tirs 2015 day 2 general toxicology buckley 2015_day 2_general... · general toxicology lorrene a...
TRANSCRIPT

Toxicology for Industrial and Regulatory ScientistsToxicology for Industrial and Regulatory Scientists
General Toxicology
Lorrene A Buckley PhD DABT ATSLorrene A Buckley, PhD, DABT, ATSSr Research Fellow
Toxicology and Drug DispositionEli Lill d C IEli Lilly and Company, Inc
28 April 201528 April 2015

Course Objectivesj
1. To describe the principles of general (descriptive) toxicology testingtoxicology testing
2. To describe how these principles are applied in nonclinical product development
3. To describe how these results support regulatory objectives
F ill b h h iFocus will be on human therapeutics• Most principles also apply to other regulated industries
(food/color additives pesticides industrial/specialty(food/color additives, pesticides, industrial/specialty chemicals, etc)
• Emphasis on US Food and Drug Administration (FDA)
Slide 2

Course Overview
• Basic Tenets of Toxicology TestingP D i C id ti• Program Design Considerations• General Regulations and Guidelines• Specific chemical/product class/use
• Study Design Considerations• General requirements: GLPs, Animal Care & Use• Specific Design ParametersSpecific Design Parameters
• Regulatory application of nonclinical data• Nonclinical safety assessment in drug development
Slide 3

Value of Nonclinical Studies
• Surrogate to characterize potential effects in humans (efficacy, safety)• Identify target organs/systemsIdentify target organs/systems• Study dose-response relationships across wide range of
doses/exposures (NOEL, NOAEL, LOAEL, MTD)• Characterize reversibility and monitorabilityCharacterize reversibility and monitorability• Identify biomarkers for diagnosis, monitoring, & understanding MoA
• Well-defined methods of investigation based on precedence & experience• Can be conducted under controlled laboratory conditions• Study effects not possible/ethical in humans (eg, systematic histopath-
ologic evaluation, embryofetal development, carcinogenic potential)g , y p , g p )• Meet requirements and expectations of regulatory authorities
N(L)O(A)EL = No (Low) Observed (Adverse) Effect Level;
Slide 4
N(L)O(A)EL = No (Low) Observed (Adverse) Effect Level;MTD = Maximum Tolerated Dose; MoA = Mechanism of Action

Basic Tenets of Toxicology Testing
General assumptions:A i l d l ill di t h ffi• Animal models will predict human efficacy and safety/toxicity
• High doses will maximize model sensitivity• High doses will maximize model sensitivity to detect effects
Slide 5

$64M Q: How Well Do Animals Predict Humans?
Olson, et al. Concordance of the Toxicity of Pharmaceuticals in Humans and in Animals Reg Tox Pharmacol 32:56 67 (2000)Pharmacol 32:56-67 (2000)
Slide 66
Courtesy of Derek Leishman

Differences Between Animals & HumansAnimals Humans
Subjects
N b L G I di id lNumber Large Groups Individual
Age Young Adult All ages
State of health Healthy Usually sick
Genetic background Homogeneous Heterogeneous
Doses
Magnitude Therapeutic to toxic Therapeutic
Schedule Usually once daily Therapeutic optimum
CircumstancesCircumstances
Housing, Nutrition Uniform, optimal Variable
Concomitant therapy Never Frequent
Diagnostic procedures
Verbal contact None Intensive
Physical exam Limited Extensive
Clinical lab Limited, standardized Individualized
Timing Predetermined Individualized
Slide 7
Timing Predetermined Individualized
Autopsy/Histopathology Always / Extensive Exceptional

Common (Human) Reactions to Drugs( ) g
Predictable from Animal Studies NOT PredictableDrowsiness Insomnia Depression NauseaSedation Constipation Appetite FatigueDry mouth Weight gain Tremor DizzinessDry mouth Weight gain Tremor DizzinessNervousness Hypotension Perspiration TinnitusVomiting Diarrhea Dermatitis HeartburnWeakness Skin rash Energy VertigoNasal Dryness of Palpitation Headachestuffiness nasopharynx Blurred visionstuffiness nasopharynx Blurred visionHypertension Anorexia Lethargy
Adapted from: M A Dorato M J Vodicnik (2008) The toxicological assessment of pharmaceutical
Slide 8
Adapted from: M.A. Dorato, M.J. Vodicnik (2008). The toxicological assessment of pharmaceutical and biotechnology products. In: Principles and Methods of Toxicology, 5th ed. (A.W. Hayes, ed.)

Concordance of Animal & Human Toxicities
•International Life Sciences Institute (ILSI) workshop (pharmaceutical survey)• 150 compounds, 221 human toxicities; 12 companies• Caveats: Retrospective survey; excluded compounds which did not
advance to clinic (eg, “killed” in preclinical development)
• 71% of the time target organSpecies % Concordance • 71% of the time, target organ toxicity seen in humans occurred in one or more animal models
• Dogs tend to be slightly more in line
pAll Species 71Rodent (rat) 43
N d t (d ) 63 with predicting human toxicity than rodents
Up to 43% of toxicities related to pharmacological action (largely anticipatable)
Non-rodent (dog) 63Other species 29
• Up to 43% of toxicities related to pharmacological action (largely anticipatable)• 94% of toxicities were detected in the 1-month studies• Highest concordance: Hematological, GI, Cardiovascular (Least = Cutaneous)
Slide 9
Olson, et al. Concordance of the Toxicity of Pharmaceuticals in Humans and in Animals. Reg Tox Pharmacol 32:56-67 (2000)

Perspective: Two different questions• Question: “Can animals predict the (human) effect?”
• Limitations to our data set Human+ -
With positive findings (Animal +), compoundnot advanced in humans (“??”)
• Clear model limitations: Animals ≠ Humans
+
nim
al +
• Room for predictive improvement Drug-Induced Liver Injury (DILI)
• Question: “Is it safe?”
An -
• Nonclinical safety assessment studies define the “space” within which a compound can be administered without unacceptable toxicityy
• Animal studies inform clinical design and monitoring• Generally, a good safety record for clinical trials in human subjects
based on nonclinical data
Slide 10

Use of High Doses in Toxicology Studies
• Nonclinical toxicology studies aim to define the bounds of what (bad) could possibly occurof what (bad) could possibly occur
• High doses to account for uncertainties in interspecies extrapolations and intra-populations susceptibilities
• Maximum Tolerated Dose (MTD) designed to provide a level of toxicity indicative of sufficient chemical challenge to allow expression of toxicity within the systemto allow expression of toxicity within the system
• Issues with excessively high doses• Extrapolation from high (animal) to low (human) doses is not p g ( ) ( )
usually linear• Distortion of normal physiologic pathways, eg, saturation of
detoxication processes
Slide 11
p
(More discussion in subsequent “Dose Selection” section)

Developing a Nonclinical Safety Evaluation Planp g y
• Compound-specific considerations that shape the plan• What do we already know?• What do we already know?
• Product type and similarity to existing agents with known safety profiles (New Molecular Entity vs Follow-on/Generic Drug)D l ff t• Drug class effects
• Pharmacology (and “exaggerated pharmacology”)• What is the molecular target or pathway?• Conserved across species? Tissue distribution?• Toxicity associated with target manipulation?
• Chemical / Metabolic stability• Metabolized similarly across species? (in vitro)
Slide 12

Developing a Nonclinical Safety Evaluation Planp g y
• For a specific therapeutic, consider use patterns:Wh i th t t h l ti ?• Who is the target human population?
• adults, infants, pregnant women, elderly, etc.• What is the proposed duration and route(s) of administration?
• Daily vs intermittent dosing; short-term vs chronic• Parenteral vs oral or dermal or inhalation dosing
• What are use pattern considerations?What are use pattern considerations?• concomitant medications, adjuvant therapy
• Dose range finding data and pilot studiesInteraction ith reg lator a thorities ill pro ide additional perspecti e /• Interaction with regulatory authorities will provide additional perspective / information
Slide 13

Developing a Nonclinical Safety Evaluation Plan
• Regulatory Guidance• ICH safety & multidisciplinary guidance documents• ICH safety & multidisciplinary guidance documents
www.ich.org• FDA Pharmacology/Toxicology
www fda gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidwww.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm065014.htm
• European Medicines Agency (EMA)www ema europa eu/emawww.ema.europa.eu/ema
• Regulatory Precedence• Reviewers’ comments regarding approved drugs
• FDA: SBA www.accessdata.fda.gov/scripts/cder/drugsatfda• EU CHMP: EPARS
• Japan PMDA: submissions are published
Slide 14
Japan PMDA: submissions are published

International Conference of Harmonization (ICH)
USAJapanMHLWUSA
FDAPhRMA Europe
EMAEFPIA
MHLWJPMA
EFPIA
• To promote uniformity in technical requirements for registration of pharmaceuticals for human use• Discuss and establish common guidelines (EU, Japan, USA) for
technical requirements• Ensure that safe, effective, and high quality medicines are developed
d i t d i th t ffi i t d t ff tiand registered in the most efficient and cost-effective manner• Prevent unnecessary duplication• Minimize the use of animal testing• Promote public health
Slide 15
• Promote public health

ICH Guidance Related to Nonclinical Safety
S1A,B,C(R2) Carcinogenicity: When Studies are Needed, Testing Methodology, Dose Selection
S2(R1) Genotoxicity StudiesS3A, B Toxicokinetics & Tissue Distribution StudiesS4 Duration of Chronic Toxicity in Rodents & NonrodentsS5(R2) Reproductive ToxicologyS5(R2) Reproductive ToxicologyS6(R1) Preclinical Safety: Biotechnology ProductsS7A, B Safety Pharmacology Studies; QT ProlongationS8 Immunotoxicology StudiesS9 Nonclinical Evaluation of Anticancer Pharmaceuticals (+Q&A)S10 Photosafety EvaluationS11 Nonclinical Testing for Pediatrics
Q3A-D Guidance on Impurities, Residual Solvents, Elemental Impurities
M3(R2)+Q&A Guidance on nonclinical safety studies for the conduct of human li i l t i l d k ti th i ti f h ti l
Slide 16
clinical trials and marketing authorization for pharmaceuticalsM7 Genotoxic Impurities

Developing a Nonclinical Safety Evaluation PlanObjectives: Early Studies (2-Week, 1-Month)Objectives: Early Studies (2 Week, 1 Month)• Identify the target organs/systems of the drug
• Gender differences? Target organs? Monitorable?g g• If recovery groups: Reversible? Progressive?• Expected? (based on pharmacology / MoA)
• Characterize the dose-response relationship of toxicity• Characterize the dose-response relationship of toxicity• Linear? Supra / sublinear?• Identify No Observed Adverse Effect Level (NOAEL)
• NOEL more appropriate for food additives, color additives, etc• Characterize plasma exposure parameters (eg, Cmax, AUC, t1/2)
• Aid in selection of doses for first-in-human (FIH) studies• NOAEL = typical push-off point for clinical dose selection
• Anticancer drugs start higher: rodent STD10 (severely toxic dose in 10%) & nonrodent HNSTD (highest non-severely toxic dose)
Slide 17
in 10%) & nonrodent HNSTD (highest non severely toxic dose) • Assist in selection of doses for longer term toxicology studies

Developing a Nonclinical Safety Evaluation PlanObjectives: Subchronic/Chronic StudiesObjectives: Subchronic/Chronic Studies
• Assessment of progressive toxicity• i.e., do toxicities seen previously occur at lower doses?
• Identification of duration-dependent or new toxicitiesS f l li i l i l• Support for longer term clinical trials
• Biomarker identification, directed toxicity evaluations (e g special histopathology CNS assessments)(e.g., special histopathology, CNS assessments)
• Support for additional toxicology studies• 13-week rodent studies support dose and design of pp g
traditional 2-year carcinogenicity studies
Slide 18

Developing a Nonclinical Safety Evaluation PlanOther Things to ConsiderOther Things to Consider
• Small Molecule vs. Biologic• Same basic principles but implementation can be very different• Same basic principles, but implementation can be very different
• Acute vs. Chronic Therapeutic Indication• The impact of Therapeutic Indication
• Risk / Benefit• Life-threatening treatment vs Long-term maintenance• Alternative treatments?
• The impact of Route of Administration• Medical device?
• The impact of Geography• ICH guidelines promote uniformity , BUT …• FDA inter-Division differences
Slide 19
• The significance of Rodent vs. Non-Rodent effects

Conduct & Design of Toxicology Studies
• Basic requirements• Animal Care and Use• Animal Care and Use• Good Laboratory Practice
• Study Design ConsiderationsStudy Design Considerations• General• Specific Route of Administration Duration of Treatment Species selection Species selection Dose selection Parameters evaluated
Slide 20

Conduct of Toxicology Studies:Laboratory Operations – Animal CareLaboratory Operations – Animal Care
• Association for Assessment and Accreditation ofLaboratory Animal Care (AAALAC) accreditationy ( )• Non-profit organization that promotes
the humane treatment of animals • Global standard for assurance of qualityG oba sta da d o assu a ce o qua ty
animal care and facility operations
• Institutional Animal Care and Use Committee (IACUC)• Institutional Animal Care and Use Committee (IACUC)• Assure that study designs and study conduct adhere to
standards• Reviews each protocol for its individual merits with an eye to• Reviews each protocol for its individual merits with an eye to
responsible animal care and use• Independent oversight with outside member• Facility review and reporting to Management
Slide 21
• Facility review and reporting to Management

Conduct of Toxicology Studies:Laboratory Operations - GLPsLaboratory Operations GLPs• Good Laboratory Practice (GLP) regulations ensure quality
and integrity of the data which must be:g y• Well-documented - every action and step is documented in detail,
signed, and dated on the day collected• Signature = you agree data is correct & you are accountable• Signature = you agree data is correct & you are accountable• Data must stand on its own without need for someone to explain
what occurredR li bl S i ti t h ld b bl t t th t d• Replicable - Scientists should be able to repeat the study• New study should produce the same results as original
• Reconstructable - Reviewers can recreate study from the data / documentation available
Slide 22
CFR Title 21 Part 58; www.fda.gov/ora/compliance;
Conduct of Toxicology Studies:Laboratory Operations - GLPsLaboratory Operations GLPs
RAWDATASOPsX, Y, Z
QA
Slide 23
QA

Design of Toxicology Studies:General ConsiderationsGeneral Considerations
• The test article (active pharmaceutical ingredient;API) should be representative of what will be usedclinically• Qualification of impurities (ICH Q3; M7)• Characterization of test and control articles
• Identity, strength, purity, and composition• Stability, homogeneityStability, homogeneity
• Considerations for inactive ingredients (excipients)and final formulation used nonclinically• e g IV polysorbate 80 causes histamine release in dogs• e.g., IV polysorbate 80 causes histamine release in dogs• “novel” excipients may require stand-alone data
Slide 24

Design of Toxicology Studies:General ConsiderationsGeneral Considerations• Two mammalian species (one nonrodent) expected (no 1 perfect model)
• Possible exceptions where one species may be sufficient:p p y• Biopharmaceuticals
• drug is pharmacologically active in only one species• similar profile in 2 species allows chronic study in one species only• similar profile in 2 species allows chronic study in one species only
due to highly targeted nature of drug (ICH S6(R1)) • Genotoxic anticancer drugs (ICH S9) – one species (rodent) may be
considered sufficient for an agent targeting rapidly dividing cellsconsidered sufficient for an agent targeting rapidly dividing cells• New route of administration for an “old” drug
• e.g., dermal formulation for oral pain medication - 1 species to assess local effects knowing systemic safety already establishedassess local effects knowing systemic safety already established
• Both genders are expected, even for drugs designed to treat one sex clinically (reproductive health/urology)
Slide 25

Design of Toxicology Studies:General ConsiderationsGeneral Considerations
Basic study designs are standardized but do differ for rodents and nonrodents• Practicalities of handling g
& cost drive smaller numbers of nonrodents
• Both young adult at i iti ti b t lifinitiation, but lifespan differences over treatment periods
• Able to more liberallyAble to more liberally sample large animals
• Toxicokinetics• Clinical pathologyp gy• Predose samples
allow within animal evaluations
Slide 26
Keller and Banks (2006)
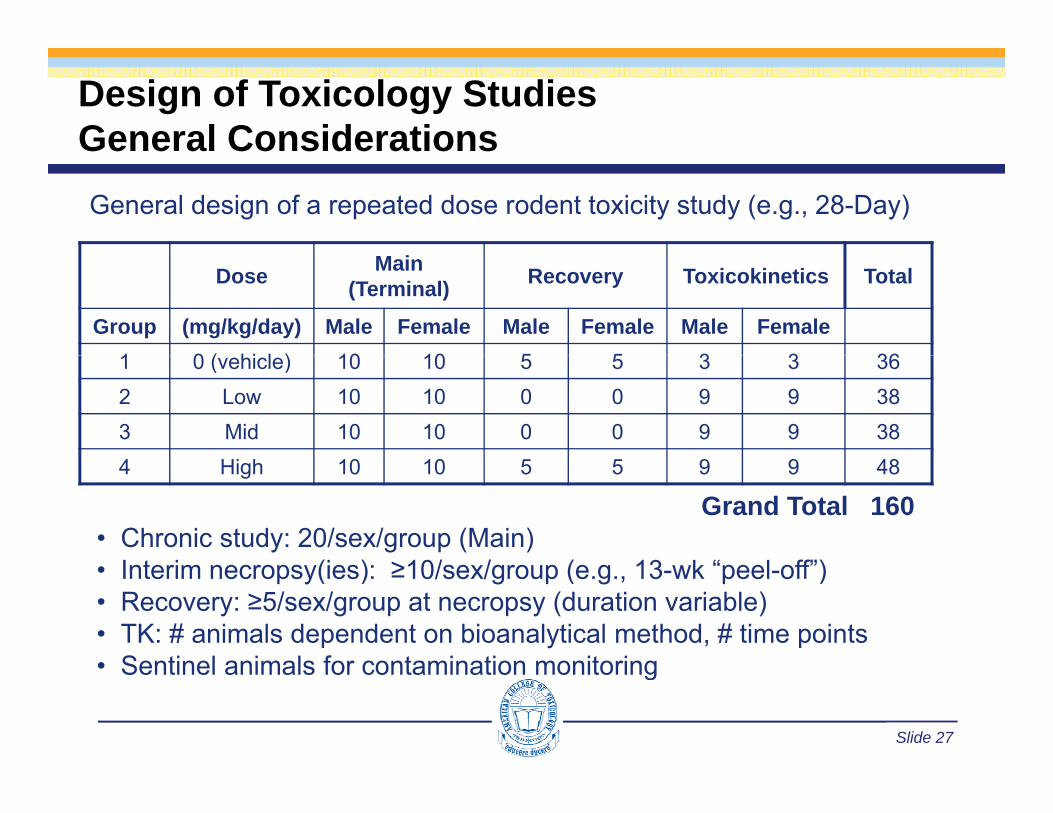
Design of Toxicology StudiesGeneral ConsiderationsGeneral ConsiderationsGeneral design of a repeated dose rodent toxicity study (e.g., 28-Day)
Dose Main (Terminal) Recovery Toxicokinetics Total
Group (mg/kg/day) Male Female Male Female Male Female1 0 ( hi l ) 10 10 5 5 3 3 361 0 (vehicle) 10 10 5 5 3 3 362 Low 10 10 0 0 9 9 383 Mid 10 10 0 0 9 9 384 High 10 10 5 5 9 9 48
Grand Total 160• Chronic study: 20/sex/group (Main)
I t i (i ) 10/ / ( 13 k “ l ff”)• Interim necropsy(ies): ≥10/sex/group (e.g., 13-wk “peel-off”)• Recovery: ≥5/sex/group at necropsy (duration variable)• TK: # animals dependent on bioanalytical method, # time points• Sentinel animals for contamination monitoring
Slide 27
• Sentinel animals for contamination monitoring

Design of Toxicology StudiesGeneral ConsiderationsGeneral ConsiderationsGeneral design of a repeated dose nonrodent toxicity study (e.g., 28-Day)
Dose Main (Terminal) Recovery Toxicokinetics Total
Group (mg/kg/day) Male Female Male Female Male Female1 0 ( hi l ) 3 3 2 2 0 0 101 0 (vehicle) 3 3 2 2 0 0 102 Low 3 3 0 0 0 0 63 Mid 3 3 0 0 0 0 64 Hi h 3 3 2 2 0 0 104 High 3 3 2 2 0 0 10
Grand Total 32• Chronic study: ≥ 4/sex/group (Main)
I t i (i ) ≥ 3/ / ( 13 k “ l ff”)• Interim necropsy(ies): ≥ 3/sex/group (e.g., 13-wk “peel-off”)• Recovery: ≥ 2/sex/group at necropsy
• duration may be substantial, eg, 3-4 mo for mAb clearance)• TK animals not required
Slide 28
• TK animals not required

Design of Toxicology Studies:Route of AdministrationRoute of Administration
• Route should be same as that intended for use in humans• Other routes may be employed if human route cannot be duplicated inOther routes may be employed if human route cannot be duplicated in
animal; must be supported by data such as:• Limited systemic exposure
• e g topical NME programs often include oral (or IV) rodent (rat)• e.g., topical NME programs often include oral (or IV) rodent (rat) + topical nonrodent (minipig) to assess both local & systemic tox
• Model limitations due to route of administrationbli l d i ( IV t l l l t l )• e.g., sublingual dosing (use IV tox + mucosal local tolerance)
• Size/physiology of the animal species may limit options• Need to understand toxicokinetic behavior of clinical vs nonclinical route• Frequency of dosing should match or exceed clinical dosing paradigm
• May increase nonclinical dosing to “cover” more rapid clearance or presence of anti-drug antibodies in animals
Slide 29
p g
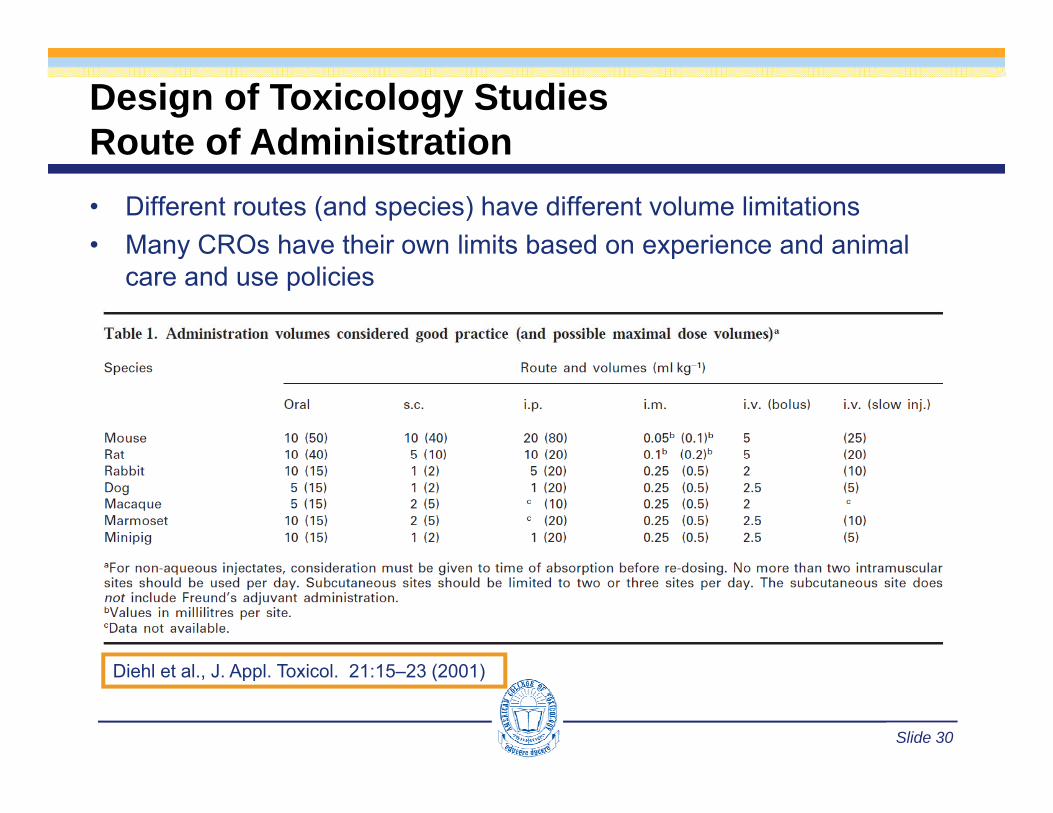
Design of Toxicology StudiesRoute of AdministrationRoute of Administration• Different routes (and species) have different volume limitations• Many CROs have their own limits based on experience and animalMany CROs have their own limits based on experience and animal
care and use policies
Slide 30
Diehl et al., J. Appl. Toxicol. 21:15–23 (2001)

Design of Toxicology Studies:Duration of TreatmentDuration of Treatment
Duration (and timing) of general toxicity studies usually related to human exposure, eg, for drugs, duration/scope of proposed clinical trials for drugsClinical Trial Duration
Toxicology duration to support Ph 1, 2 (EU) & Ph 1,
2, 3 (Japan)
Toxicology duration to support Ph 3 (EU) & Marketing
(All Regions)Rodent Non-Rodent Rodent Non-Rodent
Single dose 2 weeks 2 weeks - -< 2 weeks 2 weeks 2 weeks 1 month 1 month1 month 1 month 1 month 3 months 3 months< 3 months 3 months 3 months 6 months Chronic> 3 months 6 months 6 months> 3 months - - 6 months 6 months< 6 months 6 months 6 months - -> 6 months 6 months Chronic - -
Slide 31
ICH M3(R2) Guidance

Design of Toxicology Studies:Duration of TreatmentDuration of Treatment• Exceptions based on technical limitations (e.g., chronic intrathecal
infusions through catheters not viable due to infections, etc.)g , )• Duration of the animal toxicity studies > duration of human clinical trials
up to the maximum recommended duration• Class-related exceptions egClass related exceptions, eg,
• peroxisome proliferator-activated receptors (PPAR) agonists require 1-year nonrodent study and rodent carcinogenicity studies to assess cardiovascular safety and define safe clinicalstudies to assess cardiovascular safety and define safe clinical doses for Phase 3)
• Insulins require 1-year rodent study to investigate carcinogenic potentialpotential
Slide 32

Design of Toxicology Studies:Duration of TreatmentDuration of Treatment• Maximum recommended duration of treatment (nonrodents)
• EU: 6 months acceptable to support registrationEU: 6 months acceptable to support registration• FDA: 9-12 months
• Chronic-use drugs with short-term clinical trials (eg, AIDS, 12 mo)Fi t i Cl l l t t ith i (12 )• First-in-Class, new molecular target with no experience (12 mo)
• 6 month acceptable if:• Longer duration not feasible, eg, immunogenicity or
intolerance confounds conduct of longer term studies• Repeated short-term drug exposure for indications such as
intermittent treatment of migraine, erectile dysfunction, etc• Drugs to reduce the risk of recurrence of cancer• Drugs for indications for which life expectancy is short (3-mo
may be acceptable for advanced cancer)
Slide 33
y p )

Design of Toxicology Studies:Selection of SpeciesSelection of Species• Species selection criteria should be intentional and explicitly justified
L l b d t d i il it t h• Largely based on expected similarity to humans• Comparative pharmacology Is the (similar) target present in the animal model? Does the target have a similar function in the animal model? Is anatomy/physiology of potential target organs comparable?
• Comparative metabolic profile• In vitro screening: hepatic microsomes and/or cytosolic fractions Metabolite identification and PK profile (post Phase 1)
• Default species = rat (rodent) and dog (nonrodent)Default species rat (rodent) and dog (nonrodent)• Other factors include: Precedence with drug class (e.g., known
toxicities, mechanisms); Regulatory agency experience; Availability of historical control data; Logistical issues …
Slide 34
of historical control data; Logistical issues …

Design of Toxicology Studies:Selection of SpeciesAnimal Models of Safety Assessment
Lifespan
Selection of Species
Animal model Lifespan (years) Comment
Rodent 1 5-2
Commonly used, small/inexpensiveLarge historical database & well-characterized biology
(Rat, Mouse) 1.5-2 Highly inbred and outbred strains availableRat is preferred rodent model for general toxicology
Beagle Dog 10-15(6-8 kg)
Preferred nonrodent model for general and cardiovascular toxicology( g) gy
Cynomolgus monkey
15(3-5 kg)
Phylogenetic and physiologic homology with humansCostly and limited resource
Often employed in biotherapeutic development5-10Rabbit 5-10
(2-3 kg) Developmental & ocular toxicology; vascular irritation
MiniPig (www.rethink-eu.dk)
15-20(>13 kg) Dermal toxicology, cardiology
Transgenic
Slide 35
Transgenic Mice variable Animal models of disease; Biopharmaceuticals

Design of Toxicology Studies:Dose SelectionDose Selection
• One of the most challenging aspects of study design !• Should be intentional and explicit in justificationShould be intentional and explicit in justification
• Typically, doses selected based on pilot work, dose range finding (DRF) studies, and shorter-term GLP studies
• 7- to 10-day DRF (with no or limited histopathology) guides dose selection y ( gy) gfor 14-day or 28-day IND-enabling GLP studies
• 28-day study to select doses for 13-week study• 13-week study to select doses for chronic study (& rodent carcinogenicity)
• Typically, 3 dose levels plus control(s)• High Dose = MTD (define target organs, reversibility, etc.)
• May need to reduce or stop dosing if unexpectedly excessive toxicity• Low Dose = NOAEL and systemic exposure at clinical efficacious dose• Mid Dose = some toxicity but <MTD (understand dose-response)• Number of dose groups dependent on objectives & degree of uncertainty
Slide 36
• May have untreated (saline) control group in addition to be vehicle control

Design of Toxicology Studies:Dose SelectionDose Selection
• Five general criteria for defining the High Dose (ICH M3(R2))1. Maximum tolerated dose (MTD)1. Maximum tolerated dose (MTD)
• Dose where target organ toxicity is likely to be observed but where the dose is not so high that the study, or the interpretation of results, is jeopardised by morbidity or mortality
• MTD is usually determined by parameters such as clinical signs and reductions in body weight and food consumption
2. Limit dose• If cannot define sufficient toxicity (cannot achieve MTD)• Generally accepted as 1000 mg/kg (both rodents & nonrodents)• If 1000 mg/kg/day < 10-fold margin of exposure to clinical exposure,
d i hi h d t hi 10 f ld i OR l hi hdrive high dose to achieve 10-fold exposure margin OR employ high dose of 2000 mg/kg/day OR the Maximum Feasible Dose (MFD)
Slide 37

Design of Toxicology Studies:Dose SelectionDose Selection3. Top dose based on saturation of exposure• Toxicokinetic data indicate that absorption limits
osur
e
systemic exposure to the drug or its metabolite(s)• Acceptable to use the lowest dose which achieves
maximum exposure in the absence of other dose limiting constraintsDose
Exp
o
4. Top dose based on Maximum feasible dose (MFD)• Bound by limits of technically feasibility:
Maximum possible concentration of formulated test article
• Highly relevant for IV formulations
Maximum possible concentration of formulated test articleX
Maximum dose volume administer-able over study duration
g y• Must justify due diligence efforts to achieve best formulation5. Top dose based on 50-fold margin of exposure• Relative to maximum clinical exposure (AUC)
Slide 38
• Relative to maximum clinical exposure (AUC)

Design of Toxicology Studies:Parameters AssessedParameters Assessed
• In-life assessments• Toxicokinetics• Body weight, food consumption, clinical observations• Safety pharmacology: cardiovascular, behavior• Ophthalmology
• Clinical pathology (hematology, clinical chemistry, urinalysis)• Can be sensitive markers of alterations ↑ hepatic enzyme activity prior to morphologic changes
• Can be very specific to organ system Troponins and myocardial alteration
• Can assess functional impact of injury I t f b l i i h l h t l Impact of bone marrow lesion on peripheral hematology
• Can be evaluated over the study course (nonrodents)
Slide 39

Design of Toxicology Studies:Parameters AssessedParameters Assessed
• Postmortem Evaluations• Complete necropsy exams on all animalsComplete necropsy exams on all animals
• dying spontaneously or killed in extremis• euthanized at scheduled necropsies (not TK animals)
O /ti i h d d & i i ll i d• Organs/tissues weighed, preserved, & microscopically examined:• Comprehensive list of tissues*• Rodents: typically, histopathology performed for control and high
dose animals initially• Identified target organs from low & mid dose subsequently
evaluated• Nonrodents: all animals and tissues examined• Special stains and electron microscopy, if warranted
* Society Toxicologic Pathology: Bregman et al Toxicol Path 31:252-253 (2003);
Slide 40
Society Toxicologic Pathology: Bregman et al., Toxicol Path 31:252 253 (2003); FDA response (Jacobs et al., Toxicol Path 31:571 (2003)

Design of Toxicology Studies:Parameters AssessedParameters Assessed
• Incorporate specific assessment endpoints to address expected (based on pharmacology or compound class) or previously observed “signals”, eg:p gy p ) p y g , g
• Serum testosterone, estradiol, LH, FSH for a hormone agonist• Liver toxicity (EMA’s “Reflection Paper on Non-Clinical Evaluation of
Drug-Induced Liver Injury (DILI)”)• Markers of cell proliferation (anticipating carcinogenicity concern)• Immunotoxicity assessments (ICH S8; FDA 2002)
• Standard toxicology studies (STS) to detect immune gy ( )suppression/activation
• Hematology (eg, cytopenias, leukocytosis)• Organ weight and histopathologic examination of immune
system organs (lymph nodes, spleen, thymus)• 2nd Tier testing based on results of STS (eg, functional assays)
• T-cell dependent antibody response, lymphocyte i h t i i fl t bi k
Slide 41
immunophenotyping, inflammatory biomarkers …

Design of Toxicology Studies:A word about Large (vs Small) MoleculesA word about Large (vs Small) Molecules• Large molecules:
• Have high target selectivity and specificityHave high target selectivity and specificity Relevant animal species may be limited
• Are high-MW and complex structures Extracellular targets with limited intracellular accessExtracellular targets with limited intracellular access
• Have less potential for off-target toxicity• Are administered parenterally (SC, IV)
• Have more potential for immunogenicity• Have more potential for immunogenicity eg, anti-drug antibodies (ADAs) may reduce
exposure or effect, or may neutralize endogenous proteinsFab
Epitopes
g p• High dose selection is guided by: Saturation of pharmacodynamic
response
Fab
Fc PD E
ffect 100 mg/kg
10 mg/kg
1 mg/kg
Slide 42
Dose multiple (eg, 10x) to clinical doseTarget plasma Conc.
g g
Courtesy of Dave Clarke

Some Data with Drug A
• Drug A• S ll l l i hibit f T t A• Small molecule inhibitor of Target A• Clinical use: Daily oral dosing• Chronic indication: a non-life-threatening disease
for which other, partially-effective treatments existp y• Species selection
• Rat and dogs selected as toxicology speciesbased on similarities in:
b li fil h b li f D A metabolic profile: human metabolites of Drug Apresent in rats & dogs (in vitro)
target expression: Target A is highly conserved:mouse, rat, dog, monkey, human; although, in vitro data indicate g y gthat human Target A is ~3x more sensitive than mouse Target A
• 1-month (28-day) studies will support First-in-Human clinical trials (<1-month in duration)• I l d 1 th ibilit b ??
Slide 43
• Include 1-month reversibility groups because …??

1-Month Rat Study with Drug ADose (mg/kg) 0 0.1 1 10
N=10/sex M F M F M F M F
Mortality* 0 0 0 1 0 0 1 0
Clinical observations – mostly reversible
Soft feces* 4 2 2 2 2 6 8 12
Diarrhea* 1 2 1 2 1 3 4 6
Rough haircoat* 1 0 2 1 2 5 3 6Rough haircoat 1 0 2 1 2 5 3 6
Body weight (% change vs control) Wk 12 N/A N/A -- ↑6 ↓ 4 ↑10 ↓ 10 NR ↑15 NR
Clinical pathology (change relative to control) – all reversible
Altered electrolytes -- -- -- -- √ √ √ √
↑ ALT N/A N/A -- √ √ √ √ √
↑ AST N/A N/A -- -- -- -- √ √
↑ Total cytochrome P450 N/A N/A -- √ √ √ √ √
Morphologic pathologyMorphologic pathology
Stomach: necrosis & atrophy -- -- -- -- -- √ √ NR √ NR
Liver: Hepatocellular vacuolation -- -- -- -- √ √ √ NR √ NR
Liver: Hepatocellular necrosis -- -- -- -- -- -- √ √
Slide 44
Heart: Myocardial necrosis -- -- -- -- -- √ NR √ NR √ NR
* # of rats affected N/A = Not Applicable -- No important changes NR = Not Reversible

Some data: 1-Month Rat Study withDrug A (small molecule, oral chronic)g ( )
Dose (mg/kg) 0 (Control) 0.1 1 10
N=10/sex M F M F M F M F
Mortality* 0 0 0 1 0 0 1 0
Cli i l b ti tl iblClinical observations – mostly reversible
Soft feces* 4 2 2 2 2 6 8 12
Diarrhea* 1 2 1 2 1 3 4 6
Rough haircoat* 1 0 2 1 2 5 3 6
Body weight (% change vs control) Wk 12 N/A N/A -- ↑6 ↓ 4 ↑10 ↓ 10 NR ↑15 NR
Clinical pathology (change relative to control) – all reversible
Altered electrolytes -- -- -- -- √ √ √ √
√ √ √ √ √
>MTD
↑ ALT N/A N/A -- √ √ √ √ √
↑ AST N/A N/A -- -- -- -- √ √
↑ Total cytochrome P450 N/A N/A -- √ √ √ √ √
Morphologic pathology – NR = Not Reversiblep g p gy
Stomach: necrosis & atrophy -- -- -- -- -- √ √ NR √ NR
Liver: Hepatocellular vacuolation -- -- -- -- √ √ √ NR √ NR
Liver: Hepatocellular necrosis -- -- -- -- -- -- √ √
H M di l i √ NR √ NR √ NRN M i bl N R ibl 10
Slide 45
Heart: Myocardial necrosis -- -- -- -- -- √ NR √ NR √ NR
NOAEL √ √Not Monitorable, Not Reversible 10x
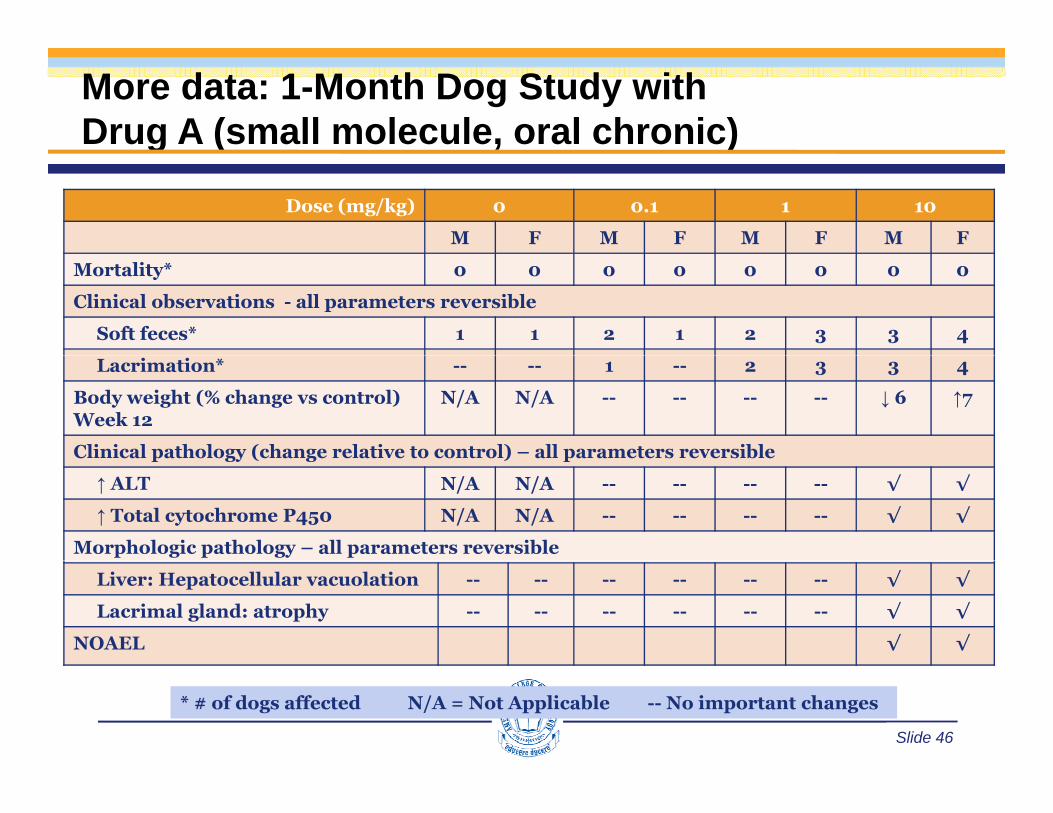
More data: 1-Month Dog Study withDrug A (small molecule, oral chronic)g ( )
Dose (mg/kg) 0 0.1 1 10
M F M F M F M F
Mortality* 0 0 0 0 0 0 0 0
Clinical observations - all parameters reversible
Soft feces* 1 1 2 1 2 3 3 4
Lacrimation* -- -- 1 -- 2 3 3 4
Body weight (% change vs control) Week 12
N/A N/A -- -- -- -- ↓ 6 ↑7
Clinical pathology (change relative to control) – all parameters reversibleC ca pat o ogy (c a ge e at ve to co t o ) a pa a ete s eve s b e
↑ ALT N/A N/A -- -- -- -- √ √
↑ Total cytochrome P450 N/A N/A -- -- -- -- √ √
Morphologic pathology – all parameters reversible
Liver: Hepatocellular vacuolation -- -- -- -- -- -- √ √
Lacrimal gland: atrophy -- -- -- -- -- -- √ √
NOAEL √ √
Slide 46
* # of dogs affected N/A = Not Applicable -- No important changes

Application of General Toxicology Studiesto Support Regulatory Requirements
for Development of Therapeutics
Slide 47
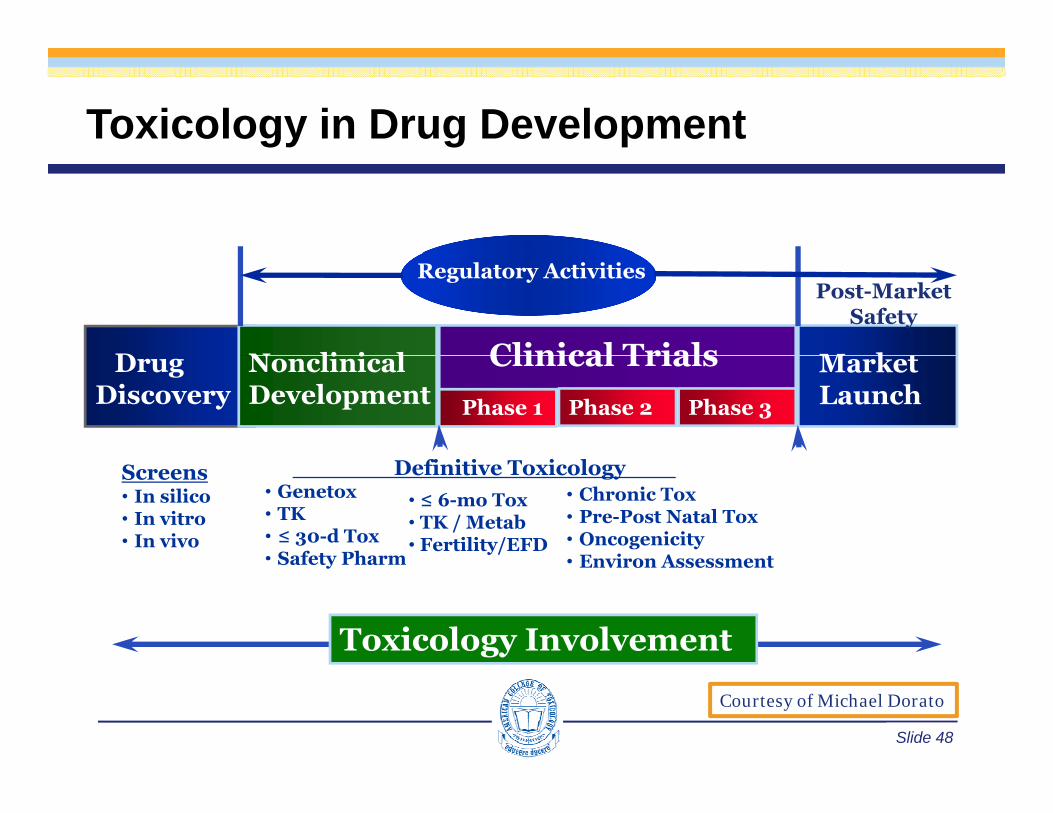
Toxicology in Drug Development
D N li i l Clinical Trials M k
Post-Market Safety
Regulatory Activities
Drug Discovery
NonclinicalDevelopment
Clinical TrialsPhase 3Phase 2Phase 1
Market Launch
• Genetox• TK• ≤ 30-d Tox• Safety Pharm
• ≤ 6-mo Tox• TK / Metab• Fertility/EFD
• Chronic Tox• Pre-Post Natal Tox• Oncogenicity• Environ Assessment
Definitive Toxicology Screens• In silico• In vitro• In vivo
Toxicology Involvement
Safety Pharm • Environ Assessment
Slide 48
Courtesy of Michael Dorato

Early Drug Development: Lead Optimization
Slide 49
Courtesy of Anja Stauber
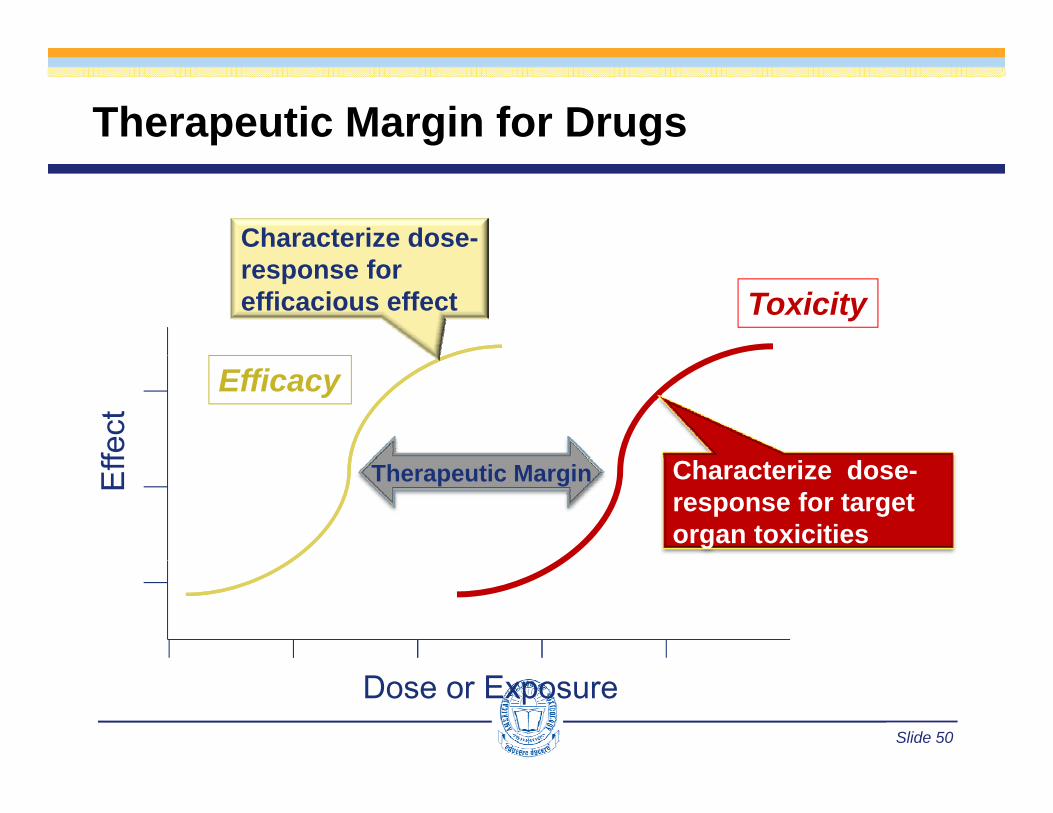
Therapeutic Margin for Drugs
Characterize dose-
Toxicityresponse for efficacious effect
fect
Efficacy
Eff Therapeutic Margin Characterize dose-
response for target organ toxicities
Slide 50
Dose or Exposure

ADME Data Supports Nonclinical Safety
• Evaluation of exposure to parent drug intoxicology speciestoxicology species• Establish dose-exposure relationship• Species or sex differences• Insight into enzyme induction• Insight into enzyme induction• Cross-species metabolism• Extent of accumulation
MOS b d
Exp
osur
e
• MOS based on exposure
MOS = NOAEL Exposure
Effect Level Exposure
Dose
Exp
osur
e
Slide 51
Dose
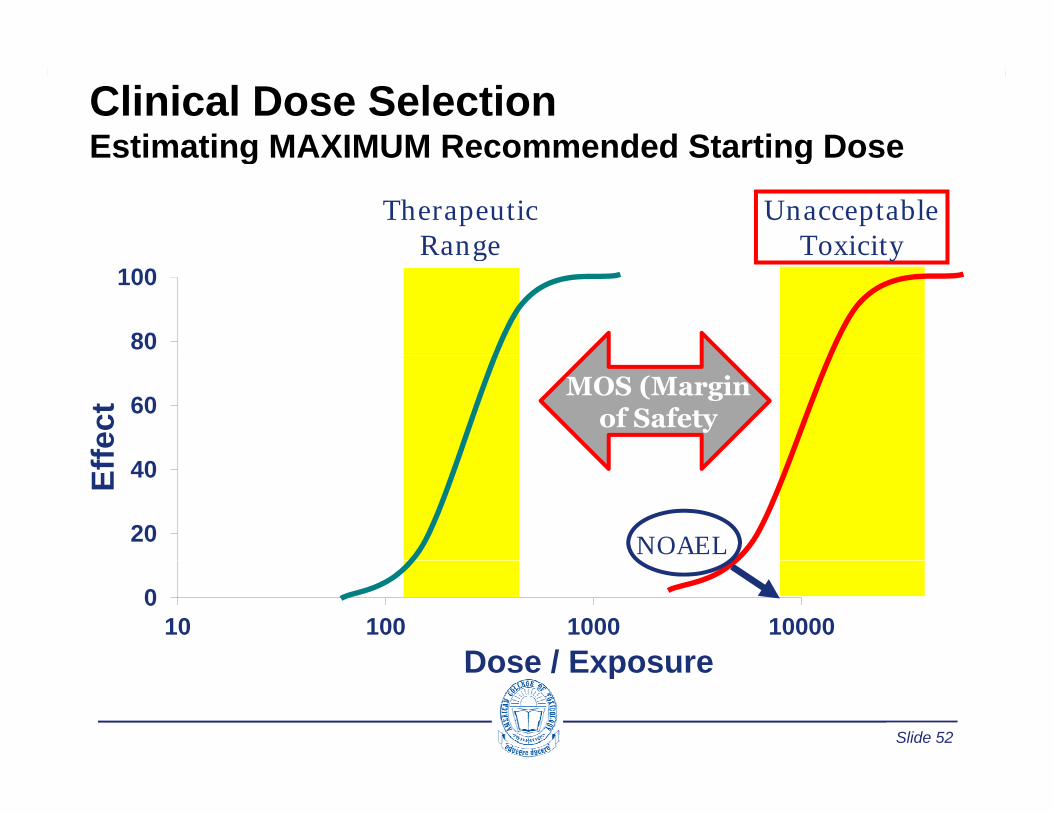
Clinical Dose SelectionEstimating MAXIMUM Recommended Starting DoseEstimating MAXIMUM Recommended Starting Dose
Therapeutic Range
Unacceptable Toxicity
80
100Range Toxicity
60
fect
MOS (Marginof Safety
20
40Eff
NOAEL
010 100 1000 10000
Dose / Exposure
Slide 52
Dose / Exposure

Clinical Dose SelectionEstimating MRSD using NOAELEstimating MRSD using NOAEL
Determine NOAELs(mg/kg) in tox studies Choose Safety Factor*
Animal NOAEL to HED justified based
MRSD = HED/SF
Yes
HED mg/kg =NOAEL (mg/kg)
jon mg/kg or other?
C t i l Consider lower dose
No
Convert animal NOAELs to HED based on BSA mg/m2
based on other factors, i.e., PAD
Select appropriate (most sensitive) species
FDA Guidance, 2005HED=Human Equivalent DoseBSA=Body Surface AreaSF=Safety Factor
Slide 53
species SF=Safety FactorPAD=Pharmacologically Active Dose

A slight digression …
Special Considerations:The NOAELThe NOAEL
Slide 54

Defining NOAEL for Pharmaceuticalsg• Highest dose/exposure that does not cause
biologically important increases in the frequency orbiologically important increases in the frequency or severity of effects between the exposed population and the appropriate control. While minimal toxic ff t b b d t thi l l th teffects may be observed at this level, they are not
considered to endanger human health, or be precursors of serious events.
The NOAEL is not Risk FreeThe NOAEL is not Risk FreeVariability in species and individual response,Variability in species and individual response,
d i f i i l id i f i i l i
M A Dorato J A Engelhardt The no observed adverse effect level in drug safety
and existence of sensitive populations,and existence of sensitive populations,increases the probability of undesired effects increases the probability of undesired effects
Slide 55
M. A. Dorato, J. A. Engelhardt. The no-observed-adverse-effect-level in drug safety evaluations: Use, issues, and definition(s). Reg. Tox. Pharm., 42(2005) 265-274.

The “Facts” About the NOAEL
• The NOAEL must be one of theexperimental doses tested and, thus,
NOAEL
is influenced by the study design
• The NOAEL does not consider the slope of the dose response curve or the nature of the effects at the next highest dosecurve or the nature of the effects at the next highest dose
• The NOAEL is sensitive to sample size with a tendency to be higher with fewer animals/dose
• The NOAEL may vary from experiment to experiment
• Risk levels may be higher than estimated by the NOAEL reflecting variability in species and/or individual responses as well as thevariability in species and/or individual responses as well as the possibility of sensitive populations (i.e., toxicology studies are conducted using “healthy” animals)
• Definition of “ad erse” can ar bet een re ie ers
Slide 56
• Definition of “adverse” can vary between reviewers

Back to the Story …
Slide 57

Clinical Dose SelectionOther Considerations (eg, Safety Factor)Other Considerations (eg, Safety Factor)
Determine NOAELs(mg/kg) in tox studies Choose Safety Factor*
Yes
Default SF = 10
HED mg/kg =NOAEL (mg/kg)or other
Animal NOAEL to HED justified based on mg/kg or other?
Convert animal NOAELs
MRSD = HED/SF
“Consider lower dose based on other factors, i.e., PAD”
No
to HED based on BSAmg/m2
Select appropriate (most sens) species
Factors which may justify need for ↑↓ MOS • Increase SF
• non-monitorablenon monitorable• steep dose/response• variable bioavailability or PK• novel therapeutic targetsp g
• Decrease SF• well-char therapeutic class• monitorable/ reversible effects
Slide 58
• predictable toxicity

Benefit – Risk Considerations
Low Risk High Risk•Well understood MOA•Life-threatening indication•Relevant nonclin test systems
•Novel or poorly understood MOA
•Human volunteersy•Useful biomarkers•Limited human dose (eg, Ph 0)
•No relevant test systems•No biomarkers
Goal: Minimize risk and escalate dose accordingly
Slide 59
After Tibbitts 2011
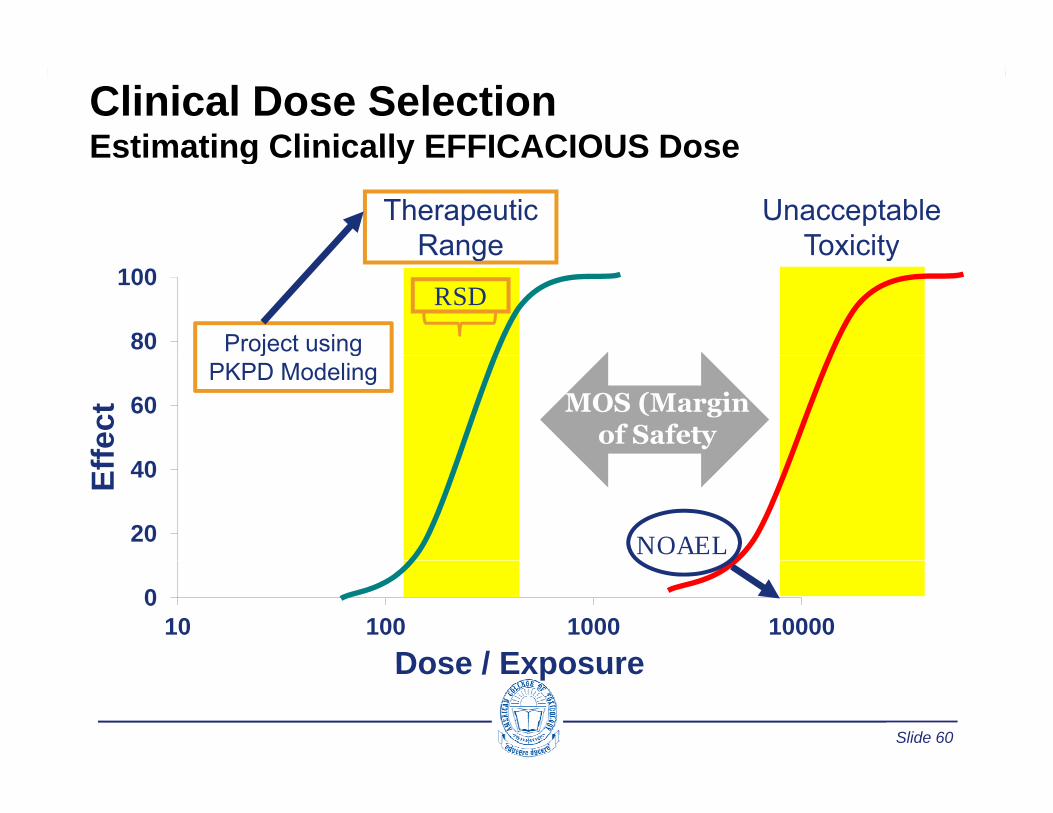
Clinical Dose SelectionEstimating Clinically EFFICACIOUS DoseEstimating Clinically EFFICACIOUS Dose
Therapeutic Range
Unacceptable Toxicity
80
100Range Toxicity
RSD
Project using
60
fect MOS (Margin
of Safety
j gPKPD Modeling
20
40Eff
NOAEL
010 100 1000 10000
Dose / Exposure
Slide 60
Dose / Exposure
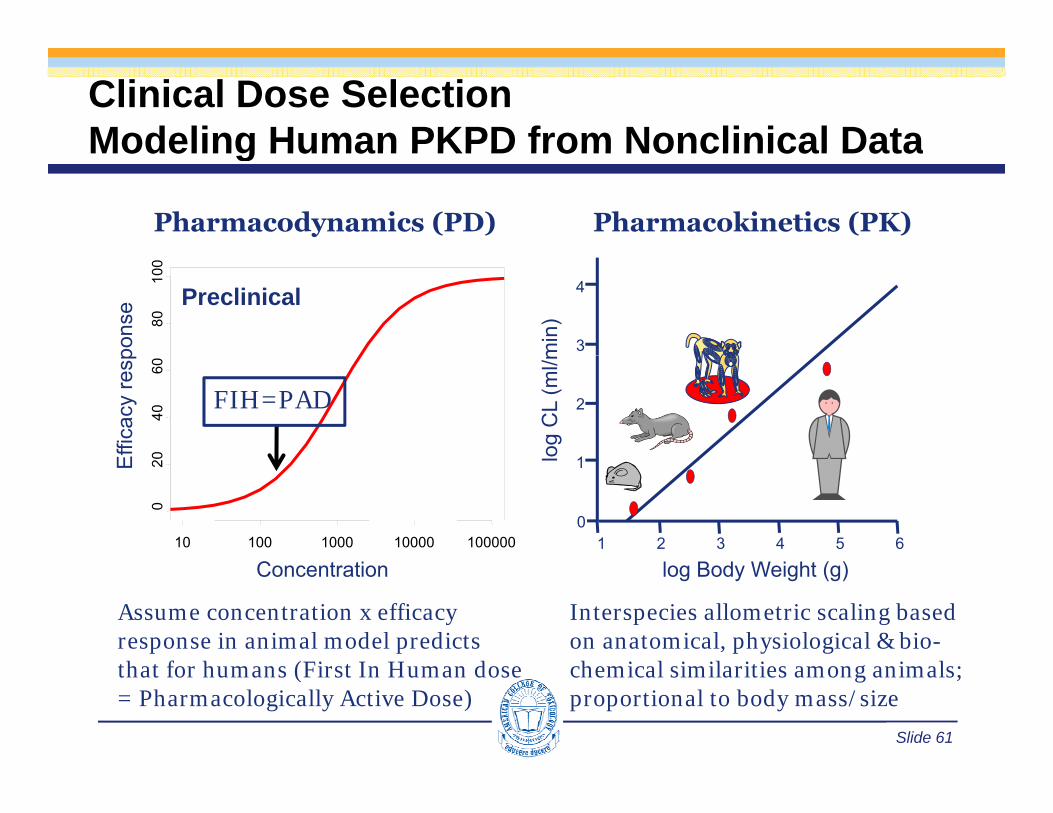
Clinical Dose SelectionModeling Human PKPD from Nonclinical DataModeling Human PKPD from Nonclinical Data
Pharmacodynamics (PD) Pharmacokinetics (PK)
min
)
3
4Preclinical
8010
0
pons
e
og C
L (m
l/m
1
2
040
60
Effi
cacy
resp
FIH=PAD
1 2 3 4 5 6l
0
1
10 100 1000 10000 100000
020E
log Body Weight (g)Concentration
Interspecies allometric scaling based on anatomical, physiological & bio-h i l i il iti i l
Assume concentration x efficacy response in animal model predicts th t f h (Fi t I H d
Slide 61
chemical similarities among animals; proportional to body mass/size
that for humans (First In Human dose = Pharmacologically Active Dose)

Clinical Dose SelectionA more conservative approach: MABELA more conservative approach: MABEL• Clinical trial doses are initially defined by nonclinical toxicity data• Typically Maximum Recommended Starting Dose (MRSD) is• Typically, Maximum Recommended Starting Dose (MRSD) is
based the NOAEL / SF• There are factors in animal studies which limit CT starting dose &
l tiescalation• “ … concerns may be derived from particular knowledge or
uncertainties on:(1) th d f ti d/(1) the mode of action, and/or(2) the nature of the target, and/or(3) the relevance of animal models”
• In these cases, ↑ Safety Factor applied to NOAEL OR• Apply Minimal Anticipated Biologic Effect Level (MABEL)
EMEA/CHMP/SWP/28367/2007 C GUIDELINE ON REQUIREMENTS FOR FIRST IN MAN CLINICAL
Slide 62
EMEA/CHMP/SWP/28367/2007 Corr.: GUIDELINE ON REQUIREMENTS FOR FIRST-IN-MAN CLINICAL TRIALS FOR POTENTIAL HIGH-RISK MEDICINAL PRODUCTS

Benefit – Risk ConsiderationsLow Risk
•Well understood MOA•Life-threatening indication•Relevant nonclin test systemsUsef l biomarkers
High Risk•Useful biomarkers•Limited human dose (eg, Ph 0)
•Novel or poorly understood MOA
•No relevant test systemsNo biomarkers•No biomarkers
•Human volunteers
Slide 63
After Tibbitts 2011

Clinical Dose Selection:MABEL and NOAEL ApproachesMABEL and NOAEL Approaches
Slide 64
after Simms, 2007

Parting Thoughts: 3Rs of Animal Testing
• The use of animal models in biomedical research has evolved to more fully embrace the 3Rsevolved to more fully embrace the 3Rs
• Introduced by Russell & Burch (1959)• Replacement, Reduction and RefinementReplacement, Reduction and Refinement
• Replacement – use of non-animal based methods or use an alternative “lower” species
• Reduction – better study designs, data sharing & inter-organisation cooperation
• Refinement – improved techniques, enrichment,Refinement improved techniques, enrichment, analgesia/anaesthesia
• Implementing 3Rs gives better science and high quality
Slide 65
data

3Rs Opportunities in Toxicology Testing
• Reducing # TK animals (fewer sample points; alternative sampling sites; improved bioanalytical techniques for microsampling)
• Incorporating male fertility assessment into 6-month rodent toxicology study
• Including fewer recovery animals (eg, control and high dose only; one study only; or no recovery control group)
• Carcinogenicity studies using: transgenic mice; one control group; rodent strain
Slide 66
Carcinogenicity studies using: transgenic mice; one control group; rodent strain with good survival

Some 3R’s examples in toxicology
• Dried blood spot sampling (reduction/refinement)• Acute studies (reduction)• Use of solid bottom cages in rats (refinement)• Environment enrichment (refinement)
• Mouse house & chew bars• Group housing, toys, forage activities for NHPs
• Judicious implementation of recovery groups (reduction)• Development of drugs depends on animal subjects
Good welfare = Better data = Better decisions
Slide 67

Toxicology for Industrial and Regulatory Scientists
THANK YOU!!
General ToxicologyLorrene A Buckley, PhD, DABT, ATS
28 April 2015

Please click on the link belo toPlease click on the link below to enter your comments on this talk
https://www.surveymonkey.com/s/VNNCM7G
Slide 69