., Moatti-Sirat, D., Poltout, V., Thévenot, D., Lemonrner, R. and Klein, J. (1992) Progress toward the Development of an Implantable Sensor for Glucose. Clin Chem, 38, 1613-1617.


summary
183
Summary
Directed enzyme-prodrug therapies aim to improve conventional
chemotherapy by activating a non toxic prodrug into a toxic drug only at the site
of the tumor. This considerably lowers the systemic toxicity associated with
conventional chemotherapy. Directed enzyme-prodrug therapy involves two
stages. In the first step the activating enzyme is directed to the tumor. In the
second step the non-toxic prodrug is systemically administered. Subsequently,
the prodrug is converted to a toxic drug by the prodrug activating enzyme
resulting in high local concentrations of an anticancer drug at the tumor site. The
targeting of the enzyme can either be mediated by antibodies, named antibody-
directed enzyme-prodrug therapy (ADEPT) (Bagshawe et al., 2004) or by a gene-
vector, named gene-directed enzyme-prodrug therapy (GDEPT) (Dachs et al.,
2005). Both ADEPT and GDEPT suffer from several shortcomings such as
immunogenicity of an antibody-enzyme conjugate in ADEPT or inefficient
transfection, unsustained gene expression, pathogenesis of viral vectors and the
risk of insertional mutagenesis in GDEPT.
Therefore we introduced a novel enzyme-prodrug strategy in which the
prodrug activating enzyme is not directly linked to a tumor targeting antibody
but encapsulated in a nanometer-sized vesicle, called nanoreactor. We first
evaluated a liposomal enzyme reactor (Huysmans et al., 2005). Nucleoside
hydrolase of Trypanosoma vivax (TvNH), was encapsulated in liposomes. TvNH is
able to hydrolyse nucleoside analogs into the respective nucleobase analogs and
ribose. The vesicles were permeabilised for substrates and products by inserting
porins of Escherichia coli (OmpF and PhoE) into the lipid membrane. By liposome
swelling assays, we could show the successful diffusion of the substrates of TvNH
across the lipid membrane. Functionalized reactors were more effective in

Summary
184
substrate hydrolysis than control reactors, however the control reactors do show
enzymatic activity which indicates that the liposomes are leaky.
Therefore we constructed nanometer-sized reactors that are composed of
PMOXA-PDMS-PMOXA triblock copolymers. These triblock copolymers are able to
self assemble into various aggregates such as vesicles, micelles, nanotubes and
free standing films in aqueous solutions. Their phase behaviour in aqueous
solutions depends on the composition, length and structure of the individual
blocks as well as on the molecular architecture of the whole polymer and the
molecular weight distribution of the individual blocks. Small changes in these
properties can lead to a different behaviour in aqueous solutions. We therefore
evaluated several PMOXA-PDMS-PMOXA polymers from different batches and
different compositions for their ability to self assemble into nanometer-sized
vesicles. We identified two batches that were able to do so: PMOXA18-PDMS72-
PMOXA18 (Ranquin et al., 2005) from the lab of Prof. W. Meier at the university in
Basel and the commercially available PMOXA21-PDMS54-PMOXA21 from Polymer
Source. Furthermore, we were able to make enzymatically active vesicles with
these polymers. To this end, TvNH was encapsulated in the polymeric vesicles
and the polymeric membrane was permeabilized by incorporation of OmpF or
Tsx. These reactors were able to hydrolyse several substrates such as adenosine,
guanosine, inosine, 2-fluoroadenosine and 6-thioguanosine. By fine tuning the
production method, enzymatically active nanoreactors with a mean radius of 75-
100 nm, as determined by DLS, were obtained. AFM measurements did not
confirm the size measured by DLS but this is attributed to the adsorption of the
reactors to the mica chip and the forces applied by the scanning tip which leads
to deformation of the reactors. On the other hand, TEM images confirmed the
size and monodispersity measured with DLS.
After a screening of several potential prodrugs, 6-thioguanosine was selected
as a good candidate. We found that the cytotoxic effect of 6-thioguanine on
neuroblastoma cells only becomes apparent after 24h which is in agreement with
the cytotoxic mechanism of 6-thioguanine (Maybaum and Mandel, 1983; Wotring
and Roti Roti, 1980). Determination of the EC50 values of 6-thioguanosine and 6-
thioguanine showed a difference in cytotoxicity between both compounds.
However, 6-thioguanosine also display toxicity at higher concentrations which is
attributed to the hydrolyses of 6-thioguanosine by human purine nucleoside

summary
185
phosphorylase (hPNP). This could lead to toxicity of the prodrug 6-thioguanosine
towards lymphocytes.
To validate the use of polymeric nanoreactors as an alternative for ADEPT and
GDEPT strategies we explored the possibility to use TvNH encapsulating
nanoreactors in combination with 6-thioguanosine as a novel enzyme-prodrug
therapy. To this end, the cytotoxicity of nanoreactors in the absence and
presence of 6-thioguanosine towards neuroblastoma cells was measured.
Although nanoreactors have a negative effect on cell growth, this is only a minor
effect and it is independent of the nanoreactor concentration. We demonstrated
that 5µM of 6-thioguanosine and 0,33 mg/ml nanoreactors are sufficient to
almost completely kill the neuroblastoma cell culture within 72 h.
Although macromolecules ranging from 10-500 nm in size can be targeted
passively to tumor tissue due to the enhanced permeation and retention effect
(EPR) (Matsumura and Maeda, 1986) (Jain, 1987), more specific targeting by
attaching ligands to the nanoreactors will improve targeting. In this study we
want to couple camelid antibodies to PMOXA-PDMS-PMOXA triblock copolymers.
As a model antibody we used cAbLys-3, an antibody directed against lysozyme.
The presence of a hydroxyl group on the polymers forced us to use the hetero
bifunctional linker PMPI which can be coupled to the polymer via its isocyanate
end and to the antibody with its maleimide group to form a thioether bond with
sulfhydryl residues. Cysteines present in cAbLys-3 are involved in disulphide
bridges and therefore unavailable for coupling.
Therefore, the solved X-ray structure of cAbLys-3 was scrutinized for possible
site-directed cysteine mutants that wouldn’t interfere with antigen binding. We
were able to construct three cAbLys-3 serine to cysteine mutants, S17C, S134C
and S141C that still can bind lysozyme with similar efficiency as the wild type
cAbLys-3. Since expression of the cAbLys-3 mutants in E. coli yields only a low
amount of protein (0.15 – 0.23 mg/l culture medium) as compared to WT
cAbLys-3 (4 mg/l), we used an eukaryotic expression host, Pichia pastoris.
Larger expression yields were obtained through high cell densities and strong
promoters. The expression yield for cAbLys-3 S17C was improved 60 fold by
using P. pastoris.
Although preliminary experiments for the coupling of TvNH-Tsx nanoreactors
to cAbLys-3 have been performed, they were unsuccessful (data not shown).

Summary
186
Much effort still has to be put into fine tuning the chemical reaction and finding
an efficient assay for the detection of nanobody-coupled nanoreactors.
References
Bagshawe, K., Sharma, S. and Begent, R. (2004) Antibody-directed enzyme prodrug therapy
(ADEPT) for cancer. Expert Opin Biol Ther, 4, 1777-1789. Dachs, G., Tupper, J. and Tozer, G. (2005) From bench to bedside for gene-directed enzyme
prodrug therapy of cancer. Anticancer drugs, 16, 349-359. Huysmans, G., Ranquin, A., Wyns, L., Steyaert, J. and Van Gelder, P. (2005) Encapsulation of
therapeutic nucleoside hydrolase in functionalised nanocapsules. J Control Release, 102, 171-179.
Jain, R.K. (1987) Transport of molecules in the tumor interstitium: a review. Cancer Res, 47, 3039-3051.
Matsumura, Y. and Maeda, H. (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res, 46, 6387-6392.
Maybaum, J. and Mandel, H.G. (1983) Unilateral chromatid damage: a new basis for 6-thioguanine cytotoxicity. Cancer Res, 43, 3852-3856.
Ranquin, A., Versees, W., Meier, W., Steyaert, J. and Van Gelder, P. (2005) Therapeutic Nanoreactors: Combining Chemistry and Biology in a Novel Triblock Copolymer Drug Delivery System. Nano Lett, 5, 2220-2224.
Wotring, L.L. and Roti Roti, J.L. (1980) Thioguanine-induced S and G2 blocks and their significance to the mechanism of cytotoxicity. Cancer Res, 40, 1458-1462.

summary
187
Samenvatting Gerichte enzym-prodrug therapieën hebben als doel de conventionele
chemotherapie te verbeteren door niet toxische prodrugs enzymatisch om te
zetten naar toxische drugs op de plaats van de tumor. Op deze manier worden
de neveneffecten, te wijten aan systemische toxiciteit van conventionele
chemotherapeutica, aanzienlijk verminderd. Gerichte enzym-prodrug therapieën
zijn opgebouwd uit twee fasen. In een eerste fase wordt het prodrug activerend
enzym naar de tumor gericht. In een tweede fase wordt de niet toxische prodrug
systemisch toegediend. Deze prodrug wordt dan enkel ter hoogte van de tumor
omgezet tot de toxische drug door het aanwezige enzym. Het enzym kan op
twee verschillende manieren naar de tumor gericht worden. Enerzijds door de
koppeling van het enzyme aan een antilichaam dat specifiek op tumor cellen
bindt (ADEPT) (Bagshawe et al., 2004) Anderzijds door het gen dat codeert voor
het enzym naar de tumor te brengen via een vector (GDEPT) (Dachs et al.,
2005). Beide systemen hebben te kampen met enkele problemen zoals
immunogeniciteit van de antilichaam-enzym conjugaten in ADEPT en een
inefficiënte gen transfectie, verlies van genexpressie, pathogenese van virale gen
vectoren en het risico op insertionele mutagenese in GDEPT.
Daarom introduceren we een nieuwe methode voor enzym-prodrug therapie
die gebaseerd is op het inkapselen van een prodrug activerend enzym in
nanometerschaal vesikels, die nanoreactoren genoemd worden.
Allereerst werden liposoom-enzym reactoren gemaakt en getest (Huysmans et
al., 2005). Het nucleoside hydrolase van Trypanosoma vivax (TvNH), dat
nucleoside analogen kan omzetten naar nucleobase analogen en ribose, werd
geïncapsuleerd in liposomen. De liposomes werden vervolgens gepermeabiliseerd
door de incorporatie van bacteriële porines afkomstig van E. coli (OmpF en PhoE)
in de lipiden membraan. Liposoom zweltesten toonden aan dat natuurlijke
substraten van dit enzym kunnen diffunderen door deze porines zodat ze kunnen
omgezet worden. Hoewel de TvNH ingekapselde liposomen die gepermeabiliseerd

Samenvatting
188
werden in staat zijn substraten te hydrolyseren, vertonen ook de niet
gepermeabiliseerd controle reactoren een zeker activiteit. Dit duid erop dat de
liposomen doorlaatbaar zijn voor het substraat of het enzyme .
Daarom werden vervolgens nanoreactoren gemaakt die opgebouwd zijn uit
PMOXA-PDMS-PMOXA triblok copolymeren. Deze polymeren zijn in staat om
spontaan aggregaten zoals vesikels, micellen, nanotubes en vrijstaande films te
vormen in waterige oplossingen. Het fasegedrag van zulke polymeren in waterige
oplossingen wordt bepaald door de compositie, de lengte en structuur van de
individuele blokken alsook de complete moleculaire architectuur en de distributie
van het moleculaire gewicht van de individuele blokken. Kleine veranderingen in
deze eigenschappen kunnen leiden tot een ander fasegedrag in waterige
oplossingen. Verschillende PMOXA-PDMS-PMOXA polymeren, afkomstig uit
verschillende synthese batchen en met verschillende composities werden daarom
gescreened. Op deze manier werden twee batchen geïdentificeerd die in staat
zijn om spontaan vesikels te vormen in waterige oplossingen. Bovendien zijn
deze vesicles actief wanneer TvNH wordt ingekapseld en de porines Tsx of OmpF
worden geïncorporeerd in het membraan. De eerste batch, PMOXA18-PDMS72-
PMOXA18 (Ranquin et al., 2005), werd ons geschonken door Prof. W. Meier van
de universiteit in Basel en de tweede batch, PMOXA21-PDMS54-PMOXA21, werd
aangekocht bij Polymer Source. Deze reactoren zijn instaat substraten als
inosine, adenosine, guanosine, 2-fluoroadenosine en 6-thioguanosine om te
zetten naar hun respectievelijke basen en ribose. Door de productie methode van
de nanoreactoren te verfijnen zijn we in staat nanoreactoren te maken met een
gemiddelde straal van 75-100 nm. De grootte van de partikels, die gemeten
werd met DLS, werd niet bevestigd door AFM metingen. Dit is te wijten aan de
adsorptie van de nanoreactoren aan de mica chip en de kracht die de naald
uitoefent op de partikels. TEM beelden daarentegen bevestigen wel de
afmetingen van de reactoren die gemeten werden via DLS. Bovendien tonen
deze beelden monodisperse, sferische vesikels.
Na screening van verschillende potentiële prodrugs, werd 6-thioguanosine
geselecteerd. In onze experimenten komt het cytotoxisch effect van de drug 6-
thioguanine op neuroblastoma cellen komt pas na 24 uur tot uiting. Deze
observatie is te verklaren door het cytotoxisch mechanisme van 6-thioguanine
dat in detail beschreven wordt door Maybaum en Mandel en Wotring en Roti Roti

samenvatting
189
(Maybaum and Mandel, 1983) (Wotring and Roti Roti, 1980). Door de EC50
waarden van zowel 6-thioguanosine als 6-thioguanine te bepalen is het duidelijk
dat 6-thioguanosine bij lage concentraties (~ 1µM) beduidend minder toxisch is
dan de drug 6-thioguanine. Bij hogere concentraties (~ 5µM) vertoont ook de
prodrug 6-thioguanosine cytotoxiciteit. Deze toxiciteit wordt veroorzaakt door de
omzetting van 6-thioguanosine naar 6-thioguanine door endogeen purine
nucleoside phosphorylase. Dat voornamelijk tot expressie komt in lymfocyten.
Om triblok copolymere nanoreactoren te valideren als alternatief voor ADEPT
en GDEPT, werd het cytotoxisch effect op neuroblastoma cellen van TvNH
ingekapselde nanoreactoren, gepermeabiliseerd door Tsx, in combinatie met 6-
thioguanosine nagegaan. Hiervoor werden twee verschillende concentraties
nanoreactoren gebruikt (0,33 mg/ml en 0,66 mg/ml). Beide reactor
concentraties vertoonden weinig effect op de celgroei, in afwezigheid van 6-
thioguanosine. Wanneer 6-thioguanosine wordt toegevoegd, volstaat 5 µM om de
volledige celpopulatie te vernietigen in 72 uur.
Macromoleculen met een grootte van 10-500 nm kunnen via het enhanced
permeation and retention effect (EPR) (Jain, 1987) (Matsumura and Maeda,
1986) op een passieve manier naar tumoren gericht worden. Het gebruik van
antilichamen gericht naar tumoren kan de accumulatie in de tumor nog
verhogen. In deze studie werden kameel antilichamen gebruikt (cAbLys-3) voor
de targeting van nanoreactoren naar tumoren. Omdat de eindstandige groep van
de PMOXA-PDMS-PMOXA polymeren bestaat uit een hydroxyl groep werden we
gedwongen de hetero bifunctionele linker PMPI te gebruiken. Deze linker kan
reageren met hydroxyl groepen op de polymeren en met thiol groepen op het
antilichaam. De 4 cysteine residu’s aanwezig in cAbLys-3, zijn betrokken bij
disulfide bruggen waardoor zij niet in aanmerking komen voor koppeling met de
nanoreactoren. Om deze reden werden er 3 extra cysteine residu’s
geïntroduceerd via gerichte mutagenese van serine residu’s. De serine residu’s
(S17, S134 en S141) werden gekozen om dat ze ver gelegen zijn van de antigen
binding site. Aangezien de expressie van de mutanten in E. coli zeer laag was
(0.15 - 0.23 mg/ml) werd een eukaryoot expressie systeem gebruikt, nl. de gist
Pichia pastoris. Op deze manier werd de expressie van cAbLys-3 S17C 60 maal
verhoogd. Hoewel er reeds preliminaire experimenten werden uitgevoerd om
cAbLys-3 mutanten te koppelen aan nanoreactoren, waren zij niet succesvol.

Samenvatting
190
Referenties
Bagshawe, K., Sharma, S. and Begent, R. (2004) Antibody-directed enzyme prodrug therapy (ADEPT) for cancer. Expert Opin Biol Ther, 4, 1777-1789.
Dachs, G., Tupper, J. and Tozer, G. (2005) From bench to bedside for gene-directed enzyme prodrug therapy of cancer. Anticancer drugs, 16, 349-359.
Huysmans, G., Ranquin, A., Wyns, L., Steyaert, J. and Van Gelder, P. (2005) Encapsulation of therapeutic nucleoside hydrolase in functionalised nanocapsules. J Control Release, 102, 171-179.
Jain, R.K. (1987) Transport of molecules in the tumor interstitium: a review. Cancer Res, 47, 3039-3051.
Matsumura, Y. and Maeda, H. (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res, 46, 6387-6392.
Maybaum, J. and Mandel, H.G. (1983) Unilateral chromatid damage: a new basis for 6- thioguanine cytotoxicity. Cancer Res, 43, 3852-3856.
Ranquin, A., Versees, W., Meier, W., Steyaert, J. and Van Gelder, P. (2005) Therapeutic Nanoreactors: Combining Chemistry and Biology in a Novel Triblock Copolymer Drug Delivery System. Nano Lett, 5, 2220-2224.
Wotring, L.L. and Roti Roti, J.L. (1980) Thioguanine-induced S and G2 blocks and their significance to the mechanism of cytotoxicity. Cancer Res, 40, 1458-1462.

Publications
191
Publications Dao-Thi MH, Van Melderen L, De Genst E, Buts L, Ranquin A,
Wyns L, Loris R. Crystallization of CcdB in complex with a GyrA
fragment. Acta Crystallogr D Biol Crystallogr. 2004 Jun;60(Pt
6):1132-4.
Ranquin A, Van Gelder P. Maltoporin: sugar for physics and
biology. Res Microbiol. 2004 Oct;155(8):611-6.
Huysmans G, Ranquin A, Wyns L, Steyaert J, Van Gelder P.
Encapsulation of therapeutic nucleoside hydrolase in functionalised
nanocapsules. J Control Release. 2005 Jan 20;102(1):171-9.
Ranquin A, Versées W, Meier W, Steyaert J, Van Gelder P.
Therapeutic nanoreactors: combining chemistry and biology in a
novel triblock copolymer drug delivery system. Nano Lett. 2005
Nov;5(11):2220-4.
Ranquin A, De Vocht C, Van Gelder P. Polymer-based
nanoreactors for medical applications. In: Polymer-based
nanostructures: Medical Applications. P Broz, Editor. Royal Society
of Chemistry. In press.
Ranquin A, De Vocht C, Wilkinson H, Steyaert J, Van Gelder P.
Nanoreactor mediated prodrug activation and killing of
neuroblastoma cells. Manuscript in preparation.
De Vocht C, Ranquin A, Van Ginderachter J, Vanhaecke T,
Versées W, Van Gelder P, Steyaert J. Polymeric enzyme-loaded
nanoreactors for enzyme replacement therapy. Manuscript in
Preparation.